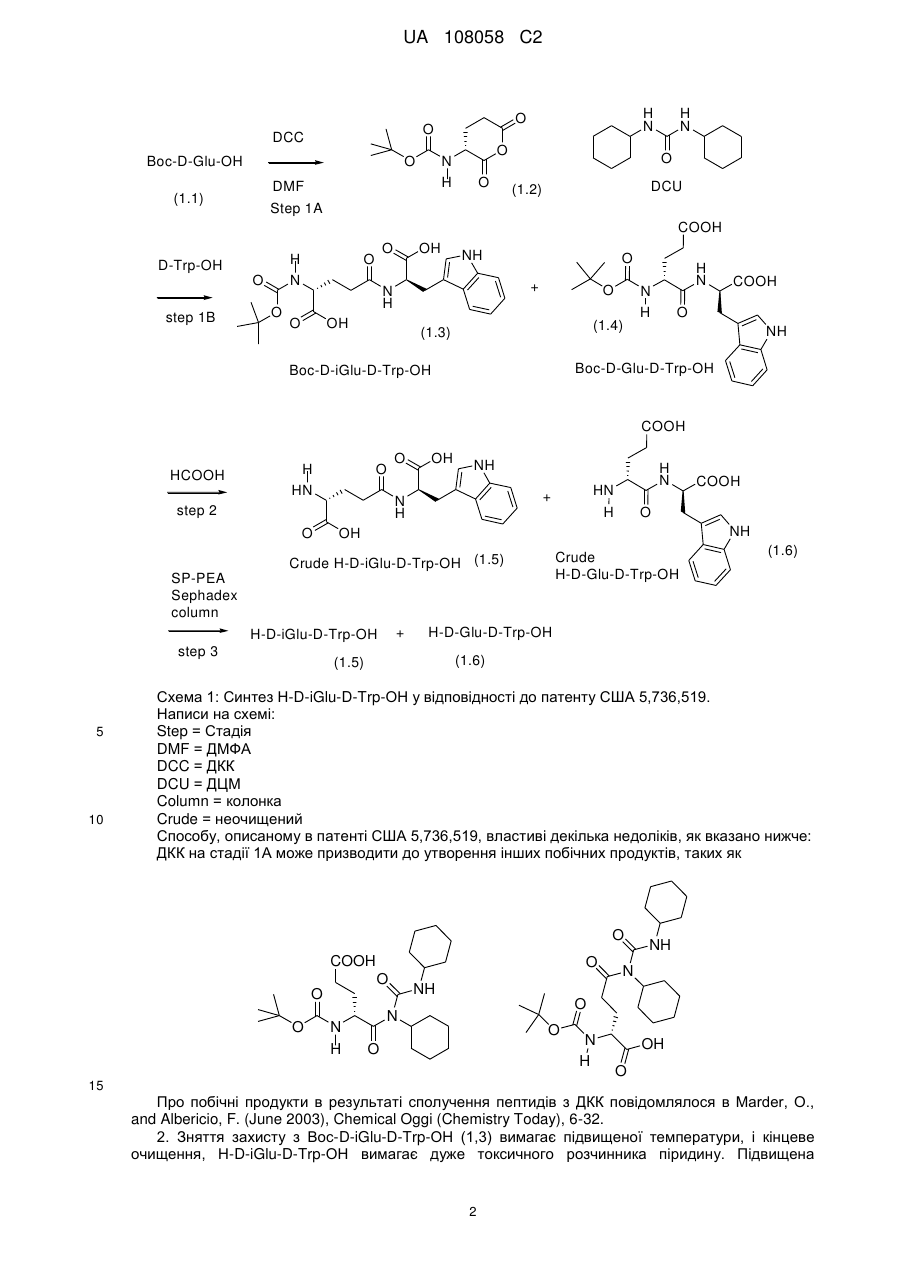
Кристалічний d-ізоглутаміл-d-триптофан і моноамонійна сіль d-ізоглутаміл-d-триптофану
Номер патенту: 108058
Опубліковано: 25.03.2015
Автори: Там Тім Фет, Жао Йанквінг, Леунг-Тоунг Рейс, Иу Лілі, Н'земба Блейз, Ванг Йінгсхенг





Формула / Реферат
1. Кристалічний D-ізоглутаміл-D-триптофан, що характеризується показниками рентгеноструктурного аналізу порошку, наведеними на фіг. 1.
2. Кристалічний D-ізоглутаміл-D-триптофан, що характеризується наступними показниками рентгеноструктурного аналізу порошку, представленими в значеннях міжплощинних відстаней d, кута Брегга 2 q і відносної інтенсивності (вираженої як відсоток по відношенню до найбільш інтенсивної лінії):
Кут [°2 q]
Значення d [Å]
Відносна інтенсивність [%]
6,67
13,239
3
11,09
7,975
4,4
11,77
7,515
1,2
13,29
6,655
4
14,26
6,205
11,3
15,58
5,685
33,3
16,81
5,269
28,9
17,27
5,13
30,4
18,35
4,832
12,2
18,87
4,7
95,8
20,05
4,424
63,6
20,9
4,247
33,2
22,03
4,032
17,1
22,88
3,884
100
23,74
3,744
97,9
24,54
3,625
41,9
25,44
3,499
20,3
25,69
3,465
12,1
26,31
3,384
16,4
27
3,3
27,4
27,75
3,212
24,9
28,18
3,164
19,3
28,79
3,099
6,8
29,13
3,063
6,2
29,91
2,985
79,2
31,04
2,879
8,6
31,49
2,839
33,7
32,54
2,749
4,4
33,29
2,689
9,3
33,97
2,637
10,5
34,99
2,562
17,3
35,54
2,524
21,8
36,14
2,483
5,1
36,74
2,444
5,9
37,35
2,406
7,7
38,31
2,348
25,6
39,01
2,307
20,3
3. Спосіб одержання у водній фазі D-ізоглутаміл-D-триптофану, вільного від неорганічних солей, який включає наступні стадії:
(a) одержання розчину кислотно-адитивної солі H-D-iGlu-D-Trp-OH у водному середовищі, істотною мірою вільному від органічного розчинника; або одержання розчину основно-адитивної солі H-D-iGlu-D-Trp-OH у водному середовищі, істотною мірою вільному від органічного розчинника;
(б) корекція pH до значень в інтервалі від приблизно 2,0 до приблизно 3,2 за допомогою розчину гідроксиду лужного металу або мінеральної кислоти для осадження H-D-iGlu-D-Trp-OH;
(в) виділення осадженого H-D-iGlu-D-Trp-OH; і
(г) вакуумне сушіння продукту, одержаного на стадії (в), з одержанням H-D-iGlu-D-Trp-OH.
4. Кристалічна амонійна сіль H-D-iGlu-D-Trp-OH (1:1), що характеризується показниками рентгеноструктурного аналізу порошку, які наведені на фіг. 2.
5. Кристалічна амонійна сіль H-D-iGlu-D-Trp-OH (1:1), що характеризується наступними показниками рентгеноструктурного аналізу порошку, представленими в значеннях міжплощинних відстаней d, кута Брегга 2 q і відносної інтенсивності (вираженої як відсоток по відношенню до найбільш інтенсивної лінії:
Кут [°2 q]
Значення d [Å]
Відносна інтенсивність [%]
9,29
9,517
4,1
12,19
7,258
4,5
13,93
6,354
76,2
15,17
5,837
27,4
16,49
5,371
9,8
17,18
5,157
3
18,56
4,778
31,6
18,88
4,696
10,5
20,02
4,431
100
22,28
3,986
3
23,31
3,814
4,6
23,66
3,757
9,8
24,03
3,7
52,9
24,37
3,649
26,3
25,07
3,549
11,4
25,61
3,475
5,6
25,96
3,43
5
27,62
3,227
29,7
28,12
3,17
55,7
28,49
3,131
12,2
29,52
3,023
23,1
30,27
2,951
3,7
30,64
2,915
7,9
31,31
2,854
11,8
31,7
2,821
29,6
32,16
2,781
19,2
32,81
2,728
16
33,78
2,652
7,4
34,14
2,625
5,1
35,76
2,509
16
36,94
2,431
11,6
37,58
2,391
25,6
38,03
2,364
12,5
39,22
2,295
1,9
6. Спосіб одержання моноамонійної солі H-D-iGlu-D-Trp-OH, вільної від неорганічних солей, який включає наступні стадії:
(а) одержання розчину кислотно-адитивної солі H-D-iGlu-D-Trp-OH у водному середовищі, істотною мірою вільному від органічного розчинника;
(б) корекція pH до значень від приблизно 3,9 до приблизно 8,6 за допомогою розчину гідроксиду металу;
(в) обробка розчину, одержаного на стадії (б), іонообмінною смолою та елюювання водою для обміну іона металу із солі в розчині на іон водню до тих пір, поки pH елюату не буде становити від приблизно 5,7 до приблизно 7,0;
(г) контакт іонообмінної смоли з регенерувальним розчином на базі аміаку, який застосовується для обміну його іонів на цільовий H-D-iGlu-D-Trp-OH, що міститься в іонообмінній смолі, таким чином, щоб утворився елюат регенерувального розчину, що містить амонійна сіль H-D-iGlu-D-Trp-OH; і
(д) випарювання розчинника з розчину, одержаного на стадії (г), з одержанням неочищеної амонійної солі;
і додатково включає наступні стадії:
(е) розчинення амонійної солі, одержаного на стадії (д), у воді з повільним додаванням ізопропанолу, таким чином, що утворюється осад моноамонійної солі; і
(є) вакуумне сушіння продукту, одержаного на стадії (е), з одержанням кристалічної форми амонійної солі H-D-iGlu-D-Trp-OH (1:1).
7. Спосіб за п. 6, який відрізняється тим, що розчин гідроксиду металу вибирається з групи, яка складається з розчину гідроксиду натрію, розчину гідроксиду літію і розчину гідроксиду калію.
8. Спосіб за п. 3 або 6, який відрізняється тим, що кислотно-адитивна сіль являє собою H-D-iGlu-D-Trp-OH гідрохлорид, який одержують за способом, що включає наступні стадії:
(i) основний гідроліз сполуки формули I:
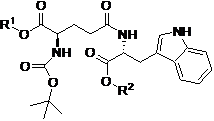 , I
, I
де R1 вибирають з групи, що складається з C1-C4алкілу і бензилу, і R2 являє собою C1-C4 алкіл, за умови, що C4 алкіл не являє собою трет-бутил,
з використанням гідроксиду металу у воді та інертному розчиннику, в присутності метанолу з одержанням, BOC-D-iGlu-D-Trp-OH, вільного від інших діастереомерів; і
(ii) зняття за допомогою хлористого водню захисту з BOC-D-iGlu-D-Trp-OH із стадії (i) в інертному органічному розчиннику; і випарювання розчинника з одержанням гідрохлориду H-D-iGlu-D-Trp-OH.
9. Спосіб за п. 3 або 6, який відрізняється тим, що кислотно-адитивна сіль являє собою H-D-iGlu-D-Trp-OH гідрохлорид, а розчин кислотно-адитивної солі, отриманої на стадії (а), одержують за способом, що включає:
(i) гідрогенізацію сполуки формули II
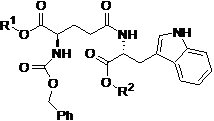 , II
, II
де R1 являє собою бензил, і R2 вибирають з групи, що складається з бензилу і водню, з використанням паладію на вугіллі в метанолі або етанолі;
(ii) очищення неочищеного H-D-iGlu-D-Trp-OH із стадії (i) за допомогою хроматографії на силікагелі, з використанням ізопропанолу і води як елюенту; і
(iii) обробка матеріалу, одержаного на стадії (ii), хлористоводневою кислотою у воді з одержанням розчину гідрохлориду H-D-iGlu-D-Trp-OH у воді.
10. Спосіб за п. 3, який відрізняється тим, що розчин основно-адитивної солі H-D-iGlu-D-Trp-OH, отриманого на стадії (а), одержують за способом, який включає:
(i) зняття за допомогою кислоти захисту з дипептиду Boc-D-Glu-(γ-D-Trp-OR2)-α-OR1 з одержанням кислотно-адитивної солі діестеру H-D-Glu-(γ-D-Trp-OR2)-α-OR1, де кожен з R1 і R2 незалежно вибирають з групи, що складається з C1-C4алкілу і бензилу;
(ii) основний гідроліз продукту із стадії (i) з використанням гідроксиду металу у воді та інертного розчинника в присутності метанолу з одержанням основно-адитивної солі H-D-iGlu-D-Trp-OH, де R1 і R2 є такими, як визначено вище, і де гідроксид металу вибраний з групи, що складається з гідроксиду натрію, гідроксиду калію і гідроксиду літію;
(iii) екстракція матеріалу із стадії (ii) розчинником, що не змішується з водою, і відокремлення водної фракції;
(iv) корекція pH водної фази із стадії (iii) до pH від приблизно 6 до приблизно 7; і
(v) випарювання розчинника з розчину із стадії (iv) з утворенням розчину, що містить співвідношення: приблизно одна частина розчиненої речовини на менш ніж приблизно 8 частин води, де розчинена речовина являє собою основно-адитивну сіль D-ізоглутаміл-D-триптофану.
11. Спосіб одержання моноамонійної солі H-D-iGlu-D-Trp-OH з кристалічного H-D-iGlu-D-Trp-OH, вільного від неорганічних солей, де спосіб включає наступні стадії:
(а) додавання кристалічного H-D-iGlu-D-Trp-OH до менш ніж приблизно одного еквівалента розчину гідроксиду амонію;
(б) корекція pH до значень в інтервалі від приблизно 6 до приблизно 7 за допомогою гідроксиду амонію;
(в) випарювання розчинника з одержанням масла; додавання ізопропанолу при перемішуванні, щоб спричинити осадження моноамонійної солі;
(г) виділення осадженої амонійної солі H-D-iGlu-D-Trp-OH; і
(д) вакуумне сушіння продукту, одержаного на стадії (в), з одержанням моноамонійної солі H-D-iGlu-D-Trp-OH.
12. Виділення кристалічного H-D-iGlu-D-Trp-OH з водного розчину H-D-iGlu-D-Trp-OH з використанням графічного методу розрахунку визначення залежності відсоткового вмісту H-D-iGlu-D-Trp-OH проти pH, де відсотковий вміст форм H-D-iGlu-D-Trp-OH в розчині становить більш ніж приблизно 50 % в розчині з pH від приблизно 2,0 до приблизно 3,2, що приводить до осадження кристалічного H-D-iGlu-D-Trp-OH.
13. Спосіб одержання вільного від неорганічних солей H-D-iGlu-D-Trp-OH за п. 3, який відрізняється тим, що він включає:
(а) одержання розчину кислотно-адитивної солі H-D-iGlu-D-Trp-OH у водному середовищі, істотною мірою вільному від органічного розчинника;
(б) корекція pH до значень від приблизно 2,0 до приблизно 3,2 за допомогою розчину гідроксиду лужного металу, щоб спричинити осадження H-D-iGlu-D-Trp-OH;
(в) виділення осадженого H-D-iGlu-D-Trp-OH; і
(г) вакуумне сушіння продукту, одержаного на стадії (c), з одержанням H-D-iGlu-D-Trp-OH.
