Спосіб одержання діарилкарбонатів, композиція твердого каталізатора та спосіб її реактивації













Формула / Реферат
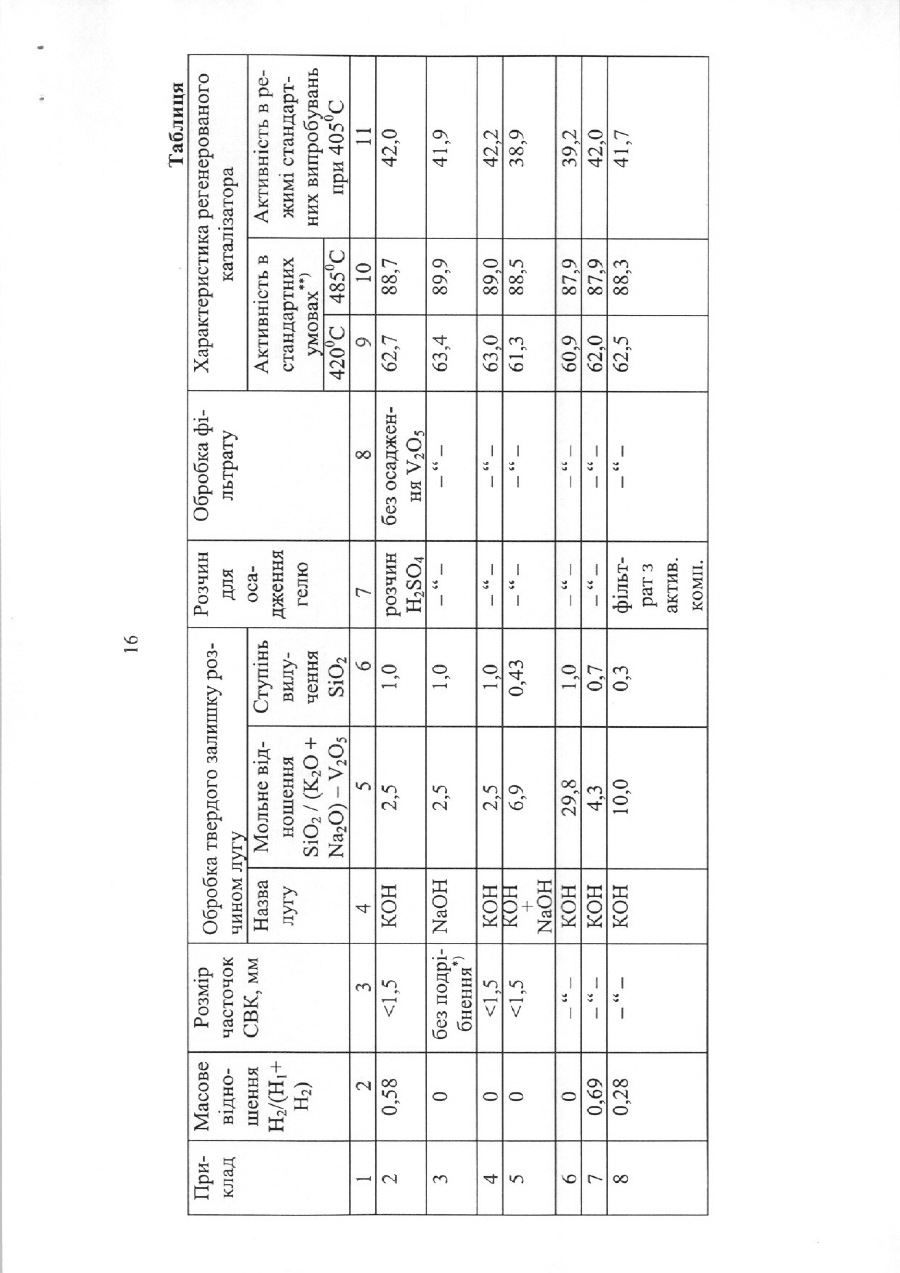
1. Спосіб одержання діарилкарбонату, який відрізняється тим, що:
(i) у первинну реакційну зону вводять діалкілкарбонат та ароматичну гідроксисполуку;
(ii) у первинній реакційній зоні підтримують двофазність та умови реакції, які ведуть до утворення алкіларилкарбонату;
(iii) трансестерифікують діалкілкарбонат ароматичною гідроксисполукою в присутності твердого каталізатора, вибраного з групи, яка складається з двох-чотирьох елементів Групи IV, V та VI Періодичної системи, на носії з пористого матеріалу, який має поверхневі гідроксильні групи;
(iv) виділяють потік двофазного продукту з первинної реакційної зони;
(v) розділяють потік двофазного продукту з (iv) для виділення пароподібного алкілового спирту та рідкого алкіларилкарбонату;
(vi) у вторинній реакційній зоні підтримують двофазність та умови реакції, які ведуть до диспропорціювання алкіларилкарбонату в діарилкарбонат;
(vii) у вторинній реакційній зоні диспропорціонують алкіларилкарбонат з (v) у присутності твердого каталізатора, вибраного з групи, яка складається з двох-чотирьох елементів Групи IV, V та VI Періодичної системи, на носії з пористого матеріалу, який має поверхневі гідроксильні групи;
(viiі) виділяють потік двофазного продукту з вторинної реакційної зони; та
(ix) розділяють потік двофазного продукту з (viii) для виділення пароподібного компоненту, який містить алкіларилкарбонат, та рідкого продукту, який містить діарилкарбонат.
2. Спосіб за п. 1, який відрізняється тим, що діалкілкарбонат являє собою діетилкарбонат (ДЕК), ароматична гідроксисполука являє собою фенол, алкіларилкарбонат являє собою етилфенілкарбонат (ЕФК), а діарилкарбонат являє собою дифенілкарбонат (ДФК).
3. Спосіб за п. 2, який відрізняється тим, що молярне співвідношення фенолу до ДФК знаходиться у межах від 0,05 до 10.
4. Спосіб за п. 1, який відрізняється тим, що твердий каталізатор в (iii) являє собою оксиди, гідроксиди, оксигідроксиди, алкоксиди або їх суміші елементів Груп IV, V та VI.
5. Спосіб за п. 4, який відрізняється тим, що носій включає оброблений кремнезем.
6. Спосіб за п. 4, який відрізняється тим, що твердий каталізатор в (vii) являє собою оксиди, гідроксиди, оксигідроксиди, алкоксиди або їх суміші елементів Груп IV, V та VI.
7. Спосіб за п. 5, який відрізняється тим, що твердий каталізатор в (iii) вибирають з групи, яка включає алкоксид металу та змішані алкоксиди металів Групи IV та V.
8. Спосіб за п. 4, який відрізняється тим, що твердий каталізатор в (iii) являє собою ТіО2/Nb2O5, на обробленому кремнеземі, де оброблений кремнезем містить менше 0,05 % мас. Na.
9. Спосіб за п. 1, який відрізняється тим, що твердий каталізатор в (vii) являє собою оксиди, гідроксиди, оксигідроксиди, алкоксиди або їх суміші елементів Груп IV, V та VI.
10. Спосіб за п. 9, який відрізняється тим, що носій включає оброблений кремнезем.
11. Спосіб за п. 10, який відрізняється тим, що твердий каталізатор в (vii) являє собою оксиди, гідроксиди, оксигідроксиди, алкоксиди або їх суміші елементів Груп IV, V та VI.
12. Спосіб за п. 11, який відрізняється тим, що твердий каталізатор в (vii) вибраний з групи, яка включає алкоксид металу та змішані алкоксиди металів Групи IV та V.
13. Спосіб за п. 9, який відрізняється тим, що твердий каталізатор в (vii) являє собою ТіО2/Nb2O5, нанесений на оброблений кремнезем, де оброблений кремнезем містить менше 0,05 % мас. Na.
14. Спосіб за п. 1, який відрізняється тим, що (vii) виконують в присутності ароматичної гідроксисполуки в молярному співвідношенні ароматичної гідроксисполуки до ДФК в межах від 0,05:1 до 10:1.
15. Спосіб за п. 14, який відрізняється тим, що (vii) виконують в присутності слідової кількості води в кількості до 0,3 % мас.
16. Спосіб за п. 1, який відрізняється тим, що (iii) виконують в присутності ароматичної гідроксисполуки в молярному співвідношенні ароматичної гідроксисполуки до ДЕК вище 0,2.
17. Спосіб за п. 1, який відрізняється тим, що вказаний носій має підвищену концентрацію поверхневих гідроксильних груп в результаті обробки розчином основи для одержання максимальної кількості гідроксильних груп без деградації фізичної цільності та міцності носія.
18. Спосіб реактивації композиції твердого каталізатора для одержання діарилкарбонатів шляхом трансестерифікації, вибраного з групи, яка включає оксиди, гідроксиди, оксигідроксиди або алкоксиди двох-чотирьох елементів Груп IV, V та VI Періодичної системи, нанесені на пористий матеріал, який має поверхневі гідроксильні групи, що була дезактивована в результаті відкладення на ній полімеру, який відрізняється тим, що приводять в контакт дезактивований каталізатор з рідиною, яка включає сполуки, що містить гідроксигрупи, у розчиннику, вибраному з групи, яка включає толуол, ксилоли, пентан, гексан, октан, декан, тетрагідрофуран та їх суміші при температурі в межах від 121,11 °С до 315,56 °С.
19. Спосіб за п. 18, який відрізняється тим, що сполуку, яка містить гідроксигрупи, вибирають з води, спирту, фенолу та їх сумішей.
20. Композиція твердого каталізатора для одержання діарилкарбонатів шляхом трансестерифікації, вибрана з групи, яка включає оксиди, гідроксиди, оксигідроксиди, алкоксиди або їх суміші двох-чотирьох елементів Груп IV, V та VI Періодичної системи, на носії з пористого матеріалу, який має поверхневі гідроксильні групи.
21. Композиція твердого каталізатора за п. 20, яка відрізняється тим, що вказаний носій додатково оброблений для підвищення числа гідроксильних груп на ньому.
22. Композиція твердого каталізатора за п. 21, яка відрізняється тим, що носій оброблений шляхом контактування з розчином основи.
23. Композиція твердого каталізатора за п. 22, яка відрізняється тим, що носій включає кремнезем.
