Спосіб одержання похідного фенотіазину
Номер патенту: 29641
Опубліковано: 15.11.2000
Автори: Походенко Віталій Дмитрович, Кошечко Вячеслав Григорович, Кіпріанова Лідія Андріївна, Калініна Людмила Іванівна



Текст
1. Способ получения производного фенотиазина, содержащего в ароматическом ядре в качестве заместителя шестичленный, гетероциклический амин, общей формулы: где Изобретение относится к органическому синтезу, в частности, к электрохимическим способам органического синтеза гетероциклических соединений и может быть использовано для получения биологически активных соединений широкого спектра. Известен способ получения производного фенотиазина замещением водорода в ароматическом кольце фенотиазина ароматическими аминами (Журн. орган. химии. - 1991. - 27, № 9. С. 2008-2014) путем окисления фенотиазина в спиртовом или ацетоновом растворе хлоридом железа (III) или гексацианоферратом (III) калия в присутствии ароматического амина. Известен также способ получения производного фенотиазина замещением водорода в ароматическом ядре фенотиазина тиоцианогруппой путем окисления фенотиазина бромом в среде уксусной кислоты в присутствии роданистого калия (Studii Si cercetari chim. Acad.RPR Fil. Cluj. - 1961. 12, № 2. - P. 309-314). Наиболее близким к предложенному способу является выбранный в качестве прототипа способ получения N-(3-фенотиазинил)-пиридиний перхлората путем окисления фенотиазина в ацетонитриле иодом в присутствии перхлората серебра с дальнейшим взаимодействием перхлората фе нотиазина с пиридином (J. Org. Chem. - 1972. - 37, № 17. - P. 2691-2697): (13) A путем окисления фенотиазина в среде апротонного растворителя и избытка шестичленного гетероциклического азотистого основания с одним гетероатомом, отличающийся тем, что окисление проводят электрохимически при комнатной температуре. 2. Способ по п. 1, отличающийся тем, что электрохимическое окисление проводят при потенциале первой волны окисления фенотиазина на платиновых электродах в присутствии фоновых электролитов - перхлоратов щелочных металлов. UA (19) Существенными недостатками этого метода являются его двухстадийность и необходимость применения взрывоопасного перхлората серебра. В основу изобретения положена задача создания способа получения производного фенотиазина, содержащего в 3-м положении ароматического ядра в качестве заместителя шестичленный гетероциклический амин, который, за счет использования электрохимического процесса окисления фенотиазина и определенных его параметров, обеспечивает получение целевого продукта в одну стадию и исключает возможность взрыва. Предлагаемый способ получения производного фенотиазина, содержащего в 3-м положении ароматического ядра в качестве заместителя шестичленный гетероциклический амин, заключается (11) 29641 где 29641 в окислении фенотиазина в среде апротонного растворителя и избытка шестичленного гетероциклического азотистого основания с одним гетероатомом, причем, согласно изобретению, окисление проводят электрохимически при комнатной температуре. Способ, согласно изобретению, осуществляют при потенциале первой волны окисления фенотиазина на платиновых электродах в присутствии фоновых электролитов - перхлоратов щелочных металлов. Предлагаемый процесс основан на эффекте электрохимической активации фенотиазина путем перевода его в активный катион-радикал, реагирующий в дальнейшем с гетероциклическим амином с образованием производного фенотиазина с азотсодержащим заместителем в 3-м положении фенильного кольца. В качестве шестичленного гетероциклического амина использовали пиридин, его производные и хинолин. Ниже представлено описание технической реализации предлагаемого способа. Электрохимически активируемое