14. Спосіб одержання вільного від неорганічних солей H-D-iGlu-D-Trp-OH за п. 3, який відрізняється тим, що він включає:
(а) одержання розчину основно-адитивної солі H-D-iGlu-D-Trp-OH у водному середовищі, по суті вільному від органічного розчинника;
(б) корекція pH до значень від приблизно 2,0 до приблизно 3,2 за допомогою розчину мінеральної кислоти, для осадження H-D-iGlu-D-Trp-OH;
(в) виділення осадженого H-D-iGlu-D-Trp-OH; і
(г) вакуумне сушіння продукту, одержаного на стадії (c), з одержанням H-D-iGlu-D-Trp-OH.
15. Спосіб за п. 13, який відрізняється тим, що кислотно-адитивна сіль на стадії (а) являє собою H-D-iGlu-D-Trp-OH гідрохлорид.
16. Спосіб за п. 13 або 14, який відрізняється тим, що pH на стадії (б) становить від приблизно 2,5 до приблизно 3,0.
17. Спосіб за п. 8, який відрізняється тим, що R1 являє собою бензил і R2 являє собою метил.
18. Спосіб за п. 8, який відрізняється тим, що інертний розчинник на стадії (з) вибирають з метанолу і метил-трет-бутилового ефіру, і гідроксид металу вибирають з групи, що складається з гідроксиду натрію, гідроксиду літію і гідроксиду калію.
19. Спосіб за п. 10, який відрізняється тим, що в сполуці формули Boc-D-Glu-(γ-D-Trp-OR2)-α-OR1 на стадії (i) R1 вибирають з групи, що складається з метилу і бензилу, і R2 вибирають з групи, що складається з метилу і етилу.
20. Спосіб за п. 10, який відрізняється тим, що інертний розчинник на стадії (ii) вибирають з групи, що складається з метанолу і метил-трет-бутилового ефіру.
21. Спосіб за п. 7, який відрізняється тим, що кислотно-адитивна сіль на стадії (а) являє собою H-D-iGlu-D-Trp-OH гідрохлорид, значення pH, що відповідає переважній формі одновалентної солі на стадії (б), становить від приблизно 5,7 до приблизно 7,0, та іонообмінна смола на стадії (в) являє собою AMBERLYST®15.
22. Фармацевтична композиція, яка відрізняється тим, що містить кристалічну сіль за п. 4 або 5 і як мінімум одну фармацевтично прийнятну допоміжну речовину.
23. Застосування ефективної кількості кристалічної солі за п. 4 або 5 як засобу проти псоріазу.
24. Застосування ефективної кількості кристалічної солі за п. 4 або 5 як імуносупресанта.
25. Застосування ефективної кількості фармацевтичної композиції за п. 22 для лікування псоріазу у суб'єкта, який потребує такого лікування.
26. Застосування ефективної кількості фармацевтичної композиції за п. 22 для імуносупресивної терапії у суб'єкта, який потребує такої терапії.
27. Кристалічний D-ізоглутаміл-D-триптофан, який характеризується піками рентгенівської порошкової дифрактограми з наступними 2q значеннями: 18.87, 20.05, 23.74 та 29.91.
28. Фармацевтична композиція, яка відрізняється тим, що містить кристалічну сіль сполуки за пп. 1, 2 або 27 і як мінімум одну фармацевтично прийнятну допоміжну речовину.
29. Застосування ефективної кількості кристалічної сполуки за пп. 1, 2 або 27 як засобу проти псоріазу.
30. Застосування ефективної кількості кристалічної сполуки за пп. 1, 2 або 27 як імуносупресанта.
31. Застосування ефективної кількості фармацевтичної композиції за п. 28 для лікування псоріазу у суб'єкта, який потребує такого лікування.
32. Застосування ефективної кількості фармацевтичної композиції за п. 28 для імуносупресивної терапії у суб'єкта, який потребує такої терапії.
.
Текст
Реферат: Винахід належить до кристалічного D-ізоглутаміл-D-триптофану та його кристалічної амонійної солі H-D-iGlu-D-Trp-OH. Також винахід розкриває спосіб одержання у водній фазі D-ізоглутамілD-триптофану, вільного від неорганічних солей, та спосіб одержання моноамонійної солі H-DiGlu-D-Trp-OH, вільної від неорганічних солей. Винахід належить також до фармацевтичної композиції, що містить кристалічну амонійну сіль H-D-iGlu-D-Trp-OH, та її застосування як засобу проти псоріазу та як імуносупресанта. UA 108058 C2 (12) UA 108058 C2 UA 108058 C2 5 10 15 20 25 30 35 40 НАЗВА ВИНАХОДУ Кристалічний D-ізоглутаміл-D-триптофан і моноамонійна сіль D-ізоглутаміл-D-триптофану ГАЛУЗЬ ВИНАХОДУ Даний винахід стосується нової стабільної кристалічної форми D-ізоглутаміл-D-триптофану та способу її виділення в чистій формі, вільній від неорганічних солей. Даний винахід також стосується нової стабільної амонійної солі D-ізоглутаміл-D-триптофану і способу її виробництва в чистій формі кристалізацією та/або традиційною хроматографією на колонці з силікагелем. ПЕРЕДУМОВИ ВИНАХОДУ D-Ізоглутаміл-D-триптофан (також відомий як H-D-iGlu-Trp-OH або тимодепресин [Thymodepressin]) являє собою синтетичний геморегуляторний дипептид, розроблений для лікування аутоімунних захворювань, в тому числі псоріазу (Sapuntsova, S. G., et al. (May 2002), Bulletin of Experimental Biology and Medicine, 133(5), 488-490). Тимодепресин вважається в Росії ефективним засобом для лікування псоріазу (патент США 5,736,519), де лікарський засіб на даний час присутній на ринку як динатрієва сіль в рідкому препараті для ін'єкційного та інтраназального введення. Він являє собою імуносупресант і селективно інгібує проліферацію клітин кісткового мозку, таким чином спричиняючи пригнічення імунітету. Відома тверда форма динатрієвої солі D-ізоглутаміл-D-триптофану являє собою аморфний порошок, що є гігроскопічним і дуже складним з технологічної точки зору. Структура динатрієвої солі тимодепресину описана в Kashirin, D. M., et al. (2000), Pharmaceutical Chemistry Journal, 34(11), 619-622. Мононатрієва сіль D-ізоглутаміл-D-триптофану ідентифікована Службою SM Chemical Abstracts (CAS) і наведене у реєстрах CAS REGISTRY , але відсутні публікації стосовно її одержання та фізичних властивостей. Порошкова або аморфна форма сполуки, такої як D-ізоглутаміл-D-триптофан, призначеної для фармацевтичного застосування, може створювати виробничі проблеми за рахунок проблем з насипною щільністю, гігроскопічністю і варіюючим вмістом води, який не може бути відрегульований сушінням під вакуумом. DІзоглутаміл-D-триптофан являє собою дипептид, і сушіння аморфної форми при підвищеній температурі, наприклад, 80–100 °C, під вакуумом не рекомендується. Єдиний шлях синтезу H-D-iGlu-D-Trp-OH, відомий з літератури, розкритий в патенті США 5,736,519. У відповідності до способу, наведеного в Прикладі 1 патенту США 5,736,519, зображеного в даному описі як Схема 1, Boc-D-Glu-OH (1,1) реагує з 1,3дициклогексилкарбодіімідом (ДКК) з утворенням циклічного ангідриду (1,2). При видаленні дициклогексилсечовини (DCU) фільтрацією, ангідрид (1,2) реагує з H-D-Trp-OH з утворенням суміші дипептиду Boc-D-iGlu-D-Trp-OH (1,3) і Boc-D-Glu-D-Trp-OH (1,4). Об'єднаний вихід неочищених Boc-D-iGlu-D-Trp-OH (1,3) і Boc-D-Glu-D-Trp-OH (1,4) становить 70 %. Однак, суміш містить тільки не більше 35 % цільової проміжної сполуки Boc-D-iGlu-D-Trp-OH (1,3). Захисну групу Boc видаляють, перемішуючи розчин (1,3) і (1,4) в мурашиній кислоті як розчиннику при температурі 40 °C протягом 1 години. Співвідношення (1,3) і (1,4) до мурашиної кислоти становить приблизно 1 г : 8 мл (мас./об.). Продукт являє собою суміш H-D-iGlu-D-Trp-OH (1,5) і H-D-Glu-D-Trp-OH (1,6). Оскільки пептиди (1,5) і (1,6) присутні в рівній кількості, очищення вимагає іонообмінної хроматографії з використанням піридинового ацетатного буфера. Вихід цільового продукту H-D-iGlu-D-Trp-OH (1,5) становить 35 % від кількості Boc-D-iGlu-D-Trp-OH (1,3). Таким чином, загальний вихід H-D-iGlu-D-Trp-OH (1,5) від кількості Boc-D-Glu-OH становить 12,25 %. 1 UA 108058 C2 DCC O Boc-D-Glu-OH O N H DMF Step 1A (1.1) H N O O O H N O DCU (1.2) COOH H N D-Trp-OH O step 1B O O O OH NH + N H O O OH O (1.4) (1.3) H N N H COOH O NH Boc-D-Glu-D-Trp-OH Boc-D-iGlu-D-Trp-OH COOH HCOOH H HN O OH NH NH Crude H-D-Glu-D-Trp-OH SP-PEA Sephadex column + H-D-iGlu-D-Trp-OH 5 10 COOH O OH Crude H-D-iGlu-D-Trp-OH (1.5) step 3 H N HN H + N H step 2 O O (1.6) H-D-Glu-D-Trp-OH (1.6) (1.5) Схема 1: Синтез H-D-iGlu-D-Trp-OH у відповідності до патенту США 5,736,519. Написи на схемі: Step = Стадія DMF = ДМФА DCC = ДКК DCU = ДЦМ Column = колонка Crude = неочищений Способу, описаному в патенті США 5,736,519, властиві декілька недоліків, як вказано нижче: ДКК на стадії 1A може призводити до утворення інших побічних продуктів, таких як O COOH O O O O N H NH N NH O N O O N H OH O 15 Про побічні продукти в результаті сполучення пептидів з ДКК повідомлялося в Marder, O., and Albericio, F. (June 2003), Chemical Oggi (Chemistry Today), 6-32. 2. Зняття захисту з Boc-D-iGlu-D-Trp-OH (1,3) вимагає підвищеної температури, і кінцеве очищення, H-D-iGlu-D-Trp-OH вимагає дуже токсичного розчинника піридину. Підвищена 2 UA 108058 C2 температура в ході зняття захисту з (1,3) може призводити до утворення N-третбутиліндольного похідного (1,7) як домішки (Löw, M., et. al. (1978), Hoppe-Seyler's Z. Physiol. Chem., 359(12):1643-51). Крім того, пептид може циклізуватися з утворенням глутаріміду (1,8) (Pandit, U.K. (1989), Pure & Appl. Chem., Vol. 61, No. 3, pp. 423-426). 5 O N O O HO N H NH2 10 15 20 O OH O O HN O O O Ph OH O (1.8). CF3COOH CH2Cl2 O L-Trp-OBzl O 2. L-Trp-OBzl NH Boc (2.1) (2.2) O NH4OOCH i-PrOH, NaHCO3 10% Pd catalyzt O L-Trp-OBzl HO NH O NH2 .CF3COOH 35 H (1.7), 1. DCC, DMF HOSu O 30 NH O O 3. В ході реакції сполучення утворюється тільки суміш Boc-D-iGlu-D-Trp-OH (1.3) і Boc-D-GluD-Trp-OH (1.4) 1:1. Максимальний вихід (1.3) не може перевищувати 50 % на стадії сполучення 1B. Суміш D-Glu-D-Trp-OH і D-iGlu-D-Trp-OH утворюється в кінці синтезу. Пептиди необхідно відокремлювати іонообмінною хроматографією і зворотно-фазовою препаративною хроматографією з високим тиском (ВТРХ). Загальний вихід H-D-iGlu-D-Trp-OH (1.5) становить 12,25 %, і очищення препаративною ВТРХ являє собою дуже тривалий та неефективний процес. Час утримання для двох подібних ізомерів, H-D-iGlu-D-Trp-OH (1.5) і H-D-Glu-D-Trp-OH (1.6) не наведений. Повторні цикли відокремлення для збільшення чистоти цільового ізомеру (1.5) є дуже неефективними. Даний спосіб не може бути адаптований до великомасштабного виробництва. 4. Протилежний діастереомер L-ізоглутаміл-L-триптофану (також відомий як H-L-iGlu-L-TrpOH або Бестим [Bestim]) являє собою імуностимулятор (див. патент США 5,774,452). Бестим застосовують в лікуванні виразок. Він зменшує запальні явища в шлунку і слизовій оболонці 12палої кишки та спричиняє регрес клінічних симптомів і рубцювання виразки (Tkacheva, A., et al. (2004), Eksp Klin Gastroenterol. (6):29-33, 163). Синтез мононатрієвої солі H-L-iGlu-L-Trp-OH (1:1) зображений на Схемі 2 (США 5,744,452). Ph 25 N H2N O O NH3+ (2.3) N H O O Na+ (2.4) [L-iGlu-L-Trp-O-] Na+ Схема 2: Синтез [L-iGlu-L-Trp-O-] Na+ у відповідності до патенту США 5,744,452. Написи на схемі: DCC = ДЦК DMF = ДМФА catalyzt = каталізатор У способі з патенту США 5,744,452 на стадії 1 як побічний продукт утворюється дициклогексилсечовина, яка повинна бути видалена фільтрацією. Заявлено, що на другій стадії трифтороцтова кислота видаляє γ-o-бензиловий естер з глутамінової кислоти (2.2). Бензиловий естер (2.3) видаляють гідрогенізацією з перенесенням за допомогою амонію форміату, паладієвого каталізатора, натрію бікарбонату в ізопропанолі при підвищеній температурі, з утворенням мононатрієвої солі H-L-iGlu-L-Trp-OH (2.4). Твердофазний синтез (2.4) також 3 UA 108058 C2 5 описаний в такому ж патенті, але триптофановий фрагмент повинен бути захищений як формамід, і пізніше захист повинен бути знятий. Інші діастереомери, L-ізоглутаміл-D-триптофан і D-ізоглутаміл-L-триптофан, також являють собою відомі сполуки (патент США 5,916,878). Синтез H-D-iGlu-L-Trp-OH, і H-L-iGlu-D-Trp-OH наведений на Схемі 3 і Схемі 4, відповідно (патент США 5,916,878). Ph HN 1. NMM, iBuO-CO-Cl, THF Ph O O OH O O HN O L-Trp-OBzl O 2. L-Trp-OBzl.HOTs NMM O O O (3.2) 62% yield (3.1) NH H2, Pd/C, MeOH O O HO NH2 (3.3) 10 Ph O Ph O N H OH H-D-iGlu-L-Trp-OH Схема 3: Синтез H-D-ізоглутаміл-L-триптофану у відповідності до патенту США 5,916,878. Написи на схемі: THF = ТГФ yield = вихід Ph O Ph HN 1. DCC, DMF HOSu O Ph HN OH O O O D-Trp-OH O 2. H-D-Trp-OH Et3N O Ph O O O (4.2) (4.1) H2, Pd/C methanol NH O O HO N H NH2 O OH (4.3) H-L-iGlu-D-Trp-OH 15 20 25 Схема 4: Синтез H-L-ізоглутаміл-D-триптофану у відповідності до патенту США 5,916,878. Написи на схемі: methanol = метанол Способи, наведені на Схемах 2, 3 і 4, можуть забезпечити синтез регіоспецифічного гамаамідного продукту (2.2), (3.2) і (4.2) без утворення альфа-амідного продукту, але вони включають стадію гідрогенізації в ході видалення бензилового естеру в сполуках (2.3), (3.2) і (4.2). Це вимагає використання великої кількості паладієвого каталізатора. Друге міркування стосується часткового відновлення індольного кільця у виробничому масштабі. Третє міркування стосується утворення глутаріміду, 2-(3-аміно-2,6-діоксо-піперидин-1-іл)-3-(1H-індол3-іл)пропіонової кислоти в процесі гідрогенізації. Четверте міркування стосується вартості. 