Спосіб одержання метакрилової кислоти та її похідних та полімери, одержані з них
Номер патенту: 110121
Опубліковано: 25.11.2015
Автори: Хаддл Томас Ендрю, Поляков Мартін, Джонсон Девід Вільям, Істхем Грехем Рональд
















Формула / Реферат
1. Спосіб одержання метакрилової кислоти шляхом каталізованого основою декарбоксилювання щонайменше однієї дикарбонової кислоти, вибраної з ітаконової, цитраконової або мезаконової кислоти або їх суміші, де декарбоксилювання здійснюють при температурі від більше ніж 240 та до 275 °C в присутності основного каталізатора.
2. Спосіб за п. 1, в якому каталізатор містить джерело OH- іонів.
3. Спосіб за п. 1 або 2, в якому декарбоксилювання здійснюють в інтервалі температур від 245 до 275 °C.
4. Спосіб за будь-яким з пп. 1-3, в якому реагенти, що представляють собою дикарбонову кислоту і переважно основний каталізатор знаходяться у водному розчині.
5. Спосіб за будь-яким з пп. 1-4, в якому декарбоксилювання здійснюють при прийнятному тиску, вище атмосферного тиску.
6. Спосіб за будь-яким з пп. 1-5, де основний каталізатор містить оксид, гідроксид, карбонат, ацетат (етаноат), алкоксид, гідрокарбонат металу або сіль здатної до розкладання дво- або три-карбонової кислоти, або одну з наведених вище сполук четвертинного амонію, або один або декілька амінів.
7. Спосіб за будь-яким з пп. 1-6, де основний каталізатор вибирають з одного або декількох наступних: LiOH, NaOH, KОН, Mg(OH)2, Ca(OH)2, Ba(OH)2, CsOH, Sr(OH)2, RbOH, NH4OH, Li2CO3, Na2CO3, K2CO3, Rb2CO3, Cs2CO3, MgCO3, CaCO3, SrCO3, BaCO3, (NH4)2CO3, LiHCO3, NaHCO3, KHCO3, RbHCO3, CsHCO3, Mg(HCO3)2, Ca(HCO3)2, Sr(HCO3)2, Ba(HCO3)2, NH4HCO3, Li2O, Na2O, K2O, Rb2O, Cs2O, MgO, CaO, SrO, BaO, Li(OR1), Na(OR1), K(OR1), Rb(OR1), Cs(OR1), Mg(OR1)2, Ca(OR1)2, Sr(OR1)2, Ba(OR1)2, NH4(OR1), де R1 являє собою будь-яку C1-C6 розгалужену, нерозгалужену або циклічну алкільну групу, необов'язково заміщену однією або декількома функціональними групами; NH4(RCO2), Li(RCO2), Na(RCO2), K(RCO2), Rb(RCO2), Cs(RCO2), Mg(RCO2)2, Ca(RCO2)2, Sr(RCO2)2 або Ba(RCO2)2, де RCO2 вибирають з цитрамалату, мезаконату, цитраконату, ітаконату, цитрату, оксалату та метакрилату; (NH4)2(CO2RCO2), Li2(CO2RCO2), Na2(CO2RCO2), K2(CO2RCO2), Rb2(CO2RCO2), Cs2(CO2RCO2), Mg(CO2RCO2), Ca(CO2RCO2), Sr(CO2RCO2), Ba(CO2RCO2), (NH4)2(CO2RCO2), де CO2RCO2 вибирають з цитрамалату, мезаконату, цитраконату, ітаконату та оксалату; (NH4)3(CO2R(CO2)CO2), Li3(CO2R(CO2)CO2), Na3(CO2R(CO2)CO2), K3(CO2R(CO2)CO2), Rb3(CO2R(CO2)CO2), Cs3(CO2R(CO2)CO2), Mg3(CO2R(CO2)CO2)2, Ca3(CO2R(CO2)CO2)2, Sr3(CO2R(CO2)CO2)2, Ba3(CO2R(CO2)CO2)2, (NH4)3(CO2R(CO2)CO2), де CO2R(CO2)CO2 вибирають з цитрату, ізоцитрату та аконітату; метиламіну, етиламіну, пропіламіну, бутиламіну, пентиламіну, гексиламіну, циклогексиламіну, аніліну; та R4NOH, де R вибирають з метилу, етилу, пропілу, бутилу.
8. Спосіб за будь-яким з пп. 1-7, в якому каталізатор може бути гомогенним або гетерогенним.
9. Спосіб за будь-яким з пп. 2-8, в якому ефективне молярне співвідношення
основа OH-:кислота, знаходиться в інтервалі між 0,001-2:1.
10. Спосіб за будь-яким з пп. 1-9, в якому продукт, що являє собою метакрилову кислоту, естерифікують з утворенням її естеру.
11. Спосіб одержання полімерів або співполімерів метакрилової кислоти або естерів метакрилової кислоти, що включає такі стадії:
(і) одержання метакрилової кислоти згідно з будь-яким з пп. 1-9;
(іі) необов'язкову естерифікацію метакрилової кислоти, одержаної на стадії (і), з утворенням естеру метакрилової кислоти;
(ііі) полімеризацію метакрилової кислоти, одержаної на стадії (і), та/або естеру, одержаного на стадії (іі), необов'язково з одним або декількома співмономерами, з утворенням їх полімерів або співполімерів.
12. Спосіб за п. 11, в якому естер метакрилової кислоти, зазначеної вище стадії (іі), вибирають з С1-С12 алкілового або С2-С12 гідроксіалкілового, гліциділового, ізоборнілового, диметиламіноетилового та трипропіленгліколевого естерів.
13. Спосіб одержання метакрилової кислоти, що включає:
одержання джерела вихідної кислоти, вибраної з аконітової, лимонної та/або ізолимонної кислоти;
здійснення декарбоксилювання та, якщо необхідно, стадії дегідратації джерела вихідної кислоти шляхом піддавання її джерела у присутності або відсутності основного каталізатора, дії температури менше 350 °C, з одержанням ітаконової, мезаконової та/або цитраконової кислоти; та
спосіб згідно з будь-яким з пп. 1-9 з утворенням метакрилової кислоти.
14. Спосіб за будь-яким з пп. 1-13, в якому концентрація реагенту(ів), що являє(ють) собою дикарбонову кислоту, становить щонайменше 0,1 М.
15. Спосіб за будь-яким з пп. 1-14, в якому концентрація каталізатора у реакційній суміші становить щонайменше 0,1 М.
16. Спосіб одержання метакрилової кислоти шляхом каталізованого основою декарбоксилювання щонайменше однієї дикарбонової кислоти, вибраної з ітаконової, цитраконової або мезаконової кислоти або їх суміші, де декарбоксилювання здійснюють в інтервалі температур між 240 та 290 °C і реагент(и), що являє(ють) собою дикарбонову(і) кислоту(и), піддають дії умов реакції протягом часу щонайменше 80 секунд в присутності основного каталізатора.
Текст
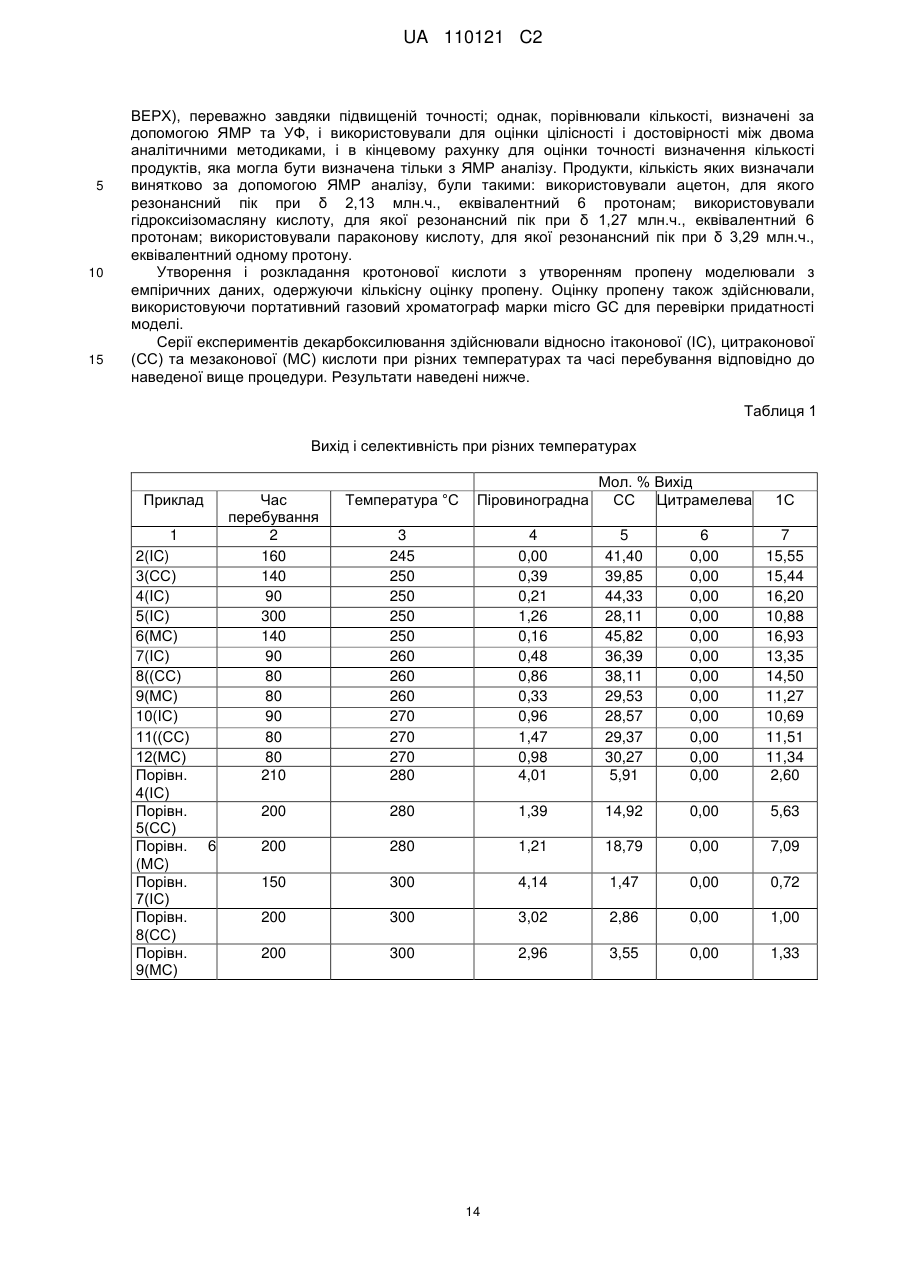
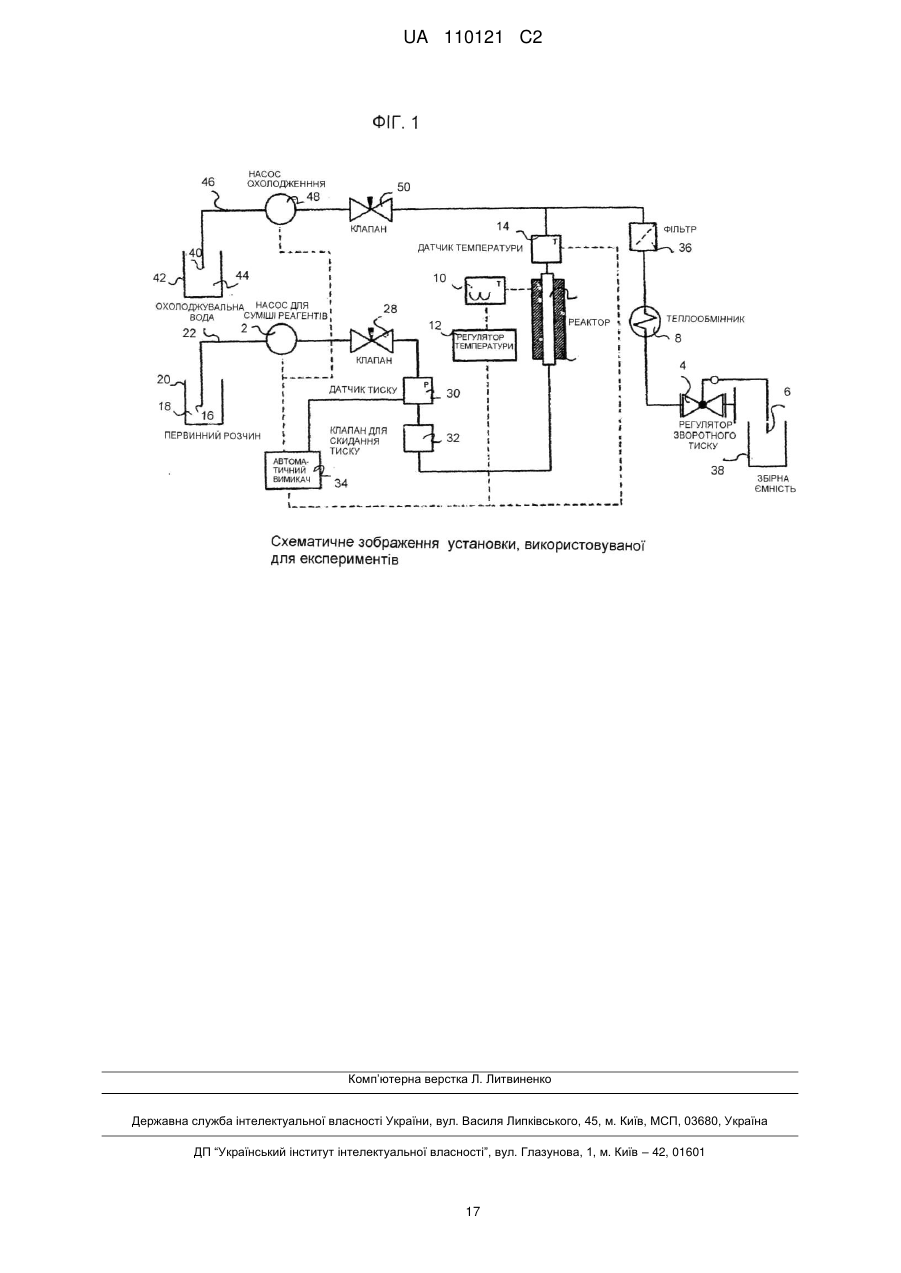
Реферат: Спосіб стосується одержання метакрилової кислоти і включає декарбоксилювання у присутності основного каталізатора щонайменше однієї або суміші дикарбонових кислот, вибраних з ітаконової, цитраконової або мезаконової кислоти при понижених температурах. UA 110121 C2 (12) UA 110121 C2 UA 110121 C2 5 10 15 20 25 30 35 40 45 50 55 60 Представлений винахід стосується способу одержання метакрилової кислоти або похідних, таких як її естери шляхом декарбоксилювання ітаконової кислоти або її джерела у присутності основних каталізаторів, зокрема, але не виключно, способу одержання метакрилової кислоти або метилметакрилату. Метакрилова кислота та її метиловий естер, метилметакрилат (ММА) є важливими мономерами у хімічній промисловості. Головне їх використання полягає в одержанні пластмас для різноманітних галузей застосування. Найбільш важливим полімеризаційним застосуванням є формування литтям, лиття під тиском або екструзійне пресування поліметилметакрилату (ПММА) для виробництва високоякісних оптично прозорих пластиків. Крім того, використовуються багато спів-полімерів, з яких важливими спів-полімерами є спів-полімери метилметакрилату з α-метилстиролом, етилакрилатом і бутилакрилатом. Насьогодні метилметакрилат, ММА (та метилакрилову кислоту, МАК) виробляють повністю з нафтохімічної сировини. Традиційно, ММА виробляють у великих промислових об’ємах за допомогою так званого шляху використання ацетонціангідрину. Спосіб є капіталоємким та передбачає одержання ММА з ацетону та ціаніду водню з високими витратами. Спосіб здійснюється шляхом утворення ацетонціаногідрину з ацетону та ціаніду водню: дегідратація цього проміжного продукту надає метакриламідсульфат, який потім гідролізують, одержуючи МАК. Проміжний ціаногідрин обробляють сірчаною кислотою, перетворюючи на сульфатний естер метакриламіду, метаноліз якого надає бісульфат амонію та ММА. Однак, цей спосіб є не тільки дорогим, окрім цього, обидва реагенти, сірчана кислота та ціанід водню потребують обережного і витратного поводження для безпечного ведення процесів виробництва, а процес призводить до одержання великого об‘єму небажаного сульфату амонію як побічного продукту. Перетворення цього сульфату амонію або на придатне для застосування добриво, або зворотно на сірчану кислоту, потребує високих капітальних витрат на обладнання та значних енерговитрат. Відомо, що у ще одному процесі, альтернативно, виходять з ізобутилену або, еквівалентно, трет-бутанолового реагента, який потім оксидують з утворенням метакролеїну, а потім з утворенням МАК. Покращений спосіб, який забезпечує високий вихід продукту і селективність, та в якому утворюється значно менше побічних продуктів, являє собою двостадійний процес, відомий як Альфа-процес. Стадію I описують у патентному документі у WO96/19434, і вона стосується застосування ліганду 1,2-біс-(ди-трет-бутилфосфінометил)бензолу на паладієвому каталізаторі у процесі карбонілювання етилену з утворенням метилпропіонату з високим виходом і селективністю. Заявник також розробив спосіб каталітичної конверсії метилпропіонату (МЕП) на ММА з використанням формальдегіду. Прийнятним каталізатором для цього способу є цезієвий каталізатор на носії, наприклад, на діоксиді кремнію. Однак, хоча цей двостадійний спосіб має значні переваги у порівнянні з існуючими конкуруючими способами, реагенти, зокрема, етилен, в основному є фракціями природної сирої нафти та природного газу, хоча біоетанол є також придатним як джерело етилену. Протягом багатьох років, як альтернативу викопним видам палива пропонували біомасу, не тільки як потенційний альтернативний енергетичний ресурс, але і як альтернативне джерело вихідної сировини для хімічного процесу. Відповідно, одним з очевидних рішень для відходу від залежності від викопних видів палива, є здійснення будь-якого з відомих процесів одержання ММА або МАК з використанням сировини, одержаної з біомаси. В цьому відношенні добре відомо, що синтетичний пальний газ (синтез-газ) (монооксид вуглецю і водень) може бути одержаний з біомаси, і в той же час із синтез-газу може бути одержаний метанол. На деяких промислових підприємствах виробляють метанол із синтез-газу на цій основі, наприклад, у Lausitzer Analytik GmbH Laboratorium für Umwelt und Brennstoffe Schwarze Pumpe у Німеччині та Biomethanol Chemie Holdings, Delfzijl, Нідерланди. Nouri і Tillman, оцінюючи застосування синтез-газу із біомаси для технологій одержання пластиків (BTP), (ESA-Report 2005:8 ISSN 1404-8167) дослідили ефективність використання метанолу, одержаного з газу дляхімічного синтезу як безпосередньої вихідної сировини для виробництва або ж для виробництва інших видів промислової сировини, такої як формальдегід. Існує також багато патентних і непатентних публікацій про одержання синтез-газу, придатного для виробництва хімічних речовин з біомаси. Виробництво етилену дегідруванням етанолу, одержаного з біомаси, також добре налагоджено на промислових підприємствах, особливо, у Бразилії. Виробництво пропіонової кислоти шляхом карбонілювання етанолу і перетворення гліцерину, одержаного з біомаси, на молекули, такі як акролеїн і акрилова кислота, також добре висвітлені у патентній літературі. 