Кристалічна сполука піридазину
Номер патенту: 99466
Опубліковано: 27.08.2012
Автори: Кент Кеннет М., Том Норма Дж., Зія Вехід, Дауді Ерік Д.



















Формула / Реферат
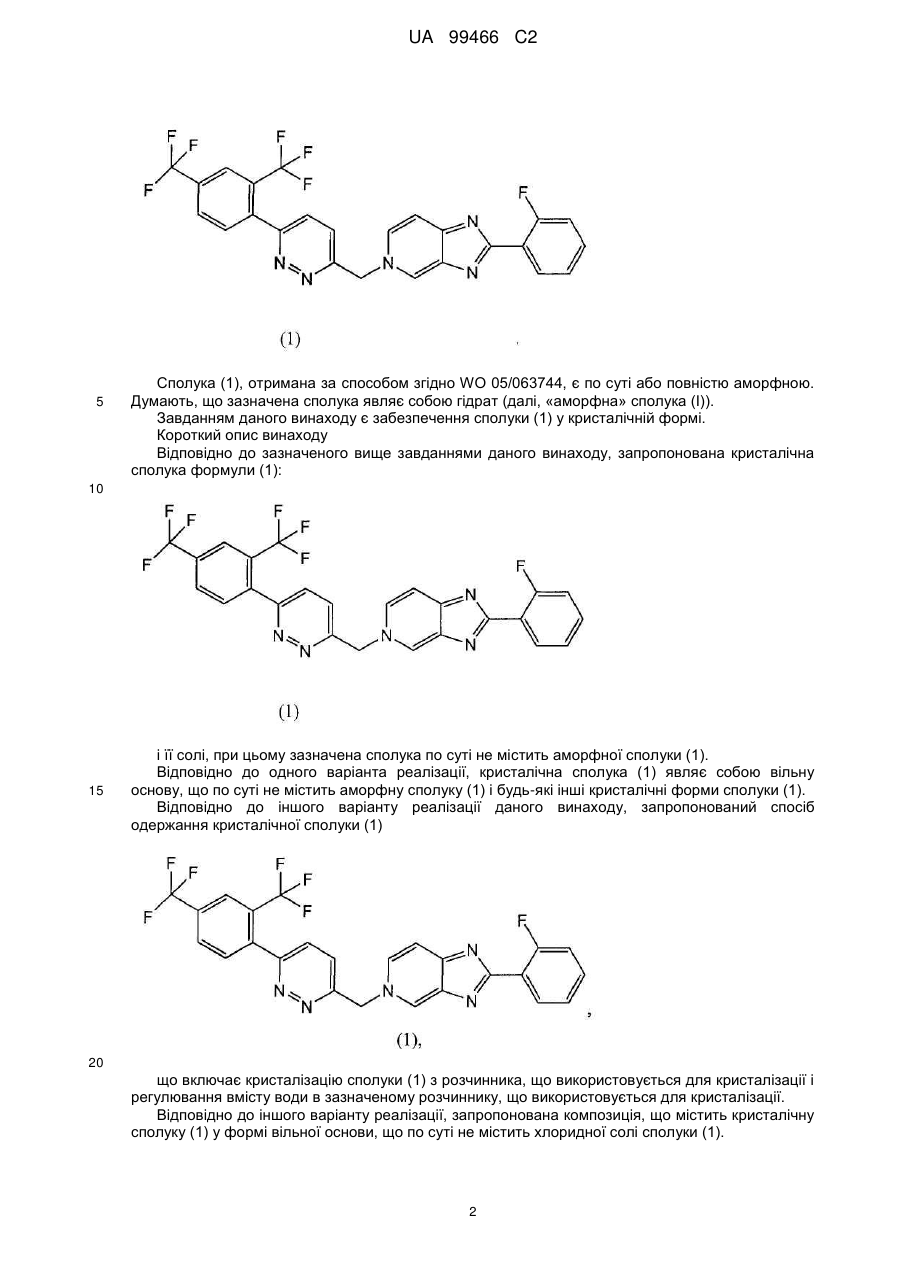
1. Кристалічна сполука формули (1)
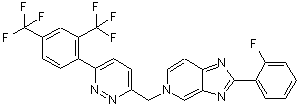 , (1)
, (1)
яка характеризується:
а) початком поглинання тепла при приблизно 235 °С на кривій диференціальної скануючої калориметрії (ДСК);
b) величиною теплоти плавлення (DНпл), що становить приблизно 81 Дж/г (42 кДж/моль); та
c) щонайменше одним піком при значенні кута дифракції 2θ приблизно 17(°) за даними рентгенівської порошкової дифрактометрії,
а також солі сполуки, причому сполука по суті не містить аморфної сполуки (1).
2. Кристалічна сполука за п. 1, яка являє собою вільну основу.
3. Кристалічна сполука за п. 1 у голчастій або стрижнеподібній формі.
4. Кристалічна сполука за п. 1, що по суті не містить хлоридної солі сполуки (1).
5. Кристалічна сполука за п. 1 у мікронізованій формі.
6. Композиція, що містить кристалічну сполуку за будь-яким з пп. 1-5, що містить менше приблизно 40 % мас. аморфної сполуки (1).
7. Композиція за п. 6, що містить менше приблизно 10 % мас. аморфної сполуки (1).
8. Композиція за п. 6, де кристалічна сполука (1) містить менше приблизно 100 ррm хлориду.
9. Кристалічна сполука (1), яка являє собою вільну основу, що по суті не містить аморфної сполуки (1) і будь-якої іншої кристалічної форми сполуки (1), причому кристалічна сполука характеризується:
а) початком поглинання тепла при приблизно 235 °С на кривій диференціальної скануючої калориметрії (ДСК);
b) величиною теплоти плавлення (DНпл), що становить приблизно 81 Дж/г (42 кДж/моль); та
c) щонайменше одним піком при значенні кута дифракції 2![]() приблизно 17(°) за даними рентгенівської порошкової дифрактометрії.
приблизно 17(°) за даними рентгенівської порошкової дифрактометрії.
10. Композиція, що містить кристалічну сполуку за будь-яким з пп. 1-5 або 9 і фармацевтично прийнятний наповнювач.
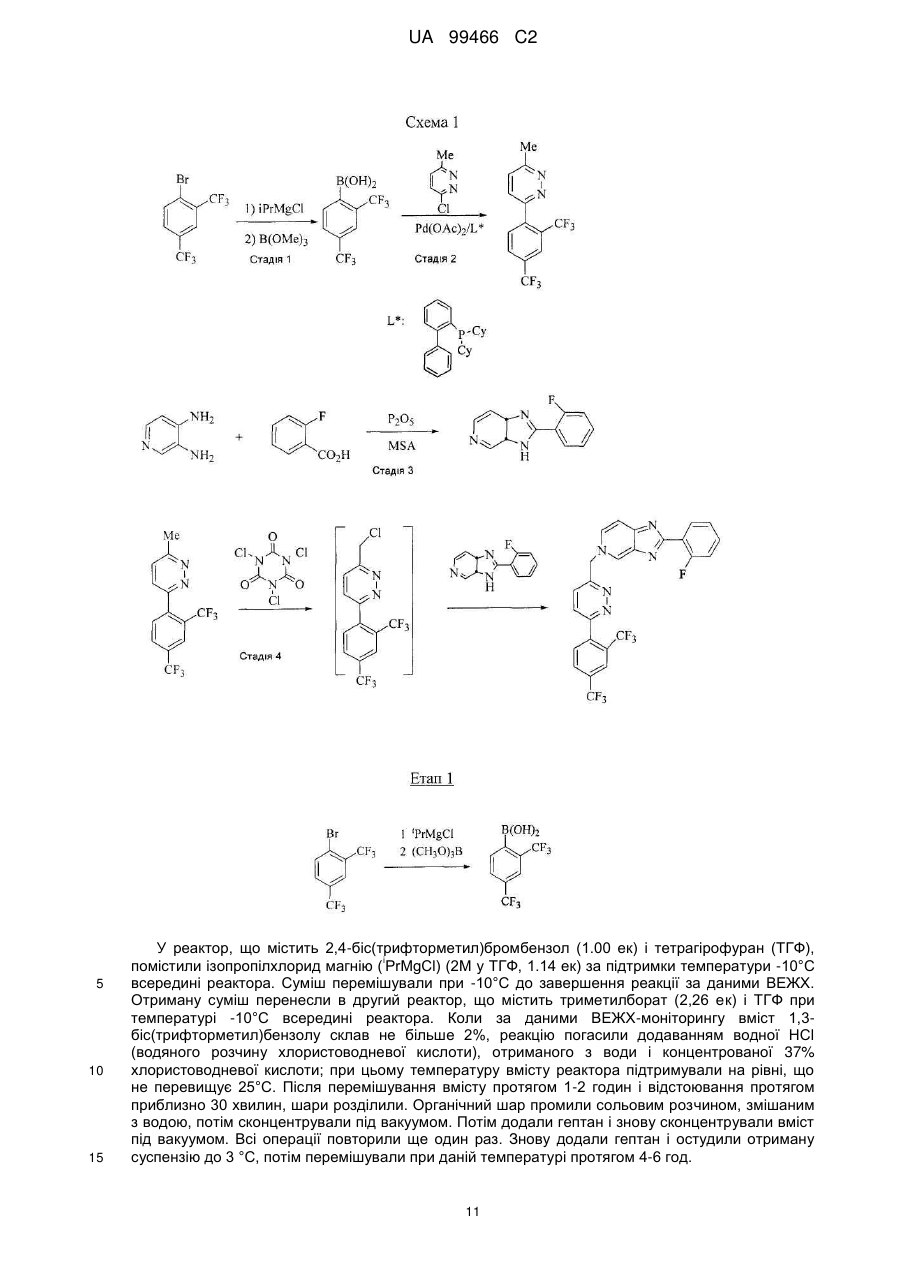
11. Спосіб одержання кристалічної сполуки (1)
,
у якому здійснюють кристалізацію сполуки (1) з розчинника, який використовується для кристалізації, і регулюють вміст води в зазначеному розчиннику, що використовується для кристалізації.
12. Спосіб за п. 11, де сполуку (1) кристалізують із розчинника, що являє собою етилацетат або суміш етилацетат/ізопропіловий спирт, з вмістом води менше приблизно 0,9 % мас.
13. Спосіб за п. 11, де вміст води регулюють таким чином, щоб у ході кристалізації осаджувалося менше приблизно 10 % мас. аморфної сполуки (1).
14. Спосіб за п. 11, де вміст води регулюють шляхом азеотропного видалення води з розчинника, який використовується для кристалізації.
15. Спосіб за п. 11, при якому у розчиннику, що використовується для кристалізації, вміст води забезпечують на рівні менше приблизно 10 %.
16. Спосіб за п. 15, що включає множину етапів кристалізації, де кристалізацію проводять із розчинників, що містять воду у послідовно спадних кількостях.
17. Спосіб за п. 15, де останній етап кристалізації проводять із розчинника, що містить менше приблизно 0,9 % води.
