Спосіб одержання лозартану калію
Номер патенту: 72803
Опубліковано: 15.04.2005
Автори: Петені Ендрене, Фішер Янош, Немеш Андраш, Фаркаш Еньоне, Хегедюш Іштван, Балло Ілдіко, Дешне Юхас Іда, Цібула Ласло, Креідл Янош, Надіне Багді Юдіт, Веркне Папп Ева



Формула / Реферат
1. Спосіб одержання лозартану калію формули (І)
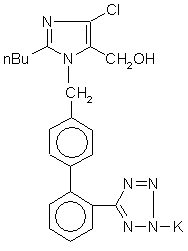 (І)
(І)
хімічна назва якого 2-н-бутил-4-хлор-1-[(2'-(тетразол-5-іл)-1,1'-біфеніл-4-іл)метил]імідазол-5-метанол калію, із використанням як вихідної сполуки 2-н-бутил-4-хлор-1-[(2'-(2-трифенілметил-2Н-тетразол-5-іл)-1,1'-біфеніл-4-іл)метил]-1Н-імідазол-4-метанолу формули (III)
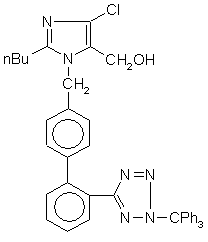 , (ІІІ)
, (ІІІ)
який відрізняється тим, що проводять реакцію сполуки формули (III) у середовищі спирту формули R-ОН (VI), де R являє собою С1-С4 лінійну алкільную групу, із 0,1-1 еквівалентом гідроксиду калію і кінцевий продукт реакції формули (І) виділяють після кристалізації, при заміні розчинника на апротонний або слабопротонний розчинник.
2. Спосіб за п. 1, який відрізняється тим, що як спирт використовують метанол.
3. Спосіб за пп. 1 або 2, який відрізняється тим, що реакцію проводять при температурі 50-80°С.
4. Спосіб за будь-яким з пп. 1, 2 і 3, який відрізняється тим, що як біполярний апротонний розчинник для кристалізації кінцевого продукту використовують ацетонітрил.
5. Спосіб за будь-яким з пп. 1, 2 і 3, який відрізняється тим, що як апротонний розчинник для кристалізації кінцевого продукту використовують лінійні або розгалужені, або циклічні аліфатичні вуглеводні.
6. Спосіб за будь-яким з пп. 1, 2 і 3, який відрізняється тим, що як протонний розчинник для кристалізації кінцевого продукту використовують втор-бутанол.
Текст
Даний винахід стосується способу синтезу відомої похідної тетразолу формули (І). Похідна тетразолу, яка тут розглядається, відома як лозартан калію, хімічна назва якої: калієва сіль 2-нбутил-4-хлор-1-[(2'-тетразол-5-іл)-1,1’-біфеніл-4-іл)метил]імідазол-5-метанол, являє собою активний інгредієнт сучасних антигіпертензивних засобів, антагоністів рецептора ангіотензина II. Відповідно до публікацій заявок РСТ WO 93/10106 і WO 95/17396, лозартан калію може бути синтезований із відповідної похідної кислоти формули (ll) у результаті реакції з гідроксидом калію. Сполука формули (II) може бути отримана детритилюванням трифенілметил (або тритил) захищеної сполуки формули (III). Відщіплення тритильної групи проводять відповідно до відомих методик детритилювання - дією сильних мінеральних кислот (хлористоводневої або сірчаної кислот). Тритиловий спирт формули (IV), що утворився, видаляють із реакційної суміші шляхом фільтрації або екстракції, перекристалізовану і виділену кислоту перетворюють у лозартан калію у водяному середовищі в присутності катіона калію (гідроксид калію або катіонообмінні смоли), і сполуку, що утворилася, перекристалізовують після обробки органічним розчинником при видаленні води азеотропною дистиляцією. Для кристалізації в якості розчинника використовують ізопропанол або суміш циклогексану і ізопропанолу. У прикладах, згаданих вище заявок на патент, детритилювання проводять із використанням водяного розчину хлористоводневої кислоти або водяного розчину сірчаної кислоти при тетрагідрофурані або ацетонітрилі. Загальний вихід