1 UA 110121 C2 5 10 15 20 25 30 35 40 45 50 55 60 Таким чином, шлях виробництва етилену, монооксиду вуглецю і метанолу з біомаси є добре налагодженими промисловими напрямками виробництва. Хімічні речовини, одержані в результаті цього процесу або реалізуються за тими ж технічними умовами, що і матеріали, одержані з нафти/газу, або використовуються в процесах, де є необхідною аналогічна чистота продукту. Таким чином, у принципі, не існує перешкоди для здійснення згаданого вище, так званого Альфа-процесу, для виробництва метилпропіонату з сировини, одержаної з біомаси. Фактично, використання в цьому процесі простої сировини, такої як етилен, моноксид вуглецю і метанол, роблять біомасу, мабуть, ідеальним кандидатом для використання. В цьому смислі, патентний документ WO2010/058119 має явне відношення до використання сировини з біомаси для описаного вище Альфа-процесу і каталітичного перетворення одержаного метилпропіонату (МЕП) на ММА, використовуючи формальдегід. Ці МЕП і формальдегід як сировина можуть бути одержані з джерела біомаси, як було зазначено вище. Однак таке рішення все ж таки передбачає значну обробку і очищення джерела біомаси для одержання промислової сировини, і ці стадії обробки передбачають значне використання викопних видів палив. Крім того, для Альфа-процесу потрібно зібрати в одній місцевості різноманітні види промислової сировини, що може призвести до проблем його придатності. У зв’язку з цим, було б вигідним, якщо б у будь-якому біохімічному способі можна було б уникнути використання різноманітних видів промислової сировини, або використовувати їх у меншій кількості. Таким чином, як і раніш, існує потреба у покращеному варіанті одержання джерела мономерів акрилату, таких як ММА та МАК із альтернативного невикопного палива. У документі PCT/GB2010/052176 описаний спосіб виробництва водних розчинів акрилатів і метакрилатів відповідно з розчинів яблучної і цитрамалевої кислот та їх солей. Carlsson et al., Ind. Eng. Chem. Res. 1994, 33, 1989-1996 розкрили декарбоксилювання ітаконової кислоти з утворенням МАК при високих температурах порядку 360 °C та з 70 % максимальним виходом. Carlsson виявив зменшення в селективності при зниженні температури від 360 до 350 °C за ідеальних умов. Загалом, для промислових процесів необхідна висока селективність для уникнення утворення небажаних побічних продуктів, що в результаті призведе до того, що безперервний процес стане неприйнятним. Для цієї цілі, особливо, для безперервного процесу, селективність для цільового продукту повинна перевищувати 90 %. Несподівано, тепер було виявлено, що висока селективність до утворення МАК понад 90 % при декарбоксилюванні ітаконової кислоти та інших ітаконових рівноважних кислот може забезпечуватись при значно нижчих температурах. Відповідно до першого аспекту представленого винаходу пропонується спосіб одержання метакрилової кислоти шляхом декарбоксилювання, у присутності основного каталізатора, щонайменше однієї дикарбонової кислоти, вибраної з ітаконової, цитраконової або мезаконової кислоти або їх сумішей, де карбоксилювання здійснюють при температурі в інтервалі більше ніж 240 до 275 °C. Реагенти, що представляють собою дикарбонову(і) кислоту(и), і основний каталізатор, не є єдиними сполуками, присутніми у реакції. Дикарбонова(і) кислота(и) разом з будь-якими іншими сполуками, присутніми у реакції, як правило, знаходяться у водному розчині протягом термічного декарбоксилювання, у присутності основного каталізатора. Переважно, здійснення декарбоксилювання при нижчих температурах запобігає утворенню значної кількості побічних продуктів, з видаленням яких можуть виникнути складнощі та можуть викликати проблеми при наступному очищенні та переробці у процесі промислового виробництва. Таким чином, спосіб забезпечує вражаюче покращену селективність у цьому інтервалі значень температур. Крім того, при декарбоксилюванні при нижчій температурі витрачається менше енергії і, таким чином, утворюється менший вуглецевий слід, ніж при декарбоксилюваннях при вищій температурі. Дикарбонові кислоти доступні з джерел невикопного палива. Наприклад, ітаконову, цитраконову або мезаконову кислоти можна було б одержувати з джерела вихідних кислот, таких як лимонна кислота або ізолимонна кислота шляхом дегідратації та декарбоксилювання при прийнятно високих температурах або із аконітової кислоти шляхом декарбоксилювання при прийнятно високих температурах. Слід прийняти до уваги, що основний каталізатор вже присутній, так, що дегідратація та/або розкладання джерела вихідної кислоти можуть бути потенційно основно-каталізовані за таких прийнятних умов. Лимонна кислота та ізолимонна кислота можуть бути одержані за допомогою відомих способів ферментації, а аконітова кислота може бути одержана з первинних кислот. Відповідно, спосіб згідно з винаходом, в тій або іншій 2 UA 110121 C2 5 10 15 20 25 30 35 40 45 50 55 60 мірі сприяє забезпеченню біологічного або частково біологічного шляху для продукування метакрилатів безпосередньо при зведенні до мінімуму залежності від викопних видів палива. Як докладно наведено вище, декарбоксилювання у присутності каталізатора щонайменше однієї дикарбонової кислоти відбувається при температурі менше ніж 270 °C, більш типово, при менше ніж 265 °C, більш переважно, при температурі до 270 °C, найбільш переважно при температурі до 265 °C. У будь-якому випадку, переважна нижча температура для способу представленого винаходу становить 245 °C, більш переважно, 250 °C, найбільш переважно, 255 °C. Переважні інтервали температури для способу представленого винаходу знаходяться в інтервалі від 245 °C та до 270 °C, більш переважно, між 250 °C і 270 °C, найбільш переважно, між 255 °C і 265 °C. Переважним чином, реакція відбувається при температурі, при якій реакційне середовище знаходиться у рідкій фазі. Як правило, реакційне середовище являє собою водний розчин. Переважним чином, декарбоксилювання у присутності основного каталізатора відбувається з реагентами дикарбонової кислоти і переважно у присутності основного каталізатора у водному розчині. Для підтримання реагентів у рідкій фазі при зазначених вище температурних умовах реакцію декарбоксилювання щонайменше однієї дикарбонової кислоти здійснюють при прийнятних величинах тиску, вище атмосферного тиску. Прийнятні значення тиску, при яких реагенти будуть підтримуватись у рідкій фазі у зазначених вище діапазонах температур складають більше, ніж 200 псі, більш прийнятно, більше, ніж 300 псі, найбільш прийнятно, більше, ніж 450 псі, у будь-якому випадку необхідний більш високий тиск, вищий ніж той, нижче якого реакційне середовище буде кипіти. Не існує верхньої границі тиску, але фахівець в даній галузі буде працювати в межах практичного обмеження і в межах можливостей апаратури, наприклад, при менш, ніж 10000 псі, більш типово, при менш, ніж 5000 псі, найбільш типово, при менш, ніж 4000 псі. Переважним чином, зазначена вище реакція відбувається при тиску між приблизно 200 і 10000 псі. Більш переважно, реакція відбувається при тиску між приблизно 300 і 5000 псі і ще більш переважніше між приблизно 450 і 3000 псі. У переважному втіленні, зазначена вище реакція відбувається при тиску, при якому реакційне середовище знаходиться у рідкій фазі. Переважним чином, умови проведення реакції включають температуру і тиск, при яких реакційне середовище знаходиться у рідкій фазі. Як зазначено вище, каталізатор є основним каталізатором. Переважним чином, каталізатор містить джерело OH іонів. Переважним чином, основний каталізатор містить оксид, гідроксид, карбонат, ацетат (етаноат), алкоксид, гідрокарбонат металу, або сіль, здатних до розкладання ди- або три-карбонової кислоти, або одну з наведених вище сполук четвертинного амонію; більш переважно оксид, гідроксид, карбонат, ацетат, алкоксид, гідрокарбонат металу Групи I або Групи II, або сіль металу ди- або три-карбонової кислоти, або метакрилової кислоти. Основний каталізатор може також містити один або більше амінів. Переважним чином, основний каталізатор вибирають з одного або декількох наступних: LiOH, NaOH, KOH, Mg(OH)2, Ca(OH)2, Ba(OH)2, CsOH, Sr(OH)2, RbOH, NH4OH, Li2CO3, Na2CO3, K2CO3, Rb2CO3, Cs2CO3, MgCO3, CaCO3, SrCO3, BaCO3, (NH4)2CO3, LiHCO3, NaHCO3, KHCO3, RbHCO3, CsHCO3, Mg(HCO3)2, Ca(HCO3)2, Sr(HCO3)2, Ba(HCO3)2, NH4HCO3, Li2O, Na2O, K2O, 1 1 1 1 1 1 Rb2O, Cs2O, MgO, CaO, SrO, BaO, Li(OR ), Na(OR ), K(OR ), Rb(OR ), Cs(OR ), Mg(OR )2, 1 1 1 1 1 Ca(OR )2, Sr(OR )2, Ba(OR )2, NH4(OR ), де R представляє собою будь-яку C1 - C6 розгалужену, нерозгалужену або циклічну алкільну групу, необов'язково заміщену однією або декількома функціональними групами; NH4(RCO2), Li(RCO2), Na(RCO2), K(RCO2), Rb(RCO2), Cs(RCO2), Mg(RCO2)2, Ca(RCO2)2, Sr(RCO2)2 або Ba(RCO2)2, де RCO2 вибирають з цитрамалату, мезаконату, цитраконату, ітаконату, цитрату, оксалату та метакрилату; (NH 