Гетероциклічний ароматичний амід, фунгіцидна суміш на його основі та спосіб контролю або попередження грибкових уражень
Номер патенту: 73519
Опубліковано: 15.08.2005
Автори: Моррісон Ірен Мае, Мейєр Кевін Джеральд, Рікс Майкл Джон, Месел Джон Луіс, Роджерс Річард Брюер, Йао Ченглін, Фітцпатрік Джина Марі, Дент Вілльям Хантер ІІІ, Найаз Нурмохамед Мохамед, Надер Бассам Салім, Гаєвскі Роберт Пітер







Формула / Реферат
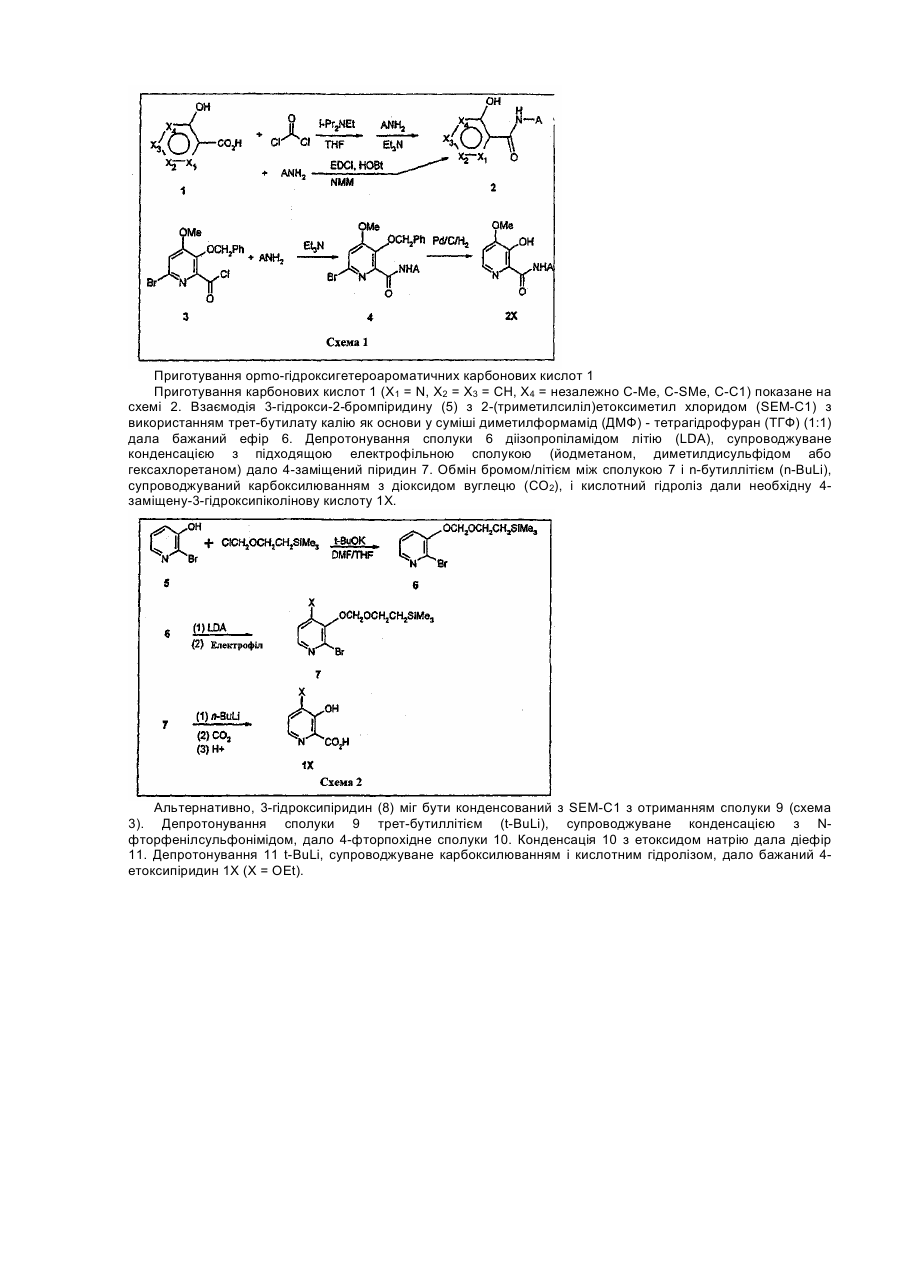
1. Гетероциклічний ароматичний амід формули І:
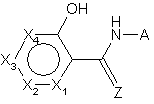 , (I)
, (I)
де
a) 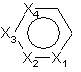 є 5- або 6-членне гетероциклічне ароматичне кільце, в якому
є 5- або 6-членне гетероциклічне ароматичне кільце, в якому
і) кожен з Х1-Х4 є незалежно О, S, NR', N, CR'' або зв'язком;
іі) не більше ніж один із Х1-Х4 є О, S або NR';
ііі) не більше ніж один із Х1-Х4 є зв'язком;
iv) коли будь-який один із Х1-Х4 є S, О або NR', один із сусідніх Х1-Х4 повинен являти собою зв'язок; і
v) щонайменше один з Х1-Х4 повинен бути О, S, NR' або N;
де
R' є Н, С1-С3 алкіл, арил; і
R'' незалежно є Н, галоген, гідрокси, С1-С3 алкіл, С1-С3 алкокси, С1-С3 алкілтіо, де сусідні R'' замісники можуть формувати кільце або сусідні R' і R'' замісники можуть формувати кільце;
b) Z є О,
c) А є
і) С2-С14 алкеніл або С2-С14 алкініл, кожен з яких може бути розгалуженим або нерозгалуженим, незаміщеним або заміщеним галогеном, ароїлом, арилокси, С1-С8 ацилокси, арилтіо, арилом, гетероарилом, гетероарилтіо, гетероарилокси, С1-С6 ацилом,
іі) С3-С14 циклоалкіл, що містить 0-3 гетероатоми і 0-2 ненасичені зв'язки, який заміщений арилокси, гетероарилокси, С1-С6 алкілтіо, арилтіо, гетероарилтіо, С1-С6 алкокси, С1-С6 галоалкокси, оксо,
ііі) С6-С14 бі- або трициклічну кільцеву систему, що містить 0-3 гетероатоми і 0-2 ненасичені зв'язки, яка може бути незаміщеною або заміщеною арилокси, С1-С6 алкокси, С1-С8 ацилокси, арилом, С1-С6 ацилом, карбоарилокси,
iv) гетероарил, який заміщений С1-С6 алкілом, С1-С6 галоалкілом, С3-С6 циклоалкілом, арилом, С1-С6 алкокси, арилтіо, карбоарилокси, карбогетероарилокси, С1-С6 карбоалкокси або амідо, незаміщеним або заміщеним однією або двома С1-С6 алкільними групами, ОС(O)арилом, в якому будь-який алкіл або циклоалкіл, що містить замісник, може бути заміщений одним або більше галогенами, і в якому будь-який арил або гетероарил, що містить замісник, може бути також незаміщеним або заміщеним галогеном, ціано, арилокси, арилом, С1-С6 галоалкілом, С1-С6 алкокси, С1-С6 галоалкокси,
v)
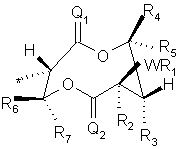 , де * = точка прикріплення,
, де * = точка прикріплення,
в якій Q1, О2 є О;
W є СН2, СНR6, або зв'язком;
R1 є С1-С8 алкілом, С2-С8 алкенілом, С2-С8 алкінілом, С3-С8 циклоалкілом, арилом або гетероарилом;
R2 є Н, С1-С3 алкілом, С2-С5 алкенілом або С2-С5 алкінілом;
R3 є Н, R1, OR1, OC(O)R1, OC(O)OR1 або OC(O)NR1R6;
R4 і R5 є незалежно Н, С1-С6 алкілом або С2-С6 алкенілом, за умови, що сума вуглеців для R4 плюс R5 становить шість або менше, і, далі, за умови, що R4 іR5 можуть бути з'єднані в С3-С6 кільце;
R6, і R7 є незалежно Н;
із застереженням, що коли
є
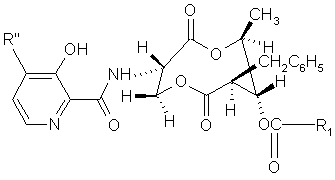 ,
,
де R'' є Н або ОСН3, тоді R1 не є С1-С8 алкілом або С2-С8 алкенілом.
2. Сполука за п. 1, яка відрізняється тим, що
є ізомером піридину, піридазину, піримідину, піразину, піразолу, тіофену, ізоксазолу, ізотіазолу та тіадіазолу.
3. Сполука за п. 2, яка відрізняється тим, що
є ізомером піридину, піридазину, піримідину, піразину, піразолу, ізотіазолу та тіадіазолу.
4. Сполука за п. 1, яка відрізняється тим, що А є С2-С14 алкенілом або С2-С14 алкінілом, кожен з яких може бути розгалуженим або нерозгалуженим, незаміщеним або заміщеним галогеном, ароїлом, арилокси, С1-С8 ацилокси, арилтіо, арилом, гетероарилом, гетероарилтіо, гетероарилокси, С1-С6 ацилом.
5. Сполука за п. 1, яка відрізняється тим, що А є С3-С14 циклоалкілом, що містить 0-3 гетероатоми і 0-2 ненасичені зв'язки, який заміщений арилокси, гетероарилокси, С1-С6 алкілтіо, арилтіо, гетероарилтіо, С1-С6 алкокси, С1-С6 галоалкокси.
6. Сполука за п. 1, яка відрізняється тим, що А є С6-С14 бі- або трициклічною кільцевою системою, що містить 0-3 гетероатоми і 0-2 ненасичені зв'язки, яка може бути незаміщеною або заміщеною арилокси, С1-С6 алкокси, С1-С8 ацилокси, арилом, С1-С6 ацилом, карбоарилокси.
7. Сполука за п. 1, яка відрізняється тим, що А є гетероарилом, який може бути незаміщеним або заміщеним С1-С6 алкілом, С1-С6 галоалкілом, С3-С6 циклоалкілом, арилом, С1-С6 алкокси, арилтіо, карбоарилокси, карбогетероарилокси, С1-С6 карбоалкокси або амідо, незаміщеним або заміщеним однією або двома С1-С6 алкільними групами, ОС(O)арилом, C(O)Ry, в якому будь-який алкіл або циклоалкіл, що містить замісник, може бути заміщений одним або більше галогенами, і в якому будь-який арил або гетероарил, що містить замісник, може бути також незаміщеним або заміщеним галогеном, ціано, арилокси, арилом, С1-С6 галоалкілом, С1-С6 алкокси, С1-С6 галоалкокси.
8. Сполука за п. 1, яка відрізняється тим, що А є
, де * = точка прикріплення,
в якій Q1, Q2 є О;
W є СН2, CHR6, або зв'язком;
R1 є С1-С8 алкілом, С2-С8 алкенілом, С2-С8 алкінілом, С3-С8 циклоалкілом, арилом або гетероарилом;
R2 є Н, С1-С3 алкілом, С2-С5 алкенілом або С2-С5 алкінілом;
R3 є Н, R1, OR1, OC(O)R1, OC(O)OR1 або OC(O)NR1R6;
R4 і R5 є незалежно Н, С1-С6 алкілом або С2-С6 алкенілом, за умови, що сума вуглеців для R4 плюс R5 становить шість або менше, і, далі, за умови, що R4 і R5 можуть бути з'єднані в С3-С6 кільце;
R6 і R7 є незалежно Н, С1-С6 алкілом, С3-С6 циклоалкілом, С2-С5 алкенілом або С2-С5 алкінілом, за умови, що щонайменше один із R6 і R7 є Н;
із застереженням, що коли
є
,
де
R'' є Н або ОСН3, тоді R1 не є С1-С8 алкілом або С2-С8 алкенілом.
9. Сполука за п. 1, яка відрізняється тим, що Z є О.
10. Сполука за п. 1, яка відрізняється тим, що Х1 є N, Х2 і Х3 є СН і Х4 є СН, або СОМе, СМе, ССl, COEt, або CSMe.
11. Сполука за п. 10, яка відрізняється тим, що Z є О і А є С2-С14 алкенілом, або С2-С14 алкінілом, кожен з яких може бути розгалуженим або нерозгалуженим, незаміщеним або заміщеним галогеном, ароїлом, арилокси, С1-С8 ацилокси, арилтіо, арилом, гетероарилом, гетероарилтіо, гетероарилокси, С1-С6 ацилом.
12. Сполука за п. 10, яка відрізняється тим, що Z є О і А є С3-С14 циклоалкілом, що містить 0-3 гетероатоми і 0-2 ненасичені зв'язки, який заміщений арилокси, гетероарилокси, С1-С6 алкілтіо, арилтіо, гетероарилтіо, С1-С6 алкокси, С1-С6 галоалкокси, карбоарилокси, карбогетероарилокси, С1-С6 карбоалкокси або амідо, незаміщеним або заміщеним однією або двома С1-С6 алкільними групами.
13. Сполука за п. 10, яка відрізняється тим, що Z є О і А є С6-С14 бі- або трициклічною кільцевою системою, що містить 0-3 гетероатоми і 0-2 ненасичені зв'язки, яка може бути незаміщеною або заміщеною арилокси, С1-С6 алкокси, С1-С8 ацилокси, арилом, С1-С6 ацилом, карбоарилокси.
14. Сполука за п. 10, яка відрізняється тим, що Z є О і А є гетероарилом, який заміщений С1-С6 алкілом, С1-С6 галоалкілом, С3-С6 циклоалкілом, арилом, С1-С6 алкокси, арилтіо, карбоарилокси, карбогетероарилокси, С1-С6 карбоалкокси або амідо, незаміщеним або заміщеним однією або двома С1-С6 алкільними групами, ОС(O)арилом, в якій будь-який алкіл або циклоалкіл, що містить замісник, може бути заміщений одним або більше галогенами, і в якій будь-який арил або гетероарил, що містить замісник, може бути також незаміщеним або заміщеним галогеном, ціано, арилокси, арилом, С1-С6 ацилом, С1-С6 галоалкілом, С1-С6 алкокси, С1-С6 галоалкокси.
15. Сполука за п. 10, яка відрізняється тим, що Z є О і А є
, де * = точка прикріплення,
в якій
Q1, Q2 є О ;
W є СН2, СНR6, або зв'язком;
R1 є С1-С8 алкілом, С2-С8 алкенілом, С2-С8 алкінілом, С3-С8 циклоалкілом, арилом або гетероарилом;
R2 є Н, С1-С3 алкілом, С2-С5 алкенілом або С2-С5 алкінілом;
R3 є Н, R1, OR1, OC(O)R1, OC(O)OR1 або OC(O)NR1R6;
R4 і R5 є незалежно Н, С1-С6 алкілом або С2-С6 алкенілом, за умови, що сума вуглеців для R4 плюс R5 становить шість або менше, і, далі, за умови, що R4 і R5 можуть бути з'єднані в С3-С6 кільце;
R6 і R7 є незалежно Н, С1-С6 алкілом, С3-С6 циклоалкілом, С2-С5 алкенілом або С2-С5 алкінілом за умови, що щонайменше один із R6 і R7 є Н;
із застереженням, що коли
є
,
де
R'' є Н або ОСН3, тоді R1 не є С1-С8 алкілом або С2-С8 алкенілом.
16. Сполука за п. 1, яка відрізняється тим, що Х1 є N, Х2 є CR'', Х3 є N, і Х4 є CR''.
17. Сполука за п. 1, яка відрізняється тим, що Х1 є N, Х2 є CR'', Х3 є CR", і Х4 є N.
18. Сполука за п. 1, яка відрізняється тим, що Х1 є N, Х2 є N, Х3 є CR", і Х4 є CR''.
19. Сполука за п. 1, яка відрізняється тим, що Х1 є N, Х2 є CR'', Х3 є О, і Х4 є зв'язком.
20. Сполука за п. 1, яка відрізняється тим, що Х1 є N, Х2 є CR'', Х3 є S, і Х4 є зв'язком.
21. Сполука за п. 1, яка відрізняється тим, що Х1 є N, Х2 є NR', Х3 є CR", і Х4 є зв'язком.
22. Сполука за п. 1, яка відрізняється тим, що Х1 є CR'', Х2 є CR'', Х3 є N, і Х4 є CR''.
23. Сполука за п. 1, яка відрізняється тим, що Х1 є N, Х2 є S, Х3 є зв'язком, і Х4 є CR''.
24. Фунгіцидна суміш, що включає сполуку за п. 1 і фітологічно прийнятний носій.
25. Суміш за п. 24, яка відрізняється тим, що включає щонайменше одну іншу сполуку, вибрану з групи, що складається з інсектицидів, фунгіцидів, гербіцидів, нематоцидів, акарицидів, артроподоцидів, бактерицидів, та їхніх комбінацій.
26. Спосіб контролю або попередження грибкових уражень, який включає застосування до вогнища грибкового ураження або до ділянки, в якій ураження повинне бути контрольоване або попереджене, фунгіцидно ефективної кількості сполуки за п. 1.
