Спосіб о-дифлуорометилування функціоналізованих аліфатичних спиртів




Формула / Реферат
1. Спосіб О-дифлуорометилування функціоналізованих аліфатичних спиртів, який відрізняється тим, що функціоналізований аліфатичний спирт обробляють дифлуоромети-льованим агентом (FSO2CF2CO2H) в пристуності солі Сu (І) в органічному розчиннику при м'якому нагріванні.
2. Спосіб за п. 1, який відрізняється тим, що як сіль міді використовують CuI або СuВr та як розчинник використовують ацетонітрил.
3. Спосіб за п. 1, який відрізняється тим, що як аліфатичний спирт може бути використаний спирт формули (І):
HO-R,
де
R являє собою С1-С6алкіл, С3-С10циклоалкіл, С6-С10арил або С2-С9гетероарил, кожен з яких може бути заміщений принаймні одним С1-С6алкілом, С3-С10циклоалкілом, С6-С10арилом, С2-С9гетероарилом, -CN, NO2, OR1, NHR2, -SC1-С6алкілом, -SOC1-С6алкілом, -SO2C1-C6алкілом, -NSO2C1-C6алкілом, -NHC1-С6алкілом, -N(C1-C6алкіл)2, -NHCOOC1-C6алкілом, -NHCONHC1C6алкілом, -CONHC1-C6алкілом, -СОС1-С6алкілом, -СООС1-С6алкілом;
R1 являє собою гідроксизахисну групу, таку як Вn або С1-С6 алкіл;
R2 являє собою амінозахисну групу, таку як Cbz або Вос.
Текст
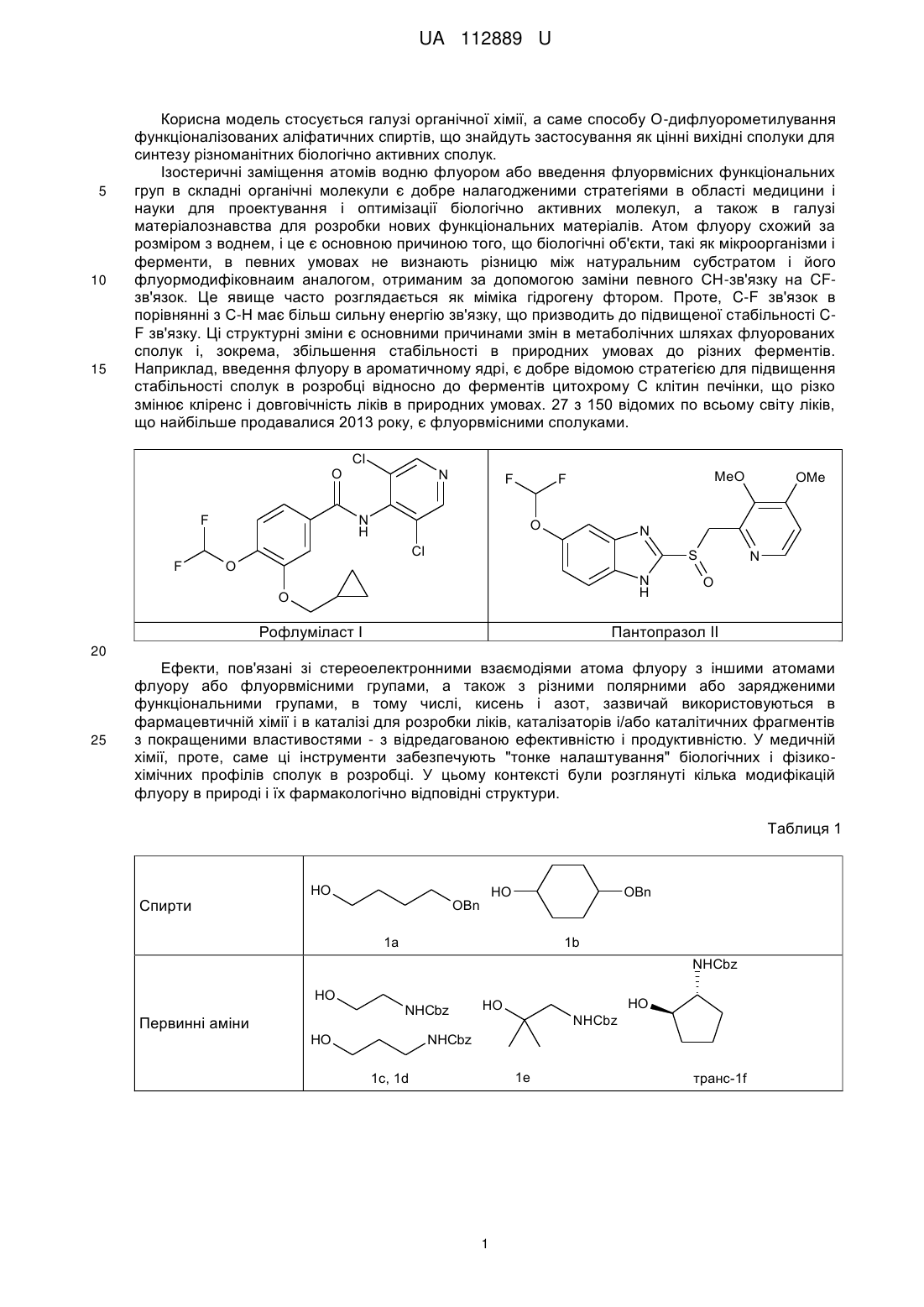
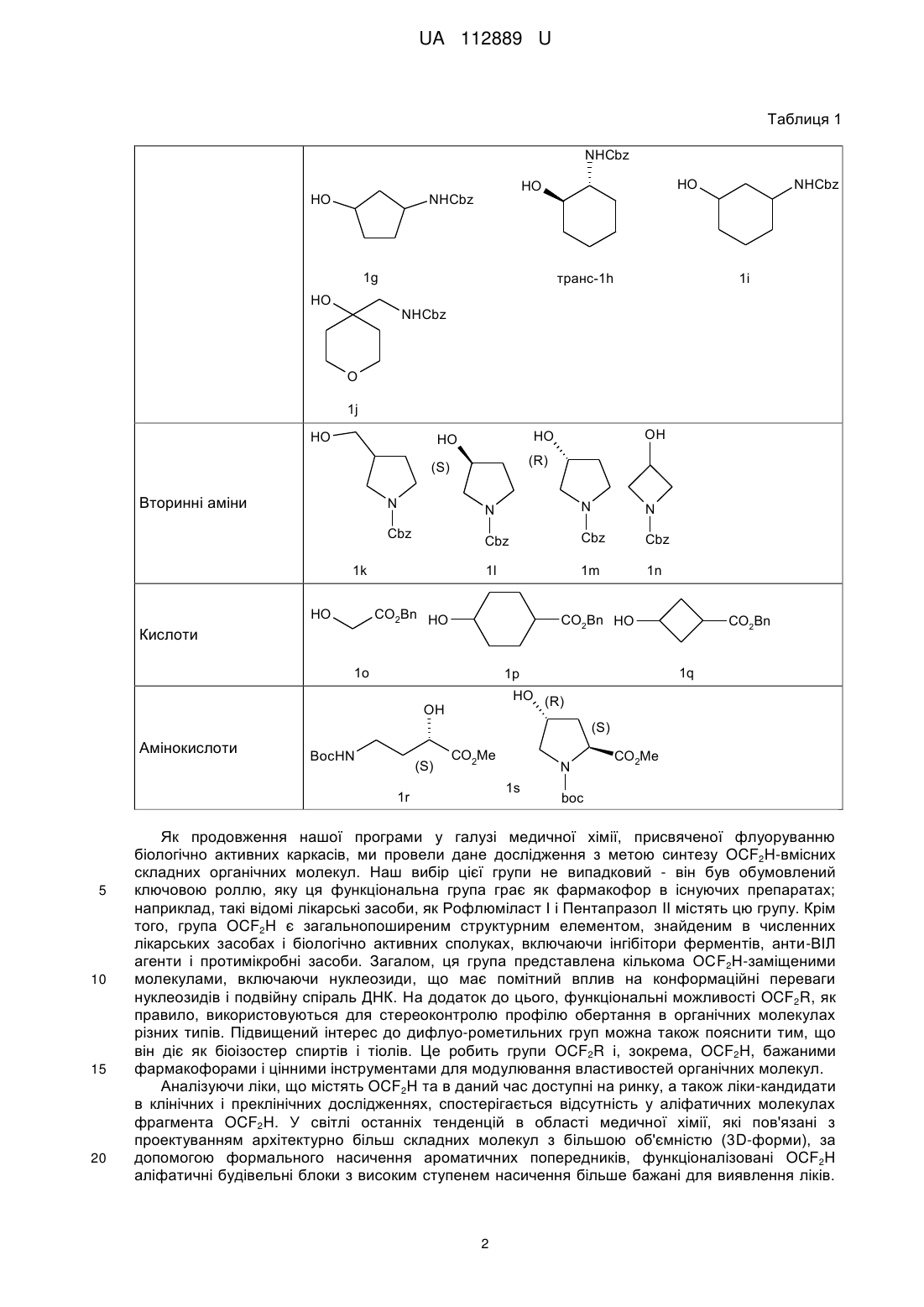
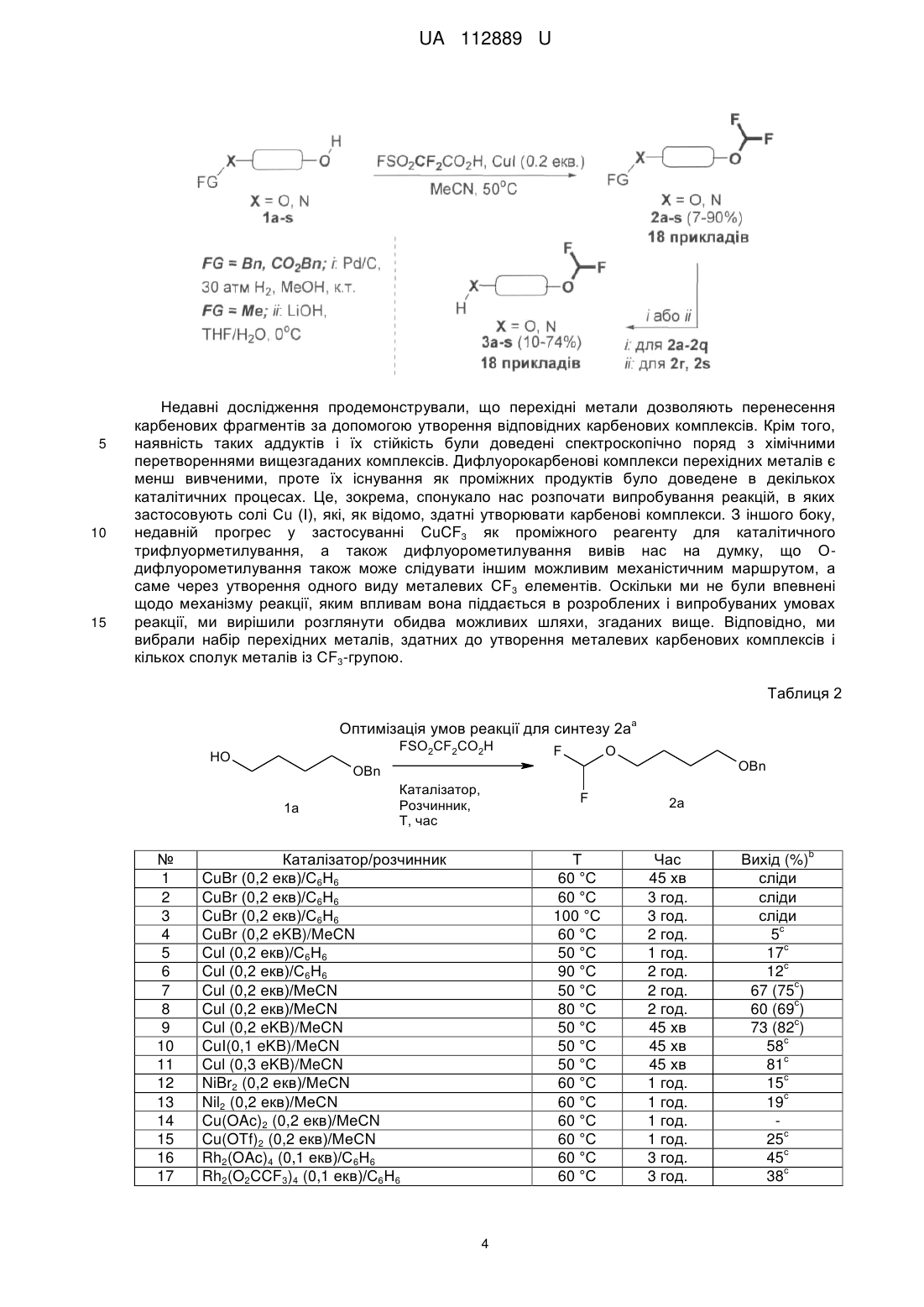
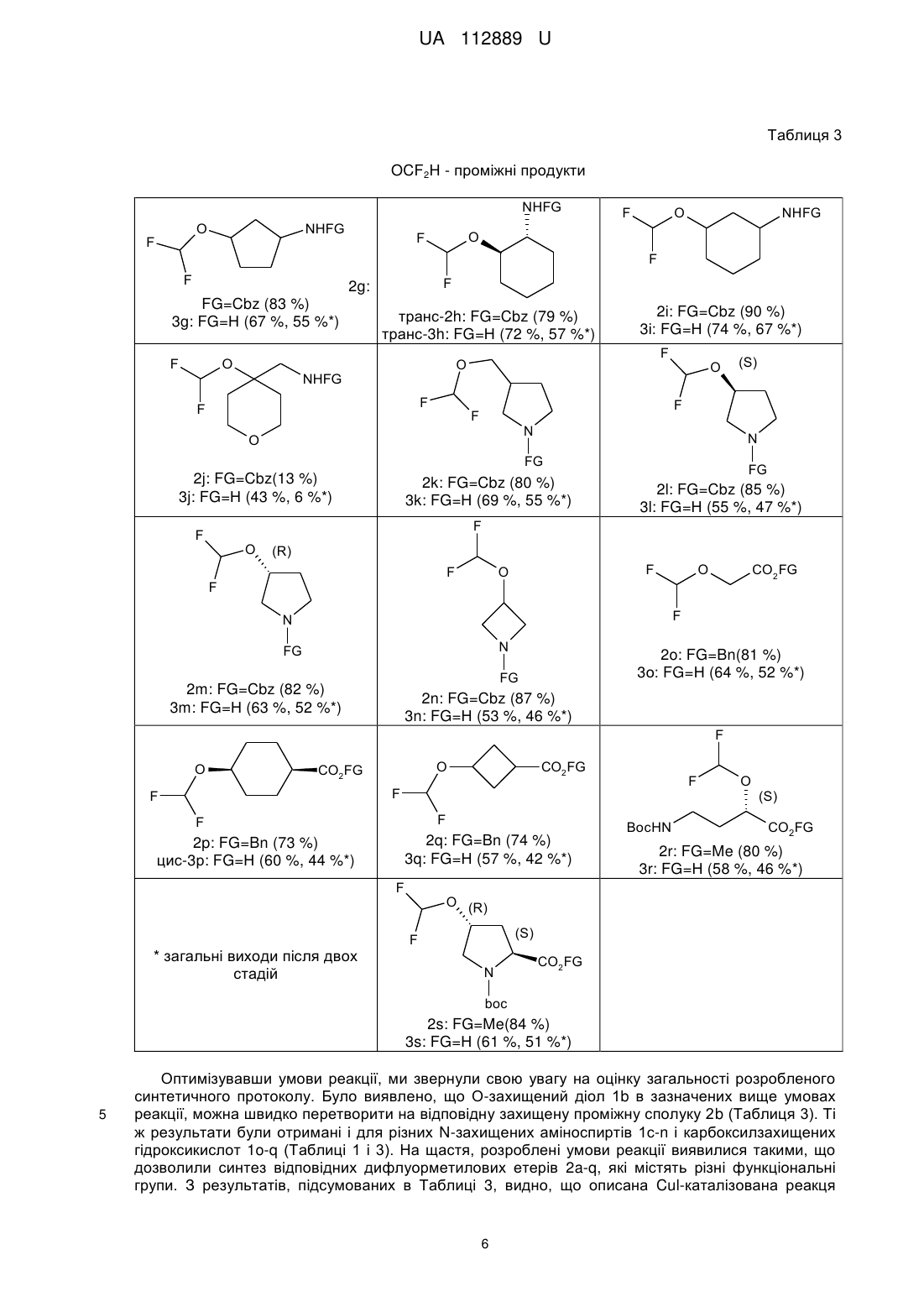
Реферат: Спосіб О-дифлуорометилування функціоналізованих аліфатичних спиртів. Функціоналізований аліфатичний спирт обробляють дифлуорометильованим агентом (FSO2CF2CO2H) в пристуності солі Сu (І) в органічному розчиннику при м'якому нагріванні. UA 112889 U (54) СПОСІБ О-ДИФЛУОРОМЕТИЛУВАННЯ ФУНКЦІОНАЛІЗОВАНИХ АЛІФАТИЧНИХ СПИРТІВ UA 112889 U UA 112889 U 5 10 15 Корисна модель стосується галузі органічної хімії, а саме способу О-дифлуорометилування функціоналізованих аліфатичних спиртів, що знайдуть застосування як цінні вихідні сполуки для синтезу різноманітних біологічно активних сполук. Ізостеричні заміщення атомів водню флуором або введення флуорвмісних функціональних груп в складні органічні молекули є добре налагодженими стратегіями в області медицини і науки для проектування і оптимізації біологічно активних молекул, а також в галузі матеріалознавства для розробки нових функціональних матеріалів. Атом флуору схожий за розміром з воднем, і це є основною причиною того, що біологічні об'єкти, такі як мікроорганізми і ферменти, в певних умовах не визнають різницю між натуральним субстратом і його флуормодифіковнаим аналогом, отриманим за допомогою заміни певного СН-зв'язку на CFзв'язок. Це явище часто розглядається як міміка гідрогену фтором. Проте, C-F зв'язок в порівнянні з С-Н має більш сильну енергію зв'язку, що призводить до підвищеної стабільності CF зв'язку. Ці структурні зміни є основними причинами змін в метаболічних шляхах флуорованих сполук і, зокрема, збільшення стабільності в природних умовах до різних ферментів. Наприклад, введення флуору в ароматичному ядрі, є добре відомою стратегією для підвищення стабільності сполук в розробці відносно до ферментів цитохрому С клітин печінки, що різко змінює кліренс і довговічність ліків в природних умовах. 27 з 150 відомих по всьому світу ліків, що найбільше продавалися 2013 року, є флуорвмісними сполуками. O F F Cl N N H MeO F F O N Cl O OMe S N H O Рофлуміласт І N O Пантопразол II 20 25 Ефекти, пов'язані зі стереоелектронними взаємодіями атома флуору з іншими атомами флуору або флуорвмісними групами, а також з різними полярними або зарядженими функціональними групами, в тому числі, кисень і азот, зазвичай використовуються в фармацевтичній хімії і в каталізі для розробки ліків, каталізаторів і/або каталітичних фрагментів з покращеними властивостями - з відредагованою ефективністю і продуктивністю. У медичній хімії, проте, саме ці інструменти забезпечують "тонке налаштування" біологічних і фізикохімічних профілів сполук в розробці. У цьому контексті були розглянуті кілька модифікацій флуору в природі і їх фармакологічно відповідні структури. Таблиця 1 Спирти HO HO OBn OBn 1a 1b NHCbz HO Первинні аміни NHCbz HO HO HO NHCbz NHCbz 1e 1c, 1d 1 транс-1f UA 112889 U Таблиця 1 NHCbz HO HO HO NHCbz 1g 1i транс-1h HO NHCbz NHCbz O 1j HO (R) (S) Вторинні аміни N N N Cbz Cbz 1l 1k N Cbz Cbz HO 1m 1n CO2Bn HO Кислоти OH HO HO CO2Bn HO 1o CO2Bn 1q 1p HO (R) OH (S) Амінокислоти BocHN (S) CO2Me 1s 1r 5 10 15 20 N CO2Me boc Як продовження нашої програми у галузі медичної хімії, присвяченої флуоруванню біологічно активних каркасів, ми провели дане дослідження з метою синтезу OCF2H-вмісних складних органічних молекул. Наш вибір цієї групи не випадковий - він був обумовлений ключовою роллю, яку ця функціональна група грає як фармакофор в існуючих препаратах; наприклад, такі відомі лікарські засоби, як Рофлюміласт І і Пентапразол II містять цю групу. Крім того, група OCF2H є загальнопоширеним структурним елементом, знайденим в численних лікарських засобах і біологічно активних сполуках, включаючи інгібітори ферментів, анти-ВІЛ агенти і протимікробні засоби. Загалом, ця група представлена кількома ОСF2Н-заміщеними молекулами, включаючи нуклеозиди, що має помітний вплив на конформаційні переваги нуклеозидів і подвійну спіраль ДНК. На додаток до цього, функціональні можливості OCF2R, як правило, використовуються для стереоконтролю профілю обертання в органічних молекулах різних типів. Підвищений інтерес до дифлуо-рометильних груп можна також пояснити тим, що він діє як біоізостер спиртів і тіолів. Це робить групи OCF2R і, зокрема, OCF2H, бажаними фармакофорами і цінними інструментами для модулювання властивостей органічних молекул. Аналізуючи ліки, що містять OCF2H та в даний час доступні на ринку, а також ліки-кандидати в клінічних і преклінічних дослідженнях, спостерігається відсутність у аліфатичних молекулах фрагмента OCF2H. У світлі останніх тенденцій в області медичної хімії, які пов'язані з проектуванням архітектурно більш складних молекул з більшою об'ємністю (3D-форми), за допомогою формального насичення ароматичних попередників, функціоналізовані OCF2H аліфатичні будівельні блоки з високим ступенем насичення більше бажані для виявлення ліків. 2 UA 112889 U 5 10 15 20 25 30 35 40 45 50 55 Дефіцит OCF2H аліфатичних структурних елементів є результатом слабкорозвинених синтетичних методик, що дозволяють отримати такі каркаси. Що стосується шляхів синтезу, відомих на сьогоднішній день для синтезу органічних сполук з групою OCF2H, існують чотири основні стратегії. А саме, (І) О-дифлуорометилування фенолів і спиртів з використанням різних джерел дифлуоркарбену; (II) С-O реакція сполучення з використанням хімічного еквівалента [ОСF2H]-синтону; (III) перетворення СО і CS груп; 13 (IV) нуклеофільний обмін інших галогенів на флуор в ОСНаl2Н групах. Що стосується стратегії (І), існує цілий ряд джерел дифлуорокарбену; наприклад такі хімічні реагенти, як HalCF2CO2Na; FSO2CF2CO2TMS; FSO2CF2CO2H; TMSCF2R (R=F, Cl, Br); (Et2O)2POCF2Br; CF3SO3CF2H і т.д. Проте, FSO2CF2CO2H через його високу ефективність, низьку ціну і синтетичну доступність часто вважається оптимальним реагентом для О-дифлуорометилування, зокрема, для великомасштабного синтезу. Незважаючи на різноманітність стратегій, відомих на сьогоднішній день, найбільш часто використовуваний синтетичний шлях в напрямку дифлуорметилових етерів зі спиртів вимагає використання озоноруйнуючого HCF2CI, так званого фреону-22. Менш шкідливі для навколишнього середовища препаративні методи, однак, часто вимагають підвищених температур, є несумісними з багатьма функціональними групами і ефективні тільки на структурно простих субстратах. Через вищезазначені обмеження використовуваних в даний час методик, розробки нових, простих і швидких методів для перетворення функціональних спиртів в дифлуорметилові етери, мають значну зацікавленість з боку науки. Відповідно, об'єктом корисної моделі є спосіб О-дифлуорометилування функціоналізованих аліфатичних спиртів, в якому функціоналізований аліфатичний спирт обробляють дифлуорометильованим агентом (FSO2CF2CO2H) в пристуності солі Сu (І) в органічному розчиннику при м'якому нагріванні. Як сіль міді може бути використана CuI або СuВr. Як розчинник може бути використаний ацетонітрил. Як функціоналізований аліфатичний спирт може бути використаний спирт формули (І): HO-R, де R являє собою С1-С6алкіл, С3-С10циклоалкіл, С6-С10арил або С2-С9 гетероарил, кожен з яких може бути заміщений, принаймні, одним С1-С6алкілом, С3-С10циклоалкілом, С6-С10 арилом, С21 2 етероарилом, -CN, NO2, OR , NHR , -SC1-C6алкілу, -SOC1-C6алкілом, -SO2C1-C6 алкілом, NSO2C1-C6алкілом, -NHC1-С6алкілом, -N(C1-C6алкілЬ, -NHCOOC1-С6алкілом, -NHCONHC1C6алкілом, -CONHC1-С6алкілом, -СОС1-С6алкілом, -СООС1-С6алкілом; 1 R являє собою гідроксизахисну группу, таку як Вn або С1-С6алкіл; 2 R являє собою амінозахисну группу, таку як Cbz або Вос. Розробка способу почалася з підготовки захищених субстратів 1a-s. Для даного проекту були вибрані О-захищені аліфатичні діоли 1а, b; N-захищені аміноспирти 1с-n; карбоксилзахищені гідроксикислоти 1m-q, а також N- і карбоксилзахищені амінокислоти, що мають вільну гідроксильну групу 1r, s (Таблиця 1). Спосіб, який описується в даній заявці є продовженням методології для нефункціоналізованих спиртів, розроблених Ченом (Chen, Q-Y.; Wu, S-W. J. Fluorine Chem. 1989, 44, 433-440.), що в нашому випадку охоплює функціоналізовані аліфатичні спирти і приводить до підвищення ефективності реакції і загальних виходів. В рамках поточної програми досліджень були розроблені різні бібліотеки складних ОСF2Н-вмісних органічних сполук, проте тут, як найбільш істотні, ми показуємо тільки з 18 прикладів (Таблиця 3). З набором вихідних сполук 1a-s (Таблиця 1) увага була приділена оптимізації стандартних умов реакції, що застосовується для цього перетворення (Схема 1, Таблиця 2). Варто відзначити, що умови запропонвоані Ченом вимагають від 2- до 3-кратного надлишку спирту разом з підвищеними температурами, і, на нашу думку, не можуть бути використані для приготування кількох грамів структурно різних OCF2H сполук. У той же час, в багатьох синтетичних методах, які відомі на сьогоднішній день, використовують сильні основи для дифлуоркарбенового синтезу. Ці методи, як правило, демонструють низьку функціональну толерантність функціональних груп і не можуть бути використані для функціоналізації складних органічних молекул на пізніх стадіях синтезу. Схема 1. Синтетична схема для одержання ОСF2Н-заміщених проміжних продуктів і кінцевих продуктів з незахищеними функціональними групами. 