Фармацевтична лікарська форма для негайного вивільнення похідної індолінону
Номер патенту: 107560
Опубліковано: 26.01.2015
Автори: Троммесхаузер Дірк, Мессершмід Роман, Лах Петер, Штопфер Петер, Зоколісс Торстен














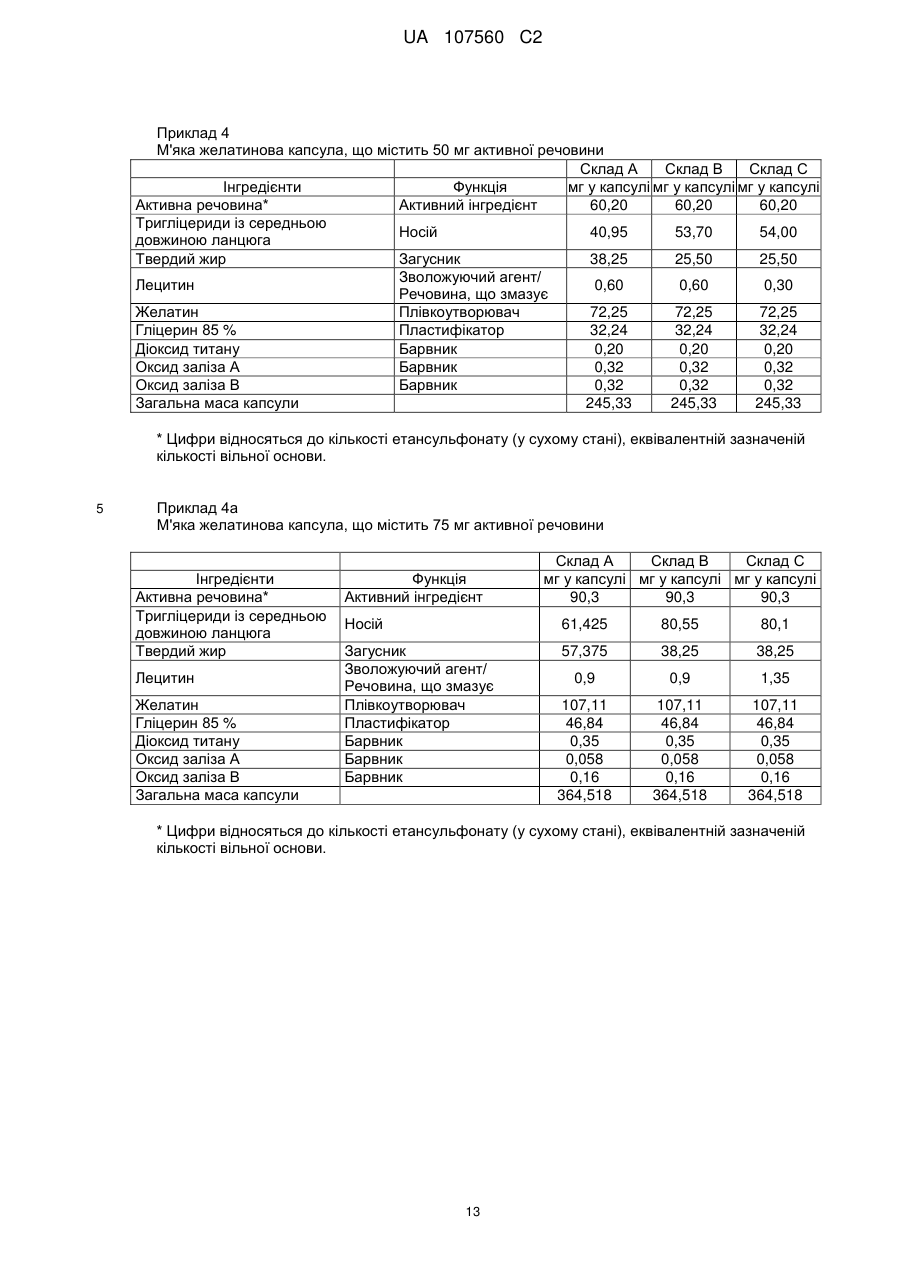
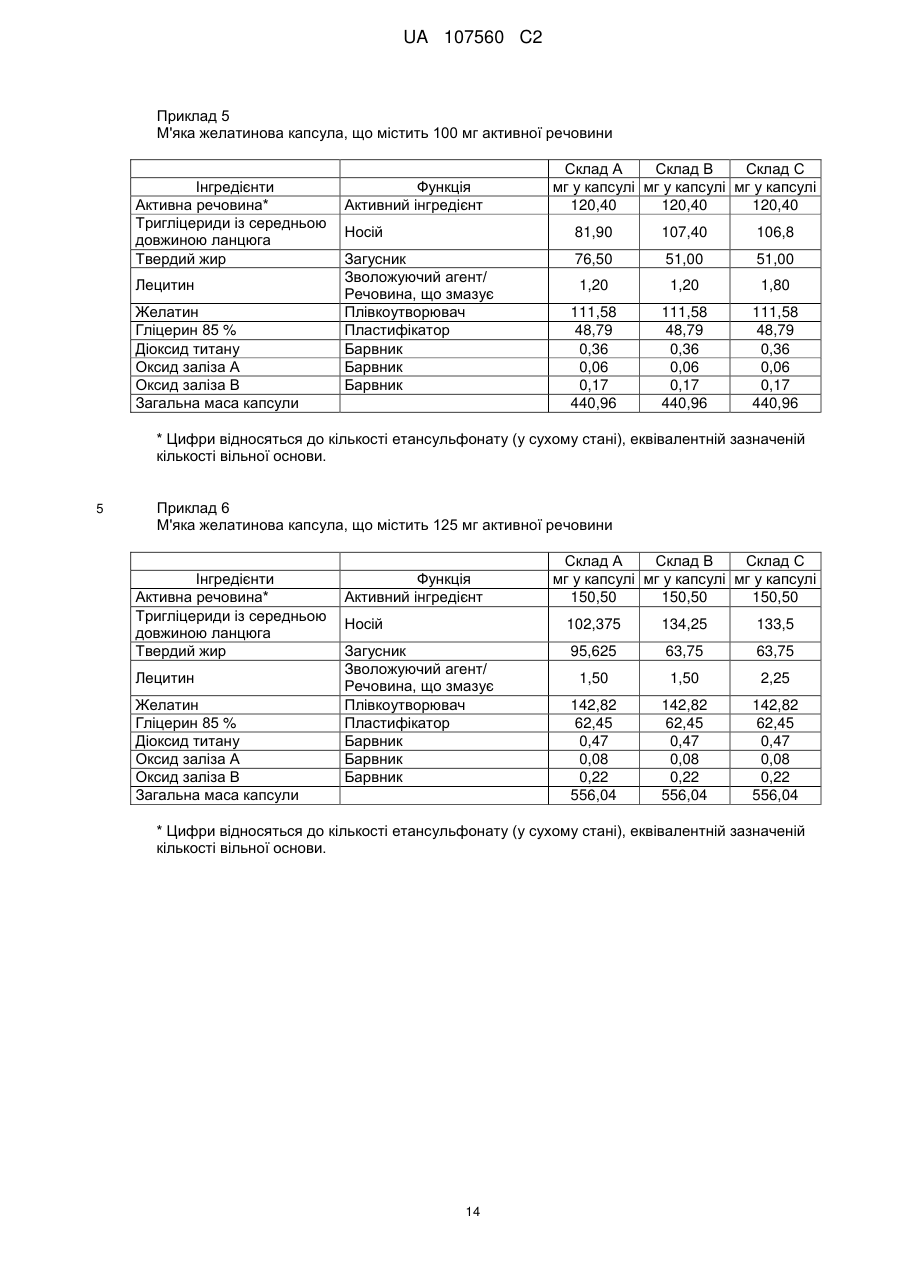
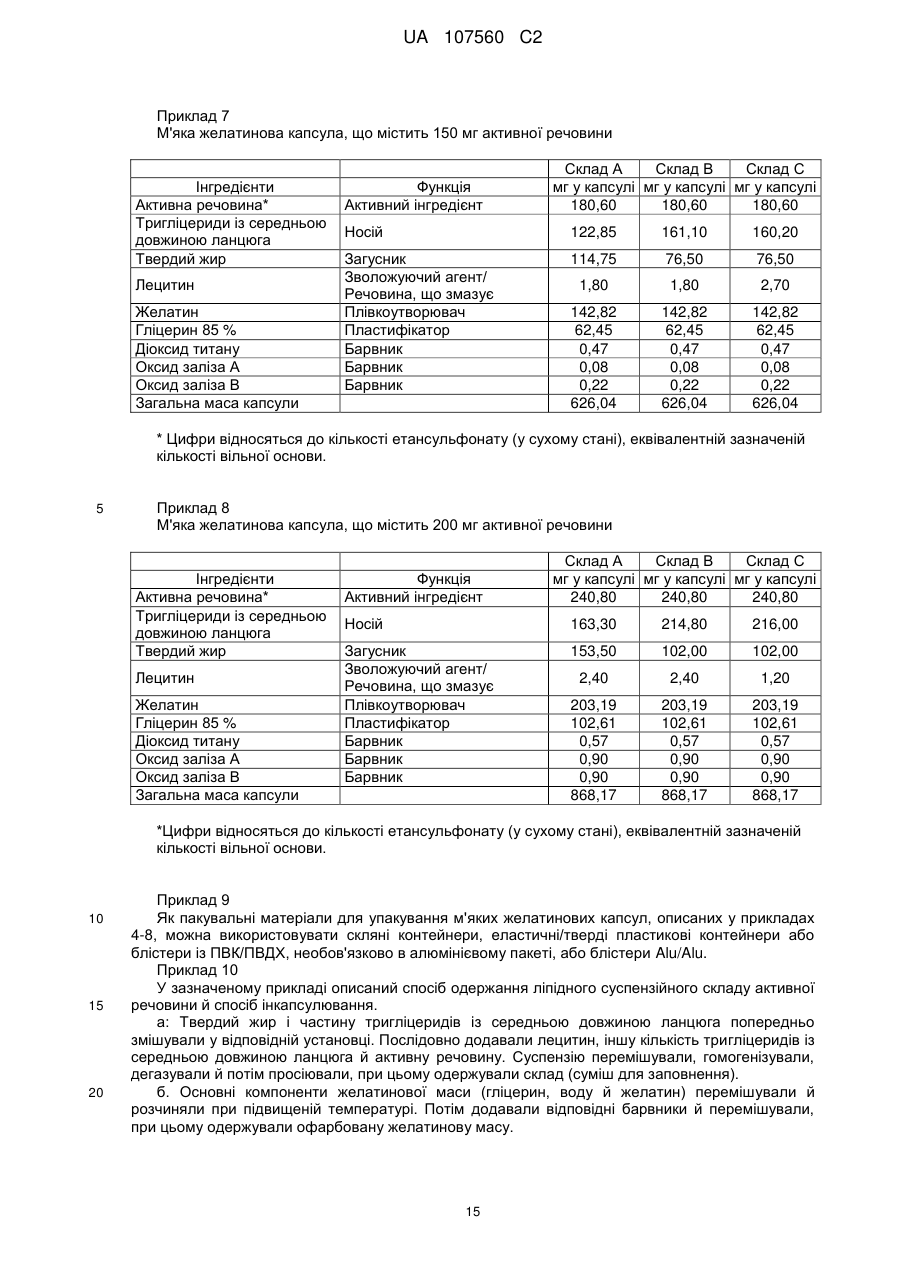
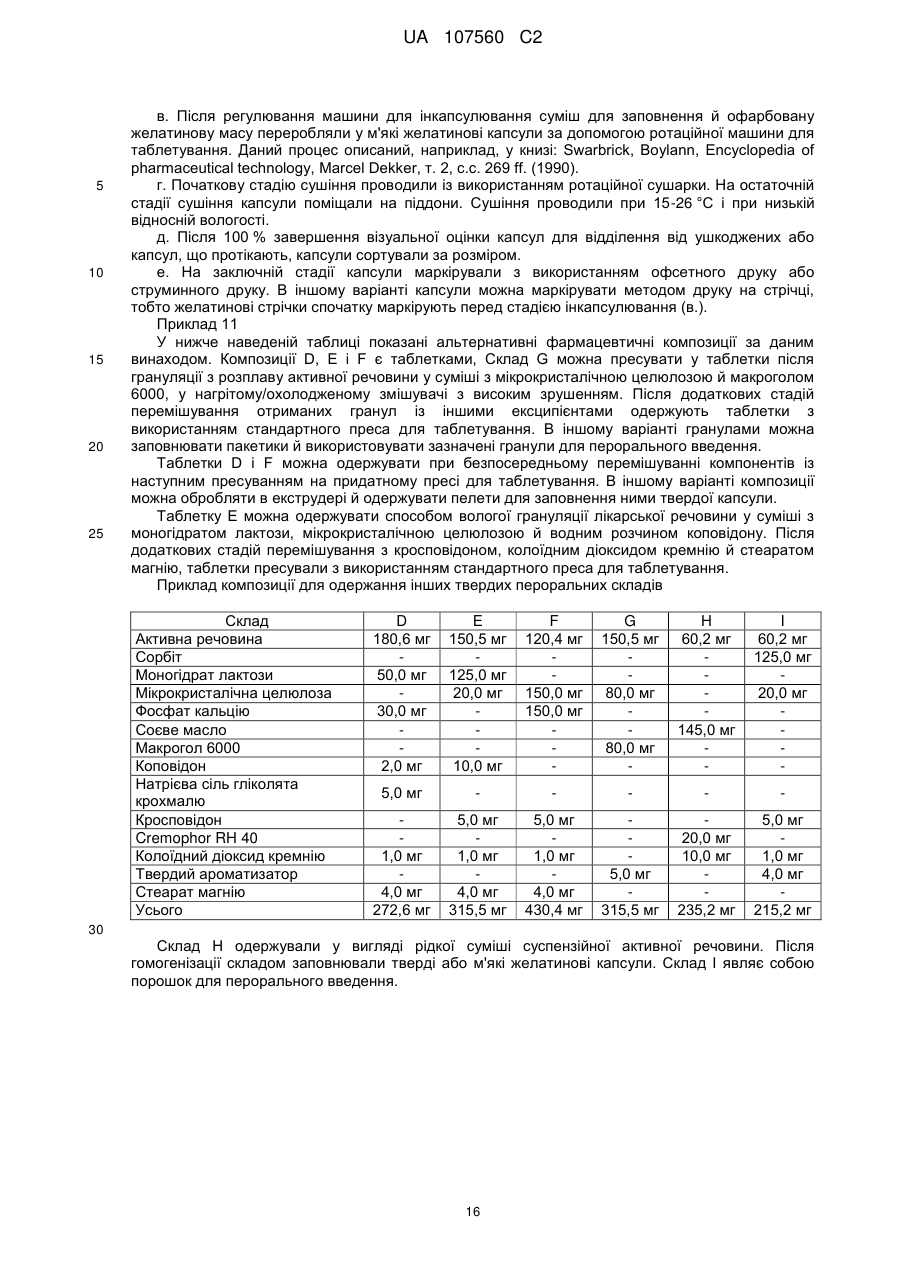


Формула / Реферат
1. Фармацевтична лікарська форма активної речовини, моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону, що забезпечує доставку активної речовини та характеризується профілем негайного вивільнення, при якому не менше 70 % (Q65 %) активної речовини розчиняється in vitro протягом 60 хв у наступних умовах розчинення in vitro, відповідно до Європейської фармакопеї, 6.2: апарат 2 (лопатева мішалка), середовище, що розчиняє 0,1 М HCl (рН 1) і швидкість перемішування від 50 до 150 об./хв, при температурі 37 °С, і яка містить склад ліпідної суспензії активної речовини, що включає в'язку суспензію активної речовини у
(і) 1-90 % тригліцеридів із середньою довжиною ланцюга;
(іі) 1-30 % твердого жиру; і
(ііі) 0,1-10 % лецитину.
