Карбоксамідин для лікування вірусного гепатиту
Номер патенту: 73761
Опубліковано: 15.09.2005
Автори: Тем Роберт, Рамасамі Канда, Лау Джонсон, Гонг Жі









Формула / Реферат
1. Застосування карбоксамідину формули 1 або його фармацевтично прийнятної солі для виготовлення лікарського засобу для лікування вірусної інфекції вірусу гепатиту С (HCV) або вірусної інфекції вірусу гепатиту В (HBV)
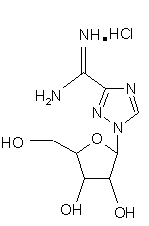 формула 1,
формула 1,
де сполука знаходиться у D-конфігурації.
2. Застосування за п. 1, де фармацевтично прийнятна сіль – це хлористоводнева сіль за формулою 1.
3. Застосування за п. 1 або п. 2, де вірусна інфекція – це інфекція вірусу гепатиту С (HCV).
4. Застосування за будь-яким з пп. 1-3, де лікарський засіб призначений для перорального введення.
5. Застосування за будь-яким з пп. 1-4, де лікарський засіб містить сполуку формули 1 у дозі між 0,1 мг на кг ваги тіла пацієнта та 40 мг на кг ваги тіла пацієнта.
6. Застосування за будь-яким з пп. 1-5, де лікарський засіб додатково містить інтерферон.
7. Застосування за п. 6, де інтерферон – це інтерферон альфа.
Текст
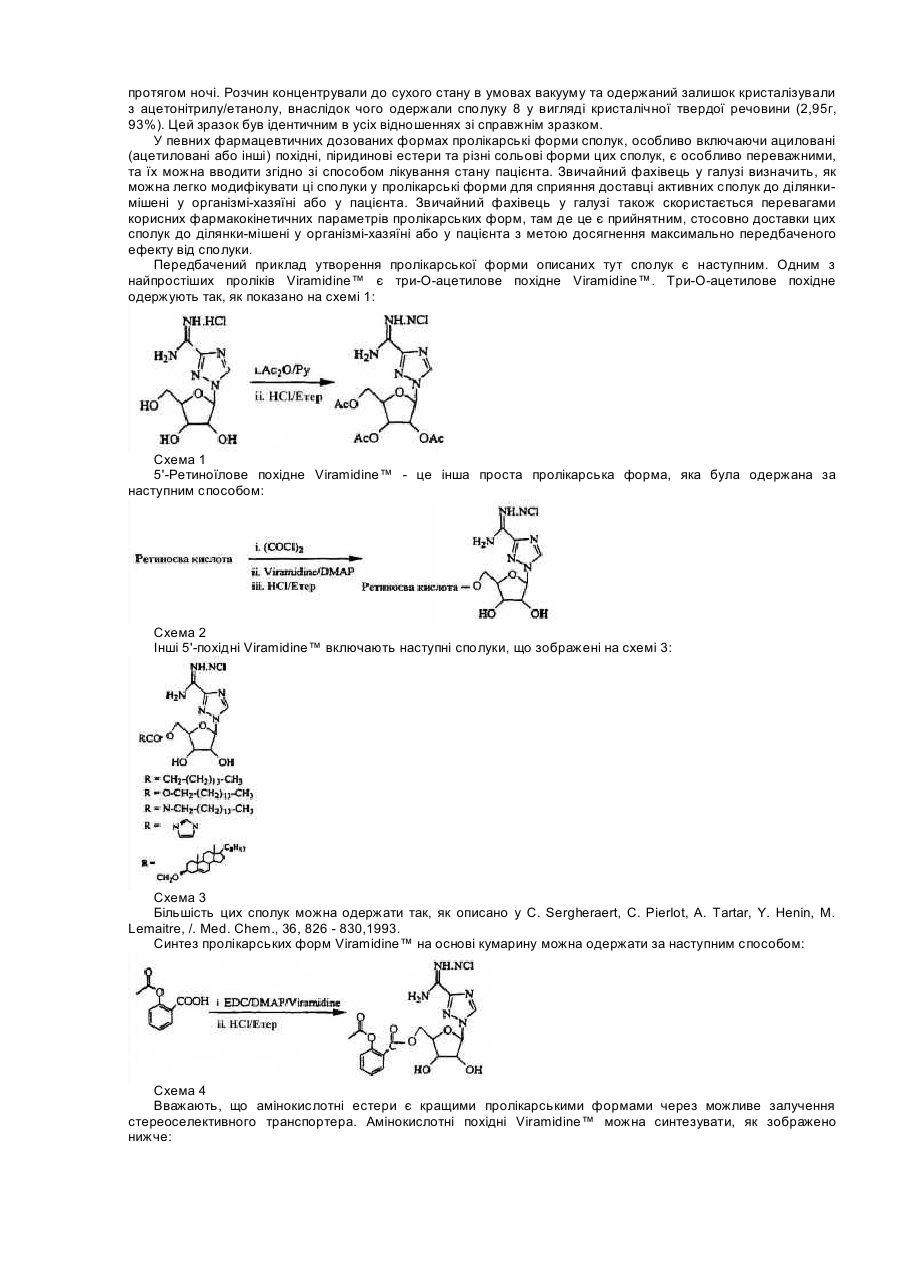
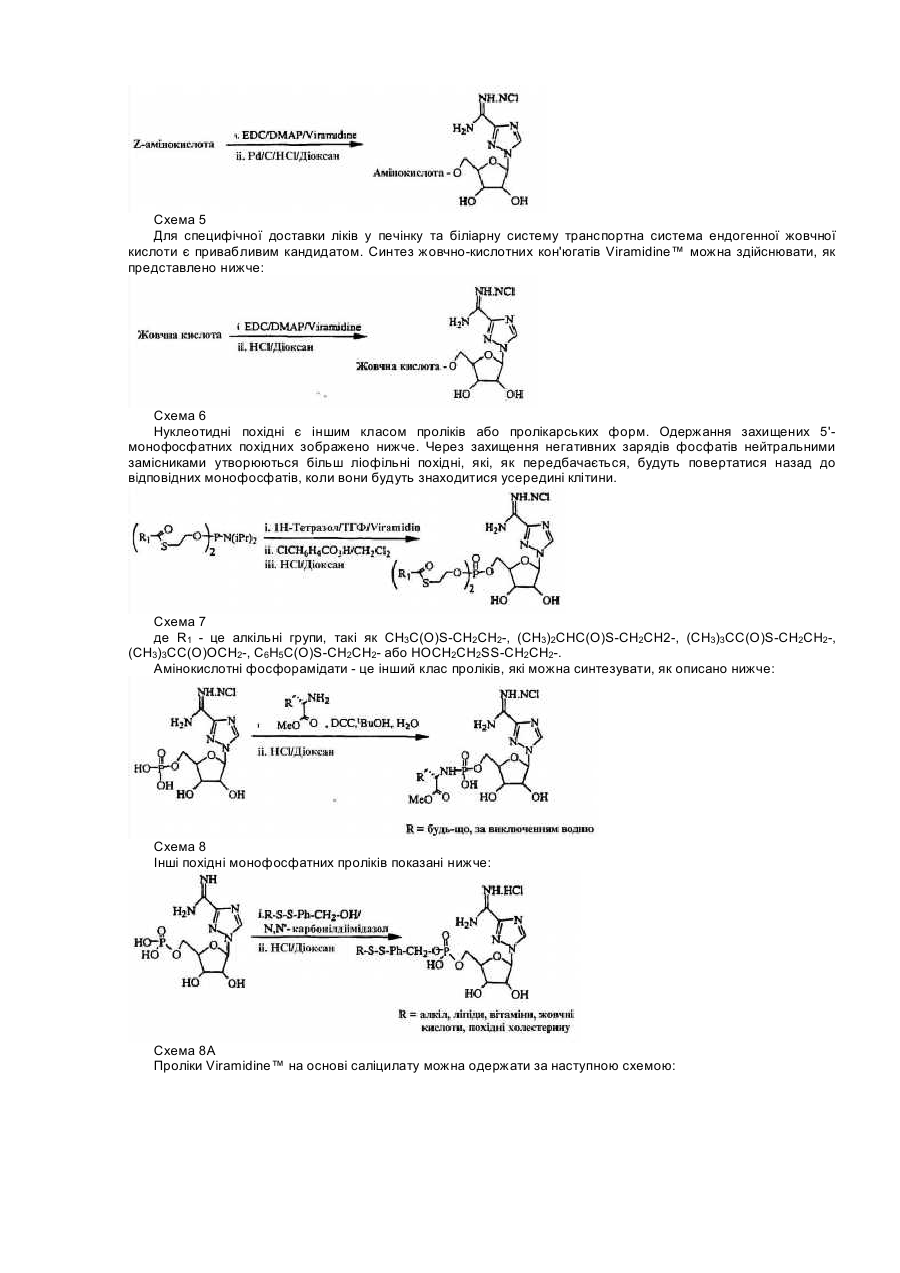
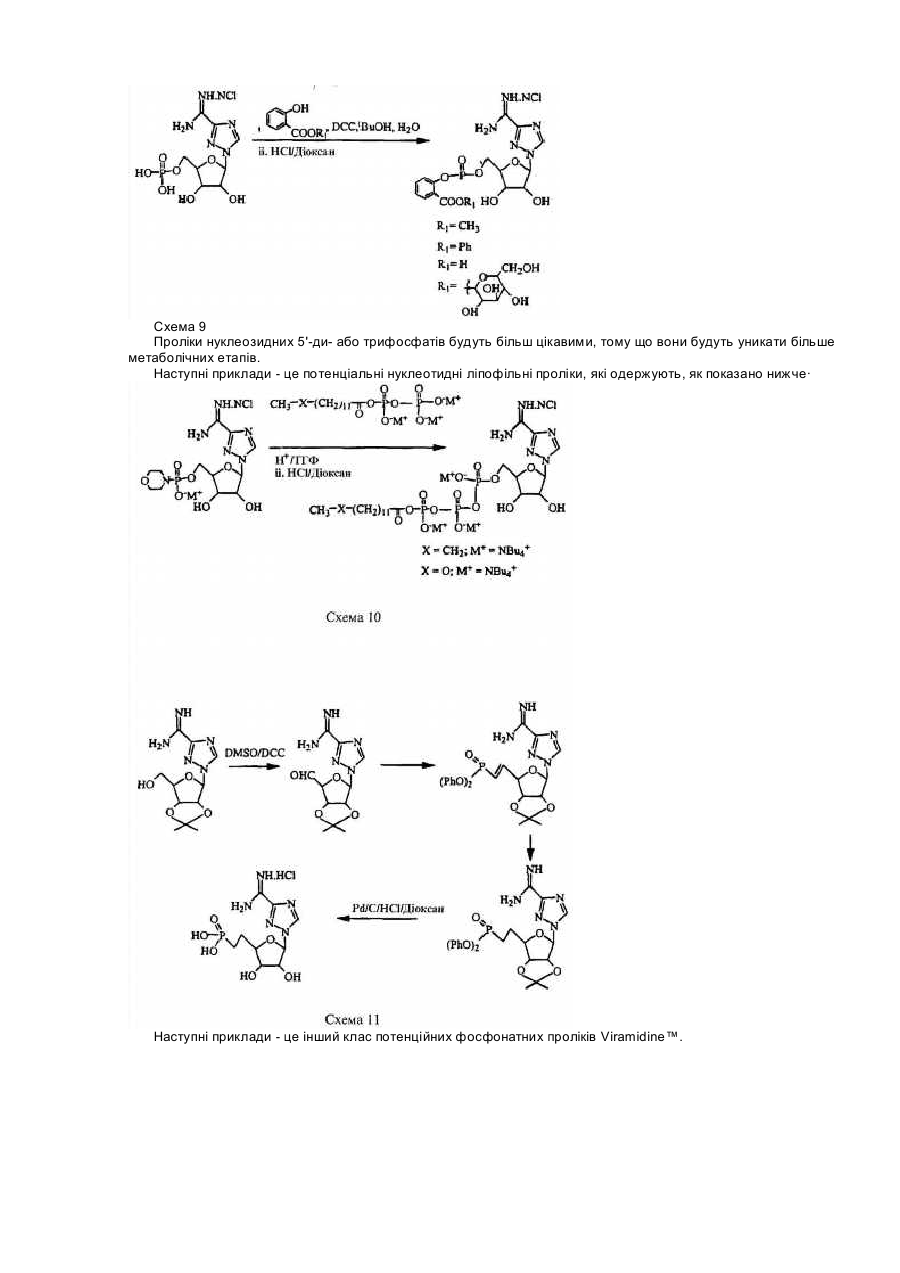
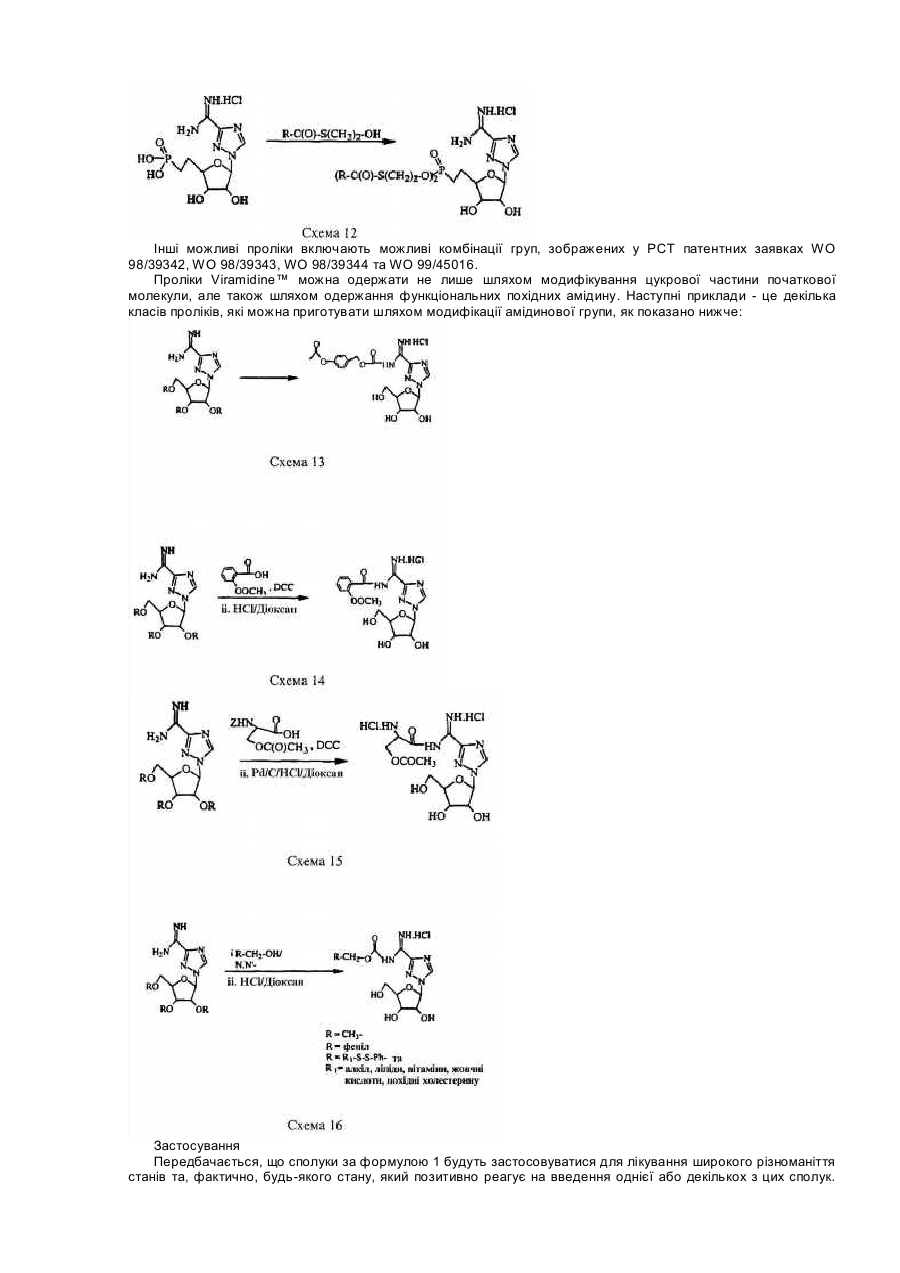
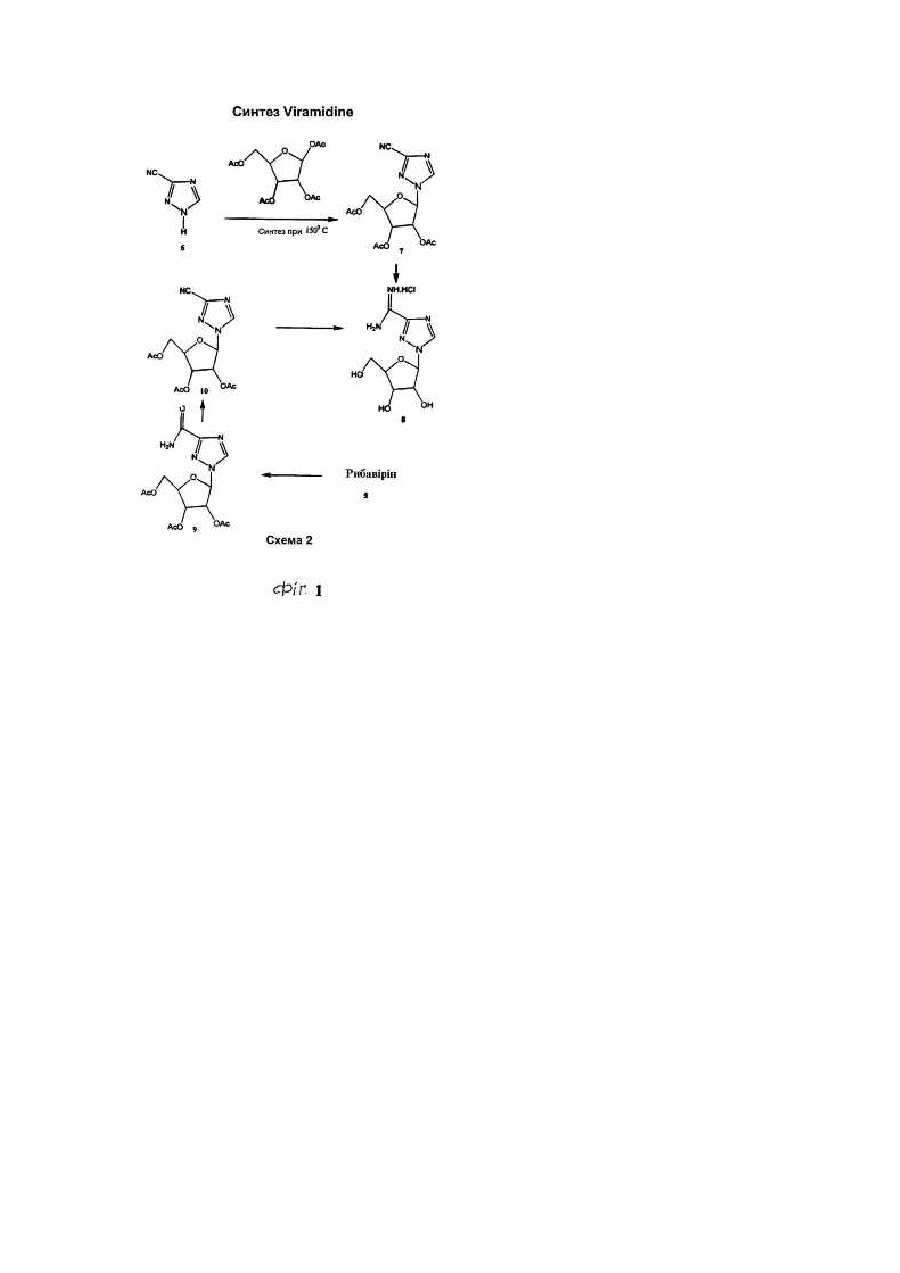
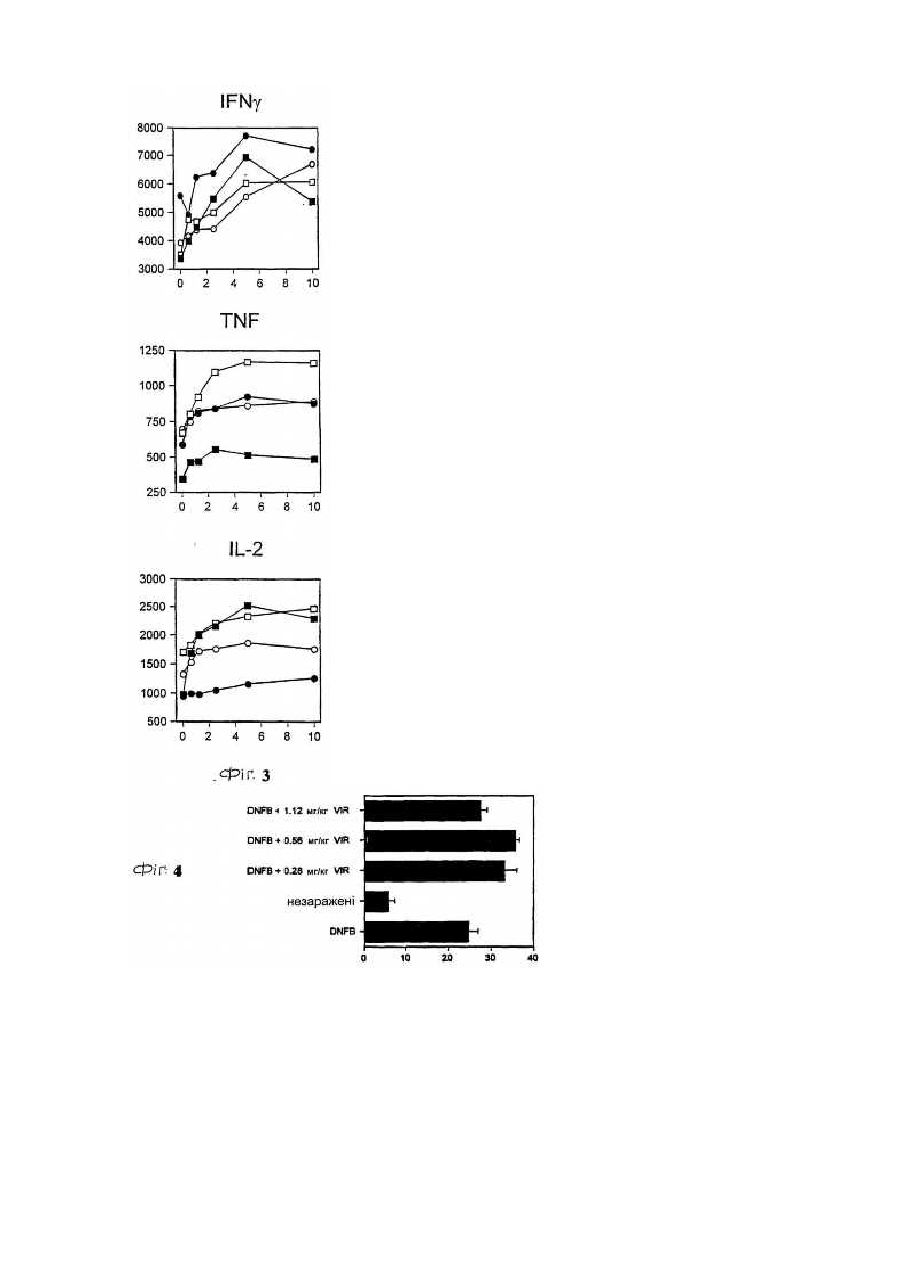
Для цієї заявки заявляється право пріоритету попередньої патентної заявки США за номером 60/182676, поданої 15 лютого 2000, та заявки США за номером 09/595365, поданої 16 червня 2000, кожну з яких у повному їхньому обсязі включено до цього опису шляхом посилання. Цей винахід відноситься до галузі нуклеозидних аналогів. Рибавірин (1-b-D-рибофуранозил-1,2,4-триазол-3-карбоксамід) - це нуклеозидний аналог, який продемонстрував ефективність у лікуванні вірусних хвороб як під час монотерапії (респіраторний синцитіальний вірус, Hall С. В., McBride J. Т., Walsh Ε. Ε., Bell D. Μ., Gala С. М., Hildreth S., Ten Eyck L. G., W. J. Hall. Aerosolized ribavirin treatment of infants with respiratory syncytial viral infection. N. Engl. J. Med. 1983, 308, 1443-1447), так і під час терапії у комбінації з інтерфероном-альфа (вірус гепатиту С, Reichard, О., Norkrans, G., Fryden, Α., Braconier, J-H., Sonnerborg, Α., Weiland, O. Randomized, double blind, placebo controlled trial of interferon alpha 2B with and without ribavirin for chronic hepatitis C. Lancet 1998, 351, 83-87). Досліди, про які повідомлялося за останній час, вказують на те, що корисність рибавірину in vivo може бути наслідком не тільки безпосереднього інгібування вірусної реплікації, але також може бути наслідком його спроможності підвищувати опосередкований Т-клітинами імунітет (Hultgren, С, Milich, D. R., Weiland, О., Sallberg, Μ. The antiviral compound ribavirin modulates the Τ helper Typel/Type 2 subset balance in hepatitis В and С virus-specific immune responses. J. Gen. Virol. 1998, 79, 2381 -2391; Ning, Q., Brown, D., Parodo, J., Cattral, M., Fung, L., Gorczynski, R., Cole, E., Fung, L., Ding, J. W., Liu M. F., Rotstein, О., Phillips Μ. J., Levy, G. Ribavirin inhibits viralinduced macrophage production of tumor necrosis factor, interleukin-1, procoagulant activity fgl2 prothrombinase and preserves Thl cytokine production but inhibits Th2 cytokine response. J. Immunol. 1998,160, 3487-3493; Martin, M. J., Navas, S., Quiroga, J. Α., Pardo, M., Carreno, V. Effects of the ribavirin-interferon alpha combination on cultured peripheral blood mononuclear cells from chronic hepatitis С patients. Cytokine 1998, 79, 2381-2391). Цей імуномодуляторний ефект рибавІрину можна продемонструвати in vitro шляхом вимірювання рівнів цитокінів Типу 1, що виробляються активованими Т-клітинами як людей, так і мишей (Tarn, R. С, Раі, В., Bard, J., Lim, С, Averett, D. R., Phan, U. Т., Malovanovic, T. Ribavirin polarizes human Τ cell responses towards a Type 1 cytokine profile. J. Hepatol. 1999, 30, 376-382), а також шляхом застосування інших способів вимірювання. Індукція зсуву цитокінів Типу 1 рибавірином є функціонально суттєвою in vivo у мишачих системах (Tarn, R. С, Lim, С, Bard, J., Раі, В. Contact hypersensitivity responses following ribavirin treatment in vivo are influenced by Type 1 cytokine polarization, regulation of IL-10 expression and costimulatory signaling. J. Immunol. 1999,163, 3709-3717). Імунні системи ссавців містять два головні класи лімфоцитів: B-лімфоцити (В-клітини), які походять з кісткового мозку, та Т-лімфоцити (Т-клітини), які походять з тимусу. B-клітини відповідають здебільшого за гуморальний імунітет (тобто, виробництво антитіл), в той час як Т-клітини відповідають здебільшого за клітина-опосередкований імунітет. Взагалі вважається, що Т-клітини розділяються на два підкласи, Т-клітини-хелпери та цитотоксичні Тклітини. Т-клітини-хелпери активують інші лімфоцити, включаючи B-клітини та цитотоксичні Т-клітини, та макрофаги шляхом виділення розчинних білкових медіаторів, які називаються цитокінами, які беруть участь у клітина-опосередкованому імунітеті. Як застосовано тут, лімфокіни - це субпопуляція цитокінів. Взагалі також вважається, що Т-клітини-хелпери розділяються на два підкласи, Тип 1 та Тип 2. Клітини Типу 1 виробляють інтерлейкін 2 (IL-2), фактор некрозу пухлини (TNFa) та інтерферон-гама (IFNg) та відповідають, перш за все, за клітина-опосередкований імунітет, такий як гіперчутливість уповільненого типу та противірусний імунітет. На відміну від цього клітини Типу 2 виробляють інтерлейкіни, IL-4, IL-5, IL-6, IL-9, IL10 та IL-13, та, перш за все, беруть участь у сприянні таким гуморальним імунним реакціям, які спостерігаються у відповідь на алергени, наприклад, перемикання ізотопу антитіла IgE та IgG4 (Mosmann, 1989, Аппи Rev Immunol, 7: 145-173). Як застосовується тут, терміни "реакції" Типу 1 та Типу 2 позначають, що вони включають повний діапазон ефектів, які є наслідками індукції лімфоцитів Типу 1 та Типу 2, відповідно. Серед іншого такі реакції включають зміни у виробництві відповідних цитокінів через транскрипцію, трансляцію, секрецію та, можливо, інші механізми, підвищену проліферацію відповідних лімфоцитів та інші ефекти, пов'язані з підвищеним виробництвом цитокінів, включаючи ефекти рухомості. Попередні заявки США (09/462714, 09/291097,09/291093, 09/471513, 60/164365, 60/164366, 60/172097, 60/175111), кожну з яких включено сюди шляхом