4 UA 108058 C2 5 10 15 20 25 30 35 40 45 50 55 Вартість похідного CBz-Glu-OBzl, такого як (3.1) і (4.1), майже вдвічі перевищує ціну відповідності Boc-Glu-OBzl при виробництві за допомогою технологій тонкого хімічного синтезу. Способи на схемах 3 і 4 вимагають очищення кінцевого продукту методом ВТРХ. Значення виходу становить 33 % і 35,9 %, відповідно. Схема 2 вимагає використання трифтороцтової кислоти, яка вводить інші домішки в реакцію. Крім того, в способі на Схемі 2 використовують дициклогексалкарбодіімід як агент пептидного сполучення. Видалення слідових кількостей домішок з цього реактиву є серйозною проблемою в хімічному виробництві. Технологія, таким чином, не може бути адаптована до промислового виробництва, і також не може бути адаптована для великомасштабного виробництва H-D-ізоглутаміл-D-триптофану. КОРОТКИЙ ОПИС ВИНАХОДУ Даний винахід стосується нової стабільної кристалічної форми D-ізоглутаміл-D-триптофану і способу виділення вказаної сполуки в чистій формі, вільній від неорганічних солей, осадженням з води, без зворотно-фазової препаративної рідинної хроматографії з високим тиском. Спосіб призначений для одержання чистого N-трет-бутоксикарбоніл-D-ізоглутаміл-D-триптофану і його діестеру, вільного від N-трет-бутоксикарбоніл-D-глутаміл-D-триптофану, і перетворення N-третбутоксикарбоніл-D-ізоглутаміл-D-триптофану і його діестеру на чистий кристалічний Dізоглутаміл-D-триптофан. Новий кристалічний D-ізоглутаміл-D-триптофан за даним винаходом легко очищувати. В порівнянні із способами з рівня техніки, описаними вище, даний винахід пропонує цілий ряд переваг, як вказано нижче. По-перше, D-ізоглутаміл-D-триптофан одержують в кристалічній формі без застосування препаративної рідинної хроматографії з високим тиском. По-друге, ключова проміжну сполуку Boc-D-iGlu-D-Trp-OH або сіль H-D-Glu-(γ-D-Trp-OMe)-αOBzl HCl одержують з високим виходом і високою чистотою. По-третє, запропоновано спосіб перетворення Boc-D-iGlu-D-Trp-OH і його діестеру або H-DGlu-(γ-D-Trp-OMe)-α-OBzl HCl солі на D-ізоглутаміл-D-триптофан з високим виходом і високою чистотою. По-четверте, чиста кристалічна форма D-ізоглутаміл-D-триптофану за даним винаходом невідома в попередньому рівні техніки. Вона може бути використана безпосередньо в рідкому препараті з корекцією pH, що усуває потребу у використанні надзвичайно гігроскопічної і нестабільної динатрієвої солі D-ізоглутаміл-D-триптофану. Даний винахід також стосується нової стабільної амонійної солі D-ізоглутаміл-D-триптофану і способу одержання її з N-трет-бутоксикарбоніл-D-ізоглутаміл-D-триптофану та виділення такої сполуки в чистій формі кристалізацією та/або традиційною хроматографією на колонці з силікагелем. Моносіль амонію D-ізоглутаміл-D-триптофан являє собою стабільну тверду є придатним для цілей відмірювання та лікарських форм. Пропонується графік ідентифікації складу для ідентифікації форм солі при різних значеннях pH. Новий спосіб одержання D-ізоглутаміл-D-триптофану і амонійної солі D-ізоглутаміл-Dтриптофану (1:1) дозволяє уникнути вищеописаних виробничих проблем і робить можливим добування та обробки тимодепресину і моноамонійної солі тимодепресину в традиційному устаткуванні для хімічних процесів. Об'єктом даного винаходу є запропонувати придатний спосіб виробництва для Dізоглутаміл-D-триптофану, який давав би субстанцію лікарського засобу, повністю вільну від інших діастереомерів, як обговорювалося вище, і здатність матеріалу до зберігання в стабільній формі протягом тривалого періоду до введення в препарат. Іншим об'єктом даного винаходу є запропонувати D-ізоглутаміл-D-триптофан, вільний від альфа-амідного ізомеру D-глутаміл-D-триптофану. Іншим об'єктом даного винаходу є запропонувати спосіб одержання чистого D-ізоглутаміл-Dтриптофану (H-D-iGlu-D-Trp-OH) з кислотно-адитивної солі, H-D-iGlu-D-Trp-OH, що дає продукт, який по суті або повністю є вільним від залишків органічних розчинників і не вимагає очищення зворотно-фазовою рідинною хроматографією з високим тиском. Твердий D-ізоглутаміл-Dтриптофан виділяють з води. Іншим об'єктом даного винаходу є запропонувати спосіб одержання чистого D-ізоглутаміл-Dтриптофан (H-D-iGlu-D-Trp-OH) з основно-адитивної солі H-D-iGlu-D-Trp-OH, який дає продукт, по суті або повністю вільний від залишків органічних розчинників і не вимагає очищення зворотно-фазовою рідинною хроматографією з високим тиском. Твердий D-ізоглутаміл-Dтриптофан виділяють з води. Іншим об'єктом даного винаходу є запропонувати спосіб, в результаті якого утворюється Dізоглутаміл-D-триптофан, по суті вільний від забруднювачів у формі неорганічних солей. 5 UA 108058 C2 5 10 15 20 25 Іншим об'єктом даного винаходу є продукування кристалічного D-ізоглутаміл-D-триптофану з показниками рентгеноструктурного аналізу порошку (РСАП), як показано на фіг. 1. Іншим об'єктом даного винаходу є продукування моноамонійної солі D-ізоглутаміл-Dтриптофану з кислотно-адитивної солі D-ізоглутаміл-D-триптофану, що дає продукт, по суті або повністю вільний від залишків органічних розчинників, і не вимагає очищення зворотно-фазовою рідинною хроматографією з високим тиском. Тверду сіль амонію D-ізоглутаміл-D-триптофан виділяють з ізопропанолу та аміаку після обробки іонообмінною смолою для видалення неорганічних солей. Іншим об'єктом винаходу є продукування моноамонійної солі D-ізоглутаміл-D-триптофану з характером показників РСАП, показаним на фіг. 2. Іншим об'єктом даного винаходу є продукування аморфної моноамонійної солі Dізоглутаміл-D-триптофану, що по суті характеризується ІЧ-спектром з Фур'е-перетворенням (ІЧФП), як показано на фіг. 5. Іншим об'єктом даного винаходу є запропонувати спосіб продукування моноамонійної солі D-ізоглутаміл-D-триптофану, по суті вільної від забруднювачів у формі неорганічних солей. Іншим об'єктом даного винаходу є запропонувати спосіб одержання кислотно-адитивної солі, H-D-iGlu-D-Trp-OH, зокрема, гідрохлориду, з чистого Boc-D-iGlu-D-Trp-OH. Іншим об'єктом даного винаходу є запропонувати спосіб одержання чистого дипептиду BocD-iGlu-D-Trp-OH без хроматографічного відокремлення. Іншим об'єктом даного винаходу є запропонувати простий спосіб хроматографічного відокремлення на колонці з силікагелем для очищення D-ізоглутаміл-D-триптофану і його моноамонійної солі. Іншим об'єктом даного винаходу є запропонувати графік ідентифікації складу для визначення інтервалу pH для відокремлення D-ізоглутаміл-D-триптофану і його моносолі одновалентної. Переважний інтервал pH для осадження D-ізоглутаміл-D-триптофану з води становить від приблизно 2,5 до приблизно 3,0. Кислотно-адитивна сіль D-ізоглутаміл-D-триптофану одержана з дипептиду Boc-D-iGlu-DTrp-OH, який одержують в результаті основного гідролізу сполуки формули I: O O R1 NH O NH NH O O O 30 35 40 45 50 O R2 I 1 2 де R вибраний з групи, що складається з C1-C4 алкілу і бензилу, і R являє собою C1-C4 алкіл, за умови, що C4 алкіл не являє собою трет-бутил, з гідроксидом металу у воді та інертному розчиннику в присутності метанолу, з утворенням, Boc-D-iGlu-D-Trp-OH, вільного від інших діастереомерів. Гідроксид металу вибраний з групи, що складається з гідроксиду літію, гідроксиду натрію і гідроксиду калію. Сполука формули I, в свою чергу, одержують в результаті пептидного сполучення Boc-D1 2 1 2 Glu(OH)-OR і D-Trp-OR , де R і R є такими, як визначено вище, з реагентами пептидного сполучення, такими як HOBt і ЕДК. Такий спосіб синтезу Boc-D-iGlu-D-Trp-OH забезпечує істотні переваги в порівнянні з попереднім рівнем техніки, представленим в патенті США 5,736,519, оскільки продукт являє собою виключно гама-пептидний продукт Boc-D-iGlu-D-Trp-OH, і альфапептидний продукт Boc-D-Glu-D-Trp-OH не може бути утворений в ході синтезу, оскільки 1 використовується Boc-D-Glu(OH) -OR . Зняття захисту з чистого дипептиду Boc-D-iGlu-D-Trp-OH за допомогою кислоти, такої як хлористоводнева кислота, трифтороцтова кислота дає кислотно-адитивну сіль. pH розчину кислотно-адитивної солі становить від приблизно 2,5 до приблизно 3,0 з одержанням тимодепресину у вигляді твердого осаду. Альтернативно, кислотно-адитивна сіль може бути перетворена на амонійну сіль шляхом обробки водного розчину матеріалу до іонообмінній хроматографії із смолою на базі сульфонової кислоти. При видаленні солі елююванням з використанням води, іонообмінну смолу промивають сумішшю аміаку та ізопропанолу для одержання неочищеної амонійної солі, яку перекристалізують з ізопропанолу та води, з одержанням чистої моноамонійної солі. Розчин основно-адитивної солі D-ізоглутаміл-D-триптофану одержують шляхом зняття захисту за допомогою кислоти, зокрема зняття захисту за допомогою HCl, із сполуки формули I. 1 2 де кожен з R і R незалежно вибраний з групи, що складається з C1-C4 алкілу і бензилу, з 6 UA 108058 C2 2 5 10 15 20 25 30 35 1 одержанням кислотно-адитивної солі діестеру H-D-Glu-(γ-D-Trp-OR )-α-OR , яку далі обробляють гідроксидом металу у воді та інертному розчиннику в присутності метанолу, з утворенням основно-адитивної солі H-D-iGlu-D-Trp-OH. Гідроксид металу вибирають з групи, що складається з гідроксиду натрію, гідроксиду літію і гідроксиду калію. Екстракція розчинником, що не змішується з водою, видаляє органічну домішку в органічну фазу; водну фракцію відокремлюють і pH становить від приблизно 6 до приблизно 7 за допомогою гідроксиду металу. Після випарювання розчинника для зменшення кількості розчинника до співвідношення розчиненої речовини до розчинника менш ніж приблизно 1:8, де розчинена речовина являє собою пептид D-ізоглутаміл-D-триптофан у формі основно-адитивної солі, pH одержаного розчину основно-адитивної солі становить від приблизно 2,5 до приблизно 3,0 за допомогою мінеральної кислоти, для осадження D-ізоглутаміл-D-триптофану. Хоча не передбачається обмеження в будь-який спосіб контексту і практики даного винаходу теорією або способом практики винаходу і його можливими поясненнями, вважається, що при pH від приблизно 2,5 до приблизно 3,0, графік ідентифікації складу на фіг. 7 і фіг. 8 показує, щоосновними формами тимодепресину є вільний пептид (H-D-iGlu-D-Trp-OH) і одновалентна сіль. Оскільки розчинність H-D-iGlu-D-Trp-OH у воді, що не містить органічних розчинників, невисока (розчинність у воді < 23 мг/мл), сполука осаджується з розчину в чистій формі. Характер показників РСАП матеріалу показаний на фіг. 1. При виробництві моноамонійної солі, розчин тимодепресину при pH від приблизно 6,0 до приблизно 8,0 очищають іонним обміном для видалення солі. Регенерувальний розчин на базі аміаку дає чисту моноамонійну сіль після кристалізації з ізопропанолу і води. Хоча можна припускати, що моноамонійна сіль є нестійкою і може знову перетворюватися на вільний дипептид, на практиці, сполука фактично зберігає стабільність протягом більш ніж двох років. Заявником винайдено моноамонійну сіль тимодепресину, яка являє собою стабільний, новий хімічний об'єкт, який може легко кристалізуватися з ізопропанолу і води. Властивості одержаного кристалічного матеріалу показані на фіг. 2. Згадані вище способи дають чистий тимодепресин і моноамонійну сіль без зворотнофазової РХВТ в промислових масштабах, але тимодепресин і моноамонійна сіль можуть бути очищені традиційною хроматографією на колонці з силікагелем, з використанням зазначених вище умов. Таким чином, замість відкидання будь-якої порції тимодепресину в матковому розчині кристалізації, за бажанням фільтрат може бути упарений і додатково очищений хроматографією на колонці з силікагелем. КОРОТКИЙ ОПИС ФІГУР Кристалічні солі за даним винаходом описані в Прикладах нижче. Фіг. 1 представляє характерний зразок РСАП кристалічного, D-ізоглутаміл-D-триптофану. Показники РСАП також можуть бути представлені в значеннях міжплощинних відстаней d, кута Брегга 2θ і відносної інтенсивності (вираженої як відсоток по відношенню до найбільш інтенсивної лінії), як показано нижче: Кут [°2θ] 6,67 11,09 11,77 13,29 14,26 15,58 16,81 17,27 18,35 18,87 20,05 20,9 22,03 22,88 23,74 24,54 25,44 25,69 26,31 Значення d [Å] 13,239 7,975 7,515 6,655 6,205 5,685 5,269 5,13 4,832 4,7 4,424 4,247 4,032 3,884 3,744 3,625 3,499 3,465 3,384 7 Відносна інтенсивність [%] 3 4,4 1,2 4 11,3 33,3 28,9 30,4 12,2 95,8 63,6 33,2 17,1 100 97,9 41,9 20,3 12,1 16,4 UA 108058 C2 Кут [°2θ] 27 27,75 28,18 28,79 29,13 29,91 31,04 31,49 32,54 33,29 33,97 34,99 35,54 36,14 36,74 37,35 38,31 39,01 5 10 Значення d [Å] 3,3 3,212 3,164 3,099 3,063 2,985 2,879 2,839 2,749 2,689 2,637 2,562 2,524 2,483 2,444 2,406 2,348 2,307 Відносна інтенсивність [%] 27,4 24,9 19,3 6,8 6,2 79,2 8,6 33,7 4,4 9,3 10,5 17,3 21,8 5,1 5,9 7,7 25,6 20,3 Порошкоподібні