Похідні 5,6-дихлорбензімідазолу, способи їх одержання, фармацевтичний склад та спосіб лікування вірусних інфекцій








Формула / Реферат
1. Соединение формулы (I):
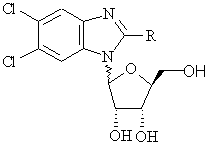 , (I)
, (I)
в которой R - водород или галоген, или –NR1R2, где
R1 и R2 одинаковы или различны и независимо выбраны из группы: водород, С1-6алкил, цианоС1-6алкил, гидроксиС1-6алкил, галогеноС1-6алкил, С3-7циклоалкил, С1-6алкилС3-7циклоалкил, С2-6алкенил, С3-7циклоалкилС1-6алкил, С2-6алкинил, арил, арилС1-6алкил, гетероциклический С1-6алкил, -СОС1-6алкил, или же
R1 и R2 вместе с атомом N, с которым они связаны, образуют 3-, 4-, 5- или 6-членное гетероциклическое кольцо,
и его фармацевтически приемлемые производные.
2. Соединение по п. 1, отличающееся тем, что имеет форму b-аномера.
3. Соединение по п. 1, отличающееся тем, что имеет форму a-аномера.
4. Соединение по п. 1, отличающееся тем, что оно соответствует формуле (Іb):
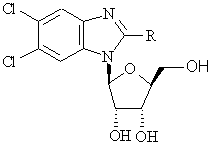 , (Ib)
, (Ib)
в которой R - галоген или –NR1R2, где
R1- водород, а
R2 выбран из группы: С1-6алкил, С1-6гидроксиалкил, С3-7циклоалкил, С1-6алкилС3-7циклоалкил, С2-6алкенил, С2-6алкинил, арил, арилалкил; или
R1 и R2, которые могут быть одинаковы или различны, оба являются С1-6алкилом; или же
R1 и R2 вместе с атомом N, с которым они связаны, образуют 3-, 4-, 5- или 6-членное гетероциклическое кольцо,
и его фармацевтически приемлемые производные.
5. Соединение по любому из пп. 1-4, отличающееся тем, что R представлено функциональной группой –NR1R2, в которой R1 - водород, a R2 выбран из группы: С1-6алкил, С3-7циклоалкил и галогеноС1-6алкил, и его фармацевтически приемлемые производные.
6. Соединение по любому из пп. 1-5, отличающееся тем, что R - изопропиламино-, изобутиламино-, втор-бутиламино-, циклопропиламино-, циклопентиламино- или 2-фтор-1-метилэтиламиногруппа, и его фармацевтически приемлемые производные.
7. Соединение по п. 1, отличающееся тем, что оно выбрано из группы: 5,6-дихлор-2-изопропиламино-1-(b-L-рибофуранозил)-1Н-бензимидазол, 2-циклопропиламино-5,6-дихлор-1-(b-L-рибофуранозил)-1Н-бензимидазол и 5,6-дихлор-2-(2-фтор-1-метилэтиламино)-1-(b-L-рибофуранозил)-1Н-бензимидазол и их фармацевтически приемлемых производных.
8. Соединение по п. 1, отличающееся тем, что оно является 5,6-дихлор-2-изопропиламино-1-(b-L-рибофуранозил)-1Н-бензимидазолом.
9. Соединение по любому из пп. 1-8, отличающееся тем, что оно пригодно для использования в терапии.
10. Соединение по любому из пп. 1-8, отличающееся тем, что оно пригодно для производства медикамента, предназначенного для лечения или профилактики вирусного заболевания.
11. Соединение по п. 10, отличающееся тем, что вирусным заболеванием является вирусный герпес.
12. Соединение по п. 11, отличающееся тем, что вирус герпеса принадлежит к группе: симплекс-вирус герпеса 1, симплекс-вирус герпеса 2, вирус опоясывающего лишая (ветряной оспы), вирус цитомегалии, вирус Эпштейна-Барра, человеческий вирус герпеса 6 и человеческий вирус герпеса 7.
13. Соединение по любому из пп. 1-8, отличающееся тем, что оно пригодно для производства медикамента, вводимого одновременно или раздельно по меньшей мере с одним другим терапевтическим агентом и предназначенного для лечения или профилактики вирусного заболевания.
14. Фармацевтически приемлемое производное соединений по любому из пп. 1-8, отличающееся тем, что оно является солью или эфиром.
15. Фармацевтически приемлемое производное по п. 14, отличающееся тем, что соль выбрана из группы: соли органических карбоновых кислот, органических сульфоновых кислот и неорганических кислот.
16. Фармацевтически приемлемое производное по п. 14, отличающееся тем, что эфир выбран из эфиров карбоновых кислот, аминокислот, сульфоновых кислот, фосфорных кислот и моно-, ди- или трифосфорных кислот.
17. Фармацевтически приемлемое производное по любому из пп. 14-16, отличающееся тем, что оно пригодно для использования в терапии.
18. Фармацевтически приемлемое производное по любому из пп. 14-16, отличающееся тем, что оно пригодно для производства медикамента, предназначенного для лечения или профилактики вирусного заболевания.
19. Фармацевтически приемлемое производное по п. 18, отличающееся тем, что вирусным заболеванием является вирусный герпес.
20. Фармацевтически приемлемое производное по п. 19, отличающееся тем, что вирус герпеса принадлежит к группе: симплекс-вирус герпеса 1, симплекс-вирус герпеса 2, вирус опоясывающего лишая (ветряной оспы), вирус цитомегалии, вирус Эпштейна-Барра, человеческий вирус герпеса 6 и человеческий вирус герпеса 7.
21. Фармацевтически приемлемое производное по любому из пп. 14-16, отличающееся тем, что оно пригодно для производства медикамента, вводимого одновременно или раздельно по меньшей мере с одним другим терапевтическим агентом и предназначенного для лечения или профилактики вирусного заболевания.
22. Фармацевтически приемлемый состав, содержащий соединение формулы (I), соответствующее пп. 1-8, или его фармацевтически приемлемое производное в сочетании с фармацевтически приемлемым носителем.
23. Фармацевтически приемлемый состав по п. 22, отличающийся тем, что он предназначен для использования в терапии.
24. Способ лечения или предотвращения симптомов или последствий заболевания инфицированного вирусом животного, включающий лечение указанного животного терапевтически эффективным количеством соединения согласно любому из пп. 1-8.
25. Способ по п. 24, отличающийся тем, что вирусным заболеванием является вирусный герпес.
26. Способ по п. 25, отличающийся тем, что вирус герпеса принадлежит к группе: симплекс-вирус герпеса 1, симплекс-вирус герпеса 2, вирус опоясывающего лишая (ветряной оспы), вирус цитомегалии, вирус Эпштейна-Барра, человеческий вирус герпеса 6 и человеческий вирус герпеса 7.
