Ліофілізований фармацевтичний препарат, який містить антитіла cetuximab або matuzumab проти рецептора egf, та спосіб його одержання
Номер патенту: 83461
Опубліковано: 25.07.2008
Автори: Крюгєр Людвіг, Мюллєр Роберт, Цобель Ханс-Петер, Малєр Ханнс-Крістіан, Бахманн Крістіане, Мартіні-Марр Ульрікє, Хаас Удо






Формула / Реферат
1. Ліофілізований фармацевтичний препарат моно- або поліклональних
антитіл, що містить цукор, амінокислоту та полісорбат, який відрізняється тим, що
антитілом є Cetuximab або Matuzumab.
2. Ліофілізований фармацевтичний препарат за п. 1, який відрізняється тим, що цукор являє собою моно-, ди- або трисахарид.
3. Ліофілізований фармацевтичний препарат за п. 2, який відрізняється тим, що цукор являє собою сахарозу, мальтозу або трегалозу.
4. Ліофілізований фармацевтичний препарат за будь-яким з пп. 1-3, який відрізняється тим, що амінокислота являє собою основну, кислу або нейтральну амінокислоту.
5. Ліофілізований фармацевтичний препарат за п. 4, який відрізняється тим, що амінокислота являє собою аргінін, лізин або орнітин.
6. Ліофілізований фармацевтичний препарат за будь-яким з пп. 1-5, який відрізняється тим, що полісорбат являє собою складний ефір жирної кислоти поліоксіетиленсорбіту, моноолеат поліоксіетилен(20)сорбіту або монолаурат поліоксіетилен(20)сорбіту.
7. Ліофілізований фармацевтичний препарат за будь-яким з пп. 1-6, який відрізняється тим, що містить ізотонічний агент у концентрації, необхідній для встановлення ізотонічності.
8. Ліофілізований фармацевтичний препарат за п. 7, який відрізняється тим,
що містить хлорид натрію як ізотонічний агент.
9. Водний фармацевтичний препарат моно- або поліклональних антитіл, отриманий шляхом відновлення ліофілізату за будь-яким з пп. 1-8 за допомогою водного розчинника.
10. Водний фармацевтичний препарат за п. 9, який відрізняється тим, що має значення рН 5-8, переважно 6-7,4.
11. Водний фармацевтичний препарат за п. 10, який відрізняється тим, що розчин має значення рН приблизно 7,2.
12. Спосіб одержання ліофілізованого фармацевтичного препарату за будь-яким з пп. 1-8, який відрізняється тим, що готують водний препарат, який містить
Cetuximab або Matuzumab як активний інгредієнт, а також цукор або аміноцукор,
амінокислоту та полісорбат як додаткові агенти, та, якщо це бажано, додаткові
фармацевтичні допоміжні речовини, та після цього розчин піддають ліофілізації.
Текст
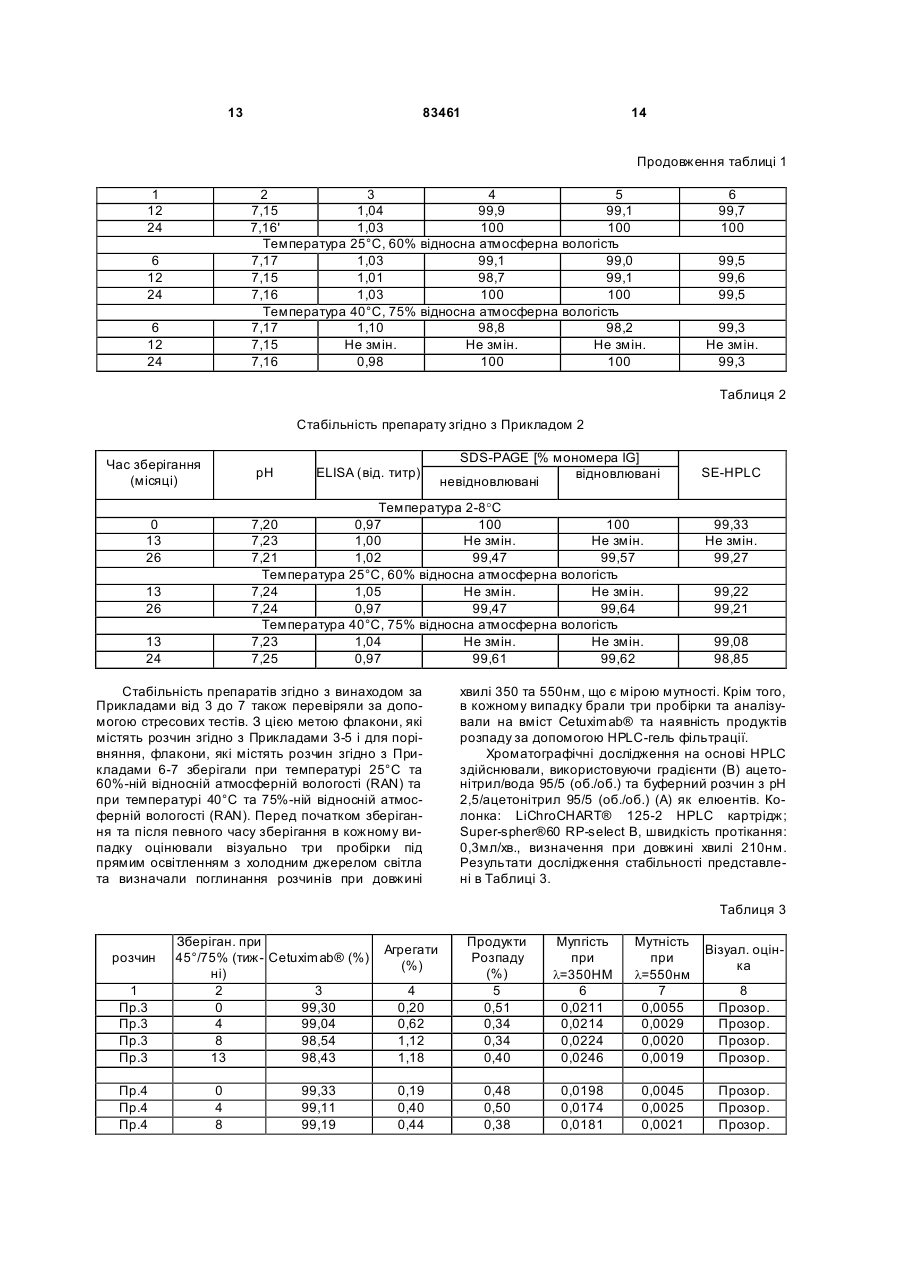
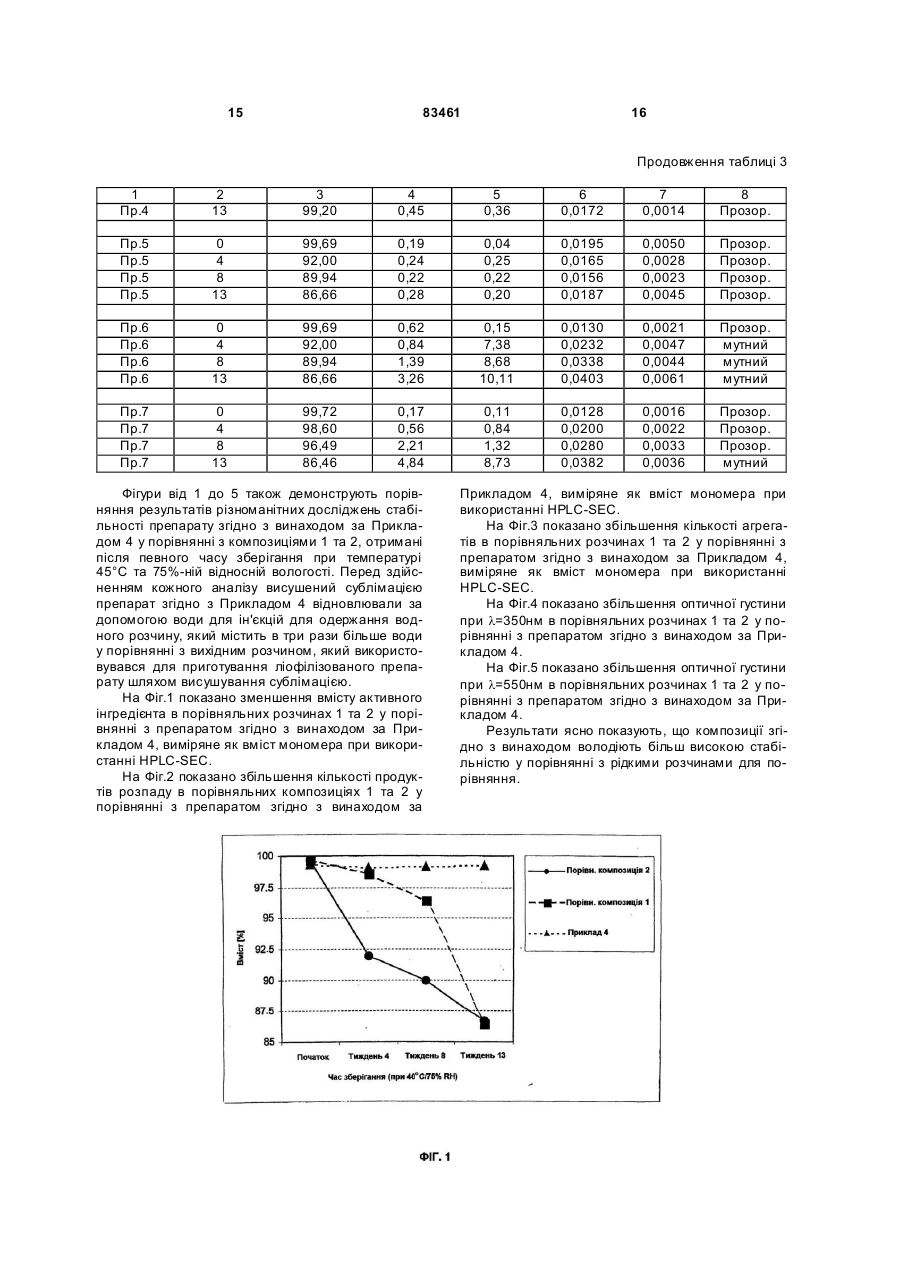
1. Ліофілізований фармацевтичний препарат моно- або поліклональних антитіл, що містить цукор, амінокислоту та полісорбат, який відрізняється тим, що антитілом є Cetuximab або Matuzumab. 2. Ліофілізований фармацевтичний препарат за п. 1, який відрізняється тим, що цукор являє собою моно-, ди- або трисахарид. 3. Ліофілізований фармацевтичний препарат за п. 2, який відрізняється тим, що цукор являє собою сахарозу, мальтозу або трегалозу. 2 (19) 1 3 83461 Дійсний винахід стосується стабільного ліофілізованого препарату, який містить антитіло, яке направлене проти рецептора епідермального фактору росту (EGFR), а також способу його отримання. Різноманітні дослідження в умовах in vitro та in vi vo показали, що блокування EGFR антитілами спричиняє направлений проти пухлин вплив на різних рівнях, наприклад, шляхом пригнічення проліферації клітин, зниження ангіогенезу, індукованого пухлиною, індукції апоптозу ракових клітин і підвищення токсичних ефектів радіотерапії та традиційної хіміотерапії. МАВ с225 (Cetuximab®) клінічно погоджене антитіло, яке зв'язується з рецептором EGF. Cetuximab® є химерним антитілом, варіабельні ділянки якого походять від миші, а постійні ділянки походять від людини, це антитіло вперше було описано Naramura et al., Cancer Immunol. Immunotherapy, 1993,37: 343-349 та у WO 96/40210 A1. МАВ 425 являє собою антитіло, що по ходить від миші, яке надмірно експресується в пухлинних клітинах і направлене проти EGFR, зокрема в клітинах карциноми А431. Воно було гуманізовано, а також були описані його химерні форми, наприклад, в ЕР 0 531 472 A1; Kettleborough et al., Protein Engineering, 1994, 4: 773-783; Bier et al., Cancer Chemother. Pharmacol., 2001,45: 519-524; Bier et al., Cancer Immonol. Immonother., 1998,48: 167-173. EMD 72000 являє собою описане антитіло (h425), яке знаходиться на клінічній фазі І/II, його постійний участок складається з к-ланцюга та g-1 ланцюга людини. Людські анти-EGFR антитіла можуть бути отримані за допомогою технології використання ксеногенних мишей, як описано у WO 91/10741 А1б WO 94/02602 А1 та WO 96/33735 А1. Специфічне антитіло, яке було отримане за допомогою цієї технології і на даний час знаходиться на клінічних випробовуваннях, являє собою ABX-EGF [Abgenix, Crit. Rev. Oncol. Hematol., 2001,38:17-23; Cancer Research 1999,59: 1236-1243]. Інші антитіла, направлені проти EGFR, також були