зразки одержані звичайною технікою фронтального пакування і проаналізовані на системі дифрактометра D8 Discovery з джерелом Cu-kα, діючим в режимі 45 кВ/45 мА. Система обладнана 2D-пропорційним детектором площі (GADDS). Експериментальні дані були одержані на двох фреймах з контактом 600 с для кожного, що охоплювало інтервал 335° (2θ). Одержані 2D дифракційні зображення далі були інтегровані для одержання стандарту, I проти 2θ, характеру дифракції. Дані обробляли за допомогою різноманітного програмного забезпечення для обробки даних Bruker AXS, в тому числі: Eva 8.0 і Topas, версія 2,1 (для аналізу апроксимації профілю та масштабування, при необхідності). Фіг. 2 представляє характерний зразок РСАП кристалічної моноамонійної солі D-ізоглутамілD-триптофану. Характер РСАП може також бути виражений в термінах міжплощинних відстаней d, кута Брегга 2θ і відносної інтенсивності (виражений як відсоток по відношенню до найбільш інтенсивного променя), як вказано нижче: Кут [°2θ] 9,29 12,19 13,93 15,17 16,49 17,18 18,56 18,88 20,02 22,28 23,31 23,66 24,03 24,37 25,07 25,61 25,96 27,62 28,12 28,49 29,52 30,27 30,64 31,31 Значення d [Å] 9,517 7,258 6,354 5,837 5,371 5,157 4,778 4,696 4,431 3,986 3,814 3,757 3,7 3,649 3,549 3,475 3,43 3,227 3,17 3,131 3,023 2,951 2,915 2,854 Відносна інтенсивність [%] 4,1 4,5 76,2 27,4 9,8 3 31,6 10,5 100 3 4,6 9,8 52,9 26,3 11,4 5,6 5 29,7 55,7 12,2 23,1 3,7 7,9 11,8 8 UA 108058 C2 Кут [°2θ] 31,7 32,16 32,81 33,78 34,14 35,76 36,94 37,58 38,03 39,22 5 10 15 20 25 Значення d [Å] 2,821 2,781 2,728 2,652 2,625 2,509 2,431 2,391 2,364 2,295 Відносна інтенсивність [%] 29,6 19,2 16 7,4 5,1 16 11,6 25,6 12,5 1,9 Спектри рентгеноструктурного аналізу порошку для D-ізоглутаміл-D-триптофану і його амонійної солі показані на фіг. 1 і фіг. 2 нижче. Слід розуміти, що значення 2θ для зразка рентгеноструктурного аналізу порошку можуть дещо варіювати від одного приладу до іншого або від одного зразка до іншого, тобто наведені значення не повинні розглядатися як абсолютні. Фіг. 3 представляє характерний зразок РСАП аморфної форми D-ізоглутаміл-D-триптофану. Фіг. 4 представляє характерний спектр поглинання в інфрачервоній (ІЧ) області кристалічної моноамонійної солі D-ізоглутаміл-D-триптофану. Фіг. 5 представляє характерний спектр поглинання в інфрачервоній (ІЧ) області аморфної моноамонійної солі D-ізоглутаміл-D-триптофану. Фіг. 6 представляє характерний спектр поглинання в інфрачервоній (ІЧ) області кристалічного D-ізоглутаміл-D-триптофану. Фіг. 7 ілюструє обчислення ідентифікації складу дипептиду H-D-iGlu-D-Trp-OH і його солі, з використанням теоретично обчислених значень pKa для кислотних і амінних груп. LH 2 являє собою форму двохосновної кислоти пептиду H-D-iGlu-D-Trp-OH, LH являє собою сіль монокарбонової кислоти H-D-iGlu-D-Trp-OH. Прикладом LH є моноамонійна сіль. L позначає форму двохосновної солі, і її прикладом є динатрієва сіль пептиду H-D-iGlu-D-Trp-OH. Фіг. 8 ілюструє обчислення ідентифікації складу дипептиду H-D-iGlu-D-Trp-OH і його солі, з використанням експериментально визначених значень pKa для кислотних і амінних груп. LH 2 являє собою форму двохосновної кислоти пептиду H-D-iGlu-D-Trp-OH, LH являє собою сіль монокарбонової кислоти H-D-iGlu-D-Trp-OH. Прикладом LH є моноамонійна сіль. L позначає форму двохосновної солі, і її прикладом є динатрієва сіль пептиду H-D-iGlu-D-Trp-OH. ДЕТАЛЬНИЙ ОПИС ВИНАХОДУ 1 В даному описі термін "Boc-D-Glu(OH)-OR " позначає структуру: O O 1 R O OH NH O O , 1 якщо R являє собою бензил, то з хімічної точки зору сполука являє собою альфабензиловий естер 2-глутамінової кислоти. 2 В даному описі термін "D-Trp-OR " позначає структуру: NH NH2 O O 30 , R2 якщо R являє собою метил, сполука являє собою метиловий естер D-триптофану. 2 1 В даному описі термін "Boc-D-Glu-(γ-D-Trp-OR )-α-OR " позначає структуру: O O 1 R NH O NH 2 NH O O O O R2 . 9 UA 108058 C2 2 1 В даному описі термін "H-D-Glu-(γ-D-Trp-OR )-α-OR " позначає структуру: O O 1 R NH O NH NH2 O O , R2 якщо R являє собою бензил, R являє собою метил, то сполука являє собою 1 2 O O O NH NH NH2 O O 5 CH3 . якщо R являє собою метил, R являє собою метил, сполука являє собою O O H3C NH O NH NH2 O 1 2 O CH3 . В даному описі термін "тимодепресин" позначає дипептид H-D-iGlu-D-Trp-OH з хімічною структурою: O O HO O HO N H NH2 10 15 20 25 30 35 NH Він також може бути позначений як H-D-Glu-(γ-D-Trp-OH)-OH. Кислотно-адитивна сіль являє собою сіль, утворену в результаті реакції аміну H-D-iGlu-DTrp-OH з неорганічними кислотами, в тому числі хлористоводневою кислотою, сірчаною кислотою, бромоводневою кислотою, фосфорною кислотою і т. п., або органічними кислотами, в тому числі мурашиною кислотою, оцтовою кислотою, пропіоновою кислотою, гліколевою кислотою, молочною кислотою, піровиноградною кислотою, щавлевою кислотою, бурштиновою кислотою, яблучною кислотою, винною кислотою, лимонною кислотою, трифтороцтовою кислотою, бензойною кислотою, саліциловою кислотою, бензолсульфоновою кислотою і толуолсульфоновою кислотою. Вона може також бути утворена в результаті зняття захисту з похідного Boc-D-iGlu-D-Trp-OH за допомогою кислоти. Основно-адитивна сіль являє собою сіль, утворену в результаті реакції карбонової кислоти H-D-iGlu-D-Trp-OH з неорганічними основами, в тому числі натрію гідроксидом, літію гідроксидом, калію гідроксидом і т. п. Даний винахід спрямований на спосіб одержання H-D-iGlu-D-Trp-OH і його амонійної солі, вільних від неорганічних солей, з кислотно-адитивних солей H-D-iGlu-D-Trp-OH, які переважно одержані з дипептиду Boc-D-iGlu-D-Trp-OH. Boc-D-iGlu-D-Trp-OH одержують з Boc-D-Glu(OH)1 2 1 2 OR і D-Trp-OR , де R вибирають з групи, що складається з бензилу і C 1-C4 алкілу, і R являє собою C1-C4 алкіл, за умови, що C4 алкіл не являє собою трет-бутил. Даний винахід також спрямований на спосіб одержання H-D-iGlu-D-Trp-OH з розчину основно-адитивної солі H-D-iGlu-D-Trp-OH, який переважно одержують з кислотно-адитивної 2 1 1 2 солі дипептиду H-D-Glu-(γ-D-Trp-OR )-α-OR , де кожний з R і R незалежно вибирають з групи, що складається з бензилу і C1-C4 алкілу. ПЕРЕВАЖНІ ВАРІАНТИ В межах декількох аспектів даного винаходу, який сформульований в Короткому описі винаходу, послідовність стадій способу і їх відносна переважність описані нижче: У варіанті даного винаходу пропонується спосіб одержання у водній фазі вільного від неорганічних солей H-D-iGlu-D-Trp-OH, який включає: 10 UA 108058 C2 5 10 15 20 25 30 35 40 45 50 55 (a) одержання розчину кислотно-адитивної солі H-D-iGlu-D-Trp-OH у водному середовищі, істотною мірою вільному від органічного розчинника; або одержання розчину основно-адитивної солі H-D-iGlu-D-Trp-OH у водному середовищі, істотною мірою вільному від органічного розчинника; (б) корекція pH до pH, що відповідає переважній формі двоосновної форми за допомогою розчину гідроксиду лужного металу або мінеральної кислоти, для осадження H-D-iGlu-D-Trp-OH; (в) виділення осадженого H-D-iGlu-D-Trp-OH; і (г) вакуумне сушіння продукту, одержаного на стадії (в), з утворенням H-D-iGlu-D-Trp-OH. В іншому варіанті даного винаходу пропонується кристалічна форма H-D-iGlu-D-Trp-OH, що являє собою D-ізоглутаміл-D-триптофан, яка характеризується зразком РСАП, представленим в описі фігур. В іншому варіанті даного винаходу пропонується кристалічний H-D-iGlu-D-Trp-OH, що являє собою D-ізоглутаміл-D-триптофан, яка характеризується зразком РСАП, проілюстрованим на фіг. 1. В іншому варіанті даного винаходу пропонується спосіб одержання моноамонійної солі H-DiGlu-D-Trp-OH, вільної від неорганічних солей, де спосіб включає наступні стадії: (а) одержання розчину кислотно-адитивної солі H-D-iGlu-D-Trp-OH у водному середовищі, істотною мірою вільному від органічного розчинника; (б) корекція pH до pH, що відповідає переважній формі одновалентної солі за допомогою розчину гідроксиду металу; (в) обробка розчину із стадії (б) іонообмінною смолою, та елюювання водою для обміну іона металу із солі в розчині на іон водню до тих пір, поки pH не буде знаходитися в інтервалі від приблизно 5,7 до приблизно 7,0; (г) контакт іонообмінної смоли з регенерувальним розчином на базі аміаку, який здійснює обмін іонів в розчині на цільовий H-D-iGlu-D-Trp-OH, що міститься в іонообмінній смолі, з утворенням таким чином регенерувального елюату, що містить амонійну сіль H-D-iGlu-D-TrpOH; (д) випарювання розчинника з розчину, одержаного на стадії (г), з одержанням неочищеної амонійної солі; (е) розчинення амонійної солі, одержаної на стадії (д), у воді з повільним додавання ізопропанолу, таким чином, що утворюється осад моноамонійної солі; і (є) вакуумне сушіння продукту, одержаного на стадії (е), з утворенням кристалічної форми амонійної солі H-D-iGlu-D-Trp-OH (1:1). Альтернативно, замість стадій (е) і (є) спосіб включає наступні стадії: (з) обробка матеріалу, одержаного на стадії (д), хроматографією на силікагелі з використанням розчину ізопропанолу та аміаку як елюенту; і (і) сушіння виморожуванням продукту, одержаного на стадії (з), з утворенням аморфної форми амонійної солі H-D-iGlu-D-Trp-OH (1:1). В іншому варіанті даного винаходу пропонується спосіб одержання моноамонійної солі H-DiGlu-D-Trp-OH з кристалічного H-D-iGlu-D-Trp-OH, вільного від неорганічних солей, де спосіб включає наступні стадії: (a) додавання H-D-iGlu-D-Trp-OH до менш, ніж одного еквіваленту розчину гідроксиду амонію; (б) корекція pH до 6–7 за допомогою гідроксиду амонію; (в) випарювання розчинника з утворенням масла; додавання ізопропанолу при перемішуванні, щоб спричинити осадження моноамонійної солі; (г) виділення осадженої амонійної солі H-D-iGlu-D-Trp-OH; і (д) вакуумне сушіння продукту, одержаного на стадії (в), з одержанням моноамонійної солі H-D-iGlu-D-Trp-OH. В іншому варіанті даного винаходу пропонується кристалічна амонійна сіль H-D-iGlu-D-TrpOH (1:1), яка характеризується показниками РСАП, представленими в описі фігур. В іншому варіанті даного винаходу пропонується кристалічна амонійна сіль H-D-iGlu-D-TrpOH (1:1), яка характеризується показниками РСАП, представленими на фіг. 2. В іншому варіанті даного винаходу пропонується аморфна амонійна сіль H-D-iGlu-D-Trp-OH (1:1), яка характеризується спектром ІЧФП (ІЧ), показаним на фіг. 5. В іншому варіанті даного винаходу пропонується спосіб одержання кислотно-адитивної солі D-ізоглутаміл-D-триптофану, де сіль являє собою H-D-iGlu-D-Trp-OH гідрохлорид, і де спосіб включає: (і) основний гідроліз сполуки формули I: 11 UA 108058 C2 O R1 O O NH NH NH O O O O R2 1 5 10 2 де R вибирають з групи, що складається з C1-C4 алкілу і бензилу, і R являє собою C1-C4 алкіл, за умови, що C4 алкіл не являє собою трет-бутил, з гідроксидом металу у воді та інертному розчиннику, в присутності метанолу, з утворенням, Boc-D-iGlu-D-Trp-OH, вільного від інших діастереомерів; (ii) зняття за допомогою хлористого водню захисту з Boc-D-iGlu-D-Trp-OH із стадії (і) в інертному органічному розчиннику; і випарювання розчинника з утворенням гідрохлориду, H-DiGlu-D-Trp-OH. В іншому варіанті даного винаходу пропонується спосіб одержання розчину кислотноадитивної солі H-D-iGlu-D-Trp-OH гідрохлориду, де спосіб включає: (а) гідрогенізацію сполуки формули II: O O R1 NH O NH NH O O O 15 20 25 30 35 40 45 O R2 II Ph 1 2 де R являє собою бензил, і R вибраний з групи, що складається з бензилу і водню, з використанням паладію на вугіллі в метанолі або етанолі; (б) очищення неочищеного H-D-iGlu-D-Trp-OH із стадії (a) хроматографією на силікагелі, з використанням ізопропанолу і води як елюенту; і (в) обробка матеріалу із стадії (б) хлористоводневою кислотою у воді, з утворенням розчину солі H-D-iGlu-D-Trp-OH гідрохлориду у воді. У згаданих вище двох способах, виготовлення кислотно-адитивної солі D-ізоглутаміл-Dтриптофану із сполуки формули I є переважним в порівнянні з одержанням із сполуки формули II з точки зору вартості проміжних хімічних сполук. В іншому варіанті даного винаходу пропонується спосіб одержання розчину основноадитивної солі, H-D-iGlu-D-Trp-OH, де спосіб включає: 2 1 (а) зняття за допомогою кислоти захисту з дипептиду N-Boc-D-Glu-(γ-D-Trp-OR )-α-OR , де 1 2 кожний з R і R незалежно вибраний з групи, що складається з C1-C4 алкілу і бензилу; (б) основний гідроліз продукту із стадії (a) за допомогою гідроксиду металу у воді та інертному розчиннику в присутності метанолу, де гідроксид металу вибраний з групи, що складається з гідроксиду натрію, гідроксиду калію і гідроксиду літію; (в) екстракція матеріалу із стадії (б) розчинником, що не змішується з водою, і відокремлення водного шару; (г) регулювання pH водної фази із стадії (в) до значень в інтервалі від приблизно 6 до приблизно 7; і (д) випарювання розчинника з розчину із стадії (г) для утворення розчину, що містить співвідношення приблизно однієї частини розчиненої речовини до менш ніж приблизно 8 частин води, де розчинена речовина являє собою основно-адитивну сіль D-ізоглутаміл-D-триптофану. В іншому варіанті даного винаходу пропонується нове діестерне похідне H-D-Glu-(γ-D-Trp2 1 1 2 OR )-α-OR діестеру гідрохлорид, де кожен з R і R незалежно вибраний з групи, що складається з бензилу і C1-C4 алкілу. 