Стійкі до спирту кишковорозчинні фармацевтичні композиції
Номер патенту: 111819
Опубліковано: 24.06.2016
Автори: Мансер Девід, Шах Хардік, Рекхі Гурвіндер Сінгх, Радді Стефен Б., Ліверсідж Гері














Формула / Реферат
1. Стійка до спирту фармацевтична композиція, яка містить:
(i) діючу речовину;
(ii) кишковорозчинну систему; і
(iii) засіб захисту від спирту, де кишковорозчинна система або засіб захисту від спирту включають ацетатфталат целюлози на органічній основі і де засіб захисту від спирту присутній в лікарській формі в кількості, яка забезпечує приріст від 10 до 500 %.
2. Композиція за п. 1, в якій або кишковорозчинне покриття, або засіб захисту від спирту необов'язково містить другий полімер, вибраний з групи, що складається з фталату гіпромелози, співпівполімерів етилакрилату-метилметакрилату, аніонних співполімерів метакрилової кислоти і етилакрилату і їх сумішей.
3. Композиція за п. 1, в якій діюча речовина вибрана з групи, яка складається з дулоксетину HCl, езомепразолу, рабепразолу натрію, мезаламіну, будесоніду, ламотриджину, декслансопразолу, панкреатину, панкреліпази, дивальпроату натрію, омепразолу, ланзопразолу, диклофенаку натрію, вальпроєвої кислоти, холіну фенофібрату, диданозину, аспірину, бісакодилу, напроксену, еритроміцину, натрій рабепразолу, аденовірусної вакцини типу 4, кальцитоніну, дарапладибу, месалзину, алендронової кислоти, епротирому, NE-F (нефритогенного фактора), глатирамеру, CH-1504, бісфосфонатної (золедронова кислота) сполуки, меркаптаміну, ларазотиду, перорального інсуліну і їх сумішей або комбінацій.
4. Композиція за п. 1, в якій кишковорозчинна система включена в композицію в формі, яка вибрана з групи, яка складається з покриття, шару, матриці та їх комбінацій.
5. Композиція за п. 1, в якій кишковорозчинна система додатково містить компоненти, що вибрані з групи, яка складається з ацетатсукцинату гідроксипропілметилцелюлози на водній і органічній основі, полівінілацетатфталату і аніонних співполімерів метакрилової кислоти і етилакрилату.
6. Композиція за п. 1, яка додатково містить розпушувач, вибраний з групи, яка складається з набухаючого матеріалу, супердезінтегранту і їх сумішей або комбінацій.
7. Композиція за п. 1, яка додатково містить бар'єрний матеріал, розташований між діючою речовиною і засобом захисту від спирту.
8. Композиція за будь-яким з попередніх пунктів, де кишковорозчинна система і засіб захисту від спирту поміщені в комбінацію матеріалів або полімерів, комбінованих в суміші ексципієнтів, або поміщені в єдину полімерну систему і поміщені в шарі, покритті або сформовані в матрицю.
9. Композиція за п. 6, де кишковорозчинна система і засіб захисту від спирту представлені як покриття з єдиною полімерною системою.
10. Композиція за будь-яким з попередніх пунктів для застосування в лікуванні захворювання, де лікування включає:
ідентифікацію пацієнта, чутливого до супутнього прийому алкоголю протягом періодів часу, коли діюча речовина буде знаходитися в шлунку пацієнта;
вибір складу з кишковорозчинним покриттям з діючою речовиною, стійкою до спирту, придатного для лікування захворювання в порівнянні з комерційно еквівалентним складом; і
введення пацієнтові, що страждає на захворювання, складу з кишковорозчинним покриттям з діючою речовиною, стійкою до спирту.
11. Композиція за будь-яким з попередніх пунктів, де діюча речовина, кишковорозчинна система і засіб захисту від спирту на органічній основі скомбіновані для утворення частинок або гранул.
Текст
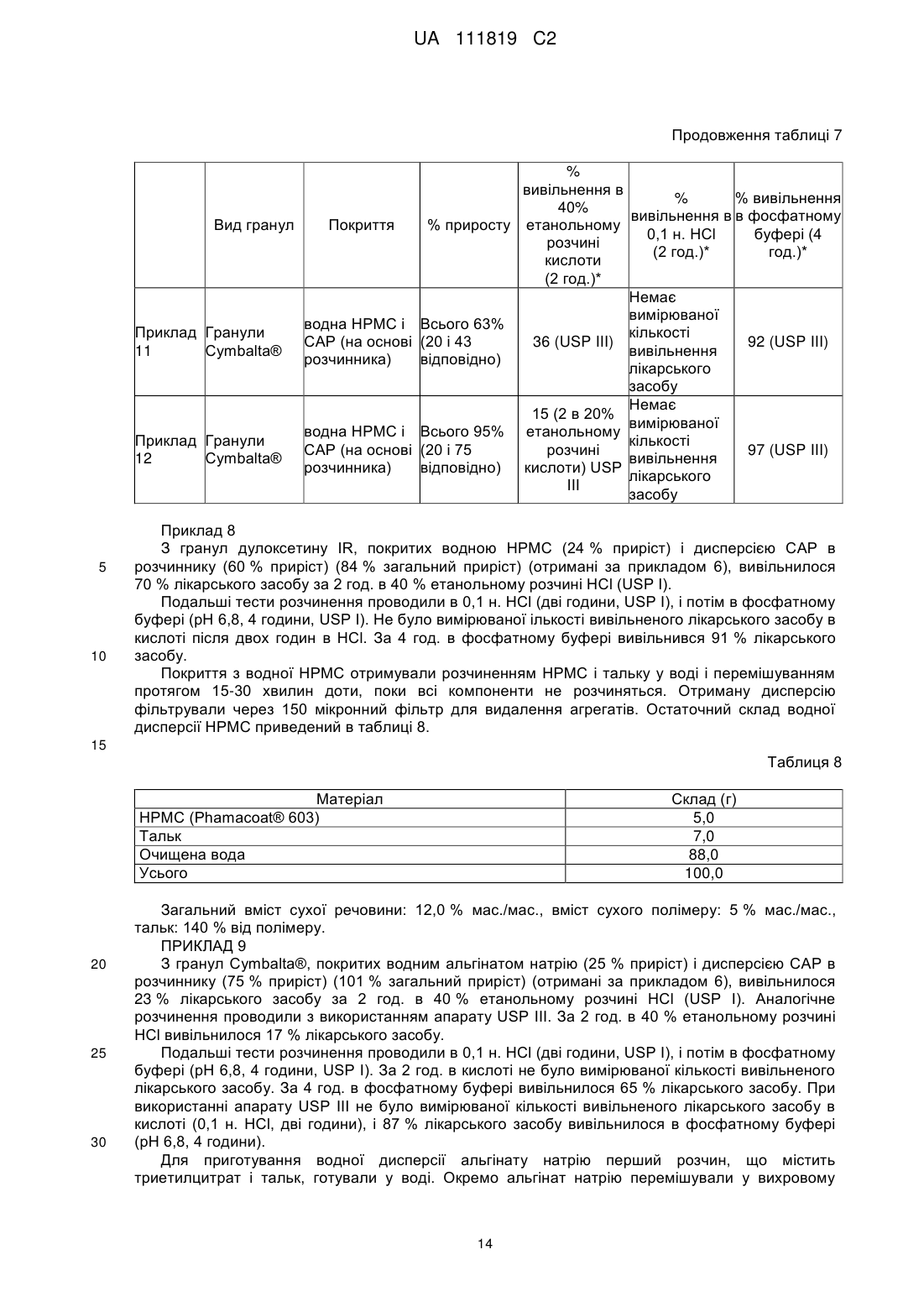
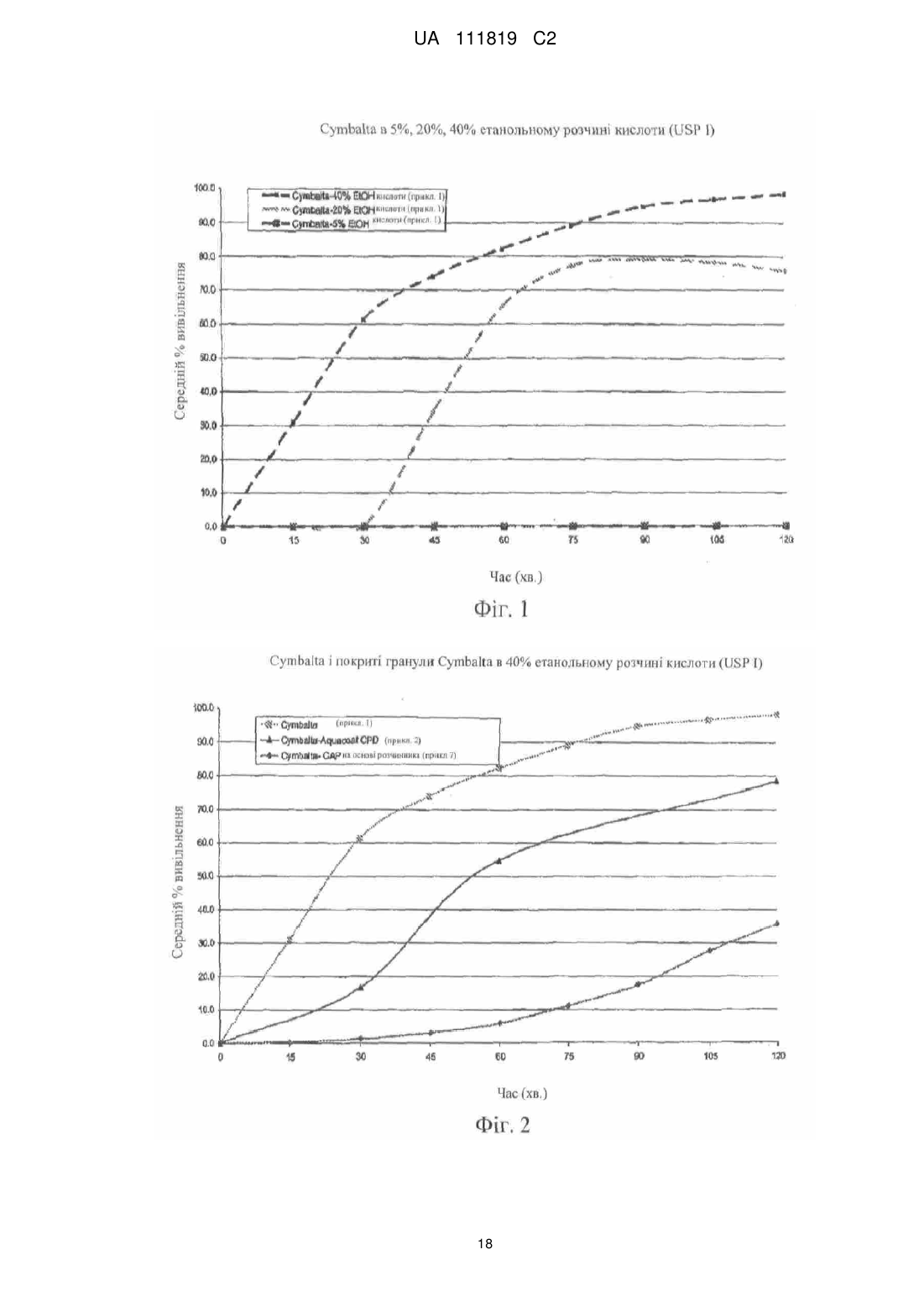
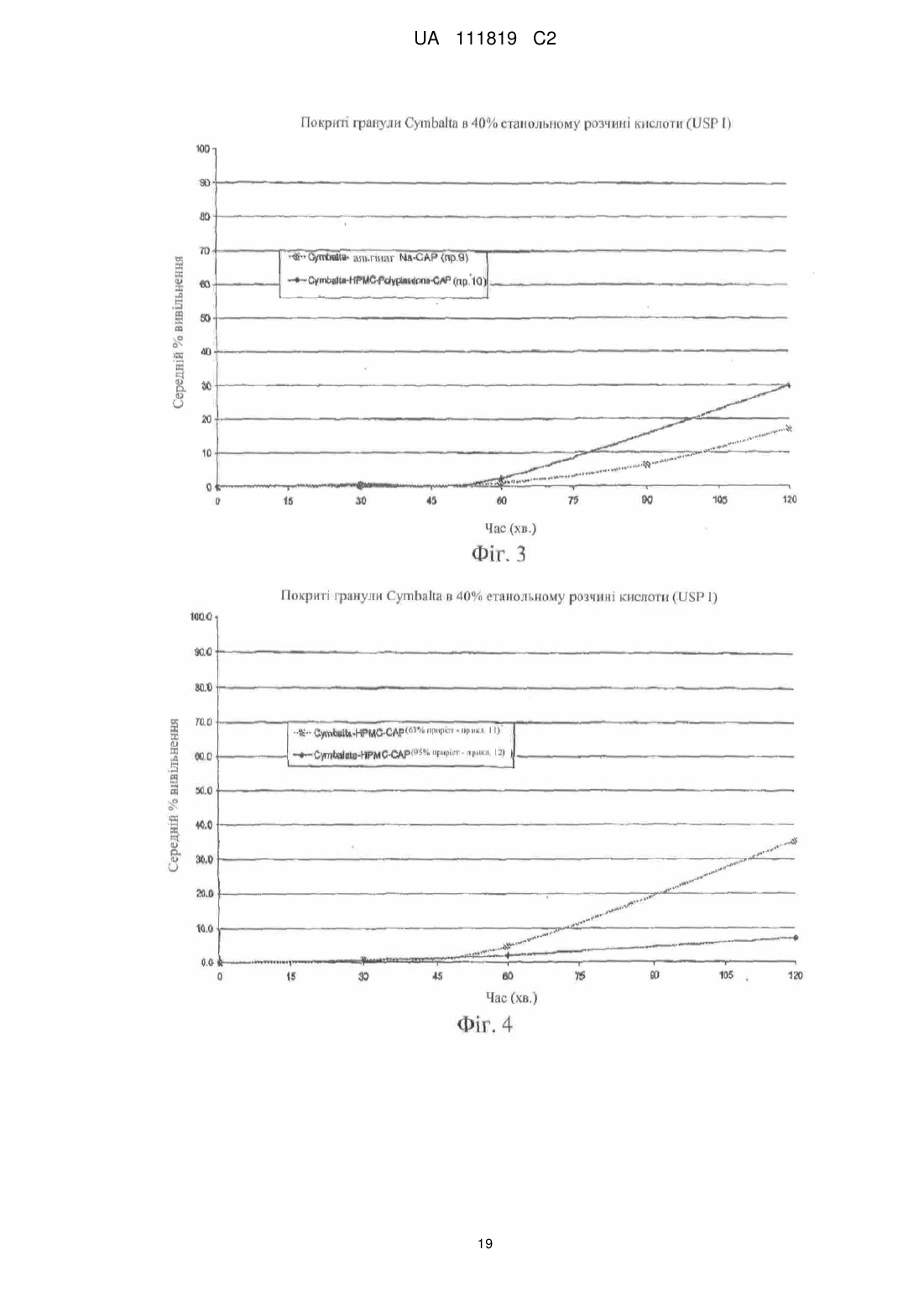
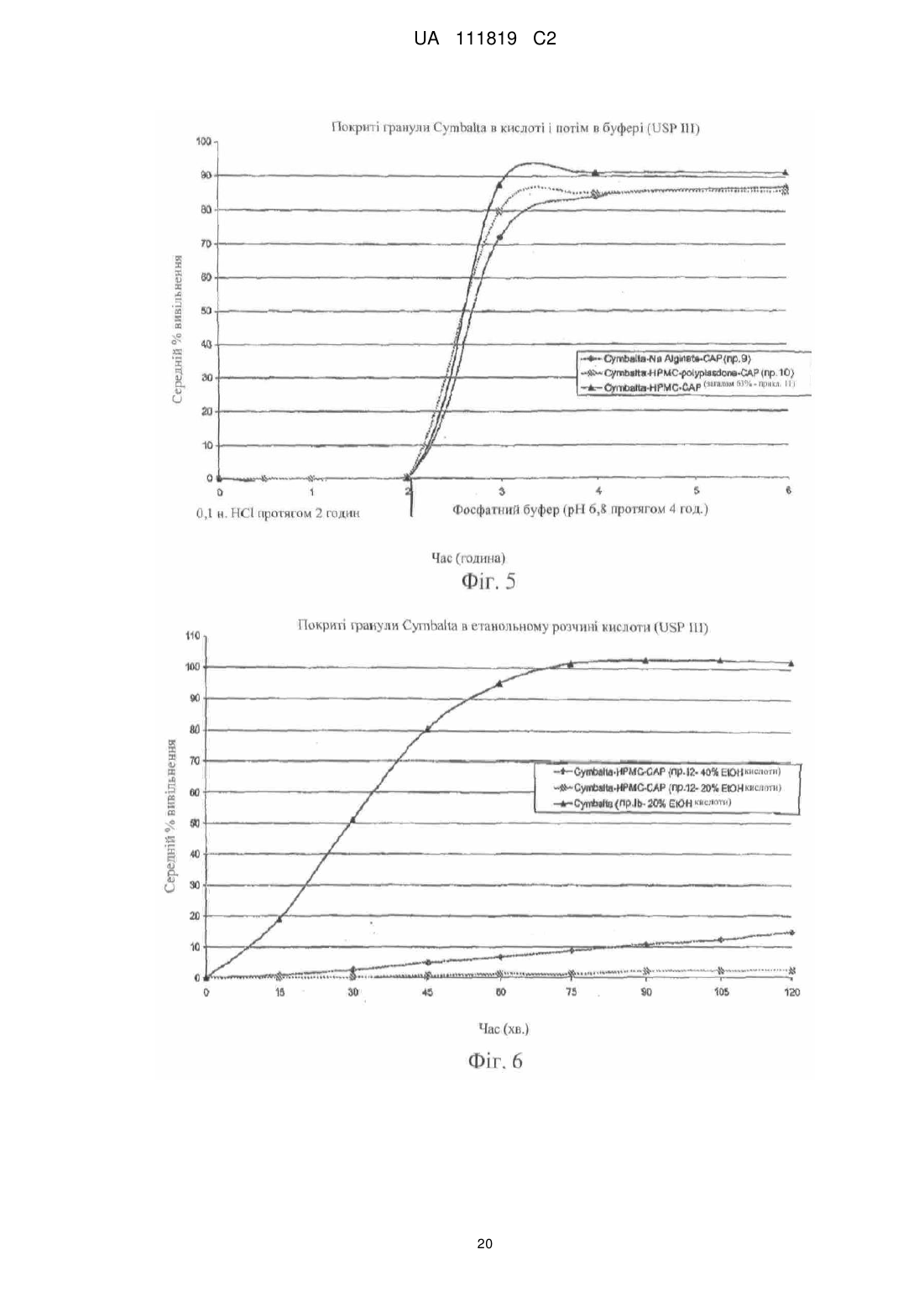
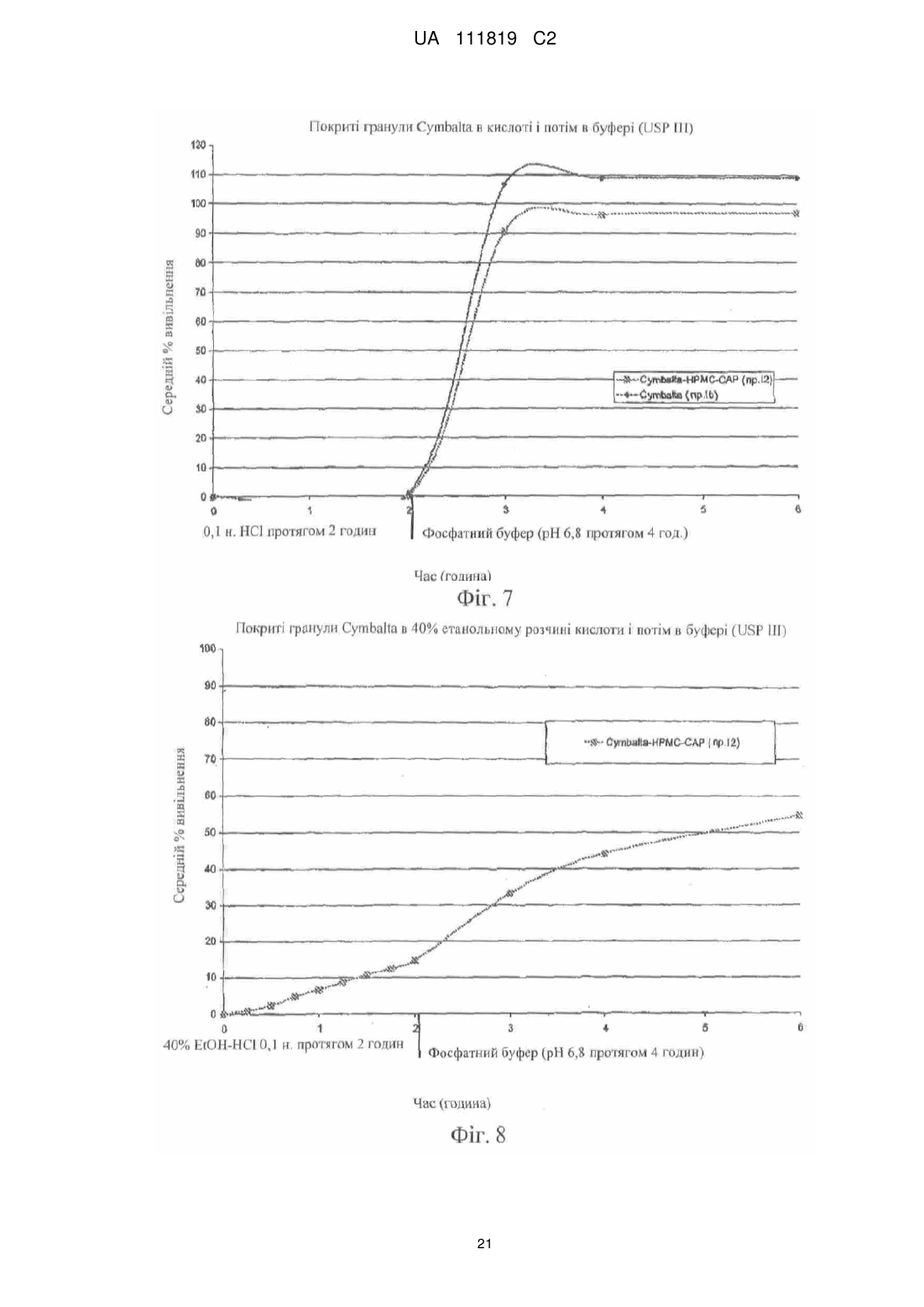
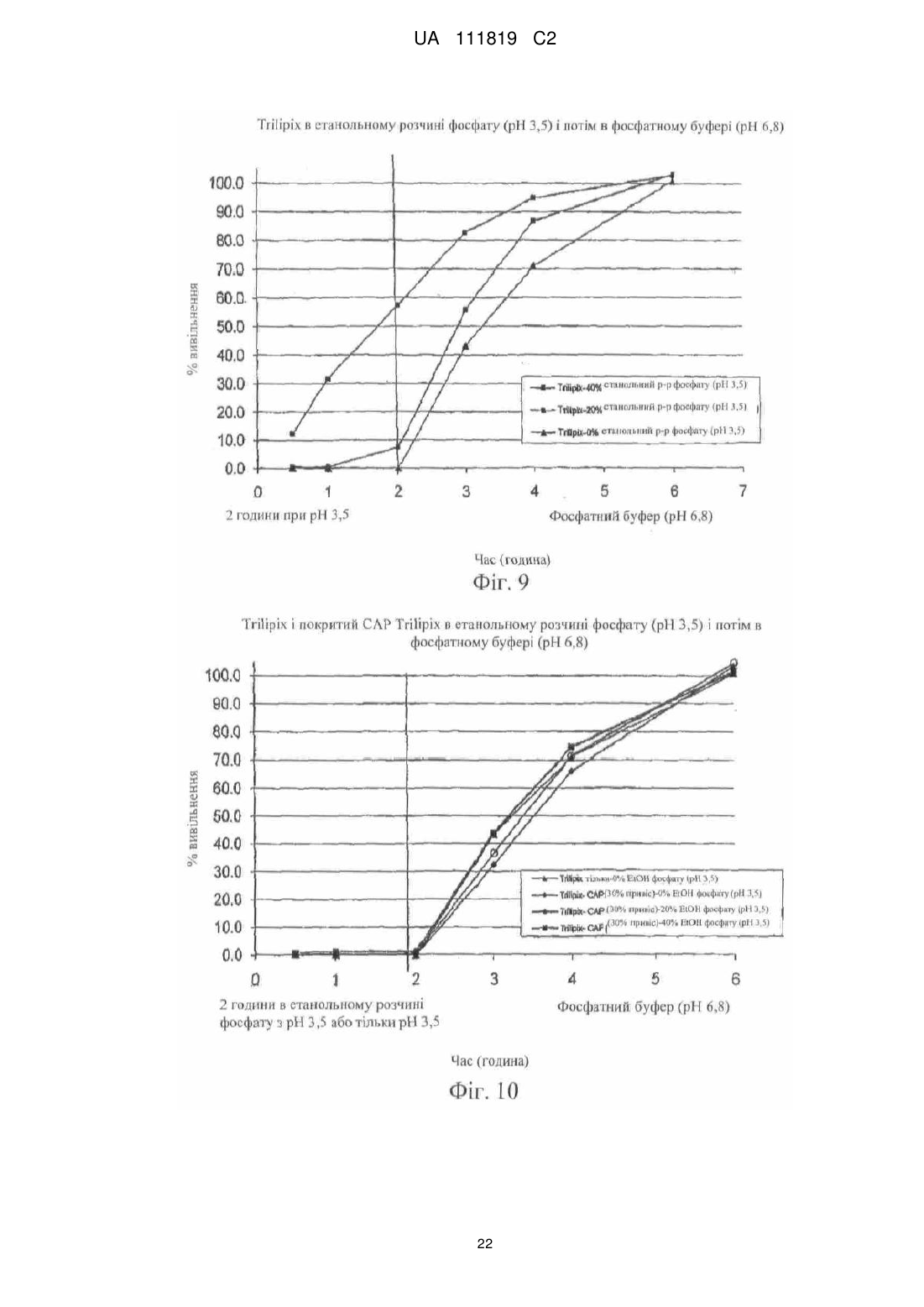
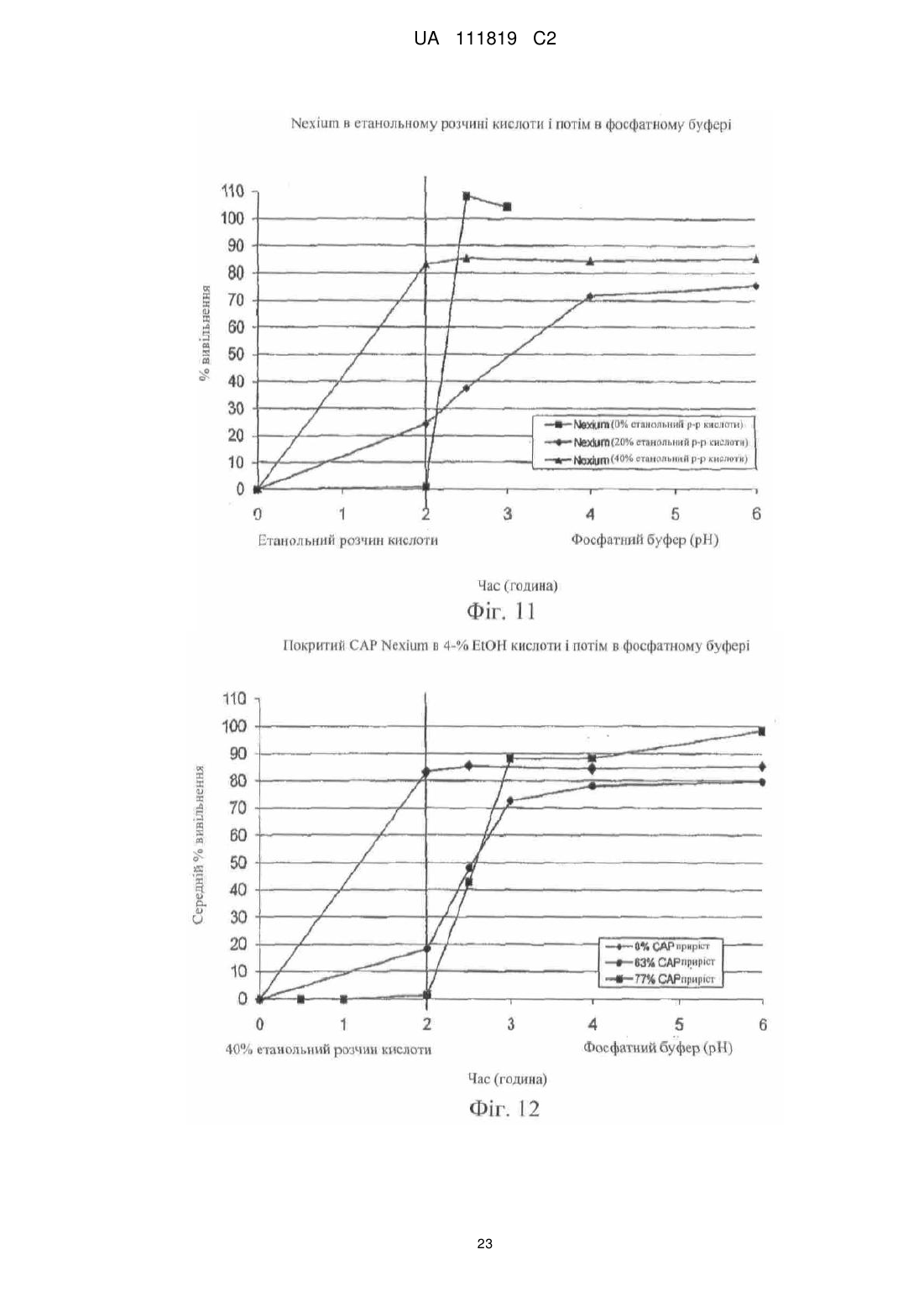
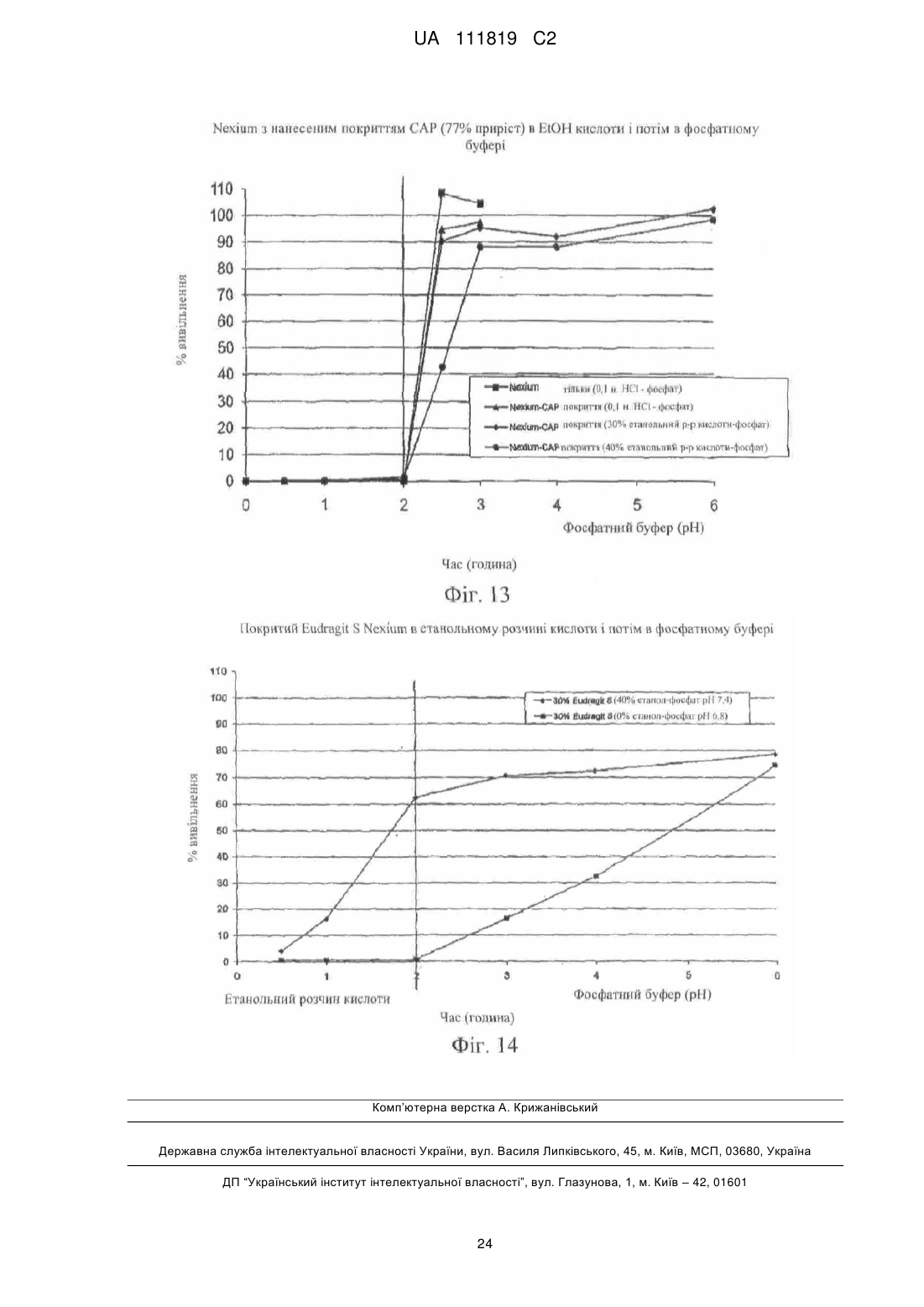
Реферат: Винахід належить до фармацевтики і медицини та стосується стійкої до спирту фармацевтичної композиції, яка містить діючу речовину, кишковорозчинну систему і засіб захисту від спирту, де кишковорозчинна система і засіб захисту від спирту включають ацетатфталат целюлози на органічній основі і де засіб захисту від спирту присутній в лікарській формі в кількості, яка забезпечує приріст від 10 % до 500 %. UA 111819 C2 (12) UA 111819 C2 UA 111819 C2 5 10 15 20 25 30 35 40 45 50 55 60 Рівень техніки Непередбачене швидке вивільнення лікарського засобу за короткий період часу повністю або значної частини лікарського засобу, що міститься в лікарській формі, називають "демпінгом дози". Демпінг дози представляє значний ризик для пацієнтів через питання безпеки і/або зменшену ефективність, особливо, в лікарській формі з контрольованим вивільненням, в якій діюча речовина препарату може бути присутньою в досить великих кількостях. У цих лікарських формах з контрольованим вивільненням ступінь лікарського засобу, що вивільнився з лікарської форми, контролюється механізмом контролю швидкості вивільнення. Звичайні механізми контролю ступеню вивільнення включають, наприклад, набухаючі полімери, гель-матриці і полімерні покриття. Порушення або неефективність механізму контролю ступеня вивільнення являє собою ймовірну причину демпінгу дози. Імовірність демпінгу дози для деяких продуктів з контрольованим вивільненням при введенні з їжею була визнана більше двадцяти років тому. Див. Hendeles L, Wubbena Р, Weinberger M. Food-induced dose dumping of once-a-day theophylline. Lancet. 22: 1471 (1984). У доповнення до їжі, присутність спирту може поставити під загрозу механізми контролю ступеня вивільнення з лікарських форм з контрольованим вивільненням. Деякі лікарські форми з контрольованим вивільненням з одними механізмами контролю ступеня вивільнення більш схильні до демпінгу дози в присутності спирту, ніж з іншими механізмами контролю ступеня вивільнення. У 2005 році Управління США по нагляду за якістю харчових продуктів і лікарських засобів (FDA) поставило вимогу виведення декількох лікарських з ринку або поставило вимогу внесення змін в попереджувальні етикетки через вплив етанолу на склади лікарського засобу з контрольованим вивільненням. Наприклад, FDA поставило вимогу від Purdue Pharma of Stamford, CT зняти капсули пролонгованого вивільнення Palladone® (гідроморфону гідрохлорид) з ринку, оскільки фармакокінетичне дослідження показало, що, коли Palladone® приймався зі спиртом, його склад з пролонгованим вивільненням порушувався, що приводило до демпінгу дози. (див. FDA прес-реліз від 13 липня 2005 року). FDA прийшло до висновку, що відношення загального ризику до профілю користі препарату Palladone® було несприятливим через його сприйнятливість до спирт-індукованого демпінгу дози. Рішення FDA було основане, зокрема, на фармакокінетичному дослідженні на здорових людях (з використанням налтрексонової блокади), яке показало, що спільний прийом Palladone® з 240 мл (8 унцій) 40% (80 встановлених градусів) спирту приводить до середньої пікової концентрації гідроморфону приблизно в шість разів більшої, ніж при прийомі з водою. Крім того, у одного пацієнта в цьому дослідженні спостерігалося 16-кратне збільшення, коли лікарський засіб приймався з 40% спиртом в порівнянні з водою. Це дослідження також показало, що 8 унцій 4% спирту (що еквівалентно 2/3 від звичайної порції пива) може у деяких пацієнтів привести майже до двократного піка концентрації гідроморфону в плазмі, ніж при прийомі препарату з водою. FDA Alert for Healthcare Professionals (July 2005): Hydromorphone Hydrochloride Extended-Release Capsules (marketed as Palladone®).http://www.fda.gov/cder/drug/InfoSheets/HCP/hydromorphoneHCP.pdf. Тест на резистентність демпінгу дози від спирту in vivo не є переважним підходом через потенційну шкоду, яку тест може представляти для людини. Переважним підходом згідно з FDA є тест розчинення in vitro в присутності 40% етанолу. На засіданні Консультативного комітету по фармації (Pharmaceutical Sciences Advisory Committee) 26 жовтня 2005 року OPS (Управління фармації) з CDER (Центр по оцінці і дослідженнях лікарських засобів) представило дані, які свідчать про те, що в сприйнятливій до спирту лікарській формі з контрольованим вивільненням більш висока концентрація етанолу (наприклад, 40%), ймовірно, спричиняє більш швидке вивільнення лікарського засобу, ніж більш низька концентрація етанолу (наприклад, 20% або 4%). Це може бути так чи ні залежно від особливостей складу з контрольованим вивільненням. (Див. Presentations at the Pharmaceutical Sciences Advisory Committee Meeting Oct. 26, 2005). Відповідно, Відділ Біоеквівалентності - 2 Управління лікарських засобів-дженериків CDER/FDA 13 травня 2009 року на семінарі AAPS, Фізичної фармації і біофармацевтики, опублікував рекомендований тест розчинення для спирт-індукованого демпінгу дози дженерикових MR пероральних лікарських засобів. Рекомендоване дослідження розчинення призначене для порівняння розчинення дженерикового (тестованого) продукту і відповідного стандартного лікарського засобу (reference listed drug). Умови для розчинення включають середовище 0,1 н. HCl з різною кількістю етанолу (об./об.), доданого до наступних процентних вмістів етанолу в середовищі: 0,0%, 5,0%, 20% і 40%. Протоколи, подібні до цих визначених дослідженням розчинення, були прийняті для встановлення надійності стійкої до спирту фармацевтичної композиції за даним винаходом. 