Текст
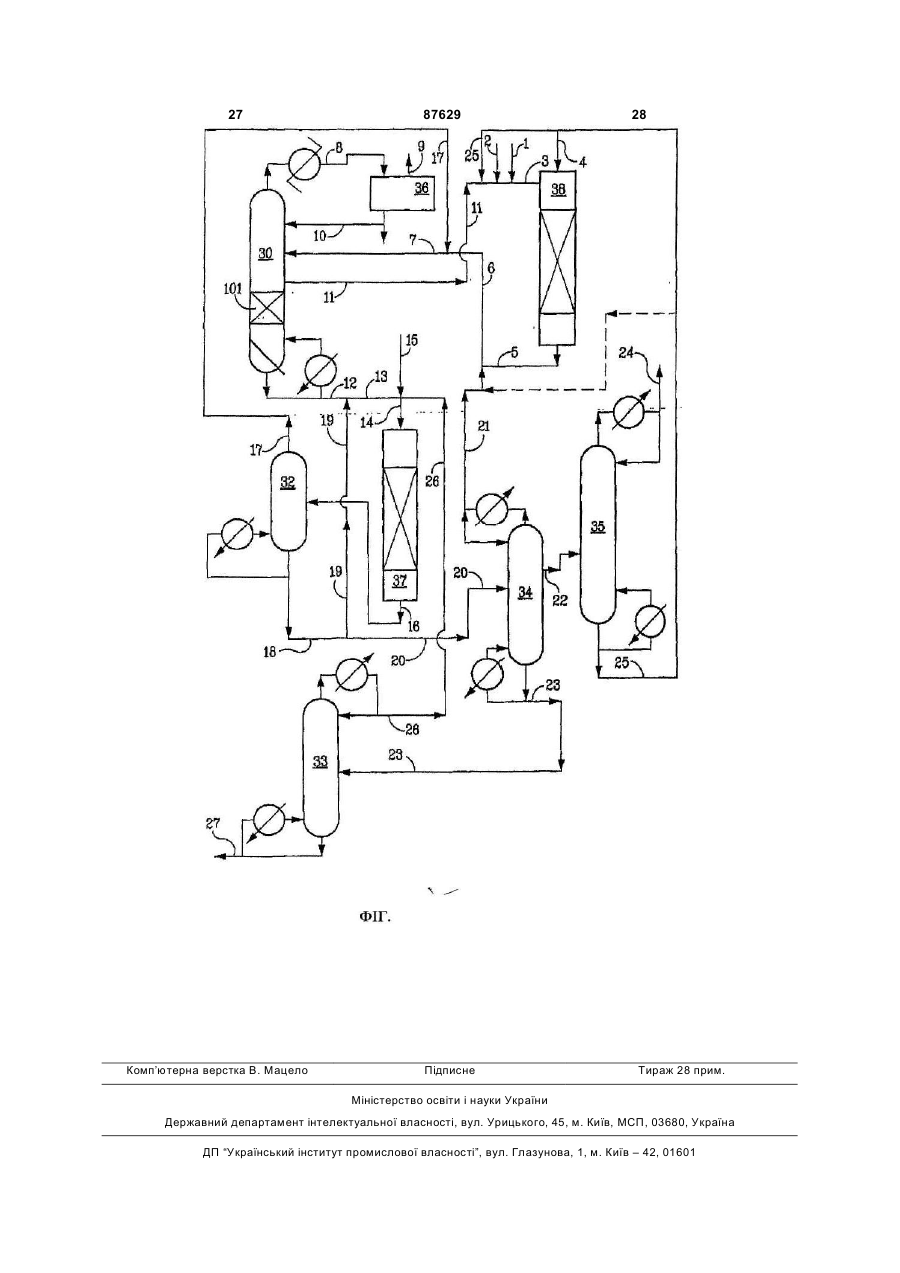
1. Спосіб одержання діарилкарбонату, який відрізняється тим, що: (i) у первинну реакційну зону вводять діалкілкарбонат та ароматичну гідроксисполуку; (ii) у первинній реакційній зоні підтримують двофазність та умови реакції, які ведуть до утворення алкіларилкарбонату; (iii) трансестерифікують діалкілкарбонат ароматичною гідроксисполукою в присутності твердого каталізатора, вибраного з групи, яка складається з двох-чотирьох елементів Групи IV, V та VI Періодичної системи, на носії з пористого матеріалу, який має поверхневі гідроксильні групи; (iv) виділяють потік двофазного продукту з первинної реакційної зони; (v) розділяють потік двофазного продукту з (iv) для виділення пароподібного алкілового спирту та рідкого алкіларилкарбонату; (vi) у вторинній реакційній зоні підтримують двофазність та умови реакції, які ведуть до диспропорціювання алкіларилкарбонату в діарилкарбонат; (vii) у вторинній реакційній зоні диспропорціонують алкіларилкарбонат з (v) у присутності твердого каталізатора, вибраного з групи, яка складається з двох-чотирьох елементів Групи IV, V та VI Періодичної системи, на носії з пористого матеріалу, який має поверхневі гідроксильні групи; (viiі) виділяють потік двофазного продукту з вторинної реакційної зони; та (ix) розділяють потік двофазного продукту з (viii) для виділення пароподібного компоненту, який містить алкіларилкарбонат, та рідкого продукту, який містить діарилкарбонат. 2 (19) 1 3 87629 4 де оброблений кремнезем містить менше 0,05 % полімеру, який відрізняється тим, що приводять в мас. Na. контакт дезактивований каталізатор з рідиною, яка 14. Спосіб за п. 1, який відрізняється тим, що (vii) включає сполуки, що містить гідроксигрупи, у розвиконують в присутності ароматичної гідроксиспочиннику, вибраному з групи, яка включає толуол, луки в молярному співвідношенні ароматичної ксилоли, пентан, гексан, октан, декан, тетрагідрогідроксисполуки до ДФК в межах від 0,05:1 до 10:1. фуран та їх суміші при температурі в межах від 15. Спосіб за п. 14, який відрізняється тим, що 121,11 °С до 315,56 °С. (vii) виконують в присутності слідової кількості во19. Спосіб за п. 18, який відрізняється тим, що ди в кількості до 0,3 % мас. сполуку, яка містить гідроксигрупи, вибирають з 16. Спосіб за п. 1, який відрізняється тим, що (iii) води, спирту, фенолу та їх сумішей. виконують в присутності ароматичної гідроксиспо20. Композиція твердого каталізатора для одерлуки в молярному співвідношенні ароматичної жання діарилкарбонатів шляхом трансестерифікагідроксисполуки до ДЕК вище 0,2. ції, вибрана з групи, яка включає оксиди, гідрокси17. Спосіб за п. 1, який відрізняється тим, що ди, оксигідроксиди, алкоксиди або їх суміші двохвказаний носій має підвищену концентрацію повечотирьох елементів Груп IV, V та VI Періодичної рхневих гідроксильних груп в результаті обробки системи, на носії з пористого матеріалу, який має розчином основи для одержання максимальної поверхневі гідроксильні групи. кількості гідроксильних груп без деградації фізич21. Композиція твердого каталізатора за п. 20, яка ної цільності та міцності носія. відрізняється тим, що вказаний носій додатково 18. Спосіб реактивації композиції твердого каталіоброблений для підвищення числа гідроксильних затора для одержання діарилкарбонатів шляхом груп на ньому. трансестерифікації, вибраного з групи, яка вклю22. Композиція твердого каталізатора за п. 21, яка чає оксиди, гідроксиди, оксигідроксиди або алкоквідрізняється тим, що носій оброблений шляхом сиди двох-чотирьох елементів Груп IV, V та VI Пеконтактування з розчином основи. ріодичної системи, нанесені на пористий матеріал, 23. Композиція твердого каталізатора за п. 22, яка який має поверхневі гідроксильні групи, що була відрізняється тим, що носій включає кремнезем. дезактивована в результаті відкладення на ній Даний винахід стосується способу одержання різних органічних карбонатів шляхом здійснення хімічних реакцій, які є обмеженими рівновагою, та розділення різних хімічних речовин, які задіяні в цих реакціях. Для кращих економічних показників є особливо бажаним досягнення вищої конверсії, ніж умова рівноваги. Даний винахід описує спосіб зсуву позиції рівноваги хімічної реакції для досягнення вищого ступеня конверсії. Діарилкарбонати, наприклад, дифенілкарбонат (ДФК), є важливим сировинним матеріалом для одержання полікарбонатів. Сучасний стан техніки пропонує одержання ДФК з диметилкарбонату (ДМК) та фенолу у дві стадії шляхом використання множини реакторів та гомогенного каталізатору, такого як алкоксид титану. В одержанні ДФК задіяні три наступні реакції. Рівновага для кожної з трьох реакцій знаходиться далеко на лівій стороні, тоді як задачею даного винаходу полягає в тому, щоб зсунути рівноваги вправо. J. L. R. Williams et al. (J. Org, Chem., 24 (1) pp.64-68, 1959) відкрив, що диспропорціювання несиметричних карбонатів, таких як метилфенілкарбонат (МФК) та симетричних карбонатів, таких як дибензилкарбонат, можна здійснювати в присутності придатного гомогенного каталізатора, наприклад, алкоксиду метала. Однак, реакція супроводжується рядом небажаних побічних реакцій, 5 87629 6 таких як розкладання карбонатних сполук до діоксполуки свинцю, сполуки міді, сполуки лужного сиду вуглецю, полімеризацією, утворенням олефіметалу, сполуки нікелю, сполуки цирконію, сполуки нів та етерів. Автори роблять висновок, що хід титану, сполуки ванадію, тощо. Побічними продукреакцій залежить від структури карбонатів та катами є СО2, анізол, бензоати та важкі матеріали. талізатора і те, що більш лужні каталізатори виUS 5426207 (1995) описує спосіб одержання діакликають більше побічних реакцій, ніж помірно рилкарбонату, такого як ДМК шляхом проведення кислий каталізатор, такий як бутоксид титану. Готрансестерифікації ДМК ароматичною гідроксиловним побічним небажаним продуктом є етер сполукою та диспропорціювання алкіларилкарбоалкілфенілу. нату в присутності гомогенного каталізатора в US 4045464 (1977) описує спосіб одержання трьох послідовних реакційних зонах. Умови вибидіарилкарбонату з фенілалкілкабонатів через реарають для максимізації утворення алкіларилкаркцію (3) в присутності гомогенних каталізаторів бонату в першій та другій реакційній зонах, в той кислоти Льюїса формули АІХ3, ТіХ3, UX4, TіX4, час як диспропорціювання проводити краще в треVOX3, VX5 та SnX4, де X явлє собою галоген, ацетій реакційній зоні. токсильну, алкоксильну чи арилоксильну групу. US 6767517 (2004) описує спосіб безперервноДФК одержували шляхом диспропорціювання етиго одержання діарилкарбонатів. Спосіб використовує три реактори реакційні дистиляційні колонкові лфенілу (ЕФК) або МФК при 180°С з 95% селектиреактори та дві ректифікаційні колонки для роздівністю в присутності гомогенних титанових каталілення проміжного продукту реакції та кінцевого заторів. продукту. US 4554110 (1985) описує поліпшений спосіб Для пом'якшення недоліків, пов'язаних з викоодержання ароматичних карбонатів з діалкілкарристанням гомогенних каталізаторів для одержанбонату та фенолу в присутності каталізатора, який ня діарилкарбонатів патенти US 5354923 (1994) та містить полімерні сполуки олова. Діарилкарбонати 5565605 (1996), WO 03/066569 (2003) описують одержують шляхом диспропорціювання алкіларигетерогенні каталізатори. лкарбонату. WO 03/066569 описує спосіб безперервного US 5210268 (1993) описує спосіб одержання одержання ароматичного карбонату, такого як діарилкарбонатів шляхом різноманітних реакцій ДФК в присутності гетерогенного каталізатора, транс етерифікації в двох реакційних зонах. Суміш одержаного шляхом нанесення діоксиду титану на ароматичного карбонату одержують за допомогою кремнезем (діаметром 3 мм) у дві стадії. реакції трансестерифікації між діалкілкарбонатом, JP 79,125617 (1979) та заявка JP NO НЕІ алкіларилкарбонатом та їх сумішшю і ароматич07[1995]-6682 описує гетерогенні каталізатори для ною гідрокси-сполукою на першому етапі. Діарилодержання дифенілкарбонату шляхом трансестекарбонат одержують шляхом здійснення диспрорифікації ДМК фенолом до МФК та диспропорціюпорціювання алкіларилкарбонат на другому етапі. вання ДМК в МФК в присутності МоО3 або V2O5, Патент описує спосіб подолання небажаної рівнонанесених на неорганічний носій, таки як кремневаги. Трансестерифікацію проводять способом зем, оксид цирконію чи діоксид титану. Трансестереакційної дистиляції шляхом введення реагентів рифікацію та диспропорціювання проводять в реау багатостадійні дистиляційні колони безперервної кторі - дистиляційній башті, яка складається з дії в присутності гомогенного каталізатора або реактора та дистиляційної башти з вилученням твердого каталізатора при безперервному вилупаралельних продуктів шляхом дистиляції. ченні одержаної суміші ароматичного карбонату як Публікація Z.