2 1 Сполуки H-D-Glu-(γ-D-Trp-OR )-α-OR невідомі в попередньому рівні техніки. Вказані сполуки можуть бути використані як проміжні сполуки для одержання дипептиду D-ізоглутаміл-D2 1 триптофану. Альтернативно, H-D-Glu-(γ-D-Trp-OR )-α-OR гідрохлорид може бути використаний у фармацевтичному препараті, де гідроліз естеру відбувається in situ, з одержанням Dізоглутаміл-D-триптофану при виготовленні препарату. У варіанті даного винаходу пропонується графік ідентифікації складу, проілюстрований на фіг. 8, для відокремлення H-D-iGlu-D-Trp-OH при pH від приблизно 2,5 до приблизно 3,0. Спосіб одержання 12 UA 108058 C2 В даному винаході пропонується, як зображено нижче на Схемі 5, надійна методика синтезу чистого N-(трет-бутоксикарбоніл)-D-ізоглутаміл-D-триптофану з високим виходом, причому зазначена надійна методика відсутня в попередньому рівні техніки (наприклад, патенті США 5,736,519). 5 O O O R1 O N H O peptide coupling N H OH D-Trp-OR2 O N H Boc-D-iGlu-(D-Trp-OR2)-OR1 O O O NH O Boc-D-Glu(OH)-OR1 base hydrolysis 2 R1 O R O O H O O H O N H NH N H O Boc-D-iGlu-D-Trp-OH 10 15 20 25 30 35 Схема 5 Написи на схемі: peptide coupling = пептидне сполучення base hydrolysis = основний гідроліз 1 1 У способі за даним винаходом, розчин Boc-D-Glu(OH)-OR , де R являє собою бензил, в інертному розчиннику реагує з N-(3-диметиламінопропіл)-N’-етилкарбодііміду гідрохлоридом (ЕДК), гідроксибензотриазолом (HOBt) і діізопропілетиламіном (ДІПЕА). Переважна температура становить від приблизно 5 до приблизно -5 °C, і переважним розчинником є дихлорметан. Після перемішування протягом від приблизно 5 до приблизно 30 хвилин, 2 2 переважно приблизно 15 хвилин, по краплях додають розчин солі D-Trp-OR HCl, де R являє собою метил, з діізопропілетиламіном (ДІПЕА). Одержаний розчин перемішують при температурі льоду, переважно від приблизно -5 до приблизно 5 °C протягом приблизно 1 години, а потім при кімнатній температурі від приблизно 12 до приблизно 20 год., переважно 2 1 приблизно 16 год. Продукт Boc-D-iGlu-(D-Trp-OR )-α-OR виділяють традиційними засобами. Сполука може бути легко кристалізована з етилацетату і гексану. Дві синтетичні домішки присутні у слідових кількостях - сполуки (A) і (B), які можуть бути видалені перекристалізацією. Вважається, що обидві сполуки утворюються з реагенту HOBt. N N N N N N N N N OH (A) (B) 2 1 Діестер Boc-D-Glu-(γ-D-Trp-OR )-α-OR в спирті змішують з розчином гідроксиду натрію. Переважна кількість гідроксиду натрію становить від приблизно 2,5 до приблизно 5 еквівалентів на еквівалент діестеру. Все ще переважно, використовують молярне співвідношення від приблизно 2 до приблизно 3,5 моль NaOH на моль діестерної сполуки. Переважне співвідношення розчинника становить приблизно 2 мл спирту на мл води, і переважне співвідношення NaOH до води становить приблизно 1 г на 20 мл. Така методика виділення включає екстракцію реакційної суміші етилацетатом, і таким чином будь-яка органічна домішка видаляється на цій стадії синтезу. Після підкислення водну фракцію екстрагують органічним розчинником, таким як етилацетат. Двоосновний N-(трет-бутоксикарбоніл)-D-ізоглутаміл-Dтриптофан (Boc-D-iGlu-D-Trp-OH) виділяють традиційними засобами у вигляді твердої речовини. Ізольований вихід для двох об'єднаних стадій становить 89 %. Такий показник 13 UA 108058 C2 5 10 15 20 25 30 35 40 45 50 55 60 перевищує показник методики з рівня техніки (патент США 5,736,519). Новий спосіб за даним винаходом додатково ілюструється в прикладах нижче. Детальне дослідження і моніторинг стадії гідролізу заявником показали, що сполука Boc-D2 1 1 2 iGlu-(D-Trp-OR )-OR , де R являє собою бензил, і R являє собою метил, спочатку перетворюється метанолом з утворенням Boc-D-iGlu-(D-Trp-OMe)-OMe, після чого сполука гідролізується до двохосновної кислоти. Заявником визначено, що метанол необхідний для швидкого гідролізу альфа-бензилового естеру. Зважаючи на великий об'єм етилацетату, необхідний для екстракції продукту, заявник винайшов двохфазну методику для ефективного 2 1 2 1 гідролізу Boc-D-iGlu-(D-Trp-OR ) -OR . Перемішують суміш Boc-D-iGlu-(D-Trp-OR )-OR в метилтрет-бутиловому ефірі (МТБЕ) і розчині гідроксиду металу, такого як розчин гідроксиду літію або 2 1 гідроксиду натрію. Переважне співвідношення гідроксиду металу до Boc-D-iGlu-(D-Trp-OR )-OR знаходиться в інтервалі приблизно 2,0-2,5 до 1. Додають метанол, і суміш енергійно перемішують протягом періоду від приблизно 1 до приблизно 6 год., переважно від приблизно 1,5 до приблизно 2,5 годин. Суміш відокремлюють від органічної фази традиційними засобами. Дана методика усуває необхідність у використанні великої кількості етилацетату для екстракції і проілюстрована в прикладах нижче. У традиційному способі знімають захист із сполуки Boc-D-iGlu-D-Trp-OH за допомогою органічних кислот, з утворенням дипептиду H-D-iGlu-D-Trp-OH, який вимагає екстенсивного очищення. Існують численні недоліки методик з рівня техніки (патент США 5,736,519), в яких використовують мурашину кислоту при температурі 40 °C для зняття захисту із суміші Boc-DiGlu-D-Trp-OH і Boc-D-Glu-D-Trp-OH, з утворенням суміші H-D-iGlu-D-Trp-OH і H-D-Glu-D-Trp-OH. Іонообмінну хроматографію та зворотно-фазову РХВТ використовують для виділення продукту. Вихід є низьким, і методика не може бути адаптована до великомасштабного виробництва. Зняття захисту з N-трет-бутоксикарбонільної групи за допомогою трифтороцтової кислоти або мурашиної кислоти утворює іон трет-бутилкарбонію, який може реагувати з індольним азотом з утворенням N-трет-бутилового продукту (Löw, M., et. al. (1978), Hoppe-Seyler's Z. Physiol. Chem., 359(12):1643-51). Іншою проблемою є утворення глутаріміду (1,8) (Pandit, U.K. (1989), Pure & Appl. Chem., Vol. 61, No. 3, pp. 423-426). Заявником було визначено, що кислотно-адитивна сіль за даним винаходом, зокрема, неочищена сіль гідрохлорид, може бути легко одержана з HCl в інертному розчиннику, такому як етилацетат, при низькій температурі, переважно від приблизно 0 °C до температури навколишнього середовища. Випарювання розчинника дає тимодепресину гідрохлорид, який використовують для одержання тимодепресину. Для прогнозу необхідного значення pH для осадження тимодепресину у формі двохосновної кислоти H-D-iGlu-D-Trp-OH, заявником було проведено теоретичне граяічне визначення складу, і було зроблено висновок про те, що тимодепресин існує в двохосновній формі при значеннях pH від приблизно 2,5 до приблизно 3,0. Даний висновок дозволив винайти спосіб виділення тимодепресину без хроматографії. Фіг. 7 ілюструє такий розрахунок. На фіг. 7, LH2 являє собою форму дикарбонової кислоти пептиду H-D-iGlu-D-Trp-OH (тобто, тимодепресину), LH являє собою форму солі монокарбонової кислоти пептиду H-D-iGlu-D-Trp-OH; один з таких прикладів являє собою моноамонійну сіль, L являє собою форму солі дикарбонової кислоти пептиду H-D-iGlu-D-Trp-OH; один з таких прикладів являє собою динатрієву сіль, і LH3 являє собою кислотно-адитивну сіль тимодепресину. Вісь X показує pH розчину. Вісь Y показує % утворення по відношенню до L (типова термінологія програмного забезпечення) і дає молярну фракцію форми, присутньої при конкретному значенні pH. При pH від приблизно 2,5 до приблизно 3,0, більша частина (80 %) дипептиду існує, як форма дикарбонової кислоти і може бути осаджена з розчину, якщо вона є нерозчинною у воді. Наше дослідження показує, що форма дикарбонової кислоти, одержана в такий спосіб виявляє розчинність у воді приблизно 23 мг/мл. При pH приблизно 7,0, 100 % форм представлені формою монокарбонової кислоти. Якщо протиіон являє собою натрій, то форма являє собою тимодепресин мононатрій. На практиці, якщо розчин гідрохлориду тимодепресину розчинений у воді, і значення pH доведено приблизно до 3,0 при перемішуванні, повільно утворюється тверда речовина, яку відфільтровують від суміші. Чистота матеріалу за даними РХ перевищує 97 % і знаходиться в межах категорії фармацевтичної чистоти як активний фармацевтичний інгредієнт. Нами визначено, що даний спосіб переважає попередній рівень техніки з точки зору виробництва і виділення чистого тимодепресину (H-D-iGlu-D-Trp-OH). Відсутня необхідність у іонообмінній і зворотно-фазовій препаративній хроматографії на колонці, оскільки єдиним побічним продуктом є натрію хлорид, розчинний у воді. Описана методика являє собою пряму методику відокремлення та очищення тимодепресину. Вона передбачає теоретичне графічне визначення 14 UA 108058 C2 5 10 15 20 25 30 35 40 45 50 складу, і спосіб переважає способи попереднього рівня техніки, які вимагають громіздкого очищення. Значення pKa кислотних і амінних груп H-D-iGlu-D-Trp-OH визначені експериментально. Графік складу дипептиду, побудований з використанням експериментально визначених значень pKa, показаний на фіг. 8. На фіг. 8, LH2 являє собою тимодепресин, LH являє собою сіль монокарбонової кислоти, L являє собою сіль дикарбонової кислоти, і LH 3 являє собою кислотноадитивну сіль тимодепресину. На осі X наведені значення pH розчину. На осі Y наведений % утворення по відношенню до L (типова термінологія програмного забезпечення) і реєструється молярна фракція форм, присутніх при конкретному значенні pH. Концентрацію 0,5 M використовують для відображення еквівалентності 1 г тимодепресину в 6 мл води для цілей відокремлення. Дана фігура показує, що приблизно 75 % тимодепресину (LH2) існує у формі дикарбонової кислоти при pH 2,7. З цієї причини, тимодепресин осаджується при pH 2,7 і може бути відфільтрований. Матковий розчин може бути упарений для одержання другої порції тимодепресину. Графік підтверджує теоретичний прогноз - форма двохосновної кислоти H-DiGlu-D-Trp-OH переважає при pH від приблизно 2,5 до приблизно 3,0; вказаний інтервал являє собою значення pH для відокремлення форми двохосновної кислоти з води у вигляді осаду. Оскільки тільки приблизно 80 % матеріалу буде осаджуватися в чистій формі, об'єм маткового розчину слід зменшити і піддати другому кругу осадження при pH від приблизно 2,5 до приблизно 3,0, переважно при pH приблизно 2,7. Зняття захисту з Boc-H-D-iGlu-D-Trp-OH за допомогою трифтороцтової кислоти в інертному розчиннику дає сіль трифтороцтової кислоти. Інертний розчинник являє собою дихлорметан, і звичайно використовують суміш трифтороцтової кислоти і дихлорметану (1:1). Випарювання розчинника дає масло, яке сушать під вакуумом для видалення залишкового розчинника. Масло диспергують у воді. При доведенні pH до приблизно 3,0 утворюється осад білої твердої речовини після перемішування протягом періоду від приблизно 12 до приблизно 16 год. При одержанні кислотно-адитивної солі, переважно використовувати HCl в інертному розчиннику для утворення гідрохлориду. Альтернативно, сіль трифтороцтоаої кислоти може бути утворена із застосуванням описаного вище способу. Використання солі хлористоводневої кислоти як кислотно-адитивної солі є переважним, оскільки зняття захисту з Boc-D-iGlu-D-TrpOH є більш ефективний при використанні HCl в інертному розчиннику, наприклад, 3 M HCl в етилацетаті. Тривалість реакції є істотно більшою при використанні трифтороцтової кислоти. Крім того, сіль трифтороцтової кислоти D-iGlu-D-Trp-OH містить декілька синтетичних домішок, які переходять до D-iGlu-D-Trp-OH у випадку осадження при pH від приблизно 2,5 до приблизно 3,0 у воді. Домішки необхідно видаляти екстенсивною перекристалізацією. Початковий матеріал Boc-D-iGlu-D-Trp-OH одержують з використанням описаної раніше методології. Кислотно-адитивну сіль необхідно сушити під вакуумом, щоб гарантувати відсутність органічних розчинників і летких домішок. У випадку осадження тимодепресину з води, одержують розчин кислотно-адитивної солі у воді. Співвідношення кислотно-адитивної солі до води знаходиться в інтервалі від приблизно 1:5 до приблизно 1:10. Все ще переважно, співвідношення кислотно-адитивної солі знаходиться в інтервалі від приблизно 1:6 до приблизно 1:8. Розчин гідроксиду металу, звичайно розчин гідроксиду натрію, використовують для осадження продукту, але гідроксид калію та розчини гідроксидів інших металів можуть бути використані. Неочищений H-D-iGlu-D-Trp-OH також може бути одержаний гідрогенізацією сполуки формули II: O O R1 NH O NH NH O O O 2 O R II Ph 1 2 де R являє собою бензил, і R вибраний з групи, що складається з бензилу і водню, з використанням паладію на вугіллі в метанолі або етанолі. Після фільтрації каталізатора фільтрат упарюють до стану масла, яке додатково очищають хроматографією на силікагелі, з використанням ізопропанолу і води як елюенту. Одержаний H-D-iGlu-D-Trp-OH може бути перетворений на сіль H-D-iGlu-D-Trp-OH гідрохлорид у воді за допомогою хлористоводневої кислоти. 