1 UA 111819 C2 5 10 15 20 25 30 35 40 45 50 55 60 Щонайменше одна спроба була здійснена для отримання складу з контрольованим вивільненням, стійкого до етанол-індукованого демпінгу дози. У опублікованій заявці на патент США № 2007/0212414, поданій Penwest Pharmaceuticals Co., °F Patterson NY (включена в даний опис за допомогою посилання), заявляється спосіб запобігання демпінгу дози лікарського засобу в присутності етанолу шляхом надання пацієнту здатності переробляти етанол під час лікування ефективною кількістю лікарського засобу в формі стійкого до етанолу складу з уповільненим вивільненням. Препарат і система доставки з уповільненим вивільненням включають щонайменше одну гетерополісахаридну смолу, щонайменше одну гомополісахаридну смолу, і щонайменше один фармацевтичний розріджувач. Цей стійкий до етанолу склад з уповільненим вивільненням заявляється по суті таким, що зберігає свій профіль розчинення з уповільненим вивільненням в присутності етанолу. Існує необхідність в даній галузі техніки в кишковорозчинних покритих фармацевтичних складах, які стійкі до етанол-індукованого демпінгу дози. Суть винаходу Винахід стосується стійкої до спирту фармацевтичної композиції, де фармацевтична композиція включає діючу речовину, що має кишковорозчинний шар, стійкий до деградації або розчинення при рН меншому ніж 5,5, і засіб захисту від спирту в кількості, достатній для запобігання істотному вивільненню діючої речовини в присутності спирту. У іншому аспекті винахід стосується композиції, що включає засіб захисту від спирту, який запобігає вивільненню діючої речовини з композиції, вміщеної в спиртове середовище, в кількості, яка менша кількості діючої речовини, що вивільняється з тієї ж композиції у відсутності засобу захисту від спирту в тому ж спиртовому середовищі. Також описаний спосіб лікування захворювання діючою речовиною шляхом введення пацієнту, який страждає на захворювання, ефективної кількості стійкої до спирту фармацевтичної композиції, що містить діючу речовину, придатну для лікування даного захворювання. У іншому аспекті винахід стосується стійкої до спирту фармацевтичної композиції, що включає діючу речовину і засіб захисту від спирту, де захищений від спирту склад має аналогічний профіль розчинення in vitro в 40% етанольному розчині кислоти (0,1 н. HCl) протягом 2 годин (USP I або III), і потім в фосфатному буфері з рН 6,8 (USP I або II) протягом 4 годин при порівнянні з комерційно еквівалентним продуктом. Ще в одному аспекті винахід стосується захищеного від спирту складу, який біоеквівалентний комерційно еквівалентному продукту. Короткий опис фігур На фіг. 1 представлений графік залежності середньої вивільненої кількості лікарського засобу, дулоксетину гідрохлориду (% вивільнення), від часу (хв.) в 5%, 20% і 40% етанольному розчині кислоти з комерційно доступних гранул Cymbalta®, які не мають покриття (приклад 1). На фіг. 2 представлений графік залежності середньої вивільненої кількості лікарського засобу, дулоксетину гідрохлориду (% вивільнення), від часу (хв.) в 40% етанольному розчині кислоти (1) з комерційно доступних гранул Cymbalta®, які не мають покриття (приклад 1); (2) гранул Cymbalta®, покритих CAP на водній основі (AQUACOAT®-CPD, що виробляється FMC Biopolymer of Philadelphia, PA) (приклад 2C); і (3) гранул Cymbalta®, покритих дисперсією CAP на органічній основі (приклад 7). На фіг. 3 представлений графік залежності вивільненої кількості лікарського засобу, дулоксетину (% вивільнення), від часу (хв.) в 40% етанольному розчині кислоти з гранул Cymbalta®, покритих водним альгінатом натрію і дисперсією CAP на органічній основі (приклад 9), і гранул Cymbalta®, покритих водним HPMC/Polyplasdone® XL і дисперсією CAP на органічній основі (приклад 10). На фіг. 4 представлений графік залежності вивільненої кількості лікарського засобу, дулоксетину (% вивільнення), від часу (хв.) в 40% етанольному розчині кислоти з гранул Cymbalta®, покритих водною HPMC і дисперсією CAP на органічній основі (приклад 11), і гранул Cymbalta®, покритих водною HPMC і дисперсією CAP на органічній основі (приклад 12). На фіг. 5 представлений графік залежності вивільненої кількості лікарського засобу, дулоксетину (% вивільнення), від часу (хв.) наступних зразків в 0,1 н. HCl (2 год.) і фосфатному буфері (рН 6,8, 4 год.) в USP III (1) гранул Cymbalta®, покритих водним альгінатом натрію і дисперсією CAP на органічній основі (приклад 9); гранул Cymbalta® покритих водним HPMC/Polyplasdone® XL і дисперсією CAP на органічній основі (приклад 10); і (3) гранул Cymbalta®, покритих водною HPMC і дисперсією CAP на органічній основі (приклад 11). На фіг. 6 представлений графік залежності для (1) комерційно доступних гранул Cymbalta®, які не мають покриття в 20% етанольному розчині кислоти в USP III (приклад 1b); (2) гранул 2 UA 111819 C2 5 10 15 20 25 30 35 40 45 50 55 60 Cymbalta®, покритих водною HPMC і дисперсією CAP на органічній основі в 20% етанольному розчині кислоти в USP III (приклад 12); (3) гранул Cymbalta®, покритих водною HPMC і дисперсією CAP на органічній основі в 40% етанольному розчині кислоти в USP III (приклад 12). На фіг. 7 представлений графік залежності вивільненої кількості лікарського засобу, дулоксетину (% вивільнення), від часу (хв.) для наступних зразків в 0,1 н. HCl (2 год.) і фосфатному буфері (рН 6,8, 4 год.) в USP III (1) комерційно доступних гранул Cymbalta®, що не мають покриття (приклад 1); і (2) гранул Cymbalta®, покритих водною HPMC і дисперсією CAP на органічній основі (приклад 12) На фіг. 8 представлений графік залежності вивільнення в % дулоксетину в 0,1 н. HCl/40% етанольному розчині кислоти (2 години), і потім в фосфатному буфері (4 години) складу, описаного в прикладі 12. На фіг. 9 представлений графік залежності вивільнення в % фенофіброєвої кислоти в етанольному розчині фосфату (рН 3,5) протягом 2 годин, і потім в фосфатному буфері (рН 6,8) з TriLipix®, описаного більш детально в прикладі 13. На фіг. 10 представлений графік залежності вивільнення в % фенофіброєвої кислоти в етанольному розчині фосфату (рН 3,5) протягом 2 годин, і потім в фосфатному буфері (рН 6,8) зі складу TriLipix®, покритого відповідно до варіанту здійснення винаходу, описаного більш детально в прикладі 13. На фіг. 11 представлений графік залежності вивільнення в % езомепразолу магнію з гранул Nexium® в 0,1 н. HCl/40% етанольному розчині кислоти (2 години), і потім в фосфатному буфері (4 години) зі складу, описаного в прикладі 13. На фіг. 12 представлений графік залежності вивільнення в % езомепразолу магнію з гранул Nexium®, покритих 63% і 77% CAP в 0,1 н. HCl/40% етанольному розчині кислоти (2 години), і потім в фосфатному буфері (4 години). На фіг. 13, представлений графік залежності вивільнення в % езомепразолу магнію з гранул Nexium® і покритих CAP гранул Nexium® в 0,1 н. HCl, і потім в фосфатному буфері (4 години). На фіг. 14 представлений графік залежності вивільнення в % езомепразолу магнію з гранул Nexium®, покритих 30% Eudragit S в 0,1 н. HCl/40% етанольному розчині кислоти (2 години), і потім в фосфатному буфері (4 години). Докладний опис винаходу FDA було приписано, що для лікарських форм з контрольованим вивільненням тест in vitro для спирт-індукованого демпінгу дози може бути рекомендованим як звичайний характеристичний тест. Ці тести будуть стосуватися не тільки опіоїдів, таких як гідроморфон морфіну, вони будуть рекомендовані для конкретних інших лікарських засобів, наприклад, але, не обмежуючись, лікарських засобів з вузьким терапевтичним індексом або лікарських засобів, для яких демпінг доза призводить до згубних наслідків з високою C max або низькою Cmin, або лікарських засобів, для яких демпінг може призвести до несприятливих токсикологічних наслідків. FDA віддає перевагу складам, стійким до етанолу по виконанню, а не просто підтверджуючим те, що в дослідженні in vivo не відбувається демпінг дози (див. Summary of FDA's position on alcohol-induced dose dumping as presented at the Pharmaceutical Sciences Advisory Committee Meeting Oct. 26, 2005). FDA запропонувало провести тести розчинення in vitro лікарських форм з контрольованим вивільненням протягом двох годин в різних концентраціях етанольного розчину HCl (0,1н.), такого як 5% етанольний розчин HCl (0,1н.), 20% етанольний розчин HCl (0,1н.) і 40% етанольний розчин HCl (0,1н.), з відбором проб кожні 15 хвилин і, коли необхідно, з подальшим витримуванням в розчині фосфатного буфера при рН 6,8 протягом чотирьох (4) годин. Умови буфера визначаються прийнятними на основі лікарської форми і включають апарат I Фармакопеї США (USP) (сито, 40 меш), швидкість лопатей 75 об/хв. (об'єм середовища: 900 мл при 37ºC), з масою, основаною на кількості, еквівалентній 60 мг діючої речовини, або USP III (40 меш), об'єм середовища 250 мл 37ºC, з масою, основаною на кількості, еквівалентній 15 мг діючої речовини. (Див. Dissolution Testing: An FDA Perspective, AAPS Workshop, Physical Pharmacy and Biopharmaceutics, Division of Bioequivalence-2, Office of Generic Drugs, CDER/FDA, 13 May 2009). Подібний тест використовувався для дослідження фармацевтичних складів за даним винаходом. Згідно з Семінаром AAPS 2009 р., FDA не вимагає профілів розчинення в декількох середовищах для DR продуктів. У одному аспекті даний винахід направлений на такі діючі речовини, для яких не треба допускати розчинення в шлунку, наприклад, через те, що вони не всмоктуються, або вони можуть піддаватися кислотній деградації, або вони можуть подразнювати шлунок, але вони розчиняються, коли лікарська форма досягає більш нейтрального рН, такий як, наприклад, в нижній або тонкій кишці. Як правило, ці