27. Способ получения соединения формулы (I) по любому из пп. 1-8, включающий взаимодействие соединения формулы (II)
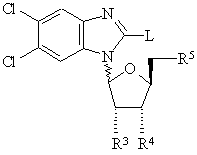 , (ІІ)
, (ІІ)
в которой L - водород, а каждый из R3, R4 и R5 представляет гидроксильную или защищенную гидроксильную группу с подходящим галогенизирующим агентом, или L - подходящий замещаемый атом или группа, a R3, R4 и R5 идентичны определенным выше,
с амином формулы H-NR1R2, где R1 и R2 идентичны определенным в п. 1, и
выполнение последовательно или одновременно одного или более следующих дополнительных действий в любом желаемом или необходимом порядке:
(і) удаление любых оставшихся защитных групп;
(ii) превращение соединения формулы (I) или его защищенной формы в последующее соединение формулы (I) или его защищенную форму;
(iii) превращение соединения формулы (I) или его защищенной формы в фармацевтически приемлемое производное формулы (I) или его защищенную форму;
(iv) превращение фармацевтически приемлемого производного соединения формулы (I) или его защищенной формы в соединение формулы (I) или его защищенную форму;
(v) превращение фармацевтически приемлемого производного соединения формулы (I) или его защищенной формы в другое фармацевтически приемлемое производное соединения формулы (I) или его защищенную форму;
(vi) разделение, когда это необходимо, α- и β-аномеров соединения формулы (I) или его защищенных производных, или фармацевтически приемлемых производных соединения формулы (I).
28. Способ получения соединения формулы (I) по любому из пп. 1-8, включающий взаимодействие соединения формулы (III)
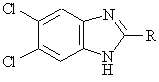 , (ІІІ)
, (ІІІ)
в которой R идентичен определенному в п. 1,
с соединением формулы (IV)
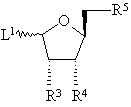 , (IV)
, (IV)
в которой каждый из R3, R4 и R5 - гидроксильная или защищенная гидроксильная группа, a L1 - подходящая отщепляемая группа в α- или β-положении, и
выполнение последовательно или одновременно одного или более следующих дополнительных действий в любом желаемом или необходимом порядке:
(і) удаление любых оставшихся защитных групп;
(ii) превращение соединения формулы (I) или его защищенной формы в последующее соединение формулы (I) или его защищенную форму;
(iii) превращение соединения формулы (I) или его защищенной формы в фармацевтически приемлемое производное формулы (I) или его защищенную форму;
(iv) превращение фармацевтически приемлемого производного соединения формулы (I) или его защищенной формы в соединение формулы (I) или его защищенную форму;
(v) превращение фармацевтически приемлемого производного соединения формулы (I) или его защищенной формы в другое фармацевтически приемлемое производное соединения формулы (I) или его защищенную форму;
(vi) разделение, когда это необходимо, α- и β-аномеров соединения формулы (I) или его защищенных производных, или фармацевтически приемлемых производных соединения формулы (I).
29. Промежуточное соединение формулы (II)
, (II)
в которой L - водород или подходящие замещаемые атом или группа, а каждый из R3, R4 и R5 - свободная или защищенная гидроксильная группа.
30. Промежуточное соединение по п. 29, отличающееся тем, что L - водород или галоген, а каждый из R3, R4 и R5 - свободная или защищенная гидроксильная группа, предпочтительно ОС(O)СН3.
31. 2-бром-5,6-дихлор-1-(2,3,5-три-O-ацетил-β-L-рибофуранозил)-1Н-бензимидазол.
32. 2-бром-5,6-дихлор-1-( β -L-рибофуранозил)-1Н-бензимидазол.
Текст
Изобретение относится к аналогам бензимидазолнуклеозида и их использованию в терапии, в частности, для лечения или профилактики вирусных инфекций, таких как вызываемые вирусами герпеса. Изобретение относится также к приготовлению аналогов нуклеозида и фармацевтических препаратов, содержащих и х. ДНК-вирусы, принадлежащие к группе герпесов, являются возбудителями наиболее распространенных вирусных заболеваний человека. Эта группа включает симплекс-вирусы герпеса типов 1 и 2 (СВГ), вирус опоясывающего лишая (ветряной оспы) (ВОЛ), вирус цитомегалии (ВЦМ), вирус Эпштейна-Барра (ВЭБ), человеческий вирус герпеса типа 6 (ЧВГ-6) и человеческий вирус герпеса типа 7 (ЧВГ-7).СВГ-1 и СВГ-2 являются наиболее распространенными возбудителями инфекции у человека. Большинство этих вирусов способны существова ть в нервных клетках носителя инфекции; заразившимся угрожают повторяющиеся клинические проявления, способные вызвать тяжелое физическое и психологическое недомогание. Характерной особенностью СВГ-инфекции часто является обширное изнуряющее поражение кожи, рта и/или гениталий. Первичное заражение может быть бессимптомным, хотя имеет тенденцию оказаться более тяжелым, чем в случаях, когда больной ранее подвергался заражению вирусом. Заражение глаз вирусом СВГ может привести к кератиту или катарактам, ставя тем самым под угрозу зрение. Заражение новорожденных, пациентов с ослабленным иммунитетом или проникновение инфекции в центральную нервную систему может оказаться смертельным. Вирус опоясывающего лишая (ветряной оспы) (ВОЛ) представляет собой вирус герпеса, вызывающий ветряную оспу и опоясывающий лишай. Ветряная оспа представляет собой первичное заболевание, возникающее при отсутствии иммунитета, и у маленьких детей протекающее слабо и сопровождающееся пузырчатой сыпью и лихорадкой. Опоясывающий лишай или зостер представляет собой возвратное заболевание, поражающее взрослых, ранее заразившихся ВОЛ. Для клинической картины опоясывающего лишая характерны невралгия и пузырчатая односторонняя сыпь, распространяющаяся в области иннервации кожи. Распространение воспаления может привести к параличу или судорогам. При поражении мозговых оболочек может наступить кома. ВОЛ представляет серьезную опасность для пациентов, принимающих иммунодепрессанты в связи с трансплантацией или злокачественной неоплазией, и создает серьезные осложнения у больных СПИД из-за того, что поражена их иммунная система. Подобно тому, как это происходит с другими вирусами герпеса, заражение ВЦМ влечет пожизненное присутствие вируса в носителе. Врожденное заражение, вызываемое заражением матери в период беременности, может дать такие клинические явления, как смерть или серьезное заболевание (микроцефалию, гепатоспленомегалию, желтуху, умственную отсталость), ретинит, ведущий к слепоте, или отставание в развитии в более или менее тяжелой форме и восприимчивость к легочным или ушным заболеваниям. ВЦМ-инфекция у пациентов, у которых иммунная система ослаблена, например, из-за злокачественной опухоли, или в результате применения иммунодепрессантов после трансплантации, или при заражении вирусом иммунодефицита человека, может вызвать ретинит, пневмонию, желудочно-кишечные и нервные расстройства. Основная болезнь, вызываемая вирусом Эпштейна-Барра, это острый или хронический инфекционный мононуклеоз (гландулярная лихорадка). Примерами других заболеваний, прямо или косвенно вызываемых ВЭБ, являются лимфопролиферативная болезнь, часто поражающая лиц с врожденным или приобретенным клеточным