2. Фармацевтична лікарська форма за п. 1, що забезпечує доставку активної речовини та характеризується профілем негайного вивільнення, при якому не менше 75 % (Q70 %) активної речовини розчиняється in vitro протягом 60 хв.
3. Фармацевтична лікарська форма за п. 1, що забезпечує доставку активної речовини та характеризується профілем негайного вивільнення, при якому не менше 85 % (Q80 %) активної речовини розчиняється іn vitro протягом 60 хв.
4. Фармацевтична лікарська форма за п. 1, що забезпечує доставку активної речовини та характеризується профілем негайного вивільнення, при якому не менше 85 % (Q80 %) активної речовини розчиняється in vitro протягом 45 хв.
5. Фармацевтична лікарська форма за п. 1, що забезпечує доставку активної речовини та характеризується профілем негайного вивільнення, при якому не менше 85% (Q80%) активної речовини розчиняється in vitro протягом 30 хв.
6. Фармацевтична лікарська форма за будь-яким із пп. 1-5, що характеризується порівнянним профілем розчинення in vitro незалежно від величини дози у діапазоні від 5 до 1000 мг активної речовини.
7. Фармацевтична лікарська форма за будь-яким із пп. 1-6, що є лікарською формою для пероральної доставки.
8. Фармацевтична лікарська форма за будь-яким із пп. 1-6, що містить активну речовину у кількості від 0,01 до 90 мас.% у розрахунку на загальну масу композиції.
9. Фармацевтична лікарська форма за будь-яким із пп. 1-6, що містить дозу у діапазоні від 5 до 1000 мг активної речовини.
Текст
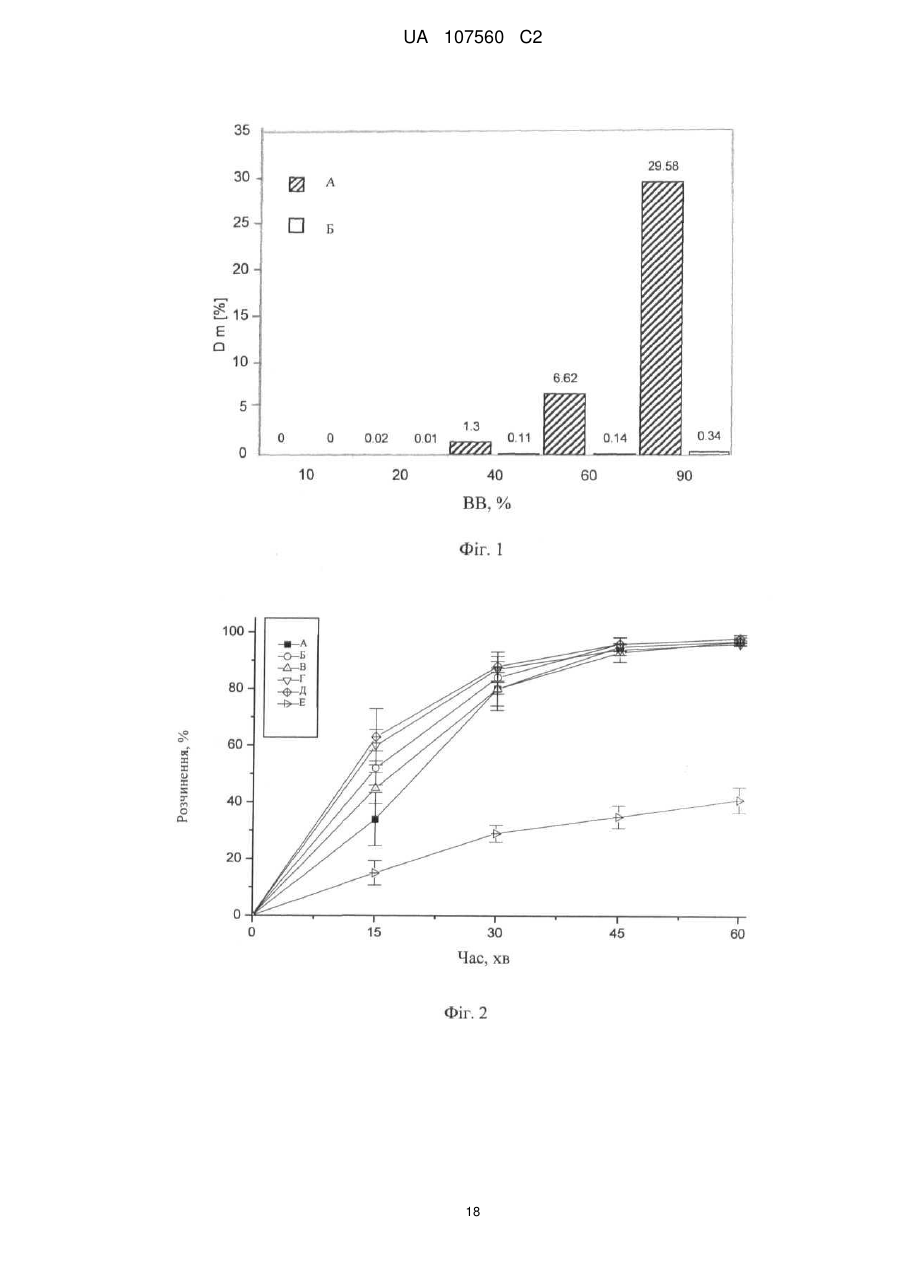
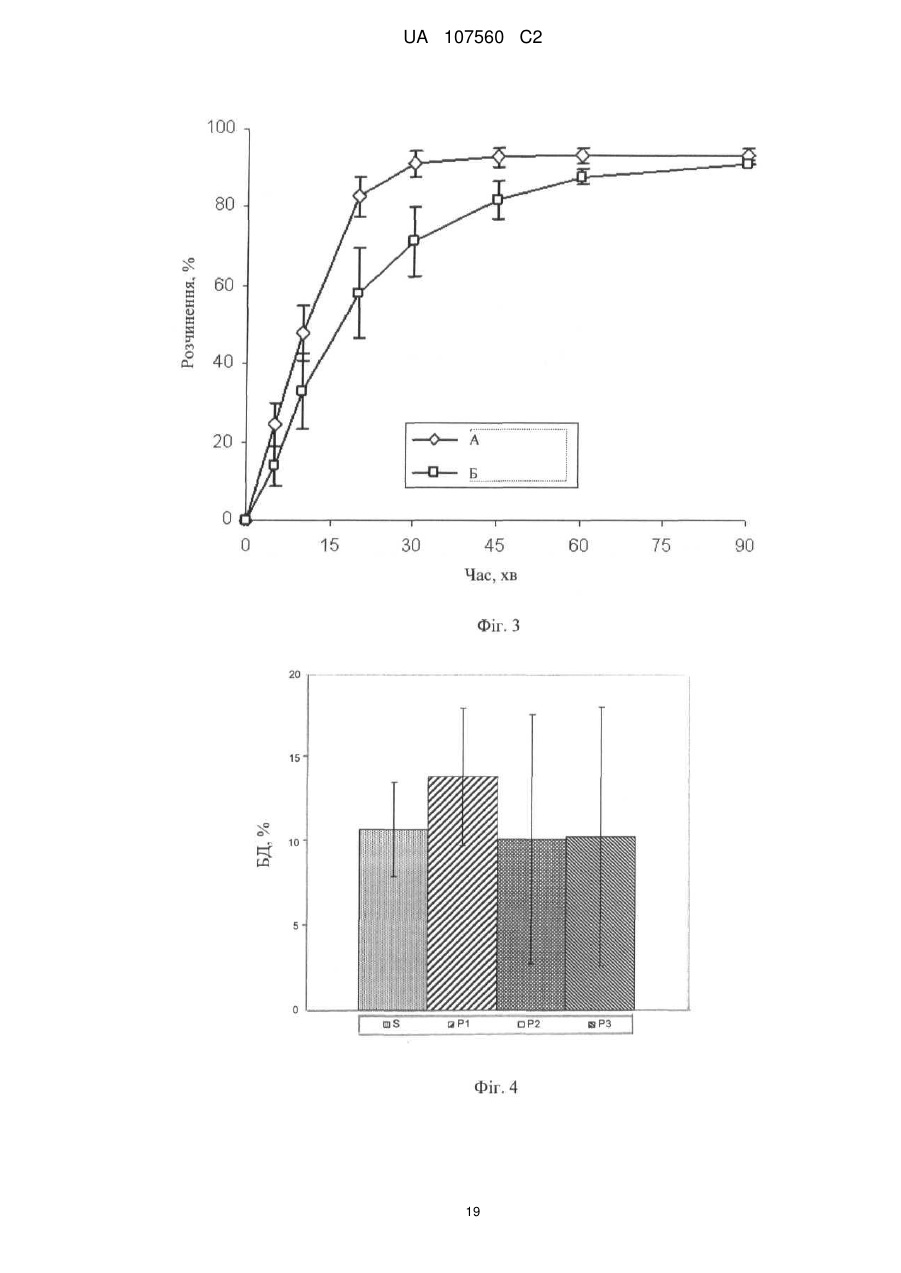
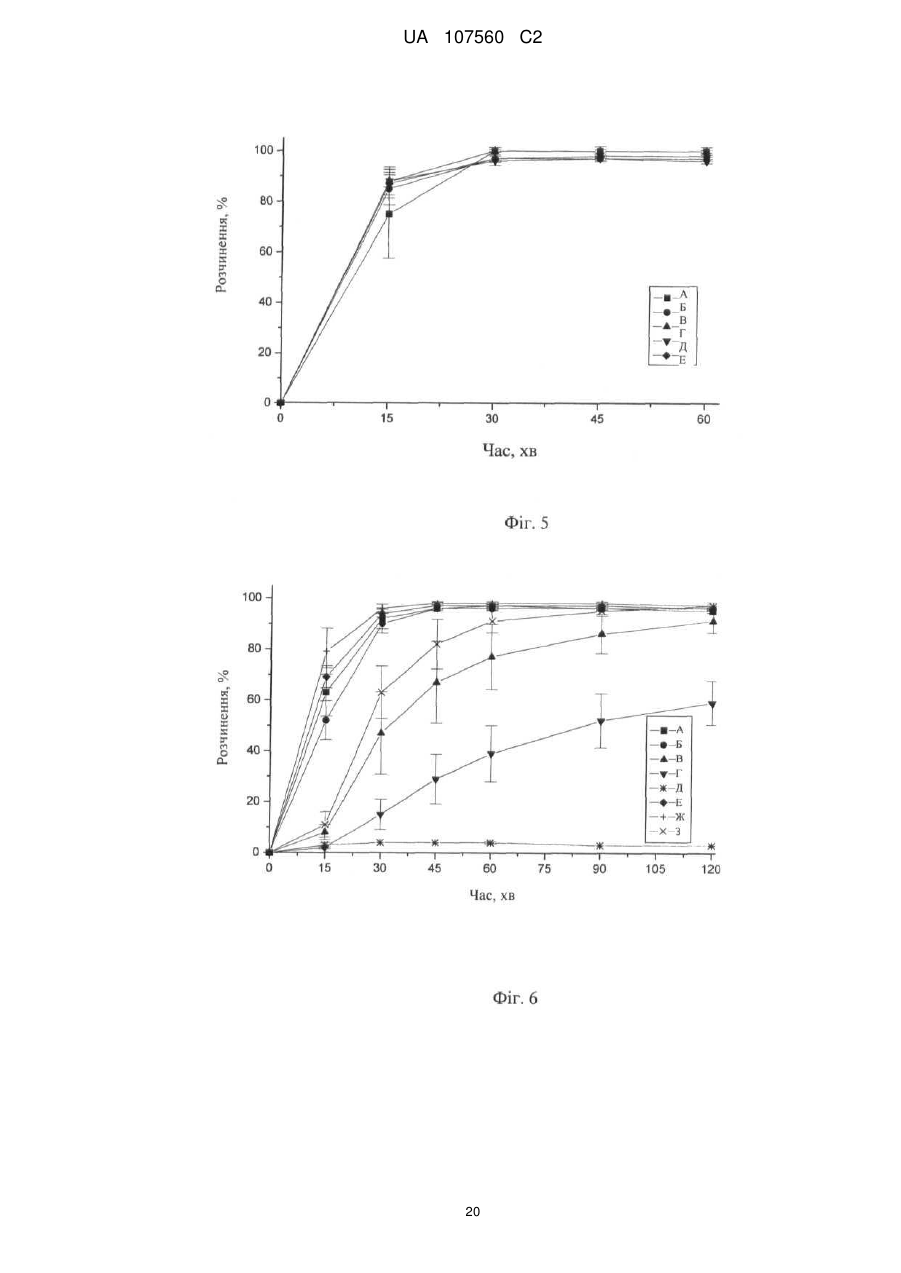
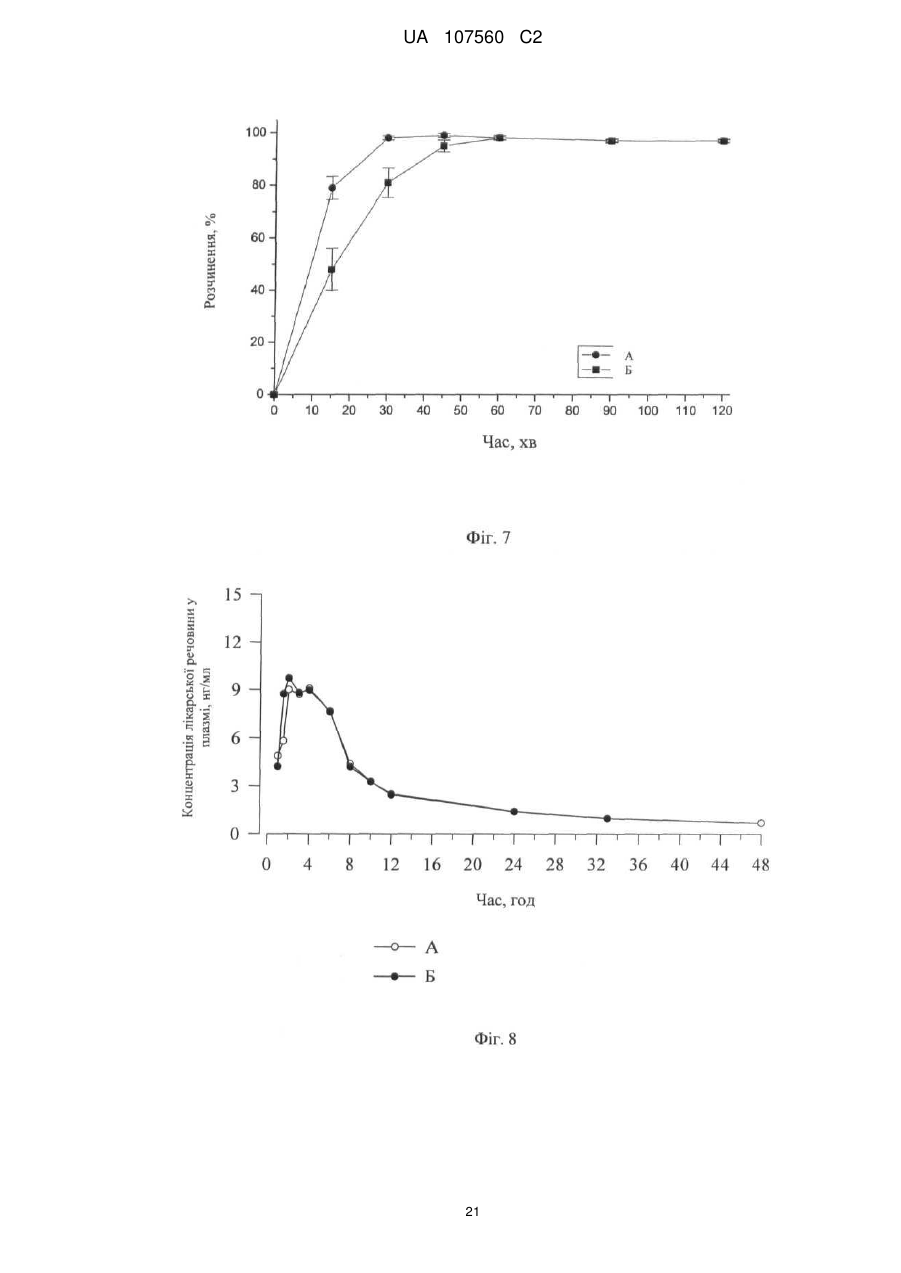
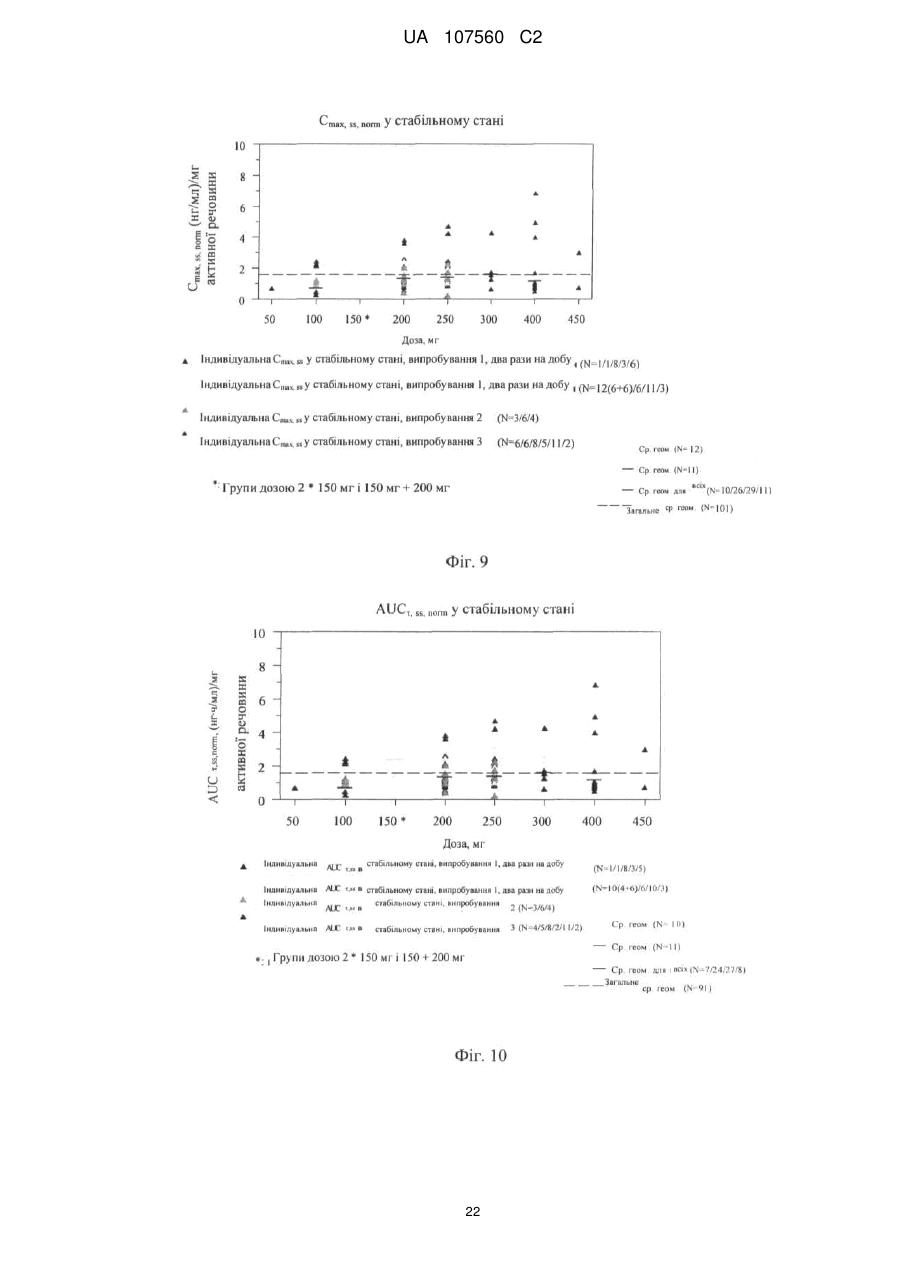
Реферат: Винахід стосується фармацевтичної лікарської форми активної речовини моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6метоксикарбоніл-2-індолінону, яка містить склад ліпідної суспензії активної речовини, що включає в'язку суспензію активної речовини у (і) 1-90 % тригліцеридів із середньою довжиною ланцюга; (іі) 1-30 % твердого жиру; і (ііі) 0,1-10 % лецитину. UA 107560 C2 (12) UA 107560 C2 UA 107560 C2 5 10 15 20 25 30 35 40 45 50 55 Даний винахід відноситься до фармацевтичної лікарської форми для доставки з негайним профілем вивільнення, що містить активну речовину, моноетансульфонат 3-Z-[1-(4-(N-((4метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл2-індолінону. Передумови створення даного винаходу Швидкість і ступінь, з якими активний інгредієнт або активний фрагмент абсорбуються з фармацевтичної лікарської форми й стають доступними у ділянці дії, називаються біодоступністю (Chen M. L. та ін., Bioavailability and bioequivalence: an FDA regulatory overview, Pharm. Res., 18: 1645-1648 (2001)). Однак практично неможливо виміряти концентрацію лікарського засобу у ділянці його дії. Отже, біодоступність оцінюють за концентраціями лікарського засобу в основному кровотоці. Системний вплив оцінюють при вимірюванні концентрації активного лікарського засобу у крові або у плазмі через різні періоди часу після введення лікарського засобу, а потім розраховують площу під кривою залежності концентрації від часу (AUC). Профілі залежності концентрації лікарського засобу у крові/плазмі від часу залежать від динаміки розчинення, розчинності, абсорбції, метаболізму, розподілу й виведення. Абсорбція лікарської речовини із твердої лікарської форми після її введення залежить від вивільнення лікарської речовини з лікарського продукту, розчинення або солюбілізації лікарської речовини у фізіологічних умовах, і крім того, від проникності через стінки кишечника у шлунково-кишковому тракті. Підвищена швидкість розчинення складу звичайно підсилює вивільнення з лікарської форми до максимального ступеня, що є необхідною умовою для задовільної біодоступності інгредієнта або активного фрагмента. У силу важливого значення цієї стадії, розчинення in vitro може мати значення для передбачення концентрацій у плазмі in vivo і, отже, біодоступності. (Guidance for Industry, Dissolution Testing of Immediate Release Solid Oral Dosage Forms, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), August 1997). Спостережуване розходження у швидкості й ступені абсорбції лікарської речовини in vivo залежить від швидкості розчинення лікарської речовини in vivo (Amidon G. L. та ін., A Theoretical Basis For a Biopharmaceutics Drug Classification: The Correlation of In Vitro Drug Product Dissolution and In Vivo Bioavailability, Pharmaceutical Research, 12: 413-420 (1995)). Виходячи із зазначених загальних міркувань, випробування на розчинення in vitro для твердих лікарських форм із негайним вивільненням, таких як таблетки й капсули, використовують для оцінки якості лікарського продукту. Продукт із негайним вивільненням забезпечує розчинення активної речовини у шлунково-кишковому тракті, не викликаючи якогонебудь запізнювання або збільшення тривалості розчинення й абсорбції лікарської речовини. Вимоги до випробувань продуктів із негайним вивільненням обговорюються у довідниках Guidance for Industry (CDER 1997), Dissolution testing for immediate release solid oral dosage forms (CDER 1997), Immediate release solid oral dosage forms-Scale up and Postapproval Changes, ICH Guidance Q6A, Specifications: Test Procedures and Acceptance Criteria For New Drug Substances And New Drug Products. Найбільше широко використовуваними методами випробування на розчинність, описаними в Європейській фармакопеї 6.2 (6-е видання), є метод обертового кошика (апарат 1) і метод з використанням лопатевої мішалки (апарат 2). Описані методи є простими, надійними, стандартизованими й широко використовуються. Вони досить універсальні й призначені для проведення випробувань по розчиненню для безлічі лікарських продуктів. Відповідно до затвердженого нормативного керівництва (наприклад, до Європейської фармакопеї 6.2, 6-е видання), рекомендується враховувати наступні параметри, що впливають на профіль розчинення, наприклад, при виборі придатних умов випробувань на розчинення in vitro для твердого перорального продукту з негайним вивільненням: апарат, швидкість перемішування, середовище, що розчиняє, й температура. Моноетансульфонат 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-Nметиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону являє собою нову речовину, що характеризується коштовними фармакологічними властивостями, насамперед для лікування онкологічних захворювань, імунологічних захворювань або патологічних станів, які включають імунологічний компонент, або фіброзних захворювань. Хімічна структура даної речовини представлена нижче формулою (I). Формула (I) 1 UA 107560 C2 O H3C N O N CH3 O N H CH3 N x H3C S O O OH N O 5 10 15 20 25 30 35 40 45 Дана речовина описана у формі основи у заявці WO 01/27081, у формі моноетансульфонату у WO 2004/013099, її застосування для лікування імунологічних захворювань або патологічних станів, які включають імунологічний компонент описане у WO 2004/017948, застосування при лікуванні онкологічних захворювань у WO 2004/096224, застосування при лікуванні фіброзних захворювань у WO 2006/067165, і інші сольові форми у WO 2007/141283. Мета даного винаходу полягає в одержанні фармацевтичної лікарської форми зазначеної вище лікарської речовини, що відповідає вимогам задовільної біодоступності, які забезпечують необхідний заданий діапазон дозування й додатково характеризуються специфічним профілем негайного вивільнення, що забезпечує прийнятний профіль залежності концентрації активного компонента у плазмі від часу. Така характеристика специфічного профілю вивільнення для даної лікарської речовини не описана у попередньому рівні техніки. Короткий виклад сутності винаходу У першому об'єкті даного винаходу пропонується фармацевтична лікарська форма активної речовини моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-Nметиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону, що забезпечує доставку активної речовини й характеризується профілем негайного вивільнення для доставки, при якому не менше 70 % (Q65 %) активної речовини розчиняється in vitro протягом 60 хв. у наступних умовах розчинення in vitro, відповідно до Європейської фармакопеї, 6.2: апарат 2 (лопатева мішалка), середовище, що розчиняє - 0,1 M HCl (pH 1) і швидкість перемішування від 50 до 150 об./хв., при температурі 37 °C. В одному об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма, що характеризується профілем негайного вивільнення для доставки, при якому у згаданих вище умовах не менше 75 % (Q70 %) активної речовини розчиняється in vitro протягом 60 хв. У ще одному об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма, яка характеризується профілем негайного вивільнення для доставки, причому у згаданих вище умовах не менше 85 % (Q80 %) активної речовини розчиняється in vitro протягом 60 хв., переважно, не менше 85 % (Q80 %) активної речовини розчиняється in vitro протягом 45 хв., найбільше переважно, не менше 85 % (Q80 %) активної речовини розчиняється in vitro протягом 30 хв. В іншому об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма, яка у згаданих вище умовах характеризується порівнянним профілем негайного вивільнення in vitro незалежно від дози у діапазоні від 5 до 1000 мг активної речовини, переважно, від 25 до 300 мг активної речовини. В одному об'єкті даного винаходу пропонується лікарська форма активної речовини, моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-Nметиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону, що забезпечує доставку активної речовини й характеризується профілем негайного вивільнення, при якому максимальна концентрація аналізованої речовини/активної речовини у плазмі у стабільному стані (Cmax, ss) зростає пропорційно дозі, переважно, якщо при цьому діапазон доз активної речовини становить від 50 до 300 мг. В одному об'єкті даного винаходу пропонується фармацевтична лікарська форма активної