3 UA 112889 U 5 10 15 Недавні дослідження продемонстрували, що перехідні метали дозволяють перенесення карбенових фрагментів за допомогою утворення відповідних карбенових комплексів. Крім того, наявність таких аддуктів і їх стійкість були доведені спектроскопічно поряд з хімічними перетвореннями вищезгаданих комплексів. Дифлуорокарбенові комплекси перехідних металів є менш вивченими, проте їх існування як проміжних продуктів було доведене в декількох каталітичних процесах. Це, зокрема, спонукало нас розпочати випробування реакцій, в яких застосовують солі Сu (І), які, як відомо, здатні утворювати карбенові комплекси. З іншого боку, недавній прогрес у застосуванні CuCF3 як проміжного реагенту для каталітичного трифлуорметилування, а також дифлуорометилування вивів нас на думку, що Одифлуорометилування також може слідувати іншим можливим механістичним маршрутом, а саме через утворення одного виду металевих CF3 елементів. Оскільки ми не були впевнені щодо механізму реакції, яким впливам вона піддається в розроблених і випробуваних умовах реакції, ми вирішили розглянути обидва можливих шляхи, згаданих вище. Відповідно, ми вибрали набір перехідних металів, здатних до утворення металевих карбенових комплексів і кількох сполук металів із СF3-групою. Таблиця 2 Оптимізація умов реакції для синтезу 2а FSO2CF2CO2H HO O F OBn OBn 1a № 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 а Каталізатор, Розчинник, Т, час F Каталізатор/розчинник CuBr (0,2 екв)/С6Н6 CuBr (0,2 екв)/С6Н6 CuBr (0,2 екв)/С6Н6 CuBr (0,2 eKB)/MeCN Cul (0,2 екв)/С6Н6 Cul (0,2 екв)/С6Н6 Cul (0,2 екв)/МеСN Cul (0,2 екв)/МеСN Cul (0,2 eKB)/MeCN CuI(0,1 eKB)/MeCN Cul (0,3 eKB)/MeCN NiBr2 (0,2 екв)/МеСN Nil2 (0,2 екв)/МеСN Cu(OAc)2 (0,2 екв)/МеСN Cu(OTf)2 (0,2 екв)/МеСN Rh2(OAc)4 (0,1 екв)/С6Н6 Rh2(O2CCF3)4 (0,1 екв)/С6Н6 Τ 60 °C 60 °C 100 °C 60 °C 50 °C 90 °C 50 °C 80 °C 50 °C 50 °C 50 °C 60 °C 60 °C 60 °C 60 °C 60 °C 60 °C 4 2a Час 45 хв 3 год. 3 год. 2 год. 1 год. 2 год. 2 год. 2 год. 45 хв 45 хв 45 хв 1 год. 1 год. 1 год. 1 год. 3 год. 3 год. Вихід (%) сліди сліди сліди с 5 с 17 с 12 с 67 (75 ) с 60 (69 ) с 73 (82 ) с 58 с 81 с 15 с 19 с 25 с 45 с 38 b UA 112889 U а Оптимізація умов реакції проводили за шкалою 10 ммоль; 1,5 еквівалента FSO2CF2CO2H. Виходи. с ГХ/МС виявлені виходи. b 5 10 15 20 25 Речовина 1а була вибрана як модельна сполука. Після вибору оптимальних речовин для реакції - зміни розчинників, кількості і типу перехідних металів, температури і часу реакції - були розроблені оптимальні умови (Таблиця 2, запис 9). В основному були зосереджені на дослідженні CuBr і CuI як каталізаторів, що дозволяють введення групи CF2H. Спочатку, досліджувався CuBr (Таблиця 2, записи 1-4); на жаль, спостерігали утворення цільового продукту, але в невеликій кількості. Кращий результат отримували за рахунок використання комбінації CuBr з MeCN як розчинника. У цих умовах продукт 2а був виявлений за допомогою ГХ/МС з виходом 5 %. Крім того, система Cul/С6Н6 кардинально поліпшила ситуацію, забезпечуючи бажаний продукт з 17 % ГХ/МС-виходом (Таблиця 2, запис 5). Проте, ці результати були недостатні, щоб застосовувати цю хімію в таких умовах реакції. Вирішальним поліпшенням з точки зору прибутковості виявилося використання MeCN як розчинника, що дозволило збільшити ГХ/МС-вихід до 75 % (Таблиця 2, запис 7). Наступні важливі параметри при оптимізації умов реакції були спостереження по часу і температурі. Під час проведення дослідів зіткнулися з проблемою теплової нестійкості продукту 2а при Cu-каталізі (таблиця 2, запис 8). Це спостереження спонукало зменшити час реакції до 45 хв, а температуру реакції до 50 °C. Нарешті, були досягнуті умови синтезу відповідного дифлуорметилового етеру 2а з 82 % ГХ/МС-виходом і 73 % виділеного продукту (Схема 1, Таблиця 2). Кращий вихід отримано з використанням 1,5 еквівалента агента дифлуорометилування (FSO2CF2CO2H) і 0,2 еквівалентів Cul, в MeCN як середовища при температурі 50 °C (Таблиця 2, запис 9). Крім того, збільшення кількості CuI до 0,3 еквівалентів не збільшувало ефективність реакції. Проте, коли використали 0,1 еквівалент Cul, ГХ/МС-вихід значно знизився (запис 10). У той же час, інші перехідні метали, такі як Ni (II) солі (Таблиця 2, запис 12, 13) і Сu (II) (Таблиця 2, запис 14, 15), не були схильні до каталізу реакції достатнім чином. Навпаки, Rh (II) солі, відомі раніше для сприяння формуванню металокомплесів карбенів, продемонстрували перспективні результати при застосуванні в основній реакції (Таблиця 2, запис 16, 17). Проте, через низьку доступність і високу загальну вартість каталізаторів на основі Rh, виключили цей варіант для подальшої оптимізації умов реакції, базуючись на меті грам-масштабного виробництва бажаних ОСF2Н-заміщених речовин. 30 Таблиця 3 OCF2H - проміжні продукти O F OFG 2a: FG=Bn (73 %) 3a: FG=Η (74 %, 54 %*) O NHFG F 2d: FG=Cbz (86 %) 3d: FG=Η (73 %, 63 %*) NHFG OFG F F F O F O F F 2b: FG=Bn (73 %) 3b: FG=Η (65 %, 47 %*) F 2c: FG=Cbz (86 %) 3c: FG=H(10 %, 9 %*) NHFG O NHFG O F F 2e: FG=Cbz(7 %) 3e: FG=Η (25 %, 2 %*) 5 F транс-2f. FG=Cbz (67 %) транс-3f. FG=Η (71 %, 47 %*) UA 112889 U Таблиця 3 OCF2H - проміжні продукти NHFG O F NHFG F O NHFG O F F F F 2g: FG=Cbz (83 %) 3g: FG=Η (67 %, 55 %*) F O транс-2h: FG=Cbz (79 %) транс-3h: FG=Η (72 %, 57 %*) 2i: FG=Cbz (90 %) 3і: FG=Η (74 %, 67 %*) F O NHFG F F O F F N O N FG 2j: FG=Cbz(13 %) 3j: FG=Η (43 %, 6 %*) (S) 2k: FG=Cbz (80 %) 3k: FG=Η (69 %, 55 %*) FG 2l: FG=Cbz (85 %) 3l: FG=Η (55 %, 47 %*) F F O (R) F F F O O CO2 FG F N N FG 2o: FG=Bn(81 %) 3о: FG=Η (64 %, 52 %*) FG 2m: FG=Cbz (82 %) 3m: FG=Η (63 %, 52 %*) 2n: FG=Cbz (87 %) 3n: FG=Η (53 %, 46 %*) F O O CO2FG CO2FG F F F F F 2q: FG=Bn (74 %) 3q: FG=Η (57 %, 42 %*) 2p: FG=Bn (73 %) цис-3р: FG=Η (60 %, 44 %*) F * загальні виходи після двох стадій BocHN O (S) CO2FG 2r: FG=Me (80 %) 3r: FG=Η (58 %, 46 %*) O (R) (S) F N CO2FG boc 2s: FG=Me(84 %) 3s: FG=Η (61 %, 51 %*) 5 Оптимізувавши умови реакції, ми звернули свою увагу на оцінку загальності розробленого синтетичного протоколу. Було виявлено, що О-захищений діол 1b в зазначених вище умовах реакції, можна швидко перетворити на відповідну захищену проміжну сполуку 2b (Таблиця 3). Ті ж результати були отримані і для різних N-захищених аміноспиртів 1с-n і карбоксилзахищених гідроксикислот 1o-q (Таблиці 1 і 3). На щастя, розроблені умови реакції виявилися такими, що дозволили синтез відповідних дифлуорметилових етерів 2a-q, які містять різні функціональні групи. З результатів, підсумованих в Таблиці 3, видно, що описана Cul-каталізована реакця 6 UA 112889 U 5 10 15 дифлуорометилювання виявилася досить загальною. Отримані сполуки були або дуже в'язкі масла або рідини; таким чином, очищення здійснювалося за допомогою перегонки у вакуумі або препаративної колонкової хроматографії на силікагелі. У разі сполук 2е, 2j та 3с ми спостерігали падіння виходу. Для 2е і 2j, швидше за все, це може бути повязано зі стерічною об'ємністю відповідних субстратів 1е, 1j. Досить низький вихід аміну 3с може бути пояснений його високою леткістю. При масштабному отриманні сполук 2d, 2l, 2m, 2r та 2s кількість FSO2CF2CO2H може бути зменшена до 1,2-1,3 еквівалентів без видимої втрати виходу. Видалення конкретної захисної групи було досягнуто відповідно стандартноих умов реакції, які зазвичай використовуються в таких випадках (Схема 1). Наприклад, залишки Вn і СО2Вn, присутні в сполуках 2a-2q, були легко видалені з відповідних ефірів і амідів за допомогою каталітичного гідрування під високим тиском. Реакцію проводили в метанолі, з використанням 5 %-Pd/C як каталізатора при 30 атм Н2 при кімнатній температурі. Таку реакцію можна легко масштабувати у грамову- і декаграмову шкалу без подальшої оптимізації. Слід зазначити, що м'якість розробленої методології дозволила синтез енантіомерно чистих сполук, 3l і 3m від відповідних (S) і (R) прекурсорів без будь-яких помітних рацемізацій. Схема 2. Наступні хімічні процеси: Синтез (S)-3-(дифлуорметокси)піролідин-2-ону 5 за допомогою циклізації з основою-промотором (8)-метил 4-аміно-2-(дифлуорметокси)-бутаноатом 4. F 1) TFA CH2Cl2 2) K2CO3 F F O BocHN 20 25 30 35 40 45 50 (S) CO2Me F F O F H2N (S) 2r (S) O 4 CO2Me N H O 5 (52%) У світлі цих результатів, ми очікували, що така реакція може бути також застосована для диверсифікації біологічно активних сполук на пізніх стадіях синтезу, наприклад, гідроксилвмісних амінокислот. Через наше зацікавлення в флуор-модифікованих природних і напівприродних продуктах, таких як амінокислоти і нуклеозиди, для цього дослідження були вибрані дві моделі амінокислот; а саме (4R)-4-гідрокси-L-пролін та (2S)-2-гідрокси GABA ((2S)-4аміно-2-гідроксибутанова кислота). Слідуючи відомим способам синтезу, ці сполуки були перетворені в захищені похідні 1r і 1s, які були придатні для подальшої СF2-функціоналізації (Таблиця 1). Використовуючи оптимальні умови, відповідні сполуки були успішно перетворені в проміжні структури 2r і 2s з хорошими виходами (80 % і 84 % відповідно). Подальше