Радіомічена сполука для утворення зображення тромбів при радіонуклідній діагностиці, радіофармацевтичний препарат та радіофармацевтична композиція на їх основі та комплект для приготування радіофармацевтичних п
Номер патенту: 32577
Опубліковано: 15.02.2001
Автори: Сворін Мічейл, Ліу Шуанг, Раджопадхі Мілінд, Баретт Джон Ендрю, Деградо Вільям Френк, Едвардс Давід Скот, Моуса Шакер Ахмед, Харріс Томас Давід




Текст
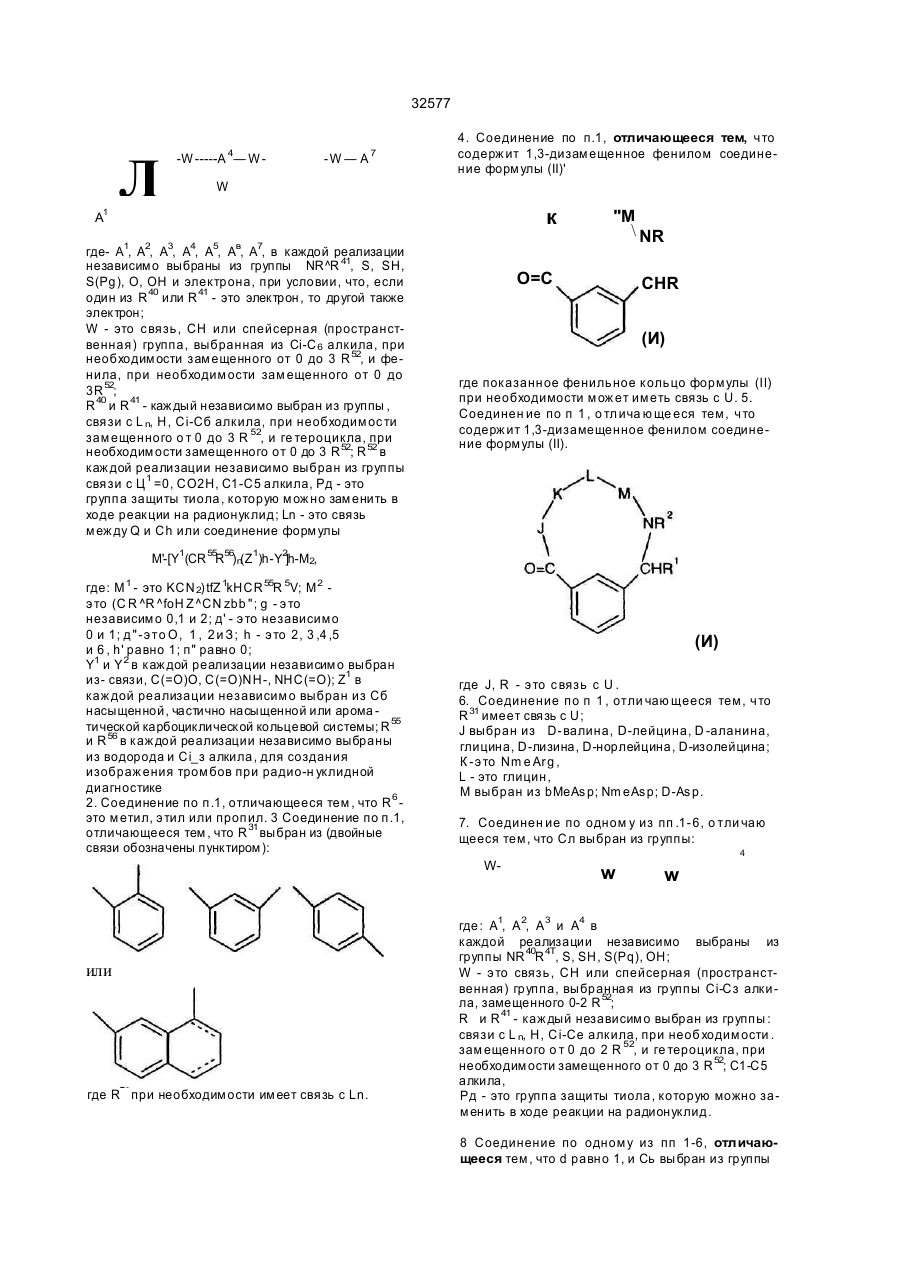
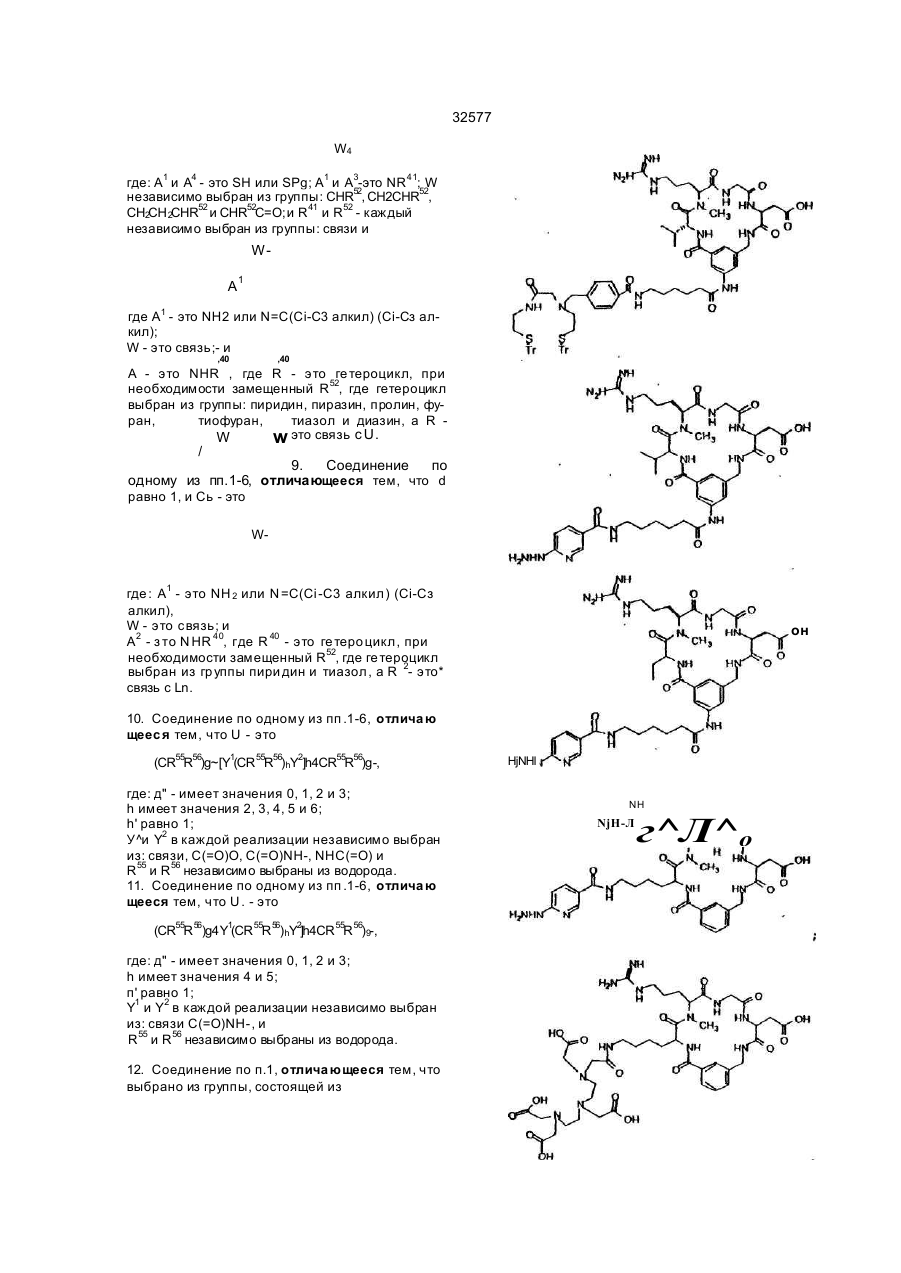
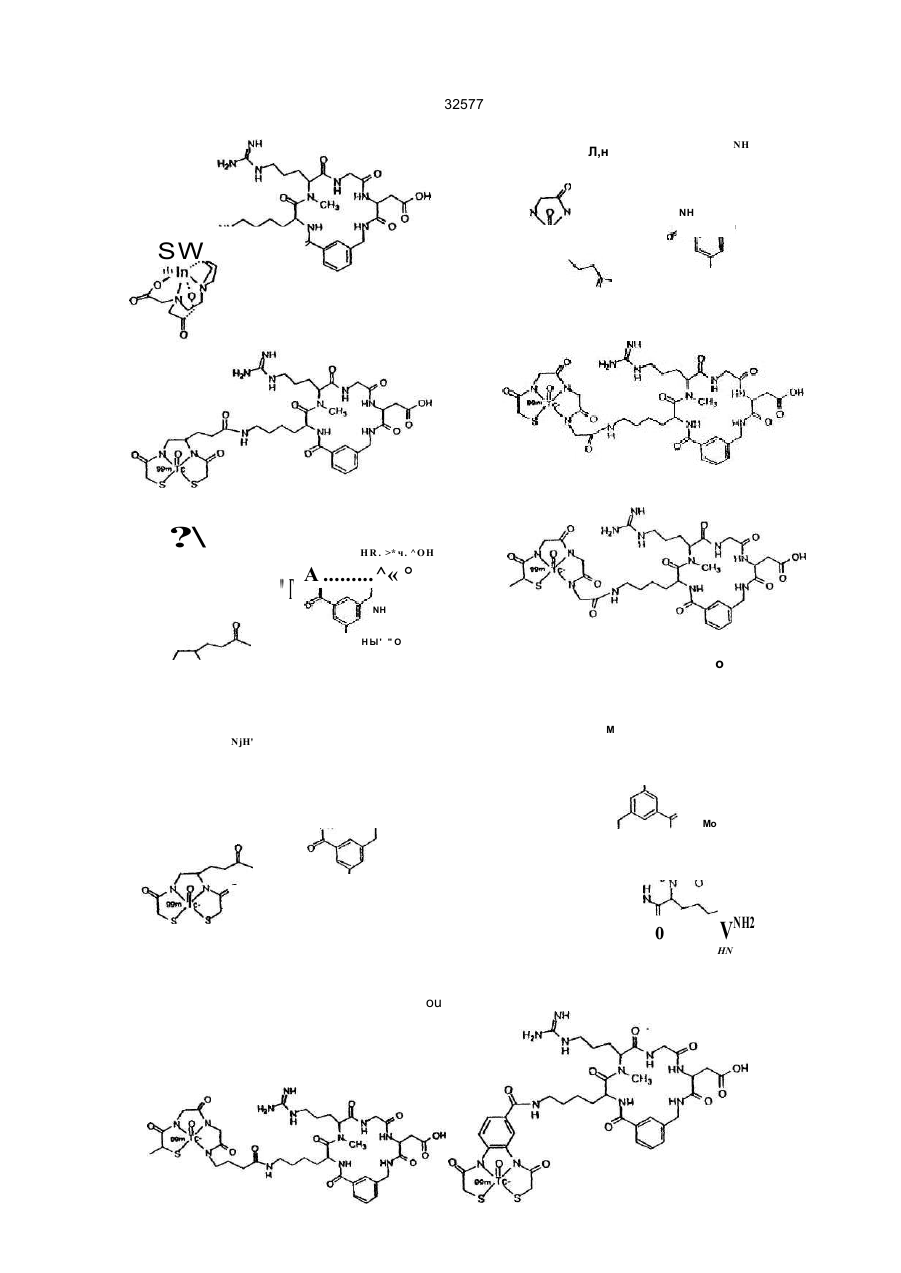
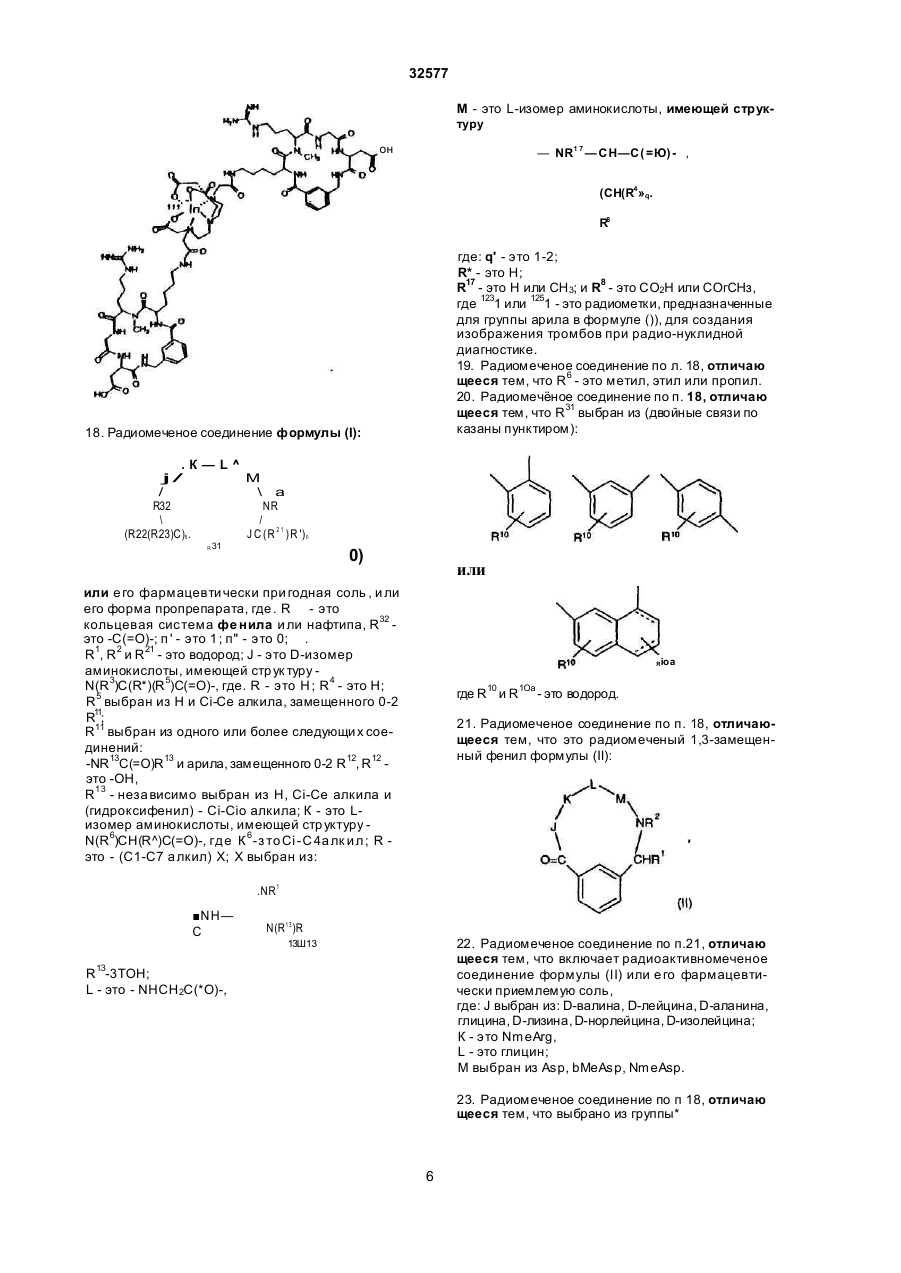
1 Радиомеченое соединение форм улы : (QU)dCh, где d равно 1-2; L n - это связующая группа , Си -. это хелатор м еталла и Q - это соединение форм улы (I) R 5 выбран из Н; Сі~Са алкил, при необходимост и замещенный от 0fl o2 R 11; связь с Ц ; К - э то L-изом ер ам инокислоты, имеющей с тр ук тур у-N(R 6)CH{R 7)C(=O)-( где . R - э то Сі- С 4 а лкил ; R - это (С 1-С 7 алкил) Х; X выбран из NR NH ------ С. N(R13)R13R 13 - это Н, L - это NHCH 2C(=O)-, M - это L-изом ер аминокислоты, имеющей стр ук тур у — N R 1 7— C H — С ( = О) , СМ (CH(R 4))q. О К j \ 2 NR R32 где: q' это 1-2; В - э то Н ; R 17 - это Н или СНз; R 8 - это СО2Н или -СО2СНз; Сь выбран из фуппы (C(R21)R1)n R31 или ее фарм ацевтически пригодная соль или ее форм а пропрепарата, где: R 31 - это кольцевая систем а фенила или нафтила, при необходимости имею щая связь с U; R 32 - это -С(О)-; л' - это 1 ; п" - э то 0 ; R 1, R 2 и R 2t - это водород; J - это D-изом ер ам инокислоты, имеющей стр уктуру Nенилацетонитрил 32577 ClaBzt CBZ, Cbz n nn Z DCC DIEA di-NMeOrn DMAP HBTU NmeArg или MeArg NmeAmf NmeAsp NmeGly или MeGly Nme-Mamb дихлорбензил довательность Связывание может выполняться как последовательное попарное соединение аминокислот, как конденсация (соединение) фрагментов (двух или нескольких аминокислот) или как комбинация обоих этих процессов либо путем твердофазного пептидного синтеза по методу Me rri field , J. Am . Chem. Sos , 85, 2149 -21 54 (1963), сущность которого здесь кратко приведена. Соединения настоящего изобретения могут1 быть также гистезированы с помощью специального автоматизированного пептидсинтезирующего оборудования. Процедуры синтеза пептидов опи саны в Stewa rt и Young "Solid Phase Peptide Synthesis", 2-ое изд., Pierce Cjemical Co., Rockford, IL (1984), Gross, Meinhofer, Udenfriend, Eds. "The Peptide: Analysis, Synthesis, Bioligy, vol. 1,2,3,5,9, Academic Press, New York, 1980-1987, Boda ns zky "Pe ptide Chemis try: A Pra ctical Textbook", Spnnger-Verlag, New York (1988) и Bo dans zky и др . "Th e P ra ctice o f Pe p tid e Synthesis", Springer-Verlag, New York (1984), суть которых здесь кратко изложена. Связывание между собой двумя аминокислотными производными, аминокислотой и пеп- • тидом, двумя пептидными фрагментами или циклизацией пептида может быть осуществлено с помощью стандартных способов связывания, таких как азидный способ, смешанный кислотноангидридный способ (на основе изобутилового эфира хлормуравьиной кислоты), карбодиимидный способ (на основе дициклогексилкарбодиимида, диизопропилкарбодиимида или водорастворимых карбодиимидов), способ активных эфиров (п-нитрофенильный эфир, N-гидроксисукцинимидный эфир), способ Вудворд реагент К, карбонилдиимидазольный способ, способ на основе фосфорсодержащих реагентов, таки х как ВОРСІ, окислительно-восстановительный способ. Некоторые из этих способов (особенно, карбодиимидный) могут быть усилены при добавлении 1-гидроксибензотриазола. Эти реакции связывания могут проводиться в любом растворе (жидкая фаза) или твердой фазе. Функциональные группы связываемых аминокислот могут быть защищены при реакциях связывания с целью избежания нежелательных связей . За щи ща ющие а ген ты , ко торые могут быть использованы, указаны в Green "Protective Group in Organic Synthesis" John Wiley & Sons, New York (1981 ) и "The Pep tid es: Anal ysis , Synthesis, Biology, vol . 3, Academic Press, New York (1981), суть которых здесь кратко изложена. а -карбоксильную гр уппу С-концевого остатка обычно защищают эфирной группой, которая может быть преобразована в карбоксильную. Эти защищающие гр уппы включают: 1) алкильные эфиры, такие как метиловый и трет-бутиловый; 2) арильные эфиры, такие как бензиловый и замещенный бензиловый; 3) эфиры, расщепляемые при слабощелоч ной обработке или обработке слабым окислите лем, таким как трихлорэтиловый и фенациловый эфиры При твердофазном синтезе аминокислота С-конца присоединяется к нерастворимому носи карбобензилокси дициклогексилкарбодиимид диизопропилэтиламин N-aMe-N-gMe-орнитин 4-диметиламинопиридин 2-(1 Н-бензотриазол-1 -ил)1,1,3,3-тетраметилурониум гексафторфосфат а -N-метиларгинин N-метиламинометилфенилаланин a-N-метиласпарагиновая кислота N-метилглицин N-метил-З-аминометилбензойная кислота NMM N-метилморфолин OcHex О-циклогексил OBzl О-бензил oSu О-сукцинимидил pNP п-нитрофенил TBTU 2-(1 Н-бензотриазол-1 -ил)1,1,3,3-тетраметилурониум Teoc 2-(триметилсилил)этилоксикарбонил Tos тозил Tr тритил Используются следующие тре хбуквенные іачения аминокислот Ala аланин Arg аргинин Asn аспарагин Asp аспарагиновая кислота Cys цистеин Gin глютамин Glu глютаминовая кислота Gly глицин His гистидиИ lie изолейцин Leu лейцин Lys лизин Met метионин Nle норлейцин Phe фенилаланин Phg фенилглицин Pro пролин Ser серии Thr треонин Thp триптофан Tyr. тирозин Val валин Соединения, относящиеся к данному изобретению, могут быть синтезированы стандартными способами, известными среднему специалисту в данной области Предпочтительные способы включают, но не ограничиваются нижеследующими Пептиды, в основном, наращивают разрушением