Експресійний вектор та способи продукування високих рівнів білків
Номер патенту: 92913
Опубліковано: 27.12.2010
Автори: Сінгх Арун К., Гоел Ашіш, Мендіратта Сенджів К.




















Формула / Реферат
1. Спосіб досягнення високої експресії потрібного білка за допомогою експресійного вектора, що складається щонайменше з наступних регуляторних елементів:
a) промотору CMV або його функціональних варіантів,
b) інтрона,
c) TPL або його функціональних варіантів,
d) генів VA або функціональних варіантів, та
e) поліаденіляційної послідовності бичачого гормону росту або функціональних варіантів.
2. Спосіб за п. 1, де потрібним білком є рекомбінантний еритропоетин.
3. Спосіб за п. 1 досягнення високої експресії злитого білка, TNFR-IgGFc.
4. Спосіб досягнення високої експресії ритуксимабу, трастузумабу, бевацузумабу або інших моноклональних антитіл за способом п. 1.
5. Спосіб за п. 1 досягнення високої експресії білка у клітині ссавця.
6. Експресійний вектор ссавця, що складається щонайменше з наступних п'яти регуляторних елементів:
a) промотору CMV або його функціональних варіантів,
b) інтрона,
c) TPL або його функціональних варіантів,
d) генів VA або функціональних варіантів, та
e) поліаденіляційної послідовності бичачого гормону росту або функціональних варіантів.
7. Вектор за п. 6, де інтроном є химерний інтрон послідовності № 1.
8. Вектор за п. 6, де TPL є регуляторний елемент послідовності № 4.
9. Вектор за п. 6, де гени VA мають послідовність № 5.
10. Новий експресійний вектор ссавця за будь-яким з попередніх пп. 6-9, що складається з наступних п'яти регуляторних елементів:
a) промотору CMV або його функціональних варіантів,
b) химерного інтрона послідовності № 1 або його функціональних варіантів,
c) TPL послідовності № 4 або його функціональних варіантів,
d) генів VA послідовності № 5 або функціональних варіантів,та
е) поліаденіляційної послідовності бичачого гормону росту або її функціональних варіантів.
11. Вектор за пп. 6-10, що додатково містить селективний та ампліфікаційний маркер, вибраний з дигідрофолатредуктази, аденозиндезамінази, орнітиндекарбоксилази, аспарагінсинтетази, глутамінсинтетази або їхніх функціональних варіантів.
12. Вектор за будь-яким з пп. 6-11, що додатково кодує ген для еритропоетину.
13. Вектор за будь-яким з пп. 6-11, що додатково кодує ген для злитого білка, TNFR-IgGFc.
14. Вектор за будь-яким з пп. 6-11, що додатково кодує гени для ритуксимабу, трастузумабу, бевацузумабу або інших моноклональних антитіл.
15. Вектор за будь-яким з пп. 6-11, що додатково кодує відповідний ген (гени), здатний (здатні) до експресії в клітинах ссавця.
16. Клітина ссавця, перетворена за допомогою експресійного вектора за пп. 6-15.
17. Вектор за пп. 6-16, де клітини ссавця для трансфекції вибираються з Cos, СHO, СНО DHFR, ВНК1 тa NSO.
18. Застосування векторів за будь-яким з попередніх пунктів для одержання, високої експресії потрібних білків.
19. Застосування векторів за п. 18, де потрібний білок вибирається з рекомбінантного еритропоетину, рекомбінантного злитого білка, TNFR-IgGFc та моноклональних антитіл, вибраних з ритуксимабу, трастузумабу й бевацузумабу.
20. Спосіб одержання високої експресії потрібного білка шляхом кодування відповідного гена в вектор за будь-яким з попередніх пп. 1-11.
Текст
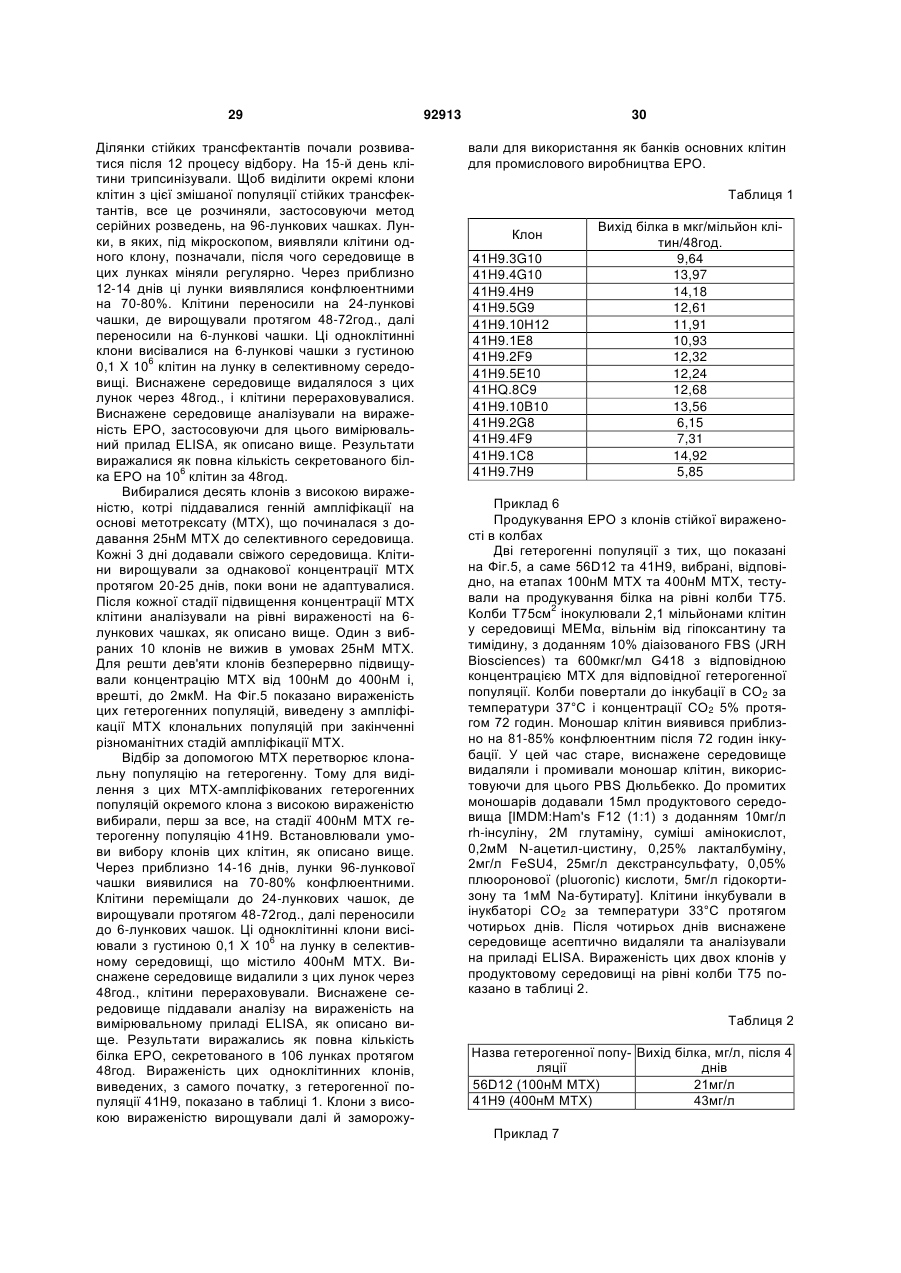
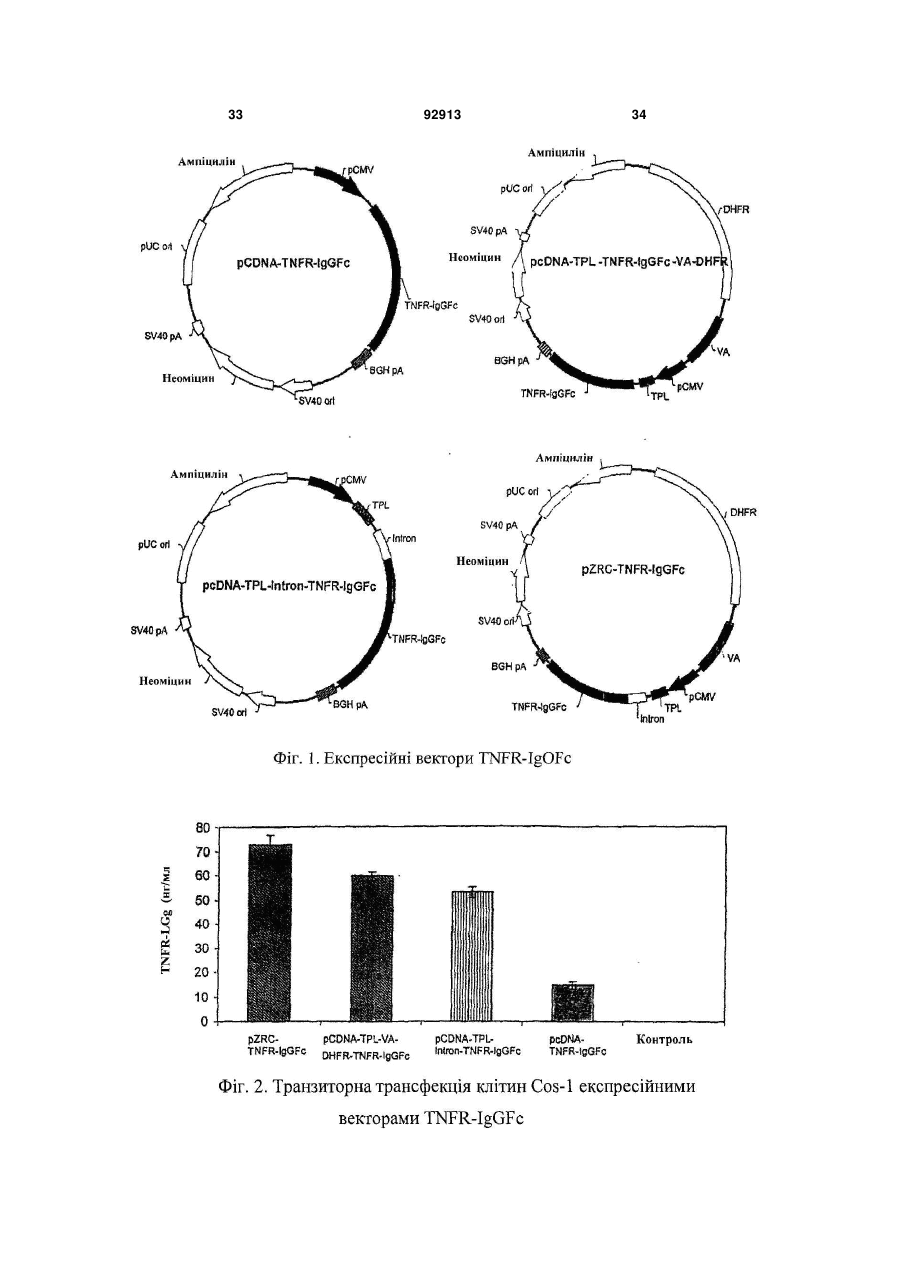
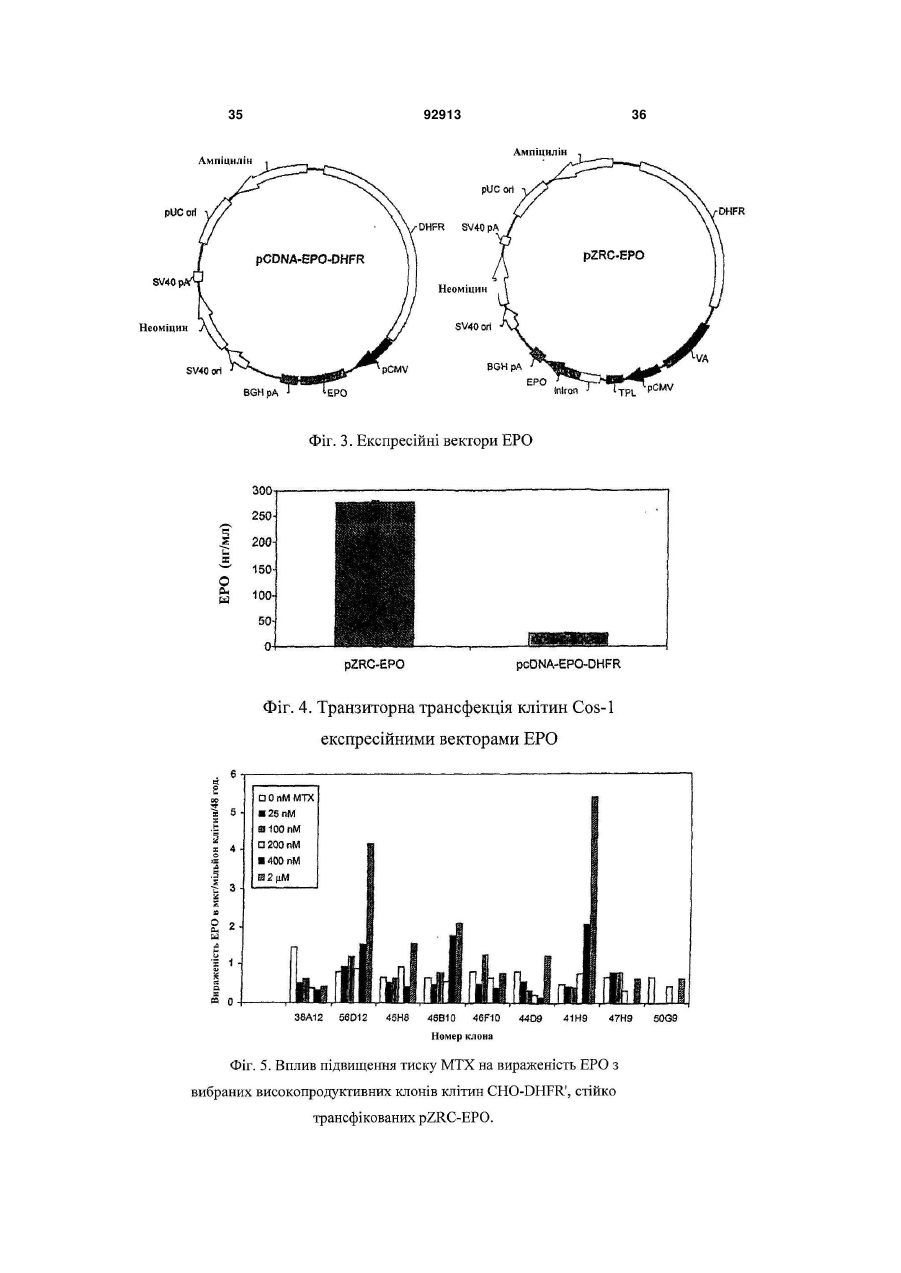
1. Спосіб досягнення високої експресії потрібного білка за допомогою експресійного вектора, що складається щонайменше з наступних регуляторних елементів: a) промотору CMV або його функціональних варіантів, b) інтрона, c) TPL або його функціональних варіантів, d) генів VA або функціональних варіантів, та e) поліаденіляційної послідовності бичачого гормону росту або функціональних варіантів. 2. Спосіб за п.1, де потрібним білком є рекомбінантний еритропоетин. 3. Спосіб за п.1 досягнення високої експресії злитого білка, TNFR-IgGFc. 4. Спосіб досягнення високої експресії ритуксимабу, трастузумабу, бевацузумабу або інших моноклональних антитіл за способом п.1. 5. Спосіб за п.1 досягнення високої експресії білка у клітині ссавця. 2 (19) 1 3 92913 4 17. Вектор за пп.6-16, де клітини ссавця для трансфекції вибираються з Cos, СHO, СНО DHFR, ВНК1 тa NSO. 18. Застосування векторів за будь-яким з попередніх пунктів для одержання, високої експресії потрібних білків. 19. Застосування векторів за п.18, де потрібний білок вибирається з рекомбінантного еритропое тину, рекомбінантного злитого білка, TNFR-IgGFc та моноклональних антитіл, вибраних з ритуксимабу, трастузумабу й бевацузумабу. 20. Спосіб одержання високої експресії потрібного білка шляхом кодування відповідного гена в вектор за будь-яким з попередніх пп.1 - 1 1 . Даний винахід стосується нових експресійних векторів для продукування високих рівнів рекомбінантних білків у клітинах ссавців та процесу продукування потрібних білків з використанням тих само експресійних векторів. Ці експресійні вектори дають на 80-100% більше вираженості рекомбінантного білка ЕРО порівняно з найкращими з опублікованих векторів. Відповідно до даних IMS (Індійська служба охорони здоров'я), передбачається зростання долі біофармацевтичних препаратів на загальному фармацевтичному ринку від 6% у 1999р. до 14% (90 мільярдів доларів США) в 2009р. Причина цього зростання попиту на біологічні препарати полягає, головним чином, у їхній точно орієнтованій дії, результатом чого є значне зниження та точне визначення ризику токсичності порівняно з ліками на основі малих молекул. Далі, використання рекомбінантних методів для продукування цих біологічних препаратів, на відміну від інших методів очищення від екстрактів тканин та рідини організму, дає змогу легко одержувати продукти високої чистоти, з чітко визначеними характеристиками безпечності та фізикохімічними характеристиками. Незважаючи на всі ці сприятливі для хворих якості, значніша частина рекомбінантних біологічних препаратів залишаються недоступними для більшості людей світу через надзвичайно високі ціни. Через це ліки, що рятують життя, на зразок еритропоетину (erythropoietin), ліки на зразок етанерцепту (etanercept), що значно поліпшують якість життя, та велика кількість протиракових засобів на зразок ритуксимабу (rituximab), трастузумабу (trastuzumab) та всіх інших моноклональних антитіл тощо доступні лише дуже невеликій частині людей, тоді як величезна більшість хворих в усьому світі не може використовувати їх в достатній кількості. Це означає, що існує нагальна необхідність зниження вартості цих ліків. Значна складова їхньої високої вартості пов'язана з виробництвом. Даний винахід пропонує вирішення цієї проблеми за рахунок експресійних векторів, що можуть забезпечити високу вираженість білка в трансфікованих ними клітинах-господарях. Останніми роками технологія рекомбінантної ДНК отримала розвиток до стану, в якому, загалом, стало можливим одержати ген, що кодує потрібний білковий продукт, котрий, в разі пода льшого використання білка як терапевтичного засобу, називається біологічним препаратом. Після одержання гена, для його вираженості можна використовувати різноманітні клітинигосподарі, для чого зазначений ген спочатку клонують в будь-який з ряду наявних експресійних векторів, властивих для відповідної клітинигосподаря, далі вносять вектор, що є носієм гена, у відповідну клітину-господаря, використовуючи різноманітні методи перетворення або трансфекції. Існують також різноманітні умови ферментації, що примушують ці клітини-господарі з перетвореними генами продукувати білок. Вибір господаря та подальші взаємозалежні вибори, поміж іншого, експресійних векторів, методів перетворень та трансфекцій, а також методологій ферментації залежать від багатьох факторів на зразок характеристик білка, що має бути продукований, кінцевого використання цього білка, необхідної його кількості, методів очищення, що можуть бути застосовані, загальної вартості, доступних технологій тощо. Наприклад, важливими складовими даного винаходу, в якому рекомбінантний білок, що продукується, має терапевтичне призначення, є первинна, вторинна та третинна структури білка, ступінь та якість його глікозиляції, чистота кінцевого продукту, кількість продукованого білка, ціна, за яку можна буде продавати ліки. Кожний з перелічених елементів вкладає свій внесок в описаний процес вибору експресійної клітини-господаря та інших взаємозалежних виборів. Одне з можливих вирішень зазначеної проблеми високої вартості виробництва біологічних препаратів полягає у вираженості їх у бактеріальних клітинах-господарях на зразок Е. coli [Marino, Μ. Η., BioPharm, 2:18-33 (1989); Georgiou G., Protein engineering: Principles and practice, Wiley Liss, New York, 101-127 (1996); Gold, L. Methods Enzymol, 185:11-14 (1990); Hodgson, J., Bio/Technology, 11:887-893 (1993); Nicaud et al., J.Biotechnol. 3:255-270 (1986); Olins, P. O., and S. С Lee, Curr. Opin. Biotechnol. 