Текст
Ця заявка вимагає пріоритет, оснований на попередній заявці 60/144, 676, поданій до Департаменту патентів та товарних знаків США 20 липня 1999p., повний опис якої введений у даний опис за допомогою посилання. Даний винахід відноситься до галузі фунгіцидних сумішей і способів. Більш детально, даний винахід стосується нових фунгіцидних гетероциклічних ароматичних амідів і способів, що включають застосування фунгіцидно ефективних кількостей таких сполук до вогнища патогенів рослин. Даний винахід також стосується способів, придатних для приготування гетероциклічних ароматичних амідів та їхніх фунгіцидних сумішей. Велика кількість протигрибкових сумішей і способів є добре відомою у даній галузі. Наприклад, антиміцин був ідентифікований як речовина природного походження з антибіотичними властивостями, що продукується Streptomyces spp., (Barrow, С. J.; et al., Journal of Antibiotics, 1997, 50(9), 729). Також було виявлено, що ці речовини є ефективними фунгіцидами (The Merck Index, Twelfth Edition, S. Budavari, Ed., Merck and Co., Whitehouse Station, N.J., 1996, p.120). WO 97/08135 описує аміди ациламіносаліцилової кислоти, які є придатними як пестициди. ЕР-А-0-661269 розкриває заміщені гетероциклічні аміди карбонової кислоти, придатні як медичні препарати. JP-A-7-233165 розкриває протигрибкові дилактони, що мають 3гідроксипіридинкарбоксильні групи з фунгіцидною дією. Ізо-бутирильні, тиглоїльні, ізо-валерильні і 2метилбутирильні похідні цих останніх сполук далі описані у наступних посиланнях: Tetrahedron 1998, 54, 12745-12774; J. Antibiot. 1997, 50(7), 551; J. Antibiot. 1996, 49(1), 639; J. Antibiot. 1996, 49(12), 1226; і Tetrahedron Lett. 1998, 39, 4363-4366. Незважаючи на це, потреба в нових фунгіцидах залишилася. Даний винахід представляє фунгіциди, які мають високу залишкову активність, більшу активність при нижчих нормах витрати, лікувальну активність та більш широкий спектр ефективності. Коротко описуючи один із аспектів даного винаходу, сполуки за даним винаходом включають гетероциклічні ароматичні аміди (ГАА) формули І: де Х1-Х4, Z і А визначені далі. Винахід також включає гідрати, солі та комплекси цих сполук. Даний винахід також представляє фунгіцидні суміші, що включають ГАА у поєднанні з фітологічно прийнятними носіями і/або розріджувачами. Також розкриті способи використання гетероциклічних ароматичних амідних сполук і сумішей. Метою даного винаходу є представити ГАА та їхні суміші, які є ефективними як протигрибкові засоби. Іншою метою даного винаходу є забезпечення методів для контролю і/або попередження грибкових заражень, методів, які включають застосування ГАА і сумішей, що їх містять. Подальші цілі і переваги даного винаходу будуть очевидні з наступного опису. Даний винахід стосується різноманітних ГАА сполук, які є активними як протигрибкові засоби. Включеними також є препаративні форми (суміші), які містять ГАА сполуки, та способи використання ГАА сполук і препаративних форм. Даний винахід також торкається способів приготування ГАА сполук і способу їхнього приготування і використання як фунгіцидів. ГАА сполуки Нові протигрибкові ГАА сполуки даного винаходу описані наступною формулою І: де: a) представляє 5- або 6-членне гетероциклічне ароматичне кільце, в якому і) кожен з Х1-Х4 є незалежно О, S, NR', N, CR" або зв'язком; іі) не більше ніж один із Х1-Х4 є О, S або NR'; ііі) не більше ніж один із Х1-Х4 є зв'язком; iv) коли будь-який один із Х1-Х4 є S, О або NR', один із сусідніх Х1-Х4 повинен представляти зв'язок; і ν) щонайменше один з Х1-Х4 повинен бути О, S, NR' або Ν; де R' представляє Н, С1-С3 алкіл, С2-С3 алкеніл, С2-С3 алкініл, гідрокси, ацилокси, С1-С6 алкоксиметил, CHF2, циклопропіл або С1-С4 алкокси; і R" незалежно представляє Н, галоген, ціано, гідрокси, С1-С3 алкіл, С1-С3 галоалкіл, циклопропіл, С1-С3 алкокси, С1-С3 галоалкокси, С1-С3 алкілтіо, арил, С1-С3 NHC(O)алкіл, NHC(O)H, С1-С3 галоалкілтіо, С2-С4 алкеніл, С2-С4 галоалкеніл, С2-С4 алкініл, С2-С4 галоалкініл або нітро, де сусідні R" замісники можуть формувати кільце або сусідні R' і R" замісники можуть формувати кільце; b) Ζ є О, S або NORz, в якому Rz є Η або С1-С3 алкілом; і c) А представляє і) С1-С14 алкіл, С2-С14 алкеніл або С2-С14 алкініл, кожен з яких може бути розгалуженим або нерозгалуженим, незаміщеним або заміщеним галогеном, гідрокси, нітро, ароїлом, арилокси, C1-C8 ацилокси, С1-С6 алкілтіо, арилтіо, арилом, гетероарилом, гетероарилтіо, гетероарилокси, С1-С6 ацилом, С1-С6 галоалкілом, С1-С6 алкокси або С1-С6 галоалкокси, іі) С3-С14 циклоалкіл, що містить 0-3 гетероатоми і 0-2 ненасичені зв'язки, який може бути незаміщеним або заміщеним галогеном, гідрокси, С1-С6 алкілом, С1-С6 галоалкілом, ціано, нітро, ароїлом, арилокси, гетероарилокси, С1-С6 алкілтіо, арилтіо, гетероарилтіо, С1-С6 алкокси, С1-С6 галоалкокси, C1-C8 ацилокси, арилом, гетероарилом, С1-С6 ацилом, карбоарилокси, карбогетероарилокси, С1-С6 карбоалкокси або амідо, незаміщеним або заміщеним однією або двома С1-6 алкільними групами, ііі) С6-С14 бі- або трициклічну кільцеву систему, що містить 0-3 гетероатоми і 0-2 ненасичені зв'язки, яка може бути незаміщеною або заміщеною галогеном, гідрокси, С1-С6 алкілом, С1-С6 галоалкілом, ціано, нітро, ароїлом, арилокси, гетероарилокси, С1-С6 алкілтіо, арилтіо, гетероарилтіо, С1-С6 алкокси, С1-С6 галоалкокси, C1-C8 ацилокси, арилом, гетероарилом, С1-С6 ацилом, карбоарилокси, карбогетероарилокси, С1-С6 карбоалкокси або амідо, незаміщеним або заміщеним однією або двома С1-С6 алкільними групами, iv) арил або гетероарил, який може бути незаміщеним або заміщеним нітро, С1-С6 алкілом, С1-С6 галоалкілом, С3-С6 циклоалкілом, С2-С6 алкенілом, С2-С6 алкінілом, арилом, гетероарилом, галогеном, гідрокси, Сі-Сб алкокси, Сі-Сб галоалкокси, карбоарилокси, карбогетероарилокси, С1-С6 карбоалкокси або амідо, незаміщеним або заміщеним однією або двома С1-С6 алкільними групами, С1-С6 алкілтіо, С1-С6 алкілсульфонілом, С1-С6 алкілсульфінілом, С1-С6 ОС(О)алкілом, ОС(О)арилом, С3-С6 ОС(О)циклоалкілом, С1С6 NНС(О)алкілом, С3-С6 NHС(О)циклоалкілом, КНС(О)арилом, NHС(О)гетероарилом, С3-С6 циклоалкілтіо, С3-С6 циклоалкілсульфонілом, С3-С6 циклоалкілсульфінілом, арилокси, гетероарилокси, гетероарилтіо, гетероарилсульфінілом, гетероарилсульфонілом, арилтіо, арилсульфінілом, арилсульфонілом, C(O)Ry, C(NORx)Ry, в якому будь-який алкіл або циклоалкіл, що містить замісник, може бути заміщений одним або більше галогенами, і в якому будь-який арил або гетероарил, що містить замісник, може бути також незаміщеним або заміщеним галогеном, ціано, нітро, ароїлом, арилокси, арилом, гетероарилом, С1-С6 ацилом, С1-С6 галоалкілом, С1-С6 алкокси, С1-С6 галоалкокси, С1-С6 карбоалкокси або амідо, незаміщеним або заміщеним однією або двома С1-С6 алкільними групами, де Ry і Rx є незалежно Н, С1-С6 алкілом, С2-С6 алкенілом, С3-С6 циклоалкілом, арилом або гетероарилом, і ν) де* = точка прикріплення в якій Q1, Q2 є О або S; W є О, СН2, CHR6, або зв'язком; R1 є С1-С8 алкілом, С2-С8 алкенілом, C2-C8 алкінілом, С3-C8 циклоалкілом, арилом або гетероарилом; R2 є Η, С1-С3 алкілом, С2-С5 алкенілом або С2-С5 алкінілом; R3 є Н, R1, OR1, OC(O)R1, OC(O)OR1 або ОС(О)NR1R6; R4 і R5 є незалежно Н, С1-С6 алкілом або С2-С6 алкенілом, за умови, що сума вуглеців для R4 плюс R5 становить шість або менше, і, далі, за умови, що R4 і R5 можуть бути з'єднані в С3-С6 кільце; R6 і R7 є незалежно Н, С1-С6 алкілом, С3-С6 циклоалкілом, С2-С5 алкенілом або С2-С5 алкінілом за умови, що, щонайменше, один із R6 і R7 є Н; із застереженням, що коли є де R" є Η або ОСН3, тоді R1 не є С1-С8 алкілом або С2-C8 алкенілом. Терміни алкіл, алкеніл, алкініл і подібні, які використовуються тут, включають у межах свого діапазону як прямі, так і розгалужені групи; терміни алкеніл, алкенілен і подібні призначені, щоб включити групи, що містять один або більше подвійних зв'язків; і терміни алкініл, алкінілен і подібні призначені, щоб включити групи, що містять один або більше потрійних зв'язків. Циклоалкіл, який використовується тут, відноситься до С3-С14 циклоалкільних груп, що містять 0-3 гетероатоми і 0-2 ненасичені зв'язки. Бі- або трициклічні кільцеві системи відносяться до С6-С14 аліфатичних кільцевих систем, що містять 0-3 гетероатоми і 0-2 ненасичені зв'язки. Згадані вище терміни далі розглядають або заміщені, або незаміщені форми. Якщо особливо не визначене інше, заміщена форма відноситься до заміщення однією або більше групами, вибраними з галогену, гідрокси, ціано, нітро, ароїлу, арилокси, арилу, арилтіо, гетероарилу, гетероарилокси, гетероарилтіо, C1-C8 ацилу, С1-С6 галоалкілу, С1-С6 алкокси, С1-С6 галоалкокси, С1-С6 алкілтіо, С1-С6 галоалкілтіо, карбоарилокси, карбогетероарилокси, С1-С6 карбоалкокси або амідо, незаміщеного або заміщеного однією або двома С1-С6 алкільними групами. Всі вищенаведені терміни і визначення припускають, що правила хімічного зв'язку і енергії деформації дотримані. Термін арил, який використовується тут, відноситься до заміщеної фенільної або нафтильної групи. Термін гетероарил відноситься до будь-якого 5- або 6-членного ароматичного кільця, що містить один або більше гетероатомів; ці гетероароматичні кільця можуть бути також об'єднані з іншими ароматичними системами. Згадані вище терміни далі розглядають або заміщені, або незаміщені форми. Заміщена форма відноситься до заміщення однією або більше групами, вибраними з нітро, С1-С6 алкілу, С1-С6 галоалкілу, С3С6 циклоалкілу, С2-С6 алкенілу, С2-С6 алкінілу, арилу, гетероарилу, галогену, гідрокси, С1-С6 алкокси, С1-С6 галоалкокси, С1-С6 алкілтіо, С1-С6 алкілсульфонілу, С1-С6 алкілсульфінілу, С1-С6 ОС(О)алкілу, ОС(О)арилу, С3-С6 ОС(О)циклоалкілу, С1-С6 NНС(О)алкілу, С3-С6 NНС(О)циклоалкілу, NHC(O)арилу, NHС(О)гетероарилу, С3-С6 циклоалкілтіо, С3-С6 циклоалкілсульфонілу, С3-С6 циклоалкілсульфінілу, арилокси, гетероарилокси, гетероарилтіо, гетероарилсульфінілу, гетероарилсульфонілу, арилтіо, арилсульфінілу, арилсульфонілу, C(O)Ry, C(NORx)Ry, де Ry і Rx є незалежно Н, С1-С6 алкілом, С2-С6 алкенілом, С3-С6 циклоалкілом, арилом або гетероарилом, в якому будь-який алкіл або циклоалкіл, що містить замісник, може бути заміщений одним або більше галогенами, і за умови, що правила хімічного зв'язку і енергії деформації дотримані. Терміни галоген і гало, які використовуються тут, включають хлор, бром, фтор і йод. Терміни галоалкіл і подібні відносяться до груп, заміщених одним або більше атомами галогену. Термін Me, який використовується тут, відноситься до метальної групи. Термін Et відноситься до етильної групи. Термін Рr відноситься до пропільної групи. Термін Вu відноситься до бутильної групи. Термін ЕtOАс відноситься до етилацетату. Термін алкокси, який використовується тут, відноситься до алкокси групи з прямим або розгалуженим ланцюгом. Термін галоалкокси відноситься до алкокси групи, заміщеної одним або більше атомами галогену. Термін гетероатом, який використовуються тут, відноситься до О, S і N. Кращі 5- або 6-членні гетероциклічні ароматичні кільця формули включають відповідні ізомери піридину, піридазину, піримідину, піразину, піролу, піразолу, імідазолу, фурану, тіофену, оксазолу, ізоксазолу, тіазолу, ізотіазолу і тіадіазолу. Найкращими гетероциклічними ароматичними кільцями є піридин, піримідин, піразин, піридазин, тіазол, ізотіазол і оксазол. Особливо кращі сполуки за формулою І основані на 2-амідо-3-гідроксипіридині, 2-амідо-3-гідрокси-4-метоксипіридині, 2-амідо3-гідроксипіразині та 4-амідо-5-гідроксипіримідині. Буде зрозумілим, що деякі комбінації груп замісників для сполук, які підпадають під визначення, дані тут, буде неможливо приготувати через стеричні і/або хімічні причини. Такі сполуки не включені в опис винаходу. Різноманітні гідрати, солі і комплекси сполук формули І можуть бути отримані загальноприйнятими способами. Наприклад, солі можуть бути утворені шляхом заміни атома водню гідроксилу на катіон, наприклад, NH4+, +N(Bu)4, К+, Na+, Ca2+, Li+, Mg2+, Fe2+, Cu2+, і т.д. Ці похідні також є придатними у відповідності з даним винаходом. У цьому документі всі температури наведені у градусах Цельсія (°С), і всі проценти є процентами за масою, якщо не зазначене інше. Термін ррm відноситься до частин на мільйон (parts per million). Термін ф/д2 відноситься до фунтів на квадратний дюйм. Термін tкип. відноситься до температури плавлення. Термін W відноситься до температури кипіння. Приготування сполук Сполуки за даним винаходом приготовані з використанням добре відомих хімічних методик. Необхідні вихідні речовини є комерційно доступними або легко синтезуються з використанням стандартних методик. Загальне приготування піридин-2-карбоксамідів Бажані ГАА (2) готують взаємодією відповідної opmo-гідроксигетероароматичної карбонової кислоти (1) з аміном у присутності сполучного реагенту (фосгену або 1-[3-диметиламінопропіл]-3-етилкарбодііміду гідрохлориду [EDCI]) плюс 1-гідроксибензотриазол (HOBt) або 1-гідрокси-7-азабензотриазол (HOAt) і кислотний поглинач, який видаляє домішки, наприклад, N-метилморфолін (NMM), триетиламін, 4(диметиламіно)піридин (DMAP) або діізопропілетиламін) (схема 1). У деяких випадках хлорангідриди із захищеними гідроксильними групами, такі як (3), могли прореагувати з відповідним аміном з одержанням проміжних амідів (4). Видалення захисних груп шляхом гідрування у присутності паладієвого (Pd) каталізатора дає бажаний продукт (2Х). Приготування opmo-гідроксигетероароматичних карбонових кислот 1 Приготування карбонових кислот 1 (Х1 = N, Х2 = Х3 = СН, Х4 = незалежно С-Ме, С-SMe, C-C1) показане на схемі 2. Взаємодія 3-гідрокси-2-бромпіридину (5) з 2-(триметилсиліл)етоксиметил хлоридом (SEM-C1) з використанням трет-бутилату калію як основи у суміші диметилформамід (ДМФ) - тетрагідрофуран (ТГФ) (1:1) дала бажаний ефір 6. Депротонування сполуки 6 діізопропіламідом літію (LDA), супроводжуване конденсацією з підходящою електрофільною сполукою (йодметаном, диметилдисульфідом або гексахлоретаном) дало 4-заміщений піридин 7. Обмін бромом/літієм між сполукою 7 і n-бутиллітієм (n-BuLi), супроводжуваний карбоксилюванням з діоксидом вуглецю (СО2), і кислотний гідроліз дали необхідну 4заміщену-3-гідроксипіколінову кислоту 1Х. Альтернативно, 3-гідроксипіридин (8) міг бути конденсований з SEM-C1 з отриманням сполуки 9 (схема 3). Депротонування сполуки 9 трет-бутиллітієм (t-BuLi), супроводжуване конденсацією з Nфторфенілсульфонімідом, дало 4-фторпохідне сполуки 10. Конденсація 10 з етоксидом натрію дала діефір 11. Депротонування 11 t-BuLi, супроводжуване карбоксилюванням і кислотним гідролізом, дало бажаний 4етоксипіридин 1Х (X = OEt). Приготування хлорангідриду 3 в загальних рисах показане на схемі 4. Так, 3-гідроксипіколінову кислоту (12) перетворювали на метиловий ефір 13 у метанолі при нагріванні зі зворотним холодильником, використовуючи трифторид бору-як каталізатор. Сполуку 13 потім бромували, використовуючи бром на водній основі для одержання диброміду 14. Після цього готували дибензиловий ефір 15 шляхом конденсації сполуки 14 з хлористим бензилом у присутності гідриду натрію. Обережний метаноліз сполуки 15 у метанолі/карбонаті калію дав похідне 4-метоксипіколінової кислоти 16. Перетворення сполуки 16 на хлорангідрид 3 було виконане з хлорангідридом щавлевої кислоти, використовуючи бензол як розчинник і каталітичні кількості ДМФ. Приготування 4-етокси-3-гідроксипіколінової кислоти (1, Χ1 = Ν, Х2 = Х3 = Н, Х4 = COEt) (див. схеми 1 і 3) a. Приготування 3-(2-(триметилсиліл)етоксиметокси)-піридину (9) До суміші ДМФ (100мл) і ТГФ (100мл), що перемішується, додавали твердий трет-бутилат калію (17,96г, 0,16моль). Після того, як вся тверда речовина розчинилася, розчин охолоджували до температури £5°С і додавали відразу весь 3-гідроксипіридин (14,25г, 0,15моль). Після перемішування протягом 10 хвилин суміш охолоджували до -10°С і по краплях додавали SEM-C1 (25г, 0,15моль) з такою швидкістю, що внутрішня температура залишалася £-5°С. Після закінчення додавання суміш перемішували при 0°С протягом 1 години, потім при кімнатній температурі протягом 2 годин. Суміш виливали у воду (600мл), і потім екстрагували