Текст
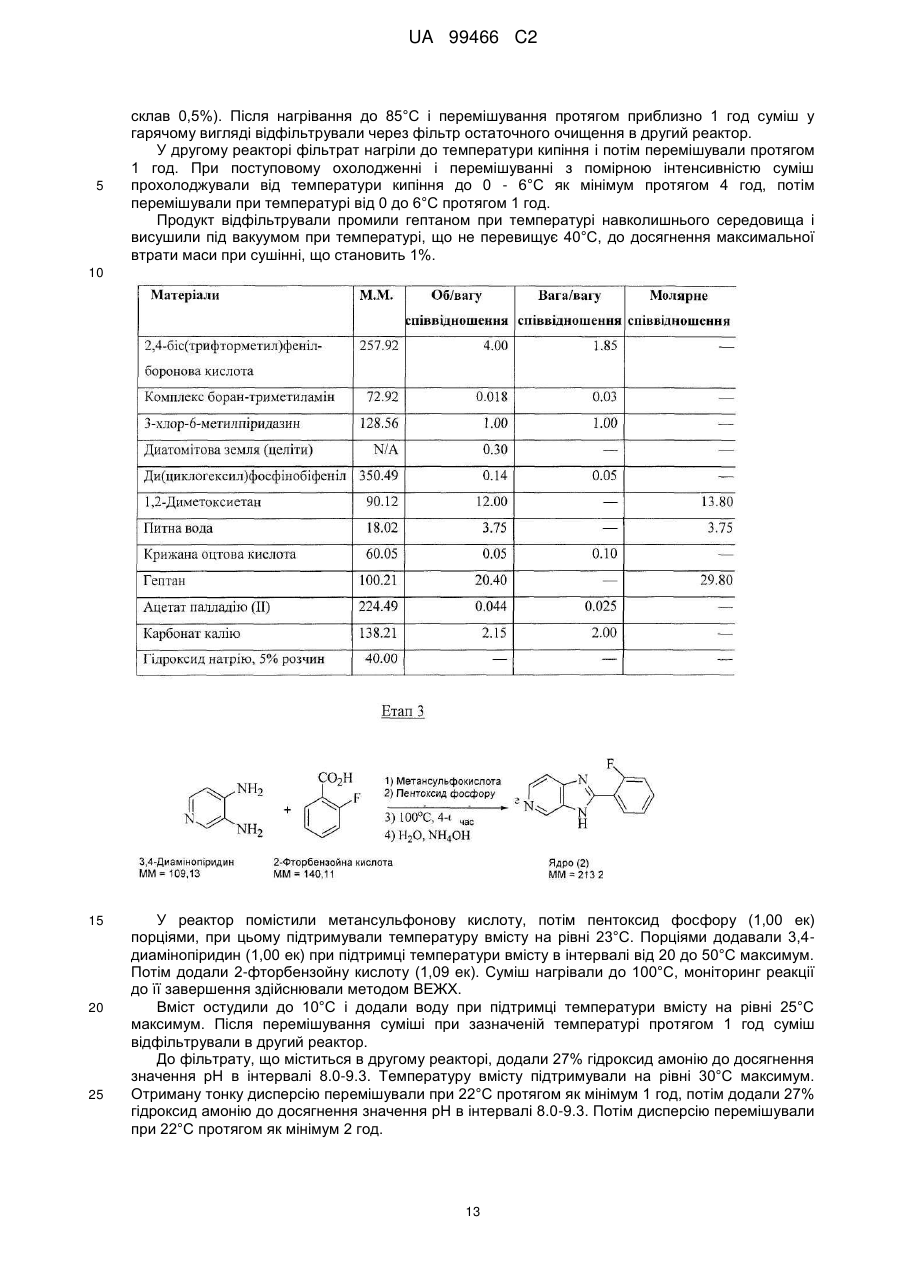
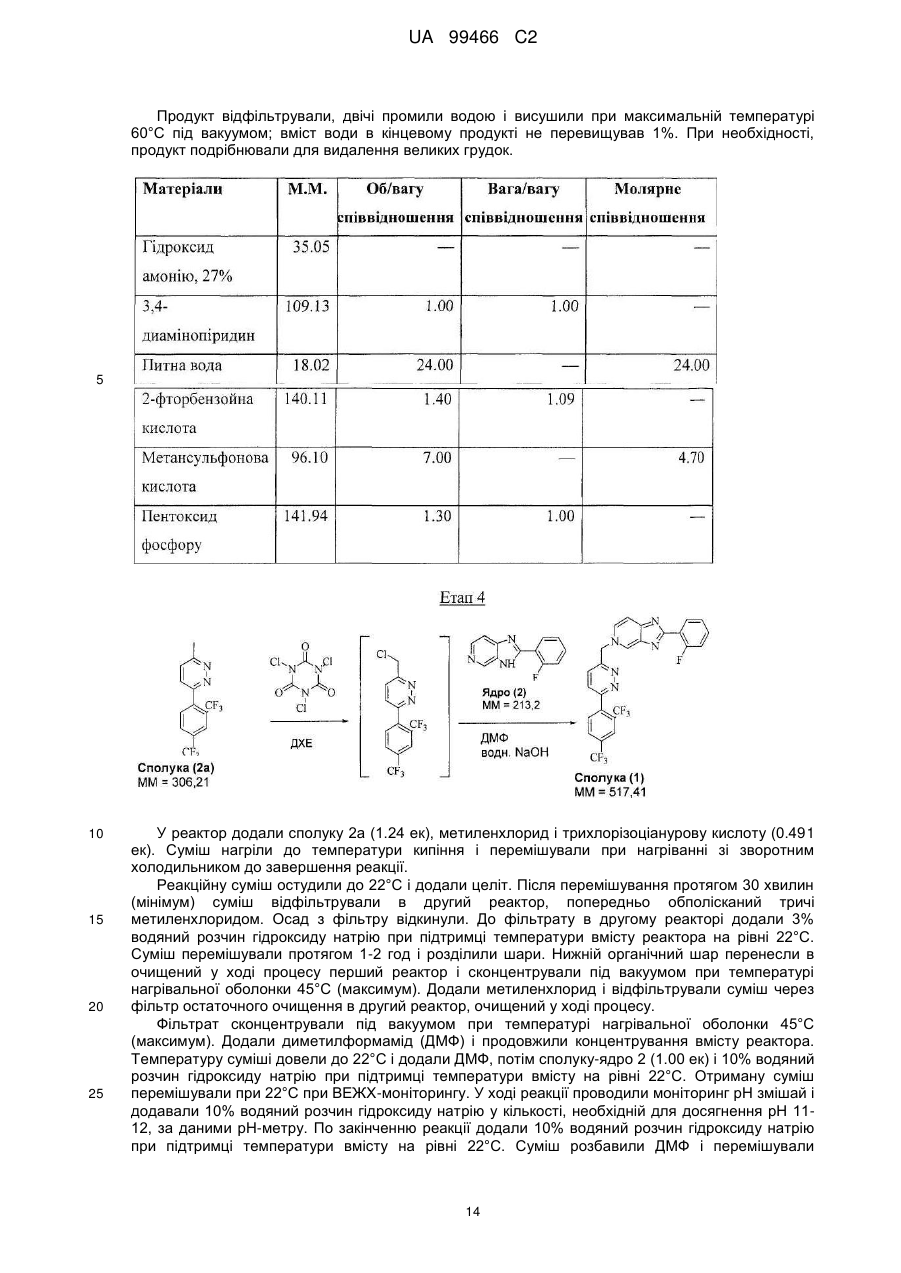
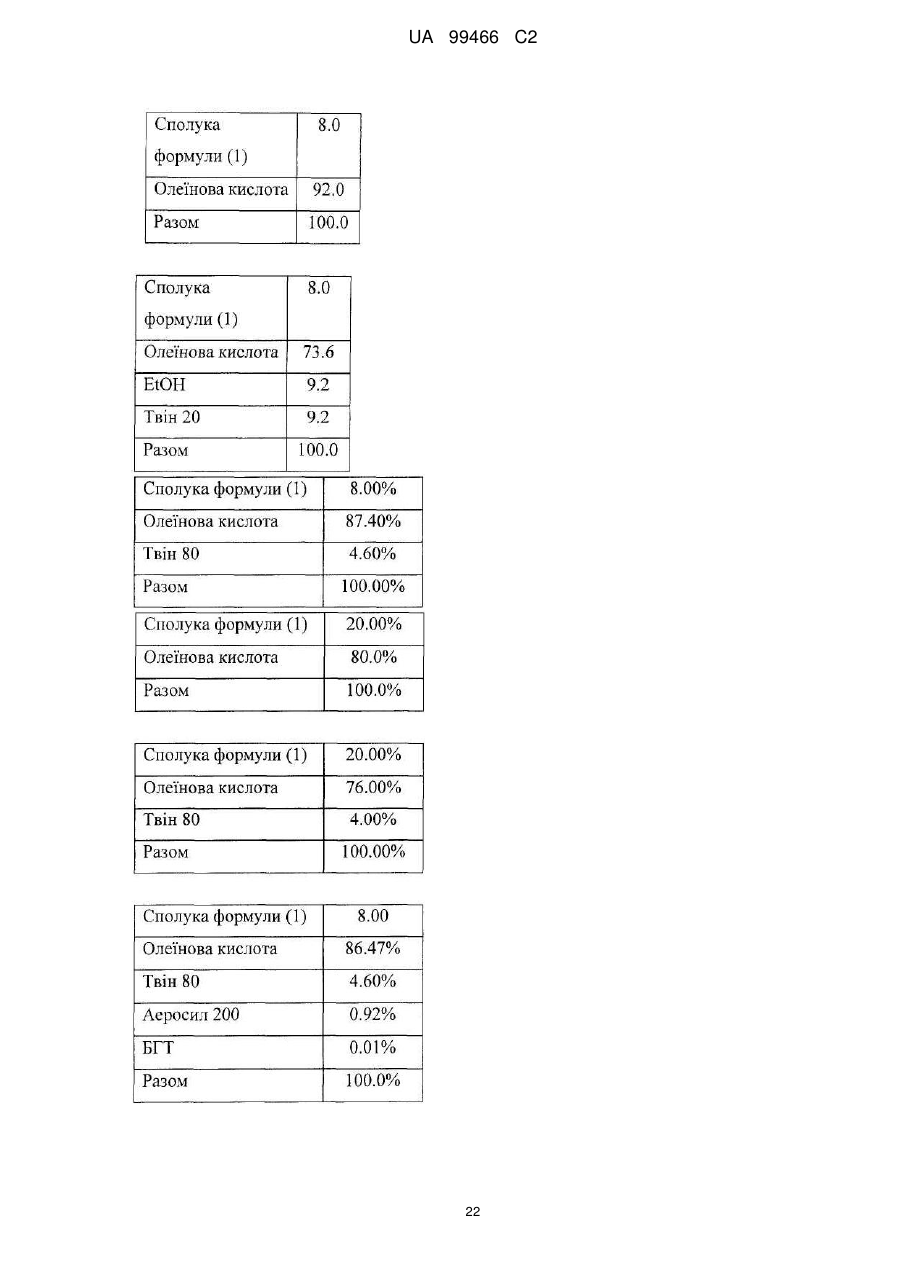
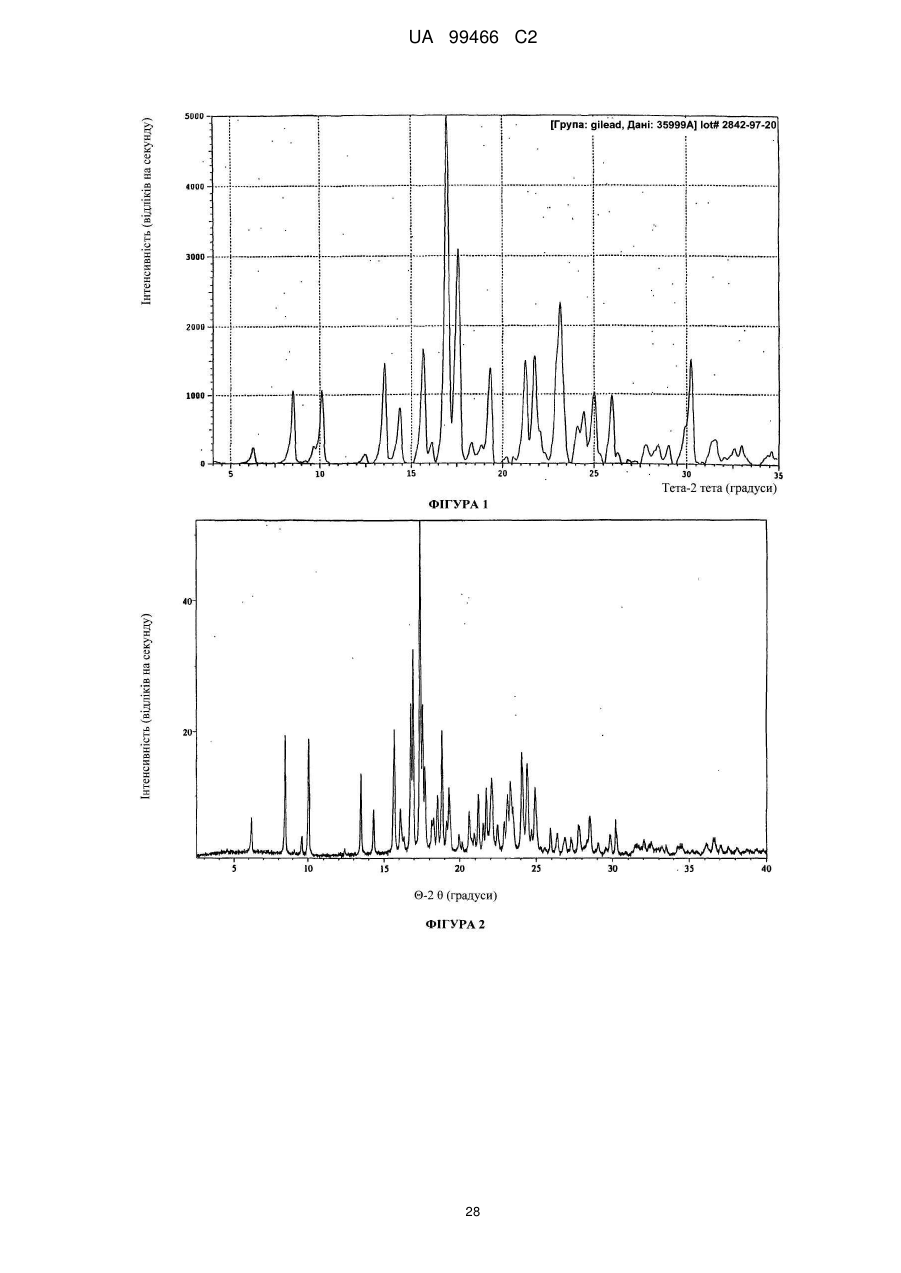
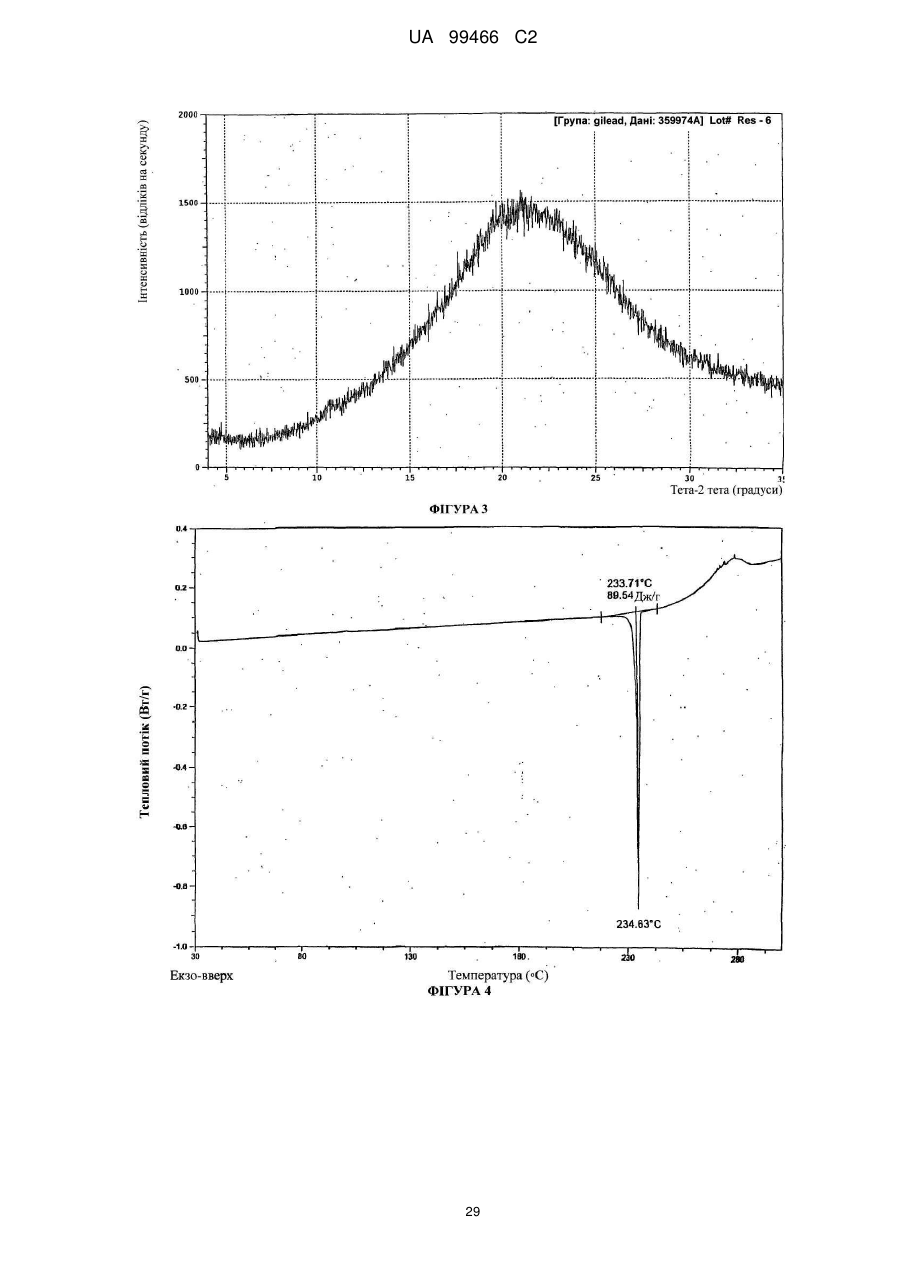
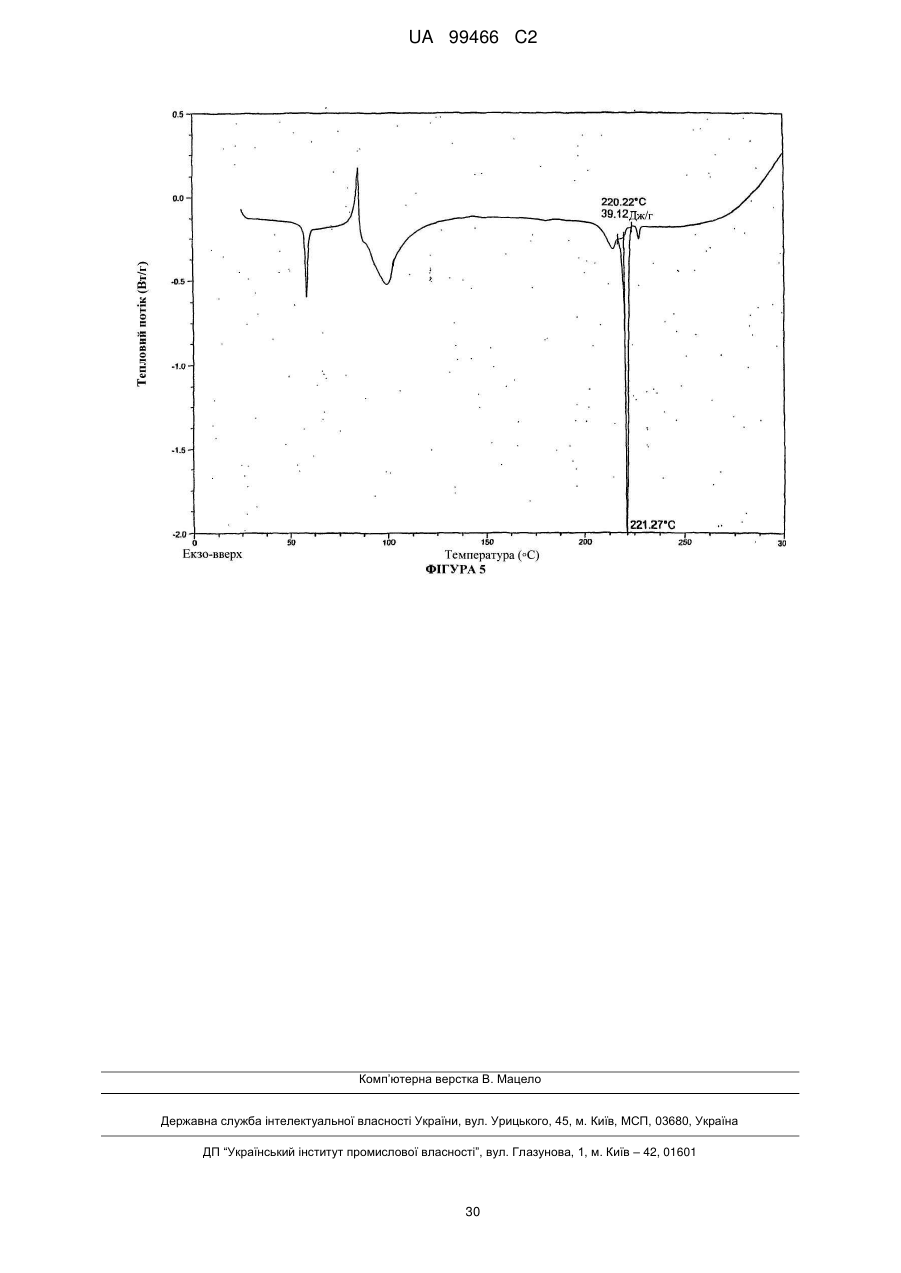
Реферат: У даному F F винаході F F запропонована кристалічна сполука формули (1) F F F N N N N N (1) і солі та сольвати зазначеної сполуки для лікування і профілактики інфекцій, що викликаються вірусом гепатиту С. Крім того, запропоновані способи одержання кристалічної сполуки (1) і сполук на основі зазначеної сполуки. UA 99466 C2 (12) UA 99466 C2 UA 99466 C2 5 10 15 20 25 30 35 40 45 50 55 60 Вірус гепатиту С являє собою оболонковий вірус із оболонкою, що містить однониткову позитивну смислову (+) РНК, що відноситься до сімейства Flaviviridae. Реплікація вірусу гепатиту С (ВГС) відбувається, головним чином, у гепатоцитах печінки. Частки ВГС у кровотоці зв'язуються з рецепторами на поверхні гепатоцитів і потім проникають у клітини. Проникнувши всередину гепатоциту, ВГС використовує внутрішньоклітинні структури, необхідні для реплікації ВГС. Lindenbach, В. Nature 436(7053): 932-8 (2005). У результаті трансляції геному ВГС утворюється єдиний білок, що включає приблизно 3011 амінокислот. Потім цей «полібілок» піддається протеолітичному процесінгу під дією вірусних і клітинних протеаз із утворенням трьох структурних (віріон-асоційованих) і семи неструктурних (NS) білків. ВГС кодує дві протеази, цистеїнову аутопротеазу NS2 і серинову протеазу NS 3-4А. Неструктурні (NS) білки далі рекрутують геном вірусу в комплекс реплікації РНК, асоційований з реаранжированими цитоплазматичними мембранами. Реплікація РНК відбувається під дією РНК-залежної РНК-полімерази NS5B, що забезпечує утворення проміжного мінус-ланцюга РНК. Далі зазначений мінус-ланцюг РНК виступає як матриця для утворення нових геномів вірусів, що складаються із плюс-ланцюга РНК. Потім геноми, що утворюються, можуть піддаватися трансляції, подальшій реплікації або упакуванню у нові вірусні частинки. Приблизно, нові вірусні частинки відпупковуються в секреторний шлях і вивільняються на поверхні клітини. ВГС має високу швидкість реплікації; в організмі інфікованого суб'єкта продукується приблизно один трильйон часток щодня. Внаслідок недостатньої корекції помилок реплікації РНК-полімеразою ВГС. ВГС має винятково високу частоту мутацій, при цьому даний фактор може сприяти використанню ВГС від імунної відповіді організму хазяїна. На підставі генетичних розходжень між ізолятами ВГС, вид вірусу гепатиту С підрозділяють на шість генотипів (1-6), при цьому кожний генотип включає кілька підтипів. Далі, на підставі генетичних розходжень підтипи підрозділяють на квазівиди. Розподіл і перевага тих або інших генотипів ВГС різна в різних регіонах світу. Наприклад, у Північній Америці переважає генотип 1а, за ним ідуть 1, 2а, 2b і 3а. У Європі переважає генотип 1, далі випливають 2а, 2b, 2с і 3а. Генотипи 4 і 5 виявляють майже винятково в Африці. Генотип має клінічну значимість із погляду оцінки потенційної відповіді на терапію інтерфероном і тривалості подібного лікування. Генотипи 1 і 4 менш чутливі до лікування інтерфероном, ніж інші генотипи (2, 3, 5 і 6). Тривалість стандартного курсу лікування інтерфероном для генотипів 1 і 4 становить 48 тижнів, у той час як повний курс лікування для генотипів 2 і 3 становить 24 тижні. За оцінкою Всесвітньої Організації Охорони здоров'я у світі від 170 до 200 мільйонів чоловік (3% населення Землі) хронічно інфіковані вірусом гепатиту С. Приблизно 75% з них хронічно інфіковані і при цьому в них у плазмі містяться кількості РНК ВГС, що виявляються. Ці хронічні носії захворювання піддаються ризику розвитку цирозу і/або раку печінки. У ході досліджень із лікарським контролем протягом 7-16 років відзначений розвиток цирозу у 7-16% пацієнтів, розвиток гепатоцелюлярної карциноми у 0,7-13% пацієнтів, при цьому 1,3-3,7% пацієнтів померло від захворювань, пов'язаних з печінкою. У цей час єдиним доступним способом лікування є терапія із застосуванням інтерферону сс2 (або його пегільованої форми), що застосовується окремо або у комбінації з рибавірином. Проте, стійку відповідь спостерігали лише приблизно у 40% пацієнтів, при цьому лікування пов'язане із серйозними несприятливими побічними ефектами. У цей час існує нагальна потреба в розробці високоефективних і селективних інгібіторів ВГС. Релевантні джерела включають патенти США 4914108; 4988707; 4990518; 5137896; 5208242; 5227384; 5302601; 5374638; 5405964; 5438063; 5486525; 6479508; і публікацію патенту США US2003/0108862 А1, патент Канади 2423800 А1, патенти Німеччини 4211474 А1, 4236026, 4309969, 4318813, Європейські патенти ЕР 0 138 552 А2, ЕР 0 706 795 А2, ЕР 1132 381 А1, патент Великобританії 2158440 А, міжнародні публікації WO 00/20416, WO 00/39127, WO 00/40583, WO 03/007945 Al, WO 03/010140 А2, WO 03/010141 А2, WO 93/02080, WO 93/14072, WO 96/11192, WO 96/12703, WO 99/27929, PCT-US2004/43112, РСТ-ВЕ2003/000117, PCTUS2005/26606, Akamatsu, et al., "New Efficient Route for Solid-Phase Synthesis of Benzimidazole Derivatives" 4: 475-483, J. COMB. CHEM., 2002, Baginski SG et al., Proc. Natl. Acad. Sci. U.S.A. 2000 Jul 5;97(14): 7981-6). Cleve et al., "Derivate des Imidazo[4.5-b]- und lmidazo[4.5-c]pyridins", 747: 158-171, JUSTUS LIEBIGS ANNALEN DER CHEMICA, 1971, Kiyama, et al., "Synthesis and Evaluation of Novel Nonpeptide Angiotensin II Receptor Antagonists: Imidazo[4, 5-c]pyridine Derivatives with an Aromatic Substituent", 43(3): 450-60, CHEM PHARM BULL, 1995, Mederski et al., "Synthesis and Structural Assignment of Some N-substituted Imidazopyridine Derivatives", 48(48): 10549-58, TETRAHEDRON, 1992, Yutilov et al., 23(1): 56-9, KH1MIKOFARMATSEVTICHESKIIZHURNAL, 1989. Крім того, див. WO 05/063744. Сполука формули (1) є об'єктом винаходу згідно WO 08/005519. 1 UA 99466 C2 5 Сполука (1), отримана за способом згідно WO 05/063744, є по суті або повністю аморфною. Думають, що зазначена сполука являє собою гідрат (далі, «аморфна» сполука (І)). Завданням даного винаходу є забезпечення сполуки (1) у кристалічній формі. Короткий опис винаходу Відповідно до зазначеного вище завданнями даного винаходу, запропонована кристалічна сполука формули (1): 10 15 і її солі, при цьому зазначена сполука по суті не містить аморфної сполуки (1). Відповідно до одного варіанта реалізації, кристалічна сполука (1) являє собою вільну основу, що по суті не містить аморфну сполуку (1) і будь-які інші кристалічні форми сполуки (1). Відповідно до іншого варіанту реалізації даного винаходу, запропонований спосіб одержання кристалічної сполуки (1) 20 що включає кристалізацію сполуки (1) з розчинника, що використовується для кристалізації і регулювання вмісту води в зазначеному розчиннику, що використовується для кристалізації. Відповідно до іншого варіанту реалізації, запропонована композиція, що містить кристалічну сполуку (1) у формі вільної основи, що по суті не містить хлоридної солі сполуки (1). 2 UA 99466 C2 5 10 15 20 25 30 35 40 45 50 55 60 Кристалічна сполука (1) підходить для застосування відповідно до способу лікування або профілактики інфекції ВГС, що включає введення суб'єктові терапевтичної або профілактичної дози кристалічної сполуки (1). Інший варіант реалізації включає застосування кристалічної сполуки (1) для одержання лікарського засобу для запобігання або лікування інфекції ВГС у ссавця (зокрема, у людини). Інший варіант реалізації даного винаходу відноситься до фармацевтичних композицій кристалічної сполуки формули (1), що містить щонайменше один фармацевтично прийнятний наповнювач. Відповідно до одного варіанту реалізації, готують склад на основі сполуки формули (1) з використанням органічної кислоти, при цьому зазначена сполука може бути отримана у вигляді дозованої фармацевтичної форми, такої як капсула. Відповідно до іншого варіанту реалізації, кристалічну сполуку (1) мікронізують і готують сполуку у вигляді суспензії. Кристалічна сполука (1) або фармацевтичні композиції згідно із даним винаходом застосовують для лікування або профілактики гепатиту С. Кристалічна сполука (1) проявляє поліпшені фармакологічні характеристики і має цінові переваги; зокрема, поліпшені такі характеристики як чистота, стабільність при зберіганні і відтворюваність при промисловому одержанні. Особливою перевагою є підвищена температура плавлення у порівнянні з аморфною формою. Інші ознаки даного винаходу, включаючи нові проміжні сполуки і композиції на основі кінцевих сполук, стануть очевидні при розгляді даної заявки в цілому. Опис креслень На Фігурі 1 представлена порошкова рентгенограма, отримана для стандартного зразка кристалічної сполуки (1), отриманої за способом згідно Прикладу 1. На Фігурі 2 представлена інша порошкова рентгенограма, отримана для кристалічної сполуки (1). На Фігурі 3 представлена порошкова рентгенограма, отримана для аморфної форми сполуки (1) Research Lot 6, отриманої за способом, описаному у Прикладі 1а в WO 08/005519. На Фігурі 4 представлена термограма диференціальної скануючої калориметрії для стандартного зразка кристалічної сполуки (1) (сканування при швидкості нагрівання 1°С/хв), отриманої за способом, описаним далі у Прикладі 1. На Фігурі 5 представлена термограма диференціальної скануючої калориметрії для аморфної форми сполуки (1) Research Lot 6 (сканування при швидкості нагрівання 5°С/хв), отриманої за способом, описаним в Прикладі 1а в WO 08/005519. Докладний опис винаходу Кристалічну сполуку (1) характеризують як тверду речовину, що містить сполуку (1), молекули якої упаковані регулярно впорядкованим чином з повторюваними фрагментами за усіма трьома напрямками у просторі. Ідентифікація кристалічної структури може бути легко проведена за допомогою ряду способів, відомих фахівцям у даній області техніки. При дослідженні випробуваної композиції під мікроскопом часто спостерігають наявність правильних форм, що дозволяєприпустити впорядкований характер внутрішньої структури. У варіанті реалізації, що відноситься до кристалічної сполуки, отриманій відповідно до Прикладу 1, зазначена правильна форма у загальному випадку являє собою стрижнеподібну або голчасту форму. Іншим способом ідентифікації кристалічної сполуки (1) є метод рентгенівської порошкової дифракції (X-ray powder diffraction, XRPD). Регулярно впорядкована структура молекул у кристалі розсіює падаючі рентгенівські промені з одержанням чіткої дифракційної картини, що відображається у вигляді спектральних піків. Зазначена картина спектральних піків для кристалічної сполуки (1) представлена на Фігурах 1 і 2. З іншого боку, на Фігурі 3 представлена порошкова рентгенограма для по суті аморфної сполуки (1), на якій відсутні явно виражені піки. У той час як піки на порошковій рентгенограмі кристалічної сполуки (1) можуть варіюватися за інтенсивністю, загальна картина, одержувана при повторному рентгенівському дифракційному аналізі, залишається такою ж самою. Порошкова рентгенограма кристалічної сполуки (1) містить основний пік (основні піки) при приблизно 17 градусах тета - 2, як правило, при 17,4 і 17,5. Під терміном «приблизно» мають на увазі «у рамках звичайних коливань значень при реєстрації піків порошкової дифракції рентгенівських променів». Подібні коливання можуть мати місце при використанні різних приладів, параметрів налаштування приладів, партій продукту, обробки продукту після кристалізації, такої як мікронізація або розмел, і різних способів підготовки зразків. У загальному випадку, «приблизно» означає ±0,5 градуса тета - 2. Приклад коливань подібного роду можна спостерігати при порівнянні Фігур 1 і 2. Зокрема, інтенсивність піку (наприклад, при приблизно 30) може залежати від орієнтації кристалів. 