взаимодействие фенотиазина с аминами проводили в стеклянной разделенной ячейке в атмосфере аргона в потенциостатическом режиме на платиновых электродах при рабочем потенциале Е=0,6 В относительно насыщенного каломельного электрода при тща тельном перемешивании магнитной мешалкой. Потенциостатический режим поддерживался с помощью потенциостата ПИ-50-1 с программатором ПР-8. В качестве апротонного растворителя использовали ацетонитрил, в качестве фоновой соли – NаСlO4, LiClO4. Способ осуществляли следующим образом: в разделенную электрохимическую ячейку в катодное отделение заливали 30-40 мл 0,1-0,25 молярного ацетонитрильного раствора фоновой соли, предварительно обезвоженной в вакууме при нагревании, а в анодное - столько же раствора фонового электролита такой же концентрации с навеской 0,5-2,5 ммоля фенотиазина, очищенного перекристаллизацией из смеси ацетонитрилгексан или возгонкой в вакууме. В течение 2030 минут барботировали инертный газ, а затем в анодное отделение добавляли 0,5-150 ммолей гетероциклического амина. После пропускания количества электричества 2,0-4,0 Ф/моль (относительно взятого в синтез фенотиазина) электролиз прекращали, ацетонитрил и избыток пиридина или хинолина отгоняли в вакууме, остаток отмывали бензолом, к нерастворимому в бензоле остатку добавляли воду и экстрагировали хлористым метиленом. Полученный после отгонки органического растворителя продукт дважды перекристаллизовывали из водного этанола (1:1). Выделенный продукт взаимодействия фенотиазина с пиридином идентифицировали сравнением это физикохимических характеристик с литературными данными (J. Org. Chem. - 1972. - 37, № 17. - Р. 26912697). Полученные нами новые соединения фенотиазина с производными пиридина и хинолином идентифицировали с помощью элементного анализа, спектроскопических методов (ИК-, электронная и масс-спектроскопия). Ниже на примерах 1-3 продемонстрирована практическая реализация предлагаемого способа получения производных фенотиазина. Пример 1. Получение N-(3-фенотиазинил)пиридиний перхлората (C17Н13ClN2O4S). В электрохимическую стеклянную разделенную ячейку с магнитной мешалкой, вводом и выводом для барботирования газов, заливали в каждое отделение по 30 мл 0,23 М раствора NaCIO4 в CH3CN. Затем в анодное отделение вносили навеску 0,2970 г (0,0015 молей) фенотиазина и навеску пиридина 9,405 г (0,1189 молей); после пропускания инертного газа в течение 20 мин. включали ток и в потенциостатическом режиме при 0,6 В при перемешивании проводили электролиз, который прекращали после пропускания 450 кулонов электричества. Для выделения целевого продукта ацетонитрил и избыток пиридина отгоняли в вакууме, остаток отмывали бензолом с целью удаления непрореагировавшего фенотиазина, к нерастворимому в бензоле остатку добавляли воду и экстрагировали хлористым метиленом. Полученный после отгонки органического растворителя продукт перекристаллизировали из водного этанола (1:1). Получили кристаллическое вещество кирпично-красного цвета. Выход выделенного N(3-фенотиазинил)-пиридиний перхлората составляет 0,2068 (72%) в пересчете на исходное количество фенотиазина, т. пл. 262°, lmax 252, 280, 412 нм (CH3CN) (J. Org. Chem. - 1972. - 37, № 17. P. 2691-2697). Примеp 2. Получение N-(3-фенотиазинил)-3карбамид пиридиний перхлората (С18Н14СlN3O5S). N-(3-фенотиазинил)-3-карбамид пиридиний перхлорат получали по методике, описанной в примере 1. При этом навеска фенотиазина составляла 0,3004 г (0,0015 моля), никотинамида 0,8548 г (0,00699 моля). При рабочем потенциале Е=0,6 В пропущено 580 кулонов электричества. После отгонки ацетонитрила остаток несколько раз промывали бонзолом, затем водой для удаления непрореагировавших фенотиазина, никотинамида и LiClO4. Из водного раствора продукт экстрагировали эфиром. Получили кристаллическое вещество коричнево-красного цвета. Вы ход выделенного N-(3-фенотиазинил)-3-карбамид пиридиний перхлората 0,395 г (62,4%) относительно взятого в реакцию фенотиазина; т. пл. 218-220°С, элементный анализ: рассчитано: С 51,49; Н 3,36; N 10,01; найдено: С 51,01; Н 3,50; N 9,90; lmax 252, 282, 438; ИК: n С=0 1700 см -1, dNH 1500 см -1; массспектроскопия: М1+ 419 а.