4)2(CO2RCO2), Li2(CO2RCO2), Na2(CO2RCO2), K2(CO2RCO2), Rb2(CO2RCO2), Cs2(CO2RCO2), Mg(CO2RCO2), Ca(CO2RCO2), Sr(CO2RCO2), Ba(CO2RCO2), (NH4)2(CO2RCO2), де CO2RCO2 вибирають з цитрамалату, мезаконату, цитраконату, ітаконату та оксалату; (NH 4)3(CO2R(CO2)CO2), Li3(CO2R(CO2)CO2), Na3(CO2R(CO2)CO2), K3(CO2R(CO2)CO2), Rb3(CO2R(CO2)CO2), Cs3(CO2R(CO2)CO2), Mg3(CO2R(CO2)CO2)2, Ca3(CO2R(CO2)CO2)2, Sr3(CO2R(CO2)CO2)2, Ba3(CO2R(CO2)CO2)2, (NH4)3(CO2R(CO2)CO2), де CO2R(CO2)CO2 вибирають з цитрату, ізоцитрату та аконітату; метиламіну, етиламіну, пропіламіну, бутиламіну, пентиламіну, гексиламіну, циклогексиламіну, аніліну; та R4NOH, де R вибирають з метилу, етилпропілу, бутилу. Більш переважно, основу вибирають з однієї або декількох наступних: LiOH, NaOH, KOH, Mg(OH) 2, Ca(OH)2, Ba(OH)2, CsOH, Sr(OH)2, RbOH, NH4OH, Li2CO3, Na2CO3, K2CO3, Rb2CO3, Cs2CO3, 3 UA 110121 C2 5 10 15 20 25 30 35 40 45 50 55 60 MgCO3, CaCO3, (NH4)2CO3, LiHCO3, NaHCO3, KHCO3, RbHCO3, CsHCO3, Mg(HCO3)2, Ca(HCO3)2, Sr(HCO3)2, Ba(HCO3)2, NH4HCO3, Li2O, Na2O, K2O, Rb2O, Cs2O,; NH4(RCO2), Li(RCO2), Na(RCO2), K(RCO2), Rb(RCO2), Cs(RCO2), Mg(RCO2)2, Ca(RCO2)2, Sr(RCO2)2 або Ba(RCO2)2, де RCO2 вибирають з ітаконату, цитрату, оксалату, метакрилату; (NH4)2(CO2RCO2), Li2(CO2RCO2), Na2(CO2RCO2), K2(CO2RCO2), Rb2(CO2RCO2), Cs2(CO2RCO2), Mg(CO2RCO2), Ca(CO2RCO2), Sr(CO2RCO2), Ba(CO2RCO2), (NH4)2(CO2RCO2), де CO2RCO2 вибирають з малату, фумарату, малеату, цитрамалату, мезаконату, цитраконату, ітаконату, оксалату; (NH4)3(CO2R(CO2)CO2), Li3(CO2R(CO2)CO2), Na3(CO2R(CO2)CO2), K3(CO2R(CO2)CO2), Rb3(CO2R(CO2)CO2), Cs3(CO2R(CO2)CO2), Mg3(CO2R(CO2)CO2)2, Ca3(CO2R(CO2)CO2)2, Sr3(CO2R(CO2)CO2)2, Ba3(CO2R(CO2)CO2)2, (NH4)3(CO2R(CO2)CO2), де CO2R(CO2)CO2 вибирають з цитрату, ізоцитрату; гідроксиду тетраметиламонію та гідроксиду тетраетиламонію. Найбільш переважно, основу вибирають з однієї або декількох наступних: NaOH, KOH, Ca(OH) 2, CsOH, RbOH, NH4OH, Na2CO3, K2CO3, Rb2CO3, Cs2CO3, MgCO3, CaCO3, (NH4)2CO3, NH4(RCO2), Na(RCO2), K(RCO2), Rb(RCO2), Cs(RCO2), Mg(RCO2)2, Ca(RCO2)2, Sr(RCO2)2 або Ba(RCO2)2, де RCO2 вибирають з ітаконату, цитрату, оксалату, метакрилату; (NH 4)2(CO2RCO2), Na2(CO2RCO2), K2(CO2RCO2), Rb2(CO2RCO2), Cs2(CO2RCO2), Mg(CO2RCO2), Ca(CO2RCO2), (NH4)2(CO2RCO2), де CO2RCO2 вибирають з цитрамалату, мезаконату, цитраконату, ітаконату, оксалату; (NH4)3(CO2R(CO2)CO2), Na3(CO2R(CO2)CO2), K3(CO2R(CO2)CO2), Rb3(CO2R(CO2)CO2), Cs3(CO2R(CO2)CO2), Mg3(CO2R(CO2)CO2)2, Ca3(CO2R(CO2)CO2)2, (NH4)3(CO2R(CO2)CO2), де CO2R(CO2)CO2 вибирають з цитрату, ізоцитрату; та гідроксиду тетраметиламонію. Каталізатор може бути гомогенним або гетерогенним. В одному втіленні, каталізатор може бути розчиненим у рідкій реакційній фазі. Однак, каталізатор може бути суспендований на твердому носії, через який може проходити реакційна фаза В такій ситуації, реакційна фаза переважно підтримується у рідкій фазі, більш переважно, у водній фазі. Переважним чином, ефективне молярне співвідношення іони OH основи:кислота становить між 0,001-2:1, більш переважно, 0,01-1,2:1, найбільш переважно, 0,1-1:1, особливо, 0,3-1:1. Під ефективним молярним співвідношенням іонів OH основи мається на увазі номінальний молярний вміст OH , одержаний від зазначених сполук. Під кислотою маються на увазі молі кислоти. Таким чином, у випадку одноосновної основи, ефективні молярні співвідношення іони OH основи:кислота будуть збігатись з співвідношеннями зазначених сполук, але у випадку дво- або триосновних основ, ефективне молярне співвідношення не буде збігатись з молярним співвідношенням зазначених сполук. Конкретніше, можна вважати, що молярне співвідношення одноосновна основа:дво- або три- карбонова кислота знаходиться переважно в інтервалі 0,001-2:1, більш переважно, 0,011,2:1, найбільш переважно, 0,1-1:1, особливо, 0,3-1:1. Оскільки в представленому винаході депротонування кислоти для утворення солі стосується тільки депротонування одної кислоти, у випадку дво- або триосновних основ, молярне співвідношення зазначеної вище основи буде відповідно відрізнятись. При необхідності, продукт метакрилової кислоти може бути естерифікований з одержанням його естеру. Потенційні естери можуть бути вибрані з C1-C12 алкілового або C2-C12 гідроксиалкілового, гліцидилового, ізоборнілового, диметиламіноетилового, трипропіленгліколевого естерів. Найбільш переважно, спирти або алкени, використовувані для утворення естерів, можуть бути одержані із біо-джерел, наприклад, біометанолу, біоетанолу, біобутанолу. Відповідно до другого аспекту представленого винаходу забезпечується спосіб одержання полімерів або cпівполімерів метакрилової кислоти або естерів метакрилової кислоти, що включає такі стадії (i) одержання метакрилової кислоти відповідно до першого аспекту представленого винаходу; (ii) необов’язкову естерифікацію метакрилової кислоти, одержаної у стадії (i), з одержанням естеру метакрилової кислоти; (iii) полімеризацію метакрилової кислоти, одержаної у стадії (i) та/або естеру, одержаного у стадії (ii), необов’язково з одним або декількома спів-мономерами, з одержанням їх полімерів або cпів-полімерів. Переважним чином, естер метакрилової кислоти, наведеної вище стадії (ii), вибирають з C 1C12 алкілового або C2-C12 гідроксиалкілового, гліцидилового, ізоборнілового, диметиламіноетилового, трипропіленгліколевого естерів, більш переважно, етилового, нбутилового, і-бутилового, гідроксиметилового, гідроксипропілового або метилметакрилатного, найбільш переважно, метилметакрилатного, етилакрилатного, бутилметакрилатного або бутилакрилатного. 4 UA 110121 C2 5 10 15 20 25 30 35 40 45 50 55 60 Переважно, такі полімери можуть складати значну частину, якщо не всі мономерні залишки, що одержують із джерела іншого, ніж джерело викопних видів палива. У будь-якому випадку, переважні спів-мономери включають, наприклад, моноетиленненасичені карбонові кислоти і дикарбонові кислоти, та їх похідні, такі як естери, аміди та ангідриди. Особливо переважними спів-мономерами є акрилова кислота, метилакрилат, етилакрилат, пропілакрилат, н-бутилакрилат, ізо-бутилакрилат, т-бутилакрилат, 2-етилгексилакрилат, гідроксиетилакрилат, ізо-борнілакрилат, метакрилова кислота, метилметакрилат, етилметакрилат, пропілметакрилат, н-бутилметакрилат, ізо-бутилметакрилат, тбутилметакрилат, 2-етилгексилметакрилат, гідроксиетилметакрилат, лаурил метакрилат, гліцидилметакрилат, гідроксипропілметакрилат, ізо-борнілметакрилат, диметиламіноетилметакрилат, трипропіленглікольдіакрилат, стирол, α-метилстирол, вінілацетат, ізоціанати, що включають толуолдіізоціанат та p,p′-метилендифенілдіізоціанат, акрилонітрил, бутадієн, бутадієн і стирол (МБС) і АБС піддають дії будь-якого з наведених вище спів-мономерів, що не є мономером, вибраним з метакрилової кислоти або естеру метакрилової кислоти в наведених вище стадіях (i) або (ii), в будь-якій наведеній спів-полімеризації згаданого кислотного мономеру в стадії (i) або згаданого естеру мономеру в стадії (ii) з одним або декількома спів-мономерами. Зрозуміло, що можливо також використовувати суміші різноманітних спів-мономерів. Самі по собі спів-мономери можуть бути одержані за допомогою такого ж процесу, що і мономери з наведених вище стадій (i) або (ii). Відповідно до ще одного аспекту представленого винаходу забезпечується поліакрилова кислота, поліметакрилова кислота, поліметилметакрилатні (ПММА) і полібутилметакрилатні гомополімери або cпів-полімери, утворені за допомогою способу відповідно до другого аспекту винаходу, наведеного в цьому документі. Відповідно до ще одного аспекту представленого винаходу забезпечується спосіб одержання метакрилової кислоти, який включає: забезпечення джерела вихідної кислоти, вибраної з аконітової, лимонної та/або ізолимонної кислоти; здійснення декарбоксилювання та, якщо необхідно, стадії дегідратації на джерелі вихідної кислоти шляхом піддавання її джерела у присутності або відсутності основного каталізатора дії достатньо високої температури, одержуючи ітаконову, мезаконову та/або цитраконову кислоту; та спосіб відповідно до першого аспекту представленого винаходу, з одержанням метакрилової кислоти. Під джерелом аконітової, лимонної та/або ізолимонної кислоти маються на увазі кислоти та їх солі, такі як їх солі металу групи I або II, та включає розчини вихідних кислот та їх солі, такі як їх водні розчини. Необов‘язково, перед, протягом або після стадії декарбоксилювання вихідної кислоти сіль може бути підкислена, для виділення вільної кислоти. Переважним чином, реагент(и), що представляють собою дикарбонову(і) кислоту(и), піддають дії умов реакції протягом періоду часу щонайменше 80 секунд. Переважним чином, реагент(и), що представляють собою дикарбонову(і) кислоту(и), або джерело їх вихідних кислот згідно з представленим винаходом піддають дії умов реакції протягом прийнятного періоду часу, викликаючи потрібну реакцію, такого як 80 секунд, як визначено в цьому документі, але більш переважно, протягом періоду часу щонайменше 100 секунд, ще більш переважніше щонайменше приблизно 120 секунд та найбільш переважно щонайменше приблизно 150 секунд. Як правило, реагент(и), що представляють собою дикарбонову(і) кислоту(и), або джерело їх вихідних кислот піддають дії умов реакції протягом періоду менше ніж приблизно 2000 секунд, більш типово менше ніж приблизно 1500 секунд, ще більш типово менше ніж приблизно 1000 секунд. Переважним чином, реагент(и), що представляють собою дикарбонову(і) кислоту(и), або джерело їх вихідних кислот, згідно з представленим винаходом, піддають дії умов реакції протягом періоду у проміжку між приблизно 75 секунд та 2500 секунд, більш переважно у проміжку між приблизно 90 секунд та 1800 секунд, і найбільш переважно у проміжку між приблизно 120 секунд та 800 секунд. Таким чином, відповідно