лозартану калію становить 72 або 80% у розрахунку на похідну кислоти формули (II), яку виділяють за допомогою складних способів. Недоліком способу який розглядають є те, що перетворення можна проводити тільки в дві стадії, відщіплення тритильної групи протікає під дією розчину сильної, що володіє корозійною дією, мінеральної кислоти - хлористоводневої або сірчаної кислоти - і бажаний лозартан калію виділяють із низьким виходом після додавання водяного розчину гідроксиду калію з використанням складних операцій: азеотропної дистиляції. Відомо, що в ході синтезу інших похідних біфенілтетразолілу, наприклад, відповідно до патенту США 5281603, відщіплення тритильної захисної групи здійснюють під дією каталітичної кількості кислоти в середовищі органічних розчинників. Відповідно до іншого відомого способу, наприклад, того, що розкритий у патенті США 5281604, тритильну груп у тетразолілхіназолінової похідної відщеплюють у результаті кип'ятіння протягом 18 годин зі зворотним холодильником із суміші метанолу і тетрагідрофурану. Очи щену кислотну похідну тетразолу, після концентрації реакційної суміші за допомогою складного методу колоночної хроматографії, одержують з низьким виходом. З такої похідної тетразолу відомими способами можуть бути отримані бажані солі. У зв'язку з вищевикладеним можна зробити висновок, що відповідно до відомих способів, лозартан калію формули (І) одержують у всі х випадках із виділеної й очищеної "лозартанової кислоти" формули (II), що одержують після детритилювання під дією каталітичної кількості кислоти. Ціль даного винаходу полягає в розробці способу, що усуває хиби відомих многостадійних способів, за допомогою якого по простій технології може бути отриманий високоякісний продукт. Автори винаходу виявили, що якщо тритил захищений 2-н-бутил-4-хлор-1-[(2'-(2-трифенілметил-2Н-тетразол-5-іл)-1,1’біфеніл-4-іл)метил]-1Н-імідазол-4-метанол формули (III) оброблюють еквімолярною кількістю гідроксиду калію в С 1-С4 спирті, то можуть бути отримані тритилалкіловий ефір формули (V), що містить алкокси групу і лозартан калію формули (І). Якщо реакцію проводять при температурі кип'ятіння зі зворотним холодильником протягом декількох годин, то бажаний продукт може бути отриманий практично з високим виходом. R=С1-С4 лінійна алкільна група. Автори винаходу несподівано виявили, що така нова, каталізована основою, реакція протікає дуже швидко, і продукт реакції може бути отриманий із високим виходом. У ході реакції детритилювання спирт утворює алкокси аніон, що забезпечує утворення тритилалкілового ефіру. Прості ефіри формули (V) мають дуже низьку розчинність у спиртах із коротким вуглецевим ланцюгом і, у зв'язку з цим, можуть бути видалені фільтрацією. Додаткові дослідження показали, що реакція протікає навіть у тому випадку, якщо похідну тритилу формули (III) оброблюють 0,1-1 еквівалентом гідроксиду калію в середовищі спирту з коротким вуглецевим ланцюгом. У випадку, щорозглядається детритилювання протікає з високим виходом - також з утворенням триалкілового ефіру - і утворенням суміші сполук, що відповідають формулам (І) і (II). Якщо реакційну суміш оброблюють спиртовим розчином, що містить еквівалентну кількість гідроксиду калію в розрахунку на сполуку формули (II), калієва сіль формули (І) утвориться негайно. Відповідно до