15 UA 108058 C2 5 10 15 20 25 30 35 40 45 50 55 60 Розчин основно-адитивної солі D-ізоглутаміл-D-триптофану одержують зняттям захисту за 2 1 1 2 допомогою кислоти з дипептиду Boc-D-Glu-(γ-D-Trp-OR )-α-OR , де кожен з R і R незалежно вибраний з групи, що складається з бензилу і C 1-C4 алкілу. Наприклад, зняття захисту за 2 1 допомогою HCl з Boc-D-Glu-(γ-D-Trp-OR )-α-OR в інертному розчиннику, такому як 2 1 . 1 дихлорметан, дає сіль H-D-Glu-(γ-D-Trp-OR )-α-OR HCl У випадку комбінації, де R являє 2 2 1 собою бензил, і R являє собою метил, продукт HCl.H-D-Glu-(γ-D-Trp-OR )-α-OR осаджується з дихлорметану і може бути видалений фільтрацією. Обробка кислотно-адитивної солі гідроксидом металу в інертному розчиннику, такому як метанол, з метою однофазного гомогенного гідролізу, абометил-трет-бутиловим ефіром з метою двохфазного гідролізу дає основно-адитивну сіль H-D-iGlu-D-Trp-OH в розчині. При екстракції реакційної суміші розчинником, що не змішується, з водою, таким як етилацетат або метил-трет-бутиловий ефір, водну фракцію нейтралізують до pH від приблизно 6 до приблизно 7, і розчин упарюють для зменшення об'єму до співвідношення менш ніж приблизно 1 частина розчиненої речовини на 8 частин води. Розчинена речовина, як прогнозується за обчисленням ідентифікації складу, як показано на фіг. 8, являє собою основно-адитивну сіль (у формі монокарбоксилату) H-D-iGlu-DTrp-OH. Якщо натрію гідроксид використовують як гідроксид металу, розчинена речовина буде являти собою мононатрієву форму H-D-iGlu-D-Trp-OH у воді. Корекція pH цього розчину до значень від приблизно 2,5 до приблизно 3,0 буде приводити до осадження твердого тимодепресину, H-D-iGlu-D-Trp-OH. Моноамонійна сіль тимодепресину може бути одержана безпосередньо з дипептиду Boc-DiGlu-D-Trp-OH. Неочищену кислотно-адитивну сіль, таку як гідрохлоридна сіль, одержану у такий спосіб, як викладено вище, обробляють іонообмінною смолою для видалення неорганічної солі. Таким чином, розчин неочищеного тимодепресину гідрохлориду розчиняють у воді, і pH становить від приблизно 6 до приблизно 8. Розчин обробляють іонообмінною смолою. Переважно смола являє собою смолу на базі сульфонової кислоти. Прикладом є AMBERLYST® 15. Неорганічну сіль видаляють промиваючи водою до pH від приблизно 5,7 до приблизно 7. Аміак використовують як регенерувальний засіб для добування амонійної солі тимодепресину із смоли. Переважно використовують концентрований розчин аміаку та ізопропанол як регенерувальний засіб. Переважне співвідношення являє собою концентрований розчин аміаку та ізопропанол в співвідношенні від приблизно 1 до приблизно 3-4, з кінцевим промивним концентрованим розчином аміаку та ізопропанолу в співвідношенні 1 частина концентрованого розчину аміаку/1 частина води/2 частини ізопропанолу. Розчин аміаку для промивання випарюють при зниженому тиску до стану масла, яке кристалізують з використанням ізопропанолу та води з одержанням моноамонійної солі у вигляді білої твердої речовини. Переважне співвідношення ізопропанолу та води для перекристалізації знаходиться в інтервалі від приблизно 5:1 до приблизно 10:1. Необхідність у колонковій хроматографії відсутня. В попередньому рівні техніки не існує способу очищення тимодепресину, окрім зворотнофазової препаративної рідинної хроматографії. Цей метод є надзвичайно тривалим і дорогим, і не може бути адаптований до великомасштабного виробництва. Заявником було визначено, що неочищений тимодепресин може бути також очищений до фармацевтичної категорії чистоти флеш-хроматографією на силікагелі з використанням ізопропанолу і води як елюенту. Переважна рухома фаза являє собою суміш ізопропанол/вода (від приблизно 10:1 до приблизно 5:1). Продукт виділяють традиційними засобами. У подібний спосіб, моноамонійна сіль також може бути очищена флеш-хроматографією на силікагелі з використанням ізопропанолу і концентрованого розчину аміаку як елюенту. Переважна рухома фаза являє собою суміш ізопропанол/розчин аміаку (від приблизно 10:1 до приблизно 5:1). Продукт виділяють традиційними засобами. Моноамонійна сіль D-ізоглутаміл-D-триптофану, одержана в результаті кристалізації з використанням ізопропанолу і води, являє собою кристалічну речовину. З іншого боку, якщо розчин моноамонійної солі D-ізоглутаміл-D-триптофану сушать виморожуванням, одержують аморфний матеріал. Обширне дослідження проводилося для підтвердження того, що рацемізація хіральних центрів в ході очищення на колонці і послідовності реакцій відсутня; деталі показані в прикладах нижче. У відповідності до даного винаходу, пропонується спосіб синтезу Boc-D-iGlu-D-Trp-OH, вільного від альфа-ізомеру аміду. Пропонується спосіб перетворення Boc-D-iGlu-D-Trp-OH на кислотно-адитивну сіль тимодепресину, зокрема, гідрохлоридну сіль. Графік ідентифікації складу надає спосіб осадження тимодепресину в чистій формі при pH приблизно 3 у воді. Крім того, пропонується спосіб очищення тимодепресину з чистотою менш ніж 97 % флешхроматографією на колонці з використанням ізопропанолу і води як елюенту. Інший аспект 16 UA 108058 C2 5 10 15 20 25 30 35 40 45 50 55 60 даного винаходу включає традиційний спосіб одержання моноамонійної солі з солі тимодепресину гідрохлориду. Неорганічну сіль видаляють іонообмінною смолою, і моноамонійну сіль виділяють з використанням регенерувального розчину на базі аміаку. Моноамонійна сіль може бути одержана кристалізацією в чистій формі. Також пропонується спосіб очищення моноамонійної солі нижчої чистоти флеш-хроматографією на колонці з силікагелем, з використанням ізопропанолу і води як елюенту. Додатково, спосіб синтезу тимодепресину, розкритий в попередньому рівні техніки (патент США 5,736,519) дає неочищений тимодепресин, який повинен бути очищений іонообмінною хроматографією і зворотно-фазовою препаративною рідинною хроматографією. Відокремлення альфа-амідного продукту H-D-Glu-D-Trp-OH від гама-амідного продукту тимодепресину D-I-DGlu-D-Trp-OH залишається найбільш серйозною проблемою виробництва. Послідовність очищення, наведена в попередньому рівні техніки, є непридатною для великомасштабного виробництва. Придатність і введення Динатрієву сіль тимодепресину застосовували для лікування псоріазу. Таким чином кристалічний тимодепресин і моноамонійна сіль тимодепресину за даним винаходом можуть бути введені в фармацевтичні композиції для введення суб'єкту в терапевтично активній кількості і в біологічно сумісній формі, придатній для введення in vivo, тобто, формі пептидів для введення, в якій терапевтичний ефект переважує будь-які токсичні ефекти. У відповідності до графіку ідентифікації складу, як показано на фіг. 8, переважними формами при нейтральному pH є монокарбоксилатна форма тимодепресину, тобто мононатрієва сіль дипептиду D-ізоглутаміл-D-триптофану, якщо протиіон являє собою натрій. Динатрієва сіль D-ізоглутаміл-D-триптофану є надзвичайно гігроскопічною і дуже складна з точки зору обробки для відмірювання. Кристалічний тимодепресин за даним винаходом виявляє такі характеристики РСАП, як детально показано на фіг. 1, і його розчинність у воді становить приблизно 20 мг/мл. Сполука є ідеальним кандидатом для заміни динатрієвої солі у виготовленні різноманітних препаратів. Хоча pH розчину D-ізоглутаміл-D-триптофану становить приблизно 3, може бути здійснена корекція значення pH за допомогою розчину гідроксиду натрію, карбонату натрію або бікарбонату натрію до pH від приблизно 7 до приблизно 7,4. Моноамонійна сіль за даним винаходом існує як в кристалічній, так і в аморфній форми. Обидві форми моноамонійної солі виявляють вкрай високу розчинність у воді. Таким чином, вони також є придатними кандидатами для рецептур. Введення нового кристалічного тимодепресину та/або його моноамонійної солі, як описано в даному описі, може бути здійснена будь-яким із загальноприйнятих способів введення для терапевтичних лікарських засобів із системною активністю. Вказані способи включають лікарські форми для перорального, парентерального та інших видів системного або місцевого застосування або застосування у формі аерозолю. В залежності від передбаченого способу введення, композиції для застосування можуть існувати у формі твердих, напівтвердих або рідких лікарських форм, таких як таблетки, супозиторії, пігулки, капсули, порошки, рідини, аерозолі, суспензії і т. п., переважно в дозованих лікарських формах, придатних для одноразового введення точних доз. Композиції будуть містити як мінімум один традиційний фармацевтичний носій або допоміжну речовину і кристалічний тимодепресину або його фармацевтично прийнятну моноамонійну сіль, і додатково можуть містити інші лікарські засоби, фармацевтичні засоби, носії, ад'юванти і т. п. Для твердих композицій можуть бути використані традиційні нетоксичні тверді носії, що включають, наприклад, маніт, лактозу, крохмаль, магнію стеарат, натрію сахаринат, тальк, целюлозу, глюкозу, сахарозу, магнію карбонат і т. п., фармацевтичної категорії. Активна сполука, як визначається вище, може бути введена у супозиторії, з використанням, наприклад, поліалкіленгліколей, наприклад, пропіленгліколю, як носія. Придатні для фармацевтичного введення рідкі композиції можуть, наприклад, бути одержані шляхом розчинення, диспергування і т. п., активної сполуки, як визначено вище, і необов'язкових фармацевтичних ад'ювантів в носії, такому як вода, сольовий розчин, водний розчин глюкози, гліцерин, етанол і т. п., з утворенням в такий спосіб розчину або суспензії. За бажанням, фармацевтична композиція для введення може також містити незначні кількості нетоксичних допоміжних субстанцій, таких як зволожувальні або емульгуючі засоби, буферизуючі засоби і т. п., наприклад, натрію ацетат, сорбітану монолаурат, натрію триетаноламіну ацетат, триетаноламіну олеат і т. п. Фактичні способи одержання таких лікарських форм відомі або будуть очевидними для фахівця в даній галузі; наприклад, див. Remington: The Science and st Practice of Pharmacy, David B. Troy (Ed.), Lipincott Williams & Wilkins, Philadelphia, PA, 21 Edition, 2006. Композиція або рецептура для введення будуть, в будь-якому випадку, містити кількість 17 UA 108058 C2 5 10 15 20 25 30 35 40 45 50 55 60 активної(и) сполуки(ук) в кількості, ефективній для полегшення симптомів у суб'єкта, що підлягає лікуванню. Парентеральне введення загалом характеризується ін'єкцією, підшкірною, внутрішньом'язовою або внутрішньовенною. Ін'єкційні засоби можуть бути виготовлені в традиційних формах, таких як рідкі розчини або суспензії, твердих формах, придатних для виготовлення розчину або суспензії в рідині перед ін'єкцією, або як емульсії. Придатними допоміжними речовинами, наприклад, є вода, сольовий розчин, глюкоза, гліцерин, етанол і т. п. Крім того, за бажанням, фармацевтичні композиції для введення можуть також містити незначні кількості нетоксичних допоміжних речовин, таких як зволожуючі або емульгуючі засоби, буферизуючі засоби і т. п., такі як натрію ацетат, сорбітану монолаурат, триетаноламіну олеат і т. п. Для тимодепресину або його моноамонійної солі переважним є пероральне або назальне (інтрабронхіальне) введення, в залежності від природи розладу, що підлягає лікуванню. Для перорального введення фармацевтично прийнятну нетоксичну композицію одержують, поєднуючи будь-яку із звичайно використовуваних допоміжних речовин фармацевтичної категорії, таку як маніт, лактоза, крохмаль, магнію стеарат, натрію сахаринат, тальк, целюлоза, глюкоза, сахароза, магнію карбонат і т. п. Такі композиції набувають форми розчинів, суспензій, таблеток, пігулок, капсул, порошків, рецептур подовженого вивільнення і т. п. Такі композиції можуть містити від приблизно 1 % до приблизно 95 % активного інгредієнта, переважно від приблизно 25 % до приблизно 70 %. Пероральне і назальне введення в легені може також здійснюватися за допомогою аерозольних лікарських форм. Для аерозольного введення, активний інгредієнт переважно поставляється в тонко подрібненій формі разом з поверхнево-активною речовиною і пропелентом. Типовий відсоток активних інгредієнтів становить від приблизно 0,01 до приблизно 20 % (мас.), переважно, від приблизно 0,04 % до приблизно 1,0 %. Поверхнево-активні речовини звичайно повинні бути нетоксичними, і переважно розчинними в пропеленті. Характерним прикладом таких засобів є естери або часткові естери жирних кислот, що містять від 6 до 22 атомів