речовини, моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-Nметиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону, що характеризується профілем негайного вивільнення, при цьому нормалізована за дозою максимальна концентрація аналізованої речовини/активної речовини у плазмі у стабільному стані (C max, ss, norm) 2 UA 107560 C2 5 10 15 20 25 30 35 40 45 50 55 60 рівна при різних дозах, переважно, якщо при цьому діапазон доз активної речовини становить від 50 до 300 мг. В іншому об'єкті даного винаходу пропонується фармацевтична лікарська форма активної речовини, моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-Nметиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону, що характеризується профілем негайного вивільнення, при цьому площа під кривою залежності концентрації аналізованої речовини/активної речовини у плазмі у стабільному стані від часу у межах інтервалу введення τ (AUCτ,ss) зростає пропорційно дозі, переважно, якщо діапазон дози активної речовини становить від 50 до 300 мг. У ще одному об'єкті даного винаходу пропонується фармацевтична лікарська форма активної речовини, моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-Nметиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону, що характеризується профілем негайного вивільнення, при цьому нормалізована за дозою площа під кривою залежності концентрації аналізованої речовини/активної речовини у плазмі у стабільному стані від часу у межах інтервалу введення τ (AUCτ,ss, norm) рівна при різних дозах, переважно, якщо при цьому діапазон доз активної речовини становить від 50 до 300 мг. В одному об'єкті даного винаходу пропонується фармацевтична лікарська форма активної речовини, моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-Nметиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону, що характеризується профілем негайного вивільнення, і при цьому досягається максимальна концентрація активної речовини у плазмі крові людини протягом від 0,75 до 6 год., переважно, протягом середнього значення 2 год. У ще одному об'єкті даного винаходу пропонується фармацевтична лікарська форма активної речовини, моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-Nметиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону, що характеризується профілем негайного вивільнення, і при цьому досягається максимальна концентрація у плазмі крові людини, принаймні, у діапазоні від 4 нг/мл до 32 нг/мл, а середнє геометричне значення становить 14 нг/мл, якщо вводять лікарську форму, що містить моноетансульфонат 3-Z-[1-(4-(N((4-метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6метоксикарбоніл-2-індолінону у дозі 150 мг (3 рази по 50 мг). В іншому об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма, що являє собою лікарську форму для пероральної доставки. В одному об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма у формі таблетки, капсули, перорального розчину, еліксиру, емульсії, пелет, порошку або гранул. В іншому об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма, що містить суспензію активної речовини. У ще одному об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма, в якій суспензія активної речовини являє собою в'язку суспензію моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6метоксикарбоніл-2-індолінону, що містить носій і загусник. В одному об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма, в якій носій являє собою ліпідний (ліпофільний) носій. В іншому об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма, при цьому у перерахованих нижче умовах розчинення in vitro відповідно до Європейської фармакопеї 6.2, ліпідна суспензія диспергована у вигляді крапель невеликого розміру: апарат 2 (лопатева мішалка), середовище, що розчиняє - 0,1 M HCl (pH 1) і швидкість перемішування від 50 до 150 об./хв., при температурі 37 °C. У ще одному об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма у формі капсули, що містить оболонку капсули й вміст капсули, при цьому вміст капсули являє собою вище згадану суспензію активної речовини. В одному об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма у формі капсули, при цьому оболонка капсули швидко розпадається in vitro, що також є необхідною умовою для вивільнення активної речовини in vivo. У ще одному об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма для застосування як лікарський засіб. В іншому об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма для застосування як фармацевтична композиція з антипроліферативною активністю. В одному об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма для лікування захворювання або стану, вибраного з онкологічних захворювань, 3 UA 107560 C2 5 10 15 20 25 30 35 40 45 50 55 імунологічних захворювань або патологічних станів, які включають імунологічний компонент, і фіброзних захворювань. У ще одному об'єкті даного винаходу пропонується застосування згаданої вище фармацевтичної лікарської форми для одержання лікарського засобу, призначеного для лікування захворювання або стану, вибраного з онкологічних захворювань, імунологічних захворювань або патологічних станів, які включають імунологічний компонент, і фіброзних захворювань. В іншому об'єкті даного винаходу пропонується спосіб лікування й/або профілактики захворювання або стану,вибраного з онкологічних захворювань, імунологічних захворювань або патологічних станів, які включають імунологічний компонент, і фіброзних захворювань, і зазначений спосіб полягає у введенні ефективної кількості вище зазначеної фармацевтичної лікарської форми перорально пацієнтові одноразово або декілька разів на день. У ще одному об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма, що містить активну речовину у кількості від 0,01 до 90 мас. %, переважно, від 0,1 до 50 мас. % у розрахунку на масу композиції. В іншому об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма, що містить дозу у діапазоні від 5 до 1000 мг активної речовини, переважно, від 25 до 300 мг активної речовини. У ще одному об'єкті даного винаходу пропонується згадана вище фармацевтична лікарська форма, що використовується при дозуванні, незалежному від маси тіла (НМТ). В іншому об'єкті даного винаходу пропонується згадана вище фармацевтична дозована форма для застосування у діапазоні доз від 0,1 мг до 20 мг активної речовини/кг маси тіла, переважно, від 0,5 мг до 5 мг активної речовини/кг маси тіла. Опис фігур Фігура 1 - Приріст маси за рахунок сорбції вологи (Dm, %) при різних значеннях відносної вологості (ВВ, %) для складів у вигляді м'якої желатинової капсули (А) і рідкої суспензії (Б). Фігура 2 - Вплив кількості використовуваного лецитину у м'якій желатиновій капсулі, що містить 150 мг активної речовини, на динаміку розчинення in vitro. Випробування на розчинення проводили з використанням апарата 2 (лопатева мішалка), 100 об./хв., 900 мл середовище, що розчиняє (0,1 M HCl), рН 1,0, 37 °C: (A) 30 % лецитину від кращої кількості, (Б) 75 % лецитину від кращої кількості, (В) 90 % лецитину від кращої кількості, (Г) краща кількість лецитину (дорівнює 100 %), (Д) 200 % лецитину від кращої кількості, (Е) 0 % лецитину. Фігура 3 - Вплив інтервалу плавлення твердого жиру на динаміку розчинення in vitro (у % речовини, що розчинилася) протягом часу (у хвилинах) м'яких желатинових капсул. Випробування на розчинення проводили з використанням апарата 2 (лопатева мішалка), 100 об./хв., 900 мл середовище, що розчиняє, рН 1,2, 37 °C: (A) інтервал плавлення 33 °C-40 °C, (Б) інтервал плавлення 40 °C-44 °C. Фігура 4 - Порівняння абсолютної біодоступності (АБ, %) за даними випробувань на пацюках протягом 24 год. у водному розчині (S) у порівнянні з різними системами носіїв (P1, P2 і P3) активної речовини - відрізки прямої лінії означають стандартні відхилення. Фігура 5 - Вплив величини дози активної речовини у діапазоні від 50 мг до 150 мг (моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-Nметиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону) на динаміку розчинення in vitro м'яких желатинових капсул. Випробування на розчинення проводили з використанням апарата 2 (лопатева мішалка), 100 об./хв., 900 мл середовище, що розчиняє (0,1 M HCl), рН 1,0, 37 °C: (A) 50 мг активної речовини, (Б) 75 мг активної речовини, (В) 100 мг активної речовини, (Г) 125 мг активної речовини, (Д) 150 мг активної речовини. Фігура 6 - Вплив рН розчинюючого середовища й присутності ПАР на динаміку розчинення in vitro м'яких желатинових капсул, що містять 150 мг активної речовини у середовищі, що розчиняє при рН 1,0 і рН 3,0, у присутності або у відсутності ПАР. Випробування на розчинення проводили з використанням апарата 2 (лопатева мішалка), 100 об./хв., 900 мл середовище, що розчиняє при рН, що змінюється у діапазоні від 1,0 до 6,8, 37 °C: (A) pH 1,0, (Б) pH 2,0, (В) pH 3,0, (Г) pH 4,0, (Д) pH 6,8, (Е) pH 1,0 і 0,5 % кремофору, (Ж) pH 1,0 і 0,5 % Твін 80, (З) pH 3,0 і 0,5 % Твін 80. Фігура 7 - Криві розчинності різних серій желатинових капсул, що містять 50 мг активної речовини, які використані при випробуваннях, описаних на фіг. 8, і характеризуються негайним і повільним профілями розчинення in vitro. Випробування на розчинення проводили з використанням апарата 2 (лопатева мішалка), 100 об./хв., 900 мл середовище, що розчиняє (0,1 M HCl) рН 1,0, 37 °C: (A) негайне вивільнення, (Б) повільне вивільнення. 