до ще одного аспекту представленого винаходу забезпечується спосіб одержання метакрилової кислоти шляхом декарбоксилювання у присутності основного каталізатора щонайменше однієї дикарбонової кислоти, вибраної з ітаконової, цитраконової або мезаконової кислоти або їх суміші, де карбоксилювання здійснюють у інтервалі температур між 5 UA 110121 C2 5 10 15 20 25 30 35 40 45 50 55 240 та 290 °C, і реагент(и), що представляють собою дикарбонову(і) кислоту(и), піддають дії умов реакції протягом періоду часу щонайменше 80 секунд. Переважно, в цьому інтервалі температур високі селективності можуть бути забезпечені при часі перебування, що достатньому для нагрівання реагентів у реакційному середовищі. Переважним чином, реагент(и), що представляють собою дикарбонову(і) кислоту(и), або джерело їх вихідних кислот представленого винаходу розчиняють у воді таким чином, що реакція відбувається у водному середовищі. З того, яким чином характеризуються наведені вище реакції, буде видно що, якщо джерело вихідної кислоти декарбоксилюється та, якщо необхідно, дегідратується у реакційному середовищі, тоді в реакційному середовищі одночасно може бути здійснено декарбоксилювання у присутності основного каталізатора щонайменше однієї дикарбонової кислоти, вибраної з ітаконової, цитраконової або мезаконової кислоти, або їх сумішей, одержаних з джерела вихідної кислоти відповідно до першого аспекту винаходу. Відповідно, декарбоксилювання і, якщо необхідно, дегідратація джерела вихідної кислоти і декарбоксилювання у присутності основного каталізатора щонайменше однієї дикарбонової кислоти можуть відбуватись у одному реакційному середовищі, тобто, два процеси можуть відбуватись як процес в одній реакційній посудині без виділення проміжних сполук. Однак, перевага надається процесу, коли джерело вихідної кислоти декарбоксилюється і, якщо необхідно, одночасно дегідратується по суті, у відсутності основного каталізатора, таким чином, що декарбоксилювання і, якщо необхідно, дегідратація джерела вихідної кислоти та декарбоксилювання у присутності основного каталізатора щонайменше однієї дикарбонової кислоти відбуваються у окремих стадіях. Переважним чином, концентрація реагенту(ів), що представляють собою дикарбонову кислоту, становить щонайменше 0,1 M, переважно у її водному джерелі; більш переважно щонайменше приблизно 0,2 M, переважно у її водному джерелі; найбільш переважно щонайменше приблизно 0,3 M, переважно у її водному джерелі, особливо, щонайменше приблизно 0,5 M. Як правило, водним джерелом є водний розчин. Переважним чином, концентрація реагента(ів), що представляє(ють) собою дикарбонову кислоту, становить менше ніж приблизно 10 M, більш переважно, менше ніж 8 M, переважно у її водному джерелі; більш переважно, менше ніж приблизно 5 M, переважно у її водному джерелі; більш переважно менше ніж приблизно 3 M, переважно у її водному джерелі. Переважним чином, концентрація реагента(ів), що представляє(ють) собою дикарбонову кислоту, знаходиться в інтервалі 0,05 M - 20, типово, 0,05-10 M, більш переважно, 0,1-5 M, найбільш переважно, 0,3-3 M. Основний каталізатор може бути розчинним у рідкому середовищі, яке може бути водою або основний каталізатор може бути гетерогенним. Основний каталізатор може бути розчинним у реакційній суміші таким чином, щоб реакція здійснювалась шляхом піддавання реагентів дії температури, вище якої, декарбоксилювання у присутності основного каталізатора реагента(ів) до утворення метакрилової кислоти та/або джерела вихідних кислот до утворення дикарбонових кислот, буде відбуватись таким чином, як відбувалось би при температурах, наведених вище. Каталізатор може бути у водному розчині. Відповідно, каталізатор може бути гомогенним або гетерогенним, але, як правило, він є гомогенним. Переважним чином, концентрація каталізатора у реакційній суміші (включаючи джерело суміші вихідних кислот, що розкладається) становить щонайменше 0,1 M або більше, переважно у її водному джерелі; більш переважно щонайменше приблизно 0,2 M, переважно у її водному джерелі; більш переважно щонайменше приблизно 0,3 M. Переважним чином, концентрація каталізатора у реакційній суміші (включаючи розкладання джерела суміші вихідної кислоти) становить менше ніж приблизно 10 M, більш переважно, менше ніж приблизно 5 M, більш переважно менше ніж приблизно 2 M та, у будь-якому випадку, переважно менше, ніж або дорівнює такій величині, яка б була у насиченому розчині, при температурі і тиску даної реакції. Переважним чином, молярна концентрація іонів OH у водному реакційному середовищі або необов‘язково джерела вихідної кислоти, що розкладається, знаходиться в інтервалі 0,05-20 M, більш переважно, 0,1-5 M, найбільш переважно, 0,2-2 M. Переважним чином, умови реакції є слабко кислотними. Переважним чином, умови проведення реакції включають значення показника pH між приблизно 2 і приблизно 9, більш переважно між приблизно 3 і приблизно 6. Для уникнення неправильного тлумачення, під терміном ітаконова кислота, мають на увазі наступну сполуку формули (i) 6 UA 110121 C2 COOH COOH (i) Для уникнення неправильного тлумачення, під терміном цитраконова кислота, мають на увазі наступну сполуку формули (ii) COOH COOH 5 (ii) Для уникнення неправильного тлумачення, під терміном мезаконова кислота, мають на увазі наступну сполуку формули (iii) HOOC COOH 10 15 20 25 30 35 40 45 50 (iii) Як зазначено вище, процес згідно з представленим винаходу може бути гомогенним або гетерогенним. Крім того, процес може бути періодичним або безперервним процесом. Переважно, один з побічних продуктів при одержанні МАК може бути гідроксиізомасляною кислотою (ГІМ), яка знаходиться в рівновазі з продуктом МАК при умовах, що використовуються для розкладання дикарбонових кислот. Відповідно, часткове або повне виділення МАК з продуктів реакції розкладання зсуває рівновагу від ГІВ до МАК, таким чином, генеруючи додаткову МАК протягом процесу або у наступній переробці розчину після виділення МАК. Як зазначено вище, джерело вихідної кислоти, таких як лимонна кислота, ізолимонна кислота або аконітова кислота переважним чином розкладається при прийнятних умовах температури і тиску, та необов‘язково у присутності основного каталізатора до утворення однієї з дикарбонових кислот винаходу. Прийнятні умови для цього розкладання складають менше ніж 350 °C, типово, менше ніж 330 °C, більш переважно, при до 310 °C, найбільш переважно при до 300 °C. У будь-якому випадку, переважна нижча температура при розкладанні становить 180 °C. Переважні інтервали температур для джерела вихідної кислоти, що розкладається, знаходяться у інтервалі між 190 та до 349 °C, більш переважно, між 200 та 300 °C, найбільш переважно, між 210 та 280 °C, особливо між 220 та 260 °C. Переважним чином, реакція розкладання джерела вихідної кислоти відбувається при температурі, при якій водне реакційне середовище знаходиться у рідкій фазі. Для підтримання реагентів у рідкій фазі при зазначених вище температурних умовах розкладання джерела вихідної кислоти, реакцію декарбоксилювання здійснюють при прийнятних величинах тиску, вище атмосферного тиску. Прийнятні значення тиску, при яких реагенти будуть підтримуватись у рідкій фазі у зазначених вище діапазонах температур, становлять більше, ніж 150 псі, більш прийнятно, більше ніж 180 псі, найбільш переважно, більше ніж 230 псі, будь-якому випадку при тиску більш вищому ніж той, нижче якого реакційне середовище буде кипіти. Не існує верхньої границі тиску, але фахівець в даній галузі буде працювати в межах практичного обмеження і в межах можливостей апаратури, наприклад, при менш, ніж 10000 псі, більш типово, при менш, ніж 5000 псі, найбільш типово, при менш, ніж 4000 псі. Переважним чином, реакція розкладання джерела вихідної кислоти відбувається при тиску в інтервалі між приблизно 150 та 10000 псі. Більш переважно, реакція відбувається при тиску в інтервалі між приблизно 180 та 5000 псі та ще більш переважніше між приблизно 230 та 3000 псі. У переважному втіленні, реакція розкладання джерела вихідної кислоти відбувається при тиску, при якому реакційне середовище знаходиться у рідкій фазі. Переважним чином, реакція розкладання джерела вихідної кислоти відбувається при температурі і тиску, при яких водне реакційне середовище знаходиться у рідкій фазі. Всі наведені тут ознаки можуть бути скомбіновані з будь-якими в наведених вище аспектах, у будь-якій комбінації. Для кращого розуміння винаходу і демонстрації яким чином можуть бути здійснені його втілення, надалі буде наведене посилання шляхом прикладу, на наступні фігури і приклади. Фігура 1 демонструє схематичне зображення реактора, використовуваного для прикладів винаходу. ПРИКЛАДИ 7 UA 110121 C2 5 10 15 20 25 30 35 40 45 50 55 60 Була проведена серія експериментів для дослідження розкладання ітаконової, цитраконової та мезаконової кислот з утворенням метакрилової кислоти при різноманітних температурах та часі перебування. Методика цих експериментів є наступною. Загальна методика Готували первинний розчин реагентів, що містить ітаконову, цитраконову або мезаконову кислоту при концентрації 0,5 M, а також гідроксид натрію при концентрації 0,5 M. Використовувані ітаконову кислоту (>=99 %) одержували від Sigma Aldrich (Каталожний номер: L2,920-4); цитраконову кислоту (98+ %) одержували від Alfa Aesar (L044178); мезаконову кислоту (99 %) одержували від Sigma Aldrich (Каталожний номер: 13,104-0). Деіонізовану воду, використовувану для сольватації вихідної кислоти/NaOH, спочатку дегазували шляхом обробки ультразвуком, використовуючи Ultrasound Bath (30 Кгц) протягом періоду 5 хвилин. Цей первинний розчин реагентів подавали до системи реактора, використовуючи насосний модуль Gilson 