омилення цих етерів амінокислот розчином LiOH в THF давало N-Вос-захищений (4R)-4(дифлуорметокси)-L-пролін 3r і N-трет-Вос-захищений (2S)-4-аміно-2-(дифлуорметокси)бутанову кислоту 3s з 61 % і 58 %-вим виходом відповідно. Вос- і трет-Вос захисні групи на функціональній аміногрупі не були видалені, оскільки відповідно до планів нашого фактичного дослідження ці будівельні блоки будуть використовуватися в подальшому для синтезу пептидів. Спроби включення синтезованого ОСF2Н-аналоаг L-проліну в оліго- і поліпептидні зв'язки в даний час проводяться в наших лабораторіях. Для того, щоб проілюструвати подальшу хімічну взаємодію, була зроблена синтетична трансформація 2r. Циклізація захищеного (2S)-2-гідрокси GABA була досягнута шляхом обробки зазначені сполуки за допомогою TFA з подальшою основною нейтралізацією K2СО3, яка призвела до фармакологічно релевантного (3S)-3-(дифлуорметокси)піролідин-2-ону 5 з 52 % виходом у вигляді чистого (3)-енантіомера (Схема 2). Рацемізація в ході реакції виявлена не була. У той же час, для того, щоб позбавитись від двох додаткових стадій, пов'язаних із захистом і зняттям захисту, ми помітили, що треба дослідити субстрати, що містять незахищені аміногрупи, наприклад гідроксилвмісні амінокислоти. На жаль, ці спроби зазнали невдачі, відповідні реакції призвели до утворення суміші неідентифікованих продуктів, що супроводжується утворенням смоли. Для підтвердження будови отриманих структурних каркасів 2, 3 і 5 ми в основному 1 13 використовували ЯМР. Характерні особливості ОСF2Н-продуктів в H і С спектрів ЯМР 1 19 рівність відповідних JC-F, JF-H констант зв'язку в H- і в F- спектрих ЯМР. Для структур з хіральним карбоном в безпосередній близькості до групи OCF2H ми спостерігали 7 UA 112889 U 5 10 15 20 25 30 35 40 45 50 55 діастереотопізм двох гемінальних атомів флуору з характерними спіновими будовами та спареними частками. Хімічні зсуви функції OCF2H відповідають раніше повідомленим даним. Таким чином, був розроблений простий спосіб О-дифлуорометилування різних функціональних спиртів FSO2CF2CO2H, який на даний момент є оптимальним реагентом дифлуорометилування. Cul-каталізовані реакції поліфункціональних спиртів забезпечують відповідні дифлуорметилові етери із виходами від помірних до високих. Крім того, цей спосіб відрізняється застосованістю до широкого кола субстратів, в тому числі аліфатичних спиртів, які мають додаткові захищені ОН, NH2, або групи СО2Н, не ставлячи під загрозу його ефективність і масштабованість. У міру вирішення такої задачі, ми бачимо розширення розробленого протоколу по синтезу інших ОСF2Н-модифікованих амінокислот, а також подальше отримання ОСF2Н-аналогів ряду фармакологічно відповідних пептидів. Експериментальна частина Розчинники очищали відповідно до стандартних процедур. Колонкову хроматографію 1 19 13 проводили з використанням Kieselgel Merck 60 (230-400 mesh) як нерухомої фази. H-, F-, СЯМР-спектри реєстрували при 499,9, 470,3 і 124,9 МГц. Хімічні зсуви наведені в м.ч. відносно 13 19 тетраметилсилану TMS (1H, С) або CFCI3 ( F) як внутрішні стандарти. ((4-(дифлуорометокси)бутокси)метил)бензен (2а). Сполуку 1а (15 г, 0,083 моль, 1 екв.) розчиняли 200 мл ацетонітрилу, до розчину додавали йодид міді (3,16 г, 0,017 моль, 0,2 екв). Суміш нагрівали до 50 °C і розчин 2-флуорсульфоніл2,2-дифлуороцтової кислоти (22,3 г, 0,125 моль, 1,5 екв) в 100 мл ацетонітрилу додавали по краплях протягом 45 хвилин. Реакційну суміш нагрівали протягом ще 30 хв при 50 °C. Потім ацетонітрил випарювали, залишок розчиняли в EtOAc і тверду фазу відфільтровували. Етилацетатний розчин концентрували у вакуумі з отриманням продукту 2а. Вихід = 73 % (14 г). Продукт являв собою прозору безбарвну рідину. 1 Н ЯМР (500 МГц, CDCl3): δ=1,71-1,74 (м, 4Н, СН2), 3,20, 3,49 (с, 2Н, СН2), 3,85, 4,18 (т, 2Н, 3 2 J=5,4 Гц, СН2), 4,49 (с, 2Н, CH2Ph), 6,16 (τ, 1H, JH-F=75,3 Гц, CF2H), 7,27-7,32 (м, 5Н, Ph). 19 2 F ЯМР (376 МГц, CDCl3): δ=-842 (д, 2F, J=76,0 Гц, CF2H). 13 1 C{ H} ЯМР (125,7 МГц, CDCl3): δ=25,9, 26,0 (CH2), 63,4 (τ, J=5,9 Гц, CH2OCF2H), 69,5 1 (CH2OCH2Ph), 72,8 (CH2Ph), 116,1 (τ, JC-F=259,0 Гц, CF2H), 127,4, 127,5, 128,3 (CHPh), 138,5 (CPh). Елементний аналіз для C12H16F2O2: С, 62,60; Η, 7,00 визначено: С, 62,74; Η, 7,07. 4-(Дифлуорометокси)бутан-1-ол (3а). Сполуку 2а (14 г, 0,061 моль, 1 екв) розчиняли в 100 мл метанолу і додавали до неї 1,5 г 5 % - Pd/C. Суміш перемішували при 30 атм Н2 протягом 96 годин при кімнатній температурі. Потім відфільтровали Pd/C і отриману суміш концентрували у вакуумі, отримуючи сполуку 3а. Вихід = 74 % (6,3 г). 1 Продукт являв собою бліду рідину. H ЯМР (500 МГц, CDCl3): δ=1,60-1,73 (м, 4Н, СН2), 2,78 3 3 2 (ш с, 1Н, ОН), 3,63 (т, 2Н, J=6,8 Гц, СН2), 3,85 (τ, 2Н, J=6,7 Гц, СН2), 6,17 (τ, 1H, JH-F=73,3 Гц, CF2H). 19 2 F ЯМР (376 МГц, CDCl3): δ=-84,5 (д, 2F, JH-F=75,1 Гц, CF2H). 13 1 С{ Н} ЯМР (125 МГц, CDCl3): δ=25,1, 29,3, 62,1 (СН2), 63,0 (τ, J=5,5 Гц, CH2OCF2H), 115,7 (τ, 1 JC-F=256,3 Гц, CF2H). Елементний аналіз для C5H10F2O2: С, 42,86; Η, 7,19. Визначено: С, 42,72; Η, 7,22. (((4-(Дифлуорометокси)циклогексил)окси)метил)бензен (2b) (суміш діастереомерів). Сполуку 1b (28,8 г, 0,14 моль, 1 екв) розчиняли в 300 мл ацетонітрилу і додавали до неї йодид міді (5,3 г, 0,028 моль, 0,2 екв). Суміш нагрівали до 50 °C, і в 100 мл ацетонітрилу додавали по краплях протягом 45 хвилин розчин 2-флуорсульфоніл-2,2-дифлуороуцтової кислоти (37,3 г, 0,21 моль, 1,5 екв). Реакційну суміш нагрівали протягом ще 30 хв при 50 °C. Потім ацетонітрил випарювали, залишок розчиняли в EtOAc і нерозчинну тверду речовину відфільтровували. Отриманий в результаті розчин EtOAc випарювали, а неочищений 2b очищали за допомогою колонкової хроматографії (EtOAc: гексан -1: 6, Rf=0,65). Вихід = 73 % (27,6 г). 1 Продукт являв собою прозоре безбарвне масло. Η ЯМР (400 МГц, CDCl3) (суміш діастереомерів): δ=1,15-1,19 (м, 2Н, СН2циклогексил), 1,69-1,72 (м, 2Н, СН2циклогексил), 1,95-1,99 (м, 2Н, СН2циклогексил), 2,07-2,14 (м, 2Н, СН2циклогексил), 3,50-3,53 (м, 1Н, СНциклогексил), 4,20-4,25 (м, 1Н, 2 СНциклогексил), 4,58 (с, 2Н, CH2Ph), 6,27 (дт, 1Н, JH-F=75,6 Гц, J=9,0 Гц, CF2H), 7,33-7,34 (м, 1 Η, Ph), 7,39-7,40 (м, 4Н, Ph). 19 2 F ЯМР (376 МГц, CDCl3): δ=-81,1 (τ, 2F, JH-F=77,0 Гц, CF2H). 8 UA 112889 U 13 5 10 15 20 25 30 35 40 45 50 55 1 С{ Н} ЯМР (100 МГц, DMSO-d6) (суміш діастереомерів): δ=27,4, 28,2, 28,4, 29,4 (СН2), 69,3, 1 69,7 (СН), 74,3, 75,0 (CH2Ph), 117,7 (дт, JC-F=258,5 Гц, J=12,2 Гц, CF2H), 127,6, 127,7, 128,6 (CHPh), 139,6, 139,6 (д, J=14,0 Гц, CPh). Елементний аналіз для C14H18F2O2: С, 65,61; Η, 7,08. Визначено: С, 65,43; Η, 7,11. 4-(Дифлуорометокси)циклогексанол (3b) (Суміш діастереомерів)· Сполуку 2b (25,5 г, 0,1 моль) розчиняли у 200 мл метанолу та додавали 3,5 г 5 %-Pd/C. Одержану суміш перемішували за кімнатної температури під 30 атм. Н2 протягом 96 годин. Потім Pd/C відфільтровували, і метанол відганяли на роторному випаровувачі. Неочищену суміш продуктів переганяли під вакуумом (1 Торр, 54 °C) і отримували 10,7 г сполуки 3b (вихід 64,7 %). 1 Продукт являв собою безбарвну речовину. H ЯМР (500 МГц, CDCl3) (суміш діастереомерів): δ=1,25-1,71 (м, 4Н, СН2циклогексил), 1,75 (с, 1Н, ОН), 1,90-2,02 (м, 4Н, СН2циклогексил), 3,73-3,75 (Μ, 2 1Η, СНциклогексил), 4,18 (М, 1H, СНциклогексил), 6,22(дт, 1Н, JH-F=75,9Гц, J=7,8Гц, СF2Н). 13 1 С{ Н} ЯМР (125 МГц, CDCl3) (суміш діастереомерів): δ=28,2, 29,3, 29,8, 31,5, 35,1, 68,2, 1 72,6 (циклогексил), 115,9 (τ, JC-F=259,6 Гц, CF2H). Елементний аналіз для C7H12F2O2: С, 50,60; Н, 7,28. Визначено: С, 50,39; Н, 7,25. Бензил-(2-(дифлуорометокси)етил)карбамат(2с). Сполуку 1с (6г, 0,0307 моль, 1 екв.) розчиняли у 60 мл ацетонітрилу та додавали CuI (1,15 г, 0,006 моль, 0,2 екв.). Реакційну суміш нагрівали до 50 °C, після чого по краплях додавали розчин 2-флуоросульфоніл-2,2-дифлуорооцтової кислоти (8,2 г, 0,046 моль, 1,5 екв) у 10 мл ацетонітрилу протягом 30 хвилин. Потім суміш перемішували ще 30 хвилин за 50 °C, після чого ацетонітрил упарювали під вакуумом. Залишок розчиняли у етилацетаті та відфільтровували тверду фазу. Ацетонітрил упарювали і одержували неочищену суміш, яку розділяли за допомогою колонкової хроматографії (гексан: EtOAc-4:1, Rf=0,65). Вихід = 86 % (6,35 г). 1 Продукт - прозора безбарвна густа олія. H ЯМР (400 МГц, CDCl3): δ=3,40 (ш с, 2Н, СН2), 2 3,86 (м, 2Н, СН2), 5,07 (с, 2Н, CH2Ph), 5,32 (ш с, 1H, NH), 6,15 (т, 1Н, JH-F=71,4 Гц, CF2H), 7,31(c, 5H, Ph). 19 2 F ЯМР (376 МГц, CDCl3): = -84,6 (д, 2F, JH-F=79,6 Гц, CF2H). 13 1 1 С{ H} ЯМР (125,7 МГц, CDCl3): =40,2, 62,5 (СН2), 66,9 (CH2Ph), 115,8 (τ, JC-F=260,0 Гц, CF2H), 127,9, 128,0, 128,4 (CHPh), 136,3 (CPh), 156,3 (C=O). Елементний аналіз для C11H13F2NO3: С, 53,88; Η, 5,34; Ν, 5,71. Визначено: С, 54,00; Η, 5,40; Ν, 5,65. 2-(Дифлуорометокси)етиламін (3с). Сполуку 2с (6,35 г, 0,026 моль, 1 екв.) розчиняли у 65 мл метанолу і додавали до розчину 0,6 г 5 %-Pd/C. Одержану суміш перемішували за кімнатної температури під 30 атм. Н 2 протягом 96 годин. Потім Pd/C відфільтровували і метанол відганяли на роторному випаровувачі. Неочищену суміш продуктів переганяли під вакуумом (18 Торр, 35 °C) і отримували 0,29 г сполуки 3с (вихід 10 %). Основна причина низького виходу - леткість продукту 3с. 1 Продукт являв собою прозору безбарвну рідину. H ЯМР (500 МГц, CDCl3): =1,22 (с, 2Н, 3 3 2 ΝΗ2), 2,85 (τ, 2Η, J=5,0 Гц, СH2NH2), 3,78 (τ, 2Η, J=5,0 Гц, СH2О), 6,17 (т, 1Н, JH-F=76,ЗГц, СF2Н). 19 2 F ЯМР (376 МГц, CDCl3): =-74,1 (д, 2F, JH-F=74,7 Гц, CF2H). 