-H. Fu et al. (J Моl. Catal. продукту з високою температурою кипіння з нижA:Chenical 118, (1997) pp.293-299) повідомляє про ньою частини дистиляційної колони та безперервсинтез дифенілкарбонату з ДМК та фенолу в приного вилучення легких паралельних продуктів, сутності різних гетерогенних каталізаторів на остаких як аліфатичний спирт або діарилкарбонат як нові оксидів металів. Повідомляється про найкрапотоку пари з верхньої частини дистиляційних кощу селективність для каталізатора на основі МоО3 лон. Диспропорціювання алкіларилкарбонату в (оптимальне завантаження 20%), нанесеного на діарилкарбонат та діалкілкарбонат також здійснюкремнезем. ють у подібний спосіб. Діарилкарбонат безперервЧерез багато недоліків, притаманним сучасно одержують шляхом здійснення трансестерифіним способам одержання ДФК, є велика потреба в кації та диспропорціювання послідовно, поліпшеному способі, який би забезпечив еконовикористовуючи багатостадійні дистиляційні коломію матеріалів, здешевив витрати на обладнання, ни. споживав менше енергії та здешевив витрати на Патенти США US 5872275 (1999) та 6262210 експлуатацію установки. Хоча в рівні техніки не (2001) описують спосіб одержання діарилкарбоназгадувалось про термін експлуатації каталізатора, ту з діалкілкарбонату та ароматичної гідроксивсі гетерогенні каталізатори дезактивуються і стасполуки в присутності рідкого гомогенного каталіють непридатними. Гетерогенні каталізатори затора та способи вилучення паралельних продукє·практичними в комерційному використанні, якщо тів з високою температурою кипіння і регенерації їх термін їх циклу та експлуатації є достатньо довкаталізатора для повторного циклу. US 6093842 гим, або, якщо каталізатори можна регенерувати (2000) описує спосіб одержання діарилкарбонату з in situ без значним фінансових витрат. Таким обдіалкілкарбонату, ароматичної гідрокси-сполуки та разом, проблема з дезактивацією каталізатора та змішаного розчину, який містить алкіларилкарбоспосіб регенерації є значними перепонами в коменат, шляхом введення потоків трьох реагентів в рційному застосуванні у рівні техніки. екстракційну/реакційну дистиляційну колону в присутності каталізатора. Прикладами каталізаторів є 7 87629 8 Даний винахід стосується способу одержання (f) розділення потоку продукту подвійної фази різноманітних органічних карбонатів шляхом прозі стадії (е) для відновлення пароподібного алкіловедення реакцій трансестерифікації та диспропорвого спирту та рідкого алкіларилкарбонату; ціювання в присутності твердого каталізатора в (g) підтримування вторинної реакційної зони в реакторі з подвійною фазою. Даний спосіб є вигідумовах реакції, сприятливих для диспропорціюним для проведення хімічних реакцій, обмежених вання алкіларилкарбонату в діарилкарбонат; рівновагою, в яких задіяні органічні карбонати, де (h) диспропорціювання алкіларилкарбонату в повинні співіснувати газова фаза та рідка фаза присутності твердого каталізатора, вибраного з для зсуву позиції рівноваги реакції з рівновагою групи, яка включає від двох до чотирьох елементів вправо, результатом чого є висока конверсія. Хіміз Групи IV, V та VI Періодичної Таблиці, нанесених чні реакції здійснюють в присутності одного чи на пористий матеріал, який має поверхневі гідробільше гетерогенних каталізаторів. ксильні групи; Кращими твердими каталізаторами, описани(і) вилучення потоку продукту подвійної фази з ми у винаході, є змішані оксидні каталізатори, які вторинної реакційної зони; складаються з двох чи більше різних елементів з (j) розділення потоку продукту подвійної фази Групи IV, V та VI Періодичної Таблиці, переважно зі стадії (і) для відновлення пароподібного компоТі, Zr, Hf, Nb, Та, Mo, V, Ві та Si, нанесених на поненту, який містить арилалкілкарбонат, та рідкого ристі матеріали, такі як кремнезем, який має повепродукту, який містить діарилкарбонат. рхневі гідроксильні групи. Нанесені на носій катаКраще, коли проводять наступні додаткові лізатори на основі алкоксиду або змішаного стадії: алкоксиду з групи алкоксидів металів IV та V Гру(k) розділення діарилкарбонату шляхом диспи, такі як алкоксиди титану, алкоксиди цирконію, тиляції; алкоксиди ванадію, алкоксиди ніобію, VO(OR)3 або (l) рециркуляцію арилалкілкарбонату у втоолігомери алкоксиду і т.п. складають переважну ринну реакційну зону. групу каталізаторів. Каталізатор трансестерифікаВажливо, щоби носій мав поверхневі гідроксиції та каталізатор диспропорціювання, які викорисльні групи. Кращим носієм є кремнезем. товують у даному способі, можуть буди однакові Кращим носієм є оброблений носій. чи різні. На Фігурі схематично представлений один ваЗагалом гетерогенні каталізатори є більш баріант втілення способу даного винаходу. жаними в порівняні з гомогенним каталізатором Були зроблені покращання у способі шляхом через труднощі, які мають місце у рециркуляції введення реакторів подвійної фази для виконання гомогенних каталізаторів. Однак усі гетерогенні трансестерифікації та диспропорціювання для каталізатори з часом дезактивуються. Дезактивоустановлення наступних умов способу одержання вані каталізатори треба замінювати свіжим каталіДФК: затором або регенерованим каталізатором in situ (1) введення потоку, в основному вільного від без особливих труднощів. етанолу, в реактор диспропорціювання; Для цілей даного винаходу термін „спосіб по(2) мінімізація ДЕК в потоці, який вводять в двійної фази" означає будь-який процес, який має реактор диспропорціювання; присутніми в реакційній зоні як рідку так і парову (3) здійснення трансестерифікації в присутносфазу незалежно від засобів досягнення парової та ті фенолу таким чином, щоби молярне співвіднорідкої фаз, включаючи „реакційну дистиляцію", шення фенолу до ЕФК було б вищим, ніж 0,2, кра„каталітичну дистиляцію", кипіння та реакцію, що ще вищим, ніж приблизно 0,3, а краще - вищим, проходить одночасно, та фракціоновану дистиляніж приблизно 0,35; цію в колонні. Термін „оброблений носій" чи „обро(4) здійснення диспропорціювання в присутноблений кремнезем" слід розуміти як носій, який сті фенолу таким чином, щоби молярне співвідномає оптимальну концентрацію поверхневих гідрошення фенолу до ДФК було б у межах від 0,05 до ксильних груп для даної площі поверхні для одер10, краще від 0,1 до 6, і молярне співвідношення жання описаних каталізаторів. фенолу до ЕФК становило б 0,01-6; В кращому варіанті втілення спосіб одержання (5) необов'язкове введення слідової кількості діарилкарбонату включає: води в каталітичну реакційну зону диспропорцію(а) множину реакційних зон, які включають певання в кількості до 0,3 мас.%, краще - до 0,1% рвинну та вторинну реакційну зону; мас; (b) введення у вказану первинну реакційну зо(6) використання вигоди від відсутності азеотну діалкілкарбонату та ароматичної гідроксиропу ДЕК/етанол для відділення ДЕК від етанолу; сполуки, (7) виключення в основному холодних місць в (c) підтримування первинної реакційної зони у каталітичних реакційних зонах; подвійній фазі та в умовах реакції, сприятливих (8) виключення рециркуляції каталізатора, для утворення алкіларилкарбонату; розділення каталізатора та безперервного дода(d) трансестерифікацію діалкілкарбонату аровання приготованого каталізатора; матичною гідрокси-сполукою в присутності твердо(9) ефективне змішування реагентів та продукго каталізатора, вибраного з групи, яка включає від тів; та двох до чотирьох елементів з Групи IV, V та VI (10) переважне підтримування присутності Періодичної Таблиці, нанесених на пористий маобох парової та рідкої фаз в реакційних зонах для теріал, який має поверхневі гідроксильні групи, вилучення продукту реакції з низькою температу(е) вилучення потоку продукту подвійної фази рою кипіння в газову фазу для отримання високої з первинної реакційної зони; конверсії, не обмеженої константою рівноваги. 9 87629 10 Реактори з нерухомими шаром працюють за ційної суміші в реакторі з подвійною фазою, як способом подвійної фази, а це означає спільне описано у данному винаході, стає відносно легко існування як пару так і рідини в реакційних зонах, створити правильний об'єм газової фази в реакщо створює умови кипіння в каталітичній реакційційній зоні при даних умовах реакції та підтримуній зоні реактора з подвійною фазою, що є бажавати стан стабільної роботи реактора. ним, не дивлячись на це робота реактора подвійСуттєвими перевагами, які одержані в порівної фази в умовах кипіння не є необхідною нянні з рівнем техніки, є такі: настільки, наскільки довго обидві газова і рідка (a) висока продуктивність бажаних продуктів; фази співіснують в реакційній зоні. (b) висока селективність бажаних продуктів; Напрямок потоку в реакторі подвійної фази (c) немає потреби відділяти каталізатор від може бути як потік знизу або потік зверху. Потік потоку продукту реакції; знизу є кращим. Також реактори з подвійною фа(d) високий строк експлуатації каталізатора; зою та нерухомим шаром мають обвідні трубопро(e) менше споживання енергії; та води для рециркуляції. Реактор з температурою (f) легка регенерація дезактивованого каталікипіння в режимі потоку вниз працює переважно заторі. таким чином, щоб він мав негативне падіння тиску, Вхідний потік в реактор трансестерифікації пещо означає нижчий тиск внизу каталітичного шару реважно включає свіжий фенол та ДЕК та змішаніж вгорі каталітичного шару. Негативне падіння ний рециркуляційний потік включає ДЕК, PhOH та тиску створюється потоком маси з високою швидЕФК. Вхідний потік, який в основному не містить кістю крізь нерухомий шар гетерогенного каталізаетанолу, в реактор трансестерифікації створює тора в каталітичній реакційній зоні. Негативне пасприяливі умови для конверсії фенолу та високої діння тиску є бажаним, але не необхідним. швидкості реакції. Це також справедливо для вхідБажаним є негативне падіння тиску становить ного потоку, який не містить ДЕК, в реактор дисприблизно 0,2 фунтів/дюйм2 на фут або більше. пропорціювання. В каталітичних реакційних зонах - транспорт маси реагентів з насипної фази в практично немає холодних місць, що є важливим частинки каталізатора-та транспорт продуктів реадля підтримування стабільних швидкостей реакції. кції зсередини пор формованих гранул каталізатоОскільки в реакторах подвійної фази трансестера в каталітичну реакційну зону; рифікації та диспропорціювання присутні обидві - бокове перемішування реагентів та продуктів парова та рідка фази, рідкі продукти реакції (етав насипній фазі; нол та ДЕК в кожній реакційній зоні) випаровують- не відбувається розвиток або відбувається ся в газову фазу, яка створює сприятливі умови незначний розвиток місць з падінням температури для високої продуктивності призначених продуктів. або гарячих місць в каталітичній реакційній зоні як Так як немає впливу небажаної високої темперадля ендо- так і для екзотермічної реакції; тури на реагенти чи продукти в присутності каталі- видалення легких продуктів реакції з рідкого затора, менше утворюється небажаних побічних реакційного середовища в газову фазу, результапродуктів, що робить більш легким та дешевим том чого є сприятливий напрямок рівноваги. очищення потоку сирого продукту ДФК. Через те, При даній температурі реакції та швидкості пощо вхідний потік в реакційну зону диспропорціютоку в каталітичній реакційній зоні умови реакції вання може складатись зі сполук з доволі високою кипіння в каталітичній реакційній зоні створюються температурою кипіння, таких як ЕФК чи МФК, азот шляхом контролю тиску, який визначається склаабо компонент з низькою температурою кипіння дом реакційного середовища. Необов'язково можвводять в реакційну зону для створення достатна вибирати чи використовувати розчинний з нижнього об'єму газової фази в реакційній зоні. Причою