3 UA 99466 C2 5 10 15 20 25 30 35 40 45 50 55 Ілюстративними прикладами інших домінантних піків на дифракційній картині кристалічної сполуки (1) є піки в положенні приблизно 8, 10, 13, 16, 19 і 24 у градусах тета - 20, як правило, 8.4, 10.0, 13.5, 15.7, 16.8, 16.9, 18.8 и 24.4°. Будь-який із цих піків, або кілька піків (особливо, при 8, 10, 15.7, 16.7 і 16.9 за наявності або відсутності піків приблизно в положенні 17°), можна застосовувати для ідентифікації порошкової рентгенограми кристалічної сполуки (1). Для ідентифікації кристалічної форми сполуки (1) не потрібно присутності якого-небудь одного або декількох основних піків, що присутні на Фігурах 1 або 2. Скоріше, присутність або відсутність основних піків, як правило, беруть до уваги поряд з іншими діагностичними характеристиками (наприклад, термограмою ДСК) при ідентифікації досліджуваного зразка як кристалічної сполуки (1). Крім того, кристалічна сполука (1) характеризується термограмою ДСК, на якій початок поглинання тепла спостерігається при температурі приблизно 235°С на кривий диференціальної скануючої калориметрії. Як правило, при проведенні даних вимірювань враховують деякі коливання результатів (звичайно 1 - 3 °С). Кристалічна сполука (1) також характеризується величиною теплоти плавлення (DH пл), що становить приблизно 81 Дж/г (42 кДж/моль). Кристалічну сполуку (1) одержують за способом, що включає розчинення сполуки (1) у розчиннику і одержання кристалів з отриманого розчину. Типовими розчинниками, що застосовуються відповідно до винаходу, є етилацетат, ізопропіловий спирт або співрозчинник, що містить етилацетат та ізопропіловий спирт. Інші підходящі розчинники підбирають за діаграмою розчинності з McConville, F.X. "Pilot Plant Real Book" (2002), де графічно представлені значення діелектричної постійної і параметра розчинності Гільдебранду для різних розчинників. Розчинники, близькі до етилового спирту або ізопропілового спирту за положенням на діаграмі (діелектрична постійна від 2,5 до 20 і параметр Гільдебранду від 15 до 24), являють собою етиловий ефір, ізобутилацетат, бутилацетат, анізол, хлорбензол, хлороформ, метилацетат, ТГФ, дихлорметан, дихлоретан, 1,2-дихлорбензол, метилізобутилкетон, метилетилкетон, циклогексанон, ацетон, 1-бутанол, 2-метоксиетанол, ізобутанол, 2-бутанол, циклогексанол, ізоаміловий спирт, піридин, метилформіат, 1-пентанол і/або 2-бутоксиетанол. Використання деяких із зазначених розчинників не є кращим внаслідок їх токсичності, однак цю проблему можна вирішити шляхом ретельного видалення розчинника із продукту. Середній фахівець у даній області техніки здатний провести лабораторне дослідження для визначення того, чи підходить розглянутий розчинник для одержання кристалічної сполуки (1). Комбінації зазначених розчинників також перебувають у рамках даноговинаходу. В основу винаходу покладена виявлена особливість, що сприяє одержанню кристалічної сполуки (1), яка полягає в тому, що для одержання і/або оптимізації процесу одержання кристалічного продукту необхідно контролювати вміст води у використовуваному для кристалізації розчиннику. Наприклад, при застосуванні етилацетату як розчинника верхньої межі вмісту води становить приблизно від 0,6% до 0,9% мас. Фактор, що додатково враховується, пов'язаний з вмістом води, полягає в можливості застосування води для видалення інших форм сполуки (1), що мають меншу розчинність в рідких ліпофільних фармацевтичних носіях у порівнянні з розчинністю кристалічної вільної основи. Наприклад, розчинність хлориду сполуки (1) менше розчинності вільної основи в розчинах жирних кислот, що застосовуються у даному описі згідно винаходу як носії. Досить високий вміст подібних солей викликає небажане помутніння фармацевтичного продукту. Завершальний етап синтезу відповідно до Прикладу 1 полягає в одержанні суміші вільної основи з малою кількістю хлориду. Хлоридну сіль, що викликає помутніння продукту, видаляють шляхом первісного розчинення продукту у розчиннику з порівняно високим вмістом води (приблизно 3-10%) у лужному середовищі. Кип'ятіння зі зворотним холодильником у цьому розчиннику забезпечує наявність води в кількості, достатній для перетворення хлориду назад у вільну основу. Потім кристалічну вільну основу кристалізують із цього розчинника. Можливо повторне проведення даного процесу зі зниженим вмістом води для поступового видалення хлоридної солі із продукту. Далі проводять завершальний етап при низькому вмісті води (зазвичай менше приблизно 0,9% води) для кристалізації вільної основи, що по суті не містить аморфної сполуки (1). У загальному випадку, помутніння фармацевтичного препарату не спостерігається, якщо вміст хлориду в кінцевому продукті становить, як правило, менше приблизно 100 ррт. Використовувана кількість води варіюється залежно від концентрації домішки хлоридної солі та інших експериментальних змінних, які можуть бути визначені фахівцем у даній області техніки. У цілому, вміст води у використовуваному для кристалізації розчиннику регулюють як для забезпечення перетворення хлоридів (або інших порівняно 4 UA 99466 C2 5 10 15 20 25 30 35 40 45 50 55 60 розчинних у воді солей сполуки (1)), так і для того, щоб уникнути утворення аморфної сполуки (1). Припустима кількість води для здійснення кожної із цих функцій розрізняється залежно від розчинника або розчинників, що застосовуються для кристалізації, концентрації сполуки (1), температури на етапі кристалізації, часу кристалізації, припустимого вмісту аморфної сполуки (1) та інших змінних. Таким чином, оптимальний вміст води, необхідний для досягнення заданого результату, визначає фахівець у даній галузі техніки, як правило, шляхом проведення звичайного дослідження із застосуванням матриці змінних. Мінімальна концентрація води, що дозволяє уникнути утворення аморфної сполуки (1), зв'язана скоріше із практичними міркуваннями економічного характеру. Наприклад, концентрація води 0,05% є прийнятною. У загальному випадку, завершальний етап кристалізації проводять у по суті безводному розчиннику. По суті безводний розчинник визначають як розчинник, що містить воду в кількості, досить малій для того, щоб кінцевий продукт містив кристалічну сполуку (1) і по суті не містив аморфної сполуки (1), тобто щоб загальний вміст аморфної сполуки становив, як правило, менше приблизно 40%, зазвичай менше приблизно 30, 20, 10, 5, 3, 2 або 1% мас. аморфної сполуки (1) від загальної маси всіх форм сполуки (1) у композиції продукту. У загальному випадку, по суті безводний розчинник містить приблизно 0,5%-0,9% мас. води відносно маси використовуваного для кристалізації розчинника. Проте, припустимий більший вміст води, якщо припустима більша частка аморфної сполуки (1) у кінцевому продукті. Однак оптимальною є відсутність кількостей аморфної сполуки, що піддаються виявленню, (1) у композиції сполуки (1). Вміст води регулюють будь-якими способами, що забезпечують бажаний вміст води на етапі кристалізації. Коли необхідно запобігти утворенню аморфної сполуки (1), придатні способи мінімізування або зниження вмісту води включають додавання осушувачів і/або азеотропне видалення води. Найбільш зручно проводити видалення води в ході розчинення сполуки (1) при кип'ятінні зі зворотним холодильником безпосередньо перед кристалізацією. Зрозуміло, регулювання вмісту води включає також додавання води, зазвичай на стадіях перетворення хлоридної солі. Можливе використання аморфної сполуки (1) як вихідної речовини для кристалізації (конверсія форм). Альтернативно, кристалізацію проводять безпосередньо з кінцевих продуктів реакції без проміжного виділення аморфної сполуки (1). Кристалізацію зазвичай проводять із розчину сполуки (1) або шляхом розчинення сполуки (1) у розчиннику або суміші розчинників при нагріванні зі зворотним холодильником (достатньому для розчинення сполуки (1), приблизно від 1 до 5 годин) з наступним охолодженням приблизно до 18-23°С протягом 4-8 годин, після чого можливе перемішування приблизно протягом 8-20 годин при 18-23°С. Перемішування не є обов'язковим, однак воно збільшує швидкість кристалізації. Нагрівання зі зворотним холодильником не є критично важливим, оскільки необхідно всього лише перевести сполуку (1) у розчин. Проте, перевага нагрівання зі зворотним холодильником складається в прискоренні розчинення сполуки (1) з одночасним азеотропним видаленням води. Вміст води регулюють до початку кристалізації або під час кристалізації, або на обох етапах, хоча в загальному випадку найкраще забезпечити зниження вмісту води нижче заданого граничного значення до того, як сполука (1) починає випадати в осад у вигляді аморфної поліморфної форми. Утворення аморфної речовини оптимізують шляхом забезпечення порівняно тривалого часу проведення кристалізації, застосування підвищених температур і знижених концентрацій сполуки (1). Визначення різних оптимальних параметрів процесу кристалізації без праці може бути здійснено середнім фахівцем у даній галузі техніки. Один з варіантів реалізації являє собою композицію, отриману шляхом об'єднання кристалічної сполуки (1) з фармацевтично прийнятним наповнювачем з наступним одержанням фармацевтичної дозованої форми, такої як таблетка або капсула. Вміст кристалічної сполуки (1) у кінцевому продукті не є обов'язковим. У той же час мається на увазі, що дозовані форми, отримані із кристалічної сполуки (1), містять тільки сполуку (1) у кристалічній формі. Проте, відповідно до деяких варіантів реалізації, кристалічна сполука (1) являє собою напівпродукт для розчинення в носії або наповнювачі. Кристалічна сполука (1) згідно із даним винаходу вводять суб'єкту, що являє собою ссавця (включаючи людину), будь-якими способами, відомими у даній галузі техніки, а саме: пероральним, інтраназальним, підшкірним, внутрішньом'язовим, інтрадермальним, внутрішньовенним, внутрішньоартеріальним, парентеральним способом або шляхом катетеризації в терапевтично ефективній кількості, тобто в кількості, що придушує ВГС або реплікацію ВГС. Думають, що зазначена кількість являє собою кількість, що забезпечує вміст у 5 UA 99466 C2 5 10 15 20 25 30 35 40 45 50 55 плазмі, що становить приблизно 100 нМ, в 3 рази перевищуючий рівень ЕС90, встановлений із застосуванням білка (protein-adjusted EC90). У загальному випадку думають, що даний рівень забезпечується шляхом щоденного перорального прийому в кількості приблизно 5 мг/кг; зазвичай приблизно від 0,7 до 2,2 мг/кг; як правило, приблизно 1,2 мг/кг маси тіла для людини. Оптимальна доза сполуки згідно із даним винаходу залежить від безлічі факторів, відомих фахівцеві в даній галузі техніки, у тому числі від біодоступності сполуки в конкретній сполуці, метаболізму і розподілу сполуки в організмі суб'єкта, прийому до або після їжі, вибору носіїв і наповнювачів, що входять до складу, та інших факторів. Як правило, придатну дозу визначає фахівець у даній галузі техніки в ході доклінічних і клінічних досліджень. Терапевтично ефективну кількість сполуки відповідно до винаходу можна розділити на декілька субодиниць для прийому протягом дня або вводити щодня або з інтервалами, що перевищують один день, залежно від природи інфекції, загального стану пацієнта і конкретного складу на основі сполуки згідно із даним винаходом. Як правило, сполуку приймають два рази в день. Сполуку згідно із даним винаходом застосовують у сполученні з іншими агентами, ефективними проти інфекцій, що викликаються ВГС. У ході лікування зазначені агенти можна вводити окремо або в комбінації зі сполукою (1) у