посилання, стосуються аспектів наших нещодавніх відкриттів, які застосовують дію різних нуклеозидів (які, щодо визначення тут, включають похідні та аналоги природних нуклеозидів) на реакції селективного модулювання лімфоцитів відносно один одного. Серед іншого, ми продемонстрували, що можна селективно пригнічувати реакцію Типу 1 або реакцію Типу 2, у той час як інша з цих двох реакцій індукується або вона відносно не зазнає впливу, та що можна селективно індукувати будьяку з реакцій Типу 1 або Типу 2, у той час як інша з цих двох реакцій пригнічується або вона відносно не зазнає впливу. Ми також відкрили той несподіваний факт, що деякі нуклеозиди, які є ефективними у селективному модулюванні реакцій Типу 1 та Типу 2 відносно одна одної, мають тенденцію спричиняти бімодальний ефект. Серед іншого, деякі нуклеозиди, що мають тенденцію взагалі пригнічувати або індукувати як активність Типу 1, так і активність Типу 2 за відносно більш високою дозою, мають тенденцію селективно модулювати Тип 1 та Тип 2 відносно один одного за відносно нижчими дозами. Було продемонстровано, що Viramidine™ (1-b-D-рибофуранозил-1,2,4-триазол-3-карбоксамідин, гідрохлорид) є активним стосовно десяти різних вірусів, що є гідним порівняння з рибавірином (J. Т. Witkowski, R. К. Robins, G. P. Khare, R. W. Sidwell, J. Med Chem , 16, 935 -937,1973; R. W. Sidwell, J. H. Huffman, D. L. Barnard, D. Y. Pifat, Antiviral Research, 10, 193 - 208,1988; B. Gabrielsen, M. J. Phelan, L. Barthel-Rosa, С See, J. W. Huggins, D. F. Kefauver, T. P. Monath, M. A. Ussery, G. N. Chmurny, E. M. Schubert, K. Upadhya, С Kwong, D. A. Carter, J. A. Secrist III, J. J. Kirsi, W. M. Shannon, R. W. Sidwell, G. D. Kini, R. K. Robins, /. Med. Chem., 35, 3231 -3238,1992). Крім того, Viramidine™, подібно до Рибавірину, є інгібітором ІМР-дегідрогенази (R. С Willis, R. К. Robins, J. E. Seegmiller, Molecular Pharmacology, 18, 287 - 295,1980). Крім того, попередні токсикологічні дослідження дозволяють припустити, що Viramidine™ є менш токсичним, ніж рибавірин (D. Y. Pifat, R. W. Sidwell, P. G. Canonico, Antiviral Research, 9, 136,1988). Також нещодавні досліди у нашій лабораторії (R. Тат, К. Ramasamy, ICN Pharmaceuticals, Inc., неопубліковані результати, 1999) виявили, що Viramidine™ та Рибавірин демонстрували подібні імуномодуляторні властивості. Ці результати разом з низькою біологічною придатністю та токсичністю рибавірину спонукають нас розробляти не тільки Viramidine™ для інших вірусних хвороб, але також готувати інші похідні Viramidine™, включаючи синтез проліків Viramidine™, та відбирати їх у якості можливих противірусних агентів. Вплив інших нуклеозидних сполук-аналогів на реакції селективного модулювання лімфоцитів відносно один одного раніше не досліджувався або він не був задокументований. Ми відкрили, що бімодальний ефект, або селективна модуляція реакцій Типу 1 та Типу 2 відносно одна одної, також виникає після введення інших нуклеозидних сполук-аналогів, таких як пролікарські форми цих сполук. Існує багато перешкод, які слід подолати на шляху перетворення біологічно активних сполук у клінічно корисні агенти. Багато ефективних біологічно активних сполук ніколи не стають клінічно корисними агентами, оскільки вони мають небажані біофармацевтичні властивості, які включають низьку біологічну придатність, яка є наслідком низької спроможності проникати крізь біологічні бар'єри, такі як гематоенцефалічний бар'єр та кишковий бар'єр. Незважаючи на те, що багато факторів впливають на біологічну придатність ліків, небажані фізико-хімічні властивості (наприклад, заряд, ліпофільність, потенціал утворення водневого зв'язку, розмір) багатьох ліків є, можливо, одним із звичайних факторів, з якими зустрічаються та які перешкоджають проникненню ліків крізь біологічні бар'єри. Отже, оптимізація фізико-хімічних характеристик (заряду, ліпофільності, потенціалу утворення водневого зв'язку, розміру) ліків є, можливо, найбільш загальною стратегією сприяння транспортуванню ліків крізь такі мембранні бар'єри. Для того, щоб оптимізувати фізико-хімічні властивості ліків, застосовується одна можлива стратегія, яка полягає у тому, щоб оптимізувати ці властивості для проліків. (Н. Bundgaard, Design ofProdrugs, Elsevier, Amsterdam, 1985, N. Bodor, L. Prokai, W. M. Wu, H. Farag, S. Jonalagadda, M. Kawamura, J. Simpkins, Science, 257. 1698-1700,1992, H. E. Taylor, K. B. Sloan, /. Pharm. Sei., 87, 5-20,1998). Термін "проліки" застосовується для того, щоб описати агент, який повинен зазнавати хімічної або ферментної трансформації в активні, або початкові, ліки після введення, так щоб продукт метаболізму або початкові ліки могли далі демонструвати бажану фармакологічну реакцію. Шляхом тимчасового та біологічно зворотного утворення певних полярних функціональних груп у дрібних органічних молекулах небажані фізико-хімічні характеристики (наприклад, заряд, потенціал утворення водневого зв'язку) цих груп вдалося "замаскувати", не змінюючи при цьому постійних фармакологічних властивостей молекул. Таку стратегію дуже успішно застосовували у випадках, коли при отриманні похідних проліків застосовується перетворення карбоксильної або гідроксильної функціональної групи в естер, який можна легко гідролізувати in vivo хімічним або ферментним способом. Пропонуючи концепцію проліків, ми передбачаємо, що введення інших складових у початкові ліки підвищить біологічну придатність, адсорбцію та противірусні ефекти. Незважаючи на існування іще невизначених механізмів, ми відкрили, що величезні потенціальні переваги можна отримати шляхом селективної модуляції реакцій Типу 1 та Типу 2 відносно одна одної. Ми зробили висновок, наприклад, що специфічна модуляція Типу 1 відносно Типу 2 може бути корисною у лікуванні різноманітних станів та хвороб - від інфекцій, інвазій, пухлин та гіперчутливості до автоімунних хвороб. Ці відкриття мають особливе значення, тому що сучасні способи лікування багатьох перелічених вище хвороб мають обмежену ефективність, значні бічні ефекти або те та інше разом. Лікування автоімунної хвороби, наприклад, часто обмежується паліативними засобами, видаленням токсичних антитіл (як у випадку з міастенією гравіс) та введенням ризикованих ліків, які включають кортикостероїди, похідні хлорохіну та протиметаболічні та протипухлинні ліки, та ліків, таких як циклоспорини, які уражають клітини імунної системи. Цей винахід спрямований на нові сполуки