иммунодефицитом, Х-связанная лимфопролиферативная болезнь, поражающая, в основном, мальчиков, связанные с ВЭБ В-клеточные опухоли, болезнь Ходжкина, карцинома носоглотки, лимфома Бэркитта, неходжкинова β-клеточная лимфома, тимомы и оральная волосковая лейкоплакия. ВЭБ-инфекция была также обнаружена в связи с различными эпителиально-клеточными опухолями верхних и нижних дыхательных путей, включая легкие. Было показано, что ЧВГ-6 является причиной infantum subitum у детей, и отторжения почки и интерстициальной пневмонии у пациентов, соответственно подвергшихся трансплантации почки или костного мозга, и может быть связан с другими заболеваниями. Имеются также свидетельства подавления роста стволовых клеток у пациентов, перенесших трансплантацию костного мозга. Этиология вызываемых ЧВГ-7 болезней не определена. Вирус гепатита В (ВГВ) представляет собой патоген огромной важности. Этот вирус этиологически связан с первичной печеночно-клеточной карциномой и, как считается, является причиной 80% случаев рака печени в мире. Клинические явления при заражении ВГВ включают головную боль, лихорадку, недомогание, тошноту, рвоту, анорексию и желудочно-кишечные боли. Репликация вируса обычно остается под контролем иммунной системы, причем период выздоровления у человека длится недели или месяцы, но заражение может оказаться более тяжелым и привести к упорной хронической болезни печени, упомянутой выше. В заявочных описаниях WO 92/07867 и WO 94/08456 приведены некоторые антивирусные аналоги полизамещенных бензимидазолнуклеозидов, включая аналоги β-D-рибофуранозилрибозида. В описании WO 93/18009 отмечены некоторые антивирусные аналоги бензимидазолнуклеозидов, в которых сахарный остаток замещен карбоциклической группой. Теперь было установлено, что некоторые описанные ниже замещенные соединения бензимидазола могут быть использованы для лечения или профилактики определенных вирусных заболеваний. В качестве первого аспекта изобретения предложены новые соединения формулы (I): в которой R - атом водорода или галогена, -NR1R2, где R1 и R2, одинаковые или различные, независимо выбраны из группы, содержащей водород, C1-6алкил, цианC1-6алкил, гидроксиC1-6алкил, галогенC1-6алкил, С37циклоалкил, C 1-6алкилС 3-7циклоалкил, С 2-6алкенил, С3-7циклоалкилC1-6алкил, С2-6алкинил, арил, арилC1-6алкил, гетероциклическийC1-6алкил, -СОС1-6алкил, или же R1R2 вместе с атомом N, с которым они связаны, образуют 3,4,5 или 6-членное гетероциклическое кольцо, и их фармацевтически приемлемые производные. Другую подходящую группу соединений формулы (I) образуют соединения формулы (la) в которой R - водород или –NR1R2, где R1 и R2, одинаковые или различные, независимо выбраны из группы, содержащей водород, C1-6алкил, цианC1-6алкил, гидроксиC1-6алкил, галоC1-6балкил, С3-7циклоалкил, С2-6алкенил, С3-7циклоалкилC1-6алкил, С2-6алкинил, арил, арилC1-6алкил. гетероциклическийC1-6алкил, -СОС1-6алкил (при условии, что R1 и R2 не являются оба водородом), или же R1R2 вместе с атомом N, с которым они связаны, образуют 3,4,5 или 6-членное гетероциклическое кольцо) и их фармацевтически приемлемые производные. Примерами соединений формулы (I) служат следующие β аномеры формулы (Ib) в которой R - атом галогена или -NR1R2, где R1 - водород, a R2, выбран из группы, содержащей С1-6алкил, гидроксиC1-6алкил, С3-7циклоалкил, C1-6алкилС 3-7циклоалкил, С2-6алкенил, С2-6алкинил, арил, арилалкил, или R1 и R2, которые могут быть одинаковыми или разными, оба являются C1-6алкилом; или же R1 и R2 вместе с атомом N, с которым они связаны, образуют 3,4,5 или 6-членное гетероциклическое кольцо и их фармацевтически приемлемые производные. Альтернативно, соединениями формулы (Ib) могут быть такие, в которых R - атом галогена или моноC1моно(C1-6гидроксиалкил)амино-, ди-C1-6алкиламино-, С3-7циклоалкиламино-, С1-6алкилС 36алкиламино-, 7циклоалкиламино-, С 2-6алкениламино-, С 2-6алкинил-амино-, ариламино-, арилалкиламино- группы или группа формулы –N(CH2)n, где n - 2,3,4 или 5 и и х фармацевтически приемлемые производные. Дальнейшими примерами соединений формулы (I) служат примеры 1-38, приведенные ниже. Здесь термин "алкил" как группа или часть группы обозначает линейную или разветвленную цепную алкильную группу. Такие алкильные группы имеют предпочтительно 1-6 атомов углерода, более предпочтительно от 1 до 4, и, в частности, содержат метил, этил, пропил, изометил, трет-бутил. Под алкенильными группами понимают группы в Е- или Z-форме или их смесь, которые могут быть разветвленными, если содержат не менее трех атомов углерода. Под галогенами имеются в виду хлор, бром, фтор и йод. Термин галогенC1-6алкил обозначает алкильную группу, в которой один или более водородных атомов замещены галогеном и которая предпочтительно содержит одну, две или три галогенные группы. Примерами таких групп служат трифторметил и фторизопропил. Арил как группа или часть группы представляет собой фенил, произвольно замещенный одним или более заместителями, выбираемыми из группы соединений, содержащей С1-6алкоксил (например, метоксил), нитро-, галогено- (например, хлор), амино-, карбокси-и гидрокси- группы. Термином "гетероциклический" обозначено насыщенное или частично насыщенное (т.е. неароматическое) 3-, 4-, 5- или 6-членное кольцо, содержащее один или более (например, от 1 до 4) гетероатомов, независимо выбираемых из группы, содержащей азот, кислород и серу. Примером таких гр упп служит пирролидин. Изобретение охватывает каждый возможный альфа и бета аномер соединений формулы (I) и их физиологически функциональные производные, существенно очищенные от другого аномера, так что содержание другого аномера не должно превышать примерно 5% по массе, предпочтительно не более примерно 2% по массе, в частности, менее 1%, а также смеси таких альфа и бета аномеров в любых пропорциях. Предпочтительны бета аномерные формы соединений формулы (I). Предпочтительны такие соединения формулы (Ib), в которых R - -NR1R2, где R1 - водород, a R2 выбран из группы, содержащей С1-6алкил, С3-7циклоалкил и галогенС 1-6алкил и их фармацевтически приемлемые производные. Особенно предпочтительны такие соединения формулы (Ib), в которых R - изопропиламин, изобутиламин, втор-бутиламин, циклопропиламин, цик-лопентиламин и 2-фтор-1-метилэтиламин и их фармацевтически приемлемые производные. Соединениями формулы (I) бета конфигурации, представляющими особый интерес благодаря антивирусному действию, являются 2-циклопропиламин-5,6-дихлор-1-(β-L-рибофуранозил)-1Н-бензимидазол, 5,6-дихлор-2-(2-фтор-1-метилэтиламин)-1-(β-L-рибофуранозил)-1Н-бензимидазол и 5,6-дихлор-2изопропиламин-1-(β-L-рибофуранозил)-1Н-бензимидазол и их фармацевтически приемлемые производные. Установлено, что 5,6-дихлор-2-изопропиламин-1-(β-L-рибофуранозил)-1Н-бензимидазол особенно эффективен при лечении цитомегаловирусных заболеваний. Соединения формулы (I), включая соединения формул (la) и (Ib) и их фармацевтически приемлемые производные в дальнейшем именуются соединениями, соответствующими изобретению. Под "фармацевтически приемлемым производным" мы понимаем любые фармацевтически или фармакологически приемлемые соль, эфир или соль такого эфира соединения, соответствующего изобретению, или любое соединение, которое, будучи введено реципиенту, способно образовывать (прямо или косвенно) соединение, соответствующее изобретению, или его метаболит или остаток, обладающие антивирусной активностью. Предпочтительные эфиры соединений, соответствующих изобретению, независимо выбраны из следующих гр упп: (1) эфиры карбоновой кислоты, получаемые этерификацией 2'-, 3'- и/или 5'-гидроксильных групп, в которых некарбонильный компонент остатка карбоновой кислоты эфирной группировки произвольно выбраны из линейных или разветвленных алкилов (например, н-пропила, трет-бутила или н-бутила), алкоксиалкила (например, метоксиме-тила), аралкила (например, бензила), арилоксиалкила (например, феноксиме-тила), арила (т.