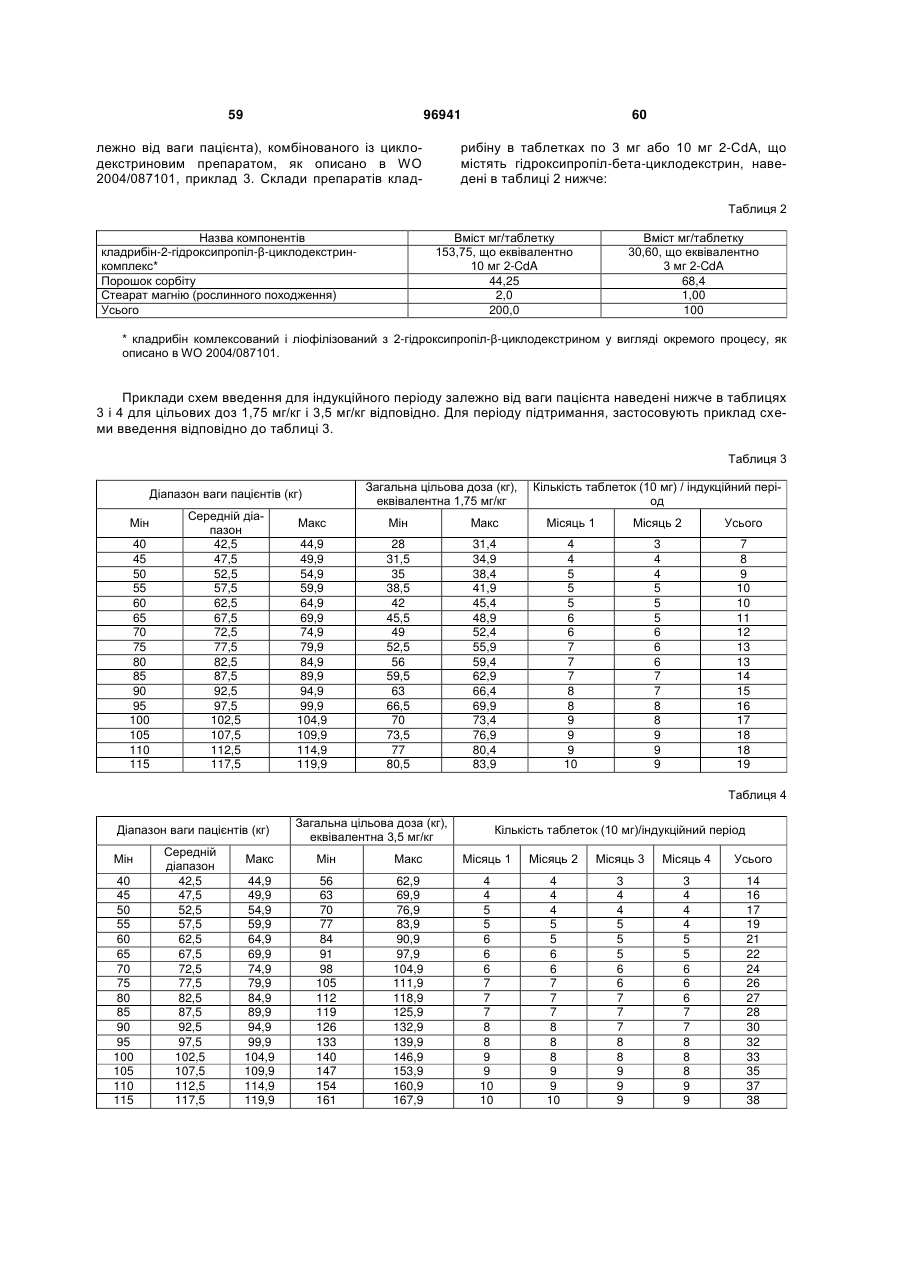
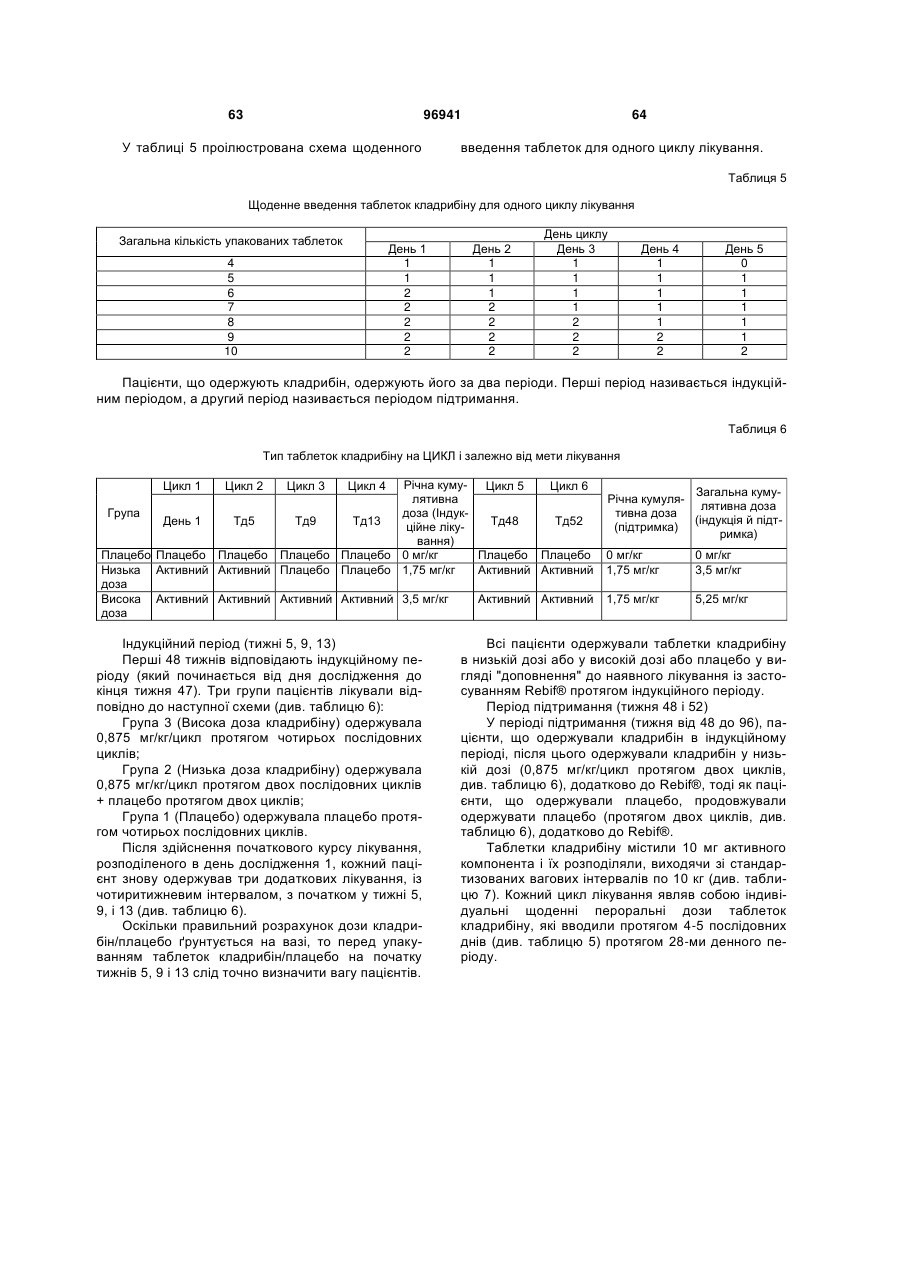
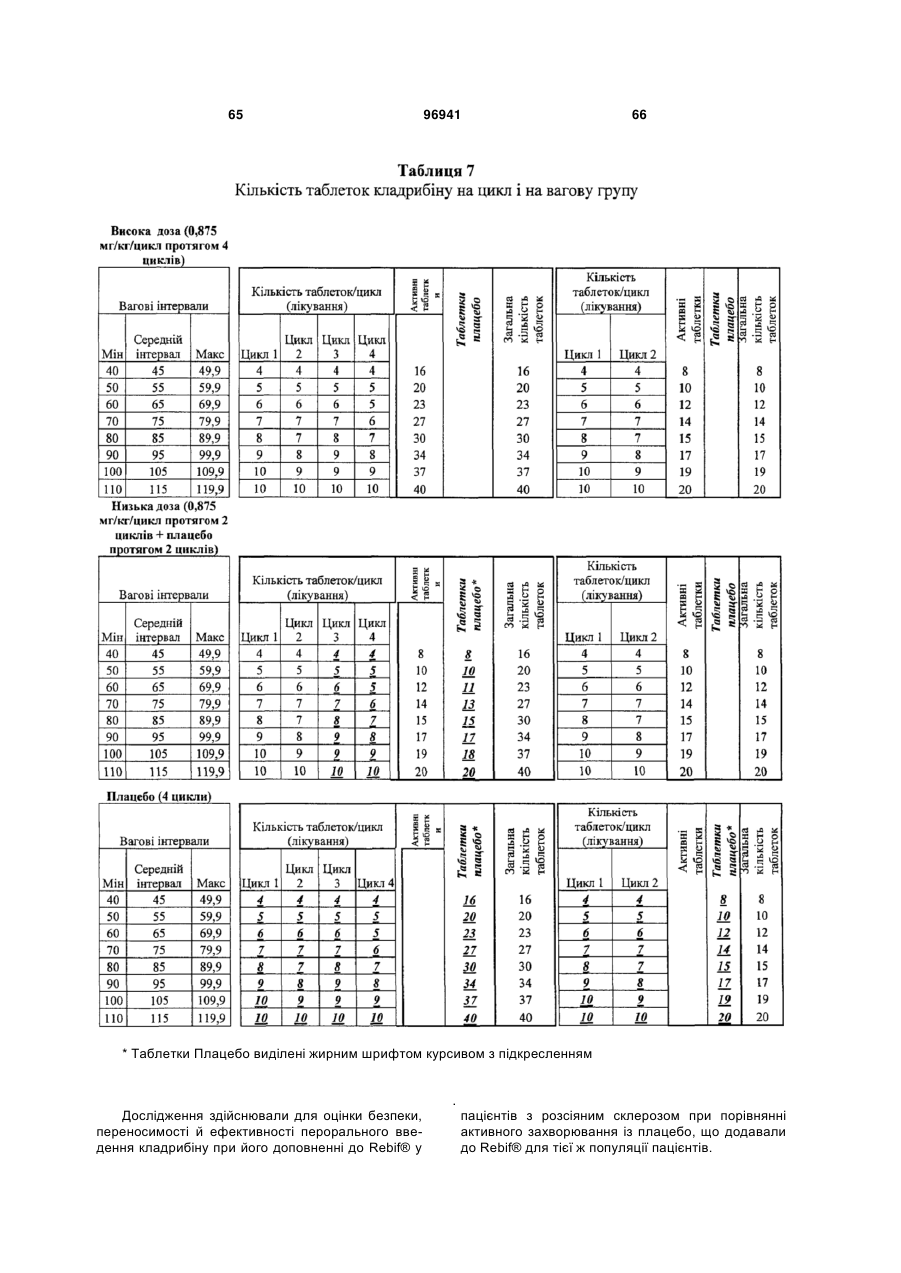
Схема дозування кладрибіну для лікування розсіяного склерозу
Номер патенту: 96941
Опубліковано: 26.12.2011
Автори: Аммоурі Назіх, Лопез-Бреснахан Марія, Бренцель Х. Джеймс Джр.































Формула / Реферат
1. Застосування комбінації кладрибіну й бета-інтерферону для приготування лікарського засобу для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, де кладрибін вводять перорально, відповідно до послідовних стадій, наведених нижче:
(і) індукційний період, у якому вводять кладрибін і в якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 1,7 мг/кг до приблизно 3,5 мг/кг;
(іі) період без кладрибіну, у якому не вводять кладрибін;
(ііі) період підтримання, у якому вводять кладрибін і в якому загальна доза кладрибіну, яку вводять протягом періоду підтримання, нижче або дорівнює загальній дозі кладрибіну, що досягається наприкінці індукційного періоду (і);
(iv) період без кладрибіну, у якому не вводять кладрибін.
2. Застосування відповідно до пункту 1, де комбінована тривалість індукційного періоду (і) з періодом без кладрибіну (іі) становить приблизно 1 рік.
3. Застосування відповідно до пункту 2, де тривалість індукційного періоду становить приблизно 4 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, або тривалість індукційного періоду становить приблизно 2 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців.
4. Застосування відповідно до будь-якого з пунктів 2, 3, де комбінована тривалість індукційного періоду (і) з періодом без кладрибіну (іі) становить приблизно 1 рік (приблизно 12 місяців) і загальна доза кладрибіну, яка досягається до закінчення цього року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг, або приблизно 3,5 мг/кг, переважно 3,5 мг/кг.
5. Застосування відповідно до будь-якого з пунктів 1-4, де комбінована тривалість періоду підтримання (ііі) з періодом без кладрибіну (iv) становить приблизно 1 рік.
6. Застосування відповідно до пункту 5, де тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців.
7. Застосування відповідно до пункту 5 або 6, де комбінована тривалість періоду підтримання (ііі) з періодом без кладрибіну (iv) становить приблизно 1 рік і загальна доза кладрибіну, що вводять протягом цього року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг.
8. Застосування відповідно до будь-якого з пунктів 1-7, де комбінована тривалість індукційного періоду (і), періоду без кладрибіну (іі), періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 2 роки.
9. Застосування відповідно до пункту 8, де:
- тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців, або
- тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців.
10. Застосування відповідно до пункту 8 або 9, де комбінована тривалість індукційного періоду (і) і періоду без кладрибіну (іі) становить приблизно 1 рік, комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік, загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг, і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг.
11. Застосування відповідно до пункту 8 або 9, де комбінована тривалість індукційного періоду (і) і періоду без кладрибіну (іі) становить приблизно 1 рік, комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік, загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 3,5 мг/кг, переважно 3,5 мг/кг, і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг.
12. Застосування відповідно до будь-якого з попередніх пунктів, де стадії (ііі) - (iv) повторюють один, два або три рази.
13. Застосування відповідно до будь-якого з попередніх пунктів, де біодоступність кладрибіну становить приблизно 40 %.
14. Застосування відповідно до будь-якого з попередніх пунктів, де загальна ефективна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 0,7 мг/кг або приблизно 1,4 мг/кг.
15. Застосування відповідно до будь-якого з попередніх пунктів, де загальна ефективна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 0,7 мг/кг.
16. Застосування відповідно до будь-якого з попередніх пунктів, де вводять кладрибін від 4 до 7 днів на місяць, переважно 4 або 5 днів на місяць, протягом індукційного періоду.
17. Застосування відповідно до пункту 16, де вводять кладрибін від дня 1 до дня 5 щомісяця протягом індукційного періоду.
18. Застосування відповідно до пункту 16 або 17, де вводять кладрибін у добовій дозі приблизно 0,175 мг/кг протягом індукційного періоду.
19. Застосування відповідно до будь-якого з попередніх пунктів, де вводять кладрибін від 4 до 7 днів на місяць, переважно 4 або 5 днів на місяць протягом періоду підтримання.
20. Застосування відповідно до пункту 19, де вводять кладрибін від дня 1 до дня 5 щомісяця протягом періоду підтримання.
21. Застосування відповідно до пункту 19 або 20, де вводять кладрибін у добовій дозі приблизно 0,175 мг/кг протягом періоду підтримання.
22. Застосування відповідно до будь-якого з попередніх пунктів, де бета-інтерферон вводять одночасно, окремо або послідовно з пероральним введенням кладрибіну.
23. Застосування відповідно до пункту 22, де бета-інтерферон вводять одночасно з пероральним введенням кладрибіну.
24. Застосування відповідно до пункту 23, де бета-інтерферон вводять протягом індукційного періоду (і), періоду підтримання (iiі) і періодів без кладрибіну (іі) і (iv).
25. Застосування відповідно до пункту 24, де бета-інтерферон вводять перед індукційним періодом (і), протягом індукційногоперіоду (і), протягом періоду підтримання (iіі), протягом періодів без кладрибіну (іі) і (iv) і після періоду без кладрибіну (iv).
26. Застосування відповідно до будь-якого з попередніх пунктів, де бета-інтерферон вибирають із групи, яка включає: Avonex® (Biogen), Rebif® (Serono), і Betaseron® (Berlex / Schering AG).
27. Застосування відповідно до пункту 26, де бета-інтерферон являє собою Rebif® (Serono).
28. Застосування відповідно до пункту 26 або 27, де бета-інтерферон вводять підшкірно або внутрішньом'язово.
29. Застосування відповідно до пункту 28, де використовуваний бета-інтерферон являє собою Rebif® (Serono) і вводять у дозі 44 мкг підшкірно три рази на тиждень.
30. Застосування відповідно до будь-якого з попередніх пунктів, де загальноприйняте лікування вибирають із групи, яка включає: лікування за допомогою бета-інтерферону, лікування за допомогою ацетату глатимареру (Copaxone®, Teva), лікування за допомогою наталізумабу (Tysabri®, Biogen/Elan) і лікування за допомогою мітоксантрону (Novantrone®, Serono).
31. Застосування відповідно до пункту 30, де загальноприйняте лікування являє собою лікування за допомогою бета-інтерферону, переважно лікування за допомогою Betaseron® (Berlex / Schering AG); Avonex® (Biogen); або Rebif® (Serono).
32. Застосування відповідно до будь-якого з попередніх пунктів, де в резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив, незважаючи на одержання принаймні одного загальноприйнятого лікування.
33. Застосування відповідно до пункту 32, де принаймні один рецидив відбувся протягом року перед початком лікування відповідно до будь-якого з попередніх пунктів.
34. Застосування відповідно до пункту 33, де у пацієнтів, яких піддають лікуванню, відбувся принаймні один рецидив протягом року, що передує початку лікування відповідно до будь-якого з попередніх пунктів і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif® три рази на тиждень.
35. Застосування відповідно до будь-якого з попередніх пунктів, де в резистентних пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС, що визначалося за допомогою методів, таких як метод МРТ, незважаючи на одержання принаймні одного загальноприйнятого лікування.
36. Застосування відповідно до пункту 35, де збільшене число уражень або збільшений об'єм мозкових уражень виникали протягом року перед початком лікування відповідно до будь-якого з попередніх пунктів.
37. Застосування відповідно до пункту 36, де у пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС протягом року, що передує початку лікування відповідно до будь-якого з попередніх пунктів і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif® три рази на тиждень.
38. Застосування відповідно до будь-якого з попередніх пунктів, де в резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив і розвивається наростаюча інвалідність внаслідок прогресуючих форм захворювання.
39. Застосування відповідно до будь-якого з попередніх пунктів, де резистентні пацієнти, яких піддають лікуванню, страждають від погіршення MS, особливо вторинного прогресуючого, прогресуючого ремітуючого або погіршення MS ремітуючого перебігу.
40. Застосування комбінації кладрибіну й бета-інтерферону для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, що включає пероральне введення кладрибіну, відповідно до послідовних стадій, наведених нижче:
(і) Індукційний період, у якому вводять кладрибін і в якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 1,5 мг/кг до приблизно 3,5 мг/кг;
(іі) Період без кладрибіну, у якому не вводять кладрибін;
(ііі) Період підтримання, у якому вводять кладрибін і в якому загальна доза кладрибіну, яку вводять протягом періоду підтримання, нижче або дорівнює загальній дозі кладрибіну, що досягається наприкінці індукційного періоду (і);
(iv) Період без кладрибіну, у якому не вводять кладрибін.
41. Застосування відповідно до пункту 40, де комбінована тривалість індукційного періоду (і) з періодом без кладрибіну (іі) становить приблизно 1 рік.
42. Застосування відповідно до пункту 41, де тривалість індукційного періоду становить приблизно 4 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, або тривалість індукційного періоду становить приблизно 2 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців.
43. Застосування відповідно до будь-якого з пунктів 41-42, де комбінована тривалість індукційного періоду (і) з періодом без кладрибіну (іі) становить приблизно 1 рік (приблизно 12 місяців) і загальна доза кладрибіну, яка досягається до закінчення цього року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг або приблизно 3,5 мг/кг, переважно 3,5 мг/кг.
44. Застосування відповідно до будь-якого з пунктів 40-43, де комбінована тривалість періоду підтримання (ііі) з періодом без кладрибіну (iv) становить приблизно 1 рік.
45. Застосування відповідно до пункту 44, де тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців.