нуклеозидних аналогів та споріднені сполуки, такі як проліки, їхнє терапевтичне застосування та синтез. В одному аспекті винаходу пропонуються сполуки нуклеозидних аналогів за формулою 1: Формула 1 - Viramidine™ (ICN 3142) Згідно з іншим аспектом винаходу фармацевтична композиція містить терапевтично ефективну кількість карбоксамідину за формулю 1, або його фармацевтично прийнятного естеру, або солі у суміші з, принаймні, одним фармацевтично прийнятним носієм. Згідно з подальшим аспектом винаходу фармацевтична композиція містить пролікарську форму карбоксамідину за формулою 1, або його фармацевтично прийнятного естеру, або солі у суміші з, принаймні, одним фармацевтично прийнятним носієм. Згідно з іще одним подальшим аспектом винаходу сполуку за формулою 1 застосовують при лікуванні будь-якого стану, який реагує позитивно на введення цієї сполуки та який за будь-яким фармацевтичним складом або протоколом введення досягає позитивної реакції. Серед іншого передбачається, що сполуки за формулою 1 можна застосовувати для лікування інфекцій, інвазій, раку, пухлин або інших неоплазм, гігантоклітинних артеріїтів або автоімунних хвороб. Фігура 1 - приклад схеми синтезу сполуки за формулою 1. Фігура 2 - це графіки, що демонструють вплив передбачених та інших сполук на синтез цитокінів Типу 1 в активованих стафілококовим ентеротоксином В (SEB) Т-клітинах людини. Фігура 3 - це графіки, що демонструють вплив концентрації 0,625-10мкМ передбаченої сполуки на синтез цитокінів Типу 1 в активованих стафілококовим ентеротоксином В Т-клітинах людини. Фігура 4 - це графік, що демонструє вплив передбаченої сполуки на реакції контактної гіперчутливості у мишей BALB/c. Фігура 5 - це графіки, що демонструють максимальну реакцію та максимальний діапазон для передбачених та інших сполук стосовно синтезу цитокінів Типу 1 в активованих стафілококовим ентеротоксином В Т-клітинах людини. Там, де наступні терміни застосовуються у цьому описі винаходу, вони застосовуються так, як визначається нижче. Терміни "нуклеозид" та "нуклеозидна сполука-аналог" ("сполука нуклеозидного аналога") є взаємозамінними та позначають сполуку, що складається з будь-якої пентозної або модифікованої пентозної складової, приєднаної до специфічного положення гетероциклу, ароматичого гетероциклу, або до природного положення пурину (9-положення) або піримідину (1-положення), або до еквівалентного положення в аналогові. Термін "нуклеотид" позначає фосфатний естер нуклеозиду, заміщений на 5¢ -позиції. Термін "гетероцикл" позначає одновалентний насичений або ненасичений карбоциклічний радикал, що має, принаймні, один гетероатом, такий як Ν, Ο або S, кожне доступне положення якого у межах кільця можна довільно замістити, незалежно, наприклад, гідрокси-, оксо-, аміно-, іміно-, нижчим алкілом, бромо-, хлорота/або ціаногрупою. У цей клас замісників включено пурини, піримідини. Термін "пурин" позначає азотисті біциклічні гетероцикли. Термін "піримідин" позначає азотисті моноциклічні гетероцикли. Термін "D-нуклеозиди" позначає нуклеозидні сполуки, що мають D-рибозну цукрову складову (наприклад, аденозин). Термін "L-нуклеозиди" позначає нуклеозидні сполуки, що мають L-рибозну цукрову складову. Терміни "L-конфігурація" та "D-конфігурація" застосовуються скрізь у цьому описі винаходу для опису хімічної конфігурації рибофуранозильної складової сполуки, яка зв'язана з піроло-піримідиновою частиною молекули. Термін "С-нуклеозиди" застосовується скрізь у цьому описі винаходу для опису типу зв'язку, що утворився між рибозною цукровою складовою та гетероциклічною основою. В С-нуклеозидах зв'язок походить з положення С-1 рибозної цукрової складової та приєднує вуглець гетероциклічної основи. Зв'язок, що утворюється в С-нуклеозидах, має вуглець-вуглецевий тип. Термін "N-нуклеозиди" застосовується скрізь у цьому описі винаходу для опису типу зв'язку, що утворився між рибозною цукровою складовою та гетероциклічною основою. У N-нуклеозидах зв'язок походить з положення С-1 рибозної цукрової складової та приєднує азот гетероциклічної основи. Зв'язок, що утворюється у N-нуклеозидах, має вуглець-азотний тип. Термін "захисна група" позначає хімічну групу, яку додають до атома кисню або азоту для запобігання його подальшого реагування під час утворення похідних інших складових в молекулі, в якій знаходиться кисень або азот. Широке різноманіття захисних груп кисню та азоту відоме для фахівців в галузі органічного синтезу. Термін "нижчий алкіл" позначає метил, етил, η-пропіл, ізопропіл, n-бутил, t-бутил, і-бутил або n-гексил. Цей термін також є прикладом циклічного, розгалуженого або прямого ланцюга з одного до шести атомів вуглецю. Термін "арил" позначає одновалентний ненасичений ароматичний карбоциклічний радикал, що має єдине кільце (наприклад, феніл) або два сконденсованих кільця (наприклад, нафтил), які можуть бути довільно заміщеними гідроксилом, нижчим алкілом, хлоро- та/або ціаногрупою. Термін "гетероцикл" позначає одновалентний насичений або ненасичений карбоциклічний радикал, що має, принаймні, один гетероатом, такий як N, О, S, Se або Р, у межах кільця, кожне доступне положення якого може, довільно, бути заміщеним або незаміщеним, незалежно, гідрокси-, оксо-, аміно-, іміно-, нижчим алкілом, бромо-, хлоро- та/або ціаногрупою. Термін "моноциклічний" позначає одновалентний насичений карбоциклічний радикал, що має, принаймні, один гетероатом, такий як О, N, S, Se або Р, у межах кільця, кожне доступне положення якого може, довільно, бути заміщеним, незалежно, цукровою складовою або будь-якими іншими групами, такими як бромо-, хлорота/або ціаногрупа, так, щоб система моноциклічного ядра зрештою ароматизувалася (наприклад, тимідин). Терміни "імуномодулятор" та "модулятор", що застосовуються тут, є взаємозамінними та позначають природні або синтетичні продукти, здатні модифікувати нормальну імунну систему або імунну систему з відхиленнями через стимулювання або пригнічення. Термін "ефективна кількість" позначає кількість сполуки за формулою 1, яка відновлює імунну функцію до нормальних рівнів або підвищує імунну функцію порівняно з нормальними рівнями з метою запобігання інфекції. Сполуки за формулою 1 можуть мати численні асиметричні центри. Отже, їх можна приготувати як в оптично активній формі, так і як рацемічну суміш. Обсяг винаходу, як його описано та заявлено у формулі винаходу, охоплює окремі оптичні ізомери та їх