е. фенила, произвольно замещенного галогеном, С1-4алкилом или С 1-4алкоксилом или аминогруппой); (2) эфиры сульфоновых кислот, такие, как алкил- или аралкилсульфонаты (например, метансульфонат); (3) эфиры аминокислот (например, L-валил или L-изолевкил); (4) Фосфонаты и (5) моно-, ди- или трифосфаты. Фосфаты могут быть далее этерифицирова-ны, например, С-].2о спиртом или его реакционноспособным производным, или 2,3-ди(С6-24)ацилглицеролом. В таких эфирах, если не обусловлено иное, любой присутствующий алкильный компонент преимущественно содержит от 1 до 18 атомов углерода, в частности, от 1 до 6, более конкретно, от 1 до 4 атомов углерода. Любой циклоалкильный компонент таких эфиров содержит преимущественно от 3 до 6 атомов углерода. Любой ариловый компонент, присутствующий в таких эфирах, преимущественно имеет фенильную группу. К предпочтительным эфирам карбоновых кислот, соответствующим изобретению, относятся ацетаты, ацетобутираты и валераты. Среди аминокислотных эфиров особенно предпочтителен L-валиновый эфир. Любое упоминание любого из вышеописанных соединений подразумевает его фармацевтически приемлемую соль. Фармацевтически приемлемые соли включают соли органических карбоновых кислот, таких, как аскорбиновая, уксусная, лимонная, молочная, винная, яблочная, малеиновая, изэтионовая, лактобионовая, паминобензойная и янтарная кислоты; органические сульфоновые кислоты, такие, как метан-сульфоновая, этансульфоновая, бензолсульфоновая и п-толуолсульфоновая, и неорганические кислоты, такие, как соляная, серная, фосфорная, сульфамидная и пирофосфорная. Фармацевтически приемлемые соли соединений формулы (I) могут быть использованы в терапии. Однако фармацевтически неприемлемые соли и основания также могут найти применение, например, при приготовлении или очистке фармацевтически приемлемого соединения. Изобретение охватывает все соли, производные (и иные) от фармацевтически приемлемой кислоты или основания. Предпочтительны соли соляной, серной, уксусной, янтарной, лимонной и аскорбиновой кислот. Другим аспектом изобретения являются соединения, соответствующие изобретению, предназначенные для лечения или профилактики вирусных заболеваний, таких, как вирусный герпес. Было показано, что соединения, соответствующие изобретению, активны при цитомегаловирусных (ЦМВ) заболеваниях, хотя ранние результаты позволяют ожидать, что соединения, соответствующие изобретению, могут оказаться активными при использовании для лечения других вир усных заболеваний герпесом, таких, как СВГ 1 и 2, ЧВГ 6 и 7, ВОЛ, ВГВ и ВЭБ. Другие вирусные заболевания, которые можно лечить в соответствии с изобретением, были рассмотрены в вводном разделе. Соединения, соответствующие изобретению особенно пригодны для лечения или профилактики заболевания ЦМВ и связанных с ним состояний. Примеры состояний, связанных с ЦМВ, поддающихся лечению согласно изобретению, приведены в вводной части. В соответствии с еще одним аспектом изобретения предлагается способ лечения или предотвращения симптомов или последствий вирусного заболевания у животных, например, млекопитающих, в том числе человека, включающий лечение животного терапевтически эффективным количеством соединения, соответствующего изобретению. Изобретение также включает способ лечения или профилактики любого из вышеупомянутых заболеваний или состояний. В соответствии с одним из воплощений данного аспекта как одно из вирусных заболеваний рассматривается герпесное вирусное заболевание, такое, как ЦМВ, СВГ 1 и 2, ВОЛ, ВЭБ, ЧВГ 6 или 7. Еще один аспект изобретения включает способ лечения или предотвращения симптомов или последствий заболевания ВГВ. Далее, в изобретении предлагается способ лечения рассмотренного во введении клинического состояния у животных, например, млекопитающих, включая человека, который предусматривает лечение животного терапевтически эффективным количеством соединения, соответствующего изобретению. Изобретение также включает способ лечения или профилактики любого из вышеупомянутых заболеваний или состояний. Еще одним аспектом изобретения является использование соединения, соответствующего изобретению, в производстве медикаментов для лечения или профилактики любого из вышеупомянутых вирусных заболеваний или состояний. Для лечения упомянутых вы ше заболеваний и состояний вышеупомянутые соединения, соответствующие изобретению, и их фармацевтически приемлемые производные могут быть использованы в сочетании с другими терапевтическими средствами. В соответствии с изобретением комбинированная терапия включает введение, по меньшей мере, одного соединения формулы (I) или его фармацевтически приемлемого производного и, по меньшей мере, одного другого фармацевтически активного ингредиента. Активные ингредиенты и фармацевтически активные соединения можно вводить одновременно в том же или ином фармацевтическом составе или в любой последовательности. Количества активных ингредиентов и фармацевтически активных соединений и выбор времени их введения следует выбирать так, чтобы достичь желаемого комбинированного лечебного эффекта. При комбинированной терапии предпочтительно вводить одно соединение, соответствующее изобретению, и одно из веществ, перечисленных ниже. Примерами таких вспомогательных терапевтических средств могут служить средства, эффективные при лечении вирусных заболеваний или связанных с ними состояний, такие, как (1 альфа, 2 бета, 3 альфа)-9-[2,3бис(гидроксиметил)циклобутил]гуанин [(-)BHCG, SQ-34514], оксетаноцин-С(3,4-бис-(гидроксиметил)-2оксетанозил)гуанин, ациклические нуклеозиды (например, ацикловир, валацикловир, фамцикловир, ганцикловир, пенцикловир), фосфонаты ациклических нуклеозидов (например, (S)-1-(3-гидрокси-2-фосфонилметоксипропил)цитозин (ГФМЦ), ингибиторы рибонуклеотидной редуктазы, такие, как 2-ацетилпиридин 5-[(2хлоранилин)тиокарбонил)тиокарбонгидразон], 3'-ацидо-3'-деоксиугимидин, другие 2',3'-дидеоксинуклеозиды, такие, как 2',3'-дидеоксицитидин, 2',3'-дидеоксиаденозин и 2',3'-дидеоксиинозин, 2',3'-дидегидротимидин, ингибиторы протеазы, такие, как