46. Застосування відповідно до пункту 44 або 45, де комбінована тривалість періоду підтримання (ііі) з періодом без кладрибіну (iv) становить приблизно 1 рік і загальна доза кладрибіну, що вводять протягом цього року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг.
47. Застосування відповідно до будь-якого з пунктів 40 - 46, де комбінована тривалість індукційного періоду (і), періоду без кладрибіну (іі), періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 2 роки.
48. Застосування відповідно до пункту 47, де:
- тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців, або
- тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців.
49. Застосування відповідно до пункту 47 або 48, де комбінована тривалість індукційного періоду (і) і періоду без кладрибіну (іі) становить приблизно 1 рік, комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік, загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг.
50. Застосування відповідно до пункту 47 або 48, де комбінована тривалість індукційного періоду (і) і періоду без кладрибіну (іі) становить приблизно 1 рік, комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік, загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 3,5 мг/кг, переважно 3,5 мг/кг і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг.
51. Застосування відповідно до будь-якого з пунктів 40-50, де стадії (ііі) - (iv) повторюють один, два або три рази.
52. Застосування відповідно до будь-якого з пунктів 40-51, де біодоступність кладрибіну становить приблизно 40 %.
53. Застосування відповідно до будь-якого з пунктів 40 - 52, де загальна ефективна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 0,7 мг/кг або приблизно 1,4 мг/кг.
54. Застосування відповідно до будь-якого з пунктів 40 - 53, де загальна ефективна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 0,7 мг/кг.
55. Застосування відповідно до будь-якого з пунктів 40-54, де вводять кладрибін від 4 до 7 днів на місяць, переважно 4 або 5 днів на місяць, протягом індукційного періоду.
56. Застосування відповідно до пункту 55, де вводять кладрибін від дня 1 до дня 5 щомісяця протягом індукційного періоду.
57. Застосування відповідно до пункту 55 або 56, де вводять кладрибін у добовій дозі приблизно 0,175 мг/кг протягом індукційного періоду.
58. Застосування відповідно до будь-якого з пунктів 40-57, де вводять кладрибін від 4 до 7 днів на місяць, переважно 4 або 5 днів на місяць протягом періоду підтримання.
59. Застосування відповідно до пункту 58, де вводять кладрибін від дня 1 до дня 5 щомісяця протягом періоду підтримання.
60. Застосування відповідно до пункту 58 або 59, де вводять кладрибін у добовій дозі приблизно 0,175 мг/кг протягом періоду підтримання.
61. Застосування відповідно до будь-якого з пунктів 40-60, де бета-інтерферон вводять одночасно, окремо або послідовно з пероральним введенням кладрибіну.
62. Застосування відповідно до пункту 61, де бета-інтерферон вводять одночасно з пероральним введенням кладрибіну.
63. Застосування відповідно до пункту 62, де бета-інтерферон вводять протягом індукційного періоду (і), періоду підтримання (iіі) і періодів без кладрибіну (іі) і (iv).
64. Застосування відповідно до пункту 63, де бета-інтерферон вводять перед індукційним періодом (і), протягом індукційного періоду (і), протягом періоду підтримання (iіі), протягом періодів без кладрибіну (іі) і (iv) і після періоду без кладрибіну (iv).
65. Застосування відповідно до будь-якого з пунктів 40-64, де бета-інтерферон вибирають із групи, яка включає: Avonex® (Biogen), Rebif® (Serono), і Betaseron® (Berlex / Schering AG).
66. Застосування відповідно до пункту 65, де бета-інтерферон являє собою Rebif® (Serono).
67. Застосування відповідно до пункту 65 або 66, де бета-інтерферон вводять підшкірно або внутрішньом'язово.
68. Застосування відповідно до пункту 67, де використовуваний бета-інтерферон являє собою Rebif® (Serono) і вводять у дозі 44 мкг підшкірно три рази на тиждень.
69. Застосування відповідно до будь-якого з пунктів 40-68, де загальноприйняте лікування вибирають із групи, яка включає: лікування за допомогою бета-інтерферону, лікування за допомогою ацетату глатимареру (Copaxone®, Teva), лікування за допомогою наталізумабу (Tysabri®, Biogen/Elan) і лікування за допомогою мітоксантрону (Novantrone®, Serono).
70. Застосування відповідно до пункту 69, де загальноприйняте лікування являє собою лікування за допомогою бета-інтерферону, переважно лікування за допомогою Betaseron® (Berlex / Schering AG); Avonex® (Biogen); або Rebif® (Serono).
71. Застосування відповідно до будь-якого з пунктів 40-70, де в резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив, незважаючи на одержання принаймні одного загальноприйнятого лікування.
72. Застосування відповідно до пункту 71, де принаймні один рецидив відбувся протягом року перед початком лікування відповідно до будь-якого з попередніх пунктів.
73. Застосування відповідно до пункту 72, де у пацієнтів, яких піддають лікуванню, відбувся принаймні один рецидив протягом року, що передує початку лікування відповідно до будь-якого з попередніх пунктів і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif®, три рази на тиждень.
74. Застосування відповідно до будь-якого з пунктів 40-73, де в резистентних пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС, що визначалося за допомогою методів, таких як метод МРТ, незважаючи на одержання принаймні одного загальноприйнятого лікування.
75. Застосування відповідно до пункту 74, де збільшене число уражень або збільшений об'єм мозкових уражень виникали протягом року перед початком лікування відповідно до будь-якого з попередніх пунктів.
76. Застосування відповідно до пункту 75, де у пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС протягом року, що передує початку лікування відповідно до будь-якого з попередніх пунктів і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif®, три рази на тиждень.
77. Застосування відповідно до будь-якого з пунктів 40-76, де в резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив і розвивається наростаюча інвалідність внаслідок прогресуючих форм захворювання.
78. Застосування відповідно до будь-якого з пунктів 40-77, де резистентні пацієнти, яких піддають лікуванню, страждають від погіршення MS, особливо вторинного прогресуючого, прогресуючого ремітуючого або погіршення MS ремітуючого перебігу.
79. Фармацевтичний набір для лікування розсіяного склерозу, який містить контейнер, що містить кладрибін, контейнер, що містить бета-інтерферон, та листок-вкладиш з інструкцією для застосування, де у листку-вкладиші вказується, що набір призначений для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, де кладрибін вводять перорально, відповідно до послідовних стадій, наведених нижче:
(і) Індукційний період, у якому вводять кладрибін і в якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 1,7 мг/кг до приблизно 3,5 мг/кг;
(іі) Період без кладрибіну, у якому не вводять кладрибін;
(ііі) Період підтримання, у якому вводять кладрибін і в якому загальна доза кладрибіну, яку вводять протягом періоду підтримання, нижче або дорівнює загальній дозі кладрибіну, що досягається наприкінці індукційного періоду (і);
(iv) Період без кладрибіну, у якому не вводять кладрибін.
80. Набір відповідно до пункту 79, де комбінована тривалість індукційного періоду (і) з періодом без кладрибіну (іі) становить приблизно 1 рік.
81. Набір відповідно до пункту 80, де тривалість індукційного періоду становить приблизно 4 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, або тривалість індукційного періоду становить приблизно 2 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців.
82. Набір відповідно до будь-якого з пунктів 80-81, де комбінована тривалість індукційного періоду (і) з періодом без кладрибіну (іі) становить приблизно 1 рік (приблизно 12 місяців) і загальна доза кладрибіну, яка досягається до закінчення цього року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг, або приблизно 3,5 мг/кг, переважно 3,5 мг/кг.
83. Набір відповідно до будь-якого з пунктів 79-82, де комбінована тривалість періоду підтримання (ііі) з періодом без кладрибіну (iv) становить приблизно 1 рік.
84. Набір відповідно до пункту 83, де тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців.
85. Набір відповідно до пункту 83 або 84, де комбінована тривалість періоду підтримання (ііі) з періодом без кладрибіну (iv) становить приблизно 1 рік і загальна доза кладрибіну, що вводять протягом цього року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг.
86. Набір відповідно до будь-якого з пунктів 79 - 85, де комбінована тривалість індукційного періоду (і), періоду без кладрибіну (іі), періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 2 роки.
87. Набір відповідно до пункту 86, де:
- тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців, або
- тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців.
88. Набір відповідно до пункту 86 або 87, де комбінована тривалість індукційного періоду (і) і періоду без кладрибіну (іі) становить приблизно 1 рік, комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік, загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг, і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг.
89. Набір відповідно до пункту 86 або 87, де комбінована тривалість індукційного періоду (і) і періоду без кладрибіну (іі) становить приблизно 1 рік, комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік, загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 3,5 мг/кг, переважно 3,5 мг/кг, і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг.
90. Набір відповідно до будь-якого з пунктів 79-89, де стадії (ііі) - (iv) повторюють один, два або три рази.
91. Набір відповідно до будь-якого з пунктів 79-90, де біодоступність кладрибіну становить приблизно 40 %.
92. Набір відповідно до будь-якого з пунктів 79-91, де загальна ефективна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 0,7 мг/кг або приблизно 1,4 мг/кг.
93. Набір відповідно до будь-якого з пунктів 79-92, де загальна ефективна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 0,7 мг/кг.
94. Набір відповідно до будь-якого з пунктів 79-93, де вводять кладрибін від 4 до 7 днів на місяць, переважно 4 або 5 днів на місяць, протягом індукційного періоду.
95. Набір відповідно до пункту 94, де вводять кладрибін від дня 1 до дня 5 щомісяця протягом індукційного періоду.
96. Набір відповідно до пункту 94 або 95, де вводять кладрибін у добовій дозі приблизно 0,175 мг/кг протягом індукційного періоду.
97. Набір відповідно до будь-якого з пунктів 79-96, де вводять кладрибін від 4 до 7 днів на місяць, переважно 4 або 5 днів на місяць протягом періоду підтримання.
98. Набір відповідно до пункту 97, де вводять кладрибін від дня 1 до дня 5 щомісяця протягом періоду підтримання.
99. Набір відповідно до пункту 97 або 98, де вводять кладрибін у добовій дозі приблизно 0,175 мг/кг протягом періоду підтримання.
100. Набір відповідно до будь-якого з пунктів 79-99, де бета-інтерферон вводять одночасно, окремо або послідовно з пероральним введенням кладрибіну.
101. Набір відповідно до пункту 100, де бета-інтерферон вводять одночасно з пероральним введенням кладрибіну.
102. Набір відповідно до пункту 101, де бета-інтерферон вводять протягом індукційного періоду (і), періоду підтримання (iіі) і періодів без кладрибіну (іі) і (iv).
103. Набір відповідно до пункту 102, де бета-інтерферон вводять перед індукційним періодом (і), протягом індукційного періоду (і), протягом періоду підтримання (iіі), протягом періодів без кладрибіну (іі) і (iv) і після періоду без кладрибіну (iv).
104. Набір відповідно до будьякого з пунктів 79-103, де бета-інтерферон вибирають із групи, яка включає: Avonex® (Biogen), Rebif® (Serono) і Betaseron® (Berlex/Schering AG).
105. Набір відповідно до пункту 104, де бета-інтерферон являє собою Rebif® (Serono).
106. Набір відповідно до пункту 104 або 105, де бета-інтерферон вводять підшкірно або внутрішньом'язово.
107. Набір відповідно до пункту 106, де використовуваний бета-інтерферон являє собою Rebif® (Serono) і вводять у дозі 44 мкг підшкірно три рази на тиждень.
108. Набір відповідно до будь-якого з пунктів 79-107, де загальноприйняте лікування вибирають із групи, яка включає: лікування за допомогою бета-інтерферону, лікування за допомогою ацетату глатимареру (Copaxone®, Teva), лікування за допомогою наталізумабу (Tysabri®, Biogen/Elan), і лікування за допомогою мітоксантрону (Novantrone®, Serono).
109. Набір відповідно до пункту 108, де загальноприйняте лікування являє собою лікування за допомогою бета-інтерферону, переважно лікування за допомогою Betaseron® (Berlex/Schering AG), Avonex® (Biogen) або Rebif® (Serono).
110. Набір відповідно до будь-якого з пунктів 79-109, де в резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив, незважаючи на одержання принаймні одного загальноприйнятого лікування.
111. Набір відповідно до пункту 110, де принаймні один рецидив відбувся протягом року перед початком лікування відповідно до будь-якого з попередніх пунктів.
112. Набір відповіднодо пункту 111, де у пацієнтів, яких піддають лікуванню, відбувся принаймні один рецидив протягом року, що передує початку лікування відповідно до будь-якого з попередніх пунктів і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif®, три рази на тиждень.
113. Набір відповідно до будь-якого з пунктів 79-112, де в резистентних пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС, що визначалося за допомогою методів, таких як метод МРТ, незважаючи на одержання принаймні одного загальноприйнятого лікування.
114. Набір відповідно до пункту 113, де збільшене число уражень або збільшений об'єм мозкових уражень виникали протягом року перед початком лікування відповідно до будь-якого з попередніх пунктів.
115. Набір відповідно до пункту 114, де у пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС протягом року, що передує початку лікування відповідно до будь-якого з попередніх пунктів, і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif®, три рази на тиждень.
116. Набір відповідно до будь-якого з пунктів 79-115, де в резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив і розвивається наростаюча інвалідність внаслідок прогресуючих форм захворювання.
117. Набір відповідно до будь-якого з пунктів 79-116, де резистентні пацієнти, яких піддають лікуванню, страждають від погіршення MS, особливо вторинного прогресуючого, прогресуючого ремітуючого або погіршення MS ремітуючого перебігу.
Текст
1. Застосування комбінації кладрибіну й бетаінтерферону для приготування лікарського засобу для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, де кладрибін вводять перорально, відповідно до послідовних стадій, наведених нижче: (і) індукційний період, у якому вводять кладрибін і в якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 1,7 мг/кг до приблизно 3,5 мг/кг; (іі) період без кладрибіну, у якому не вводять кладрибін; (ііі) період підтримання, у якому вводять кладрибін і в якому загальна доза кладрибіну, яку вводять протягом періоду підтримання, нижче або дорівнює загальній дозі кладрибіну, що досягається наприкінці індукційного періоду (і); (iv) період без кладрибіну, у якому не вводять кладрибін. 2. Застосування відповідно до пункту 1, де комбінована тривалість індукційного періоду (і) з періодом без кладрибіну (іі) становить приблизно 1 рік. 3. Застосування відповідно до пункту 2, де тривалість індукційного періоду становить приблизно 4 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, або тривалість індукційного періоду становить приблизно 2 місяці й 2 (19) 1 3 періоду без кладрибіну (іі) становить приблизно 1 рік, комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік, загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг, і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. 11. Застосування відповідно до пункту 8 або 9, де комбінована тривалість індукційного періоду (і) і періоду без кладрибіну (іі) становить приблизно 1 рік, комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік, загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 3,5 мг/кг, переважно 3,5 мг/кг, і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. 12. Застосування відповідно до будь-якого з попередніх пунктів, де стадії (ііі) - (iv) повторюють один, два або три рази. 13. Застосування відповідно до будь-якого з попередніх пунктів, де біодоступність кладрибіну становить приблизно 40 %. 14. Застосування відповідно до будь-якого з попередніх пунктів, де загальна ефективна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 0,7 мг/кг або приблизно 1,4 мг/кг. 15. Застосування відповідно до будь-якого з попередніх пунктів, де загальна ефективна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 0,7 мг/кг. 16. Застосування відповідно до будь-якого з попередніх пунктів, де вводять кладрибін від 4 до 7 днів на місяць, переважно 4 або 5 днів на місяць, протягом індукційного періоду. 17. Застосування відповідно до пункту 16, де вводять кладрибін від дня 1 до дня 5 щомісяця протягом індукційного періоду. 18. Застосування відповідно до пункту 16 або 17, де вводять кладрибін у добовій дозі приблизно 0,175 мг/кг протягом індукційного періоду. 19. Застосування відповідно до будь-якого з попередніх пунктів, де вводять кладрибін від 4 до 7 днів на місяць, переважно 4 або 5 днів на місяць протягом періоду підтримання. 20. Застосування відповідно до пункту 19, де вводять кладрибін від дня 1 до дня 5 щомісяця протягом періоду підтримання. 21. Застосування відповідно до пункту 19 або 20, де вводять кладрибін у добовій дозі приблизно 0,175 мг/кг протягом періоду підтримання. 22. Застосування відповідно до будь-якого з попередніх пунктів, де бета-інтерферон вводять одночасно, окремо або послідовно з пероральним введенням кладрибіну. 23. Застосування відповідно до пункту 22, де бетаінтерферон вводять одночасно з пероральним введенням кладрибіну. 24. Застосування відповідно до пункту 23, де бетаінтерферон вводять протягом індукційного періоду 96941 4 (і), періоду підтримання (iiі) і періодів без кладрибіну (іі) і (iv). 25. Застосування відповідно до пункту 24, де бетаінтерферон вводять перед індукційним періодом (і), протягом індукційного періоду (і), протягом періоду підтримання (iіі), протягом періодів без кладрибіну (іі) і (iv) і після періоду без кладрибіну (iv). 26. Застосування відповідно до будь-якого з попередніх пунктів, де бета-інтерферон вибирають із групи, яка включає: Avonex® (Biogen), Rebif® (Serono), і Betaseron® (Berlex / Schering AG). 27. Застосування відповідно до пункту 26, де бетаінтерферон являє собою Rebif® (Serono). 28. Застосування відповідно до пункту 26 або 27, де бета-інтерферон вводять підшкірно або внутрішньом'язово. 29. Застосування відповідно до пункту 28, де використовуваний бета-інтерферон являє собою Rebif® (Serono) і вводять у дозі 44 мкг підшкірно три рази на тиждень. 30. Застосування відповідно до будь-якого з попередніх пунктів, де загальноприйняте лікування вибирають із групи, яка включає: лікування за допомогою бета-інтерферону, лікування за допомогою ацетату глатимареру (Copaxone®, Teva), лікування за допомогою наталізумабу (Tysabri®, Biogen/Elan) і лікування за допомогою мітоксантрону (Novantrone®, Serono). 31. Застосування відповідно до пункту 30, де загальноприйняте лікування являє собою лікування за допомогою бета-інтерферону, переважно лікування за допомогою Betaseron® (Berlex / Schering AG); Avonex® (Biogen); або Rebif® (Serono). 32. Застосування відповідно до будь-якого з попередніх пунктів, де в резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив, незважаючи на одержання принаймні одного загальноприйнятого лікування. 33. Застосування відповідно до пункту 32, де принаймні один рецидив відбувся протягом року перед початком лікування відповідно до будь-якого з попередніх пунктів. 34. Застосування відповідно до пункту 33, де у пацієнтів, яких піддають лікуванню, відбувся принаймні один рецидив протягом року, що передує початку лікування відповідно до будь-якого з попередніх пунктів і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif® три рази на тиждень. 35. Застосування відповідно до будь-якого з попередніх пунктів, де в резистентних пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС, що визначалося за допомогою методів, таких як метод МРТ, незважаючи на одержання принаймні одного загальноприйнятого лікування. 36. Застосування відповідно до пункту 35, де збільшене число уражень або збільшений об'єм мозкових уражень виникали протягом року перед початком лікування відповідно до будь-якого з попередніх пунктів. 37. Застосування відповідно до пункту 36, де у пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС протягом року, що пере 5 дує початку лікування відповідно до будь-якого з попередніх пунктів і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif® три рази на тиждень. 38. Застосування відповідно до будь-якого з попередніх пунктів, де в резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив і розвивається наростаюча інвалідність внаслідок прогресуючих форм захворювання. 39. Застосування відповідно до будь-якого з попередніх пунктів, де резистентні пацієнти, яких піддають лікуванню, страждають від погіршення MS, особливо вторинного прогресуючого, прогресуючого ремітуючого або погіршення MS ремітуючого перебігу. 