нерацемічні суміші, а також рацемічні форми сполук за формулою 1. Терміни "a" та "β" вказують на специфічну стереохімічну конфігурацію замісника на асиметричному атомі вуглецю в хімічній структурі, як зображено. Термін "енантіомери" позначає пару стереоізомерів, які є не супернеможливими дзеркальними відбиттями один одного. Суміш пари енантіомерів у відношенні 1:1 є "рацемічною" сумішшю. Термін "ізомери" позначає різні сполуки, які мають однакову формулу. "Стереоізомери" - це ізомери, які різняться лише влаштуванням атомів у просторі. "Фармацевтично прийнятні солі" можуть бути будь-якими солями, що походять з неорганічних та органічних кислот або лугів. Сполуки Нуклеозидні сполуки-аналоги цього винаходу взагалі описуються формулою 1: Формула 1 - Viramidine™ (ICN 3142) де хімічна конфігурація сполуки - це L-конфігурація або D-конфігурація. Приклад синтезу передбачених сполук (тут: Viramidine™) може виконуватися згідно зі способом, який описано нижче та зображено на Фігурі 1. 3-Ціано-1-(2,3,5-три-O-ацетил-b-В-рибофуранозил)-1,2,4-триазол (7) Суміш 3-ціано-1,2,4-триазолу (18,8г, 200ммоль) (6), 1,2,3,5-тетра-O-ацетил-b-D-рибофуранози (63,66г, 200ммоль) та біс(р-нітрофеніл)фосфату (1г) розташували у круглодонній колбі (500мл). Колбу розташували на попередньо нагрітій масляній бані при 165-175°С в умовах вакууму, що утворюється водоструминним насосом, та перемішували протягом 25 хвилин. Витиснену оцтову кислоту збирали охолоджуваною льодом пасткою, розташованою між водоструминним насосом та круглодонною колбою. Колбу вилучили з масляної ванни та дозволили їй охолодитися. Коли температура колби досягла приблизно 60-70°С, ввели EtOAc (300мл) та насичений NaHCO3 (150мл) та екстрагували EtOAc. Водний шар знов екстрагували EtOAc (200мл). Комбінований екстракт EtOAc промили насиченим NaHCO3 (300мл), водою (200мл) та соляним розчином (150мл). Органічний екстракт висушили над безводним Na2SO4, профільтрували та фільтрат випарили до сухого стану. Залишок розчинили в ефірі (100мл), внаслідок чого після охолодження при 0°С протягом 12 годин одержали безбарвні кристали. Тверду речовину відфільтрували, промили мінімальною кількістю холодного EtOH (20мл) та висушили в умовах високого вакууму над твердим NaOH. Вихід: 56,4г (80%). Т.пл. 96-97°С. 1 HΜΡ (CDCl3): δ 2,11 (с, 3Н, СОСН3), 2,13 (с, 3Н, СОСН3), 2,14 (с, 3Н, СОСН3), 4,22 (дд, 1H), 4,46 (м, 2Н), 5,52 (т, 1H, J = 6,0 Гц), 5,70 (м, 1Н), 6,01 (д, 1H, С1Н, J = 3,6 Гц) та 8,39 (с, 1Н, С5Н). Елементний аналіз. Обчислено для C14H16N4O7 (352,30): С, 47,73; Н, 4,58; Ν, 15,90. Знайдено: С, 47,70; Н, 4,63; Ν, 16,01. Гідрохлорид 1-b-D-рибофуранозил-1,2,4-триазол-3-карбоксамідину (Viramidine™) (8) Суміш сполуки 7 (14,08г, 40,0ммоль), NH4CI (2,14г, 40,0ммоль) та безводного аміаку (150мл) нагрівали у сталевій бомбі при 85°С протягом 18 годин. Сталеву бомбу охолодили, відкрили та її вміст випарили до сухого стану. Залишок кристалізували з MeCN-EtOH, внаслідок чого отримали 10,6г (95%) сполуки 8. Т.пл. 177-179°С. 1 HΜΡ (ДМСО-d6): δ 3,44-4,2 (м, 3Н), 4,40 (м, 2Н), 5,04 (т, 1Н), 5,29 (м, 1H), 5,74 (м, 1Н), 5,87 (д, 1Н, С1Н), 8,96 (ш.с., 3Н) та 9,17 (с, 1Н, С5Н). Елементний аналіз. Обчислено для C8H14ClN5O4 (279,68): С, 34,35, Η, 5,05, Ν, 25,04, СІ, 12,69. Знайдено: С, 34,39, Η, 5,10, Ν, 25,14, СІ 12,71. Альтернативно, синтез можна виконувати з комерційно доступного Ribavirin™, згідно з наступним способом. 2¢,3¢,5'-Три-ацетил-1-b-D-рибофуранозил-1,2,4-триазол-3-карбоксамід(9) Суспензію 1-b-D-рибофуранозил-і ,2,4-триазол-3-карбоксаміду (Ribavirin™) (28,4г, 116,4ммоль) (5) в ацетангідриді (200мл) та піридині (50мл) перемішували при кімнатній температурі протягом ночі. Одержаний прозорий розчин концентрували в умовах вакууму та отримали прозору піну (43,1г, кількісний). Ця піна була гомогенною на тонкошаровій хроматографії, та її застосовували безпосередньо для наступного етапу без попереднього очищення. Невелику кількість очистили шляхом флеш-хроматографії та отримали аналітичний зразок. 1 H ЯМР (300 МГц, ДМСО-d6) δ 2,01, 2,08, 2,09 (3с, 9Н, СОСН3), 4,10 (м, 1Н), 3,52 (м, 2Н), 5,58 (т, Ш), 5,66 (м, 1H), 6,33 (д, 1Н, J = 3,0 Гц, С1Н), 7,73, 7,92, (2с, 2Н, CONH2), 8,86 (с, 1Н, С5Н триазол). Елементний аналіз (C10H18 N4O8) С, Н, N. 3-Ціано-2',3',5'-три-O-ацетил-1-b-D-рибофуранозил-1,2,4-триазол (10) У розчин сполуки 9 (43,1г, 116,4ммоль) у хлороформі (500мл) додали триетиламін (244мл) та суміш охолодили до 0°С на льодо-сольовій бані. Хлорангідрид фосфорної кислоти (30,7мл, 330ммоль) додавали краплями під час перемішування та дозволили розчину нагрітися до кімнатній температури. Після того, як суміш перемішували при кімнатній температурі протягом 1 години, тонкошарова хроматографія (гексан/ацетон, 3:1) вказувала на повне зникнення початкового матеріалу. Коричневу реакційну суміш концентрували до сухого стану в умовах вакууму та залишок розчинили у хлороформі (500мл). Органічний розчин промили насиченим водним бікарбонатом натрію (3´200мл), висушили над безводним сульфатом натрію та концентрували в умовах вакууму. Залишок зазнав хроматографії на силікагелі (флешхроматографія), 20% ацетону у гексані, внаслідок чого отримали 33,14г (81% від рибавірину) чистої сполуки 10 у вигляді аморфної твердої речовини. Ця тверда речовина була ідентичною в усіх відношеннях зі справжнім зразком: Т.пл. 101-103°С, нерозчинний залишок (бромід калію) n 2250 (CN), 1750 (С=О), см-1; 1H ЯМР (300 МГц, CDCl3) δ 2,04, 2,06, 2,07 (3с, 9Н, ацетилметили), 4,15 (дд, 1Н), 4,40 (м, 1H), 5,47 (т, 1Н), 5,63 (дд, 1H), 5,95 (д, 1H, J = 3,2 Гц, С1Н), 8,34 (с, 1H, С5Н триазол). Гідрохлорид 1-b-D-рибофуранозил-1,2,4-триазол-3-карбоксамідину (8) До суспензії сполуки 10 (4,0г, 11,4ммоль) у метанолі (100мл) додали молярний розчин метанолового метоксиду натрію (12мл) та суміш перемішували при кімнатній температурі протягом ночі. Розчин підкислили до pH 4 промитим метанолом полімером Dowex H+, полімер відфільтрували та фільтрат концентрували до сухого стану в умовах вакууму. Залишок розчинили у мінімальній кількості метанолу (15мл) та перенесли до склянки під тиском. Додали хлорид амонію (0,61г, 11,4ммоль) та розчин метанолу, насиченого при 0°С сухим аміачним газом (75мл), склянку герметично закрили та розчин перемішували при кімнатній температурі протягом ночі. Розчин концентрували до сухого стану в умовах вакууму та одержаний залишок кристалізували з ацетонітрилу/етанолу, внаслідок