Н-трет-бутил-дегидро-2-[-2(R)-гидрокси-4-фенил-3(S)]-[[N-(2-хинолилкарбонил)-L-аспаргинил]бутил]-(4аS,8аS)-изохинолин-3(S)-карбоксамид (Ro 31-8959), аналоги оксатиоланнуклеозида, такие, как (-)-цис-1-(2-(гидроксиметил)-1,3-оксатиолан-5-ил)-цитозин (ЗТС) или цис-1(2-(гидроксиметил)-1,3-оксатиолан-5-ил)-5-фторцитозин (FTC), 3'-деокси-3'-фтортимидин, 5-хлор-2',3'дидеокси-3'-фторуридин, (-)-цис-4-[2-амин-6-(циклопропиламин)-9Н-пурин-9-ил]-2-циклопентен-1-метанол, рибавирин, 9-[4-гидрокси-2-(гидроксиметил)бут-1-ил-]-гуанин (H2G), тат ингибиторы, такие, как 7-хлор-5-(2пиррил)-3Н-1,4-бензодиазепин-2(Н)-он (Ro5-3335), или 7-хлор-1,3-дигидро-5-(1Н-пиррол-2-ил)-3Нбензодиазепин-2-амин (Ro24-7429), интерфероны, такие, как α-интерферон, ингибиторы почечных выделений, такие, какпробеницид, ингибиторы переноса нуклеозидов, такие, как дипиридамол; пентоксифиллин, N-Ацетилцистеин (NAC), Процистеин, α-трихосантин, фосфономуравьиная кислота, а также иммуномодуляторы, такие, как интерлевкин II или тимозин, факторы колонностимуляции макрофага гранулоцитов, эритропоэтин, растворимый CD4 и его производные, полученные путем генной инженерии, или ингибиторы обратной транскриптазы, такие, как невирапин (BI-RG-587), ловирид (α-АРА) и делавуридин (ВНАР). При комбинированной терапии более предпочтительно вводить один из вышеперечисленных препаратов и композицию соединений из предпочтительных или наиболее предпочтительных подгрупп соединений формулы (I), как описано выше. Наиболее предпочтительно при комбинированной терапии использовать один из вышеупомянуты х препаратов совместно с одним из соединений формулы (I) перечисленных здесь. Далее, изобретение включает использование соединений, соответствующи х изобретению, при производстве медикаментов для одновременного или раздельного ввода, содержащих, по меньшей мере, одно иное терапевтическое средство из перечисленных выше. Соединения, соответствующие изобретению, называемые здесь также активными ингредиентами, с терапевтическими целями можно вводить любым подходящим способом, включая оральные, ректальные, назальные, местные (включая введение сквозь кожу, подбородок и под язык), вагинальные и парентеральные (включая введение подкожно, внутримышечно, внутривенно и в кожу). Целесообразно выбирать предпочтительный способ в зависимости от состояния и возраста пациента, вида заболевания и выбранного активного ингредиента. В общем случае доза для каждого из вышеупомянутых состояний должна быть в пределах от 0,01 до 250мг на кг массы тела реципиента (т.е. человека) в день, предпочтительно от 0,1 до 100мг, более предпочтительно от 0,5 до 30мг, в частности, от 1 до 20мг на кг массы тела в день. (Если не обусловлено иное, все массы активного ингредиента вычисляют для соединения основной формулы (I); для солей и эфиров этого соединения массы следует увеличивать в соответствующей пропорции). Желаемую дозу предпочтительно разбить на две, три, четыре, пять, шесть или более поддоз, вводимых через соответствующие интервалы в течение дня. В некоторых случаях желаемую дозу можно вводить не каждый день. Эти поддозы можно вводить в виде единиц дозировки, например, содержащих от 10 до 1000мг, предпочтительно от 20 до 500мг, и наиболее предпочтительно от 100 до 400мг активного ингредиента на комплект единиц дозировки. В идеальном случае активный ингредиент следует вводить так, чтобы достичь его максимальной концентрации в плазме в пределах примерно от 0,025 до 100мкМ, предпочтительно примерно от 0,1 до 70мкМ, наиболее предпочтительно примерно от 0,25 до 50мкМ. Этого можно достичь, например, внутривенной инъекцией 0,1-5%-го раствора активного ингредиента, при желании солевого, или при оральном введении в виде болюсов, содержащих примерно от 0,1 до 250мг/кг активного ингредиента. Желаемый уровень в крови можно поддерживать непрерывным вливанием, получая таким образом примерно от 0,01 до 5,0мг/кг/час, или разовыми вливаниями, содержащими активный ингредиент в количестве примерно от 0,4 до 15мг/кг. Хотя активный ингредиент можно вводит одиночно, его предпочтительно сформировать как фармацевтический состав. Составы, соответствующие изобретению, содержат, по меньшей мере, один активный ингредиент, определенный выше, вместе с одним или более приемлемыми его носителями и, при желании, с другими терапевтическими препаратами. Каждый носитель должен быть "приемлемым" в смысле совместимости с другими ингредиентами состава и безвредности для пациента. В число составов входят пригодные для орального, ректального, назального, локального (включая черезкожное, подбородочное и подъязычное), вагинального, парентерального (включая подкожное, внутримышечное, внутривенное, внутрикожное и внутривитреальное) введения. Составы удобно готовить в виде единиц дозировки любыми способами, применяемыми в фармакологии. Эти способы включают стадию ассоциирования активного ингредиента с носителем, который представляет один или более вспомогательных ингредиентов. Обычно составы готовят путем однородного и тесного ассоциирования активного ингредиента с жидким носителем или тонкодисперсным твердым носителем или обоими с последующей формовкой. Изобретение далее включает фармацевтические составы, как они определены выше, в которых соединение формулы (I) или его фармацевтически приемлемое производное и, по меньшей мере, один иной фармацевтический препарат представлены по отдельности и в виде набора составных частей. Композиции для чрезкожного введения могут быть представлены в виде отдельных накладок, приспособленных для длительного тесного контакта с эпидермисом реципиента. Такие накладки содержат активное соединение 1) в виде произвольно буферированного водного раствора, или 2) растворенным и/или диспергированным в клее, или 3) диспергированным в полимере. Подходящая концентрация активного вещества составляет примерно от 1 до 25%, предпочтительно примерно от 3 до 15%. Следует отметить возможность введения активного соединения с накладки электропереносом или ионофо-резом, как это в общей форме описано в Pharmaceutical Research, 3(6), 318 (1986). Композиции для орального введения могут быть представлены в виде отдельных единиц, таких, как капсулы, крахмальные облатки или таблетки, содержащих каждая определенное количество активного ингредиента; в виде порошка или гранул; в виде раствора или суспензии в воде или иной жидкости или в виде водомасляной или масловодяной жидкой эмульсии. Активный ингредиент может быть также в виде болюса, сиропа или пасты. Таблетка может быть изготовлена прессованием или формовкой, при желании, с одним или более вспомогательными ингредиентами. Прессованные таблетки изготавливают прессованием в соответствующей установке активного ингредиента в сыпучем виде, например, в виде порошка или гранул, при желании, в смеси со связующим ве ществом (например, повидоном, желатином, гидроксипропилметилцеллюлозой), лубрикантом, инертным разбавителем, консервантом, дизинтегрантом (например, крахмальным гликоллатом натрия, структурированным