40. Застосування комбінації кладрибіну й бетаінтерферону для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, що включає пероральне введення кладрибіну, відповідно до послідовних стадій, наведених нижче: (і) індукційний період, у якому вводять кладрибін і в якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 1,5 мг/кг до приблизно 3,5 мг/кг; (іі) період без кладрибіну, у якому не вводять кладрибін; (ііі) період підтримання, у якому вводять кладрибін і в якому загальна доза кладрибіну, яку вводять протягом періоду підтримання, нижче або дорівнює загальній дозі кладрибіну, що досягається наприкінці індукційного періоду (і); (iv) період без кладрибіну, у якому не вводять кладрибін. 41. Застосування відповідно до пункту 40, де комбінована тривалість індукційного періоду (і) з періодом без кладрибіну (іі) становить приблизно 1 рік. 42. Застосування відповідно до пункту 41, де тривалість індукційного періоду становить приблизно 4 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, або тривалість індукційного періоду становить приблизно 2 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців. 43. Застосування відповідно до будь-якого з пунктів 41-42, де комбінована тривалість індукційного періоду (і) з періодом без кладрибіну (іі) становить приблизно 1 рік (приблизно 12 місяців) і загальна доза кладрибіну, яка досягається до закінчення цього року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг або приблизно 3,5 мг/кг, переважно 3,5 мг/кг. 44. Застосування відповідно до будь-якого з пунктів 40-43, де комбінована тривалість періоду підтримання (ііі) з періодом без кладрибіну (iv) становить приблизно 1 рік. 45. Застосування відповідно до пункту 44, де тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. 46. Застосування відповідно до пункту 44 або 45, де комбінована тривалість періоду підтримання (ііі) з періодом без кладрибіну (iv) становить приблизно 1 рік і загальна доза кладрибіну, що вводять 96941 6 протягом цього року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. 47. Застосування відповідно до будь-якого з пунктів 40-46, де комбінована тривалість індукційного періоду (і), періоду без кладрибіну (іі), періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 2 роки. 48. Застосування відповідно до пункту 47, де: - тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців, або - тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. 49. Застосування відповідно до пункту 47 або 48, де комбінована тривалість індукційного періоду (і) і періоду без кладрибіну (іі) становить приблизно 1 рік, комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік, загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. 50. Застосування відповідно до пункту 47 або 48, де комбінована тривалість індукційного періоду (і) і періоду без кладрибіну (іі) становить приблизно 1 рік, комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік, загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 3,5 мг/кг, переважно 3,5 мг/кг і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. 51. Застосування відповідно до будь-якого з пунктів 40-50, де стадії (ііі) - (iv) повторюють один, два або три рази. 52. Застосування відповідно до будь-якого з пунктів 40-51, де біодоступність кладрибіну становить приблизно 40 %. 53. Застосування відповідно до будь-якого з пунктів 40 - 52, де загальна ефективна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 0,7 мг/кг або приблизно 1,4 мг/кг. 54. Застосування відповідно до будь-якого з пунктів 40 - 53, де загальна ефективна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 0,7 мг/кг. 55. Застосування відповідно до будь-якого з пунктів 40-54, де вводять кладрибін від 4 до 7 днів на місяць, переважно 4 або 5 днів на місяць, протягом індукційного періоду. 56. Застосування відповідно до пункту 55, де вводять кладрибін від дня 1 до дня 5 щомісяця протягом індукційного періоду. 7 57. Застосування відповідно до пункту 55 або 56, де вводять кладрибін у добовій дозі приблизно 0,175 мг/кг протягом індукційного періоду. 58. Застосування відповідно до будь-якого з пунктів 40-57, де вводять кладрибін від 4 до 7 днів на місяць, переважно 4 або 5 днів на місяць протягом періоду підтримання. 59. Застосування відповідно до пункту 58, де вводять кладрибін від дня 1 до дня 5 щомісяця протягом періоду підтримання. 60. Застосування відповідно до пункту 58 або 59, де вводять кладрибін у добовій дозі приблизно 0,175 мг/кг протягом періоду підтримання. 61. Застосування відповідно до будь-якого з пунктів 40-60, де бета-інтерферон вводять одночасно, окремо або послідовно з пероральним введенням кладрибіну. 62. Застосування відповідно до пункту 61, де бетаінтерферон вводять одночасно з пероральним введенням кладрибіну. 63. Застосування відповідно до пункту 62, де бетаінтерферон вводять протягом індукційного періоду (і), періоду підтримання (iіі) і періодів без кладрибіну (іі) і (iv). 64. Застосування відповідно до пункту 63, де бетаінтерферон вводять перед індукційним періодом (і), протягом індукційного періоду (і), протягом періоду підтримання (iіі), протягом періодів без кладрибіну (іі) і (iv) і після періоду без кладрибіну (iv). 65. Застосування відповідно до будь-якого з пунктів 40-64, де бета-інтерферон вибирають із групи, яка включає: Avonex® (Biogen), Rebif® (Serono), і Betaseron® (Berlex / Schering AG). 66. Застосування відповідно до пункту 65, де бетаінтерферон являє собою Rebif® (Serono). 67. Застосування відповідно до пункту 65 або 66, де бета-інтерферон вводять підшкірно або внутрішньом'язово. 68. Застосування відповідно до пункту 67, де використовуваний бета-інтерферон являє собою Rebif® (Serono) і вводять у дозі 44 мкг підшкірно три рази на тиждень. 69. Застосування відповідно до будь-якого з пунктів 40-68, де загальноприйняте лікування вибирають із групи, яка включає: лікування за допомогою бета-інтерферону, лікування за допомогою ацетату глатимареру (Copaxone®, Teva), лікування за допомогою наталізумабу (Tysabri®, Biogen/Elan) і лікування за допомогою мітоксантрону (Novantrone®, Serono). 70. Застосування відповідно до пункту 69, де загальноприйняте лікування являє собою лікування за допомогою бета-інтерферону, переважно лікування за допомогою Betaseron® (Berlex / Schering AG); Avonex® (Biogen); або Rebif® (Serono). 71. Застосування відповідно до будь-якого з пунктів 40-70, де в резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив, незважаючи на одержання принаймні одного загальноприйнятого лікування. 72. Застосування відповідно до пункту 71, де принаймні один рецидив відбувся протягом року перед початком лікування відповідно до будь-якого з попередніх пунктів. 96941 8 73. Застосування відповідно до пункту 72, де у пацієнтів, яких піддають лікуванню, відбувся принаймні один рецидив протягом року, що передує початку лікування відповідно до будь-якого з попередніх пунктів і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif®, три рази на тиждень. 74. Застосування відповідно до будь-якого з пунктів 40-73, де в резистентних пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС, що визначалося за допомогою методів, таких як метод МРТ, незважаючи на одержання принаймні одного загальноприйнятого лікування. 75. Застосування відповідно до пункту 74, де збільшене число уражень або збільшений об'єм мозкових уражень виникали протягом року перед початком лікування відповідно до будь-якого з попередніх пунктів. 76. Застосування відповідно до пункту 75, де у пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС протягом року, що передує початку лікування відповідно до будь-якого з попередніх пунктів і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif®, три рази на тиждень. 77. Застосування відповідно до будь-якого з пунктів 40-76, де в резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив і розвивається наростаюча інвалідність внаслідок прогресуючих форм захворювання. 78. Застосування відповідно до будь-якого з пунктів 40-77, де резистентні пацієнти, яких піддають лікуванню, страждають від погіршення MS, особливо вторинного прогресуючого, прогресуючого ремітуючого або погіршення MS ремітуючого перебігу. 79. Фармацевтичний набір для лікування розсіяного склерозу, який містить контейнер, що містить кладрибін, контейнер, що містить бетаінтерферон, та листок-вкладиш з інструкцією для застосування, де у листку-вкладиші вказується, що набір призначений для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, де кладрибін вводять перорально, відповідно до послідовних стадій, наведених нижче: (і) індукційний період, у якому вводять кладрибін і в якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 1,7 мг/кг до приблизно 3,5 мг/кг; (іі) період без кладрибіну, у якому не вводять кладрибін; (ііі) період підтримання, у якому вводять кладрибін і в якому загальна доза кладрибіну, яку вводять протягом періоду підтримання, нижче або дорівнює загальній дозі кладрибіну, що досягається наприкінці індукційного періоду (і); (iv) період без кладрибіну, у якому не вводять кладрибін. 80. Набір відповідно до пункту 79, де комбінована тривалість індукційного періоду (і) з періодом без кладрибіну (іі) становить приблизно 1 рік. 9 81. Набір відповідно до пункту 80, де тривалість індукційного періоду становить приблизно 4 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, або тривалість індукційного періоду становить приблизно 2 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців. 82. Набір відповідно до будь-якого з пунктів 80-81, де комбінована тривалість індукційного періоду (і) з періодом без кладрибіну (іі) становить приблизно 1 рік (приблизно 12 місяців) і загальна доза кладрибіну, яка досягається до закінчення цього року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг, або приблизно 3,5 мг/кг, переважно 3,5 мг/кг. 83. Набір відповідно до будь-якого з пунктів 79-82, де комбінована тривалість періоду підтримання (ііі) з періодом без кладрибіну (iv) становить приблизно 1 рік. 84. Набір відповідно до пункту 83, де тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. 85. Набір відповідно до пункту 83 або 84, де комбінована тривалість періоду підтримання (ііі) з періодом без кладрибіну (iv) становить приблизно 1 рік і загальна доза кладрибіну, що вводять протягом цього року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. 86. Набір відповідно до будь-якого з пунктів 79-85, де комбінована тривалість індукційного періоду (і), періоду без кладрибіну (іі), періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 2 роки. 87. Набір відповідно до пункту 86, де: - тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців, або - тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. 88. Набір відповідно до пункту 86 або 87, де комбінована тривалість індукційного періоду (і) і періоду без кладрибіну (іі) становить приблизно 1 рік, комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік, загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг, і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. 89. Набір відповідно до пункту 86 або 87, де комбінована тривалість індукційного періоду (і) і періоду без кладрибіну (іі) становить приблизно 1 рік, комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік, загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 96941 10 3,5 мг/кг, переважно 3,5 мг/кг, і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. 90. Набір відповідно до будь-якого з пунктів 79-89, де стадії (ііі) - (iv) повторюють один, два або три рази. 91. Набір відповідно до будь-якого з пунктів 79-90, де біодоступність кладрибіну становить приблизно 40 %. 92. Набір відповідно до будь-якого з пунктів 79-91, де загальна ефективна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 0,7 мг/кг або приблизно 1,4 мг/кг. 93. Набір відповідно до будь-якого з пунктів 79-92, де загальна ефективна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 0,7 мг/кг. 94. Набір відповідно до будь-якого з пунктів 79-93, де вводять кладрибін від 4 до 7 днів на місяць, переважно 4 або 5 днів на місяць, протягом індукційного періоду. 95. Набір відповідно до пункту 94, де вводять кладрибін від дня 1 до дня 5 щомісяця протягом індукційного періоду. 96. Набір відповідно до пункту 94 або 95, де вводять кладрибін у добовій дозі приблизно 0,175 мг/кг протягом індукційного періоду. 97. Набір відповідно до будь-якого з пунктів 79-96, де вводять кладрибін від 4 до 7 днів на місяць, переважно 4 або 5 днів на місяць протягом періоду підтримання. 98. Набір відповідно до пункту 97, де вводять кладрибін від дня 1 до дня 5 щомісяця протягом періоду підтримання. 99. Набір відповідно до пункту 97 або 98, де вводять кладрибін у добовій дозі приблизно 0,175 мг/кг протягом періоду підтримання. 100. Набір відповідно до будь-якого з пунктів 7999, де бета-інтерферон вводять одночасно, окремо або послідовно з пероральним введенням кладрибіну. 101. Набір відповідно до пункту 100, де бетаінтерферон вводять одночасно з пероральним введенням кладрибіну. 102. Набір відповідно до пункту 101, де бетаінтерферон вводять протягом індукційного періоду (і), періоду підтримання (iіі) і періодів без кладрибіну (іі) і (iv). 103. Набір відповідно до пункту 102, де бетаінтерферон вводять перед індукційним періодом (і), протягом індукційного періоду (і), протягом періоду підтримання (iіі), протягом періодів без кладрибіну (іі) і (iv) і після періоду без кладрибіну (iv). 104. Набір відповідно до будь-якого з пунктів 79103, де бета-інтерферон вибирають із групи, яка включає: Avonex® (Biogen), Rebif® (Serono) і Betaseron® (Berlex/Schering AG). 105. Набір відповідно до пункту 104, де бетаінтерферон являє собою Rebif® (Serono). 106. Набір відповідно до пункту 104 або 105, де бета-інтерферон вводять підшкірно або внутрішньом'язово. 107. Набір відповідно до пункту 106, де використовуваний бета-інтерферон являє собою Rebif® 11 96941 12 (Serono) і вводять у дозі 44 мкг підшкірно три рази на тиждень. 108. Набір відповідно до будь-якого з пунктів 79107, де загальноприйняте лікування вибирають із групи, яка включає: лікування за допомогою бетаінтерферону, лікування за допомогою ацетату глатимареру (Copaxone®, Teva), лікування за допомогою наталізумабу (Tysabri®, Biogen/Elan), і лікування за допомогою мітоксантрону (Novantrone®, Serono). 109. Набір відповідно до пункту 108, де загальноприйняте лікування являє собою лікування за допомогою бета-інтерферону, переважно лікування за допомогою Betaseron® (Berlex/Schering AG), Avonex® (Biogen) або Rebif® (Serono). 110. Набір відповідно до будь-якого з пунктів 79109, де в резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив, незважаючи на одержання принаймні одного загальноприйнятого лікування. 111. Набір відповідно до пункту 110, де принаймні один рецидив відбувся протягом року перед початком лікування відповідно до будь-якого з попередніх пунктів. 112. Набір відповіднодо пункту 111, де у пацієнтів, яких піддають лікуванню, відбувся принаймні один рецидив протягом року, що передує початку лікування відповідно до будь-якого з попередніх пунктів і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif®, три рази на тиждень. 113. Набір відповідно до будь-якого з пунктів 79112, де в резистентних пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС, що визначалося за допомогою методів, таких як метод МРТ, незважаючи на одержання принаймні одного загальноприйнятого лікування. 114. Набір відповідно до пункту 113, де збільшене число уражень або збільшений об'єм мозкових уражень виникали протягом року перед початком лікування відповідно до будь-якого з попередніх пунктів. 115. Набір відповідно до пункту 114, де у пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС протягом року, що передує початку лікування відповідно до будь-якого з попередніх пунктів, і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif®, три рази на тиждень. 116. Набір відповідно до будь-якого з пунктів 79115, де в резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив і розвивається наростаюча інвалідність внаслідок прогресуючих форм захворювання. 117. Набір відповідно до будь-якого з пунктів 79116, де резистентні пацієнти, яких піддають лікуванню, страждають від погіршення MS, особливо вторинного прогресуючого, прогресуючого ремітуючого або погіршення MS ремітуючого перебігу. Даний винахід стосується застосування багаторазових доз кладрибіну в комбінації з бета інтерфероном для лікування розсіяного склерозу у пацієнтів, які резистентні принаймні до одного загальноприйнятого лікування MS. Розсіяний склероз (MS) є найбільш відомим хронічним запальним демієлінізуючим захворюванням центральної нервової системи. Захворювання звичайно починається у віці від 20 до 40 років. Жінки хворіють у два рази частіше, ніж чоловіки. Згодом, MS може приводити до нагромадження різних видів неврологічної інвалідності. Клінічна недієздатність при MS розвивається в результаті повторного запального ушкодження з наступною втратою мієліну й аксонів, що приводить до атрофії тканини. MS проявляється фізичними симптомами (загостреннями й прогресуванням захворювання), запальними процесами в центральній нервовій системі (ЦНС), атрофією головного мозку й порушенням пізнавальних процесів. Наявні симптоми включають вогнищеві сенсорні недостатності, сенсорну слабість, порушення зору, порушення рівноваги й втому. Можуть зустрічатися погіршення статевої функції й порушення функції сфінктера. Приблизно в половини пацієнтів з MS можуть проявлятися порушення пізнавальних процесів або депресія. Зараз MS розглядають як багатофазне захворювання, у якому періоди клінічного спокою (ремісії) чергуються із загостреннями. Тривалість ремісій може бути різною й вони можуть тривати декілька років, рідко є постійними. Розрізняють чотири періоди розвитку захворювання: розсіяний склероз ремітуючого перебігу (RRMS), вторинний прогресуючий розсіяний склероз (SPMS), первинний прогресуючий розсіяний склероз (PPMS) і прогресуючий рецидивуючий розсіяний склероз (PRMS). У понад 80% пацієнтів з MS спочатку проявляється RRMS період із клінічним загостренням неврологічних симптомів, з наступним відновленням, що може бути повним або неповним (Lublin і Reingold, Neurology, 1996, 46:907-911). При RRMS, кумуляція інвалідності відбувається в результаті неповного відновлення після рецидивів. Через десять років після початку захворювання, у половини пацієнтів з RRMS розвивається прогресуючий перебіг, що називається SPMS. При SP фазі, погіршення інвалідності відбувається внаслідок нагромадження залишкових симптомів після нападів, а також у результаті прихованого прогресування між нападами (Lublin і Reingold вище). 