аминогруппы С-конца и связыванием со следующей соотве тствующей защи щенной аминогруппой с помощью пептидной связи. Такая процедура сня тия защи ты повторяе тся до те х пор, пока не будет достигн ута желаемая после 18 32577 телю (обычно полистирол) Эти нерастворимые носители содержат группу, способную реагировать с карбоксильной группой с образованием связи, устойчивой в условия х наращивания пептидной цепочки и легко расщепляемой после этого Примерами таковых являются оксим-смола (DeGrado и Kaiser (1980) J Огд Спет 45, 12951300), хлор- или бромметилсмола, гидроксиметил-смола и аминометил-смола Многие из эти х смол с уже включенной коммерчески требуемой С-концевой аминокислотой вполне доступны а-амино гр уппа каждой амин окислоты должна быть защищена Для этого могут использоваться любые известные защищающие группы Примерами таковых являются 1) аиилы, например, формил, трифторацетил, фталил, п-толуолсульфонил. 2) ароматические карбаматы, такие как бензилоксикарбонил (Cbz) и замещенные бензилоксикарбонилы, 1 -(п-бифенил)-1 -метилэтоксикарбовил и 9-фторанилметилоксикарбонил (Fmoc), 3) алифатические карбаматы,.такие как третбутилоксикарбонил (Вое), этоксикарбонил, диизопролилметоксикарбонил и аллилоксикарбонил, 4) циклические алкильные карбаматы, нап ример, циклопертилоксикарбонил и адамантилоксикарбонил, 5) алкильные, такие как трифенилметил -и бензил, 6) триалкилсиланы, например, триметилсилан, 7) тиолсодержащие, например, фенилтиокарбонил и дитиосукцинол Предпочтительными а-аминозащищающими группировками являются "Вое" и "Fmoc" Многие производные аминокислот, соответственно защищенные для синтеза пептидов, являются доступными а-аминозащищающая группа удаляется перед взаимодействием со следующей аминокислотой При использовании Вос-группы предпочтительными являются трифторуксусная кислота, чистая или в дихлорметане, либо - HCI в диоксане Образовавшаяся соль аммония затем нейтрализуется (или перед связыванием, или в процессе его) щелочными растворами, например, водными буферными растворами или растворами третичных аминов в дихлорметане или диметилформамиде При использовании Fmoc-группы предпочтительными реагентами являются пиперидин или замещенные пиперидины в диметилформамиде, однако может быть использован любой вторичный амин или водные щелочные растворы Удаление защищающей группы проводится при температуре от 0°С до комнатной Любая аминокислота, несущая функциональные группы в боковой цепи, должна быть защи щена в процессе синтеза пептида с помощью упомянутых защи щающи х гр упп Специапист учтет тот факт, что выбор и использование соответствующи х защи щающи х гр упп боковых цепей будет зависеть от самой аминокислоты и наличия (присутствия) др уги х за щищающи х групп в пептиде Избрание такой защищающей группировки является важным по той причине, что она не должна быть удалена при снятии защиты и связывании а-аминогруппы Например, если выбрана Вое для за щи ты а -аминогруппы, то приемлемыми защищающими группами являются следующие п-толуолсульфонил (тозил) колец и нитрогруппа для аргинина, бензилоксикарбонил, замещенные бензилоксикарбонилы, тозил или трифторацетил для лизина, бензил или алкильные эфиры, такие как циклопентил для глутаминовои и аспарагиновои кислот, бензильные эфиры для серина и треонина, бензиловые эфиры, замещенные бензиловые эфиры или 2-бромбензилоксикарбонил для тирозина, п-метилбензил, n-метоксибензил, ацетамидометил, бензил, или трет бутилсуль фонил для цистеина, а индольное кольцо триптофана может быть оставлено незамещенным или защищенным формальной группой Если для а-аминозащиты выбран Fmoc, обычно применяют защищающие группы на основе третбутильного радикала Например, Вое может быть использован для лизина, третбутиловый эфир - для серина, треонина и тирозина, и третбутиловый эфир - для глутаминовои и аспарагиновои кислот Когда наращивание и циклизация пептида закончены, все защищающие гр уппировки удаляются При жидкофазном синтезе защищающие группировки удаляются любым путем, который зависит от природы самих удаляемых групп Эти способы хорошо знакомы специалистам При использовании твердофазного синтеза пептид должен быть удален с подложки без одновременного снятия защиты с функциональных групп, чтобы не помешать процессу циклизации пептида Таким образом, е сли пеп тид должен быть подвергнут циклизации, условия отщепления защитных гр упп нужно выбирать таким образом, чтобы свободная - карбоксилатная группа и свободная «-аминогруппа возникли без одновременного удаления защищающи х гр упп Например, пептид^ может быть удален со смолы с помощью так называемого "гидразинолиза" и затем присоединен азидным методом Другой подходящий способ представляет собой синтез пептидов на оксимовой смоле, который протекает за счет внутримолекулярных нуклеофильных перемещений со смолы, в результате че го си н тезир уе тся цик ли ческий пеп ти д (Osapa y, Pro fit, fnd Ta ylo r (1990) Te trah edron Letters 43, 6121-6124) При использовании оксимовой смолы схема Вое защиты особенно предпочтительна Далее предпочтительный способ снятия защитных гр упп боковых цепей, в основном, включает обработку ангидридом HF, содержащим добавки диметилсульфонида, анизола, тиоанизола или п-крезола при 0°С Расщепление защитных гр упп пептида может быть также завершено другими кислотными реагентами, такими как смесь трифторметансульфокислоты и трифтор уксусной кислоты Редкие аминокислоты, используемые в настоящем изобретении, могут бы ть синтезированы стандартными способами, описанными в р або та х по данн ой обла сти [Th e Pep ti des Anal ysis, Synthesis, Biology, Vo 5, hh 342-449, Academic Preaa, New York (1981)], N-алкилсодержащие аминокислоты могут быть получены с помощью способов, описанных ранее [Cheum et al, 19 32577 (1977 ) Can J Chem . 