вуглецю, такі як капронова, каприлова, лауринова, пальмітинова, стеаринова, лінолева, ліноленова, олестеаринова та олеїнова кислоти з багатоатомним аліфатичним спиртом або його циклічним ангідридом, таким як етиленгліколь, гліцерин, еритритол, арабітол, маніт, сорбіт, ангідриди гекситолу, одержані з сорбіту (естери сорбітану, присутні на ринку під назвою SPAN®) і поліоксиетиленові та поліоксипропіленові похідні вказаних естерів. Можуть використовуватися змішані естери, такі як змішані або природні гліцериди. Переважними поверхнево-активними агентами є олеати або сорбітан, наприклад, присутні на ринку під назвами ARLACEL®C (сорбітану сесквіолеат), SPAN®80 (сорбітану моноолеат) і SPAN®85 (сорбітану триолеат). Вміст поверхнево-активної речовини може складати від з приблизно 0,1 % до приблизно 20 % (мас.) композиції, переважно, від приблизно 0,25 % до приблизно 5 %. Решта композиції звичайно являє собою пропелент. Зріджені пропеленти звичайно є газоподібними в умовах навколишнього середовища і конденсуються під тиском. Серед придатних зріджених пропелентів - нижчі алкани, що містять до п'яти атомів вуглецю, такі як бутан і пропан; і, переважно, фторовані або фторхлоровані алкани, такі як присутні на ринку під назвою FREON®. Суміші вищезгаданих компонентів також можуть бути використані. У виробництві аерозолю, ємність, обладнану придатним клапаном, заповнюють відповідним пропелентом, що містить тонко подрібнений активний інгредієнт і поверхнево-активну речовину. Таким чином, інгредієнти утримуються при підвищеному тиску до викиду при спрацьовуванні клапана. Для місцевого застосування такі композиції містять ефективну кількість сполуки даного класу в суміші як мінімум з одним фармацевтично прийнятним нетоксичним носієм. Придатний інтервал композиції становить від приблизно 0,1 % до приблизно 10 % активного інгредієнта, і решта являє собою носії; переважно, від приблизно 1 % до приблизно 2 % активного інгредієнта. Концентрація активного інгредієнта у фармацевтичних композиціях, придатних для місцевого застосування, буде варіювати в залежності від конкретної активності сполуки, яку використовують в зв'язку з присутнім станом і суб'єктом, який підлягає лікуванню. Придатні носії або розчинники лікарського засобу для місцевого застосування вказаних сполук включають креми, мазі, лосьйони, емульсії, розчини і т. п. Наприклад, придатна мазь для місцевого застосування сполук за даним винаходом містить від приблизно 15 до приблизно 45 % насиченого жирного спирту, що містить 16–24 атоми вуглецю, такого як цетиловий спирт, стеариловий спирт, бегеніловий спирт і т. п., і від приблизно 45 до приблизно 85 % (мас.) гліколевого розчинника, такого як пропіленгліколь, 18 UA 108058 C2 5 10 15 поліетиленгліколь, дипропіленгліколь і суміші вказаних компонентів. Мазь може також містити від приблизно 0 до приблизно 15 % (мас.) пластифікатора, такого як поліетиленгліколь, 1,2,6гексантріол, сорбіт, гліцерин і т. п.; від приблизно 0 до приблизно 15 % (мас.) зв'язувального засобу, такого як насичені жирні кислоти, що містять від 16 до 24 атомів вуглецю, наприклад, стеаринова кислота, пальмітинова кислота, бегенова кислота, аміди жирних кислот, наприклад, олеамід, пальмітамід, стеарамід, бегенамід, і естери жирних кислот, що містить 16–24 атоми вуглецю, такі як сорбіту моностеарат, поліетиленгліколю моностеарат, поліпропіленгліколь, або відповідний моноестер інших жирних кислот, таких як олеїнова кислотна і пальмітинова кислота; і від приблизно 0 до приблизно 20 % (мас.) засоби, що підсилює проникнення, такого як диметилсульфоксид або диметилацетамід. Терапевтично активна кількість кристалічного тимодепресину або його амонійної солі може варіювати у відповідності до таких факторів, як патологічний стан, вік, стать і маса тіла індивідуума. Схема лікування може бути змінена таким чином, щоб забезпечити оптимальну терапевтичну відповідь. Загалом, схема щоденного введення повинна включати дозу в інтервалі від приблизно 1 до приблизно 200 мг пептиду. Далі наведені приклади характерних рецептур, які жодним чином не обмежують контекст виготовленні різноманітних фармацевтичних композицій: Інгредієнти Активний інгредієнт Лактоза, висушена розпиленням Крохмаль кукурудзяний Магнію стеарат 20 Кількість на таблетку (мг) 25 20 153 2 Наведені вище інгредієнти ретельно змішують і пресують в таблетки з однією рискою. Інгредієнти Активний інгредієнт Лактоза, висушена розпиленням Магнію стеарат Кількість на таблетку (мг) 100 148 2 Наведені вище інгредієнти змішують і вводять в тверду желатинову капсулу. Інгредієнти Активний інгредієнт Лактоза Крохмаль кукурудзяний Магнію стеарат Кількість на таблетку (мг) 200 145 50 5 25 Наведені вище інгредієнти ретельно змішують і пресують в таблетки з однією рискою. Інгредієнти Активний інгредієнт Лактоза Крохмаль кукурудзяний Магнію стеарат Кількість на таблетку (мг) 108 15 25 2 Наведені вище інгредієнти змішують і вводять в тверду желатинову капсулу. 30 Інгредієнти Активний інгредієнт Лактоза Кількість на таблетку (мг) 150 92 Наведені вище інгредієнти змішують і вводять в тверду желатинову капсулу. Ін'єкційний препарат, буферизований до pH приблизно 7, одержують з наступним складом: 19 UA 108058 C2 Інгредієнти Активний інгредієнт KH2PO4 KOH (1 н розчин) Вода (дистильована, стерильна) 0,2 г 2 мл необхідна кількість до pH 7 необхідна кількість до 20 мл Ін'єкційний препарат, буферизований до pH приблизно 7, одержують з наступним складом: Інгредієнти Активний інгредієнт Вода (дистильована, стерильна) NaOH (0,2 н) 0,01 г необхідна кількість до 1 мл необхідна кількість до pH 7 5 Суспензію для перорального застосування одержують з наступним складом: Інгредієнти Активний інгредієнт Фумарова кислота Метилпарабен Цукор Сорбіт (70 % розчин) VEEGUM® K (Vanderbilt Co.) Ароматизатор Барвники Вода дистильована 0,1 г 0,5 г 2,0 г 0,1 г 25,5 г 12,85 г 1,0 г 0,035 мл необхідна кількість до 100 мл Препарат для місцевого застосування: 10 Інгредієнти Активна сполука SPAN® 60 TWEEN® 60 Мінеральне масло Вазелін Метилпарабен Пропілпарабен БГА (бутильований гідроксианізол) Вода дистильована 15 20 25 30 Грами 0,2–2 2 2 5 10 0,15 0,05 0,01 необхідна кількість до 100 мл Всі наведені вище інгредієнти, окрім води, поєднують і нагрівають до приблизно 45 °C при перемішуванні. Далі додають достатню кількість води з температурою приблизно 45 °C при енергійному перемішуванні для емульгації інгредієнтів, і далі додають необхідну кількість води до 100 г. Нижче даний винахід пояснений детально з посиланням на Приклади, але даний винахід не обмежується ними в будь-якій спосіб. Приклад 1 Одержання N-α-трет-бутоксикарбоніл-γ-D-глутаміл(α-бензиловий естер)-D-триптофану метилового естеру або (2R)-трет-бутоксикарбоніламіно-(4R)-[2-(1H-індол-3-іл)-1метоксикарбоніл-етилкарбамоїл]-масляної кислоти бензилового естеру або N-трет-Boc-D-Glu-(γD-Trp-OMe)-α-OBzl). Методика 1A: Одержання чистого довідкового зразка N-трет-Boc-D-Glu-(γ-D-Trp-OMe)-α-OBzl з очищенням хроматографією на силікагелі. При перемішуванні до охолодженого на льоду розчину Boc-D-Glu-OBzl (6,00 г, 17,8 ммоль) в CH2Cl2 (70 мл) послідовно додають ЕДК (5,11 г, 26,6 ммоль), HOBt (3,60 г, 26,6 ммоль) та ДІПЕА (4,60 мл, 26,6 ммоль). Далі, по краплях додають розчин H-D-Trp-OMe.HCl (6,77 г, 26,6 ммоль) та ДІПЕА (4,60 мл, 26,6 ммоль) в CH2Cl2 (50 мл). Одержану суміш перемішують при температурі льоду (від -3 °C до 0 °C) протягом 1 год., після чого дозволяють нагрітися до кімнатної 20 UA 108058 C2 5 10 15 20 25 30 35 40 45 50 55 температури та перемішують протягом 16 год. Реакційну суміш упарюють до сухого стану. Залишок розподіляють між EtOAc та насиченим розчином NaHCO 3. Органічну фракцію збирають, промивають 10 % розчином лимонної кислоти, і далі сольовим розчином. Органічну фракцію сушать над натрію сульфатом, фільтрують та упарюють до одержання густого масла. Залишок очищують колонковою хроматографією на силікагелі з використанням градієнту суміші розчинників гексан/EtOAc (співвідношення 8:2, 7:3 і 3:7, об./об.) як елюенту, з одержанням 1 названого в заголовку продукту (9,40 г, 98 %) у вигляді білої твердої речовини. H ЯМР (ДМСОd6) δ проміле 10,86 (с, 1H), 8,31 (д, J=7,4 Гц, 1H), 7,49 (д, J=7,7 Гц, 1H), 7,31-7,35 (м, 7H), 7,14 (д, J=2,0 Гц, 1H), 7,06 (т, J=7,9 Гц, 1H), 6,98 (т, J=6,8 Гц, 1H), 5,12 (к, J=5,9 Гц, 2H), 4,5 (к, J=6,5 Гц, 1H), 3,96-4,03 (м, 1H), 3,55 (с, 3H), 2,98-3,16 (м, 2H), 2,18-2,24 (м, 2H), 1,86-1,95 (м, 1H), 1,71-1,80 13 (м, 1H), 1,37 (с, 9H); C ЯМР (ДМСО-d6) δ проміле: 172,4 (C), 172,3 (C), 171,4 (C), 155,6 (C), 136,1 (C), 136,0 (C), 128,4 (CH), 127,9 (CH), 127,7 (CH), 127,1 (C), 123,6 (CH), 120,9 (CH), 118,4 (CH), 117,9 (CH), 111,4 (CH), 109,5 (C), 78,2 (C), 65,8 (CH 2), 53,3 (CH), 53,16(CH), 51,7 (CH3), 31,3 + (CH2), 28,2 (CH3), 27,1 (CH2), 26,4 (CH2); МС (співвідношення маси до заряду) 538 [M+1] ; аналітично обчислено для C29H35N3O7, 0,5H2O: C, 63,72; H, 6,64; N, 7,69; знайдено: C, 63,79; H, 6,06; N, 7,65. Методика 1B: Одержання чистого зразка N-трет-Boc-D-Glu-(γ-D-Trp-OMe)-α-OBzl перекристалізацією. Суспензію Boc-D-Glu-OBzl (60,26 г, 178,6 ммоль) в CH2Cl2 (335 мл) охолоджують приблизно до -1 °C та перемішують протягом 15 хв. Далі, послідовно додають ДІПЕА (46,70 мл, 268,0 ммоль), HOBt (36,20 г, 268,0 ммоль), ЕДК (51,38 г, 268,0 ммоль). Після цього, по краплях додають розчин H-D-Trp-OMe.HCl (68,25 г, 268,0 ммоль) та ДІПЕА (46,70 мл, 268,0 ммоль) в CH2Cl2 (187 мл). Одержану суміш перемішують при температурі льоду (від -1 °C до -5 °C) протягом 2 год., після чого дозволяють нагрітися до кімнатної температури та перемішують протягом ночі в атмосфері азоту. Реакційну суміш упарюють до сухого стану. Залишок розподіляють між EtOAc (200 мл), насиченим розчином Na2CO3 (100 мл) та H2O (150 мл). Водну фракцію знову екстрагують етилацетатом (200 мл). Органічні фракції збирають, промивають H 2O (100 мл), 10 % розчином лимонної кислоти (2 × 200 мл) і сольовим розчином (60 мл). Органічну фракцію сушать над натрію сульфатом, фільтрують та упарюють до сухого стану. Залишок розчиняють в етилацетаті (141 мл), потім додають гексан (106 мл). Одержану суспензію перемішують протягом 6 год. та фільтрують. Тверду речовину ретельно промивають гексаном (100 мл), потім сушать під вакуумом в печі при 40 °C, витримуючи при цій температурі протягом ночі. 1 Одержують тверду речовину практично білого кольору (81,67 г, 85 %). Дані H ЯМР підтверджують структуру (див. Приклад 1, Методика 1A). Методика 1C: Одержання чистого зразка N-трет-Boc-D-Glu-(γ-D-Trp-OMe)-α-OBzl без хроматографічного очищення та визначення синтетичних домішок. Boc-D-Glu-OBzl (48,0 г, 142,2 ммоль) розчиняють в 270 мл дихлорметану і далі охолоджують до 0-5 °C з використанням льодяної бані. Додають HOBt (23,8 г, 156,4 ммоль) з наступним додаванням ДІПЕА (27,0 мл, 156,4 ммоль) та перемішують протягом 10 хв. ЕДК (38 г, 199,1 ммоль) та попередньо змішаний розчин H-D-Trp-OMe (виготовлений з H-D-Trp-OMe.HCl [39,7 г, 156,4 ммоль] та ДІПЕА [27,0 мл, 156,4 ммоль] в 150 мл дихлорметану, перемішують при кімнатній температурі протягом 20 хв.) послідовно додають до розчину. Реакційну суміш перемішують при 0 °C, витримуючи при цій температурі протягом 2 год., і потім протягом ночі при кімнатній температурі. Реакційну суміш виливають на 250 мл дистильованої води та екстрагують. Органічну фракцію промивають 250 мл 10 % розчину лимонної кислоти, 2 × 5 % розчином NaHCO3 і сольовим розчином. Органічну фракцію сушать над натрію сульфатом та упарюють під вакуумом з одержанням блідо-жовтої, пінистої твердої речовини. Тверду речовину розчиняють в приблизно 250 мл етилацетату та упарюють до сухого стану. Операцію здійснюють двічі з утворенням воскоподібної твердої речовини. До твердого матеріалу додають 100 мл етилацетату та перемішують при кімнатній температурі. Суміш перемішують з помірною або великою швидкістю до утворення суспензії з консистенцією рідкого тіста - цей процес займає приблизно 45 хв. (перемішування протягом тривалого періоду часу може призводити до затвердіння розчину у вигляді желатиноподібної субстанції). Далі, додають 75 мл гексану, і суміш перемішують ще протягом 10 хв. В цій точці додають ще 20 мл етилацетату, і суспензію фільтрують негайно з одержанням пухнастої твердої речовини блідорожевого кольору. Тверду речовину негайно промивають тричі по 30 мл гексану, що допомагає видалити рожевувате забарвлення. Фільтрат збирають та дозволяють стояти нерухомо 21 UA 108058 C2 5 10 15 протягом 40 хв. Гранульована тверда речовина осаджується з фільтрату. Суміш фільтрують, і тверду речовину тричі промивають по 10 мл гексану. Фільтрат збирають та упарюють до твердого стану. Тверду речовину розчиняють в 20 мл етилацетату та перемішують до утворення суспензії з консистенцією рідкого тіста. Далі, додають 40 мл гексану, і суміш перемішують протягом 5 хв. Суміш фільтрують і зібрану тверду речовину промивають гексаном. Об'єднані тверді речовини сушать протягом ночі в печі (35 °C) під вакуумом до постійної маси. Таким чином, одержують 59,0 г (77,2 %) названої сполуки. Тпл: 1 83,1-87,5 °C. Дані H ЯМР були ідентичними описаним у Прикладі 1, Методика 1A. Чистота за даними РХВТ (% площі піку): 97,2 %; час утримання: 7,56 хв.; умови РХВТ: колонка Waters Symmetry C 18, 3,9 × 150 мм, 5 мкм; рухома фаза: 0,035 % HClO4, pH 2/CH3CN, градієнт (хв.