діючі речовини вимагають фармацевтичного складу, 3 UA 111819 C2 5 10 15 20 25 30 35 40 45 50 55 який запобігає розчиненню в шлунку – які звичайно називаються кишковорозчинними складами ("EC") або складами з відстроченим вивільненням ("DR"). На відміну від цих складів інші склади називаються продуктами "з пролонгованим вивільненням ER або XR", "з контрольованим вивільненням CR", "однократними щоденними" або "один раз на день" (див., наприклад, COREG® CR (один раз на день карведилолу фосфат, GlaxoSmithKline) і ADDERALL® XR (змішані солі амфетаміну, декстроамфетаміну, Shire US Inc.)). Ці кишковорозчинні склади спеціально розроблені для того, щоб вивільняти частину діючої речовини в шлунку, а також вивільняти діючу речовину в тонкому кишечнику в контрольованому режимі. Незважаючи на те, чи називається продукт "з контрольованим вивільненням", "пролонгованої дії", "однократним щоденним" або "один раз на день" для цілей даного винаходу, критичним визначенням є те, чи дозволяє чи ні фармацевтичний склад вивільнення діючої речовини в шлунку. Відповідно до одного звичайного варіанту здійснення даний винахід направлений на такі діючі речовини, які не повинні значно розчинятися в шлунку. Термін "демпінг", що використовується в даному описі, описує як катастрофічне вивільнення діючої речовини, так і вивільнення, яке не буде біоеквівалентним відповідно до стандартів FDA для параметрів Cmax, Tmax і/або AUC. Управління США по нагляду за якістю харчових продуктів і лікарських засобів (FDA) визначило біоеквівалентність як "відсутність істотної відмінності в швидкості і ступені, з якими діюча речовина або її активний компонент в фармацевтичних еквівалентах або фармацевтичних альтернативах стає доступною в місці дії лікарського засобу при введенні в тій же молярній дозі в тих же умовах в належним чином спланованому дослідженні". (FDA, 2003) Іншими словами, FDA вважає два продукти біоеквівалентними, якщо 90% CI кожного або всіх відносно середніх Cmax, AUC(0-t) і AUC(0-∞) тестованого складу по відношенню до еталонного складу повинно бути в межах від 80,00% до 125,00%. Якщо дослідження біоеквівалентності не можуть бути виконані через можливу шкоду пацієнту, тоді тест розчинення in vitro тестованого складу порівнюється з еталонним складом (наприклад, комерційно еквівалентним продуктом). Це є відповідним визначенням FDA того, чи є тестований склад (наприклад, захищений від спирту склад за даним винаходом) еквівалентним еталонному складу (наприклад, комерційно еквівалентному продукту). При порівнянні тестованого і еталонного складів профілі розчинення повинні бути порівняні з використанням коефіцієнта подібності (f2). Коефіцієнт подібності являє собою логарифм оберненого квадратного кореня суми квадратів розходжень і є ступенем подібності розчинення в процентах (%) між двома кривими. Два профілі розчинення вважаються «подібними», коли значення f2≥50. Див. Waiver of In vivo Bioavailability and Bioequivalence Studies for ImmediateRelease Solid Oral Dosage Forms Based on а Biopharmaceutics Classification System, U.S. Department of Health and Human Services, Food and Drug Administration Center for Drug Evaluation and Research (CDER), August 2000. Існує ряд відомих складів для запобігання вивільненню діючої речовини зі складу під час її проходження через шлунок. Приклади включають склади, розкриті в патентах США 7011847, 6159501, 5273760; і опублікованих патентних заявках США 2008/0085304, 2004/0170688 і 2008/0226711, включених в даний опис за допомогою посилання. Використовувані в цих системах матеріали включають, наприклад, жирні кислоти, віск, шелак і пластики. Як правило, матеріали, які створюють такі системи, розділяються на дві групи: на водній основі і на основі систем розчинників. Більшість кишковорозчинних систем працює шляхом надання поверхні, яка є стабільною при надто кислому рН шлунка, але швидко розпадається при менш кислому (відносно більш основному) рН. Наприклад, кишковорозчинні системи не будуть розчинятися в кислих шлункових соках (близько рН 3), але вони будуть розчинятися при більш високому рН (приблизно вище рН 5, такому як 5,5) середовища, присутнього в тонкій кишці. Будь-яка система, яка запобігає розчиненню діючої речовини в шлунку, включаючи, але, не обмежуючись ними, приведені вище, на яку посилаються в даному описі, називається узагальнено "кишковорозчинні системи". Необмежувальні приклади кишковорозчинних систем включають HPMC-АС на водній і органічній основі: ацетатсукцинат гідроксипропілметилцелюлози-HF (AQOAT, що продається Shin-Etsu Chemical Co., Ltd. of Japan); PVAP: полівінілацетатфталат (SURETERIC® від Colorcon, Inc., Harleysville, PA); CAP на водній основі: ацетатфталат целюлози (AQUACOAT®-CPD від FMC Biopolymer of Philadelphia, PA); CAP на органічній основі: ацетатфталат целюлози (Eastman C-A-P, Eastman Co.), аніонні співполімери метакрилової кислоти і етилакрилату, що продаються під торговою маркою EUDRAGIT® категорій L, S і FS (Evonik Degussa, Darmstadt, DE). 4 UA 111819 C2 5 10 15 20 25 30 35 40 45 50 55 60 Кишковорозчинні системи наносяться на лікарську форму у вигляді шару або покриття, або знаходяться в формі матриці. Кишковорозчинна система являє собою один матеріал або комбінацію матеріалів. Звичайні комерційно доступні фармацевтичні склади, в яких використовується кишковорозчинна система в формі покриття або шару для забезпечування діючої речовини від розчинення в шлунку, включають CYMBALTA® (дулоксетину HCl, Lilly USA, LLC); Nexium® (езомепразол, AstraZeneca LP); ACIPHEX® (рабепразол натрію, Eisai Inc. і Ortho-McNeil-Janssen Pharmaceuticals, Inc.); ASACOL® HD (мезаламін, Procter & Gamble Pharmaceuticals, Inc); LIALDA® (мезаламін, Shire US Inc.); PENTASA® (мезаламін, Shire US Inc); ENTECORT® EC (будесонід в капсулах, AstraZeneca LP); LAMICTAL® XR (ламотриджин в таблетках, GlaxoSmithKline); KAPIDEX® (декслансопразол, Takeda Pharmaceuticals North America, Inc); Creon® (панкреатин в капсулах, Solvay S.A.); ULTRASE® (панкреліпаза в капсулах, Axcan Pharma US); PROTONIX® (пантопразол, Pfizer Inc.); DEPAKOTE® (дивальпроат натрію, Abbott Laboratories); PROLOSEC® (омепразол, AstraZeneca LP); PREVACID® (ланзопразол, Novartis Consumer Health, Inc); ARTHOTEC® (диклофенак натрію, Pfizer Inc.); STAVZOR® (вальпроєва кислота, Noven Therapeutics LLC); TriLipix® (капсули фенофіброєвої кислоти з відстроченим вивільненням, Abbott Laboratories) і VIDEX® EC (диданозин, Bristol-Myers Squibb). Звичайні діючі речовини (як в наявних в продажу продуктах, так і ні), для яких використовується або може використовуватися кишковорозчинний шар для забезпечування діючої речовини від розчинення в шлунку, включають аспірин, бісакодил, напроксен, еритроміцин, рабепразол натрію, аденовірусну вакцину типу 4, кальцитонін, дарапладиб, месалзин, алендронову кислоту, епротиром, NE-F (нефритогенний фактор), глатирамер, CH1504 (неметаболізований антифолат від Chelsea Therapeutics International, Ltd.), ORAZOL® (бісфосфонатна (золедронова кислота) сполука, Merrion Pharmaceuticals), меркаптамін, ларазотид і пероральний інсулін. Даний винахід не обмежується кишковорозчинними лікарськими формами, що продаються в цей час, і передбачається для використання з діючою речовиною, яка сприйнятлива до етаноліндукованого демпінгу. У прикладі варіанту здійснення стійкої до спирту фармацевтичної композиції за даним винаходом використовується "засіб захисту від спирту" для запобігання або сповільненню етанол-індукованого демпінгу діючої речовини з лікарської форми. Засіб захисту від спирту може бути одним матеріалом, наприклад, полімером, або комбінацією матеріалів, наприклад, комбінацією полімерів в розчині ексципієнта. Засіб захисту від спирту міститься в шарі або покритті, або він знаходиться в формі матриці в альтернативних варіантах здійснення. Прийнятні матеріали засобу захисту від спирту включають, але, не обмежуючись ними, ацетатфталат целюлози на органічній основі, співполімери метакрилату амоній, співполімери ефіру метакрилату, співполімери метакрилової кислоти, природний і синтетичний крохмаль, поліалкіленоксиди і природна і синтетична целюлоза, включаючи модифіковану целюлозу, таку як гідроксипропілметилцелюлоза (HPMC), гідроксипропілцелюлоза (HPC) гідроксиметилцелюлоза (HMC), метилцелюлоза (VC), гідроксіетилцелюлоза (HEC) і карбоксиметилцелюлоза (CMC), віск, такий як віск комах і тварин, рослинний віск, мінеральний віск, нафтовий віск і синтетичний віск. У прикладі варіанту здійснення засіб захисту від спирту являє собою ацетатфталат целюлози на органічній основі, що продається під торговою маркою Eastman С-А-Р® або Cellacefate, NF компанією Eastman Chemical Company, Kingsport, TN USA. Засіб захисту від спирту може бути присутнім в складі в кількості, достатній для придання стійкості до спирту в даній концентрації етанолу. Згідно з одним аспектом винаходу засіб захисту від спирту додають до комерційно еквівалентного складу в кількості 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 100%, 150%, 200%, 250%, 300%, 350%, 400%, 450% і 500% по приросту. Фармацевтична композиція за даним винаходом є стійкою до спирту, що основано на відносинах між процентом вивільнення діючої речовини з лікарської форми в спиртовому середовищі або в безспиртовому середовищі після того, як лікарська форма піддається впливу спиртового середовища. У інших прикладах варіантів здійснення даний винахід являє собою стійку до спирту фармацевтичну композицію, яка забезпечує стійкість до етанол-індукованого демпінгу і є біоеквівалентною комерційно еквівалентному складу діючої речовини. Як обговорювалося раніше, з метою кількісного визначення стійкості до етанол-індукованого демпінгу тест розчинення проводили в 5%, 20% і 40% етанольному розчині HCl (див. Посібник FDA, який обговорювався вище) протягом двох годин. Автори додавали також концентрації етанолу 30% і 35%. 