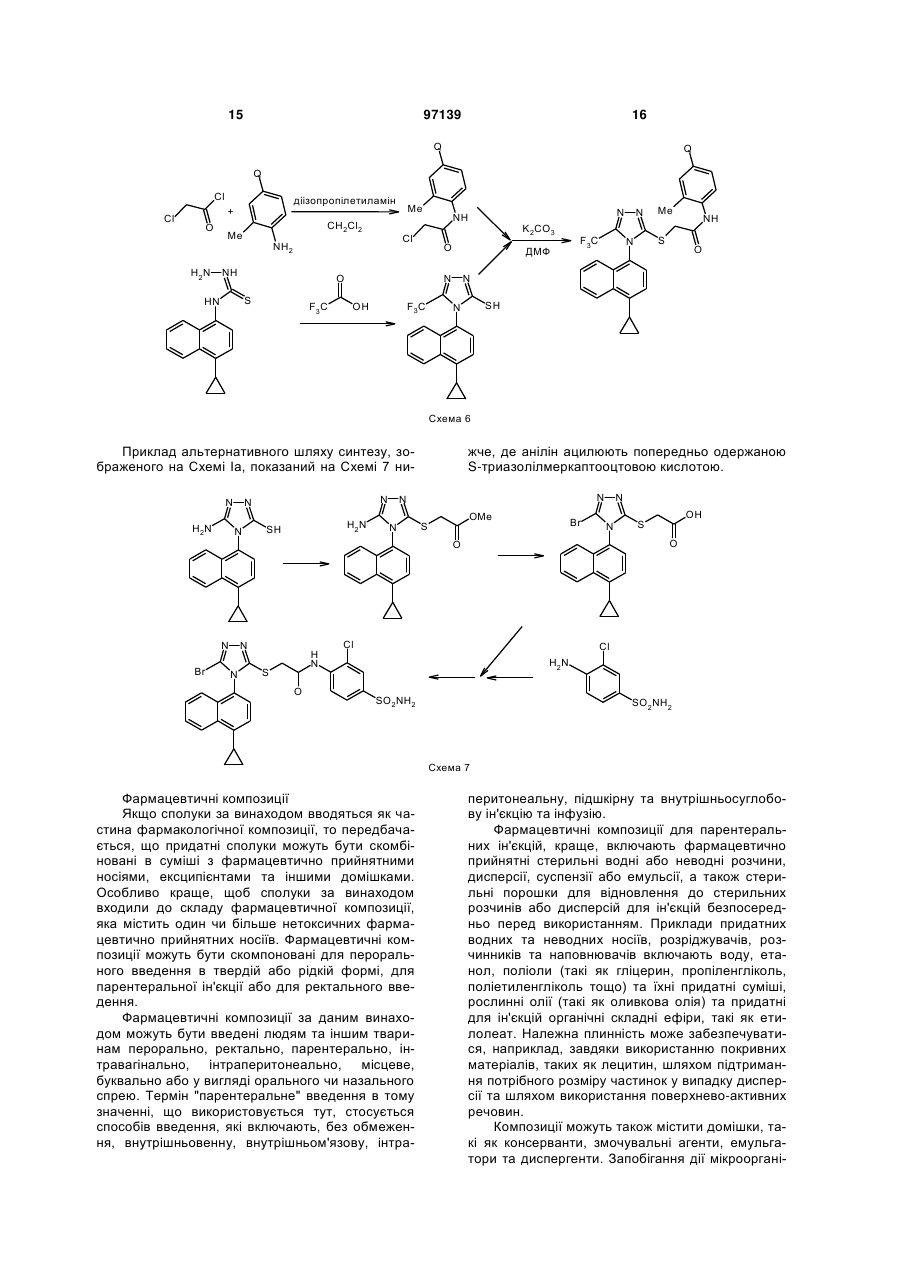
2-(5-бром-4-(4-циклопропілнафталін-1-іл)-4н-[1,2,4]-триазол-3-ілсульфаніл)оцтова кислота та її метиловий ефір



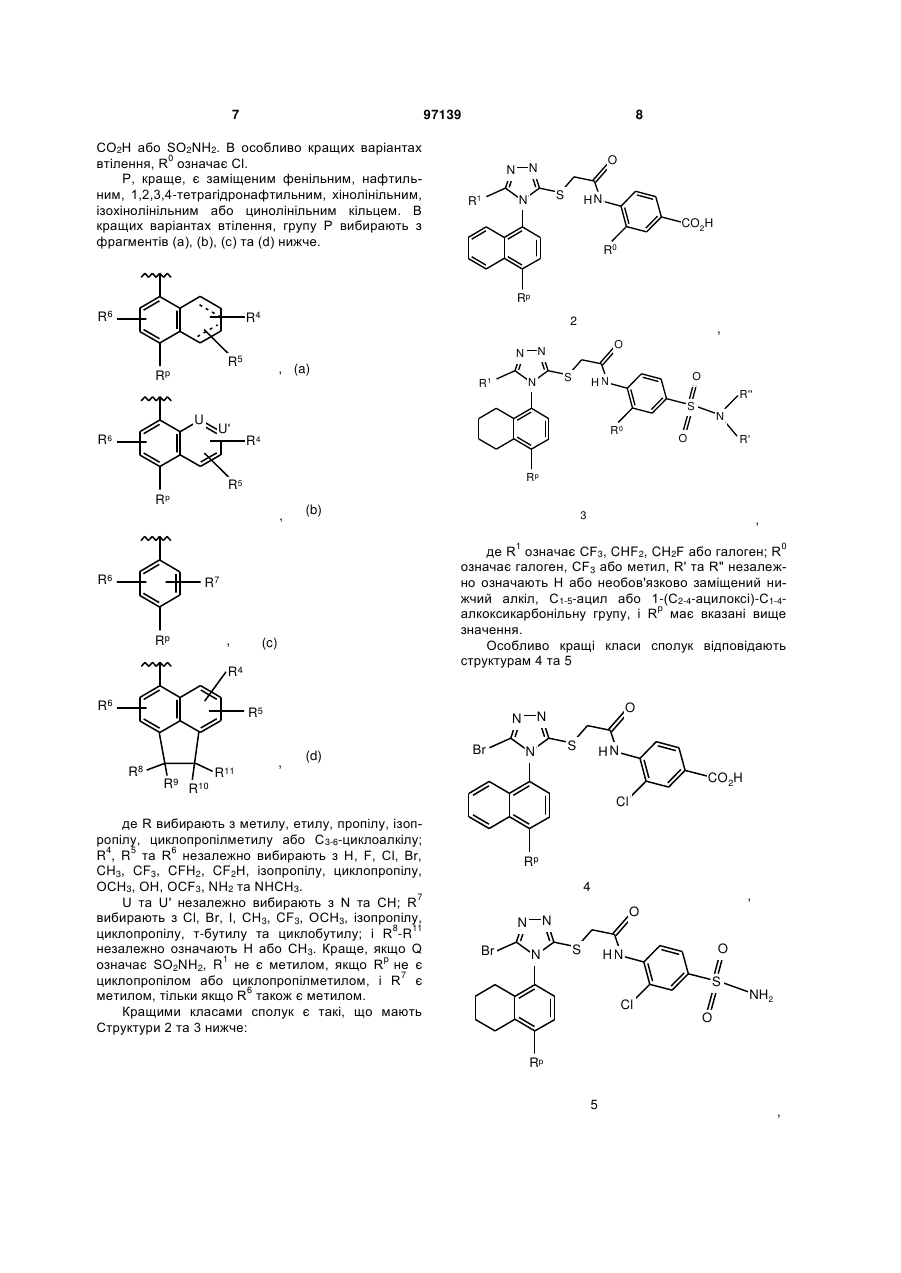
Формула / Реферат
1. Сполука наведеної нижче формули:
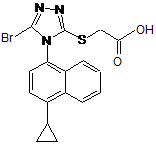 .
.
2. Сполука наведеної нижче формули:
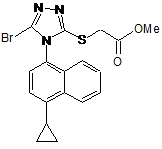 .
.
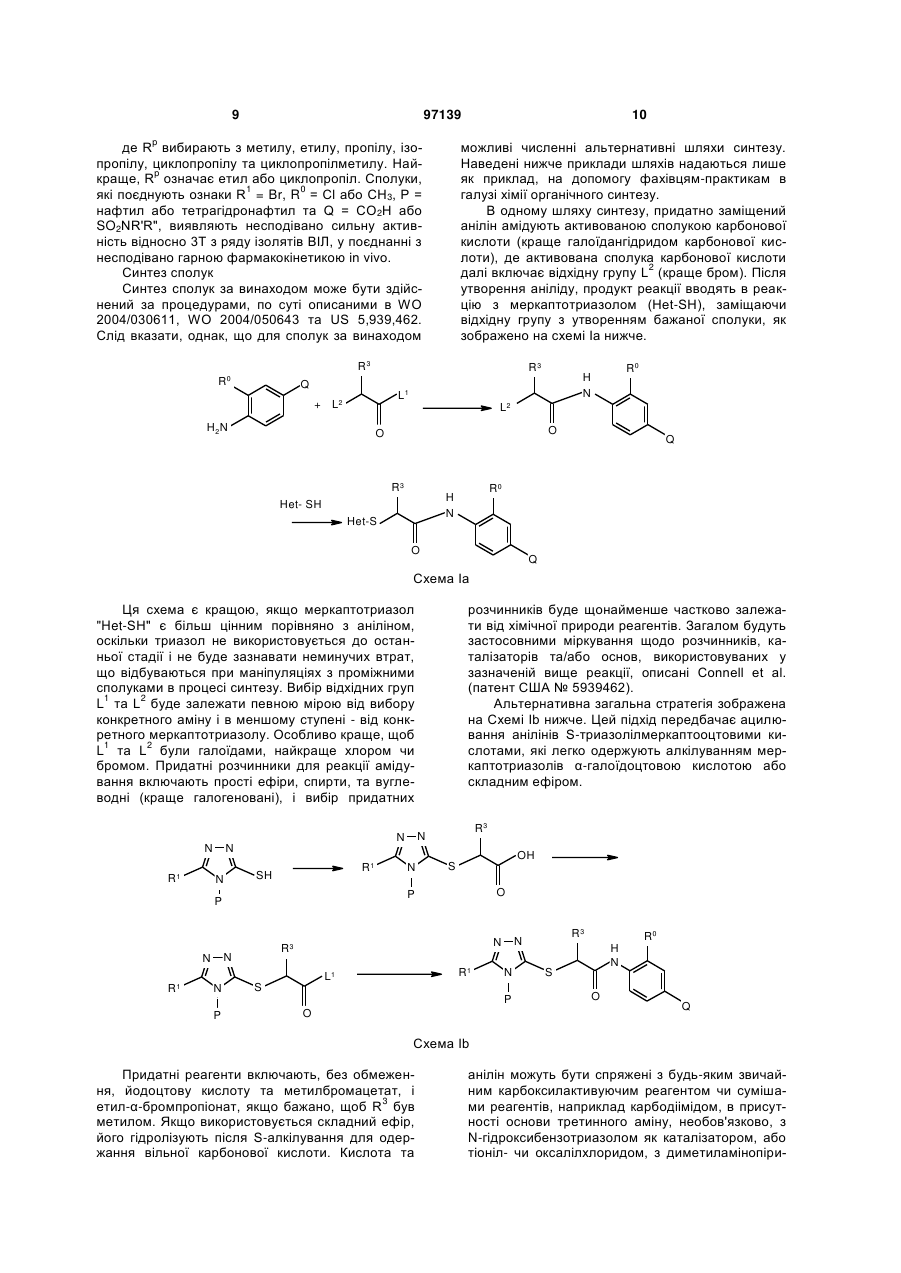
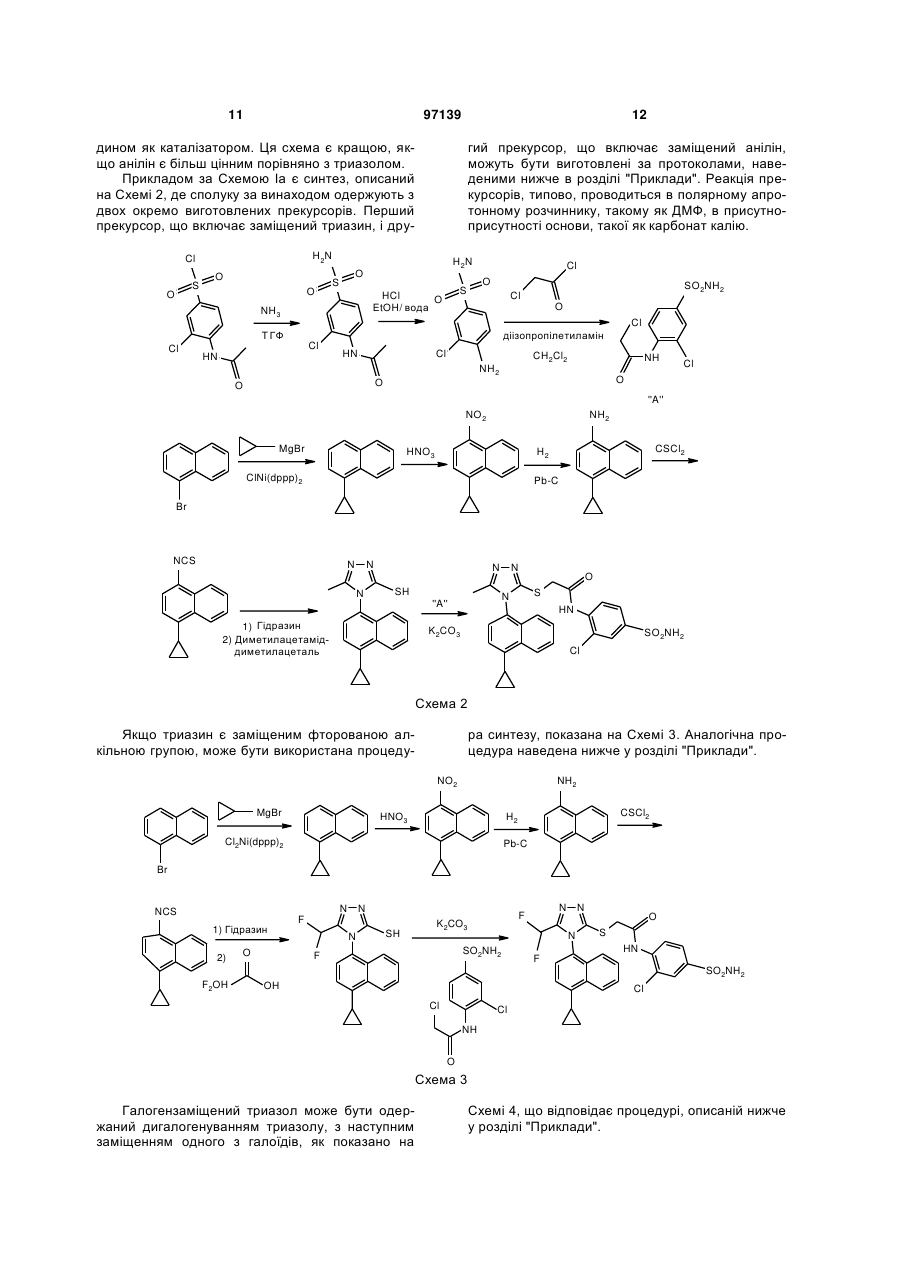
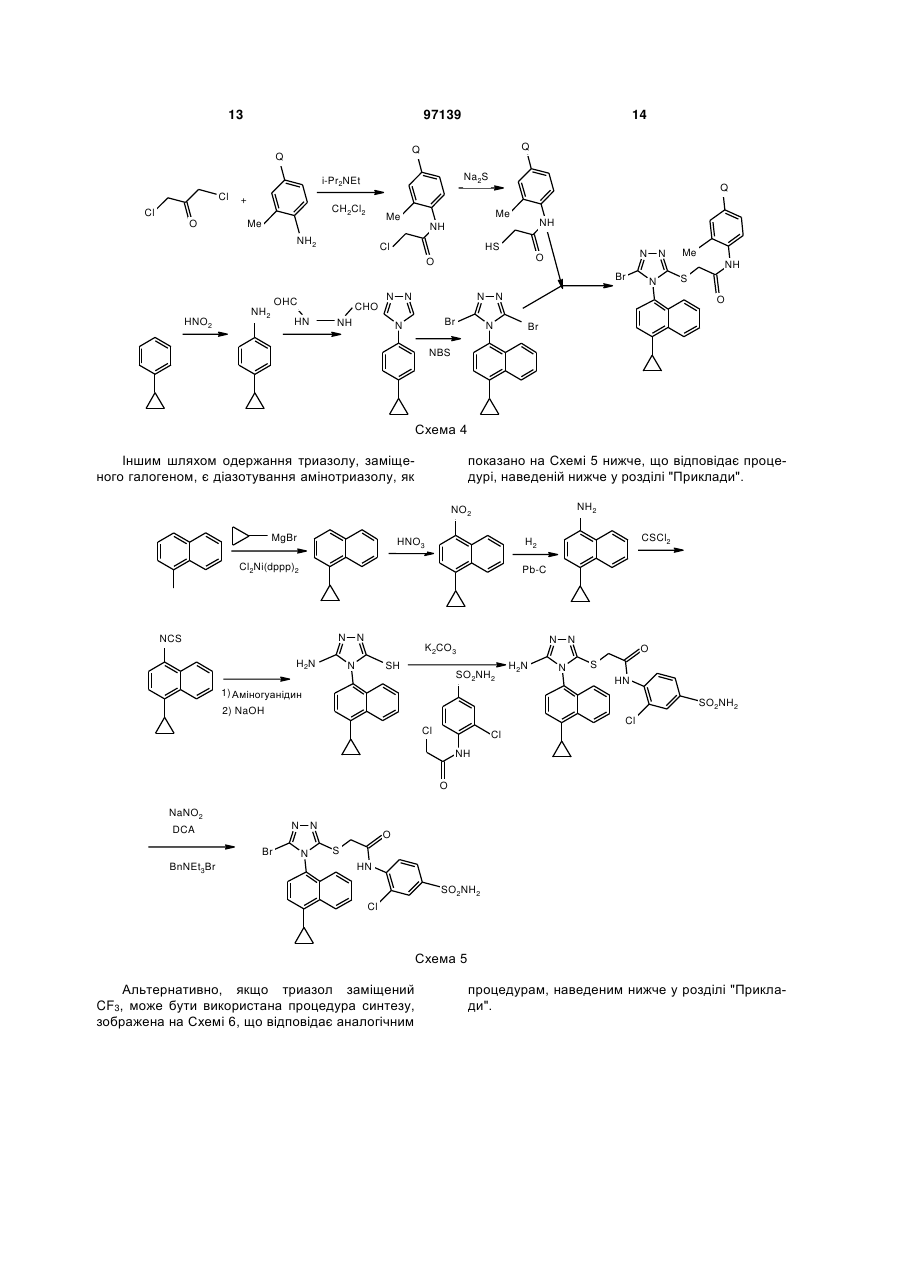
Текст
1. Сполука наведеної нижче формули: N N Br N OH S O . 2. Сполука наведеної нижче формули: N N Br N OMe S C2 O (13) (21) a200909742 (22) 25.08.2005 (24) 10.01.2012 (31) 60/604,219 (32) 25.08.2004 (33) US (31) 60/604,220 (32) 25.08.2004 (33) US (31) 60/686,351 (32) 31.05.2005 (33) US (62) a 200702491, 25.08.2005 (46) 10.01.2012, Бюл.№ 1, 2012 р. (72) ЖІРАРДЕ ЖАН-ЛЮК, FR/US, КОХ ЮНГ-ХІО, KR/US (73) АРДЕА БІОСАЄНСІЗ, ІНК., US (56) WO 2004/030611 A3, 15.04.2004, 70 арк. UA 82048, C2, 11.03.2008, 11 арк. UA 7333, C2, 15.07.2005, 3 арк. WO 2004/050643, A2, 17.06.2004, 102 арк. US 5939462, A, 17.08.1999, 18 арк. 2 (11) UA сполук. Для подолання резистентності, щонайменше в деякому ступені, можуть бути введені нові інгібітори типу нуклеозидів (самі або в комбінації з іншими інгібіторами типу нуклеозидів), і приклади альтернативних лікарських засобів включають ставудин (d4T), диданозин (ddl), комбівір (Combivir™) (фірмова назва для комбінації ламівудину та зидовудину) та тризивір (Trizivir™) (фірмова назва для комбінації 3ТС, AZT та абакавіру). На жаль, виникнення резистентності до одного інгібітора нуклеозидного типу часто супроводжується виникненням певного ступеня резистентності до іншого інгібітора нуклеозидного типу, що часто призводить до необхідності переходу до іншого класу лікарських засобів. У таких випадках, пацієнт може одержати інгібітор протеази (наприклад, саквінавір, індинавір, нелфінавір і т. д.), типово, в комбінації з іншими антиретровірусними агентами. Однак, відносно складна схема введення таких (19) Ця заявка претендує на пріоритет тимчасових заявок США 60/604219, поданої 25 серпня 2004 p., 60/604220, поданої 25 серпня 2004 р., та 60/686351, поданої 31 травня 2005 p., обсяг кожної з яких цілком включений сюди за посиланням. Винахід стосується інгібіторів ферментів та використання інгібіторів ферментів для лікування хвороби. Більш конкретно, винахід стосується in vitro та in vivo інгібування зворотної транскриптази ВІЛ як способу лікування інфекції ВІЛ. Фахівцям відомі численні методи лікування ВІЛ, і, поміж інших фармацевтично активних сполуки, інгібітори зворотної транскриптази забезпечують значний терапевтичний ефект у багатьох ВІЛ-інфікованих пацієнтів. Наприклад, ламівудин (3ТС) або зидовудин (AZT) є відносно добре переносимими антиретровірусними лікарськими засобами. Однак, останнім часом виникли численні вірусні штами із значною резистентністю до цих 97139 . 3 комбінацій часто створює організаційні та фінансові проблеми для багатьох пацієнтів, і їхнє дотримання часто є меншим, ніж бажано. Нещодавно, лікування ВІЛ було сфокусоване на комбінаційних терапіях, які включають введення нуклеозидних інгібіторів зворотної транскриптази з інгібіторами протеази і з ненуклеозидними інгібіторами зворотної транскриптази та потрійних комбінацій нуклеозидних інгібіторів зворотної транскриптази, ненуклеозидних інгібіторів зворотної транскриптази та інгібіторів протеази. На жаль, комбіновані терапії інгібіторами протеази з нуклеозидними інгібіторами зворотної транскриптази часто погано переносяться та часто призводять до дострокового припинення терапії. Тому, найсучасніші комбінаційні лікування включають комбінацію нуклеозидних інгібіторів зворотної транскриптази та ненуклеозидних інгібіторів зворотної транскриптази. Інгібітори ненуклеозидного типу (наприклад, невірапін, делавірдин та ефавіренц) є структурно неоднорідною групою сполук, які гадано зв'язуються з ненуклеозидним карманом зворотних транскриптаз. Вони істотно підвищують антивірусну ефективність при спільному введенні з інгібіторами нуклеозидного типу. Хоч інгібітори ненуклеозидного типу вважаються перспективним новим класом антивірусних лікарських засобів, у них залишається кілька недоліків. Вартість відомих зараз інгібіторів ненуклеозидного типу є відносно високою, а одна мутація вірусних зворотних транскриптаз може індукувати крос-резистентність відносно великого класу ненуклеозидних інгібіторів зворотної транскриптази. Тому існує потреба в створенні нових ненуклеозидних інгібіторів зворотної транскриптази, які б мали сильний антивірусний ефект, особливо проти мутантних штамів ВІЛ, що виявляють резистентність відносно відомих зараз ненуклеозидних інгібіторів зворотної транскриптази. Вірус ВІЛ має відносно високу частоту мутацій, що часто призводить до лікарської резистентністі відносно сучасних методів лікування. Проводилися дослідження з метою ідентифікації спектра мутацій в білках зворотної транскриптази (RT) вірусів, виділених у пацієнтів з невдалими результатами терапії з використанням щонайменше одного ненуклеозидного інгибітора зворотної транскриптази (NNRTI), і результати показали, що мутант K103N був домінуючиму пацієнтів, які приймали ефавіренц, у той час як Y181C був домінуючим у пацієнтів, які приймали невірапін. Інші одиничні мутації включали K01Е, G190S/A/E та Y188L/C. Деякі з найбільш поширених подвійних мутацій у пацієнтів з невдалою терапією з використанням ефавіренцу включають K103N-P225H, K103N-V108I, K103N-K101Q, K103N-L100I, K103NF227L, V106I-Y188L, K103N-Y188L та K103NG190A. Існує потреба у створенні нових композицій та методів інгібування цих та інших мутантів зворотних транскриптаз. Дана заявка пов'язана з роботами, раніше розкритими в заявках PCT/US02/26186, поданій 23 серпня 2002 p., неопублікованій, та PCT/US03/27433, поданій 22 серпня 2003 p., опублікованій як WO 2004/030611 15 квітня 2004 p., які 97139 4 знаходяться у суспільній власності. Патент США 5939462, виданий на ім'я Connell et al., розкриває велику кількість заміщених гетероциклів, придатних для застосування як антагоністів рецептора NPY5, деякі з яких є Sтриазолілмеркаптоацетанілідами, подібними до загальної структури 1, наведеної нижче. Simoneau et al., у міжнародній патентній публікації WO 2004/050643, розкривають тетразоли та кілька триазолів, що мають структури, подібні до описаних в даному винаході, які виявляють активність інгібіторів зворотної транскриптази. Автори винаходу знайшли, що зворотна транскриптаза (RT) ВІЛ може бути інгібована вибраним класом S-триазоліл-α-меркаптоацетанілідів, представлених загальною структурою 1. Несподівано, деякі з цих сполук були здатними інгібувати різні мутовані 3Т, включаючи K103N, Y181C та Y188L. R0 R3 N N R1 N S H N O P Q , I 1. 1 У формулі 1, R означає галоген, нижчий алкіл, нижчий алкеніл або нижчий алкініл, де нижчі алкільні, нижчі алкенільні або нижчі алкінільні групи можуть необов'язково бути заміщеними, краще, 3 одним чи більше галогенами. R означає Н або 0 метил, і замісник R означає Н, галоген, CF3, нижчу алкокси- або нижчу алкілтіогрупу. Q означає СО2Н, SO3H, CONR'R" або SO2NR'R", де R' та R" незалежно означають Н, нижчий алкіл або нижчий алкіл, заміщений однією чи більше OR, CO2R, NHR, NR2 або CF3 групами, де R означає Н чи нижчий алкіл, або R' та R" разом з атомом азоту, до якого вони приєднані, утворюють 4-, 5- або 6членне гетероциклічне кільце. Р означає ароматичне або гетероароматичне кільце, що має замісники, як більш детально описано далі. Відповідно, даний винахід пропонує сполуки, які інгібують зворотну транскриптазу ВІЛ in vitro та in vivo. Винахід також передбачає фармацевтичні композиції, які включають одну чи більше сполук за винаходом, використання сполук за винаходом для виготовлення фармацевтичних композицій для лікування ВІЛ та методи лікування пацієнта, інфікованого ВІЛ, шляхом введення терапевтично ефективної кількості однієї чи більше сполук за винаходом або їхньої фармацевтично прийнятної солі. Термін "алкіл" в тому значенні, що використовується тут, стосується циклічного, розгалуженого або лінійного вуглеводневого радикала, у якому всі вуглець-вуглецеві зв'язки є простими зв'язками, а термін "нижчий алкіл" стосується алкільних груп, що містять від одного до десяти атомів вуглецю. Термін "циклоалкіл" в тому значенні, що використовується тут, стосується циклічної або поліциклічної алкільної групи, що містить від 3 до 15 атомів 5 вуглецю. Циклоалкільна група може включати численні сконденсовані кільця, причому одне з дистальних кілець може бути ароматичним (наприклад, індан-2-іл, тетрагідронафт-1-ил і т. д.). Аналогічно, термін "алкеніл" в тому значенні, що використовується тут, стосується циклічного, розгалуженого або лінійного вуглеводневого радикала, у якому один чи більше вуглець-вуглецевих зв'язків є подвійними зв'язками, а термін "нижчий алкеніл" стосується алкенільної групи, що містить від одного до десяти атомів вуглецю. Термін "циклоалкеніл" в тому значенні, що використовується тут, стосується циклічної або поліциклічної алкільної групи, що містить 3-15 атомів вуглецю, у якій один чи більше з вуглець-вуглецевих зв'язків є подвійними зв'язками. Циклоалкенільна група може включати численні конденсовані кільця, причому одне з дистальних кілець може бути ароматичним (наприклад, інден-2-іл, 1,2-дигідронафт-1-ил і т.д.). Так само, термін "алкініл" в тому значенні, що використовується тут, стосується алкільної або алкенільної групи, що визначені вище, у яких щонайменше один вуглець-вуглецевий зв'язок був заміщений потрійним зв'язком. Термін "нижчий алкініл", таким чином, включає алкінільні групи, що містять від одного до десяти атомів вуглецю. В тому значенні, що використовується тут, термін "алкокси" стосується групи -OR, де R означає нижчий алкіл, нижчий алкеніл, нижчий алкініл, арил-нижчий алкіл, гетероарил-нижчий алкіл або гетероцикло-нижчий алкіл. Аналогічно, термін "арилокси" стосується групи -ОАr, де Аr означає арильну або гетероарильну групу. Терміни "арил" та "Аr" використовуються тут взаємозамінно і стосуються моноциклічної або поліциклічної вуглеводневої групи з 6-14 атомами вуглецю, яка має щонайменше одне ароматичне кільце, що утворює точку приєднання групи. Поліциклічні арильні групи можуть мати ізольовані кільця (наприклад, біфеніл) або конденсовані кільця, у яких щонайменше одне кільце є ароматичним (наприклад, 1,2,3,4-тетрагідронафт6-ил, нафтил, антрил або фенантрил). Терміни "гетероцикл" або "гетероциклічне кільце" використовуються тут взаємозамінно і стосуються насиченої, частково нанасиченої або ароматичної циклоалкільної або арильної групи, що має одне кільце (наприклад, морфоліно, піридил чи фурил) або численні сконденсовані кільця (наприклад, нафтиридил, хіноксалініл, хінолініл чи індолізиніл), у якій щонайменше один атом вуглецю в кільці був заміщений гетероатомом. Термін "гетероатом" в тому значенні, що використовується тут, стосується атома, який не є вуглецем (типово S, О, Р або N). Терміни "гетероарил" та "гетероароматичний" стосуються гетероциклів, у яких щонайменше одне гетероциклічне кільце є ароматичним. Далі, термін "необов'язково заміщений" в тому значенні, що використовується тут, означає, що один чи більше атомів водню, ковалентно зв'язаних з групою або замісником, визначеними вище, або вільною електронною парою на атомі