описані, наприклад, в ЕР 0 586 002 В1 та в J. Natl. Cancer Inst, 1993, 85: 27-33 (МАВ 528). Подібно до інших антитіл, EGFR антитіла також використовуються парентерально як розчин для терапевтичного застосування. Особливою проблемою розчинів, що містять такі антитіла, є їх схильність до агрегації та утворення білкових мультимерів. У випадку мультимерів, що розпадаються, це може бути приписано випадковому утворенню внутрішньомолекулярних дисульфідних містків при взаємодії між сусідніми групами. Гідрофобні взаємодії та наступне утворення мультимерів, які не розпадаються, також є можливими. Крім того, можуть також відбуватися реакції дезамідування, які в подальшому призводять до реакцій розкладу білків. Описані реакції денатурації виникають зокрема підчас збереження при підвищеній температурі або підчас напруги при зрушенні, які виникають, наприклад, при транспортуванні. В загальному випадку рідкі препарати, таким чи 4 ном, мають більш низьку придатність як лікарські форми для широкого застосування. Звичайний процес, направлений на стабілізацію антитіл, являє собою висушування сублімацією розчинів, які містять антитіла і допоміжні речовини. Видалення» води знижує утворення продуктів розпаду і агрегатів [Hsu et al., Dev. Biol. Stand., 1991, 74: 255-267 та Pical et al., Dev. Biol. Stand., 1991,74: 21-27]. WO 93/00807 A1 описує ліофілізовані препарати білків, які з метою стабілізації містять поліетиленгліколі і цукор. Однак поліетиленгліколі є сумнівними з токсикологічної точки зору і, таким чином, їх, по можливості, слід уникати в складі лікарських засобів, якщо вони призначені для парентерального введення. WO 98/22136 А2 розкриває ліофілізований препарат, який містить антитіло, аміноцукор, амінокислоту та поверхнево активну речовину. Не дивлячись на те, що препарат заявлений для антитіл взагалі, тільки препарати, розкриті як робочі приклади, є такими, що включають моноклональні антитіла, направлені проти вірусу гепатиту В (АК HBV), при цьому в кожному випадку препарат містить антитіло проти Lселектину (анти-L-селектин) та антитіло проти рецептора неаврального фактора росту L (антиLNGFR). Тоді як препарати, що містять АК HBV та анти-L-селектин були приготовані із розчинів, які мають максимальну концентрацію антитіла 8мг/мл та 7мг/мл, відповідно, препарат, який містить антитіло, направлене проти фактора росту антиLNGFR, готували при використанні розчину, який містив лише 0,25мг/мл антитіла, але щодо решти мав ідентичний якісний та кількісний склад допоміжних агентів. Не дивлячись на те, що препарат, який містить анти-LNGFR, таким чином, має більш ніж в двадцять разів менший вміст антитіла, і відповідно більш низький вміст продуктів розпаду, які можна було би очікувати не було надано ніяких даних про їх стабільність, на відміну від препаратів, які містять інші антитіла. Задачею дійсного винаходу є одержання стабілізованого препарату антитіл, направлених проти EGFR. Препарат не повинен містити будь-яких токсично неприйнятних допоміжних агентів, повинен бути стабільним протягом відносно довгого періоду часу в умовах підвищеної напруги, таких, як висока температура та атмосферна вологість, повинен відновлюватись за допомогою водного розчинника для одержання готового до вживання розчину з високим вмістом активного інгредієнта. Несподівано було виявлено, що препарат, який відповідає цим вимогам, отримують шляхом висушування сублімацією водного розчину, який, крім цих антитіл до EGFR, також містить цукор або аміноцукор, амінокислоту та поверхнево активну речовину. Дійсний винахід, таким чином, стосується стабільних ліофілізованих препаратів моно- або поліклональних антитіл, при цьому вказані препарати містять цукор або аміноцукор, амінокислоту та поверхнево активну речовину і характеризується тим, що антитіло є антитілом, направленим проти рецептора епідермального фактора росту 5 83461 (EGFR). Антитіло, яке може міститися, являє собою будь-яке антитіло, яке направлене проти епідермального фактора росту, зокрема мишачі гуманізовані або химерні антитіла, згадані на початку, та антитіла до EGFR, які були приготовані і можуть бути приготовані шляхом вказаної технології на основі ксеногенних мишей. Антитіло до EGFR, яке присутнє в препараті згідно з винаходом, переважно являє собою Cetuximab® або EMD 72000, а також мишачі, гуманізовані або химерні аналоги антитіл, які їм відповідають. Особлива перевага віддається препаратам, які містять Cetuximab® або EMD 72000 як антитіло. Препарат згідно з винаходом є фізіологічно добре переносимим, може бути легко приготованим, точно дозованим, а також є стабільним щодо продуктів розпаду та агрегатів при зберіганні та під час повторюваних процесів заморожування та від танення. Він є стабільним при зберіганні протягом періоду, який складає, принаймні, від трьох місяців до одного-двох років при зберіганні в умовах холодильника (2-8°С) і при кімнатній температурі (2327°С, 60%-ній відносній атмосферній вологості (RAH)). Несподівано було виявлено, що препарат згідно з винаходом є стабільним при зберіганні протягом вказаного періоду при підвищених температурах і більш високій відносній атмосферній вологості, наприклад при температурі 40°С та 75%-ній відносній атмосферній вологості. Ліофілізований препарат може бути відновлений простим способом для одержання готового до вживання розчину, який не містить видимих частинок, а саме шляхом додавання водного розчинника, наприклад, води для ін'єкцій або для одержання ізотонічного водного розчину. Відновлений розчин є стабільним протягом періоду часу приблизно 5 днів, але, зокрема, особливо переважно використати його протягом чотирьох днів. Відновлення препарату згідно з винаходом за допомогою водних розчинників переважно дозволяє одержувати препарат розчинів, які містять антитіло та мають значення рН в межах від 5 до 8, переважно таких, які мають значення рН від 6,0 до 77,4, особливо переважним значенням рН є приблизно 7,2, та осмолярність від 250 до 350мОсмол/кг. Відновлений препарат може, таким чином, вводитися безпосередньо внутрішньовенно, внутрішньоартеріально, а також підшкірно, без суттєвого болю. Крім того, препарат може також додаватися до розчинів для ін фузії, таких, як, наприклад, розчин глюкози, ізотонічний фізіологічний розчин або розчин Зінгера, який також може містити додаткові інгредієнти, так, що дозволяє вводити відносно великі кількості активного інгредієнта. Згідно з переважним втіленням винаходу ліофілізований фармацевтичний препарат суттєво складається з антитіла, цукру та аміноцукру, амінокислоти, буфера та поверхнево активної речовини. Препарат згідно з винаходом дозволяє одержувати препарат розчинів антитіла, який відповідає за своєю концентрацією клінічним потребам. Перевага віддається розчинам антитіла, які мають концентрацію антитіла від приблизно 0,5 до 25мг/мл, особливо переважно від 5 до 20мг/мл, дуже переважно від 10 до 15мг/мл. Препарат згід 6 но з винаходом, таким чином, дозволяє одержувати препарати, готові для використання, які мають суттєво більш високу концентрацію, ніж описано для препаратів згідно з WO 98/22136 Ф2. Цукор, який використовується в препараті згідно з винаходом, може являти собою моно-, диабо трисахариди. Прикладом моносахаридів, які можуть бути придатними, є глюкоза, маноза, галактоза, фруктоза та сорбоза, прикладами дисахаридів, які можуть бути придатними, є сахароза, лактоза, мальтоза та трегалоза, а прикладом трисахариду, який може бути придатним, є рафіноза. Перевага віддається сахарозі, лактозі, мальтозі і трегалозі, особливо переважною є сахароза. Також можливою є присутність аміноцукрів, тобто моносахаридів, які містять первинну, вторинну або третинну аміногрупу або ацильовану аміногрупу (-NH-CO-R) замість гідроксильної групи. Для цілей дійсного винаходу особлива перевага віддається глюкозаміну, N-метилглюкозаміну, галактозаміну та нейраміновій кислоті. Цукор/аміноцукор міститься в препараті згідно з винаходом в такій кількості, щоб він був присутній в заключному розчині після відновлення за допомогою запропонованого об'єму розчинника в концентрації від приблизно 1 до 200мг/мл. Цукор переважно міститься у відновленому розчині в концентрації від 30 до 65мг/мл. Придатні амінокислоти, які використовуються згідно з винаходом, являють собою основні амінокислоти, такі, як, наприклад, аргінін, гістидин, орнітин, лізин, серед інших, при цьому амінокислоти бажано використовувати у вигляді їх неорганічних солей (переважно в формі солей хлористоводневої кислоти, тобто, в формі гідрохлоридів). У випадку, коли використовуються вільні амінокислоти, бажане значення рН встановлюють шля хом додавання придатних фізіологічно переносимих буферних речовин, таких, як, наприклад, органічні або неорганічні кислоти, такі, як лимонна кислота або фосфорна кислота, сірчана кислота, оцтова кислота, мурашина кислота та або їх солі. Перевага віддається цитратам та фосфатам, з якими одержують особливо стабільні ліофілізати. Переважними амінокислотами є аргінін, лізин та орнітин. Крім того, також є можливим використання кислих амінокислот, таких, як, наприклад, глютамінова кислота та аспарагінова кислота, або нейтральних амінокислот, таких,-як, наприклад, ізолейцин, лейцин та аланін, або ароматичних амінокислот, таких, як, наприклад, фенілаланін, тирозин або триптофан. Вміст амінокислот в препараті згідно з винаходом складає від 1 до 100мг/мл, переважно від 1 до 50мг/мл, особливо переважно 3-30мг/мл (базуючись в кожному випадку на відновленому розчині). Використовуваними поверхнево активними речовинами є всі сурфактанти, які зазвичай використовуються в фармацевтичних препаратах, преважно полісорбати і полімери поліоксиетиленуполіоксипропілену. Особлива перевага віддається монолаурату поліоксиетилен(20)сорбіту та моноолеату поліоксиетилен(20)сорбіту. Згідно з винаходом препарат містить від 0,001 до 1мас.