5 UA 111819 C2 5 10 15 20 25 30 35 40 45 50 55 60 У іншому експерименті для кількісного визначення стійкості до етанол-індукованого демпінгу проводили два окремих тести розчинення, один в 0,1 н. HCl (2 години, як описано вище), потім інший (з використанням іншого зразка) в фосфатному буфері з рН 6,8 (4 години). Далі були проаналізовані профілі розчинення кожного. У ще одній схемі експерименту для кількісного визначення стійкості до етанол-індукованого демпінгу проводили послідовне розчинення одного і того ж зразка. Цей тест розчинення включав розчинення в етанольному розчині кислоти (2 години), і потім в фосфатному буфері з рН 6,8 (4 години). Послідовні етанольний розчин кислоти і фосфатний буфер призначені для імітації in vivo умов всмоктування людиною спирту одночасно з введенням лікарської форми. Лікарська форма спочатку проходить через який містить спирт/кислий шлунок (середній час знаходження в шлунково-кишковому тракті - 2 год.), і потім проходить через тонкий кишечник, в якому більш нейтральний рН (середній час знаходження в шлунково-кишковому тракті ~ 4 год.). Етанол не вважається присутнім в нижніх відділах кишечнику, оскільки швидко всмоктується в шлунку. Дослідження розчинення проводилися з використанням апарату I Фармакопеї США (сита, 40 меш) при 75 об/хв. [об'єм середовища: 900 мл при 37ºC] з масою 60 мг, еквівалентною діючою; і апарату III Фармакопеї США (40 меш) [об'єм середовища: 250 мл при 37ºC] з масою 15 мг, еквівалентною діючій. Було б небажано, щоб кишковорозчинне покриття складу, який містить діючу речовину, відому як утворюючу токсичні продукти розпаду в шлунку, порушилося при впливі спиртового середовища. Одним з таких продуктів, який має подібну поведінку, є CYMBALTA® (дулоксетин HCl з кишковорозчинним покриттям), що продається Lilly, Inc. Як повідомлялося в The Rearrangement of Duloxetine Under Mineral Acid Conditions, RJ Bopp, AP Breau, TJ Faulkinbury, PC Heath, С Miller, 206th Natl. Am. Che. M. Soc. Meeting; Mar 13 1993, Abstract# 111, дулоксетину HCl швидко піддається сольволізу і перегрупуванню у водній HCl з отриманням 1-(2тієніл)карбінолу, нафтолу і 1-(2-тієніл) 2- і 4-заміщених нафтолів. У цей час вважають, що покритий кишковорозчинним покриттям склад, що містить діючу речовину, про яку невідомо, чи викликає вона токсичні ефекти, коли розчиняється або навіть демпінгує дозу в шлунку, але швидше, наслідком демпінгу дози є субтерапевтичний ефект діючої речовини. Одним з таких прикладів є TriLipix® (фенофіброєва кислота, також стосується холінфенофібрату), виробництва Abbott Laboratories of North Chicago, IL. Abbott провели ряд досліджень, що демонструють, що таблетки фенофіброєвої кислоти з негайним вивільненням мали значно більше (1,4 рази) Cmax, менший (0,67 рази) T max, і варіабельність годування/голодування в порівнянні з Tricor®-145 (фенофібрат). Їх регіоспецифічне дослідження привело до висновку, що для розробки біоеквиваленту складу комерційно доступної таблетки фенофібрату, профіль вивільнення складу, що містить фенофіброєву кислоту (наприклад, TriLipix®), необхідно сповільнити, щоб відповідати більш повільним властивостям поглинання фенофібрату (Tricor®-145) в ШК тракті. Див. TriLipix® SBA Study K LF178P 03 03 KH 05 02 (regiospecific study) page 43. Маючи це на увазі, відповідно до Короткого Обґрунтування Твердження, таблиця 7.2.1.D Медичного огляду TriLipix В демографічних і основних характеристиках для дослідження M05-758 визначено 52,3% цільових пацієнтів TriLipix® як "що п'ють", 7,2% як "що кинули пити", 40,5% як «що не п'ють». Таким чином, якщо дозволити фенофіброєвій кислоті вивільнитися з TriLipix® в шлунку, то наслідком буде етанол-індукований демпінг, що приведе до більш високого Cmax і більш короткого Tmax діючого інгредієнта. У одному варіанті здійснення даний винахід запобігає або сповільнює етанол-індукований демпінг діючої речовини складу до ступеня, коли немає вимірного вивільнення діючої речовини при вміщенні дозованої форми в 40% етанол. Відповідно, засіб захисту від спирту додає стійкості до етанол-індукованого демпінгу, коли не більше ніж 1%, 2%, 5%, 8%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% або 99% діючої речовини вивільняється з лікарської форми в 40% етанолі після 0,25, 0,5, 0,75, 1, 1,25, 1,5, 1,75 або 2 год. Крім того, засіб захисту від спирту додає стійкості до етанол-індукованого демпінгу, коли не більше ніж близько 1%, 2%, 5%, 8%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% або 99% діючої речовини вивільняється з лікарської форми в 35% етанолі після 0,25, 0,5, 0,75, 1, 1,25, 1,5, 1,75 або 2 год. Ще в доповнення, засіб захисту від спирту додає стійкості до етанол-індукованого демпінгу, коли не більше ніж близько 1%, 2%, 5%, 8%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% або 99% діючої речовини вивільняється з лікарської форми в 30% етанолі після 0,25, 0,5, 0,75, 1, 1,25, 1,5, 1,75 або 2 год. Ще засіб захисту від спирту додає стійкості до етанол-індукованого демпінгу, коли не більше ніж близько 1%, 2%, 5%, 8%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 6 UA 111819 C2 5 10 15 20 25 30 35 40 45 50 80%, 85%, 90%, 95% або 99% діючої речовини вивільняється з лікарської форми в 20% етанолі після 0,25, 0,5, 0,75, 1, 1,25, 1,5, 1,75 або 2 год. Ще далі засіб захисту від спирту додає стійкості до етанол-індукованого демпінгу, коли не більше ніж близько 1%, 2%, 5%, 8%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% або 99% діючої речовини вивільняється з лікарської форми в 5% етанолі після 0,25, 0,5, 0,75, 1, 1,25, 1,5, 1,75 або 2 год. У іншому варіанті здійснення винахід стосується складу, який спрямований на запобігання або сповільнення етанол-індукованого демпінгу діючої речовини, де діюча речовина вивільняється в меншій кількості, ніж кількість діючої речовини, що вивільняється з комерційної еквівалентного складу. Під "комерційно еквівалентним складом або продуктом" розуміється те, що склад діючої речовини схвалений FDA для використання, але не має ознаки засобу захисту від спирту за даним винаходом. Наприклад, згідно з даним варіантом здійснення винахід направлений на склад, в якому деяка кількість діючої речовини вивільняється в присутності спирту, але ця кількість менша, ніж кількість, яка вивільняється з комерційно еквівалентного складу. Відповідно, засіб захисту від спирту додає стійкості до етанол-індукованого демпінгу, коли не більше ніж 1%, 2%, 5%, 8%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% або 99% діючої речовини вивільняється з лікарської форми в 40% етанолі після 0,25, 0,5, 0,75, 1, 1,25, 1,5, 1,75 або 2 год. в порівнянні з кількістю діючої речовини, що вивільняється з комерційно еквівалентного складу в тій же концентрації етанолу за той же час. Крім того, засіб захисту від спирту додає стійкості до етанол-індукованого демпінгу, коли не більше ніж близько 1%, 2%, 5%, 8%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% або 99% діючої речовини вивільняється з лікарської форми в 35% етанолі після 0,25, 0,5, 0,75, 1, 1,25, 1,5, 1,75 або 2 год. в порівнянні з кількістю діючої речовини, що вивільняється з комерційно еквівалентного складу в тій же концентрації етанолу за той же час. Ще крім того, засіб захисту від спирту додає стійкості до етанол-індукованого демпінгу, коли не більше ніж близько 1%, 2%, 5%, 8%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% або 99% діючої речовини вивільняється з лікарської форми в 30% етанолі після 0,25, 0,5, 0,75, 1, 1,25, 1,5, 1,75 або 2 год. в порівнянні з кількістю діючої речовини, що вивільняється з комерційно еквівалентного складу в тій же концентрації етанолу за той же час. Ще засіб захисту від спирту додає стійкості до етанол-індукованого демпінгу, коли не більше ніж близько 1%, 2%, 5%, 8%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% або 99% діючої речовини вивільняється з лікарської форми в 20% етанолі після 0,25, 0,5, 0,75, 1, 1,25, 1,5, 1,75 або 2 год. в порівнянні з кількістю діючої речовини, що вивільняється з комерційно еквівалентного складу в тій же концентрації етанолу за той же час. Крім того, засіб захисту від спирту додає стійкості до етанол-індукованого демпінгу, коли не більше ніж близько 1%, 2%, 5%, 8%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% або 99% діючої речовини вивільняється з лікарської форми в 5% етанолі після 0,25, 0,5, 0,75, 1, 1,25, 1,5, 1,75, або 2 год. в порівнянні з кількістю діючої речовини, що вивільняється з комерційно еквівалентного складу в тій же концентрації етанолу за той же час. У іншому аспекті винахід стосується складів, в яких доза не демпінгується в спиртовому середовищі, і при подальшому приміщенні в фосфатний буфер (для симуляції змін в рН травного тракту нижче шлунка) мають по суті той же профіль вивільнення в порівнянні з тим же складом при розчиненні в фосфатному буфері, де склад не піддався перед цим впливу етанольного розчину кислоти. У цьому аспекті винаходу склад за винаходом має швидкість вивільнення в фосфатному буфері, яка по суті не залежить від попереднього впливу середовища спирту. У таблиці А приведені деякі комерційно доступні лікарські форми (наприклад, комерційно еквівалентні лікарські форми), які виявилися стійкими в середовищі етанольного розчину кислоти, але при подальшому випробуванні в фосфатному буфері показують зміну в їх швидкості розчинення. 