ефіром (3x150мл). Ефірні екстракти об'єднували, послідовно промивали 2 н. NaOH (100мл), водою (50мл) і насиченим розчином NaCl (100мл), сушили (MgSO4) і концентрували з одержанням рідини коричневого кольору. Перегонка дала бажаний ефір 9 у вигляді безбарвної рідини (20,8г), tкип. 95-99°C при 0,03мм рт. ст. b. Приготування 4-фтор-3-(2-Триметилсиліл)етоксиметокси) піридину (10) До розчину, що перемішується, сполуки 9 (12,39г, 0,055моль) в ефірі (200мл), охолодженому до £-70°С в атмосфері аргону, повільно додавали t-BuLi (40мл, 1,5Μ розчин у пентані). Під час додавання температуру реакції підтримували на рівні £-68°С. Після закінчення додавання суміш додатково перемішували протягом 60 хвилин при £-70°С, потім переносили за допомогою канюлі до розчину, що перемішується, Νфторбензилсульфоніміду (18,92г) у зневодненому ТГФ (200мл), який також охолоджували до £-70°С в атмосфері аргону. Після закінчення додавання охолоджувальну баню видаляли, і реакційній суміші давали нагрітися до кімнатної температури. Додавали воду (100мл) і відділяли органічну фазу, сушили (MgSO4) і концентрували з одержанням масла коричневого кольору. Хроматографія (силікагель, гексан-ацетон, 9:1) давала бажаний продукт 10 у вигляді масла оранжевого кольору (7,5г), яке містило близько 15% вихідної речовини. Цю сиру суміш використовували безпосередньо в наступній реакції. с. Приготування 4-етокси-3-(2-(триметилсиліл)етоксиметокси)піридину (11) До розчину, що перемішується, етилату натрію (0,9г, 13ммоль) в етанолі (10мл) додавали відразу всю сполуку 10 (1,07г, 4,4ммоль). Отриману в результаті суміш перемішували при кімнатній температурі протягом 48 годин, потім виливали у воду (100мл). Отриману у результаті суміш екстрагували ефіром (3x50мл). Ефірні екстракти об'єднували, сушили (MgSO4) і концентрували. Отримане у результаті масло бурштинового кольору хроматографували (силікагель, гексан-ацетон, 4:1) з одержанням 11 у вигляді масла жовтого кольору (0,6г). d. 4-Етокси-3-гідроксипіридин-2-карбонова кислота (1, Х1 = Ν, Χ2 = Χ3 = СН, Х4 = СОЕt) Розчин, що перемішується, сполуки 11 (2,9г) у ТГФ (50мл) в атмосфері аргону охолоджували до £-70°С. До нього повільно додавали t-BuLi (8мл, 1,5Μ розчин у пентані), зберігаючи температуру реакції на рівні £ 66°С. Після закінчення додавання суміш перемішували при £-70°С протягом 45 хвилин і потім виливали у рідкий розчин подрібненого сухого льоду в ефірі. Отриману в результаті суміш перемішували доти, поки вона не досягала кімнатної температури, потім розчинники упарювали. До сухого залишку додавали ТГФ (25мл) і 4 н. HCI (15мл), і отриману в результаті суміш перемішували при кімнатній температурі протягом двох годин. У кінці цього періоду нерозчинні речовини відфільтровували, промивали невеликим об'ємом ТГФ і сушили на повітрі, одержуючи зазначену у заголовку сполуку у вигляді твердої речовини білого кольору (1,05г). Приготування 6-бром-3-бензилокси-4-метоксипіридин-2-карбонової кислоти (16) та її хлорангідриду(3) (див. схему 4) а. Приготування метил 4,6-дибром-3-гідроксипіридин-2-карбоксилату (14) У 3-горлу колбу місткістю 2л, оснащену краплинною лійкою і механічною мішалкою, додавали воду (800мл) і метил 3-гідроксипіридин-2-карбоксилат (15,3г). До цього розчину, що перемішується, повільно додавали бром (32г). По мірі розвитку реакції тверді частинки відділялися від розчину, і реакційну суміш ставало важко перемішувати. Після закінчення додавання суміш енергійно перемішували до зникнення забарвлення брому. 1Н-ЯМР (CDCІ3) невеликого зразка сирого продукту показало, що це була суміш у співвідношенні приблизно 3:1 від моно- до дибромованого продукту. До реакційної суміші обережно додавали карбонат натрію (31,8г), і потім по краплях додавали додаткову кількість брому (12г). Після зникнення забарвлення брому реакційну суміш доводили до рН 5 (приблизно) за допомогою концентрованої НСІ, і отриману у результаті суміш екстрагували СН2СІ2 (3х150мл). Органічні екстракти об'єднували, сушили (MgSO4) і концентрували з утворенням твердої речовини оранжевого кольору (14г). Цю речовину можна перекристалізувати з метилциклогексану (після обробки активованим вугіллям) з. отриманням сполуки 14 у вигляді твердої речовини білого кольору з tпл.. 181-183°С. b. Приготування метил 4,6-дибром-3-бензилоксипіридин-2-карбоксилату (15) До розчину, що перемішується, гідриду натрію (0,6г) у ДМФ (50мл) повільно додавали сполуку 14 (7,1г). Після закінчення додавання суміш перемішували при кімнатній температурі протягом 15 хвилин, потім додавали відразу весь бензилхлорид (3,05г). Після цього суміш нагрівали при температурі 90°С протягом шести годин, охолоджували, виливали у воду (500мл) і екстрагували ефіром (2x200мл). Ефірні екстракти об'єднували, промивали 2 н. NaOH (50мл), сушили (MgSO4) і розчинник упарювали з отриманням сполуки 15 у вигляді твердої речовини світло-жовтого кольору (8,3г). Перекристалізація з невеликим об'ємом метанолу дала аналітичний зразок з tпл. 75-76°C. c. 6-Бром-3-бензилокси-4-метоксипіридин-2-карбонова кислота (16) Суміш сполуки 15 (25,5г), карбонату калію (75г) і метанолу (300мл), що енергійно перемішувалася, нагрівали зі зворотним холодильником протягом 30 годин. Суміш охолоджували, вливали у воду (800мл) і доводили рН розчину до 2 шляхом додавання концентрованої НСІ. Отриману у результаті суміш екстрагували СН2СІ2 (3x150мл). Органічні екстракти об'єднували, сушили (MgSO4) і розчинник упарювали з отриманням майже безбарвного масла (20,5г), яке повільно тверднуло при стоянні. Його перекристалізовували з суміші метанол (125мл)/вода (40мл) з одержанням бажаної кислоти 16 (11,6г), tпл. 134-135°С. d. Приготування 6-бром-3-бензилокси-4-метоксипіридин-2-карбоніл хлориду (3) До суміші, що перемішується, сполуки 16 (2,54г, 7,5ммоль) у бензолі (30мл), який містить ДМФ (3 краплі), додавали хлорангідрид щавлевої кислоти (1,90г, 15ммоль) (за один раз). Після того, як виділення газу припинилося (приблизно через 45 хвилин), тепер уже гомогенний розчин перемішували ще 15 хвилин, після чого розчинник упарювали. Додавали 1,2-дихлоретан (30мл), і розчинник знову упарювали, отримуючи кількісний вихід сполуки 3 у вигляді майже безбарвного масла. Цю речовину розчиняли у СН2СІ2 (10мл) або ТГФ (10мл) і використовували безпосередньо у послідуючих реакціях зв'язування. 6-Бромо-3-гідроксипіколінова кислота (17) До розчину, що механічно перемішується, метил 3-гідроксипіколінату (30,6г) у воді (800мл) повільно додавали бром (32г) протягом 30-хвилинного періоду. Після закінчення додавання перемішування продовжували протягом ще однієї години. Додавали ефір (300мл) і продовжували перемішування доти, поки всі тверді частинки не розчинилися. Органічний шар відділяли, і водну фазу екстрагували ефіром (200мл). Органічні фази об'єднували, сушили (MgSO4), і упарювали розчинник, одержуючи 32,8г метил 6-бром-3гідроксипіколінату у вигляді твердої речовини не зовсім білого кольору. Перекристалізація з суміші метанол/вода дала аналітичний зразок з tпл. 115-117°С. До розчину, що перемішується, цього складного ефіру (2,32г) у ТГФ (15мл) додавали відразу весь розчин LіОН×Н2О (1г) у воді (7мл). Отриману у результаті суміш перемішували протягом 2 годин при кімнатній температурі і потім виливали у воду (100мл). рН розчину доводили до приблизно 3 за допомогою 1 н. НСl, після чого суміш екстрагували СН2СІ2 (3x100мл). Органічний екстракт сушили (MgSO4), фільтрували і концентрували з одержанням 2,0г твердої речовини білого кольору, чиї 1Н-ЯМР і мас-спектр були сумісними з бажаною кислотою 17, зазначеною у заголовку. 3-Бензилокси-6-метоксипіколінова кислота (18) Розчин метил 3-бензилоксипіколінату (4,86г) і 3-хлорпероксибензойної кислоти (5,75г, 60% перкислоти) у СН2Сl2 (100мл) перемішували при кімнатній температурі протягом 40 годин. Потім реакційну суміш екстрагували 5% розчином бісульфату натрію (100мл), а далі - 0,5 н. розчином NaOH (150мл). Після висушування (MgSO4) розчинник упарювали з отриманням 4,9г метил 3-бензилоксипіколінат-1-оксиду у вигляді твердої речовини білого кольору. Перекристалізація із суміші метилциклогексан/толуол дала кристалічну тверду речовину з tпл. 104-106°С. Розчин цієї сполуки (16,1г) в оцтовому ангідриді (80мл) перемішували і нагрівали на масляній бані при температурі 125°С протягом 3 годин. Надлишок оцтового ангідриду видаляли на ротаційному випарнику і залишок переводили у метанол (200мл). Додавали концентровану сірчану кислоту (1мл) і отриману у результаті суміш нагрівали зі зворотним холодильником протягом 90 хвилин. Розчинник упарювали, і потім до залишку додавали насичений бікарбонат натрію. Отриману у результаті суміш екстрагували CH2Cl2 (3x100мл). Органічні фракції об'єднували, сушили (MgSO4) і розчинник упарювали з одержанням 15,5г метил 3-бензилокси-6-гідроксипіколінату у вигляді твердої речовини жовтого кольору. Перекристалізація з толулолу дала тверду речовину блідо-жовтого кольору з tпл. 91-92°С. До розчину, що перемішується, цієї сполуки (10,25г) у толуолі (125мл), нагрітого на масляній бані до 60°С, додавали карбонат срібла (16,6г), і потім метилйодид (8,52г). Отриману у результаті суміш перемішували і нагрівали протягом 3 годин при 60°С. Після охолодження суміш фільтрували крізь Celite® і розчинник упарювали, одержуючи масло жовтого кольору. Хроматографія на силікагелі (гексан-ацетон, 4:1) дала майже безбарвне масло, дані 1Н-ЯМР і мас-спектр якого були сумісними з метил 3-бензилокси-6-метоксипіколінатом. Гідроліз цього складного ефіру до зазначеної у заголовку кислоти 18 був виконаний з LiОН×Н2О, як описано вище для споріднених складних ефірів. 4-Гідроксипіримідин-5-карбонова кислота (19) Етил 4-гідроксипіримідин-5-карбоксилат можна приготувати, дотримуючись методики М. Pesson et al., Eur. J. Med. Chem. - Chim. Then 1974, 9, 585. Розчин цього складного ефіру (500мг, 3ммоль) у ТГФ (10мл) і МеОН (5мл) обробляли LiОН×Н2О (373мг, 8,9ммоль) і перемішували протягом ночі. Суміш гасили концентрованою НСl (1мл) і екстрагували ЕtOАс (2x20мл). Об'єднаний органічний екстракт сушили (MgSO4) і концентрували з одержанням 260мг зазначеної у заголовку сполуки 19 у вигляді твердої речовини оранжевого кольору з tпл. 220°C (з розкладом). 4-Гідрокси-2-метилпіримідин-5-карбонова кислота (20) Етил 4-гідрокси-2-метилпіримідин-5-карбоксилат готували, дотримуючись методики Geissman et al., J. Org. Chem., 1946, 11, 741. Розчин цього складного ефіру (750мг, 4,11ммоль) у ТГФ (10мл) і МеОН (5мл) обробляли LiОН×Н2О (431мг, 10,3ммоль) і перемішували протягом ночі. Суміш гасили концентрованою НСl (1мл) і екстрагували ЕtOАс (2x20мл). Об'єднаний органічний екстракт сушили (MgSO4) і концентрували з одержанням 155мг зазначеної у заголовку сполуки 20 у вигляді твердої речовини білого кольору з tпл. 180°С (з розкладом). 5,6-Дихлор-3-гідроксипіразин-2-карбонова кислота (21) Метил 3-аміно-5,6-дихлорпіразин-2-карбоксилат (5,0г, 23ммоль) перемішували у концентрованій сірчаній кислоті (140мл) і охолоджували до 0°С. Повільно додавали нітрит натрію, підтримуючи температуру близькою до 0°С. Після витримування протягом додаткових 30 хвилин при 0°С суміші давали нагрітися до кімнатної температури і перемішували протягом 3 годин. Після цього суміш виливали у 500г льоду, що призводило до барботування і спінювання. Через 30 хвилин суміш екстрагували тричі ЕtOАс. Об'єднаний органічний екстракт сушили (MgSO4), фільтрували і концентрували. Тверду речовину жовтого кольору, яка залишилася, промивали водою і сушили на повітрі, отримуючи 5,0г твердої речовини жовтого кольору з tпл. 114-116°С, 13СЯМР спектр якої був сумісний з метиловим ефіром зазначеної у заголовку сполуки. Цю тверду речовину (5,0г) обробляли 1 н. NaOH (20мл), і суміш нагрівали при 90°С протягом 1,5 години. Після охолодження суміш підкислювали концентрованою НСl, після чого екстрагували тричі ЕtOАс. Висушування (MgSO4), фільтрування і концентрування дозволили отримати 0,48г твердої речовини темножовтого кольору, 1Н-ЯМР і мас-спектри якої співпадали з кислотою 21, зазначеною у заголовку. 6-Хлор-3-гідрокси-5-метоксипіразин-2-карбонова кислота (22) Суміш, що перемішується, метил 3-аміно-5,6-дихлорпіразин-2-карбоксилату (5,0г, 23ммоль) і метилату натрію (3,6г, 67,5ммоль) в абсолютному МеОН (50мл) нагрівали зі зворотним холодильником протягом 2 годин, потім давали охолонути і підкисляли концентрованою НСl. Осад збирали шляхом фільтрації, промивали водою і сушили на повітрі, отримуючи 3,6г твердої речовини коричневого кольору. Перекристалізація з суміші гексан-ЕtOАс (1:1) дала змогу отримати 2,6г твердої речовини блідо-жовтого кольору, спектр якої співпадав з метил 3-аміно-6-хлор-5-метоксипіразин-2-карбоксилатом. Цю сполуку (1г, 4,6ммоль) переводили у концентровану сірчану кислоту, охолоджували до 0°С, і повільно обробляли нітритом натрію (0,5г, 6,9ммоль). Через 30 хвилин перебування при 0°С суміш виливали у 300г льоду/води, що призводило до спінювання. Перемішування продовжували протягом 30 хвилин, потім тверду речовину збирали шляхом фільтрації і промивали водою. Вологу тверду речовину переводили у EtOAc, сушили (MgSO4), фільтрували і концентрували. Це давало 0,95г не зовсім білої твердої речовини з tпл.,. 180182°С, ЯМР спектри якої були сумісними з метил 6-хлор-3-гідрокси-5-метоксипіразин-2-карбоксилатом. Цю тверду речовину (0,9г, 4,1ммоль) обробляли 1 н. NaOH (60мл), і суміш перемішували протягом 1 години, після чого підкисляли концентрованою НСІ. Осад збирали шляхом фільтрування і промивали водою, потім розчиняли у EtOAc, сушили (MgSO4), фільтрували і концентрували. Це давало 0,62г твердої речовини блідо-жовтого кольору з tпл. 170-173°C, спектри якої були сумісними з бажаною кислотою 22, зазначеною у заголовку. 4-Гідроксіізотіазол-3-карбонова кислота (23) Цю кислоту отримували, дотримуючись методики, показаної на схемі 5. Так, до розчину, що перемішується, твердого КОН (88%, 6,98г, 0,11моль) у 75мл EtOH у колбі, яку промивали потоком азоту, додавали тіооцтову кислоту (8,36г, 0,11моль), промиту 25мл EtOH. Суміш перемішували в атмосфері азоту протягом 5 хвилин у колбі, закритій пробкою. До неї додавали 0,1моль сирої сполуки брому (свіжо приготованої згідно з М. Hatanaka and Т. Ishimaru, J. Med. Chem., 1973, 16, 798). Колбу промивали потоком азоту і закривали пробкою. Суміш перемішували на водяній бані при кімнатній температурі протягом 3 годин, після чого виливали у 300мл СН2СІ2 і 1000мл води. Водний шар екстрагували чотири рази по 200мл СН2СІ2. Об'єднані органічні екстракти промивали 100мл холодної води і насиченим соляним розчином і сушили. Сиру суміш фільтрували і концентрували. Отримане у результаті масло хроматографували на силікагелі, використовуючи як елюент діетиловий ефір, з отриманням 13г масла світложовтого кольору, яке тверднуло при стоянні до клейкої твердої речовини. Спектральні дані були сумісними з етил 2-ацетиламіно-4-ацетилтіо-3-оксобутаноатом. До розчину, що швидко перемішується, цієї сполуки (12,95г) у 450мл хлороформу, охолодженого на льодяній бані до температури нижче 5°С, протягом 45 хвилин додавали по краплях бром (15,8г, 2 еквіваленти) у 50мл хлороформу. Перемішування продовжували на льодяній бані протягом додаткових 45 хвилин, а потім при кімнатній температурі протягом 30 годин. Після цього суміш промивали 200мл води, а потім іншими 100мл води. Об'єднані водні фази реекстрагували 100мл хлороформу. Об'єднані хлороформні розчини промивали насиченим соляним розчином і сушили над MgSO4. Розчин фільтрували і концентрували до сирого масла. Його хроматографували на силікагелі, використовуючи послідовний градієнт від петролейного ефіру-СН2СІ2 (3:1) до СН2СІ2, одержуючи спочатку 0,79г етил 5-бром-4-гідроксіізотіазол-3карбоксилату, і потім 3,40г етил 4-гідроксіізотіазол-3-карбоксилату у вигляді безбарвних кристалів з tпл. 4447°С, сумісних по мас-спектрам і 1Н-ЯМР. До 710мг останнього складного ефіру у 30мл ТГФ додавали 370мг LiOH×Н2O (2,2 еквіваленти) у 10мл води. Суміш перемішували протягом 3 годин при кімнатній температурі, потім охолоджували у холодильнику. Тверду речовину, яка випала в осад, збирали шляхом фільтрації з одержанням 710мг дилітієвої солі карбонової кислоти. Цю сіль переводили у 7мл води, охолоджували на льодяній бані, і доводили рН до 1 шляхом додавання 2 н. НСІ. Отриманий у результаті розчин екстрагували тричі по 50мл EtOAc. Об'єднані екстракти промивали 5мл соляного розчину і сушили (Na2SO4), фільтрували і фільтрат поміщали у холодильник. Охолоджений розчин знову фільтрували і фільтрат концентрували з одержанням 230мг безбарвної твердої речовини з tпл. 185-189°С, 1Н-ЯМР і 13С-ЯМР спектри якої були сумісними із зазначеною у заголовку сполукою 23. 