10% MS пацієнтів мають PPMS, що характеризується прихованим прогресуванням симптомом від початку захворювання. У менше 5 % пацієнтів спостерігається PRMS і часто для них розгляда 13 ється такий же прогноз, що й для PPMS. Вважають, що для різних підгруп пацієнтів задіяні різні патогенні механізми й вони спричиняються істотні ускладнення для класифікації захворювання (Lassmann та ін., 2001, Trends Моl. Med., 7, 115121; Lucchinetti та ін., Curr. Opin. Neurol, 2001, 14, 259-269). Початок MS визначають за проявом перших неврологічних симптомів порушення функції ЦНС. Успіхи в дослідженні спинномозкової рідини (CSF) і магнітно-резонансної візуалізації (МРТ) спростили встановлення діагнозу й сприяли ранній діагностиці (Noseworthy та ін., The New England Journal of Medicine, 2000, 343, 13, 938-952). Опубліковано міжнародну панель для діагностики MS, у якій переглянуті критерії, що полегшують діагностику MS, і вони включають МРТ разом із клінічними й параклінічними діагностичними методами (Me Donald та ін., 2001, Ann. Neurol., 50:121-127). Сучасні підходи лікування MS являють собою лікування, що модифікують захворювання, тобто модифікують перебіг MS, модулюють або пригнічують імунну систему. FDA (комісія з контролю за ліками й живильними речовинами) дозволила чотири імуномодулуючі засоби для лікування RRMS: три бета інтерферони (Betaseron®, Berlex; Avonex®, Biogen; Rebif®, Serono) і ацетат глатимареру (Copaxone®, Teva). Також FDA дозволила застосування одного імуносупресора при погіршенні MS, мітоксантрон (Novantrone®, Serono). Також використовуються інші імуносупресори, хоча вони не схвалені FDA. Одним із цих засобів є кладрибін, хлорований аналог пурину 2-хлор-2'дезоксіаденозин (2-CdA), який, як вважають, корисний для лікування MS (ЕР 62685ЗВ1 і US 5,506,214). У деяких клінічних дослідженнях кладрибіну у пацієнтів з розсіяним склерозом аналізували в/в і п/ш застосування кладрибіну при MS. Здійснювали два подвійні анонімні дослідження у фазі II із плацебо як контролем відповідно для лікування хронічного прогресуючого MS (Selby та ін., 1998, Can. J. Neurol. Sci., 25:295-299) і MS ремітуючого перебігу відповідно (Romine та ін., 1999, Proceedings of the Association of American Physicians, 111, 1, 35-44). В першому дослідженні, використовували кладрибін у дозі 0,1 мг/кг/добу протягом 7 днів, що вводили шляхом безперервної в/в інфузії. Лікування повторювали протягом 4 послідовних місяців. У другому клінічному дослідженні, використовували кладрибін у дозі 0,07 мг/кг/добу протягом 5 днів, що вводили шляхом підшкірної ін'єкції. Лікування повторювали протягом 6 послідовних місяців. Додатково, здійснювали дослідження у фазі III із плацебо як контролем у пацієнтів з первинним прогресуючим (РР) або вторинним прогресуючої (SP) розсіяним склерозом (Rice та ін., 2000, Neurology, 54, 5, 1145-1155). У цьому дослідженні, дві групи пацієнтів одержували кладрибін шляхом підшкірної ін'єкції в дозі 0,07 мг/кг/добу. Лікування повторювали або протягом 2 місяців або 6 місяців. Клінічні дослідження у фазі II забезпечили дані позитивної дії кладрибіну у пацієнтів з MS, виходя 96941 14 чи з оцінки відповідно до шкали Kutzke Extended Disability Status Scale (EDSS), Scripps Neurologic rating Scale (SNRS) і даних ядерної магнітної візуалізації (MPT) (Beutler та ін., 1996, Proc. Nat. Acad. Sci. USA, 93, 1716-1720; Romine та ін., 1999 вище). Результати дослідження у фазі III, були переконливими щодо істотного зменшення уражень головного мозку, виміряних шляхом MPT (Rice та ін., 2000, вище). Для найбільш високих доз спостерігали деякі побічні дії (АЕ), такі як збільшений відсоток інфекції щодо порушеної імунної функції або мієлосупресії (Selby та ін., 1998, вище; Beutler та ін., 1994, Acta hematol, 91:10-15). Внаслідок незначної різниці безпеки між ефективною дозою й дозою, при якій проявляються АЕ, дотепер, всі клінічні дослідження для кладрибіну при розсіяному склерозі здійснювали або шляхом в/в або шляхом п/ш введення. У результаті цього, Beutler та ін. (Beutler та ін., 1996, Seminars in Hematology, 33, 1(S1), 45-52) виключили пероральний шлях введення для лікування розсіяного склерозу за допомогою кладрибіну. Grieb та ін. описали невелике дослідження, проведене на 11 пацієнтах з розсіяним склерозом ремітуючого перебігу (Grieb та ін., 1995, Archivum Immunologiae et Therapiae Experimentalis, 43 (5-6), 323-327), у якому кладрибін вводили перорально протягом 6 місяців 5-ти денним курсом у сумарній дозі приблизно 4-5,7 мг/кг (вага пацієнтів приблизно 52 і приблизно 75 кілограм, відповідно) тобто сумарна ефективна доза становила 2-2,85 мг/кг. Для деяких пацієнтів, проводили одиничне повторне лікування протягом 5 днів з кумулятивною дозою 0,4-0,66 мг/кг після періоду без кладрибіну протягом 3 або 6 місяців. Побічні дії, які спостерігалися при вищеописаній схемі лікування, були менш важкими в порівнянні з побічними діями, які спостерігалися у пацієнтів, що страждають від хронічного прогресуючого розсіяного склерозу, яких лікували за допомогою в/в інфузії кладрибіну (Sipe та ін., 1994, Lancet, 344, 9-13), але однаково вони мали місце. Додатково, терапевтична ефективність вищеописаної пероральної схеми лікування відносно інфузійної терапії є спірною (Grieb та ін., 1995, вище) і була ідентифікована група, що "не відповідає " (Stelmasiak тa iн., 1998, Laboratory Investigations, 4(1), 4-8). Отже, є бажаним наявність способу лікування розсіяного склерозу, який включає пероральне введення кладрибіну, що буде забезпечувати таку ж або поліпшену дію на MS ураження зі зниженою частотою та/або тяжкістю побічних дій. Додатково, оскільки MS являє собою хронічне захворювання, то буде бажаним знижувати частоту та/або тяжкість побічних дій таким чином, щоб було можливим повторне лікування. Також є бажаним тривалий сприятливий вплив лікування за допомогою кладрибіну між періодами лікування. Також є бажаним наявність способу лікування розсіяного склерозу, що буде дозволяти здійснювати лікування пацієнтів, які резистентні принаймні до одного загальноприйнятого лікування. Даний винахід стосується застосування кладрибіну в комбінації з бета інтерфероном для при 15 готування фармацевтичного препарату для лікування розсіяного склерозу, де препарат кладрибіну вводять перорально. Зокрема, винахід стосується застосування кладрибіну в комбінації з бета інтерфероном для приготування лікарського засобу для лікування пацієнтів, які резистентні принаймні до одного загальноприйнятого лікування. Один варіант здійснення винаходу забезпечує поліпшену схему дозування для кладрибіну в комбінації з бета інтерфероном для лікування розсіяного склерозу. Додатковий варіант здійснення винаходу забезпечує застосування кладрибіну в комбінації з бета інтерфероном для приготування фармацевтичного препарату для лікування розсіяного склерозу, де зменшуються побічні дії, що дозволяє подальше застосування кладрибіну. В одному варіанті здійснення, винахід забезпечує: 1. Застосування комбінації кладрибіну й бетаінтерферону для приготування лікарського засобу для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, де кладрибін вводять перорально, відповідно до послідовних стадій, наведених нижче: (і) Індукційний період, у якому вводять кладрибін і в якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 1,7 мг/кг до приблизно 3,5 мг/кг; (іі) Період без кладрибіну, у якому не вводять кладрибін; (ііі) Період підтримання, у якому вводять кладрибін і в якому загальна доза кладрибіну, яку вводять протягом періоду підтримання, нижче або дорівнює загальній дозі кладрибіну, що досягається наприкінці індукційного періоду (і); (iv) Період без кладрибіну, у якому не вводять кладрибін. В іншому варіанті здійснення, винахід забезпечує: 2. Застосування комбінації кладрибіну й бетаінтерферону для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, що включає пероральне введення кладрибіну, відповідно до послідовних стадій, наведених нижче: (і) Індукційний період, у якому вводять кладрибін і в якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 1,5 мг/кг до приблизно 3,5 мг/кг; (іі) Період без кладрибіну, у якому не вводять кладрибін; (ііі) Період підтримання, у якому вводять кладрибін і в якому загальна доза кладрибіну, яку вводять протягом періоду підтримання, нижче або дорівнює загальній дозі кладрибіну, що досягається наприкінці індукційного періоду (і); (iv) Період без кладрибіну, у якому не вводять кладрибін. В іншому варіанті здійснення, винахід забезпечує: 3. Продукт, що містить кладрибін і бетаінтерферон у вигляді комбінованого препарату для 96941 16 одночасного, окремого або послідовного застосування для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, де кладрибін вводять перорально, відповідно до послідовних стадій, наведених нижче: (і) Індукційний період, у якому вводять кладрибін і в якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 1,7 мг/кг до приблизно 3,5 мг/кг; (іі) Період без кладрибіну, у якому не вводять кладрибін; (ііі) Період підтримання, у якому вводять кладрибін і в якому загальна доза кладрибіну, яку вводять протягом періоду підтримання, нижче або дорівнює загальній дозі кладрибіну, що досягається наприкінці індукційного періоду (і); (iv) Період без кладрибіну, у якому не вводять кладрибін. В іншому варіанті здійснення, винахід забезпечує: 4. Застосування або продукт відповідно до абзацу 1, 2 або 3 у даній заявці вище, де індукційний період триває аж до 4 місяців, або аж до 3 місяців, або аж до 2 місяців. В іншому варіанті здійснення, винахід забезпечує: 5. Застосування або продукт відповідно до будь-якого абзацу 1 - 4 у даній заявці вище, де індукційний період триває аж до 2 місяців і загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. В іншому варіанті здійснення, винахід забезпечує: 6. Застосування або продукт відповідно до будь-якого абзацу 1 - 4 у даній заявці вище, де індукційний період триває аж до 4 місяців і загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 3,5 мг/кг, переважно 3,5 мг/кг. В іншому варіанті здійснення, винахід забезпечує: 7. Застосування або продукт відповідно до будь-якого абзацу 1 - 5 у даній заявці вище, де період без кладрибіну (іі) триває аж до 10 місяців, або аж до 9 місяців, або аж до 8 місяців. В іншому варіанті здійснення, винахід забезпечує: 8. Застосування або продукт відповідно до абзацу 7 у даній заявці вище, де період без кладрибіну (іі) триває 8 місяців. В іншому варіанті здійснення, винахід забезпечує: 9. Застосування або продукт відповідно до будь-якого абзацу 1 - 8 у даній заявці вище, де період без кладрибіну (iv) триває аж до 10 місяців. В іншому варіанті здійснення, винахід забезпечує: 10. Застосування або продукт відповідно до будь-якого абзацу 1 - 9 у даній заявці вище, де комбінована тривалість індукційного періоду (і) з періодом без кладрибіну (іі) становить приблизно 1 рік (приблизно 12 місяців). 17 В іншому варіанті здійснення, винахід забезпечує: 11. Застосування або продукт відповідно до абзацу 10 у даній заявці вище, де тривалість індукційного періоду становить приблизно 4 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, або тривалість індукційного періоду становить приблизно 3 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, або тривалість індукційного періоду становить приблизно 2 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців. В іншому варіанті здійснення, винахід забезпечує: 12. Застосування або продукт відповідно до будь-якого абзацу 1 - 9 у даній заявці вище, де комбінована тривалість індукційного періоду (і) з періодом без кладрибіну (іі) становить приблизно 1 рік (приблизно 12 місяців) і загальна доза кладрибіну, яка досягається до закінчення цього року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг або приблизно 3,5 мг/кг, переважно 3,5 мг/кг. В іншому варіанті здійснення, винахід забезпечує: 13. Застосування або продукт відповідно до абзацу 12 у даній заявці вище, де тривалість індукційного періоду становить приблизно 4 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, або тривалість індукційного періоду становить приблизно 3 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, або тривалість індукційного періоду становить приблизно 2 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців. В іншому варіанті здійснення, винахід забезпечує: 14. Застосування або продукт відповідно до будь-якого абзацу 1 - 13 у даній заявці вище, де період підтримання триває аж до 4 місяців, або аж до 3 місяців або аж до 2 місяців. В іншому варіанті здійснення, винахід забезпечує: 15. Застосування або продукт відповідно до будь-якого абзацу 1 - 14 у даній заявці вище, де період підтримання триває аж до 2 місяців і загальна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. В іншому варіанті здійснення, винахід забезпечує: 16. Застосування або продукт відповідно до будь-якого абзацу 1 - 15 у даній заявці вище, де комбінована тривалість періоду підтримання (ііі) з періодом без кладрибіну (iv) становить приблизно 1 рік (приблизно 12 місяців). В іншому варіанті здійснення, винахід забезпечує: 17. Застосування або продукт відповідно до абзацу 16 у даній заявці вище, де тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців, або тривалість періоду підтримання (ііі) становить приблизно 3 місяці й три 96941 18 валість періоду без кладрибіну (iv) становить приблизно 9 місяців. В іншому варіанті здійснення, винахід забезпечує: 18. Застосування або продукт відповідно до абзацу 16 у даній заявці вище, де тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В іншому варіанті здійснення, винахід забезпечує: 19. Застосування або продукт відповідно до абзацу 16 у даній заявці вище, де комбінована тривалість періоду підтримання (ііі) з періодом без кладрибіну (iv) становить приблизно 1 рік (приблизно 12 місяців) і загальна доза кладрибіну, яку вводять протягом цього року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. В іншому варіанті здійснення, винахід забезпечує: 20. Застосування або продукт відповідно до абзацу 19 у даній заявці вище, де тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців, або тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців. В іншому варіанті здійснення, винахід забезпечує: 21. Застосування або продукт відповідно до абзацу 19 у даній заявці вище, де тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В іншому варіанті здійснення, винахід забезпечує: 22. Застосування або продукт відповідно до будь-якого абзацу 1 - 21 у даній заявці вище, де комбінована тривалість індукційного періоду (і), періоду без кладрибіну (іі), періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 2 роки (приблизно 24 місяця). В іншому варіанті здійснення, винахід забезпечує: 23. Застосування або продукт відповідно до абзацу 22 у даній заявці вище, де: - тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців або; - тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців, або; - тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців, або; 19 - тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців, або; - тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців, або; - тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців, або; - тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду безкладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців, або; - тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців, або; - тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В іншому варіанті здійснення, винахід забезпечує: 24. Застосування або продукт відповідно до абзацу 22 у даній заявці вище, де комбінована тривалість індукційного періоду (і) і періоду без кладрибіну (іі) становить приблизно 1 рік (12 місяців), комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік (приблизно 12 місяців), загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. В іншому варіанті здійснення, винахід забезпечує: 25. Застосування або продукт відповідно до абзацу 24 у даній заявці вище, де: - тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців або; - тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблиз 96941 20 но 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців, або; - тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців, або; - тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців, або; - тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців, або; - тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців, або; - тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців, або; - тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців, або; - тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В іншому варіанті здійснення, винахід забезпечує: 26. Застосування або продукт відповідно до абзацу 22 у даній заявці вище, де комбінована тривалість індукційного періоду (і) і періоду без кладрибіну (іі) становить приблизно 1 рік (12 місяців), комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік (приблизно 12 місяців), загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 3,5 мг/кг, переважно 3,5 мг/кг і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. В іншому варіанті здійснення, винахід забезпечує: 27. Застосування або продукт відповідно до абзацу 26 у даній заявці вище, де: - тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без клад 21 рибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців або; - тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців, або; - тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців, або; - тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців, або; - тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців, або; - тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців, або; - тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців, або; - тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців, або; - тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В іншому варіанті здійснення, винахід забезпечує: 28. Застосування або продукт відповідно до будь-якого абзацу 1 - 27 у даній заявці вище, де стадії (ііі) - (iv) повторюють один, два або три рази. В іншому варіанті здійснення, винахід забезпечує: 29. Застосування або продукт відповідно до будь-якого абзацу 1 - 28 у даній заявці вище, де індукційний період триває протягом 2 місяців, загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить 1,75 мг/кг, періоду без кладрибіну (іі) триває протягом 10 місяців, пе 96941 22 ріоду підтримання триває протягом 2 місяців і загальна доза кладрибіну, яку вводять протягом періоду підтримання, становить 1,75 мг/кг В іншому варіанті здійснення, винахід забезпечує: 30. Застосування або продукт відповідно до абзацу 29 у даній заявці вище, де період без кладрибіну (іν) триває протягом 10 місяців. В іншому варіанті здійснення, винахід забезпечує: 31. Застосування або продукт відповідно до будь-якого абзацу 1 - 28 у даній заявці вище, де індукційний період триває протягом 4 місяців, загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить 3,5 мг/кг, періоду без кладрибіну (іі) триває протягом 8 місяців, періоду підтримання триває протягом 2 місяців і загальна доза кладрибіну, яку вводять протягом періоду підтримання, становить 1,75 мг/кг, період без кладрибіну (iv) триває протягом 10 місяців. В іншому варіанті здійснення, винахід забезпечує: 32. Застосування або продукт відповідно до абзацу 31 у даній заявці вище, де період без кладрибіну (iv) триває протягом 10 місяців. В іншому варіанті здійснення, винахід забезпечує: 33. Застосування або продукт відповідно до будь-якого абзацу 1 - 32 у даній заявці вище, де стадії (ііі) - (іν) повторюють один раз. В іншому варіанті здійснення, винахід забезпечує: 34. Застосування або продукт відповідно до будь-якого абзацу 1 - 33 у даній заявці вище, де біодоступність кладрибіну становить приблизно 40%. В іншому варіанті здійснення, винахід забезпечує: 35. Застосування або продукт відповідно до будь-якого абзацу 1 - 34 у даній заявці вище, де загальна ефективна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 0,7 мг/кг до приблизно 1,4 мг/кг. В іншому варіанті здійснення, винахід забезпечує: 36. Застосування або продукт відповідно до будь-якого абзацу 1 - 35 у даній заявці вище, де загальна ефективна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 0,7 мг/кг або приблизно 1,4 мг/кг. В іншому варіанті здійснення, винахід забезпечує: 36. Застосування або продукт відповідно до будь-якого абзацу 1 - 36 у даній заявці вище, де загальна ефективна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 0,7 мг/кг. В іншому варіанті здійснення, винахід забезпечує: 37. Застосування або продукт відповідно до будь-якого абзацу 1 - 36 у даній заявці вище, де вводять кладрибін від 4 до 7 днів на місяць, переважно 4 або 5 днів на місяць, протягом індукційного періоду. 23 В іншому варіанті здійснення, винахід забезпечує: 38. Застосування або продукт відповідно до абзацу 37 у даній заявці вище, де вводять кладрибін від дня 1 до дня 5, або від дня 1 до дня 4, щомісяця протягом індукційного періоду. В іншому варіанті здійснення, винахід забезпечує: 39. Застосування або продукт відповідно до абзацу 37 або 38 у даній заявці вище, де вводять кладрибін у добовій дозі приблизно 0,175 мг/кг протягом індукційного періоду. В іншому варіанті здійснення, винахід забезпечує: 40. Застосування або продукт відповідно до абзацу 37, 38 або 39 у даній заявці вище, де вводять кладрибін декілька разів на добу протягом індукційного періоду, переважно два або три рази на добу, більш переважно два рази на добу. В іншому варіанті здійснення, винахід забезпечує: 41. Застосування або продукт відповідно до будь-якого абзацу 1 - 40 у даній заявці вище, де вводять кладрибін від 4 до 7 днів на місяць, переважно 4 або 5 днів на місяць, протягом періоду підтримання. В іншому варіанті здійснення, винахід забезпечує: 42. Застосування або продукт відповідно до абзацу 41 у даній заявці вище, де вводять кладрибін від дня 1 до дня 5, або від дня 1 до дня 4 щомісяця протягом періоду підтримання. В іншому варіанті здійснення, винахід забезпечує: 43. Застосування або продукт відповідно до абзаців 41 або 42 у даній заявці вище, де вводять кладрибін у добовій дозі приблизно 0,175 мг/кг протягом періоду підтримання. В іншому варіанті здійснення, винахід забезпечує: 44. Застосування або продукт відповідно до абзацу 41, 42 або 43 у даній заявці вище, де вводять кладрибін декілька разів на добу протягом періоду підтримання, переважно два або три рази на добу, більш переважно два рази на добу. В іншому варіанті здійснення, винахід забезпечує: 45. Застосування або продукт відповідно до будь-якого абзацу 1 - 44 у даній заявці вище, де бета-інтерферон вводять одночасно, окремо або послідовно з пероральним введенням кладрибіну. В іншому варіанті здійснення, винахід забезпечує: 46. Застосування або продукт відповідно до абзацу 45 у даній заявці вище, де бета-інтерферон вводять перед індукційним періодом (і), та/або після періоду підтримання (ііі). В іншому варіанті здійснення, винахід забезпечує: 47. Застосування або продукт відповідно до абзацу 45 у даній заявці вище, де бета-інтерферон вводять протягом періоду без кладрибіну (іі) та/або (iv). В іншому варіанті здійснення, винахід забезпечує: 96941 24 48. Застосування або продукт відповідно до абзацу 45 у даній заявці вище, де бета-інтерферон вводять одночасно з пероральним введенням кладрибіну. В іншому варіанті здійснення, винахід забезпечує: 49. Застосування або продукт відповідно до абзацу 48 у даній заявці вище, де бета-інтерферон вводять протягом індукційного періоду (і) та/або періоду підтримання (iv). В іншому варіанті здійснення, винахід забезпечує: 50. Застосування або продукт відповідно до абзацу 49 у даній заявці вище, де бета-інтерферон вводять протягом індукційного періоду (і), періоду підтримання (iv) і періодів без кладрибіну (іі) і (iv). В іншому варіанті здійснення, винахід забезпечує: 51. Застосування або продукт відповідно до абзацу 50 у даній заявці вище, де бета-інтерферон вводять перед індукційним періодом (і), протягом індукційного періоду (і), протягом періоду підтримання (iv), протягом періодів без кладрибіну (іі) і (iv) і після періоду без кладрибіну (iv). В іншому варіанті здійснення, винахід забезпечує: 52. Застосування або продукт відповідно до будь-якого абзацу 1 - 51, де бета-інтерферон вибирають із групи, яка включає: бета-інтерферон 1а і бета-інтерферон 1b. В іншому варіанті здійснення, винахід забезпечує: 53. Застосування або продукт відповідно до абзацу 52 у даній заявці вище, де бета-інтерферон вибирають із групи, яка включає: Avonex® (Biogen), Rebif® (Serono), і Betaseron® (Berlex / Schering AG). В іншому варіанті здійснення, винахід забезпечує: 54. Застосування або продукт відповідно до абзацу 53 у даній заявці вище, де бета-інтерферон являє собою Rebif® (Serono). В іншому варіанті здійснення, винахід забезпечує: 55. Застосування або продукт відповідно до будь-якого абзацу 52 - 54 у даній заявці вище, де бета-інтерферон вводять системно. В іншому варіанті здійснення, винахід забезпечує: 56. Застосування або продукт відповідно до абзацу 55 у даній заявці вище, де бета-інтерферон вводять підшкірно або внутрішньом'язево. В іншому варіанті здійснення, винахід забезпечує: 57. Застосування або продукт відповідно до абзацу 56 у даній заявці вище, де бета-інтерферон вводять у дозі принаймні 44 мкг підшкірно на введення. В іншому варіанті здійснення, винахід забезпечує: 58. Застосування або продукт відповідно до абзацу 56 у даній заявці вище, де схему дозування бета-інтерферону вибирають із групи, яка включає: 12 ММЕ (44 мкг) бета-інтерферону три рази 25 на тиждень, 12 ММЕ (44 мкг) щодня, 24 ММЕ (88 мкг) три рази на тиждень і 24 ММЕ (88 мкг) щодня. В іншому варіанті здійснення, винахід забезпечує: 59. Застосування або продукт відповідно до абзацу 56 у даній заявці вище, де використовуваний бета-інтерферон являє собою Rebif® (Serono) і його вводять у дозі 44 мкг підшкірно три рази на тиждень. В іншому варіанті здійснення, винахід забезпечує: 60. Застосування або продукт відповідно до будь-якого абзацу 1 - 59 у даній заявці вище, де загальноприйняте лікування вибирають із групи, яка включає: лікування за допомогою бета інтерферону, лікування за допомогою ацетату глатимареру (Copaxone®, Teva), лікування за допомогою наталізумабу (Tysabri®, Biogen/Elan), і лікування за допомогою мітоксантрону (Novantrone®, Serono). В іншому варіанті здійснення, винахід забезпечує: 61. Застосування або продукт відповідно до абзацу 60 у даній заявці вище, де загальноприйняте лікування являє собою лікування за допомогою бета інтерферону, переважно лікування за допомогою Betaseron® (Berlex / Schering AG); Avonex® (Biogen); або Rebif® (Serono). В іншому варіанті здійснення, винахід забезпечує: 62. Застосування або продукт відповідно до абзацу 61 у даній заявці вище, де загальноприйняте лікування являє собою лікування за допомогою Rebif® (Serono). В іншому варіанті здійснення, винахід забезпечує: 63. Застосування або продукт відповідно до абзацу 60 або 61 у даній заявці вище, де пацієнти, яких піддають лікуванню, резистентні до одного, двох, трьох або чотирьох загальноприйнятих лікувань. В іншому варіанті здійснення, винахід забезпечує: 64. Застосування або продукт відповідно до будь-якого абзацу 1 - 63 у даній заявці вище, де в резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив, незважаючи на одержання принаймні одного загальноприйнятого лікування. В іншому варіанті здійснення, винахід забезпечує: 65. Застосування або продукт відповідно до абзацу 64 у даній заявці вище, де принаймні один рецидив відбувся протягом року перед початком лікування відповідно до будь-якого з попередніх пунктів. В іншому варіанті здійснення, винахід забезпечує: 66. Застосування або продукт відповідно до абзацу 65 у даній заявці вище, де у пацієнтів, яких піддають лікуванню, відбувся принаймні один рецидив протягом року, що передує початку лікування відповідно до будь-якого з попередніх пунктів і їх піддавали лікуванню за допомогою Rebif® 96941 26 (Serono), особливо 12 ММЕ (44 мкг) Rebif® три рази на тиждень. В іншому варіанті здійснення, винахід забезпечує: 67. Застосування або продукт відповідно до будь-якого з абзаців 1 - 66 у даній заявці вище, де в резистентних пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС, що визначалося за допомогою методів, таких як метод МРТ, незважаючи на одержання принаймні одного загальноприйнятого лікування. В іншому варіанті здійснення, винахід забезпечує: 68. Застосування або продукт відповідно до абзацу 67 у даній заявці вище, де збільшене число уражень або збільшений об'єм мозкових уражень виникали протягом року перед початком лікування відповідно до будь-якого з попередніх пунктів. В іншому варіанті здійснення, винахід забезпечує: 69. Застосування або продукт відповідно до пункту 68 у даній заявці вище, де у пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС протягом року, що передує початку лікування відповідно до будь-якого з попередніх пунктів і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif® три рази на тиждень. В іншому варіанті здійснення, винахід забезпечує: 70. Застосування або продукт відповідно до будь-якого абзацу 1 - 69 у даній заявці вище, де в резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив, і розвивається наростаюча інвалідність внаслідок прогресуючих форм захворювання. В іншому варіанті здійснення, винахід забезпечує: 71. Застосування або продукт відповідно до будь-якого абзацу 1 - 70 у даній заявці вище, де резистентні пацієнти, яких піддають лікуванню, страждають від погіршення MS, особливо вторинного прогресуючого, прогресуючого ремітуючого або погіршення MS ремітуючого перебігу. Докладний опис винаходу Визначення Термін "розсіяний склероз" у межах значень даного винаходу може бути визначений згідно із DSM-IV класифікацією (Diagnosis and Statistical Manual of Inflammatory CNS Disorders, Fourth Edition, American Psychiatric Association, Washington D.C., 1994). "Загальна доза" або "кумулятивна доза" стосується загальної дози кладрибіну, яку вводять протягом лікування, тобто дози, яка досягається при закінченні лікування, що розраховується шляхом додавання щоденних доз. Наприклад, загальна доза кладрибіну, що відповідає лікуванню 0,7 мг/кг кладрибіну на добу протягом 5 днів, становить 3,5 мг/кг або загальна доза кладрибіну, що відповідає лікуванню 0,35 мг/кг кладрибіну на добу протягом 5 днів, становить 1,7 мг/кг. 27 "Загальна ефективна доза" або "кумулятивна ефективна доза" стосується біодоступної дози кладрибіну після даного періоду введення, тобто біодоступної дози, яка досягається при закінченні лікування, що розраховується шляхом додавання щоденних доз, зменшених на коефіцієнт біодоступності. Наприклад, загальна ефективна доза кладрибіну, що відповідає лікуванню 0,7 мг/кг кладрибіну на добу протягом 5 днів, де біодостушгість кладрибіну дорівнює приблизно 40%, становить 1,4 мг/кг або загальна ефективна доза кладрибіну, що відповідає лікуванню 0,35 мг/кг кладрибіну на добу протягом 5 днів, де біодоступність кладрибіну дорівнює приблизно 40%, становить 0,7 мг/кг. Звичайно, біодоступність кладрибіну або препаратів кладрибіну, які використовують у контексті даного винаходу, становить від приблизно 30% до приблизно 90%, переважно від приблизно 40% до приблизно 60%, наприклад, приблизно 50%. "Тиждень" стосується періоду часу приблизно 5, приблизно 6 або приблизно 7 днів. "Місяць" стосується періоду часу приблизно 28, приблизно 29, приблизно 30 або приблизно 31 днів. "Лікування" включає послідовне здійснення "індукційного лікування" і принаймні "підтримуючого лікування". Звичайно, лікування відповідно до винаходу включає "індукційне лікування" і приблизно одне або приблизно два або приблизно три підтримуючих лікування. Звичайно, лікування відповідно до винаходу становить приблизно 2 роки (приблизно 24 місяця) або приблизно 3 роки (приблизно 36 місяців) або приблизно 4 роки (приблизно 48 місяців). "Індукційне лікування" включає послідовне здійснення (і) індукційного періоду, де кладрибін або фармацевтичний препарат кладрибіну за винаходом вводять перорально й (іі) період без кладрибіну. Індукційний період триває аж до приблизно 4 місяців або аж до приблизно 3 місяців або аж до приблизно 2 місяців. Наприклад, індукційний період триває від приблизно 2 до приблизно 4 місяців. Індукційний період включає пероральне введення кладрибіну або його фармацевтичного препарату протягом від приблизно 1 до приблизно 7 днів щомісяця. "Період без кладрибіну" являє собою період, у якому пацієнтові не вводять кладрибін. Протягом періоду без кладрибіну, пацієнт може не одержувати ніякого препарату або йому можуть вводити плацебо або інший засіб, що не містить лікарського засобу. Період без кладрибіну триває аж до приблизно 10 місяців або аж до 9 місяців або аж до приблизно 8 місяців. Наприклад, період без кладрибіну триває від приблизно 8 до приблизно 10 місяців, звичайно принаймні приблизно 8 місяців. "Підтримуюче лікування" включає послідовне здійснення (і) періоду підтримання, у якому кладрибін або фармацевтичний препарат кладрибіну з а винаходом вводять перорально в більш низькій або еквівалентній дозі відносно дози кладрибіну, що вводять перорально протягом індукційного лікування, і (іі) період без кладрибіну. Період підтримання триває протягом аж до приблизно 4 міся 96941 28 ців, або аж до приблизно 3 місяців, або аж до приблизно 2 місяців, переважно аж до приблизно 2 місяців. Наприклад, період підтримання триває протягом приблизно 2 до приблизно 4 місяців, переважно протягом приблизно 2 місяців. Період підтримання включає пероральне введення кладрибіну або його фармацевтичного препарату протягом від приблизно 1 до приблизно 7 днів щомісяця. У контексті даного винаходу, сприятлива дія, включаючи, але не обмежуючись тільки ними, ослаблення, зменшення, зниження або скорочення патологічного розвитку після початку захворювання, може проявлятися після одного або декількох "лікувань", після "індукційного лікування", після "підтримуючого лікування" або протягом періоду без кладрибіну. "Добова доза" стосується загальної дози кладрибіну, яку вводять пацієнтові перорально в кожен день введення. Добової дози можна досягти за допомогою одиничного або декількох введень на добу, таких як, наприклад, один раз на добу, два рази на добу або три рази на добу. Дозування, яке вводять, у вигляді одиничної або багаторазової доз, індивідуумові, буде відрізнятися залежно від багатьох факторів, включаючи фармакокінетичні властивості, стани й характеристики пацієнта (стать, вік, вага тіла, стан здоров'я, розмір), інтенсивність симптомів, що супроводжують лікування, частоту лікування й бажаний результат. Пацієнти, що страждають від MS, можуть бути визначені, наприклад, як такі, що мають клінічно встановлений або лабораторно встановлений MS відповідно до критерію Schumacher або Poser (Schumacher та ін., 1965, Ann. NY Acad. Set 1965; 122:552-568; Poser та ін., 1983, Ann. Neurol. 13(3): 227-31). "Рецидиви" охоплюють неврологічні проблеми, які розвиваються протягом короткого періоду часу, звичайно днів, але в деяких випадку більш короткого, такого як години або навіть хвилини. Ці напади найбільш часто задіють рухові, чутливі, зорові або координаційні порушення, що виявляються раніше при захворюванні. Пізніше також можуть проявлятися порушення сечового міхура, кишечнику, статеві розлади й порушення пізнавальної функції. Іноді початок нападу проявляється через декілька тижнів. Звичайно рецидив MS задіює період погіршення, з неврологічними недостатностями, потім плато, у якому пацієнт не почуває себе ні гірше, ні краще, з наступним періодом відновлення. Відновлення звичайно починається протягом декількох тижнів. "Ефективність" лікування відповідно до винаходу можна визначити на основі змін перебігу захворювання у відповідь на застосування відповідно до винаходу. Наприклад, ефективність лікування MS можна оцінити за допомогою частоти рецидивів в RRMS і наявності або відсутності нових уражень у ЦНС, що визначали за допомогою методів, таких як метод MPT (Miller та ін., 1996, Neurology, 47(Suppl 4): S217; Evans та ін., 1997, Ann. Neurology, 41:125-132). 29 Спостереження за зменшенням та/або пригніченням МРТ Т1 гадоліній-розширених ушкоджень (які, як вважають, характеризують ділянки активного запалення) дозволяє одержати первинну змінну ефективності. Вторинні змінні ефективності включають МРТ Т1 збільшений об'єм мозкових уражень, МРТ Т1 збільшену кількість уражень, МРТ Т2 об'єм уражень (які, як вважають, характеризують загальне навантаження захворювання, тобто демієлінізацію, гліоз, запалення й руйнування аксонів), МРТ Т1 збільшений об'єм гіпоінтенсивних уражень (які, як вважають, характеризують первинну демієлінізацію й руйнування аксонів), час до прогресування MS, частоту й тяжкість загострень і час до загострення, оцінку згідно із Expanded Disability Status Scale і оцінку згідно із Scripps Neurologic Rating Scale (SNRS) (Sipe та ін., 1984, Neurology, 34, 1368-1372). Методи ранньої й точної діагностики розсіяного склерозу й наступного прогресування захворювання описані в Mattson, 2002, Expert Rev. Neurotherapeutics, 319-328. Ступінь інвалідності пацієнтів з MS, можна визначити, наприклад, за допомогою шкали оцінки Kurtzke Expanded Disability Status Scale (EDSS) (KurtzL·, 1983, Neurology, 33, 1444-1452). Звичайне зниження кількості балів згідно із EDSS відповідає поліпшенню захворювання й навпаки, підвищення кількості балів згідно із EDSS відповідає погіршенню захворювання. Кладрибін (2-CdA) 2-CdA і його фармакологічно прийнятні солі можуть застосовуватися при здійсненні даного винаходу. Кладрибін може бути приготовлений у вигляді будь-якого фармацевтичного препарату, який підходить для перорального введення. Типові пероральні препарати 2-CdA описані в (WO 96/19230; WO 96/19229; US 6,194,395; US 5,506,214; WO 2004/087100; WO 2004/087101), зміст цих джерел повністю включено в дану заявку як посилання. Приклади компонентів для пероральних препаратів наведені нижче. Способи приготування 2-CdA добре відомі в даній галузі техніки. Наприклад, приготування 2CdA описане в (ЕР 173,059; WO 04/028462; WO 04/028462; US 5,208,327; WO 00/64918) і Robins та ін., J. Am. Chem. Soc., 1984, 106: 6379. Альтернативно, фармацевтичні препарати 2-CdA можуть бути отримані від Bedford Laboratories, Bedford, Ohio. Пероральне введення кладрибіну може здійснюватися у формі капсули, таблетки, пероральної суспензії або сиропу. Таблетка або капсули можуть містити приблизно від 3 до 500 мг кладрибіну. Переважно, вони можуть містити від приблизно 3 до приблизно 10 мг кладрибіну, більш переважно приблизно 3, приблизно 5 або приблизно 10 мг кладрибіну. Капсули можуть являти собою желатинові капсули й можуть містити, додатково до кладрибіну в кількості, вказаній вище, невелика кількість, наприклад, менше 5 мас.%, стеарату магнію або іншого наповнювача. Таблетки можуть містити вищевказану кількість сполуки й сполучне, яке може являти собою желатиновий розчин, кро 96941 30 хмальну пасту у воді, полівініл-полівініловий спирт у воді й т.д. зі звичайним цукром для покриття. У переважному варіанті здійснення даного винаходу, кладрибін готують у вигляді таблеток. Переважно вказані таблетки містять 3 мг або 10 мг 2CdA. Ще більш переважно вказані таблетки являють собою таблетки, описані в таблиці 2 у даній заявці нижче (3 мг або 10 мг 2-CdA на таблетку). Композиції кладрибіну Композиції згідно із даним винаходом додатково можуть містити один або декілька фармацевтично прийнятних додаткових компонентів, таких як квасці, стабілізатори, протимікробні засоби, буфери, барвники, ароматизатори, ад'юванти й інших. Композиції згідно із даним винаходом можуть знаходитися у формі таблеток або ромбоподібних таблеток, приготовлених відповідно до загальноприйнятих способів. Наприклад, таблетки й капсули для перорального введення можуть містити загальноприйняті наповнювачі, включаючи, але не обмежуючись тільки ними, зв'язувальні речовини, наповнювачі, ковзаючі речовини, дезінтегратори й змочувальні речовини. Зв'язувальні речовини включають, але не обмежуючись тільки ними, патоку, акацію, желатин, сорбіт, трагакант, рослинний клей крохмалю й полівінілпіролідон. Наповнювачі включають, але не обмежуючись тільки ними, лактозу, цукор, мікрокристалічну целюлозу, кукурудзяний крохмаль, фосфат кальцію й сорбіт. Ковзні речовини включають, але не обмежуючись тільки ними, стеарат магнію, стеаринову кислоту, тальк, поліетиленгліколь і діоксид кремнію. Дезінтегратори включають, але не обмежуючись тільки ними, картопляний крохмаль і натрійкрахмал гліколят. Змочувальні речовини включають, але не обмежуючись тільки ними, лаурилсульфат натрію). Таблетки можуть бути покриті відповідно до методів, добре відомих в даній галузі техніки. Композиції згідно із даним винаходом також можуть бути представлені у вигляді рідких препаратів, включаючи, але не обмежуючись тільки ними, водні або масляні суспензії, розчини, емульсії, сиропи й еліксири. Композиції також можуть бути приготовлені у вигляді безводного продукту для відновлення водою або іншим підходящим наповнювачем перед застосуванням. Такі рідкі препарати можуть містити додаткові речовини, включаючи, але не обмежуючись тільки ними, суспендуючі речовини, емульсифікатори, неводні наповнювачі й консерванти. Суспендуючий засіб включає, але не обмежуючись тільки ними, сироп сорбіту, метилцелюлозу, глюкозу/цукровий сироп, желатин, гідроксіетилцелюлозу, карбоксиметилцелюлозу, гель алюміній-стеарат і гідровані харчові жири. Емульсификатори включають, але не обмежуючись тільки ними, лецитин, моноолеат сорбіту й гуміарабік. Неводні наповнювачі включають, але не обмежуючись тільки ними, харчові масла, мигдальну олію, фракціоновану кокосову олію, жирні складні ефіри, пропіленгліколь і етиловий спирт. Консерванти включають, але не обмежуючись тільки ними, метил або пропіл парагідроксибензоат і сорбінову кислоту. Бета-інтерферон (інтерферон-β або IFN-β), 31 Відповідно до даного винаходу, кладрибін вводять у комбінації з терапевтично ефективною кількістю бета-інтерферону. Бета-інтерферон вводять перед, одночасно або послідовно із кладрибіном. Активні речовини, які вводять одночасно з іншими терапевтичними засобами, можна вводити в тих же або різних композиціях і тими ж або іншими шляхами введення. Термін "бета-інтерферон" (інтерферон-β або IFN-β), як використовується в даному винаході, охоплює інтерферон фібробластів людини, який може бути нативним, тобто очищеним із природного джерела, або отриманий за допомогою методик рекомбінатної ДНК із прокаріотичних джерел (наприклад, Escherichia coli, Е. соlі) або з еукаріотичних клітин-хазяїв, наприклад, із дріжджів або клітин ссавців. Клітини ссавців, такіяк клітини яєчників китайського хом'ячка (СНО) або клітини людини є переважним хазяїном для одержання рекомбінантного бета-інтерферону. Бетаінтерферон може бути глікозилованим або неглікозилованим. Якщо бета-інтерферон, використовуваний відповідно до даного винаходу, є неглікозилованим (наприклад, отриманий в Е. соlі), то переважним є введення підвищених кількостей бета-інтерферону для одержання біологічної або фармакологічної дії, порівнянної з дією глікозилованого бета-інтерферону. Наприклад, кількість неглікозилованого бета-інтерферону, що приблизно в 10 разів вище за кількості глікозилованого бета-інтерферону, переважно вводять для одержання порівнянних активностей. Термін "бета-інтерферон", як використовується в даній заявці, також охоплює функціональні похідні, мутеїни, аналоги, і фрагменти, або злиті білки бета-інтерферону. Таким чином, терміни "інтерферон (IFN)" і "бета-інтерферон (IFN-бета)", як використовується в даній заявці, охоплюють інтерферон фібробластів, особливо людського походження, отриманий шляхом виділення з біологічних рідин, або отриманий за допомогою методик рекомбінантної ДНК із прокаріотичних або еукаріотичних клітин-хазяїв, а також його солі, функціональні похідні, варіанти, аналоги й активні фрагменти. Застосування інтерферону людського походження є переважним відповідно до даного винаходу. В одному варіанті здійснення даного винаходу використовуваний білок бета-інтерферон являє собою бета-інтерферон 1а, такий як, наприклад, Avonex® (Biogen) або Rebif® (Serono). В іншому варіанті здійснення даного винаходу білок бетаінтерферон являє собою бета-інтерферон 1b, такий як, наприклад, Betaseron® (Berlex / Schering AG). Переважно, бета-інтерферон, використовуваний у рамках даного винаходу, являє собою Betaseron® (Berlex / Schering AG); Avonex® (Biogen); або Rebif® (Serono). Більше кращий бета-інтерферон являє собою Rebif® (Serono). Rebif® (інтерферон бета-1a) являє собою очищений глікопротеїн, який складається з 166 амінокислот, з молекулярною вагою приблизно 22,500 кДа. Його одержують за допомогою методики рекомбінантної ДНК, використовуючи генети 96941 32 чно сконструйовані клітини яєчників китайського хом'ячка, у які вбудований ген бета-інтерферону людини. Амінокислотна послідовність Rebif® ідентична до послідовності бета-інтерферону людини, що має походження із природного фібробласта. Природний бета-інтерферон і інтерферон бета-1a (Rebif®) глікозиловані в кожній вуглеводній частині, що містить одиничний N-зв'язаний комплекс. Використовуючи порівняльний стандарт, відкалібрований відносно стандартного природного