температурою кипіння для створення кращих кладами таких компонентів з низькою температуумов кипіння в каталітичній реакційній зоні, коли рою кипіння є етиловий етер, пропіловий етер, дим температура кипіння реагенту чи продукту близька етиловий етер, метан, етан, пропан, бутан, гексан, або вища за передбачувану температуру реакції. гептани, толуол та ксилени. Даний винахід особливо є корисним для реакКраще, коли всі чи принаймні частина компоцій, де температура кипіння продукту реакції є винентів з низькою температурою кипіння вводять в щою за температуру реакції, або дуже близькою реакційну зону як дуже нагрітий газ. Оскільки етадо температури реакції, таким чином, що не буде нол та ДЕК не утворюють азеотропу, відділення зовсім чи буде недостатній об'єм газової фази в ДЕК для рециркуляції приводить до меншого спореакційній зоні в умовах реакції. Приклади такої живанні енергії та менших витрат на обладнання у реакції включають одержання дифенілкарбонату порівнянні з існуючими комерційними способами, шляхом диспропорціювання етилфенілкарбонату де ДФК одержують з ДМК та фенолу. або метилфенілкарбонату, або одержання біс-(2Є дві або три реакційні зони. В першій реакетил-1-гексил) карбонату шляхом реакції трансесційній зоні алкіларилкарбонат, такий як ЕФК одертерифікації алкілкарбонату 2-етил-1-гексанолом. жують шляхом транс етерифікації ДЕК фенолом. В Краща температура реакції знаходиться в межах другій реакційній зоні Одержують ДФК шляхом від приблизно 200° до 400°F. Оскільки ДМК та ДЕК диспропорціювання ЕФК. Реакційні зони включакиплять при температурах вищих, ніж приблизно ють два реактори з нерухомим шаром. Необов'яз305°F, утворення парової фази у великому комерково три реакційні зони можуть включати три реакційному реакторі з нерухомим шаром стає важкою тори з нерухомим шаром, або два реактори з задачею при відносно великих швидкостях потоку нерухомим шаром та каталітичний дистиляційний рідини. Шляхом контролювання потоку об'єму пару колонковий реактор. Реактори завантажують одв реактор при даній швидкості потоку рідкої реак 11 87629 12 ним або двома різними гетерогенними каталізатошляхом спільного осадження чи просочування у рами. порошкоподібній формі чи формованій формі, а Прикладами органічних карбонатів одержаних потім випалити від 200°С до приблизно 750°С, згідно з даним винаходом є ДФК, ЕФК, МФК, ДЕК, краще - від приблизно 250°С до приблизно 600°С ДМК, біс-(2-етилгексил) карбонат та карбонати на повітрі. Спільне осадження чи просочення можмоногліцериду жирної кислоти. Даний винахід є на проводити у водній фазі або у органічній фазі, особливо корисним в одержанні дифенілкарбонату такій як вуглеводні, етери, кетони, спирти та їх (ДФК) з ДЕК та фенолу. Хоча ДФК є кращим діалсуміші. Коли осадження проводять в органічній кілкарбонатом для одержання ДФК; цілком зрозуфазі, краще використовувати органометалеві споміло, що спосіб є також корисним для одержання луки. Наприклад, два різних розчини різних оргаДФК шляхом використання ДМК або будь-якого нометалевих сполук додають в придатному оргаіншого алкілкарбонату або алкіларилкарбонату. нічному розчиннику в одночасно при інтенсивному Для запобігання перегріву ребойлера при проперемішуванні в умовах осадження при придатних веденні трансестерифікації та диспропорціювання температурах. Інколи при перемішуванні чи після в каталітичному дистиляційному колонковому реанього необхідним є третій розчин для гелеутвокторі безпосередньо в ребойлер можна прокачурення чи осадження. Наприклад, третім розчином вати компонент з низькою температурою кипіння може бути вода, основний чи кислотний водний або суміш компонентів з низькою температурою розчин в придатному органічному розчиннику, такипіння. кому як спирт, кетон, органічний естер чи їх суміші. Таким чином, дана технологія є частиною даДругим необов'язковим способом є одночасне ного винаходу для одержання різних органічних додавання першого органометалевого розчину та карбонатів як для твердих каталізаторів, так і для третього розчину до другого органометалевого гомогенного каталізатора, який використовують у розчину при інтенсивному перемішуванні. При неспособі. Дана технологія не була описана у рівні обхідності осади піддають старінню при придатній техніки для одержання органічних карбонатів, татемпературі від приблизно 25°С до приблизно ких як ДФК, ДЕМ та ДЕК. 200°С протягом періоду від 30 хвилин до приблизКращими гетерогенними каталізаторами є но 30 годин в придатному середовищі. Іноді спільзмішані оксиди, гідроксиди та алкоксиди елементів но осаджений продукт у водному розчині піддають IV, V та VI Групи на носіях, які осаджені на пористі старінню в нейтральному, слабо кислотному чи основи. Ці елементи осаджують на оксидні, гідроосновному органічному середовищі. ксидні або оксигідроксидні форми пористого носія, Середовище, яке використовують для старінтакого як кремнезем, оксид цирконію чи діоксид ня, може чи не може містити невелику кількість титану. За формою носії можуть являти собою води в залежності від природи матеріалу, який пелети, гранули, екструдати, сфери і т.п. розміром піддають старінню. Середовище, яке використоприблизно 1 - 5 мм. Осаджена можна проводити у вують для старіння, може бути слабокислим, слаодностадійний чи багатостадійний спосіб. Приклабоосновним чи нейтральним. Продукт після стадами змішаних оксидних каталізаторів є: Nb2O3ріння висушують при температурі від приблизно ТіО2, V2O3-ТіО2, МоО3-ТіО2, ТіО2-ZrO2, Nb2O5-V2O3, 100°С до приблизно 400°С, а потім випалюють при MoO3-V2O5, MoО3-ZrO2, TiO2-ZrO2-SiO2, TiO2-Nb2O5температурі від приблизно 250°С до приблизно SiO2, MoO3-Nb2O5-TiO2, V2O5 Nb2O5-TiO2, MoO3750°С. При необхідності просочування одним чи Nb2O5-SiO2, TiO2-Bi2O3-SiO2, MoO3-Nb2O5-ZrO2, двома елементами на придатному носії провоTiO2-Nb2O5-Bi2O3, MоO3-V2OS-TiO2, TiO2-Bi2O3-SiO2, дять, використовуючи органічний розчин, який місMoO3-Bi2O3-SiO2, TiO2-ZrO2 -Bi2O3-SiO2 та TiO2тить одну чи дві органометалеві сполуки, або водZrO2-Nb2O5-Bi2O3-SiO2. ний розчин, який містить одну чи дві сполуки. Загальна методика одержання цих змішаних Необов'язково можна здійснювати багаторазові оксидних каталізаторів включає просочування та просочування, використовуючи різні розчини. спільне осадження, або комбінацію цих двох споОднак, фахівець може вибрати для викориссобів, які виконують як одностадійний чи багатотання будь-який гетерогенний каталізатор, описастадійний. Фахівець може виконати просочування ний в рівні техніки в залежності від того, наскільки одного, двох чи трьох металевих компонентів на каталізатор придатний для реактора з нерухомим пористий носій чи носій зі змішаного оксиду, одершаром для роботи великого комерційного реактожаних спільним осадженням. Просочування можна ра. Прикладами гетерогенних каталізаторів, опивиконувати у одностадійний чи багатостадійний саних в рівні техніки є оксид титану, TS-1, Ті-МСМспосіб. 41, оксид молібдену, оксид ванадію, оксид свинцю Продукти, одержані спільним осадженням чи та змішаний оксид MgLa як такі та краще нанесені просоченням, які одержані у порошкоподібній фона носій, як описано тут. рмі, піддають тепловій обробці при температурах Важливо, щоби носій мав поверхневі гідроксивід приблизно 150°С до приблизно 600°С. Порошльні групи. Кремнезем є кращим носієм. Термін коподібним матеріалам придають форму зручного „оброблений носій" чи „оброблений кремнезем" розміру від приблизно 1 мм до приблизно 5 мм слід розуміти як носій, що має оптимізовану кондля використання у реакторі з нерухомим шаром. центрацію гідроксильних груп для даної питомої Формовані матеріали випалюють при температурі поверхні для приготування описаних тут каталізавід 200°С до приблизно 750°С, краще - від приблиторів. В залежності від способу приготування крезно 250°С до приблизно 600°С на повітрі. Необомнезему, кремнезем може не мати достатнього в'язково один чи два металеві компоненти можна числа поверхневих гідроксильних груп для даної осадити на формований матеріал, одержаний питомої поверхні. Для такого кремнезему кремне 13 87629 14 зем оброблюють для введення додаткових поверди ніобію і т.п. Алкоксиди металів V Групи вклюхневих гідроксильних груп за допомогою водного чають алкоксид нижчої валентності, такий як тетосновного розчину, а потім ретельно промивають раалкоксид та окситриалкоксид, такий як VО(OR)3 водою, а після піддають випалюванню при темпеабо олігомери чи оксоалкоксид. Прикладами краратурі від 280°С до 650°С перед використанням. щих носіїв є кремнезем, оксид цирконію, діоксид Необов'язково комерційно доступні кремнеземні титану, діоксид титану - кремнезем, кремнезем носії фахівець може піддавати регідратації. Регідоксид алюмінію та кремнезем - оксид цирконію. ратований кремнезем випалюють при температурі Застосування обробленого кремнезему є особлипри температурі від 280°С до 650°С для оптимізаво важливим для приготування каталізаторів на ції концентрації щільності поверхневих гідроксильоснові алкоксиду металу на носії. Каталізатор на них груп перед використанням. Таким чином, краоснові алкоксиду металу на носії може мати один щим кремнеземним носієм, який використовують чи два алкоксиди різних металів. для приготування твердого каталізатора, є „оброГетерогенні каталізатори на основі алкоксидів блений кремнезем". До класу кращих носіїв, зокметалів одержують шляхом контактування розчину рема кремнеземних носіїв, належать такі, у яких алкоксиду металу або змішаного розчину двох концентрація поверхневих гідроксильних груп була алкоксидів різних металів з носієм, таким як кремзбільшена шляхом обробки основним розчином, як незем при температурі від приблизно 20° до приописано, для одержання максимального числа близно 400°F, краще - від приблизно 40° до пригідроксильних груп без деградації фізичної цільноблизно 300°F. Розчин алкоксиду одержують сті та міцності носія. Контроль за вмістом натрію шляхом розчинення одного чи двох різних алкокна кремнеземному носії є дуже важливим для сидів металів у розчиннику. Розчинник не повинен одержання ароматичних карбонатів, таких як ЕФК у будь-який спосіб впливати на реакцію утворення та ДФК, оскільки основні домішки, такі як оксид кисневого містка. Прикладами таких розчинників є лужного металу