3-Бензилокси-і-метилпіразол-4-карбонова кислота (24) і 5-бензилокси-1-метилпіразол-4-карбонова кислота (25) Суміш етил 3-гідрокси-1-метилпіразол-4-карбоксилату і етил 5-гідрокси-1-метилпіразол-4-карбоксилату (одержану за методикою Y. Wang, et al., Zhejiang Gongxueyuan Xuebao, 1994,2, 67) бензилювали відповідно до методики S. Yamamoto, et al.,. Japanese Patent JP 62148482, 1987, і суміш розділяли за допомогою колонкової хроматографії, використовуючи як елюент гексани-ЕtOАс (3:1), з одержанням етил 3-бензилокси1-метилпіразол-4-карбоксилату і етил 5-бензилокси-1-метилпіразол-4-карбоксилату, чистота яких була доведена за допомогою 1H-ЯМР. Етил 3-бензилокси-і-метилпіразол-4-карбоксилат (283мг, 1,08ммоль) у ТГФ (10мл), МеОН (2мл) і воді (5мл) обробляли LiOH×Н2O (91мг, 2,17ммоль) і перемішували протягом ночі. Суміш гасили концентрованою НСl (1мл) і екстрагували EtOAc (2x20мл). Об'єднані органічні шари сушили (MgSO4) і концентрували, одержуючи тверду речовину білого кольору (227мг) з tпл. 169-172°С, спектри якої співпадали з 3-бензилокси-1метилпіразол-4-карбоновою кислотою (24). Етил 5-бензилокси-1-метилпіразол-4-карбоксилат (755мг, 2,9ммоль) гідролізували аналогічно, використовуючи LiOH×Н2O (243мг, 5,8ммоль) у ТГФ (20мл), МеОН (4мл) і воді (10мл), що дозволило отримати 608мг 5-бензилокси-1-метил-4-карбонової кислоти (25) у вигляді твердої речовини білого кольору з tпл. 117122°С. Приготування інших гетероароматичних карбонових кислот 4-Гідроксинікотинову кислоту готували за методикою М. Mittelbach et al., Arch. Pharm. (Weinheim, Germany)1985, 313, 481-486. 2-Гідрокси-6-метилнікотинову кислоту можна приготувати, дотримуючись методу A. Dornow, Chem. Ber. 1940, 73, 153. 4,6-Диметил-2-гідроксинікотинову кислоту можна приготувати, дотримуючись методу R. Mariella and E. Belcher, J. Am. Chem. Soc, 1951, 73, 2616. 5-Хлор-2-гідрокси-6метилнікотинову кислоту можна приготувати за методикою A. Cale et. al., J. Med. Chem., 1989, 32, 2178. 2,5Дигідроксинікотинову кислоту можна приготувати за методом P. Nantka-Namirski and A Rykowski, Chem. Abstr., 1972, 77, 114205. З-Гідроксіізонікотинову кислоту готували згідно з методом J. D. Crum and С. Η. Fuchsman, J. Heterocycl. Chem. 1966, 3, 252-256. 3-Гідроксипіразин-2-карбонову кислоту можна приготувати згідно методу A. P. Krapcho et al., J. Heterocycl. Chem. 1997, 34, 27. 5,6-Диметил-3-гідроксипіразин-2-карбонову кислоту можна приготувати шляхом гідролізу відповідного етилового ефіру, синтез якого описаний С І. Зав'яловим і А. Г. Завозіним, Изв. Акад. Наук СССР, 1980, (5), 1067-1070. 4-Гідроксипіридазин-3-карбонову кислоту готували за методом І. Ichimoto, К. Fujii, and С. Tatsumi, Agric. ВіоІ. Chem., 1967, 31, 979. 3,5-Дигідрокси-1,2, 4-триазин6-карбонову кислоту готували за методом Е. Falco, Ε. Pappas, and G. Hitchings, J. Am. Chem. Soc, 1956, 78, 1938. 5-Гідрокси-3-метилтіо-1,2,4-триазин-6-карбонову кислоту готували, дотримуючись методу R. Barlow and A. Welch, J. Am. Chem, Soc, 1956, 78, 1258. Гідроксіізотіазол-, гідроксіізоксазол- і гідроксипіразол-карбонові кислоти готували за методом Τ. Μ. Willson et al., Bioorg. Med. Chem. Lett., 1996, 6, 1043. 3-Гідрокси-1, 2,5тіадіазол-4-карбонову кислоту готували за методом J. М. Ross et al., J. Am. Chem. Soc, 1964, 86, 2861. 3Гідроксіізоксазол-4-карбонову кислоту одержували, дотримуючись методики, описаної К. Bowden et al., J. Chem. Soc. (C), 1968,172. 3-Гідрокси-1-фенілпіразол-4-карбоксилат одержували відповідно до методу A. W. Taylor and R. Т. Cook, Tetrahedron, 1987, 43, 607. 3-Бензилоксихінолін-2-карбонову кислоту готували, дотримуючись методики D. L. Boger and J. Η. Chen, J. Org. Chem., 1995, 60,7369-7371. Загальне приготування проміжних амінів і анілінів Синтез циклічних, нециклічних і бензиламінів проводили шляхом відновлення відповідних оксимів, використовуючи або гідриди металів, або реакції розчинення металу, як проілюстровано R. О. Hutchins і М. К. Hutchins у Comprehensive Organic Synthesis; В. Μ. Trost, Ed.; Pergamon Press: Oxford, 1991; Vol. 8, p. 65; або J. W. Huffinan у Comprehensive Organic Synthesis; B. M. Trost, Ed.; Pergamon Press: Oxford, 1991; Vol 8, p. 124. Альтернативно, ці аміни можна приготувати безпосередньо з необхідних кетонів і альдегідів через реакцію Leuckart, як проілюстровано R. Carlson, Т. Lejon, T. Lunstedt and Ε. LeClouerec, Ada Chem. Scand. 1993, 47, 1046. Аніліни в загальному готували каталітичним відновленням відповідних нітроароматичних сполук, використовуючи як каталізатори паладій на вугіллі або сульфідовану платину на вугіллі. Такі методики є добре задокументованими, як, наприклад, у R. L. Augustine, Catalytic Hydrogenation, Marcel Decker, Inc., New York, 1965. Аміни 49, які є 9-членними дилактонними кільцевими системами, готували відповідно до методик М. Shimano, N. Kamei, Т. Shibata, К. bioguchi, N. Itoh, Т. Ikari and Η. Senda, Tetrahedron, 1998, 54, 12745, або шляхом модифікацій цих методик. Така .модифікація показана на схемі 6. Так, сполуку 26 (з вищенаведеного посилання) відновлювали боргідридом літію, і одержаний у результаті первинний спирт блокували триізопропілсиланом (TIPS) з отриманням сполуки 27. Вільна гідроксильна група 27 реагувала з 1-бром-2метил-2-пропеном, що супроводжувалося каталітичним відновленням подвійного зв'язку з утворенням сполуки 28. Селективне видалення пара-метоксибензильної (РМВ) групи, що блокує, супроводжуване конденсацією з N-t-BOC-O-бензил-L-серином, давало сполуку 29. Видалення TIPS групи, супроводжуване окисленням отриманої в результаті гідроксильної групи, давало сполуку 30. Цю речовину (30) послідовно перетворювали на амін 31, використовуючи методики, описані у вищенаведеному посиланні. Подібним чином, синтез амінодилактонів 38 і 48, у яких відсутня функціональність екзоциклічного ефіру, в загальних рисах показаний на схемах 7 і 8, відповідно. Приготування сполуки 27 (див. схему 6) До розчину боргідриду літію (2,0Μ у ТГФ, 7,5мл, 15ммоль) у 7,5мл зневодненого ТГФ додавали 0,1мл триметилбораху. Цю суміш охолоджували в атмосфері азоту до -30°С. До цього розчину по краплях додавали розчин сполуки 26 (4,58г, 10ммоль) у 10мл ТГФ протягом 10-хвилиного періоду. Розчин перемішували протягом 1 години при -30°С, потім протягом 5 годин при 0°С. Після цього по краплях додавали насичений розчин хлориду амонію (10мл), суміш перемішували протягом 10 хвилин, і фази відділяли. Водну фазу екстрагували ЕtOАс (2x25мл), а об'єднані органічні фази промивали насиченим соляним розчином, сушили над сульфатом натрію і упарювали до сухого залишку. Сирий продукт хроматографували, одержуючи 2,1г твердої речовини білого кольору. Зразок, перекристалізований із суміші гексан-EtOAc, давав тонкі білі голчасті кристали з tпл. 91-93°С. [a]D25 = +31,9° (С = 1,04, СНСl3). Цей діол (2,04г, 6,22ммоль) розчиняли у 4мл зневодненого ДМФ і додавали імідазол (680мг, 10ммоль). Розчин охолоджували на льодяній бані, а потім протягом 2 хвилин додавали триізопропілхлорсилан (1,39мл, 6,5ммоль). Суміш перемішували при кімнатній температурі протягом 4 годин, потім виливали у суміш лід-вода, і екстрагували 20% ефіром в гексанах (3x15мл). Об'єднані органічні фази промивали соляним розчином, сушили і фільтрували крізь невисокий шар силікагелю, який промивали 20мл того ж самого розчинника. Розчинник упарювали, одержуючи 2,77г сполуки 27 у вигляді блідо забарвленого в'язкого масла, яке було дуже чистим (за 1Н-ЯМР). Приготування сполуки 28 (див. схему 6) Гідрид натрію (60% дисперсія у маслі, 400мг, 10ммоль) вносили у колбу місткістю 50мл і тричі промивали гексанами. Додавали ДМФ (15мл) і суспензію перемішували, оскільки протягом 15 хвилин по краплях додавали сполуку 27 (2,53г, 5,19ммоль) у 5мл зневодненого ДМФ. Реакційну суміш перемішували протягом 15 хвилин, потім охолоджували до температури нижче 10°С і протягом 5 хвилин додавали 1-бром-2-метил-2пропен (1мл, 10ммоль), після чого суміш перемішували протягом 2 годин при кімнатній температурі. Суміш розділяли між гексанами/льодяним розчином хлориду амонію, обробляли як у приготуванні сполуки 27, і сирий продукт хроматографували з одержанням 2,20г безбарвного масла, чистота якого була доведена за допомогою 1H-ЯМР та елементного аналізу. Цю речовину (2,38г, 4,4ммоль) розчиняли в 50мл ЕtOАс у колбі Мортона місткістю 100мл в атмосфері азоту. Додавали 150мг 5% платини на вугіллі, і. суміш перемішували при 1 атмосфері водню протягом 20 хвилин. Каталізатор видаляли фільтруванням, і розчинник упарювали з одержанням 2,35г сполуки 28 у вигляді безбарвного масла, чистота якого була доведена за допомогою 1НЯМР. Приготування сполуки 29 (див. схему 6) У колбу місткістю 50мл, оснащену магнітною мішалкою, вносили розчин ефіру 28 (2,0г, 3,68ммоль) у 40мл СН2Сl2 і 2мл води. Розчин розмішували в атмосфері азоту і охолоджували на льодяній бані до температури 96% чистоти за результатами ГРХ. Дані 1Н-ЯМР (CDCl3) і ГРХ/мас-спектрометрії (m/е = 219, 221) співпадали з 3-(3-хлорфенокси)аніліном. 3-(4-Трифторметилфенокси)анілін До розчину, що перемішується, 3-гідроксианіліну (6,55г) і 4-фторбензотрифториду (9,85г) у ДМСО (50мл) додавали за один раз трет-бутилат калію (7,86г). Отриманий у результаті темний розчин нагрівали протягом 4 годин при 95°С, охолоджували до кімнатної температури, після чого виливали у воду (600мл). Суміш екстрагували ефіром (3x125мл). Органічну фазу промивали 2 н. гідроксидом натрію (2x75мл) і водою (100мл), сушили (MgSO4), і розчинник упарювали з одержанням темного масла. Це масло переганяли з одержанням зазначеного у заголовку аніліну у вигляді безбарвного масла (8,7г) з tкип. 110-112°С при 0,15мм рт. ст. 4-(4-Трифторметилфенілтіо)анілін До розчину, що перемішується, 4-фторбензотрифториду (9,85г) і 4-амінотіофенолу (7,51г) у ДМСО (60мл), охолоджуваного на льодяній бані, додавали за один раз трет-бутилат калію (6,73г). Отриману у результаті суміш перемішували протягом 10 хвилин при 0°С, і потім при 60°С протягом ночі. Після охолодження суміш виливали у воду (600мл), і отриману у результаті суміш екстрагували ефіром (2x200мл). Органічну фазу промивали 2 н. гідроксидом натрію (50мл), і потім водою (50мл). Після сушіння (MgSO4), розчинник упарювали з одержанням твердої речовини коричневого кольору. Перекристалізація з гексану дала зазначений у заголовку анілін у вигляді твердої речовини жовтого кольору з tкип. 97-99°C. 4-(3-Трифторметилбензил)анілін Реактив Гріньяра (Grignard) готували додаванням розчину 4-бром-N,N-біс-(триметилсиліл)аніліну (9,48г) у зневодненому ТГФ (75мл) до суміші, що перемішується, стружки магнію (1,09г) у зневодненому ТГФ (10мл). Другий розчин каталізатора, Li2СuСІ4 (0,33г), готували додаванням СuСl2 (0,20г) і LiCl (0,13г) до зневодненого ТГФ (25мл) і перемішуванням до одержання гомогенного розчину. Потім цей розчин каталізатора додавали до розчину 3-трифторметибензил броміду (7,17г) у зневодненому ТГФ (75мл). Оранжево-червоний розчин охолоджували на льодяній бані (в атмосфері азоту), і в нього через канюлю швидко переносили вищезазначений розчин Гріньяра (попередньо охолоджений на льодяній бані). Після перемішування протягом 15 хвилин при 0°С суміш перемішували протягом ночі при кімнатній температурі. Реакційну суміш гасили додаванням насиченого розчину NH4CI (25мл). Органічну фазу відділяли, сушили (MgSO4) і розчинник упарювали з одержанням темного масла (11г). До цього масла додавали 4 н. HCI (50мл), і отриману у результаті суміш перемішували при кімнатній температурі протягом 3 годин. Суміш робили основною шляхом обережного додавання твердого карбонату натрію, а потім екстрагували ефіром (3x100мл). Органічну фазу сушили (MgSO4) і розчинник упарювали. Додавали EtOAc (100мл), і розчин декантували з невеликої кількості нерозчинної речовини. Знову упарювали розчинник і залишок хроматографували (силікагель, гексан/ЕtOАс (3:1)). Другий елюат збирали з одержанням масла оранжевого кольору, яке швидко темніло. Дані ЯМР (CDCl3) і ГРХ/мас-спектрометрії (m/е = 251) були сумісними із зазначеною у заголовку сполукою. Цю речовину перетворювали на сіль HCI з одержанням твердої речовини коричневого кольору. 4-(3-Трифторметилбензоїл)анілін Розчин, що перемішується, 4-бром-N,N-біс-(триметилсиліл)аніліну (9,24г) у зневодненому ТГФ (100мл) охолоджували до -78°С в атмосфері аргону. До нього повільно додавали 2,5Μ розчин н-бутиллітію у гексані (12мл). Після закінчення додавання реакційну суміш перемішували при -78°С протягом 10 хвилин, а потім по краплях додавали розчин N-метил-N-метокси-3-фторметилбензаміду (6,8г) у зневодненому ТГФ (25мл). Після закінчення додавання суміш перемішували при -78°С протягом 1 години, потім охолоджувальну баню видаляли і давали реакційній суміші нагрітися до 10°С. Реакцію гасили додаванням насиченого розчину NH4Cl (50мл), а потім води (10мл). Органічну фазу відділяли, сушили (MgSO4) і розчинник упарювали з одержанням рідини жовтого кольору (12г). Її переводили у ефір (100мл) і додавали 4 н. HCI (100мл). Отриману у результаті суміш перемішували протягом 30 хвилин при кімнатній температурі, протягом цього часу відділялася тверда речовина. Цю тверду речовину відфільтровували, промивали декількома порціями ефіру, потім обережно додавали до насиченого розчину NaHCO3 (100мл), який перемішується. Отриману у результаті суміш екстрагували ефіром (2x100мл), органічну фазу сушили (MgSO4), і розчинник упарювали з одержанням твердої речовини жовто-білого кольору (5,7г). Перекристалізація з суміші метанол/вода дала тверду речовину білого кольору з tпл. 130-131°С. Спектральні дані були сумісними зі сполукою, зазначеною у заголовку. Етил 2-аміно-5-(4-трифторметилфенокси)бензоат До розчину, що механічно перемішується, трет-бутилату калію (15,71г) у ДМСО (75мл) додавали за один раз 5-гідроксіантранілову кислоту (10,2г). Суміш перемішували при кімнатній температурі в атмосфері аргону протягом 10 хвилин, потім додавали 4-фторбензотрифторид (11,16г), і отриману у результаті суміш перемішували і нагрівали при 75-80°С протягом ночі. Після охолодження суміш виливали у воду (600мл) і доводили рН розчину до приблизно 2,5. Отриману у результаті тверду речовину відфільтровували, промивали декількома порціями води, і потім перекристалізовували з суміші метанол/вода (вугілля) з одержанням твердої речовини жовтувато-коричневого кольору (13,5г) з tпл. 165-167°С. Цю тверду речовину переводили у безводний етанол (250мл) і обережно додавали концентровану сірчану кислоту (15мл). Отриману у результаті суміш нагрівали зі зворотним холодильником протягом 24 годин, після чого більшу частину етанолу упарювали. Залишок обережно додавали в льодяну воду (600мл), отриману у результаті суміш робили основною шляхом повільного додавання 50% розчину NaOH, і потім екстрагували ефіром (2x150мл). Органічну фазу промивали водою (100мл), потім насиченим розчином NaCl (50мл). Після сушіння (MgSO4) розчинник упарювали з одержанням масла жовтого кольору приблизно 98% чистоти (за даними ГРХ). ГРХ/мас-спектрометрія показала вихідну сполуку для іона m/е = 325, яка є сумісною із зазначеною у заголовку сполукою. 