4:520-525 (1993); Shatzman, A. R, Curr. Opin. Biotechnol, 6:491-493 (1995)]. Широко використовуваним бактеріальним господарем є Escherichia coli, що використовується для продукування рекомбінантних білків і широко застосовується в промисловому виробництві. Його численні переваги включають лег 5 кість культивації, низьку вартість та високий виробничий потенціал [Shuhua Taa et al., Protein Expression and Purification, 25:430-436 (2002); Cornelia Rossmann et al. Protein expression and Purification 7:335-342 (1996)]. Однак бактеріальні господарі не є, загалом, ідеальним засобом, оскільки не мають необхідного механізму глікозиляції білків [Old R W, and Primrose S.B., Principles of Gene Manipulations, An introduction to genetic Engineering, Blackwell science, United Kingdom. (1994)], а більшість терапевтичних білків для ссавців не можуть діяти на повну силу без належної глікозиляції. Крім того, відсутність механізму секреції, необхідного для ефективного виділення білка в культурне середовище, обмежена здатність сприяти потужному утворенню дисульфідних зв'язків, неналежне згортання, руйнування білка протеазами клітин-господарів, значні відмінності у використанні кодонів, інші модифікації на зразок глікацій - все це разом робить бактеріальні системи значно менш привабливими порівняно з ссавцевими [Fuh, G. et al., J. Biol. Chem, 265:3111-3115 (1990); Liang et al., Biochem. J, 229:429-439 (1985); Sarmientos et al., Bio/Technology, 7:495-501 (1989); Sawas C. Makrides, Microbiological Reviews, Sept. 1996. 512538; N. Jenkins and E. M. Curling, Enzyme Microb. Technol, 16:354-364 (1994)]. Тому звичайно виразити терапевтичні білки в бактеріях не виходить, і в біологічних препаратах для вираженості терапевтичних білків найчастіше використовуються еукаріотичні клітини-господарі, незважаючи навіть на те, що це призводить до підвищення вартості виробництва внаслідок нижчих рівнів вираженості рекомбінантного білка, більш жорстких вимог до вирощування культури, уповільненого росту тощо [Cornelia Rossmann, Protein Expression and Purification, 7:335-342 (1996); Geoff T. Yarranton, Current Opinion in Biotechnology, 1:133-140 (1990)]. Оскільки ссавцеві системи клітин-господарів мають велику перевагу в продукуванні терапевтичного білка, дуже важливою є проблема високої вартості використання їх у виробничому процесі. Суттєвого пониження вартості виробництва можна досягти за рахунок підвищення продуктивності, тому було докладено значних зусиль для збільшення виробництва продукції на основі цих клітин-господарів. Фактори, що звичайно впливають на кількість продукту, продукованого клітиною-господарем, можуть включати як такі, що є зовнішніми для цієї клітини - наприклад, стан культури, - так і внутрішні для клітин, більшість з яких містять фактори, що регулюють ефективність та якість транскрипції [Foecking and Hofstetter, Gene, 45:101-105 (1986); Kaufman et al., Journal of molecular Biology, 159:601-621 (1985); Wurm et al., PNAS, 1983:5414-5418 (1986); Reiser and Hauser, Drug Research, 37:482-485 (1987); Zettlmeissl et al., Biotechnology, 5:720-725 (1987)] та трансляції [(R. Grabherr and K. Bayer. Food Technol. Biotechnol. 39 (4) 265-269 (2001); Randal J. Kaufman et al. Molecular Biotechnology, 16 (2), 151-160, (2000); Juraj Hlavaty etal., Urology 341, 1-11,(2005); С Μ. Stenstrom et al., Gene. 92913 6 273(2), 259-65, (2001). M. Ibba and D. Soil, Science, 186, 1893, (1999)]. Крім того, ці фактори залежать переважно від структури самого експресійного вектора. Незважаючи на те, що існує чимало публікацій про спроби підвищення продуктивності клітин-господарів через покращення умов вирощування культури [Palermo D. P. et al., Journal of Biotechnology, 19:35-48 (1991); Birch and Fraud, Biologicals, 22:127-133 (1994); Osman et al., Biotechnology and Bioengineering, 77:398407 (2003); Dezengotita et al., Biotechnology and Bioengineering, 77:369-380 (2002); Schmelzer & Miller, Biotechnology Prog., 18:346-353 (2002); Dezengotita et al., Biotechnology and Bioengineering, 78:741-752 (2002); Sun et al., Biotechnology Prog., 20:576-589 (2004)], покращення цих зовнішніх факторів дає змогу домогтися лише обмеженого поліпшення вираженості й не забезпечує комерційної ефективності без попередньої оптимізації експресійного вектора для одержання ідеального базового рівня вираженості. Відомо чимало публікацій про дослідження внутрішніх факторів з метою покращення вираженості генів. Внутрішні фактори, що їх описано нижче, відомі також як регуляторні елементи, що регулюють вираженість генів у багато способів. З минулих досліджень відомо, що для вираженості потрібного гена в експресійному векторі, його потрібно помістити під контроль відповідної 5'- та 3'- фланкуючої послідовності, що дасть змогу генові транскрибуватися в mRNA, а далі точно транслюватися в білок. Існують публікації про численні 5'- та 3'- фланкуючі послідовності на зразок ТАТА-боксів [Boshart, Μ. et al., Cell, 41:521530 (1985); Browning, K.S. et al. J. Biol. Chem., 263:9630-9634 (1998); Dorsch-Hasler, K. et al., PNAS, 82:8325-8329 (1985)], промотори на зразок вірусних - наприклад, миттєвий ранній промотор CMV, ранні або пізні промотори SV40, основний пізній аденовірусний промотор [Luigi R., Gene, 168:195-198 (1996); Pizzomo, M.C. et al., J. Virol., 62:1167-1179 (1988); Okayama and Berg, Мої. Cell. Biol., 2:161-171 (1982); Wong et al., Science, 228:810-815 (1985); Foecking and Hofsteffer, Gene, 45:101-105 (1986)], а також ссавцеві промотори на зразок промотору металотіоніну мишей, бетаактиновий промотор мишей [Nicole Israel et al., Gene, 51:197-204 (1987); Karin, M. and Richards, Nature, 299:797-802 (1982); Miyazaki et al., Proc. Natl. Acad. Sci. USA, 83:9537-9541 (1986)], енхансери на зразок миттєвого раннього енхансера CMV [Cockett, M.I. et al., Nucleic Acids Research, 19:319-325 (1996)], кодони початку та закінчення трансляції [Lehninger et al., Principles of Biochemistry - 3rd edition, Worth Publishers, Chapter 27, p 1025], сайта поліаденилювання на зразок бичачого гормону росту (BGH) та сайти поліаденилювання SV40 [Carsweil, S. and Alwine, J.C., Моl. Cell. Biol. 9:4248-4258 (1989)]. Ще один внутрішній фактор - інтрони, що звичайно формують інтегральні частини евкаріотичних генів як проміжні послідовності між екзонами і точно видаляються з первинного транскрипту в процесі, відомому як сплайсинг РНК, з формуванням зрілої 7 mRNA. У багатьох дослідженнях було показано, що сплайсинг РНК відповідає за стійкість mRNA [Buchman et al. Моl, Cell Biol. 8:4395-404 (1988); Peterson et al., Proc. Natl. Acad Sci. USA, 83:888387 (1986)], та регулювання вираженості генів [Brinster et al., Proc. Natl. Acad. Sci. USA, 85:83640 (1988); Dynan, W.S. and Tjian, R., Nature, 316:774-778 (1985)]. Винайдено також синтетичні химерні інтрони, на зразок опублікованих в роботі Huang et al., що складаються з 5'-донорного сайту аденовірусного основного пізнього транскрипту та 3'-сплайсингового сайту імуноглобуліну миші [Huang et al., Nucleic Acids Res., 18:937-47 (1990)]. Такі химерні інтрони краще за інші, широко використовувані інтрони підтримують вираженість гетерологічних генів [Huang et al., Nucleic Acids Res., 18:937-47 (1990); Ted Choi et al., Molecular and Cellular Biology, 11 (6): 3070-3074(1991)]. Широко застосовуваним джерелом високоефективних внутрішніх факторів є віруси. Відомо, що віруси - найефективніші паразити в природі, які використовують власні внутрішні фактори для маніпуляції вираженістю генів господаря й вірусу з метою поширення й виживання. Крім того, проводились численні дослідження ролі вірусів в утворенні експресійних векторів, що мали на меті суттєве поліпшення продукування білка. Деякі з найефективніших промоторів, відомих у молекулярній біології, виводяться з вірусів [Luigi R., Gene, 168:195-19S (1996); Pizzomo, Μ. С. et al., J. Virol, 62:1167-1179 (1988); Okayama and Berg, Моl. Cell. Biol., 2:161-171 (1982); Wong et al., Science, 228:810-815 (1985); Foecking and Hofsteffer, Gene, 45:01-105 (1986)]. Численні віруси ретельно досліджувались на генетичному рівні з визначенням окремих послідовностей, що можуть змінювати ядерний та цитоплазмічний метаболізм mRNA в клітинах-господарях. Одним з таких елементів є потрійний провідний елемент аденовірусу (TPL) (GI: 209811) [Akcgarvi G. et al., J. Моl Biol, 134(1): 143-58 (1979)], що, як відомо, покращує трансляцію навіть не вірусної РНК, в разі приєднання до неї, в інфікованих вірусами клітинах [Berkner K.L. et al., Nucleic Acids Res, I3G;:841-57 (1985)]. Всі mRNA, закодовані головним блоком основного пізнього транскрипту, мають цю 5' некодовану область. Цей елемент може скорочувати період напіввиведення транскриптів з ядра [Huang et al. J Virol, 2(1):22535 (1998)]. Відомо також, що цей елемент покращує трансляцію mRNA [Kaufman R. J. et al., Proc Natl Acad Sci U S A., 82(3):689-93 (1985)]. Інший елемент - аденовірусні вірусасоційовані гени І та II РНК (GI:-209811) або їхній функціональний варіант. Показано, що вірусасоційовані гени І та II РНК (VA-гени) підвищують ефективність трансляції гена, що містить TPL-послідовність [Kaufman R. J. et al., Proc Na:: Acad Sci USA, 82/3):689-93 (1985)]. VA-ген I PHK бере участь у дефосфориляції EIF2a, чим підвищує інтенсивність синтезу білка [O'Malley et al., Cell, 44:391400 (1986); Thimmapayya В, et al., Cell, 31:543-551 (1982)]. Ще одним широко використовуваним внутрішнім фактором є число копій гена, що складає 92913 8 найбільш привабливий варіант підвищення вираженості генів [Kaufman and Sharp, Journal of molecular Biology, 159:601-602 (1982); Pendse G.J. et al., Biotechnology and Bioengineering, 40:119129 (1992); Schimke, R.T. (Ed.), Gene Amplification. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, 1982]. Найбільш широкого застосування набув метод збільшення числа копій гена шляхом вибору клітин для ампліфікації гена. В цьому підході - наприклад, як описано в патентах ЕР0045809 та US4634665, - клітина-господар трансформується парою генів. Перший ген цієї пари кодує потрібний білок, другий - селективний маркер (наприклад, дигідрофолатредуктазу) (DHFR) [Alt, F.W. et al., J. Biol. Chem, 253:13371370 (1978)]. Ці два гени присутні на одному експресійному векторі або на двох окремих. Після трансфекції цією парою генів - в разі застосування методу з використанням гена DHFR як селективного маркера, дія якого зводиться до нуля продуктом гена селективного маркера, - клітини культивуються в умовах підвищення концентрації токсичного агента на зразок метотрексату. Було виявлено, що лінії клітин, котрі виживали при високих концентраціях токсичного агента, мали більше число копій як гена селективного маркера, так і гена потрібного продукту. Вибрана клітинагосподар, що має підвищене число копій відповідного гена, може продукувати більшу кількість потрібного білка порівняно з попередньою лінією клітин. В аналогічній стратегії ампліфікації генів використовувались інші селективні маркери на зразок аденозиндезамінази (ADA), орнітиндекарбоксилази (ODC), аспарагінсинтетази (AS) [Chiang, T. and McConlogue, Моl. Cell. Biol. 764769 (1988); Cartier, M. et al., Моl. Cell. Biol, 7:16231628 (1987); Germann, U.A. et al., J. Biol. Chem. 264:7418-7424 (1989); Mkeille Cartier and Clifford P. Stanners, Gene, 95:223-230 (1990); Wood C.R. et al., J Immunol, 145:3011-3016 (1990); Kellems R. E. et al., In Genetics and Molecular Biology of Industrial Microorganisms, American Society for Microbiology, Washington, 215-225 (1989)], та глютамінсинтетази (GS) [Catherine W-H. et al., J. Biol. Chem., 276:43, 39577-39585 (2001); Bebbington, et al., Biotechnology, 10:169-175 (1992); Bebbington, C.R., Monoclonal antibodies: the next generation, Zola, H, fed). Bios Scientific, Oxford, 65-181 (1995); Wilson, R.H., In "Gene Amplification in Mammalian Cells" ed Kellens, R.E., Marcel Dekker Inc., New York, 301-311(1993)]. Ці регуляторні елементи та внутрішні фактори, що їх описано вище, загалом не здатні поодинці забезпечити вираженість генів, тому мають застосовуватися в сполученнях. Однак якщо самі елементи вивчено досить добре, то ефективність їхніх сполучень, стосовно сильної вираженості, не є абсолютно передбачуваною. Певні сполучення дають дуже слабку вираженість порівняно з іншими. Наприклад, у роботі Jang et al використання експресійного вектора, що складається з сполучення SR-альфа промотора, AMV-провідної послідовності РНК та DHFR, дало змогу отримати вираженість еритропоетину (ЕРО) лише в 45 міжнародних одиниць/мл (еквівалентно 0,346мкг/мл) 9 [Jang Η Ρ et al., Biotechnol. Appl. Biochem, 32:167172, (2000)]. Водночас, у патенті US5955422 повідомляється про рівні ЕРО від 750 до 1470 одиниць/ мільйон клітин / 48год. (або від 375 до 735одиниць/ мільйон клітин/ 24год.), досягнуті з використанням експресійного вектора, складеного з іншого сполучення елементів - промотора SV40, послідовності поліаденилової кислоти та DHFR. Ще один експресійний вектор описано в патенті US5888774. Він складається з сполучення промотора EFI та елементів ароВ SAR і дає вираженість від 1500 до 1700 міжнародних одиниць ЕРО/ мільйон клітин/ 24год. Стосовно інших рекомбінантних білків, на зразок TNFR-IgGFc (Enbrel), існує публікація про експресійний вектор, що складається зі сполучення промотора CMV, TPL, VA-генів І та II, а також DHFR [US 5605690, Cindy A Jacobs and Craig A Smith]. Попри всі описані вище переваги рекомбінантних біологічних препаратів, великою проблемою залишається висока вартість їх виробництва особливо тих, в яких використовуються експресійні системи ссавців. Тому, незважаючи на численні публікації методів підвищення вираженості білка через модуляцію експресійного вектора, досі існує потреба в розробці нових експресійних векторів для подальшого підвищення продуктивності евкаріотичних клітин-господарів. Як не дивно, незважаючи на величезний об'єм знань, накопичених у цій царині за останні два десятиліття, на сьогодні фахівець не в змозі просто вибрати сполучення внутрішніх факторів і сконструювати експресійний вектор з гарантовано високою вираженістю. Додавання до сполу 92913 10 чення певного елемента може не справляти значного адитивного чи синергетичного впливу на можливості вираженості вектора. Тому процес розробки нового експресійного вектора, що забезпечував би високий рівень вираженості білка, досі потребує емпіричної перевірки великого числа можливих варіантів. Ми винайшли новий експресійний вектор, котрий, після стійкої трансфекції в клітинах CHO-DHFR', дає вираженість в 11 830 міжнародних одиниць/мл (91мкг/мл) в 168годинній культурі, що рівнозначно від 2366 до 3549 міжнародних одиниць/ 106 клітин/ 24год. або від 18,2 до 27,3мкг/106 клітин/ 24год. Як не дивно, цей рівень вираженості ЕРО є на 80-100% вищим за деякі з найкращих опублікованих векторів. Цей новий експресійний вектор суттєво знизить вартість виробництва ЕРО та інших рекомбінантних біологічних препаратів. Даний винахід вирішує проблему, описану вище, в розділі "Відомий рівень техніки", застосуванням нових експресійних векторів для значного покращення продукування рекомбінантних білків у клітинах ссавців. Крім того, винахід включає спосіб одержання таких векторів та спосіб їх застосування для забезпечення високого рівня вираженості білків. Таким чином, однією з основних цілей даного винаходу є створення вдосконаленого способу одержання високих рівнів вираженості потрібних білків шляхом використанням нового вектора, що описаний в даному документі. В даному винаході використано наступні послідовності: 11 92913 12 13 92913 14 15 В даному винаході використано нове сполучення п'яти елементів, одержаних з вірусів та різноманітних інших векторних джерел. Це дало змогу створити новий вектор, що забезпечує синергетичний вплив зазначених елементів у формі підвищення вираженості потрібних білків у клітинах-господарях ссавців. Зокрема, ці експресійні вектори складаються з нового сполучення таких п'яти елементів: миттєвого раннього промотора CMV, потрійного провідного елемента аденовіру 92913 16 су (TPL) (GI:209811), гібридного (химерного) інтрона (послідовність №1), генів І та II аденовірусної вірус-асоційованої РНК (GI:209811) та послідовність поліадениляції бичачого гормону росту. Якщо використовувати це нове сполучення п'яти елементів у поєднанні з додатковими елементами у базовому векторі, що необхідні для його функціонування як вектора, генеровані таким чином нові вектори проявляють синергетично більш високу вираженість порівняно з іншими 17 92913 векторами, що містять лише деякі з зазначених п'яти елементів у поєднанні з базовим вектором. Стійка трансфекція нашого нового експресійного вектора, що містить також ампліфікаційний маркер DHFR, дає вираженість ЕРО, вищу на 80100% порівняно з найкращими з досі опублікованих векторів. У кращому варіанті, нові експресійні вектори складаються з миттєвого раннього промотора CMV, потрійного провідного елемента аденовірусу (TPL) (послідовність №4), гібридного (химерного) інтрона (послідовність №1), генів І та II аденовірусної вірус-асоційованої РНК (послідовність №5), сайту клонування, селективного маркера клітин ссавців, селективного маркера прокаріотних клітин, ампліфікаційного/селективного маркера та сайту поліадениляції бичачого гормону росту. Все це було в наявності у відповідних позиціях зазначених векторів. Білки, які можна продукувати за допомогою цих векторів, включають гормони на зразок FSH (фолікулостимулюючий гормон), антитіла, химерні білки на зразок етанерцепту, компоненти крові на зразок фактора VII, фактори росту на зразок 18 еритропоетину, цитокіни на зразок інтерферонів, TNF (фактор некрозу пухлин) тощо. Фіг.1 - схематичне представлення різноманітних експресійних векторів TNFR-IgGFc включно з новим експресійним вектором pZRC-TNFRIgGFc. Фіг.2 - експресійний білок TNFR-IgGFc з клітин Cos-1, транзиторно трансфікований різноманітними експресійними векторами TNFR-IgGFc включно з новим експресійним вектором pZRCTNFR-IgGFc. Фіг.3 - схематичне представлення різноманітних експресійних векторів ЕРО включно з новим експресійним вектором pZRC-EPO. Фіг.4 - порівняння вираженості ЕРО в клітинах Cos-1, транзиторно трансфікованих новим експресійним вектором pZRC-EPO та іншим комерційним експресійним вектором pcDNA3.1, що кодує ЕРО. Фіг.5 - вплив підвищення тиску МТХ на вираженість ЕРО з вибраних високопродуктивних клонів клітин CHO-DHFR' стійко трансфікованих pZRC-EPO. Джерело елементів конструювання нового експресійного вектора Основний остов експресійного вектора П'ять регуляторних елементів Промотор CMV Химерний інтрон TPL Гени VAІ та II Послідовність поліадениляції BGH Мініген DHFR 1 2 pcDNA3. 