4 UA 107560 C2 5 10 15 20 25 30 35 40 45 50 55 60 Фігура 8 - Профілі залежності середнього геометричного концентрації у плазмі від часу для складів, що містять моноетансульфонат 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-Nметиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону при повільному (A, 3 м'які желатинові капсули, що містять 50 мг активної сполуки) і більш швидкому вивільненні in vitro (B, 3 м'які желатинові капсули, що містять 50 мг активної речовини) у випробуваннях на біодоступність в організмі людини. Концентрація у плазмі відноситься до моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6метоксикарбоніл-2-індолінону. Фігура 9 - Індивідуальні й середні геометричні нормалізовані за дозою максимальні концентрації у плазмі при устояних значеннях для моноетансульфонату 3-Z-[1-(4-(N-((4метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл2-індолінону, отримані у трьох різних серіях випробувань фази I за участю ракових пацієнтів після введення активної речовини у лікарській формі у вигляді м'яких желатинових капсул. Фігура 10 - Індивідуальні й середні геометричні нормалізовані за дозою значення площі під кривою (AUC) у стабільному стані для моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону, отримані у трьох різних серіях випробувань фази I за участю ракових пацієнтів після введення активної речовини у лікарській формі у вигляді м'яких желатинових капсул. Докладний опис винаходу Методи вимірювання швидкості розчинення за даним винаходом відповідають Європейській фармакопеї 6.2 і описані нижче. У випробуваннях на розчинення використовували апарат 2 (лопатева мішалка) відповідно до Європейської фармакопеї 6.2, при швидкості обертання мішалки 100 об./хв. і середовище, що розчиняє 0,1 M HCl, рН 1,0, без добавок, при 37 °C. У випробуваннях можна змінювати об'єм середовища, що розчиняє. У додаткових методах швидкість перемішування також можна змінювати у діапазоні від 50 до 150 об./хв. при використанні апаратів 1 і 2 відповідно до Європейської фармакопеї 6.2, рН середовища, що розчиняє від 1 до 6.8, об'єм від 500 до 2000 мл, необов'язково використовували грузила, необов'язково у присутності ПАР й/або ферментів, і, необов'язково, у присутності органічних розчинників або стандартних замінників кишкових або шлункових рідин. В інших умовах, таких як змінене значення рН середовища, що розчиняє, як показано на фіг. 6, швидкість розчинення може змінюватися. Таким чином, відповідно до результатів, представлених на фіг. 6, швидкість розчинення може зменшуватися зі збільшенням рН, що можна пояснити зміною рН-залежної розчинності активної речовини. Крім того, у присутності ПАР швидкість розчинення може зростати. Додаткові варіанти умов випробувань на розчинність, такі як температура, швидкість обертання, об'єм або тип апарата, також можуть впливати на швидкість розчинення. Згідно з даним винаходом, випробування на розчинення з використанням апарата 2 (лопатева мішалка), при 100 об./хв., у 900 мл середовища, що розчиняє (0,1 M HCl), рН 1,0 і при 37 °C свідчать про те, що присутність лецитину у складі може підвищувати швидкість розчинення, на відміну від складу, що не містить лецитин (фіг. 2). Крім того, згідно з даним винаходом було встановлено, що профіль розчинення лікарського продукту не залежить від величини дози активної речовини, як показано на фіг. 5 для лікарської форми у вигляді м'якої желатинової капсули. Крім того, порівняння профілів розчинення м'яких желатинових капсул у середовищі, що розчиняє при pH 1,0 і pH 3,0 у присутності або під час відсутності ПАР свідчить про те, що розчинення сполук, що містять дану активну речовину, може бути поліпшене у присутності ПАР (фіг. 6). У деяких варіантах швидкості розчинення, вимірювані з використанням апарата 2 (лопатева мішалка), при 100 об./хв. у 900 мл середовища, що розчиняє (0,1 M HCl), рН 1,0 і при 37 °C, можуть значно відрізнятися при розчиненні різних серій фармацевтичних лікарських форм у вигляді м'яких желатинових капсул, як показано на фіг. 7 для двох різних серій, використаних у фазі I випробувань на біодоступність в організмі людини (фіг. 8). Як було встановлено для вимірюваних значень часу вивільнення аж до 60 хв., серія А характеризується більше швидким вивільненням, ніж серія Б. Однак, таке розходження у профілях розчинення для різних серій аж до часу вивільнення лікарської речовини протягом 60 хв, що спостерігається при 100 об./хв., не має значення для фармакокінетичного профілю активної речовини in vivo у випадку використання сполуки із негайним вивільненням, як показано на фіг. 8. У фазі I випробувань (дивися фіг. 9) концентрації активної речовини у плазмі визначали у декількох пацієнтів уже через 0,5 год. після введення лікарської речовини й у більшості пацієнтів через 1 год. після введення лікарського засобу. Концентрація активної речовини у 5 UA 107560 C2 5 10 15 20 25 30 35 40 45 50 55 60 плазмі зростала протягом, приблизно, 2-4 год. після введення до 9 нг/мл (срг значення означає середнє геометричне значення) при введенні у дозі 150 мг здоровим пацієнтам. У деяких пацієнтів відзначали повторне зростання або вихід на плато концентрації активної речовини у плазмі протягом, приблизно, 4-6 год. Потім концентрація у плазмі знижувалася, принаймні, за біекспоненціальним законом. Концентрація активної речовини у плазмі склала приблизно 15 % від максимальної концентрації у плазмі через 24 год. після введення й приблизно 7-8 % через 48 год. після введення. У приблизно 2/3 пацієнтів вимірювані концентрації активної речовини у плазмі спостерігалися через 48 год. після введення лікарського засобу. Розходження у концентраціях активної речовини у плазмі через різні проміжки часу зростали через 2 год. (середнє геометричне коефіцієнта варіації: 100-250 %), але зменшувалися через більше тривалі проміжки часу (середнє геометричне коефіцієнта варіації: 30-45 %). Таким чином, концентрація активної речовини у плазмі у різних пацієнтів характеризується високою варіабельністю фармакокінетичних параметрів у всіх випробуваннях, що не дозволяло проводити формальну статистичну оцінку залежності від дози. Однак, за даними трьох серій випробувань у фазі I у страждаючих раком пацієнтів із різними прогресуючими солідними пухлинами, при візуальному огляді не спостерігали відхилення від пропорційного дозі підвищення величин AUC і Cmax активної речовини ні після введення однократної дози, ні у стабільному стані після введення одноразової і дворазової щоденних доз (фіг. 9 і 10). Як наслідок, у ракових пацієнтів середні геометричні значення C max, ss і AUCτ,ss для активної речовини зростали пропорційно дозі після однократного введення й у стабільному стані при введенні один раз на день і два рази на день. При візуальному огляді у різних клінічних випробуваннях у ракових пацієнтів не виявлено відхилення від пропорційної дози концентрації лікарської речовини в плазмі перед введенням у стабільному стані (C pre, ss). Крім того, у двох серіях випробувань у фазі 2 монотерапії моноетансульфонатом 3-Z-[1-(4(N-((4-метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6метоксикарбоніл-2-індолінону, після введення лікарської форми у вигляді м'яких желатинових капсул, у пацієнтів із недрібноклітинним раком легенів (НДКРЛ) або гормоностійким раком передміхурової залози не спостерігається помітне розходження у пропорційній дозі при збільшенні концентрації активної речовини у плазмі до введення в обох випробуваних групах, яким вводили по 150 і 250 мг активної речовини двічі на день у стабільному стані. Придатні форми для фармацевтичної лікарської форми за даним винаходом включають, наприклад, таблетки, капсули або пероральні розчини, еліксири, емульсії, пелети, порошки або гранули. Вміст фармацевтично активної сполуки/активної речовини, тобто моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6метоксикарбоніл-2-індолінону, повинен перебувати у діапазоні від 0,01 до 90 мас. %, переважно від 0,1 до 50 мас. % у розрахунку на масу композиції, тобто вміст повинен забезпечувати дозу, необхідну для прояву терапевтичного ефекту. При необхідності визначені дози можна вводити декілька разів на день. Придатні таблетки можна одержати, наприклад, при змішуванні активної речовини із відомими ексципієнтами, наприклад, інертними розріджувачами, такими як карбонат кальцію, фосфат кальцію або лактоза, дезінтегруючими агентами, такими як кукурудзяний крохмаль або альгінова кислота, зв'язувальними речовинами, такими як крохмаль або желатин, речовинами, що змазують, такими як стеарат магнію або тальк і/або агентами для вповільнення вивільнення, такими як карбоксиметилцелюлоза, фталат-ацетат целюлози або полівінілацетат. Таблетки можуть також містити декілька шарів. Таблетки з покриттям можна одержати при нанесенні покриття на ядра таблетки способом, аналогічним нанесенню покриття на таблетки з використанням речовин, які звичайно використовуються для нанесення покриття на таблетки, наприклад, колідон або шелак, аравійська камедь, тальк, діоксид титану або цукор. Для вповільнення вивільнення або запобігання несумісності ядро також може містити декілька шарів. Аналогічним чином покриття таблетки може містити декілька шарів для вповільнення вивільнення, при цьому можна використовувати ексципієнти, згадані вище при описі таблеток. Сиропи або еліксири, що містять активну речовину за даним винаходом, тобто моноетансульфонат 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)1-фенілметилен]-6-метоксикарбоніл-2-індолінону, можуть додатково містити підсолоджувач, такий як сахарин, цикламат, гліцерин або цукор, а також ароматизатор, наприклад, ароматичну речовину, таку як ванілін або апельсиновий екстракт. Вони також можуть містити ад'юванти або загусники для суспензій, такі як натрієва сіль карбоксиметилцелюлози, зволожуючі агенти, такі як, наприклад, продукти конденсації жирних спиртів з етиленоксидом або консерванти, такі як пара-гідроксибензоати. 