13 1 1 С{ H} ЯМР (125 МГц, CDCl3): =40,8, 65,5 (СН2), 115,7 (τ, JC-F=258,8 Гц, CF2H). Елементний аналіз для C3H7F2NO: С, 32,43; Η, 6,35; Ν, 12,61. Визначено: С, 32,22; Η, 6,28; Ν, 12,50. Бензил-(3-(дифлуорометокси)пропіл)карбамат(2а). Сполуку 1 d (60 г, 0,287 моль, 1 екв.) розчиняли у 600 мл ацетонітрилу та додавали CuI (10,95 г, 0,058 моль, 0,2 екв.). Реакційну суміш нагрівали до 50 °C, після чого по краплях додавали розчин 2-флуоросульфоніл-2,2-дифлуорооцтової кислоти (61,3 г, 0,344 моль, 1,5 екв) у 100 мл ацетонітрилу протягом 45 хвилин. Потім суміш перемішували ще 30 хвилин за 50 °C, після чого ацетонітрил упарювали під вакуумом. Залишок розчиняли у етилацетаті та відфільтровували тверду фазу. Ацетонітрил упарювали і одержували неочищену суміш, яку розділяли за допомогою колонкової хроматографії (гексан: EtOAc-4: 1, Rf=0,65). Вихід = 86 % (63 г). 1 Продукт являв собою прозору безбарвну густу олію. H ЯМР (400 МГц, CDCl3): =1,85-1,87 (м, 2Н, СН2), 3,31 (ш с, 2Н, СН2), 3,91 (м, 2Н, СН2), 4,99 (ш с, 1Н, NH), 5,10 (с, 2Н, CH2Ph), 6,20 (τ, 2 1H, JH-F=72,4 Гц, CF2H), 7,36 (с, 5Н, Ph). 19 2 2 F ЯМР (376 МГц, CDCl3): =-81,8 (дд, F, JH-F=76,8 Гц, CF2H), -81,0 (дд, F, JH-F=76,8 Гц, CF2H). 9 UA 112889 U C{ H} ЯМР (125,7 МГц, CDCl3): =29,4, 38,00, 61,4 (CH2), 66,8 (CH2Ph), 116,2 (τ, JC-F=261,3 Гц, CF2H), 128,2, 128,6 (CHPh), 136,7 (CPh), 156,7 (C=O). Елементний аналіз для C12H15F2NO3: С, 55,59; Η, 5,83; Ν, 5,40. Визначено: С, 55,75; Η, 5,87; Ν, 5,31. 3-(Дифлуорорметокси)пропан-1-амін (3d). Сполуку 2d (63 г, 0,243 моль, 1 екв.) розчиняли у 300 мл метанолу і додавали до розчину 5 г 5 %-Pd/C. Одержану суміш перемішували за кімнатної температури під 30 атм. Н 2 протягом 96 годин. Потім Pd/C відфільтровували і метанол відганяли на роторному випаровувачі. Неочищену суміш продуктів переганяли під вакуумом (18 Торр, 43 °C) і отримували 22,1 г сполуки 3d (вихід 72,6 %). 1 Продукт являв собою прозору безбарвну рідину. H ЯМР (400 МГц, CDCl3): =1,42 (с, 2Н, 3 ΝΗ2), 1,77 (квінт, 2Н, V=6,5 Гц, СH2СН2NH2), 2,81 (τ, 2Η, J=7,0 Гц, СН2СH2NH2), 3,92 (т, 2Н, 3 2 J=6,0 Гц, СH2О), 6,18 (т, 1Н, JH-F=75,2 Гц, CF2H). 19 2 F ЯМР (376 МГц, CDCl3): =-84,6 (д, 2F, JH-F=76,1 Гц, CF2H). 13 1 1 С{ H} ЯМР (100 МГц, CDCl3): =32,6, 38,6, 61,4 (СН2), 116,0 (τ, JC-F=272,0 Гц, CF2H). Елементний аналіз для C4H9F2NO: С, 38,40; Η, 7,25; Ν, 11,19. Визначений: С, 38,55; Η, 7,19; Ν, 11,13. Бензил-(2-гідрокси-2-метилпропіл)карбамат (1e). До перемішуваного розчину аміноспирту (49,6 г, 0,557 моль, 1 екв.) та карбонату калію (153,8 г, 1,115 моль, 2 екв) у суміші ТГФ-вода (800:400, мл:мл) при температурі 0 °C по краплинах додавали бензил хлороформіат (94,95 г, 0,557 моль, 1 екв). Після цього реакційну суміш перемішували при кімнатній температурі протягом 18 годин. По закінченні реакції, ТГФ упарювали на роторному випаровувачі, а залишок екстрагували тричі за допомогою дихлорометану. Органічну фазу сушили над сульфатом натрію, і після фільтрування випарювали на роторному випаровувачі. Неочищену суміш продуктів розділяли за допомогою колонкової хроматографії (етлацетат-петролейний ефір 1:3). Вихід - 71,9 % (89,3 г). 1 Продукт являв собою прозору жовтувату олію. H ЯМР (500 МГц, CDCl3): =1,19 (с, 6Н, 2хСН3), 2,66 (с, 1Н, ОН), 3,17 (с, 2Н, NHCH2), 5,09 (ш с, 2Н, CH2Ph, ), 5,45 (с, 1Н, NHCH2), 7,33 (ш с, 5Н, Ph). 13 1 С{ H } ЯМР (125 МГц, CDCl3): =27,1 (СН3), 51.8, 67,0 (СН2), 71,0 (С), 128,2, 128,3,128,6 (CHPh), 136,6 (CPh), 157,5 (С=О). Елементний аналіз для C12H17NO3: С, 64,55; Η, 7,67; Ν, 6,27; О, 21,50. Визначено: С, 64,34; Η, 7,79; Ν, 6,18. Бензил-(2-(дифлуорометокси)-2-метилпропіл)карбамат (2е). Сполуку 1e (89,3 г, 0,4 моль, 1 екв.) розчиняли у 750 мл ацетонітрилу та додавали CuI (15,25 г, 0,08 моль, 0,2 екв.). Реакційну суміш нагрівали до 50 °C, після чого по краплях додавали розчин 2-флуоросульфоніл-2,2-дифлуорооцтової кислоти (106,85 г, 0,6 моль, 1,5 екв) у 200 мл ацетонітрилу протягом 45 хвилин. Потім суміш перемішували ще 30 хвилин за 50 °C, після чого ацетонітрил упарювали під вакуумом. Залишок розчиняли у етилацетаті та відфільтровували тверду фазу. Ацетонітрил упарювали і одержували неочищену суміш, яку розділяли за допомогою колонкової хроматографії (гексан: EtOAc - 3: 1, Rf=0,6). Вихід = 6,8 % (7,4 г). 1 Продукт являв собою прозору безбарвну рідину. H ЯМР (400 МГц, CDCl3): =1,34 (с, 6Н, 3 2 2хСН3), 3,32 (д, 2H, J=6,7 Гц, NHCH2), 5,10 (ш с, 3Н, CH2Ph, NHCH2), 6,25 (τ, 1Н, JH-F=76,4 Гц, CF2H), 7,34 (ш с, 5Н, Ph). 19 2 F ЯМР (376 МГц, CDCl3): = -79,3 (д, 2F, JH-F=76,5 Гц, CF2H). 13 1 1 С{ H} ЯМР (125 МГц, CDCl3): =24,0 (Me), 50,3 (С), 67,0 (CH2Ph), 80,3 (CH2N), 115,6 (τ, JCF=250,8 Гц, CF2H), 128,2, 128,3,128,7 (CHPh), 136,6 (CPh), 157,0 (C=O). Елементний аналіз для C13H17F2NO3: С, 57,14; Η, 6,27; Ν, 5,13. Визначено: С, 56,98; Η, 6,30; Ν, 5,20. 2-(Дифлуорометокси)-2-метилпропан-1-амін (3е). Сполуку 2е (7,4 г, 0. 027 моль, 1 екв.) розчиняли у 100 мл метанолу і додавали до розчину 1 г 5 %-Pd/C. Одержану суміш перемішували за кімнатної температури під 30 атм. Н 2 протягом 48 годин. Потім Pd/C відфільтровували і метанол відганяли на роторному випаровувачі. Неочищену суміш продуктів переганяли під вакуумом (18 Торр, 32 °C) і отримували 0,95 г сполуки 3е (вихід 25,2 %). 1 Продукт являв собою прозору безбарвну рідину. H ЯМР (500 МГц, CDCl3): =1,33 (с, 6Н, 2 2хСН3), 1,37 (с, 2Н, СН2), 2,73 (с, 2Н, ΝΗ2), 6,35 (τ, 1Η, JH-F=75,9 Гц, CF2H). 19 2 F ЯМР (376 МГц, CDCl3): = -76,9 (д, 2F, JH-F=77,1 Гц, CF2H). 13 1 1 С{ H} ЯМР (125 МГц, CDCl3): =23,6 (СН3), 51,6 (СН2), 80,3 (С), 115,1 (т, JC-F=250,0 Гц, CF2H). 13 5 10 15 20 25 30 35 40 45 50 55 60 1 1 10 UA 112889 U 5 10 15 20 25 30 35 40 45 50 55 60 Елементний аналіз для C5H11F2NO: С, 43,16; Η, 7,97; Ν, 10,07. Визначено: С, 43,08; Η, 8,02; Ν, 10,01. Бензил-транс-2-(Дифлуорометокси)циклопентил)карбамат (2f). Сполуку 1f (41,6 г, 0,177 моль, 1 екв.) розчиняли у 400 мл ацетонітрилу та додавали CuI (6,7 г, 0,035 моль, 0,2 екв.). Реакційну суміш нагрівали до 50 °C, після чого по краплях додавали розчин 2-флуоросульфоніл-2,2-дифлуорооцтової кислоти (47,2 г, 0,265 моль, 1,5 екв) у 200 мл ацетонітрилу протягом 45 хвилин. Потім суміш перемішували ще 30 хвилин за 50 °C, після чого ацетонітрил упарювали під вакуумом. Залишок розчиняли у етилацетаті та відфільтровували тверду фазу. Ацетонітрил упарювали і одержували неочищену суміш, яку розділяли за допомогою колонкової хроматографії (гексан: EtOAc-2: 1, Rf=0,6). Вихід = 67 % (33,6 г). 1 Продукт являв собою прозору безбарвну густу олію. H ЯМР (500 МГц, CDCl3): =1,49-2,03 (м, 6Н, СН2циклопентил), 3,97 (ш с, 1Н, СНциклопентил), 4,39 (ш с, 1Н, СНциклопентил), 5,10 (ш с, 3Н, CH2Ph 2 і NH), 6,32 (τ, 1Н, JH-F=75,2 Гц, CF2H), 7,34 (ш с, 5Н, Ph). 19 F ЯМР (376 МГц, CDCl3): = -81,7 (м, 2F, CF2H). 13 1 С{ H} ЯМР (125 МГц, CDCl3): =29,3, 30,1, 53,1, 57,3, 66,3 (циклопентил), 80,1 (CH2Ph), 1 116,0 (τ, JC-F=255,9 Гц, CF2H), 127,7, 127,8, 128,1 (CHPh), 136,0 (CPh), 155,6 (C=O). Елементний аналіз для C14H17F2NO3: С, 58,94; Η, 6,01; Ν, 4,91. Визначено: С, 58,81; Η, 6,05; Ν, 4,82. транс-2-(Дифлуорометокси)циклопентанамін (3f). Сполуку 2f (33,6 г, 0,1177 моль, 1 екв.) розчиняли у 300 мл метанолу і додавали до розчину 4 г 5 %-Pd/C. Одержану суміш перемішували за кімнатної температури під 30 атм. Н2 протягом 36 годин. Потім Pd/C відфільтровували і метанол відганяли на роторному випаровувачі. Неочищену суміш продуктів переганяли під вакуумом (18 Торр, 67 °C) і отримували 12,6 г сполуки 3f (вихід 71 %). 1 Продукт являв собою прозору безбарвну рідину. H ЯМР (500 МГц, CDCl3): =1,21-1,27 (м, 3Н, ΝΗ2, СН2циклопентил), 1,60-1,66 (м, 3Н, ΝΗ2, СН2циклопентил), 1,88-1,98 (м, 2Н, СН2циклопентил), 3,162 3,20 (м, 1Н, СНциклопентил), 4,01-4,03 (м, 1Н, СН циклопентил), 6,16 (τ, 1Η, JH-F=76,3 Гц, CF2H). 19 2 2 F ЯМР (376 МГц,CDCl3): = -81,5 (дд, F, JF-F=164,6 Гц, JH-F=75,4 Гц, CF2H), -80,9 (дд, F, 2 2 JF-F=160,4 Гц, JH-F=76,0 Гц, CF2H). 13 1 С{ H} ЯМР (125 МГц, CDCl3): =19,8, 30,0, 31,3 (СН2циклопентил), 57,5, 83,5 (СНциклопентил), 115,8 1 (τ, JC-F=257,7 Гц, CF2H). Елементний аналіз для C6H11F2NO: С, 47,68; Η, 7,34; Ν, 9,27. Визначено: С, 47,80; Η, 7,38; Ν, 9,30. Бензил-(3-(дифлуорометокси)циклопентил)карбамат (2g) (суміш діастереомерів). Сполуку 1g (35,7г, 0,152 моль, 1 екв.) розчиняли у 350 мл ацетонітрилу та додавали CuI (5,8 г, 0,03 моль, 0,2 екв.). Реакційну суміш нагрівали до 50 °C, після чого по краплях додавали розчин 2-флуоросульфоніл-2,2-дифлуорооцтової кислоти (40,5 г, 0,03 моль, 1,5 екв) у 100 мл ацетонітрилу протягом 45 хвилин. Потім суміш перемішували ще 30 хвилин за 50 °C, після чого ацетонітрил упарювали під вакуумом. Залишок розчиняли у етилацетаті та відфільтровували тверду фазу. Ацетонітрил упарювали і одержували неочищену суміш, яку розділяли за допомогою колонкової хроматографії (гексан: EtOAc-2: 1, Rf=0,6). Вихід = 83 % (36 г). 1 Продукт являв собою прозору безбарвну олію. H ЯМР (400 МГц, CDCl3) (суміш діастереомерів): =1,66-1,93 (м, 2Н, СН2циклопентил), 1,96-2,21 (м, 4Н, СН2циклопентил), 4,20 (ш с, 1Н, 2 СНциклопентил), 4,68 (м, 1Н, СНциклопентил), 5,05 (ш с, 1Н, ΝΗ), 5,10 (с, 2Н, CH2Ph), 6,18 (дт, 1H, JHF=74,6 Гц, J=6,7 Гц, CF2H), 7,33-7,36 (м, 5Н, Ph). 19 F ЯМР (376 МГц, CDCl3): = -82,4 - -81,9 (м, 2F, CF2H). 13 1 С{ H} ЯМР (125 МГц, CDCl3) (суміш діастереомерів): =31,1, 31,5, 40,1, 50,5, 53,0, 60,0, 66,3 1 (циклопентил), 115,6 (τ, JC-F=259,6 Гц, CF2H), 127,8, 128,2, 136,1 (CHPh), 144,5 (СPh), 155,3 (C=O). Елементний аналіз для C14H17F2NO3: С, 58,94; Η, 6,01; Ν, 4,91. Визначено: С, 58,81; Η, 6,06; Ν, 4,87. 3-(Дифлуорометокси)циклопентиламін (3g) (суміш діастереомерів). Сполуку 2g (36г, 0,1261 моль, 1 екв.) розчиняли у 300 мл метанолу і додавали до розчину 4 г 5 %-Pd/C. Одержану суміш перемішували за кімнатної температури під 30 атм. Н2 протягом 36 годин. Потім Pd/C відфільтровували і метанол відганяли на роторному випаровувачі. Неочищену суміш продуктів переганяли під вакуумом (18 Торр, 65 °C) і отримували 12,7 г сполуки 3g (вихід 66,6 %). 