азоту або фосфору, може бути заміщений ковалентноз 97139 6 в'язаним неводневим замісником, вибраним з групи, що складається з R, Аr, арил-нижчий алкіл, ОН, SH, OR, SR, OAr, SAr, S(=O)R, S(=O)Ar, SO2R, SO2Ar, галоген, CF3, OCF3, SCF3, NH2, NHR, NR2, + NR3 , NHCOR, NHCOAr, NHS(=O)R, NHS(=O)Ar, NHSO2R, NHSO2Ar, NO2, CN, CO2R, CONH2, CONHR, CONR2, C(=O)R, гетероарил та гетероарил-нижчий алкіл. У зазначених вище замісниках, R означає нижчий алкіл, нижчий алкеніл, нижчий алкініл, арил-нижчий алкіл, гетероарил-нижчий алкіл або гетероцикліл-нижчий алкіл. Термін "проліки" в тому значенні, що використовується тут, стосується модифікації сполуки за винаходом, у якій модифікована сполука виявляє меншу фармакологічну активність (порівняно з немодифікованою сполукою) і у якій модифікована сполука перетворюється назад на немодифіковану форму in vivo, краще, у клітині-мішені (наприклад, Т-клітині або гепатоциті) або органі-мішені (наприклад, лімфатичному вузлі або селезінці). Перетворення сполуки за винаходом на форму проліків може бути корисним, якщо активний лікарський засіб є занадто токсичним для безпечного системного введення, якщо немодифікована сполука погано всмоктується з травного тракту або якщо організм має тенденцію до руйнування немодифікованої сполуки до того, як вона досягне своєї мішені. Термін "інгібування зворотної транскриптази" стосується прямого чи опосередкованого зменшення утворення ДНК з матричної РНК або ДНК зворотною транскриптазою. Пряме інгібування включає самогубне, конкурентне та неконкурентне інгібування, алостеричне інгібування або зв'язування інгібітора в ненуклеозидному кармані. Приклади опосередкованого інгібування включають виснаження нуклеозидів для синтезу ДНК, індукування або участь у конформаційних змінах і т. д. В тому значенні, що використовується тут, термін "зменшення вірусного розмноження" означає, що титр вірусу в зразку знижується. Зниження може бути досягнуте різними способами, включаючи часткове чи повне інгібування вірусної реплікації, часткове або повне інгібування процесингу або складання вірусного білка, інгібування вірусного входження до, або виходу з, інфікованої клітини та/або виведення вірусу із системи за рахунок імунної відповіді на вірус. Винахід пропонує, поміж іншого, сполуки такої структури: R0 R3 N N R1 N S H N Q O P , I ,І 1 3 0 де Р, Q, R , R та R мають вказані вище зна1 чення. В кращих варіантах втілення, R вибирають 3 з Сl, Br, I, CH3, CF3, CHF2 та CH2F; R означає Н; 0 R вибирають з Сl, Br, CF3 та СН3; і Q означає 7 97139 СО2Н або SO2NH2. В особливо кращих варіантах 0 втілення, R означає Сl. Р, краще, є заміщеним фенільним, нафтильним, 1,2,3,4-тетрагідронафтильним, хінолінільним, ізохінолінільним або цинолінільним кільцем. В кращих варіантах втілення, групу Р вибирають з фрагментів (а), (b), (с) та (d) нижче. 8 R1 N R1 S N O N N O N N S HN HN CO2H R R0 Rp Rp R6 R4 2 R5 N N Rp U S N R1 U' R6 , (a) S N R1 O HN HN R'' S CO2H R4 R0 N O R0 R' Rp R5 Rp Rp N N O 3 , O , 2 (b) 3 , 1 R6 R7 Rp , (c) R4 R6 0 де R означає CF3, CHF2, CH2F або галоген; R означає галоген, CF3 або метил, R' та R" незалежно означають Н або необов'язково заміщений нижчий алкіл, С1-5-ацил або 1-(С2-4-ацилоксі)-С1-4p алкоксикарбонільну групу, і R має вказані вище значення. Особливо кращі класи сполук відповідають структурам 4 та 5 R5 Br R8 R9 R10 , R11 (d) Br S N N N O N N N HN CO2H Cl де R вибирають з метилу, етилу, пропілу, ізопропілу, циклопропілметилу або С3-6-циклоалкілу; 4 5 6 R , R та R незалежно вибирають з Н, F, Сl, Br, CH3, CF3, CFH2, CF2H, ізопропілу, циклопропілу, ОСН3, ОН, OCF3, NH2 та NHCH3. 7 U та U' незалежно вибирають з N та СН; R вибирають з Сl, Br, I, CH3, CF3, ОСН3, ізопропілу, O 8 11 циклопропілу, т-бутилу та циклобутилу; і R -R N N незалежно означають Н або СН3. Краще, якщо Q 1 p означає SO2NH2, N не S метилом, якщо R не є R є HN Br 7 циклопропілом або циклопропілметилом, і R є 6 CO2H метилом, тільки якщо R також є метилом. Кращими класами сполук є такі, що мають Cl Структури 2 та 3 нижче: Rp Rp 4 N N Br , O N S O HN S Cl NH2 O Rp Rp 4 5 , S 9 97139 p де R вибирають з метилу, етилу, пропілу, ізопропілу, циклопропілу та циклопропілметилу. Найp краще, R означає етил або циклопропіл. Сполуки, 1 0 які поєднують ознаки R = Br, R = Сl або СН3, Р = нафтил або тетрагідронафтил та Q = СО2Н або SO2NR'R", виявляють несподівано сильну активність відносно 3Т з ряду ізолятів ВІЛ, у поєднанні з несподівано гарною фармакокінетикою in vivo. Синтез сполук Синтез сполук за винаходом може бути здійснений за процедурами, по суті описаними в WO 2004/030611, WO 2004/050643 та US 5,939,462. Слід вказати, однак, що для сполук за винаходом 10 можливі численні альтернативні шляхи синтезу. Наведені нижче приклади шляхів надаються лише як приклад, на допомогу фахівцям-практикам в галузі хімії органічного синтезу. В одному шляху синтезу, придатно заміщений анілін амідують активованою сполукою карбонової кислоти (краще галоїдангідридом карбонової кислоти), де активована сполука карбонової кислоти 2 далі включає відхідну групу L (краще бром). Після утворення аніліду, продукт реакції вводять в реакцію з меркаптотриазолом (Het-SH), заміщаючи відхідну групу з утворенням бажаної сполуки, як зображено на схемі Іа нижче. R3 R0 R3 Q L1 + L2 R0 H N L2 H2 N O O R3 R0 H N Het- SH Het-S Q O Q Схема Іа Ця схема є кращою, якщо меркаптотриазол "Het-SH" є більш цінним порівняно з аніліном, оскільки триазол не використовується до останньої стадії і не буде зазнавати неминучих втрат, що відбуваються при маніпуляціях з проміжними сполуками в процесі синтезу. Вибір відхідних груп 1 2 L та L буде залежати певною мірою від вибору конкретного аміну і в меншому ступені - від конкретного меркаптотриазолу. Особливо краще, щоб 1 2 L та L були галоїдами, найкраще хлором чи бромом. Придатні розчинники для реакції амідування включають прості ефіри, спирти, та вуглеводні (краще галогеновані), і вибір придатних N R1 N OH R1 SH N N N O R1 N S P P O R3 N N R3 L1 R1 S P P N R3 N N N розчинників буде щонайменше частково залежати від хімічної природи реагентів. Загалом будуть застосовними міркування щодо розчинників, каталізаторів та/або основ, використовуваних у зазначеній вище реакції, описані Connell et al. (патент США № 5939462). Альтернативна загальна стратегія зображена на Схемі Іb нижче. Цей підхід передбачає ацилювання анілінів S-триазолілмеркаптооцтовими кислотами, які легко одержують алкілуванням меркаптотриазолів α-галоїдоцтовою кислотою або складним ефіром. R0 H N S O Q Схема Іb Придатні реагенти включають, без обмеження, йодоцтову кислоту та метилбромацетат, і 3 етил-α-бромпропіонат, якщо бажано, щоб R був метилом. Якщо використовується складний ефір, його гідролізують після S-алкілування для одержання вільної карбонової кислоти. Кислота та анілін можуть бути спряжені з будь-яким звичайним карбоксилактивуючим реагентом чи сумішами реагентів, наприклад карбодіімідом, в присутності основи третинного аміну, необов'язково, з N-гідроксибензотриазолом як каталізатором, або тіоніл- чи оксалілхлоридом, з диметиламінопіри 11 97139 12 дином як каталізатором. Ця схема є кращою, якщо анілін є більш цінним порівняно з триазолом. Прикладом за Схемою Іа є синтез, описаний на Схемі 2, де сполуку за винаходом одержують з двох окремо виготовлених прекурсорів. Перший прекурсор, що включає заміщений триазин, і друH2N Cl S O гий прекурсор, що включає заміщений анілін, можуть бути виготовлені за протоколами, наведеними нижче в розділі "Приклади". Реакція прекурсорів, типово, проводиться в полярному апротонному розчиннику, такому як ДМФ, в присутноприсутності основи, такої як карбонат калію. O O S H2 N O S HCl O EtOH/ вода NH3 Cl O O Cl діізопропілетиламін T ГФ Cl SO2NH2 Cl Cl HN HN Cl CH2Cl2 NH NH2 O Cl O O ''A'' NH2 NO2 MgBr CSCl2 H2 HNO3 ClNi(dppp)2 Pb-C Br NCS N N N N N SH N ''A'' 1) Гідразин 2) Диметилацетаміддиметилацеталь O S HN K2CO3 SO2NH2 Cl Схема 2 Якщо триазин є заміщеним фторованою алкільною групою, може бути використана процеду ра синтезу, показана на Схемі 3. Аналогічна процедура наведена нижче у розділі "Приклади". NH2 NO2 MgBr CSCl2 H2 HNO3 Cl2Ni(dppp)2 Pb-C Br N N NCS 1) Гідразин 2) O F N SH N SO2NH2 F N N F K2CO3 O S HN F SO2NH2 F2OH OH Cl Cl Cl NH O Схема 3 Галогензаміщений триазол може бути одержаний дигалогенуванням триазолу, з наступним заміщенням одного з галоїдів, як показано на Схемі 4, що відповідає процедурі, описаній нижче у розділі "Приклади". 13 97139 Q Q Q Na2S i-Pr2NEt Cl + CH2Cl2 Cl O 14 Me Me Me NH2 Q NH NH Cl HS N N O O NH Br OHC HNO2 NH2 CHO HN N N NH N N N Br N N Me S O Br NBS Схема 4 Іншим шляхом одержання триазолу, заміщеного галогеном, є діазотування амінотриазолу, як показано на Схемі 5 нижче, що відповідає процедурі, наведеній нижче у розділі "Приклади". NH2 NO2 MgBr CSCl2 H2 HNO3 Cl2Ni(dppp)2 Pb-C N NCS N N K2CO3 H2N N SH SO2NH2 H2N N N O S HN 1) Аміногуанідин SO2NH2 2) NaOH Cl Cl Cl NH O NaNO2 N DCA Br BnNEt3Br N N O S HN SO2NH2 Cl Схема 5 Альтернативно, якщо триазол заміщений CF3, може бути використана процедура синтезу, зображена на Схемі 6, що відповідає аналогічним процедурам, наведеним нижче у розділі "Приклади". 15 97139 16 Q Q Q Cl Cl діізопропілетиламін + O H2N CH2Cl2 Me Cl NH2 NH Me S F3C ДМФ N F3C OH K2CO3 O O HN N NH F3C N Me S N NH O N SH N Схема 6 Приклад альтернативного шляху синтезу, зображеного на Схемі Іа, показаний на Схемі 7 ниN H2N N N N H2N SH жче, де анілін ацилюють попередньо одержаною S-триазолілмеркаптооцтовою кислотою. N N N OMe S Br N N O O N Br N N H N S O OH S Cl Cl H2N SO2NH2 SO2NH2 Схема 7 Фармацевтичні композиції Якщо сполуки за винаходом вводяться як частина фармакологічної композиції, то передбачається, що придатні сполуки можуть бути скомбіновані в суміші з фармацевтично прийнятними носіями, ексципієнтами та іншими домішками. Особливо краще, щоб сполуки за винаходом входили до складу фармацевтичної композиції, яка містить один чи більше нетоксичних фармацевтично прийнятних носіїв. Фармацевтичні композиції можуть бути скомпоновані для перорального введення в твердій або рідкій формі, для парентеральної ін'єкції або для ректального введення. Фармацевтичні композиції за даним винаходом можуть бути введені людям та іншим тваринам перорально, ректально, парентерально, інтравагінально, інтраперитонеально, місцеве, буквально або у вигляді орального чи назального спрею. Термін "парентеральне" введення в тому значенні, що використовується тут, стосується способів введення, які включають, без обмеження, внутрішньовенну, внутрішньом'язову, інтра перитонеальну, підшкірну та внутрішньосуглобову ін'єкцію та інфузію. Фармацевтичні композиції для парентеральних ін'єкцій, краще, включають фармацевтично прийнятні стерильні водні або неводні розчини, дисперсії, суспензії або емульсії, а також стерильні порошки для відновлення до стерильних розчинів або дисперсій для ін'єкцій безпосередньо перед використанням. Приклади придатних водних та неводних носіїв, розріджувачів, розчинників та наповнювачів включають воду, етанол, поліоли (такі як гліцерин, пропіленгліколь, поліетиленгліколь тощо) та їхні придатні суміші, рослинні олії (такі як оливкова олія) та придатні для ін'єкцій органічні складні ефіри, такі як етилолеат. Належна плинність може забезпечуватися, наприклад, завдяки використанню покривних матеріалів, таких як лецитин, шляхом підтримання