на кремнеземі спричинює небавуглеводні, етери, кетони, спирти та їх суміші. Кожані побічні реакції та призводить до нестабільноли два різних алкоксиди металів наносять на ності каталізатора. Лужний метал на кремнеземному сій, необов'язково готують два розчини алкоксидів носії призводить до нестабільності роботи каталірізних металів і вводять в реакцію з носієм послізатора та небажаних побічних реакцій для трансдовно. естерифікації та диспропорціювання, які продукуM(OR)n + x ОН (на поверхні носія) = > (RO)nють алкіларилкарбонат та діарилкарбонат. x(O-)x (на каталізаторі) + x ROH Кращий „оброблений кремнеземний" носій буде де мати менше ніж приблизно 0,05% Na, краще - меn = 4 або 5, X = 1, 2, 3 або 4 та R = алкільна чи нше ніж приблизно 0,03% Na. Обробка кремнезему арильна група водним розчином лужного металу дає додаткову Алкоксид титану на носії являє собою кислотперевагу, яка стосується розширення пор. Однак, ний каталізатор. Вища активність каталізатора на вилуговування надто багато кремнезему з кремнеоснові алкоксиду титану на кремнеземному носії у земного носія під час обробки розчином лужного порівнянні з гомогенним каталізатором на основі металу призводить до виникнення проблеми, поалкоксиду титану відноситься до вищої кислотносв'язаної зі збереженням фізичної цільності та міцті Ті+4 на носії. Кислотність каталізатора відіграє ності. важливу роль для реакцій трансестерифікації та Іншими каталізаторами, описаними в даному диспропорціювання, які каталізуються кислотою, в винаході, є каталізатори на основі алкоксиду меодержанні ароматичних карбонатів. талу чи змішаних алкоксидів металів, нанесених Каталізатор на основі алкоксиду металу на нона носії, які одержують шляхом зв'язування алкоксії можна приготувати in situ, що являє собою несидів металів з пористими носіями через кисневі обов'язковий спосіб. Реактори завантажують обмісткові зв'язки. Кращим носієм є оброблений креробленим носієм. Розчини алкоксиду металлу мнезем, який має менше ніж приблизно 0,05% Na, циркулюють в реакторі при температурі від кімнаткраще - менше ніж приблизно 0,03% Na. Оптиміної до приблизно 400°F. Після утворення каталізазація концентрації поверхневих гідроксильних груп тора на основі алкоксиду металу на носії будьна кремнеземному носії є дуже важливою для який розчинник, що залишився, з реакторів вилустворення стабільних, міцно з'єднаних активних чають. Після промивання реакторів придатним центів алкоксидів металів, оскільки для з'єднання розчинником, таким як етанол, пентан чи толуол, активних атомів металів з поверхнею кремнезему та після необов'язкової теплової обробки каталізачерез зв'язки М- О - Si не використовують випалюторів в потоці інертного газу, такого як азот, при вання при високій температурі. Особливо бажаною температурі від приблизно 80° до приблизно є одержання максимальної кількості зв'язків М-О400°F, краще від приблизно 100° до приблизно Si. 350°F каталізатори готові для трансестерифікації Таким чином, оптимальна концентрація гідрота диспропорціювання. Для приготування каталіксильних груп для даної поверхні включає максизаторів на основі металу чи змішаних алкоксидів мальну кількість гідроксильних груп, яку можна металів на носії особливо важливо використовуваодержати для даної питомої поверхні у межах бати „оброблений носій". Прикладом обробленого лансу між домішками лужного металу і міцністю носія є оброблений кремнезем, як описано вище. носія, як описано вище. У даному винаходу спостерігали дезактивацію Кращими алкоксидами металів є алкоксиди гетерогенних каталізаторів. Це особливо проявляметалів Групи IV та V, такі як алкоксиди титану, лось в реакції диспропорціювання алкіларилкаралкоксиди цирконію, алкоксиди ванадію, алкоксибонатів, таких як МФК, ЕФК і т.п. Виявили, що при 15 87629 16 чиною дезактивації каталізатора є відкладення Несподівано було виявлено, що таку дезактиважких полімерів на твердому каталізаторі, який вацію каталізатора можна пом'якшити шляхом блокує активні центри каталізатора і заповнює проведення реакції диспропорціювання в присутпори каталізатора. Дезактивований каталізатор ності ароматичної гідрокси-сполуки, такої як фестає темно-коричневим чи чорним в залежності від нол. Більш того, є додаткова вигода від проведенступеня осадження полімеру. Швидкість дезактиня диспропорціювання в присутності слідової вації для даного гетерогенного каталізатора значкількості води в каталітичній реакційній зоні в кільно швидша для реакції диспропорціювання, ніж кості до 0,3% мас, краще - до 0,10% мас. Виявлядля реакції трансестерифікації. Було винайдено, ється, що значна частина важких полімерів, осащо дезактивований каталізатор можна регенеруджених на каталізаторах, є полікарбонати. вати in situ шляхом деполімеризації полімерів, Результатом проведення диспропорціювання в осаджених на каталізатор, шляхом контактування присутності ароматичної гідроксильної сполуки, дезактивованого каталізатора зі сполукою яка місабо як ароматичної гідроксильної сполуки та і слітить гідроксильну групу, такою як пара, метанол, дової кількості води в каталітичній реакційній зоні є етанол, фенол або суміші гідрокси-сполук при підприйнятно стабільна робота каталізатора. Однак, вищеній температурі. Необов'язково можна викослід розуміти, що надто велика кількість ароматиристовувати розчин гідрокси-сполуки у розчиннику. чної гідрокси-сполуки призводить до неприйнятно Кращими розчинниками є бензол, толуол, ксилени, низької швидкості одержання ДФК через рівновагу пентан, гексан, октан, декан, ТГФ або будь-які суреакції трансестерифікації. Таким чином, підтриміші розчинників. Використовувати розчинник для мання молярного співвідношення ароматичної деполімеризації полімерів, осаджених на дезактигідрокси-сполуки до ДФК від 0,05 до 10, краще - від вований каталізатор, можна лише за бажанням і 0,1 до 6 в реакційній зоні диспропорціювання є це не є обов'язковим. Регенерований каталізатор важливим як для забезпечення довгого прийнятнокраще висушувати при температурі від 200° до го циклу роботи каталізатора так і для хорошої приблизно 500°F в потоці інертного газу (наприпродуктивності ДФК. Для реакції транс етерифікаклад, азоту) перед застосуванням. Регенерацію ції молярне співвідношення фенолу до ДФК підкаталізатора сполуками, які містять гідрокси-групи, тримують на рівні вище 0,2, краще - вище 0,3, а ще здійснювали шляхом обробки дезактивованих какраще - вище 0,35. талізаторів in situ в потоці спирту або розчину Приклад схематичної схеми виробничого проспирт-вода, чи паром при температурі від 250° до цесу одержання ДФК наводиться на Фігурі. Там 600°F, краще - від 270° до 450°F. Обробку дезакзображені два нерухомих шари, реактори подвійтивованих каталізаторів можна здійснювати спільної фази та п'ять дистиляційних колон. Перший но розчином спирту та паром. Наприклад, дезакреактор подвійної фази 38 в першу чергу признативований каталізатор можна обробити спочатку чений для реакції трансестерифікації для одерспиртом чи розчином спирту, а потів паром, чи жання ЕФК з ДЕК та фенолу. Другий реактор понавпаки. Регенерацію каталізатора краще проводвійної фази 37 в першу чергу призначений для дити при достатньому тиску, щоби, принаймні, реакції диспропорціювання для одержання ДФК з деяка кількість водної фази залишилась в шарі ЕФК. Необов'язково можна ввести додаткові реаккаталізатора. Кращим спиртом є метанол, етанол тори з нерухомим шаром (не показані) послідовно або суміші цих двох спиртів. Необов'язково розчин між реакторами 38 та 37. Головною задачею цього спирту може містити воду в кількості до 80% мас, додаткового реактора є одержання додаткового краще - до 20% мас, а ще краще - до 5% мас. ПоЕФК. Вхідний потік свіжого фенолу 1 та вхідний тік з реактора деполімеризації містить фенол, ДЕК, потік свіжого ДЕК 2 змішують з рециркульованими ЕФК та слідові кількості фенетолу та важких спопотоками 11 та 25 і потім вводять в реактор полук як продуктів реакції деполімеризації. двійної фази 38 через трубопровід 3. Необов'язкоДля використання гліколевого етеру для виво в 38 вводять азот через трубопровід 4. Одермивання полімерів на каталізаторі и покращання жують продукт ЕФК та паралельний продукт селективності розчинник, як описано вище, ввоетанол шляхом проведення реакції трансестеридять як компонент реакційної суміші в каталітичну фікації в 38. В реакторі 38 паралельний продукт реакційну зону та виділяють з реакційної суміші етанол випаровують у газову фазу. Потік 5, що для рециркуляції. Прикладами таких етерів як розвиходить з реактора, вводять в першу дистиляційчинників є діетиловий етер етиленгліколю, димену колону 30 через трубопроводи 5, 6 та 7. Етанол тиловий етер етиленгліколю, діетиловий етер діевідганяють в колоні 30 у верхній потік 8 з невелитиленгліколю, диметиловий етер діетиленгліколю і кою кількістю ДЕК. Потік 8 частково охолоджують і т.п. потім вводять в барабан-сепаратор „газ-рідина" 36 Методика регенерації каталізатора, описана в і потік газу 9 рециркулюють в реактор 38 і 37. Потік цьому винаході, може бути використана у будьрідини 10 (етанол) з 36 рециркулюють в установку якому способі одержання ароматичних карбонатів, одержання ДЕК для одержання ДЕК. Боковий потік де використовують гомогенний каталізатор. Для 11 з колони 30, який складається з ДЕК, фенолу та регенерації гомогенної каталітичної системи розЕФК, рециркулюють знову в реактор 38 через тручин спирту повинен бути достатньо сухим, щоби бопровід 3. Необов'язково за бажанням можна вміст води не перевищував приблизно 0,2% мас. встановити каталітичний шар 101 в нижній секції Таким чином, методика регенерації каталізатора, колони 30 нижче точки відходу бокового потоку описана в цьому винаході, є придатною для будьдля додаткової конверсії фенолу в ЕФК. Дистиляякого способу одержання органічних карбонатів. ційна колона 30 сконструйована та працює таким чином, що боковий потік 11 в основному не містить 17 87629 18 етанолу. Рециркуляційний контур для реактора продукт являв собою білосніжну гранулу. Загальна подвійної фази 38 включає трубопроводи 5, 6 та 7, вага каталізатора становила 9,67 г. Це - каталізаколону 30 та трубопроводи 11 і 3. Нижній потік 12 з тор А. реактора 30 вводять в реактор подвійної фази 37 Приклад 2 через трубопроводи 13 і 14. Потік 12 містить ЕФК Каталізатор на основі змішаного оксиду ніота фенол. Але він також містить невеликі кількості бію/титану на кремнеземному носії приготували ДФК та побічний продукт - фенетол. Колона 30 згідно з винаходом. 