1 (Invitrogen1) pcDNA3.1 (Invitrogen) pIREShyg3 (Clontech2) pCAV-NOT-TNFR (ATCC 68088) pCAV-NOT-TNFR (ATCC 68088) pcDNA3.1 (Invitrogen) pDHFR2.9 (ATCC 37165) Продукт компанії Invitrogen Life Technologies, США Продукт компанії Clontech Laboratories Inc., США Використано скорочення: CMV TPL Гени VA І та II BGH ЕРО TNFR-IgGFc цитомегаловірус трикомпонентний лідер гени І та II вірус-асоційованої РНК бичачий гормон росту еритропоетин химерний білок рецептора фактора некрозу пухлин та частина Fc імуноглобуліну G Даний винахід вирішує проблему високої вартості виробництва, описану вище, в розділі "Відомий рівень техніки", застосуванням нових експресійних векторів для значного покращення продукування рекомбінантних білків у клітинах ссавців. В цих нових векторах використано давно відомі з публікацій регуляторні елементи, але застосовані в унікальних сполученнях, що синергетично забезпечують несподівано високу вираженість. Крім того, винахід включає спосіб застосування таких векторів для забезпечення високого рівня вираженості білків. Нове сполучення елементів, використаних у даному винаході, складається з: a) миттєвого раннього промотора CMV, b) потрійного провідного елемента аденовірусу (TPL) (послідовність №4 та Gl:209811), c) гібридного (химерного) інтрона (послідовність №1), d) генів І та II аденовірусної вірусасоційованої РНК (послідовність №5 та GI:209811), e) сайту поліадениляції бичачого гормону росту. Для розробки цих нових векторів, базові експресійні вектори, що їх можна додавати до нового сполучення елементів, описаного вище, складаються з: a) Сайту клонування. 19 b) Відповідного селективного маркера клітин ссавців, вибраного з групи, що складається з ліко-стійких маркерів на зразок неоміцину, гідроміцину, пуроміцину тощо, а також інших маркерів. c) Відповідного селективного маркера прокаріотних клітин, вибраного з групи, що складається з маркерів, стійких до антибіотиків, на зразок ампіциліну, канаміцину тощо, а також інших маркерів. d) Ампліфікаційного/селективного маркера, вибраного з групи, що складається з дигідрофолатредуктази, аденозиндезамінази, орнітиндекарбоксилази, аспарагінсинтетази, глутамінсинтетази тощо. Спосіб одержання потрібних векторів включає виділення різноманітних елементів вектора з інших векторних джерел або з їхніх біологічних джерел та введення їх в інший вектор, що утворює остов плазміди. Методи, застосовані для створення цих конструкцій, виводяться з опублікованих векторних конструкцій [Sambrook J et al., Molecular Cloning - A laboratory Manual, Cold Spring Harbor Laboratory Press, New York, (1989)], за необхідності - з відповідною модифікацією. Для продукування потрібного білка, перш за все, одержують кодування відповідних генів цього білка. Для одержання потрібного гена звичайно використовують наступні методи - поодинці або в сполученні: (1) виділення матричної РНК (mRNA) потрібного гена та використання її як матриці для продукування комплементарної ДНК (cDNA) через зворотну транскрипцію; (2) виділення природного гена з бібліотек геномних ДНК з використанням сполучень відповідних гібридизаційних зондів та рестрикційних ферментів, характерних для генів; (3) виділення гена застосуванням особливої ампліфікації певних генних фрагментів за допомогою ланцюгової реакції полімерази (PCR) з використанням однієї або декількох пар характерних для гена праймерів; (4) хімічний синтез гена з його складових нуклеотидів. Одержаний ген клонується на сайті клонування нового вектора з застосуванням методів, відомих фахівцям. Одержана в результаті структура перетворюється на відповідну ссавцеву клітину-господаря. Ссавцева клітина-господар, що може бути використана в цьому способі, вибирається з групи, що включає лінії клітин СНО та СНО DHFR', BHKL NSO, COS тощо. Перетворені клітини вибираються з огляду на їхню здатність рости у відповідному середовищі, що містить антибіотики, а також на здатність до вираження потрібного білка. Далі вибраний клон вирощується в умовах відповідної культури, що забезпечує ріст і продукування високих рівнів потрібних білків. Число копій гена і, як наслідок, його вираженість можна збільшувати, вибираючи клітини з великим числом копій генів в умовах підвищення селективного тиску особливого цитотоксичного лікарняного засобу, дію якого можна нейтралізувати лише збільшенням числа копій селективного маркера. Оскільки ампліфікація генів цитотоксич 92913 20 ним селективним тиском часто призводить до перетворення клона в гетерогенну популяцію клітин зі змінним числом копій потрібних генів, кінцевий високопродуктивний клон вибирається з гетерогенної популяції рестрикційним розщепленням. Приклад 1 Структура експресійних векторів TNFR-IgGFc Як базовий остовний для всіх наших векторів, було взято ссавцевий експресійний вектор pcDNA 3.1 (Invitrogen Life Technologies). Цей вектор складається з миттєвого раннього промотора CMV та поліадениляційної сигнальної послідовності BGH для транскрипційного контролю гена. Це два елементи з п'яти, що утворюють наше нове сполучення. Далі, цей вектор складається з певної кількості сайтів клонування (MCS), початкової форми pUC реплікації бактерій, гена, стійкого до ампіциліну, для селекції в Е. соlі, та гена, стійкого до неоміцину, для селекції в ссавцевих клітинах. Ще три елементи з нового сполучення п'яти елементів, а саме аденовірусна трикомпонентна провідна послідовність, гібридний (химерний) інтрон, що складається з 5'-донорського сайту аденовірусного основного пізнього транскрипту, 3'-сплайсингового сайту імуноглобуліну миші та аденовірусних генів РНК VA І і II, вводили в цей остов pcDNA 3.1 в різноманітних сполученнях для пошуку того, в якому всіелементи працюватимуть синергетично, забезпечуючи дуже високу вираженість. cDNA, що кодують злитий ген TNFR-IgGFc (послідовність №2) та ген еритропоетину людини (послідовність №3), використовувались як репортери для вивчення впливу сполучень цих елементів на вираженість білка. На Фіг.1 показано експресійні вектори, розроблені для TNFR-IgGFc, на Фіг.3 - такі само вектори для ЕРО. TNFR-IgGFc - це злитий білок, що містить 75 кілодальтонів TNFR (1-235а.а.) та фрагмент Fc IgGl (що містить СН2, СН3 та шарнірну область) [US5605690]. Цей злитий білок продається як лікарський засіб під назвою Enbrel (Amgen) для лікування ревматоїдного артриту. Його дія полягає в видаленні з кровообігу запального цитокіну TNF-α. Для клонування TNFR I cDNA як матрицю було використано тотальну РНК плаценти людини (Clontech) для зворотної реакції транскриптази-полімерази (RT-PCR) на основі клонування потрібної послідовності. Було синтезовано двониткову cDNA з тотальної РНК плаценти людини з використанням зворотної транскриптази MMLV (МВІ Fermentas, USA) через праймерування залежно від різновиду гена [Maniatis et al., Molecular cloning; A Laboratory Manual (Cold Spring harbor Laboratory Press, Cold Spring Harbor, N.Y.), (1990)]. Далі цю cDNA було піддано 40 циклам ампліфікації PCR з використанням 100 пікомолів вироджених олігонуклеотидних праймерів, залежно від різновиду гена, в об'ємі 100мкл, що складають 50мМ Tris-Cl (pH8,3), 2,5мМ MgCl2, по 200мкМ 4 dNTP та 2,5 одиниць Pfu полімерази. Кожний цикл ампліфікації PCR складався з інкубацій за температури 94°С протягом 30с (денатурація), 58°С протягом 30с (відпал) та 72°С протя 21 гом 1хв. (розтягнення). Ампліфікований продукт реакції PCR розчиняли в 1-процентному розчині агарозного гелю. Потрібний фрагмент розміром приблизно у 705 основних пар виділяли з гелю й очищали, використовуючи для цього комплект виділення Qiagen Gel. Цей очищений фрагмент ДНК лігували в MCS pcDNA 3.1 після рестрикційного розщеплення як вектора, так і очищеного продукту PCR з використанням для цього EcoRI та PvuII (МВІ Fermentas, USA). Продукт лігації перетворювали на Е. coli DH5α, після чого трансформанти оцінювали стосовно стійкості до ампіциліну. Плазмі дну ДНК, виділену приблизно з 20 таких колоній, аналізували на наявність TNFR I cDNA рестрикційним розщепленням з використанням різноманітних рестрикційних ферментів. Послідовність одного з таких плазмідів було визначено за допомогою автоматичного секвенсера ДНК (АВІ). Виявилося, що він має правильні интеграцію та послідовність TNFR I cDNA в векторі pcDNA 3.1. Ця плазмідна ДНК отримала назву pcDNA-TNFR. Аналогічно, послідовність IgGIFc також виділяли, використовуючи як матрицю клонування на основі RT-PCR тотальну РНК плаценти людини (Clonetech). PCR визначали за допомогою пари олігонуклеотидних праймерів, залежно від різновиду гена, що відповідали області кодування послідовності TgGlFc (699bp). Кожний цикл ампліфікації PCR складався з інкубацій за температури 94°С протягом 30с (денатурація), 58°С протягом 30с (відпал) та 72°С протягом 1хв. (розтягнення). Ампліфікований продукт реакції PCR розчиняли в 1-процентному розчині агарозного гелю. Потрібний фрагмент розміром приблизно у 725 основних пар виділяли з гелю й очищали, використовуючи для цього комплект виділення Qiagen Gel. Цей очищений фрагмент ДНК лігували в вектор pTZ57R (МВІ Fermentas) після лінеаризації