бета-інтерферону В 003 (Другий міжнародний стандарт для інтерферону, фібробласти людини GB 23 902 531), Rebif® має питому активність приблизно 270 мільйонів міжнародних одиниць (ММЕ) антивірусної активності на мг інтерферону бета1а, визначену в біологічному дослідженні цитопатогенної дії в умовах in vitro, використовуючи клітини WISH і вірус везикулярного стоматиту. Таблиця переходу для ММЕ й мкг бетаінтерферону ММЕ мкг 3 11 12 44 18 66 24 88 Rebif® 44 мкг містить приблизно 12 ММЕ антивірусної активності, як визначено за допомогою цього методу. Rebif® (рекомбінантний бета-інтерферон людини) являє собою найбільш сучасний розроблений підхід для інтерферонотерапії розсіяного склерозу (MS) і він характеризує істотний прогрес у лікуванні. Rebif® являє собою інтерферон (IFN)бета 1a, який продукується клітинними лініями ссавців. Встановлено, що даний інтерферон бета1а при підшкірному введенні три рази на тиждень ефективний для лікування розсіяного склерозу ремітуючого перебігу (RRMS). Інтерферон бета-1а може мати позитивну дію при довгостроковому курсі лікування MS шляхом зменшення кількості й тяжкості рецидивів і зменшення тяжкості захворювання й перебігу хвороби, що визначається за допомогою МРТ. Відповідно до даного винаходу, де бетаінтерферон являє собою рекомбінантний бетаінтерферон 1b, який продукується в Е. соlі, комерційно доступний під торговельною маркою Betaseron, його переважно можна вводити підшкірно через день у дозі приблизно від 250 до 300 мкг або від 8 ММЕ до 9,6 ММЕ на пацієнта. Відповідно до даного винаходу, де бетаінтерферон являє собою рекомбінантний бетаінтерферон 1а, який продукується клітинами яєчника китайського хом'ячка (клітинами СНО), комерційно доступний під торговельною маркою Avonex, його переважно можна вводити внутрішньом'язево один раз на тиждень у дозі приблизно від 30 мкг до 33 мкг або від 6 ММЕ до 6,6 ММЕ на пацієнта. Відповідно до даного винаходу, де бетаінтерферон являє собою рекомбінантний бетаінтерферон 1а, який продукується клітинами яєчника китайського хом'ячка (клітинами СНО), комерційно доступний під торговельною маркою Rebif, його переважно можна вводити підшкірно три рази на тиждень (TIW) у дозі від 22 до 44 мкг або від 6 33 ММЕ до 12 ММЕ на пацієнта. Переважно, на введення вибирають дозу 44 мкг або 12 ММЕ. Білки бета-інтерферону відповідно до даного винаходу можуть включати функціональні похідні, варіанти й мутеїни бета-інтерферону. "Функціональні похідні", як використовується в даній заявці, охоплюють похідні бета-інтерферону, і їх варіанти або мутеїни й злиті білки, які можуть бути отримані з функціональних груп, які знаходяться у вигляді бокових ланцюгів на залишках або N- або С-кінцевих груп, як добре відомо в даній галузі. Ці функціональні похідні охоплюються даним винаходом доти, поки вони залишаються фармацевтично прийнятними, тобто вони не руйнують активність білка, що істотно подібна, або краща, ніж активність бета-інтерферону, і не надають токсичних властивостей композиції, які їх містять. Ці похідні можуть включати, наприклад, бокові ланцюги поліетиленгліколю, які можуть поліпшувати інші властивості білка, такі як стабільність, період напіввиведення, біодоступність, переносимість організмом людини або імуногенність. Для досягнення цієї мети, бета-інтерферон може бути зв'язаний, наприклад, з поліетиленгліколем (ПЕГ). Пегілювання може здійснюватися за допомогою відомих методів, описаних, наприклад, у міжнародній заявці WO 92/13095. Особливо, ПЕГінтерферон може бути отриманий відповідно до відомостей, розкритих в WO 99/55377. Таким чином, у переважному варіанті здійснення, функціональне похідне бета-інтерферону містить принаймні один компонент, приєднаний до однієї або декількох функціональних груп, які представлені у вигляді однієї або декількох бокових ланцюгів на амінокислотних залишках. Надзвичайно переважним є варіант здійснення винаходу, у якому компонент являє собою поліетиленглікольний (ПЕГ) компонент. Відповідно до даного винаходу, до бета-інтерферону також можуть бути приєднані декілька ПЕГ компонентів. Інші похідні включають модифіковані білки бета-інтерферону, такі як форми бета-інтерферону із тривалою дією. Особливо, бета-інтерферон із тривалою дією може бути вибраний з пегілованого бета-інтерферону, злитих білків бета-інтерферонHAS, і злитих білків бета-інтерферон-Fc. Іншими похідними є складні аліфатичні ефіри карбоксильних груп, аміди карбоксильних груп, отримані шляхом взаємодії з аміаком або з первинними або вторинними амінами, N-ацильні похідні вільних амінокислотних груп амінокислотних залишків, утворені з ацильними компонентами (наприклад, алканоїльні або карбоциклічні ароїльні групи) або О-ацильні похідні вільних гідроксильних груп (наприклад, такі похідні залишків серилу або треонілу), утворені з ацильними компонентами. "Варіанти" або "мутеїни", як використовується в рамках даного винаходу, стосуються аналогів бета-інтерферону, у яких один або декілька амінокислотних залишків природного бета-інтерферону замінені іншими амінокислотними залишками, або вилучені, або один або декілька амінокислотних залишків додані до природної послідовності бетаінтерферону, без значного зниження активності 96941 34 отриманих продуктів у порівнянні з бетаінтерфероном дикого типу. Ці мутеїни одержують за допомогою відомого синтезу та/або за допомогою методик сайт-спрямованого мутагенезу, або за допомогою інших відомих підходящих методик. "Варіант" або "мутеїн" відповідно до даного винаходу охоплює білки, які кодуються нуклеїновою кислотою, такою як ДНК або РНК, яка гібридизується із ДНК або РНК, що кодує бета-інтерферон, як описано, наприклад, в US 4,738,931 у строгих умовах. Термін "строгі умови" стосується умов гібридизації й наступного промивання, які для середнього фахівця в даній галузі техніки умовно розглядаються як "строгі". Див. Ausubel та ін., Current Protocols in Molecular Biology, Interscience, N.Y., §§6.3 і 6.4 (1987, 1992). Без обмежень, прикладами строгих умов є умови промивання на 1220°С нижче за розраховану Тm гібриду в дослідженні, наприклад, 2 × SSC і 0,5% SDS протягом 5 хвилин, 2 × SSC і 0,1% SDS протягом 15 хвилин; 0,1 × SSC і 0,5% SDS при 37°С протягом 30 60 хвилин і потім, 0,1 × SSC і 0,5 % SDS при 68°С протягом 30 60 хвилин. Для фахівця в даній галузі техніки також є очевидним, що строгість умов також залежить від довжини послідовностей ДНК, олігонуклеотидних зондів (таких як 10-40 основ) або змішаних олігонуклеотидних зондів. Якщо використовуються змішані зонди, то кращим є застосування хлориду тетраметиламонію (ТМАС) замість SSC. Див. Ausubel, вище. Ідентичність вказує на подібність між двома або більше поліпептидними послідовностями або двома або більше полінуклеотидними послідовностями, визначену шляхом порівняння послідовностей. У цілому, ідентичність стосується точної відповідності нуклеотиду до нуклеотиду або амінокислоти до амінокислоти для двох полінуклеотидних або двох поліпептидних послідовностей, відповідно, по всіх довжині порівнюваних послідовностей. Для послідовностей, які не характеризуються точною відповідністю, можна визначити "% ідентичності". У цілому, дві послідовності, що підлягають порівнянню, вирівнюють для одержання максимальної кореляції між послідовностями. Воно може включати вставку "гепів" у будь-яку одну або обидві послідовності, для посилення ступеня вирівнювання. % ідентичності можна визначити для повної довжини кожної з послідовностей, що підлягають порівнянню (так зване глобальне вирівнювання), яке переважно придатне для послідовностей такої ж або дуже подібної довжини, або для більш коротких, визначених довжин (так зване локальне вирівнювання), що є більш підходящим для послідовностей неоднакової довжини. Способи порівняння ідентичності й гомології двох або більше послідовностей добре відомі в даній галузі. Відповідно, програми, доступні, наприклад, в Wisconsin Sequence Analysis Package, версія 9.1 (Devereux J та ін., 1984), наприклад, програми BESTFIT і GAP, можна використовувати для визначення % ідентичності між двома полінуклеотидами й % ідентичності й % гомології між двома поліпептидними послідовностями. В BESTFIT використають алгоритм "локальної гомо 35 логії" Smith і Waterman (1981) і виявляють найкращу одиничну ділянку подібності між двома послідовностями. Також у даній галузі відомі інші програми для визначення ідентичності та/або подібності між послідовностями, наприклад, сімейство програм BLAST (Altschul S F та ін., 1990, Altschul S F та ін., 1997, доступні на домашній сторінці NCBI за адресою www.ncbi.nlm.nih.gov) і FASTA (Pearson W R, 1990). "Варіант" або "мутеїн" відповідно до даного винаходу охоплює білки, що мають амінокислоти, у достатній мірі дублюючі такі бета-інтерферону, таким чином, що вони мають практично таку ж активність, що й бетаінтерферон. Функціональне дослідження для оцінки того, чи має будь-який варіант або мутеїн аналогічну активність, що й бета-інтерферон, являє собою, наприклад, визначення активності інтерферону при цитопатогенній дії вірусу везикулярного стоматиту в клітинах WISH, наприклад, описане Youcefi та ін., 1985. Отже, представляється можливим за допомогою звичайного досліду визначити, чи має будь-якої даний мутеїн по суті таку ж активність, що бета-інтерферон. У переважному варіанті здійснення, будь-який такий варіант або мутеїн має принаймні 40% ідентичність або гомологію з послідовністю бетаінтерферону, як описано, наприклад, в US 4,738,931. Більш переважно, він має принаймні 50%, принаймні 60%, принаймні 70%, принаймні 80% або, найбільш переважно, принаймні 90% ідентичність або гомологію. Мутеїни бета-інтерферону, які можна використовувати відповідно до даного винаходу, або нуклеїнової кислоти, яка їх кодує, включає кінцеву множину істотно відповідних послідовностей у вигляді замінених пептидів або полінуклеотидів, що можуть бути отримані за допомогою загальноприйнятих методів середнім фахівцем у даній галузі техніки, не потребуючих надмірного досвіду, на основі знань і правил, розкритих у даній заявці. Кращими змінами для мутеїнів відповідно до даного винаходу є зміни, відомі як "консервативні" заміни. Консервативні амінокислотні заміни поліпептидів бета-інтерферону можуть включати синонімічні амінокислоти в межах груп, які мають істотно подібні фізико-хімічні властивості, що заміна між представниками групи буде зберігати біологічну функцію молекули (Grantham, 1974). Очевидно, що вставки й делеції амінокислот також можуть бути здійснені у вищевизначених послідовностях без впливу на їх функцію, особливо, якщо інсерції або делеції зачіпають тільки декілька амінокислот, наприклад, до тридцяти, і переважно до десяти, і не видаляють або витісняють амінокислоти, які є критичними для функціональної конформації, наприклад, цистеїнові залишки. Білки й мутеїни, одержувані за допомогою таких делецій та/або інсерцій, підпадають під обсяг даного винаходу. Переважно, синонімічні групи амінокислот являють собою групи, наведені в таблиці І. Більш переважно, синонімічні групи амінокислот являють собою групи, наведені в таблиці II; і найбільш переважно синонімічні групи амінокислот являють собою групи, наведені в таблиці III. 96941 36 37 Прикладами одержання амінокислотних замін у білках, які можна використовувати для одержання мутеїнів бета-інтерферону для застосування в даному винаході включає будь-які відомі стадії способи, такі як описані в патентах US 4,959,314, 4,588,585 і 4,737,462, на ім'я Mark та ін.; 5,116,943 на ім'я Koths та ін., 4,965,195 на ім'я Namen та ін.; 4,879,111 на ім'я Chong та ін.; і 5,017,691 на ім'я 96941 38 Lee та ін.; і білки із заміненим лізином, описані в патенті US 4,904,584 (Shawта ін.). "Варіант" або "мутеїн" відповідно до даного винаходу охоплює також так звані "консенсусні інтерферони". "Консенсусні інтерферони" являють собою варіанти IFN, які не зустрічаються в природі (US 6,013,253). Консенсусні інтерферони також можуть використовуватися відповідно до винаходу. Відповідно до даного винаходу, сіль бетаінтерферону також може використовуватися для лікування розсіяного склерозу. Термін "солі" у даній заявці стосується як солей карбоксильних груп, так і солей приєднання кислоти аміногруп білків, описаних вище, або їх аналогів. Солі карбоксильної групи можуть утворюватися за допомогою методів, відомих у даній галузі, і включають неорганічні солі, наприклад, солі натрію, кальцію, амонію, тривалентного заліза або цинку, і подібні, і солі з органічними основами, які при цьому утворюються, наприклад, з амінами, такими як триетаноламін, аргінін або лізин, піперидин, прокаїн і інших. Солі приєднання кислоти включають, наприклад, солі з мінеральними кислотами, такими як, наприклад, соляна кислота або сірчана кислота, і солі з органічними кислотами, такими як, наприклад, оцтова кислота або щавлева кислота. Безсумнівно, будь-які такі солі повинні зберігати біологічну активність бета-інтерферону, що може бути виміряна, наприклад, у біологічному аналізі, описаному вище. Термін "злитий білок" стосується поліпептиду, що містить бета-інтерферон, або його варіант або мутеїн або фрагмент, злитий з іншим білком, який, наприклад, має подовжений час утримання в рідинах організму. Отже, бета-інтерферон може бути злитий з іншим білком, поліпептидом або подібним, наприклад, імуноглобуліном або його фрагментом. Таким чином, в подальшому варіанті здійснення, бета-інтерферон містить злиті імуноглобулін, тобто бета-інтерферон являє собою злитий білок, що містить всю або частину бета-інтерферону, злиту з усім або частиною імуноглобуліну. Методи одержання злитих білків імуноглобуліну добре відомі в даній галузі техніки, такі як, наприклад, описані в міжнародній заявці WO 01/03737. Середньому фахівцю у даній галузі техніки слід взяти до уваги, що отриманий злитий білок за винаходом зберігає біологічну активність бетаінтерферону. Злиття може бути безпосереднім або за допомогою короткого лінкерного пептиду, який може бути дуже коротким, наприклад, довжиною від 1 до 3 амінокислотних залишків, або більш довгим, наприклад, довжиною 13 амінокислотних залишків. Вказаний лінкер може являти собою трипептид з послідовністю E-F-M (Glu-Phe-Met), наприклад, або лінкер з 13 амінокислот з послідовністю, що містить Glu-Phe-Gly-Ala-Gly-Leu-ValLeu-Gly-Gly-Gln-Phe-Met, або лінкер, збагачений Gly-Ser, вставлений між послідовністю бетаінтерферону й послідовністю, що має походження з імуноглобуліну. Отриманий злитий блок має поліпшені властивості, такими як подовжений час утримання в рідинах організму (період напіввиве 39 дення), підвищена питома активність, підвищений рівень експресії, або полегшується очищення злитого білка. В подальшому переважному варіанті здійснення, бета-інтерферон злитий з константною ділянкою молекули Ig, яку часто називають Fc частиною імуноглобуліну. Переважно, він злитий з ділянками важкого ланцюга, наприклад, такими як домени СН2 і СН3 IgG1 людини. Інші ізоформи молекул Ig також придатні для одержання злитих білків відповідно до даного винаходу, таких як ізоформи IgG2, IgG3 або IgG4, або інші класи Ig, такі як, наприклад, IgM або IgA. Злиті білки можуть бути мономерними або мультимерними, гетероабо гомомультимерними. Методи одержання злитих білків імуноглобуліну відомі в даній галузі, наприклад, з ЕР 526 452 або з US 5,155,027. Ig злиті білки, що містять компоненти бета-інтерферону, описані, наприклад, в ЕР 227 110, US 5,541,087, WO 97/24137 або WO 00/23472. "Фрагмент" відповідно до даного винаходу стосується будь-якого піднабору бетаінтерферону, що являє собою більш короткий пептид, який зберігає бажану біологічну активність, що може бути виміряно, наприклад, у біологічному дослідженні, описаному вище. Фрагменти легко можуть бути отримані шляхом видалення амінокислот з будь-якого кінця молекули й тестування отриманого фрагмента щодо його властивостей як агоніста рецептора. Відомі протеази для видалення однієї амінокислоти за один раз з будь-якого Nкінця або С-кінця поліпептиду, і в такий спосіб визначені фрагменти, які зберігають бажану біологічну активність, можуть бути ідентифіковані, наприклад, у тесті, описаному Youcefi та ін., 1985, і задіють тільки загальноприйняті методики. Незважаючи на те, що даний винахід забезпечує рекомбінантні методи для одержання вищеописаних похідних, такі похідні також можуть бути отримані шляхом загальноприйнятих методів синтезу білка, які добре відомі середньому фахівцеві в даній галузі техніки. Бета-інтерферон, або його варіант/мутеїн, функціональне похідне, активний фрагмент або злитий білок, що має активність бета-інтерферону, переважно вводять системно, і переважно підшкірно або внутрішньом'язево. Також даним винаходом охоплюються внутрішньошкірний, трансдермальний (наприклад, у препаратах з уповільненим вивільненням), внутрішньовенний, пероральний, внутрішньочерепний, епідуральний, місцевий, ректальний і інтраназальний шляхи. Також можна використовувати будь-які інші терапевтично ефективні шляхи введення, наприклад, абсорбцію через епітеліальні або ендотеліальні тканини, або шляхом генної терапії, у якій молекулу ДНК, яка кодує бета-інтерферон, вводять пацієнтові (наприклад, за допомогою вектора), що викликає експресію й секрецію бетаінтерферону в умовах in vivo. Бета-інтерферон може бути приготовлений у вигляді фармацевтичної композиції, тобто разом з фармацевтично прийнятним носієм, наповнювачами або іншими речовинами. Визначення "фармацевтично прийнятний" охоплює будь-який носій, який не перешкоджає прояву ефективності біоло 96941 40 гічної дії активного компонента й не токсичний для хазяїна, якому його вводять. Наприклад, для парентерального введення активний білок або активні білки можуть бути приготовлені в одиничній дозованій формі для ін'єкції в наповнювачах, таких як сольовий розчин, розчин декстрози, сироватковий альбумін і розчин Рінгера. Відповідно до даного винаходу, якщо бетаінтерферон являє собою рекомбінантний бетаінтерферон 1а, який продукується у клітинах яєчника китайського хом'ячка (клітинах СНО), комерційно доступний під торговельною маркою Rebif, то він переважно може бути приготовлений у вигляді препарату без HSA (сироватковий альбумін людини) (утримуючого рекомбінантний інтерферон бета-1а і наповнювачі), як описано в WO 2004/096263. Дозування, яке вводять, у вигляді одиничної або багаторазової доз, індивідуумові, буде відрізнятися залежно від багатьох факторів, включаючи фармакокінетичні властивості бета-інтерферону, шлях введення, стани й характеристики пацієнта (стать, вік, вага тіла, стан здоров'я, розмір), інтенсивність симптомів, що супроводжують лікування, частоту лікування. Коректування й доробка певних дозових інтервалів можуть бути здійснені середнім фахівцем у даній галузі. В одному варіанті здійснення даного винаходу бета-інтерферон вводять у дозі принаймні 44 мкг підшкірно на введення. Переважні дози й схеми введення відповідно до даного винаходу вибирають із групи, яка включає: 12 ММЕ (44 мкг) бетаінтерферону три рази на тиждень, 12 ММЕ (44 мкг) щодня, 24 ММЕ (88 мкг) три рази на тиждень, 24 ММЕ (88 мкг) щодня. Ці дози переважно вводять підшкірно. В одному особливо переважному варіанті здійснення бета-інтерферон вводять у дозі 44 мкг підшкірно три рази на тиждень. Також кращим є введення бета-інтерферону в дозі 100 мкг (приблизно 27 ММЕ) один раз на тиждень внутрішньом'язево. Добові дози також можна вводити у вигляді розділених доз або у формі з уповільненим вивільненням, ефективної для одержання бажаних результатів. Друге або наступне введення можна здійснювати в дозі, що є такою ж, або менша або більша за початкову або попередню дозу, введену індивідуумові. Пацієнти Пацієнти, яких піддають лікуванню, відповідно до способу за даним винаходом, є пацієнтами, що страждають від розсіяного склерозу, які резистентні принаймні до одного загальноприйнятого лікування MS. Переважно резистентних пацієнтів піддають лікуванню за допомогою загальноприйнятої терапії MS, що не є клінічно адекватною для полегшення одного або декількох симптомів, зв'язаних з таких порушенням. Звичайно такі пацієнти страждають від розсіяного склерозу, але вони відчувають погіршення або стагнацію симптомів захворювання, незважаючи на лікування MS за допомогою такої загальноприйнятої терапії й мають потребу в додатковій терапії для поліпшення симптомів, зв'язаних з таким порушенням. 