305 аналітичної ВЕРХ, оснащений насосною насадкою Gilson 10 SC. Швидкість, при якій робочий розчин реагентів нагнітали до системи реактора, залежала від необхідного часу перебування та об’єму реактора. Швидкість подавання також залежала від густини реакційного середовища, яка у свою чергу залежала від температури реакційної суміші. Робочий розчин реагентів нагнітали до реактора через трубопровід з нержавіючої сталі (SS 316) (Sandvik), з зовнішнім діаметром 1/16”. Реактор складається з прямолінійного відрізку труби 1/2” SS 316, поміщеного до алюмінієвого блоку, оснащеного двома патронними електричними нагрівальними елементами потужністю 800 Вт фірми Watlow. Перехід трубопроводу SS 316 з діаметра 1/16” на 1/2” забезпечується перехідними муфтами з нержавіючої сталі Swagelok SS 316 та необхідним проміжним ступенем труби діаметром 1/8” (тобто, перехід трубопроводу з діаметру 1/16” на трубопровід діаметром 1/8” та на трубопровід діаметром 1/2”). Об’єм реактора вираховували теоретично і вірність результату підтверджували різницею в масі між реактором, наповненим водою, та порожнім; для описаних експериментів, об’єм 3 реактора становив 19,4 см . Після труби діаметром 1/2”, що представляє собою ’реактор’, за допомогою перехідних муфт, діаметр трубопроводу зворотно змінювали на 1/16”, перед з’єднанням з трубною хрестовиною діаметром 1/16” Swagelok SS 316. На цій хрестовині встановлювали термопару (типу K), для контролю температури сировинного потоку на виході. Об’єм реактора (використовуваний для забезпечення часу перебування) визначали як об’єм секції трубопроводу діаметром 1/2” між двома перехідними муфтами для переходу з діаметру 1/2” на 1/8”, розташованими безпосередньо перед та після алюмінієвого блоку. Суміш продуктів зрештою проходила через теплообмінник (відрізок труби діаметром 1/8”, який знаходиться всередині труби діаметром 1/4”, через яку протитечією проходить охолоджувальна вода), і регулятор зворотного тиску Tescom з ручним керуванням, за допомогою якого утворюється зворотний тиск (тиск всієї системи від даної точки до точки виходу з насосу): для всіх описаних експериментів, використовуваний тиск становив 3000 псі. Перед підготуванням до аналізу, зразки збирали у пробірки. Потрібну для здійснення реакції температуру забезпечували за допомогою термостату, оснащеного регулятором температури Gefran (800 P), який регулював споживану потужність двох патронних електричних нагрівальних елементів Watlow. Кожна серія експериментів включала в себе роботу при визначеній температурі, при цьому час перебування між прогонами змінювали. На насосному модулі Gilson була встановлена потрібна швидкість потоку для першого прогону. Насос залишали включеним протягом близько 20 хвилин, нагнітаючи тільки деіонізовану воду, для того, щоб стабілізувати теплопередачу в алюмінієвому блоці. Вважалась, що теплопередача досягла рівноваги, коли температура розчину, зафіксована термопарою (з точністю до 1 °C), встановленою на виході з реактора, не змінювалась протягом часу більше ніж 5 хвилин. На цій стадії вхід насосу переносили від ємності деіонізованої води до ємності приготованої реакційної суміші. Загальний об’єм установки (включаючи реактор) становив приблизно в два рази більше ніж об’єм реактора; це було попередньо визначено експериментально. Для конкретної швидкості потоку, реакційну суміш нагнітали приблизно в три рази довше, ніж період часу необхідний для того, щоб суміш почала з‘являтися з кінцевого випускного отвору, для того щоб переконатись, що досягнутий стабільний стан реакції. Після цього для аналізу відбирали в кількості 20 мл зразок розчину на виході з установки. Обидві швидкості, як швидкість розчину, відібраного на виході, так і швидкість, при якій первинний розчин подається, реєстрували в залежності від часу для того, щоб контролювати стабільність роботи насосу. Після відбору зразка з конкретного прогону, вхід насосу перемикали зворотно на ємність деіонізованоїводи, і підвищували швидкість потоку до її максимального значення протягом приблизно 10 хвилин для гарантії, що весь матеріал, що залишився від попереднього 8 UA 110121 C2 5 10 15 20 25 30 35 40 45 50 55 прогону, був видалений з системи. Цю процедуру потім повторювали для дослідження відповідного часу перебування. Аналіз Кількісний аналіз продуктів здійснювали, використовуючи систему ВЕРХ, оснащену багатохвильовим УФ детектором серії Agilent 1200. Продукти розділяли, використовуючи + колонку, заповнену Rezex RHM-моносахаридом H (8 %) (Phenomenex), захищену запобіжною колонкою, підтримуючи при температурі 75 °C. Використовували ізократичний спосіб, який здійснювався з використанням водної 0,005 M H2SO4 рухомої фази, з швидкістю потоку 0,4 -1 млхв. . Було виявлено, що сполуки, які містяться у зразках продукту, мають оптимальне значення коефіцієнта поглинання УФ при найкоротшій довжині хвилі 210 нм, яку здатний сприйняти багатохвильовий детектор (ширина смуги 15 нм). Всі сполуки продукту калібрували для їх УФ детектування, шляхом кореляції їх УФ коефіцієнта поглинання в залежності від меж діапазону значень концентрацій. Визначали діапазони лінійної характеристики для кожної сполуки, та найбільш прийнятний діапазон концентрацій, знайдений для всіх сполук, що -3 -3 представляють інтерес, знаходився в інтервалі між 5 10 M та 1 10 M. Таким чином, належне кількісне виявлення більшості продуктів забезпечували в інтервалі від 1 до 100 розведень зразків, одержаних в установці перед аналізом ВЕРХ (розведення від 1 до 100 означає, що, коли починають з 0,5 M сировинного розчину, будь який продукт, утворений з виходом між 20-100 %, потрапляв би в межі діапазону лінійної характеристики концентрацій). Якщо сполуки опинялися за межами цього діапазону лінійної характеристики (наприклад, вихід менше ніж 20 %), здійснювали другий аналіз ВЕРХ, використовуючи розведення в інтервалі від 1 до 10. Деякі зразки, які не були точно визначені з використанням методики розведень в інтервалі від 1 до 10, вважалися слідами в концентрації і, таким чином, не приймались у розрахунках до уваги. Процедура Здійснювали наступну процедуру. Спочатку одержували суміш реагентів, що містить вихідну кислоту і гідроксид натрію. Потрібну швидкість потоку для забезпечення часу перебування обчислювали, використовуючи об‘єм реактора і густину води (обчислену, виходячи з температури). Фігура 1 ілюструє схематичне зображення установки згідно з представленим винаходом. Первинний розчин 18 завантажували до ємності 20, яка була приєднана до вихідного патрубка 16. Вихідний патрубок приєднувався через контур потоку 22 до насосу 2 для суміші реагентів, який призначений нагнітати розчин 18 до реактора 24, камера якого розміщена у патронному електричному нагрівальному елементі 26, який охоплює вздовж по колу реактор 24. Контур потоку 22 між насосом 2 та реактором 24 проходив від насосу через клапан 28 для здійснення контролю подачі, датчик тиску 30 та клапан для скидання тиску 32. Крім того, до датчика тиску 30, насосу для суміші реагентів 2 та датчика температури 14 був під’єднаний автоматичний вимикач 34. Датчик температури 14 розташовувався на контурі потоку 22, безпосередньо після реактора 24 та перед вихідним патрубком 6. Крім того, після датчика температури 14, контур потоку проходить до вихідного патрубка через фільтр 36, теплообмінник 8 та регулятор зворотного тиску 4. У точці виходу 6, продукт збирали до збірної ємності 38. На реакторі 24 також був встановлений блок контролю температури 10, 12 для контролю температури в реакторі 24. До установки також входить охолоджувальна система, що складається з окремого вхідного патрубка 40 для охолоджувальної води 44 в резервуарі для охолоджувальної води 42. Вхідний патрубок 40 з’єднаний з вихідним патрубком 6 через контур 46, на якому встановлений окремий насос контуру охолодження 48, а за ним клапан 50 для контролю подачі охолоджувальної води. Безпосередньо після датчика температури 14 реактора 24, та перед фільтром 36 для припинення будь-якої реакції після реактора, контур охолоджувальної води 46 змішувався з контуром реакційної суміші 22. Насос охолоджувального контуру 48 та блок контролю температури 10, 12 також приєднані до автоматичного вимикача 34 для необхідного вимикання, якщо виконуються умови вимикання. Включали насос 2 та у систему нагнітали деіонізовану воду. Регулятор зворотного тиску 4 поступово доводили до потрібного тиску (3000 псі). -1 Коефіцієнт корисної дії насосу перевіряли при швидкості 5 млхв. шляхом реєстрації часу, витраченого для відбору 20 мл води на виході 6 із системи. Коефіцієнт корисної дії, що становив > 90 %, вважався прийнятним. Потім на насосі встановлювали таку швидкість потоку, яка була необхідна для здійснення прогону. 