2-Амінобензонорборнан До розчину, що перемішується, бензонорборнену (2,84г) у зневодненому ТГФ (8мл), охолодженого до 0°С в атмосфері аргону, швидко додавали 1Μ розчин борану в ТГФ (6,7мл). Розчин перемішували протягом 10 хвилин при 0°С, а потім при кімнатній температурі протягом 90 хвилин. Реакційну суміш знову охолоджували до 0°С і за один раз додавали гідроксиламін-О-сульфокислоту (1,58г). Льодяну баню видаляли і реакційну суміш перемішували при кімнатній температурі протягом 2 годин. Додавали 1 н. HCI (25мл) і ефір (20мл), і продовжували перемішування протягом 10 хвилин. Фази відділяли і органічну фазу відкидали. Водну фазу робили основною шляхом обережного додавання 50% розчину NaOH, і потім екстрагували ефіром (3x30мл). Органічну фазу сушили (MgSO4) і розчинник упарювали з одержанням рідини жовтого кольору (1,35г), яка мала 98% чистоту, як оцінювалось ГРХ. Дані ЯМР (CDCl3) і ГРХ/мас-спектрометрії (m/е = 159) були сумісними зі сполукою, зазначеною у заголовку. Приготування суміші (3-трифторметилбензилоксиметил)норборниламінів 53 Приготування цієї суміші зображене на схемі 9. Так, суміш екзо- і ендо-норборненкарбонових кислот 49 (у співвідношенні ~1:4) (7,0г), 2-йодпропану (12,8г) і карбонату калію (10,4г) у ДМСО (40мл) перемішували і нагрівали при 55°С протягом ночі. Після охолодження суміш розводили водою (125мл), і потім екстрагували пентаном. Органічну фазу сушили (MgSO4) і розчинник упарювали з одержанням безбарвного масла (8,2г). Це масло додавали до розчину 2-пропілату натрію (3,6г) у 2-пропанолі (100мл), і отриману у результаті суміш нагрівали зі зворотним холодильником протягом 16 годин. Видалення 2-пропанолу, розведення водою (200мл) і екстракція пентаном давали ізопропіловий ефір норборнену 50 у вигляді суміші екзо і ендо (52:48).Суміш розділяли на чисті ізомери за допомогою хроматографії (силікагель, гексан/ЕtOАс, 95:5). Екзо ізомер сполуки 50 (4,0г) розчиняли в ефірі (50мл), охолоджували до 0°С, і повільно додавали 1Μ розчин гідриду літію алюмінію в ефірі (14мл). Після закінчення додавання суміш нагрівали зі зворотним холодильником протягом 1 години. Після охолодження реакцію гасили послідовним додаванням води (0,53мл), 15% розчину NaOH (0,53мл), і знову води (1,59мл). Отриману у результаті суміш сушили (MgSO4), фільтрували, і розчинник упарювали з одержанням екзо-спирту 51 (2,7г) у вигляді безбарвної рідини. Дані ГРХ/мас-спектрометрії (m/е = 124) були сумісними із зазначеною структурою. До суміші, що перемішується, гідриду калію (1,0г) у зневодненому ТГФ (25мл) обережно додавали розчин сполуки 51 (2,7г) у ТГФ (10мл). Після закінчення додавання суміш перемішували при кімнатній температурі протягом 30 хвилин, і потім додавали відразу весь 3-трифторметилбензилбромід (5,98г) (екзотермічна реакція). Реакційну суміш нагрівали зі зворотним холодильником протягом 2 годин, охолоджували і потім виливали у воду (150мл). Екстракція ефіром (2x75мл), сушіння (MgSO4) і упарювання розчинника дали масло жовтого кольору, яке очищали за допомогою хроматографії (силікагель, гексан/ацетон, 97:3), одержуючи чисту сполуку 52 у вигляді безбарвного масла (5,2г). Дані ЯМР (CDCl3) і ГРХ/мас-спектрометрії (m/е = 282) були сумісними зі структурою сполуки 52. Перетворення сполуки 52 на діастереомерну суміш амінів 53 здійснювали за методикою з боран/гідроксиламін-О-сульфокислотою, описаною раніше (20% вихід). 3-(3-Піридил)-1-пропанамін Цей амін отримували, спочатку перетворюючи 3-(3-піридил)-1-пропанол на відповідний хлорид, згідно з методикою В. Jursic et al., Synthesis, 1988, (11), 868, а потім перетворюючи цей хлорид на амін відповідно до методики D. J. Dumas et al., J. Org. Chem., 1988, 53, 4650. 3-[[5-(Трифторметил)-2-піридил]окси]-1-пропанамін 2-Фтор-5-трифторметилпіридин (1,831г, 11ммоль) розчиняли у безводному ТГФ (15мл), перемішуючи в атмосфері азоту, і охолоджували до 0°С на льодяній бані. До суміші протягом 30 хвилин по краплях додавали розчин 3-аміно-1-пропанолу (0,76мл, 10ммоль) у безводному ТГФ (15мл) і 1Μ трет-бутилат калію у ТГФ (10мл, 10ммоль). Розчину жовтого кольору давали перемішатися і нагрітися до кімнатної температури на льодяній бані протягом ночі. Реакційну суміш виливали у воду (75мл) і екстрагували ефіром (2x50мл). Органічну фазу промивали соляним розчином (50мл), сушили (Nа2SО4) фільтрували та упарювали під вакуумом до рідини жовтого кольору, яка була майже чистою за результатами ЯМР і мас-спектрометрії, і була використана як така без подальшого очищення. (+)-транс-1-Гідрокси-2-аміноциклопентангідробромід (±)-транс-1-Бензилокси-2-аміноциклопентан гідробромід (8,2г, 42,8ммоль) обробляли 40% НВr (60мл). Після перемішування протягом 3 днів розчин концентрували під вакуумом із одержанням 7,09г (91%) бромистоводневої солі у вигляді твердої речовини оранжевого кольору, яка була чистою за даними 1H-ЯМР (ДМСО-d6). 2,3-Дигідро-2,2-диметил-1Н-інден-1-амін Цей амін готували відповідно до методики міжнародного патенту WO 9927783. 10-Аміно-ендо-2,5-метанбіцикло [4.4.0] дек-3-ен (56) Цю сполуку готували, як показано на схемі 10. Так, хлорид алюмінію (700мг, 5,2ммоль) додавали до розчину 2-циклогексен-1-ону (2,0г, 20,8ммоль) у толуолі (200мл). Через 40 хвилин додавали недавно дистильований циклопентадієн (13,7г, 208ммоль) і суміш нагрівали до 100°С протягом 2 годин. Після охолодження суміш розводили Еt2О (300мл) і промивали насиченим NaHCO3 (2x150мл) та соляним розчином (100мл). Об'єднані органічні шари сушили (MgSO4), фільтрували і концентрували. Залишок очищали за допомогою флеш-хроматографії з використанням в якості елюенту суміші гексани/Et2О (50:1), одержуючи ендо (1,74г) і екзо (943мг) ізомери 2,5-метанбіцикло[4.4.0]дек-3-ен-10-ону (54), які були чистими за даними 1НЯМР і ГРХ/мас-спектрометрії. Ацетат натрію (1,79г, 21,8ммоль) додавали порціями до розчину ендо-2,5-метанбіцикло[4.4.0]дек-3-ен-10ону (54) (1,61г, 9,9ммоль) і гідроксиламіну гідрохлориду (758мг, 10,9ммоль) у метанолі (33мл), і перемішували протягом ночі при кімнатній температурі. Реакційну суміш гасили Н2О і екстрагували ефіром (2x50мл). Об'єднані органічні шари сушили (MgSO4), фільтрували і концентрували з одержанням оксиму ендо-2,5метанбіцикло[4.4.0]дек-3-ен-10-ону (55) у вигляді в'язкого залишку, чистого за даними 1Н-ЯМР і ГРХ/масспектрометрії. Оксим ендо-2,5-метанбіцикло[4.4.0]дек-3-ен-10-ону (55) (500мг, 2,79ммоль) розчиняли у ЕtOАс (25мл) і додавали 10% паладій на вугіллі (50мг). Після 3 годин під тиском Н2 (40ф/д2), суспензію фільтрували крізь Celite® і концентрували. Отриманий у результаті залишок розчиняли у ЕtOН (25мл) і вносили Raney®-Ni (1,0г). Суспензію насичували NH3 і тримали під тиском Н2 (45ф/д2). Через 6 годин суспензію фільтрували крізь Celite®, розводили EtOAc (100мл) і промивали насиченим NaHCO3 (100мл). Об'єднані органічні шари сушили над MgSO4, фільтрували і концентрували. 1H-ЯМР і ГРХ/мас-спектрометрія виявили зазначений у заголовку амін 56 у вигляді суміші (2:1) діастереоізомерів (418мг). 10-Аміно-4-(4'-метилпент-3'-еніл)-біцикло[4.4.0]дек-3-ен (59) Приготування цієї сполуки проводили, як показано на схемі 11. Так, хлорид алюмінію (700мг, 5,2ммоль) додавали до розчину 2-циклогексен-1-ону (2,0г, 20,8ммоль) у толуолі (100мл). Через 40 хвилин додавали мірцен (17г, 125ммоль) і суміш нагрівали до 100°С протягом 2 годин. Після охолодження суміш розводили Et2O (300мл) і промивали насиченим NaHCO3 (2x150мл) і соляним розчином (100мл). Об'єднані органічні шари сушили над MgSO4, фільтрували і концентрували. Залишок очищали за допомогою флешхроматографії з використанням в якості елюенту суміші гексаниЛйгО (50:1), одержуючи 4-(4'-метилпент-3'еніл)-біцикло[4.4.0]дек-3-ен-10-он (57) (2,55г), який був чистим за даними 1Н-ЯМР і ГРХ/мас-спектрометрії. Ацетат натрію (1,73г, 21ммоль) додавали порціями до розчину 4-(4'-метилпент-3'-еніл)-біцикло[4.4.0]дек3-ен-10-ону (57) (2,23г, 9,6ммоль) і гідроксиламіну гідрохлориду (733мг, 10,5ммоль) у метанолі (32мл), і перемішували протягом ночі при кімнатній температурі. Реакційну суміш гасили Н2О і екстрагували ефіром (2x50мл). Об'єднані органічні шари сушили над MgSO4, фільтрували і концентрували. Це давало оксим 4-(4' метилпент-3'-еніл)-біцикло[4.4.0]дек-3-ен-10-ону (58) у вигляді в'язкого залишку, чистого за даними 1Н-ЯМР і ГРХ/мас-спектрометрії. Оксим 4-(4'-метилпент-3'-еніл)-біцикло[4.4.0]дек-3-ен-10-ону (600мг, 2,42ммоль) розчиняли у ЕtOН (25мл) і вносили Raney®-Ni (1,0г). Суспензію насичували NH3 і тримали під тиском Н2 (45ф/д2). Через 6 годин суспензію фільтрували крізь Celite®, розводили ЕtOАс (100мл) і промивали насиченим NаНСО3 (100мл). Об'єднані органічні шари сушили над MgSO4, фільтрували і концентрували. 1Н-ЯМР і ГРХ/мас-спектрометрія показали зазначений у заголовку чистий амін (550мг). 2-Аміно-7-фурил-3-метил-4-хроманон гідрохлорид (63) Цю хлористоводневу амінну сіль готували, як показано на схемі 12. Так, 7-трифторметансульфонат-3метил-4-хроманон (3,0г, 9,7ммоль) (приготований згідно з методикою К. Koch and M. S. Biggers, J. Org. Chem. 1994, 59, 1216) додавали до розчину 2-(трибутилстаніл)фурану (3,79г, 10,6ммоль), Pd(PPh3)4 (223мг, 0,19ммоль), LiCl (1,23г, 29,0ммоль) і двох кристалів 2,6-ди-t-бутил-4-метилфенолу у 1,4-діоксані (50мл), і нагрівали зі зворотним холодильником протягом 12 годин. Після охолодження суміш гасили насиченим NH4CI (40мл) і екстрагували Et2O (2x50мл). Об'єднані органічні шари сушили над MgSO4, фільтрували і концентрували. Залишок очищали за допомогою флеш-хроматографії з використанням в якості елюенту суміші гексани/EtOAc (20:1), одержуючи 7-фурил-3-метил-4-хроманон (60) (1,78г) у вигляді твердої речовини жовтого кольору з tпл. 94-95°С. Ацетат натрію (395мг, 4,82ммоль) додавали порціями до розчину 7-фурил-3-метил-4-хроманону (60) (500мг, 2,19ммоль) і гідроксиламінугідрохлориду (167мг, 2,41ммоль) у метанолі (5мл), і перемішували протягом ночі при кімнатній температурі. Реакційну суміш гасили Н2О і екстрагували ефіром (2x25мл). Об'єднані органічні шари сушили над MgSO4, фільтрували і концентрували з одержанням оксиму 7-фурил-3метил-4-хроманону (61) у вигляді білої твердої речовини з tпл. 175-177°C. Толуолсульфоніл хлорид (397мг, 2,08ммоль) додавали до охолодженого до 0°С розчину оксиму 7-фурил3-метил-4-хроманону (61) (461мг, 1,89ммоль) і піридину (0,5мл) у СН2СІ2 (10мл). Через 6 годин суміш розводили СН2СІ2 (30мл) і промивали 5% HCI (20мл). Органічний шар сушили над MgSO4, фільтрували і концентрували. Залишок очищали за допомогою флеш-хроматографії з використанням в якості елюенту суміші гексани/ЕtOАс (5:1), одержуючи 7-фурил-3-метил-4-хроманон-O-(толуолсульфоніл)-оксим (62) (429мг) у вигляді твердої речовини рожевого кольору з tпл. 163-164°С (з розкладом). Етанольний розчин етилату натрію (0,35мл, 2,87М, 1,0ммоль) додавали до розчину, що перемішується, 7фурил-3-метил-4-хроманон-О-(толуолсульфоніл)-оксиму (62) (410мг, 1,0ммоль) у бензолі (4мл). Через 18 годин додавали 3 н. HCI (6мл) і шари відділяли. Органічну фазу далі екстрагували 3 н. HCI (2x10мл), і об'єднані водні екстракти концентрували з одержанням сирої сполуки 63, зазначеної у заголовку, у вигляді твердої речовини оранжевого кольору (388мг), яку використовували такою як є, без подальшого очищення. 2-Аміно-7-(3'-метоксипропініл)-3-метил-4-хроманон гідрохлорид (65) Цей гідрохлорид аміну готували, як показано на схемі 13. Так, 7-трифторметансульфонат-3-метил-4хроманон (3,10г, 10ммоль) (приготований згідно з методикою К. Koch and M. S. Biggers, J. Org. Chem. 1994, 59, 1216) додавали до розчину метилпропаргілового ефіру (1,05г, 15ммоль), (Ph3P)4Pd (210мг, 0,30ммоль) і Et3N (6мл) у ДМФ (30мл), і нагрівали при 70°С протягом 1 години. Після охолодження суміш гасили насиченим NH4CI (40мл) і екстрагували Et2O (2x50мл). Об'єднані органічні шари сушили над MgSO4, фільтрували і концентрували. Залишок очищали за допомогою флеш-хроматографії з використанням в якості елюенту суміші гексани/ЕtOАс (9:1), одержуючи 7-(3'-метоксипропініл)-3-метил-4-хроманон (64) (1,40г) у вигляді твердої речовини білого кольору з tпл. 60-63°С. Перетворення сполуки 64 на зазначену у заголовку сполуку 65 виконували у такий же спосіб, як описано вище для 2-аміно-7-фурил-3-метил-4-хроманону гідрохлориду. 2-Аміно-a-тетралон гідрохлорид (66) Цю сполуку одержували з α-тетралону, як показано на схемі 14, за тією ж методикою, яка описана вище для 2-аміно-7-фурил-3-метил-4-хроманон гідрохлориду. 2-Аміно-ендо-6,9-етанбіцикло [4.4.0] дек-7-енон гідрохлорид (70) Цей гідрохлорид аміну готували, як показано на схемі 15. Так, хлорид алюмінію (700мг, 5,2ммоль) додавали до розчину 2-циклогексен-і-ону (2,0г, 20,8ммоль) у толуолі (100мл). Через 40 хвилин додавали циклогексадієн (8,3г, 104ммоль) і суміш нагрівали до 100°С протягом 2 годин. Після охолодження суміш розводили Εt2O3 (300мл) і промивали насиченим NaHCO3 (2x150мл) та соляним розчином (100мл). Об'єднані органічні шари сушили над MgSO4, фільтрували і концентрували. Залишок очищали за допомогою флешхроматографії з використанням в якості елюенту суміші гексани-Еt2О (50:1), одержуючи ендо-2,5етанбіцикло[4.4.0]дек-7-ен-10-он (67) (2,77г), який був чистим за даними 1Н-ЯМР і ГРХ/мас-спектрометрії. Розчин ендо-2,5-етанбіцикло[4.4.0]дек-7-ен-10-ону (67) (2,17г, 12,3ммоль) у ТГФ (20мл) додавали до охолодженого до -78°С розчину діізопропіламіду літію (LDA) (6,7мл, 2,0Μ у ТГФ, 13,5ммоль) у ТГФ (30мл). Через 45 хвилин додавали триметилсиліл хлорид (2,0г, 18,5ммоль), і суміш повільно нагрівали до 0°С. Суміш розводили насиченим розчином NaHCO3 (30мл), екстрагували Еt2О (2x30мл), сушили (MgSO4) і концентрували. Залишок розчиняли у ТГФ (60мл), і порціями додавали N-бромсукцинімід (2,6г, 14,7ммоль). Через 30 хвилин суміш розводили насиченим розчином NH4CI (30мл) і екстрагували Et2O (2x40мл). Об'єднані органічні шари сушили (MgSO4) і концентрували. Залишок очищали за допомогою флеш-хроматографії з використанням в якості елюенту суміші гексани-Еt2О (33:1), одержуючи 2-бром-ендо-6,9етанбіцикло[4.4.0]дек-7-енон (68) (1,44г) у вигляді масла світло-жовтого кольору, яке було чистим за даними 1 Н-ЯМР і ГРХ/мас-спектрометрії. Азид натрію (280мг, 4,3ммоль) додавали до розчину 2-бром-ендо-6,9-етанбіцикло[4.4.0]дек-7-енону (68) (850мг, 3,9ммоль) у ДМФ (20мл). Через 2 години суміш розводили водою (30мл) і екстрагували Еt2О (2x40мл). Об'єднані органічні шари сушили (MgSO4) і концентрували. Залишок очищали за допомогою флешхроматографії з використанням в якості елюенту суміші гексани-Еt2О (20:1), одержуючи 2-азидо-ендо-6,9етанбіцикло[4.4.0]дек-7-енон (69) (469мг) у вигляді масла, яке було чистим за даними 1Н-ЯМР. Трифенілфосфін (486мг, 1,85ммоль) додавали до розчину 2-азидо-ендо-6,9-етанбіцикло[4.4.0]дек-7-енону (69) (310мг, 1,42ммоль) у ТГФ (10мл) і воді (1мл). Після перемішування протягом 12 годин суміш розводили 6 н. HCI (10мл), і шари відділяли. Органічну фазу екстрагували 6 н. HCI (2x5мл), і об'єднані водні шари концентрували до сухого залишку з одержанням бажаної зазначеної у заголовку сполуки 70 у вигляді густого масла оранжевого кольору (500мг), дані 1Н-ЯМР (ДМСО-d6) якого були сумісними із зазначеною структурою. Ізопропіл ендо-2-амінонорборнан-5-карбоксилат (71) та ізопропіл ендо-2-амінонорборнан-6-карбоксилат (72) Ці аміни готували з ізопропіл норборн-2-ен-5-карбоксилату у такий самий спосіб, як описано вище (див. схему 9). Загальна методика для відновного амінування кетонів до амінів Кетон (1ммоль), ацетат амонію (20ммоль) і 3А молекулярні сита (2,8 еквівалентів за масою) змішували у безводному метанолі у сухій колбі в атмосфері азоту. Додавали ціаноборгідрид натрію (4ммоль) і отриману у результаті суміш перемішували при кімнатній температурі до зникнення вихідного кетону, яке показував аналіз методом ТШХ. Із реакційної суміші під вакуумом видаляли метанол, і залишок розчиняли у 6 н. HCI. Після перемішування протягом 15 хвилин не основні речовини видаляли екстракцією з діетиловим ефіром. рН водної фази обережно підвищували до ~8, використовуючи 50% водний розчин NaOH, і амін екстрагували EtOAc (тричі). ЕtOАс екстракти об'єднували, промивали соляним розчином, сушили (Na2SO4), фільтрували і концентрували з одержанням відповідного аміну. Сирий амін був у цілому чистим і використовувався без подальшого очищення. Загальна методика для видалення захисної ВОС-групи амінів До льодяного розчину аміну із захисною ВОС-групою (1ммоль) у зневодненому СН2СІ2 (1мл) додавали триетилсилан (0,5мл) і трифтороцтову кислоту (1мл). Розвиток реакції контролювали за зникненням вихідної речовини (від 5 хвилин до 1,5 години). Реакційну суміш розводили толуолом і концентрували. Залишок розчиняли у воді (10мл) і EtOAc (20мл), рН доводили до ~8 (водним розчином NaHCO3), і органічну фазу відділяли. Водну фазу екстрагували ЕtOАс (2x15мл). Органічні фази об'єднували, промивали соляним розчином, сушили (Na2SO4), фільтрували і концентрували з одержанням аміну. Приготування амінів 73 і 74 Ці аміни готували з відповідних відомих кетодилактонів (J. Org. Chem. 1998, 63, 9889-94) у стандартних умовах відновного амінування, описаних вище. 1Н, 13С ЯМР та 14 спектри були сумісними із зазначеними структурами. Приготування амінів 77 і 78 Приготування цих амінів показане на схемі 16. Макродилактон 75 готували відповідно до методики в J. Org. Chem. 1998, 63, 9889-94. Так, N-t-BOC-аспарагінова кислота (2,33г) реагувала з 2-хлорметил-3хлорпропеном (1,25г) і Cs2CO3 (7,0г) у ДМФ (1000мл) при стандартних умовах макролактонізації, зазначених у вищенаведеному посиланні, з одержанням 1,12г (40% вихід) сполуки 75 у вигляді склоподібної твердої речовини. Мас-спектр (ЕІ-) показав [М-1]+ при (m/е) 284, тоді як 1Н, 13С ЯМР та ІЧ спектри були сумісними зі структурою 75. До розчину алкену 75 (288мг, 1,01ммоль) у зневодненому ЕtOАс (6мл) додавали 10% паладій на вугіллі (60мг). Отриману у результаті суміш продували азотом і перемішували протягом 2,5 годин у апараті Парра для гідрування при тиску водню 45ф/д2. Реакційну суміш продували азотом, фільтрували і концентрували. Залишок, після очищення за допомогою колонкової флеш-хроматографії (силікагель, суміш гексан-EtOAc, 7:3), давав 91мг (32% вихід) відновленого продукту 76.1H, 13С-ЯМР та ІЧ спектри були сумісними зі структурою 76. Видалення захисної ВОС-групи зі сполук 75 і 76, з дотриманням описаної вище загальної методики видалення захисної ВОС-групи, дало відповідні аміни 77 і 78. 