потрібного розміру частинок у випадку дисперсії та шляхом використання поверхнево-активних речовин. Композиції можуть також містити домішки, такі як консерванти, змочувальні агенти, емульгатори та диспергенти. Запобігання дії мікрооргані 17 змів може бути забезпечене включенням різних антибактеріальних та протигрибкових агентів, наприклад парабену, хлорбутанолу, фенолу, сорбінової кислоти тощо. Може також бути бажаним включення ізотонічних агентів, таких як цукри, хлорид натрію тощо. Уповільнене всмоктування фармацевтичної форми для ін'єкцій може бути забезпечене включенням агентів, що уповільнюють всмоктування, таких як алюмініймоностеарат та желатин. Для пролонгації ефекту сполук за винаходом, може бути бажаним уповільнювання всмоктування лікарського засобу, введеного підшкірною або внутрішньом'язовою ін'єкцією. Це може бути досягнуте використанням рідкої суспензії кристалічного або аморфного матеріалу з поганою розчинністю у воді. Швидкість всмоктування сполуки тоді буде залежати від її швидкості розчинення, яка, у свою чергу, може залежати від розміру кристалів та кристалічної форми. Альтернативно, уповільнене всмоктування парентерально введених сполук за винаходом може бути досягнуте шляхом розчинення або суспендування лікарського засобу в масляному носії. Форми депо для ін'єкцій виготовляють шляхом створення унітарної або мікродисперсної матриці сполуки за винаходом в біодеградуючих полімерах, включаючи, без обмеження, полілактид-полігліколід, полі(складні ортоефіри) та поліангідриди. Швидкість вивільнення лікарського засобу можна контролювати шляхом зміни співвідношення лікарського засобу та полімеру і природи конкретного використовуваного полімеру. Композиції депо для ін'єкцій можуть бути також виготовлені шляхом включення сполуки до ліпосом або мікроемульсій, сумісних з тканинами організму. Тверді дозовані форми для перорального введення включають, без обмеження, капсули, таблетки, пілюлі, порошки, драже та гранули. В таких твердих дозованих формах активну сполуку змішують із щонайменше одним інертним фармацевтично прийнятним ексципієнтом або носієм, таким як цитрат натрію або дикальційфосфат та/або (а) наповнювачами або розріджувачами, такими як крохмалі, лактоза, сахароза, глюкоза, маніт та кремнекислота, (b) зв'язуючими, такими як карбоксиметилцелюлоза, альгінати, желатин, полівінілпіролідон, сахароза та гуміарабік, (с) зволожувачами, такими як гліцерин, (d) агентами, що викликають дезінтеграцію, такими як агар, карбонат кальцію, картопляний або маніоковий крохмаль, альгінова кислота, певні силікати та карбонат натрію, (е) агентами уповільнення розчинення, такими як парафін, (f) прискорювачами всмоктування, такими як четвертинні амонієві сполуки, (g) змочувальними агентами, такими як цетиловий спирт та гліцеринмоностеарат, (h) абсорбентами, такими як каолін та бентонітова глина, та (і) змащувальними речовинами, такими як тальк, стеарат кальцію, стеарат магнію, тверді поліетиленгліколі, лаурилсульфат натрію, та їхніми сумішами. Тверді дозовані форми можуть також включати буферні агенти. 97139 18 Тверді композиції можуть також бути використані як наповнювачі у наповнених м'яких та твердих желатинових капсулах з використанням таких ексципієнтів, як лактоза або молочний цукор, а також високомолекулярні поліетиленгліколі тощо. Тверді дозовані форми можуть бути виготовлені з покриттями та оболонками, такими як ентеросолюбільні покриття та інші покриття, добре відомі фахівцям з приготування фармацевтичних композицій. Вони можуть необов'язково містити агенти забезпечення непрозорості і можуть також мати такий склад, щоб вони вивільняли активний інгредієнт(и) тільки або краще у певній частині кишкового тракту, необов'язково уповільнено. Активні сполуки можуть також бути в мікрокапсульованій формі. Рідкі дозовані форми для перорального введення включають фармацевтично прийнятні емульсії, розчини, суспензії, сиропи та еліксири. На додаток до активних сполук, рідкі дозовані форми можуть містити інертні розріджувачі, звичайно використовувані фахівцями, такі як вода або інші розчинники, солюбілізатори та емульгатори, такі як етиловий спирт, ізопропіловий спирт, етилкарбонат, етилацетат, бензиловий спирт, бензилбензоат, пропіленгліколь, 1,3-бутиленгліколь, диметилформамід, олії (зокрема бавовняна, арахісова, кукурудзяна, зародкова, оливкова, рицинова та кунжутна олії), гліцерин, тетрагідрофурфуриловий спирт, поліетиленгліколі та складні ефіри жирних кислот та сорбіту, та їхні суміші. Пероральні рідкі композиції можуть також включати ад'юванти, такі як змочувальні агенти, емульгувальні та суспендувальні агенти, барвні, підсолоджувальні, смакові та ароматизуючі агенти. Композиції для ректального або вагінального введення є, краще, супозиторіями, які можуть бути приготовлені шляхом змішування сполук за даним винаходом з придатними неподразливими ексципієнтами або носіями, такими як какаова олія, поліетиленгліколь або інші воски для супозиторіїв, що є твердими при кімнатній температурі, але рідкими при температурі тіла і тому плавляться у порожнині прямої кишки або піхви та вивільняють активну сполуку. Сполуки за даним винаходом можуть також бути введені в формі ліпосом. Як відомо фахівцям, ліпосоми загалом одержують з фосфоліпідів або інших ліпідних речовин. Ліпосоми, типово, утворюють з моно- або багатоламелярних гідратованих рідких кристалів, диспергованих у водному середовищі. Можуть бути використані будьякі нетоксичні, фізіологічно прийнятні ліпіди, здатні утворювати ліпосоми. Композиції у формі ліпосом можуть містити, на додаток до сполуки за даним винаходом, стабілізатори мембран, консерванти, ексципієнти тощо. Кращими ліпідами є фосфоліпіди та фосфатидилхоліни (лецитини), як природні, так і синтетичні. Способи утворення ліпосом є відомими фахівцям. Див., наприклад, Prescott, Ed., Methods in Cell Biology, Volume XIV, Academic Press, New York, N.Y. (1976), p. 33 et seq. Сполуки за даним винаходом можуть бути використані у формі фармацевтично прийнятних 19 солей, утворених неорганічними або органічними кислотами. Під фармацевтично прийнятною сіллю розуміються такі солі, які, в межах обґрунтованої медичної оцінки, є придатними для використання в контакті з тканинами людей та нижчих тварин без небажаної токсичності, подразнення, алергічних реакцій тощо, і мають прийнятне співвідношення корисний ефект/ризик. Фармацевтично прийнятні солі є добре відомими фахівцям. Наприклад, S.M. Berge et al. докладно описують фармацевтично прийняти солі у J. Pharmaceutical Sciences, 1977, 66:1 et seq. Солі можуть бути одержані in situ під час кінцевого виділення та очищення сполук за винаходом або окремо шляхом проведення реакції вільної основної форми з придатною кислотою. Приклади кислотноадитивних солей включають, без обмеження, ацетат, адипат, альгінат, цитрат, аспартат, бензоат, бензолсульфонат, бісульфат, бутират, камфорат, камфорсульфонат, цитрат, глюконат, глутамат, гліцерофосфат, гемісульфат, гептаноат, гексаноат, фумарат, гідрохлорид, гідробромід, гідройодид, 2-оксіетансульфонат (ізетіонат), лактат, малеат, метансульфонат, нікотинат, 2нафталінсульфонат, оксалат, памоат, пектинат, 3-фенілпропіонат, фосфат, півалат, пропіонат, сукцинат, сульфат, тартрат, бікарбонат, птолуолсульфонат та ундеканоат. Основні азотовмісні групи можуть також бути кватернізовані такими агентами, як нижчий галоїдалкіл, такий як хлор-, бром- та йодметили, -етили, -пропіли та бутили; діалкілсульфати, такі як диметил-, діетил-, дибутил- та діамілсульфати; довголанцюгові галоїдні сполуки, такі як хлор-, бром- та йоддецил, -лаурил, -міристил та -стеарили; галоїдарилалкіл, такий як бромбензили і фенетили та інші. При цьому одержують водо- чи маслорозчинні чи -дисперговані продукти. Солі приєднання основи можуть бути одержані in situ під час кінцевого виділення та очищення сполук за даним винаходом або згодом, шляхом проведення реакції фрагмента, який включає групу карбонової кислоти, з придатною основою, такою як гідроксид, карбонат або бікарбонат фармацевтично прийнятного катіону металу, або з аміаком чи органічним первинним, вторинним чи третинним аміном. Фармацевтично прийнятні солі включають, без обмеження, солі лужних та лужноземельних металів, таких як літій, натрій, калій, кальцій, магній та алюміній тощо, і нетоксичні солі четвертинного амонію та амінів, включаючи амоній, тетраметиламоній, тетраетиламоній, метиламін, диметиламін, триметиламін, триетиламін, діетиламін, етиламін тощо. Інші приклади органічних амінів, придатних для утворення солей приєднання кислоти, включають етилендіамін, етаноламін, діетаноламін, піперидин, піперазин, глюкозамін, лейцин тощо. Фактичні рівні доз активних інгредієнтів у фармацевтичній композиції за даним винаходом можуть змінюватися таким чином, щоб одержати кількість активної сполуки (сполук), що є ефективною для досягнення бажаної терапевтичної відповіді для конкретних пацієнта, композиції та способу введення. Вибраний рівень доз буде 97139 20 залежати від активності конкретної сполуки, шляху введення, схеми дозування, тяжкості стану, лікування якого проводиться, та стану і медичної історії пацієнта, що лікується. Дослідження з визначення доз є рутинними, і фахівці в даній галузі є здатними почати з доз сполуки на рівнях, нижчих ніж потрібні для досягнення бажаного терапевтичного ефекту, та поступово підвищувати дозу до досягнення бажаного ефекту. Загалом, пацієнту-ссавцю вводять перорально дози на рівні від приблизно 0,1 до приблизно 100, краще від приблизно 5 до приблизно 50 мг активної сполуки на кілограм ваги тіла за добу. Якщо бажано, ефективна добова доза може бути розділена на кратні дози з метою введення, наприклад від двох до чотирьох окремих доз за добу. Сполуки за винаходом можуть бути введені самі або в комбінації з іншими агентами для лікування ВІЛ. Особливо передбачувані додаткові сполуки включають інгібітори зворотної транскриптази нуклеозидного типу (наприклад, ламівудин, зидовудин, ставудин, абакавір, тенофовір або диданозин), ненуклеозидні інгібітори зворотної транскриптази (наприклад, невірапін, делавірдин, ефавіренц), інгібітори протеази (наприклад, ритонавір, саквінавір, індинавір, нелфінавір), інгібітори злиття (наприклад, енфувіртид), антагоністи CCR5, імунотерапевтичні агенти (наприклад, рибавірин, IL-2), і активні, пасивні та/або терапевтичні вакцини. Комбіновані терапії за даним винаходом включають введення щонайменше однієї сполуки за даним винаходом або її функціонального похідного та щонайменше одного іншого фармацевтично активного інгредієнта. Активний інгредієнт(и) та фармацевтично активні агенти можуть бути введені окремо або разом і при введені окремо це може відбуватися одночасно або окремо в будь-якому порядку. Кількості активного інгредієнта(ів) та фармацевтично активного агента(ів) і відносний час введення слід вибирати таким чином, щоб досягнути бажаного комбінованого терапевтичного ефекту. Таким чином, даний винахід пропонує фармацевтичні композиції, які включають одну чи більше сполук, що мають структуру відповідно до будь-якої з формул 1-5, визначених вище, де сполука або сполуки є присутніми в концентрації, що ефективно інгібує зворотну транскриптазу та/або реплікацію ВІЛ у клітині пацієнта при введенні композиції пацієнту. В кращих варіантах втілення, фармацевтична композиція за винаходом включає одну чи більше сполук відповідно до однієї