0,592 г Nb(OC4H9-n) розчинили також сконструйована і працює для мінімізації ДЕК в 80 мл толуолу. Гранульований кремнезем, виков нижньому потоці 12. Потік 12 комбінують з рециристаний в Контрольному Прикладі 1, висушували ркульованим потоком 19 в потік 13. Потік 13 комбіпри 320°С протягом 2 годин на повітрі. 25 мл (9,27 нують з потоком газу - азоту 15 в потік 14, який г) цього сухого кремнезему кип'ятили зі зворотним вводять в 37. В реакторі 37 одержують продукт холодильником з вищевказаним розчином бутокДФК та паралельний продукт ДЕК шляхом провесиду ніобію. Після кип'ятіння протягом 5,5 годин за дення диспропорціювання ЕФК. ДЕК в реакторі 37 тією ж методикою, що і в Контрольному Прикладі випаровують у газову фазу. Потік 16, який вихо1, надлишок толуолу випарили з колби. Розчин дить з реактора вводять в другу дистиляційну ководи в метанолі, одержаний шляхом змішування лону 32. Потік 16 складається головним чином з 0,209 г води з 120 мл метанолу, вводили в колбу і ДЕК, ЕФК, фенолу, ДФК та невеликих кількостей потім розчин кип'ятили зі зворотним холодильниетанолу і побічних продуктів. Етанол та ДЕК в поком в кип'ячому метанолі протягом години. Надтоці 16 відганяють разом з парою в колону 32 як лишок метанолу злили з колби. Розчин тетрабутоверхній потік 17, який вводять в першу колону 30 ксиду титану, приготований шляхом розчинення через трубопровід 7. Нижній потік 18 з колони 32 3,56 г бутоксиду титану в 90 мл толуолу, влили у розділяється на два потоки 19 і 20. Потік 19 рецирколбу і потім вміст колби кип'ятили зі зворотним кулюють знову в другий реактор 37 через трубохолодильником в кип'ячому метанолі протягом 5,5 проводи 13 і 14. годин. Надлишок метанолу випарили з колби. МаРециркуляційний контур для другого реактора теріал, який знаходився у колбі, вилучили з колби подвійної фази 37 включає трубопровід 16, колону та висушили при 120°С у вакуумній печі протягом 32 та трубопроводи 18, 19, 13 і 14. Другий потік 20 1,5 години. Висушений кремнезем випалювали вводять в третю дистиляційну колону 34, де ДЕК, при 500°С протягом 2 годин Зовнішній вигляд вищо залишився в потоці, виділяють як верхній потік паленого продукту відрізнявся від каталізатору в 21, який направляють в першу колону 30 через Контрольному Прикладі 1. Він нагадував більше трубопроводи 6 та 7. Боковий потік 22 з 34 вводять гранульований кремнеземний носій ніж білосніжв четверту дистиляційну колону 35 для видалення ний каталізатор в Контрольному Прикладі 1. Загапобічного продукту - фенетолу як верхній потік 24. льна вага каталізатора становила 10,28 г. Це Нижній потік 25 з колони 34 рециркулюють в 38. каталізатор В. Необов'язково нижній потік 25 з 35 можна рецирПриклад 3 кулювати в першу колону 30 через трубопроводи В цьому прикладі оброблений кремнезем ви31, 21, 5 та 6. Нижній потік 23 з 34 вводять в дискористовували для приготування каталізатора на тиляційну колону 33 для добування продукту ДФК. основі оксиду титану/ніобію, нанесеного на кремВерхній потік 26, який складається головним чинезем. Каталізатор на основі змішаного оксиду ном з ЕФК, рециркулюють в реактор диспропорцініобію/титану на обробленому кремнеземі готуваювання 37 через трубопровід 14. Колона 33 прали згідно з винаходом. Для приготування обробцює при тиску нижче атмосферного. Нижній потік леного кремнезему згідно з винаходом використа27 з 33 являє собою потік сирого ДФК. Є можлили той же гранульований кремнезем, що і в вість вибору у застосуванні іншого матеріалу, таКонтрольному Прикладі 1. Гранульований кремнекого як діетиловий етер, диметиловий етер, ізопезем (40,56 г) обробляли розчином гідроксиду нантан чи бутан замість азоту чи часткової заміни трію, одержаного шляхом розчинення 8,059 г азоту для роботи двох реакторів подвійної фази 38 NaOH в 226 г води при кімнатній температурі прота 37. тягом 7 хвилин при перемішуванні. Оброблений Контрольний приклад 1 кремнезем декілька раз ретельно промивали хоЗгідно з рівнем техніки приготували каталізалодною водою, а потім - гарячою водою (приблизтор на основі оксиду титану (9,2 % мас), нанесено 65°С) для вилучення слідів натрію на кремнений на кремнеземний носій (WO 03/066569). 3,839 земі. Оброблений кремнезем висушували при г Ti(OC4Hg-n)4 розчинили в 90 мл сухого толуолу. 150°С протягом годин, а потім випалювали при Гранульований кремнезем (+8 меш, 655 ч. на млн. 325°С протягом 2 годин. Цей випалений кремнеNa за масою, 300 м2/г БЕТ SA та 1 см3/г PV) зазем містив 300 ч. на млн. Na за масою. Розчин здалегідь висушили при 330°С протягом 2 годин алкоксиду ніобію готували шляхом розчинення на повітрі. Розчин бутоксиду титану кип'ятили зі 0,844 г Nb(OC4H9-n) в 80 мл толуолу. 8,46 г обробзворотним холодильником з 25 мл (9,159) сухих леного кремнезему кип'ятили зі зворотним хологранул кремнезему в кип'ячому розчині толуолу в дильником у вищевказаному розчині бут оксиду колбі з конденсатором. Після кип'ятіння протягом ніобію протягом 3 годин у колбі з конденсатором, приблизно 6 годин, надлишок толуолу з колби виякий охолоджувався водою. Після охолодження парили. Бутоксид титану на кремнеземному носії надлишок розчину вилучали з колби. Готували вилучили з колби та висушили при 120°С у вакуусуміш метанол-вода шляхом перемішування 0,645 мній печі протягом 1, 5 год. Висушений кремнезем г води з 90 мл метанолу. Цю суміш метанол-вода випалили при 500°С протягом 2 годин. Випалений вливали у колбу і вміст колби знову кип'ятили зі 19 87629 20 зворотним холодильником. Після кип'ятіння зі звоного потоку на вершині нерухомого каталітичного ротним холодильником протягом години надлишок шару, температуру якого контролювали незалежрозчину вилучали з колби. Розчин тетрабутоксиду но. Реактор з нерухомим шаром працював у ретитану, одержаний шляхом розчинення 3,67 г бужимі потоку вниз. Дистиляційна колона складалася токсиду титану в 80 мл толуолу, вливали у колбу і з ребойлера ємністю 2 літри та колони 42" х 1" OD вміст колби кип'ятили зі зворотним холодильником (0,870" ID). В колоні знаходились три датчики тиспротягом 1 години 45 хвилин. Надлишок толуолу у ку для контролю та реєстрації тиску угорі колони, колбі з колби вилучали. Матеріал у колбі виймали на вершині та внизу каталітичного шару. Ребойлер з колби і висушували при 120°С у вакуумній печі також мав датчик рівня рідини. Нерухомий шар протягом 1 години. Висушений кремнезем випамав рециркуляційний контур; потік в реактор з рідлювали при 500°С протягом 2 годин. Зовнішній кого середовища в ребойлері прокачували крізь вигляд каталізатора більше нагадував гранульореактор в режимі потоку вниз, а потім повертали в ваний кремнеземний носій. Це - каталізатор С. ребойлер. Свіжий вхідний потік ДЕК прокачували в Приклад 4 рециркуляційний контур перед подачею в реактор. Каталізатор на основі алкоксиду титану, нанеСвіжий вхідний потік розчину фенолу окремо просеного на носій з обробленого кремнезему, готукачували в рециркуляційний контур перед подавали згідно з винаходом. Той же гранульований чею в реактор. При необхідності в систему вводикремнезем (80,55г), що і в Контрольному Прикладі ли азот. Пару з дистиляційної колони 1, обробляли розчином гідроксиду натрію, одерконденсували конденсували та вилучали як верхжаного шляхом розчинення 8,3 г NaOH в 580 мл ній потік рідини. Газ, який не конденсувався, вивадеіонізованої води при кімнатній температурі пронтажували з реактора. тягом 8 хвилин при перемішуванні. Оброблений Для безперервного протікання трансестерифікремнезем промивали холодною водою, а потім кації вхідний розчини фенолу вводили безперервгарячою водою. Промитий кремнезем обробляли но при заданій швидкості при цьому потік продукту розчином нітрату амонію, одержанного шляхом 99 безперервно вилучали з ребойлера з заздалегідь г нітрату амонію в 2 літрах деіонізованої води при встановленою постійною швидкістю та безперерв80°С протягом 2 годин. Обробку кремнезему розно вилучали продукт вгорі. Швидкість введення чином нітрату амонію повторювали 13 разів. В ДЕК є каскадною для підтримування постійного кінці кремнезем промивали деіонізованою водою рівня рідини в ребойлері. при кімнатній температурі. Промитий кремнезем Тест 1 висушували при 110°С протягом години, а після Задачею цього прогону є демонстрація роботи цього випалювали при 370°С протягом 1,5 годин, а реактора подвійної фази з нерухомим шаром для потім - при 375°С протягом 30 хвилин. Випалений реакції трансестерифікації. Подвійну фазу в катакремнезем 23 ч. на млн. ΝΑ за масою та мав 2,9% літичній реакційній зоні створили за допомогою мас.% втрати маси при випалюванні при 550°С. киплячої суміші парової фази та рідкої фази в каТетрабутоксид титану готували шляхом розчиненталітичній реакційній зоні. ня 4,74 г тетрабутоксиду титану в 70 мл толуолу. Каталізатор А (25 мл; 9,67 г) завантажили в 10,44 г (31 мл) обробленого кремнезему кип'ятили реактор. В ребойлер завантажили 167,6 г фенолу зі зворотним холодильником у розчині тетрабутокта 737,4 г. ДЕК. сиду титану протягом 6 годин, а потім надлишок Тестування проводили за наступних умов: розчину титану у колбі вилучали. Вилучений розТиск угорі колони: 18 (надлишковий тиск у фучин титану містив 0,50% мас. Ni. Кремнезем пронтах/дюйм2) мивали 90 мл толуолу при кімнатній температурі Температура ребойлера: 335°F Промитий продукт висушували при 170°С протяТемпература дистиляційної колони: 300 гом 3 годин у вакуумній печі. Зовнішній вигляд го310°F тового каталізатора нагадував оброблені гранули Швидкість рециркуляції: 66 мл/хв. кремнезему. Невелику частину цього готового каТиск на вершині реактора з нерухомим шаром: талізатора випалювали при 500°С протягом 2 го24,6 (надлишковий тиск у фунтах/дюйм2) дин на повітрі для визначення вмісту Ті на каталіТиск в нижній частині реактора з нерухомим заторі. Вміст Ті випаленого калатізатора становить шаром: 20,5 (надлишковий тиск у фунтах/дюйм ) 3,25%. Зовнішній вигляд випаленого каталізатора Температура реактора з нерухомим шаром: нагадував оброблені гранули кремнезему. Цей 338 - 342°F експеримент показує успішне закріплення алкокШвидкість потоку азоту в ребойлер: 60 мл/хв. сиду титану на обробленому кремнеземі. 