%, переважно від 0,005 до 0,1мас.% і особливо 7 83461 переважно приблизно 0,01мас.% (базуючись в кожному випадку на відновленому розчині). Якщо препарат згідно з винаходом містить буферні розчини, то вони повинні, в основному, бути фізіологічно прийнятними речовинами, які прийнятні для встановлення бажаного значення рН. Кількість буферних речовин вибирають таким чином, щоб після відновлення ліофілізованного препарату, наприклад, за допомогою води для ін'єкцій, отриманий водний розчин мав буферну концентрацію від 5ммол/л до 20ммол/л, переважно приблизно 10ммол/л. Переважними буферними розчинами є цитратний буферний розчин або фосфатний буферний розчин. Придатні фосфатні буферні розчини являють собою розчини солей фосфорної кислоти моно- та/або динатрію і -калію, таких, як гідрофосфат динатрію або дигідрофосфат калію, а також суміші солей натрію і калію, такі, як, наприклад, суміші гідрофосфату динатрію та дигідрофосфату калію. Якщо відновлений розчин не є ізотонічним внаслідок осмотичних властивостей антитіла, то допоміжні речовини, які використовуються для стабілізації, ізотонічний агент, переважно фізіологічно прийнятна сіль, така, як, наприклад, хлорид натрію або хлорид калію, або фізіологічно переносимий поліол, такий, як, наприклад, глюкоза або гліцерин, можуть також бути присутніми в концентрації, необхідній для встановлення ізотонічності. Крім того, ліофілізати згідно з винаходом можуть містити фізіологічно прийнятні допоміжні речовини, такі, як, наприклад, антиоксиданти, такі, як аскорбінова кислота або глютатіон, консерванти, такі, як фенол, крезол, метил- або пропілпарабен, хлорбутанол, тіомерсал або хлорид бензалконію, поліетиленгліколі (PEG), такі, як PEG 3000, 3350, 4000 або 6000, або циклодекстрини, такі, як гідроксипропіл-b-циклодекстрин, сульфобутилетил-b-циклодекстрин або gциклодекстрин. Препарат згідно з винаходом може бути приготований шляхом одержання водного препарату, що містить Cetuximab® або EMD 72000 як активний інгредієнт, а також цукор, аміноцукор, амінокислоту та поверхнево активну речовину як добавки, та, якщо це бажано, фармацевтичні допоміжні речовини, з наступною ліофілізацією розчину. Водний препарат може бути приготований шляхом додавання вказаних допоміжних агентів до розчину, який містить Cetuximab® або EMD 72000. З цією метою визначені кількості маточних розчинів, які містять вказані допоміжні агенти в певних концентраціях, переважно додають до розчину, який має певну концентрацію Cetuximab® або EMD 72000, як одержано з його препарату, та суміш, якщо це є бажаним, розводять до попередньо розрахованої концентрації водою. Альтернативно, допоміжні речовини можуть також додаватись як тверді речовини до початкових розчинів, які містять Cetuximab®. Якщо Cetuximab® або EMD 72000 знаходяться в твердій фазі, наприклад, у формі ліофілізату, то препарат згідно з винаходом може бути приготований спочатку розчиненням відповідних антитіл у воді або у водному розчині, який містить однин або більше допоміжних агентів, з наступним додаванням необхідних в кожному 8 випадку кількостей маточних розчинів, що містять додаткові допоміжні речовини, додаткових допоміжних речовин в твердій формі та/або води. Cetuximab® або EMD 72000 також можуть переважно бути розчиненими безпосередньо в розчині, який містить всі додаткові допоміжні речовини. Один або більше допоміжних агентів, які містяться в препараті згідно з винаходом, можуть переважно бути доданими під час або в кінці процесу приготування препарату конкретного антитіла до EGFR. Це переважно виконують шляхом розчинення Cetuximab® або EMD 72000 безпосередньо у водному розчині, який містить одну, більше, ніж одну, або всі допоміжні речовини на заключному етапі очистки, який здійснюють після його приготування. Для цього, щоб отримати препарат, відповідний(і) додатковий(і) інгредієнт(и), який(і) необхідний(і), додають в меншій кількості в кожному випадку та/або не додають взагалі. Особливо переважно для відповідного інгредієнта розчиняти його безпосередньо у водному розчині, який містить всі додаткові допоміжні речовини на кінцевому етапі очистки, яку виконують після його приготування, безпосередньо одержуючи розчин, який піддають ліофілізації. Розчин, який містить відповідне антитіло і допоміжні речовини, доводять до значення рН від 5 до 8, стерильно фільтрують і піддають висушуванню сублімацією. Приклади пояснюють винахід, але не обмежують його. Якщо використовують 10ммол/л буфера на основі фосфату натрію або фосфату калію зі значенням рН 7,2, то він містить 2,07г/л 7-гідрату фосфату динатрію та 0,31г/л моногідрату дигідрофосфату натрію або 1,220г/л гідрофосфату дикалію та 0,4050г/л дигідрофосфату калію. Приклад 1 Ліофілізат отримували з водного розчину, який містить: 10мг/мл EMD 72000, 10ммол/л буфера на основі фосфату калі з рН 7,2, 17мол/л аргініну, 3мас.