згаданих вище фактів, цей винахід відноситься до синтезу лозартану калію формули (І), що має хімічну назву 2-н-бутил-4-хлор-1-[(2'-(2-тетразол-5-іл)-1,1’-біфеніл-4-іл)метил]імідазол-5-метанол калію, із 2-н-бутил-4-хлор-1-[(2'-(2-трифенілметил-2Н-тетразол-5-іл)-1,1’-біфеніл-4-іл)метил]-1Н-імідазол-4метанол формули (III), по реакції сполуки (III) у середовищі спирту формули (VI), - де R являє собою С1-С4 лінійну алкильну груп у, із 0,1-1 еквівалентом гідроксиду калію, причому кінцевий продукт реакції, що відповідає формулі (І), виділяють після кристалізації при заміні використовуваного розчинника на апротонний або слабопротонний розчинник. R-OH VI У способі, відповідно до даного винаходу, у якості спирту переважно використовують метанол. Реакцію переважно проводять при 20-100°С, більш переважно, при 50-80°С. Апротонний біполярний розчинник, який використовують для кристалізації кінцевого продукту, переважно, являє собою ацетонітрил, або в якості апротонного розчинника можна використовувати аліфатичні вугле водні з лінійним або розгалуженим вуглецевим ланцюгом або циклічні аліфатичні вуглеводні, так само як в інших випадках, у якості протонного розчинника можна використовувати вторинний бутиловий спирт. Дану реакцію можна проводити в будь-якому С 1-С4 спирті нормальної будівлі, проте, якщо вуглецевий ланцюг більш довгий, то для детритилювання потрібна більша кількість часу і досягається менший вихід цільового продукту. Найкращі умови проведення реакції досягаються при використанні метанолу. У цьому випадку через кілька годин проведення реакції може бути досягнутий вихід порядку 95%. Якщо н-бутанол використовують у реакції перетворення (III)®(І) при 80°С протягом 15-20 годин, то може бути досягнутий ви хід цільового продукту ви ще 80%. Неполярний тритилалкіловий ефір формули (V), отриманий у якості побічного продукту, має низьку розчинність у використовуваному спирті і, внаслідок цього, може бути видалений із реакційного середовища, головним чином, шляхом фільтрації. Лозартан калію високої чистоти може бути виділений із спиртового фільтрата в результаті заміни розчинника з високим виходом. Після випарювання спирту шляхом дистиляції для перекристалізації також можуть бути використані апротонні неполярні розчинники (наприклад, циклогексан, гептан), слабопротонні вторинні спирти, такі як втор-бутанол і, що є несподіваним, апротонний біполярний ацетонітрил. Вихідний матеріал, 2-н-бутил-4-хлор-1-[(2'-(2-трифенілметил-2Н-тетразол-5-іл)-1,1'-біфеніл-4-іл)метил]1Н-імідазол-4-метанол формули (III) може бути синтезований у відповідності зі способом, описаним у літературі: J. Med. Chem. 1991, 34, 2525-2547, і J. Org. Chem. 1994, 59, 6391-6394. Переваги способу, відповідно до даного винаходу, можуть бути підсумовані таким чином: тритиловий спирт формули (IV), що утворюється в якості побічного продукту у відомій реакції кислотного детритилювання у водяному середовищі, являє собою полярний продукт і тому може бути відділений від також полярного лозартану калію лише при істотних втратах бажаної сполуки. У відомих способах для відділення сполуки формули (II), що утворилася, від тритилового спирту необхідно використовувати операції (екстракції, фільтрації), які є трудомісткими. Відповідно до заявленого способу, можна уникнути проведення трудомісткої