1 Продукт являв собою прозору безбарвну олію. H ЯМР (400 МГц, CDCl3) (суміш діастереомерів): =1,24 (с, 2Н, ΝΗ2), 1,42-1,69 (м, 2Н, СН2циклопентил), 1,75-1,87 (м, 2Н, 3 СН2циклопентил), 1,91-2,15 (м, 2Н, СН2циклопентил), 3,24, 3,48 (квінтеt, 1H, J=6,0 Гц, СНциклопентил), 4,502 64 (м, 1H, СНциклопентил), 6,10 (дт, 1H, JH-F=75,7 Гц, J=13,7 Гц, CF2H). 11 UA 112889 U F ЯМР (376 МГц, CDCl3): = -82,2- -81,7 (м, 2F, CF2H). 1 С{ H} ЯМР (125 МГц, CDCl3) (суміш діастереомерів): =31,2, 31,5, 33,2, 33,8, 42,7, 42,8, 1 50,5, 51,1, 76,1, 76,2 (циклопентил), 115,8 (τ, JC-F=258,1 Гц, J=12,8 Гц, CF2H). Елементний аналіз для C6H11F2NO: С, 47,68; Η, 7,34; Ν, 9,27. Визначено: С, 47,81; Η, 7,40; Ν, 9,23. Бензил-(транс-2-(дифлуорометокси)циклогексил)карбамат(2b). Сполуку 1h (39 г, 0,156 моль, 1 екв.) розчиняли у 350 мл ацетонітрилу та додавали CuI (6г, 0,032 моль, 0,2 екв.). Реакційну суміш нагрівали до 50 °C, після чого по краплях додавали розчин 2-флуоросульфоніл-2,2-дифлуорооцтової кислоти (41,8 г, 0,235 моль, 1,5 екв) у 100 мл ацетонітрилу протягом 45 хвилин. Потім суміш перемішували ще 30 хвилин за 50 °C, після чого ацетонітрил упарювали під вакуумом. Залишок розчиняли у етилацетаті та відфільтровували тверду фазу. Ацетонітрил упарювали і одержували неочищену суміш, яку розділяли за допомогою колонкової хроматографії (гексан: EtOAc-2: 1, Rf=0,6). Вихід = 78,6 % (36,8 г). 1 Продукт являв собою прозору безбарвну густу олію. H ЯМР (500 МГц, CDCl3): =1,28-1,75 (м, 6Н, СН2циклогексил), 2,04-2,08 (м, 2Η, СН2циклогексил), 3,56 (ш с, 1Н, СНциклогексил), 3,92 (м, 1H, 2 СНциклогексил), 4,97 (ш с, 1Н, ΝΗ), 5,11 (с, 2Н, CH2Ph), 6,22 (τ, 1Η, JH-F=74,6 Гц, CF2H), 7,30-7,34 (м, 5Н, Ph). 19 F ЯМР (376 МГц, CDCl3): = -81,0 (м, 2F, CF2H). 13 1 С{ H} ЯМР (125 МГц, CDCl3): =23,5, 31,2, 31,9, 53,3 (СН2циклогексил), 60,0, 66,2 (СHциклогексил), 1 75,6 (CH2Ph), 115,6 (τ, JC-F=257,3 Гц, CF2H), 127,6, 128,1 (CHPh), 136,3 (CPh), 155,7 (C=O). Елементний аналіз для C15H19F2NO3: С, 60,19; Η, 6,40; Ν, 4,68. Визначено: С, 60,23; Η, 6,35; Ν, 4,60. транс-2-(Дифлуорометокси)циклогексанамін (3h). Сполуку 2h (30 г, 0,1 моль, 1 екв.) розчиняли у 300 мл метанолу і додавали до розчину 4 г 5 %-Pd/C. Одержану суміш перемішували за кімнатної температури під 30 атм. Н 2 протягом 36 годин. Потім Pd/C відфільтровували, і метанол відганяли на роторному випаровувачі. Неочищену суміш продуктів переганяли під вакуумом (18 Торр, 75 °C) і отримували 12 г сполуки 3h (вихід 72,5 %). 1 Продукт являв собою прозору безбарвну рідину. H ЯМР (500 МГц, CDCl3): =0,96-1,14 (м, 4Η, ΝΗ2, СН2циклогексил), 1,19-1,25 (м, 4Н, СН2циклогексил), 1,49-1,57 (м, 2Н, СН2циклогексил), 1,73-1,87 (м, 2 2Н, СН2циклогексил), 2,47-2,53 (м, 1Н, СНциклогексил), 3,43-3,48 (м, 1H, СНциклогексил), 6,14 (τ, 1Η, JHF=75,0 Гц, CF2H). 19 2 2 F ЯМР (376 МГц, CDCl3): - -80,7 (дд, F, JF-F=161,0 Гц, JH-F=70,0 Гц, CF2H), -79,0 (дд, 2F, 2 2 JF-F=155,0 Гц, JH-F=75,0 Гц, CF2H). 13 1 С{ H} ЯМР (125 МГц, CDCl3): =23,7, 23,8, 31,3, 33,0 (СН2циклогексил), 53,6, 81,5 (СНциклогексил), 1 116,1 (τ, JC-F=258,9 Гц, CF2H). Елементний аналіз для C7H13F2NO: С, 50,90; Η, 7,93; Ν, 8,48. Визначено: С, 50,79; Η, 7,96; Ν, 8,53. Бензил-(3-(дифлуорометокси)циклогексил)карбамат (2і) (Суміш діастереомерів). Сполуку 1і (49,8 г, 0,2 моль, 1 екв.) розчиняли у 450 мл ацетонітрилу та додавали CuI (7,6 г, 0,04 моль, 0,2 екв.). Реакційну суміш нагрівали до 50 °C, після чого по краплях додавали розчин 2-флуоросульфоніл-2,2-дифлуорооцтової кислоти (53,4 г, 0,3 моль, 1,5 екв) у 100 мл ацетонітрилу протягом 45 хвилин. Потім суміш перемішували ще 30 хвилин за 50 °C, після чого ацетонітрил упарювали під вакуумом. Залишок розчиняли у етилацетаті та відфільтровували тверду фазу. Ацетонітрил упарювали і одержували неочищену суміш, яку розділяли за допомогою колонкової хроматографії (гексан: EtOAc-2: 1, Rf=0,6). Вихід = 90,3 % (54г). 1 Продукт являв собою прозору безбарвну густу олію. H ЯМР (500 МГц, CDCl3) (суміш діастереомерів): =1,16-2,26 (м, 6Н, СН2циклогексил), 3,62, 3,90 (ш с, 1Н, СНциклогексил), 4,11, 4,48 (м, 2 1Н, СНциклогексил), 5,01-5,18 (м, 3Н, СН2циклогексил, ΝΗ), 5,26 (с, 2Н, CH2Ph), 6,20 (дт, 1H, JH-F=75,3 Гц, J=5,4 Гц, CF2H), 7,31-7,34 (м, 5Н, Ph). 19 2 2 F ЯМР (376 МГц, CDCl3): = -81,8 (д, F, J=74,0 Гц, CF2H), -81,0 (д, F, J=74,0 Гц, CF2H). 13 1 C{ H} ЯМР (125 МГц, CDCl3) (суміш діастереомерів): =18,9, 20,2, 30,3, 31,5, 37,3, 38,7, 1 45,5. 47,8 (СН2циклогексил), 53,1, 66,1 (СНциклогексил), 70,5, 72,3 (CH2Ph), 115,9 (дт, JC-F=258,1 Гц, J=10,6 Гц, CF2H), 127,7, 128,1 (CHPh), 136,3 (CPh), 155,2 (C=O). Елементний аналіз для C15H19F2NO3: С, 60,09; Η, 6,40; Ν, 4,68. Визначено: С, 60,22; Η, 6,42; Ν, 4,76. 3-(Дифлуорометокси)циклогексанамін (3і) (Суміш діастереомерів). Сполуку 2h (30 г, 0,1 моль, 1 екв.) розчиняли у 300 мл метанолу і додавали до розчину 4 г 5 %-Pd/C. Одержану суміш перемішували за кімнатної температури під 30 атм. Н 2 протягом 36 годин. Потім Pd/C відфільтровували і метанол відганяли на роторному випаровувачі. 19 1З 5 10 15 20 25 30 35 40 45 50 55 60 12 UA 112889 U 5 10 15 20 25 30 35 40 45 50 55 60 Неочищену суміш продуктів переганяли під вакуумом (18 Торр, 77 °C) і отримували 12,3 г сполуки 3h (вихід 74,3 %). 1 Продукт являв собою прозору безбарвну рідину H ЯМР (400 МГц, CDCl3) (суміш діастереомерів): =0,97-1,46 (м, 4Н, СН2циклогексил), 1,52 (с, 2Н, ΝΗ2), 1,49-1,57 (м, 2Н, СН2циклопентил), 1,62-2,18 (м, 2Н, СН2циклогексил), 2,68-3,11 (м, 1Н, СНциклогексил), 4,02-4,49 (м, 1H, 2 СНциклогексил), 6,19 (дт, 1Н, JH-F=76,0 Гц, J=4,6 Гц, CF2H). 19 F ЯМР (376 МГц, CDCl3): = -81,4 - -81,0 (м, 2F, CF2H). 13 1 С{ H} ЯМР (100 МГц, CDCl3) (суміш діастереомерів): =19,2, 21,7, 30,8, 32,2, 35,1, 35,3, 1 40,8, 43,2, 45,4, 49,0, 71,6, 73,0 (циклогексил), 116,1, 116,4 (τ, JH-F=253,4 Гц, CF2H). Елементний аналіз для C15H19F2NO3: С, 60,19; Η, 6,40; F, 12,69; Ν, 4,68. Визначено: С, 60,15; Η, 6,42; F, 12,65; Ν, 4,65. Бензил-((4-гідрокситетрагідро-2H-піран-4-іл)метил)карбамат(1]). До перемішуваного розчину аміноспирту (31,5 г, 0,516 моль, 2екв.) та карбонату калію (66,4 г, 0,481 моль, 1,9 екв) у суміші ТГФ-вода (400:200, мл:мл) при температурі 0 °C по краплинно додавали бензил хлороформіат (43 г, 0,252 моль, 1 екв). Після цього реакційну суміш перемішували при кімнатній температурі протягом 18 годин. По закінченні реакції, ТГФ упарювали на роторному випаровувачі, а залишок екстрагували тричі за допомогою дихлорометану. Органічну фазу сушили над сульфатом натрію і після фільтрування випарювали на роторному випаровувачі. Неочищену суміш продуктів розділяли за допомогою колонкової хроматографії (етлацетат-петролейний ефір 1:3). Вихід - 75,1 % (47,85 г). 1 Продукт являє білу тверду речовину, Тпл = 143-146 °C. H ЯМР (500 МГц, CDCl3): =1,481,51 (м, 2Η, СН2тетрагідропіран), 1,59-1,64 (м, 2Н, СН2 тетрагідропіран), 2,72 (с, 1Н, ОН), 3,21 (ш с, 2Н, СН 2 тетрагідропіран), 3,72 (ш с, 4Н, СН2 тетрагідропіран), 5,09 (с, 2Н, СН2Рh), 5,41 (ш с, 1Η,ΝΗ), 7,34 (с, 5Н, Ph). 13 1 С{ H} ЯМР (125 МГц, CDCl3): =35,5, 51,3, 63,7, 67,2 (тетрагідропіран), 69,2 (CH2Ph), 128,2, 128,4, 128,7 (CHPh), 136,4 (CPh), 157,7 (C=O). Елементний аналіз для C1H19NO4: С, 63,38; Η, 7,22; Ν, 5,28. Визначено: С, 63,22; Η, 7,27; Ν, 5,25. Бензил-((4-(дифлуорометокси)тетрагідро-2H-піран-4-іл)метил)карбамат (2j). Сполуку 1j (47,85 г, 0,18 моль, 1 екв.) розчиняли у 450 мл ацетонітрилу та додавали CuI (6,9 г, 0,036 моль, 0,2 екв.). Реакційну суміш нагрівали до 50 °C, після чого по краплях додавали розчин 2-флуоросульфоніл-2,2-дифлуорооцтової кислоти (48,3 г, 0,271 моль, 1,5 екв) у 100 мл ацетонітрилу протягом 45 хвилин. Потім суміш перемішували ще 30 хвилин за 50 °C, після чого ацетонітрил упарювали під вакуумом. Залишок розчиняли у етилацетаті та відфільтровували тверду фазу. Ацетонітрил упарювали і одержували неочищену суміш, яку розділяли за допомогою колонкової хроматографії (гексан: EtOAc-2:1, Rf=0,65). Вихід =12,8 % (7,3 г). 1 Продукт являв собою прозору безбарвну густу олію. H ЯМР (500 МГц, CDCl3): =1,70-1,83 (м, 4Η, СН2 тетрагідропіран), 3,44-3,46 (м, 2Н, СН2 тетрагідропіран), 3,74 (ш с, 4Н, NHСH2, СН2 тетрагідропіран), 2 5,11 (с, 2Н, CH2Ph), 5,23 (ш с, 1Н, NH), 6,36 (т, 1H, JH-F=76,0 Гц, CF2H), 7,35 (с, 5Н, Ph). 19 2 F ЯМР (376 МГц, CDCl3): = -77,6 (д, 2F, JH-F=77,7 Гц, CF2H). 13 1 С{ H} ЯМР (125 МГц, CDCl3): =33,3, 47,6, 63,3, 67,3 (тетрагідропіран), 78,8 (CH2Ph), 114,7 1 (τ, JC-F=264,6 Гц, CF2H), 127,7, 127,8, 128,2 (CHPh), 135,8 (CPh), 156,4 (C=O). Елементний аналіз для C15H19F2NO4: С, 57,14; Η, 6,07; Ν, 4,44. Визначено: С, 57,01; Η, 6,13; Ν, 4,41. (4-(Дифлуорометокси)тетрагідро-2H-піран-4-іл)метанамін (3j). Сполуку 2j (7,3 г, 0,232 моль, 1 екв.) розчиняли у 100 мл метанолу і додавали до розчину 1 г 5 %-Pd/C. Одержану суміш перемішували за кімнатної температури під 30 атм. Н 2 протягом 48 годин. Потім Pd/C відфільтровували і метанол відганяли на роторному випаровувачі. Неочищену суміш продуктів переганяли під вакуумом (18 Торр, 44 °C) і отримували 1,8 г сполуки 3h (вихід 42,9 %). 1 Продукт являв собою прозору безбарвну рідину H ЯМР (400 МГц, CDCl3): =1,24 (м, 2Η, ΝΗ2), 1,64-1,86 (м, 4Η, СН2тетрагідропіран), 2,87 (с, 2Η, NH2CH2), 3,74-3,76(м, 4Н, СН2тетрагідропіран), 6,54 2 (τ, 1Н, J=77,8 Гц, CF2H). 19 2 F ЯМР (376 МГц, CDCl3): = -77,2 (д, 2F, JH-F=84,7 Гц, CF2H). 