6 UA 107560 C2 5 10 15 20 25 30 35 40 45 50 55 60 Капсули, що містять активну речовину за даним винаходом, тобто моноетансульфонат 3-Z[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6метоксикарбоніл-2-індолінону, можна одержати, наприклад, при змішуванні активної речовини із інертними носіями, такими як лактоза або сорбіт, з наступним заповненням сумішшю желатинових капсул. Ексципієнти, які можна використовувати, включають, наприклад, воду, фармацевтично прийнятні органічні розчинники, такі як вуглеводні (наприклад, нафтові фракції), рослинні масла (наприклад, арахісове або кунжутне масло), одно- або багатоатомні спирти (наприклад, етанол або гліцерин), носії, такі як, наприклад, природні мінеральні порошки (наприклад, каоліни, глини, тальк, крейда), синтетичні мінеральні порошки (наприклад, високодисперсну кремнієву кислоту або силікати), цукри (наприклад, тростинний цукор, лактозу й глюкозу), емульгатори (наприклад, лігнін, сульфіт-спиртову барду, метилцелюлозу, крохмаль і полівінілпіролідон) і мастильні речовини (наприклад, стеарат магнію, тальк, стеаринову кислоту й лаурилсульфат натрію). Таблетки для перорального введення можуть містити, крім вище згаданих носіїв, добавки, такі як цитрат натрію, карбонат кальцію й гідрофосфат кальцію у суміші з різними добавками, такими як крохмаль, переважно, картопляний крохмаль, желатин тощо. Крім того, мастильні речовини, такі як стеарат магнію, лаурилсульфат натрію й тальк, можна одночасно використовувати у процесі одержання таблеток. У випадку водних суспензій активну речовину можна комбінувати з різними ароматизаторами або барвниками, у суміші з вище згаданими ексципієнтами. Доза, призначена для перорального введення людині, становить від 5 до 1000 мг на введення, переважно від 25 до 300 мг, при однократному або більше введень на день. Однак, іноді може виникати необхідність у зміні зазначеної дози залежно від маси тіла, способу введення, індивідуальної реакції на активну речовину, природи складу й часу або інтервалу, протягом якого вводять активну речовину. Таким чином, у деяких випадках може виявитися достатнім використання дози менше зазначеного нижнього значення, причому в інших випадках можна перевищити верхню межу. При введенні великих кількостей можна рекомендувати поділ дози на декілька менших доз, що вводяться протягом дня. Наступні додаткові приклади фармацевтичних лікарських форм представлені для ілюстрації даного винаходу й не обмежують його обсяг. Активні інгредієнти/речовини або активні компоненти можна вводити відповідним способом у рідкій формі або у вигляді ліпофільних або гідрофільних систем-носіїв, або у вигляді розчину або суспензії, змішаної з індивідуальним ексципієнтом-носієм або змішаної з середовищемносієм складного складу, отриманої з декількох компонентів. Інкапсулювання таких рідких складів у капсули, як у м'які желатинові, так і у тверді желатинові капсули, являє собою надзвичайно надійний спосіб введення таких фармакологічно активних речовин. Розчини Для одержання систем на основі розчинів необхідно, щоб активна речовина розчинялася у носії. Підвищену абсорбцію погано абсорбуємого лікарського засобу у шлунково-кишковому тракті (ШКТ) можна забезпечити при збільшенні швидкості розчинення лікарського засобу у присутності жовчних кислот. У шлунково-кишковому тракті солі жовчних кислот проявляють властивості біологічних ПАР й утворюють при змішуванні з фосфоліпідами термодинамічно стабільні змішані міцели. У багатьох випадках вибір складу обмежений властивостями розчинника, в інших випадках лікарський засіб виявляється недостатньо розчинним у будьякому ліпідному складі. Можна створити середовище носія таким чином, щоб у шлунку спонтанно утворювалася емульсія або мікроемульсія, при цьому прискорюється абсорбція фармакологічно активної сполуки. Зазначені системи відомі як само(мікро)- емульгуючі системи для доставки лікарських засобів. При одержанні таких систем необхідно точно дотримуватися співвідношення компонентів, тому що навіть при незначній зміні складу відбувається необоротне ушкодження системи й зниження її переваг. Наприклад, активна сполука може осаджуватися зарахунок зміни солюбілізуючих властивостей капсульованого складу. Зазначений процес осадження може бути необоротним і може приводити до зниження дози при введенні пацієнтові. Емульгуючі властивості капсульованого складу також можуть змінюватися й після введення у шлунку емульсія може не утворюватися. У результаті порушується швидкість і відтворюваність абсорбції фармакологічно активної сполуки. Суспензії Тому що суспензії є термодинамічно нестійкими багатофазними системами, при їхній розробці варто брати до уваги різні характеристики таких систем. Фізична стабільність суспензійного складу повинна виключати ріст часток, а також перекристалізацію у поліморфну 7 UA 107560 C2 5 10 15 20 25 30 35 40 45 50 55 60 форму, розчинність якої відрізняється, або осадження, що супроводжується грудкуванням осаду. Такі фактори можуть впливати на вивільнення лікарської речовини із лікарської форми й, отже, змінювати ступінь впливу на пацієнта після зберігання продукту протягом його строку придатності. Отже, розчинність активної речовини в одному ексципієнті-носії або у системі-носії не є обов'язковою умовою фізичної стабільності системи. Ліпофільні системи-носії Ліпофільні ексципієнти широко використовуються як системи захисту від вологи для збереження хімічно нестабільних речовин. Для цієї мети можуть застосовуватися різні типи жирів або восків у твердих лікарських формах або у проміжних продуктах при їхньому одержанні для запобігання міграції водяної пари або кисню, що містяться у навколишньому середовищі, й для підвищення хімічної стабільності активної речовини. Включення лікарської речовини у ліпофільні сполучні агенти із розплаву також може запобігти контактуванню з вологою. Тому що тверді гідрофобні системи характеризуються низьким розпадом, вивільнення лікарської речовини у таких системах уповільнюється, на відміну від вивільнення лікарської речовини з рідких ліпідних складів із низькою в'язкістю. Таке вповільнене вивільнення лікарської речовини проявляється у вигляді характерних профілів концентрації активної речовини у плазмі з модифікованої системи доставки лікарської речовини (Ritschel W. та ін., Die Tablette, 2-е вид., ECV, Aulendorf, с. 267f (2002)). Отже, в'язкість рідких систем є важливим параметром і її варто суворо контролювати для забезпечення відповідного вивільнення лікарської речовини. Використовувані на практиці ліпофільні або ліпідні склади являють собою групу різних складів, що мають ряд різноманітних властивостей, який є наслідком змішування до п'яти класів ексципієнтів, починаючи від чистих тригліцеридних масел до змішаних гліцеридів, ліпофільних ПАР, гідрофільних ПАР й водорозчинних співрозчинників. Оцінка якості Якість складу можна оцінити, вимірюючи його відносну біодоступність, тобто порівнюючи його біодоступність з біодоступністю водного розчину активної речовини. Таким чином, ліпідні суспензії також можуть впливати на пацієнта завдяки відповідній розчинності активної речовини у фізіологічних умовах. Іншими словами, якщо спостерігається збільшення ступеня вивільнення лікарської речовини із лікарського продукту у присутності ПАР, можна чекати, що вивільнення лікарської речовини із лікарського продукту також збільшиться в умовах in vivo, тобто у присутності ПАР у шлунковокишковому тракті. М'яка желатинова капсула, що містить рідкий склад, який включає в'язку суспензію моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-Nметиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону у тригліцеридах із середньою довжиною ланцюга, твердих жирах і лецитині, відповідає вимогам задовільної біодоступності у необхідному діапазоні доз при лікуванні лікарською речовиною, моноетансульфонатом 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-Nметиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону. Дана рідка композиція включає ліпідну суспензію активної речовини. Цей склад також відповідає певному діапазону профілів негайного вивільнення, що забезпечує придатний профіль залежності концентрації активного агента у плазмі від часу, що є метою даного винаходу. Перевага такої м'якої желатинової капсули, що містить ліпідну суспензію, полягає у тому, що всмоктування води складом капсули практично виключено. Лікарська форма розділена на три різних компоненти, а саме: гідрофільну оболонку капсули (а) і гідрофобну систему-носій (б), в якій суспендований слабко гігроскопічний порошок активної речовини (в). Внаслідок наявності вологи у навколишньому середовищі, вміст води у цих різних відділеннях може відрізнятися. Волога може мігрувати за рахунок дифузії до досягнення рівноважного стану. Вміст води може впливати на різні властивості лікарського продукту, такі як хімічна стабільність активної речовини (переважно внаслідок гідролізу), розчинення активної речовини або еластичність оболонки капсули. Вода у даній системі насамперед всмоктується оболонкою капсули, що можна встановити за даними експерименту по сорбції водяної пари, а також при кореляції приросту маси зі ступенем розм'якшення капсули (як показано на фіг. 1). При цьому поглинання води не впливає на хімічну стабільність лікарської речовини, що підтверджується результатами випробування на стабільність систем за даним винаходом у жорстких умовах, наприклад, при зберіганні протягом 1 місяця при 70 °C, а також протягом тривалого періоду (протягом 3 років) і прискорених випробуваннях (протягом 6 місяців) на стабільність. Крім того, у результаті випробувань було встановлено, що не відбувається помітного приросту маси, а також не виникають проблеми злипання капсул за даним винаходом, якщо зберігати капсули у щільно впакованому вигляді при температурі нижче 30 °C. Таким чином, 8 UA 107560 C2 5 10 15 20 25 30 35 40 45 50 55 рекомендовані умови упакування таких капсул являють собою, наприклад, скляні контейнери або еластичні/тверді пластикові контейнери (наприклад, сулії ПЕВП), алюмінієві блістери (наприклад, alu/alu блістери), пластикові блістери (наприклад, ПВК, ПВДХ або Aclar®), необов'язково додатково упаковані в алюмінієвий пакет або двошаровий полімерний пакет. Як правило, м'які желатинові капсули включають оболонку капсули з желатину, один або більше пластифікаторів, насамперед гліцерин, необов'язково допоміжні матеріали, такі як барвники, пігменти, ароматизатори, цукор, олігосахариди або полісахариди, і вміст