з формул 2-5. Особливо передбачається, що множина сполук може бути включена до однієї фармацевтичної композиції, з метою забезпечення інгібування в широкому діапазоні множини мутантних ферментів 3Т. Що стосується придатних концентрацій передбачуваних сполук у фармацевтичних композиціях, слід розуміти, що пересічний фахівець в даній галузі може легко відрегулювати кількість сполуки для забезпечення інгібування зворотної транскриптази та/або реплікації ВІЛ. Наприклад, інгібування реплікації ВІЛ у клітині (типово Тклітинах, інфікованих вірусом ВІЛ) можна контро 21 97139 лювати in vitro з використанням гемокультури та системи аналізу на основі люциферази, як описано нижче. Альтернативно, інгібування зворотної транскриптази може контролюватися in vivo з використанням полімеразної ланцюгової реакції зі зворотною транскриптазою (RT-PCR) для визначення кількості копій вірусної ДНК та/або РНК у крові або лімфатичних вузлах (що містять ВІЛінфіковані клітини). Загалом передбачається, що придатні концентрації будуть забезпечувати концентрацію у сироватці від 1 нМ до 100 мкМ, і у деяких випадках - від 0,01 нМ до 1 нМ). Приклади Наступні експерименти наведені лише для прикладу і не повинні розглядатися як такі, що обмежують обсяг винаходу. Сполуки за винаходом 2-[5-Бром-4-(4-циклопропілнафталін-1-іл)-4Н[1,2,4]триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід (Метод А) O N N Br N S HN Cl O S NH 2 O 1-Циклопропілнафталін Циклопропілмагнійбромід (150 мл, 0,5М у тетрагідрофурані) повільно додають до розчину 1бромнафталіну (10 г, 50 ммоль) і [1,3біс(дифенілфосфіно)-пропан]дихлорнікелю(ІІ) в тетрагідрофурані (10 мл) і перемішують при 0 °С. Реакційну суміш перемішують при кімнатній температурі протягом 16 годин і розчинник випарюють при зниженому тиску. Додають ЕtOAс та хлорид амонію у воді. Після екстракції, органічний шар осушують над сульфатом натрію, фільтрують та концентрують при зниженому тиску. Залишок очищають хроматографією на силікагелі, одержуючи 1-циклопропілнафталін (6,4 г, 76 %). 1-Циклопропіл-4-нітронафталін Нітрит натрію (30 мл) повільно додають (протягом 2 годин) до 1-циклопропілнафталіну (6,4 г, 38 ммоль), перемішуваного при 0 °С. Реакційну суміш перемішують при 0 °С протягом ще 30 хв., а потім повільно виливають на лід. Додають воду, а потім ЕtOAс. Після екстракції органічний шар промивають 1 % водним розчином NaOH, потім промивають водою, осушують над сульфатом натрію, фільтрують та концентрують при зниженому тиску. Залишок очищають хроматографією на силікагелі, одержуючи 1-циклопропіл4-нітронафталін (5,2 г, 64 %). 1-Аміно-4-циклопропілнафталін Розчин 1-циклопропіл-4-нітронафталіну (5 г, 23 ммоль) в етанолі (200 мл) перемішують під воднем в присутності Pd/C (загалом 10 %, 1,8 г). Реакційну суміш струшують протягом ночі, потім фільтрують через целіт. Розчинник випарюють і 22 залишок очищають хроматографією на силікагелі, одержуючи 1-аміно-4-циклопропілнафталін (3,1 г, 73 %). 1-Циклопропіл-4-ізотіоціанатонафталін Тіофосген (1,1 г, 9,7 ммоль) додають до розчину 1-аміно-4-циклопропілнафталіну (1,8 г, 9,7 ммоль) та діізопропілетиламіну (2 екв.) в дихлорметані (50 мл), перемішуваному при 0 °С. Реакційну суміш перемішують протягом 5 хв. при цій температурі, потім додають 1 % розчин НСl у воді та органічний шар відокремлюють, промивають розсолом, осушують над сульфатом натрію, фільтрують і розчинник випарюють при зниженому тиску. Додають гексан і осад, що утворюється, збирають на фільтрі. Розчинник випарюють, одержуючи 1-циклопропіл-4-ізотіоціанатонафталін (1,88 г, 86 %). 5-Аміно-4-(4-циклопропілнафталін-1-іл)-4Н[1,2,4]триазол-3-тіол Суміш аміногуанідину гідрохлориду (3,18 г, 29 ммоль), 1-циклопропіл-4-ізотіоціанатонафталіну (3,24 г, 14 ммоль) та діізопропілетиламіну (3 екв.) в ДМФ (20 мл) перемішують при 50 °С протягом 15 годин. Розчинник випарюють, додають толуол і розчинник випарюють знову. Додають 2,0М водний розчин гідроксиду натрію (30 мл) і реакційну суміш нагрівають при 50 °С протягом 60 годин. Реакційну суміш фільтрують, і фільтрат нейтралізують 2,0М водним розчином НСl. Знов фільтрують, потім випарюють розчинник та очищають залишок хроматографією на силікагелі, одержуючи 5-аміно-4-(4-циклопропілнафталін-1-іл)-4Н[1,2,4]триазол-3-тіол (2,0 г, 49 %). 2-[5-Аміно-4-(4-циклопропілнафталін-1-іл)4Н-[1,2,4]триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід До розчину 5-аміно-4-(4циклопропілнафталін-1-іл)-4Н-[1,2,4]триазол-3тіолу (708 мг, 2,5 ммоль), K2СО3 (380 мг, 2,5 ммоль) в ДМФ (20 мл) додають 2-хлор-N-(2-хлор4-сульфамоїлфеніл)ацетамід (710 мг, 2,5 ммоль). Реакційну суміш перемішують при кімнатній температурі протягом ночі. Після закінчення реакції розчинник випарюють. Залишок очищають хроматографією на силікагелі, одержуючи 2-[5-аміно4-(4-циклопропілнафталін-1-іл)-4Н-[1,2,4]триазол3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід (1,26 г, 95 %). 2-[5-Бром-4-(4-циклопропілнафталін-1-іл)-4Н[1,2,4]триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід Дихлороцтову кислоту (180 мкл, 2,2 ммоль) додають до суспензії 2-[5-аміно-4-(4циклопропілнафталін-1-іл)-4Н-[1,2,4]триазол-3ілсульфаніл]-N-(2-хлор-4-сульфамоїлфеніл)ацетаміду (0,59 г, 1,1 ммоль), нітриту натрію (1,5 г, 22 ммоль) та ВТЕАВr (0,91 г, 3,3 ммоль) в дибромметані (30 мл). Реакційну суміш перемішують при кімнатній температурі протягом 4 годин, потім екстрагують дихлорметаном та бікарбонатом натрію у воді. Органічний шар осушують над сульфатом натрію, фільтрують та концентрують при зниженому тиску. Залишок очищають хроматографією на силікагелі, одержуючи 2-[5-бром-4(4-циклопропілнафталін-1-іл)-4Н-[1,2,4]триазол-3 23 97139 ілсульфаніл]-N-(2-хлор-4-сульфамоїлфеніл)ацетамід (224 мг, 31 %). N 2-[5-Бром-4-(4-циклопропілнафталін-1-іл)-4Н[1,2,4]триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід (Метод В) N N N N N N H2 N 24 SH H2 N OCH3 Cl N OCH3 S NaNO2 Cl2CCO2H O O K2CO3 ДМФ кімн.темп., 24 год Br OCH3 S N O BnEt3NBr CHBr3 кімн.темп., 3 год LiOH THF-EtOH-H2O 00С, 1 год. Cl N N Br N H N S O Метиловий складний ілсульфаніл]оцтової кислоти Матеріали тіотриазол метилхлорацетат карбонат калію диметилформамід H2N SO2NH2 Cl ефір SO2NH2 Br N OH S O POCl3 піридин 00С, 1 год. 2-[5-аміно-4-(4-циклопропілнафталін-1-іл)-4Н-[1,2,4]триазол-3Кількість 2,24 0,73 мл 1,21 г 40 мл Процедура: До суспензії тіотриазолу та карбонату калію в ДМФ додають метилхлорацетат по краплях при кімнатній температурі протягом 5 хв. Реакцію перемішують при кімнатній температурі протягом 24 год. і повільно виливають в перемішуваний охолоджений льодом водний розчин. Осад кориМатеріали тіотриазол L10183-58 бромоформ нітрит натрію бензилтриетиламонійбромід дихлороцтова кислота N N Кількість 709 мг 10 мл 2,76г 1,63г 0,33 мл Процедура: До розчину метилового складного ефіру 2-[5аміно-4-(4-циклопропілнафталін-1-іл)-4Н[1,2,4]триазол-3-ілсульфаніл]оцтової кислоти та бензилтриетиламонійхлориду в бромоформі додають нітрит натрію. До суміші додають дихлороцтову кислоту і реакційну суміш перемішують при кімнатній температурі протягом 3 год. Суміш без Мол. вага 282,36 108,52 138,21 ммоль 7,9 8,3 (1,05екв.) 8,7 (1,1 екв.) (5 мл/ммоль) чнювато-жовтого кольору збирають вакуумфільтрацією та сушать під високим вакуумом при 50 °С протягом 16 год. в присутності Р2O5, одержуючи 2,24 г (80 %) названої в заголовку сполуки. Метиловий складний ефір 2-[5-бром-4-(4циклопропілнафталін-1-іл)-4Н-[1,2,4]триазол-3ілсульфаніл]оцтової кислоти Мол. вага 354,43 69,00 272,24 128,94 ммоль 2,0 (5 мл/ммоль) 40 (20 екв.) 6,0 (3 екв.) 4,0 (2 екв.) посередньо завантажують до 7-дюймової колонки силікагелю, заповненої СН2Сl2. Колонку спочатку елююють СН2Сl2 до повного закінчення елюції СНВr3, а потім елююють сумішшю ацетон/СН2Сl2 (5:95), одержуючи 713 мг (85 %) названої в заголовку сполуки. 2-[5-Бром-4-(4-циклопропілнафталін-1-іл)-4Н[1,2,4]триазол-3-ілсульфаніл]оцтова кислота 25 Матеріали метиловий складний ефір тіотриазолу тетрагідрофуран етанол вода гідроксид літію Процедура: До розчину метилового складного ефіру 2-[5бром-4-(4-циклопропілнафталін-1-іл)-4Н[1,2,4]триазол-3-ілсульфаніл]оцтової кислоти в суміші THF та ЕtOН при 0 °С додають розчин LiOH в Н2О по краплях протягом 5 хв. Реакція завершується після перемішування при 0 °С протягом ще 45 хв. Реакцію нейтралізують до рН 7 додаванням 0,5Н розчину НСl при 0 °С і одержану суміш концентрують під вакуумом до 1/5 її вихідного об'єму. Суміш розводять H2O (~20 мл) та підкислюють до рН 2-3 шляхом додавання 0,5Н Матеріали тіотриазолкарбонова кислота 4-аміно-3-хлорфенілсульфонамід піридин оксихлорид фосфору Процедура: До розчину карбонової кислоти та аніліну, вказаних вище, у піридині при 0 °С, додають РОСl3 по краплях протягом 5 хв. Реакція завершується після перемішування при 0 °С протягом ще 50 хв. Реакційну суміш гасять додаванням H2O (1 мл), потім концентрують під вакуумом до світло-коричневої маслянистої рідини, яку розводять CH2Cl2 (200 мл). Органічний шар промивають H2O (1х50 мл), насиченим розчином NаНСО3 (1х50 мл), а потім розсолом (1х50 мл). Органічний розчин осушують над Na2SO4 та концентрують до сухого залишку. Одержану маслянисту рідину розтирають з ЕtOН, одержуючи світло 97139 26 Кількість 1,14г 10 мл 10 мл 10 мл 98 мг Мол. вага 418,31 23,95 ммоль 2,7 (~3 мл/ммоль) (~3 мл/ммоль) (~3 мл/ммоль) 4,1 (1,5екв.) НСl до утворення липкої твердої речовини. (Якщо продукт утворюється у вигляді маслянистої рідини при підкислюванні, то рекомендується екстракція CH2Cl2.) Тверду речовину жовтуватокоричнюватого кольору збирають вакуумфільтрацією та сушать під високим вакуумом при 50 °С протягом 16 год. в присутності Р2О5, одержуючи 1,02 г (93 %) названої в заголовку сполуки. 2-[5-Бром-4-(4-циклопропілнафталін-1-іл)-4Н[1,2,4]-триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід Кількість 884 мг 452 мг 22 мл 0,24 мл Мол. вага 404,28 206,65 153,33 ммоль 2,2 2,2 (10 мл/ммоль) 2,6 (1,2 екв.) жовту тверду речовину. До суміші додають Н2О, щоб зібрати додаткову кількість твердої речовини. Світло-жовту тверду речовину збирають вакуум-фільтрацією та сушать під високим вакуумом протягом 16 год., одержуючи 930 мг (72 %) продукту. Додатковий продукт (132 мг, 10 %) виділяють екстракцією фільтрату CH2Cl2, а потім хроматографією на колонці з використанням суміші ацетон/СН2Сl2 (20:80). 