стадії азеотропної дистиляції, що проводиться після одержання солі калію у водяному середовищі. Інша перевага заявленого способу полягає в тому, що після каталізованої основою реакції детритилювання, що протікає в середовищі спиртів із коротким ланцюгом, переважно, у метанолі, практично з кількісним виходом, со значенням розчинності, яке відрізняється майже на порядок, між отриманим тритилалкіловим ефіром і полярним лозартаном калію, у обраному належним чином, апротонному розчиннику, роблять можливим виділення чистої нерозчинної сполуки формули (І) із високим виходом без утворення сполуки формули (II). Даний винахід ілюструється прикладами, що не обмежують область застосування винаходу: Приклад 1 Синтез лозартану калію формули (І) У 500мл колбі в атмосфері азоту протягом 30 хвилин при температурі кипіння зі зворотним холодильником нагрівали суміш 175мл сухого метанолу, 20г (0,026моля) сольвату 2-н-бутил-4-хлор-1-[(2'(2-трифенілметил-2Н-тетразол-5-іл)-1,1’-біфеніл-4-іл)метил]-1Н-імідазол-4-метанолу і метил-ізобутилкетону і 1,46г (0,026моля) гідроксиду калію в 25мл метанолу. Після кип'ятіння зі зворотним холодильником протягом 4 годин реакційну суміш охолоджували до кімнатної температури, оброблювали 0,6г деревного вугілля і фільтрували. Отриманий фільтрат концентрували до об'єму 30-35мл при зниженому тиску і після додавання 85мл ацетонітрилу знову концентрували до об'єму 30-35мл. Після додавання ще 85мл ацетонітрилу отриманий розчин концентрували до об'єму 60-65мл. Суспензію перемішували при температурі 0-(+)2°С протягом 2 годин, осаджені кристали відфільтровували, тричі промивали 30мл холодного ацетонітрилу і сушили при 70°С і одержували 11,5г (94%) вказаної вище сполуки. Т.пл.: 262264°С. Приклад 2 Синтез лозартану калію формули (І) У 500мл колбі в атмосфері азоту протягом 3 годин при температурі кипіння зі зворотним холодильником, нагрівали суміш 180мл сухого метанолу, 20г (0,026моля) сольвату 2-н-бутил-4-хлор-1-[(2'(2-трифенілметил-2Н-тетразол-5-іл)-1,1'-біфеніл-4-іл)метил]-1Н-імідазол-4-метанолу і метил-ізобутилкетону і 0,1г (0,00178моля) гідроксиду калію. Реакційну суміш о холоджували до кімнатної температури і після додавання 1,35г (0,0241моля) гідроксиду калію в 10мл метанолу оброблювали 0,5г деревного вугілля й отриману суміш фільтрували. Фільтрат концентрували до об'єму 30мл при зниженому тиску і після додавання 80мл ацетонітрилу знову концентрували до об'єму 35мл. Після додавання додаткової кількості ацетонітрилу (85мл) отриману суспензію охолоджували до 0°С, осаджені кристали відфільтровували після 1 годинного перемішування, двічі промивали 30мл холодного ацетонітрилу і сушили при 70°С і одержували 11,3г (93,4%) вказаної вище сполуки. Т.пл.: 261-263°С. Приклад 3 Синтез лозартану калію формули (І) Суміш, що містить 200мл сухого етанолу, 20г (0,026моля) сольвату 2-н-бутил-4-хлор-1-[(2'-(2трифенілметил-2Н-тетразол-5-іл)-1,1'-біфеніл-4-іл)метил]-1Н-імідазол-4-метанолу і метилізобутилкетону і 1,45г (0,026моля) гідроксиду калію, протягом 9 годин кип'ятили зі зворотним холодильником у колбі ємністю 500мл, оброблювали 0,5г деревного вугілля і фільтрували. Отриманий фільтрат концентрували до об'єму 30мл при зниженому тиску і після додавання 150мл ацетонітрилу знову концентрували до об'єму 60мл. Отриману суспензію перемішували при 0°С протягом 1 години, осаджені кристали відфільтровували, двічі промивали 25мл холодного ацетонітрилу і сушили при 70°С і одержували 10,6г (88%) вказаної вище сполуки. Т.