капсули, що містить розчинник, ад'юванти й одну або більше фармакологічно активних речовин. Термін желатин, що використовується у даному контексті, означає не тільки немодифікований желатин, відповідно до Європейської Фармакопеї, але також модифікований желатин, такий як, наприклад, сукцинований желатин. Згаданий вище склад на основі ліпідної суспензії активної речовини, моноетансульфонату 3Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6метоксикарбоніл-2-індолінону, містить в'язку суспензію моноетансульфонату 3-Z-[1-(4-(N-((4метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл2-індолінону у ліпідному носії, загусник і речовину, що змазує / солюбілізуючий агент. Кількість моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-Nметиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону, переважно, становить величину у діапазоні від 1 до 90 мас. % у розрахунку на масу складу ліпідної суспензії, більше переважно від 10 до 50 мас. %. Щоб виключити вище згадані проблеми, пов'язані з фізичною стабільністю, такі як перекристалізація або ріст часток, активна речовина повинна або повністю не розчинятися, або розчинятися у носії. Аналіз розчинності ліпофільних, гідрофільних і амфіфільних ексципієнтів і сумішей вказує на можливість використання різних носіїв для одержання складу згаданої вище ліпідної суспензії. Таким чином, придатні носії або компоненти носія для активної речовини, моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-Nметиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону, включають ацетиловані моногліцериди, гліцериди кукурудзяного масла, етилолеат, моно/діолеат гліцерину, монолінолеат гліцерину, каприлокапрат макроголгліцерину, лінолеат макроголгліцерину, частково заміщені гліцериди із середньою довжиною ланцюга, тригліцериди із середньою довжиною ланцюга, тригліцериди каприлової/капронової кислоти, тригліцериди каприлової/капронової/лінолевої кислоти, тригліцериди каприлової/капронової/бурштинової кислоти, дикаприлат/дикапрат пропіленгліколя, поліоксиловане гідроване касторове масло, олеїнова кислота, поліоксиловане касторове масло, монокаприлат пропіленгліколя, монолаурат пропіленгліколя, рафіноване масло тваринного походження, рафіноване соєве масло, рафіноване рослинне масло, моностеарат сорбіту, триацетин, триетилцитрат або їх суміші. Проблеми, пов'язані зі стабільністю, такі як гідролітичне розкладання активної речовини, також можуть бути викликані компонентами ліпофільного носія. У наслідок цього системи-носії на основі гідрофільних поліетиленгліколей будуть в основному проявляти знижену стабільність у порівнянні з більше гідрофобними носіями, такими як ліпідні носії. Для згаданого вище складу ліпідної суспензії найбільше кращим ліпідним носієм є тригліцериди із середньою довжиною ланцюга. Їхній вміст перебуває у діапазоні від 1 до 90 мас. % у розрахунку на масу ліпідної суспензії, переважно від 10 до 70 мас. %. Придатні тригліцериди із середньою довжиною ланцюга можна вибрати із ряду комерційних продуктів Miglyol 812®, Miglyol 810®, Miglyol 818®, Miglyol 829® або Miglyol 840®. Загусник регулює в'язкість суспензії. Він стабілізує суспензійну систему, забезпечує оптимальні умови при переробці й гарантує необхідну якість капсули, насамперед відносно однорідності досліджуваного складу або профілю розчинності. Придатними загусниками, які використовують при одержанні вище згаданого суспензійного складу, є утворюючі олеогелі ексципієнти, такі як колоїдний діоксид кремнію або бентоніт, або ліпофільні або амфіфільні ексципієнти із високою в'язкістю, такі як бджолиний мед, моностеарат гліцерину, гідроване рослинне масло, частково гідроване рослинне масло або тверді жири. Для згаданого вище суспензійного складу найбільше кращим загусником є твердий жир. Переважно, його вміст становить від 1 до 30 мас. % у розрахунку на масу суспензійного складу, більше переважно від 10 до 30 мас. %. Найбільше придатні тверді жири характеризуються температурою плавлення у діапазоні від 30 °C до 44 °C, найбільше переважно, температура плавлення перебуває у діапазоні від 33 °C до 40 °C. Придатними комерційними продуктами є Gelucire® 33/01, Witepsol® W35 або Softisan® 378. Визначення найбільше придатного діапазону 9 UA 107560 C2 5 10 15 20 25 30 35 40 температур плавлення твердих жирів можна проводити, як показано на фіг. 3, при оцінці впливу діапазону плавлення твердого жиру на профіль розчинення in vitro протягом часу. Лецитин є стандартним ексципієнтом для систем-носіїв у м'яких желатинових капсулах. Його використовують як речовину, що змазує, у висококонцентрованій суспензії при інкапсулюванні, для запобігання блокування шлангів і насосів і для забезпечення високої масової однорідності інкапсульованого складу. Крім того, лецитин є ПАР, що може поліпшити розподіл крапель складу при проведенні випробувань на розчинення in vitro (див. фіг. 2), а також при усмоктуванні лікарської речовини in vivo. Лецитин також може поліпшити змочування кристалів активної речовини. Придатним лецитином є комерційний продукт Topcithin®. Лецитин можна використовувати аж до певного вмісту, для поліпшення профілю розчинення готових капсул. Надлишкові кількості не приводять до додаткової переваги при випробуваннях на розчинення in vitro, як показано на фіг. 2. У згаданому вище суспензійному ліпідному складі вміст лецитину перебуває у діапазоні від 0,1 до 10 мас. % у розрахунку на масу ліпідної суспензії, найбільше переважно від 0,25 до 2,5 %. В іншому варіанті склад ліпідної суспензії активної речовини, моноетансульфонату 3-Z-[1-(4(N-((4-метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6метоксикарбоніл-2-індолінону, містить в'язку суспензію моноетансульфонату 3-Z-[1-(4-(N-((4метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл2-індолінону у середовищі тригліцеридів із середньою довжиною ланцюга, твердого жиру, лецитину й одного або більше макроголгліцеринів, таких як, наприклад, гідроксистеарат макроголгліцерину (випускається, наприклад, за назвою Eumulgin HRE 40 PH) або рицинолеат макроголгліцерину (інша назва поліоксиловане касторове масло або випускається, наприклад, під назвами Cremophor EL, Cremophor® RH40 або Eumulgin RO 35 PH). У згаданому вище суспензійному ліпідному складі кількість макроголгліцерину(ів) перебуває у діапазоні від 0,1 до 50 мас. % у розрахунку на масу ліпідної суспензії, найбільше переважно від 0,3 до 10 %. Три системи-носія (ліпофільна P3, ліпофільна P1 і ліпофільна P2, що містить ПАР, для напівтвердих суспензійних складів, описаних вище) випробовували на біодоступність у неклінічних умовах, при цьому було встановлено, що всі вони придатні як варіанти при одержанні пероральної лікарської форми активної речовини, моноетансульфонату 3-Z-[1-(4-(N((4-метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6метоксикарбоніл-2-індолінону. Однак, у зв'язку з біодоступністю результати, показані на фіг. 4, свідчать про те, що кращі ліпідні (ліпофільні) суспензійні склади, що містять в'язку суспензію моноетансульфонату 3-Z-[1(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6метоксикарбоніл-2-індолінону у середовищі тригліцеридів із середньою довжиною ланцюга, твердого жиру й лецитину. Таким чином, на фіг. 4 показані результати порівняння абсолютної біодоступності (АБ, %) за даними випробувань на пацюках протягом 24 год. для водного розчину (S) і різних систем-носіїв (P1, P2 і P3) активної речовини, моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону. Експеримент докладно описаний нижче. У нижчеподаній таблиці представлений склад досліджених систем-носіїв (напівтвердих суспензійних складів). 45 Склад Інгредієнти Активна речовина Тригліцериди із середньою довжиною ланцюга Твердий жир Cremophor RH40 Лецитин Гліцерин 85 % Очищена вода Макрогол 600 Макрогол 4000 P1 43,48 37,83 18,26 -0,43 ---- P2 [%]* 42,19 41,77 12,66 2,95 0,42 ---- P3 31,75 ----3,17 4,76 58,10 2,.22 невеликі відхилення наведених кількостей від 100 % можуть бути пов'язані з помилками при округленні. 10 UA 107560 C2 5 10 15 20 25 30 35 40 45 50 55 Напівтвердими суспензіями наповнюють тверді желатинові капсули (Capsugel, № Y0303490). Кожна капсула містить, приблизно, від 15 до 20 мг сполуки. Капсули вводили пацюкам за допомогою спеціального пристрою типу шлункового зонда. Для порівняння через зонд вводили водний розчин, що містить 0,5 % Natrosol 250 HX. Для розрахунку абсолютної біодоступності додатковій групі пацюків внутрішньовенно вводили речовину, яка розчинена у 5 % розчині глюкози (водний розчин (S)). Використовували по 5 самців пацюків лінії Han Wistar (клон CrlGlxBrlHan:WI) у кожній групі. Зразки крові відбирали через 0,5 год., 1 год., 2 год., 4 год., 8 год., 24 год. після введення дози й плазму аналізували стандартним методом РХВР/МС/МС (з тандемною мас-спектрометрією). Площу під кривою залежності концентрації у плазмі від часу (AUC) розраховували лінійним методом трапецій. Для розрахунку абсолютної біодоступності значення нормалізованих за дозою AUC для перорального складу ділили на значення нормалізованих за дозою AUC для внутрішньовенного складу. Як можна бачити із результатів експерименту, показаних на фіг. 4, рівні величини біодоступності спостерігалися для водного розчину (S: 11 %) і для різних систем-носіїв активної речовини (P1:14 %, P2:10 % і P3:10 %), однак варіабельність величин для окремих особ (стандартне відхилення біодоступності) менше для водного розчину (S) і системи-носія (P1) у порівнянні з системами-носіями (P2) і (P3) (2,8 і 4,1 проти 7,4 і 7,1), що вказує на практично повну відносну біодоступність досліджених складів (P1, P2 і P3) у порівнянні з розчином (S), але також на більше високу варіабельність для систем-носіїв (P2) і (P3). Ліпідний суспензійний склад, описаний у даному контексті, можна включати у капсульовану фармацевтичну лікарську форму, що містить оболонку капсули й склад капсули (або вміст капсули), при цьому склад капсули (або вміст