2-[5-Бром-4-(4-циклопропіл-7метоксинафталін-1-іл)-4Н-[1,2,4]-триазол-3ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід 27 97139 28 O NH2 O O HN OH O HN OH O S O a b O HN O O HN O O OH S O O Br O O O HN O Br c Br e d NH2 NH2 O O Br f 1-Аміно-4-циклопропіл-7-метоксинафталін До перемішуваного розчину 8-аміно-2нафтолу (5 г, 31,4 ммоль) в суміші тетрагідрофурану (50 мл) та дихлорметану (100 мл) додають ди-т-бутилдикарбонат (6,86 г, 31,4 ммоль). Суміш перемішують при 70 °С протягом 18 годин. Після охолодження суміші до кімнатної температури, додають насичений водний карбонат натрію і продукт екстрагують дихлорметаном. Органічний шар промивають водою та розсолом, осушують над сульфатом натрію, фільтрують та концентрують при зниженому тиску. Одержаний залишок очищають хроматографією на колонці із силікагелем (дихлорметан:етилацетат, 9:1), одержуючи N-ВОС-похідне а (4,85г, вихід 60 %). До суміші N-ВОС-похідного а (4,85 г, 18,7 ммоль) та триетиламіну (3,91, 28,1 ммоль) в дихлорметані (170 мл) додають метансульфоновий ангідрид (3,58 г, 20,6 ммоль) при 0 °С. Суміш перемішують протягом 30 хв. та виливають в насичений водний розчин бікарбонату натрію. Органічний шар екстрагують дихлорметаном, осушують над сульфатом натрію, фільтрують та концентрують при зниженому тиску, одержуючи складний метансульфонатний ефір b (6,22 г, кількісний вихід). До розчину метансульфонату b (6,12 г, 18,1 ммоль) в 150 мл оцтової кислоти додають Nбромсукцинімід (3,39 г, 19 ммоль). Суміш перемішують протягом 2 год. і додають воду та дихлорметан. Водний шар доводять до рН 7 додаванням 10Н водного гідроксиду натрію. Органічний шар екстрагують дихлорметаном, осушують над сульфатом натрію, фільтрують та концентрують при зниженому тиску, одержуючи сирове 5-бромпохідне с (7,6 г, кількісний вихід). Суміш с (7,72 г, 18,5 ммоль) та 10 % водного розчину гідроксиду натрію (370 мл) в тетрагідрофурані (220 мл) перемішують при 50 °С протягом g 5 днів. Суміш охолоджують до 0 °С та нейтралізують концентрованою хлористоводневою кислотою. Суміш концентрують при зниженому тиску та продукт екстрагують етилацетатом. Органічний шар осушують над сульфатом натрію, фільтрують та концентрують, одержуючи нафтол d (5,87 г, вихід 94 %). Суміш нафтолу d (3,53 г, 10,4 ммоль), метилйодиду (0,65 мл, 10,4 ммоль) та гідроксиду натрію (417 мг, 10,4 ммоль) в ацетоні (25 мл) перемішують при кімнатній температурі протягом 4 годин. Одержану суміш концентрують та залишок очищають хроматографією на колонці (85 % гексан/15 % етилацетат), одержуючи 2,39 г, вихід 65 %, простого метилового ефіру е. Суміш простого метилового ефіру е (3,25 г, 9,22 ммоль) в 4Н НСl в 1,4-діоксані (92 мл) перемішують при кімнатній температурі протягом 1 години. Суміш концентрують при зниженому тиску та додають етилацетат і насичений розчин бікарбонату натрію. Екстрагований органічний шар промивають водою та розсолом, осушують над сульфатом натрію та концентрують при зниженому тиску, одержуючи 2-метокси-5-бром-8амінонафталін f (2,14 г, вихід 92 %). До розчину амінонафталіну f (1 г, 4,0 ммоль), циклопропілборонової кислоти (438 мг, 5,1 ммоль), фосфату калію (2,97 г, 14 ммоль) та трициклогексилфосфіну (112 мг, 0,4 ммоль) в толуолі (21 мл) та воді (0,8 мл) під атмосферою азоту додають ацетат паладію (45 мг, 0,2 ммоль) при енергійному перемішуванні. Суміш нагрівають до 100 °С протягом 3 год., а потім охолоджують до кімнатної температури. Додають воду і суміш екстрагують етилацетатом, осушують над сульфатом натрію та концентрують. Очищення хроматографією на колонці (50 % гексан/50 % етилацетат) дає названу в заголовку сполуку д (699 мг, вихід 82 %). 29 97139 30 O N N NCS NH2 H2N O O O g h N N H2 N N S O O H2 N S O j N Br O O S N S N Cl H N O O O O Cl + i Cl H N Cl HN SH N S NH2 NH2 k I Сполуку g (699 мг, 3,28 ммоль) розчиняють в 18 мл дихлорметану. Додають бікарбонат натрію (9 мл, насич. розчин) та тіофосген (0,25 мл, 3,28 ммоль) і суміш перемішують при кімнатній температурі протягом 1 год. Органічний шар відокремлюють, осушують над сульфатом натрію та концентрують, одержуючи 819 мг, вихід 98 %, сполуки h, яку використовують на наступній стадії без додаткового очищення. Сполуку h (819 мг, 3,21 ммоль) розчиняють в 6 мл диметилформаміду, додають аміногуанідину гідрохлорид (532 мг, 4,8 ммоль) та діізопропілетиламін (0,84 мл, 4,8 ммоль) і суміш перемішують при 50 °С протягом 18 годин. Суміш потім концентрують та до залишку додають 2М водний розчин гідроксиду натрію (10 мл). Суміш перемішують при 50 °С протягом 18 годин, а потім охолоджують до кімнатної температури. Одержану суміш потім нейтралізують водним 1Н НСl і осад збирають, одержуючи сполуку і (200 мг, вихід 25 %). Сполуки і (63 мг, 0,2 ммоль) та j (57 мг, 0,2 ммоль) розчиняють в ДМФ (2 мл) і додають карбонат калію (30 мг, 0,2 ммоль). Суміш перемішують при кімнатній температурі протягом 18 годин. Потім додають до суміші воду і утворений осад збирають, одержуючи 70 мг (57 %) сполуки k. Дихлороцтову кислоту (0,05 мл, 0,226 ммоль) додають до суміші сполуки j (63 мг, 0,113 ммоль), бензилтриетиламонійброміду (93 мг, 0,34 ммоль) та нітриту натрію (156 мг, 2,26 ммоль) в дибромметані (5 мл). Суміш перемішують при кімнатній температурі протягом 18 годин в темряві. Реакційну суміш потім концентрують та утворений залишок очищають препаративною ТШХ (95 % дихлорметану/5 % метанолу), одержуючи 13,8 мг сульфонової кислоти та 2 мг названої в заголовку сполуки I. 2-[5-Бром-4-(4-циклопропіл-2-метилнафталін1-іл)-4Н-[1,2,4]-триазол-3-ілсульфаніл]-N-(2-хлор4-сульфамоїлфеніл)ацетамід NH2 NH2 a Br NCS NH2 b c N H2N N Cl HN SH H2N Cl + N N N H N S O N H N S Cl O S NH2 g Cl O S NH2 h N O O H2 N S O f e Br d O N N O O O 31 До перемішуваного розчину 2-метил-1амінонафталіну а (7,5 г, 47,7 ммоль) в тетрагідрофурані (225 мл) додають N-бромсукцинімід (10 г, 56,2 ммоль) при 0 °С. Суміш перемішують при кімнатній температурі протягом 4 годин. Додають до суміші воду і продукт екстрагують етилацетатом. Органічний шар промивають водою та розсолом, осушують над сульфатом натрію, фільтрують та концентрують при зниженому тиску. Одержаний залишок очищають хроматографією на колонці (75 % гексану/25 % етилацетату), одержуючи 4,73 г, вихід 42 %, сполуки b. До розчину b (1 г, 4,24 ммоль), циклопропілборонової кислоти (472 мг, 5,5 ммоль), фосфату калію (3,14 г, 14,8 ммоль) та трициклогексилфосфіну (118 мг, 0,42 ммоль) в толуолі (22 мл) та воді (0,85 мл) під атмосферою азоту додають ацетат паладію (47 мг, 0,21 ммоль). Суміш нагрівають до 100 °С протягом 3 год., а потім охолоджують до кімнатної температури. Додають воду і суміш екстрагують етилацетатом, осушують над сульфатом натрію та концентрують. Очищення хроматографією на колонці (90 % гексану/10 % етилацетату) дає сполуку с (728 мг, вихід 87 %). Сполуку с (728 мг, 3,7 ммоль) розчиняють в 18 мл дихлорметану. Додають бікарбонат натрію (9 мл, насич. розчин) та тіофосген (0,28 мл, 3,7 ммоль) і суміш перемішують при кімнатній температурі протягом 1 год. Потім органічний шар відокремлюють, осушують над сульфатом натрію та концентрують, одержуючи 877 мг, вихід 99 %, сполуки d, яку використовують на наступній стадії без додаткового очищення. 97139 32 Сполуку d (877 мг, 3,7 ммоль) розчиняють в 6 мл диметилформаміду, додають хлористоводневу сіль аміногуанідину (608,5 мг, 5,5 ммоль) та діізопропілетиламін (1,0 мл, 5,5 ммоль) і суміш перемішують при 50 °С протягом 18 годин. Суміш концентрують і до одержаного залишку додають 2М водний розчин гідроксиду натрію (15 мл). Суміш перемішують при 50 °С протягом 18 годин, а потім охолоджують до кімнатної температури. Одержану суміш потім нейтралізують водним 1Н НСl і осад збирають, одержуючи сполуку е (472 мг, вихід 50 %). Сполуки е (100 мг, 0,34 ммоль) та f (96 мг, 0,34 ммоль) розчиняють в ДМФ (2 мл) і додають карбонат калію (51 мг, 0,37 ммоль). Суміш перемішують при кімнатній температурі протягом 18 годин. Потім додають до суміші воду і осад, що утворився, збирають та очищають препаративною ТШХ (90 % дихлорметану/10 % метанолу), одержуючи 83 мг, вихід 45 %, сполуки g. Дихлороцтову кислоту (0,03 мл, 0,31 ммоль) додають до суміші сполуки g (83 мг, 0,15 ммоль), бензилтриетиламонійброміду (125 мг, 0,46 ммоль) та нітриту натрію (211 мг, 3,06 ммоль) в дибромметані (5 мл). Суміш перемішують при кімнатній температурі протягом 18 годин в темряві. Реакційну суміш потім концентрують і одержаний залишок очищають препаративною ТШХ (95 % дихлорметану/5 % метанолу), одержуючи 55,7 мг сульфонової кислоти та 7 мг названої в заголовку сполуки h. 2-[5-Бром-4-(2-хлор-4-циклопропілфеніл)-4Н[1,2,4]-триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід 33 97139 34 O NH2 O HN NH2 HN Cl Cl Br Cl Cl Br a b d c N NCS H2N O N N Cl Cl HN SH Cl Cl + O H2N S O e N H2 N N N H N S Cl g f N Cl Br O O S O N N H N S Cl O Сполуку а (1 г, 4,8 ммоль) розчиняють в 10 мл безводного метиленхлориду. До цієї суміші додають триетиламін (0,68 мл, 4,8 ммоль) і реакцію перемішують при кімнатній температурі протягом 5 хв. Потім додають ацетилхлорид (0,5 мл, 7,2 ммоль) при 0 °С і суміш перемішують при кімнатній температурі протягом 2 годин. Додають воду та дихлорметан і шари розділяють. Органічний шар потім осушають над сульфатом натрію та концентрують, одержуючи 1,11 г, вихід 92 %, сполуки b. До розчину b (500 мг, 2,01 ммоль), циклопропілборонової кислоти (225 мг, 2,62 ммоль), фосфату калію (1,49 г, 7,04 ммоль) та трициклогексилфосфіну (56 мг, 0,2 ммоль) в толуолі (10 мл) та воді (0,4 мл) під атмосферою азоту додають ацетат паладію (23 мг, 0,1 ммоль). Суміш нагрівають до 100 °С протягом 3 год., а потім охолоджують до кімнатної температури. Додають воду і суміш екстрагують етилацетатом, осушують над сульфатом натрію та концентрують, одержуючи 550 мг сирового продукту с, який використовують на наступній стадії без додаткового очищення. Сполуку с (500 мг, 2,4 ммоль) розчиняють в 4 мл етанолу. Додають водну 1Н НСl (4 мл) і суміш перемішують при кип'ятінні зі зворотним холодильником протягом 8 годин. Розчинник видаляють під вакуумом, одержуючи 440 мг сполуки d, яку використовують на наступній стадії без додаткового очищення. Сполуку d (440 мг, 2,6 ммоль) розчиняють в 14 мл дихлорметану. Додають бікарбонат натрію (7 мл, насич. розчин) та тіофосген (0,2 мл, 2,6 O S O NH2 NH2 h Cl i ммоль) і суміш перемішують при кімнатній температурі протягом 1 год. Потім органічний шар відокремлюють, осушують над сульфатом натрію та концентрують, одержуючи 877 мг, вихід 99 %, сполуки е, яку використовують на наступній стадії без додаткового очищення. Сполуку е (447 мг, 2,1 ммоль) розчиняють в 3 мл диметилформаміду, додають хлористоводневу сіль аміногуанідину (355 мг, 3,2 ммоль) та діізопропілетиламін (0,56 мл, 3,2 ммоль) і суміш перемішують при 50 °С протягом 18 годин. Суміш потім концентрують та до одержаного залишку додають 2М водний розчин гідроксиду натрію (10 мл). Суміш перемішують при 50 °С протягом 18 годин, а потім охолоджують до кімнатної температури. Одержану суміш потім нейтралізують водною 1Н НСl і осад (продукт) збирають, одержуючи сполуку f (240 мг, вихід 44 %). Сполуки f (89 мг, 0,33 ммоль) та g (94 мг, 0,33 ммоль) розчиняють в ДМФ (1,5 мл) і додають карбонат калію (51 мг, 0,37 ммоль). Суміш перемішують при кімнатній температурі протягом 18 годин. Потім додають до суміші воду і утворений осад збирають та очищають препаративною ТШХ (90 % дихлорметану/10 % метанолу), одержуючи 116 мг, вихід 68 %, сполуки h. Дихлороцтову кислоту (0,04 мл, 0,46 ммоль) додають до суміші сполуки h (116 мг, 0,23 ммоль), бензилтриетиламонійброміду (183 мг, 0,68 ммоль) та нітриту натрію (304 мг, 4,6 ммоль) в дибромметані (5 мл). Суміш перемішують при кімнатній температурі протягом 18 годин в темряві. Реакційну суміш потім концентрують та оде 35 97139 ржаний залишок очищають препаративною ТШХ (95 % дихлорметану/5 % метанолу), одержуючи 99,10 мг сульфонової кислоти та 17,90 мг названої в заголовку сполуки i. NH2 36 4-(2-(5-Бром-4-(2-хлор-4-циклопропіл-6метилфеніл)-4Н-1,2,4-триазол-3-ілтіо)ацетамідо)3-хлорбензойна кислота NCS NH2 Cl Cl Cl Br 1 N H2 N 3 2 O N N N N Cl HN SH Cl H2N Cl + N H N S Cl O Cl COOH COOH 6 5 4 N N Br N H N S Cl Cl O COOH 7 До розчину 1 (1 г, 4,5 ммоль), циклопропілборонової кислоти (506 мг, 5,9 ммоль), фосфату калію (3,34 г, 15,8 ммоль) та трициклогексилфосфіну (126 мг, 0,45 ммоль) в толуолі (20 мл) та воді (0,76 мл) під атмосферою азоту додають ацетат паладію (51 мг, 0,23 ммоль). Суміш нагрівають