55 , 90 6; Freidi nger et al , (1982 ) J Org Chem 48 ,77 (19 82)] Соединения настоящего изобретения могут быть получены нижеописываемыми методами Исходные материалы и методики для получения соединений будут описаны ниже дополнительно Процедура твердофазного пептидного синтеза выполнялась в 25-миллилитровых полипропиленовых фильтрационных тр убках о т BioRad Inc. или в 60-миллилитровых стеклянных сосудах о т Peptides International Оксимовая смола (уровень замещения = 0,96 ммоль/r) была получена в соответствии с методикой, описанной (Deg ra do a nd Kaise r (1980 ) J Org Ch em, 4 5 , 1295) или закуплена у Novabiochem (уровень замещения = 0,62 ммоль/г) Все реагенты и растворители использовались без предварительной очистки Трет-бутилоксикарбонил (Вое) аминокислоты и другие исходные аминокислоты могут быть получены на коммерческой основе у фирм Bachem Inc , Bachem Biosciences Inc , (Филадельфия, PA) Ad vanced Chem Tech (Луневилль, KY), Peminsula Laboratories (Belmont, Ca) или Sigma (St Louis, МО), 2-(1Н-Бензотриазол-1-ил)-1,1,3,3тетраметилурониум гексафторфосфат (НВТИ) и ТВТИ были закуплены у Ad vanced Chem Tach NМетилморфолин (МММ) м-крезол, D-2-аминомасляная кислота (Abu), триметилацетилхлорид, диизопропилэтиламин (DIEA), 3-цианобензойная кислота и [2-(трет-бутилоксикарбонилоксилимино)-фенилацетонитрил](Вос-0№) были приобретены у Aldnch Chemical Company Диметилформамид (DMF), этилаце тат, хлоро форм (СНС Із), метанол (Ме ОН), пиридин и соляная кислота (HCI) были получены от Baker Ацетонитрил, дихлорметан (DCM), уксусная кислота (НОАс), трифторуксусная кислота (TFA), сложный этиловый эфир, триэтиламин, ацетон и сульфат магния были получены и ЕМ Scince Палладий на угле - катализатор (10% Pd) был приобретен у Fluka Chemical Company Абсолютный этанол был получен от Quantum Chemical Corporation Тонкослойная хроматография выполнялась на силикагельных 60 F254 TCL пластинках (толщина слоя 0 ,2 мм ) ко то р ые бы ли з ак уп л е ны у ЕМ Separations TCL визуализация была выполнена с использованием ультрасвета, впрыскивания йода, нингидрина и/или с помощью впрыскивателя Sakaguchi Точки замерзания определялись с помощью аппаратуры Thomas Hoover или Electothermal 9200 melting point и не проверялись HPLC-анализы были про деланы с помо щью Hewlett Packard 1090, Waters Delta Prep 3000, Rainin или Du Pont 8800 систем Спектры ЯМР были получены на 300 MHZ General Electric QE300, Vanan 300 или Vanan 400 - спектрометрах Масс-спектрометрия ("бомбардировка быстрым атомом") (FAB-MS) была выполнена на VG ZabЕ-масс-спектрометре с двойной фокусировкой с использованием Xenon FAB-пушки как источника ионов или Finnigan MAT 8230 BOC-D-2-аминомасляная кислота (ВОСD-Abu) была получена модификацией метода, ранее описанного в литературе (Itoh , Hagiwara, and Kamiya (1975) Tett Lett, 4393), как показано ниже на схеме BOC-ON рН х 9, Et, N тель), которая была использована далее без дополнительной о чистки 1HN MR (CDCb) 0 ,98 (t, ЗН), 1 ,45 (s, 9Н), 1 ,73 (т, 1Н), 1 ,90 (т, 1Н), 4 ,29 (т, 1Н), 5 ,05 (т, 1Н ) Синтез R31 циклизирующи х разъясняет синтез определенных циклизирующи х, которые являются промежуточными от группы R31 до Q Другие разделы поясняют синтез других циклизирующи х Синтез производных ВОС-аминометилбензойной кислоты, ВОС-аминофенилуксусной кислоты и ВОС-аминометилфенилуксусной кислоты Производные ВОС-аминометилбензойной кислоты, используемые как циклизующие в синтезе соединений данного изобретения, получают известными способами, например, описанными в Tell Lett, 4393 (1975), Modem Synthetic Reactions, H O. House (1972 ), или Ha lting e t al , J Am Ch em So c, 50 3370 (1928) и как показано на схеме ниже D-2-аминомасляная кислота D-2-аминомасляную кислоту (1,0 г, 9,70 ммоль) растворяли в 20 мл НгО и добавляли раствор Вое ON (2,62 г, 10,6 ммоль) в 20 мл ацетона Получившийся белый осадок растворяли прибавлением триэтиламина (3,37 мл, 24,2 ммопь) до образования бледно-желтого раствора (рН 9, на влажной рН-бумаге) При комнатной температуре раствор перемешивали в течение ночи, при этом под вакуумом был отогнан ацетон Оставшуюся водную фазу трижды экстрагировали эфиром, подкисляли до рН 2 концентрированной HCI и затем экстрагировали трижды этилацетатом Собранный органический слой высушивали над безводным сульфатом магния и подвергали выпариванию при пониженном давлении, чтобы получить т-бутилоксикарбонил-0-2-аминомасляную кислоту, напоминающую масло*(2,05 г, более чем количественный выход, содержит раствори NH—-ВОС 20 32577 Гидрохлорид 3-аминометилбензойной кислоты." 3-циан обензойн ую кисло ту (10 ,0 г, 68 ммоль) растворяли в 200 мл этанола при нагревании на во дяной бане при темпера туре 3550°С. Добавляли концентрированную HCI (6,12 мл, -73 ммоль) и полученный раствор перенесли в 500-миллиметровую обработанную азотом круглодонную колбу, содержащую катализатор - палладий на угле (1,05 г, 10% Pd/C). С успензию перемешивали в атмосфере водорода в течение 38 часов, фильтровали через стеклянную воронку и тщательно промывали водой Этанол удалялся при пониженном давлении и оставшийся водный слой, который содержал белый осадок, растворяли в добавленных сюда же 250 мл воды. Диэтиловый эфир (250 мл) добавлялся и суспензию переносили на фильтр При энергичном встряхивании все твердые частицы растворялись и водный слой был затем дважды промыт эфиром, упарен при пониженном давлении до-150 мл и лиофилизирован до получения выше указанного соединения (хлоргидрат 3-аминометилбензойной кислоты) (8,10 г, 64%) в виде осадка бежевого цвета. 