% сахарози, 0,01мас.% монолауратуполіоксиетилен(20)сорбіту, 0,4мас.% PEG 6000. Приготування здійснювали шляхом перемішування певних об'ємів водних розчинів, які містять відповідні допоміжні речовини у визначених концентраціях. Використовували наступні розчини: Розчин А (розчин активного інгредієнта), який містить: 20мг/мл EMD 72000, 10ммол/л буфера на основі фосфа ту калію з рН 7,2. Розчин В (буферно-сольовий розчин): 10ммол/л буфера на основі фосфа ту калію з рН 7,2, 6мас.% сахарози, 0,02мас.% монолаурату поліоксиетилен(20)сорбіту, 34ммол/л аргініну, 0,8мас.% поліетиленгліколю 6000. 9 Для приготування препарату згідно з винаходом об'єднували один з одним рівні об'єми розчину А та розчину В. Приготований розчин стерильно фільтрували перед запаковуванням. Кожний флакон наповнювали 2мл розчину. Після цього флакон частково герметизували за допомогою пробок та піддавали ліофілізації. Після висушування сублімацією флакони герметично закривали. Приклад 2 Ліофілізат одержували з водного розчину, який містить: 10мг/мл EMD 72000, 10ммол/л буфера на основі фосфату калі з рН 7,2, 14ммол/л аргініну, 3мас.% сахарози, 0,01мас.% монолаурату поліоксиетилен(20)сорбіту. Приготування здійснювали шляхом перемішування певних об'ємів водних розчинів, які містять відповідні допоміжні речовини у визначених концентраціях. Використовували наступні розчини: Розчин А (розчин активного інгредієнта), який містить: 20мг/мл EMD 72000, 10ммол/л буфера на основі фосфа ту калію з рН7,2. Розчин В (буферно-сольовий розчин): 10ммол/л буфера на основі фосфа ту калію з рН 7,2, 6мас.% сахарози, 0,02мас.% монолаурату поліоксиетилен(20)сорбіту, 34ммол/л аргініну. Приготований розчин стерильно фільтрували перед запаковуванням. Кожний флакон наповнювали 20мл розчину. Після цього флакон частково герметизували за допомогою пробок та піддавали ліофілізації. Після висушування сублімацією флакони герметично закривали. Приклад 3 Ліофілізат одержували з водного розчину, який містить: 15мг/мл Cetuximab®, 5ммол/л цитрату, 100ммол/л аргініну, 4мас.% манітолу, 0,01мас.% моноолеату поліоксиетилен(20)сорбіту. Приготування здійснювали шляхом перемішування певних об'ємів водних розчинів, які містять відповідні допоміжні речовини у визначених концентраціях. Використовували наступні розчини: Розчин А (розчин активного інгредієнта), який містить: 19мг/мл Cetuximab®, 5ммол/л цитрату, 127ммол/л аргініну, 0,01мас.% моноолеату поліоксиетилен(20)сорбіту. Розчин В (буферно-сольовий розчин): 5ммол/л цитрату, 19,05мас.% манітолу, 83461 10 0,01мас.% моноолеату поліоксиетилен(20)сорбіту. Для приготування препарату згідно з винаходом 7,9мл розчину А та 2,1мл розчину В об'єднували один з одним. Приготований розчин фільтрували перед запаковуванням, використовуючи стерильний фільтр. Кожний флакон наповнювали 2мл розчину за допомогою піпетки. Після цього флакон частково герметизували за допомогою пробок та піддавали ліофілізації. Після висушування сублімацією флакони герметично закривали пробками. Приклад 4 Ліофілізат одержували з водного розчину, який містить: 15мг/мл Cetuximab®, 5ммол/л цитрату, 100ммол/л аргініну, 1,5мас.% сахарози, 0,01мас.% монолаурату поліоксиетилен(20)сорбіту. Приготування здійснювали шляхом перемішування певних об'ємів водних розчинів, які містять відповідні допоміжні речовини у визначених концентраціях. Використовували наступні розчини: Розчин А (розчин активного інгредієнта), який містить: 19мг/мл Cetuximab®, 5ммол/л цитрату, 127ммол/л аргініну, 0,01мас.% моноолеату поліоксиетилен(20)сорбіту. Розчин В (буферно-сольовий розчин): 5ммол/л цитрату, 7,1мас.% сахарози, 0,01мас.% моноолеату поліоксиетилен(20)сорбіту. Для приготування препарату згідно з винаходом 7,9мл розчину А та 2,1мл розчину В об'єднували один з одним. Приготований розчин фільтрували перед запаковуванням, використовуючи стерильний фільтр. Кожний флакон наповнювали 2мл розчину за допомогою піпетки. Після цього флакон частково герметизували за допомогою пробок та піддавали ліофілізації. Після висушування сублімацією флакони герметично закривали пробками. Приклад 5 Ліофілізат одержували з водного розчину, який містить: 15мг/мл Cetuximab®, 10ммол/л буфера на основі фосфа ту калію з рН 7,2, 14ммол/л гідро хлориду L-аргініну, 88ммол/л сахарози, 0,01мас.% монолаурату поліоксиетилен(20)сорбіту, значення рН доводили до 7,5, використовуючи фосфорну кислоту. Приготування здійснювали шляхом перемішування певних об'ємів водних розчинів, які містять відповідні допоміжні речовини у визначених концентраціях. Використовували наступні розчини: Розчин А (розчин активного інгредієнта), який містить: 25мг/мл Cetuximab®, 11 83461 10ммол/л буфера на основі фосфа ту калію з рН 7,2, воду для ін'єкцій. Розчин В (буферно-сольовий розчин): 10ммол/л буфера на основі фосфа ту калію з рН 7,2, 35,6ммол/л гідрохлориду L-аргініну, 219ммол/л сахарози, 0,025мас.