до 100 °С протягом 3 год., а потім охолоджують до кімнатної температури. Додають воду і суміш екстрагують етилацетатом, осушують над сульфатом натрію та концентрують, одержуючи 775 мг сирового 2-хлор-4-циклопропіл-6метилбензоламіну (2), який використовують на наступній стадії без додаткового очищення. Сполуку 2 (775 мг, 4,3 ммоль) розчиняють в 9 мл дихлорметану. Додають бікарбонат натрію (4,5 мл, насич. розчин) та тіофосген (0,33 мл, 4,3 ммоль) і суміш перемішують при кімнатній температурі протягом 1 год. Потім органічний шар відокремлюють, осушують над сульфатом натрію та концентрують, одержуючи 935 мг 1-хлор-5циклопропіл-2-ізотіоціанато-3-метилбензолу (3), який використовують на наступній стадії без додаткового очищення. Сполуку 3 (935 мг, 4,2 ммоль) розчиняють в 5 мл диметилформаміду, додають хлористоводневу сіль аміногуанідину (695 мг, 6,3 ммоль) та діізопропілетиламін (1,1 мл, 6,3 ммоль) і суміш перемішують при 50 °С протягом 18 годин. Суміш потім концентрують і до одержаного залишку додають 2М водний розчин гідроксиду натрію (20 мл). Суміш перемішують при 50 °С протягом 18 годин, а потім охолоджують до кімнатної температури. Одержану суміш потім нейтралізують водною 1Н НСl і осад (продукт) збирають, одержуючи 5-аміно-4-(2-хлор-4-циклопропіл-6метилфеніл)-4Н-1,2,4-триазол-3-тіол (4) (780 мг, вихід 66 %). Сполуку 4 (100 мг, 0,36 ммоль) та 3-хлор-4(2-хлорацетамідо)бензойну кислоту (5) (88 мг, 0,36 ммоль) розчиняють в ДМФ (2 мл) і суміш перемішують при 50 °С протягом 18 годин. Потім додають воду і суміш екстрагують етилацетатом. Органічний шар відокремлюють, осушують над сульфатом натрію та концентрують, одержуючи 192 мг сирової 4-(2-(5-аміно-4-(2-хлор-4циклопропіл-6-метилфеніл)-4Н-1,2,4-триазол-3ілтіо)ацетамідо)-3-хлорбензойної кислоти (6), яку використовують на наступній стадії без додаткового очищення. Дихлороцтову кислоту (0,065 мл, 0,78 ммоль) додають до суміші сполуки 6 (192 мг, 0,39 ммоль), бензилтриетиламонійброміду (318 мг, 1,17 ммоль) та нітриту натрію (538 мг, 7,8 ммоль) в дибромметані (10 мл). Суміш перемішують при кімнатній температурі протягом 18 годин в темряві. Реакційну суміш потім концентрують і одержаний залишок очищають препаративною ТШХ (95 % дихлорметану/5 % метанолу), одержуючи 88 мг, вихід 42 %, 4-(2-(5-бром-4-(2-хлор-4 37 97139 циклопропіл-6-метилфеніл)-4Н-1,2,4-триазол-3ілтіо)ацетамідо)-3-хлорбензойної кислоти (7). 38 4-[2-(5-Бром-4-(4-циклопропілнафталін-1-іл)4Н-1,2,4-триазол-3-ілтіо)ацетамідо]-3хлорбензойна кислота NH2 NH2 NCS Br 1 2 N N H2N N 3 SH Cl N N O Cl HN H2N H N S N O Cl COOH + COOH 4 6 5 Cl N N Br N H N S O COOH 7 До розчину 1 (500 мг, 2,01 ммоль), циклопропілборонової кислоти (225 мг, 2,62 ммоль), фосфату калію (1,49 г, 7,04 ммоль) та трициклогексилфосфіну (56 мг, 0,2 ммоль) в толуолі (10 мл) та воді (0,4 мл) під атмосферою азоту додають ацетат паладію (23 мг, 0,1 ммоль). Суміш нагрівають до 100 °С протягом 3 год., а потім охолоджують до кімнатної температури. Додають воду і суміш екстрагують етилацетатом, осушують над сульфатом натрію та концентрують, одержуючи 550 мг сирового 4-циклопропілнафталін-1-аміну (2), який використовують на наступній стадії без додаткового очищення. Сполуку 2 (440 мг, 2,6 ммоль) розчиняють в 14 мл дихлорметану. Додають бікарбонат натрію (7 мл, насич. розчин) та тіофосген (0,2 мл, 2,6 ммоль) і суміш перемішують при кімнатній температурі протягом 1 год. Потім органічний шар відокремлюють, осушують над сульфатом натрію та концентрують, одержуючи 877 мг, вихід 99 %, 1циклопропіл-4-ізотіоціанатонафталіну (3), який використовують на наступній стадії без додаткового очищення. Сполуку 3 (447 мг, 2,1 ммоль) розчиняють в 3 мл диметилформаміду, додають хлористоводневу сіль аміногуанідину (355 мг, 3,2 ммоль) та діізопропілетиламін (0,56 мл, 3,2 ммоль) і суміш перемішують при 50 °С протягом 18 годин. Суміш потім концентрують та до одержаного залишку додають 2М водний розчин гідроксиду натрію (10 мл). Суміш перемішують при 50 °С протягом 18 годин, а потім охолоджують до кімнатної температури. Одержану суміш потім нейтралізують водною 1Н НСl і осад (продукт) збирають, одержуючи 5-аміно-4-(4-циклопропілнафталін-1-іл)4Н-1,2,4-триазол-3-тіол (4) (240 мг, вихід 44 %). Сполуку 4 (789 мг, 2,79 ммоль) та 3-хлор-4(2-хлорацетамідо)бензойну кислоту (5) (693 мг, 2,79 ммоль) розчиняють в ДМФ (6 мл) і суміш перемішують при 50 °С протягом 18 годин. Потім додають воду і суміш екстрагують етилацетатом. Органічний шар відокремлюють, осушують над сульфатом натрію та концентрують, одержуючи 1,04 г, вихід 75 %, 4-(2-(5-аміно-4-(4циклопропілнафталін-1-іл)-4Н-1,2,4-триазол-3ілтіо)ацетамідо)-3-хлорбензойної кислоти (6). Дихлороцтову кислоту (0,35 мл, 4,2 ммоль) додають до суміші сполуки 6 (1,04 г, 2,1 ммоль), бензилтриетиламонійброміду (1,65 г, 6,1 ммоль) та нітриту натрію (2,9 г, 42,1 ммоль) в дибромметані (44 мл). Суміш перемішують при кімнатній температурі протягом 18 годин в темряві. Реакційну суміш потім концентрують і одержаний залишок очищають хроматографією на колонці (95% дихлорметану/5 % метанолу), одержуючи 393 мг, вихід 34 %, 4-(2-(5-бром-4-(4циклопропілнафталін-1-іл)-4Н-1,2,4-триазол-3ілтіо)ацетамідо)-3-хлорбензойної кислоти (7). 4-(2-(5-Бром-4-(7-метокси-4-метилнафталін1-іл)-4Н-1,2,4-триазол-3-ілтіо)ацетамідо)-3хлорбензойна кислота 39 97139 40 Ph N NH2 NH2 OH OH 1 Bn OMe 2 Bn Bn N OMe CHO N N N MeO Bn NH2 N NCS OMe OMe 4 H2N 3 5 6 O SH OMe Cl HN 7 Cl N N H2 N + N H N S O MeO COOH COOH 8 10 9 N Br Cl N H N S N O MeO COOH 11 Суміш 8-аміно-2-нафтолу 1 (8,2 г, 52 ммоль), бензальдегіду (16 мл, 156 ммоль) та сульфату натрію (41,3 г, 291 ммоль) в ТГФ (100 мл) перемішують при кипінні зі зворотним холодильником протягом ночі. Суміш охолоджують до кімнатної температури, фільтрують та концентрують при зниженому тиску. Одержаний залишок очищають хроматографією на колонці (гексан/етилацетат/триетиламін 75/23/2), одержуючи 12,65 г неочищеного (Е)-8(бензиліденаміно)нафталін-2-олу (2), який використовують на наступній стадії без додаткового очищення. Суміш 2 (12,65 г, 51,2 ммоль), Mel (6,4 мл, 102 ммоль) та NaOH (6,14 г, 153 ммоль) в ацетоні (125 мл) перемішують при кімнатній температурі протягом 2 годин. Одержану суміш концентрують та залишок розчиняють в простому ефірі, промивають водою та розсолом і концентрують. Одержаний залишок розчиняють в 2Н НСl-ТГФ (780 мл, 2:1) та перемішують при кімнатній температурі протягом 1,5 год. Одержаний розчин промивають ефіром, водний шар підлуговують Na2CO3 та екстрагують ефіром. Органічний шар промивають розсолом, осушують над сульфатом натрію та концентрують. Одержаний залишок очищають хроматографією на колонці (Нех/ЕtOАс 3:1), одержуючи 6,94 г, вихід 78 %, 7метоксинафталін-1-аміну (3). До перемішуваної суміші 3 (6,94 г, 40 ммоль) та карбонату калію (16,6 г, 120 ммоль) в ацетоні (100 мл) додають бензилбромід (19,0 мл, 160 ммоль) при 0 °С. Суміш кип'ятять під зворотним холодильником протягом 3 днів і охолоджують до кімнатної температури. Осад видаляють і фільтрат концентрують. Одержаний залишок очищають хроматографією на колонці (гекс. 100 %) для видалення бензилброміду, що не прореагував, а потім етилацетатом (100 %), одержуючи 11,75 г, вихід 83 %, N,N-дибензил-7-метоксинафталін-1аміну (4). До перемішуваного розчину ДМФ (30 мл) додають РОСl3 (10,65 мл, 116 ммоль) протягом 30 хвилин при 0 °С. Суміш потім перемішують при 0°С протягом 30 хвилин та додають сполуку 4 (11,75 г, 33,2 ммоль) в ДМФ (120 мл). Суміш перемішують при кімнатній температурі протягом шести днів та виливають у воду з льодом. Суміш продукту екстрагують дихлорметаном і органічний шар промивають водою, водним бікарбонатом натрію та розсолом, осушують над сульфатом натрію та концентрують, одержуючи 13,58 г 4-(дибензиламіно)-6-метокси-1-нафтальдегіду (5), який використовують на наступній стадії без додаткового очищення. Суміш 5 (5,0 г, 13,1 ммоль) та Pd/вугілля (812мг) в метанолі (150 мл) перемішують під атмосферою водню (276 кПа, 40 PSI) протягом 18 41 97139 годин. Суміш пропускають через целіт та концентрують. Одержаний залишок очищають хроматографією на колонці (Нех/ЕtOАс 3:1), одержуючи 826 мг, вихід 35 %, 7-метокси-4-метилнафталін-1аміну (6).Сполуку 6 (826 мг, 4,4 ммоль) розчиняють в 25 мл дихлорметану. Додають бікарбонат натрію (15 мл, насич. розчин) та тіофосген (0,34 мл, 4,4 ммоль) і суміш перемішують при кімнатній температурі протягом 1 год. Потім органічний шар відокремлюють, осушують над сульфатом натрію та концентрують, одержуючи 1,9 г, вихід 99 %, 4-ізотіоціанато-6-метокси-1метилнафталіну (7), який використовують на наступній стадії без додаткового очищення. Сполуку 7 (1,0 г, 4,4 ммоль) розчиняють в 10мл диметилформаміду, додають хлористоводневу сіль аміногуанідину (723 мг, 6,5 ммоль) та діізопропілетиламін (1,14 мл, 6,5 ммоль) і суміш перемішують при 50 °С протягом 18 годин. Суміш потім концентрують і до одержаного залишку додають 2М водний розчин гідроксиду натрію (10 мл). Суміш перемішують при 50 °С протягом 18 годин, а потім охолоджують до кімнатної температури. Одержану суміш потім нейтралізують водною 1Н НСl і осад (продукт) збирають, одержуючи 5-аміно-4-(7-метокси-4-метилнафталін-1іл)-4Н-1,2,4-триазол-3-тіол (8) (1,14 мг, вихід 91%).Сполуку 8 (200 мг, 0,7 ммоль) та 3-хлор-4(2-хлорацетамідо)бензойну кислоту (9) (174 мг, 0,7 ммоль) розчиняють в ДМФ (3 мл) і суміш перемішують при 50 °С протягом 18 годин. Потім додають воду та суміш екстрагують етилацетатом. Органічний шар відокремлюють, осушують над сульфатом натрію та концентрують, одержуючи 304 мг 4-(2-(5-аміно-4-(7-метокси-4метилнафталін-1-іл)-4Н-1,2,4-триазол-3ілтіо)ацетамідо)-3-хлорбензойної кислоти (10), яку використовують на наступній стадії без додаткового очищення. Дихлороцтову кислоту (0,1 мл, 1,2 ммоль) додають до суміші сполуки 10 (304 мг, 0,6 ммоль), бензилтриетиламонійброміду (492 мг, 1,8 ммоль) та нітриту натрію (828 мг, 12 ммоль) в дибромметані (10 мл). Суміш перемішують при кімнатній температурі протягом 18 годин в темряві. Реакційну суміш потім концентрують і одержаний залишок очищають хроматографією на колонці (95 % дихлорметану/5 % метанолу), одержуючи 80 мг, вихід 24 %, 4-(2-(5-бром-4-(7метокси-4-метилнафталін-1-іл)-4Н-1,2,4-триазол3-ілтіо)ацетамідо)-3-хлорбензойної кислоти (11). 2-[5-Бром-4-(4-циклопропілнафталін-1-іл)-4Н[1,2,4]триазол-3-ілсульфаніл]-N-(2-хлор-4-Nпропіонілсульфамоїлфеніл)ацетамід Cl N N Br N NH S O O O S NH O 42 До круглодонної колби на 50 мл завантажують 2-[5-бром-4-(4-циклопропілнафталін-1-іл)-4Н[1,2,4]триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід (45 мг, 0,076 ммоль), EDC (29 мг, 0,15 ммоль) та пропіонову кислоту (6,7 мкл, 0,09 ммоль) в суміші 5 мл ТГФ та 5 мл метиленхлориду. До суміші додають DMAP (18,3 мг, 0,15 ммоль) однією порцією. Реакційну суміш перемішують при кімнатній температурі протягом 14 год. Розчинники випаровують при зниженому тиску, одержуючи в'язкий маслянистий залишок. Залишок знов розчиняють в 20 мл метиленхлориду, потім промивають 20 мл 2,0М водного розчину НСl. Органічний шар осушують над Na2SO4. Розчинник видаляють за допомогою роторного випарника, одержуючи маслянистий залишок. Залишок очищають хроматографією на колонці силікагелю сумішшю метанолу та метиленхлориду (1:9). Одержують 18,5 мг (38 %) бажаного продукту у вигляді білої твердої речовини. 