9 UA 110121 C2 5 10 15 20 25 30 35 Подача води (не зображено) до теплообмінника 8 була встановлена на нижчому рівні помірного протікання потоку, в залежності від температури реакції та швидкості потоку на насосі для експерименту. Термостат нагрівача 10, оснащений регулятором температури 12, налагоджували на температуру, необхідну для здійснення прогону. Як тільки досягалась потрібна температура (яка фіксувалась термостатом 10), температуру на виході із реактора контролювали за допомогою датчика температури у реакторі 14 до тих пір, доки спостережуване значення температури (з точністю до 1 °C) не зберігалось постійним протягом щонайменше 5 хвилин (на це, як правило, витрачалось приблизно 20 хвилин). Вхід до насосу 16 перемикали з ємності для деіонізованої води (не зображено) на ємність для приготованої суміші реагентів 18 (ця операція вимагає зупинки нагнітання потоку протягом декількох секунд). Реєстрували початковий об’єм суміші реагентів в ємності 18. Обчислення можуть вказати період, після якого розчин продукту почне з’являтись з вихідного патрубка 6 системи. Однак, на практиці, це підтверджувалось візуально і по наявності звуку бульбашок газу, що виходять з установки (утвореного в результаті розкладання реагентів). Це дозволяло продовжити роботу протягом періоду, який становив у три рази довше ніж період, протягом якого з’являвся розчин продукту. Це підтверджувало, що суміш продукту є гомогенною. На виході 6 з системи, відбирали 20 мл розчину продукту і реєстрували час, витрачений для цього відбору. Кінцевий час і об’єм були також зафіксовані для суміші реагентів. Після збирання продукту, вхід насосу переносили до ємності для деіонізованої води і насос налагоджували на “prime mode” (максимальну швидкість потоку), і проганяли протягом приблизно 10 хвилин. Швидкість потоку на насосі потім налагоджували на потрібну величину для наступного прогону. Знову контролювали температуру на виході з реактора і вважали її усталеною, коли її значення не змінювалось протягом щонайменше 5 хвилин (зазвичай на це витрачалось приблизно 10 хвилин). Цю методику проведення експерименту повторювали до тих пір, поки не здійснювали всі необхідні для експерименту прогони. Після завершення всіх прогонів, до системи насосом при роботі у режимі максимальної швидкості потоку нагнітали деіонізовану воду і вимикали нагрівач (термостат). Якщо температура суміші на виході із реактора падала нижче 80° C, вимикали насос і припиняли також подачу води до теплообмінника. Виходи продукту виражені як абсолютні мольні проценти (100 молів продукт/молів завантажених реагентів) Приклад 1 Розкладання ітаконової кислоти Вихідна речовина Основа Конц. основи Температура Тиск 0,5 M Ітаконова кислота NaOH 0,5 M 250 °C 3000 псі Час перебування МАК Вихід PY Вихід CC Вихід IC Вихід (секунди) 540 19,25 0,36 36,76 16,35 HIB/PC Вихід 11,26 MC Вихід CT Вихід 15,18 0,07 40 45 50 Селективність = Вихід МАК/(1-(Вихід IC + Вихід CC + Вихід MC + Вихід HIB/PC )) Селективність = 94,13 % Де МАК Метакрилова кислота PY піровиноградна кислота CC цитраконова кислота CM цитрамалева кислота IC ітаконова кислота HIB гідроксиізомасляна кислота MC мезаконова кислота CT кротонова кислота PC параконова кислота 10 UA 110121 C2 Порівняльний приклад 1 Розкладання ітаконової кислоти Вихідна речовина Основа Конц. основи Температура Тиск 0,5 M Ітаконова кислота NaOH 0,5 M 310 °C 3000 псі Час перебування МАК Вихід 540 5 58,36 PY Вихід CC Вихід IC Вихід 2,69 0,50 0,84 HIB/PC Вихід 22,12 MC Вихід CT Вихід 0,64 0,74 Селективність = Вихід МАК/(1-(Вихід IC + Вихід CC + Вихід MC + Вихід HIB/PC)) Селективність = 76,89 % Порівняльний приклад 2 Розкладання ітаконової кислоти Вихідна речовина Основа Конц. основи Температура Тиск Час перебування 480 0,5 M Ітаконова кислота NaOH 0,5 M 330 °C 3000 псі МАК Вихід 56,74 PY Вихід CC Вихід IC Вихід 2,63 0 0 HIB/PC Вихід 19,33 MC Вихід CT Вихід 0,08 0,53 10 Селективність = Вихід МАК/(1-(Вихід IC + Вихід CC + Вихід MC + Вихід HIB/PC)) Селективність = 70,41 % Порівняльний приклад 3 Розкладання ітаконової кислоти Вихідна речовина Основа Конц. основи Температура Тиск 0,5 M Ітаконова кислота NaOH 0,5 M 350 °C 3000 псі 15 Час перебування 180 20 25 30 МАК Вихід PY Вихід CC Вихід 54,42 2,67 0,26 IC Вихід 0,16 HIB/PC Вихід 13,07 MC Вихід CT Вихід 0,30 0,63 Селективність = Вихід МАК/(1-(Вихід IC + Вихід CC + Вихід MC + Вихід HIB/PC)) Селективність = 63,12 % Приклади 2-12 та Порівняльні приклади 4-9 Загальна процедура Готували робочий розчин реагентів, що містить ітаконову, цитраконову або мезаконову кислоту при концентрації 0,5 M і гідроксид натрію при концентрації 0,5 M. Використовувані ітаконову кислоту (>=99 %) одержували від Sigma Aldrich (Каталожний номер: L2,920-4); цитраконову кислоту (98+ %) одержували від Alfa Aesar (L044178); мезаконову кислоту (99 %) одержували від Sigma Aldrich (Каталожний номер: 13,104-0). Цей робочий розчин реагентів подавали до системи реактора, використовуючи насосний модуль Gilson 205 аналітичної ВЕРХ, оснащений насосною насадкою Gilson 10 SC. Витрату потоку насосу контролювали з комп‘ютера з програмним забезпеченням Gilson Unipoint. Робочий розчин реагентів нагнітали до реактора через трубопровід з нержавіючої сталі (SS 316) (Sandvik), з зовнішнім діаметром 1/16”. Реактор складається з відрізка трубчастого зміяку діаметром 1/18” SS 316, який охоплює циліндричну алюмінієву основу, орбітальна поверхня якої має нарізану різьбу під розміри труби діаметром 1/8”, забезпечуючи велику площу контакту між 11 UA 110121 C2 5 10 15 20 25 30 35 40 45 50 55 60 основою та трубою. В серцевині цієї циліндричної основи розміщався патронний електричний нагрівальний елемент потужністю 1 квт фірми Watlow, який передає через теплопроводність тепло зсередини основи. Зовні, трубчастий зміяк був також розміщений всередині манжетного електричного нагрівача, потужністю 1 квт фірми Watlow. Роздільний шар, виготовлений з латуні був поміщений між манжетним нагрівачем та зовнішньою лицьовою поверхнею трубчастого зміяку, з нарізкою під внутрішню поверхню (знаходячись в контакті з трубою), для забезпечення гарного контакту площі поверхні, і як результат забезпечення теплопередачі від манжетного нагрівача до труби. Використовуваний для реактора трубопровід діаметром 1/8”, по обох його кінцях, оснащували перехідними муфтами ss 316 фірми Swagelok для переходу з діаметру 1/16” на 1/18”. Перехідна муфта на виході з реактора знаходилася безпосередньо перед з‘єднанням з трубною хрестовиною діаметром 1/16” Swagelok ss 316, яка впускала другий контур охолоджувальної води, забезпечуючи можливість вимірювати температуру за допомогою термопари діаметром 1/16” типу K (Radio Spares), і забезпечуючи шлях виходу для потоку охолодженого продукту. Вся система реактора, включаючи складові елементи перехідних муфт, до самої трубної хрестовини діаметром 1/16”, була термоізольована шарами із скловати, алюмінієвої фольги та стрічки з скловолокна; це сприяло мінімізації градієнтів температури на вході до реактора та виході із реактора, між нагрівачем та трубною хрестовиною діаметром 1/16”. Для дослідження різних значень інтервалів часу перебування використовували два об‘єми реактора, а об‘єм коригували шляхом зменшення кількості витків навколо алюмінієвої основи реактора. У кожному випадку вважали, об‘єм реактора представляє собою об‘єм між перехідною муфтою на вході до реактора, та точкою підмішування охолоджувального контуру на трубній хрестовині діаметром 1/16” на виході реактора. В обох випадках передбачуваний об‘єм реактора визначали шляхом нагнітання точно відміряної кількості води до порожніх комунікаційних елементів реактора, які попередньо висушували при підвищеній температурі з наступною продувкою газоподібним азотом; цей процес повторювали декілька разів у випадку виникнення розбіжності. Після охолодження, суміш продукту зрештою проходила через теплообмінник, який складався з відрізку труби діаметром 1/16” довжиною приблизно 1,5 м, потім зону охолоджування, проходячи через відрізок труби ss 316 діаметром 1/14” такої ж довжини, через який вода могла б проходити протитечією, для видалення тепла, що залишилося від потоку суміші продукту; розміри труби для цієї системи теплообмінника, вибрані для мінімізації загального об’єму установки. Зрештою, потік продукту проходив через регулятор зворотного тиску з ручним керуванням Tescom, за допомогою якого утворюється зворотний тиск (тиск всієї системи від даної точки до виходу з насосу): для всіх описаних експериментів, використовуваний тиск становив 3000 псі. Зразки для кожного дослідженого часу перебування збирали до пробірок автоматично, за допомогою колектора фракцій Gilson 201, яким також керували з комп‘ютера з програмним забезпеченням Unipoint. Програми, здійснені в Unipoint, використовували в своїй роботі наступний протокол: швидкість потоку повинна узгоджуватись з тим значенням, яке необхідне для конкретного часу перебування, як було попередньо обчислено; потім насос мав продовжувати нагнітати при цій швидкості потоку до тих пір, поки об’єм у три рази більший, ніж об’єм всієї установки, не проходив через усю систему, забезпечуючи прийнятний час для встановлення рівноваги як для нагрівача так і суміші потоку продукту; колектор фракцій потім переносили до точки виходу продукту із системи в заданих місцях фракції та збирали заданий об’єм водного продукту; колектор фракцій зрештою переносили до виходу потоку із системи до ємності для відходів, і швидкість потоку приводили до значення, необхідного для наступного значення часу перебування, що представляє інтерес. Потрібну температуру для кожного експерименту досягали, за допомогою термостату, оснащеного регулятором температури Gefran (800 P), який регулював споживану потужність, як для патронного електричного нагрівального елементу Watlow, так і для манжетного нагрівача Watlow. Досягнену температуру контролювали за допомогою термостату через термопару 1/16” типу K, розташовану у отворі діаметром 1/16”, який просвердлювали у верхній частині алюмінієвої основи, поблизу від місця контакту з трубчастим зміяком. Другу термопару розташовували у другому отворі у безпосередній близькості до першого, яка контролювала температуру на незалежному модулі відображення температури; цей модуль з’єднували з електронним вимикачем ланцюга, який забезпечував можливість припинення подавання електроенергії до всіх електронних пристроїв у випадку виникнення температури перегріву, а також використовували для перевірки узгодженості між зафіксованою ним температурою та тою температурою, яка була виміряна термостатом. Для кожного експерименту, термостат 12 UA 110121 C2 5 10 15 20 25 30 35 40 45 50 55 60 налагоджували