1HNMR (D2O), 4,27 (s, 2Н), 7,60 (t, 1H), 7,72 (d, 1H ),8 ,06(d,2H ). Т-бутилоксикарбонил-3-аминометилбензойная кислота (Boc-Mamb) Указанное соединение получают в' соответствии с модифицированными стандартными процедурами, описанными в литературе (itoh, Hagiwara, and Kamiya (1975) Tett., Lett., 4393). 3NH4OH CO2H F«SO4-7H2O Аминометилбензойную кислоту (в форме гидрохлоридной соли) (3,0 г, 16,0 ммоль) растворяли в 60 мл НгО. Сюда же добавляли раствор Вое ON (4,33 г, 17 ,6 ммоль) в 60 мл ацетона, после чего триэтиламин (5,56 мл, 39,9 ммоль). Раствор становился желтым и рН достигал 9 (на влажной рНбумаге) при добавлении 1,0 мл (7,2 ммоль) триэтиламина. Раствор перемешивался в течение ночи при комнатной температуре, в процессе чего ацетон отгонялся при пониженном давлении, а оставшийся водный слой промывали трижды эфиром. Во дный слой затем подкисляли до рН 2 2Н HCI и затем трижды экстрагировали этилацетатом. Объединенные органические слои трижды промывали водой, высушивали над безводным сульфатом магния и выпаривали под вакуумом до сухо го состояния Продукт был перекристаллизован из смеси этилацетат/гексан и получали указанное вещество дважды (2,58 г, 64%) в виде бело го поро шка tnn 123-125°C. ^N MR (CDCI3) 1,47 (s, 9H), 4,38 (br s, 2H), 4,95 (br s,1H), 7,45 (t, 1H), 7,55 (d, 1H), 8,06 (d, 2H). Синтез m-бутилоксикарбонил-З-аминофенилуксусной кислоты Т-бутилоксикарбонил-3-аминофенилуксусные кислоты, используемые как промежуточные соединения в синтезе соединений настоящего изобретения, получают известными способами, например, описанным Collman and Groh (1982), J. Am Chem, Soc , 104, 1391, и как показано на следующей схеме СО2Н t-BuOjCOCO 2t-Bu 5ТЁА Т-бутилоксикарбонил-3-аминофенилуксусная кислота Раствор 3-аминофенилуксусной кислоты (Aldrich, 10 г, 66 ммоль), ди-трет-бутилдикарбонат (15,8 г, 72 ммоль) и DIEA (8,6 г, 66 моль) в 50 мл дихлорметана перемешивают при комнатной температуре в течение ночи Реакционную смесь концентрируют, при разделении между дихлорметаном и водой, водный слой отделяют, подкисляют 1Н HCI до рН 3 и экстрагируют дихлорметаном Экстракты промывают водой, раствором соли, высушивают над безводным сульфатом натрия и окончательно высушивают при лониженном давлении. Полученный материал был очищен перекристаллизацией из гептана, в результате было получе CO2N * но вышеуказанное соединение (3,7 г, 22%) в виде порошка белого цвета, точка плавления 105°С, 1 HNMR (CDCb) 7,35 (s, 1H), 7,25 (m, ЗН), 6,95 (m, 1Н), 6,60 (br s, 1H), 3,65 (s, 2H), 1,50 (s, 9H). Синтез гидрохлорида 2-аминометилбензойной кислоты, и гидрохлорида 2-аминометилфенилуксусной кислоты Гидрохлорид 2-аминометилбензойной кислоты и гидрохлорид 2-аминометилфенилуксусной кислоты, используемые как промежуточные 'соединения в синтезе заявленных соединений, получают известными методами, например, как описано Naito et a!., J. Antibiotics, ЗО: 698 (1977); или Young and Sweet J. Am Chem. Soc, 80: 800 (1958), и как показано ниже 21 32577 лоты. d-Лактам 2-аминометилфенилуксусной кис Т. пл. 233°С (dec); 'HNMR (De-DMSO), 13,40 (br s, 1H), 8,35 (br s, 3H), 8,05 (d, 1H), 7,60 (m, 3H), 4,35 (br s, 2H). Синтез промежуточных циклических соединений. Этот раздел раскрывает синтез определенных промежуто чны х циклически х соединений, они выполняют роль предшественника Q группы в заявленных соединениях, (QLn)dCn; (Q)d, U-Ch. Эти соединения могут быть непосредственно мечены радиоизотопами или модифицированы присоединением линкерной группы (групп) или хелатора (хелаторов). Т-Бутилоксикарбонил-3-аминометилбензойная кислота (Boc-Mamb) связывается с оксимовой смолой с помощью модифицированного метода, описанного DeGrado and Kaiser (1980) J. Org. Chem. 45, 1295, при использовании 1 эквивалента 3-аминометилбензойной кислоты (с учетом степени .замещения смолы, 1 эквивалента HBTU и 3 эквивалентов NMM. Boc-Mamb (1 эквивалент) может быть присоединен к оксимовой смоле пр и и спользо вани и 1 эк ви вале н та ОСС и DMAP (каждого из них) в метиленхлориде. Время связывания от 15 до 96 часов Степень связывания затем определяется или тестом пикриновой кислоты (Sann, Rent, Tam and Memfield (1981), Anal. Biochem. 117, 145-157) или количественным нингидриновым анализом (Gisin (1972) Anal Chim., Acta, 58, 248-249). Непрореагировавшие оксимгр уппы блоки р уют 0 ,5 триме тилаце ти лхлоридом/0,5 М диизопропилэтиламином в D MF в течение 2 часов Снятие Вое защищающей гр уппы осуществляют 25% TFA в DCM в течение 30 мин. Оставшиеся аминокислоты или производные аминокислот связываются при 2-10 кратном избытке (связанном с "нагруженностью" первой аминокислоты и аминокислотного производного) соответствующей аминокислоты или аминокислотных производных и HBTU в приблизительно 8 мл DMF. Смолу затем нейтрализуют 3 эквивалентами NMM (в соответствии с количеством используемой аминокислоты) и продолжительность связывания составляет о т 1 часа до нескольких дней. Полнота связывания контролируется качественным нингидриновым анализом или тестом пикриловой кислоты в случаях, когда аминокислота взаимодействовала с вторичным амином. Отсутствие связывания аминокислот, если необходимо, определяли по этим же результатам. После того, как линейный пептид был собран, N-концевая группа Вое удалялась обработкой 25% TFA в DCM в течение 30 мин. Смолу затем нейтрализовали обработкой 10% DIEA в DCM. Циклизацию с сопутствующим расщеплением пептида выполняли с помощью метода Osapay и Taylor (1990) J. Am. Chem. Soc. 112, 6046) суспендированием смолы в приблизительно 10 мл/г D MF, до бавляя 1 эк вивален т НОАс (в соотве тствии с загруженностью первой аминокислоты) и перемешивая при 50-60°С в течение от 60 до 72 часов. Затем фильтровали через стеклянную воронку, 