е.м. и М2+ 321 а.е.м. Пример 3. Получение N-(3-фенотиазинил)хинолиний перхлората (C21H15ClN2O4S). Методика получения и выделения продукта аналогична описанной в примере 1, при этом количества исходных компонентов составляли: NaClO4 - 2,2648 г (0,25 М), фенотиазина - 0,2890 г (0,0014 моля), хинолина - 1,0950 г (0,0085 моля). Получено кристаллическое вещество коричневого цвета. Выход N-(3-фенотиазинил)-хинолиний перхлората 0,3412 г (55,11%), т. пл. 252-255°С; элементный анализ: рассчитано: С 59,08; Н 3,55; N 6,56; измерено: С 59,83; Н 3,05; N 6,84; lmax 238, 258, 435; масс-спектроскопия: M1+ 328 а.е.м. и М2+ 427 а.е.м. 2 29641 Сравнение известного и предлагаемого способа получения C 17H13ClN2O4S Известный способ Условия проведения синтеза: 1-я стадия - получение перхлората фенотиазина из фенотиазина, I2 и AgClO4 2-я стадия - взаимодействие перхлората фенотиазина с пиридином Предлагаемый способ Электрохимическое окисление фенотиазина в присутствии пиридина - одностадийный процесс при комнатной температуре __________________________________________________________ ДП "Український інститут промислової власності" (Укрпатент) Україна, 01133, Київ-133, бульв. Лесі Українки, 26 (044) 295-81-42, 295-61-97 __________________________________________________________ Підписано до друку ________ 2002 р. Формат 60х84 1/8. Обсяг ______ обл.-вид. арк. Тираж 35 прим. Зам._______ ____________________________________________________________ УкрІНТЕІ, 03680, Київ-39 МСП, вул. Горького, 180. (044) 268-25-22 ___________________________________________________________ 3
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparation of phenothiazine derivative
Автори англійськоюPokhodenko Vitalii Dmytrovych, Koshechko Viacheslav Hryhorovych, Kiprianova Lidiia Andriivna, Kalinina Liudmyla Ivanivna
Назва патенту російськоюСпособ получения производного фенотиазина
Автори російськоюПоходенко Виталий Дмитриевич, Кошечко Вячеслав Григорьевич, Киприанова Лидия Андреевна, Калинина Людмила Ивановна
МПК / Мітки
МПК: C25B 3/00, C07D 417/02
Мітки: одержання, фенотіазину, спосіб, похідного
Код посилання
<a href="https://ua.patents.su/3-29641-sposib-oderzhannya-pokhidnogo-fenotiazinu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання похідного фенотіазину</a>
Спосіб одержання похідного сульфонаміду

Номер патенту: 6154
Опубліковано: 29.12.1994
Автори: Пітер Едвард Кросс, Джоффрі Ноель Томас, Джон Едмунд Ерроусміт
МПК: C07C 313/00, A61K 31/18, A61K 31/5375, C07C 323/49, C07C 217/14, C07C 323/36, A61K 31/165, C07C 323/62, A61K 31/535, C07C 301/00, C07C 237/30, A61K 31/395, A61K 31/13, C07C 67/00, C07C 211/29, C07C 213/00, C07C 235/46, A61P 9/06, C07C 217/18, C07C 323/25, C07C 311/08, C07C 217/84, A61K 31/135, C07C 231/00, C07D 295/18, C07C 209/00
Мітки: сульфонаміду, похідного, одержання, спосіб
Формула / Реферат:
1. Способ получения производного сульфонамида формулыотличающийся тем, что соединение общей формулыгде R1 - NН2; или NНSО2СН3; R2 - NH2 или NНSО2СН3, при условии, что хотя бы один из R1 и R2 - NН2, подвергают ацилированию соединением CH3SO2Cl или (CH3SO2)2O и полученный продукт выделяют в свободном виде.2. Способ по п. 1, отличающийся тем, что процесс осуществляют в присутствии акцептора...