0 рефлюксу з барабану для рефлюксу Проведення трансестерифікації та диспропорПрогін, який тривав години, почали, коли темціювання пература реактора досягла цільової температури Каталізатори тестували в установці, яка мала 340°F. Під час прогону безперервно закачували реактор з нерухомим шаром, дистиляційну колону ДЕК, щоб підтримувати постійний рівень рідини. та барабан для рефлюксу. Реактор з нерухомим Після 45 годин в систему завантажили 23 г фенолу шаром мав діаметр 1/2 дюйми та довжину 15 у формі 20,92% мас. розчину фенолу протягом дюймів. Він мав три термопари для моніторингу періоду 55 хвилин при швидкості потоку 2 мл/хв. температури в трьох позиціях вище каталітичного Прогін продовжували 77 годин на робочому циклі. шару, посередині каталітичного шару та нижче Середня швидкість потоку рідини угорі становила каталітичного шару. Верхня половина та нижня приблизно 0,3 мл/хв. Потік угорі складався голополовина температур реактора контролювались вним чином з ДЕК та невеликої кількості етанолу. незалежно. Установка також мала підігрівач вхідДля аналізу брали зразки з ребойлера та потоку 21 87629 22 угорі. Після закінчення 77 годинного циклу 64,8 фази. Прогін, який тривав години, почали, коли моль% фенолу, завантаженого в установку, було температура реактора досягла цільової темпераконвертовано. Вихід становив 63,1 моль% ЕФК; тури 340°F. Тест проводили з використанням того 1,52 моль% ДФК та 0,26 моль% побічних продуктів ж каталізатор, що і в Тесті 2. Під час прогону безза загальною масою фенолу, завантаженого в сиперервно закачували толуол для підтримування стему. Головним побічним продуктом був фенепостійного рівня рідини і швидкість потоку вгорі тол, який відповідав за 33,6 моль% побічних простановила 7 мл/хв. Потік угорі складався головним дуктів. Середня продуктивність становила 1,81 чином з ДЕК та слідової кількості етанолу. Загальм/год./кг каталізатора для ЕФК; 0,041 м/год./кг кана маса вхідного потоку в ребойлер становила талізатора для ДФК та 0,01 м/год./кг каталізатора 635,3 г. Склад вхідного потоку був таким: 44,19% для побічних продуктів. Продуктивність ЕФК в мас. толуолу; 8,42 %мас. ДЕК; 0,07 % мас. фенецьому прикладі вища за способи, описані в рівні толу; 0,29 % мас. побічних продуктів; 11,21 % мас. техніки, що показує кращу продуктивність способу, фенолу; 31,88 % мас. ЕФК та 4,94 % мас. ДФК. описаного у даному винаході. Тестування проводили за таких умов: Тест 2 Тиск угорі колони: 16,3 (надлишковий тиск у Задачею цього прогону є демонстрація роботи фунтах/дюйм) реактора подвійної фази з нерухомим шаром та Температура ребойлера: 311°F каталізатора на основі змішаного каталізатора на Температура дистиляційної колони: 295 основі змішаного оксиду ніобію та титану згідно з 305°F винаходом для реакції трансестерифікації. ПоШвидкість рециркуляції: 67 мл/хв. двійну фазу в каталітичній реакційній зоні створиТиск на вершині реактора з нерухомим-шаром: ли за допомогою киплячої суміші парової фази та 32,8 (надлишковий тиск у фунтах/дюйм2). рідкої фази в каталітичній реакційній зоні. Тиск в нижній частині реактора з нерухомим Каталізатор В (24 мл; 9,172 г) завантажили в шаром: 18,5 (надлишковий тиск у фунтах/дюйм2) реактор. Цей тест проводили за умов, аналогічних Температура реактора з нерухомим шаром: умовам в Тесті 1. 167,6 г фенолу та 737,4 г ДЕК 327 - 330°F завантажили в ребойлер. Прогін, який тривав гоШвидкість потоку азоту в ребойлер: 50 мл/хв. дини, почали, коли температура реактора досягла 0 рефлюксу з барабану для рефлюксу цільової температури 340°F. Під час прогону безПісля 6 годин роботи аналіз продукту з ребойперервно закачували ДЕК, щоб підтримувати полера показав 5,8 моль % конверсії ЕФК з 92,5% стійний рівень рідини. Після 22 годин в систему селективністю до ДФК. Продуктивність ДФК станозавантажили 91,62 г фенолу у формі 31,28 % мас. вила 0,614 м/год./кг каталізатора. розчину фенолу протягом періоду 2 годин 22 хвиТест 4 лин при швидкості потоку 2 мл/хв. Прогін продовЦей тест проводили для демонстрації безпежували 73 години на робочому циклі. Середня рервного прогону для одержання ЕФК в реакторі з швидкість потоку рідини угорі становила приблизнерухомим шаром, який працював в режимі темно 0,3 мл/хв. Потік угорі складався головним чиператури кипіння реактора за умов стабільно станом з ДЕК та невеликої кількості етанолу. Для ну. аналізу брали зразки з ребойлера та потоку угорі. Приготували іншу партію каталізатора, ідентиПісля закінчення 73 годинного циклу 64,8 моль% чного каталізатору В в Прикладі 2. 9,04 г (приблифенолу, завантаженого в установку, було конверзно 25 мл) каталізатора завантажили в реактор. В товано. Вихід становив 63,1 моль% ЕФК; 1,4 ребойлер для безперервного прогону завантажили моль% ДФК та 0,27 моль% побічних продуктів за 200 г фенолу та 610 г ДЕК. Прогін, який тривав загальною масою фенолу, завантаженого в систегодини, почали, коли температура реактора досягму. Головним побічним продуктом був фенетол, ла цільової температури 340°F. Під час прогону який відповідав за 55,9 моль% побічних продуктів. безперервно закачували ДЕК для підтримування Середня продуктивність становила 2,567 м/год./кг постійного рівня рідини. Після прогону протягом каталізатора для ЕФК; 0,059 м/год./кг каталізатора 23,25 год. в систему завантажили 85,58 г фенолу у для ДФК та 0,022 м/год./кг каталізатора для побічвигляді 31,28% мас. розчину фенолу протягом 2 них продуктів. Продуктивність ЕФК в цьому пригод. 15 хв. при швидкості потоку 2 мл/хв. Безперекладі вища за каталізатор, описаний в рівні техніки рвний прогін за умов стабільного стану здійснювата Тесті 1. ли шляхом безперервного закачування 32,2 мас. Тест 3 % розчину фенолу в ДЕК при швидкості 0,18 Цей тест проводили для демонстрації диспромл/хв. та безперервного вилучення потоку продукпорціювання ЕФК в ДФК в серійному виробництві. ту при швидкості 0,15 мл/хв. Закачували ДЕК для Реактор з нерухомим шаром працював в режимі підтримування постійного 81% рівня рідини в рекипіння, створеного з використанням того ж облабойлері. Після 242,5 год. прогону за умов стабільднання, яке описано раніше. ного стану одержали наступний результат: Потік сировини готували як в Тесті 2 і потім Час прогону: 242,5 год. надлишок ДЕК в потоці сировини відганяли для Тиск угорі колони: 19,6 (надлишковий тиск у концентрування ЕФК та додавали толуол. Задафунтах/дюйм2) чею додавання толуолу до концентрованого потоТемпература ребойлера: 354°F ку сировини було проведення диспропорціювання Температура дистиляційної колони: 330°F в режимі подвійної фази в реакторі, який працює в Швидкість рециркуляції: 67 мл/хв. режимі потоку вниз. В реакційній зоні утворили Тиск на вершині реактора з нерухомим шаром: киплячу реакційну суміш парової фази та рідкої 20,8 (надлишковий тиск у фунтах/дюйм2) 23 87629 24 Тиск в нижній частині реактора з нерухомим кість дезактивації, вища активність каталізатора С шаром: 19,2 (надлишковий тиск у фунтах/дюйм ) та показана регенерація шляхом деполімеризації Температура реактора з нерухомим шаром: дезактивованого каталізатора С. Тест на звичай341°F ному каталізаторі на основі оксиду титану на креШвидкість потоку азоту в ребойлер: 60 мл/хв. мнеземі (Контрольний Приклад 1) був проведений Рефлюкс з барабану для рефлексу: 0 в Тесті 6А. Тестування каталізатора на основі зміРівень рідини в ребойлері: 81,04% шаного оксиду Ti/Nb на обробленому кремнеземі Швидкість введення вхідного розчину 32,2% (каталізатор С в Прикладі 3) було проведено в PhOH/ДЕК: 0,15 мл/хв. Тесті 6В. Швидкість потоку ДЕК: 0,28 мл/хв. Тест 6А Швидкість потоку вгорі: 0,269 мл/хв. Приготували інший каталізатор на основі оксиду Ті, подібний каталізатору А в Контрольному Швидкість потоку внизу: 0,185 мл/хв. Прикладі 1. Єдиною відмінністю від Контрольного Склад потоків (%мас.) Прикладу 1 було випалювання того ж кремнезему Компонент Угорі Унизу при 340°С протягом 4 годин на повітрі. 25 мл (9,0 Етанол 3,5793 0,0448 г) каталізатора завантажили в описаний реактор з ДЕК 96,1344 62,5443 нерухомим шаром. Зовнішній вигляд каталізатор Фенетол 0,0176 являв собою гранули кремнеземи, покриті білим цукром як у каталізатора з Контрольного Прикладу Фенол 0,2208 17,5226 ЕФК 0,0655 19,2401 1. В ребойлер завантажили 285 г фенолу та 530 г ДЕК. ДФК 0,6306 Конверсія фенолу Тестування проводили за наступних умов: (моль%) 39,5% Тиск угорі колони: 19-21 (надлишковий тиск у Вихід ЕФК: 37,6 Селективність ЕФК: 95,04 фунтах/дюйм2) моль% моль% Температура ребойлера: 345 - 347°F Вихід ДФК: 1,9моль% Селективність ДФК: 4,87 Температура дистиляційної колони: 270 моль% 285°F Селективність фенетолу: Швидкість рециркуляції: 66 - 67 мл/хв. 0,05% моль% Тиск на вершині реактора з нерухомим шаром: 20 - 22 (надлишковий тиск у фунтах/дюйм2) Продуктивність ЕФК: 1,50 м/год./кг каталізатоТиск в нижній частині реактора з нерухомим шаром: 19-22 (надлишковий тиск у фунтах/дюйм2) ра Фенетол являв собою єдиний побічний проТемпература реактора з нерухомим шаром: дукт, який було визначено в потоці продукті за 332 - 337°F допомогою газової хроматографії та газової хроШвидкість потоку азоту в ребойлер: 60 мл/хв. матографії-мас-спектрометрії. Рефлюкс з барабану для рефлексу: 0 Тест 5 Швидкість введення вхідного розчину Цей тест було проведено для демонстрації PhOH/ДЕК: 0,17-0,18 мл/хв. безперервного прогону для реакції трансестерифіШвидкість потоку внизу: 0,18-0,19 мл/хв. кації між карбонатом пропілену та етанолом для Під час цього прогону потік ДЕК вводили у касодержання ДЕК та паралельних продуктів пропікадному режимі до рівня рідини 88% (випадкова ленгліколю, використовуючи реактор з нерухомим шкала) для підтримання постійного рівня рідини. шаром в режимі температури кипіння. Прогін проводили шляхом постійного дренування Приготували іншу партію каталізатора, ідентиреакційної суміші в ребойлері при закачуванні 40,4 чного каталізатору В в Прикладі 2. 9,6 г (приблизмас.% розчину фенолу в ДЕК при постійній швидно 25 мл) каталізатора завантажили в реактор. 