7 UA 111819 C2 Таблиця А Продукт лікарського засобу Дві стадії із середовищем (0Єдина стадія із 2) год. в 0,1 н. середовищем HCl, 40% спирту (0-2) год. в 0,1 н. (2-4) год. в HCl і 40% фосфатному буфері, pH Aciphex DR таблетки (рабепразол натрію) Kapidex DR капсули (декслансопразол) 5 10 15 20 25 30 Піків не виявлено Піків не виявлено Покриття (і неактивні компоненти) Цукрові сфери, карбонат магнію, сукроза, низькозаміщена гідроксипропілцелюлоза, діоксид Вивільнення титану, гідроксипропілцелюлоза, лікарського гіпромелоза 2910, тальк, співполімер засобу на 10 хв. метакрилової кислоти, раніше після поліетиленгліколь 8000, обробки 40% триетилцитрат, полісорбат 80 і спиртом, ніж колоїдний діоксид кремнію. тільки в 0,1 н. Гранула 1: Eudragit L30 D-55 або HCl Eudragit L100-55 Гранула 2: Суміш Eudragit S100 і Eudragit L-100 Колоїдний діоксид кремнію; Істотна кросповідон; гідрована рицинова олія; відмінність в гіпромелоза; лактоза; магнію стеарат; швидкостях співполімер метакрилової кислоти; вивільнення мікрокристалічна целюлоза; повідон лікарського (полівідон) К-30; натрію гідроксид; засобу крохмаль (кукурудзяний); тальк; триетилцитрат Відповідно до цього варіанту винаходу в складі за винаходом доза не демпінгується в спиртовому середовищі, і при подальшому приміщенні в фосфатний буфер демонструє по суті той же in vivo біоеквівалентний фармакокінетичний профіль і/або подібний in vitro профіль розчинення в порівнянні з таким же складом в фосфатному буфері, але які раніше не були схильні до дії середовища спирту. Відповідно, засіб захисту від спирту додає стійкості до етанол-індукованого демпінгу, коли після 2 годин в етанольному розчині кислоти (40% етанолу в 0,1 н. HCl) відсутня вимірювана кількість вивільненої діючої речовини і різниця між кількістю діючої речовини, вивільненою із захищеного від спирту складу за винаходом, і її кількістю, вивільненою з комерційно еквівалентного складу, коли обидва склади потім вміщені в фосфатний буфер з рН 6,8 (4 години), становить 1%, 2%, 5%, 8%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% або 99%. Засіб захисту від спирту додає стійкості до етанол-індукованого демпінгу, коли після 2 годин в етанольному розчині кислоти (35% етанолу в 0,1 н. HCl) відсутня вимірювана кількість вивільненої діючої речовини і різниця між кількістю діючої речовини, вивільненою із захищеного від спирту складу за винаходом, і її кількістю, вивільненою з комерційно еквівалентного складу, коли обидва склади потім вміщені в фосфатний буфер з рН 6,8 (4 години), становить 1%, 2%, 5%, 8%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% або 99%. Засіб захисту від спирту додає стійкості до етанол-індукованого демпінгу, коли після 2 годин в етанольному розчині кислоти (30% етанолу в 0,1 н. HCl) відсутня вимірювана кількість вивільненої діючої речовини і різниця між кількістю діючої речовини, вивільненою із захищеного від спирту складу за винаходом, і її кількістю, вивільненою з комерційно еквівалентного складу, коли обидва склади потім вміщені в фосфатний буфер з рН 6,8 (4 години), становить 1%, 2%, 5%, 8%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% або 99%. Засіб захисту від спирту додає стійкості до етанол-індукованого демпінгу, коли після 2 годин в етанольному розчині кислоти (20% етанолу в 0,1 н. HCl) відсутня вимірювана кількість вивільненої діючої речовини і різниця між кількістю діючої речовини, вивільненою із захищеного від спирту складу за винаходом, і її кількістю, вивільненою з комерційно еквівалентного складу, 8 UA 111819 C2 5 10 15 20 25 30 35 40 45 50 55 60 коли обидва склади потім вміщені в фосфатний буфер з рН 6,8 (4 години), становить 1%, 2%, 5%, 8%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% або 99%. Засіб захисту від спирту додає стійкості до етанол-індукованого демпінгу, коли після 2 годин в етанольному розчині кислоти (5% етанолу в 0,1 н. HCl) відсутня вимірювана кількість вивільненої діючої речовини і різниця між кількістю діючої речовини, вивільненою із захищеного від спирту складу за винаходом, і її кількістю, вивільненою з комерційно еквівалентного складу, коли обидва склади потім вміщені в фосфатний буфер з рН 6,8 (4 години), становить 1%, 2%, 5%, 8%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% або 99%. Відповідно до одного прикладу варіанту здійснення стійкої до спирту фармацевтичної композиції за винаходом, де лікарська форма являє собою множину частинок, засіб захисту від спирту наноситься у вигляді шару або покриття при виготовленні лікарської форми. Не має значення, що покриття або шар, сформований із засобом захисту від спирту, може мати невеликі або мікроскопічні отвори, тріщини, щілини або дірки. Навпаки, визначальною ознакою є те, чи додає покриття або шар складу стійкості до етанол-індукованого демпінгу дози. У варіанті здійснення, де засіб захисту від спирту являє собою шар або покриття, засіб захисту від спирту знаходиться зовні від діючої речовини, незалежно від того, чи є діюча речовина частиною ядра, шару або диспергована в матриці. Наприклад, в одному варіанті здійснення засіб захисту від спирту може бути нанесений у вигляді покриття безпосередньо на діючу речовину в об'ємній формі. Наприклад, конкретний лікарський засіб в об'ємній формі має розмір частинок більший ніж 10 мкм. Ці об'ємні частинки лікарського засобу можуть бути безпосередньо покриті засобом захисту від спирту, і потім спресовані в таблетку, на яку наноситься кишковорозчинне покриття. Альтернативно, захищені від спирту покриті частинки лікарського засобу можуть бути вміщені в матрицю, яка виконана з кишковорозчинного матеріалу або яка сама по собі покрита кишковорозчинним матеріалом. У подальшому варіанті здійснення матеріал, який включає засіб захисту від спирту, не є шаром або покриттям, але спільно змішаний, доданий, сполучений або перемішаний з діючою речовиною в лікарській формі. У деяких варіантах здійснення здатність забезпечувати діючу речовину від демпінгу дози в присутності спирту і здатність забезпечувати діючу речовину від розчинення в кислому середовищі шлунка включені в комбінації матеріалів або полімерів, об'єднаних в ексципієнтну суміш, або включені в одній полімерній системі і розташовані в шарі, покритті або сформовані в матриці. Для цілей даного винаходу потрібно розуміти, що коли мова йде про засіб захисту від спирту, передбачається, що він може мати кишковорозчинні властивості. Крім того, потрібно розуміти, що коли мова йде про кишковорозчинний матеріал, передбачається, що він може гальмувати етанол-індукований демпінг дози. У варіанті здійснення, де лікарська форма являє собою гранулу з множини частинок, для того щоб нанести захисний від спирту шар на гранулу з множини частинок, гранули (30-50 г) покривають з використанням пристрою з псевдозрідженим шаром для нанесення покриття (Mini Vector, MFL 01). Кількість засобу захисту від спирту (і розпушувача, описаного нижче), включеного в стійку до спирту фармацевтичну композицію за даним винаходом, визначається в процентах приросту. Наприклад, у варіанті здійснення, де лікарська форма являє собою гранулу з множини частинок, гранула, що покривається, має масу 10 г, і повинно бути нанесено 10% по масі шару засобу захисту від спирту, тоді достатня кількість захищаючого від спирту шару розпилюється на гранулу, так що загальна маса гранули збільшується до 11 г. Математично (1 г доданого засобу захисту від спирту/10 г вихідної гранули)*100%=10% приросту). У іншому прикладі, якщо потрібно додати розпушувач (описаний більш детально нижче) в гранулу з 20% приростом, то можна розпилювати достатню кількість матеріалу розпушувача на гранулу шаром або покриттям для додавання 2 г маси гранулі. Якщо бажано додати засіб захисту від спирту на цю гранулу (яка тепер має загальну масу 12 г) з 50% приростом, то можна розпилювати достатню кількість матеріалу засобу захисту від спирту і довести загальну масу гранули до 18 г ((6 г матеріалу засобу захисту від спирту/12 г гранула)*100% становить 50% приросту). Матеріал засобу захисту від спирту присутній в лікарській формі в кількості, яка забезпечує процент приросту в діапазоні від 20% до 80%, від 30% до 70%, від 40%, до 60% або від 45% до 55%. Альтернативно, засіб захисту від спирту присутній в лікарській формі в кількості, яка забезпечує процент приросту близько 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75% або 80%. У подальшому варіанті здійснення даний винахід включає розпушувач, який складається з набухаючого матеріалу і/або супердезінтегранта. 