чого одержали сполуку 8 у вигляді кристалічної твердої речовини (2,95г, 93%). Цей зразок був ідентичним в усіх відношеннях зі справжнім зразком. У певних фармацевтичних дозованих формах пролікарські форми сполук, особливо включаючи ациловані (ацетиловані або інші) похідні, піридинові естери та різні сольові форми цих сполук, є особливо переважними, та їх можна вводити згідно зі способом лікування стану пацієнта. Звичайний фахівець у галузі визначить, як можна легко модифікувати ці сполуки у пролікарські форми для сприяння доставці активних сполук до ділянкимішені у організмі-хазяїні або у пацієнта. Звичайний фахівець у галузі також скористається перевагами корисних фармакокінетичних параметрів пролікарських форм, там де це є прийнятним, стосовно доставки цих сполук до ділянки-мішені у організмі-хазяїні або у пацієнта з метою досягнення максимально передбаченого ефекту від сполуки. Передбачений приклад утворення пролікарської форми описаних тут сполук є наступним. Одним з найпростіших проліків Viramidine™ є три-О-ацетилове похідне Viramidine™. Три-О-ацетилове похідне одержують так, як показано на схемі 1: Схема 1 5'-Ретиноїлове похідне Viramidine™ - це інша проста пролікарська форма, яка була одержана за наступним способом: Схема 2 Інші 5'-похідні Viramidine™ включають наступні сполуки, що зображені на схемі 3: Схема 3 Більшість цих сполук можна одержати так, як описано у С. Sergheraert, С. Pierlot, A. Tartar, Υ. Henin, Μ. Lemaitre, /. Med. Chem., 36, 826 - 830,1993. Синтез пролікарських форм Viramidine™ на основі кумарину можна одержати за наступним способом: Схема 4 Вважають, що амінокислотні естери є кращими пролікарськими формами через можливе залучення стереоселективного транспортера. Амінокислотні похідні Viramidine™ можна синтезувати, як зображено нижче: Схема 5 Для специфічної доставки ліків у печінку та біліарну систему транспортна система ендогенної жовчної кислоти є привабливим кандидатом. Синтез жовчно-кислотних кон'югатів Viramidine™ можна здійснювати, як представлено нижче: Схема 6 Нуклеотидні похідні є іншим класом проліків або пролікарських форм. Одержання захищених 5'монофосфатних похідних зображено нижче. Через захищення негативних зарядів фосфатів нейтральними замісниками утворюються більш ліофільні похідні, які, як передбачається, будуть повертатися назад до відповідних монофосфатів, коли вони будуть знаходитися усередині клітини. Схема 7 де R1 - це алкільні групи, такі як CH3C(O)S-CH2CH2-, (CH3)2CHC(O)S-CH2CH2-, (CH3)3CC(O)S-CH2CH2-, (СН3)3СС(О)ОСН2-, C6H5C(O)S-CH2CH2- або HOCH2CH2SS-СН2СН2-. Амінокислотні фосфорамідати - це інший клас проліків, які можна синтезувати, як описано нижче: Схема 8 Інші похідні монофосфатних проліків показані нижче: Схема 8А Проліки Viramidine™ на основі саліцилату можна одержати за наступною схемою: Схема 9 Проліки нуклеозидних 5'-ди- або трифосфатів будуть більш цікавими, тому що вони будуть уникати більше метаболічних етапів. Наступні приклади - це потенціальні нуклеотидні ліпофільні проліки, які одержують, як показано нижче· Наступні приклади - це інший клас потенційних фосфонатних проліків Viramidine™. Інші можливі проліки включають можливі комбінації груп, зображених у РСТ патентних заявках WO 98/39342, WO 98/39343, WO 98/39344 та WO 99/45016. Проліки Viramidine™ можна одержати не лише шляхом модифікування цукрової частини початкової молекули, але також шляхом одержання функціональних похідних амідину. Наступні приклади - це декілька класів проліків, які можна приготувати шляхом модифікації амідинової групи, як показано нижче: Застосування Передбачається, що сполуки за формулою 1 будуть застосовуватися для лікування широкого різноманіття станів та, фактично, будь-якого стану, який позитивно реагує на введення однієї або декількох з цих сполук. Серед іншого особливо передбачається, що сполуки за винаходом можна застосовувати для лікування інфекцій, інвазій, раку, або пухлин, або автоімунних хвороб. Далі передбачається, що сполуки за винаходом можна застосовувати для лікування станів або хвороб в специфічних органах пацієнта, таких як печінка або серце. Інфекції, які передбачається лікувати сполуками цього винаходу, включають респіраторний синцитіальний вірус (RSV), вірус гепатиту В (HBV), вірус гепатиту С (HCV), простий герпес типу 1 та 2, генітальний герпес, герпетичний кератит, герпетичний енцефаліт, оперізувальний герпес, вірус імунодефіциту людини (ВІЛ), вірус грипу А, вірус hantann (геморагічна гарячка), вірус папіломи людини (HPV), кір та грибки. Інвазії, які передбачається лікувати сполуками цього винаходу, включають інвазії найпростішими, а також гельмінтні інвазії та інвазії іншими паразитами. Типи раку або пухлин, які передбачається лікувати, включають такі типи, що спричиняються вірусом, та ефект може включати інгібування трансформації інфікованих вірусом клітин до неопластичного стану, інгібування розповсюдження вірусів з трансформованих клітин до інших нормальних клітин та/або припинення росту трансформованих вірусом клітин. Автоімунні та інші хвороби, які передбачається лікувати, включають артрит, псоріаз, кишкову хворобу, діабет підлітків, вовчак, розсіяний склероз, подагру та подагричний артрит, ревматоїдний артрит, відторгнення трансплантату, гігантоклітинний артеріїт, алергію та астму. Іще інші передбачені варіанти застосування сполук згідно з цим винаходом включають застосування їх як посередників у хімічному синтезі інших нуклеозидних або нуклеотидних аналогів, які, у свою чергу, є корисними як терапевтичні агенти або для інших цілей. Згідно з іще іншим аспектом, спосіб лікування ссавця містить введення терапевтично та/або профілактично ефективної кількості фармацевтичного засобу, який містить сполуку цього винаходу. У цьому аспекті ефект може стосуватися модулювання деякої частини імунної системи ссавця, особливо модулювання лімфокінних профілів Типу 1 та Типу 2 відносно один одного. Там, де виникає модуляція лімфокінів Типу 1 та Типу 2, особливо передбачається, що модуляція може включати пригнічення як Типу 1, так і Типу 2, та більш переважно стимуляцію лімфокінів Типу 1, або підвищення реакції Типу 1 відносно реакції Типу 2. Особливо передбачається, що Viramidine™ (1,39мкг/мл) підвищує експресію та синтез цитокінів Типу 1 у (переважно активованих) Т-лімфоцитах, та результати різних експериментів зображено на Фігурах 2-5. Фігура 2 зображує вплив 5мкМ вірамідину (сполуки за формулою 1), рибавірину та левовірину на синтез цитокінів Типу 1 в активованих стафілококовим ентеротоксином В Т-клітинах людини (n = 5 донорів), при цьому вірамідин демонструє очевидне підвищення у реакції Типу 1, порівняно з контрольними експериментами з триазолом. Фігура 3 - це графіки залежності ефект-доза впливу вірамідину в діапазоні 0,625-10мкМ на синтез цитокінів Типу 1 в активованих стафілококовим ентеротоксином В Т-клітинах людини (дані репрезентують 4 окремих донорів). Ефект in vivo підвищеної реакції Типу 1 в аналізі контактної гіперчутливості (CHS) вірамідину очевидно продемонстровано на Фігурі 4, а Фігура 5 демонструє порівняння між вірамідином та левовірином/рибавірином стосовно концентрації нуклеозиду для максимальної реакції та максимальний діапазон реакцій (вісь "у" зображує кількість пацієнтів, які реагували, у певному експерименті). Приготування Т-клітин людини та активація in vitro Мононуклеари периферійної крові здорових донорів або пацієнтів з ревматоїдним артритом виділили шляхом центрифугування у градієнті щільності з наступним збагаченням Т-клітинами із застосуванням засобу Lymphokwik (One Lambda, Canoga Park CA). Забруднювальні моноцити видалили завдяки прилипанню до пластика. Очищені Т-клітини були більш ніж на 99% CD2+, менше ніж на 1% HLA-DR+ та менше ніж на 5% CD25+, та їх зберігали у RPMI-AP5 (середовище RPMI-1640, що містить 20мМ буферу HEPES, pH 7,4, 5% автологічної плазми, 1% L-глутаміну, 1% пеніциліну/стрептоміцину та 0,05% 2-меркаптоетанолу). Для визначення рівнів білка цитокінів Т-клітини (1´106 клітин в об'ємі 1мл) активували шляхом додавання 10нг РМА (ФМА) та 0,5мкг іономіцину (обидва отримали у Calbiochem, La Jolla, CA) та інкубували у планшетах з 24 лунками у присутності 0-20мкМ нуклеозиду протягом аж до 48 годин при температурі 37°С та 5% СО2 у зволоженому інкубаторі. Після активації супернатанти проаналізували на виробництво клітино-похідних цитокінів. Для досліджень проліферації та життєздатності вищеописаний метод модифікували до формату планшетів з 96 лунками, застосовуючи 0,2´106 клітин в об'ємі 0,2мл та виконували активацію 2нг РМА (ФМА) та 0,1мкг іноміцину. В окремих експериментах 5´106 клітин у 2мл активували 20нг ФМА та 1мкг іономіцину. Альтернативно, клітини можна активувати in vitro стафілококовим ентеротоксином В згідно з опублікованими процедурами. При цьому усю РНК виділили з Т-клітин після інкубації протягом 6-24 годин та проаналізували шляхом ПЛР ревертази з метою визначення рівнів мРНК різних цитокінів та запальних посередників. Також в окремих експериментах Т-клітини людини очистили далі (застосовуючи реагенти, що збагачують клітини, які отримали від Stem Cell Technologies, Vancouver, ВС) і отримали чисті популяції CD4+ (менше ніж на 1% CD8+, застосовуючи реагент для виділення Т-клітин людини CD4+ від RosetteSep) та субпопуляції Т-клітин CD8+ (менше ніж на 1% CD4+, застосовуючи реагент для виділення Т-клітин людини CD4+ від RosetteSep), після чого 1´106 клітин на 1мл активували ФМА та іономіцином, як в усіх експериментах з Т-клітинами. Аналізи зовнішньоклітинних цитокінів Рівні цитокінів людини визначили у клітинних супернатантах після відповідного розрідження, застосовуючи набори для твердофазного імуноферментного аналізу (ELISA), що є специфічними для IL-2, IFNy, TNFot, IL-4 та IL-5 (Biosource International, Camarillo, CA). Рівні цитокінів мишей визначили, застосовуючи набори для твердофазного імуноферментного аналізу, які є специфічними для IFNy та IL-4 мишей (R and D Systems, Minneapolis, MN). Усі результати твердофазного імуноферментного аналізу виразили у пг/мл. Деякі дані зображено як відсоткове відношення до активованих контрольних клітин, яке підрахували як відношення рівнів цитокінів активованих Т-клітин у присутності нуклеозиду тесту до рівня цитокінів необроблених активованих Тклітин, помножене на 100%. Нульовий вплив на рівні цитокінів нуклеозидів тесту дасть відсоткове значення активованих контрольних клітин - 100%. Альтернативні дані було продемонстровано як зміну відсоткового значення від активованих контрольних клітин ([(пг/мл тесту - пг/мл активованих контрольних)/пг/мл активованих контрольних] χ 100%). Нульовий вплив на рівні цитокінів нуклеозидів тесту становив би 0%. Контактна гіперчутливість (CHS) Реактивність до контактного алергену, DNFB, визначили у мишей BALB/c, як описано раніше (Ishii, Ν., Κ. Takahashi, H. Nakajima, S. Tanaka, P. W. Askenase. 1994. DNFB contact sensitivity (CS) in BALB/c and C3H/He mice. J. Invest. Dermatol. 102:321). Стисло, мишей сенсибілізували шляхом застосування 20мкл 0,3% DNFB в ацетоні: оливковій олії, 4:1, на поголених черевах незаражених мишей. Для оптимального визначення контактної гіперчутливості мишам ввели з обох боків кожного вуха 20мкл 0,12% DNFB через 5 днів після сенсибілізації. Несенсибілізовані миші також зазнали алергізації, та їх застосовували як контрольні у кожному експерименті. Через 24 години здійснили вимірювання товщини вуха та проаналізували реакцію на DNFB шляхом віднімання значень після алергізації від значень до алергізації. Там, де вказано, 7-b-Dрибофуранозил-4-оксопіроло[2,3-d]піримідин-5-карбоксамідин у дозі 6,2мкг у 50мкл фізіологічного розчину з фосфатним буфером (0,3мг/кг) або 12,4мкг у 100мкл фізіологічного розчину з фосфатним буфером (0,6мг/кг), вводили шляхом внутрішньочеревинної ін'єкції під час алергізації DNFB. Ці дози 7-b-D-рибофуранозил-4оксопіроло[2,3-d]піримідин-5-карбоксамідину дали максимальний ефект у попередніх дослідах оптимізації. Після останніх вимірювань товщини вуха мишей умертвили шляхом зміщення шийного відділу хребта та видалили пазушні/бічні пазушні лімфатичні вузли. Після виділення загальної клітинної РНК з клітин виділених лімфатичних вузлів виконували аналіз ПЛР ревертази та саузерн-блотинг для визначення рівнів мРНК IFNg, IL-2 та IL-10. Подальші експерименти Взагалі передбачається, що зсув імунної реакції до реакції Типу 1 є переважним. Отже, передбачається, що сполуки згідно з суттю винаходу можуть бути особливо корисними у лікуванні вірусних хвороб (переважно вірусних інфекцій, у яких реакція типу 1 знижується або пригнічується). З метою підтвердження ефективності модулювання імунної реакції здійснили різні експерименти, та наступний опис є прикладом суті декількох експериментів, виконаних з передбаченими сполуками: In vitro - вірамідин інгібував інфекцію вірусу