повидоном, структурированной натрийкарбоксиметилцеллюлозой), поверхностноактивным или диспергирующим реагентом). Формованные таблетки могут быть изготовлены формованием в соответствующей установке смеси порошкообразного соединения с инертным жидким разбавителем. Таблетка может, при желании, иметь покрытие или надрезы; ее состав подбирают так, чтобы после приема обеспечить медленное или контролируемое высвобождение активного ингредиента, используя для этого гидроксипропилметилцеллюлозу в различной пропорции, добиваясь этим нужного режима высвобождения. При желании, таблетки могут быть снабжены брюшным покрытием, обеспечивающим высвобождение в кишечнике, а не в желудке. Составы, предназначенные для локального введения во рту, включают лепешки, содержащие активный ингредиент с корригентом, обычно сахарозой с аравийской камедью или трагакантом, пастилки с активным ингредиентом в инертной основе, такой, как желатин с глицерином или сахароза с аравийской камедью, и полоскания для рта, содержащие активный ингредиент с подходящим жидким носителем. Составы для ректального введения могут быть в виде суппозитория на подходящей основе, содержащей, например, масло какао или салицилат. Составы для вагинального могут быть изготовлены в виде пессариев, тампонов, кремов, желе, паст, пены или спринцеваний, содержащих, помимо активного ингредиента, носители, применяемые в таких случаях. Фармацевтические составы для ректального введения с твердым носителем наиболее предпочтительно изготавливать в виде дозированных суппозиториев. В качестве носителя можно использовать масло какао или другие вещества, обычно используемые для этой цели. Необходимую форму суппозиторию можно придать путем формования смеси активного ингредиента с размягченным или расплавленным носителем (носителями) с последующим охлаждением. Составы для парентерального введения включают водные и неводные изотонические стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатический фактор и растворенные вещества, делающие состав изотоничным по отношению к крови предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут содержать суспендирующие и сгущающие добавки. Составы могут быть представлены в единичных дозах или многодозных запечатанных упаковках, например, ампулах или флаконах, и их можно хранить лиофилизированными, добавляя стерильный жидкий носитель, например, воду для инъекций непосредственно перед использованием. Растворы и суспензии для срочных инъекций могут быть приготовлены из стерильных порошков, гранул и таблеток, подобных описанным выше. Предпочтительная дозировка составов должна обеспечивать наличие в нем дневной дозы или единицы или дневной поддозы (как это было описано выше) активного ингредиента. Очевидно, что, помимо активных ингредиентов, указанных выше, составы, соответствующие изобретению, могут содержать другие обычно применяемые препараты, соответствующие данному составу, например, составы для орального введения могут содержать такие добавки, как корригирующие и придающие сладость и густо ту. Далее, изобретение включает следующие способы приготовления соединений формулы (I) и их производных: (А) реакцию соединения формулы (II) в которой L - водород, а каждый из R3,R4 и R5 представляет гидроксильную или защищенную гидроксильную группу с подходящим галогенизатором, таким, как N-бромсукцинамид, или L - подходящий отщепляемый атом или группа, например, атом галогена, такого, как бром, или группа, такая, как органо- (например, алкил) суль-фоновая, или органо- (например, алкил или аралкил) сульфатная, такие, как метилсульфоновая (MeS(О)2), метилсульфонатная (MeS(O)2O) или тозилатная (4-MePhS(O)2O) группа, a R3,R4 и R5 идентичны определенным выше с амином формулы H-NR1R2 (где R1 и R2 идентичны определенным ранее), или (Б) реакция соединения формулы (III) где R идентичен определенному ранее, с соединением формулы (IV) в которой каждый из R3,R4 и R5 - гидроксильная или защищенная гид-роксильная группа, a L1 - подходящая отщепляемая группа в а- или β-позиции, например, галоген (например, фтор, хлор или бром), алкил- или арилтио (например, фенилтио), или арил или алифатическая эфирная группа, такая, как бензоат или ацетат; после этого или одновременно с этим возможно выполнение следующи х дополнительных действий в любом желаемом порядке: (і) удаление любых оставши хся защитны х гр упп; (ii) превращение соединения формулы (I) или его защищенной формы в последующее соединение формулы (I) или его защи щенную форму; (iii) превращение соединения формулы (I) или его защищенной формы в фармацевтически приемлемое производное формулы (I) или его защищенную форму; (iv) превращение фармацевтически приемлемого производного соединения формулы (I) или его защищенной формы в соединение формулы (I) или его защищенную форму; (ν) превращение фармацевтически приемлемого производного соединения формулы (I) или его защищенной формы в другое фармацевтически приемлемое производное соединения формулы (I) или его защищенную форму; (vi) отделение во всех случаях, когда это необходимо, альфа и бета аномеров соединения формулы (I) или его защищенных производных или фармацевтически приемлемых производных соединения формулы (I). Способ А удобно использовать для приготовления соединения формулы (I), когда R - галоген; при этом Такие соединения можно получать вводом в реакцию соединения формулы (II), в которой L - водород, a Rd,R4 и R5 - защищенные гидроксильные группы, предпочтительно ОС(О)СН3, с галогенизатором. Галогенизацию можно проводить обычным способом, например, бромированием с использованием такого броминатора, как N-бромсукцинимид (NBS) в апротонном растворителе, таком, как тетрагидрофуран или, предпочтительно, в 1,4 диоксане, подогретом до 60-100°С, предпочтительно 100°С. Соединения формулы (I), в которых R - -NR1R2 (где R1 и R2 идентичны определенным выше) преимущественно получают из соединений формулы (II), в которых L - атом галогена, такого, как бром или хлор, реакцией с амином H-NR1R2 (где R1 и R2 идентичны определенным выше). Реакция преимущественно проводят при повышенной температуре, например, 70-80°С в органическом растворителе, таком, как этанол или диметилсульфоксид. Защитная группа может быть удалена обычными способами, хорошо известными специалистам в данной области. Соединения формулы (II), в которых каждый из R3,R4 и R5 представляет гидроксильную группу, могут быть приготовлены, например, из соответствующего соединения формулы (II), в котором каждый из R 3,R4 и R5 представляет защищенную гидроксильную гр уппу. Для R3 ,R4 и R5 могут быть использованы обычные защитные группы. Преимущественно могут быть использованы эфирные группы, подобные описанным выше эфирам в отношении соединений формулы (I). Эти защитные группы можно удалить либо обычными способами, как, например, с использованием карбоната натрия в метаноле, либо используя энзимы, например, энзимы печени свиньи. С другой стороны, R3,R4 и R5 могут иметь силильные защитные группы, такие, как трет-бутилди