2-[5-Бром-4-(4-циклопропілнафталін-1-іл)-4Н[1,2,4]триазол-3-ілсульфаніл]-N-(2-хлор-4пропіонілсульфамоїлфеніл)лізинамід Cl N N Br N H N S O O O O S NH + NH3Cl NH3 Cl + До круглодонної колби на 25 мл завантажують 2-[5-бром-4-(4-циклопропілнафталін-4-іл)-4Н[1,2,4]триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід (50 мг, 0,085 ммоль), EDC (35 мг, 0,18 ммоль), Boc-Lys(Boc)-OH DCHA (47 мг, 0,09 ммоль) в суміші 5 мл ТГФ та 5 мл метиленхлориду. До суміші додають DMAP (16 мг, 0,13 ммоль) однією порцією. Реакційну суміш перемішують при кімнатній температурі протягом 14 год. Розчинники випарюють при зниженому тиску, одержуючи в'язкий маслянистий залишок. Залишок розчиняють в 5 мл 4,ОМ НСl в діоксані. Реакцію перемішують при кімнатній температурі протягом 14 год. Розчинник випарюють при зниженому тиску, одержуючи в'язкий маслянистий залишок. Залишок промивають по черзі 10 мл метиленхлориду та 10 мл ефіру, одержуючи названу в заголовку сполуку у вигляді світло-жовтої твердої речовини (44 мг, 65 %). Реагенти 1-Метил-4-нітронафталін 43 NO2 97139 44 H2 N HN До 1-метилнафталіну (8,0 г, 56 ммоль) в круглодонній колбі при 0 °С додають азотну кислоту (26 мл) по краплях. (ПРИМІТКА: Повільне додавання азотної кислоти є дуже важливим для запобігання утворенню інших регіоізомерів). Після перемішування реакційної суміші протягом ще 15хв. при 0 °С її виливають в 65 мл Н2О. Водний розчин двічі екстрагують бензолом і об'єднаний бензольний розчин промивають 10 % розчином NaOH, осушають Na2SO4 та концентрують. Хроматографія на силікагелі (ЕtOАс:гексани = 5:95) дає продукт, який містить ще кілька процентів іншого регіоізомеру. Його перекристалізовують з ЕtOАс/гексанів, одержуючи 9,0 г (43 %) 1. 4-Метилнафталін-1-іламін NH2 До розчину 1-метил-4-нітронафтиламіну (4,0 г, 21 ммоль) в етанолі (300 мл) додають нікель Ренея (4 човника для зважування). Суміш перемішують під Н2 (1 атм) протягом 16 год. Реакцію фільтрують крізь шар целіту та концентрують. Очищення флеш-хроматографією на колонці із силікагелем (ЕtOАс:гексани = 15:85) дає продукт (3,2 г, 75 %). 4-Етил-5,6,7,8-тетрагідронафталін-1-іламін S До розчину тіофосгену (0,33 мл, 4,3 ммоль) в безводному метиленхлориді (5 мл) при 0 °С додають по краплях розчин 4-метилнафтиламіну (671 мг, 4,3 ммоль) та діізопропілетиламіну (1,5 мл, 8,6 ммоль) в безводному метиленхлориді (5 мл). Після перемішування реакційної суміші протягом ще 10 хв. при 0 °С, її промивають 1 % розчином НСl, а потім Н2О, осушують Na2SO4 та концентрують, одержуючи темно-коричневу маслянисту рідину. Маслянисту рідину розчиняють в гексанах (15 мл) і одержану коричневу суспензію фільтрують. Фільтрат концентрують, одержуючи чистий тіоізоціанат. До розчину тіоізоціанату в безводному ацетонітрилі (20 мл) додають гідразин (0,13 мл, 4,3 ммоль) при кімнатній температурі. Після перемішування при кімнатній температурі протягом 20 хв. суміш концентрують. Одержану маслянисту рідину жовтого кольору розтирають з ЕtOАс:гексанами (1:1), одержуючи (701 мг, вихід 7 1%) продукту у вигляді білуватої твердої речовини. 5-Дифторметил-4-(4-метилнафталін-1-іл)-4Н[1.2.4]триазол-3-тіол N N F2HC N SH NH2 Процедура є по суті ідентичною шляху для 4метилнафталін-1-іламіну, описаному вище, однак, починається з розчину 5-етил-8-нітро-1,2,3,4тетрагідронафталіну (795 мг, 3,95 ммоль). 4-Метилнафталін-1-ілтіосемікарбазид Розчин 4-метилнафтилтіосемікарбазиду (180 мг, 0,78 ммоль) в дифтороцтовій кислоті (2 мл) нагрівають при 100 °С протягом 4 год. Коли суміш охолоджують до кімнатної температури, з реакційної суміші кристалізується біла тверда речовина. Щоб зібрати більшу кількість продукту, до суміші додають 2 мл гексанів. Фільтрація дає продукт (179 мг, вихід 79 %) у вигляді білої твердої речовини. 5-Фторметил-4-(4-метилнафталін-1-іл)-4Н[1.2.4]триазол-3-тіол 45 97139 46 N N F2HC N Cl N N SH F2 HC N H N S O До розчину 4-метилнафтилтіосемікарбазиду (158 мг, 0,68 ммоль) в МеОН (10 мл) та 4,37М NaOMe (0,23 мл, 1,02 ммоль) додають етилфторацетат (0,13 мл, 1,37 ммоль) та перемішують при кімнатній температурі протягом 17 год. Реакційну суміш концентрують, додають воду і промивають діетиловим ефіром. У водному шарі встановлюють рН за допомогою НСl та збирають на фільтрі продукт у вигляді білої твердої речовини 1 (78 мг, вихід 42 %). Н ЯМР (ДМСО, 300 МГц) 14,26 (с, 1Н), 8,14 (д, J=8,4 Гц, 1Н), 7,67-7,52 (м, 4Н), 7,26 (д, J=8,4 Гц, 1Н), 5,20 (дд, J=12,0, 21,0 Гц, 1Н), 5,03 (дд, J=12,0, 20,4 Гц, 1Н), 2,74 (с, 3Н). 2-[5-Дифторметил-4-(4-метилнафталін-1-іл)4Н-[1,2,4]триазол-3-ілсульфаніл]-N-(2-метил-4сульфамоїлфеніл)ацетамід N N F2 HC N H N S O До розчину 5-дифторметил-4-(4-етил-5,6,7,8тетрагідронафталін-1-іл)-4Н-[1,2,4]триазол-3тіолу (85 мг, 0,28 ммоль), K2СО3 (41,8 мг, 0,30 ммоль) в ДМФ (2,0 мл) додають 2-хлор-N-(2метил-4-сульфамоїлфеніл)ацетамід (77,8 мг, 0,28 ммоль). Реакційну суміш перемішують при кімнатній температурі протягом ночі. Після закінчення реакції, додають до реакції МеОН та перемішують до утворення осаду і збирають продукт на 1 фільтрі (71,0 мг, вихід 46 %). Н ЯМР (ДМСО, 300 МГц) 5 10,14 (с, 1Н), 8,03 (д, J=8,1 Гц, 1Н), 7,88 (д, J=2,4 Гц, 1Н), 7,74 (дд, J = 2,1, 8,4 Гц, 1Н), 7,46 (шир.с, 2Н), 7,34-7,00 (м, 3Н), 4,33 (уявн. кв, J = 15,6 Гц, 2Н), 2,71 (т, J = 5,7 Гц, 2Н), 2,62 (кв, J=7,5 Гц, 2Н), 2,28-2,08 (м, 2Н), 1,72-1,60 (м, 4Н), 1,19 (т, J=7,5 Гц, 3Н). N-(2-хлор-4-сульфамоїлфеніл)-2-[5дифторметил-4-(4-метилнафталін-1-іл)-4Н[1,2,4]триазол-3-ілсульфаніл]ацетамід N SO2 NH2 F2 HC Cl N N H N S O До розчину 5-дифторметил-4-(4метилнафталін-1-іл)-4Н-[1,2,4]триазол-3-тіолу (53 мг, 0,18 ммоль), K2СО3 (27,0 мг, 0,20 ммоль) в ДМФ (1,5 мл) додають 2-метил-N-(2-метил-4сульфамоїлфеніл)ацетамід (47 мг, 0,18 ммоль). Реакційну суміш перемішують при кімнатній температурі протягом 16 год. Після завершення реакції додають до реакції Н2О (4,0 мл) і перемішують до утворення осаду та збирають продукт на 1 фільтрі (77,0 мг, вихід 83%). Н ЯМР (ДМСО, 300 МГц) 9,84 (шир.с, 1Н), 8,18 (д, J=8,0 Гц, 1Н), 7,70-7,53 (м, 7Н), 7,18 (т, J=51,5 Гц, 1Н), 7,11 (д, J=8,0 Гц, 1Н), 4,26 (с, 2Н), 2,82 (с, 3Н), 2,27 (с, 3Н). N-(2-хлор-4-сульфамоїлфеніл)-2-[5дифторметил-4-(4-етил-5,6,7,8тетрагідронафталін-1-іл)-4Н-[1,2,4]триазол-3ілсульфаніл]ацетамід SO2NH2 SO2 NH2 До розчину 5-дифторметил-4-(4метилнафталін-1-іл)-4Н-[1,2,4]триазол-3-тіолу (59 мг, 0,20 ммоль), K2СО3 (30,0 мг, 0,22 ммоль) в ДМФ (1,5 мл) додають 2-хлор-N-(2-метил-4сульфамоїлфеніл)ацетамід (57 мг, 0,20 ммоль). Реакційну суміш перемішують при кімнатній температурі протягом 16 год. Після завершення реакції додають до реакції Н2О (4,0 мл) та перемішують до утворення осаду і збирають продукт на 1 фільтрі (77,0 мг, вихід 71 %). Н ЯМР (ДМСО, 300 МГц) 10,11 (шир.с, 1Н), 8,18 (д, J=10,0 Гц, 1Н), 8,01 (д, J=10,0 Гц, 1Н), 7,87 (с, 1Н), 7,75-7,54 (м, 5Н), 7,46 (шир.с, 2Н), 7,18 (т, J=50,0 Гц, 1Н), 7,11 (д, J=10,0 Гц, 1Н), 4,32 (с, 2Н), 2,27 (с, 3Н). 2-[5-Фторметил-4-(4-метилнафталін-1-іл)-4Н[1,2,4]триазол-3-ілсульфаніл]-N-(2-метил-4сульфамоїлфеніл)ацетамід 47 N F2 HC 97139 N S N F2 HC SO2 NH2 До розчину 5-фторметил-4-(4метилнафталін-1-іл)-4Н-[1,2,4]триазол-3-тіолу (89 мг, 0,33 ммоль), K2СО3 (50,0 мг, 0,36 ммоль) в ДМФ (2,0 мл) додають 2-хлор-N-(2-метил-4сульфамоїлфеніл)ацетамід (87 мг, 0,33 ммоль). Реакційну суміш перемішують при кімнатній температурі протягом 16 год. Після завершення реакції додають до реакції H2O (2,0 мл) та перемішують до утворення осаду і фільтрують. Очищення оберненофазовою ВЕРХ дає продукт у вигляді твердої речовини (53,3 мг, вихід 50 1 %). Н ЯМР (ДМСО, 300 МГц) 9,84 (шир.с, 1Н), 8,18 (д, J=8,4 Гц, 1Н), 7,71-7,53 (м, 7Н), 7,26 (с, 2Н), 7,10 (д, J=8,7 Гц, 1Н), 5,34 (дд, J=12,0, 27,3 Гц, 1Н), 5,18 (дд, J=12,3, 26,4 Гц, 1Н), 4,22 (с, 2Н), 2,75 (с, 3Н), 2,25 (с, 3Н). N-(2-хлор-4-сульфамоїлфеніл)-2-[5фторметил-4-(4-метилнафталін-1-іл)-4Н[1,2,4]триазол-3-ілсульфаніл]ацетамід N Cl N N Cl N N H N O F2 HC 48 H N S O SO2 NH2 До розчину 5-фторметил-4-(4метилнафталін-1-іл)-4Н-[1,2,4]триазол-3-тіолу (89 мг, 0,33 ммоль), K2СО3 (50,0 мг, 0,36 ммоль) в ДМФ (2,0 мл) додають 2-хлop-N-(2-хлор-4сульфамоїлфеніл)ацетамід (93 мг, 0,33 ммоль). Реакційну суміш перемішують при кімнатній температурі протягом 16 год. Після завершення реакції додають до реакції Н2О (2,0 мл) та перемішують до утворення осаду і фільтрують, одержуючи тверду речовину (126,8 мг, вихід 74 1 %). Н ЯМР (ДМСО, 300 МГц) 10,12 (шир.с, 1Н), 8,18 (д, J = 8,7 Гц, 1Н), 8,04 (дд, J = 4,8, 8,7 Гц), 7,87 (с, 1Н), 7,76-7,52 (м, 5Н), 7,46 (с, 2Н), 7,11 (д, J = 8,7 Гц, 1Н), 5,35 (дд, J = 12,3, 26,7 Гц, 1Н), 5,19 (дд, J = 11,7, 25,8 Гц, 1Н), 4,26 (с, 2Н), 2,75 (с, 3Н). N-(2-хлор-4-сульфамоїлфеніл)-2-[4-(4-етил5,6,7,8-тетрагідронафталін-1-іл)-5-фторметил-4Н[1,2,4]триазол-3-ілсульфаніл]ацетамід N H N S O SO2NH2 До розчину 4-(4-етил-5,6,7,8тетрагідронафталін-1-іл)-5-фторметил-4Н[1,2,4]триазол-3-тіолу (85 мг, 0,29 ммоль), K2СО3 (44,4 мг, 0,32 ммоль) в ДМФ (2,0 мл) додають 2хлор-N-(2-хлор-4-сульфамоїлфеніл)ацетамід (82,6 мг, 0,29 ммоль). Реакційну суміш перемішують при кімнатній температурі протягом 16 год. Після завершення реакції додають до реакції Н2О (2,0 мл) та перемішують до утворення осаду і фільтрують, одержуючи тверду речовину (73,0 1 мг, вихід 47 %). Н ЯМР (ДМСО, 300 МГц) 5 10,15 (с, 1Н), 8,05 (д, J=8,4 Гц, 1Н), 7,87 (с, 1Н), 7,74 (д, J=8,4 Гц, 1Н), 7,46 (с, 2Н), 7,21-7,06 (м, 2Н), 5,26 (д, J = 48,0 Гц. 2Н), 4,29 (уявн.кв, J=15,6 Гц, 2Н), 2,71-2,58 (м, 3Н), 2,25 (с, 1Н), 2,25-2,09 (м, 2Н), 1,72-1,59 (м, 4Н), 1,19 (т, J = 7,5Гц, 3Н). З використанням відповідних вихідних матеріалів, наступні сполуки були одержані за процедурами, аналогічними описаним вище методам: 2-[5-бром-4-(2-хлор-4(циклопропілметил)феніл)-4Н-[1,2,4]-триазол-3ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід, 2-[5-бром-4-(2-хлор-4-циклобутилфеніл)-4Н[1,2,4]-триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід, 2-[5-бром-4-(2-хлор-4(циклопропілметил)нафталін-1-іл)-4Н-[1,2,4]триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід, 2-[5-бром-4-(2-хлор-4-циклопропілфеніл)-4Н[1,2,4]-триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід, 2-[5-трифторметил-4-(2-хлор-4циклопропілнафталін-1-іл)-4Н-[1,2,4]-триазол-3ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід, 2-[5-бром-4-(4-циклопропіл-5,6,7,8тетрагідронафталін-1-іл)-4Н-[1,2,4]-триазол-3ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід, 2-[5-бром-4-(4-етилнафталін-1-іл)-4Н-[1,2,4]триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід, 2-[5-бром-4-(4-етил-5,6,7,8тетрагідронафталін-1-іл)-4Н-[1,2,4]-триазол-3ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід, 2-[5-бром-4-(5-циклопропілхінолін-8-іл)-4Н[1,2,4]-триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід, 2-[5-бром-4-(5-циклопропілізохінолін-8-іл)-4Н[1,2,4]-триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід, 49 2-[5-бром-4-(5-циклопропілхінолін-8-іл)-4Н[1,2,4]-триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід, 2-[5-бром-4-(1-метилаценафтен-5-іл)-4Н[1,2,4]-триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід, 2-[5-бром-4-(2-метилаценафтен-5-іл)-4Н[1,2,4]-триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід, 2-[5-бром-4-(1,1-диметилаценафтен-5-іл)-4Н[1,2,4]-триазол-3-ілсульфаніл]-N-(2-хлор-4сульфамоїлфеніл)ацетамід. Інгібування зворотної транскриптази ВІЛ-1 Проводили скринінг сполук на інгібуючу активність проти вірусу імунодефіциту людини типу 1 (ВІЛ-1) за