пл.: 262-264°С. Приклад 4 Синтез лозартану калію формули (І) Суміш, що містить 100мл н-бутанолу, 7,64г (0,01моля) сольвату 2-н-бутил-4-хлор-1-[(2'-(2трифенілметил-2Н-тетразол-5-іл)-1,1'-біфеніл-4-іл) метил]-1Н-імідазол-4-метанолу і метилізобутилкетону і 0,56г (0,01моля) гідроксиду калію, протягом 20 годин перемішували при 80°С у колбі ємністю 250мл, оброблювали 0,5г деревного вугілля і фільтрували. Отриманий фільтрат концентрували до об'єму 10мл при зниженому тиску і після додавання 100мл ацетонітрилу знову концентрували до об'єму 60мл. Отриману суспензію перемішували при 0°С протягом 1 години, осаджені кристали відфільтровували, двічі промивали 25мл холодного ацетонітрилу і сушили при 70°С і одержували 3,78г (82%) вказаної вище сполуки. Т.пл.: 263-265°С. Приклад 5 Синтез лозартану калію формули (І) Суміш, що містить 200мл сухого метанолу, 20г (0,026моля) сольвату 2-н-бутил-4-хлор-1-[(2'-(2трифенілметил-2Н-тетразол-5-іл)-1,1'-біфеніл-4-іл)метил]-1Н-імідазол-4-метанолу і метилізобутилкетону і 1,45г (0,026моля) гідроксиду калію, протягом 3 годин кип'ятили зі зворотним холодильником у колбі ємністю 500мл, оброблювали 0,4г деревного вугілля і фільтрували при кімнатній температурі. Отриманий фільтрат концентрували до об'єму 30мл при зниженому тиску і після додавання 160мл гептана знову концентрували до об'єму 130мл. Отриману суспензію перемішували при 0°С протягом 2 годин, осаджені кристали відфільтровували, промивали холодним гептаном і сушили при 70°С і одержували 11,3г (92,5%) вказаної вище сполуки. Т.пл.: 263-265°С. Приклад 6 Синтез лозартану калію формули (І) Метанольний фільтрат, отриманий за методикою приклада 5, концентрували до об'єму 30мл при зниженому тиску і після додавання 150мл гексану, знову концентрували до об'єму 100мл. Отриману суспензію перемішували протягом 1 години при 0°С, осаджені кристали фільтрували, промивали холодним гексаном і сушили і отримували 11,5г (94,1%) вказаної вище сполуки. Т.пл.: 262-264°С.
ДивитисяДодаткова інформація
Назва патенту англійськоюA method for the preparation of losartan potassium
Автори англійськоюFischer Janos, Czibula, Laszlo, Werkne Papp Eva
Назва патенту російськоюСпособ получения лозартана калия
Автори російськоюФишер Янош, Веркне Папп Эва
МПК / Мітки
МПК: A61P 9/12, C07D 403/10
Мітки: лозартану, спосіб, калію, одержання
Код посилання
<a href="https://ua.patents.su/3-72803-sposib-oderzhannya-lozartanu-kaliyu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання лозартану калію</a>
Спосіб одержання калію 3-метил-1,2,4-триазоліл-5-тіоацетату, який виявляє антиаритмічну активність

Номер патенту: 14702
Опубліковано: 04.02.1997
Автори: Панасенко Олександр Іванович, Книш Євген Григорович
МПК: A61K 31/41
Мітки: 3-метил-1,2,4-триазоліл-5-тіоацетату, одержання, спосіб, калію, виявляє, антиаритмічну, активність
Формула / Реферат:
Спосіб одержання калію 3-метил-1,2,4-триа-золіл-5-тіоацетату, який виявляє антиаритмічну активність, відрізняється тим, що 3-метил-1,2,4-триазоліл-5-тіон вводять в реакцію з хлорацетатом калію в лужному середовищі, причому вихідні продукти реакції беруться в еквімолекулярних співвідношеннях.