1Н, 13С-ЯМР і ІЧ були сумісними із зазначеними структурами. Синтез феніл дилактону 81 До льодяного (0°С) розчину, що добре перемішується, фенілбурштинової кислоти (0,923г, 5,2ммоль) і DMAP (0,064г, 0,52ммоль) у зневодненому СН2СІ2 (55мл) протягом 30 хвилин в атмосфері азоту по краплях додавали розчин ВОС-серинолу (Synthesis 1998, 1113-1118) (1,0г, 5,2ммоль). Отриману у результаті суміш повільно нагрівали до кімнатної температури, перемішували протягом додаткових 12 годин, розводили СН2Сl2 (40мл) і екстрагували насиченим водним розчином бікарбонату натрію (3x10мл). Основні екстракти об'єднували, обережно підкислювали 2 н. HCI і екстрагували ЕtOАс (3x20мл). Об'єднаний ЕtOАс екстракт промивали соляним розчином, сушили (Na2SO4), фільтрували і концентрували з одержанням білої пінистої речовини (1,7г). 1Н-ЯМР показав суміш діастереомерів (1:1) кислот 79. До льодяної суспензії, що добре перемішується, кислот 79 (1,00г, 2,72ммоль) і трифенілфосфіну (786мг, 3,0ммоль) у зневодненому ТГФ (122мл) по краплях протягом 3 годин додавали розчин діетил азодикарбоксилату (0,52г, 3,0ммоль) у ТГФ (55мл). Отриману у результаті суміш повільно нагрівали до кімнатної температури, перемішували протягом додаткових 5 годин, і концентрували до приблизно 5мл. Залишкову суміш розводили ЕtOАс (50мл) і водою (20мл). Органічну фазу відділяли, промивали водним розчином NаНСО3 (10мл), соляним розчином (10мл), сушили (Na2SO4), фільтрували і концентрували з одержанням маслянистого залишку. Очищення за допомогою флеш-хроматографії (силікагель, гексани) дало 228мг (22% вихід) суміші (1:1) дилактонів 80 з tпл. 161-162°С. Мас-спектр (ЕІ) показав М+ при m/е 349. Видалення захисної ВОС-групи при стандартних умовах видалення захисних ВОС-груп, описаних вище, дало амін 81. Синтез дилактонамінів 84 і 85 До розчину, що перемішується, серинолу (3,0г, 15,7ммоль), піридину (1,24г, 0,98моль) і DMAP (0,19г, 1,57ммоль) у зневодненому СН2Сl2 (140мл) по краплях додавали розчин N-CBz аспарагінового ангідриду (3,52г, 14,13ммоль) у зневодненому ТГФ (20мл). Після перемішування протягом 2 годин при кімнатній температурі реакційну суміш концентрували до об'єму приблизно 10мл і розводили ЕtOАс (100мл) і водою (30мл). рН суміші доводили до 8,5 (водним розчином NаНСО3), і водну фазу відділяли, підкисляли 2 н. HCI до рН 3, і екстрагували EtOAc (3x20мл). Об'єднаний органічний екстракт промивали соляним розчином, сушили (Na2SO4), фільтрували і концентрували з одержанням 5,8г сполуки 82 у вигляді пінистої речовини білого кольору. 1Н-ЯМР спектри показали, що вона була досить чистою і містила суміш діастереомерів. До розчину трифенілфосфіну (3,60г, 13,75ммоль) і 1,3-діізопропілкарбодііміду (2,80г, 13,75ммоль) у зневодненому ТГФ (1,15л) протягом 3 годин по краплях додавали розчин кислоти 82 (5,5г, 12,5ммоль) у зневодненому ТГФ (100мл). Отриману у результаті суміш перемішували протягом додаткових 6 годин, концентрували під вакуумом до об'єму приблизно 20мл, і розводили ефіром (200мл) та водою (100мл). Органічну фазу відділяли і промивали 5% водним розчином NаНСО3 та соляним розчином, сушили (Nа2SО4), фільтрували і концентрували під вакуумом. Маслянистий залишок очищали за допомогою колонкової флешхроматографії з одержанням 1,3г (23% вихід) бажаних дилактонів 83. Мас-спектр (ES-) показав при m/е 421 (М-1)+. 1Н, 13С-ЯМР та 14 спектри були сумісними зі структурою 83. Дилактон 83 піддавався видаленню захисних груп при стандартних умовах видалення захисних ВОСгруп, з одержанням аміну 84. До розчину дилактону 83 із захисною N-CBz-групою (200мг, 0,47ммоль) у EtOAc (10мл) додавали 10% паладій на вугіллі (40мг), і отриману у результаті суміш перемішували протягом 12 годин під тиском балонного газоподібного водню. Реакційну суміш продували N2, фільтрували крізь лійку з матованого скла, і концентрували з одержанням аміну 85 (126мг). Цей сирий амін використовували без подальшого очищення. Приготування амінів 86 і 88 Синтез 2,6,6-триметил-2,4-циклогептадієніламіну (86) і 2,3,6,6-тетраметил-3-циклогептенону (87), який є попередником аміну 88, показані на схемі 19. Так, еукарвон (Can. J. Chem. 1974, 52, 1352) легко перетворювали на відповідний амін 86, використовуючи методику ізопропоксид титанy/NaBН4/Et3Nопосередкованого відновного амінування, описану в Synlett 1999, 1781. Додавання триметилалюмінію до еукарвону, каталізоване Сu (l) (реакція Міхаеля), з використанням методики, описаної у Tetrahedron 1995, 51, 743-754, дало 2,3,5,5-тетраметил-3-циклогептенон (87). Останній перетворювали на 2,3,5,5-тетраметил-2циклогептеніламін (88) відповідно до загальної методики міжнародного патенту WO 9927783. N-Метил-N-(2-фенілетил)-(1,5,5-триметил-3-аміноциклогексил) карбамід (89) 1,5,5-Триметил-3-оксо-1-циклогексилкарбонову кислоту (M.S. Ziegler and R.Μ. Herbst, J. Org. Chem. 1951, 16, 920) приєднували до N-метил-2-фенілетиламіну, використовуючи стандартні HOAt, EDCI і DMAPопосередковані умови зв'язування з одержанням [N-метил-N-(2-фенілетил)]-1,5,5-триметил-3-оксо-1циклогексил-карбоксаміду у вигляді олії блідо-жовтого кольору. Мас-спектр виявив вихідний іон при m/е 301. 1 H і 13С-ЯМР спектри були сумісними з цією структурою. Амін 89 готували з цього кетону відповідно до загальної методики міжнародного патенту WO 9927783, шляхом перетворення на відповідний N-гідроксіоксим, супроводжуваного гідруванням у присутності Raney®Ni. 1Н-ЯМР аміну виявив суміш (1:1) діастереомерів. 3-(3,3-Диметилбутоксикарбоніл)-3,5,5-триметилциклогексиламін (90) 1,5,5-Триметил-3-оксо-1-циклогексилкарбонову кислоту (3,0г) (М. S. Ziegler and R. Μ. Herbst, J. Org. Chem. 1951, 16, 920) обробляли 3,3-диметилпентанолом (1,84г), DMAP (2,21г) і 1,3-діізопропілкарбодіімідом (2,17г) у СН2СІ2 (80мл) при стандартних умовах зв'язування з одержанням 2,41г (55% вихід) 3-(3,3диметилбутоксикарбоніл)-3,5,5-триметилциклогексанону. Мас-спектр (ЕІ) виявив вихідний іон при m/е 268. Цей кетон перетворювали на зазначений у заголовку амін 90 відповідно до загальної методики міжнародного патенту WO 9927783, шляхом перетворення на відповідний оксим, супроводжуваного гідруванням у присутності Raney®-Ni. 1Н-ЯМР аміну 90 виявив суміш (1:1) діастереомерів. 4-(4,6-біс-трифторметил-2-піридил)окси-3,3,5,5-тетраметилциклогексиламін(93) Синтез цього аміну показаний на схемі 20. Так, 4-гідрокси-3,3,5,5-тетраметилциклогексил-1,1етиленгліколь ацеталь (900мг, 4,2ммоль) розчиняли у зневодненому ДМФ (8,4мл), суміш охолоджували до 0°С, і додавали 35% (за масою) масляну суспензію КН (591мг, 5,04ммоль). Після перемішування суміші протягом 1 години по краплях додавали розчин 2-хлор-4,6-біс-трифторметил-2-піридину (1,48г, 6,3ммоль) у ДМФ (2мл). Суміш перемішували при 0°С протягом 1 години, потім при кімнатній температурі протягом 12 годин, і обережно гасили хлоридом амонію. Додавали діетиловий ефір (100мл), і органічну фазу відділяли, промивали соляним розчином, сушили (MgSO4) і концентрували до твердої речовини темно-коричневого кольору. Перекристалізація з гарячого гексану дала 950мг (53% вихід) 4-(4,6-біс-трифторметил-2піридил)окси-3,3,5,5-тетраметилциклогексил-1,1-етиленгліколь ацеталь (91) з tпл. 105-106°С. Ацеталь 91 (900мг) розчиняли у 30мл суміші ТГФ, діоксану і 2 н. HCI (1:1:1), і отриманий у результаті розчин перемішували при кімнатній температурі протягом 12 годин, коли ГРХ виявила повне зникнення вихідної речовини. Суміш розводили водою і діетиловим ефіром (50мл кожного), органічну фазу відділяли, промивали соляним розчином, сушили (Nа2SО4) і концентрували з одержанням маслянистого залишку. Цей залишок хроматографували на силікагелі (гексан-ЕtOАс, 5:1) з одержанням 712мг (96% вихід) кетону 92 у вигляді безбарвного масла. Мас-спектр (ЕІ) виявив вихідний іон m/е 383. Відновне амінування сполуки 92 до зазначеного у заголовку аміну 93 виконували відповідно до загальної методики міжнародного патенту WO 9927783. 3-(2,3-Дихлорпропілокси)метил-3,5,5-триметилциклогексиламін (97) Синтез аміну 97 показаний на схемі 21. Дихлорування алкену 94, відповідно до методики Tetrahedron Lett. 1991, 32, 1831-4, дало ацеталь 95. Останній (500мг) розчиняли у суміші ТГФ і 2 н. HCI (1:1). Отриманий у результаті розчин перемішували при кімнатній температурі протягом 1 години, коли ТШХ виявила, що вихідна речовина зникла. Суміш розводили ЕtOАс і водою (30мл кожного), і органічну фазу відділяли та промивали соляним розчином, сушили (Nа2SО4), фільтрували і концентрували з одержанням 383 мг кетону 96 у вигляді масла. 1H-ЯМР був сумісним із діастереомерною сумішшю ізомерів. Відновне амінування з дотриманням стандартної методики, описаної вище, дало зазначений у заголовку амін 97. 3-Бензоїл-3,5,5-триметилциклогексиламін (100) Приготування цього аміну показане на схемі 22. 3-Ціано-3,5,5-тетраметилциклогексил-1,1етиленглікольацеталь (98) (світовий патент WO 9927783) при реакції з феніллітієм, супроводжуваній кислотним гідролізом, давав дикетон 99, який перетворювали на зазначений у заголовку амінокетон 100 відповідно до методики вищезгаданого патенту. 5b-(2-фенілетил)-3b-метокси-4b-метил-4-нітро-циклогексиламін 105) Приготування аміну 105 показане на схемі 23. Конденсація нітроетану з дигідрокоричним альдегідом, відповідно до методики Bull. Chem. Soc. Jap. 1968, 41, 1441, дала відповідний нітроспирт 101. Дегідратація сполуки 101, відповідно до методики Synthesis, 1982, 1017, яка супроводжувалась підтримуваною полімером трифенілфосфін-опосередкованою ізомеризацією (Tetrahedron Lett. 1998, 39, 811-812), дала алкен 103. Циклоприєднання Дільса-Альдера сполуки 103 до дієну Данішевського, відповідно до методики Tetrahedron Lett. 2000, 41, 1717, дало кетон 104. Кетон 104 перетворювали на амін 105 відповідно до стандартної методики міжнародного патенту WO 9927783. 3-Ціано-3,5,5-триметилциклогексиламін (106) Цю сполуку одержували (схема 24) шляхом відновного амінування 3-ціано-3,5,5-триметилциклогексанону відповідно до стандартної методики відновного амінування, описаної вище. Мас-спектр (ЕІ) виявив вихідний іон m/е 167. 3-Аміно-5-фенілтіопіран (107) Цю сполуку готували, як показано на схемі 25. Так, до 0,96г (5ммоль) 5-феніл-3-тіопіранону (Р. Т. Lansbury, et al., J. Am. Chem. Soc. 1970, 92, 5649) у 50мл безводного метанолу додавали 7,7г (100ммоль) ацетату амонію і 6,5г 3А молекулярних сит. Після перемішування протягом 30 хвилин при кімнатній температурі до суміші порціями додавали 1,25г (20ммоль) ціаноборгідриду натрію. Після перемішування протягом 16 годин суміш фільтрували самопливом і метанол упарювали під вакуумом. Залишок розділяли між льодом/НСІ і ефіром. Кислу водну фазу ще двічі екстрагували ефіром, потім її робили основною за допомогою льоду і 50% водного розчину NaOH. Суміш екстрагували СН2Сl2, сушили (MgSO4) і упарювали з одержанням 0,19г (20%) зазначеної у заголовку сполуки. ГРХ/мас-спектрометрія показала 100% чистоту з молекулярним іоном 193. 4-(4-Трифторметил)феноксициклогексиламін (109) Цю сполуку готували відповідно до схеми 26. До розчину, що перемішується, гідриду натрію (1,2г, 0,05моль) у 50мл ДМФ протягом 10 хвилин по краплях додавали розчин 1,4-діоксастро[4.5]декан-8-олу (7,5г, 0,047моль) у 15мл ДМФ. Суміш перемішували при кімнатній температурі протягом 30 хвилин. До неї додавали відразу весь 4-фторбензотрифторид (7,71г, 0,047моль), і реакційну суміш перемішували при кімнатній температурі протягом 2 годин, і після цього протягом ночі при 70°С. Реакційну суміш виливали у холодну воду (700мл), і розчин злегка підкисляли, додаючи 1 н. HCI. Суміш фільтрували і водний фільтрат екстрагували гексаном (2x150мл). Відфільтровані тверді частинки розчиняли у гексанових екстрактах і промивали водою (50мл): Розчин сушили над MgSO4, фільтрували і концентрували з одержанням білої твердої речовини. Цю тверду речовину перекристалізовували із суміші метанол/вода, одержуючи чистий кеталь (8,6г, 61%). Силікагель (30г) суспендували у 150мл СН2Сl2. До цієї суспензії протягом 5 хвилин по краплях додавали 7мл 12% розчину HCI у воді. Суміш енергійно перемішували, щоб уникнути утворення грудочок. До неї додавали розчин вищезгаданого кеталю (8,0г, 26,49ммоль), розчиненого у 75мл СН2СІ2, і реакційну суміш перемішували протягом 3 годин. Після цього суміш фільтрували і шар силікагелю промивали 500мл СН2СІ2. Розчинник упарювали з одержанням 5,8г (86%) 4-(4-трифторфенокси)циклогексанону (108). Відновне амінування кетону 108 відповідно до стандартної методики відновного амінування, описаної вище, дало зазначену у заголовку сполуку 109. 4-Бензоїлокси-3,3,5,5-тетраметилциклогексиламін (111) Цю сполуку готували, дотримуючись методики за схемою 27. До розчину, що перемішується, 7,7,9,9тетраметил-1,4-діоксаспіро[4.5]декан-8-олу (0,37г, 1,73ммоль) у 6мл ТГФ, охолодженого до 0°С, по краплях додавали n-BuLi (2,5Μ у гексанах, 1,73ммоль, 0,7мл). Реакційну суміш перемішували протягом 10 хвилин. Після цього додавали бензоїл хлорид (1,73ммоль, 0,2мл), і суміші давали нагрітися до кімнатної, температури та перемішували протягом ночі. Реакційну суміш виливали у 50мл 0,5 н. NaOH і екстрагували ефіром (3x20мл). Ефірний шар сушили над MgSO4, фільтрували і концентрували. Залишок очищали радіальною хроматографією з використанням у якості елюенту суміші гексан-ЕtOАс (4:1). Таким чином було отримано 0,55г (~100%) бензоїлоксикеталю. Силікагель (2,2г) суспендували у 10мл СН2Сl2. До цієї суспензії протягом 5 хвилин по краплях додавали 0,5мл 12% розчину HCI у воді. Суміш енергійно перемішували, щоб уникнути утворення грудочок. До неї додавали розчин вищезгаданого бензоїлоксикеталю, розчиненого у 5мл СН2СІ2, і реакційну суміш перемішували протягом 3 годин. Після цього суміш фільтрували, і шар силікагелю промивали 100мл СН2Сl2. Розчинник упарювали з одержанням 0,46г (90%) бензоїлоксициклогексанону 110 у вигляді прозорого масла. До розчину, що перемішується, бензоїлоксициклогексанону 110 (0,46г, 1,68ммоль) у 4мл метанолу додавали відразу весь розчин гідроксиламіну гідрохлориду (0,23г, 3,25ммоль) і ацетату калію (0,32г, 3,25ммоль) у 4мл води. Реакційну суміш перемішували при кімнатній температурі протягом ночі. Додавали 20мл води і отриману у результаті суміш екстрагували ефіром (3x10мл). Ефірні екстракти об'єднували, промивали насиченим розчином NаНСО3 (1x20мл) і соляним розчином (1x15мл). Ефірний шар сушили над MgSO4, фільтрували і концентрували з одержанням бажаного оксиму (0,39г, 80%) у вигляді суміші Ε і Ζ ізомерів. Нікель Raney® (0,8г у вологому стані, Aldrich Chemical Co.) у товстостінній склянці Парра місткістю 500мл для реакцій під тиском промивали водою (3x20мл), потім етанолом (3x20мл), кожного разу декантуючи розчинник, яким промивали. До цього промитого каталізатора додавали розчин оксиму (0,39г, 1,35ммоль) у безводному етанолі (30мл). Для розчинення було потрібно дещо нагріти цей розчин. Отриману у результаті суміш насичували аміаком, барботуючи газоподібний аміак крізь розчин протягом 1 хвилини. Цей розчин поміщали в атмосферу водню (початковий тиск водню = 50ф/д2) в апарат Парра для струшування і струшували протягом 7 годин. Після цього реакційну суміш фільтрували крізь шар Celite® і розчинник упарювали з одержанням майже безбарвної рідини (0,37г, кількісний вихід). Протонний ЯМР і ГРХ/масспектрометрія були сумісними з цією речовиною, яка є діастереомерною сумішшю (у відношенні 4:1) зазначеного у заголовку аміну 111. Цю речовину використовували такою як є, без додаткового очищення. 