% монолаурату поліоксиетилен(20)сорбіту, значення рН доводили до 7,5, використовуючи фосфорну кислоту. Для приготування препарату згідно з винаходом 6 мл розчину А та 4мл розчину В об'єднували один з одним. Приготований розчин фільтрували перед запаковуванням, використовуючи стерильний фільтр. Кожний флакон наповнювали 2мл розчину за допомогою піпетки. Після цього флакон частково герметизували за допомогою пробок та піддавали ліофілізації. Після висушування сублімацією флакони герметично закривали пробками. Приклад 6 (препарат для порівняння 1) Водний розчин, який містить: 5мг/мл Cetuximab®, 10ммол/л буфера на основі фосфа ту калію з рН 7,2, 145ммол/л хлориду натрію. Розчин отримували шляхом елюювання активного інгредієнта із колонки при використанні розчину В на кінцевому етапі хроматографічної очистки активного інгредієнта, який здійснювали після приготування. Розчин В (буферно-сольовий): відповідає розчину, але не містить активного інгредієнта. Для приготування препарату для порівняння 10 мл кожного з розчинів А та В об'єднували один з одним. Приклад 7 (препарат для порівняння 2) Водний розчин, який містить: 5мг/мл Cetuximab®, 10ммол/л буфера на основі фосфа ту калію з рН 7,2, 45ммол/л хлориду натрію, 0,01мас.% моноолеату поліоксиетилен(20)сорбіту. Приготування здійснювали шляхом перемішування певних об'ємів водних розчинів, які містять відповідні допоміжні речовини у визначених концентраціях. Використовували наступні розчини: Розчин А (розчин активного інгредієнта), який містить: 9,7мг/мл Cetuximab®, 12 10ммол/л буфера на основі фосфа ту калію з рН 7,2, 145ммол/л хлориду натрію. Розчин отримували шляхом елюювання активного інгредієнта із колонки при використанні розчину В на кінцевому етапі хроматографічної очистки активного інгредієнта, який здійснювали після приготування. Розчин В (буферно-сольовий): відповідає розчину, але не містить активного інгредієнта. Розчин С (розчин складного ефіру жирної кислоти поліоксиетиленсорбіту): відповідає розчину В, але додатково містить 1мас.% моно олеату поліоксиетилен(20)сорбіту. Для приготування препарату для порівняння 10мл розчину А та 9,8мл розчину В, а також 0,2мл розчину С об'єднували один з одним. Приготований розчин фільтрували перед запаковуванням, використовуючи стерильний фільтр. Кожний флакон наповнювали 2мл розчину за допомогою піпетки. Після цього флакони герметично закривали пробками. Дослідження стабільності препаратів Стабільність препаратів згідно з винаходом, отриманих в Прикладах 1 та 2, досліджували в умовах напруги. Для цього приготовані ліофілізати зберігали при температурі 40°С та відносні атмосферній вологості (RAN) 75%. Препарати зберігали до певного моменту часу та аналізували, використовуючи придатні аналітичні способи. Можлива нестабільність препаратів антитіл проявлялась, головним чином, утворенням агрегатів та утворенням продуктів розпаду. Продукти розпаду переважно визначали за допомогою електрофорезу в гелі (SDS-PAGE) та ізоелектричного фокусування (IEF), в той час, як візуальне спостереження та вимірювання мутності використовували для визначення видимих агрегатів, витіснювальну хроматографію на основі розмірів (HPLC-SEC) здійснювали для визначення розчинних агрегатів. Подібно до цього, тест ELIS А (твердо фазовий імуноферментний аналіз), використовуваний для оцінки препаратів, служив для перевірки цілісності та здатності зв'язуватися з рецептором. Результати, представлені в Таблицях 1 та 2, ясно підтверджують кількісний та якісний склад приготованих препаратів навіть при підвищених температурах зберігання та підвищеній відносній атмосферній вологості (40°С/75% RAN). Таблиця 1 Стабільність препарату згідно з Прикладом 1 Час зберігання (місяці) 1 0 6 рН 2 7,11 7,14 SDS-PAGE [% мономера IG] невідновлювані відновлювані 3 4 5 Температура 2-8°С 1,00 100 100 1,01 99,3 100 ELISA (від. титр) SE-HPLC 6 100 99,6 13 83461 14 Продовження таблиці 1 1 12 24 2 3 4 5 7,15 1,04 99,9 99,1 7,16' 1,03 100 100 Температура 25°С, 60% відносна атмосферна вологість 7,17 1,03 99,1 99,0 7,15 1,01 98,7 99,1 7,16 1,03 100 100 Температура 40°С, 75% відносна атмосферна вологість 7,17 1,10 98,8 98,2 7,15 Не змін. Не змін. Не змін. 7,16 0,98 100 100 6 12 24 6 12 24 6 99,7 100 99,5 99,6 99,5 99,3 Не змін. 99,3 Таблиця 2 Стабільність препарату згідно з Прикладом 2 Час зберігання (місяці) рН ELISA (від. титр) SDS-PAGE [% мономера IG] відновлювані невідновлювані Температура 2-8°С 7,20 0,97 100 100 7,23 1,00 Не змін. Не змін. 7,21 1,02 99,47 99,57 Температура 25°С, 60% відносна атмосферна вологість 7,24 1,05 Не змін. Не змін. 7,24 0,97 99,47 99,64 Температура 40°С, 75% відносна атмосферна вологість 7,23 1,04 Не змін. Не змін. 7,25 0,97 99,61 99,62 0 13 26 13 26 13 24 Стабільність препаратів згідно з винаходом за Прикладами від 3 до 7 