вигляді єдиної дозованої форми, такої як таблетка, розчин для внутрішньовенного введення або капсула. Зазначені інші агенти включають, наприклад, інтерферон-альфа, рибавірин і/або сполуки згідно ЕРІ 162196, WO 03/010141, WO 03/007945, WO 00/204425 і/або WO 03/010140 (та інших заявок, що входять у сімейство патентів-аналогів для зазначених патентних документів). Інші агенти, що підходять для введення в ході лікування разом зі сполукою згідно із даним винаходом, включають сполуки, що проходять у цей час клінічні випробування, зокрема, інгібітори протеази ВГС, такі як VX-950 (Vertex Pharmaceuticals), SCH 5030347 (Schering Plough) і BILN-2061 (Boehringer Ingelheim), нуклеозидні інгібітори ВГС, такі як NM283, NM107 (обоє Idenix/Novartis) і R1626 (Hoffmann-LaRoche), а також ненуклеозидні інгібітори ВГС, включаючи ВГС-086 і -796 (обоє ViroPharma/Wyeth). Додаткові антивірусні агенти застосовують у традиційних кількостях. Якщо ефект від застосування сполуки відповідно до винаходу і додаткової сполуки є адитивним, то можливе пропорційне зменшення кількості кожного активного агента, тим більше якщо агенти діють синергічно. Проте, у загальному випадку агенти застосовують у звичайних активних кількостях у складі єдиних комбінованих композицій. Агенти, що вводяться спільно, як правило, вводять до складу єдиних композицій разом зі сполукою відповідно до винаходу, якщо зазначені агенти і сполука хімічно сумісні та призначені для введення однаковим шляхом. Якщо це не так, то зазначені агенти і сполуку можна забезпечити у вигляді медичного набору або упаковки, що містять два агенти в різних контейнерах або відділеннях. Як правило, сполуку відповідно до винаходу забезпечують у вигляді вільної основи, однак можливе одержання її у вигляді солі. Солі зазвичай одержують шляхом додавання органічних і/або неорганічних кислот до вільної основи. Приклади включають (1) неорганічні кислоти, такі як галогеноводневі кислоти, наприклад, хлористоводнева або бромистоводнева кислота, сірчана кислота, азотна кислота, фосфорна кислота і сульфамінові кислоти; або (2) органічні кислоти, такі як оцтова, пропіонова, гідрооцтова (гліколева), бензойна, 2-гідроксипропіонова, 2оксопропіонова, молочна, фумарова, винна, піровиноградна, малеїнова, малонова, яблучна, саліцилова (наприклад, 2-гідроксибензойна), п-аміносаліцилова, ізетіонова, лактобіонова, бурштинова, щавлева та лимонна кислоти; органічні сульфокислоти, такі як метансульфонова, етансульфонова, бензосульфонова, п-толуолсульфонова, С1-С6 алкилсульфонові, бензолсульфонові, п-толуолсульфонові і циклогексансульфамінова кислоти. Типові солі являють собою хлориди, сульфати, бісульфати, мезилати, безилати, езилати, фосфати, оксалати, малеати, сукцинати, цитрати, малонати і/або фумарати. Крім того, даний винахід включає солі, утворені сполукою згідно із даним винаходом і однією або декількома амінокислотами, зазвичай природними амінокислотами, такими як амінокислоти, що містяться в білках. Кислотний противоіон бажано є фізіологічно безпечним і нетоксичним або ж фармацевтично прийнятним, якщо тільки сіль не є напівпродуктом, що застосовується для одержання сполук, коли токсичність не має істотного значення. Звичайна сполуку (1) вводять у формі вільної основи, однак придатні солі включають мезилати (солі метансульфокислоти) і НСl. Сполуки відповідно до винаходу включають сольвати, утворені сполукою згідно із даним винаходом або його солям, такі як, наприклад, гідрати, алкоголяти і т.п. Склади на основі фармацевтичної сполуки відповідно до винаходу можна готувати із застосуванням традиційних фармацевтичних носіїв і наповнювачів, що обираються відповідно 6 UA 99466 C2 5 10 15 20 25 30 35 40 45 50 55 60 до звичайної практики. Таблетки містять ексципієнти; агенти, що сприяють ковзанню; наповнювачі; сполучні агенти і т.п. Водні сполуки готують у вигляді стерильних форм, при цьому якщо зазначені сполуки призначені для доставки шляхом, відмінним від перорального, то в загальному випадку вони повинні бути ізотонічними. Сполуки можуть містити наповнювачі, наприклад, запропоновані в "Handbook of Pharmaceutical Excipients" (2005), включаючи аскорбінову кислоту та інші антиоксиданти; хелатуючі агенти, такі як ЕДТА; вуглеводи, такі як декстрин, гідроксиалкілцелюлоза, гідроксиметилцелюлоза і/або органічні кислоти, такі як олеїнова або стеаринова кислота. Термін «фармацевтично прийнятний носій» у даному описі позначає будь-який матеріал або речовину, введену до складу разом з активним інгредієнтом для полегшення його одержання і/або його застосування або поширення до місця, що піддається лікуванню. Придатні фармацевтичні носії для застосування у сполуках згідно із даним винаходом добре відомі фахівцям у даній галузі техніки. Такі носії включають допоміжні речовини, такі як зволожуючі агенти; диспергуючі агенти; адгезиви; емульгатори; розчинники; агенти, що полегшують ковзання; матеріали покриття; антибактеріальні та протигрибкові агенти (наприклад, фенол, сорбінова кислота, хлорбутанол) та ізотонічні агенти (такі як цукри або хлористий натрій), за умови, що ці речовини застосовуються у фармацевтичній практиці, тобто не є токсичними для ссавців. Фармацевтичні композиції згідно із даним винаходом одержують будь-якими придатними способами, наприклад, шляхом гомогенного змішування, нанесення покриття і/або здрібнювання активних інгредієнтів в один етап або кілька етапів з обраною речовиною-носієм і, там, де це доцільно, з іншими допоміжними речовинами, такими як поверхнево-активні речовини. Композиції, що містять сполуку згідно із даним винаходом, приготовлену у вигляді мікросфер (зазвичай діаметр мікросфери становить приблизно від 1 до 10 мкм (gm)), підходять як сполуки з уповільненим або контрольованим вивільненням. В одному з можливих складів сполуку (1) здрібнено до тонкоздрібненої форми, при цьому зазвичай середній розмір часток у будь-якій точці варіюється в інтервалі приблизно від 1 до 20 мікронів. Продукт відповідно до Прикладу 1 являє собою голчасті або стрижнеподібні кристали, довжина яких варіюється в інтервалі приблизно від 25 до 40 мікронів. Зазначені кристали можна мікронізувати на вихровому млині Jet mill-00 приблизно при 60-80 psi (413, 7-551,6 кПа) для одержання часток розміром приблизно 3-4 мікрони, площа поверхні яких становить 7-8 кв.м/г. Проте, вихідні розміри кристалів різні в різних партіях і ступінь їх здрібнювання є предметом вибору. Відповідно, мікронізовану кристалічну сполуку (1) визначають попросту як кристалічну або аморфну сполуку (1), що піддавалася процедурі мікронізації, таку як сполука, представлена в даному описі як приклад. Ні розмір, ні площа поверхні отриманих часток не має істотного значення. Мікронізовану сполуку (1) суспендують у водяному розчині; можливо, додаючи до нього суспендуючий агент, емульгатори та поверхнево-активні речовини, як описано далі. Зазвичай фармацевтична сполука являє собою солюбілізовану форму сполуки (1), де кристалічну сполуку (1) розчинено у відповідному розчиннику або солюбілізуючій речовині, або їх комбінації. Кристалічну сполуку (1) солюбілізують у фармацевтично прийнятному наповнювачі для введення з терапевтичною або профілактичною метою. Придатні розчини сполуки (1) для фармацевтичних препаратів включають воду разом з різними органічними кислотами (зазвичай С4 - С24), як правило, з жирними кислотами, такими як капринова, олеїнова, лауринова, пальмітинова і/або міристинова кислота. Жирні кислоти можуть бути насиченими або ненасиченими або ж являти собою суміші насичених і ненасичених кислот. Крім того, на додаток до органічних кислот або замість них можна використовувати поліетиленгліколі (ПЕГи) і/або моно-, ди- або тригліцериди з коротким, середнім або довгим вуглецевим ланцюгом. Можливо також використання в такий же спосіб пегільованих жирних кислот з коротким, середнім або довгим вуглецевим ланцюгом. Найчастіше органічні кислоти є карбоновими кислотами, кислотні властивості яких пов'язані з карбоксильною групою -СООН. Сульфонові кислоти, що містять групу OSO 3H, є порівняно сильними для застосування відповідно до винаходу. У загальному випадку, кислота бажано містить ліпофільний домен. Придатними є моно- і дикарбонові кислоти. Придатні поверхнево-активні речовини можна використовувати у всіх сполуках згідно із даним винаходом (будь-яку або декілька з нижчеперелічених речовин, як правило, кожну із зазначених речовин). Такі речовини відомі також як емульгатори або емульсифікатори і підходять для застосування у фармацевтичних композиціях згідно із даним винаходом. Зазначені речовини є неіонними, катіонними і/або аніонними речовинами, що мають придатні емульгуючі, диспергуючі і/або змочувальні властивості. Придатні аніонні поверхнево-активні 7 UA 99466 C2 5 10 15 20 25 30 35 40 45 50 55 60 речовини включають водорозчинні мила і водорозчинні синтетичні поверхнево-активні речовини. Придатні мила являють собою солі лужних або лужноземельних металів, незаміщені або заміщені солі амонію і вищих жирних кислот (С10-С22), наприклад, натрієві або калієві солі олеїнової або стеринової кислоти, або суміш солей природних жирних кислот, отриманих з кокосової олії або тваринного жиру (tallow oil). Синтетичні поверхнево-активні речовини включають натрієві або кальцієві солі поліакрилових кислот; сульфати та сульфонати жирів; сульфоновані похідні бензімідазолу та алкіларилсульфонати. Сульфати або сульфонати жирів зазвичай присутні у формі солей лужних або лужноземельних металів, незаміщених солей амонію або солей амонію, заміщених алкілом або арилом, що містить від 8 до 22 атомів вуглецю, наприклад, у вигляді натрієвої або кальцієвої солі лігносульфонової кислоти або додецилсульфонової кислоти або у вигляді суміші сульфатів жирних спиртів, отриманих із природних жирних спиртів, солей лужних або лужноземельних металів складних ефірів сірчаної або сульфонової кислот (таких як лаурилсульфат натрію) і сульфонових кислот із продуктами приєднання жирних спиртів / окису етилену. Придатні сульфоновані похідні бензімідазолу краще містять від 8 до 22 атомів вуглецю. Приклади алкіларилсульфонатів включають солі натрію, кальцію і додецилбензосульфонової кислоти або дибутилнафталінсульфонової кислоти, або продукт конденсації нафталінсульфонової кислоти і формальдегіду. Крім того, придатними є відповідні фосфати, наприклад, солі складних ефірів фосфорної кислоти, і продукти приєднання п-нонілфенолу та оксиду етилену і/або оксиду пропілену, або фосфоліпіди. Придатні для застосування з даною метою фосфоліпіди являють собою природні (вироблені клітинами організму рослини або тварини) або синтетичні фосфоліпіди типу цефаліну або лецитину, такі як, наприклад, фосфатидилетаноламін, фосфатидилсерин, фосфатидилгліцерин, лізолецитин, кардіоліпін, диоктанілфосфатидил-холін, дипальмітоїлфосфатидил-холін та їх суміші. Водні емульсії, що містять такі речовини, також перебувають у рамках даного винаходу. Придатні неіонні поверхнево-активні речовини включають поліетоксильовані та поліпропоксильовані похідні алкілфенолів, жирні спирти, жирні кислоти, аліфатичні аміни або аміди, що містять щонайменше 12 атомів вуглецю в молекулі, алкіларенсульфонаті і диалкілсульфосукцинати, такі як похідні ефіру полігліколю та аліфатичних і циклоаліфатичних спиртів, насичених і ненасичених жирних кислот і алкілфенолів, при цьому зазначені похідні краще містять від 3 до 10 гліколевих ефірних груп і від 8 до 20 атомів вуглецю в (аліфатичному) вуглеводневому фрагменті алкілфенолу. Далі, придатними неіонними поверхнево-активними речовинами є водорозчинні продукти приєднання поліетиленоксиду і поліпропіленгліколю, етилендиамінопропіленгліколь, що