Спосіб одержання сульфамоілзаміщеного похідного фенетіламіна і його кислотної солі

Номер патенту: 5981
Опубліковано: 29.12.1994
Автори: Сінаті Хасімото, Казуо Імаї, Такасі Фудзікура, Тоїті Такєнака, Куніхіро Ніїгата
Мітки: одержання, солі, кислотної, похідного, сульфамоілзаміщеного, спосіб, фенетіламіна
Формула / Реферат:
(57) Способ получения сульфамоилза-мещенного производного фенэтиламинаобщей формулыгде R1 – аминогрупа,R2 - низший алкильный радикал,R3 - иод, фенилсульфинильная группа, низшая алкоксигруппа, фенилтиогруппа, R4 – R 8-водород,R9 - водород или низшая алкоксигруппа,у - атом кислорода, а таже его кислотной соли, о т л ичающийся тем, что соединение общей формулыгде R1, R2, R4, R5 – R9...
Спосіб одержання похідного альфа-нафтохінону

Номер патенту: 21654
Опубліковано: 20.01.1998
Автори: Юнаков Михайло Леонідович, Мисак Анатолій Євтіхійович, Айзенберг Вікторія Леонідівна, Новак Ян, Мисак Оксана Анатоліївна
МПК: C07C 7/00, C07C 50/00, C07C 46/00
Мітки: одержання, похідного, спосіб, альфа-нафтохінону
Формула / Реферат:
Способ получения производного альфа - нафтохинона общей формулы С10Н5О2 где R = ОН,включающий окисление 1,5-диоксинафталина хромовой смесью, фильтрование, промывание и высушивание образующегося осадка, а также последующее экстрагирование и кристаллизацию целевого продукта, отличающийся тем, чтоокислению хромовой смесью подвергают неочищенный...
Спосіб одержання похідного ерголіну
Номер патенту: 26400
Опубліковано: 30.08.1999