Punta Того LLC-MK2 (ниркові клітини макаки резус з ЕС50 8мг/мл (штам Adames), та 12мг/мл (штам Balliet) - СС50 становив 320мг/мл (1,0-1,2 рейтинг вірусу). In vivo - підшкірне або пероральне введення вірамідину стало причиною 100% виживаності (10 C57BL/6 мишей/групу) мишей, яким підшкірно вводили вірус Punta Того (штам Adames). Протягом 24 годин після інфекції вірусом Punta Того in vivo мінімальна ефективна пішкірна доза становила 32мг/кг для рибавірину, а для вірамідину вона становила 96мг/кг за умовою підшкірного введення b. і. d. протягом 5 днів. Протягом 24 годин після інфекції вірусом Punta Того in vivo мінімальна ефективна пероральна доза становила 20мг/кг для рибавірину, а для вірамідину вона становила 40мг/кг за умовою перорального введення b. і. d. протягом 5 днів. Взагалі, найбільш переважні варіанти застосування згідно з цим винаходом - це ті варіанти застосування, у яких активні сполуки є відносно менш цитотоксичними до клітин хазяїна, які не є мішенями, та відносно більш активними для мішені. У цьому відношенні переважним також може бути те, що L-нуклеозиди можуть мати підвищену стійкість порівняно з D-нуклеозидами, що може стати причиною кращої фармакокінетики. Цього результату можна досягти, тому що L-нуклеозиди можуть не розпізнаватися ферментами та, отже, можуть мати триваліший напіврозпад. Передбачається, що сполуки згідно з цим винаходом будуть вводитися in vivo, in vitro або ex vivo у будьякому відповідному фармацевтичному складі та за будь-яким відповідним протоколом. Отже, введення можна здійснювати перорально, парентерально (включаючи підшкірні ін'єкції, внутрішньовенні ін'єкції, внутрішньом'язові ін'єкції, внутрішньогрудинні ін'єкції або способами інфузії), шляхом інгаляції або ректально, місцево тощо, та у складах дозованих форм, які містять звичайні нетоксичні фармацевтично прийнятні носії, наповнювачі та ад'юванти. Заради наведення прикладу, передбачається, що до сполук згідно з цим винаходом можна додати фармацевтично прийнятний носій. Наприклад, сполуки цього винаходу можна вводити перорально, як фармацевтично прийнятні солі. Оскільки сполуки за цим винаходом є здебільшого водорозчинними, то їх можна вводити внутрішньовенно у фізіологічному сольовому розчині (наприклад, забуференими до pH від приблизно 7,2 до приблизно 7,5). Для цього можна застосовувати звичайні буфери, такі як фосфати, бікарбонати або цитрати. Звичайно, будь-який фахівець у галузі зможе модифікувати фармацевтичні склади згідно з рекомендаціями опису для одержання численних складів для певного способу введення, але не за рахунок нестабільності складів цього винаходу та їхньої терапевтичної активності. Зокрема, модифікацію цих сполук, з метою зробити їх більш розчинними у воді або іншому наповнювачі, можна легко здійснити шляхом незначних модифікацій (сольовий склад, естерифікація тощо), що є звичайною практикою у галузі. Звичайним у галузі є також модифікування способу введення та режиму доз певної сполуки для надання фармакокінетиці цих сполук максимально корисного ефекту у пацієнта. Крім того, сполуки згідно з цим винаходом можна вводити окремо або у комбінації з іншими агентами для лікування перелічених вище інфекцій або станів. Комбінована терапія згідно з цим винаходом містить введення, принаймні, однієї сполуки цього винаходу або її функціонального похідного та, принаймні, одного іншого фармацевтично активного інгредієнта. Активний(-і) інгредієнт(-и) та фармацевтично активні агенти можна вводити окремо або разом, і коли їх вводять окремо, введення можна виконувати одночасно або окремо у будь-якій черзі. Кількість активного інгредієнта(-ів) та фармацевтично активного агента(-ів) та відносний вибір певного часу введення буде обиратися з метою досягти бажаного * комбінованого терапевтичного ефекту. Переважно, комбінована терапія містить введення однієї сполуки цього винаходу або її фізіологічно функціонального похідного та одного з агентів, про який згадується нижче у цьому описі. Прикладами інших ліків або активних інгредієнтів, які, як передбачається, є ефективними у комбінації з модулятором за формулою 1, є противірусні агенти, такі як інтерферон, включаючи, проте не обмежуючись ними, інтерферон a та g, рибавірин, ацикловір та ΑΖΤ™, протигрибкові агенти, такі як толнафтат, Fungizone™, Lotrimin™, Mycelex™, ністатин та амфотерацин, протипаразитарні препарати, такі як Mintezol™, Niclocide™, Vermox™ та Flagyl™, кишкові агенти, такі як Immodium™, Lomotil™ та Phazyme™, протипухлинні агенти, такі як інтерферон α та γ, Adriamycin™, Cytoxan™, Imuran™, метотрексат, Mithracin™, Tiazofurin™, Taxol™, дерматологічні агенти, такі як Aclovate™, Cyclocort™, Denorex™, Florone™, Oxsoralen™, вугільний дьоготь та саліцилова кислота, антимігреневі препарати, такі як сполуки ерготаміну, стероїди та імуносупресори, що не перелічені вище, які включають циклоспорини, Diprosone™, гідрокортизон, Floron™, Lidex™, топікорт та валізон, та метаболічні агенти, такі як інсулін, та інші ліки, які можуть не підпадати під вище перелічені категорії, до яких належать цитокіни, такі як IL2, IL4, IL6, IL8, IL10 та IL12. Особливо переважними первинними ліками є AZT, 3ТС, 8-заміщені гуанозинові аналоги, 2,3-дидезоксинуклеозиди, інтерлейкін II, інтерферони, такі як ΙαΒ-інтерферони, тукарезол, левамізоль, ізопринозин та циклолігнани. Приклади таких подальших терапевтичних агентів включають агенти, що є ефективними для модулювання імунної системи або пов'язаних станів, такі як AZT, 3ТС, 8-заміщені гуанозинові аналоги, 2',3'дидезоксинуклеозиди, інтерлейкін II, інтерферони, такі як a-інтерферон, тукарезол, левамізоль, ізопринозин та циклолігнани. Певні сполуки згідно з цим винаходом можуть бути ефективними для підсилення біологічної активності певних агентів згідно з цим винаходом шляхом зниження метаболізму або дезактивації інших сполук та у такій якості вводяться разом для досягнення цього певного ефекту. Стосовно дози, звичайний фахівець у галузі зрозуміє, що терапевтично ефективна кількість буде різнитися для певної інфекції або стану, який слід лікувати, та буде залежати від їхньої суворості, режиму лікування, який слід застосовувати, фармакокінетики агента, який застосовується, а також від пацієнта (тварина або людина), якого слід лікувати. Передбачається, що різні альтернативні дози є також прийнятними, включаючи дози між 0,5мг/кг та 0,1мг/кг або менше, а також дози від 0,5мг/кг до 