містить від 1 до 10 атомів вуглецю у вуглецевому ланцюзі, при цьому зазначені продукти приєднання включають від 20 до 250 ефірних груп етиленгліколю і/або від 10 до 100 ефірних груп пропіленгліколю. Подібні сполуки зазвичай містять від 1 до 5 одиниць етиленгліколю на одиницю пропіленгліколю. Репрезентативні приклади неіонних поверхнево-активних речовин являють собою нонілфенол-поліетоксиетанол, полігліколеві ефіри касторової олії, продукти приєднання поліпропілен/поліетиленоксиду, трибутилфеноксиполіетоксиетанол, поліетиленгліколь і октилфеноксиполіетоксиетанол. Крім того, як неіонні поверхнево-активні речовин підходять ефіри жирних кислот поліетиленсорбітану (такі як поліоксиетилен сорбітану триолеат), гліцерол, сорбітан, сахароза і пентаеритрит. Придатні катіонні поверхнево-активні речовини включають четвертинні амонієві солі, зокрема, галоїди, що містять 4 вуглеводневих радикали, що можливо містять як замісників галоген, феніл, заміщений феніл або гідрокси; наприклад, четвертинні амонієві солі, що містять як замісник у атома N щонайменше один алкільний радикал С 8 - С22 (наприклад, цетил, лаурил, пальмітил, містрил і олеїл) і, як інші замісники, незаміщений або галогенований нижчий алкіл, бензил і/або гідрокси-нижчий алкіл. Ще докладніший опис поверхнево-активних речовин, що підходять для застосування з даною метою, див. в "McCutcheon's Detergents and Emulsifiers Annual" (MC Publishing Crop., Ridgewood, New Jersey, 1981), "Tensid-Taschenbucw", 2nd ed. (Hanser Verlag, Vienna, 1981) і "Encyclopedia of Surfactants," (Chemical Publishing Co., New York, 1981). Сполуку згідно із даним винаходом вводять будь-яким шляхом, що підходить при лікуванні конкретного стану, наприклад, пероральним, ректальним, назальним, місцевим (включаючи окулярний, букальний і сублінгвальний), вагінальним і парентеральним (включаючи підшкірний, внутрішньом'язовий, внутрішньовенний, внутрішньошкірний, інтратекальний і епідуральний). Кращі шляхи введення можуть бути різними і залежать, наприклад, від стану суб'єкта; однак, як правило, введення здійснюють пероральним шляхом. Склади на основі сполуки згідно із даним винаходом, призначені для перорального прийому, зазвичай представлені у вигляді дискретних одиниць, таких як капсули, облатки або таблетки, кожна з яких містить певну кількість активного інгредієнта; у порошкоподібній або гранульованій 8 UA 99466 C2 5 10 15 20 25 30 35 40 45 50 55 60 формі; у вигляді розчину або суспензії у водній або неводній рідині; або у вигляді рідкої емульсії типу «олія у воді» або «вода в олії». Сполука відповідно до винаходу може бути представлена у вигляді болюсу, електуарію або пасти. Таблетки виготовляють шляхом пресування або формування, можливо із застосуванням одного або декількох допоміжних інгредієнтів. Пресовані таблетки виготовляють шляхом пресування в придатній машині сполуки відповідно до винаходу у вільнотекучій формі, такій як порошок або гранули, можливо в суміші з наповнювачем, змазуючою речовиною, інертним розріджувачем, консервантом, поверхнево-активною речовиною і/або диспергуючим агентом. Формовані таблетки зазвичай виготовляють шляхом формування у придатній машині суміші порошкоподібної сполуки, змоченої інертним рідким розріджувачем. На таблетки можна нанести покриття або ділильну насічку, або ж виготовити їх так, щоб забезпечити вповільнене або контрольоване вивільнення активних інгредієнтів, що містяться в них. Можливо місцеве застосування сполук у вигляді кремів і мазей, що містять активний інгредієнт (інгредієнти) у кількості, що становить, наприклад, від 0,075 до 20% вага/вагу (включаючи активний інгредієнт (інгредієнти) в інтервалі від 0,1% до 20% із кроком 0,1% вага/вагу, наприклад, 0,6% вага/вагу, 0.7% вага/вагу, і т.д.), краще від 0,2 до 15% вага/вагу, і найкраще від 0,5 до 10% вага/вагу. У складі мазі сполуку використовують разом з парафіновою або водорозчинною мазевою основою. Альтернативно, сполуку вводять до складу крему разом із кремовою основою типу «вода в олії». При бажанні водна фаза кремової основи може містити, наприклад, щонайменше 30% вага/вагу багатоатомного спирту, тобто спирту, що містить дві або більше гідроксильні групи, такого як пропіленгліколь, бутан-1,3-діол, манітол, сорбітол, гліцерин і поліетиленгліколь (включаючи ПЕГ400) і їх суміші. При бажанні склади для місцевого застосування можуть включати сполуку, що збільшує абсорбцію або проникнення активного інгредієнта через шкіру та інші ушкоджені ділянки. Приклади подібних агентів, що підсилюють шкірне проникнення, включають диметилсульфоксид і відповідні аналоги. Олійна фаза емульсій згідно із даним винаходом складається із традиційних інгредієнтів, з'єднаних відомими способами. У той час як дана фаза може містити лише емульгатор (що називається також емульгентом), бажане включення до складу суміші щонайменше одного емульгатора з жиром або олією, або і з жиром, і з олією. Гідрофільний емульгатор можна ввести до складу разом з ліпофільним емульгатором, що виступає як стабілізатор. Крім того, краще спільне введення до складу і олії, і жиру. Емульгатор (емульгатори) разом зі стабілізатором (стабілізаторами) утворюють так званий емульгуючий віск, а віск разом з олією і жиром утворюють так звану емульгуючу мазеву основу, що утворює олійну дисперсну фазу сполуки крему. Підбор придатних олій і жирів для сполуки заснований на досягненні заданих косметичних характеристик. Таким чином, зазначений крем може являти собою нежирний продукт, що не залишає плям та легко змивається придатної консистенції, який не витікає з туб та інших видів контейнерів. Можливе застосування одно- або двоосновних алкілових складних ефірів з лінійним або розгалуженим ланцюгом, таких як диізоадипат, ізоцетилстеарат, пропіленгліколевий диефір жирних кислот кокосового горіха, ізопропілміристат, децилолеат, ізопропілпальмітат, бутилстеарат, 2-етилгексилпалмітат або суміш складних ефірів з розгалуженим ланцюгом, відома як Кродамол КАП, причому останні три речовини є кращими складними ефірами. Їх можна застосовувати окремо або в комбінації, залежно від необхідних властивостей. Як альтернативу можна використовувати ліпіди з високою температурою плавлення, такі як білий м'який парафін і/або рідкий парафін, або інші мінеральні олії. Сполуки, що підходять для місцевого офтальмологічного застосування, включають також очні краплі, у яких активний інгредієнт розчинений або суспендований у відповідному носії, зокрема, у водному розчиннику, що підходить для розчинення активного інгредієнта. Активний інгредієнт може міститися в подібних сполуках у концентрації від 0,5 до 20%, краще від 0,5 до 10%, зокрема, у концентрації приблизно 1,5 % в/в. Сполуки, що підходять для місцевого застосування в ротовій порожнині, включають коржі, що містять активний інгредієнт в ароматизованій основі, у загальному випадку представляє собою сахарозу і гуміарабік або трагакант; пастилки, що містять активний інгредієнт в інертній основі, такий як желатин і гліцерин або сахароза і гуміарабік; рідина для полоскання рота, що містить активний інгредієнт у придатному рідкому носії. Сполуки для ректального застосування можуть мати форму супозиторіїв з придатною основою, що містить, наприклад, кокосову олію або саліцилат. Композиції, що підходять для інтраназального введення, у яких носій є твердою речовиною, включають крупнодисперсний порошок з розміром частинок в інтервалі, наприклад, 20 - 500 мкм (включає розмір частинок в інтервалі від 20 до 500 мікронів із кроком в 5 мікрон, наприклад, 30 мікрон, 35 мікрон і т.д.), що 9 UA 99466 C2 5 10 15 20 25 30 35 вводиться за допомогою аерозольного або порошкового інгалятора, при цьому велика кількість прикладів таких інгаляторів є в продажі. Придатні сполуки, у яких носієм є рідина, для застосування, наприклад, у вигляді назального спрея або краплі для носа, включають водні або олійні розчини активного інгредієнта. Сполуки, що підходять для вагінального введення, можуть бути представлені у вигляді песаріїв, тампонів, кремів, гелів, паст, пін або аерозолів, що містять поряд з активним інгредієнтом носії, добре відомі фахівцям у даній галузі техніки. Сполуки, що підходять для введення парентеральним шляхом, включають водні і неводні стерильні розчини для ін'єкцій, які можуть містити антиоксиданти, буферні речовини, бактеріостати і розчинені речовини, які надають сполуці ізотонічні стосовно крові передбачуваного реципієнта властивості; а також водні і неводні стерильні суспензії, що можливо містять суспендуючі агенти та загущувачі. Ці сполуки представлені в контейнерах, що містять одиничну дозу або безліч доз, наприклад, герметично закриті ампули і пухирці, і можуть зберігатися в сублімованому (ліофілізованому) вигляді, коли безпосередньо перед використанням необхідно лише додати до продукту стерильний рідкий носій, наприклад, воду для ін'єкцій. Розчини і суспензії для ін'єкцій, призначені для негайного застосування, можна приготувати зі стерильних порошків, гранул і таблеток, подібних вищеописаним. Сполука згідно із даним винаходом може бути отримана у вигляді композиції з контрольованим вивільненням, у якій вивільнення сполуки контролюють і регулюють із метою зменшення частоти прийомів дозованої сполуки або для поліпшення фармакокінетичного і токсичного профілю сполуки відповідно до винаходу. Композиції з контрольованим вивільненням одержують традиційними способами, багато хто з яких включає введення активної сполуки до складу разом з одним або декількома полімерними носіями, такими як поліефір, поліамінокислоти, полівінілпіролідон, співполімер етилену з вінілацетатом, метилцелюлоза, карбоксиметилцелюлоза і/або протамінсульфат. Швидкість вивільнення ліків і тривалість дії можна контролювати шляхом поміщення активного інгредієнта всередину частинок, наприклад, у мікрокапсули, отримані з полімерних матеріалів, таких як гідрогелі, полімолочна кислота, гідроксиметилцелюлоза, поліметилметакрилат та інші вищеописані полімери. Крім того, придатними є колоїдні системи доставки лікарського препарату, такі як ліпосоми, мікросфери, мікроемульсії, наночастинки, нанокапсули і т.