фениловые, трет-бутилдиметиловые, триизопропропилсилильные группы, которые могут быть удалены путем использования подходящего источника фтора, например, HF/Пиридина, н-Bu4NF или Et4NF или циклического ацеталя или кеталя, такого, как бензилидиновая или изопропилидиновая группы, которые могут быть удалены в кислотной среде, с использованием, например, тозиловой кислоты и метанола. С другой Стороны, соединение формулы (II), где R3,R4 и R5 - защи щенные гидроксильные группы, может быть введено в реакцию с реагентом при таких условиях, что о тщепляемая группа L будет преобразована в желаемую группу R одновременно с удалением защитной группы. Примерами таких реагентов могут служить циклопропиламин и другие первичные и вторичные амины при условии, что они достаточно нуклеофильны и не являются стерически заторможенными. Соединения формулы (I) и соединения формулы (II), в которых R и L определены выше, могут быть приготовлены с помощью реакции соединения формулы (V) (где X эквивалентно R или L, определенным выше) с соединением формулы (IV) (где каждый из R3,R4 и R5 - гидроксильная группа или защищенная гидроксильная группа, a L1 определен выше). соединения формулы (VI) с реагентом или реагентами, способными циклизировать диамин в бензимидазол. Соединения формулы (I) обычно могут реагировать с изотиоцианатом формулы (VII): S=C=NR 1R2 (VII) в которой R1 и R2 идентичны определенным выше. Реакция может быть осуществлена в присутствии карбодиимида, такого, как дициклогексилкарбодиимид или 1-циклогексил-3-(2-морфолинэтил)карбодиимид мето-п-толуол-сульфонат обычно в присутствии апротонного ароматического растворителя, такого, как толуол и, что наиболее предпочтительно, пиридин, при повышенной температуре, предпочтительно, 75-150°С. Соединения формулы (V), в которой X - водород, могут быть куплены, или приготовлены реакцией соединения формулы (VI) с формамидином в водно-кислотной среде при температуре от комнатной до 80°С. Соединения формул (VI) и (VII) могут быть приготовлены способами, хорошо известными специалистам и описанными в литературе, или куплены. Эфиры, соответствующие изобретению, могут быть получены способами, хорошо известными специалистам в этой области, например, соединение формулы (I) может быть превращено в фармацевтически приемлемый эфир реакцией с подходящим этерификатором, например, галогенангидридом или ангидридом подходящей кислоты.. Соединение формулы (I) может быть превращено в соответствующий фармацевтически приемлемый эфир формулы (I) реакцией с алкилирующим реагентом обычным способом. Реакцию между соединениями (IV) и (V) можно осуществить, используя кислоту Льюиса, такую, как, например, триметилсилилтрифлат, тетра хлорид олова или трифторид бора, причем предпочтителен первый. Реакцию обычно проводят в апротонном растворителе при повышенной температуре, например, в ацетонитриле при 15-30°C или 1,2-дихлорэтане при 70-90°С. Для описанной выше процедуры соединение формулы (V) с целью улучшения растворимости преимущественно триметилсилилируют в позиции N1, обрабатывая, например, триметилсилилхлоридом, гексаметилдизилазаном или, наиболее предпочтительно, Ν,Ο-бис-триметилсилилацетамидом (BSA). Такая силилация может быть проведена в растворителе, в качестве которого предпочтительны 1,2-дихлорэтан или ацетонитрил, предпочтительно при 70-80°С. По завершении реакции силилации можно добавить кислоту Льюиса с последующим добавлением соединения формулы (IV). Соединения формулы (IV) могут быть приготовлены способами, хорошо известными специалистам в этой области, например, способом, аналогичным применяемому для производных D-рибозы, или способами, известными из литературы, например, описанными в работе Acton et al. J. Am. Chem. Soc, 1964, 86, 5352. Предпочтительным соединением формулы (IV) является такое, в котором R3,R4,R5 и L1 представляет каждое группу ОС(О)СН 3. Это соединение может быть также приготовлено способом, аналогичным разработанному для D-рибозы (R.D.Guthrie and S.С.Smith, Chemistry and Industry, 1968, pp.547-548) преимущественно с последующей рекристаллизацией из этанола. Соединения формулы (V), в которых X - L или группа -NR1R2 (где L,R1 и R2 идентичны определенным выше), могут быть получены, следуя способу, приведенному в описании РСТ WO92/07867, включенному сюда ссылкой. С другой стороны соединения формулы (V), в которой X-R, a R- -NR1R2. где R1 и R2 идентичны определенным выше, можно получить реакцией. Соединения формулы (I), включая их эфиры, могут быть превращены в их фармацевтически приемлемые соли обычными способами, например, путем обработки подходящей кислотой. Эфир или соль эфира формулы (I) можно превратить в исходное соединение, например, гидролизом. Альфа и бета аномеры могут быть разделены и выделены в чистом виде с помощью хроматографии на силикагеле, с использованием одиночного растворителя или комбинации растворителей, такой, как метанолдихлорметан в пропорции 1:20. Далее, изобретение включает соединения формулы (II), как они определены выше, как новые промежуточные соединения. Предпочтительные соединения формулы (II) включают такие, в которых L - атом водорода или галогена, предпочтительно хлора или брома, a R3,R4 и R5 - гидроксильная или защищенная гидроксильная группы, предпочтительно, ОС(О)СН3Особенно предпочтительными соединениями формулы (II) являются 2-бром-5,6-дихлор-1-(2,3,5-три-Оацетил-β-L-рибофурано-зил)-1Н-бензимидазол и 2-бром-5,6-дихлор-1-(β-L-рибофуранозил)-1Н-бензимидазол. Изобретение включает также промежуточные соединения формулы (V), в которой X-R, a R- гр уппа NR1R2, где R1 и R2 идентичны определенным выше при условии, что R1 и R2 не являются оба водородом или метилом. Нижеследующие примеры предназначены только для иллюстрации, и ни в какой мере не ограничивают рамок изобретения. Термин "активный ингредиент", используемый в фармацевтических примерах означает соединение формулы (I) или его фармацевтическм приемлемое производное. Этот термин включает также соединение формулы (I) или его фармацевтически приемлемое производное в комбинации с одним или более терапевтическими агентами. Пример 1. 2-бром-5,6-дихлор-1-(2,3,5-три-О-ацетил-β-L-рибофуранозил)-1Н-бензимидазол. 2-бром-5,6-дихлорбензимидазол (1,0г, 3,8ммолей), N,O-бис(триметилсилил)ацетамид (Aldrich, 0,94мл, 3,8ммолей) и ацетонитрил (Aldrich Sure Seal, 25мл) были смешаны и выдержаны с обратным холодильником в атмосфере азота в течение 1ч. Раствор был охлажден до комнатной температуры и в него был добавлен триметилсилилтрифлат (Aldrich, 1,5мл, 7,6ммолей). Через 15мин. был добавлена твердая 1,2,3,4-тетра-Оацетил-L-рибофураноза (1,2г, 3,8ммолей), полученная способом Гатри-Смита (Chemistry and industry, 1968, pp. 547-548), за исключением того, что в качестве сырья использована L-рибоза. Раствор перемешивали в азотной атмосфере при комнатной температуре в течение 18ч., затем влили в 10%-й водный бикарбонат натрия (100мл) и экстрагирован дихлорметаном (2x150мл). Органические слои были высушены безводным сульфатом магния, отфильтрованы и выпарены. Твердый остаток был очищен на силикагелевой колонне (5x20см, 230-400меш) смесью ацетона и CH2CI2 (пропорция 1:30), в результате чего был получен 2-бром-5,6дихлор-1-(2,3,5-три-О-ацетил-β-L-рибофуранозил)-1Н-бензимидазол (1,2г, 2,2ммолей, 60%); t° плавления 142°С; [a]20D=(+)87,4 (с=0,5 ДМФА); УФ1мах(е): рН = 7,0: 298нм (7.600), 289 (7.400), 254 (8.800); 0,1 N NaOH: 298нм (7.600), 289 (7.400), 256 (7.300); МС(ЭИ): м/з (относительная интенсивность) 524 (0,15, М+); 1Н ЯМР (ДМСО-d6) d 8,08 (s,1H,Ar-H), 8,01 (s,1H,Ar-H), 6,22 (d,1H,H-1', J=7,1 Гц), 5,56 (dd,1H,H-2', J=7,1 Гц, J=7,2Гц, 5,45 (dd,1H,H-3', J=7,2 Гц, J=4,5 Гц), 4,55-4,47 (m,2H,H-4' и 5'), 4,37 (d,1H,H-5'), J=9,7 Гц), 2,15 (s,3H,OAc), 2,14 (s,3H,OAc), 2,01 (s,3H,OAc). Анал. вычисл. для C18H17N2O 7Cl2Br: C-41,25; H-3,27; N-5,34. Получено: C-41,16; H-3,39; N-5,20. Кроме того, было получено небольшое количество альфа аномера (2-бром-5,6-дихлор-1-(2,3,5-три-Оацетил-α-L-рибофурано-зил)-1Н-бензимидазол) (0,11г, 0,22ммолей, 6%); t° плавления