на необхідну температуру, що залежала від найбільшої швидкості потоку, яка може потребуватись, тобто, для випадку, якщо нагрівач буде потребувати максимальної потужності. Налагоджуючи в такій спосіб термостат, підтверджували мінімальний час, необхідний для встановлення рівноваги електроенергії, підводжуваної до нагрівача, де швидкість потоку поступово знижувалась, якщо досліджувалися триваліші проміжки часу перебування у кожному експерименті. У випадку досліджень всіх проміжків часу перебування, оскільки швидкість потоку змінювали на початковому етапі, було помічено, що реактор налаштовувався на початкову температуру (з точністю до 1 °C) протягом незначної частини загального часу, що надає можливість для встановлення рівноваги. У всіх цих експериментах, охолоджувальний потік скрізь задавали таким, що дорівнював первинному потоку через реактор; це надавало можливість забезпечення значного рівня стабільності охолодження, в той час як швидкість потоку первинного розчину, і як наслідок час перебування, змінювався. Безпосередньо перед початком подачі електроенергії на термостат (і таким чином, на нагрівачі реактора), зразки вихідної речовини збирали при різних швидкостях потоку, необхідних при здійсненні експериментів, при швидкості охолоджувального потоку, що у кожному випадку дорівнювала швидкості потоку вихідної речовини. Ці зразки вихідної речовини могли б бути порівняні зі зразками продукту протягом аналізу для того, щоб визначити виходи продукту та масові баланси. Збирання зразків вихідної речовини в такий спосіб допомагає розрахувати варіювання коефіцієнту корисної дії між двома насосами при різних швидкостях потоку, що представляють інтерес. Аналіз Кількісний аналіз більшості продуктів здійснювали, з використанням рідинного хроматографа серії Agilent 1200 системи ВЕРХ, оснащеної багатохвильовим УФ детектором. + Продукти розділяли, використовуючи колонку, заповнену Rezex RHM-моносахаридом H (8 %) (Phenomenex), захищену запобіжною колонкою, підтримуючи при температурі 75 °C. Використовували ізократичний спосіб, який здійснювався з використанням водної 0,005 M -1 H2SO4 рухомої фази, з швидкістю потоку 0,4 млхв. . Було виявлено, що сполуки, які містяться у зразках продукту, мають оптимальне значення коефіцієнта поглинання УФ при найкоротшій довжині хвилі 210 нм, яку здатний сприйняти багатохвильовий детектор (ширина смуги 15 нм). Всі сполуки продукту калібрували для їх УФ детектування, шляхом кореляції їх коефіцієнта поглинання УФ в залежності від меж діапазону значень концентрацій. Визначали діапазони лінійної характеристики для кожної сполуки, та найбільш прийнятний діапазон концентрацій, -3 знайдений для всіх сполук, що представляють інтерес, знаходився в інтервалі між 5 10 M та -3 1 10 M. Таким чином, належне кількісне виявлення більшості продуктів забезпечували в інтервалі від 1 до 100 розведень зразків, одержаних в установці перед аналізом ВЕРХ (розведення від 1 до 100 означає, що, коли починають з 0,5 M сировинного розчину, будь який продукт, утворений з виходом між 20-100 %, потрапляв би в межі діапазону лінійної характеристики концентрацій). Якщо сполуки опинялися за межами цього діапазону лінійної характеристики (наприклад, вихід менше ніж 20 %), здійснювали другий аналіз ВЕРХ, використовуючи розведення в інтервалі від 1 до 10. Деякі зразки, які не були точно визначені з використанням методики розведень в інтервалі від 1 до 10, вважалися слідами в концентрації і, таким чином, не приймались у розрахунках до уваги. Незначна кількість компонентів продукту не могла бути визначена за допомогою ВЕРХ з УФ детектуванням, або через їх низький коєфіцієнт УФ поглинання, або через проблеми сумісного елюювання під час хроматографічного розділення. Визначення цих компонентів, натомість, 1 здійснювали за допомогою аналізу спектрів ядерного магнітного резонансу ( H ЯМР), з використанням спектрометра ЯМР Bruker dpx 300 Мгц. Зразки продукту аналізували у водній формі, у тому вигляді як продукт виходив після установки (одержуючи сумарну концентрацію продукту приблизно 0,25 M після розведення охолоджувальним потоком), розводили у D 2О (Aldrich, 99,98 %) у співівдношенні 1:2 відповідно. Внутрішній стандарт не додавався, і замість цього концентрації різних речовин стандартизували по відношенню до чітко виділеного резонансного піку компонента, концентрація якого була достовірно відома з ВЕРХ аналізу. Для цієї цілі, вибирали або пік протонного резонансу для нетермінальної CH2 групи ітаконової кислоти, δ 3,18 млн.ч. (еквівалентний 2 протонам), або пік протонного резонансу термінальної метилової CH3 групи метакрилової кислоти, δ 1,79 млн.ч. (еквівалентний 3 протонам), в залежності від того, який з них був ширший. Інтеграли всіх інших резонансних піків у спектрі могли би використовуватись для визначення кількості всіх інших хімічних речовин суміші продукту на основі концентрації ітаконової або метакрилової кислоти, як було попередньо визначено УФ детектуванням; однак, коли визначення кількості компонентів можливо було здійснити за допомогою і УФ, і ЯМР детектування, вибирали УФ детектування (за допомогою 13 UA 110121 C2 5 10 15 ВЕРХ), переважно завдяки підвищеній точності; однак, порівнювали кількості, визначені за допомогою ЯМР та УФ, і використовували для оцінки цілісності і достовірності між двома аналітичними методиками, і в кінцевому рахунку для оцінки точності визначення кількості продуктів, яка могла бути визначена тільки з ЯМР аналізу. Продукти, кількість яких визначали винятково за допомогою ЯМР аналізу, були такими: використовували ацетон, для якого резонансний пік при δ 2,13 млн.ч., еквівалентний 6 протонам; використовували гідроксиізомасляну кислоту, для якої резонансний пік при δ 1,27 млн.ч., еквівалентний 6 протонам; використовували параконову кислоту, для якої резонансний пік при δ 3,29 млн.ч., еквівалентний одному протону. Утворення і розкладання кротонової кислоти з утворенням пропену моделювали з емпіричних даних, одержуючи кількісну оцінку пропену. Оцінку пропену також здійснювали, використовуючи портативний газовий хроматограф марки micro GC для перевірки придатності моделі. Серії експериментів декарбоксилювання здійснювали відносно ітаконової (IC), цитраконової (CC) та мезаконової (MC) кислоти при різних температурах та часі перебування відповідно до наведеної вище процедури. Результати наведені нижче. Таблиця 1 Вихід і селективність при різних температурах Приклад 1 2(IC) 3(СС) 4(IС) 5(IС) 6(МС) 7(IС) 8((СС) 9(МС) 10(IС) 11((СС) 12(МС) Порівн. 4(ІС) Порівн. 5(СС) Порівн. (MC) Порівн. 7(ІС) Порівн. 8(СС) Порівн. 9(МС) Мол. % Вихід СС Цитрамелева Час перебування 2 160 140 90 300 140 90 80 80 90 80 80 210 Піровиноградна 3 245 250 250 250 250 260 260 260 270 270 270 280 4 0,00 0,39 0,21 1,26 0,16 0,48 0,86 0,33 0,96 1,47 0,98 4,01 5 41,40 39,85 44,33 28,11 45,82 36,39 38,11 29,53 28,57 29,37 30,27 5,91 6 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 7 15,55 15,44 16,20 10,88 16,93 13,35 14,50 11,27 10,69 11,51 11,34 2,60 200 6 Температура °С 280 1,39 14,92 0,00 5,63 200 280 1,21 18,79 0,00 7,09 150 300 4,14 1,47 0,00 0,72 200 300 3,02 2,86 0,00 1,00 200 300 2,96 3,55 0,00 1,33 14 1С UA 110121 C2 Продовження Таблиці 1 8 НІВ 1,70 1,38 1,01 5,36 1,10 2,23 1,72 0,48 4,12 4,28 1,6 17,55 10,37 8,18 20,27 15,66 15,2 5 10 15 9 MC 15,48 17,61 13,99 16,76 13,42 15,98 16,21 46,37 16,10 16,45 36,77 5,72 9,78 12,49 1,68 1,72 2,29 10 МАК 16,15 16,18 14,30 29,72 11,63 22,36 20,64 8,12 32,23 31,67 16,39 57,08 46,79 42,74 63,13 55,85 56,42 Мол. % Вихід 11 12 Кротонова Параконова 0,05 9,62 0,02 9,02 0,00 9,91 0,00 7,30 0,00 10,94 0,20 8,17 0,25 7,27 0,00 3,70 0,32 5,25 0,63 3,72 0,33 2,01 1,01 1,22 0,71 2,64 0,64 3,29 1,05 1,17 1,14 2,92 1,15 2,86 13 Пропенова 0,05 0,11 0,06 0,61 0,10 0,85 0,44 0,12 1,76 0,89 0,31 4,91 7,78 5,57 6,31 16,03 14,24 14 Селективність 99,38 96,88 98,21 94,08 98,64 93,62 93,01 93,71 91,37 91,35 91,01 85,19 82,58 85,21 84,53 73,44 75,46 Слід зазначити, що всі наукові статті і документи, які реєструвались одночасно або до цього опису в зв'язку з цією заявкою, та до яких є доступ до публічного ознайомлення з цим описом, включені у даний винахід як посилання. Всі ознаки винаходу, розкриті в цьому описі (включаючи будь-які супроводжувальні пункти формули, реферат та фігури), та/або всі стадії будь-якого способу або процесу розкриті так, що можуть бути поєднані в будь-яку комбінацію, за винятком комбінацій, в яких щонайменше деякі з таких ознак та/або стадій, є взаємовиключальними. Кожна ознака, розкрита в цьому описі (включаючи будь-які супроводжувальні пункти формули, реферат та фігури) може бути замінена альтернативними ознаками, що виконують ті ж функції еквівалентного або аналогічного призначення, якщо не вказане інше. Таким чином, якщо не вказане інше, то кожна описана ознака є одним із прикладів тільки характерного ряду еквівалентних або подібних ознак. Винахід не обмежений деталями вищезгаданого(вищезгаданих) втілення(втілень). Винахід поширюється на будь-яку одну нову ознаку, або на будь-яку нову комбінацію ознак, розкритих у цьому описі (включаючи будь-які супроводжувальні пункти формули, реферат та фігури), або на будь-яку одну нову стадію, або будь-яку нову комбінацію стадій будь-якого способу або процесу, розкритого тут. 20 ФОРМУЛА ВИНАХОДУ 25 30 35 1. Спосіб одержання метакрилової кислоти шляхом каталізованого основою декарбоксилювання щонайменше однієї дикарбонової кислоти, вибраної з ітаконової, цитраконової або мезаконової кислоти або їх суміші, де декарбоксилювання здійснюють при температурі від більше ніж 240 та до 275 °C в присутності основного каталізатора. 2. Спосіб за п. 1, в якому каталізатор містить джерело OH іонів. 