9 UA 111819 C2 5 10 15 20 25 30 Звичайні набухаючі матеріали включають, але, не обмежуючись ними, агар, альгінову кислоту, карбомери, карагенан, ацетат целюлози, хітозан, гуарову камедь, гідроксипропілцелюлозу, гіпромелозу, ацетатсукцинат гіпромелози, фталат гіпромелози, метилцелюлозу, полоксамер, полікарбофіл, поліетиленоксид, повідон, гіалуронат натрію, ксантанову камедь і зеїн. Набухаючий матеріал присутній в розпушувачі в кількості від близько 1%, 2%, 3%, 5%, 7%, 9%, 10%, 12%, 14%, 15%, 17%, 19%, 20%, 22%, 23%, 24%, 25%, 27%, 29%, 30%, 32%, 35%, 38%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80, 85%, 90%, 95%, 98%, 99% або 100% (коли розпушувач повністю є набухаючим матеріалом). Звичайні супердезінтегранти включають, але, не обмежуючись ними, Polyplasdone® XL або XL-10 (1-етенилпіролідин-2-он, ISP Pharmaceuticalsis, Columbia, MD); альгінат кальцію, кальційкарбоксиметилцелюлозу, натрійкарбоксиметилцелюлозу, целюлозу, хітозан, колоїдний діоксид кремнію, натрійкроскармелозу, кросповідон, докузат натрію, гуарову камедь, гідроксипропілцелюлозу, алюмосилікат магнію, метилцелюлозу, мікрокристалічну целюлозу, полакрилін калію, повідон, альгінат натрію, натрійкарбоксиметилкрохмаль і крохмаль. Супердезінтегрант присутній в розпушувачі в кількості від близько 1%, 2%, 3%, 5%, 7%, 9%, 10%, 12%, 14%, 15%, 17%, 19%, 20%, 22%, 23%, 24%, 25%, 27%, 29%, 30%, 32%, 35%, 38%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80, 85%, 90%, 95%, 98%, 99% або 100% (коли розпушувач повністю є супердезінтегрантом). Розпушувач (чи складається виключно з супердезінтегранта або з комбінації супердезінтегранта і набухаючого матеріалу) присутній в лікарській формі в кількості, яка забезпечує процент приросту в межах від близько 20% до 80%, від 30% до 70%, від 40% до 60% або від 45% до 55%. Альтернативно, розпушувач присутній в лікарській формі в кількості, яка забезпечує процент приросту 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75% або 80%. При певних обставинах, засіб захисту від спирту може взаємодіяти з діючою речовиною і впливати на розчинення/вивільнення діючої речовини. Відповідно, в ще одному варіанті здійснення стійка до спирту фармацевтична композиція включає бар'єрний матеріал, розташований між діючою речовиною і засобом захисту від спирту. Приклади Наступні приклади приведені для ілюстрації даного винаходу. Однак потрібно розуміти, що винахід не обмежується конкретними умовами або деталями, описаними в цих прикладах. У таблицю 1 зведені дані досліджень, проведених на комерційно доступних капсулах негайного вивільнення дулоксетину HCl Cymbalta®. Таблиця 1 % % вивільнення вивільнення в % в 40% % 20% вивільнення в % етанольному вивільнення Вид гранул Покриття етанольному фосфатному приросту розчині в 0,1 н. HCl розчині буфері (4 кислоти (2 год.)* кислоти год.)* (2 год.)* (2 год.)* немає вимірюваної Приклад гранули немає Н.Д. 80 (USP I) >98 (USP I) кількості >99 (USP I) 1а Cymbalta® лікарського засобу немає вимірюваної гранули не 1b немає Н.Д. >99 (USP III) кількості >99 (USP III) Cymbalta® досліджували лікарського засобу 35 40 Приклад 1 (а і b) З 60 мг комерційно доступного Cymbalta® (дулоксетину HCl), капсули відстроченого вивільнення (які називаються в даному описі "гранули Cymbalta®"), вивільнилося 80% лікарського засобу за 2 год. в 20% етанольному розчині кислоти (USP I) і практично весь лікарський засіб вивільнилося за 2 год. в 40% етанольному розчині кислоти (USP I). З гранули 10 UA 111819 C2 5 Cymbalta® вивільнився практично весь лікарський засіб за 2 год. в 20% етанольному розчині кислоти при використанні USP III. Подальші тести розчинення проводили в 0,1 н. HCl (дві години, USP I і USP III), і потім в фосфатному буфері (рН 6,8, 4 год., USP I і USP III). У кислоті не вивільнилося вимірюваної кількості лікарського засобу і практично весь лікарський засіб вивільнився в фосфатному буфері через 4 год. У таблицю 2 зведені результати, описані в прикладах 2-5. Таблиця 2 Вид гранул Приклад 2а 2b 2с гранули Cymbalta® гранули Cymbalta® гранули Cymbalta® Приклад 3 гранули Cymbalta® Приклад 4 гранули дулоксетину IR гранули дулоксетину IR гранули дулоксетину IR Приклад 5 10 15 20 25 гранули дулоксетину IR % вивільнення % вивільнення в 20% в 40% % етанольному етанольному приросту розчині кислоти розчині кислоти (2 год.)* (2 год.)* Покриття водний ацетатсукцинат гідроксипропілметил целюлози-HF водний полівінілацетатфталат 15 15 10 і 50 водний CAP полімер етилакрилат, метилметакрилат 50:50 на органічній основі полімер етилакрилат, метилметакрилат 50:50 на органічній основі полімер етилакрилат, метилметакрилат 40:60 на органічній основі полімер етилакрилат, метилметакрилат 60:40 на органічній основі полімер етилакрилат, метилметакрилат 50:50 на органічній основі, вміщений в V-Caps® 60 не досліджували не досліджували >99 >90 >99 >99 40 99 30 >90 не досліджували 30 >99 не досліджували 30 >99 не досліджували 50 4 39 Приклад 2 (а, b і с) З гранул Cymbalta®, покритих кишковорозчинними дисперсіями на водній основі, такими як ацетатсукцинат гідроксипропілметилцелюлози-HF (AQOAT, що продається Shin-Etsu Chemical Co., Ltd. of Japan), полівінілацетатфталат (SURETERIC® від Colorcon, Inc., Harleysville, PA)і ацетатфталат целюлози на водній основі (AQUACOAT®-CPD від FMC Biopolymer of Philadelphia, PA) вивільнився практично весь лікарський засіб за 2 год. в 40% етанольному розчині кислоти. Приклад 3 З гранул Cymbalta®, покритих Eudragit® RS і Eudragit® L (50:50) (етилакрилат, метилметакрилат полімери, Evonik Industries, Essen GE) (цільовий 40% приріст) вивільнилося менш ніж 20% лікарського засобу за 2 год. в 20% етанольному розчині HCl (USP I), і вивільнився практично весь лікарський засіб за 2 год. в 40% етанольному розчині HCl. Суміш етилакрилату, метилметакрилату отримували розчиненням полімеру Eudragit® RS в денатурованому дегідратованому спирті в змішувачі з малими зсувними зусиллями. Полімер Eudragit® L додавали в розчин до повного розчинення. Триетилцитрат і тальк додавали в розчин і перемішували до повного диспергування. Остаточний склад суміші етилакрилату, метилметакрилату, яким були покриті гранули Cymbalta®, наведений в таблиці 3. 11 UA 111819 C2 Таблиця 3 Матеріал Eudragit® RS PO Eudragit® L 100 55 Триетилцитрат Тальк Денатурований дегідратований спирт, USP (SDA-3C) Очищена вода Усього 5 Склад (г) 3,5 3,5 1,4 3,5 83,2 4,9 100,0 Повний вміст сухої речовини: 11,9% мас./мас., вміст сухого полімеру: 7,0% мас./мас. Приклад 4 Гранули дулоксетину з негайним вивільненням ("IR") виготовляли нанесенням дисперсії дулоксетину (таблиця 4) на цукрові кульки (Surespheres®, цукрові сфери 30/35, Colorcon Ltd.) з використанням розпилювальної сушарки з киплячим шаром (Glatt 1.1). Таблиця 4 Матеріал Дулоксетину HCl Гідроксипропілметилцелюлоза Очищена вода Усього 10 15 Склад (г) 7,0 5,0 88,0 100,0 З гранул дулоксетину IR, покритих Eudragit® RS і Eudragit® L (етилакрилат, метилметакрилат полімери, Evonik Industries, Essen GE) (50:50, 40:60 і 60:40) (30%-42% цільовий приріст), вивільнився практично весь лікарський засіб за 2 год. в 20% етанольному розчині HCl (USP I). Приклад 5 З гранул Cymbalta®, покритих Eudragit® RS і Eudragit® L (50:50) (етилакрилат, метилметакрилат полімери, Evonik Industries, Essen GE) (цільовий 50% приріст), вміщені в Vcaps® (капсули гідроксипропілметилцелюлози з двох частин від Capsugel® of Greenwood, SC), вивільнилося 4% лікарського засобу за 2 год. в 20% етанольному розчині кислоти (USP I) і вивільнилося 39% лікарського засобу за 2 год. в 40% етанольному розчині кислоти (USP III). 20 Таблиця 5 % % % вивільнення вивільнення в % вивільнення в 20% 40% % вивільнення в Вид гранул Покриття етанольному етанольному приросту в 0,1 н. HCl фосфатному розчині розчині (2 год.)* буфері (4 кислоти кислоти год.)* (2 год.)* (2 год.)* Гранули CAP (на Приклад не Дулоксетину основі 50 25 1,5 60 6 досліджували IR розчинника) Немає 7 в 35% вимірюваної CAP (на 36 і 31 (USP I 65 і 94 (USP Приклад Гранули етанольному кількості основі 42 і III I і III 7 Cymbalta® розчині вивільнення розчинника) відповідно) відповідно) кислоти лікарського засобу 25 Приклад 6 У таблицю 5 зведені дані про вивільнення 25% лікарського засобу за 2 год. в 20% етанольному розчині HCl (USP I) гранул дулоксетину IR, покритих дисперсією ацетатфталату целюлози (CAP) в розчиннику (50% цільовий приріст). Розчинення також проводили в 0,1 н. HCl 12 UA 111819 C2 5 (дві години, USP I), і потім в фосфатному буфері (рН 6,8, 4 години, USP I). За 2 год. в 0,1 н. HCl вивільнилося 1,5% лікарського засобу. За 4 год. в фосфатному буфері вивільнилося 60% лікарського засобу. Дисперсію CAP в розчиннику отримували розчиненням CAP в ізопропіловому спирті і воді. До отриманого розчину додавали триетилцитрат і тальк. Розчин перемішували протягом 12-15 хвилин. Остаточний склад дисперсії CAP в розчиннику наведений в таблиці 6. Таблиця 6 Матеріал Ацетатфталат целюлози (Eastman® CAP) Триетилцитрат Тальк Очищена вода Ацетон Ізопропіловий спирт (IPA) Усього 10 15 20 Склад (г) 8,6 1,7 1,7 2,0 43,0 43,0 100,0 Загальний вміст сухої речовини: 12% мас./мас., вміст сухого полімеру: 8,6% мас./мас., пластифікатор: 19,77% від полімеру. Приклад 7 З гранул Cymbalta®, покритих дисперсією CAP в розчиннику (42% приріст) (отримані за прикладом 6), вивільнилося 7% лікарського засобу за 2 год. в 35% етанольному розчині кислоти (USP I) і 36% лікарського засобу за 2 год. в 40% етанольному розчині HCl (USP I) (31% при використанні апарату USP III) (див. таблицю 5). Подальші тести розчинення проводили в 0,1 н. HCl (дві години, USP I), і потім в фосфатному буфері (рН 6,8, 4 години, USP I). За 2 год. в кислоті не було вимірюваної кількості вивільненого лікарського засобу. За 4 год. в фосфатному буфері вивільнилося 65% лікарського засобу (USP I). При використанні апарату USP III не було вимірюваної кількості вивільненого лікарського засобу в кислоті (0,1 н. HCl, дві години), і 74% лікарського засобу вивільнилося в фосфатному буфері (рН 6,8, 4 години). Приклади 8-12, зведені в таблицю 7, ілюструють варіанти здійснення винаходу, що включають розпушувач, який містить набухаючу речовину і/або супердезінтегрант. Таблиця 7 % вивільнення в % % вивільнення 40% вивільнення в вфосфатному Вид гранул Покриття % приросту етанольному 0,1 н. HCl буфері (4 розчині (2 год.)