4>ильтрат DMF выпаривали, перерастворяли в НОАс или смеси ацетонитрил: НгО (1:1) и лиофилизовали до получения Данное соединение получают при модификации метода, описанного ранее в литературе (Naito et al., 1977) J. Antibiotics, 30: 698). К охлажденной льдом суспензии 2-инданона (10,8 г, 82 ммоля) и азидотриметилсилана (9,4 г, 82 ммоля) в 115 мл хлороформа приливали 25 мл концентрированной серной кислоты таким образом, чтобы температура удерживалась в интервале 3040°С. После выдерживания в течение 3-х часов реакционная смесь выливалась на лед и водный слой подщелачивали, концентрированным гидроксидом аммония, хлороформный слой отделяли, промывали водой, солевым раствором, высушивали над безводным сульфатом магния и досушивали в вакууме. Продукт очищали сублимацией (145°С, < 1 мм), после чего подвергали перекристаллизации из бензола до получения выше указанного соединения (5,4 г, 45%) в виде бледно-желтых кристаллов. * Т. пл. 149-150°С; 1HNMR (CDCI3) 7 ,20 (m, 5Н), 4,50 (s, 2H), 3,60 (s, 2H). Гидрохлорид 2-аминометилфенилуксусной кислоты Указанное соединение получают при модификации метода, ранее описанного в литературе (Naito et al., 1977) J Antibiotics, 30: 698). Смесь dлактама 2-аминометилфенилуксусной кислоты (6,4 г, 44 ммоля) и 21 мл 6Н HCI нагревали с обратным холоди льником 4 часа. Реакционную смесь обраба ты ва ли ак ти виро ванным углем (Nont А), фильтровали, высушивали, а к убовые остатки растирали с ацетоном. В результате фильтрации получали вышеназванное вещество (5,5 г, 62%) в виде бесцветных, кристаллов. Т. пл. 168°С (dec): 'HNMR (D 6-DMSO) 12,65 (br s, 1H), 8,35 (br s, 3H), 7,50 (m, 1H),-7,35 (m, 3H), 4,05 (ABq, 2H), 3,80 (s, 2H). d-Лактам 2-аминометилбензойной кислоты Указанное соединение получают при модификации метода, ранее описанного в литературе (Danishefsky et ah, (1975) J. Org. Chem. 40: 796). Смесь о-метилового эфира толуиловой кислоты (45 г, 33 моля), N-бромсукцинимида (57 г, 32 моля) и дибензоилпероксид (0,64 г) в 175 мл четыреххлористого углерода нагревали с обратным холодильником 4 часа. Охлажденную реакционную смесь фильтровали, высуши вали при пониженном давлении вновь растворяли в 250 мл метанола, концентрировали добавлением гидроксида аммония (75 мл, 1,11 моль). Реакционную смесь затем кипятили с обратным холодильником в течение 5 часов, концентрировали, фильтровали, осадок промывали водой, затем эфиром. Полученный продукт очищали перекристаллизацией из воды и получали вышеназванное вещество (11,0 г, 26%) в виде белого осадка. Т. пл. 150°С; 'HNMR (CDCJ3) 7,90 (d, 1H), 7,60 (t, 1H), 7,50 (t, 2H), 7,00 (br s, 1H), 4,50 (s, 2H). Гидрохлорид 2-аминометилбензойной кислоты Данное соединение получают по способу, описанному для гидрохлорида 2-аминометилфенилуксусной кислоты. Лактам (3,5 г, 26 ммоль) был превращен в указанное соединение (2,4 г, 50%) в виде бесцветных кристаллов. 22 3 2577 защищенного циклического продукта. Он затем обрабатывается с помощью стандартных процедур безводной HF (Stewart and Joung (1984) "Твердо фазный пеп тидный син тез " 2 изд-е, Pierce Chemical Co., 85), содержащей 1 мл/г мкрезола или анизола как поглотителя при 0°С в течение 20-60 мин ут для удаления за щитны х групп боковых цепей. Неочищенный продукт можно очистить обратно-фазовой HPLC хроматографией , использ уя 2,5 см препаративн ую Vydaq С18 колонку с линейным градиентом ацетонитрила, содержащим 0,1% TFA до получения чистого циклического продукта. Следующие N-a-Boc-защи щенные аминокислоты могут использоваться для синтеза: Boc-Arg(Tos), Boc-N-a-Me Arg(Tos), Boc-Gly, Вое-Asp (OcHex), ВОС-3-аминометил-Ниодо-бензойная кислота, Boc-D-Ke, Boc-NmeAsp (OcHex), Boc-NMe-Mamb, Boc-D-Phg, Boc-D-Asp (OBzl), Boc-L-Asp (OcHex), Boc-aMe-Asp (OcHex), Boc-bMe-Asp (OcHex), Boc-L-AIa, Boc-L-Pro, BocD-Nie, Boc-d-Leu, Boc-D-Val, Boc-D-2-аминомасляная кислота (Boc-D-Abu), Boc-Phe , Boc-DSer(Rzl), Boc-D-AIa, Вос-3-аминометилбензойная кислота (Boc-Mamb), Boc-D-Lys (2-Clz), Boc-d-Ala, Boc-D-Рго, Boc-D-Phe, Boc-D-Tyr (CbBzl), BocNMe-Amf (CBZ), Вос-аминотетралинкарбоксильная кислота, Вос-аминометилнафтойная кислота, Вос-4-аминометилбензойнэя кислота или ВосNMeGty. Предпочтительные N-a-Boc-защищенные аминокислоты, используемые в этом синтезе: Boc-Arg(Tos}, Boc-N-a-Me Arg(Tos), Boc-Gi y, BocAsp (OcHex), Boc-D-Leu, Boc-D-Val, Boc-D-2-аминомасляная кислота (Boc-D-Abu), Boc-Phe, BocD-Ser(Bzl), Boc-D-AIa, ВОС-3-аминометилбензойная кислота (Boc-Mamb), Boc-D-Lys (2-Glz) BocAla, Boc-D-Pro или Boc-NMeGJy. Синтез заявленных соединений ниже проиллюстрирован. Нижеследующие таблицы дают представление о соединениях, защищаемых в настоящем изобретении. Циклическое соединение - Фрагмент 1 (промежуточное № 1) !4nKno-(Gly-NMeArg-Gly-Asp-Mamb); соединение формулы II, где J = Gty, К = NmeArg, L = =Gly, М = Asp, R 1 = R2 = H. Указанное соединение получают, используя основной метод, описанный ниже для цикло(D-Val-NMe Arg-Gl y-Asp-Mamb). Пептид получали исходя из расчета 0,336 ммоля, для того, чтобы получить защищенный циклопептид в количестве 218 мг, 84%. Пептид (200 мг) в 200 мл м-крезола были обработаны безводной HF при 0°С в течение 1 часа. Неочищенный материал осаждали эфиром, перерастворяли в водной НОАс, лиофилизировали и получали указанное соединение в виде бледно-желтого осадка (158 мг, больше чем количественный выход вычислено через ацетатную соль). Очистка завершалась обращеннофазовым методом ВЭЖХ с использованием препаративной Vydac С18 колонки (2,5 см) и 0,23%/мин градиента 2-11% ацетони трила, содержаще го 0,1% TFA, и лиофилизацией до получения TFA соли указанного соединения в виде пушистого белого осадка (21%-выделено, окончательный выход 