цього вектора й розщеплення очищеного продукту PCR за допомогою EcoRI та Not I (MBI Fermentas, USA). Продукт лігації перетворювали на Е. coli DH5α, після чого трансформанти оцінювали стосовно стійкості до ампіциліну. Плазмідну ДНК, виділену приблизно з 20 таких колоній, аналізували на наявність IgGIFc cDNA рестрикційним розщепленням з використанням різноманітних рестрикційних ферментів. Послідовність одного з таких плазмідів було визначено за допомогою автоматичного секвенсера ДНК (АВІ). Виявилося, що він має правильні інтеграцію та послідовність IgGl Fc cDNA в векторі pTZ57R. Ця плазмідна ДНК отримала назву pTZ57R-IgGlFc. pcDNA-TNFR-IgGFc Злиття фрагментів генів TNFR І і IgGIFc проводилося таким способом. Клонований фрагмент TNFR І виділяли з pcDNA-TNFR за допомогою повного розщеплення з використанням EcoRI, та PvuII. Фрагмент IgGIFc виділяли з структури pTZ57R-IgGlFc розщепленням її з застосуванням Pvu II та Not І. Ці фрагменти виділяли з агарозного гелю за допомогою комплекту виділення Qiagen gel. Оба фрагменти ДНК змішували з лінеаризованим вектором pcDNA3.1, попередньо розщепленим за допомогою EcoRI та NotI в три 92913 22 компонентній реакції лігації. Продукт лігації перетворювали на Ε coli DH5α, і трансформанти оцінювали стосовно стійкості до ампіциліну. Плазмідну ДНК, виділену приблизно з 10 таких колоній, аналізували на наявність злитого продукту TNFRIgGFc рестрикційним розщепленням з використанням різноманітних рестрикційних ферментів. Послідовність одного з таких плазмідів було визначено за допомогою автоматичного секвенсера ДНК (АВІ). Виявилося, що він має правильні интеграцію та послідовність гібридного продукту TNFR-IgGFc. Ця векторна структура отримала назву pcDNA-TNFR-IgGFc (Фіг.1) і містить злитий ген з приєднаним промотором CMV на кінці 5', а також поліадениляційну послідовність BGH на кінці 3'. pcDNA-TPL-TNFR-IgGFc-VA DHFR Для створення цього вектора, перш за все, вставляли аденовірусний елемент TPL в нижню частину промотора CMV та у верхню частину гена TNFR-IgGlFc в pcDNA-TNFR-IgGFc. Це досягалося повним розщепленням pCAV/NOT-TNFR (АТСС 68088) за допомогою Kpnl and Nde І, що давало фрагмент 700bр, котрий містить TPL. Його далі очищали, використовуючи для цього комплект виділення Qiagen Gel. Цей фрагмент 700bр лігували у векторі pcDNA-TNFR-IgGFc, розщепленому з використанням KрnІ та Nde І. Продукт лігації перетворювали на Е. соlі DH5α, і трансформанти оцінювали стосовно стійкості до ампіциліну. Плазмідну ДНК аналізували на наявність і орієнтацію TPL в одержаному плазміді рестрикційним розщепленням з використанням різноманітних рестрикційних ферментів. Одну з таких позитивних плазмідних ДНК, що отримала назву plnt-TNFR-IgG-1 було далі використано для вставляння двох елементів - гена РНК 1000bp VA, виділеного з pCAV/NOT-TNFR подвійним розщепленням за допомогою EcoRI та Not І, і міні-гена DHFR, виділеного з плазміду pDHFR 2.9 (АТСС 37165) [Crouse GF et аl., Моl. Cell. Biol, 3:257-266 (1983)] повним розщепленням за допомогою ВатНІ та Sspl. Ці два фрагменти - міні-гена DHFR та гена VA РНК, - перемішували з plnt-TNFR-IgG-1, попередньо розщепленим за допомогою Muni & BgllI, в трикомпонентній реакції лігації. Продукт лігації перетворювали на Е. соlі DH5α, і трансформанти оцінювали стосовно стійкості до ампіциліну. Плазмідну ДНК аналізували на наявність і орієнтацію гена VA РНК та міні-гена DHFR в одержаному плазміді рестрикційним розщепленням з використанням різноманітних рестрикційних ферментів. Ця векторна структура отримала назву pcDNA-TPL-TNFR-IgGlFc-VA-DHFR (Фіг.1). pcDNA-TPL-Intron-TNFR-lgGFc Експресійний вектор pcDNA-TPL-Intron-TNFRlgGFc було генеровано вставлянням химерного інтрона в нижню частину аденовірусного елемента TPL та верхню частину гена TNFR-IgGFc у векторі plnt-TNFR-IgG-1. Це було досягнуто повним розщепленням pIREShyg3 (Clontech) за допомогою BstXI та EcoRI з одержанням фрагмента 350bр химерного інтрона, котрий далі очищали, використовуючи для цього комплект виділення Qiagen Gel. Цей фрагмент 350bр лігували у век 23 торну структуру plnt-TNFR-IgG-1, попередньо розщеплену за допомогою BstXI та EcoRI. Продукт лігації перетворювали на Е. соіі DH5cc, і трансформанти оцінювали стосовно стійкості до ампіциліну. Плазмідну ДНК аналізували на наявність і орієнтацію TPL, інтрона та гена TNFRIIgGIFc в одержаному плазміді рестрикційним розщепленням з використанням різноманітних рестрикційних ферментів. Послідовність одного з таких плазмідів було визначено за допомогою автоматичного секвенсера ДНК (АВІ). Виявилося, що він має правильну інтеграцію TPL, химерного інтрона та гена TNFRI-IgGIFc у векторі. Ця векторна структура отримала назву pCDNA-TPL-IntronTNFR-IgGFc (Фіг.1). pcDNA-TPL-Intron-TNFR-IgGFc-VADHFR(pZRC-TNFR-IgGFc) Описану вище плазмідну ДНК pcDNA-TPLIntron-TNFR-IgGFc було використано для вставляння двох елементів - гена РНК 1000bp VA, виділеного з pCAV/NOT-TNFR подвійним розщепленням за допомогою EcoRI та Not І, та міні-гена DHFR, виділеного з плазміду pDHFR 2.9 повним розщепленням за допомогою BamHI and Sspl. Фрагменти для міні-гена DHFR та гена VA PHK лігували в описаному вище pcDNA-TPL-IntronTNFR-lgGFc, за допомогою Muni & BgIII, у трикомпонентній реакції лігації. Продукт лігації перетворювали на Е. соlі DH5α, і трансформанти оцінювали стосовно стійкості до ампіциліну. Плазмідну ДНК аналізували на наявність і орієнтацію гена VA РНК та міні-гена DHFR в одержаному плазміді рестрикційним розщепленням з використанням різноманітних рестрикційних ферментів. Крім того, перевіряли інтеграцію та підтверджували її визначенням послідовностей ДНК. Цю векторну структуру було названо pcDNA-TPLIntron-TMFR-IgGFc-VA-DHFR, інша назва - pZRCTNFR-IgGFc (Фіг.1). Приклад 2 Порівняння впливу різноманітних експресійних векторів TNFR-IgGFc на вираженість методом транзиторної трансфекції Фібробластові клітини нирки африканської зеленої мавпи Cos 1 (АТСС №CRL-1650) було використано для експерименту з транзиторної трансфекції. Ці клітини регулярно поміщали в середовище повного росту (модифікація Дюльбекко (Dulbecco) середовища Ігла (Eagle) з 4мМ Lглутаміну, доведеного до вмісту 1,5г/л бікарбонату натрію , 4,5г/л глюкози, 10% ембріональної коров'ячої сироватки (FBS) та антибіотиків) за температури 37°С в атмосфері 5-процентного двоокису вуглецю (СО2). За день до трансфекції клітини, що були у середній фазі логарифмічного росту, трипсинізували й висіяли в чашки Петрі з 6 лунками, з густиною 0,2 мільйона клітин на лунку в 3мл середовища повного росту й інкубували за температури 37°С та концентрації 5% СО2. Трансфекцію проводили за допомогою реактиву Qiagen Polyfect, з використанням відомих протоколів. На день трансфекції трансфекційну суміш було приготовано наступним чином. 1,5мг ДНК, розчиненої у воді, що не містила ендотоксинів, пропустили через фільтрувальний шприц 0,2мкМ. 92913 24 Для найбільшої структури (pZRC-TNFR-IgGFc) використали, як такий, вектор 1,5мкг, тоді як для інших, менших структур еквімолярну кількість (стосовно гена TNFR-IgGFc) кожної структури змішували з порожнім вектором, що давало 1,5мкг ДНК. Цю ДНК спочатку розчиняли в середовищі росту клітин (модифікація Дюльбекко середовища Ігла) з 4мМ L-глутаміну, доведеного до вмісту 1,5г/л бікарбонату натрію, 4,5г/л глюкози, без сироватки антибіотиків - всього 100мкл. Все це інтенсивно перемішували, далі прокручували протягом декількох секунд, щоб прибрати краплі зі стінок та верхньої частини пробірки. Далі до цього розчину ДНК додавали 10мкл реактиву PolyFect Transfection. Проводили ретельне перемішування пропусканням через піпетку від 5 до 7 разів. Після цього зразки інкубували за кімнатної температури (20-25°С) протягом 10 хвилин для комплексоутворення. Під час комплексоутворення все середовище росту обережно відсмоктували з 6-лункової чашки Петрі. Висіяні клітини промивали в 3мл стерильного буферованого соляного розчину 1Х (PBS). Після цього додавали 1,5мл свіжого повного середовища росту. Після 10 хвилин комплексоутворення 0,6мл повного середовища росту додавали до пробірки, що містила трансфекційні комплекси. Далі чашки обережно прокручували для ретельного перемішування й інкубували за температури 37°С, в атмосфері 5% СО2. Через 40 та 60год. трансфекції виснажене середовище видаляли з лунок та піддавали аналізу на вираженість TNFR-IgGFc за допомогою апаратури ELISA, як описано далі. Потрібне число лунок у 96-лунковій титровій чашці (Nunc Maxisorp) покрили в об'ємі у 100мкл на лунку антитілом Goat anti-Human IgGFc Fragment (Calbiochem, Cat no 401439) концентрації 2мкг/мл в PBS (pH7,3). Чашки інкубували за температури 4°С до ранку, в умовах підвищеної вологості для ефективного покриття. Через 12-18 годин незв'язане антитільне покриття знімали, а лунки блокували блокувальним буфером (1% BSA, 0,05% Tween 20, 5% знятого молока в 1Х PBS, рН7,2) та інкубували протягом 1 години за температури 37°С. Після 1 години блокування чашки тричі промивали буфером PBST (по 250мкл кожного разу) (1Х PBS, 0,1% BSA, 0,05% Tween 20). Далі зразки по 100мкл (тестові та контрольні, відповідно розріджені в 1Х PBS, що містить 1% BSA) додавали до лунок у двох екземплярах. Як контрольний