* год.)* кислоти (2 год.)* Немає вимірюваної Гранули водна HPMC і Всього 84% Приклад кількості Дулоксетину CAP (на основі (24 і 60 70 (USP I) 91 (USP I) 8 вивільнення IR розчинника) відповідно) лікарського засобу Немає водний альгінат вимірюваної Всього 101% Приклад Гранули натрію і CAP 23 і 17 (USP I і кількості 65 і 87 (USP I і (25 і 75 9 Cymbalta® (на основі III відповідно) вивільнення III відповідно) відповідно) розчинника) лікарського засобу Немає водна HPMC/ вимірюваної Polyplasdone® Всього 69% Приклад Гранули 35 і 30 (USP I і кількості 61 і 86 (USP I і XL і CAP (на (9 і 60 10 Cymbalta® III відповідно) вивільнення III відповідно) основі відповідно) лікарського розчинника) засобу 13 UA 111819 C2 Продовження таблиці 7 Вид гранул Покриття Приклад Гранули 11 Cymbalta® Приклад Гранули 12 Cymbalta® 5 10 водна HPMC і CAP (на основі розчинника) водна HPMC і CAP (на основі розчинника) % вивільнення в % % вивільнення 40% вивільнення в в фосфатному % приросту етанольному 0,1 н. HCl буфері (4 розчині (2 год.)* год.)* кислоти (2 год.)* Немає вимірюваної Всього 63% кількості (20 і 43 36 (USP III) 92 (USP III) вивільнення відповідно) лікарського засобу Немає 15 (2 в 20% вимірюваної Всього 95% етанольному кількості (20 і 75 розчині 97 (USP III) вивільнення відповідно) кислоти) USP лікарського III засобу Приклад 8 З гранул дулоксетину IR, покритих водною HPMC (24 % приріст) і дисперсією CAP в розчиннику (60 % приріст) (84 % загальний приріст) (отримані за прикладом 6), вивільнилося 70 % лікарського засобу за 2 год. в 40 % етанольному розчині HCl (USP I). Подальші тести розчинення проводили в 0,1 н. HCl (дві години, USP I), і потім в фосфатному буфері (рН 6,8, 4 години, USP I). Не було вимірюваної ількості вивільненого лікарського засобу в кислоті після двох годин в HCl. За 4 год. в фосфатному буфері вивільнився 91 % лікарського засобу. Покриття з водної HPMC отримували розчиненням HPMC і тальку у воді і перемішуванням протягом 15-30 хвилин доти, поки всі компоненти не розчиняться. Отриману дисперсію фільтрували через 150 мікронний фільтр для видалення агрегатів. Остаточний склад водної дисперсії HPMC приведений в таблиці 8. 15 Таблиця 8 Матеріал HPMC (Phamacoat® 603) Тальк Очищена вода Усього 20 25 30 Склад (г) 5,0 7,0 88,0 100,0 Загальний вміст сухої речовини: 12,0 % мас./мас., вміст сухого полімеру: 5 % мас./мас., тальк: 140 % від полімеру. ПРИКЛАД 9 З гранул Cymbalta®, покритих водним альгінатом натрію (25 % приріст) і дисперсією CAP в розчиннику (75 % приріст) (101 % загальний приріст) (отримані за прикладом 6), вивільнилося 23 % лікарського засобу за 2 год. в 40 % етанольному розчині HCl (USP I). Аналогічне розчинення проводили з використанням апарату USP III. За 2 год. в 40 % етанольному розчині HCl вивільнилося 17 % лікарського засобу. Подальші тести розчинення проводили в 0,1 н. HCl (дві години, USP I), і потім в фосфатному буфері (рН 6,8, 4 години, USP I). За 2 год. в кислоті не було вимірюваної кількості вивільненого лікарського засобу. За 4 год. в фосфатному буфері вивільнилося 65 % лікарського засобу. При використанні апарату USP III не було вимірюваної кількості вивільненого лікарського засобу в кислоті (0,1 н. HCl, дві години), і 87 % лікарського засобу вивільнилося в фосфатному буфері (рН 6,8, 4 години). Для приготування водної дисперсії альгінату натрію перший розчин, що містить триетилцитрат і тальк, готували у воді. Окремо альгінат натрію перемішували у вихровому 14 UA 111819 C2 змішувачі з високим зсувом. Альгінат натрію потім додавали до першого розчину триетилцитрату і тальку при постійному помішуванні протягом щонайменше 30 хвилин. Остаточний склад водної дисперсії альгінату натрію наведений в таблиці 9. Таблиця 9 Матеріал Склад (г) 0,85 0,1 0,45 98,6 100,0 Натрію альгінат Триетилцитрат Тальк Очищена вода Усього 5 10 15 20 Загальний вміст сухої речовини: 1,4 % мас./мас., вміст сухого полімеру: 0,85 % мас./мас., пластифікатор: 11,7 % від сухого полімеру, тальк: 52,9 % від сухого полімеру. Приклад 10 З гранул Cymbalta®, покритих водним HPMC/Polyplasdone® XL (9 % приріст) і дисперсією CAP в розчиннику (60 % приріст) (69 % загальний приріст) (отримані за прикладом 6), вивільнилося 35 % лікарського засобу за 2 год. в 40 % етанольному розчині HCl (USP I). Аналогічне розчинення було проведене з використанням апарату USP III. За 2 год. в 40 % етанольному розчині кислоти вивільнилося 30 % лікарського засобу (USP III). Подальші тести розчинення проводили в 0,1 н. HCl (дві години, USP I), і потім в фосфатному буфері (рН 6,8, 4 години, USP I). Не було вимірюваної кількості вивільненого лікарського засобу в кислоті. За 4 год. в фосфатному буфері вивільнився 61 % лікарського засобу (USP I). При використанні апарату USP III не було вимірюваного вивільнення в кислоті (0,1 н. HCl, дві години), і 86 % лікарського засобу вивільнилося в фосфатному буфері (рН 6,8, 4 години). Для отримання дисперсії HPMC/Polyplasdone® XL перший розчин HPMC готували у воді. Окремо, кросповідон і тальк перемішували у вихровому змішувачі з високим зсувом. Дисперсію кросповідону і тальку додавали в розчин HPMC при постійному перемішуванні протягом 30 хвилин. Остаточний склад дисперсії HPMC/Polyplasdone® XL наведений в таблиці 10. Таблиця 10 Матеріал HPMC (Phamacoat 603) Тальк Кросповідон (Polyplasdone XL) Очищена вода Усього 25 30 35 40 Склад (г) 5,0 2,5 0,5 92,0 100,0 Загальний вміст сухої речовини: 8,0 % мас./мас., вміст розпушувача: 0,5 % мас./мас. Приклад 11 З гранул Cymbalta®, покритих водною HPMC (20 % приріст) і дисперсією CAP в розчиннику (43 % приріст) (63 % загальний приріст) (отримані за прикладом 6), вивільнилося 36 % лікарського засобу за 2 год. в 40 % етанольному розчині HCl (USP III). Подальші тести розчинення проводили в 0,1 н. HCl (дві години, USP III), і потім в фосфатному буфері (рН 6,8, 4 години, USP III). За 2 год. в кислоті не було вимірюваної кількості вивільненого лікарського засобу. За 4 год. в фосфатному буфері вивільнилося 92 % лікарського засобу (USP III). Приклад 12 З гранул Cymbalta®, покритих водним HPMC (20 % приріст) і дисперсією CAP в розчиннику (75 % приріст) (95 % загальний приріст) (отримані за прикладом 6), вивільнилося 15 % лікарського засобу за 2 год. в 40 % етанольному розчині HCl (USP III) і 2 % лікарського засобу за 2 год. в 20 % етанольному розчині HCl (USP III). Гранули після дослідження в 40 % етанольному розчині кислоти вивчали при розчиненні в фосфатному буфері (рН 6,8, 4 години, USP III), і вони вивільнили 55 % лікарського засобу. Подальші тести розчинення проводили в 0,1 н. HCl (дві години, USP III), і потім в фосфатному буфері (рН 6,8, 4 години, USP III). За 2 год. в кислоті не було вимірюваної кількості вивільненого лікарського засобу. За 4 год. в фосфатному буфері вивільнилося 97 % лікарського засобу. 15 UA 111819 C2 5 10 15 20 25 30 35 40 Характеристики розчинення прикладу 12 були також вивчені в дещо інших умовах. Композицію вміщували в 0,1 н. HCl/40 % етанольний розчин кислоти (2 години), і потім в фосфатний буфер (4 години) (USP III). Результати цього послідовного тесту розчинення показані на фіг. 8. Приклад 13 Були вивчені характеристики розчинення TriLipix® (капсули холіну фенофібрату з відстроченим вивільненням для перорального введення). Кожна капсула з відстроченим вивільненням містила кишковорозчинні покриті міні-таблетки, що складаються з холінфенофібрату. Фенофіброєва кислота, активний метаболіт холінфенофібрату, має більш високу розчинність у воді, ніж фенофібрат при лужному pH. FDA і Abbott прийшли до згоди, що проведення тестів розчинення/вивільнення в кислоті (рН 3,5) є більш інформативним для активності лікарського засобу. Див. NDA 22-224 Clinical Pharmacology and Biopharmaceutics section 2.6, pages 46-48. Відповідно, з комерційно доступного TriLipix® (капсули з відстроченим вивільненням) вивільнялося близько 8 % лікарського засобу за 2 год. в 20 % етанольному розчині кислоти (рН 3,5) (апарат USP II), і вивільнялося більше 58 % лікарського засобу за 2 год. в 40 % етанольному розчині кислоти (рН 3,5) (апарат USP II). Див. фіг. 9. Подальше розчинення в фосфатному буфері (рН 6,8) показало, що 100 % лікарського засобу вивільнилося зі складу з відстроченим вивільненням через 6 годин. Міні-таблетки TriLipix® покривали дисперсією ацетатфталату целюлози (CAP) в розчиннику в кількості близько 30 % приросту. На фіг. 10 показано розчинення і вивільнення фенофіброєвої кислоти для цього складу покриття. Не було вимірюваної кількості вивільненого лікарського засобу за 2 год. в 0 %, 20 % і 40 % етанольному розчині кислоти (рН 3,5) (апарат USP II). Подальше розчинення в фосфатному буфері (рН 6,8) показало, що 100 % лікарського засобу вивільнилося зі складу з відстроченим вивільненням через 6 годин. Приклад 14 Гранули Nexium® були вивчені в 20 % і 40 % етанольному розчині кислоти (фіг. 11), і повний демпінг дози спостерігався в 40 % етанольному розчині кислоти. Гранули Nexium® покривали аналогічною дисперсією ацетатфталату целюлози в розчиннику (63 % приріст), як описано в прикладі 6. Із цього складу вивільнялося 20 % лікарського засобу в 40 % етанольному розчині HCl і 80 % лікарського засобу в фосфатному буфері з рН 6,8 (фіг. 12). Гранули Nexium® також покривали дисперсією ацетатфталату целюлози в розчиннику з отриманням 77 % приросту, які вивільняли 1,5 % лікарського засобу в 40 % етанольному розчині HCl і вивільняли 90 % лікарського засобу в фосфатному буфері з рН 6,8 (див. також фіг. 