1,0мг/кг або більше. Далі передбачається, що, незважаючи на те, що успішного лікування можна досягти з деякими вірусними інфекціями при відносно низьких концентраціях у плазмі сполук за формулою 1, інші вірусні інфекції можуть потребувати відносно високих доз. Передбачається, проте, що відповідний режим можна розробити шляхом введення невеликої кількості, а потім підвищення кількості, доки бічні ефекти не стануть шкідливими або до досягнення бажаного ефекту. Введення активної сполуки може різнитися від тривалого введення (внутрішньовенна крапельниця) до декількох пероральних введень на день (наприклад, Q.I.D) та може включати, серед інших способів введення, пероральне, місцеве, парентеральне, внутрішньом'язове, внутрішньовенне, підшкірне, трансдермальне (яке може містити агент, що підсилює проникання), трансбукальне введення та введення супозиторіїв. Для того, щоб приготувати композиції згідно з цим винаходом, терапевтично ефективну кількість однієї або більше сполук згідно з цим винаходом переважно ретельно змішують з фармацевтично прийнятним носієм згідно з традиційними способами приготування фармацевтичних сполук з метою отримання дози. Носій може мати широке різноманіття форм залежно від форми препарату, який необхідно вводити, наприклад, перорально або парентерально. Для приготування фармацевтичних композицій у дозованій формі для перорального введення можна застосовувати будь-які звичайні фармацевтичні середовища. Отже, для рідких пероральних препаратів, таких як суспензії, еліксири та розчини, можна застосовувати підхожі носії та добавки, які включають воду, гліколі, олії, спирти, ароматизатори, консерванти, барвники та їм подібні речовини. Для твердих пероральних препаратів, таких як порошки, таблетки, капсули, та для твердих препаратів, таких як супозиторії, можна застосовувати підхожі носії та добавки, які включають крохмалі, цукровий носій, такий як декстроза, маніт, лактоза та споріднені носії, розріджувачі, гранулятори, мастильні речовини, зв'язувальні речовини, розщеплювальні агенти та їм подібні речовини. Якщо бажано, таблетки або капсули можуть мати ентеросолюбільне покриття або бути пролонгованої дії за стандартними способами. Для парентеральних фармацевтичних складів носій буде звичайно включати стерильну воду або водний розчин хлориду натрію, проте можна включити інші інгредієнти, що сприяють дисперсії. Звичайно, там, де слід застосовувати стерильну воду та зберігати її стерильною, склади та носії слід також стерилізувати. Також можна приготувати суспензії для ін'єкцій, у випадку чого можна застосовувати відповідні рідкі носії, суспендувальні агенти тощо.
ДивитисяДодаткова інформація
Назва патенту англійськоюCarboxamidine for treatment of viral hepatitis
Назва патенту російськоюКарбоксамидин для лечения вирусного гепатита
МПК / Мітки
МПК: A61P 31/12, A61K 31/70
Мітки: лікування, вірусного, гепатиту, карбоксамідин
Код посилання
<a href="https://ua.patents.su/15-73761-karboksamidin-dlya-likuvannya-virusnogo-gepatitu.html" target="_blank" rel="follow" title="База патентів України">Карбоксамідин для лікування вірусного гепатиту</a>
Спосіб лікування вірусного гепатиту

Номер патенту: 51575
Опубліковано: 15.11.2002
Автори: Сенченко Ольга Валентинівна, Зубов Олександр Дем'янович
МПК: A61K 35/407
Мітки: спосіб, гепатиту, вірусного, лікування
Формула / Реферат:
Спосіб лікування вірусного гепатиту, який відрізняється тим, що у ворітну вену печінки черезшкірно пункційно вводять завись фетальних гепатоцитів при концентрації 5-6,0.105 кл/мл в дозі 5,0-15,0 мл 1 раз на місяць триразово, при цьому черезшкірне пункційне введення здійснюють під контролем ультразвукового дослідження з кольоровим доплерівським картуванням.
Спосіб лікування гострого вірусного гепатиту с

Номер патенту: 57374
Опубліковано: 16.06.2003
Автор: Фролов Валерій Мітрофанович
МПК: A61K 33/06, A61K 45/00
Мітки: гепатиту, лікування, спосіб, гострого, вірусного
Формула / Реферат:
1. Спосіб лікування гострого вірусного гепатиту С, що включає введення препаратів -інтерферону та циклоферону, який відрізняється тим, що додатково вводять гепатопротекторний препарат антраль по 0,2 г 3-4 рази на добу протягом 10-15 діб поспіль.2. Спосіб за п.1, який відрізняється тим, препарати
Спосіб лікування хронічного вірусного гепатиту

Номер патенту: 38888
Опубліковано: 15.05.2001
Автори: Архій Емілія Йосипівна, Дербак Марія Антонівна
МПК: A61K 35/64, A61P 1/16, A61K 31/51, A61K 31/4415
Мітки: гепатиту, хронічного, вірусного, спосіб, лікування
Формула / Реферат:
Спосіб лікування хронічного вірусного гепатиту, що включає введення хворому гепатопротекторів, вітамінів В1, В6, С та сорбентів, який відрізняється тим, що додатково вводять трофосан 2.1 у кількості 5-7 г два рази на день протягом 20 днів.
Спосіб лікування гострого вірусного гепатиту в

Номер патенту: 69734
Опубліковано: 15.09.2004
Автор: Фролов Валерій Мітрофанович
МПК: A61K 31/08, A61K 31/195, A61K 31/00
Мітки: спосіб, гепатиту, гострого, вірусного, лікування
Формула / Реферат:
1. Спосіб лікування гострого вірусного гепатиту В (ГВГВ), що включає введення лаферону внутрішньом'язово по 3 млн МО на добу (тричі по 1 млн МО з інтервалом 8 годин між введенням препарату) при середньотяжкому перебігу ГВГВ та по 6 млн МО на добу (тричі по 2 млн МО) при тяжкому перебігу гепатиту і одночасно циклоферону у вигляді 12,5 % розчину по 2 мл через день при середньотяжкому та по 2 мл щоденно при тяжкому перебігу ГВГВ, всього на...
Спосіб лікування загострень хронічного вірусного гепатиту с

Номер патенту: 56508
Опубліковано: 15.05.2003
Автор: Фролов Валерій Мітрофанович
МПК: A61K 45/00, A61K 35/28
Мітки: вірусного, спосіб, гепатиту, лікування, загострень, хронічного
Формула / Реферат:
Спосіб лікування загострень хронічного вірусного гепатиту С, що включає введення хворим індуктора інтерфероногенезу циклоферону, який відрізняється тим, що додатково вводять препарати a-інтерферону по 1 млн. ОД 3 рази на добу внутрішньом'язово перший тиждень лікування, потім по 1 млн. ОД 2-3 рази на тиждень протягом 2-3 місяців поспіль, в залежності від досягнутого ефекту; спленін по 2 мл 2 рази на добу внутрішньом'язово протягом перших 10-15...
Попередній патент: Повітророзподільник
Наступний патент: Піримідинові сполуки, спосіб їх одержання, фармацевтична композиція та їх використання
Випадковий патент: Спосіб одержання цифрових зображень плоских непрозорих об'єктів