допомогою високопродуктивного клітинного аналізу з використанням ВІЛ-1, що експресує люциферазу світлячка як репортерний ген, псевдотипованого оболонковим глікопротеїном вірусу езикулярного стоматиту (VSV-G). Експериментальні процедури були по суті такими, як описано Connor et al., Journal of Virology (1996), 70:5306-5311 (Characterization of the functional properties of env genes from long-term survivors of human immunodeficiency virus type 1 infection), та Popik et al., Journal of Virology (2002), 76:47094722 (Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors + for productive entry into CD4 T cells). Слід особливо зазначити, що вірус містить дві інтродуковані мутації гена 3Т (K103N та Y181C, створені ПЛР-мутагенезом), які роблять вірус високорезистентним відносно сучасних ненуклеозидних лікарських засобів проти ВІЛ-1. Вихідний вірусний матеріал був одержаний котрансфекцією плазмідної ДНК, що кодує VSV-G, з вектором pNL43Env(-)Luc(+) в клітини 293Т. Через шістдесят чотири години після трансфекції, вірусовмісне середовище збирають центрифугуванням та зберігають замороженим при -80 °С. Клітини HeLa інфікують VSV-G псевдотипованим вірусом в присутності діагностичної сполуки у форматі 384-ямкового мікротитрувального планшета. Через сорок вісім годин після первинного інфікування додають до клітин лізисний буфер та реагент для аналізу люциферази (Promega) і визначають люциферазну активність підрахунком створеної люмінесценції за допомогою люмінометра LJL. Оскільки ген люциферази переноситься у вірусному геномі, рівень його експресії безпосередньо відображає рівень реплікації вірусу в присутності сполуки. Для оцінки активності сполук проти ВІЛ-1 дикого типу використовували клітинну лінію HeLaJC53, що експресує високі рівні CD4 та CCR5 (Platt et al., Journal of Virology (1998), 72:28552864: Effect of CCR5 and CD4 cell surface concentrations on infection by macrophagetropic isolates of human immunodeficiency virus type 1). Клітинна лінія була модифікована шляхом виділення стабільної клітинної лінії, що експресує люциферазу під контролем промотору ВІЛ-1 (довгий термінальний повтор, тобто ДТП). ВІЛ-1 інфекція цієї клітинної лінії стимулює транскрипцію люциферази з промотору ВІЛ-1 і рівень експресії 97139 50 гена люциферази є пропорційним рівню реплікації вірусу (Harrington et al. у Journal of Virology Methods (2000), 88:111-115: Direct detection of nfection of HIV-1 in blood using a centrifugationindicator cell assay; та Roos et al. у Virology (2000), 273: 307-315: LuSIV cells: a reporter cell line for the detection and quantitation of a single cycle of HIV та SIV replication). Процедури вірусної інфекції, тестування сполук та визначення люциферазної активності були такими самими, як для VSV-G псевдотипованого ВІЛ-1. Були використані два підходи для оцінки цитотоксичності позитивних сполук, виявлених шляхом проведення аналізів на вірус ВІЛ-1. Перший підхід використовував іншу модифіковану клітинну лінію HeLa-JC53, яка конститутивно експресує високий рівень люциферази без вірусної інфекції. Рівень експресії люциферази в цих клітинах служив індикатором реплікації клітин в присутності сполук. Процедури тестування сполук та визначення люциферазної активності були такими саме, як у тестах вірусної інфекції. Інший аналіз токсичності використовував клітини HeLeJC53 та комерційно доступний аналітичний набір MTS (Promega) для вимірювання мітохондріальної функції клітин. З використанням методів, аналогічних описаним вище, були синтезовані 2-[5бром-4-(4-циклопропілнафталін-1-іл)-4Н[1,2,4]триазол-3-ілсульфаніл]-N-(2-метил-4сульфамоїлфеніл)ацетамід та 2-[5-бром-4-(4етилнафталін-1-іл)-4Н-[1,2,4]триазол-3ілсульфаніл]-N-(2-хлор-4-сульфамоїлфеніл)ацетамід, а також N-4-карбамільний аналог, 2-[5бром-4-(4-етилнафталін-1-іл)-4Н-[1,2,4]триазол-3ілсульфаніл]-N-(2-хлор-4карбамоїлфеніл)ацетамід, та N-4-карбоксильний аналог. Кожну сполуку тестували з використанням панелі мутантних зворотних транскриптаз ВІЛ, включаючи 20 з 22 мутантів, які зустрічаються у приблизно 2 % чи більше зразків пацієнтів, резистентних до найбільш широко застосовуваного ненуклеозидного інгібітора 3Т ВІЛ ефавіренцу ((4S)-6-хлор-4-(циклопропілетиніл)-1,4-дигідро4-(трифторметил)-2Н-3,1-бензоксазин-2-ону). Для кожного з 20 тестованих мутантів, що часто зустрічаються, щонайменше одна з цих сполук була більше ніж у 20 разів більш дієвою, ніж ефавіренц, або мала ЕС50 менше ніж 1 нМ. В більшості випадків задовольнялися обидва критерії. В більшості випадків всі три сполуки були більш дієвими, ніж ефавіренц. Сполуки порівнювали на активність відносно мутантних зворотних транскриптаз дикого типу, Y181C та Y188L. Обидва аміди були значно кращими, ніж карбонова кислота, відносно всіх трьох ферментів. Результати Сполуки за винаходом тестували на зворотних транскриптазах ВІЛ дикого типу та чотирьох мутантних. Результати наведені в Таблиці 1 як ЕС50 (нМ) та IC50 (нМ). У Таблиці, А означає значення 100 нМ. Н.в. - не визначено. Кращими сполуками в цьому винаході є ті, що виявляють активність відносно дикого типу (WT) та резистентних мутантів нижче 50 нМ як для ЕС50, так і для IC50 51 97139 52 Таблиця 1 R2 N N O 1 №R А S N R1 Ar A 2 ЕС50WT ЕС50Y181C ЕС50Y188L ІС50WTRT ІС50Y181C ІС50Y188L R (нМ) (нМ) (нМ) (нМ) (нМ) (нМ) Аr HN Н Me S С С С А С С Н А А А А А С Н O 1 CF2H А В С А С С OH O Me HN 2 CF2H O Cl OH Me HN 3 CF2H O Me OH Me 1 № R A 2 EC50 Ar R WT EC50 EC50 IC50 WT IC50 Y181C IC50 Y188L (нМ) Y181C (нМ) Y188L (нМ) RT (нМ) (нМ) (нМ) HN 4 CF2H O Cl H A A A A A C H A C C A C C H A A C A B C A A A A A C OH Me HN 5 CF2H Me O OH Me HN 6 Br Me O OH Me HN 7 Br Me O H OH Me HN 8 Br Cl O OH Me H 53 97139 54 HN 9 Br O H A A A O H Cl A A A A A C H A A A A A B H A A A A A B H A A A A A C H A A A A A C H A B C A A B H A B C A A C H A A C A A C H A A A A A A H B B C A A B O-Na+ Me HN 10 Br Cl OH HN 11 Br O Cl O-Na+ HN 12 Br O Cl O-K+ HN OMe 13 Br O Cl OH HN 14 CF2H O Cl OH HN Me O 15 Br Cl S OH O HN 16 CF3 O Cl OH Me HN 17 CH2F O Cl OH Me HN OMe 18 Br O Cl OH Me OMe 19 Br HN O Cl S O OH 55 97139 56 HN O 20 Br Br S H C C C A A B H B C C A A C H A A B A A B H A A A A A C H C C C A B C H A A A A A B H A A A A A C H A A C A B C H A A A A A C H A A C A B C OH O Me Cl HN O 21 Br Cl S OH O HN 22 Br O Br OH Me HN Me 23 Br O Cl OH F3 C HN O O 24 Br Cl S O OH HN Cl 25 Br O Cl OH HN 26 Br O Cl OH HN 27 Br Cl OH OMe Me 28 Br Cl O HN O Cl OH HN Me 29 Br Cl O OH 57 F3C 97139 58 HN 30 Br H A A C A B C H A A C A A C H C C C C C C H A A C A A C H A A A H A H A A A A A C H O Cl A A A A A B H A A A A A C H A A A A A B OH HN Cl 31 Br O Cl OH HN 32 Br O Cl OH O Me Me 33 Br HN O Cl OH OMe 34 Br HN O Cl OH HN 35 Br O Me A OH HN 36 CF2H O Cl S NH2 O HN 37 Br O Cl S O NH2 HN 38 Br O Me S O NH2 HN 39 Br O Cl S O NH2 59 97139 60 HN O 40 Br Me S H A A A A A C H A A C A C C H A A A A A C H A A A A B C H A A A A A C H A A B A C C H A A A A B C H A A A A A C H A A B A A C H A A C A B C NH2 O HN O 41 CH3 Me S NH2 O Me HN O 42 Br Cl S NH2 O HN O 43 CH3 Cl S NH2 O Me HN O 44 CF2H Cl S NH2 O HN O 45 CH3 Me S NH2 O Me HN O 46 CH3 Cl S NH2 O Me HN O 47 CF2H Cl S O NH2 Me HN O 48 CF2H Me S O NH2 Me HN O 49 CFH2 Me S O Me NH2
ДивитисяДодаткова інформація
Назва патенту англійськоюNormal;heading 1;heading 2;heading 3;2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio)acetic acid and methyl ester
Автори англійськоюGirardet Jean-luc, Koh Yung-Hyo
Назва патенту російською2-(5-бром-4-(4-циклопропилнафталин-1-ил)-4н-[1,2,4]-триазол-3-илсульфанил)уксусная кислота и ее метиловый эфир
Автори російськоюЖирарде Жан-Люк, Кох Юнг-Хио
МПК / Мітки
МПК: A61K 31/4196
Мітки: метиловий, кислота, ефір, 2-(5-бром-4-(4-циклопропілнафталін-1-іл)-4н-[1,2,4]-триазол-3-ілсульфаніл)оцтова
Код посилання
<a href="https://ua.patents.su/42-97139-2-5-brom-4-4-ciklopropilnaftalin-1-il-4n-124-triazol-3-ilsulfaniloctova-kislota-ta-metilovijj-efir.html" target="_blank" rel="follow" title="База патентів України">2-(5-бром-4-(4-циклопропілнафталін-1-іл)-4н-[1,2,4]-триазол-3-ілсульфаніл)оцтова кислота та її метиловий ефір</a>
Метиловий ефір 2-тіазоліламіду цитраконової кислоти, який проявляє аналгетичну активність

Номер патенту: 48067
Опубліковано: 15.08.2002
Автори: Дроговоз Світлана Мефодіївна, Кабачний Володимир Іванович, Макаренко Ольга Миколаївна, Яковлєва Лариса Василівна, Томаровська Тетяна Олександрівна
МПК: A61P 23/00, C07D 277/46, A61K 31/426
Мітки: проявляє, кислоти, 2-тіазоліламіду, метиловий, анальгетичну, цитраконової, активність, ефір
Формула / Реферат:
Метиловый эфир 2-тиазолиламида цитраконовой кислоты формулы,проявляющий анальгетическую активность.
Метиловий ефір 3,5-дибром-4-амінобензолсульфонілоксамінової кислоти, який проявляє аналгетичну та протизапальну активність

Номер патенту: 48005
Опубліковано: 15.08.2002
Автори: Банний Іван Прокопович, Дроговоз Світлана Мефодіївна, Снітковський Євген Львович, Зупанець Ігор Альбертович, Черних Валентин Петрович
МПК: A61P 29/00, A61P 25/04, A61K 31/63, C07C 211/46
Мітки: кислоти, проявляє, метиловий, 3,5-дибром-4-амінобензолсульфонілоксамінової, ефір, анальгетичну, протизапальну, активність
Формула / Реферат:
Метиловый эфир 3,5-дибром-4-аминобензолсульфонилоксаминовой кислоты формулыпроявляющий анальгетическую и противовоспалительную активность.
Метиловий ефір 2-лейцино-3-хлор-1,4-нафтохінону, який має властивості фунгіцида, бактерицида та регулятора росту рослин

Номер патенту: 23440
Опубліковано: 02.06.1998
Автори: Марінцова Наталя Генадіївна, Новіков Володимир Павлович, Карабанов Юрій Вікторович, Дяк Юрій Платонович, Картофліцька Алевтина Павлівна, Мусич Олена Григорівна
МПК: A01P 21/00, C07C 50/00, A01P 3/00, A01N 29/00
Мітки: рослин, властивості, метиловий, бактерицида, має, ефір, росту, фунгіцида, 2-лейцино-3-хлор-1,4-нафтохінону, регулятора
Формула / Реферат:
Метиловый эфир 2-лейцино-3-хлор-1,4-нафтохинона, формулы:обладающий свойствами фунгицида, бактерицида и регулятора роста растений.
Спосіб одержання 3-арил-n-(2н-1,2,4-триазол-3-іл)-3-(2н-1,2,4-триазол-3-іламіно)пропіонамідів

Номер патенту: 58768
Опубліковано: 15.08.2003
Автори: Широбокова Марія Георгіївна, Десенко Сергій Михайлович, ЛІПСОН ВІКТОРІЯ ВІКТОРІВНА, КАРНОЖИЦЬКА ТЕТЯНА МИХАЙЛІВНА
МПК: C07C 231/00, C07D 249/08
Мітки: одержання, спосіб, 3-арил-n-(2н-1,2,4-триазол-3-іл)-3-(2н-1,2,4-триазол-3-іламіно)пропіонамідів
Формула / Реферат:
Спосіб одержання 3-арил-N-(2Н-1,2,4-триазол-3-іл)-3-(2Н-1,2,4-триазол-3-іламіно)пропіонамідів шляхом взаємодії 2,2-диметил-1,3-діоксан-4,6-діону, 3-аміно-1,2,4-триазолу та бензальдегідів, який відрізняється тим, що реакцію проводять при температурі 100-130°С у відсутності розчинника до утворення кристалічного осаду.
5-бром-n-(2′-карбокси-4′-бромфеніл)антранілова кислота, яка проявляє анальгетичну, протизапальну, діуретичну та протигрибкову активність

Номер патенту: 79903
Опубліковано: 25.07.2007
Автори: Чикіна Олена Леонідівна, Ісаєв Сергій Григорович, Жегунова Галина Петрівна, Кобзар Наталія Петрівна, Бевз Наталія Юріївна
МПК: C07C 229/18, A61K 31/185, C07C 229/00
Мітки: проявляє, протизапальну, протигрибкову, яка, анальгетичну, 5-бром-n-(2'-карбокси-4'-бромфеніл)антранілова, активність, діуретичну, кислота
Формула / Реферат:
5-бром-N-(2'-карбокси-4'-бромфеніл)антранілова кислота формули, яка проявляє анальгетичну, протизапальну, діуретичну та протигрибкову активність.