16,3%). Масс-спектр: М+Н=533,26. Циклический фрагмент 2. ^Kno-(D-Ala-NMeArg-G)y-Asp-Mamb); соединение форм улы II, где J = D-Ala , К = NmeArg, L s Gly, M = Asp, R1=R2=H. Указанное соединение полумают используя основной метод, описанный ниже для цикло-(ОVal-NMeArg-G(y-Asp-Mamb). Установлено, что отсоединение остатка Boc-N-MeArg(Tos) необходимо. Пептид получали исходя из расчета 0,244 ммоля для то го, чтобы получить защи щенный циклопептид (в количестве 117 мг; 61%). Пептид (110 мг) и 100 мл м-крезола обрабатывали безводной HF при 0°С в течение 1 часа. Неочищенный материал осаждали эфиром, перерастворяли в водной НОАс, лиофилизировали и получали указанное соединение в виде бледно-желтого осадка. Очистку завершали обращеннофазоаым методом ВЭЖХ в обратной фа з е на пре пар а ти вно й Vyd a c C 18 ко лонке (2,5 см) с использованием 0,25%/мин градиента 2-11% ацетонитрила, содержащего 0,1% TFA соли указанного соединения 8 виде пушистого белого осадка Масс-спектр: М+Н=547,33. Циклическое промежуточное соединение 3. ^wio-(D-Abu-NMe Arg-Gl y-Asp-Mamb); соединение формулы II, где J = D-Abu, К = NmeArg, L = Gl y, M = Asp, R 1=R=H. Указанное соединение получают, используя основной метод, описанный ниже для циклического соединения N 4. Пептид получают исходя из расчета 0,101 ммоля для того, чтобы получить защи щенный циклопептид (51 мг, 63%) Пепти д (43 мг) и 50 мкл м-крезола обрабатывались безводной HF при 0°С в течение 30 ч. Неочищенный продукт осаждали эфиром, перерастворяли в водной НОАс, лиофилизировзли. чтобы получить указанное соединение в виде бледно-желтого осадка (23 мг, 68,7 % вычислено как ацетатная соль). Очистку заканчивали обращеннофазовым методом ВЭЖХ на препаративной Vydac C18 колонке с использованием 0,23%/мин градиента 714% ацетонитрила, содержащего 0,1% трифторуксусной кислоты и затем лиофилизировали до получения TFA сопи указанного соединения в виде пушистого белого осадка (31% - выделено); к он е чны й вы хо д 12 ,4 % ). Ма сс-сп ек тр : М+Н=561,46. Циклическое промежуточное соединение За ^Kno-(Abu-NMeArg-Gly-Asp-Mamb). соединение формулы II, где J = Abu, К = NmeArg , L = Gl y, M = Asp, R'=H ; R=H . Указанное соединение получают, используя основной метод, описанный для цикло-(О-Уа!NMeArg-Gl y-Asp-Mamb) (Циклическое промежуточное соединение 4). DCC/DMAP использовался для присоединения Boc-Mamb к оксилмовой смоле. TBTU используется как связывающий агент. Пептид получают исходя из расчета 0,596 ммоль, чтобы получи ть защи щенный циклопептид (182 мг, 38,4%). Пептид (176 мг) и 0,176 мл анизола обрабатывали безводной HF при 0°С в течение 20 мин. Неочищенный материал осаждали эфиром, растворяли в водном ацетонитриле, лиофилизировали и попучали указанное соединение (116 мг, 90 ,4% вычислено на основе фтористой 23 32577 соли). Очистку заканчивали ВЭЖХ хроматографией (обращеннофазовый метод) на препаративной Vydac C18 колонке (2,5 см) с использованием 0,45%/мин градиента 9-27% ацетонитрила, содержащего 0,1% TFA и затем лиофилизировали до получения TFA соли указанного соединения в виде пушисто го белого осадка (выделено 1,92%, конечный выход 0,574%); FAB-MS. Массспектр. М+Н=561,39. Циклическое промежуточное соединение 4. l4nwio-(D-Val-NMe Arg-Gl y-Asp-Mamb); соединение формулы II, где J = D-Val, К = NmeArg, L = Gly, М = Asp, R 1 = R2 = H. В 25-миллиметровую полипропиленовую пробирку, снабженную стеклянной фриттой, добавляли Boc-Mamb (0,126 г, 0,5 ммоля) и 6 мл DMF. Сюда же добавляли HBTU (0,194 г, 0,5 ммоля), оксиловую смолу (0,52 г, степень замеще ния = 0 ,96 ммо ль /г) и N-ме тилмор фолин (0,165 мл, 1,50 ммоль). Суспензию перемешивали при комнатной температуре в течение 24 часов. За тем смолу тща тельно промывают (10-12 объемами); DMF (ЗХ), Ме ОН (1 Х), DCM (ЗХ), МеОН (2 Х) и DC M (ЗХ) Количественным нингидриновым тестом был определен уровень замещения: 0,389 ммоль/г. Непрореагировавшие оксимгруппы были блокированы в результате обработки 0,5 М триметилацетилхлорид/0,5 М DIEA в DMF в течение 2 часов. Дальнейшее выполнялось следующим образом: (Стадия 1). Смолу промывали DMF(3 X), МеОН (1 Х), DCM (ЗХ), Ме ОН(2Х) и DCM (ЗХ) (Стадия 2). С третичных Вос-гр упп была снята защита 25% TFA в DC M в течение 30 мин. (Стадия 3). Смолу промывали DCM (ЗХ), МеОН (1Х), DCM (2X), МеОН (ЗХ) и DMF (ЗХ) (Стадия 4) ВосAsp (OcHex) (0,613 г, 1,94 ммоль), HBTU (0,753 г, 1,99 ммоль), 8 мл D MF и N -мети лмор фолин (0,642 мл, 5,84 ммоль) были добавлены к смоле и реакция продолжалась в течение 2,5 часов (Стадия 5). Завершение реакции связывания контролировалось качественным нингидриновым тестом. Стадии 1-5 повторяли до те х пор, пока не получали желаемую последовательность. Связывание Boc-D-Val с NMeArg контролировалось тестом пикриновой кислоты После того как линейный пептид был собран, N-терминальная Т-Вос группа удалялась обработкой 25% TFA в D MM (30 мин). Смолу тщате льно промы вали D C M (ЗХ), Ме ОН(2 Х) и DCM(3 X) и за тем нейтра лизова ли 10% DIEA в ОС М (2 x1 мин). Смолу тщате льно промывали DC M (ЗХ) и МеОН(ЗХ) и вы суши вала сь Половин у смолы (0,101 ммоль) подвергали "сши вке" (циклизо ва ли ) в рез ульта те обр або тки 6 мл DMF, содержа щим НОАс (5,8 мл, 0,101 ммоль) и нагре ван ия при 50°С в те чени е 72 ча со в. Смолу затем отфи льтровы вали через стеклянный фи льтр и тща те льно промывали D MF. DMF-филь трат выпаривали в масле, перерастворяли в смеси аце тонитри ла: Н гО (1 :1) лиофилизировали до получения желаемого циклопептида (49 мг, 60%). Пептид (42 мг) обраба ты вали без водной HF при 0°С в присутствии 50 мл м-крезола (в качестве поглотителя) в течение 30 мин для удаления защищающи х гр упп боковых цепей Неочи щенный материал осаждали