також перевіряли за допомогою стресових тестів. З цією метою флакони, які містять розчин згідно з Прикладами 3-5 і для порівняння, флакони, які містять розчин згідно з Прикладами 6-7 зберігали при температурі 25°С та 60%-ній відносній атмосферній вологості (RAN) та при температурі 40°С та 75%-ній відносній атмосферній вологості (RAN). Перед початком зберігання та після певного часу зберігання в кожному випадку оцінювали візуально три пробірки під прямим освітленням з холодним джерелом світла та визначали поглинання розчинів при довжині SE-HPLC 99,33 Не змін. 99,27 99,22 99,21 99,08 98,85 хвилі 350 та 550нм, що є мірою мутності. Крім того, в кожному випадку брали три пробірки та аналізували на вміст Cetuximab® та наявність продуктів розпаду за допомогою HPLC-гель фільтрації. Хроматографічні дослідження на основі HPLC здійснювали, використовуючи градієнти (В) ацетонітрил/вода 95/5 (об./об.) та буферний розчин з рН 2,5/ацетонітрил 95/5 (об./об.) (А) як елюентів. Колонка: LiChroCHART® 125-2 HPLC картрідж; Super-spher®60 RP-select В, швидкість протікання: 0,3мл/хв., визначення при довжині хвилі 210нм. Результати дослідження стабільності представлені в Таблиці 3. Таблиця 3 розчин 1 Пр.3 Пр.3 Пр.3 Пр.3 Пр.4 Пр.4 Пр.4 Зберіган. при 45°/75% (тиж- Cetuximab® (%) ні) 2 3 0 99,30 4 99,04 8 98,54 13 98,43 0 4 8 99,33 99,11 99,19 4 0,200,62 1,12 1,18 Продукти Розпаду (%) 5 0,51 0,34 0,34 0,40 Мупгість при l=350ΗΜ 6 0,0211 0,0214 0,0224 0,0246 Мутність при l=550нм 7 0,0055 0,0029 0,0020 0,0019 0,19 0,40 0,44 0,48 0,50 0,38 0,0198 0,0174 0,0181 0,0045 0,0025 0,0021 Агрегати (%) Візуал. оцінка 8 Прозор. Прозор. Прозор. Прозор. Прозор. Прозор. Прозор. 15 83461 16 Продовження таблиці 3 1 Пр.4 2 13 3 99,20 4 0,45 5 0,36 6 0,0172 7 0,0014 8 Прозор. Пр.5 Пр.5 Пр.5 Пр.5 0 4 8 13 99,69 92,00 89,94 86,66 0,19 0,24 0,22 0,28 0,04 0,25 0,22 0,20 0,0195 0,0165 0,0156 0,0187 0,0050 0,0028 0,0023 0,0045 Прозор. Прозор. Прозор. Прозор. Пр.6 Пр.6 Пр.6 Пр.6 0 4 8 13 99,69 92,00 89,94 86,66 0,62 0,84 1,39 3,26 0,15 7,38 8,68 10,11 0,0130 0,0232 0,0338 0,0403 0,0021 0,0047 0,0044 0,0061 Прозор. мутний мутний мутний Пр.7 Пр.7 Пр.7 Пр.7 0 4 8 13 99,72 98,60 96,49 86,46 0,17 0,56 2,21 4,84 0,11 0,84 1,32 8,73 0,0128 0,0200 0,0280 0,0382 0,0016 0,0022 0,0033 0,0036 Прозор. Прозор. Прозор. мутний Фігури від 1 до 5 також демонструють порівняння результатів різноманітних досліджень стабільності препарату згідно з винаходом за Прикладом 4 у порівнянні з композиціями 1 та 2, отримані після певного часу зберігання при температурі 45°С та 75%-ній відносній вологості. Перед здійсненням кожного аналізу висушений сублімацією препарат згідно з Прикладом 4 відновлювали за допомогою води для ін'єкцій для одержання водного розчину, який містить в три рази більше води у порівнянні з вихідним розчином, який використовувався для приготування ліофілізованого препарату шляхом висушування сублімацією. На Фіг.1 показано зменшення вмісту активного інгредієнта в порівняльних розчинах 1 та 2 у порівнянні з препаратом згідно з винаходом за Прикладом 4, виміряне як вміст мономера при використанні HPLC-SEC. На Фіг.2 показано збільшення кількості продуктів розпаду в порівняльних композиціях 1 та 2 у порівнянні з препаратом згідно з винаходом за Прикладом 4, виміряне як вміст мономера при використанні HPLC-SEC. На Фіг.3 показано збільшення кількості агрегатів в порівняльних розчинах 1 та 2 у порівнянні з препаратом згідно з винаходом за Прикладом 4, виміряне як вміст мономера при використанні HPLC-SEC. На Фіг.4 показано збільшення оптичної густини при l=350нм в порівняльних розчинах 1 та 2 у порівнянні з препаратом згідно з винаходом за Прикладом 4. На Фіг.5 показано збільшення оптичної густини при l=550нм в порівняльних розчинах 1 та 2 у порівнянні з препаратом згідно з винаходом за Прикладом 4. Результати ясно показують, що композиції згідно з винаходом володіють більш високою стабільністю у порівнянні з рідкими розчинами для порівняння. 17 83461 18 19 Комп’ютерна в ерстка О. Рябко 83461 Підписне 20 Тираж 26 прим. Міністерство осв іт и і науки України Держав ний департамент інтелектуальної в ласності, вул. Урицького, 45, м. Київ , МСП, 03680, Україна ДП “Український інститут промислов ої в ласності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюFreeze-dried pharmaceutical containing cetuximab or matuzumab antibodies against egf receptor and method for its production
Автори англійськоюMahler Hanns-Christian, Zobel Hans-Peter, Mueller Robert, Bachmann Christiane, Haas Udo, Krueger Ludwig, Martini-Marr Ulrike
Назва патенту російськоюЛиофилизованный фармацевтический препарат, содержащий антитела cetuximab или matuzumab против рецептора egf, и способ его получения
Автори російськоюМалер Ханнс-Кристиан, Цобель Ганс-Петер, Мюллер Роберт, Бахманн Кристиане, Хаас Удо, Крюгер Людвиг, Мартини-Марр Ульрике
МПК / Мітки