41 У варіанті здійснення даного винаходу, загальноприйняте лікування вибирають із групи, яка включає: лікування за допомогою бета інтерферону, лікування за допомогою ацетату глатимареру (Copaxone®, Teva), лікування за допомогою наталізумабу (Tysabri®, Biogen/Elan), або лікування за допомогою мітоксантрону (Novantrone®, Serono). У переважному варіанті здійснення, загальноприйняте лікування являє собою лікування за допомогою бета інтерферону, переважно лікування за допомогою Betaseron® (Berlex / Schering AG); Avonex® (Biogen); або Rebif® (Serono). У переважному варіанті здійснення, загальноприйняте лікування являє собою лікування за допомогою Rebif® (Serono). У варіанті здійснення даного винаходу, пацієнти, яких піддають лікуванню відповідно до способу за даним винаходом резистентні до одного, двох, трьох або чотирьох загальноприйнятих лікувань, описаних у даній заявці. У переважному варіанті здійснення, у резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив, особливо один, два, три, чотири або п'ять рецидивів, незалежно від прогресування інвалідності, незважаючи на одержання принаймні одного загальноприйнятого лікування. Переважно, принаймні один рецидив, особливо один, два, три, чотири або п'ять рецидивів, відбувалися протягом року перед початком лікування відповідно до даного винаходу. У переважному варіанті здійснення, у пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив, особливо один, два, три, чотири або п'ять рецидивів, протягом року, що передує початку лікування за даним винаходом і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif® три рази на тиждень. У переважному варіанті здійснення, у резистентних пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив, особливо один, два, три, чотири або п'ять рецидивів, і розвивається наростаюча інвалідність внаслідок прогресуючих форм захворювання, незважаючи на одержання принаймні одного загальноприйнятого лікування. Ступінь інвалідності пацієнтів MS можна оцінити, наприклад, за допомогою шкали Kurtzke Expanded Disability Status Scale (EDSS) (Kurtzke, 1983, Neurology, 33, 1444-1452). Звичайне зниження кількості балів згідно із EDSS відповідає поліпшенню захворювання й навпаки, підвищення кількості балів згідно із EDSS відповідає погіршенню захворювання. У переважному варіанті здійснення, принаймні один рецидив, особливо один, два, три, чотири або п'ять рецидивів, відбувалися протягом року перед початком лікування відповідно до даного винаходу. У переважному варіанті здійснення, у пацієнтів, яких піддають лікуванню, проявляється принаймні один рецидив, особливо один, два, три, чотири або п'ять рецидивів, протягом року, що передує початку лікування даного винаходу і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif® три рази на тиждень. У переважному варіанті здійснення, у резистентних пацієнтів, яких піддають лікуванню, розвива 96941 42 ється наростаюча інвалідність внаслідок прогресуючих форм захворювання, незважаючи на одержання принаймні одного загальноприйнятого лікування. Ступінь інвалідності пацієнтів MS можна оцінити, наприклад, за допомогою шкали Kurtzke Expanded Disability Status Scale (EDSS) (Kurtzke, 1983, Neurology, 33, 1444-1452). Звичайне зниження кількості балів згідно із EDSS відповідає поліпшенню захворювання й навпаки, підвищення кількості балів згідно із EDSS відповідає погіршенню захворювання. У переважному варіанті здійснення, у пацієнтів, яких піддають лікуванню, проявляється підвищення балів відповідно до шкали EDSS на 0,5; 1; 1,5; 2; 2,5; 3; 3,5; 4; 4,5; 5; 5,5; 6; 6,5; 7; 7,5; 8 або 8,5; незважаючи на одержання принаймні одного загальноприйнятого лікування. Переважно, підвищення балів відповідно до шкали EDSS становить 0,5; 1; 1,5; 2; 2,5; 3; 3,5; 4; 4,5 або 5 незважаючи на одержання принаймні одного загальноприйнятого лікування. Ще більш переважно, підвищення балів відповідно до шкали EDSS становить 0,5; 1; 1,5 або 2; 2,5 незважаючи на одержання принаймні одного загальноприйнятого лікування. Найбільш переважно, підвищення балів відповідно до шкали EDSS становить 0,5, 1 або 1,5 (переважно 1) незважаючи на одержання принаймні одного загальноприйнятого лікування. У переважному варіанті здійснення вказане підвищення балів відповідно до шкали EDSS відбувається протягом одного, двох, трьох, чотирьох, п'яти, шести, семи, восьми або десяти років перед початком лікування відповідно до даного винаходу. Переважно підвищення балів відповідно до шкали EDSS відбувається протягом одного, двох, трьох, чотирьох або п'яти, років перед початком лікування відповідно до даного винаходу. Ще більш переважно, підвищення балів відповідно до шкали EDSS відбувається протягом одного, двох або трьох років перед початком лікування відповідно до даного винаходу. Найбільш переважно, підвищення балів відповідно до шкали EDSS відбувається протягом одного або двох років, переважно одного року, перед початком лікування відповідно до даного винаходу. У переважному варіанті здійснення даного винаходу, у пацієнтів, яких піддають лікуванню, проявляється підвищення їх балів відповідно до шкали EDSS, пов'язане із принаймні одним рецидивом, особливо одним, двома, трьома, чотирма, п'ятьма, шістьома, сімома або десятьма рецидивами, переважно одним, двома, трьома, чотирма або п'ятьма рецидивами, ще більш переважно одним, двома, або трьома рецидивами, найбільш переважно одним або двома (переважно одним) рецидивами. У переважному варіанті здійснення, у пацієнтів, яких піддають лікуванню, проявляється підвищення їх балів відповідно до шкали EDSS, як розкрито в даній заявці вище, незважаючи на лікування за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif® три рази на тиждень. У переважному варіанті здійснення, у резистентних пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС, що визначалося за допомогою методів, таких як метод MPT (Miller та 43 ін., 1996, Neurology, 47(Suppl 4): S217; Evans та ін., 1997, Ann. Neurology, 41:125-132), незважаючи на одержання принаймні одного загальноприйнятого лікування. У переважному варіанті здійснення, збільшене число уражень або збільшений об'єм мозкових уражень виникали протягом року перед початком лікування відповідно до даного винаходу. У переважному варіанті здійснення, у пацієнтів, яких піддають лікуванню, розвивалося збільшене число уражень або збільшений об'єм мозкових уражень у ЦНС протягом року, що передує початку лікування даного винаходу і їх піддавали лікуванню за допомогою Rebif® (Serono), особливо 12 ММЕ (44 мкг) Rebif® три рази на тиждень. У варіанті здійснення даного винаходу, резистентні пацієнти, яких піддають лікуванню, страждають від погіршення MS, особливо вторинного прогресуючого, прогресуючого ремітуючого або погіршення MS ремітуючого перебігу, незважаючи на лікування за допомогою загальноприйнятої терапії. У варіанті здійснення даного винаходу, резистентними пацієнтами, яких піддавали лікуванню відповідно до способу за даним винаходом, є пацієнти, що страждають від прогресуючого рецидивуючого розсіяного склерозу (PRMS) або первинного прогресуючого розсіяного склерозу (PPMS). У варіанті здійснення винаходу, пацієнтів вибирають із чоловіків або жінок у віці від 18 до 55 років. Комбінована терапія шляхом перорального введення кладрибіну й бета-інтерферону (IFNбета або IFN-β) відповідно до даного винаходу Відповідно до даного винаходу, кладрибін вводять у комбінації з терапевтично ефективною кількістю бета-інтерферону. Бета-інтерферон вводять до, одночасно та/або послідовно із кладрибіном. Активні речовини, які вводяться одночасно з іншими терапевтичними засобами, можуть вводити в тих самих або різних композиціях і однаковими або різними шляхами введення. Об'єктом даного винаходу є застосування комбінації кладрибіну й бета-інтерферону для приготування лікарського засобу для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, де кладрибін вводять перорально, відповідно до послідовних стадій, наведених нижче: (і) Індукційний період, у якому вводять кладрибін і в якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 1,7 мг/кг до приблизно 3,5 мг/кг; (іі) Період без кладрибіну, у якому не вводять кладрибін; (ііі) Період підтримання, у якому вводять кладрибін і в якому загальна доза кладрибіну, яку вводять протягом періоду підтримання, нижче або дорівнює загальній дозі кладрибіну, що досягається наприкінці індукційного періоду (і); (iv) Період без кладрибіну, у якому не вводять кладрибін. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де 96941 44 індукційний період триває аж до приблизно 4 місяців або аж до приблизно 3 місяців або аж до приблизно 2 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де індукційний період триває аж до приблизно 2 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де індукційний період триває аж до приблизно 4 місяців. В подальшому переважному варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, у якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. У переважному варіанті здійснення, загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг, і індукційний період триває аж до приблизно 2 місяців. В подальшому переважному варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, у якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 3,5 мг/кг, переважно 3,5 мг/кг. У переважному варіанті здійснення, загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 3,5 мг/кг, переважно 3,5 мг/кг, і індукційний період триває аж до приблизно 4 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де період без кладрибіну (іі) триває аж до приблизно 10 місяців, або аж до приблизно 9 місяців або аж до приблизно 8 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де період без кладрибіну (іі) триває аж до приблизно 8 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де період без кладрибіну (іі) триває принаймні приблизно 8 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де період без кладрибіну (іі) триває аж до приблизно 10 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де комбінована тривалість індукційного періоду (і) з періодом без кладрибіну (іі) становить приблизно 1 рік (приблизно 12 місяців). Переважно тривалість індукційного періоду становить приблизно 4 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, або тривалість індукційного 45 періоду становить приблизно 3 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, або тривалість індукційного періоду становить приблизно 2 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де комбінована тривалість індукційного періоду (і) з періодом без кладрибіну (іі) становить приблизно 1 рік (приблизно 12 місяців) і загальна доза кладрибіну, яка досягається до закінчення цього року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг або приблизно 3,5 мг/кг, переважно 3,5 мг/кг. Переважно тривалість індукційного періоду становить приблизно 4 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, або тривалість індукційного періоду становить приблизно 3 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, або тривалість індукційного періоду становить приблизно 2 місяці й тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де період без кладрибіну (iv) триває аж до приблизно 10 місяців, або аж до приблизно 9 місяців або аж до приблизно 8 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де період без кладрибіну (iv) триває аж до приблизно 10 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де період без кладрибіну (iv) триває принаймні приблизно 8 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де періоди без кладрибіну (іі) та/або (іν) тривають від приблизно 8 до приблизно 10 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де плацебо-таблетки вводять протягом періоду без кладрибіну. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де в період без кладрибіну не здійснюють ніякого введення засобів. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де період підтримання триває аж до приблизно 4 місяців, або аж до приблизно 3 місяців, або аж до приблизно 2 місяців, переважно аж до приблизно 2 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, у якому загальна доза кладрибіну, яку вводять про 96941 46 тягом періоду підтримання, (ііі) становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. В подальшому переважному варіанті здійснення, загальна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг, і періоду підтримання триває аж до приблизно 2 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де комбінована тривалість періоду підтримання (ііі) з періодом без кладрибіну (iv) становить приблизно 1 рік (приблизно 12 місяців). Переважно тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців, або тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців. Ще більш переважно, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де комбінована тривалість періоду підтримання (ііі) з періодом без кладрибіну (iv) становить приблизно 1 рік (приблизно 12 місяців) і загальна доза кладрибіну, яку вводять протягом цього року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. Переважно тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців, або тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців. Ще більш переважно, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де комбінована тривалість індукційного періоду (і), періоду без кладрибіну (іі), періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 2 роки (приблизно 24 місяця). У варіанті здійснення винаходу, тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В 47 іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно З місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де комбінована тривалість індукційного періоду (і) і періоду без кладрибіну (іі) становить приблизно 1 рік (12 місяців), комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік (приблизно 12 місяців), загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. У варіанті здійснення винаходу, тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно З місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) 96941 48 становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де комбінована тривалість індукційного періоду (і) і періоду без кладрибіну (іі) становить приблизно 1 рік (12 місяців), комбінована тривалість періоду підтримання (ііі) і періоду без кладрибіну (iv) становить приблизно 1 рік (приблизно 12 місяців), загальна доза кладрибіну, яку вводять протягом першого року лікування, становить приблизно 3,5 мг/кг, переважно 3,5 мг/кг і загальна доза кладрибіну, яку вводять протягом другого року лікування, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. У варіанті здійснення винаходу, тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно З місяці й 49 тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 4 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 8 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 9 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 3 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 9 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 4 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 8 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 3 місяці й тривалість періоду без кладрибіну (іν) становить приблизно 9 місяців. В іншому варіанті здійснення, тривалість індукційного періоду становить приблизно 2 місяці, тривалість періоду без кладрибіну (іі) становить приблизно 10 місяців, тривалість періоду підтримання (ііі) становить приблизно 2 місяці й тривалість періоду без кладрибіну (iv) становить приблизно 10 місяців. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де стадії (ііі) - (iv) повторюють принаймні один, два або три рази. Інший кращий варіант здійснення даного винаходу забезпечує застосування комбінації кладрибіну й бета-інтерферону для приготування лікарського засобу для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, де кладрибін вводять перорально, відповідно до послідовних стадій, наведених нижче: (і) Індукційний період, у якому вводять кладрибін і в якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 1,7 мг/кг до приблизно 3,5 мг/кг; (іі) Період без кладрибіну, у якому не вводять кладрибін; 96941 50 (ііі) Період підтримання, у якому вводять кладрибін і в якому загальна доза кладрибіну, яку вводять протягом періоду підтримання, нижче або дорівнює загальній дозі кладрибіну, що досягається наприкінці індукційного періоду (і) (iv) Період без кладрибіну, у якому не вводять кладрибін; де індукційний період триває аж до приблизно 4 місяців, або аж до приблизно З місяців, або аж до приблизно 2 місяців; періоду без кладрибіну (іі) триває аж до приблизно 10 місяців, або аж до приблизно 9 місяців, або аж до приблизно 8 місяців; періоду підтримання (ііі) триває аж до приблизно 2 місяців; періоду без кладрибіну (iv) триває аж до приблизно 10 місяців; загальна доза кладрибіну, яку вводять на протязі періоду підтримання, становить приблизно 1,7 мг/кг і стадії (ііі) - (iv) здійснюють один, два або три рази. В іншому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бетаінтерферону для приготування лікарського засобу для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, де кладрибін вводять перорально, відповідно до послідовних стадій, наведених нижче: (і) Індукційний період, у якому вводять кладрибін і в якому загальна ефективна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 0,7 мг/кг до приблизно 1,4 мг/кг; (іі) Період без кладрибіну, у якому не вводять кладрибін; (ііі) Період підтримання, у якому вводять кладрибін і в якому загальна ефективна доза кладрибіну, яку вводять протягом періоду підтримання, (ііі) нижче або дорівнює загальній ефективній дозі кладрибіну, що досягається наприкінці індукційного періоду, (і); (iv) Період без кладрибіну, у якому не вводять кладрибін. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону для приготування лікарського засобу для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, де кладрибін вводять перорально, відповідно до послідовних стадій, наведених нижче: (і) Індукційний період, у якому вводять кладрибін і в якому загальна ефективна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 0,7 мг/кг до приблизно 1,4 мг/кг; (іі) Період без кладрибіну, у якому не вводять кладрибін; (ііі) Період підтримання, у якому вводять кладрибін і в якому загальна ефективна доза кладрибіну, яку вводять протягом періоду підтримання, нижче, ніж загальна ефективна доза кладрибіну, що досягається наприкінці індукційного періоду, (і); (iv) Період без кладрибіну, у якому не вводять кладрибін; де індукційний період триває аж до приблизно 4 місяців, або аж до приблизно З місяців, або аж 51 до приблизно 2 місяців; періоду без кладрибіну (іі) триває аж до приблизно 10 місяців, або аж до приблизно 9 місяців, або аж до приблизно 8 місяців; періоду підтримання (ііі) триває аж до приблизно 2 місяців; періоду без кладрибіну (іі) триває аж до приблизно 10 місяців; загальна ефективна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 0,7 мг/кг і стадії (ііі) (iv) здійснюють один, два або три рази. В іншому переважному варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону для приготування лікарського засобу для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, де кладрибін вводять перорально, відповідно до послідовних стадій, наведених нижче: (і) Індукційний період, у якому вводять кладрибін і в якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 1,7 мг/кг до приблизно 3,5 мг/кг; (іі) Період без кладрибіну, у якому не вводять кладрибін; (ііі) Період підтримання, у якому вводять кладрибін і в якому загальна доза кладрибіну, яку вводять протягом періоду підтримання, нижче, ніж загальна доза кладрибіну, що досягається наприкінці індукційного періоду (і); (iv) Період без кладрибіну, у якому не вводять кладрибін; де індукційний період триває аж до приблизно 4 місяців, або аж до приблизно З місяців, або аж до приблизно 2 місяців; періоду без кладрибіну (іі) триває аж до приблизно 10 місяців, або аж до приблизно 9 місяців, або аж до приблизно 8 місяців; періоду підтримання (ііі) триває аж до приблизно 2 місяців; періоду без кладрибіну (iv) триває аж до приблизно 10 місяців; загальна доза кладрибіну, яку вводять на протязі періоду підтримання, становить приблизно 1,7 мг/кг і стадії (ііі) - (iv) здійснюють один, два або три рази. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально в добовій дозі приблизно від 3 до 30 мг кладрибіну, переважно від 5 до 20 мг кладрибіну, найбільш переважно 10 мг кладрибіну. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, у якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 3,5 мг/кг і загальна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де загальна ефективна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 1,4 мг/кг і загальна ефективна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 0,7 мг/кг. 96941 52 В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально один раз на день протягом індукційного періоду. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально декілька разів на добу протягом індукційного періоду, переважно два або три рази на добу, більш переважно два рази на добу. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, відповідно до чого кладрибін вводять перорально приблизно 1 до приблизно 7 днів на місяць, переважно від приблизно 4 до приблизно 7 днів на місяць протягом індукційного періоду, і більш переважно 4 або 5 днів на місяць протягом індукційного періоду. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, відповідно до чого кладрибін вводять перорально приблизно 0,02 днів/кг до приблизно 0,08 днів/кг на місяць протягом індукційного періоду. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, відповідно до чого кладрибін вводять перорально приблизно 0,02 днів/кг до приблизно 0,08 днів/кг на місяць протягом періоду підтримання. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально в добовій дозі приблизно 10 мг кладрибіну від дня 1 до приблизно дня 2 щомісяця протягом індукційного періоду. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально в добовій дозі приблизно 10 мг кладрибіну від дня 1 до приблизно дня З щомісяця протягом індукційного періоду. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально в добовій дозі приблизно 10 мг кладрибіну від дня 1 до приблизно дня 4 щомісяця протягом індукційного періоду. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально в добовій дозі приблизно 10 мг кладрибіну від дня 1 до приблизно дня 5 щомісяця протягом індукційного періоду. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально в добовій дозі приблизно 10 мг кладрибіну від дня 1 до приблизно дня 6 щомісяця протягом індукційного періоду. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й 53 бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально в препараті, описаному в WO 2004/087101 або в WO 2004/087100. Препарат кладрибіну особливо переважно являє собою препарат, описаний в WO 2004/087101, приклад 3 або в таблиці 2 приклади 1 дійсної заявки. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально в добовій дозі приблизно 10 мг кладрибіну від дня 1 до приблизно дня 4 щомісяця протягом індукційного періоду й де фармацевтичний препарат являє собою фармацевтичний препарат, описаний в WO 2004/087101 або в WO 2004/087100. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально в добовій дозі приблизно 10 мг кладрибіну від дня 1 до приблизно дня 5 щомісяця протягом індукційного періоду й де фармацевтичний препарат являє собою фармацевтичний препарат, описаний в WO 2004/087101 або в WO 2004/087100. Як описано в даній заявці вище, кладрибін вводять у комбінації з терапевтично ефективною кількістю бета-інтерферону. Введення перорально кладрибіну й бета-інтерферону може бути одночасним, розділеним або послідовним. Отже, об'єктом даного винаходу є будь-яке застосування комбінації кладрибіну й бета-інтерферону, описане в даній заявці, де кладрибін вводять перорально відповідно до послідовних етапів, описаних у даній заявці, і, де бета-інтерферон вводять одночасно, окремо або послідовно з пероральним введенням кладрибіну. Іншим об'єктом даного винаходу є застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально відповідно до послідовних етапів, описаних у даній заявці, і де бета-інтерферон вводять перед індукційним періодом (і), та/або після періоду підтримання (ііі). Іншим об'єктом даного винаходу є застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально відповідно до послідовних етапів, описаних у даній заявці, і де бета-інтерферон вводять протягом періоду без кладрибіну (іі) та/або (iv). У переважному варіанті здійснення даного винаходу бета-інтерферон вводять одночасно з пероральним введенням кладрибіну. Отже, іншим об'єктом даного винаходу є застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально відповідно до послідовних етапів, описаних у даній заявці, і де бета-інтерферон вводять протягом індукційного періоду (і) та/або періоду підтримання (iv). Ще більш переважно, бета-інтерферон вводять протягом індукційного періоду (і), періоду підтримання (iv) і періодів без кладрибіну (іі) і (iv). Ще більш переважно, бета-інтерферон вводять перед індукційним періодом (і), протягом індукційного періоду (і), протягом періоду підтримання (iv), про 96941 54 тягом періодів без кладрибіну (іі) і (iv) і після періоду без кладрибіну (iv). Як описано в даній заявці вище, у переважному варіанті здійснення даного винаходу білок бета-інтерферону вибирають із групи, яка включає: бета-інтерферон 1а, такий як, наприклад, Avonex® (Biogen) або Rebif® (Serono), і бета-інтерферон 1b, такий як, наприклад, Betaseron® (Berlex / Schering AG). Переважно, бета-інтерферон, який використовують згідно із даним винаходом, являє собою Rebif® (Serono). Бета-інтерферон переважно вводять системно, і переважно підшкірно або внутрішньом'язево. В одному варіанті здійснення даного винаходу бета-інтерферон вводять у дозі принаймні 44 мкг підшкірно на введення. Кращі дози й схеми відповідно до даного винаходу вибирають із групи, яка включає: 12 ММЕ (44 мкг) бета-інтерферону три рази на тиждень, 12 ММЕ (44 мкг) щодня, 24 ММЕ (88 мкг) три рази на тиждень, 24 ММЕ (88 мкг) щодня. Ці переважно вводять підшкірно. В одному особливо переважному варіанті здійснення бетаінтерферон вводять у дозі 44 мкг підшкірно три рази на тиждень, переважно в цьому випадку використовуваний бета-інтерферон являє собою Rebif® (Serono). Способи лікування з використанням комбінованої терапії перорально введеного кладрибіну й бета-інтерферону (IFN-бета або IFN-β) відповідно до даного винаходу В іншому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бетаінтерферону для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, що включає пероральне введення кладрибіну, відповідно до послідовних стадій, наведених нижче: (і) Індукційний період, у якому вводять кладрибін і в якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 1,5 мг/кг до приблизно 3,5 мг/кг; (іі) Період без кладрибіну, у якому не вводять кладрибін; (ііі) Період підтримання, у якому вводять кладрибін і в якому загальна доза кладрибіну, яку вводять протягом періоду підтримання, нижче або дорівнює загальній дозі кладрибіну, що досягається наприкінці індукційного періоду (і); (iv) Період без кладрибіну, у якому не вводять кладрибін. У переважному варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, що включає пероральне введення кладрибіну, відповідно до послідовних стадій, наведених нижче: (і) Індукційний період, у якому вводять кладрибін і в якому загальна ефективна доза кладрибіну, що досягається наприкінці індукційного періоду, становить від приблизно 0,7 мг/кг до приблизно 1,4 мг/кг; 55 (іі) Період без кладрибіну, у якому не вводять кладрибін; (ііі) Період підтримання, у якому вводять кладрибін і в якому загальна ефективна доза кладрибіну, яку вводять протягом періоду підтримання, нижче або дорівнює загальній ефективній дозі кладрибіну, що досягається наприкінці індукційного періоду, (і); (iv) Період без кладрибіну, у якому не вводять кладрибін. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де стадії (ііі) - (iv) повторюють принаймні один або два рази. У переважному варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону для лікування пацієнтів, що страждають від розсіяного склерозу, і які резистентні принаймні до одного загальноприйнятого лікування розсіяного склерозу, що включає пероральне введення кладрибіну, відповідно до послідовних стадій, наведених нижче: (і) Введення кладрибіну таким чином, щоб загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становила від приблизно 1,7 мг/кг до приблизно 3,5 мг/кг; (іі) Відсутність введення кладрибіну протягом періоду без кладрибіну; (ііі) Введення кладрибіну таким чином, щоб загальна доза кладрибіну, яку вводять протягом періоду підтримання, була нижче або дорівнює загальній дозі кладрибіну, що досягається наприкінці індукційного періоду (і); (iv) І необов'язково, період без кладрибіну, у якому не вводять кладрибін. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де індукційний період триває аж до приблизно 4 місяців, або аж до приблизно 3 місяців, або аж до приблизно 2 місяців. В подальшому переважному варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, у якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. У переважному варіанті здійснення, загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг, і індукційний період триває аж до приблизно 2 місяців. В подальшому переважному варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, у якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 3,5 мг/кг, переважно 3,5 мг/кг. У переважному варіанті здійснення, загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 3,5 мг/кг, переважно 3,5 мг/кг, і індукційний період триває аж до приблизно 4 місяців. В подальшому переважному варіанті здійснення, винахід забезпечує застосування комбінації 96941 56 кладрибіну й бета-інтерферону, як описано в даній заявці, де загальна ефективна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 1,4 мг/кг. В подальшому переважному варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де період без кладрибіну триває аж до приблизно 10 місяців, або аж до приблизно 9 місяців, або аж до приблизно 8 місяців. В подальшому переважному варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де період підтримання триває аж до приблизно 4 місяців, або аж до приблизно 3 місяців або аж до приблизно 2 місяців. В подальшому переважному варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, у якому загальна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де загальна ефективна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 0,7 мг/кг. В подальшому переважному варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де період підтримання здійснюють після періоду без кладрибіну. В подальшому переважному варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, у якому загальна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 3,5 мг/кг і загальна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 1,7 мг/кг, переважно 1,75 мг/кг. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де загальна ефективна доза кладрибіну, що досягається наприкінці індукційного періоду, становить приблизно 1,4 мг/кг і загальна ефективна доза кладрибіну, яку вводять протягом періоду підтримання, становить приблизно 0,7 мг/кг. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально в добовій дозі приблизно 3 до приблизно 30 мг. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально в добовій дозі приблизно 10 мг. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально приблизно 1 до приблизно 7 днів на місяць протягом індукційного 57 періоду, переважно від приблизно 4 до приблизно 7 днів на місяць протягом індукційного періоду, і більш переважно 4 або 5 днів на місяць протягом індукційного періоду. В подальшому варіанті здійснення, винахід забезпечує застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де стадії (ііі) - (iv) повторюють принаймні один або два рази. Як описано в даній заявці вище, кладрибін вводять у комбінації з терапевтично ефективною кількістю бета-інтерферону. Введення перорально кладрибіну й бета-інтерферону може бути одночасним, окремим або послідовним. Таким чином, об'єктом даного винаходу є будь-яке застосування комбінації кладрибіну й бета-інтерферону, описане в даній заявці, де кладрибін вводять перорально відповідно до послідовних етапів, описаних у даній заявці, і де бета-інтерферон вводять одночасно, окремо або послідовно з пероральним введенням кладрибіну. Іншим об'єктом даного винаходу є застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально відповідно до послідовних етапів, описаних у даній заявці, і де бета-інтерферон вводять перед індукційним періодом (і), та/або після періоду підтримання (ііі). Іншим об'єктом даного винаходу є застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально відповідно до послідовних етапів, описаних у даній заявці, і де бета-інтерферон вводять протягом періоду без кладрибіну (іі) та/або (iv). У переважному варіанті здійснення даного винаходу бета-інтерферон вводять одночасно з пероральним введенням кладрибіну. Отже, іншим об'єктом даного винаходу є застосування комбінації кладрибіну й бета-інтерферону, як описано в даній заявці, де кладрибін вводять перорально відповідно до послідовних етапів, описаних у даній заявці, і де бета-інтерферон вводять протягом індукційного періоду (і) та/або періоду підтримання (iv). Ще більш переважно, бета-інтерферон вводять протягом індукційного періоду (і), періоду підтримання (iv) і періодів без кладрибіну (іі) і (iv). Ще більш переважно, бета-інтерферон вводять перед індукційним періодом (і), протягом індукційного періоду (і), протягом періоду підтримання (iv), протягом періодів без кладрибіну (іі) і (iv) і після періоду без кладрибіну (iv). Як описано в даній заявці вище, у переважному варіанті здійснення даного винаходу білок бета-інтерферону вибирають із групи, яка включає: бета-інтерферон 1а, такий як, наприклад, Avonex® (Biogen) або Rebif® (Serono), і бета-інтерферон 1b, такий як, наприклад, Betaseron® (Berlex / Schering AG). Переважно, бета-інтерферон, використовуваний згідно із даним винаходом, являє собою Rebif® (Serono). Бета-інтерферон переважно вводять системно, і переважно підшкірно або внутрішньом'язево. В одному варіанті здійснення даного винаходу бета-інтерферон вводять у дозі принаймні 44 мкг підшкірно на введення. Кращі дози й схеми відпо 96941 58 відно до даного винаходу вибирають із групи, яка включає: 12 ММЕ (44 мкг) бета-інтерферону три рази на тиждень, 12 ММЕ (44 мкг) щодня, 24 ММЕ (88 мкг) три рази на тиждень, 24 ММЕ (88 мкг) щодня. Ці дози переважно вводять підшкірно. В одному особливо переважному варіанті здійснення бета-інтерферон вводять у дозі 44 мкг підшкірно три рази на тиждень, переважно в цьому випадку використовуваний бета-інтерферон являє собою Rebif® (Serono). Вся патентна й непатентна література, процитована в даній заявці, у такий спосіб повністю включена в даний винахід як посилання. Подальші аспекти й переваги даного винаходу будуть розкриті в наступних прикладах, які повинні розглядатися тільки як ілюстративні, і які не обмежують обсягу даної заявки. Приклади Наступні скорочення ставляться відповідно до нижчеподаних визначень: кг (кілограм), мкг (мікрограм), мг (міліграм), АЕ (побічні дії), ЦНС (Центральна нервова система), CSF (Спинномозкова рідина), EDSS (Expanded Disability Status Scale шкала стану поширення інвалідності, SNRS (Scripps Neurologic Rating Scale, неврологічна шкала оцінки згідно Scripps), IFN (інтерферон), в/в (внутрішньовенний), ММЕ (мільйон міжнародних одиниць), MS (розсіяний склероз), МРТ (магніторезонансна томографія), п/о (перорально), PPMS (Первинний прогресуючий розсіяний склероз), PRMS (Прогресуючий рецидивуючий розсіяний склероз), RRMS (Розсіяний склероз ремітуючого перебігу), SPMS (Вторинний прогресуючий розсіяний склероз), п/ш (підшкірно), TIW (Три рази на тиждень), 2-CdA (2-хлор-2'дезоксіаденозин або кладрибін), МО (міжнародна одиниця). Ефективність і безпека перорального введення кладрибіну, в остаточному підсумку багатодозового введення, і введення в комбінації з бетаінтерфероном, відповідно до винаходу можуть бути оцінені, наприклад, відповідно до нижчеописаних протоколів: Приклад 1: Пероральне введення кладрибіну для лікування рецидивуючих форм MS Дослідження проводили на шістдесяти пацієнтах з рецидивуючими формами клінічно встановленого розсіяного склерозу. Спочатку кожного пацієнта досліджували щодо нормальної функції печінки, нирок і кісткового мозку для визначення вихідних значень. Пацієнтів вибирали серед чоловіків або жінок у віці від 18 до 55 років, у яких проявлявся один або декілька рецидивів протягом попередніх 12 місяців. Пацієнти жіночої статі не були вагітними. Пацієнтів випадково розділяли на групи для лікування, наведені в таблиці 1 нижче: Таблиця 1 Група 1 2 3 2-CdA 1,75 мг/кг 3,5 мг/кг Кожний пацієнт у групах 2 і 3 одержував 3 мг або 10 мг 2-CdA (1,2 або 3 введення на добу за 59 96941 лежно від ваги пацієнта), комбінованого із циклодекстриновим препаратом, як описано в WO 2004/087101, приклад 3. Склади препаратів клад 60 рибіну в таблетках по 3 мг або 10 мг 2-CdA, що містять гідроксипропіл-бета-циклодекстрин, наведені в таблиці 2 нижче: Таблиця 2 Назва компонентів кладрибін-2-гідроксипропіл-β-циклодекстринкомплекс* Порошок сорбіту Стеарат магнію (рослинного походження) Усього Вміст мг/таблетку 153,75, що еквівалентно 10 мг 2-CdA 44,25 2,0 200,0 Вміст мг/таблетку 30,60, що еквівалентно 3 мг 2-CdA 68,4 1,00 100 * кладрибін комлексований і ліофілізований з 2-гідроксипропіл-β-циклодекстрином у вигляді окремого процесу, як описано в WO 2004/087101. Приклади схем введення для індукційного періоду залежно від ваги пацієнта наведені нижче в таблицях 3 і 4 для цільових доз 1,75 мг/кг і 3,5 мг/кг відповідно. Для періоду підтримання, застосовують приклад схеми введення відповідно до таблиці 3. Таблиця 3 Загальна цільова доза (кг), еквівалентна 1,75 мг/кг Діапазон ваги пацієнтів (кг) Мін 40 45 50 55 60 65 70 75 80 85 90 95 100 105 110 115 Середній діапазон 42,5 47,5 52,5 57,5 62,5 67,5 72,5 77,5 82,5 87,5 92,5 97,5 102,5 107,5 112,5 117,5 Кількість таблеток (10 мг) / індукційний період Макс Мін Макс Місяць 1 Місяць 2 Усього 44,9 49,9 54,9 59,9 64,9 69,9 74,9 79,9 84,9 89,9 94,9 99,9 104,9 109,9 114,9 119,9 28 31,5 35 38,5 42 45,5 49 52,5 56 59,5 63 66,5 70 73,5 77 80,5 31,4 34,9 38,4 41,9 45,4 48,9 52,4 55,9 59,4 62,9 66,4 69,9 73,4 76,9 80,4 83,9 4 4 5 5 5 6 6 7 7 7 8 8 9 9 9 10 3 4 4 5 5 5 6 6 6 7 7 8 8 9 9 9 7 8 9 10 10 11 12 13 13 14 15 16 17 18 18 19 Таблиця 4 Діапазон ваги пацієнтів (кг) Мін 40 45 50 55 60 65 70 75 80 85 90 95 100 105 110 115 Середній діапазон 42,5 47,5 52,5 57,5 62,5 67,5 72,5 77,5 82,5 87,5 92,5 97,5 102,5 107,5 112,5 117,5 Загальна цільова доза (кг), еквівалентна 3,5 мг/кг Кількість таблеток (10 мг)/індукційний період Макс Мін Макс Місяць 1 Місяць 2 Місяць 3 Місяць 4 Усього 44,9 49,9 54,9 59,9 64,9 69,9 74,9 79,9 84,9 89,9 94,9 99,9 104,9 109,9 114,9 119,9 56 63 70 77 84 91 98 105 112 119 126 133 140 147 154 161 62,9 69,9 76,9 83,9 90,9 97,9 104,9 111,9 118,9 125,9 132,9 139,9 146,9 153,9 160,9 167,9 4 4 5 5 6 6 6 7 7 7 8 8 9 9 10 10 4 4 4 5 5 6 6 7 7 7 8 8 8 9 9 10 3 4 4 5 5 5 6 6 7 7 7 8 8 9 9 9 3 4 4 4 5 5 6 6 6 7 7 8 8 8 9 9 14 16 17 19 21 22 24 26 27 28 30 32 33 35 37 38
ДивитисяДодаткова інформація
Назва патенту англійськоюCladribine regimen for treating multiple sclerosis
Автори англійськоюBrentzel H James Jr., Lopez-Bresnahan Maria, Ammoury Nazih
Назва патенту російськоюСхема дозирования кладрибина для лечения рассеянного склероза
Автори російськоюБренцель Х. Джеймс Джр., Лопез-Бреснахан Мария, Аммоури Назих
МПК / Мітки
МПК: A61K 31/7056, A61K 38/21, A61P 25/00
Мітки: схема, дозування, розсіяного, лікування, кладрибіну, склерозу
Код посилання
<a href="https://ua.patents.su/34-96941-skhema-dozuvannya-kladribinu-dlya-likuvannya-rozsiyanogo-sklerozu.html" target="_blank" rel="follow" title="База патентів України">Схема дозування кладрибіну для лікування розсіяного склерозу</a>
Режим застосування кладрибіну для лікування розсіяного склерозу

Номер патенту: 96260
Опубліковано: 25.10.2011
Автори: Ітьє Арно, Лопес-Бреснахан Марія, Мюнафо Ален, Де Лука Джампьєро
МПК: A61P 25/00, A61K 31/7076, A61K 38/21
Мітки: склерозу, кладрибіну, режим, розсіяного, застосування, лікування
Формула / Реферат:
1. Застосування кладрибіну для приготування фармацевтичної композиції для лікування розсіяного склерозу, де ця композиція має бути застосована перорально з дотриманням наведених нижче послідовних етапів: (і) індуктивного періоду, який триває від приблизно 2 місяців до приблизно 4 місяців, де вводять вказану фармацевтичну композицію кладрибіну, і де загальна доза кладрибіну, що досягається наприкінці індуктивного періоду, становить від...
Спосіб лікування розсіяного склерозу

Номер патенту: 38961
Опубліковано: 15.05.2001
Автори: Віничук Степан Мілентійович, Мяловицька Олена Анатоліївна
МПК: A61K 38/43
Мітки: склерозу, розсіяного, лікування, спосіб
Формула / Реферат:
(21) 2000127056 (54) (57)Дата прийняттярішення28.03.2001 р.Спосіб-лікування розсіяного склерозу шляхом застосування лікарських препаратів, який відрізняється тим, що на тлі базисної терапії призначають флогензим по 3 таблетки тричі на добу протягом трьох тижнів, а потім флогензим застосовують впродовж 12 місяців.
Спосіб лікування початкових проявів розсіяного склерозу

Номер патенту: 60047
Опубліковано: 15.09.2003
Автор: Мяловицька Олена Анатоліївна
МПК: A61K 38/21
Мітки: склерозу, проявів, розсіяного, початкових, спосіб, лікування
Формула / Реферат:
Спосіб лікування початкових проявів розсіяного склерозу, що включає медикаментозну терапію, який відрізняється тим, що внутрішньом’язово призначають лаферон під час першого загострення по 1 млн. міжнародних одиниць 2 рази на добу протягом 10 діб на тлі базисної терапії, а у наступні 6 місяців лаферон вводять по 1 млн. міжнародних одиниць 2 рази на тиждень.
Спосіб лікування розсіяного склерозу

Номер патенту: 2825
Опубліковано: 16.08.2004
Автори: Прокопів Марія Мирославівна, Віничук Степан Мілентійович, Матюшко Микола Григорович
МПК: A61K 45/00
Мітки: розсіяного, лікування, спосіб, склерозу
Формула / Реферат:
Спосіб лікування розсіяного склерозу шляхом застосування лікарських препаратів, який відрізняється тим, що через один місяць після курсу пульс-терапії кортикостероїдних препаратів у разі загострення додатково призначають внутрішньовенне крапельне введення імуноглобуліну нормального людського у дозі 25-50мл (у розрахунку 0,2-0,4г на 1кг ваги) протягом 3-5 днів, а у наступні 12-24 місяців – 25мл (1,25г) один раз на місяць.
Спосіб лікування розсіяного склерозу

Номер патенту: 2826
Опубліковано: 16.08.2004
Автори: Віничук Степан Мілентійович, Прокопів Марія Мирославівна, Матюшко Микола Григорович
МПК: A61K 38/43
Мітки: розсіяного, склерозу, лікування, спосіб
Формула / Реферат:
Спосіб лікування розсіяного склерозу, що включає застосування лікарських препаратів, який відрізняється тим, що на тлі базисної терапії призначають Вобензим по 3 капсули тричі на добу за 30 хвилин до їди, запиваючи їх 250мл води впродовж 12 місяців.
Попередній патент: Композиція, яка містить агент для стабілізації активних інгредієнтів у питній воді і шипучу суміш, спосіб виготовлення такої композиції та її використання
Наступний патент: Похідні амінопіперидину як інгібітори бпхе (білка-переносника холестерилового ефіру)
Випадковий патент: Наземний транспортний засіб "рекламобіль"