эфиром, перерастворяли в водной Н ОАс и лио филизировали до получения указанного соединения в виде слабожелтого осадка (23 мг, 70%, в расчете на ацетатную соль). Очистка производилась обращеннофазовым методом ВЭЖХ в обратной фазе на препаративной Vydac C18 колонке (2,5 см) и при 0,23%/мин градиенте 7-18% ацетони трила, содержащего 0,1% трифтор уксусной кислоты, и получали трифторацетат вышеназванного соединения в виде пушистого белого осадка (выделено 24%, окончательный выход 9,4%), FAB-MS: [М+Н]=575,45. Жидкофазный синтез циклического промежуточного соединения 4 Следующие аббревиатуры используются далее для TLC систем растворителей: хлороформ/метанол 95:5=СМ; хлороформ/уксусная кислота 95:5=СА; хлороформ/метанол/ уксусная кислота 95:5=СМА. BocNMe Afg(Tos)-43ly-OBzl - 25 ммоль BocNMe Arg(Tos) (11,07 г, ВасНет), 30 ммоль GlyOBzl тозилата (10,10 г, Bachem), 25 ммоль HBTU (О-бензотриазол-N, N, N', N'-тетраметил-урониумгексафторфосфат, 9,48 r, Advanced Chemtech), и 75 ммоль DIEA (диизопропилэтиламин Aldrich) были растворены в 25 мл СНгЗД. Реакция протекала 1 час, растворитель отгонялся под вакуумом в виде сиропа, который затем растворяли в 400 мл этилацетата Полученный раствор подвергали экстракции (порциями по 150 мл) 2x5% лимонной кислотой tx водой, 2х насыщенным раствором №НСОз, 1х насыщенным раствором NaCI. Органический слой высушивали над MgSO*. а растворитель отгонялся под вакуумом. Получившийся масляный остаток растирали с петролейным эфиром и высушивали в глубоком вакууме минимум в течение 1 часа, вы ход 14,7 г (99,5%) TLC Rf(cw) = 0,18 R I(CA) = 0,10; NMR соответствует структуре, FABMS М+Н+=590,43 (Теоретически ожидаемое 590,26). NMe Arg (Tos)-Gly-OBzl - 14 ммоль (BocNMa Arg)Tos)-Giy-OBzl (24,5 ммоль) растворяли в 30 мл TFA, продолжительность взаимодействия 5 мин , за тем ра створи тель о тго няли • при комнатной температуре и остаточном давлении 1 мм. Получившийся сироп растворяли в 400 мл ледяного этилацетата и экстрагировали 100 мл ледяного насыщенного раствора №НСОз, водную фазу экстрагировали дважды 200 мл этилацетата, а смесь органических фаз экстрагировали 25 мл насыщенного раствора NaCI. Растворитель отгонялся при пониженном давлении до получения вязкого кубового остатка, который растирали с 300 мл эфира. Получившийся осадок растирали эфиром, получая гигроскопичное вещество, которое высушивали в вакууме: выход 10,33 г (86,2%) TLC Rf(CM) = 0,03 R f(cMA) = 0,20; NMR соответствует структуре; FABMS M+H+= =490,21 (Теоретически 490,20). Boc-D-Val-N Me Arg(Tos)-Gl y-OBzl - 9,80 ммоль, NMeArg(Tos)-Gly-OBzl (4,80 г), 9,82 ммоль Boc-D-Val (2,13 г, Bachem) и 10,0 ммоль HBTU (3,79 г) растворяли в 10 мл метиленхлориде. Колбочку помещали на ледяную ванночку и добавляли 20 ммоль DIEA (3,48 мл) Реакция шла 24 32577 Выход 7,51 г (79%); TLC Rr(CM, = 0,41; () = 0 ,66 ; N MR соо тве тствуе т струк туре; FABMS М+Н+=476,30 (Теоретически 476,18). TFA-Mamb-Ox-BocMamb-Ox, 7,4 г (15,5 ммоль) растворяли в 30 мл метиленхлорида + 10 мл TFA (25% TFA). Реакция шла при комнатной температуре 1 час, растворите ль выпарива ли при пониженном давлении 20 мин при комнатной температуре, затем 15 мин при 40°. Получившийся сироп растирали с эфиром (200 мл) при 5°. Спустя час образовавшиеся кристаллы отфильтровывали и хорошо промывали эфиром вы ход 7,22 г (95%); RUCMA) = 0,25; NMR соответствует структуре, FABMS M+H += 376,22 (Теоретически 376,12). Boc-Asp(OcHex)-Mamb-Ox - 20 ммоль ВосAsp(OcHe x), (6,308 г Bachem) и 44 ммоль DIEA (7,66 ммоль) растворяли в 20 мл DMF. Добавляли 20 ммоль HBTU (7,58 г Aldrich Chtm Tech) и реакция шла 2 мин при энергичном встря хивании Добавляли TFA-Mamb-Ox (7,13 г, 15 ммоль) и продолжали реакцию в течение ночи при комнатной температуре. Растворитель выпаривали при пониженном давлении до маслообразного остатка, который затем растворяли в 500 мл этилацетата и этот раствор экстрагировали порциями по 150 мл: дважды 5% лимонной кислотой, 1 раз водой, дважды насы щенным раствором №НСОз, раз насыщенным раствором NaCI Органический слой высушивали над MgSO4, растворитель отгоняли при пониженном давлении. Образовавшийся остаток растирали с петролейным эфиром и высуши вали в глубоком вакууме: выход 9,76 г (97%), TCL RHCM) = 0,55, NMR соответствуе т стр уктуре, FABMS М+Н+= 673,45 (Теоретически 673,23). TFA Asp(OcHex)-Mamb-Ox - 15 ммоль ВосAsp(OcHex)-Mamb-Ox было растворено в 50 мл 35% в СН2С1г и оставлено для реакции в течение 90 мин. Растворитель отгоняли при пониженном давлении при комнатной температуре в течение 10 мин. Затем при 40° в течение 15 мин. Для удаления следов TFA было добавлено 25 мл DMF и растворитель отгоняли при 50°. Оставшийся сироп растирали с эфиром (200 мл), затем высушивали в глубоком вакууме: вы хо д 9,61 г (93%); RI (CMA) = 0,45; NMR соответствуе т стр уктуре; FABMS M+H += 573,56 (Теоретически 573,23). Boc-D-Val-N Me Arg (Tos)-Gl y-Asp(OcHe x)Mamb-Ox 10 ммоль TFA Asp(OcHex)-Mamb-Ox + 10 ммоль Boc-D-Val-NMeArg(Tos)-Gly +1 0 ммоль HBTU + ЗО ммоль DIEA растворяли в 20 мл DMF. Спустя 4 часа растворитель отгоняли при пониженном давлении, а остаток переносили в 600 мл' этилацетата, после чего экстрагировали 300 мл порциями: 5% лимонной кислоты, воды и насыщенного раствора NaCI. Ор ганический слой высушивали над MgSO 4, выпаривали при пониженном давлении растирали с эфиром и высуши вали в вакууме: выход 9,90 г (86%); Rf
ДивитисяДодаткова інформація
Назва патенту англійськоюRadiolabeled compound as imaging agents for the diagnosis of arterial and venous thrombi, radiopharmaceuticals and radiopharmaceutical composition and kits for its preparation