12). Гранули Nexium® і гранули Nexium® з САР-покриттям (77 % приросту) не вивільняли вимірюваної кількості лікарського засобу в 0,1 н. HCl і вивільняли 90 % лікарського засобу в фосфатному буфері з рН 6,8 (фіг. 13). Гранули Nexium® з САР-покриттям (77 % приросту) не вивільняли вимірюваної кількості лікарського засобу в 30 % етанольному розчині HCl і вивільняли 90 % лікарського засобу в фосфатному буфері рН 6,8 (фіг. 13). Гранули Nexium®, покриті дисперсією Eudragit® S в розчиннику (до 30 % приросту) вивільняли 60 % лікарського засобу в 40 % етанольному розчині HCl (фіг. 14). Для отримання дисперсії Eudragit® S матеріали, вказані в таблиці 11, перемішували в змішувачі з низьким зсувом. Повільно додавали воду і IPA доти, поки суміш не розчиниться. Додавали триетилцитрат і тальк і перемішували протягом 12-15 хв. Таблиця 11 Матеріал Склад (г) 7,5 0,8 3,7 3,0 34,0 Eudragit® S Триетилцитрат Тальк Очищена вода Ацетон 45 Гранули покривали, використовуючи пристрій з псевдозрідженим шаром для нанесення покриття. ФОРМУЛА ВИНАХОДУ 50 1. Стійка до спирту фармацевтична композиція, яка містить: (i) діючу речовину; 16 UA 111819 C2 5 10 15 20 25 30 35 40 (ii) кишковорозчинну систему; і (iii) засіб захисту від спирту, де кишковорозчинна система або засіб захисту від спирту включають ацетатфталат целюлози на органічній основі і де засіб захисту від спирту присутній в лікарській формі в кількості, яка забезпечує приріст від 10 до 500 %. 2. Композиція за п. 1, в якій або кишковорозчинне покриття, або засіб захисту від спирту необов'язково містить другий полімер, вибраний з групи, що складається з фталату гіпромелози, співпівполімерів етилакрилату-метилметакрилату, аніонних співполімерів метакрилової кислоти і етилакрилату і їх сумішей. 3. Композиція за п. 1, в якій діюча речовина вибрана з групи, яка складається з дулоксетину HCl, езомепразолу, рабепразолу натрію, мезаламіну, будесоніду, ламотриджину, декслансопразолу, панкреатину, панкреліпази, дивальпроату натрію, омепразолу, ланзопразолу, диклофенаку натрію, вальпроєвої кислоти, холіну фенофібрату, диданозину, аспірину, бісакодилу, напроксену, еритроміцину, натрій рабепразолу, аденовірусної вакцини типу 4, кальцитоніну, дарапладибу, месалзину, алендронової кислоти, епротирому, NE-F (нефритогенного фактора), глатирамеру, CH-1504, бісфосфонатної (золедронова кислота) сполуки, меркаптаміну, ларазотиду, перорального інсуліну і їх сумішей або комбінацій. 4. Композиція за п. 1, в якій кишковорозчинна система включена в композицію в формі, яка вибрана з групи, яка складається з покриття, шару, матриці та їх комбінацій. 5. Композиція за п. 1, в якій кишковорозчинна система додатково містить компоненти, що вибрані з групи, яка складається з ацетатсукцинату гідроксипропілметилцелюлози на водній і органічній основі, полівінілацетатфталату і аніонних співполімерів метакрилової кислоти і етилакрилату. 6. Композиція за п. 1, яка додатково містить розпушувач, вибраний з групи, яка складається з набухаючого матеріалу, супердезінтегранту і їх сумішей або комбінацій. 7. Композиція за п. 1, яка додатково містить бар'єрний матеріал, розташований між діючою речовиною і засобом захисту від спирту. 8. Композиція за будь-яким з попередніх пунктів, де кишковорозчинна система і засіб захисту від спирту поміщені в комбінацію матеріалів або полімерів, комбінованих в суміші ексципієнтів, або поміщені в єдину полімерну систему і поміщені в шарі, покритті або сформовані в матрицю. 9. Композиція за п. 6, де кишковорозчинна система і засіб захисту від спирту представлені як покриття з єдиною полімерною системою. 10. Композиція за будь-яким з попередніх пунктів для застосування в лікуванні захворювання, де лікування включає: ідентифікацію пацієнта, чутливого до супутнього прийому алкоголю протягом періодів часу, коли діюча речовина буде знаходитися в шлунку пацієнта; вибір складу з кишковорозчинним покриттям з діючою речовиною, стійкою до спирту, придатного для лікування захворювання в порівнянні з комерційно еквівалентним складом; і введення пацієнтові, що страждає на захворювання, складу з кишковорозчинним покриттям з діючою речовиною, стійкою до спирту. 11. Композиція за будь-яким з попередніх пунктів, де діюча речовина, кишковорозчинна система і засіб захисту від спирту на органічній основі скомбіновані для утворення частинок або гранул. 17 UA 111819 C2 18 UA 111819 C2 19 UA 111819 C2 20 UA 111819 C2 21 UA 111819 C2 22 UA 111819 C2 23 UA 111819 C2 Комп’ютерна верстка А. Крижанівський Державна служба інтелектуальної власності України, вул. Василя Липківського, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 24
ДивитисяДодаткова інформація
Назва патенту англійськоюAlcohol resistant enteric pharmaceutical compositions
Автори англійськоюLiversidge, Gary, Manser, David, Shah, Hardik, Ruddy, Stephen B., Rekhi, Gurvinder, Singh
Автори російськоюЛиверсидж Гери, Мансер Дэвид, Шах Хардик, Радди Стэфэн Б., Рекхи Гурвиндер Сингх
МПК / Мітки
МПК: A61K 9/52, A61K 9/22, A61K 9/28, A61K 47/34, A61K 47/38, A61K 9/30
Мітки: кишковорозчинні, стійкі, композиції, фармацевтичні, спирту
Код посилання
<a href="https://ua.patents.su/26-111819-stijjki-do-spirtu-kishkovorozchinni-farmacevtichni-kompozici.html" target="_blank" rel="follow" title="База патентів України">Стійкі до спирту кишковорозчинні фармацевтичні композиції</a>
Пінотвірні композиції з високим вмістом спирту з сурфактантами на основі силіконів, концентрат композиції, способи утворення та дозування піни із застосуванням композиції, негерметизований дозатор, спосіб утвор

Номер патенту: 89077
Опубліковано: 25.12.2009
Автори: Фернандес де Кастро Марія Тереза, Муньос Франсіско, Койвісто Брюс Майкл
МПК: A01N 31/02, A01N 55/10, C11D 17/00, C11D 3/48, A01N 25/16
Мітки: дозування, вмістом, концентрат, силіконів, утвор, піни, негерметизований, застосуванням, пінотвірні, спосіб, утворення, високим, основі, спирту, композиції, сурфактантами, способи, дозатор
Формула / Реферат:
1. Здатна до утворення піни спиртовмісна композиція, яка містить:a) С1-4 спирт або його суміші у кількості більше 40 об. % усієї композиції;b) засіб піноутворення, що містить ефективний фізіологічно прийнятний сурфактант, що містить ліпофільний ланцюг на основі силіконових сполук, для змочування та піноутворення, у кількості принаймні 0,01 мас. % усієї композиції, де вказаний засіб піноутворення вибраний так, щоб при дозуванні...
Стійкі композиції вітаміну с

Номер патенту: 100841
Опубліковано: 11.02.2013
Автор: Томас Ісаак
МПК: A61Q 19/00, A61K 8/67, A61K 31/375, A61P 17/16, A61K 8/891
Мітки: композиції, вітаміну, стійкі
Формула / Реферат:
1. Безводна композиція, що включає:(a) 5-15 % за вагою порошкової аскорбінової кислоти,(b) від 20 до 30 % за вагою сполуки, що містить силікон, і(c) від 40 до 50 % за вагою ефірного масла,причому ефірним маслом є кунжутне масло,а частинки аскорбінової кислоти суспендовані у сполуці, що містить силікон,в якій принаймні 50 % вихідної кількості аскорбінової кислоти залишається стабільною при зберіганні...
Фармацевтичні композиції модафінілу, спосіб одержання композиції модафінілу та пероральна дозована лікарська форма

Номер патенту: 88256
Опубліковано: 12.10.2009
Автори: Хікок Крейг, Пейтел Піюш Р., Парікх Алпа
МПК: A61K 9/16, A61K 31/165, A61K 9/14
Мітки: спосіб, композиції, одержання, дозована, модафінілу, лікарська, фармацевтичні, форма, пероральна
Формула / Реферат:
1. Фармацевтична композиція, яка містить дві або більше фракцій твердих частинок модафінілу з вихідної партії модафінілу, де кожна фракція має обмежений діапазон розмірів частинок, і де один або декілька діапазонів розмірів частинок, присутніх у вихідній партії, відсутні у фармацевтичній композиції, де більше ніж приблизно 5 % частинок у композиції мають розмір більший ніж приблизно 200 мікрон.2. Композиція за п. 1, де модафініл являє...
Рідкі фармацевтичні композиції палоносетрону

Номер патенту: 90449
Опубліковано: 11.05.2010
Автори: Канелла Роберта, Бралья Ріккардо, Бралья Енріко, Кальдерарі Джорджо, БОНАДЕО Даніеле
МПК: A61P 1/08, A61K 31/473, A61K 9/08
Мітки: рідкі, фармацевтичні, палоносетрону, композиції
Формула / Реферат:
1. Фармацевтично стабільний ізотонічний внутрішньовенний розчин палоносетрону гідрохлориду для профілактики або зменшення блювання, що включає:a) від 0,03 мг/мл до 0,2 мг/мл палоносетрону гідрохлориду в розрахунку на масу основи палоносетрону; іb) стерильний фармацевтично прийнятний водний носій, що містить маніт в кількості, ефективній для регуляції тонічності, при рН від 4,0 до 6,0, іс) EDTA в кількості від 0,005 мг/мл...
Фармацевтичні композиції, призначені для лікування порушень внутрішнього вуха

Номер патенту: 89858
Опубліковано: 10.03.2010
Автор: Мейєр Томас
МПК: A61K 31/137, A61P 27/16
Мітки: внутрішнього, вуха, фармацевтичні, порушень, лікування, призначені, композиції
Формула / Реферат:
1. Застосування композиції, яка містить (і) фармацевтичну діючу речовину, вибрану з групи, що включає арилциклоалкіламін або його похідну або фармацевтично активну сіль, де арилциклоалкіламін являє собою антагоніст рецептора NMDA, і (іі) біосумісний полімер або комбінацію біосумісних полімерів для приготування лікарського засобу для лікування захворювань внутрішнього вуха. 2. Застосування композиції за п. 1, де композиція є твердою,...
Попередній патент: Антитіло проти csf-1r
Наступний патент: Модифікований зв’язуючий протеїн, що інгібує взаємодію vegf-а-рецептора
Випадковий патент: П'єзоелектричний перетворювач механічних величин