13 1 С{ H}ЯМР (100 МГц, CDCl3): =33,2, 49,3 (СН2тетрапдропіран), 63,1 (Стетрапдропіран), 79,4 (CH2NH2), 1 115,2 (τ, JC-F=231,2 Гц, CF2H). Елементний аналіз для C7H13F2NO2: С, 46,40; Η, 7,23; Ν, 7,73. Визначено: С, 46,53; Η, 7,27; Ν, 7,68. Бензил-3-((дифлуорометокси)метил)піролідин-1-карбоксилат (2k). Сполуку 1k (40г, 0,17 моль, 1 екв) розчиняли в 400 мл ацетонітрилу і до неї додавали йодид міді (6,5 г, 0,034 моль, 0,2 екв). Суміш нагрівали до 50 °C і в 100 мл ацетонітрилу додавали по 13 UA 112889 U 5 10 15 20 25 30 35 40 45 50 55 60 краплях протягом 45 хвилин розчин 2-флуорсульфонілової-2,2-дифлуороцтової кислоти (45,4 г, 0,2549 моль, 1,5 екв). Реакційну суміш нагрівали протягом ще 30 хв при 50 °C. Далі, ацетонітрил випарювали, залишок розчиняли в EtOAc і відфільтровували нерозчинну тверду речовину. Отриманий в результаті розчин EtOAc випарювали, а неочищений 2к очищали за допомогою колонкової хроматографії (гексан: EtOAc-7: 1, Rf=0,6). Вихід = 80 % (39 г). 1 Продукт являв собою прозоре густе масло. H ЯМР (400 МГц, CDCl3): =1,67 (м, 1Н, СН2піролідин), 1,99 (ш с, 1Н, пік під сигналом Ме-групи від етилацетату, СН2піролідин), 2,48 (м, 1Н, СНпіролідин), 3,14-3,37 (м, 2Н, СН2піролідин), 3,49-3,81 (м, 4Н, ОСН2, СН2піролідин), 5,08 (с, 2Н, CH2Ph), 2 6,16 (τ, 1H, JH-F=74,7 Гц, CF2H), 7,28-7,33 (м, 5Н, Ph). 19 2 2 F ЯМР (376 МГц, CDCl3): = -85,0 (д, F, JH-F=74,2 Гц, CF2H), -84,9 (д, F, JH-F=74,2 Гц, CF2H). 13 1 C{ H} ЯМР (125,7 МГц, CDCl3): =27,5, 28,3, 37,6, 38,5, 45,2, 45,6, 48,5, 48,9 (піролідин), 1 63,3 (CH2O), 66,9 (CH2Ph), 115,9 (τ, JC-F=262,3 Гц, CF2H), 128,0, 128,1, 128,6 (CHPh), 137,1 (CPh), 154,9 (C=O). Елементний аналіз для C14H17F2NO3: С, 58,99; Η, 6,01; Ν, 4,91. Визначено: С, 58,93; Η, 6,07; Ν, 4,83. 3-((Дифлуорометокси)метил)піролідин (3k). Сполуку 2k (30 г, 0,105 моль, 1 екв) розчиняли в 150 мл метанолу і додавали до нього 3 г 5 %-Pd/C %. Суміш перемішували при 30 атм Н2 протягом 36 год. при кімнатній температурі. Після завершення реакції Pd/C відфільтровували і реакційну суміш концентрували у вакуумі. Продукт 3k очищали перегонкою під вакуумом (18 Торр, 57 °C). Вихід -68,6 % (10,9 г). 1 Продукт являв собою прозору безбарвну рідину. H ЯМР (400 МГц, CDCl3): =1,15-1,20 (м, 1H, СН2піролідин), 1,61-1,70 (м, 1Н, СН2піролідин), 1,75 (с, 1Н, ΝΗ), 2,11-2,18 (м, 1Н, СНпіролідин), 2,412 2,45 (м, 1Н, СН2піролідин), 2,59-2,80 (м, 3Н, СН2піролідин), 3,46-3,56 (м, 2Н, ОСН2), 5,95 (т, 1H, JHF=75,1 Гц, CF2H). 19 2 F ЯМР (376 МГц, CDCl3): = -84,5 (д, 2F, JH-F=77,0 Гц, CF2H). 13 1 С{ H} ЯМР (125 МГц, CDCl3): =28,4, 37,8, 46,2, 49,5 (СН2), 65,5 (τ, J=4,7 Гц, CH), 115,4 (τ, 1 JC-F=258,8 Гц, CF2H). Елементний аналіз для C6H11F2NO: С, 47,68; Η, 7,34; Ν, 9,27. Визначено: С, 47,81; Η, 7,29; Ν, 9,21. (5)-бензил 3-(дифлуорометокси)піролідин-1-карбоксилат (2l). Сполуку 1l (50 г, 0,226 моль, 1 екв) розчиняли в 400 мл ацетонітрилу і додавали до неї йодид міді (8,6 г, 0,045 моль, 0,2 екв). Суміш нагрівали до 50 °C і додавали по краплях протягом 45 хвилин розчин 2-флуорсульфоніл-2,2-дифлуороцтової кислоти (52,3 г, 0,294 моль, 1,3 екв) в 100 мл ацетонітрилу. Реакційну суміш нагрівали протягом ще 30 хв при 50 °C. Далі, ацетонітрил випарювали, залишок розчиняли в EtOAc і нерозчинні тверді речовини відфільтровували. Отриманий в результаті розчин EtOAc випарювали, а неочищений 2l очищали за допомогою колонкової хроматографії (гексан: EtOAc-7: 1, Rf=0,6). Вихід = 84,8 % (52 г). 1 Продукт являв собою прозоре безбарвне масло. H ЯМР (400 МГц, CDCl3, Етил ацетат всередині): =2,06 (ш с, 2Н, СН2піролідин), 3,50-3,58 (м, 4Н, СН2піролідин), 4,79 (м, 1H, СНпіролідин), 5,11 2 (с, 2Н, CH2Ph), 6,20 (τ, 1H, JH-F=74,8 Гц, CF2H), 7,28-7,34 (м, 5Н, Ph). 19 2 F ЯМР (376 МГц, CDCl3): = -83,1 (д, 2F, JH-F=72,5 Гц, CF2H). 13 1 С{ H} ЯМР (125,7 МГц, CDCl3): =31,6, 32,4, 43,6, 44,0, 52,0, 52,3 (СН2піролідин), 66,9 (CH2Ph), 1 72,5, 73,1 (СНпіроліднн), 115,8 (τ, JC-F=273,0 Гц, CF2H), 127,9, 128,0, 128,5 (CHPh), 136,8 (СPh), 154,8 (C=O). Елементний аналіз для С13Н15F2NO3: С, 57,56; Η, 5,57; Ν, 5,16. Визначено: С, 57,70; Η, 5,61; Ν, 5,11. (5)-3-(біфлуорометокси)піролідин (3l). Сполуку 2l (52 г, 0,192 моль, 1 екв) розчиняли в 300 мл метанолу і до нього додавали 5 г 5 % -Pd/С. Суміш перемішували при 30 атм Н2 протягом 36 год. при кімнатній температурі. Потім Pd/C відфільтровували і реакційну суміш концентрували у вакуумі. Продукт 3l очищали перегонкою під вакуумом (18 Торр, 51 °C). Вихід = 55,2 % (14,5 г). 1 Продукт являв собою прозору безбарвну рідину. [α] D = -9,03 (EtOH, c=72,925 ммоль/л). H ЯМР (400 МГц, CDCl3): =1,87-2,00 (м, 3Н, ΝΗ, СН2піролідин), 2,81-3,11 (м, 4Н, СН2піролідин), 4,71 (ш 2 с, 1H, СНпіролідин), 6,17 (т, 1H, JH-F=74,6 Гц, CF2H). 19 2 F ЯМР (376 МГц, CDCl3): = -82,1 (д, 2F, JH-F=74,1 Гц, CF2H). 13 1 1 С{ H} ЯМР (100 МГц, CDCl3): =33,6, 45,5, 54,0 (СН2піролідин), 75,8 (СНпіролідин), 116,0 (τ, JCF=258,4 Гц, CF2H). Елементний аналіз для C5H9F2NO: С, 43,79; Η, 6,62; Ν, 10,21. Визначено: С, 43,65; Η, 6,65; Ν, 10,15. 14 UA 112889 U 5 10 15 20 25 30 35 40 45 50 55 60 (R)-бензил 3-(дифлуорометокси)піролідин-1-карбоксилат (2m). Сполуку 1m (40 г, 0,181 моль, 1екв) розчиняли в 400 мл ацетонітрилу і додавали до неї йодид міді (6,9 г, 0,036 моль, 0,2 екв). Суміш нагрівали до 50 °C і додавали по краплях протягом 45 хвилин розчин 2-флуорсульфоніл-2,2-дифлуороцтової кислоти (41,9 г, 0,235 моль, 1,3 екв) в 100 мл ацетонітрилу. Реакційну суміш нагрівали протягом ще 30 хв при 50 °C. Потім ацетонітрил випарювали, залишок розчиняли в EtOAc, а нерозчинні тверді речовини фільтрували. Отриманий в результаті розчин EtOAc випарювали, а неочищений 2m очищали за допомогою колонкової хроматографії (гексан: EtOAc-7: 1, Rf-0,6). Вихід = 82,6 % (40,5 г). 1 Продукт являв собою прозоре безбарвне масло. H ЯМР (500 МГц, DMSO-d6): =2,08 (ш с, 2Н, СН2піролідин), 3,55-3,57 (м, 4Н, СН2піролідин), 4,79 (м, 1Н, СНпіролідин), 5,12 (с, 2Н, CH2Ph), 6,21 (τ, 2 1Н, JH-F=74,4 Гц, CF2H), 7,29-7,35 (м, 5Н, Ph). 19 2 F ЯМР (376 МГц, CDCl3): = -83,0 (д, 2F, JH-F=74,5 Гц, CF2H). 13 1 С{ H} ЯМР (125 МГц, CDCl3): =31,2, 31,9, 43,2, 43,5, 51,5, 51,9 (СН2піролідин), 66,5, 72,2 1 (СНпіролідин), 72,9 (CH2Ph), 115,5 (τ, JC-F=261,9 Гц, CF2H), 127,5, 128,1 (CHPh), 136,4 (CPh), 155,3 (C=O). Елементний аналіз для С13Н13F2NO3: С, 57,56; Η, 5,57; Ν, 5,16. Визначено: С, 57,41; Η, 5,52; Ν, 5,13. (R)-3-(Дифлуорометокси)піролідин (3m). Сполуку 2m (40,5 г, 0,149 моль, 1 екв) розчиняли в 300 мл метанолу і до неї додавали 5 г 5 % - Pd/C. Суміш перемішували при 30 атм Н2 протягом 36 год. при кімнатній температурі. Потім Pd/C відфільтровували і отриману суміш концентрували у вакуумі. Продукт 3m очищали перегонкою під вакуумом (18 Торр, 51 °C). Вихід = 63,5 % (13 г). 1 Продукт являв собою прозору безбарвну рідину. [α]D = +6,31 (EtOH, c=72,925 mmol/L). H ЯМР (500 МГц, CDCl3): =1,95-2,05 (м, 2Н, СН2піролідин), 2,92-3,14 (м, 3Н, СН2піролідин, СНпіролідин), 2 3,43 (с, 2Н, СН2піролідин), 4,75 (с, 1Н, ΝΗ), 6,20 (τ, 1Η, JH-F=74,4 Гц, CF2H). 19 2 F ЯМР (376 МГц, CDCl3): = -82,3 (д, 2F, JH-F=74,5 Гц, CF2H). 13 1 1 С{ H} ЯМР (125 МГц, CDCl3): =33,3, 45,1, 53,5 (СН2), 75,4 (СН), 116,0 (т, JC-F=295,8 Гц, CF2H). Елементний аналіз для C5H9F2NO: С, 43,79; Η, 6,62; Ν, 10,21. Визначено: С, 43,72; Η, 6,70; Ν, 10,15. Бензил 3-(дифлуорометокси)азетидин-1-карбоксилат (2n). Сполуку 1n (44,7г, 0,218 моль, 1 екв) розчиняли в 400 мл ацетонітрилу і додавали до неї йодид міді (8,2 г, 0,043 моль, 0,2 екв). Суміш нагрівали до 50 °C, і додавали по краплях протягом 45 хвилин розчин 2-флуорсульфоніл-2,2-дифлуороцтової кислоти (57,65 г, 0,324 моль, 1,5 екв) в 100 мл ацетонітрилу. Реакційну суміш нагрівали протягом ще 30 хв при 50 °C. Потім ацетонітрил випарювали, залишок розчиняли в EtOAc і нерозчинну тверду речовину відфільтровули. Після цього отриманий розчин етилацетату випарювали і неочищений 2n очищали за допомогою колонкової хроматографії (гексан: EtOAc-7: 1, Rf=0,6). Вихід = 86,5 % (48 г). 1 Продукт являв собою прозоре жовтувате масло. H ЯМР (400 МГц, CDCl3): =4,06-4,09 (м, 2Η, СН2азетидин), 4,26-4,31 (м, 2Н, СН2азетидин), 4,93-4,94 (м, 1Н, СНазетидин), 5,11 (с, 2Н, CH2Ph), 6,24 2 (τ, 1H, JH-F=73,0 Гц, CF2H), 7,36 (ш с, 5Н, Ph). 19 2 F ЯМР (376 МГц, CDCl3): = -84,9 (д, 2F, JH-F=74,9 Гц, CF2H). 13 1 С{ H} ЯМР (125 МГц, CDCl3): =56,8 (СН2), 62,1 (τ, 66,3 J=6,5 Гц, СН2), 66,8 (СH), 115,3 (τ, 1 JC-F=260,6 Гц, CF2H), 127,9, 128,0, 128,4 (CHPh), 136,3 (CPh), 156,1 (C=O). Елементний аналіз для C12H13F2NO3: С, 56,03; Η, 5,09; Ν, 5,45. Визначено: С, 55,89; Η, 5,16; Ν, 5,39. 3-(Дифлуорометокси)азетидин (3n). Сполуку 2n (48 г, 0,187 моль, 1 екв) розчиняли в 300 мл метанолу і до неї додавали 5 г 5 % Pd/C. Суміш перемішували при 30 атм Н 2 протягом 36 год. при кімнатній температурі. Потім Pd/C відфільтровували і реакційну суміш концентрували у вакуумі. Продукт 3n очищали перегонкою під вакуумом (18 Торр, 47 °C). Вихід = 53,5 % (12,3 г). 1 Продукт являв собою прозору безбарвну рідину. H ЯМР (500 МГц, CDCl3): =2,08 (с, 1Н, 3 2 NH), 3,70-3,71 (м, 4H, СН2азетидин), 4,91 (квінтет, 1H, J=7,0 Гц, СН азетидин), 6,21 (τ, lH, JH-F=73,9ru, CF2H). 19 2 F ЯМР (376 МГц, CDCl3): = -84,1 (д, 2F, JH-F=74,4 Гц, CF2H). 13 1 1 С{ H} ЯМР (125 МГц, CDCl3): =54,3 (CH2), 66,3 (CH), 115,4 (τ, JC-F=261,1 Гц, CF2H). Елементний аналіз для C4H7F2NO: С, 39,03; Η, 5,73; Ν, 11,38. Визначено: С, 39,11; Η, 5,79; Ν, 11,34. Бензил 2-(дифлуорометокси)ацетат (2о). 15 UA 112889 U 5 10 15 20 25 30 35 40 45 50 55 60 Сполуку 1ο (50,15 г, 0,302 моль, 1 екв) розчиняли в 500 мл ацетонітрилу і додавали до неї йодид міді (11,5 г, 0,06 моль, 0,2 екв). Суміш нагрівали до 50 °C і додавали по краплях протягом 45 хвилин розчин 2-флуорсульфоніл-2,2-дифлуороцтової кислоти (80,65 г, 0,453 моль, 1,5 екв) в 100 мл ацетонітрилу. Реакційну суміш нагрівали протягом ще 30 хв при 50 °C. Потім ацетонітрил випарювали, залишок розчиняли в EtOAc і тверду фазу відфільтровали. Отриманий в результаті розчин EtOAc випарювали і неочищений 2о очищали за допомогою колонкової хроматографії (гексан: EtOAc-1: 7, Rf=0,7). Вихід = 81,2 % (53 г). 1 Продукт являв собою прозору безбарвну рідину. H ЯМР (400 МГц, CDCl3): =4,44 (с, 2Н, 2 СН2СО), 5,21 (с, 2Н, CH2Ph), 6,34 (τ, 1H, JH-F=73,9 Гц, CF2H), 7,36 (ш с, 5Н, Ph). 