Автори англійськоюDegrado William Frank, Mousa Shaker Ahmed, Sworin Michael, Barrett John Andrew, Edwards David Scott, Harris Thomas David, Rajopadhye Milind, Liu Shuang
Назва патенту російськоюРадиомеченое соединение для образования изображения тромбов для радионуклидной диагностики, радиофармацевтический препарат и радиофармацевтическая композиция на их основе, комплект для приготовления радиофармацевтических препаратов
Автори російськоюДеградо Вильям Френк, Моуса Шакер Ахмед, Сворин Мичейл, Баретт Джон Эндрю, Эдвардс Давид Скот, Харрис Томас Давид, Раджопадхи Милинд, Лиу Шуанг
МПК / Мітки
МПК: A61K 51/06
Мітки: діагностиці, радіофармацевтичних, радіофармацевтичний, утворення, комплект, основі, препарат, приготування, тромбів, зображення, радіомічена, композиція, радіонуклідній, сполука, радіофармацевтична
Код посилання
<a href="https://ua.patents.su/105-32577-radiomichena-spoluka-dlya-utvorennya-zobrazhennya-trombiv-pri-radionuklidnijj-diagnostici-radiofarmacevtichnijj-preparat-ta-radiofarmacevtichna-kompoziciya-na-kh-osnovi-ta-komplekt.html" target="_blank" rel="follow" title="База патентів України">Радіомічена сполука для утворення зображення тромбів при радіонуклідній діагностиці, радіофармацевтичний препарат та радіофармацевтична композиція на їх основі та комплект для приготування радіофармацевтичних п</a>
Комплексні сполуки похідних діетилентриамінпентаоцтової кислоти як контрастні засоби у діагностиці, що дає зображення

Номер патенту: 27275
Опубліковано: 15.09.2000
Автори: Гріс Хайнц, Фоглер Хуберт, Платцек Йоханнес, Вайнманн Ханнс-Йоахім, Шуманн-Гіампірі Габріеле, Шмітт- Вілліх Херіберт
МПК: A61K 49/00, C07F 5/00, C07C 229/16, C07F 15/00, C07F 9/94, A61K 49/06, C07C 227/00, A61K 49/04, C07C 229/76
Мітки: похідних, діетилентриамінпентаоцтової, діагностиці, засоби, дає, кислоти, контрастні, зображення, сполуки, комплексні
Текст:
...кислоты 2,57 г (5 ммол) описанной в примере 5b) комплексообразующей кислоты аналогично указанной для примера 1с) инструкции превращают до 3,30 г (98% от теории) заглавного соединения. Получают бесцветное хлопьевидное твердое вещество, Тпл. = 207оС. Вычислено: С 39,57, Н 4,23, N 6,29, Cd 23,55 Найдено: С 39,51, Н 4,26, N 6,35, Cd 23,27 Т1-релаксация (1/ммол.сек.) составляет в во де 4,39±0,12 в плазме 6,31±0,15. Пример 6. а)...
Сполука для утворення пінокислоти

Номер патенту: 14313
Опубліковано: 25.04.1997
Автори: Лісовий Георгій Антонович, Балакіров Юрій Айрапетович
МПК: E21B 43/27
Мітки: сполука, утворення, пінокислоти
Формула / Реферат:
Состав для образования пенокислоты, содержащей соляную и уксусную кислоты, отличающийся тем, что, с целью снижения его коррозионной активности, он дополнительно содержит серную кислоту и пятипроцентный водный раствор поверхностно-активного вещества при следующем соотношении компонентов, мас.%:Соляная кислота 50-60Уксусная кислота 10-20Серная кислота...
Фармацевтична композиція, що призначена для одержання порошків або шипучих таблеток, яка містить ефективну кількість ібупрофену, фармацевтичний препарат на її основі та спосіб його одержання

Номер патенту: 27058
Опубліковано: 28.02.2000
Автори: Брю-Ман'єз Ніколь, Кордоліані Жан-Франсуа, Друєн Жан Ів, Товєн Жерар
МПК: A61K 31/19, A61K 9/46
Мітки: основі, призначена, шипучих, одержання, ібупрофену, фармацевтичний, яка, спосіб, порошків, таблеток, препарат, містить, кількість, фармацевтична, композиція, ефективну
Формула / Реферат:
1. Фармацевтическая композиция, предназначенная для получения порошков или шипучих таблеток, содержащая эффективное количество ибупрофена или одной из его фармацевтически приемлемых солей в качестве активного ингредиента, фармацевтически приемлемую систему газирования, содержащую, по крайней мере, один щелочной карбонат и, по крайней мере, одну органическую кислоту, отличающаяся тем, что она дополнительно содержит, по крайней мере, один...
Комплексна сполука міді з трис(оксиметил)амінометаном як антибактеріальний препарат та спосіб її одержання

Номер патенту: 22930
Опубліковано: 05.05.1998
Автори: Гогуля Сергій Степанович, Люлько Олексій Володимирович, Мартємянов Владімір Владіміровіч, Рильцев Владімір Валєнтіновіч, Коваленко Алла Леонідівна, Філатов Владімір Міхайловіч
МПК: A61K 33/34, C08L 73/00
Мітки: комплексна, одержання, трис(оксиметил)амінометаном, антибактеріальний, препарат, спосіб, сполука, міді
Формула / Реферат:
1. Комплексное соединение меди с трис(оксиметил)аминометаном со структурной формулойв качестве антибактериального препарата. 2. Способ получения комплексного соединения по п.1, включающий обработку трис(оксиметил)аминометана раствором хлорида меди с последующим выделением целевого продукта, отличающийся тем, что процесс синтеза ведут в водном растворе при комнатной температуре и мольном соотношении хлорид меди:...
Інсектицідний препарат на основі піретроїдів

Номер патенту: 3469
Опубліковано: 27.12.1994
Автори: Іштван Секей, Рудольф Шоош, Тодор Пфлігель, Дьордь Кьормьоці, Йожеф Дукаі, Агостон Давид, Йожеф Боззаі
МПК: A01N 53/00, A01P 7/04
Мітки: основі, піретроїдів, інсектицидний, препарат
Формула / Реферат:
Инсектицидный препарат на осново пиретроидов, содержащий активное соединение и целевые добавки: синергист - пиперонил-бутоксид, поверхностно-активное вещество и носитель, отличающийся тем, что, с целью повышения активности препарата он дополнительно содержит в качестве целевых добавок средство для повышения проницаемости — бензиловый спирт, антиокислитель — бутилокситолуол, пропилгаллат, в качестве активного соединения состав содержит...
Попередній патент: Жаростійка бетонна суміш
Наступний патент: Засіб для лікування паралітичної непрохідності кишечнику
Випадковий патент: Турбомашина, що містить сопловий апарат (варіанти)