капсули) містить ліпідний суспензійний склад, як визначено вище. Капсульована фармацевтична лікарська форма може являти собою м'яку желатинову капсулу, тверду желатинову капсулу або капсулу з гідроксипропілметилцелюлози (ГПМЦ), полівінілового спирту або пулулану. У випадку твердої желатинової капсули або капсули з гідроксипропілметилцелюлози (ГПМЦ), полівінілового спирту або пулулану, вміст у капсулі можна додатково запечатати або запаяти. Переважно, капсула є м'якою желатиновою капсулою, що складається з оболонки капсули, яка включає желатин, один або більше пластифікаторів і, необов'язково, додаткові допоміжні матеріали, а також вміст капсули відрізняється тим, що вміст капсули містить ліпідний суспензійний склад, як описано вище. Капсульовану лікарську фармацевтичну форму, насамперед м'які желатинові капсули, можна зберігати у придатних скляних контейнерах або в еластичних/твердих пластикових контейнерах, переважно не на основі ПВХ-матеріалів, або у пластикових (наприклад, ПВХ, ПВДХ, або Aclar®) блістерах, не обов'язково упакованих в алюмінієвий матеріал (алюмінієві пакети) або в алюмінієві блістери, що містять, наприклад, нижній шар фольги з поліаміду/Al/ПВХ і покриття з алюмінієвої фольги, при цьому зазначене покриття забезпечує найкращий захист від води. Отже, можна створити контейнери таким чином, щоб забезпечити надійний захист капсульованої фармацевтичної лікарської форми, і насамперед м'яких желатинових капсул, наприклад, для захисту їх від впливу світла, кисню або води. Еластичні пластикові контейнери можуть містити додатковий захист, наприклад, у вигляді додаткового алюмінієвого упакування. Капсульовану фармацевтичну лікарську форму можна виготовити стандартними методами одержання капсул. М'яку желатинову капсулу можна виготовити стандартними методами одержання м'яких желатинових капсул, такими як, наприклад, з використанням "ротаційної машин для таблетування", як описано, наприклад, у книзі Swarbrick, Boylann: Encyclopedia of pharmaceutical technology, Marcel Dekker, т. 2, с.с. 269 ff (1990) або у книзі Lachmann та ін., "The Theory and Practice of Industrial Pharmacy", 2-е вид., с.с. 404-419 (1976) або іншими методами, такими як процедури, описані, наприклад, у статті Jimerson R. F. та ін., "Soft gelatin capsule update", Drug Dev. Ind. Pharm., т 12, No. 8-9, с.с. 1133-1144 (1986). Ліпідний суспензійний склад можна одержати стандартними методами для одержання складів, наприклад, при змішуванні інгредієнтів при попередньо визначеній температурі у попередньо визначеному порядку, при цьому одержують гомогенізовану суспензію. В іншому варіанті ліпідний суспензійний склад можна одержати, як описано у прикладі 10. Ліпідний суспензійний склад, що містить активну речовину, готові м'які желатинові капсули, що містять зазначений склад, і пакувальні матеріали, призначені для упакування готових м'яких желатинових капсул, описані у наступних прикладах і фігурах. Приклади представлені винятково для ілюстрації винаходу й не обмежують обсяг даного винаходу. 11 UA 107560 C2 5 Приклади систем-носіїв (складів), м'яких желатинових капсул, матеріалів для упакування й способу одержання ліпідного суспензійного складу активної речовини Як активну речовину у всіх прикладах використовували моноетансульфонат 3-Z-[1-(4-(N-((4метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл2-індолінону. Приклад 1 Система-носій на основі ліпідів Склад Інгредієнти Активна речовина Тригліцериди із середньою довжиною ланцюга Твердий жир Лецитин A 43,48 28,70 27,39 0,43 B [%]* 43,48 37,83 18,26 0,43 C 43,48 38,045 18,26 0,215 * невеликі відхилення наведених кількостей від 100 % можуть бути пов'язані з помилками при округленні. 10 Приклад 2 Система-носій на основі ліпідів з додатковою ПАР Інгредієнти Активна речовина Тригліцериди із середньою довжиною ланцюга Твердий жир Cremophor RH40 Лецитин [%]* 42,19 41,77 12,66 2,95 0,42 * невеликі відхилення наведених кількостей від 100 % можуть бути пов'язані з помилками при округленні. 15 Приклад 3 Гідрофільна система-носій Інгредієнти [%]* 31,75 3,17 4.76 58,10 2,22 Активна речовина Гліцерин 85 % Очищена вода Макрогол 600 Макрогол 4000 * невеликі відхилення наведених кількостей від 100 % можуть бути пов'язані з помилками при округленні. 12 UA 107560 C2 Приклад 4 М'яка желатинова капсула, що містить 50 мг активної речовини Склад A Склад B Склад C Інгредієнти Функція мг у капсулі мг у капсулі мг у капсулі Активна речовина* Активний інгредієнт 60,20 60,20 60,20 Тригліцериди із середньою Носій 40,95 53,70 54,00 довжиною ланцюга Твердий жир Загусник 38,25 25,50 25,50 Зволожуючий агент/ Лецитин 0,60 0,60 0,30 Речовина, що змазує Желатин Плівкоутворювач 72,25 72,25 72,25 Гліцерин 85 % Пластифікатор 32,24 32,24 32,24 Діоксид титану Барвник 0,20 0,20 0,20 Оксид заліза A Барвник 0,32 0,32 0,32 Оксид заліза B Барвник 0,32 0,32 0,32 Загальна маса капсули 245,33 245,33 245,33 * Цифри відносяться до кількості етансульфонату (у сухому стані), еквівалентній зазначеній кількості вільної основи. 5 Приклад 4а М'яка желатинова капсула, що містить 75 мг активної речовини Інгредієнти Активна речовина* Тригліцериди із середньою довжиною ланцюга Твердий жир Лецитин Желатин Гліцерин 85 % Діоксид титану Оксид заліза A Оксид заліза B Загальна маса капсули Функція Активний інгредієнт Склад A Склад B Склад C мг у капсулі мг у капсулі мг у капсулі 90,3 90,3 90,3 Носій 61,425 80,55 80,1 Загусник Зволожуючий агент/ Речовина, що змазує Плівкоутворювач Пластифікатор Барвник Барвник Барвник 57,375 38,25 38,25 0,9 0,9 1,35 107,11 46,84 0,35 0,058 0,16 364,518 107,11 46,84 0,35 0,058 0,16 364,518 107,11 46,84 0,35 0,058 0,16 364,518 * Цифри відносяться до кількості етансульфонату (у сухому стані), еквівалентній зазначеній кількості вільної основи. 13 UA 107560 C2 Приклад 5 М'яка желатинова капсула, що містить 100 мг активної речовини Інгредієнти Активна речовина* Тригліцериди із середньою довжиною ланцюга Твердий жир Лецитин Желатин Гліцерин 85 % Діоксид титану Оксид заліза A Оксид заліза B Загальна маса капсули Функція Активний інгредієнт Склад A Склад B Склад C мг у капсулі мг у капсулі мг у капсулі 120,40 120,40 120,40 Носій 81,90 107,40 106,8 Загусник Зволожуючий агент/ Речовина, що змазує Плівкоутворювач Пластифікатор Барвник Барвник Барвник 76,50 51,00 51,00 1,20 1,20 1,80 111,58 48,79 0,36 0,06 0,17 440,96 111,58 48,79 0,36 0,06 0,17 440,96 111,58 48,79 0,36 0,06 0,17 440,96 * Цифри відносяться до кількості етансульфонату (у сухому стані), еквівалентній зазначеній кількості вільної основи. 5 Приклад 6 М'яка желатинова капсула, що містить 125 мг активної речовини Інгредієнти Активна речовина* Тригліцериди із середньою довжиною ланцюга Твердий жир Лецитин Желатин Гліцерин 85 % Діоксид титану Оксид заліза A Оксид заліза B Загальна маса капсули Функція Активний інгредієнт Склад A Склад B Склад C мг у капсулі мг у капсулі мг у капсулі 150,50 150,50 150,50 Носій 102,375 134,25 133,5 Загусник Зволожуючий агент/ Речовина, що змазує Плівкоутворювач Пластифікатор Барвник Барвник Барвник 95,625 63,75 63,75 1,50 1,50 2,25 142,82 62,45 0,47 0,08 0,22 556,04 142,82 62,45 0,47 0,08 0,22 556,04 142,82 62,45 0,47 0,08 0,22 556,04 * Цифри відносяться до кількості етансульфонату (у сухому стані), еквівалентній зазначеній кількості вільної основи. 14 UA 107560 C2 Приклад 7 М'яка желатинова капсула, що містить 150 мг активної речовини Інгредієнти Активна речовина* Тригліцериди із середньою довжиною ланцюга Твердий жир Лецитин Желатин Гліцерин 85 % Діоксид титану Оксид заліза A Оксид заліза B Загальна маса капсули Функція Активний інгредієнт Склад A Склад B Склад C мг у капсулі мг у капсулі мг у капсулі 180,60 180,60 180,60 Носій 122,85 161,10 160,20 Загусник Зволожуючий агент/ Речовина, що змазує Плівкоутворювач Пластифікатор Барвник Барвник Барвник 114,75 76,50 76,50 1,80 1,80 2,70 142,82 62,45 0,47 0,08 0,22 626,04 142,82 62,45 0,47 0,08 0,22 626,04 142,82 62,45 0,47 0,08 0,22 626,04 * Цифри відносяться до кількості етансульфонату (у сухому стані), еквівалентній зазначеній кількості вільної основи. 5 Приклад 8 М'яка желатинова капсула, що містить 200 мг активної речовини Інгредієнти Активна речовина* Тригліцериди із середньою довжиною ланцюга Твердий жир Лецитин Желатин Гліцерин 85 % Діоксид титану Оксид заліза A Оксид заліза B Загальна маса капсули Функція Активний інгредієнт Склад A Склад B Склад C мг у капсулі мг у капсулі мг у капсулі 240,80 240,80 240,80 Носій 163,30 214,80 216,00 Загусник Зволожуючий агент/ Речовина, що змазує Плівкоутворювач Пластифікатор Барвник Барвник Барвник 153,50 102,00 102,00 2,40 2,40 1,20 203,19 102,61 0,57 0,90 0,90 868,17 203,19 102,61 0,57 0,90 0,90 868,17 203,19 102,61 0,57 0,90 0,90 868,17 *Цифри відносяться до кількості етансульфонату (у сухому стані), еквівалентній зазначеній кількості вільної основи. 10 15 20 Приклад 9 Як пакувальні матеріали для упакування м'яких желатинових капсул, описаних у прикладах 4-8, можна використовувати скляні контейнери, еластичні/тверді пластикові контейнери або блістери із ПВК/ПВДХ, необов'язково в алюмінієвому пакеті, або блістери Alu/Alu. Приклад 10 У зазначеному прикладі описаний спосіб одержання ліпідного суспензійного складу активної речовини й спосіб інкапсулювання. а: Твердий жир і частину тригліцеридів із середньою довжиною ланцюга попередньо змішували у відповідній установці. Послідовно додавали лецитин, іншу кількість тригліцеридів із середньою довжиною ланцюга й активну речовину. Суспензію перемішували, гомогенізували, дегазували й потім просіювали, при цьому одержували склад (суміш для заповнення). б. Основні компоненти желатинової маси (гліцерин, воду й желатин) перемішували й розчиняли при підвищеній температурі. Потім додавали відповідні барвники й перемішували, при цьому одержували офарбовану желатинову масу. 15 UA 107560 C2 5 10 15 20 25 в. Після регулювання машини для інкапсулювання суміш для заповнення й офарбовану желатинову масу переробляли у м'які желатинові капсули за допомогою ротаційної машини для таблетування. Даний процес описаний, наприклад, у книзі: Swarbrick, Boylann, Encyclopedia of pharmaceutical technology, Marcel Dekker, т. 2, с.