д. Залежно від шляху введення фармацевтична композиція, наприклад, таблетки, може вимагати нанесення захисної оболонки. Для забезпечення ще повнішого розуміння даного винаходу далі наведені приклади, які є винятково ілюстративними і не обмежують даний винахід. Процентні вмісти компонентів композиції є ваговими відсотками, якщо з контексту явно не випливає інше. Приклад 1 Синтез кристалічного 5-((6-(2,4-біс(трифторметил)феніл]піридазин-3-іл)метил)-2-(2фторфеніл)-5H-імідазо[4,5-с]піридину 40 10 UA 99466 C2 5 10 15 У реактор, що містить 2,4-біс(трифторметил)бромбензол (1.00 ек) і тетрагірофуран (ТГФ), i помістили ізопропілхлорид магнію ( PrMgCl) (2M у ТГФ, 1.14 ек) за підтримки температури -10°С всередині реактора. Суміш перемішували при -10°С до завершення реакції за даними ВЕЖХ. Отриману суміш перенесли в другий реактор, що містить триметилборат (2,26 ек) і ТГФ при температурі -10°С всередині реактора. Коли за даними ВЕЖХ-моніторингу вміст 1,3біс(трифторметил)бензолу склав не більше 2%, реакцію погасили додаванням водної НСl (водяного розчину хлористоводневої кислоти), отриманого з води і концентрованої 37% хлористоводневої кислоти; при цьому температуру вмісту реактора підтримували на рівні, що не перевищує 25°С. Після перемішування вмісту протягом 1-2 годин і відстоювання протягом приблизно 30 хвилин, шари розділили. Органічний шар промили сольовим розчином, змішаним з водою, потім сконцентрували під вакуумом. Потім додали гептан і знову сконцентрували вміст під вакуумом. Всі операції повторили ще один раз. Знову додали гептан і остудили отриману суспензію до 3 °С, потім перемішували при даній температурі протягом 4-6 год. 11 UA 99466 C2 Продукт відфільтрували промили гептаном двічі і висушили під вакуумом, максимальна температура при сушінні склала 40°С. 5 10 15 20 25 3-хлор-6-метилпіридазин (1.00 ек), 2-(дициклогексилфосфіно)біфеніл (0.05 ек), 2,4біс(трифторметил)фенілборонову кислоту (1.85 ек), 1,2-диметоксиетан і водяний розчин карбонату калію помістили в реактор. Після трикратного дегазування азотом додали ацетат палладію (0,025 ек) і нагрівали вміст реактора зі зворотним холодильником при перемішуванні до передбачуваного завершення реакції. Реакційну суміш остудили до 22°С. Додали гептан, потім целіт. Після приблизно 30 хвилинного перемішування при 22°С суміш відфільтрували в перший реактор, попередньо обполісканий сумішшю 1,2-диметоксиетану і гептану. Шари фільтрату розділили. До органічного шару додали комплекс боран-триметиламін (0.03 ек), воду і оцтову кислоту. Отриману суміш з рН не вище 4 перемішували протягом 1-2 год при 22°С, потім нагрівали зі зворотним холодильником при температурі близько 80°С протягом 2-3 год. Після повторного охолодження до 22°С, рН суміші довели до 10-11 додаванням 5% водного гідроксиду натрію, підтримуючи температуру вмісту на рівні 22°С, потім перемішували суміш протягом 1-2 год. Суміш відфільтрували і розділили шари. Водний шар відкинули, а органічний шар відфільтрували через картриджі ZetaCarbon у перший реактор, що очищається в ході процесу, попередньо обполісканий 1,2-диметоксиетаном через вугільні картриджі. Фільтрат сконцентрували під вакуумом при максимальній заданій температурі нагрівальної оболонки 60°С. Додали гептан, потім сконцентрували вміст під вакуумом при максимальній заданій температурі нагрівальної оболонки 60°С. До концентрату додали додаткова кількість гептану і за допомогою ЯМР перевірили вміст 1,2-диметоксиетану (ДМЕ) у суміші (максимальний вміст 12 UA 99466 C2 5 склав 0,5%). Після нагрівання до 85°С і перемішування протягом приблизно 1 год суміш у гарячому вигляді відфільтрували через фільтр остаточного очищення в другий реактор. У другому реакторі фільтрат нагріли до температури кипіння і потім перемішували протягом 1 год. При поступовому охолодженні і перемішуванні з помірною інтенсивністю суміш прохолоджували від температури кипіння до 0 - 6°С як мінімум протягом 4 год, потім перемішували при температурі від 0 до 6°С протягом 1 год. Продукт відфільтрували промили гептаном при температурі навколишнього середовища і висушили під вакуумом при температурі, що не перевищує 40°С, до досягнення максимальної втрати маси при сушінні, що становить 1%. 10 15 20 25 У реактор помістили метансульфонову кислоту, потім пентоксид фосфору (1,00 ек) порціями, при цьому підтримували температуру вмісту на рівні 23°С. Порціями додавали 3,4диамінопіридин (1,00 ек) при підтримці температури вмісту в інтервалі від 20 до 50°С максимум. Потім додали 2-фторбензойну кислоту (1,09 ек). Суміш нагрівали до 100°С, моніторинг реакції до її завершення здійснювали методом ВЕЖХ. Вміст остудили до 10°С і додали воду при підтримці температури вмісту на рівні 25°С максимум. Після перемішування суміші при зазначеній температурі протягом 1 год суміш відфільтрували в другий реактор. До фільтрату, що міститься в другому реакторі, додали 27% гідроксид амонію до досягнення значення рН в інтервалі 8.0-9.3. Температуру вмісту підтримували на рівні 30°С максимум. Отриману тонку дисперсію перемішували при 22°С протягом як мінімум 1 год, потім додали 27% гідроксид амонію до досягнення значення рН в інтервалі 8.0-9.3. Потім дисперсію перемішували при 22°С протягом як мінімум 2 год. 13 UA 99466 C2 Продукт відфільтрували, двічі промили водою і висушили при максимальній температурі 60°С під вакуумом; вміст води в кінцевому продукті не перевищував 1%. При необхідності, продукт подрібнювали для видалення великих грудок. 5 10 15 20 25 У реактор додали сполуку 2а (1.24 ек), метиленхлорид і трихлорізоціанурову кислоту (0.491 ек). Суміш нагріли до температури кипіння і перемішували при нагріванні зі зворотним холодильником до завершення реакції. Реакційну суміш остудили до 22°С і додали целіт. Після перемішування протягом 30 хвилин (мінімум) суміш відфільтрували в другий реактор, попередньо обполісканий тричі метиленхлоридом. Осад з фільтру відкинули. До фільтрату в другому реакторі додали 3% водяний розчин гідроксиду натрію при підтримці температури вмісту реактора на рівні 22°С. Суміш перемішували протягом 1-2 год і розділили шари. Нижній органічний шар перенесли вочищений у ході процесу перший реактор і сконцентрували під вакуумом при температурі нагрівальної оболонки 45°С (максимум). Додали метиленхлорид і відфільтрували суміш через фільтр остаточного очищення в другий реактор, очищений у ході процесу. Фільтрат сконцентрували під вакуумом при температурі нагрівальної оболонки 45°С (максимум). Додали диметилформамід (ДМФ) і продовжили концентрування вмісту реактора. Температуру суміші довели до 22°С і додали ДМФ, потім сполуку-ядро 2 (1.00 ек) і 10% водяний розчин гідроксиду натрію при підтримці температури вмісту на рівні 22°С. Отриману суміш перемішували при 22°С при ВЕЖХ-моніторингу. У ході реакції проводили моніторинг рН змішай і додавали 10% водяний розчин гідроксиду натрію у кількості, необхідній для досягнення рН 1112, за даними рН-метру. По закінченню реакції додали 10% водяний розчин гідроксиду натрію при підтримці температури вмісту на рівні 22°С. Суміш розбавили ДМФ і перемішували 14 UA 99466 C2 5 10 15 20 25 протягом 2 год. Протягом мінімум 1 години суміш відфільтрували в перший реактор, очищений у ході процесу, що містить воду, при підтримці температури вмісту на рівні 16°С, з наступним промиванням ДМФ. Отриману суспензію перемішували протягом 1-3 год при 22°С. Неочищений продукт відфільтрували промили водою і метил-трет-бутиловим ефіром (МТБЕ). Неочищений вологий продукт із фільтру перенесли в перший реактор, додали етилацетат (ЕtOАс). Суміш нагріли до температури кипіння і перемішували при нагріванні зі зворотним холодильником до повного розчинення твердих речовин. Вміст води не повинне бути менше 6.0%. При поступовому охолодженні температуру вмісту довели до 22°С протягом 4 годин як мінімум. Продукт, що кристалізувався, відфільтрували і промили ЕtOАс, потім знову перенесли в перший реактор. Додали етилацетат (ЕtOАс). Суміш нагріли до температури кипіння і перемішували при нагріванні зі зворотним холодильником до повного розчинення твердих речовин. Вміст води не повинне бути менше 1.0%. Суміш відфільтрували в другий реактор (попередньо оброблений ЕtOАс) у гарячому вигляді через фільтр для остаточного очищення, промивши ЕtOАс. Продукт сконцентрували при атмосферному тиску. Після доведення температури до 65°С і додавання ЕtOАс, вміст посудини нагріли до температури кипіння і перемішували при температурі кипіння протягом приблизно 30 хвилин. Перевіряли вміст води і, якщо вміст води перевищував 0,2%, повторювали цей же цикл. У випадку, якщо вміст води не перевищувало 0,2%, вміст реактора нагрівали до температури кипіння, потім перемішували при температурі кипіння протягом 1-3 годин. Вміст реактора поступово прохолоджували до 22°С протягом 4 годин як мінімум, потім перемішували при даній температурі протягом 8 годин як мінімум. Продукт відфільтрували, промили ЕtOАс і висушили під вакуумом при температурі 60°С максимум. Потім продукт подрібнили. 15 UA 99466 C2 1 5 19 Спектри ядерного магнітного резонансу ( Н-, С- и F-ЯМР) Спектри ядерно-магнітного резонансу (ЯМР) сполуки (1) відповідають його передбачуваній 13 19 1 структурі. С, F і H-NMR спектри сполуки (1) у ДМСО-d6 були отримані на спектрометрі Varian UnityInova-400 FT-NMR. Спектри представлені в нижченаведеній Таблиці. Величини хімічного зрушення в спектрах ЯМР були встановлені в ході 2D кореляційних експериментів (COSY, HSQC, НМВС і HSQCTOCSY). 1 13 Н- і С-ЯМР величини хімічного зрушення для Сполуки (1) щодо еталонного стандарту. 10 16 UA 99466 C2 5 10 Диференціальна скануюча калориметрія Зразки аморфної сполуки (1), позначені як «Research lot 6» були отримані за способом, наведеним в Прикладі 1a WO 08/005519, включеним у даний опис у всій повноті за допомогою посилання. Інші зразки являли собою кристалічну сполуку (1). Вимірювання проводили за допомогою приладу для диференціальної скануючої калориметрії (ДСК) (DSC2010, виробництво ТА Instruments Corporation), в атмосфері азоту, вага зразка 5 ±1 мг, швидкість підйому температури: 1 °С у хв, 5°С у хв або 10°С у хв, відкритий алюмінієвий тигель та індій як реперна речовина. На кривій ДСК визначали ентальпію, екстрапольовану температуру початку піку і температуру, що відповідає вершині ендотермічного піку. Зведені результати ДСК для Research lot 6 і репрезентативних зразків кристалічної сполуки (1) у формі вільної основи (для різних партій) представлені в Таблиці 1 і на Фігурах 4 і 5, відповідно. При ДСК-скануванні кристалічної форми сполуки (1) зі швидкістю 1°С/хв ентальпія ендотермічного піку склала 17 UA 99466 C2 приблизно 80 Дж/г, екстрапольована температура початку піку була рівна 233,2°С ±2.0°С. Температура, що відповідає вершині ендотермічного піку, склала 233,9°С ±3,0°С. Таблиця 1 Приклади значень параметрів ДСК, отриманих для різних партій Сполуки (1) Примітки: усі в °С за виключенням ентальпії ** 5° С/хв. Вказано для Lot 6 Нед. пік - недозволений пік 5 10 15 20 25 30 35 Рентгенівська порошкова дифрактометрія - Дослідження 1 Зразки, отримані відповідно до Прикладу 1a WO 05/063744 і за способом згідно із даним винаходом, аналізували у тому вигляді, у якому вони були отримані, лише перемішавши зразки шпателем перед аналізом. Зразок зафіксували в алюмінієвому осередку і провели вимірювання за допомогою рентгенівського порошкового дифрактометра (XRD-6000, Shimadzu Lab X, виробництво Shimadzu Corporation, джерело рентгенівського випромінювання: Сu—К1 ray, напруга на трубці: 35 кВ, електричний струм у трубці: 40 м, швидкість сканування: 2° у хв, безперервне сканування, крок сканування: 0.02°, інтервал сканування: 4 - 35°, обертання по осі : 60 про/хв). На порошковій рентгенограмі, отриманій на немікронізованих голчастих кристалах сполуки (1), присутні характеристичні дифракційні піки при кутах дифракції 2 (°) 13.46, 15.59, 16.90, 17.48, 23.05 і 30.15; вимірювання проводили на рентгенівському порошковому дифрактометрі (Фігура 1). Було відзначено, що немікронізовані «тугоплавкі» (235°С) кристалічні форми сполуки (1), досліджувані в даному прикладі, проявляють деякі ефекти, обумовлені кращою орієнтацією і розміром частинок. Внаслідок цього рентгенограму, представлену на Фігурі 1, варто розглядати лише як приклад, тому що при зміні розміру кристала і його орієнтації змінюється величина піків на рентгенограмі. Крім того, можлива невелика помилка при вимірюванні величини дифракційного піку при вищезгаданому куті дифракції 29 (°), обумовлена вимірювальною апаратурою або умовами вимірювань і т.