19 2 F ЯМР (376 МГц, CDCl3): = -86:6 (д, 2F, JH-F=73,6 Гц, CF2H). 13 1 1 С{ H} ЯМР (100 МГц, CDCl3): =59,9, 67,3 (СН2), 115,3 (τ, JC-F=272,0 Гц, CF2H), 128,5, 128,8 (CHPh), 134,9 (CPh), 167,6 (C=O). Елементний аналіз для C10H10F2O3: С, 55,56; Η, 4,66; F, 17,58. Визначено: С, 55,67; Η, 4,71; F, 17,52. 2-(Дифлуорометокси)оцтова кислота (3о). Сполуку 2о (53 г, 0,245 моль, 1 екв) розчиняли в 300 мл метанолу і додавали до неї 7 г 5 % Pd/C. Суміш перемішували при 30 атм ГІ2 протягом 72год. при кімнатній температурі. Після завершення реакції Pd/C відфільтровували і реакційну суміш концентрували у вакуумі. Продукт 3о розчиняли в конц. розчині бікарбонату натрію і отриманий розчин тричі промивали EtOAc. Потім розчин обережно підкислювали концентрованим розчином соляної та лимонної кислоти і тричі екстрагували за допомогою ЕtOАс. Об'єднаний органічний шар промивали двічі насиченим розчином солі, сушили над сульфатом магнію і концентрували у вакуумі, отримуючи продукт 3о. Вихід = 64,1 % (19,8 г). 1 Продукт являв собою тверду речовину білого кольору. Тпл. = 46-48 °C. H ЯМР (400 МГц, 2 CDCl3): =4,51 (с, 2Н, СН2), 6,36 (т, 1H, JH-F=75,9 Гц, CF2H), 10,62 (ш с, 1Н, СООН). 19 2 F ЯМР (376 МГц, CDCl3): = -86,8 (д, 2F, JH-F=87,9 Гц, CF2H). 13 1 1 С{ H} ЯМР (100 МГц, CDCl3): =59,1 (СН2), 115,2 (τ, JC-F=285,6 Гц, CF2H), 173,7 (С=О). Елементний аналіз для C3H4F2O3: С, 28,58; Η, 3,20. Визначено: С, 28,52; Η, 3,10. 1,4-бензил 4-(дифлуорометокси)циклогексанкарбоксилат (2р) (Суміш діастереомерів). Сполуку 1р (у вигляді суміші цис- і транс-ізомерів, 7: 3) (42 г, 0,179 моль, 1 екв) розчиняли в 500 мл ацетонітрилу і додавали до неї йодид міді (6,85 г, 0,36 моль, 0,2 екв). Суміш нагрівали до 50 °C і додавали по краплях протягом 45 хвилин розчин 2-флуорсульфоніл-2,2-дифлуороцтової кислоти (47,9 г, 0,269 моль, 1,5 екв) в 100 мл ацетонітрилу. Реакційну суміш нагрівали протягом ще 30 хв при 50 °C. Потім ацетонітрил випарювали, залишок розчиняли в EtOAc і нерозчинні тверді речовини відфільтровували. Отриманий в результаті розчин EtOAc випарювали і неочищений 2р очищали за допомогою колонкової хроматографії (гексан: EtOAc-1: 7, Rf=0,6). Вихід = 73,4 % (37,4 г). 1 Продукт являв собою прозоре безбарвне густе масло. H ЯМР (400 МГц, CDCl3): =1,47-2,42 (м, 8Н, СН2циклогексил), 4,06, 4,33 (с, 1H, СНциклогексил), 5,10 (ш с, 1H, СНциклогексил), 5,26 (с, 2Н, 2 CH2Ph), 6,21 (τ, 1H, JH-F=73,9 Гц, CF2H), 7,34 (ш с, 5Н, Ph). 19 2 2 F ЯМР (376 МГц, CDCl3): = -83,6 (д, 2F, JH-F=73,7 Гц, CF2H), -83,1 (д, 2F, JH-F=73,7 Гц, CF2H). 13 1 С NMR{ H} (100 МГц, DMSO-d6) (Суміш діастереомерів): =14,0, 22,1, 23,2, 26,3, 29,3, 31,3, 1 1 40,8 (циклогексил), 65,5 (СНСО), 74,3 (CH2Ph), 117,2 (τ, JC-F=253,0 Гц, CF2H), 117,4 (τ, JCF=253,0 Гц, CF2H), 127,8, 128,0, 128,5 (CHPh), 136,3, 136,4 (CPh), 174,2, 174,2 (C=O). Елементний аналіз для C15H18F2O3: С, 63,37; Η, 6,38. Визначено: С, 63,22; Η, 6,43. 1,4-цис-(дифлуорометокси)циклогексанкарбонова кислота (3р). Сполуку 2р (30 г, 0,106 моль, 1 екв) розчиняли в 300 мл метанолу і додавали 6,5 г 5 % -Pd/C. Суміш перемішували при 30 атм Н2 протягом 72 год. при кімнатній температурі. Потім Pd/C відфільтровували і реакційну суміш концентрували у вакуумі. Сирий продукт розчиняли в конц. розчині бікарбонату натрію і отриманий розчин тричі промивали EtOAc. Потім розчин обережно підкислювали концентрованим розчином соляної та лимонної кислоти і тричі екстрагували за допомогою EtOAc. Об'єднаний органічний шар промивали двічі насиченим розчином солі, сушили над сульфатом магнію і концентрували у вакуумі і перекристалізували з петролейного ефіру - етилацетат (10: 1) з отриманням продукту 3р. Вихід = 60 % (12,3 г). Сполуку Зр отримували у вигляді чистого цис-ізомеру. 1 Продукт являв собою тверду речовину білого кольору. Тпл. = 117-119 °C. H ЯМР (400 МГц, CDCl3): =1,45-1,61 (м, 4Н, СН2циклогексил), 2,07-2,10 (м, 4Н, СН2циклогексил), 2,32-2,37 (м, 1H, 2 СНциклогексил), 4,09 (ш с, 1H, СНциклогексил), 6,23 (т, 1H, JH-F=74,6 Гц, CF2H), 11,0 (ш с, 1Н, СООН). 19 2 F ЯМР (376 МГц, CDCl3): = -81,2 (д, 2F, JH-F=76,4 Гц, CF2H). 16 UA 112889 U С{ H} ЯМР (100 МГц, CDCl3): =28,3, 31,6 (СН2циклогексил), 41,4, 73,0 (СНциклогексил), 116,2 (т, JC-F=258,4 Гц, CF2H), 181,6 (СО2Н). Елементний аналіз для C8H12F2O3: С, 49,48; Η, 6,23; F, 19,57. Визначено: С, 49,33; Η, 6,19. Бензил 3-(дифлуорометокси)циклобутанкарбоксилат (2q) (Суміш діастереомерів). Сполуку 1q (12,2 г, 0,059 моль, 1 екв) розчиняли в 200 мл ацетонітрилу і додавали до неї йодид міді (2,25 г, 0,012 моль, 0,2 екв). Суміш нагрівали до 50 °C і додавали по краплях протягом 45 хвилин розчин 2-флуорсульфоніл-2,2-дифлуороцтової кислоти (15,8 г, 0,089 моль, 1,5 екв) в 100 мл ацетонітрилу. Реакційну суміш нагрівали протягом ще 30 хв при 50 °C. Потім ацетонітрил випарювали, залишок розчиняли в EtOAc і нерозчинну тверду речовину відфільтровули. Отриманий в результаті розчин EtOAc випарювали і неочищений 2q очищали за допомогою колонкової хроматографії (гексан: EtOAc-1: 7, Rf=0,6). Вихід = 73,9 % (11,2 г). 1 Продукт являв собою прозоре безбарвне масло. H ЯМР (400 МГц, CDCl3) (Суміш діастереомерів): =2,40-2,47 (м, 2Н, СН2циклобутил), 2,57-2,59 (м, 2Н, СН2циклобутил), 2,71-2,75 (м, 1H, 2 СНциклобутил), 4,51-4,57 (м, 1Н, СНциклобутил), 5,11 (с, 2Н, CH2Ph), 6,13 (τ, 1H, JH-F=74,0 Гц, CF2H), 7,34 (ш с, 5Н, Ph). 19 2 F ЯМР (376 МГц, CDCl3): = -83,6 (д, 2F, JH-F=72,0 Гц, CF2H). 13 1 1 С{ H}ЯМР (125,7 МГц, CDCl3): =29,9, 34,4, 63,9 (циклобутил), 66,7 (CH2Ph), 115,7 (τ, JCF=259,8 Гц, CF2H), 128,3, 128,4, 128,7 (CHPh), 136,0 (CPh), 173,7 (C=O). Елементний аналіз для C13H14F2О3: С, 60,93; Η, 5,51. Визначено: С, 61,03; Η, 5,45. 3-(Дифлуорометокси)циклобутанкарбонова кислота (3q) (Суміш діастереомерів). Сполуку 2q (3,5 г, 0,014 моль, 1 екв) розчиняли в 100 мл метанолу і додавали до неї 0,9 г 5 % Pd/C. Суміш перемішувалипри 30 атм Н2 протягом 36 год. при кімнатній температурі. Потім Pd/C відфільтровували і реакційну суміш концентрували у вакуумі. Продукт 3q розчиняли в конц. розчині бікарбонату натрію і отриманий розчин тричі промивали EtOAc. Потім розчин обережно підкислювали розчином концентрованої соляної кислотт та лимонної кислоти і тричі екстрагували за допомогою EtOAc. Об'єднаний органічний шар промивали двічі насиченим розчином солі, сушили над сульфатом магнію і концентрували у вакуумі з отриманням продукту 3q. Вихід = 57,3 % (1,3 г). 1 Продукт являв собою прозоре безбарвне масло. H ЯМР (400 МГц, CDCl3) (Суміш діастереомерів): =2,43-2,48 (м, 2Н, СН2циклобутил), 2,61-2,63 (м, 2Н, СН2циклобутил), 2,71-2,76 (м, 1H, 2 СНциклобутил), 4,55-4,59 (м, 1Н, СНциклобутил), 6,15 (τ, 1Η, JH-F=74,0 Гц, CF2H), 12,0 (с, 1H, СООН). 19 2 F ЯМР (376 МГц, CDCl3): = -83,9 (д, 2F, JH-F=71,6 Гц, CF2H). 13 1 1 С{ H} ЯМР (125 МГц, CDCl3): =29,3, 31,6, 33,7, 63,1 (τ, J=5,6 Гц), 115,1 (τ, JC-F=261,2 Гц, CF2H), 181,1 (CO2H). Елементний аналіз для C6H8F2O3: С, 43,38; Η, 4,85. Визначено: С, 43,49; Η, 4,91. Терт-бутил (S-3-(метоксикарбоніл)-3-(дифлуорометокси)пропілкарбамат (2r). Сполуку 1r (3,2 г, 0,014 моль, 1 екв) розчиняли в 50 мл ацетонітрилу і додавали до неї йодид міді (0,5 г, 0,0026 моль, 0,2 екв). Суміш нагрівали до 50 °C і додавали по краплях протягом 30 хвилин розчин 2-флуорсульфоніл-2,2-дифлуороцтової кислоти (2,95 г, 0,017 моль, 1,2 екв) в 20 мл ацетонітрилу. Реакційну суміш нагрівали протягом ще 30 хв при 50 °C. Потім ацетонітрил випарювали, залишок розчиняли в EtOAc і нерозчинні тверді речовини відфільтровували. Отриманий в результаті розчин EtOAc випарювали і неочищений 2r очищали за допомогою колонкової хроматографії (гексан: EtOAc-3: 1, Rf=0,65). Вихід = 79,8 % (3,1 г). 1 Продукт являв собою прозоре безбарвне масло. H ЯМР (400 МГц, CDCl3): =1,45 (с, 9Н, tBu), 1,99-2,08 (м, 2Н, СН2), 3,27 (ш с, 2Н, СН2), 3,79 (с, 3Н, ОМе), 4,65-4,68 (м, 2Н, NH, СН), 6,36 2 (т, 1Н, JH-F=74,6 Гц, CF2H). 19 2 2 F ЯМР (376 МГц, CDCl3): = -85,0 (дд, F, JF-F=160,2 Гц, JH-F=73,4 Гц, CF2H), -83,9 (дд, F, 2 2 JF-F=158,5 Гц, JH-F=71,8 Гц, CF2H). 13 1 1 C{ H} ЯМР (100 МГц, CDCl3): =27,7, 28,4 (t-Bu), 32,4, 52,6, 69,8, 80,0, 115,5 (τ, JC-F=272,0 Гц, CF2H), 155,8, 170,7 (C=O). Елементний аналіз для C11H19F2NO5: С, 46,64; Η, 6,76; Ν, 4,94. Визначено: С, 46,78; Η, 6,70;Ν, 5,02. (2S)-2-(дифлуорометокси)-4-[[(1,1-диметилетокси)карбоніл]аміно]-бутанова кислота (3r). Сполуку 2r (1 г, 0,0035 моль, 1 екв) розчиняли в 20 мл THF і додавали відразу розчин LiOHH2O (0,178 г, 0,0042 моль, 1,2 екв) в 5 мл води при 0 °C. Реакційну суміш нагрівали до кімнатної температури і перемішували протягом 4 год. Потім реакційну суміш розбавляли 25 мл етилацетату і 20 мл води. Органічний шар відокремлювали, двічі промивали 20 мл води. Об'єднаний водний розчин промивали один раз 25 мл EtOAc, обережно підкислювали 20 мл конц. розчину лимонної кислоти і тричі екстрагували 30 мл EtOAc. Об'єднаний органічний шар 13 1 1 5 10 15 20 25 30 35 40 45 50 55 17 UA 112889 U 5 10 15 20 25 30 35 40 45 50 55 промивали насиченим розчином солі, сушили над сульфатом магнію і концентрували у вакуумі з отриманням сполуки 3r. Вихід = 57,9 % (0,55 г). Продукт являв собою тверду речовину білого кольору. Тпл. = 96-99 °C, []D = -21,3 (МеОН, 1 с= 37,14 ммоль/л). H ЯМР (500 МГц, DMSO-d6): =1,37 (с, 9Н, t-Вu), 1,76-1,91 (м, 2Н, СН2), 3,01 2 (ш с, 2Н, СН2), 4,50-4,52 (м, 1Н, СН), 6,70 (τ, 1Η, JH-F=76,0 Гц, CF2H), 6,87 (ш с, 1H, NH), 13,20 (ш с, 1H, СООН). 19 2 F ЯМР (376 МГц, DMSO-d6: = -82,3 (д, 2F, JH-F=75,5 Гц, CF2H). 