280 кості приблизно 0,19 мл/хв. Прогін продовжували г карбонату пропілену та 635 г етанолу завантапротягом 115 годин без перерви. Верхній потік жили в ребойлер для безперервного прогону. Проскладався головним чином з ДЕК, етанолу та негін, який тривав години, почали, коли температура великих кількостей фенолу та ЕФК. Для аналізу реактора досягла цільової температури 335°F. Під відбирали зразки з ребойлера та верхнього поточас прогону потік продукт постійно вилучали з реку. Єдиним побічним продуктом був фенетол в бойлера при постійній швидкості 0,14 мл/хв. Для нижньому потоці з ребойлера. Результати навопідтримання постійного рівня рідини 85% в реакдяться в Таблиці 1. тор у каскадному режимі постійно закачували 23,1 мас.% розчину карбонату пропілену в етанолі. Таблиця 1 Прогін продовжували 162 години. Під час прогону конверсія карбонату пропілену була майже постійГодини прогону 31 61 85 115 ною. Середня конверсія пропілену протягом цього Конверсія фенолу 17,8 20,8 17,7 11,9 прогону становила 14,5 моль%. Продуктивність (моль%) ДФК становила 1,81 моль/год./кг каталізатора. Продуктивність ЕФК 0,771 0,869 0,754 0,502 Тест 6 (моль/год./кг Задачею цього експерименту була демонПродуктивність ДФК 0,008 0,013 0,013 0,0092 страція переваг каталізатора на основі змішаного (моль/год./кг оксиду, нанесеного на носій з обробленого кремПродуктивність фене- 0,00160,00180,00180,0017 незему, відносно звичайного каталізатора, описатолу (моль/год./кг ного в рівні техніки. Були виявлені менша швид 25 87629 26 Тест 6В рвного 234-годинного потокового прогону навоТестували поведінку каталізатора С, приготодиться в Таблиці 2. Верхній потік складався голованого у Прикладі 3, у трансестерифікації ДЕК вним чином з ДЕК, етанолу та невеликих фенолом. 8,4 г (25 мл) каталізатора С завантажукількостей фенолу та ЕФК. Для аналізу відбирали вали в реактор з нерухомим шаром. В ребойлер зразки з ребойлера та верхнього потоку. Єдиним завантажували 287 г фенолу та 530 г ДЕК. Теступобічним продуктом був фенетол в нижньому повання проводили за наступних умов: тоці з ребойлера. Результат цього прогону в ТабТиск угорі колони: 17-19 (надлишковий тиск у лиці 2 чітко показує повільнішу дезактивацію та фунтах/дюйм2) вищу активність ніж каталізатор А в Тесті 6А. Температура ребойлера: 345 - 348°F Результат цього прогону в Таблиці 2 чітко поТемпература дистиляційної колони: 270 казує повільнішу дезактивацію та вищу активність 285°F каталізатора С ніж каталізатор А. Швидкість рециркуляції: 66 - 67 мл/хв. Прогін продовжували 360 годин у потоковому Тиск на вершині реактора з нерухомим шаром: режимі. Спостерігали постійну дезактивацію ката19 - 21 (надлишковий тиск у фунтах/дюйм2) лізатора. Після 360 годин роботу реактора припиТиск в нижній частині реактора з нерухомим нили для регенерації каталізатора. Регенерацію шаром: 19-22 (надлишковий тиск у фунтах/дюйм ) каталізатора проводили при 340°F та надлишкоТемпература реактора з нерухомим шаром: вому тиску 230 фунтів/дюйм2 шляхом де полімери331 - 335°F зації полімерів на каталізаторі шляхом циркуляції Швидкість потоку азоту в ребойлер: 60 мл/хв. етанолу в реакторі протягом 17 годин. Основним Рефлюкс з барабану для рефлексу: 0 продуктами деполімеризації етанолом були феШвидкість введення вхідного розчину нол, ДЕК та неідентифіковані побічні продукти. PhOH/ДЕК: 0,18-0,19 мл/хв. Прогін поновили з регенерованим каталізатором. Швидкість потоку внизу: 0,18 - 0,20 мл/хв. Результати тесту регенерованого каталізатора Під час цього прогону потік ДЕК вводили у каснаводяться в Таблиці 3. кадному режимі до рівня рідини 88% (випадкова Результат в Таблиці 3 чітко показує, що каташкала) для підтримання постійного рівня рідини. лізатор можна регенерувати шляхом здійснення Прогін проводили шляхом постійного дренування деполімеризації полімерів, відкладених на каталіреакційної суміші в ребойлері при закачуванні 36,5 заторі. мас.% розчину фенолу в ДЕК. Результат безпереТаблиця 2 Години прогону Конверсія фенолу (моль%) Продуктивність ЕФК (моль/год./кг) Продуктивність ДФК (моль/год./кг) Продуктивність фенетолу (моль/год./кг) 33 23,5 0,686 0,008 0,0018 56 38,5 1,119 0,020 0,0019 94 34,7 1,027 0,019 0,0014 160 31,9 0,976 0,019 0,011 216 29,1 0,556 0,012 0,0006 234 25,0 0,510 0,012 0,0006 Таблиця 3 Години прогону 2 Тиск угорі колони (надлишковий тиск у фунтах/дюйм ) Температура ре бойлера (°F) Температура дистиляційної колони (°F) Швидкість рециркуляції (мл/хв.) Тиск на вершині реактора з нерухомим шаром (надлишковий тиск у фунтах/дюйм2) Тиск в нижній частині реактора з нерухомим шаром (надлишковий тиск у фунтах/дюйм2) Температура реактора з нерухомим шаром (°F) Швидкість введення вхідного розчину PhOH/ДЕК (мл/хв.) Швидкість потоку внизу (мл/хв.) Конверсія фенолу (моль%) Вихід ЕФК (моль%) Вихід ДФК (моль%) Селективність ЕФК (моль%) Селективність ДФК (моль%) Селективність фенетолу (моль%) Продуктивність ЕФК (моль/год./кг) Продуктивність ДФК (моль/год./кг) 360 Регенерація каталізатора 18,2 347 393 428 65 19,5 344 273286 66 18,1 344 271285 67 20 22,2 20,4 19,2 22,1 20 330331 0,18 0,18 23,4 22,8 0,610 97,3 2,60 0,13 0,681 0,009 331330 0,18 0,18 27,2 26,2 1,017 96,3 3,73 0,10 0,830 0,016 273-285 330-330 0,18 0,16 17,5 16,47 0,95 94,35 5,44 0,21 0,452 0,013 27 Комп’ютерна верстка В. Мацело 87629 Підписне 28 Тираж 28 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for producing organic carbonates
Автори англійськоюRyu Yong J.
Назва патенту російськоюСпособ получения диарилкарбонатов, композиция твердого катализатора и способ ее реактивации
Автори російськоюРью Йонг Дж.
МПК / Мітки
МПК: C07C 69/96, B01J 21/06, B01J 23/16
Мітки: каталізатора, реактивації, одержання, твердого, спосіб, композиція, діарилкарбонатів
Код посилання
<a href="https://ua.patents.su/14-87629-sposib-oderzhannya-diarilkarbonativ-kompoziciya-tverdogo-katalizatora-ta-sposib-reaktivaci.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання діарилкарбонатів, композиція твердого каталізатора та спосіб її реактивації</a>
Спосіб одержання каталізатора конверсії so2 в so3 з використанням спрацьованого ванадієвого каталізатора
Номер патенту: 70330
Опубліковано: 15.10.2004
Автори: Малкіман Веніамін, Манаєва Любовь
МПК: B01J 38/00, B01J 23/16
Мітки: спрацьованого, одержання, використанням, каталізатора, ванадієвого, спосіб, конверсії
Формула / Реферат:
1. Спосіб одержання каталізатора конверсії SO2 в SO3 з використанням спрацьованого ванадієвого каталізатора, який включає вилуговування активних компонентів у пульпі, відокремлення їх з фільтратом від твердого залишку, обробку фільтрату та твердого залишку, змішування останнього з активними компонентами з фільтрату, введення компонентів свіжої контактної маси, сушіння, формування та термообробку, який відрізняється тим, що твердий залишок...
Композиція каталізатора для окислення етану і/або етилену до оцтової кислоти, спосіб її приготування та спосіб одержання оцтової кислоти

Номер патенту: 82367
Опубліковано: 10.04.2008
Автори: Розен Брюс І., Джордж Річард Дж., Бразділ Джеймс Франк
МПК: B01J 37/02, C07C 51/25, B01J 23/54, C07C 51/215, B01J 23/16
Мітки: приготування, спосіб, етану, каталізатора, етилену, кислоти, оцтової, одержання, композиція, окислення
Формула / Реферат:
1. Композиція каталізатора для окиснення етану і/або етилену до оцтової кислоти, яка містить, у сполученні з киснем, елементи молібден, ванадій, ніобій та титан відповідно до емпіричної формули:MoaWbTicVdNbeYf (І),у якій Y означає один або декілька елементів, вибраних із групи, яка включає Сr, Та, В, Аl, Ga, In, Pt, Zn, Cd, Bi, Ce, Co, Rh, Ir, Cu, Ag, Fe, Ru, Os, K, Rb, Cs, Mg, Ca, Sr, Ba, Zr, Hf, Ni, P, Pb, Si, Sn, Tl, U, Re,...
Композиція каталізатора гідрокрекінгу, спосіб його одержання та процес перетворення вуглеводневої сировини з його використанням

Номер патенту: 86193
Опубліковано: 10.04.2009
Автори: Оувеханд Корнеліс, Крейгтон Едвард Джуліус
МПК: C10G 47/00, B01J 29/00, B01J 35/00
Мітки: спосіб, гідрокрекінгу, композиція, процес, одержання, перетворення, каталізатора, сировини, використанням, вуглеводневої
Формула / Реферат:
1. Композиція каталізатора гідрокрекінгу, яка містить металевий компонент гідрогенізації, нанесений на носій, який є цеолітом зі структурою фоязиту, що має розмір елементарної комірки в інтервалі від 24,10 до 24,40 Å, об'ємне співвідношення між діоксидом кремнію й оксидом алюмінію - SAR більше 12 і питому поверхню принаймні 850 м2/г, виміряну БЕТ-методом у відповідності зі стандартною методикою ASTM D 4365-95 із застосуванням адсорбції...
Спосіб одержання каталізатора реакції виділення кисню

Номер патенту: 68287
Опубліковано: 15.07.2004
Автори: Стадник Ольга Олександрівна, Іванова Наталія Дмитрівна, Болдирєв Євген Іванович
МПК: B01J 27/06, C25B 11/00, B01J 23/75
Мітки: кисню, каталізатора, реакції, одержання, виділення, спосіб
Формула / Реферат:
Спосіб одержання каталізатора реакції виділення кисню шляхом синтезу оксидної сполуки кобальту, який відрізняється тим, що каталізатор одержують електролізом водного розчину, внаслідок чого каталізатор містить ОН- -групу, електроліз проводять з розчину, що містить, г/л: сульфат кобальту CoSO4.7H2O 10-30 фторид амонію NH4F 20-50 вода H2O ...
Спосіб виготовлення каталізатора для одержання вінілацетату у псевдозрідженому шарі, каталізатор для одержання вінілацетату і спосіб одержання вінілацетату з використанням каталізатора

Номер патенту: 65569
Опубліковано: 15.04.2004
Автори: Бейкер Майкл Джеймс, Салем Джордж Фредерік
МПК: C07C 67/04, B01J 8/20, B01J 37/02, B01J 37/00, B01J 35/00, C07C 67/055, C07C 69/15, B01J 33/00, C07B 61/00, B01J 23/54
Мітки: шарі, вінілацетату, спосіб, каталізатора, одержання, псевдозрідженому, виготовлення, використанням, каталізатор
Формула / Реферат:
1. Спосіб виготовлення каталізатора, використовуваного з метою одержання вінілацетату в реакторі з псевдозрідженим шаром, який включає:(І) введення попередньо сформованих пористих мікросферичних частинок носія у контакт із розчинами паладієвої сполуки і принаймні однієї сполуки металу, що виявляє спорідненість, таким чином, що паладій і метал, який виявляє спорідненість, тонко диспергують у мікросферичних частинках носія,(II)...
Попередній патент: Ущільнювач субстрату
Наступний патент: Спосіб переробки органічної сировини в термохімічному реакторі
Випадковий патент: Спосіб діагностики реакцій психічної дезадаптації