Автори: Бедескі Анжело, Кабрі Уолтер, Кандіані Іларіа, Заріні Франко
МПК: A61P 5/08, C07D 457/00, A61P 25/16, A61K 31/48
Мітки: спосіб, похідного, ерголіну, одержання
Формула / Реферат:
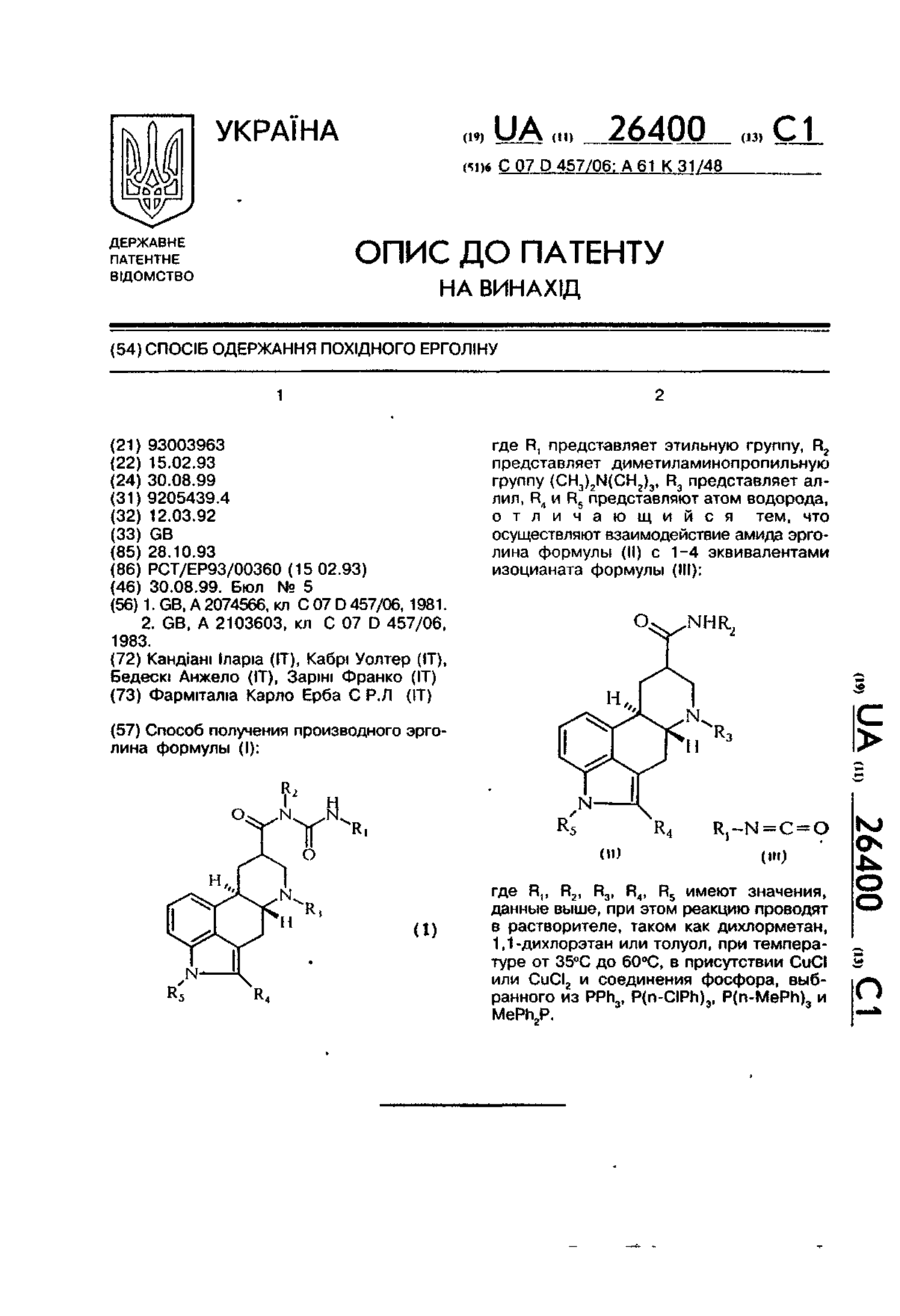
Способ получения производного эрголина формулы (l):где R1 представляет этильную группу, R2 представляет диметиламинопропильную группу (CH3)2N(CH2)3, R3 представляет аллил, R4 и R5 представляют атом водорода, отличающийся тем, что осуществляют взаимодействие амида эрголина формулы (ll) с 1 - 4 эквивалентами изоцианата формулы (lll):где R1, R2, R3, R4, R5 имеют значения, данные выше, при этом реакцию проводят в...
Спосіб одержання 23-(с1-с6-алкілоксимів)-ll-f28249

Номер патенту: 26907
Опубліковано: 29.12.1999
Автори: Молдінг Дональд Рой, КУМАР Аніл
Мітки: одержання, спосіб, 23-(с1-с6-алкілоксимів)-ll-f28249
Формула / Реферат:
Способ получения 23-(С3-С6-алкилоксимов)-LL-F28249, отличающийся тем, что в соединении LL-F28249 защищают 5-гидроксигруппу п-нитробензоилхлоридом с получением 5-0-(п-нитробензоил)-LL-F28249 соединения, окисляют полученное соединение до 5-0(п-нитробензоил)-23-оксо-LL-Р28249 производного в кристаллическом состоянии, которое оксимируют в любой последовательности, выполняя взаимодействие указанного производного с С1-С6-алкоксиамином или его...
Попередній патент: Протипухлинний засіб
Наступний патент: Планетарна передача
Випадковий патент: Пристрій для зчитування інформації з магнітного носія