13 1 1 С{ H} ЯМР (125 МГц, DMSO-d6): =28,2 (t-Bu), 32,0, 36,0, 71,5, 77,7, 116,9 (τ, JC-F=280,5 Гц, CF2H), 155,6, 171,3 (C=O). Елементний аналіз для C10H17F2NO5: С, 44,61; Η, 6,36; Ν, 5,20. Визначено: С, 44,72; Η, 6,30; Ν, 5,17. (2S,4R)-1-терт-бутил 2-метил 4-(дифлуорометокси)піролідин-1,2-дикарбоксилат (2s). Сполуку 1s (7,5 г, 0,031 моль, 1 екв) розчиняли в 100 мл ацетонітрилу і додавали до неї йодид міді (1,18г, 0,0062 моль, 0,2 екв). Суміш нагрівали до 50 °C і додавали по краплях протягом 30 хвилин розчин 2-флуорсульфоніл-2,2-дифлуороцтової кислоти (6,6 г, 0,037 моль, 1,2 екв) в 30 мл ацетонітрилу. Реакційну суміш витримували протягом ще 30 хвилин при 50 °C. Потім ацетонітрил випарювали, залишок розчиняли в EtOAc і нерозчинні тверді речовини відфільтровували. Отриманий в результаті розчин EtOAc випарювали і неочищений 2s очищали за допомогою колонкової хроматографії (гексан: EtOAc-1: 4, Rf=0,7). Вихід = 84,2 % (7,4 г). 1 Продукт являв собою прозоре безбарвне масло. H ЯМР (500 МГц, CDCl3): =1,46 (с, 3Н, tВu), 1,51 (с, 6Н, t-Bu), 2,21-2,26 (м, 1H, СН2піролідин), 2,45-2,50 (м, 1H, СН2піролідин), 3,59-3,78 (м, 2Н, 2 СН2піролідин), 3,79 (с, 3Н, ОМе), 4,39-4,50 (м, 1H, СНпіролідин), 4,90 (с, 1H, СНпіролідин), 6,28 (т, 1H, JHF=74,8 Гц, CF2H). 19 F ЯМР (376 МГц, CDCl3): = -84,0- -82,8 (м, 2F, CF2H). 13 1 С{ H} ЯМР (125 МГц, CDCl3): =28,2, 28,3 (t-Вu), 36,2, 37,2, 52,1, 52,3, 71,2, 71,9, 80,5 1 (піролідин), 115,6 (τ, JC-F=262,8 Гц, CF2H), 153,4, 154,1, 172,6 (C=O). Елементний аналіз для C12H19F2NO5: С, 48,83; Η, 6,49; Ν, 4,74. Визначено: С, 48,68; Η, 6,45; Ν, 4,69. (2S,4R)-1-(терт-бутоксикарбоніл)-4-(дифлуорометокси)піролідин-2-карбоксилова кислота (3s). Сполуку 2s (2,75 г, 0,0093 моль, 1 екв) розчиняли в 40 мл THF і додавали відразу розчин LiOHH2O (0,47 г, 0,0112 моль, 1,2 екв) в 10 мл води при 0 °C. Реакційну суміш нагрівали до кімнатної температури і перемішували протягом 4 год. Потім реакційну суміш розбавляли 75 мл етилацетату і 40 мл води. Органічний шар відокремлювали, двічі промивали 20 мл води. Об'єднаний водний розчин промивали один раз 50 мл EtOAc, обережно підкислювали 35 мл конц. розчину лимонної кислоти і тричі екстрагували 40 мл EtOAc. Об'єднаний органічний шар промивали насиченим розчином солі, сушили над сульфатом магнію і концентрували у вакуумі і отримували цільову сполуку 3s. Вихід = 61,1 % (1,6 г). Продукт являв собою тверду речовину білого кольору. Тпл. = 70-75 °C, []D = -36,6 (ЕtOН, с = 1 35,55 ммоль/л). H ЯМР (400 МГц, CDCl3): =1,14 (с, 3Н, t-Bu), 1,45 (с, 6Н, t-Bu), 2,23-2,47 (м, 2Н, СН2піролідин), 3,56-3,68 (м, 2Н, СН2піролідин), 4,35-4,42 (м, 1Н, СНпіролідин), 4,83 (с, 1H, СНпіролідин), 6,21 2 (τ, 1H, JH-F=73,5 Гц, CF2H), 8,70 (ш с, 1Н, СООН). 19 F ЯМР (376 МГц, CDCl3): = -84,0 - -82,9 (м, 2F, CF2H). 13 1 С{ H} ЯМР (125 МГц, CDCl3): =28,2, 28,3 (t-Вu), 35,5, 37,2, 52,1, 52,5, 71,2, 71,5, 81,0, 81,8 1 (піролідин), 115,6 (τ, JC-F=262,6 Гц, CF2H), 153,6, 155,7, 177,8 (C=O). Елементний аналіз для C11H17F2NO5: С, 46,97; Η, 6,09; Ν, 4,98. Визначено: С, 47,09; Η, 6,15; Ν, 4,91. (S)-3-(дифлуорометокси)піролідин-2-он (5). Сполуку 2r (1,5 г, 0,0053 моль, 1 екв) розчиняли в 5 мл DCM і охолоджували до 0 °C за допомогою бані з льодом. Потім відразу додавали трифлуороцтову кислоту (5 мл, 0,0653 моль, 12,3 екв). Реакційну суміш перемішували протягом 30 хвилин при 0 °C і потім додавали конц. розчин карбонату калію для лужного середовища. Отриману суспензію тричі екстрагували 30 мл EtOAc. Об'єднаний органічний шар промивали насиченим розчином солі, сушили над сульфатом магнію і концентрували у вакуумі. Сирий продукт очищали за допомогою флешхроматографії (гексан: EtOAc-5: 1), отримуючи сполуку 5. Вихід = 52,5 % (0,42 г). = Продукт являв собою тверду речовину білого кольору. Тпл. = 57-61 °C, []D -72,9 (МеОН, с= 1 66,18 ммоль/л). H ЯМР (400 МГц, CDCl3): =2,16-2,53 (м, 2Н, СН2), 3,28-3,45 (м, 2Н, СН2), 4,67 3 2 (т, 1H, J=8,1 Гц, СН), 6,48 (τ, 1Н, JH-F=75,8 Гц, CF2H), 7,50 (ш с, 1Н, NH). 18 UA 112889 U F ЯМР (376 МГц, CDCl3): = -84,3 (дд, F, JH-F=158,5 Гц, JH-F=75,7 Гц, CF2H), -83,4 (дд, F, 2 JF-F=158,5 Гц, JH-F=73,5 Гц, CF2H). 13 1 1 С{ H} ЯМР (100 МГц, CDCl3): =28,7, 38,8 (CH2), 70,8 (CH), 115,8 (τ, JC-F=258,4 Гц, CF2H), 173,7 (C=O). Елементний аналіз для C5H7F2NO2: С, 39,74; Η, 4,67; Ν, 9,27. Визначено: С, 39,80; Η, 4,71; Ν, 9,33. 19 2 2 2 5 ФОРМУЛА КОРИСНОЇ МОДЕЛІ 10 15 20 25 1. Спосіб О-дифлуорометилування функціоналізованих аліфатичних спиртів, який відрізняється тим, що функціоналізований аліфатичний спирт обробляють дифлуорометильованим агентом (FSO2CF2CO2H) в пристуності солі Сu (І) в органічному розчиннику при м'якому нагріванні. 2. Спосіб за п. 1, який відрізняється тим, що як сіль міді використовують CuI або СuВr та як розчинник використовують ацетонітрил. 3. Спосіб за п. 1, який відрізняється тим, що як аліфатичний спирт може бути використаний спирт формули (І): HO-R, де R являє собою С1-С6алкіл, С3-С10циклоалкіл, С6-С10арил або С2-С9гетероарил, кожен з яких може бути заміщений принаймні одним С1-С6алкілом, С3-С10циклоалкілом, С6-С10арилом, С21 2 С9гетероарилом, -CN, NO2, OR , NHR , -SC1-С6алкілом, -SOC1-С6алкілом, -SO2C1-C6алкілом, NSO2C1-C6алкілом, -NHC1-С6алкілом, -N(C1-C6алкіл)2, -NHCOOC1-C6алкілом, NHCONHC1C6алкілом, -CONHC1-C6алкілом, -СОС1-С6алкілом, -СООС1-С6алкілом; 1 R являє собою гідроксизахисну групу, таку як Вn або С1-С6 алкіл; 2 R являє собою амінозахисну групу, таку як Cbz або Вос. Комп’ютерна верстка Л. Литвиненко Державна служба інтелектуальної власності України, вул. Василя Липківського, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 19
ДивитисяДодаткова інформація
МПК / Мітки
МПК: C08F 210/00
Мітки: аліфатичних, о-дифлуорометилування, спосіб, функціоналізованих, спиртів
Код посилання
<a href="https://ua.patents.su/21-112889-sposib-o-difluorometiluvannya-funkcionalizovanikh-alifatichnikh-spirtiv.html" target="_blank" rel="follow" title="База патентів України">Спосіб о-дифлуорометилування функціоналізованих аліфатичних спиртів</a>
Спосіб одержання аліфатичних спиртів та метанолу

Номер патенту: 56369
Опубліковано: 15.05.2003
Автори: Лунев Микола Кирилович, Чистосердов Володимир Михайлович
МПК: B01J 23/00, C07C 29/15, C07C 29/16, C07C 31/00, B01J 21/00
Мітки: спосіб, метанолу, одержання, спиртів, аліфатичних
Формула / Реферат:
1. Спосіб одержання аліфатичних спиртів та метанолу із синтез-газу шляхом взаємодії оксидів вуглецю та водню при підвищених температурі та тиску, в присутності каталізатора, який відрізняється тим, що процес проводять каскадно в присутності різних каталізаторів у два ступені, причому на першому ступені використовують каталізатор, до складу якого входять (мас. %): ZnO - 22,5-62,5, Сr2О3 - 21,0-45,0, V2O5 - 0,5-8,0, K2O - 0,5-6,0, CuO -...
Спосіб одержання метанолу і аліфатичних спиртів

Номер патенту: 28613
Опубліковано: 15.05.2002
Автори: Блізніченко Сергій Костянтинович, Соміков Анатолій Платонович, Балахнічева Людмила Миколаєвна, Леонов Валерій Євгенійович
МПК: C07C 31/00, C07C 29/48
Мітки: метанолу, спиртів, одержання, спосіб, аліфатичних
Формула / Реферат:
1. Спосіб одержання метанолу і аліфатичних спиртів шляхом газофазного неповного окислення вуглеводневих газів кисневмісним газом при підвищених температурі та тиску, який відрізняється тим, що одержаний проміжний продукт оксидат - сирець піддають каталітичному гідрокарбонілюванню в присутності водню, оксидів вуглецю, вуглеводневих газів і азоту при температурі 90-300°С і тиску 0,25-9,5 МПа, співвідношенні Н2:СО=2.2.Спосіб по п.1, який...
Спосіб отримання метанолу та інших аліфатичних спиртів

Номер патенту: 59190
Опубліковано: 15.08.2003
Автори: Головко Дмитро Миколайович, Целіщев Олексій Борисович, Лорія Марина Геннадіївна, Мілоцький Вадим Вадимович, Єрмоленко Володимир Васильович
МПК: C07C 29/48, C07C 31/00, C07C 7/00
Мітки: спиртів, спосіб, інших, метанолу, отримання, аліфатичних
Формула / Реферат:
1. Спосіб отримання метанолу та інших аліфатичних спиртів, що включає газофазну взаємодію вуглеводневих газів з водяною парою, який відрізняється тим, що метанол і інші аліфатичні спирти отримують прямим гідроксилюванням вуглеводневого газу або суміші газів водяною парою, для чого вихідний вуглеводневий газ і пару, або суміш газів і пару, у розрахунковому співвідношенні подають у реакційний апарат, де реакційну масу піддають дії...
Спосіб очистки води від аліфатичних спиртів

Номер патенту: 33908
Опубліковано: 25.07.2008
Автори: Солодовнік Тетяна Володимирівна, Унрод Володимир Ізяславович, Тимчук Алла Федорівна, Сазонова Валентина Федорівна, Кожемяк Марина Анатоліївна
МПК: C02F 1/24
Мітки: очистки, аліфатичних, спиртів, спосіб, води
Формула / Реферат:
Спосіб очистки стічних вод від аліфатичних спиртів адсорбцією, який відрізняється тим, що використовують адсорбент хітозан у кількості 300 мг на 1 л розчину, адсорбцію проводять при рН 4-8, для відділення адсорбенту використовують флотацію.
Спосіб одержання аліфатичних спиртів для виробництва моторного палива

Номер патенту: 4647
Опубліковано: 28.12.1994
Автори: Зубілін Іван Георгієвич, Павлій Людмила Василівна, Тараканов Анатолій Олексійович, Леонов Валерій Євгенович, Францєнюк Іван Васільєвіч, Успенський Сергій Константинович
МПК: C07C 29/151, C07C 31/00
Мітки: спосіб, спиртів, виробництва, аліфатичних, палива, моторного, одержання
Формула / Реферат:
1. Способ получения алифатических спиртов для производства моторных топлив из синтез-газа при повышенной температурена цинк-хром-калий-ванадиевом катализаторе, отличающийся тем, что в зону катализа вместе с синтез-газом вводят этиленсодержащий газ с концентрацией его в газовой фазе в пределах 0,05-3,00 об. %, причем соотношение Н2/СО в синтез-газе составляет 1,8-2,2, и процесс ведут при температуре 378-400°С.2. Способ по п. 1,...
Попередній патент: Спосіб паралельного синтезу аліфатичних оксамідів
Наступний патент: Протикумулятивний екран для бойової і транспортної техніки
Випадковий патент: Спосіб інформаційного забезпечення маневрування морського судна