ДивитисяДодаткова інформація
Назва патенту англійською5,6-dichlorobenzimidazole derivatives, methods for the preparation thereof, pharmaceutical composition and method for treatment of viral infections
Назва патенту російськоюПроизводные 5,6-дихлорбензимидазола, способы их получения, фармацевтический состав и способ лечения вирусных инфекций
МПК / Мітки
МПК: C07H 19/052, A61P 31/12, C07D 235/04, A61K 31/7052
Мітки: склад, вірусних, 5,6-дихлорбензімідазолу, лікування, спосіб, інфекцій, фармацевтичний, похідні, способи, одержання
Код посилання
<a href="https://ua.patents.su/18-66744-pokhidni-56-dikhlorbenzimidazolu-sposobi-kh-oderzhannya-farmacevtichnijj-sklad-ta-sposib-likuvannya-virusnikh-infekcijj.html" target="_blank" rel="follow" title="База патентів України">Похідні 5,6-дихлорбензімідазолу, способи їх одержання, фармацевтичний склад та спосіб лікування вірусних інфекцій</a>
Похідні та аналоги 2-дезокси-2,3-дидегідро-n-ацетилнеурамінової кислоти, спосіб їх одержання, фармацевтична композиція, спосіб лікування вірусних інфекцій

Номер патенту: 41252
Опубліковано: 17.09.2001
Автори: Данілек Базіль, Ву Вен-Янг, Іцштейн Лоренс Марк фон, Фан То Ван, Джін Бетті
МПК: C07D 257/00, C07C 317/12, C07D 239/26, A61K 31/505, A61P 31/00, A61K 31/435, C07D 409/04, C07D 309/30, A61K 31/34, C07D 211/84, C07D 309/26, A61K 31/382, C07D 309/28, C07D 211/74, C07D 401/14, C07D 211/72, C07D 211/78, C07C 47/38, C07D 309/22, C07D 211/70, C07D 405/14, A61P 31/12, C07C 321/00, C07C 225/00, A61K 31/41, C07C 47/20, A61K 31/00, C07D 409/14, C07C 309/00, C07D 401/04, A61K 31/38, C07D 335/00, C07D 309/20, C07D 405/04
Мітки: кислоти, похідні, композиція, вірусних, аналоги, 2-дезокси-2,3-дидегідро-n-ацетилнеурамінової, інфекцій, лікування, одержання, фармацевтична, спосіб
Формула / Реферат:
1. Производные и аналоги 2-дезокси-2,3-дидегидро-N-ацетил-неураминовой кислоты формулы (I)где в общей формуле (I) А представляет кислород, углерод или серу,R1 обозначает СООН, Р(О)(ОН)2, NО3, SOOH, SО3Н, тетразол, СH2СНО, СНО или CH(CHO)2,R2 обозначает Н, OR6, F, Cl, Br, CN, NHR6, SR6 или CH2X, где Х представляет NHR6, галоген или OR6, иR6 представляет водород; ацильную группу, имеющую от 1 до 4 атомов...
Похідні триазолу, спосіб їх одержання, фармацевтичний препарат, спосіб лікування або профілактики грибкових інфекцій та проміжна сполука

Номер патенту: 46828
Опубліковано: 17.06.2002
Автори: Грін Стюарт, Мертіашоу Чарльз В., Стефенсон Пітер Т.
МПК: A61P 31/00, C07F 9/6518, A61K 31/00, A61P 31/10, C07F 9/6558, A61K 31/675
Мітки: фармацевтичний, сполука, препарат, похідні, профілактики, триазолу, спосіб, грибкових, проміжна, інфекцій, одержання, лікування
Формула / Реферат:
1. Производные триазола формулы I:R1-ОР(О)(ОН)2 , Iгде R1 представляет собой группу формулы IагдеR2 представляет фенил, замещенный одним или несколькими атомами галогена;R3 представляет Η или СН3;R3a представляет Η или вместе с R3 может представлять =СН2; и R4 представляет 5- или 6-членное азотсодержащее гетероциклическое кольцо, которое произвольно замещено одной группой или более,...
Лікарський засіб та пептид для лікування вірусних інфекцій, сніду та/або снід-асоційованого комплексу

Номер патенту: 41938
Опубліковано: 15.10.2001
Автори: Енгел Юрген, Кутчер Бернхард, Бернд Міхаель, Німейер Ульф
МПК: A61P 31/18, A61K 38/08, A61K 38/02
Мітки: засіб, лікування, лікарський, пептид, вірусних, снід-асоційованого, комплексу, інфекцій, сніду
Формула / Реферат:
1. Лекарственое средство для лечения вирусных инфекций, СПИДа и/или СПИД-ассоциированного комплекса, содержащее активное вещество, отличающееся тем, что в качестве активного вещества оно содержит фармацевтически эффективное количество по крайней мере одного соединения с аминокислотной последовательностью общей формулы IAc-D-Nal(2)-D-Phe-(4-CI)-xxx-A-B-yyy-zzz-Arg-C-D-AIa-NH2,где ххх = D-Pal(3), D-Phe(4-CI), D-Trp,ууу =...
Похідні імідазопіридинів, спосіб їх одержання, фармацевтичний склад та спосіб його одержання

Номер патенту: 41358
Опубліковано: 17.09.2001
Автори: Медерскі Вернер, МІНК Клаус-Отто, Дорш Дітер, Луес Інгеборг, Оссвальд Матіас, Шеллінг П'єр, Байєр Норберт
МПК: A61P 27/02, A61P 25/20, A61P 25/24, A61K 31/435, C07D 471/04, A61K 31/495, A61P 25/26, A61P 25/08, A61P 17/00, A61K 31/535, A61P 25/28, A61P 43/00, A61P 9/12, A61P 9/10, A61P 9/08, A61P 15/00, A61P 13/02, A61P 9/00
Мітки: одержання, імідазопіридинів, похідні, спосіб, склад, фармацевтичний
Формула / Реферат:
1. Производные имидазопиридина формулы (I)где RR1 означает F;R2 – SO2NHCOR5;R3 -A;R4 - группа формулы CnH2nR9;R5 - А, - CtH2t(C3-C8 циклоалкил), CtH2t -Аг;R9 - COOA, COOH, Ar-CO-NR6R7, -CO-R8, -CO-Ar, R6 и R7 соответственноH, A, ArCnH2n- или R6 и R7 вместе означают алкиленовую цепочку с С-5, R8 означает С1С5 - алкил, А – С1-С6 - алкил, Аг - незамещенная группа фенила, t...
Фармацевтичний склад, спосіб його одержання, спосіб лікування психотичних станів та гіперактивності

Номер патенту: 50772
Опубліковано: 15.11.2002
Автори: Тімко Роберт Джозеф, Еддікс Уільям Джозеф, Парікх Бхавніш Вінод
МПК: A61K 9/20, A61K 31/55, A61P 25/18
Мітки: психотичних, станів, лікування, склад, гіперактивності, фармацевтичний, одержання, спосіб
Формула / Реферат:
1. Фармацевтический состав замедленного выделения, включающий гелеобразующий агент и 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b,f][1,4]тиазепин или его фармацевтически приемлемую соль, вместе с одним или более фармацевтически приемлемыми наполнителями.2. Фармацевтический состав замедленного выделения по п. 1, отличающийся тем, что гелеобразующий агент является гидроксипропилметилцеллюлозой.3. Фармацевтический...
Попередній патент: Заміщені ізоксазоліни, спосіб їх одержання, засіб на їх основі та спосіб захисту рослин
Наступний патент: Інгібітори інтерлейкін-1b-перетворюючого ферменту, фармацевтична композиція, спосіб лікування та профілактики захворювань
Випадковий патент: Система фільтрації