с. 269 ff. (1990). г. Початкову стадію сушіння проводили із використанням ротаційної сушарки. На остаточній стадії сушіння капсули поміщали на піддони. Сушіння проводили при 15-26 °C і при низькій відносній вологості. д. Після 100 % завершення візуальної оцінки капсул для відділення від ушкоджених або капсул, що протікають, капсули сортували за розміром. е. На заключній стадії капсули маркірували з використанням офсетного друку або струминного друку. В іншому варіанті капсули можна маркірувати методом друку на стрічці, тобто желатинові стрічки спочатку маркірують перед стадією інкапсулювання (в.). Приклад 11 У нижче наведеній таблиці показані альтернативні фармацевтичні композиції за даним винаходом. Композиції D, E і F є таблетками, Склад G можна пресувати у таблетки після грануляції з розплаву активної речовини у суміші з мікрокристалічною целюлозою й макроголом 6000, у нагрітому/охолодженому змішувачі з високим зрушенням. Після додаткових стадій перемішування отриманих гранул із іншими ексципієнтами одержують таблетки з використанням стандартного преса для таблетування. В іншому варіанті гранулами можна заповнювати пакетики й використовувати зазначені гранули для перорального введення. Таблетки D і F можна одержувати при безпосередньому перемішуванні компонентів із наступним пресуванням на придатному пресі для таблетування. В іншому варіанті композиції можна обробляти в екструдері й одержувати пелети для заповнення ними твердої капсули. Таблетку E можна одержувати способом вологої грануляції лікарської речовини у суміші з моногідратом лактози, мікрокристалічною целюлозою й водним розчином коповідону. Після додаткових стадій перемішування з кросповідоном, колоїдним діоксидом кремнію й стеаратом магнію, таблетки пресували з використанням стандартного преса для таблетування. Приклад композиції для одержання інших твердих пероральних складів Склад Активна речовина Сорбіт Моногідрат лактози Мікрокристалічна целюлоза Фосфат кальцію Соєве масло Макрогол 6000 Коповідон Натрієва сіль гліколята крохмалю Кросповідон Cremophor RH 40 Колоїдний діоксид кремнію Твердий ароматизатор Стеарат магнію Усього D 180,6 мг 50,0 мг 30,0 мг 2,0 мг E 150,5 мг 125,0 мг 20,0 мг 10,0 мг F 120,4 мг 150,0 мг 150,0 мг G 150,5 мг 80,0 мг 80,0 мг H 60,2 мг 145,0 мг I 60,2 мг 125,0 мг 20,0 мг 5,0 мг 1,0 мг 4,0 мг 272,6 мг 5,0 мг 1,0 мг 4,0 мг 315,5 мг 5,0 мг 1,0 мг 4,0 мг 430,4 мг 5,0 мг 315,5 мг 20,0 мг 10,0 мг 235,2 мг 5,0 мг 1,0 мг 4,0 мг 215,2 мг 30 Склад H одержували у вигляді рідкої суміші суспензійної активної речовини. Після гомогенізації складом заповнювали тверді або м'які желатинові капсули. Склад I являє собою порошок для перорального введення. 16 UA 107560 C2 ФОРМУЛА ВИНАХОДУ 5 10 15 20 25 30 35 1. Фармацевтична лікарська форма активної речовини, моноетансульфонату 3-Z-[1-(4-(N-((4метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл2-індолінону, що забезпечує доставку активної речовини та характеризується профілем негайного вивільнення, при якому не менше 70 % (Q65 %) активної речовини розчиняється in vitro протягом 60 хв у наступних умовах розчинення in vitro, відповідно до Європейської фармакопеї, 6.2: апарат 2 (лопатева мішалка), середовище, що розчиняє 0,1 М HCl (рН 1) і швидкість перемішування від 50 до 150 об./хв, при температурі 37 °С, і яка містить склад ліпідної суспензії активної речовини, що включає в'язку суспензію активної речовини у (і) 1-90 % тригліцеридів із середньою довжиною ланцюга; (іі) 1-30 % твердого жиру; і (ііі) 0,1-10 % лецитину. 2. Фармацевтична лікарська форма за п. 1, що забезпечує доставку активної речовини та характеризується профілем негайного вивільнення, при якому не менше 75 % (Q70 %) активної речовини розчиняється in vitro протягом 60 хв. 3. Фармацевтична лікарська форма за п. 1, що забезпечує доставку активної речовини та характеризується профілем негайного вивільнення, при якому не менше 85 % (Q80 %) активної речовини розчиняється іn vitro протягом 60 хв. 4. Фармацевтична лікарська форма за п. 1, що забезпечує доставку активної речовини та характеризується профілем негайного вивільнення, при якому не менше 85 % (Q80 %) активної речовини розчиняється in vitro протягом 45 хв. 5. Фармацевтична лікарська форма за п. 1, що забезпечує доставку активної речовини та характеризується профілем негайного вивільнення, при якому не менше 85% (Q80%) активної речовини розчиняється in vitro протягом 30 хв. 6. Фармацевтична лікарська форма за будь-яким із пп. 1-5, що характеризується порівнянним профілем розчинення in vitro незалежно від величини дози у діапазоні від 5 до 1000 мг активної речовини. 7. Фармацевтична лікарська форма за будь-яким із пп. 1-6, що є лікарською формою для пероральної доставки. 8. Фармацевтична лікарська форма за будь-яким із пп. 1-6, що містить активну речовину у кількості від 0,01 до 90 мас.% у розрахунку на загальну масу композиції. 9. Фармацевтична лікарська форма за будь-яким із пп. 1-6, що містить дозу у діапазоні від 5 до 1000 мг активної речовини. 17 UA 107560 C2 18 UA 107560 C2 19 UA 107560 C2 20 UA 107560 C2 21 UA 107560 C2 22 UA 107560 C2 Комп’ютерна верстка Л. Литвиненко Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 23
ДивитисяДодаткова інформація
Назва патенту англійськоюPharmaceutical dosage form for immediate release of an indolinone derivative
Автори російськоюMesserschmid, Roman, Lach, Peter, Sokoliess, Torsten, Stopfer, Peter, Trommeshauser, Dirk
МПК / Мітки
МПК: A61K 9/48, A61K 47/44, A61P 35/00, A61P 37/00, A61K 9/10, A61K 31/404
Мітки: вивільнення, форма, похідної, негайного, лікарська, фармацевтична, індолінону
Код посилання
<a href="https://ua.patents.su/25-107560-farmacevtichna-likarska-forma-dlya-negajjnogo-vivilnennya-pokhidno-indolinonu.html" target="_blank" rel="follow" title="База патентів України">Фармацевтична лікарська форма для негайного вивільнення похідної індолінону</a>
Капсульована лікарська форма, що містить суспензійну композицію похідної індолінону

Номер патенту: 104590
Опубліковано: 25.02.2014
Автори: Брокс Вернер, Мессершмід Роман, Біндер Рудольф, Бок Томас
МПК: A61K 47/44, A61K 9/48, A61K 31/404, A61P 37/00, A61K 9/10, A61P 35/00
Мітки: похідної, форма, капсульована, індолінону, суспензійну, композицію, містить, лікарська
Формула / Реферат:
1. Композиція активної сполуки, моноетансульфонату 3-Z-[1-(4-(N-((4-метилпіперазин-1-іл)метилкарбоніл)-N-метиламіно)аніліно)-1-фенілметилен]-6-метоксикарбоніл-2-індолінону, що являє собою в'язку ліпідну суспензію активної сполуки у тригліцеридах із середньою довжиною ланцюга, твердому жирі та лецитині.2. Капсула, що включає оболонку капсули й капсульований склад, причому капсульований склад містить композицію за п. 1.3. Капсула...
Фармацевтична лікарська форма антиепілептичних препаратів

Номер патенту: 19486
Опубліковано: 15.12.2006
Автор: Шатілло Андрій Валерійович
МПК: A61K 41/00, A61P 25/08
Мітки: форма, препаратів, антиепілептичних, фармацевтична, лікарська
Формула / Реферат:
Фармацевтична лікарська форма антиепілептичних препаратів пролонгованої дії, яка відрізняється тим, що діюча речовина в складі лікарської форми є електролітом, а допоміжні речовини не є електролітами.
Лікарська форма ранолазину пролонгованої дії

Номер патенту: 67793
Опубліковано: 15.07.2004
Автори: Вольф Ендрю А., Лангрідж Джон, Бейкер Фіона
МПК: A61K 9/20, A61K 31/495
Мітки: дії, лікарська, ранолазину, форма, пролонгованої
Формула / Реферат:
1. Фармацевтична дозована лікарська форма пролонгованої дії, яка містить щонайменше приблизно 50% (мас.) ранолазину та щонайменше одну рН-залежну в'яжучу речовину, що уповільнює виділення ранолазину з цієї дозованої лікарської форми пролонгованої дії у разі, коли вона зазнає впливу водного середовища, що має рН шлунка, та сприяє вивільненню терапевтичної кількості ранолазину у водному розчині, рівень рН якого перевищує приблизно 4,5.2....
Фармацевтична лікарська форма, що швидко тане в роті (варіанти) та спосіб формування гранул

Номер патенту: 63993
Опубліковано: 16.02.2004
Автори: Санджеєв Х. Котарі, Дів'якант С. Десаі
МПК: A61K 47/00, A61K 9/22
Мітки: формування, форма, роті, фармацевтична, гранул, варіанти, спосіб, лікарська, швидкої, тане
Формула / Реферат:
1. Фармацевтична лікарська форма, що швидко тане в роті, яка відрізняється тим, що містить лікарську речовину і комбінацію з чотирьох ексципієнтів – супердезінтегранту, диспергатора, речовини, що сприяє розподілу інгредієнтів (розподілювача), і зв'язувальної речовини.2. Фармацевтична лікарська форма за п. 1, яка відрізняється тим, що вона містить не більше, ніж приблизно 30% (мас.) лікарської речовини і не більше, ніж приблизно 85%...
Лікарська форма уповільненого вивільнення глюкозаміну

Номер патенту: 105513
Опубліковано: 26.05.2014
Автори: Срінівасан Р, Т Махеш Кумар, Ботхра Пукхрадж Чханданмал, Самбасіва Рао Марам, Нсв Раджу, Кандарапу Рагхупатхі
МПК: A61P 19/00, A61K 9/22, A61K 31/726
Мітки: глюкозаміну, лікарська, вивільнення, уповільненого, форма
Формула / Реферат:
1. Лікарська форма уповільненого вивільнення глюкозаміну та його солей, що містить комбінацію гідроксипропілметилцелюлози з молекулярною масою 100000 та гідроксипропілметилцелюлози з молекулярною масою 200000 як засіб, що уповільнює вивільнення, і один або більше фармацевтично прийнятних наповнювачів, де глюкозамін та його солі присутні частково у внутрішньогранулярній формі та частково у позагранулярній формі.2. Лікарська форма...
Попередній патент: Спосіб приготування гідролізату
Наступний патент: Поліморфні форми ацилсульфонамідів
Випадковий патент: Пристрій для табличної реалізації арифметичних операцій множення та додавання чисел за модулем mi класу лишків