зразок TNFR-IgGFc, використовували багаторазову пробірку Enbrel (етанерцепт), наповнену 25мг/мл білка Enbrel (виробництва Immunex Corporation, США, продається компанією Amgen and Wyeth Pharmaceuticals). Концентрації контрольних зразків, що використовувалися для побудови контрольної кривої, дорівнювали 1нг/мл, 2нг/мл, 5нг/мл, 10нг/мл, 20нг/мл, 40нг/мл і 80нг/мл. Після інкубації протягом 90 хвилин, що давало змогу утворитися зв'язкам антигенів з антитілами, чашки тричі промивали буфером PBST (по 250мкл кожного разу), і до лунок додавали 100мкл детекційного антитіла Goat anti-Human IgGFc-HRP (Pierce, Cat No.31416) розрідженого до співвідношення 25 1:20000 в 1Х PBS, що містить 1% з подальшою інкубацією за температури 37°С протягом 1год. Чашки тричі промивали для видалення незв'язаного кон'югату антитіло-HRP. 100мкл розчинупідкладки, що містить 8мг порошку OPD [офенилендіамін (1,2-бензолдіамін) дигідрохлорид] (Sigma cat No P-1526) та додавали 10мкл перекису водню, одержаного в цитрат-фосфатному буфері (122,8мг безводної лимонної кислоти та 188мг Na2HPO4 в 20мл дистильованої води, доведені до рН від 4,8 до 5,00) на лунку з подальшою інкубацією протягом 30хв. в темряві, за температури 37°С. Реакцію припиняли додаванням 50 p.1 1N сірчаної кислоти на лунку. Вимірювали оптичну густину, використовуючи вимірювальний прилад ELISA, на довжині хвилі 490нм. Значення для тестових зразків виводилися за допомогою програмного забезпечення SoftMax Pro (Molecular Devices) з використанням 4-параметричної стандартної кривої, отриманої з рішення рівняння Лебера-Марквардта (Leveberg-Marquardt). Результати: На Фіг.2 показано репрезентативні дані одного такого експерименту. Найвищу вираженість було отримано на pZRC-TNFR-IgGFc (72,5±4,3нг/мл), що на 21,97% вище порівняно з pcDNA-TPL-TNFR-IgGFc-VA-DHFR (59,44±1,8нг/мл), на 36,66% вище порівняно з pcDNA-TPL-Intron-TNFR-IgGFc (53,05±2,2нг/мл) і на 395,55% вище порівняно з pcDNA-TNFR-IgGFc (I4,63±1,4нг/мл). Синергетична дія п'яти елементів ще більше посилюється завдяки тому, що найбільший плазмід pZRC-TNFR-IgGFc, що мав би відповідати найнеефективнішій трансфекції, все ж забезпечує найвищу вираженість. Здатність нового сполучення елементів pZRC-TNFR-IgGFc забезпечувати високу вираженість підтверджується наступними прикладами. Приклад 3 Структура експресійних векторів ЕРО Винайдено два експресійних вектори ЕРО. Новий експресійний вектор pZRC-EPO є аналогічним pZRC-TNFR-IgGFc - новому експресійному вектору, якому раніше віддавали перевагу за високу вираженість в експериментах з транзиторною трансфекцією, - і кодує не TNFR-IgGFc, а ЕРО. Другий вектор - pcDNA3.1, - це комерційний експресійний вектор, що кодує ЕРО і також має ген для DHFR. pcDNA-EPO-DHFR Щоб одержати еритропоетин cDNA людини, тотальну РНК, виведену з нирки людини (Clontech), використали як матрицю для RT-PCR. Двониткову cDNA синтезували з тотальної РНК за допомогою зворотної транскриптази MMLV (МВІ Fermentas, USA), праймінгом залежно від різновиду гена [Maniatis et al., Molecular cloning; A Laboratory Manual (Cold Spring harbor Laboratory Press, Cold Spring Harbor, N.Y.), 1990]. Далі cDNA нирки піддавали 35 циклам ампліфікації PCR з використанням 100 пікомолів вироджених олігонуклеотидних праймерів, в залежності від різновиду гена, в об'ємі 100мкл, що містять 50мМ TrisCl (pH8,3), 2,5мМ MgCl2, по 200мкМ dNTP та 2,5 одиниці Pfu полімерази. Кожний цикл ампліфікації 92913 26 PCR складався з інкубації за температури 94°С протягом 30с (денатурація), 61°С протягом 1хв. (відпал) та 72°С протягом 1хв. (розтягнення). Ампліфікований продукт реакції PCR розчиняли в 1процентному розчині агарозного гелю. Потрібний фрагмент розміром приблизно у 590 основних пар виділяли з гелю й очищали, використовуючи для цього комплект виділення Qiagen Gel. Цей очищений фрагмент ДНК лігували в MCS pcDNA 3.1 після обмеження розщеплення як вектора, так і очищеного продукту PCR за допомогою EcoRI and Xbal (МВІ Fermentas, USA) що створює клейкі кінці для направленого клонування. Продукт лігації перетворювали на Е. coli DH5α, і трансформанти оцінювали стосовно стійкості до ампіциліну. Плазмідну ДНК, виділену приблизно з 20 таких колоній, аналізували на наявність ЕРО cDNA рестрикційним розщепленням з використанням різноманітних рестрикційних ферментів. Послідовність одного з таких плазмідів було визначено за допомогою автоматичного секвенсера ДНК (АВІ). Виявилося, що він має правильні інтеграцію та послідовність ЕРО cDNA у векторі pcDNA 3.1. Цей плазмід, pcDNA-EPO, використали для вставляння міні-гена DHFR, виділеного з pDHFR 2.9 повним розщепленням за допомогою Hind III. Міні-ген 2.9 Kb DHFR затупили з кінців, використавши для цього полімеразу Кленова (Klenow Polymerase) (MBI Fermentas, USA), та лігували в pcDNA-EPO після лінеаризації за допомогою Mlu I (MBI Fermentas, USA) і затуплення кінців за допомогою полімерази Кленова (MBI Fermentas, USA). Продукт лігації перетворили на Е. соlі DH5α, і трансформанти оцінювали стосовно стійкості до ампіциліну. Плазмідну ДНК pcDNA-EPODHFR (Фіг.3) аналізували на наявність і орієнтацію мінігена DHFR в одержаному плазміді рестрикційним розщепленням з використанням різноманітних рестрикційних ферментів. pcDNA-TPL-Intron-EPO-VA-DHFR(pZRC-EPO) Цей експресійний вектор було генеровано вставлянням аденовірусного TPL в нижню частину промотора CMV та верхню частину химерного інтрона, який далі вставляли у верхню частину ЕРО cDNA в структурі pCDNA-EPO. Крім того, в цей вектор, між геном, стійким до ампіциліну і промотором CMV, вставляли ген VA РНК та мініген DHFR. Для цього спочатку розщеплювали вектор pcDNA-TPL-Intron-TNFR-IgGFc, застосовуючи для цього EcoRI та Хbаl, щоб видалити фрагмент TNFR-IgGFc. Генерований таким чином фрагмент вектора 6kb очищали, використовуючи для цього комплект виділення Qiagen Gel, та лігували геном ЕРО з кінцями EcoRI та Хbаl. Продукт лігації перетворювали на Е. coli DH5α, і трансформанти оцінювали стосовно стійкості до ампіциліну. Плазмідну ДНК аналізували на наявність і орієнтацію TPL, химерного інтрона та ЕРО cDNA в одержаному плазміді рестрикційним розщепленням з використанням різноманітних рестрикційних ферментів. Послідовність одного з таких плазмідів було визначено за допомогою автоматичного секвенсера ДНК (АВІ). Виявилося, що він має правильну интеграцію TPL, химерного інтрона та гена ЕРО у векторі. Цю плазмідну ДНК, 27 що отримала назву plnt-EPO-1, використовували для вставляння гена 1000bp VA PHK, виділеного з pCAV/NOT-TNFR подвійним розщепленням, використовуючи для цього EcoRI та Not І, та мінігена DHFR, виділеного з плазміду pDHFR 2.9 повним розщепленням за допомогою ВаmНІ & Ssp1. Ці два фрагменти було ліговано за допомогою plnt-EPO-1, що попередньо розщепили, використавши для цього Muni & BgIII, у трикомпонентній лігації. Продукт лігації перетворили на Е. coli DH5α, і трансформанти оцінювали стосовно стійкості до ампіциліну. Плазмі дну ДНК аналізували на наявність і орієнтацію гена VA РНК та мінігена DHFR в одержаному плазміді рестрикційним розщепленням з використанням різноманітних рестрикційних ферментів. Крім того, перевіряли та підтверджували інтеграцію аналізом послідовностей ДНК. Ця векторна структура отримала назву pCDNA-TPL-Intron-EPO-VA-DHFR або pZRC-EPO (Фіг.3). Приклад 4 Порівняння ефективності вираженості різноманітних експресійних векторів ЕРО за транзиторною трансфекцією Для транзиторної трансфекції було вирощено та приготовано, як описано вище, клітини Cos 1. Як описано в попередньому прикладі, були приготовані також трансфекційні суміші. Процедура трансфекції також проводилась відповідно до опису, даного в попередньому прикладі, і виснажене середовище прибирали від клітин через 40год. та 64год. трансфекції та передавали на вимірювальний прилад ELISA для аналізу вираженості ЕРО, як описано далі. Передусім, 96-лункові мікротитрові чашки (Nunc Maxisorp) покривали моноклональним антитілом миші, в об'ємі 50мкл на лунку, поверх рекомбінантного ЕРО людини (R&D Systems AntihEPO Purified Mouse Mab, Clone 9C21D11, Cat# MAB287) за концентрації 2нг/мл у карбонатному буфері, рН9,6. Ці чашки інкубували за температури 4°С до ранку в умовах підвищеної вологості для ефективного покриття. Через 12-18 годин покриття з антитіла знімали і лунки блокували блокувальним буфером, як описано вище. Після 1 години блокування чашки промивали тричі (по 250нл) буфером PBST. Далі зразки об'ємом по 50нл, розріджені до потрібної концентрації в 1Х PBS з вмістом 1% BSA, додавали до лунок у двох екземплярах. До контрольних ЕРО застосовували Ерrех 4000 (рекомбінантний ЕРО людини, 4000 міжнародних одиниць/0,4мл), що виробляється компанією Cilag AG Scbaffhausen, Швейцарія. Концентрації контрольних зразків, використаних для побудови контрольної кривої, дорівнювали: 4 міжнародних одиниць/мл, 2 міжнародних одиниць/мл, 1 міжнародних одиниць/мл, 0,4 міжнародних одиниць/мл, 0,2 міжнародних одиниць/мл та 0,1 міжнародних одиниць/мл. Після інкубації протягом 90 хвилин, що давало змогу утворитися зв'язкам антигенів з антитілами, чашки тричі промивали буфером PBST (по 250мкл кожного разу). Після цього додавали 50мкл первинного антитіла (rabbit antihuman ЕРО, очищений IgG, виробництво R&D 92913 28 Systems, Cat # AB-286-NA) за концентрації 2мкг/мл м 1Х PBS з вмістом 1% BSA, після чого відбувалася інкубація протягом 1 години за температури 37°С. Після 1 години інкубації чашки тричі промивали буфером PBST (по 250мкл кожного разу) і додавали 50мкл детекційного/вторинного антитіла (goat anti-rabbit IgG-HRP, Bangalore Genei, Cat # HPO020), розчиненого у співвідношення 1:8000 в 1X PBS з вмістом 1% BSA, pH7,2, після чого відбувалася інкубація протягом 1год. за температури 37°С. Чашки тричі промивали буфером PBST (по 250мкл кожного разу) для видалення незв'язаного кон'югату. Далі додавали підкладку, для чого використовували 100мкл розчину-підкладки (приготованого відповідно до даного вище опису) на лунку, і інкубували в темряві протягом 30хв. за температури 37°С. Реакцію припиняли доданням 50мкл p.1 1N сірчаної кислоти на лунку. Вимірювали оптичну густину, використовуючи вимірювальний прилад ELISA, на довжині хвилі 490нм. Значення для тестових зразків виводилися за допомогою програмного забезпечення SoftMax Pro (Molecular Devices) з використанням 4-параметричної стандартної кривої, отриманої з рішення рівняння Лебера-Марквардта (Leveberg-Marquardt). Результати: На Фіг.4 показано репрезентативні дані одного такого експерименту. Найвищу вираженість було отримано на pZRC-EPO (276,53±3,09нг/мл), що на 984,85% вище порівняно з pcDNA-EPODHFR (25,49±2,38нг/мл). Ці дані підтверджують висновки прикладу 3 про те, що новий експресійний вектор, який складається з п'яти елементів, забезпечує дуже високу вираженість різноманітних репортерних генів. Чудову властивість нового сполучення елементів pZRC-EPO підтверджено стійкою трансфікацією клітин, як показано далі, у прикладі 7. Приклад 5 Стійка трансфекція клітин СНО DHFR за допомогою pZRC-EPO Клітини СНО DHFR (клітини яєчника китайського хом'яка з мутованим кодуванням дигідрофолатредуктази) регулярно тримали в повному середовищі (середовище MEM α (Sigma), доповнене 10% FBS (Нуclone) та сумішшю гіпоксантину й тимідину). Під час трансфекції клітини, що перебували в середині логарифмічної фази росту, трипсинізували з колби 25см2, промивали один раз у 5мл повного середовища і повторно суспензували в PBS. До 1 Χ 106 клітин додавали п'ятнадцять мікрограмів pZRC-Epo, лінеаризованого рестрикційним ферментом Sspl і електропорували (350В) за допомогою електропоратора Stratagene Eectroporator 1000. Після короткого періоду відновлення у повному середовищі, що тривало 48год., клітини піддавали подвійному відбору, помістивши їх у селективне середовище (10% діалізованого FBS служили додатком до середовища МЕМα без додання суміші гіпоксантину й тимідину, плюс 500мкг/мл G418 (Sigma)). Далі клітини повертали в інкубатор з середовищем 5% СО2, де залишали рости протягом двох тижнів. Середовище змінювалося кожні 3 дні. 29 Ділянки стійких трансфектантів почали розвиватися після 12 процесу відбору. На 15-й день клітини трипсинізували. Щоб виділити окремі клони клітин з цієї змішаної популяції стійких трансфектантів, все це розчиняли, застосовуючи метод серійних розведень, на 96-лункових чашках. Лунки, в яких, під мікроскопом, виявляли клітини одного клону, позначали, після чого середовище в цих лунках міняли регулярно. Через приблизно 12-14 днів ці лунки виявлялися конфлюентними на 70-80%. Клітини переносили на 24-лункові чашки, де вирощували протягом 48-72год., далі переносили на 6-лункові чашки. Ці одноклітинні клони висівалися на 6-лункові чашки з густиною 0,1 X 106 клітин на лунку в селективному середовищі. Виснажене середовище видалялося з цих лунок через 48год., і клітини перераховувалися. Виснажене середовище аналізували на вираженість ЕРО, застосовуючи для цього вимірювальний прилад ELISA, як описано вище. Результати виражалися як повна кількість секретованого білка ЕРО на 106 клітин за 48год. Вибиралися десять клонів з високою вираженістю, котрі піддавалися генній ампліфікації на основі метотрексату (МТХ), що починалася з додавання 25нМ МТХ до селективного середовища. Кожні 3 дні додавали свіжого середовища. Клітини вирощували за однакової концентрації МТХ протягом 20-25 днів, поки вони не адаптувалися. Після кожної стадії підвищення концентрації МТХ клітини аналізували на рівні вираженості на 6лункових чашках, як описано вище. Один з вибраних 10 клонів не вижив в умовах 25нМ МТХ. Для решти дев'яти клонів безперервно підвищували концентрацію МТХ від 100нМ до 400нМ і, врешті, до 2мкМ. На Фіг.5 показано вираженість цих гетерогенних популяцій, виведену з ампліфікації МТХ клональних популяцій при закінченні різноманітних стадій ампліфікації МТХ. Відбір за допомогою МТХ перетворює клональну популяцію на гетерогенну. Тому для виділення з цих МТХ-ампліфікованих гетерогенних популяцій окремого клона з високою вираженістю вибирали, перш за все, на стадії 400нМ МТХ гетерогенну популяцію 41Н9. Встановлювали умови вибору клонів цих клітин, як описано вище. Через приблизно 14-16 днів, лунки 96-лункової чашки виявилися на 70-80% конфлюентними. Клітини переміщали до 24-лункових чашок, де вирощували протягом 48-72год., далі переносили до 6-лункових чашок. Ці одноклітинні клони висіювали з густиною 0,1 X 106 на лунку в селективному середовищі, що містило 400нМ МТХ. Виснажене середовище видалили з цих лунок через 48год., клітини перераховували. Виснажене середовище піддавали аналізу на вираженість на вимірювальному приладі ELISA, як описано вище. Результати виражались як повна кількість білка ЕРО, секретованого в 106 лунках протягом 48год. Вираженість цих одноклітинних клонів, виведених, з самого початку, з гетерогенної популяції 41Н9, показано в таблиці 1. Клони з високою вираженістю вирощували далі й заморожу 92913 30 вали для використання як банків основних клітин для промислового виробництва ЕРО. Таблиця 1 Клон 41H9.3G10 41H9.4G10 41Н9.4Н9 41H9.5G9 41Н9.10Н12 41Н9.1Е8 41H9.2F9 41Н9.5Е10 41HQ.8C9 41H9.10B10 41H9.2G8 41H9.4F9 41H9.1C8 41H9.7H9 Вихід білка в мкг/мільйон клітин/48год. 9,64 13,97 14,18 12,61 11,91 10,93 12,32 12,24 12,68 13,56 6,15 7,31 14,92 5,85 Приклад 6 Продукування ЕРО з клонів стійкої вираженості в колбах Дві гетерогенні популяції з тих, що показані на Фіг.5, а саме 56D12 та 41Н9, вибрані, відповідно, на етапах 100нМ МТХ та 400нМ МТХ, тестували на продукування білка на рівні колби Т75. Колби Т75см2 інокулювали 2,1 мільйонами клітин у середовищі МЕМα, вільнім від гіпоксантину та тимідину, з доданням 10% діаізованого FBS (JRH Biosciences) та 600мкг/мл G418 з відповідною концентрацією МТХ для відповідної гетерогенної популяції. Колби повертали до інкубації в СО2 за температури 37°С і концентрації СО2 5% протягом 72 годин. Моношар клітин виявився приблизно на 81-85% конфлюентним після 72 годин інкубації. У цей час старе, виснажене середовище видаляли і промивали моношар клітин, використовуючи для цього PBS Дюльбекко. До промитих моношарів додавали 15мл продуктового середовища [IMDM:Ham's F12 (1:1) з доданням 10мг/л rh-інсуліну, 2Μ глутаміну, суміші амінокислот, 0,2мМ N-ацетил-цистину, 0,25% лакталбуміну, 2мг/л FeSU4, 25мг/л декстрансульфату, 0,05% плюоронової (pluoronic) кислоти, 5мг/л гідокортизону та 1мМ Na-бутирату]. Клітини інкубували в інукбаторі СО2 за температури 33°С протягом чотирьох днів. Після чотирьох днів виснажене середовище асептично видаляли та аналізували на приладі ELISA. Вираженість цих двох клонів у продуктовому середовищі на рівні колби Т75 показано в таблиці 2. Таблиця 2 Назва гетерогенної попу- Вихід білка, мг/л, після 4 ляції днів 56D12 (100нM MTX) 21мг/л 41Н9 (400нМ МТХ) 43мг/л Приклад 7 31 92913 Ефективне продукування ЕРО в роллерфлаконах Одну з репрезентативних гетерогенних популяцій, описаних вище (41Н9), вже адаптовану до 2мкМ МТХ, використали для дослідження в роллер-флаконах для аналізу можливості продукування білка у більших кількостях. Клітини 41Н9 додатково вирощувались у колбах Т75 та Т175, щоб одержати досить клітин для посіву в роллерфлакони. 5 роллер-флаконів інокулювали 30 мільйонами клітин - в кожному випадку в середовище МЕМос (Sigma) з доданням 2мкМ МТХ, без вмісту гіпоксантину та тимідину, з вмістом 10% діалізованого (JRH Biosciences) та 600мкг/мл G418. Після перемішування клітин з середовищем, роллер-флакони повертали до призначеного для них інкубатора, де витримували, для росту, за температури 37°С та швидкості обертання близько 0,35об/хв. Перевірка клітин через 24год. показала значний ріст. Флакони знов повернули до інкубатора для росту й перевіряли під мікроскопом кожні 24год. Після 96 годин інокуляції моношари клітин були конфлюентні приблизно на 80-85%. В цей час виснажене середовище видаляли і додавали 210мл/флакон продуктового середовища [IMDM:Ham's F12 (1:1) з додаванням 10мг/л rhінсуліну, 2мМ глутаміну, суміші амінокислот, 0,2мМ N-ацетил-цистину, 0,25% лакталбуміну, 2мг/л FeSO4, 25мг/л декстрансульфату, 0,05% плюоронової кислоти, 5мг/л гідокортизону та 1мМ Na-бутирату]. Клітини інкубували в інкубаторах для роллер-флаконів протягом семи днів. Після цього виснажене середовище асептично видаляли з роллер-флаконів, аналізували на приладі ELISA, як описано вище, і заносили результати до таблиці 3. Таблиця 3 Назва гетерогенної популяції 41Н9(2мкММТХ) Вихід білка в мг/л через сім днів 91мг/л Цей рівень продуктивності гетерогенної, стійко трансфікованої та МТХ-ампліфікованої популяції, з якої ще не вибрано окремі клони, проявляє значно кращий рівень вираженості порівняно з опублікованими в літературі для окремих стійких клонів. Наприклад, стійкі клони, генеровані трансфекцією та ампліфікацією генів до 20нМ МТХ з використанням вектора, що містить промотор SRα, провідну послідовність AMV РНК 4, DHFR та резистентність зеоцину, могли б дати клон у 45 міжнародних одиниць/мл (еквівалентно 0,346мкг/мл) [Jang Η Ρ et al., Biotechnol. Appl. Biochem, 32:167-172, (2000)]. 