3. Спосіб за п. 1 або 2, в якому декарбоксилювання здійснюють в інтервалі температур від 245 до 275 °C. 4. Спосіб за будь-яким з пп. 1-3, в якому реагенти, що представляють собою дикарбонову кислоту і переважно основний каталізатор знаходяться у водному розчині. 5. Спосіб за будь-яким з пп. 1-4, в якому декарбоксилювання здійснюють при прийнятному тиску, вище атмосферного тиску. 6. Спосіб за будь-яким з пп. 1-5, де основний каталізатор містить гідроксид, карбонат, ацетат (етаноат), алкоксид, гідрокарбонат металу або сіль здатної до розкладання дво- або три 15 UA 110121 C2 5 10 15 20 25 30 35 40 45 50 55 карбонової кислоти, або одну з наведених вище сполук четвертинного амонію, або один або декілька амінів. 7. Спосіб за будь-яким з пп. 1-6, де основний каталізатор вибирають з одного або декількох наступних: LiOH, NaOH, KОН, Mg(OH)2, Ca(OH)2, Ba(OH)2, CsOH, Sr(OH)2, RbOH, NH4OH, Li2CO3, Na2CO3, K2CO3, Rb2CO3, Cs2CO3, MgCO3, CaCO3, SrCO3, BaCO3, (NH4)2CO3, LiHCO3, NaHCO3, KHCO3, RbHCO3, CsHCO3, Mg(HCO3)2, Ca(HCO3)2, Sr(HCO3)2, Ba(HCO3)2, NH4HCO3, Li2O, Na2O, 1 1 1 1 1 1 K2O, Rb2O, Cs2O, MgO, CaO, SrO, BaO, Li(OR ), Na(OR ), K(OR ), Rb(OR ), Cs(OR ), Mg(OR )2, 1 1 1 1 1 Ca(OR )2, Sr(OR )2, Ba(OR )2, NH4(OR ), де R являє собою будь-яку C1-C6 розгалужену, нерозгалужену або циклічну алкільну групу, необов'язково заміщену однією або декількома функціональними групами; NH4(RCO2), Li(RCO2), Na(RCO2), K(RCO2), Rb(RCO2), Cs(RCO2), Mg(RCO2)2, Ca(RCO2)2, Sr(RCO2)2 або Ba(RCO2)2, де RCO2 вибирають з цитрамалату, мезаконату, цитраконату, ітаконату, цитрату, оксалату та метакрилату; (NH4)2(CO2RCO2), Li2(CO2RCO2), Na2(CO2RCO2), K2(CO2RCO2), Rb2(CO2RCO2), Cs2(CO2RCO2), Mg(CO2RCO2), Ca(CO2RCO2), Sr(CO2RCO2), Ba(CO2RCO2), (NH4)2(CO2RCO2), де CO2RCO2 вибирають з цитрамалату, мезаконату, цитраконату, ітаконату та оксалату; (NH4)3(CO2R(CO2)CO2), Li3(CO2R(CO2)CO2), Na3(CO2R(CO2)CO2), K3(CO2R(CO2)CO2), Rb3(CO2R(CO2)CO2), Cs3(CO2R(CO2)CO2), Mg3(CO2R(CO2)CO2)2, Ca3(CO2R(CO2)CO2)2, Sr3(CO2R(CO2)CO2)2, Ba3(CO2R(CO2)CO2)2, (NH4)3(CO2R(CO2)CO2), де CO2R(CO2)CO2 вибирають з цитрату, ізоцитрату та аконітату; метиламіну, етиламіну, пропіламіну, бутиламіну, пентиламіну, гексиламіну, циклогексиламіну, аніліну; та R4NOH, де R вибирають з метилу, етилу, пропілу, бутилу. 8. Спосіб за будь-яким з пп. 1-7, в якому каталізатор може бути гомогенним або гетерогенним. 9. Спосіб за будь-яким з пп. 2-8, в якому ефективне молярне співвідношення, основа OH :кислота, знаходиться в інтервалі між 0,001-2:1. 10. Спосіб за будь-яким з пп. 1-9, в якому продукт, що являє собою метакрилову кислоту, естерифікують з утворенням її естеру. 11. Спосіб одержання полімерів або співполімерів метакрилової кислоти або естерів метакрилової кислоти, що включає такі стадії: (і) одержання метакрилової кислоти згідно з будь-яким з пп. 1-9; (іі) необов'язкову естерифікацію метакрилової кислоти, одержаної на стадії (і), з утворенням естеру метакрилової кислоти; (ііі) полімеризацію метакрилової кислоти, одержаної на стадії (і), та/або естеру, одержаного на стадії (іі), необов'язково з одним або декількома співмономерами, з утворенням їх полімерів або співполімерів. 12. Спосіб за п. 11, в якому естер метакрилової кислоти, зазначеної вище стадії (іі), вибирають з С1-С12 алкілового або С2-С12 гідроксіалкілового, гліциділового, ізоборнілового, диметиламіноетилового та трипропіленгліколевого естерів. 13. Спосіб одержання метакрилової кислоти, що включає: одержання джерела вихідної кислоти, вибраної з аконітової, лимонної та/або ізолимонної кислоти; здійснення декарбоксилювання та, якщо необхідно, стадії дегідратації джерела вихідної кислоти шляхом піддавання її джерела у присутності або відсутності основного каталізатора, дії температури менше 350 °C, з одержанням ітаконової, мезаконової та/або цитраконової кислоти; та спосіб згідно з будь-яким з пп. 1-9 з утворенням метакрилової кислоти. 14. Спосіб за будь-яким з пп. 1-13, в якому концентрація реагенту(ів), що являє(ють) собою дикарбонову кислоту, становить щонайменше 0,1 М. 15. Спосіб за будь-яким з пп. 1-14, в якому концентрація каталізатора у реакційній суміші становить щонайменше 0,1 М. 16. Спосіб одержання метакрилової кислоти шляхом каталізованого основою декарбоксилювання щонайменше однієї дикарбонової кислоти, вибраної з ітаконової, цитраконової або мезаконової кислоти або їх суміші, де декарбоксилювання здійснюють в інтервалі температур між 240 та 290 °C і реагент(и), що являє(ють) собою дикарбонову(і) кислоту(и), піддають дії умов реакції протягом часу щонайменше 80 секунд в присутності основного каталізатора. 16 UA 110121 C2 Комп’ютерна верстка Л. Литвиненко Державна служба інтелектуальної власності України, вул. Василя Липківського, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 17
ДивитисяДодаткова інформація
Назва патенту англійськоюNormal;heading 1;heading 2;heading 3;process for the production of methacrylic acid and its derivatives and polymers produced therefrom
Автори англійськоюJohnson, David William, Eastham, Graham Ronald, Poliakoff, Martyn, Huddle, Thomas Andrew
Назва патенту російськоюСпособ получения метакриловой кислоты и ее производных и полимеры, полученные из них
Автори російськоюДжонсон Дэвид Вильям, Истхем Грэхем Рональд, Поляков Мартин, Хаддл Томас Эндрю
МПК / Мітки
МПК: C07C 51/38, C08L 33/06, C07C 57/04
Мітки: похідних, спосіб, метакрилової, полімери, кислоти, них, одержані, одержання
Код посилання
<a href="https://ua.patents.su/19-110121-sposib-oderzhannya-metakrilovo-kisloti-ta-pokhidnikh-ta-polimeri-oderzhani-z-nikh.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання метакрилової кислоти та її похідних та полімери, одержані з них</a>
Спосіб одержання акрилової та метакрилової кислоти

Номер патенту: 107815
Опубліковано: 25.02.2015
Автори: Істхем Грехем Рональд, Хаддл Томас Ендрю, Джонсон Девід Вільям, Поляков Мартін
МПК: C07C 69/54, C07C 57/04, C07C 67/08, C07C 51/38
Мітки: одержання, кислоти, спосіб, акрилової, метакрилової
Формула / Реферат:
1. Спосіб одержання сполуки формули (і): , формула (і)в якій R = Н або СН3,де спосіб включає піддавання джерела сполуки формули (іі) реакції, в умовах температури і тиску: , формула (іі)в якій R має значення, як визначено вище,де, коли R = СН3, джерело сполуки...
Спосіб одержання гетероарилфенілборонової кислоти як проміжної сполуки для одержання похідних фенілпіридинсульфонаміду та сполуки, одержані за цим способом

Номер патенту: 83418
Опубліковано: 10.07.2008
Автори: Хоуган Філіп Джон, Меудт Андреас, Батлін Роджер Джон, Батлін Маргарет Енн
МПК: C07F 5/00, C07D 401/14
Мітки: проміжної, одержані, цим, способом, похідних, гетероарилфенілборонової, сполуки, спосіб, одержання, фенілпіридинсульфонаміду, кислоти
Формула / Реферат:
1. Спосіб одержання сполуки формули Іу якій,Х1 вибирають із О, NR1 або S; іХ2 вибирають із СН або N;де R1 являє собою азотозахисну групу,в якому проводять: послідовну взаємодію сполуки формули IIз(і) метил- або необов'язково заміщеним...
Спосіб експериментальної терапії дисфункції слинних залоз при дії на органи ротової порожнини метилового ефіру метакрилової кислоти

Номер патенту: 101142
Опубліковано: 25.08.2015
Автори: Міщенко Артур Володимирович, Соловйова Наталія Веніамінівна, Нагорняк Іван Васильович, Костенко Віталій Олександрович, Денисенко Софія Валеріївна
МПК: A61K 47/18, A61K 6/00
Мітки: кислоти, залоз, ротової, слинних, дисфункції, метакрилової, терапії, дії, метилового, порожнини, ефіру, експериментальної, спосіб, органі
Формула / Реферат:
Спосіб експериментальної терапії дисфункції слинних залоз при дії на органи ротової порожнини метилового ефіру метакрилової кислоти, що включає застосування під час моделювання в експерименті на лабораторних тваринах (білих щурах) дисфункції слинних залоз (аплікації 1 % розчину метилового ефіру метакрилової кислоти на слизову оболонку порожнини рота протягом 30 діб) фармакологічних засобів, який відрізняється тим, що як фармакологічні...
Спосіб одержання ефірів циклопропілкарбонової кислоти та сполуки, одержані цим способом

Номер патенту: 79924
Опубліковано: 10.08.2007
Автори: Кларк Адріан, Джоунс Елфін, Мінідіс Анна, Ларссон Ульф
МПК: C07C 67/347, C07C 69/017, C07C 233/58, C07C 69/003, C07C 69/96, C07C 69/743, C07C 61/00, C07C 69/753
Мітки: цим, одержані, сполуки, ефірів, кислоти, циклопропілкарбонової, способом, спосіб, одержання
Формула / Реферат:
1. Спосіб одержання проміжної сполуки формули (І): ,(I)в якій:R представляє феніл, заміщений одним або декількома галогенами;Y представляє OR1, де R1 являє собою лінійний алкіл, розгалужений алкіл, циклоалкіл або заміщений біциклогептил, який включає:взаємодію солі триметилсульфоксонію з твердим гідроксидом металу в диметилсульфоксиді при...
Спосіб одержання похідних карбонової кислоти

Номер патенту: 50739
Опубліковано: 15.11.2002
Автор: Фукс Еберхард
МПК: C07C 231/00, C07D 213/81, C07C 67/22, C07D 213/84, C07C 233/02, C07D 213/80, C07B 61/00, C07C 69/34, C07D 213/79, C07D 213/803
Мітки: спосіб, одержання, карбонової, похідних, кислоти
Формула / Реферат:
1. Спосіб одержання похідних карбонової кислоти загальної формули (І):в якій означають:Х - OR2 або NH2,R1 - алкіл з 1-20 атомами вуглецю, гідроксиалкіл з 1-20 атомами вуглецю, циклоалкіл з 3-12 атомами вуглецю, алкілциклоалкіл з 4-12 атомами вуглецю, циклоалкілалкіл з 4-12 атомами вуглецю, алкілциклоалкілалкіл з 5-20 атомами вуглецю, арил,...
Попередній патент: Вакуумно-дугове джерело плазми
Наступний патент: Біциклічні похідні як анальгетики, фармацевтична композиція на їх основі та спосіб усунення болю
Випадковий патент: Лабораторна установка для насичення особливо щільно сформованої раціонально підібраної суміші