Спосіб одержання калію 3-метил-1, 2, 4-триазоліл-5-тіоацетату, який виявляє антиаритмічну активність

Номер патенту: 22471
Опубліковано: 03.03.1998
Автори: Панасенко Олександр Іванович, Книш Євген Григорович
МПК: A61K 31/41
Мітки: спосіб, активність, одержання, 4-триазоліл-5-тіоацетату, 3-метил-1, виявляє, антиаритмічну, калію
Формула / Реферат:
Спосіб одержання калію 3-метил-1,2,4-триазоліл-5-тіоацетату, який виявляє антиаритмічну активність, який відрізняється тим, що 3-метил-1,2 5,4-триазоліл-5-тіон вводять в реакцію с хлороцтовою, бромоцтовою або иодоцтовою кислотою в присутності лужного компоненту (калію гідроксиду, калію гідрокарбонату або калію карбонату), причому на 1 еквівалент 3-метил-1,2,4-триазоліл-5-тіону і 1 еквівалент галогеноцтової кислоти беруть 2 еквіваленти лужного...
Спосіб одержання калію азотнокислого

Номер патенту: 26469
Опубліковано: 30.08.1999
Автори: Федоров Володимир Іванович, Маслов Володимир Дмитрович, Білоконь Євген Миколайович, Пісний Василь Михайлович, Дубров Леонід Васильович, Калініченко Вадім Іванович, Мачула Сергій Леонідович
МПК: C01D 9/00
Мітки: спосіб, азотнокислого, одержання, калію
Формула / Реферат:
1. Спосіб одержання калію азотнокислого шляхом взаємодії хлориду калію з азотною кислотою, виділення із реакційної суміші цільового продукту, який відрізняється тим, що взаємодію хлориду калію з азотною кислотою проводять в присутності оксидів марганцю MnOn (при n = 1,5 - 2,0) при співвідношенні KCl : HNO3 : MnOn рівному 1 : (0,8 - 1,2) : (0,9 - 1,2) від стехіометрично необхідного.2. Спосіб одержання калію азотнокислого по п.1, який...
Спосіб кристалізації лозартану

Номер патенту: 57045
Опубліковано: 16.06.2003
Автори: Епштейн Альберт Д., Кеннеді Майкл Т., Дайєнманн Ерік А., Ларсон Карен А., Махадеван Гарі, Брін Патрік
МПК: C07D 403/10
Мітки: спосіб, кристалізації, лозартану
Формула / Реферат:
1. Спосіб кристалізації калійлозартану, при якому проводять такі стадії:а) перегонка суміші ізопропанол-вода, що містить калійлозартан, до вмісту води від приблизно 2,4 до приблизно 2,8%;б) охолодження суміші до температури від приблизно 65°С до приблизно 70°С;в) додавання в ємність приблизно 0,5 мас. % суспензії тонкорозмеленого калійлозартану в циклогексані при температурі від приблизно 60°С до приблизно 65°С із...
Спосіб одержання ( 1,1-діоксотіолан-3-іл)-дитіокарбамату калію

Номер патенту: 30599
Опубліковано: 15.01.2003
Автори: Даниленко Валерій Васильович, Шкарапута Леонід Миколайович, Кухар Валерій Павлович, Язловицький Анатолій Васильович
МПК: A01N 43/10, A01P 3/00, C07D 333/48
Мітки: 1,1-діоксотіолан-3-іл)-дитіокарбамату, калію, одержання, спосіб
Формула / Реферат:
Спосіб одержання (1,1-діоксотіолан-3-іл)-дитіокарбамату калію шляхом одночасного дозування розчину гідроксиду калію в етиловому спирті та (1,1-діоксотіолан-3-іл)-аміну (ЗАС) в молярному співвідношенні (1,2...1,3) : 1,0 в розчин сірковуглецю в етиловому спирті при температурі 25-35 до встановлення молярного співвідношення завантажених ЗАС та сірковуглецю (0,08...0,13) :...
Попередній патент: Спосіб автоматичного регулювання парової турбіни
Наступний патент: Спосіб збереження та введення гонадотропних гормонів тваринам
Випадковий патент: Полімінеральний склад у вигляді розсолу і спосіб його виробництва