32 У патенті США №5 955 422 показано, що клони, генеровані з клітин СНО DHFR, котрансфікованих вектором, що містить геномну копію гена ЕРО під контролем промотора SV40, послідовність поліаденилової кислоти та вектор, що містить DHFR, після ампліфікації гена МТХ давали вихід від 750 до 1470 одиниць/ мільйон клітин/ 48год. (або від 375 до 735 одиниць/ мільйон клітин/ 24год.) у продуктовому середовищі без сироватки, в роллер-флаконах. В іншому патенті, US5888774, повідомляється про клон ЕРО, генерований з клітин СНО -K1, трансфікованих вектором, що містить ЕРО cDNA, стимульовану промотором EF1 з елементами ароВ SAR та резистивністю до неоміцину, що може продукувати від 1500 до 1700 міжнародних одиниць ЕРО/ мільйон клітин/ 24год. (в разі аналізу між третім та четвертим днями використання культури). Навпаки, семиденний репрезентативний зразок середовища нашої гетерогенної популяції, 41Н9, попередньо адаптований до 2мкМ МТХ, як виявилося, містить 11830 міжнародних одиниць/мл у роллер-флаконі, як показали вимірювання ELISA. Виходячи з оцінки густини клітин від 108 до 1,5 X 108 клітин на роллер-флакон, швидкість продукування ЕРО в 7-денній культурі об'ємом 210мл становила від 2366 до 3549 міжнародних одиниць/106 клітин/ 24год., що значно перевищує описані вище рівні, досягнуті з іншими опублікованими векторами, у яких використовуються інші різноманітні сполучення регуляторних елементів. Аналогічно, в роботі Sung Kwan Yoon et al [Biotechnology and Bioengineering. 82(3):2S9-298, (2003)] повідомляється про повністю оптимізований процес продукування ЕРО з використанням лінії клітин, що розвилися в клітини СНО DHFR через процес ампліфікації генів до тиску вибору МТХ у 5мкМ. Цей процес дав вихід приблизно 50мкг/мл після визрівання в культурі протягом близько 200год. за температури 33°С. Наша стійко трансфікована гетерогенна популяція 41Н9 на основі pZRC-EPO, адаптована до порівняно більш низьких рівнів МТХ (2мкМ), виявилася здатною продукувати 11,830 міжнародних одиниць/мл (91мкг/мл) у 168-годинній культурі, як свідчать результати вимірювань на ELISA. Ці рівні вираженості ЕРО з новим експресійним вектором pZRC-EPO виявилися на 81-100% вищими за найкращі з векторів, досі опублікованих в літературі. Метод продукування ЕРО, що його описано вище для випадку гетерогенної популяції 41Н9, можна застосувати також до стійких клонів, що згадуються в прикладі 5, Таблиця 1. Новий вектор даного винаходу поміщено до ІМТЕСН, м. Чандігар, Індія, що є визнаним депозиторієм за Будапештським договором. Очікується присвоєння інвентарного номера. 33 92913 34 35 92913 36 37 92913 38 39 92913 40 41 92913 42 43 92913 44 45 Комп’ютерна верстка Т. Чепелева 92913 Підписне 46 Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюExpression vector and methods for production of high levels of proteins
Автори англійськоюSingh Arun K., Goel Ashysh, Mendiratta Sendzhyv K.
Назва патенту російськоюЭкспрессионный вектор и способы продуцирования высоких уровней белков
Автори російськоюСингх Арун К., Гоел Ашиш, Мендиратта Сенджив К.
МПК / Мітки
МПК: C12N 15/09
Мітки: білків, високих, вектор, експресійний, рівнів, способи, продукування
Код посилання
<a href="https://ua.patents.su/23-92913-ekspresijjnijj-vektor-ta-sposobi-produkuvannya-visokikh-rivniv-bilkiv.html" target="_blank" rel="follow" title="База патентів України">Експресійний вектор та способи продукування високих рівнів білків</a>
Продукування високоманозних білків у рослинних культурах

Номер патенту: 89944
Опубліковано: 25.03.2010
Автори: Бартфелд Деніел, Баум Гідеон, Левковіч Айяла, Шаалтіел Йозеф, Хашмуелі Шарон
МПК: A61K 48/00, C12P 21/00, C12N 15/52
Мітки: високоманозних, білків, продукування, рослинних, культурах
Формула / Реферат:
1. Виділена молекула нуклеїнової кислоти, яка має нуклеотидну послідовність, що кодує лізосомальний білок глюкоцереброзидазу людини, неперервно зв'язаний із С-кінцевим вакуолярним націлювальним сигналом і N-кінцевим сигнальним пептидом ендоплазматичного ретикулуму. 2. Виділена молекула нуклеїнової кислоти за п. 1, де вказаний вакуолярний націлювальний сигнал являє собою вакуолярний націлювальний сигнал основного гена хітинази А...
Послідовність днк, рекомбінантна молекула днк, експресійний вектор днк, клітина-хазяїн, стабільно трансформована послідовністю днк, рослина та її потомство, стабільно трасформовані послідовністю днк, та спосіб

Номер патенту: 86341
Опубліковано: 27.04.2009
Автори: Лігон Хоуп Томпсон, Хон Томас, де Хаан Петрус Теодорус, Кононова Марія, Ставолоне Лівія
МПК: C12N 15/09, C12N 15/82, C07K 14/01
Мітки: стабільної, експресійний, потомство, спосіб, рослина, клітина-хазяїн, трансформована, днк, трасформовані, молекула, рекомбінантна, послідовність, вектор, послідовністю
Формула / Реферат:
1. Послідовність ДНК, яка має здатність забезпечувати експресію зв'язаної з нею нуклеотидної послідовності, де послідовність ДНК включає нуклеотидну послідовність, представлену в SEQ ID NO:1.2. Послідовність ДНК за п. 1, де послідовність ДНК включає нуклеотидну послідовність, представлену в SEQ ID NO:2.3. Послідовність ДНК за п. 1, де послідовність ДНК включає нуклеотидну послідовність, представлену в SEQ ID NO:3.4....
Гуманізовані антитіла, способи їх продукування, спосіб лікування хвороби крона, спосіб лікування розсіяного склерозу, фармацевтична композиція

Номер патенту: 29494
Опубліковано: 15.11.2000
Автор: ЛІН Августін І-Зарн
МПК: C12P 21/08, C07K 16/28, C12N 5/10, A61K 39/395, C12N 15/09, A61P 37/00, C12N 15/13
Мітки: лікування, склерозу, продукування, фармацевтична, гуманізовані, розсіяного, хвороби, композиція, спосіб, способи, антитіла, крона
Текст:
...Настоящее изобретение относится к гуманизированным антителам, имеющим избирательную специфику саязызания по отношению к определенным субпопуляциям Т-клеток, при этом указанные антитела обладают высокой чувствительностью в своем связывании с этими субпопуляциями и проявляют специфичность и аффинность в ходе этого связывания, сопоставимые, если даже не превосходящие эти же характеристики мАт-прототипов, являющихся источниками происхождения...
Гібридні білки, фармацевтична композиція, що містить гібридний білок, ізольована молекула днк, що містить гібридний білок, вектор експресії, клітина-хазяїн, спосіб продукування гібридного білка, спосіб індукува

Номер патенту: 52646
Опубліковано: 15.01.2003
Автори: Чаппел Скотт С., Кампбелл Роберт К., Джеймсон Брендфорд А.
МПК: C07K 14/59, C07K 14/715
Мітки: композиція, експресії, фармацевтична, молекула, днк, гібридні, гібридний, містить, гібридного, індукува, білка, продукування, ізольована, білок, спосіб, білки, вектор, клітина-хазяїн
Формула / Реферат:
1. Гібридний білок, який містить дві різні коекспресовані амінокислотні послідовності, що утворюють гетеродимер, кожна з яких містить:(a) щонайменше одну амінокислотну послідовність, вибрану з групи, що складається з ланцюга гомомерного рецептора, ланцюга гетеромерного рецептора, ліганду, відмінного від гонадотропіну, фрагмента згаданого ланцюга згаданого гомогенного рецептора, згаданого ланцюга згаданого гетеромерного рецептора або...
Поліпептид ліганду mpl (варіанти), ізольована молекула нуклеїнової кислоти, що кодує поліпептид ліганду mpl, вектор експресії, клітина-хазяїн, спосіб продукування поліпептиду ліганду mpl (варіанти), композиція,

Номер патенту: 72869
Опубліковано: 16.05.2005
Автори: де Совідж Фредерік, Ітон Дан І.
МПК: C12P 21/02, A61K 38/22, C12N 15/19, C12P 21/08, A61K 38/00, C07H 21/04, A61P 7/02, A61P 7/00, C07K 14/52, C07K 19/00, C12N 15/16, C12N 15/09, C12N 15/12, C12N 5/10, C12Q 1/68
Мітки: ліганду, кодує, ізольована, експресії, спосіб, композиція, вектор, клітина-хазяїн, нуклеїнової, кислоти, молекула, варіанти, поліпептид, продукування, поліпептиду
Формула / Реферат:
1. Полипептид лиганда mpl, получаемый по способу, включающему:(і) просеивание человеческой геномной библиотеки с олигонуклеотидом на основе геномной последовательности, представленной на фиг. 9, для изоляции геномной ДНК, которая включает кодирующую последовательность экзона лиганда mpl, представленную на фиг. 9, вместе с остальными экзонами гена,(ii) вставку ДНК в вектор экспрессии,(iii) трансфекцию клеток млекопитающих...
Попередній патент: Спосіб лікування хронічного гепатиту с
Наступний патент: Гетероциклічні сполуки як позитивні алостеричні модулятори метаботропних глутаматних рецепторів
Випадковий патент: Спосіб виробництва пектиновмісного згущеного молока з цукром