МПК: A61K 47/34, A61K 47/26, A61K 39/395, A61K 47/18, A61P 35/00
Мітки: ліофілізований, фармацевтичний, рецептора, містить, одержання, препарат, cetuximab, антитіла, спосіб, matuzumab
Код посилання
<a href="https://ua.patents.su/10-83461-liofilizovanijj-farmacevtichnijj-preparat-yakijj-mistit-antitila-cetuximab-abo-matuzumab-proti-receptora-egf-ta-sposib-jjogo-oderzhannya.html" target="_blank" rel="follow" title="База патентів України">Ліофілізований фармацевтичний препарат, який містить антитіла cetuximab або matuzumab проти рецептора egf, та спосіб його одержання</a>
Фармацевтичний препарат, який містить ібупрофен та кодеїн, і спосіб його приготування

Номер патенту: 42781
Опубліковано: 15.11.2001
Автор: Холмс Брайен
МПК: A61K 9/20, A61K 31/19, A61K 31/485
Мітки: фармацевтичний, містить, приготування, препарат, спосіб, кодеїн, ібупрофен
Формула / Реферат:
1. Фармацевтический препарат, содержащий ибупрофен или его фармацевтически приемлемую соль и кодеин или его фармацевтически приемлемую соль в сочетании с фармацевтически приемлемым носителем, где указанный носитель содержит лубрикант, дезинтегрант и разбавитель, отличающийся тем, что этот лубрикант по существу свободен от стеариновой кислоты и ионов стеарата и выбран из гидрогенизированных растительных масел.2. Препарат по п.1,...
Фармацевтичний препарат, який містить вориконазол

Номер патенту: 57083
Опубліковано: 16.06.2003
Автор: Хардінг Валері Деніс
МПК: A61K 47/40
Мітки: фармацевтичний, вориконазол, містить, препарат
Формула / Реферат:
1. Фармацевтичний препарат, який містить вориконазол або його фармацевтично прийнятне похідне та похідне циклодекстрину формули (І),де R1a-g, R2a-g і R3a-g незалежно представляють ОН або О(СH2)4SO3Н,за умови, принаймні, що один з R1a-g представляє O(СН2)4SО3Н або його фармацевтично прийнятну сіль.2. Препарат згідно з п. 1, де середня кількість груп O(СН2)4SО3Н на одну молекулу формули (І) знаходиться в межах...
Фармацевтична композиція, що призначена для одержання порошків або шипучих таблеток, яка містить ефективну кількість ібупрофену, фармацевтичний препарат на її основі та спосіб його одержання

Номер патенту: 27058
Опубліковано: 28.02.2000
Автори: Кордоліані Жан-Франсуа, Брю-Ман'єз Ніколь, Друєн Жан Ів, Товєн Жерар
МПК: A61K 31/19, A61K 9/46
Мітки: композиція, порошків, спосіб, фармацевтична, одержання, містить, призначена, шипучих, фармацевтичний, ібупрофену, препарат, яка, кількість, ефективну, таблеток, основі
Формула / Реферат:
1. Фармацевтическая композиция, предназначенная для получения порошков или шипучих таблеток, содержащая эффективное количество ибупрофена или одной из его фармацевтически приемлемых солей в качестве активного ингредиента, фармацевтически приемлемую систему газирования, содержащую, по крайней мере, один щелочной карбонат и, по крайней мере, одну органическую кислоту, отличающаяся тем, что она дополнительно содержит, по крайней мере, один...
Стабілізований фармацевтичний препарат, який містить гормон росту і гістидин, кристали гормону росту, що містять гістидин, та спосіб їх одержання

Номер патенту: 41502
Опубліковано: 17.09.2001
Автори: Хоелгард Анні Рассінг, Скрівер Ларс, Соренсен Ханс Холмегард
МПК: A61K 38/27
Мітки: фармацевтичний, одержання, гістидин, гормон, кристали, містять, гормону, спосіб, препарат, містить, росту, стабілізований
Формула / Реферат:
1. Фармацевтический препарат, содержащий гормон роста или производное гормона роста и гистидин или производное гистидина, отличающийся тем, что он содержит гистидин или производное гистидина в количестве от 0,1 до 12 мг на 1 мг гормона роста.2. Фармацевтический препарат по п.1, отличающийся тем, что он содержит гормон роста в форме буферного водного раствора гормона роста, забуференного гистидиновым буфером в концентрации...
Фармацевтичний препарат моксифлоксацину та спосіб його одержання

Номер патенту: 72483
Опубліковано: 15.03.2005
Автори: Вайземанн Клаус, Малер Ханс-Фрідріх, Боше Патрік
МПК: A61K 47/28, A61K 9/20, A61K 31/4709, A61P 31/04
Мітки: препарат, одержання, фармацевтичний, спосіб, моксифлоксацину
Формула / Реферат:
1. Фармацевтичний препарат для орального застосування, який містить моксифлоксацин або його сіль і/або гідрат, мінімум один сухий зв'язувальний засіб, мінімум один допоміжний засіб, який сприяє розпаду, і мінімум один засіб, який змазує, який відрізняється тим, що препарат містить від 2,5 % до 25 % лактози.2. Фармацевтичний препарат для орального застосування згідно з п. 1, який відрізняється тим, що одинична доза препарату містить від...
Попередній патент: Редуктор універсальний для тістомісильних машин і мішалок
Наступний патент: Спосіб демонтажу ушкодженої промислової установки, об’єкт укриття ушкодженої промислової установки та спосіб його зведення
Випадковий патент: Спосіб оцінки процесів репарації при протезуванні різними видами імплантатів