п. Як правило, помилка вимірювань потрапляє в інтервал ±0.3. У специфікації до Shimadzu XRD-6000 зазначений інтервал ±0.04. Крім того, можна чекати деяких відхилень положення піків, обумовлених змінами властивостей продукту і експериментальних умов; таким чином, дані за положенням піків варто вважати приблизними. Порошкова рентгенограма для «низькоплавкої» (220°С) твердої форми сполуки (1), що входить до складу продукту, отриманого відповідно до способу, описаного в Прикладі 1а (або ж наведеному в даному описі способу, до етапу повторного суспендування), відповідає аморфній речовині (Фігура 3). Сполука (1) згідно із даним винаходом зазвичай проявляє дійсну розчинність 0,7 мкг/мл, значення рКа, рівне 5.8, значення log P, рівне 2.8; і середньогеометричний профіль «рНрозчинність» (по 3 партіям), де рН 2 відповідає розчинність 458 мкг/мл, а рН 7.3 відповідає розчинність 0.7 мкг/мл. Розчинність (середнє геометричне значення для 3 партій) у штучних інтестінальних рідинах (натще: рН 6.4, 0.75 м лецитин, 3 м таурохолат натрії, 270 мОсмоль; після прийому їжі: рН 5.0, 3.75 м лецитин, 15 м таурохолат натрію, 635 мОсмоль) склала 19,1 мкг/мл (натще) і 122 мкг/мл (після прийому їжі). 18 UA 99466 C2 5 10 15 20 Обмірювані параметри варіюються в різних партіях; таким чином, всі представлені далі параметри, за винятком молекулярної маси, варто вважати приблизними. При титруванні з кислотами виявили ще вищу розчинність у випадку мезилату (>20 мг/мл) у порівнянні із хлоридом (приблизно 0.6 мг/мл) або сульфатом (приблизно 0.5 мг/мл) як протиіоном. Рентгенівська порошкова дифрактометрія - Дослідження 2 Інший зразок кристалічної сполуки (1), отриманий за способом згідно із даним винаходом, проаналізували за способом, аналогічному наведеному в Дослідженні 1, з тією різницею, що вимірювання проводили на рентгенівському порошковому дифрактометрі PANalytical X'Pert Pro MPD PW3040 Pro, виробництво PANalytical Inc., із джерелом рентгенівського випромінювання: Cu-К ray (1.54059 А), напругою трубки: 45 кВ, силою струму: 40 мА, інтервалом сканування: 1 55 °2, величиною кроку: 0.008 °2, часом нагромадження даних вимірювань: 3373 с, швидкість сканування: 0.9° у хв, щілина дифрактометра : DS: 1/2°, SS: 1/4°, час одного обороту: 0.5 с, режим: пропущення (transmission). Результати представлені на Фігурі 2. Приклад 2 Готування композицій із застосуванням Сполуки (1) Кристалічну сполуку (1) застосовують як напівпродукт для одержання фармацевтично прийнятних розчинів. У представлених нижче прикладах сполука композицій є такою, що вміст активного інгредієнта становить 10% вага/вагу. Нижче наведені приклади кількісних сполук на основі сполуки (1) у вигляді капсул по 20 мг і 40 мг, необхідних для одержання 12 кг розчину. Кількісна сполука композиції капсул, що включають Сполуку(і), 20 мг і 40 мг 19 UA 99466 C2 5 10 15 20 25 30 35 - Ємність/посудина: 12 кг, нержавіюча сталь - У наступному порядку зважують: - 0.012 кг бутилгідрокситолуолу (0.10%) - 0.035 кг бутилгідроксианізолу (0.35%) - 1.2 кг Сполуки (1) у формі вільної основи (10%). - 0.6 кг Полісорбату 80 (5%) - 10.153 кг олеїнової кислоти (еквівалентно 84.55 г (84.55%)) Капсули, що містять солюбілізовану Сполуку (1) у кількості 20 мг або 40 мг, одержують у ході послідовностей одиничних етапів процесу. Лікарська речовина на основі Сполуки (1), олеїнову кислоту, полісорбат 80, бутилгідрокситолуол (БГТ), і бутилгідроксианізол (БГА) змішували до одержання розчину. Розчином заповнювали тверді желатинові капсули, що складаються із двох частин. Потім закриті капсули герметизували водяним розчином спирту, що випаровується в процесі герметизації. Перед упакуванням герметичні капсули перевіряли на протікання у ході випробування під вакуумом. Альтернативні сполуки Можливе застосування кристалічної сполуки формули (1) як напівпродукт для готування солюбілізованої форми разом з наступними агентами: - Жирні кислоти (з коротким, середнім або довгим вуглецевим ланцюгом, насичені або ненасичені), зазвичай від С4 до С22. Типовими жирними кислотами є линолева кислота, лаурилова кислота, капринова кислота або олеїнова кислота. - Спирти, такі як етанол, бензиловий спирт, гліцерин, поліетиленгліколь 200, поліетиленгліколь 300, поліетиленгліколь 400. - Поверхнево-активні речовини, включаючи іонні та неіонні поверхнево-активні речовини. Приклади неіонних поверхнево-активних речовин включають ефіри жирних кислот і поліоксиетиленсорбіту, ефіри сорбіту та жирної кислоти, похідні поліетоксиетиленкасторової олії, поліоксиетиленгліцерину оксистеарат, поліетиленгліколь 60, гірогенізована касторова олія і/або блок-співполімери етиленоксиду та пропіленоксиду. - Антиоксиданти, наприклад, бутилгідроксианізол (БГА), бутилгідрокситолуол (БГТ), аскорбілпальмітат, вітамін Е і/або ПЕГ 1000 сукцинат вітаміну Е для хімічної стабільності. - Підсилювач в'язкості (діоксид кремнію, поліетиленгліколі, оксид титану і т.п.). - А також суміші перерахованих вище компонентів. Можливо інкапсулювання сполуки у капсулу з м'якого еластичного желатину або твердого желатину або твердої гідроксипропілметилцелюлози. Рідка сполука (у вигляді розчину або інкапсульованого розчину) забезпечує поліпшену біодоступність при пероральному введенні. 20 UA 99466 C2 5 10 15 20 Заповнення капсул Композиції та способи одержання м'яких еластичних желатинових капсул добре відомі фахівцям у даній галузі техніки. Композиція зазвичай містить від 30-50% вага/вагу желатину, 1040% вага/вагу пластифікатора або суміші пластифікаторів і приблизно 25-40% вага/вагу води. Пластифікатор може являти собою гліцерин, сорбіт або похідні сорбіту, пропіленгліколь і т.п. або їх комбінації. Для одержання і заповнення м'яких еластичних желатинових капсул можна застосовувати різні способи, такі як спосіб із застосуванням ротаційної машини, машини для запечатування (liner machine), машина для виготовлення капсул Accogel (accogel machine) і т.п. Тверді желатинові капсули або капсули з гідроксипропілметилцелюлози (ГПМЦ) можна придбати в Capsugel, Greenwood, S.C. і в інших виробників. Капсули заповнюють вручну або за допомогою машини для заповнення капсул. Одержання сполук У загальному випадку, композиції згідно із даним винаходом можна одержати у такий спосіб. Інгредієнти змішують у посудині придатного розміру за допомогою міксера, розташованого зверху (можливе продування ємності, у якій здійснюють змішування, азотом). Фармацевтично прийнятні жирні кислоти і фармацевтично прийнятні антиоксиданти змішують при кімнатній температурі. (Якщо буде потреба розчин можна нагріти, наприклад, приблизно до 45°С у випадку лаурилової кислоти, для переводу жирної кислоти у рідкий стан). Додають сполуку формули (1) і перемішують до розчинення. Фармацевтично прийнятну поверхнево-активну речовину додають при перемішуванні. Придатними навішеннями отриманої суміші заповнюють тверді желатинові капсули. Додаткові сполуки композицій 25 21 UA 99466 C2 22 UA 99466 C2 5 10 15 Приклад 2а Мікронізований склад Сполуки (1) Мікронізовану лікарську речовину (Jet mill-00 при 60-80 psi; середній розмір частинок 3-4 мікрони, приблизно 7-8 кв.м/г) методом сухого змішування перемішали з лактозою, мікрокристалічною целюлозою, кроскармелозой натрію, лаурилсульфатом натрію, винною кислотою і гідроксипропілцелюлозой. Суміш гранулювали шляхом розпилення розчину суміші. Гранули висушили в псевдозрідженому шарі. Висушені гранули подрібнили в млині, потім змішали з додатковою кількістю мікрокристалічної целюлози і кроскармелозою натрію. До порошкоподібної суміші додали змазуючий агент - стеарат магнію, потім суміш спресували в таблетки за допомогою ротаційного таблеткового преса. Потім таблетки покрили плівковою оболонкою. У таблиці, наведеній нижче, представлені сумарні результати випробувань різних складів на собаках, що одержували по 40 мг сполуки (1), що відповідає приблизно 4 мг/кг. Таблиця демонструє поліпшені характеристики солюбілізованого сполук, що містять сполуку (1). СУМАРНІ РЕЗУЛЬТАТИ ДОСЛІДЖЕНЬ IN-VIVO 23 UA 99466 C2 5 10 15 20 25 У таблиці: АРІ - активний фармакологічний інгредієнт; Capric acid - капринова кислота; Laurie acid - лауринова кислота; Oleic acid - олеїнова кислота; SLS only - тільки лаурилсульфат натрію; SLS & tartaric - лаурилсульфат натрію і винна кислота; ППК24 - площа під фармакокінетичною кривою (24 години); ВСВ - відносне стандартне відхилення. Приклад 3 Противірусна активність Сполуки (І) Сполука згідно із даним винаходом проявляє активність проти реплікону ВГС (дослідження описане в WO 05/063744) у відношенні обох генотипів 1а і 1, надзвичайно низьку цитотоксичність ((>50000 нМ у клітинах Huh-7, Hep2 і МТ4) і досить сприятливий коефіцієнт селективності. Сполука проявляє істотно меншу активність проти генотипу 2а. Активність Сполуки 1 проти репліконів ВГС Генотипу 1 і 1а Клітини, що містять реплікони ВГС Генотипу 1 (Con-1/lucneo) і 1а (Н77/nео) інкубували із серійними розведеннями сполуки (1), 2'С-метил аденозином (2'СМе) або IFN протягом 3 днів у присутності або під час відсутності 40 мг/мг сироваткового альбуміну людини (HSA). Після інкубації вміст РНК репліконів в оброблених клітинах визначали або шляхом дослідження репортера люциферази (1 реплікон), або кількісної ПЦР у режимі реального часу (1а реплікон); отримані дані використовували для розрахунку значень ЕС50 (50% ефективної інгібуючої концентрації) для інгібіторів. Показано, що Сполука (1) інгібує реплікони генотипу 1 і генотипу 1а зі значеннями ЕС50 0,6 і 3,6 нМ, відповідно (Таблиця 1). У присутності сироваткового альбуміну людини значення ЕС50 для Сполуки (1) підвищилося до 11 нМ. 24 UA 99466 C2 Таблиця А: Активність Сполуки (1) проти репліконів ВГС Генотипів 1а і 1 5 10 15 20 Середнє значення ЕС50 і стандартну помилку розраховували за даними щонайменше 4 незалежних експериментів. Активність Сполуки (1) проти реплікону ВГС Генотипу 1а і вірусу Противірусну активність сполуки (1) у відношенні ВГС генотипу 2а протестували на клітинах, хронічно інфікованих вірусом генотипу 2а, а також на клітинах, що репліціюють субгеномний реплікон 2а. Клітини лінії Huh-7, що містять хронічно репліціюючий вірус ВГС генотипу 2а (J6/ JFH-Rluc) або субгеномні реплікони, культивували зі Сполукою (1) або 2'СМеА протягом 3 днів під час відсутності сироваткового альбуміну людини. По закінченні культивування визначили кількість люциферази в клітинах, що містять вірус 2а, і активність протеази NS3 ВГС у клітинах, що містять реплікон 2а, шляхом аналізу люциферази (Promega) і нового дослідження флуоресценції з тимчасовим дозволом, відповідно. Противірусна активність Сполуки (1) значно знизилася в обох моделях клітинних культур, хронічно інфікованих ВГС-2а (ЕС50 = 2.9 мкМ), і в моделі субгеномного реплікону 2а (ЕС 50 = 21.9 мкМ) у порівнянні із клітинами Huh-7, що репліціюють субгеномний реплікон ВГС-1 (ЕС50 = 0.0006 мкМ) (Таблиця 2). Аналіз даних результатів дозволяє припустити, що зниження противірусного впливу Сполуки (1) на ВГС генотипу 2а можливо пояснюється генотипними розходженнями між генотипом 1 і генотипом 2 ВГС. Таблиця В: Активність Сполуки (1) проти ВГС Генотипів 1а і 2а 25 30 35 A Середнє значення ЕС50 і стандартну помилку розраховували за даними щонайменше 4 незалежних експериментів. Провели оцінку цитотоксичності Сполуки (1) для ряду типів клітин, включаючи лінії клітин, що містять реплікон ВГС (Huh-7, SL3 і МН4), і ліній клітин, що не містять реплікону (HepG2, MT4), шляхом люмінесцентного аналізу життєздатності клітин із застосуванням CellTiter-Glo Luminescence Cell Viability assay (Promega). У жодній із клітинних ліній не було відзначено токсичного впливу при максимальних концентраціях досліджуваної речовини (50 мкМ) (Таблиця 3). Ці результати у сполученні з ефективним противірусним впливом (ЕС 50 = 0.62-3.6 нМ) на реплікони ВГС-1 і ВГС-1a указують на високий коефіцієнт селективності (СС50/ ЕС50>1300080000) для Сполуки (1). Таблиця С: Цитотоксичність Сполуки (1) у лініях клітин, що містять реплікон ВГС 25 UA 99466 C2 5 10 15 20 25 30 35 40 45 а Середнє значення ЕС50 і стандартну помилку розраховували за даними щонайменше 4 незалежних експериментів, b лінії клітин, що містять реплікон ВГС Активність Сполуки (1) у комбінації