4-Аміно-2,2,6,6-тетраметилциклогексил-6-хлор-2-піридинкарбоксилат (113) Цю сполуку синтезували, як показано на схемі 28. До розчину, що перемішується, 7,7,9,9-тетраметил-1,4діоксаспіро[4.5]декан-8-олу (0,32г, 1,50ммоль) у 5мл ТГФ, охолодженого до 0°С, по краплях додавали n-BuLi (2,5Μ у гексанах, 1,50ммоль, 0,6мл). Суміш перемішували протягом 10 хвилин. Після цього додавали 6хлорпіколіноїл хлорид (1,50ммоль, 0,26г) у вигляді розчину в 1мл ТГФ, і реакційній суміші давали нагрітися до кімнатної температури. Розчин загусав, тому додавали ще 5мл ТГФ і реакційну суміш перемішували протягом ночі. Реакційну суміш виливали у 40мл 0,5 н. NaOH і екстрагували ефіром (3x20мл). Ефірний шар сушили над MgSO4, фільтрували і концентрували. Протонний ЯМР виявив очікуваний продукт разом із вихідною речовиною у співвідношенні 1,6:1. Ці сполуки не можна було розподілити хроматографією на силікагелі, тому суміш переносили на наступну стадію, де її очищали. Силікагель (1,4г) суспендували у 10мл СН2Сl2. До цієї суспензії протягом 5 хвилин по краплях додавали 0,3мл 12% розчину HCI воді. Суміш енергійно перемішували, щоб уникнути утворення грудочок. До неї додавали розчин вищезгаданої суміші, розведеної у 5мл СН2Сl2, і реакційну суміш перемішували протягом 3 годин. Після цього суміш фільтрували, і шар силікагелю промивали 100мл СН2Сl2. Розчинник упарювали з одержанням масла. Осадження бажаного піколінового ефіру 112 здійснювали додаванням 10мл суміші гексан-EtOAc (4:1). Отриману у результаті тверду речовину відфільтровували і промивали 10мл суміші гексан-ЕtOАс (4:1). Змиви гексан-ЕtOАс об'єднували і упарювали з одержанням масла. Вищенаведену процедуру повторювали тричі, одержуючи піколіновий ефір 112 у вигляді твердої речовини білого кольору (214мг, 46% для двох стадій). Протонний ЯМР і ГРХ/мас-спектрометрія показали бажаний продукт >95% чистоти. Суміш цього складного ефіру (200мг, 0,65ммоль), ізопропілату титану (IV) (1,30ммоль, 0,38мл), хлориду амонію (1,30ммоль, 70мг) і триетиламіну (1,30ммоль, 0,18мл) в абсолютному етанолі (10мл) перемішували в атмосфері азоту при кімнатній температурі протягом 12 годин. Потім додавали боргідрид натрію (0,97ммоль, 40мг) і отриману у результаті суміш перемішували протягом додаткових 8 годин при кімнатній температурі. Після цього реакцію гасили, виливаючи суміш у водний розчин аміаку (20мл, 2,0М), і отриманий у результаті розчин екстрагували ефіром (3x20мл). Об'єднані ефірні екстракти екстрагували 2 н. HCI (2x20мл), щоб відділити не основні речовини. Кислотний розчин промивали один раз ефіром (20мл), а потім обробляли водним розчином гідроксиду натрію (2 н.) до рН 10-12, і екстрагували ЕtOАс (3x20мл). Об'єднані EtOAc екстракти сушили над MgSO4, фільтрували і концентрували з одержанням масла. Цей продукт співпадав із діастереомерною сумішшю (6:1) зазначених у заголовку циклогексиламінів. Протонний ЯМР і ГРХ/масспектрометрія показали бажаний продукт ~75% чистоти. Цю суміш амінів використовували такою як є, без подальшого очищення. транс-2-Тіометилциклогексиламін Цей амін готували із циклогексену, використовуючи техніку азасульфенілування за В. М. Trost and T. Shibata, J. Am. Chem. Soc. 1982,104, 3225. 4-Фенілтіоциклогексиламін (115) Цю сполуку готували, дотримуючись методики, показаної на схемі 29. До розчину, що перемішується, 4фенілтіоциклогексанону (V. К. Yadav and D. A. Jeyaraj, /. Org. Chem. 1998, 63, 3474) (1,20г, 5,83ммоль) у 20мл метанолу додавали відразу весь розчин бензилоксіаміну гідрохлориду (1,80г, 11,22ммоль) і ацетату калію (1,10г, 11,22ммоль) у 20мл води. Реакційну суміш перемішували при кімнатній температурі протягом ночі. Додавали 60мл води і отриману у результаті суміш екстрагували ефіром (3x40мл). Ефірні екстракти об'єднували, промивали насиченим NаНСО3 (1x50мл) і соляним розчином (1x40мл). Ефірний шар сушили над MgSO4, фільтрували і концентрували з одержанням масла. Цю речовину очищали за допомогою радіальної хроматографії (гексан-EtOAc, 9:1) з одержанням відповідного О-бензилоксиму 114 (1,72г, 95%) у вигляді суміші Ε і Ζ ізомерів. Гідрид літію алюмінію (5,08ммоль, 0,19г) суспендували у 10мл безводного ефіру і охолоджували до 0°С. По краплях додавали О-бензилоксим 114, розчинений у 5мл ефіру, давали реакційній суміші нагрітися до кімнатної температури і перемішували протягом 4 годин. Надлишок гідриду літію алюмінію руйнували шляхом обережного одночасного додавання води (0,2мл) і 1 н. NaOH (0,2мл). Суміш фільтрували і солі промивали 50мл ефіру. Розчинник упарювали з одержанням 0,62г (93%) зазначеного у заголовку аміну 115 у вигляді масла. Протонний ЯМР і ГРХ/мас-спектрометрія виявили продукт у вигляді суміші діастереомерів амінів (у співвідношенні 1,3:1) >95% чистоти. 3-{[3-(Трифторметил)-2-піридиніл]сульфаніл}-циклогексиламін(117) Цей амін готували за методом, показаним на схемі 30. До розчину, що перемішується, 2-циклогексен-1ону (0,44мл, 4,58ммоль) і 2-меркапто-5-трифторметилпіридину (0,82г, 4,58ммоль) у 20мл СН2Сl2 при кімнатній температурі додавали трихлорид вісмуту (60мг, 0,18ммоль). Реакційну суміш перемішували при кімнатній температурі протягом ночі і концентрували. Залишок очищали за допомогою радіальної хроматографії з використанням у якості елюенту суміші гексан-ЕtOАс (4:1), одержуючи 1,12г (89%) кон'югата продукту приєднання 2-(3-оксо-циклогексилтіо)-5-трифторметилпіридину (116). До розчину, що перемішується, сполуки 116 (0,26г, 0,95ммоль) у 3мл метанолу додавали відразу весь розчин бензилоксіаміну гідрохлориду (0,29г, 1,83ммоль) і ацетату калію (0,18г, 1,83ммоль) у 3мл води. Реакційну суміш перемішували при кімнатній температурі протягом ночі. Додавали 10мл води і отриману у результаті суміш екстрагували ефіром (3x10мл). Ефірні екстракти об'єднували, промивали насиченим розчином NaHCO3 (1x15мл) і соляним розчином (1x15мл). Ефірний шар сушили над MgSO4, фільтрували і концентрували з одержанням масла. Цю речовину очищали за допомогою радіальної хроматографії (гексанЕtOАс, 9:1) з одержанням відокремлених оксимів (0,32г, 89%). Ε-ізомер (Rf = 0,33) і Z-ізомер (Rf = 0,25) показали сумісні спектральні характеристики (за даними протонного ЯМР і ГРХ/мас-спектрометрії). Гідрид літію алюмінію (1,33ммоль, 50мг) суспендували у 3мл безводного ефіру і охолоджували до 0°С. По краплях додавали об'єднані оксими, розчинені у 1мл ефіру, реакційній суміші давали нагрітися до кімнатної температури і перемішували протягом 4 годин. Надлишок гідриду літію алюмінію руйнували шляхом обережного одночасного додавання води (50мкл) і 1 н. NaOH (50мкл). Суміш фільтрували і солі промивали ефіром до об'єму 100мл. Ефірний розчин екстрагували 2 н. HCI (2x50мл), щоб відділити не основні речовини. Кислотний водний розчин промивали один раз ефіром (50мл), а потім обробляли водним розчином гідроксиду натрію (2М) до рН 10-12, і екстрагували ефіром (3x50мл). Ефірний шар сушили над MgSO4, фільтрували і концентрували з одержанням 121мг (52%) бажаного зазначеного у заголовку аміну 117 у вигляді масла. Протонний ЯМР і ГРХ/мас-спектрометрія виявили продукт у вигляді суміші діастереомерів амінів (у співвідношенні 1,3:1) >95 % чистоти. 1-(5-Аміно-1,3,3-триметилциклогексил)-4-феніл-1-бутанон (120) Синтез цього аміну виконували за методом, зображеним на схемі 31. Суспензію нафталіну (1,23г, 9,57ммоль) і гранул літію (67мг, 9,57ммоль) у 10мл ТГФ перемішували в атмосфері азоту при кімнатній температурі протягом ночі. Цей розчин нафталіду літію охолоджували до -60°С і додавали феніл 3фенілпропіл сульфід (1,1г, 4,78ммоль). Реакційну суміш нагрівали до -20°С, щоб забезпечити необоротність реакції, і потім знову охолоджували до -60°С. До неї додавали розчин 7-ціано-7,9,9-триметил-1,4діоксаспіро[4.5]декану (0,5г, 2,39ммоль) у 5мл ТГФ, і розчин нагрівали до 0°С та перемішували при цій температурі протягом 2 годин. Реакційну суміш гасили шляхом додавання 10мл насиченого розчину хлориду амонію, а потім обробляли 2 н. HCI до рН ~4 і перемішували при кімнатній температурі протягом ночі. Суміш екстрагували ефіром (3x30мл), сушили над MgSO4, фільтрували та упарювали. Залишок очищали за допомогою радіальної хроматографії з використанням в якості елюенту суміші гексан-ЕtOАс (6:1). Таким чином отримали суміш (1:3) 3-(2-оксо-4-фенілбутил)-3,5,5-триметилциклогексанону 118 (136мг, Rf = 0,18) і його кеталю (509 мг, Rf = 0,33), продукту неповного гідролізу. Загальний вихід для приєднання 1-літій-3фенілпропану до нітрилу був розрахований як 85%. Силікагель (1,82г) суспендували у 10мл СН2Сl2. До цієї суспензії протягом 5 хвилин по краплях додавали 0,41мл 12% розчину HCI у воді. Суміш енергійно перемішували, щоб уникнути утворення грудочок. До неї додавали розчин вищезгаданого кеталю, розведеного у 2мл СН2Сl2, і реакційну суміш перемішували протягом 3 годин. Після цього суміш фільтрували і шар силікагелю промивали 50мл СН2Сl2. Розчинник упарювали з одержанням 0,48г (100%) 3-(1-оксо-4-фенілбутил)-3,5,5-триметилциклогексанону (118) у вигляді безбарвного масла, відповідного 118 за своїми ЯМР і ГРХ/МС властивостями. До розчину, що перемішується, цього біс-кетону (0,62г, 2,17ммоль) у 7мл метанолу додавали відразу весь розчин гідроксиламіну гідрохлориду (0,16г, 2,28ммоль) і ацетату натрію (0,25г, 3,03ммоль) у 7мл води. Реакційну суміш перемішували при кімнатній температурі протягом 1 години. Додавали 20мл води і отриману у результаті суміш екстрагували ефіром (3x20мл). Ефірні екстракти об'єднували, промивали насиченим розчином NaHCO3 (1x20мл) і соляним розчином (1x20мл). Ефірний шар сушили над MgSO4, фільтрували і концентрували з одержанням бажаного моно-оксиму 119 (0,57г, 87%) у вигляді суміші Ε і Ζ ізомерів. Нікель Raney® (0,8г у вологому стані, Aldrich Chemical Co.) у товстостінній склянці Парра місткістю 500мл для реакцій під тиском промивали водою (3x20мл), потім етанолом (3x20мл), кожного разу декантуючи розчинник, яким промивали. До цього промитого каталізатора додавали розчин оксиму 119 (0,57г, 1,89ммоль) у безводному етанолі (40мл). Отриману у результаті суміш насичували аміаком, барботуючи газоподібний аміак крізь розчин протягом 1 хвилини. Цей розчин поміщали в атмосферу водню (початковий тиск водню = 50ф/д2) в апарат Парра для струшування і струшували протягом 7 годин. Після цього реакційну суміш фільтрували крізь шар Celite® і розчинник упарювали з одержанням масла (0,43г, 80%). Аналіз методом ГРХ/мас-спектрометрії показав суміш діастереомерів (1:1) зазначених у заголовку амінів 120, поряд із незначною кількістю не ідентифікованого побічного продукту. Цю суміш амінів використовували безпосередньо як вона є, без подальшого очищення. 2-Бензил-6-метил-4-піраніламін (122) Цей амін одержували відповідно до схеми 32. До 0,37г (1,8ммоль) 2-бензил-6-метил-4-піранону (G. Piancatilli, et. al., Synthesis, 1982, 248) додавали 0,22г (3,1ммоль) гідроксиламіну гідрохлориду і 0,16г (2ммоль) ацетату натрію у 10мл метанолу. Після перемішування протягом ночі суміш розділяли між СН2Сl2 і водою. Органічну фазу сушили та упарювали. Маслянистий залишок тверднув при стоянні при кімнатній температурі, даючи 0,4г (99%) бажаного оксиму 121 у вигляді суміші (1:1) Ζ і Ε ізомерів (за даними ГРХ/мас-спектрометрії з молекулярним іоном 219), яку використовували такою, як вона є, у нижченаведеній реакції відновлення. До 0,4г 2-бензил-6-метил-4-піранон оксиму (121) (1,8ммоль) у 50мл 95% етанолу додавали 0,8г (маса у вологому стані) нікелю Raney®, який тричі промивали водою і тричі - етанолом. Суміш поміщали під тиском водню 41ф/д2 в апарат Парра для струшування, де струшували протягом 32 годин. Після продування суміш фільтрували самопливом і упарювали під вакуумом. Залишок розділяли між CH2Cl2 і водним розчином карбонату натрію. Органічну фазу сушили та упарювали під вакуумом із одержанням 0,19г суміші бажаного зазначеного у заголовку аміну 122 плюс оксиму 121 у співвідношенні 2:1, за даними ГРХ/масспектрометричного аналізу. Суміш використовували такою, як вона є, без подальшого розділення. 1-Бензоїл-4-амінопіперидин Цю сполуку одержували за методикою Bhattacharyya, et al., SynLett, 1999,11,1781. 1-(4-Метилбензил)-4-піперидиніламін (125) Синтез цієї сполуки виконували відповідно до схеми 33. До 5,05г (50ммоль) 4-гідроксипіперидину і 7,08г (50ммоль) р-метилбензил хлориду у 25мл трет-бутанолу додавали надлишок твердого карбонату калію, і суміш нагрівали на паровій бані протягом 3 годин. Суміш охолоджували до кімнатної температури і розділяли між ефіром і водою. Органічну фазу екстрагували холодною розведеною HCI, і кислотну водну фазу двічі екстрагували ефіром. Водну фазу робили основною за допомогою льоду і 50% водного розчину NaOH і екстрагували ефіром. Ефірну фазу промивали розведеним водним розчином бікарбонату натрію, соляним розчином, сушили та упарювали під вакуумом із одержанням 5,3г (52%) 1-(4-метилбензил)-4гідроксипшеридину (123) у вигляді масла. ГРХ/мас-спектрометрія показала 100% чистоту з молекулярним іоном 205. До 2,8мл (32ммоль) оксаліл хлориду у 75мл СН2Сl2 при -78°С додавали 4,6мл (64ммоль) ДМСО. До цієї суміші додавали 5,3г (26ммоль) 1-(4-метилбензил)-4-піперидинолу 123 у 10мл СН2Сl2, і суміш перемішували протягом 5 хвилин на холоді. Суміш гасили 18мл (129ммоль) триетиламіну, давали нагрітися до кімнатної температури, і додавали насичений водний розчин хлориду амонію. Органічну фазу промивали водою і соляним розчином, сушили та упарювали з одержанням 4,27г (81%) 1-(4-метилбензил)-4-піперидинону (124), який використовували таким як є, без подальшого очищення. ГРХ/мас-спектрометрія показала 100% чистоту з молекулярним іоном 203. До 4,25г (21ммоль) 1-(4-метилбензил)-4-піперидинону 124 у 200мл безводного метанолу додавали 32,2г (420ммоль) ацетату амонію і 25г 3А молекулярних сит. Після перемішування протягом 30 хвилин, до суміші порціями додавали 5,25г (84ммоль) ціаноборгідриду натрію. Після перемішування протягом 16 годин, суміш фільтрували самопливом і метанол упарювали під вакуумом. Залишок розділяли між ефіром і льодом/НСІ. Кислотний водний шар двічі екстрагували ефіром, робили основним за допомогою 50% водного розчину NaOH і льоду, та екстрагували СН2Сl2 з одержанням 2,1г (48%) зазначеного у заголовку аміну 125 у вигляді густого масла. ГРХ/мас-спектрометрія показали молекулярний іон 204. Продукт використовували таким, як є, без подальшого очищення. 1-(3-Трифторметилбеюил)-4-піперидиніламін (127) Сполуку готували відповідно до схеми 34. До 0,8г (3,1ммоль) 1-(3-трифторметилбензил)-4-піперидону [приготованого у такий же спосіб, як і 1-(4-метилбензил)-4-піперидинон) 123] у 7мл піридину додавали 0,22г (3,1ммоль) гідроксиламіну гідрохлориду, і суміш перемішували протягом ночі. Суміш упарювали під вакуумом і залишок розділяли між ефіром і розведеним водним розчином бікарбонату натрію. Органічну фазу сушили та упарювали під вакуумом із одержанням 0,52г (62%) оксиму у вигляді масла, яке використовували таким, як є, далі на стадії гідрування. ГРХ/мас-спектрометрія виявила молекулярний іон 272. До 0,5г (2ммоль) цього оксиму у 75мл етанолу додавали 0,5г (маса у вологому стані) нікелю Raney®, який тричі промивали водою і тричі - етанолом. У суміш протягом кількох хвилин барботували газоподібний аміак, і все поміщали в апарат Парра для струшування під тиском водню 45ф/д2, де струшували протягом 7 годин. Із посудини видаляли газ (вентилювали) і суміш фільтрували самопливом. Залишок розчиняли в ефірі, фільтрували та упарювали з одержанням 0,43г (81%) зазначеного у заголовку аміну 127, який використовували таким як є, без подальшого очищення. ГРХ/мас-спектрометрія виявила єдиний пік з молекулярним іоном 258. цис/транс-2-Метил-3-тетрагідрофуриламін (128) Цей амін одержували, дотримуючись методики за схемою 35. До 1,15г (10ммоль) 2метилтетрагідрофуран-3-он оксиму (приготованого за допомогою стандартних методик із комерційно доступного 2-метилтетрагідрофуран-3-ону) у 50мл метанолу додавали 1г (маса у вологому стані) нікелю Raney®, який тричі промивали водою і тричі - етанолом, і поміщали в апарат Парра для струшування під тиском водню 44ф/д2. Через 18 годин суміш продували, видаляючи газ, і фільтрували самопливом. Метанол упарювали під вакуумом, а залишок переносили в ефір і сушили. Ефірну фазу упарювали під вакуумом із одержанням 0,6г (59%) зазначеного у заголовку аміну 128 у вигляді суміші цис/транс ізомерів. ГРХ/масспектрометрія виявила 41% із молекулярним іоном 101 і 59% із молекулярним іоном 101. Суміш аміну використовували такою, як є, без подальшого очищення. 2-Бензил-2,6-диметил-4-піраніламін (133) Цей амін одержували, дотримуючись методики, відображеної на схемі 36. До 4,88г (19,7ммоль) триметилсилілового ефіру 3-триметилсилілоксимасляної кислоти у 40мл СН2Сl2 при -78°С додавали 2,4г (18ммоль) фенілацетону і 1 краплю триметилсиліл трифлату. Суміші давали постояти на холоді протягом 2 днів, після чого гасили 0,5мл піридину і давали нагрітися до кімнатної температури. Органічну фазу промивали розведеним водним розчином бікарбонату натрію, сушили та упарювали під вакуумом. Залишок переганяли під вакуумом із одержанням 2,89г (67%) 2-бензил-2,6-диметил-4-метилен-1,3-діоксан-4-ону (129) з tкип. 125-132°С при 0,6мм рт. ст. ГРХ/мас-спектрометрія виявила два ізомери, кожен із основним піком 134 (фенілацетон). До 1,5г (6,8ммоль) 2-бензил-2,6-диметил-4-метилен-1,3-діоксан-4-ону (129) в атмосфері азоту додавали 2,9г (13,9ммоль) біс-(циклопентил)-біс-метил титаноцену у 20мл зневодненого ТГФ. Суміш нагрівали зі зворотним холодильником протягом 16 годин. Реакційну суміш охолоджували до кімнатної температури і гасили надлишком ефіру. Всю суміш фільтрували крізь шар силікагелю з ефіром у якості елюенту. Фільтрат упарювали і хроматографували на силікагелі з ЕtOАс і гексаном (1:4), що містить 0,2% триетиламіну. Фракції, що містили продукт, упарювали і розводили у петролейному ефірі, та фільтрували під вакуумом із одержанням 1,2г твердої речовини. ГРХ/мас-спектрометрія виявила суміш (у співвідношенні приблизно 3:1) 2бензил-2,6-диметил-4-метилен-1,3-діоксану (130) із молекулярним іоном 218, і вихідної речовини 129. Суміш використовували такою, як є, у нижченаведеній реакції перегрупування. До 1,2г (5,5ммоль) цієї суміші у 5мл толуолу в атмосфері азоту додавали 10,99мл (11ммоль) гідриду триізобутил алюмінію при -78°С. Реакційній суміші давали постояти на холоді протягом 16 годин, і потім реакцію гасили кількома краплями води. Суміші давали нагрітися до кімнатної температури і додавали надлишок насиченого водного розчину хлориду амонію. Суміш екстрагували надлишком СН2Сl2 (важке розділення від солей алюмінію). Органічний шар сушили та упарювали з одержанням 1,1г (90%) 2-бензил-2,6-диметил-4гідроксипіранолу (131) у вигляді суміші ізомерів (75:25) (за даними ГРХ/мас-спектрометрії). До 1,1г (5ммоль) сполуки 131 у 10мл СН2СІ2 порціями додавали 1,6г (7,5ммоль) хлорхромату піридинію, перемішуючи за допомогою магнітної мішалки. Через 1 годину при кімнатній температурі додавали ефір і суміш фільтрували крізь шар силікагелю та промивали ефіром. Фільтрат упарювали з одержанням 0,88г (80%) 2-бензил-2,6-диметил-4-піранону (132). ГРХ/мас-спектрометрія показала 99% чистоти з основним піком 127 (М-бензил). Суміш ізомерів використовували такою, як є, у нижченаведеній реакції відновного амінування. До 0,88г (4ммоль) сполуки 132 у 40мл безводного метанолу додавали 6,16г (80ммоль) ацетату амонію і 5г 3А молекулярних сит. Після перемішування протягом 45 хвилин при кімнатній температурі, порціями додавали 1,02г (16ммоль) ціаноборгідриду натрію, перемішуючи за допомогою магнітної мішалки. Суміш фільтрували самопливом, і метанол упарювали під вакуумом. Залишок розділяли між ефіром і розведеною холодною HCI. Водну фазу двічі екстрагували ефіром, потім її робили основною за допомогою льоду і 50% водного розчину NaOH. Продукт екстрагували СН2Сl2, сушили та упарювали з одержанням 0,43г (49%) двокомпонентної суміші ізомерів зазначеного у заголовку аміну 133. ГРХ/мас-спектрометрія показала 58% із молекулярним іоном 128 і 42% із молекулярним іоном 128. 1-(3-Фенілпропіоніл)-4-амінопіперидин (136) Цей амін синтезували відповідно до методу, представленого на схемі 37. До 4г (40ммоль) 4гідроксипіперидину у 20мл толуолу додавали фенілпропіоніл хлорид (отриманий із 6г (40ммоль) фенілпропіонової кислоти при надлишку тіоніл хлориду). До суміші додавали надлишок 2 н. водного розчину NaOH. Після перемішування протягом 24 годин толуоловий шар відкидали і водну фазу екстрагували CH2Сl2, сушили та упарювали під вакуумом із одержанням 3,63г (39%) 1-(3-фенілпропіоніл)-4-гідроксипіперидину (134). ГРХ/мас-спектрометрія показала 100% чистоту з молекулярним іоном 233. До 1,68мл оксаліл хлориду (19,2ммоль) у 35мл СН2СІ2 при -78°С додавали 2,73мл (38,5ммоль) зневодненого ДМСО у 5мл СН2Сl2. Після цього додавали 3,6г (15,4ммоль) 1-(3-фенілпропіоніл)-4гідроксипіперидину 134 у 5мл СН2СІ2, і суміш перемішували протягом 5 хвилин на холоді. Потім додавали 10,73мл (77ммоль) триетиламіну у 5мл СН2СІ2, і суміші давали нагрітися до кімнатної температури. Суміш гасили насиченим водним розчином хлориду амонію. Органічну фазу двічі промивали водою, промивали насиченим соляним розчином, сушили та упарювали під вакуумом із одержанням 3,2г (89%) 1-(3фенілпропіоніл)-4-кетопіперидину (135). ГРХ/мас-спектрометрія показала 100% чистоту з молекулярним іоном 231. До 3,2г (13,8ммоль) сполуки 135 у 125мл безводного метанолу додавали 21,3г ацетату амонію і 20г 3А молекулярних сит. Після перемішування протягом 30 хвилин, порціями при перемішуванні додавали 3,47г (55,2ммоль) ціаноборгідриду натрію. Через 3 години суміш фільтрували самопливом, і метанол упарювали під вакуумом. Залишок розділяли між льодом/НСІ і ефіром. Кислотну водну фазу екстрагували ще двічі ефіром. Водну фазу робили основною за допомогою льоду і 50% водного розчину NaOH. Суміш екстрагували СН2Сl2, сушили та упарювали під вакуумом із одержанням 1,5г (47%) зазначеного у заголовку аміну 136. ГРХ/мас-спектрометрія виявила 100% чистоту з молекулярним іоном 232. Приготування аміну 139 Синтез цього аміну показаний на схемі 38. У тефлонову пробірку з пробкою, що загвинчується, вносили сполуку 137 (М. Shimano et al., Tetrahedron, 1998, 54, 12745) (0,80г, 1,21ммоль) і 6мл піридину. Розчин охолоджували до 0°С, обробляли 1,1мл комплексу HF-піридин і нагрівали до кімнатної температури та перемішували протягом 17 годин. Після цього додавали ще 1,1мл HF-піридину і реакційну суміш перемішували протягом додаткових 30 годин. Цю суміш виливали у льодяний розчин, що перемішується, 40мл 1 н. HCI і 20мл суміші (1:1) гексан-діетиловий ефір. Шари відділяли, і водний шар екстрагували сумішшю (1:1) гексан-діетиловий ефір (2x20мл). Об'єднані органічні шари промивали льодяною 1 н. HCI (1x20мл) і соляним розчином (1x20мл). Розчин сушили над MgSO4, фільтрували і концентрували. Сирий продукт очищали за допомогою радіальної хроматографії (гексан-ЕtOАс, 3:1) з одержанням 282мг гідроксіефіру (плюс незначні домішки), який переносили безпосередньо на наступну стадію. До розчину, що перемішується, сирого гідроксіефіру (282мг, 0,48ммоль) у піридині, охолодженого до 0°С, по краплях додавали ізобутирил хлорид (0,2мл, 1,92ммоль). Охолоджувальну баню видаляли і суміш перемішували протягом 5 годин. Додавали 2мл води і суміш перемішували протягом додаткових 30 хвилин. Розчин екстрагували ефіром (3x10мл). Ефірний шар послідовно промивали льодяною 1 н. HCI (2x10мл), насиченим розчином NaHCO3 (1x10мл) і соляним розчином (1x10мл). Розчин сушили над MgSO4, фільтрували і концентрували. Сирий продукт очищали за допомогою радіальної хроматографії (гексан-ЕtOАс, 4:1) з одержанням 171мг ізобутирилового ефіру 138 (23% загалом для двох стадій). ВОС-групу цього ефіру видаляли, дотримуючись стандартних умов видалення захисних ВОС-груп, описаних раніше, з одержанням бажаного аміну 139. Приготування аміну 145 Цей амін готували, як зображено на схемі 39. Гідроксіефір 140 (М. Shimano et al., Tetrahedron, 1998, 54,12745) (6,27ммоль) розчиняли у 15мл ДМФ і охолоджували до 0°С. До цього розчину послідовно додавали DMAP (1,53г, 12,53ммоль), EDCI (1,8г, 9,40ммоль) і N-BOC-O-Bn-(L)-тpeoнiн (2,52г, 8,15ммоль). Реакційну суміш нагрівали до кімнатної температури і перемішували протягом ночі. Розчин виливали у суміш, що швидко перемішується, 30мл льодяної 0,5 н. HCI і 50мл суміші (4:1) гексан-ефір. Шари відділяли, і водний шар екстрагували сумішшю (4:1) гексан-ефір (1x30мл). Об'єднані органічні шари промивали 0,5 н. HCI (1x20мл) і соляним розчином (2x20мл). Розчин сушили над MgSO4, фільтрували і концентрували. Сиру речовину хроматографували на силікагелі (150г) з використанням 1,25л суміші (3:1) СН2Сl2-гексани для елюювання анізальдегіду, супроводжуваного використанням суміші (65:10:25) СН2Сl2-ефір-гексани для елюювання зв'язаного продукту 141 (3,95г, 88%). Суміш бензилового ефіру 141 (1,32г, 1,84моль) і 200мг 10% паладію на вугіллі у 25мл ЕtOАс струшували в апараті Парра для струшування під тиском водню 50ф/д2 протягом 5 годин. Суміш фільтрували крізь шар Celite® і концентрували з одержанням гідроксикислоти 142 (680мг, 70%), цілком чистої за даними ЯМР аналізу. До розчину, що перемішується, гідроксикислоти 142 (1,54г, 2,86ммоль) і бензилброміду (1,5мл, 12,29ммоль) у 7мл ДМФ додавали твердий бікарбонат натрію (1,2г, 14,27ммоль). Суміш перемішували при кімнатній температурі протягом 24 годин, після чого розділяли між 25мл води і 10мл суміші (4:1) гексани-ефір. Шари відділяли і водний шар екстрагували сумішшю (4:1) гексани-ефір (2x10мл). Об'єднані органічні шари промивали 0,1 н. NaOH (1x10мл) і водою (1x10мл). Розчин сушили над MgSO4, фільтрували і концентрували. Сиру речовину очищали за допомогою радіальної хроматографії (гексан-ЕtOАс, 4:1) з одержанням 1,04г (60%) гідроксибензилового ефіру 143. До розчину, що перемішується, ефіру 143 (840мг, 1,34ммоль) і оцтового ангідриду (1,0мл, 10,68ммоль) у 7мл піридину додавали DMAP (40мг, 0,67ммоль). Реакційну суміш перемішували при кімнатній температурі протягом 4 годин і розводили 80мл EtOAc. Цей розчин послідовно промивали насиченим CuSO4 (3x30мл), 1 н. HCI (1x30мл), насиченим NаНСО3 (1x30мл) і соляним розчином (1x30мл). Розчин сушили над MgSO4, фільтрували і концентрували з одержанням 0,9г (100%) ацетату 144, досить чистого за даними спектрального аналізу. Ацетат 144 перетворювали за допомогою стадій, подібних до описаних раніше, одержуючи амін 145.
ДивитисяДодаткова інформація
Назва патенту англійськоюHeterocyclic aromatic amide, fungicide mixture based thereon and a method for control or prevention of fungous affection
Назва патенту російськоюГетероциклический ароматический амид, фунгицидная смесь на его основе и способ контроля или предупреждения грибковых поражений
МПК / Мітки
МПК: C07D 239/28, C07D 285/00, A01P 3/00, C07D 401/12, C07D 493/08, A01N 43/72, C07D 241/24, C07D 213/82, C07D 249/10, C07D 495/08, C07D 213/81, C07D 405/14, A01N 43/40, A01N 43/02, C07D 275/00, C07D 405/12
Мітки: основі, фунгіцидна, ароматичний, спосіб, амід, грибкових, уражень, контролю, попередження, гетероциклічний, суміш
Код посилання
<a href="https://ua.patents.su/85-73519-geterociklichnijj-aromatichnijj-amid-fungicidna-sumish-na-jjogo-osnovi-ta-sposib-kontrolyu-abo-poperedzhennya-gribkovikh-urazhen.html" target="_blank" rel="follow" title="База патентів України">Гетероциклічний ароматичний амід, фунгіцидна суміш на його основі та спосіб контролю або попередження грибкових уражень</a>
Фунгіцидна композиція та спосіб обробки сільськогосподарських культур для усунення або профілактики грибкових уражень

Номер патенту: 58490
Опубліковано: 15.08.2003
Автори: Ауберт Кароле, Дуверт Патріс
МПК: A01N 43/653, A01N 43/647, A01P 3/00
Мітки: культур, сільськогосподарських, обробки, композиція, уражень, профілактики, фунгіцидна, грибкових, спосіб, усунення
Формула / Реферат:
1. Фунгіцидна композиція, яка відрізняється тим, що вона містить бромуконазол (В) та інший триазол (А), вибраний з тебуконазолу та епоксиконазолу.2. Фунгіцидна композиція за п. 1, яка відрізняється тим, що вагове співвідношення В/А складає від 0,1 до 20, переважно від 0,5 до 10.3. Фунгіцидна композиція за п. 1 або 2, яка відрізняється тим, що як триазол А використовують тебуконазол та вагове співвідношення В/А складає від 0,5 до...
Фунгіцидна суміш на основі амідних сполук

Номер патенту: 65599
Опубліковано: 15.04.2004
Автори: Ейккен Карл, Лоренц Гізела, Хампель Манфред, Шелбергер Клаус, Аммерманн Еберхард, Штратманн Зігфрид, Шерер Марія
МПК: A01N 47/12, A01N 47/34, A01N 43/40, A01N 43/08, A01N 37/18, A01P 3/00
Мітки: основі, фунгіцидна, суміш, амідних, сполук
Формула / Реферат:
1. Фунгіцидна суміш, яка містить у функції активних компонентівa) амідну сполуку формули IbдеR4 означає галоген іR11 означає феніл, заміщений галогеном,б) карбоксіамід II, вибраний із групи сполук IIa та IIb,
Фунгіцидна суміш на основі амідних сполук і похідних піридину

Номер патенту: 68368
Опубліковано: 16.08.2004
Автори: Ейккен Карл, Аммерманн Еберхард, Шелбергер Клаус, Лоренц Гізела, Штратманн Зігфрид, Хампель Манфред, Шерер Марія
МПК: A01N 43/40, A01N 43/50, A01P 3/00, A01N 43/38, A01N 43/76
Мітки: амідних, сполук, фунгіцидна, суміш, піридину, основі, похідних
Формула / Реферат:
1. Фунгіцидна суміш, яка містить як активні компонентиа) амідну сполуку формули Іb, (Ib)деR4 означає галоген іR11 означає феніл, заміщений галогеном,iб) фунгіциди з групи дикарбоксімідівабов) похідну піримідину формули III,, (III)де R означає метил, пропін-1-іл або циклопропіл,абог) принаймні один активний інгредієнт формул IV або...
Фунгіцидна суміш на основі амідних сполук і похідних піридину

Номер патенту: 65600
Опубліковано: 15.04.2004
Автори: Лоренц Гізела, Шелбергер Клаус, Ейккен Карл, Аммерманн Еберхард, Штратманн Зігфрид, Хампель Манфред, Шерер Марія
МПК: A01N 47/12, A01N 57/12, A01N 43/40, A01N 37/18, A01N 43/76, A01N 47/14, A01N 37/20, A01N 37/36, A01N 43/50
Мітки: сполук, піридину, фунгіцидна, амідних, похідних, основі, суміш
Формула / Реферат:
1. Фунгіцидна суміш, яка містить у функції активних компонентіва) амідну сполуку формули Іb, (Ib)деR4 означає галоген іR11 означає феніл, заміщений галогеном,iб) дитіокарбамат(ІІ), вибраний із групи, яка містить- етиленбіс(дитіокарбамат) (цинковий комплекс) марганцю (IIа),- етиленбіс(дитіокарбамат) марганцю (ІІb),- аміачний етиленбіс(дитіокарбамат) цинку (IIc) і-...
Насіння рослин, спосіб захисту насіння від грибкових захворювань і фунгіцидна композиція для захисту насіння від грибкових захворювань

Номер патенту: 39171
Опубліковано: 15.06.2001
Автори: Шазале Моріс, Мюгн''є Жак
МПК: A01C 1/08, A01N 59/26
Мітки: спосіб, композиція, захворювань, насіння, фунгіцидна, захисту, рослин, грибкових
Формула / Реферат:
1. Семена растений, содержащие на поверхности или внутри семян эффективное количество фунгицидно действующего начала, отличающиеся тем, что фунгицидно действующее начало выбирают из группы, состоящей из фосфористой кислоты или ее солей, а указанное фунгицидно действующее начало присутствует в количестве от 1 г/100 кг до 1 кг/100 кг.2. Семена растений по п. 1, отличающиеся тем, что фунгицидно действующее начало присутствует в количестве...
Попередній патент: Похідні n-(індолкарбоніл)піперазину
Наступний патент: Фільтрувальний елемент
Випадковий патент: Світловий інформаційний пристрій для керування дорожнім рухом