з IFN In Vitro проти ВГС Пегільований інтерферон- (ПЕГ-IFN-) у комбінації з рибавірином застосовують у сучасній стандартній терапії пацієнтів з інфекцією ВГС. Дослідження in vitro комбінації Сполуки (1) і IFN- проводили на клітинах, що містять реплікони. Дані аналізували за допомогою моделі MacSynergy template, розробленої Причардом (Prichard) і Шипманом (Shipman). Результати даного дослідження дозволяють припустити наявність адитивної взаємодії між Сполукою (1) і IFN-. Приклад 4 Противірусна активність, фармакокінетика і безпека сполуки (1) заданими Фази 1 дослідження, проведеного серед суб'єктів, інфікованих ВГС генотипу 1 (уперше на людині) Рандомізоване подвійне сліпе дослідження із плацебо-контролем спрямовано на виявлення безпеки/переносимості, фармакокінетики і противірусної активності разових (частина А) і множинних (частина В) доз сполуки (1) (у розчині олеїнової кислоти, як описано вище) у суб'єктів, хронічно інфікованих ВГС генотипу 1 (GT-1) без декомпенсованого цирозу. У дослідженні брали участь суб'єкти у віці 18-60 років, що не одержували раніше лікарської терапії, стан здоров'я яких у цілому було хорошим. У завершеній частині дослідження (Частина А) п'ять послідовних груп з 6 суб'єктів були рандомізовані (5:1) для одержання разових зростаючих доз Сполуки 1 (40, 120, 240, 240-разом з їжею або 480 мг) або плацебо. У поточній частині дослідження (Частина В) чотири послідовних групи з 12 суб'єктів були рандомізовані (10:2) для одержання множинних зростаючих доз Сполуки 1 (40 мг BID (два рази на добу), 120 мг BID, 240 мг QD, 240 мг BID) або плацебо протягом 8 днів. Середній вік суб'єкта (з 31 суб'єкта, що приймали участь у частині А) становив 43,6 років, краще в групу входили чоловіки (20/31), білої раси (25/31), інфіковані або ВГС Генотип-1а (24) 10 або 1 (6). Середній (діапазон) базовий рівень вірусного навантаження ВГС склав 6.6 Log PHK ME (міжнародних одиниць)/мл (5.2-7.3). Була відзначена хороша переносимість разових доз сполуки (1) при відсутності серйозних або перешкоджаючих лікуванню несприятливих явищ (НЯ). Найчастішим НЯ був головний біль. Всі НЯ за ступенем важкості оцінили як легкі, за винятком одного епізоду помірного головного болю. Пов'язаних з лікуванням відхилень 3 і 4 ступеня в лабораторних показниках відзначено не було. Середнє значення періоду напіввиведення сполуки (1) із плазми варіювалося від 10 до 15 годин за групами. Системний вплив препарату підвищувався приблизно у два рази у випадках введення сполуки (1) разом з їжею з високим вмістом жиру. Середня концентрація сполуки (1) через 24 години після введення натще дози 240 мг приблизно в 7 разів перевищувала значення ЕС50, розраховане за зв'язуванням білка реплікону ВГС GT-1 in vitro. Після введення разової дози максимальний противірусний ефект спостерігали через 24 години, з відхиленням від 10 медіани в інтервалі від 0.46 до 1.49 Log ВГС РНК МО (міжнародних одиниць)/мл за групами. Індивідуальні відхилення за РНК ВГС серед всіх реципієнтів сполуки (1) варіюються від 0,19 до 10 2.54 log МО/мл після введення разової дози. Це дослідження являє собою першу клінічну демонстрацію противірусної активності сполуки (1). При впливі разової дози сполуки (1) відзначали хорошу переносимість, сприятливі фармакокінетичні властивості і високу противірусну активність сполуки (1). ФОРМУЛА ВИНАХОДУ 50 1. Кристалічна сполука формули (1) 26 UA 99466 C2 F F F F F F F N N 5 10 15 20 25 N N N , (1) яка характеризується: а) початком поглинання тепла при приблизно 235 °С на кривій диференціальної скануючої калориметрії (ДСК); b) величиною теплоти плавлення (DНпл), що становить приблизно 81 Дж/г (42 кДж/моль); та c) щонайменше одним піком при значенні кута дифракції 2 приблизно 17(°) за даними рентгенівської порошкової дифрактометрії, а також солі сполуки, причому сполука по суті не містить аморфної сполуки (1). 2. Кристалічна сполука за п. 1, яка являє собою вільну основу. 3. Кристалічна сполука за п. 1 у голчастій або стрижнеподібній формі. 4. Кристалічна сполука за п. 1, що по суті не містить хлоридної солі сполуки (1). 5. Кристалічна сполука за п. 1 у мікронізованій формі. 6. Композиція, що містить кристалічну сполуку за будь-яким з пп. 1-5, що містить менше приблизно 40 % мас. аморфної сполуки (1). 7. Композиція за п. 6, що містить менше приблизно 10 % мас. аморфної сполуки (1). 8. Композиція за п. 6, де кристалічна сполука (1) містить менше приблизно 100 ррm хлориду. 9. Кристалічна сполука (1), яка являє собою вільну основу, що по суті не містить аморфної сполуки (1) і будь-якої іншої кристалічної форми сполуки (1), причому кристалічна сполука характеризується: а) початком поглинання тепла при приблизно 235 °С на кривій диференціальної скануючої калориметрії (ДСК); b) величиною теплоти плавлення (DНпл), що становить приблизно 81 Дж/г (42 кДж/моль); та c) щонайменше одним піком при значенні кута дифракції 2 приблизно 17(°) за даними рентгенівської порошкової дифрактометрії. 10. Композиція, що містить кристалічну сполуку за будь-яким з пп. 1-5 або 9 і фармацевтично прийнятний наповнювач. 11. Спосіб одержання кристалічної сполуки (1) F F F F F F F N N 30 35 40 N N N , у якому здійснюють кристалізацію сполуки (1) з розчинника, який використовується для кристалізації, і регулюють вміст води в зазначеному розчиннику, що використовується для кристалізації. 12. Спосіб за п. 11, де сполуку (1) кристалізують із розчинника, що являє собою етилацетат або суміш етилацетат/ізопропіловий спирт, з вмістом води менше приблизно 0,9 % мас. 13. Спосіб за п. 11, де вміст води регулюють таким чином, щоб у ході кристалізації осаджувалося менше приблизно 10 % мас. аморфної сполуки (1). 14. Спосіб за п. 11, де вміст води регулюють шляхом азеотропного видалення води з розчинника, який використовується для кристалізації. 15. Спосіб за п. 11, при якому у розчиннику, що використовується для кристалізації, вміст води забезпечують на рівні менше приблизно 10 %. 16. Спосіб за п. 15, що включає множину етапів кристалізації, де кристалізацію проводять із розчинників, що містять воду у послідовно спадних кількостях. 17. Спосіб за п. 15, де останній етап кристалізації проводять із розчинника, що містить менше приблизно 0,9 % води. 27 UA 99466 C2 28
ДивитисяДодаткова інформація
Назва патенту англійськоюCrystalline pyridazine compound
Автори англійськоюDowdy, Eric, D., Kent, Kenneth, M., Tom, Norma, J., Zia, Vahid
Назва патенту російськоюКристаллическое соединение пиридазина
Автори російськоюДауди Эрик Д., Кент Кеннет М., Том Норма Дж., Зия Вехид
МПК / Мітки
МПК: C07D 471/04, A61P 31/12, A61K 31/4353
Мітки: піридазину, сполука, кристалічна
Код посилання
<a href="https://ua.patents.su/32-99466-kristalichna-spoluka-piridazinu.html" target="_blank" rel="follow" title="База патентів України">Кристалічна сполука піридазину</a>
Кристалічна форма eto2c-ch2-(r)cgl-aze-pab-oh

Номер патенту: 66869
Опубліковано: 15.06.2004
Автори: Лундблад Аніта, Едвардссон Даніель, Гедстрем Лена, Петтерссон Урсула
МПК: C07K 5/06, A61P 7/02, C07K 5/065, A61K 38/00, A61P 43/00
Мітки: кристалічна, форма, eto2c-ch2-(r)cgl-aze-pab-oh
Формула / Реферат:
1. Кристалічна по суті форма EtО2C-CH2-(R)Cgl-Aze-Pab-OH чи її фармацевтично прийнятна сіль.2. Сполука за п. 1, яка відрізняється тим, що має форму ангідрату.3. Сполука за п. 2, яка відрізняється тим, що не має форму солі.4. Сполука за будь-яким з пп. 1-3, яка відрізняється тим що має масову частку води не більше 2%.5. Сполука за будь-яким з пп. 2-4, яка відрізняється тим, що виявляє на кривій диференційної...
Кристалічна форма похідних 1-[2-(2,4-диметилфенілсульфаніл)-феніл]піперазину як сполука з комбінованою активністю стосовно зворотного захоплення серотоніну, 5-ht3 та 5-ht1a для лікування когнітивних порушень

Номер патенту: 93250
Опубліковано: 25.01.2011
Автори: Мьорк Арне, Банг-Андерсен Бенні, Рок Майкл Харольд, Мур Ніколас, Мілі Майкл Дж., Фалт Андре, Холм Рене, Лопес де Дієґо Хейді, Бродерсен Йорґен, Рінґор Лоне Мунк, Стенсбьол Тіне Брайан, Йорґенсен Мортен
МПК: A61P 25/28, C07D 295/08, A61K 31/495
Мітки: захоплення, активністю, 1-[2-(2,4-диметилфенілсульфаніл)-феніл]піперазину, порушень, форма, когнітивних, стосовно, кристалічна, лікування, 5-ht1a, комбінованою, серотоніну, похідних, сполука, зворотного, 5-ht3
Формула / Реферат:
1. Кристалічна форма 1-[2-(2,4-диметилфенілсульфаніл)феніл]піперазину і його фармацевтично прийнятних солей, гідратів та сольватів, вибрана з кристалічної основи, гідроброміду (α), гідроброміду (β), гідроброміду (γ), гідроброміду (гідрату), гідроброміду (етилацетатного сольвату), гідрохлориду, гідрохлориду (моногідрату), мезилату, солі фумарової кислоти, солі малеїнової кислоти, солі мезовинної кислоти, солі L-(+)-винної...
Похідні піридазину

Номер патенту: 94052
Опубліковано: 11.04.2011
Автори: Хунцікер Даніель, Майвег Александер В., Кун Бернд, Найдхарт Вернер, Амрайн Курт
МПК: C07D 237/08, C07D 237/26, C07D 237/28, A61K 31/502
Мітки: піридазину, похідні
Формула / Реферат:
1. Сполуки формули, (I) у якій R1 означає циклоалкіл, арилалкіл або арилоксіалкіл; R2 означає циклоалкіл, арилалкіл або арилоксіалкіл; або R1 і R2 разом з атомами вуглецю Сa і Сb, до яких вони приєднані, утворюють фрагменти
Похідне 3-арил-2-гідроксипропіонової кислоти, спосіб його отримання (варіанти), проміжна сполука, фармацевтична композиція (варіанти) та спосіб профілактики та/або лікування асоційованих з резистентністю до інс

Номер патенту: 71912
Опубліковано: 17.01.2005
Автор: Андерссон К'єлль
МПК: A61P 43/00, C07C 217/76, A61P 3/06, C07C 69/734, C07C 311/13, C07C 317/22, A61K 31/216, C07D 263/04, C07C 311/16, C07C 311/08, C07C 233/29, C07C 235/42, C07C 303/00, C07C 271/44, C07C 275/34, C07C 323/56, C07C 309/00, C07C 323/19, A61P 3/10, C07C 271/58, C07C 233/75, A61P 9/10, C07C 271/38, A61K 31/255, C07C 323/44, A61P 9/12, C07C 317/28, C07C 335/00, C07C 271/28, C07C 233/25, A61K 31/192, C07C 255/54, A61P 3/04
Мітки: профілактики, лікування, резистентністю, варіанти, похідне, кислоти, інс, проміжна, сполука, спосіб, отримання, композиція, фармацевтична, асоційованих, 3-арил-2-гідроксипропіонової
Формула / Реферат:
1. Похідне 3-арил-2-гідроксипропіонової кислоти формули (I) Iта його фармацевтично прийнятні солі, сольвати та кристалічні форми.2. Сполука за п. 1, яка відрізняється тим, що призначена для використання в терапії.3. Сполука за п. 1, яка відрізняється тим, що її використовують у виробництві лікувального засобу для профілактики та/або лікування асоційованих з резистентністю до інсуліну клінічних станів.4....
Поверхнево-активна сполука, спосіб її одержання, композиція, пестицидна композиція та спосіб захисту сільськогосподарських культур

Номер патенту: 69429
Опубліковано: 15.09.2004
Автори: Мур Керолайн Естеп, Чоу Віктор Шіу-Чіу, Шаннон Теммі Тайлер, Хопкінсон Майкл Джеймс
МПК: A01P 7/04, A01P 3/00, A01N 41/00, A01P 13/00
Мітки: композиція, спосіб, культур, поверхнево-активна, захисту, сполука, пестицидна, сільськогосподарських, одержання
Формула / Реферат:
1. Сполука формули (Н-В)+A-,деА- означає:,де кожен R1 незалежно означає С2-С4алкілен з прямим або розгалуженим ланцюгом,n означає число від 1 до 50 включно,(H-B)+ означає катіон формули:,де R2 вибирають із групи, яка включає водень, С1-С24алкіл і...
Попередній патент: Метаболіти (r)-3-(4-(7н-піроло[2,3-d]піримідин-4-іл)-1н-піразол-1-іл)-3-циклопентилпропаннітрилу, інгібітору janus кіназ
Наступний патент: Солі інгібітора янус-кінази (r)-3-(4-(7н-піроло[2,3-d]піримідин-4-іл)-1н-піразол-1-іл)-3-циклопентилпропаннітрилу
Випадковий патент: Лабораторний стенд для визначення критичного (гасячого) діаметра