- % CH3CN) 0–35, 10–90, 12–90; швидкість потоку: 1 мл/хв.; λ: 230, 260, 280 нм. Аналіз домішок в маточному розчині. Результати аналізу маточного розчину методом ТШХ (суміш етилацетат/гексан 50:50) показані в таблиці нижче. Обидві плями A та B флуоресціюють в УФ-світлі, але дають негативний результат нінгідринових тестів. Плями продукту і початкова лінія дають позитивні результати нінгідринових тестів. Зразки A та B виділяють колонковою хроматографією на 1 силікагелі, і їх структура з'ясована за даними H ЯМР та МС/МС. Структура A та B показана нижче. Значення Rf в суміші етилацетат/гексан 50:50: 20 Пляма A B Продукт Значення Rf 0,80 0,60 0,39 N N N N N N N N N OH Spot A 25 30 35 40 45 Spot B Написи на схемі: Spot = Пляма Плями A та B відповідають домішкам, пов'язаним з HOBt. Домішки можуть бути видалені перекристалізацією. Будь-які слідові домішки можуть бути видалені на наступній стадії гідролізу. Приклад 2 Одержання N-α-трет-бутоксилкарбоніл-D-ізоглутаміл-D-триптофану або (2R)-третбутоксикарбоніламіно-(4R)-[1-карбокси-2-(1H-індол-3-іл)-етилкарбамоїл]-масляної кислоти або N-трет-α-Boc-D-iGlu-D-Trp-OH. Методика 2A: Однофазний гідроліз за допомогою NaOH. При перемішуванні до розчину N-трет-Boc-D-Glu-(γ-D-Trp-OMe)-α-OBzl (3,7 г, 6,9 ммоль) з Прикладу 1, Методика 1A, в метанолі (40 мл), додають розчин NaOH (1,0 г, 25 ммоль) в H 2O (20 мл). Одержаний розчин перемішують при кімнатній температурі протягом ночі. Реакційну суміш виливають в 1 н розчин NaOH (100 мл), і водну суміш промивають етилацетатом (2 100 мл). Водну фракцію підкислюють 3 н розчином HCl, після чого екстрагують етилацетатом (2 50 мл). Органічні фракції об'єднують, сушать над натрію сульфатом та упарюють до сухого стану при 1 зниженому тиску. Одержують тверду речовину білого кольору (2,7 г, 91 %). Тпл: 148-158 °C; H ЯМР (ДМСО-d6) δ проміле: 12,47 (ш, 2H), 10,82 (с, 1H), 8,21 (д, J=7,8 Гц, 1H), 7,53 (д, J=7,8 Гц, 1H), 7,34 (д, J=8,1 Гц, 1H), 7,13 (д, J=2,0 Гц, 1H), 7,06 (т, J=7,5 Гц, 2H), 6,98 (т, J=7,4 Гц, 1H), 4,46 (к, J=5,3 Гц, 1H), 3,88-3,83 (м, 1H), 3,17-2,97 (дд, J=5,2 та 8,4 Гц, 2H), 2,23-2,10 (м, 2H), 1,90-1,82 13 (м, 1H), 1,75-1,68 (м, 1H), 1,38 (с, 9H); C ЯМР (ДМСО-d6) δ проміле: 173,9 (C), 173,4 (C), 171,5 (C), 155,6 (C), 136,1 (C), 127,2 (C), 123,5 (CH), 120,9 (CH), 118,4 (CH), 118,2 (CH), 111,4 (CH), 109,9 (C), 78,0 (C), 53,1 (CH), 52,9 (CH), 31,7 (CH 2), 28,2(CH3), 27,2 (CH2), 26,7 (CH2); ІЧ-ФП (KBr) -1 ν: 3415, 3338, 2986, 1719, 1686, 1654, 1534, 1424, 1366, 1252, 1169, 1069, 744, 634, 429 см ; МС + (співвідношення маси до заряду) 434 [M+1] . 22 UA 108058 C2 5 10 15 20 25 30 35 40 45 50 55 60 Методика 2B: Однофазний гідроліз за допомогою LiOH. При перемішуванні до охолодженого на льоді (від 0 °C до 5 °C) розчину Boc-D-Glu-(γ-D-TrpOCH3)-α-OBzl (46,06 г, 85,68 ммоль) в метанолі (200 мл) додають розчин LiOH (10,78 г, 257,0 ммоль) в H2O (136 мл). Одержаний розчин перемішують та утримують при температурі від 0 °C до 10 °C, витримуючи при цій температурі протягом 3 год. Реакційну суміш виливають у насичений розчин Na2CO3 (100 мл) та H2O (150 мл), водну суміш промивають етилацетатом (2 150 мл). Водну фракцію підкислюють до pH=2–3 за допомогою 3 н розчину HCl, після чого екстрагують етилацетатом (2 200 мл). Органічні фракції об'єднують, сушать над натрію сульфатом та упарюють до сухого стану при зниженому тиску. Одержують тверду речовину 1 білого кольору (36,65 г, 98,7 %). Дані H ЯМР та МС/МС підтверджують структуру (див. Приклад 2, Методика 2A). Методика 2C: Одержання Boc-D-iGlu-D-Trp-OH без хроматографічного очищення з використанням двохфазного процесу гідролізу. Літію гідроксид (4,1 г, 97,7 ммоль) розчиняють в 35 мл дистильованої води. Далі, додають 65 мл метил-трет-бутилового ефіру (МТБЕ) з наступним додаванням дипептиду Boc-D-Glu-(γ-DTrp-OCH3)-α-OBzl, (25 г, 46,5 ммоль), одержаного, як описано у Прикладі 1. Негайно утворюється дуже густа суспензію, і 15 мл метанолу та 15 МТБЕ додають при енергійному перемішуванні. Додають ще 2 мл метанолу, і тверду речовину повільно розчиняють протягом приблизно 5 хв. Одразу після розчинення матеріалу розчин набуває жовтого/зеленого кольору, тоді як верхня органічна фракція набуває блідо-зеленого кольору, і водна фракція - жовтого кольору. Реакційну суміш енергійно перемішують при кімнатній температурі протягом 80 хв., і в цій точці часу в органічній фракції не залишається початкового матеріалу, та водна фракція містить продукт (контроль методом ТШХ: суміш EtOAC/гексан [1:1], об./об.). Розчин виливають у ділильну лійку, і 2 фракції розділяють. Органічну фракцію промивають 15 мл води. Органічна фракція набуває рожевого кольору при промиванні водою. Об'єднану водну фракцію промивають двічі по 30 мл етилацетату. Водну фракцію підкислюють приблизно до pH 2 додаванням по краплях 16,6 мл 6 н кислоти хлористоводневої при кімнатній температурі. Водну фракцію екстрагують двічі по 50 мл етилацетату. Максимальну кількість метанолу додають в ході другої екстракції для сприяння розчинності продукту в органічній фракції. Об'єднані органічні фракції сушать над натрію сульфатом та упарюють під вакуумом з жовтої рідини з одержанням твердої речовини білого кольору. Тверду речовину сушать протягом ночі в печі (28 °C) під вакуумом до постійної маси. 1 Таким чином одержують 18,6 г (вихід 92 %) названої сполуки. Тпл: 179,0-184,6 °C; Дані H ЯМР були ідентичними описаним у Прикладі 2A; чистота за даними РХВТ (% площі піку): 98,3 %; час утримання: 5,33 хв.; умови РХВТ: колонка Waters Symmetry C 18, 3,9 × 150 мм, 5 мкм; рухома фаза: 0,035 % HClO4, pH 2/CH3CN, градієнт (T, хв.- % CH3CN) 0-20, 10-90, 12-90; швидкість потоку: 1 мл/хв.; λ:230, 260, 280 нм. Контроль вищезгаданої реакції може бути здійснений за допомогою РХВТ. В Методиці 2C вище, присутність бензилового спирту, що утворюється в результаті гідролізу фрагменту бензилового естеру, можна спостерігати за допомогою ТШХ (суміш EtOAC/гексан [1:1, об./об.] як елюент). Бензиловий спирт утворюється в результаті гідролізу фрагменту бензилового естеру. Домішка, що відповідає першій плямі, є такою ж, як у Прикладі 1, Методика 1C. Контроль за допомогою РХВТ та аналіз методом РХ/МС показують, що Boc-D-Glu-(γ-D-Trp-OCH3)-α-OBzl спочатку реагує з основою і метанолом з утворенням Boc-D-Glu-(γ-D-Trp-OCH3)-α-OCH3, який далі швидко гідролізується з одержанням двохосновної кислоти, Boc-D-Glu-(γ-D-Trp-OH)-OH або Boc-D-iGlu-D-Trp-OH. Приклад 3: Одержання D-ізоглутаміл-D-триптофану. Методика 3A: Одержання D-ізоглутаміл-D-триптофану та його очищення перекристалізацією. Boc-D-iGlu-D-Trp-OH (20,0 г, 46,14 ммоль, з Прикладу 2) вміщують до 3-горлої круглодонної колби об'ємом 1 л, обладнаної магнітною мішалкою. Додають етилацетат (300 мл) і одержану суспензію охолоджують до -10 °C в льодяній бані. Газоподібний HCl пропускають крізь холодну суспензію. Інтервал температури від -4 °C до -10 °C підтримують в ході реакції, і прогрес реакції контролюють за допомогою РХВТ. У визначеній точці гетерогенна реакційна суміш перетворюється на прозорий світло-рожевий гомогенний розчин. Після перетворення початкового матеріалу реакційна суміш знову перетворюється на суспензію. Леткі матеріали далі видаляють під вакуумом з одержанням твердої речовини світло-рожевого кольору. Тверду 23 UA 108058 C2 5 10 15 20 25 30 35 40 45 50 55 60 речовину розчиняють в 60 мл води деіонізованої, і одержаний розчин промивають дихлорметаном (2 × 25 мл). pH водного розчину далі доводять приблизно до 3,0 додаванням NaOH (10 M, приблизно 3,6 мл) при охолодженні. Одержаний розчин фільтрують для видалення будь-яких залишкових частинок твердої речовини. Фільтрат збирають та енергійно перемішують. Тверду речовину відокремлюють фільтрацією. Фільтрат залишають для подальшого використання. Тверду речовину далі вміщують знову до круглодонної колби і додають 30 мл демінералізованої води. Суміш енергійно перемішують, і тверду речовину відокремлюють фільтрацією. Фільтрат залишають для подальшого використання. Тверду речовину далі промивають льодяно-холодною демінералізованою водою (4 × 15 мл). Третій промивний водний розчин не містить хлоридів, що підтверджується негативним результатом випробування з використанням AgNO3 (4 % розчин). Тверду речовину сушать у повітрі, після чого вміщують до вакуумної печі при 36 °C, витримуючи при цій температурі протягом ночі з одержанням 8,5 г (чистота за даними РХВТ, % площі піку: 98,3 %). Фільтрати з описаних вище стадій об'єднують і здійснюють таку ж процедуру перекристалізації з одержанням ще 3,2 г продукту (чистота за даними РХВТ, % площі піку: 98,7 %). Об'єднаний вихід для 2 порцій становить 11,7 г (75 %). Додаткова обробка кінцевого фільтрату дає третю порцію (1,0 г, чистота за даними РХВТ, % площі піку: 82,0 %). 1 H ЯМР (D2O-NaOD, pH 7,0) δ проміле: 7,64 (д, J=7,9 Гц, 1H), 7,43 (д, J=8,1 Гц, 1H), 7,19-7,16 (м, 2H), 7,10 (т, J=7,4 Гц, 1H), 4,52-4,48 (м, 1H), 3,48 (т, J=6,1 Гц, 1H), 3,34-3,29 (м, 1H), 3,08-3,02 (м, 1H), 2,30-2,17 (м, 2H), 1,92-1,75 (м, 2H). Спектр рентгеноструктурного аналізу порошку для одержаного матеріалу показано на фіг. 1. Метод РХВТ: колонка XTerra МС C18; 5 мкм, 4,6 × 250 мм; рухома фаза: A = водна фракція: 4 мМ Трис, 2 мМ ЕДТА, pH 7,4; B = органічна фракціа: CH3CN; програма градієнта: B %: 0 хв. 5 %, 15 хв. 55 %, 30 хв. 55 %, 32 хв. 5 %, 40 хв. 5 %. Швидкість потоку = 1 мл/хв.; об'єм ін'єкції = 5 мкл; λ: 222, 254, 282, 450 нм; час утримання продукту = 6,4 хв. Методика 3B: Етилацетат (250 мл), попередньо охолоджений до 0 °C, насичують газоподібним HCl протягом 25 хв. Додають Boc-D-iGlu-D-Trp-OH (15,0 г, 34,6 ммоль) з утворенням суспензії. Розчин перемішують протягом 90 хв. при температурі льодяної бані. Розчинник випарюють під вакуумом з утворенням білої твердої речовини. Тверду речовину розчиняють в 35 мл дистильованої води з утворенням густого розчину світло-коричневого кольору. Водну фракцію промивають по 30 мл дихлорметану, після чого переносять в хімічний стакан об'ємом 100 мл. З використанням pH-електроду для контролю кислотності, pH доводять до значень від 1,28 до 2,96 з використанням 3,2 мл 10 н розчину NaOH. Розчин перемішують протягом 1 год. при кімнатній температурі, повільно утворюється білий осад. Тверду речовину відокремлюють вакуумною фільтрацією і ретельно промивають водою. Неочищену тверду речовину суспендують в 20 мл дистильованої води, перемішуючи протягом 2 год. при кімнатній температурі. Суміш фільтрують, тверду речовину збирають і сушать до постійної маси в печі під вакуумом протягом ночі (40 °C). Таким чином, одержують 8,6 г (вихід 74,5 %) названої сполуки. чистота за даними РХВТ (% площі піку): 98,8 %. Час утримання: 4,21 хв.; умови РХВТ: колонка Waters Symmetry C 18, 3,9 × 150 мм, 5 мкм; рухома фаза: 0,035 % HClO4, pH 2/CH3CN, градієнт 1 (T, хв.- % CH3CN) 0-10, 10-90, 12-90; швидкість потоку: 1 мл/хв.; λ: 230, 260, 280 нм; дані H ЯМР відповідають структурі. Приклад 4: Синтез моноамонійної солі D-ізоглутаміл-D-триптофану (1:1) та її виділення за допомогою очищення колонковою хроматографією. Методика 4A: Газоподібний HCl пропускають при перемішуванні крізь охолоджений на льоду (0–5 °C) розчин Boc-D-Glu-D-Trp-OH (2,5 г, 5,8 ммоль) в етилацетаті (60 мл) протягом 2,5 год. Реакційну суміш далі упарюють до сухого стану. Очищення залишку колонковою хроматографією на силікагелі з використанням градієнту суміші розчинника ізопропанолу та гідроксиду амонію (28– 30 % NH4OH) (співвідношення 8:2 і 7:3, об./об.) як елюенту дає названий в заголовку продукт (1,8 г, 84,7 %) у вигляді білої твердої речовини після випарювання розчинника. Тпл: 124–128 °C; 1 H ЯМР (ДМСО-d6) δ проміле: 10,98 (с, 1H), 8,25 (д, J=5,9 Гц, 1H), 7,53 (7,8 Гц, 1H), 7,30 (д, J=8,0 Гц, 1H), 7,17 (с, 1H), 7,00 (т, J=7,7 Гц, 1H), 6,92 (т, J=7,2 Гц, 1H), 4,28 (м, 1H), 3,22-3,31 (м, 2H), 13 2,90-2,96 (м, 1H), 2,23-2,25 (м, 2H), 1,97-1,98 (м, 1H), 1,84-1,86 (м, 1H); C ЯМР (ДМСО-d6) δ проміле: 175,5 (C), 171,6 (C), 171,4 (C), 136,0 (C), 127,6 (C), 123,5 (CH), 120,5 (CH), 118,3 (CH), 14 117,9 (CH), 111,5 (C), 111,3 (CH), 55,3 (CH), 53,7 (CH), 32,5 (CH2), 27,8 (CH2), 27,4 (CH2); N ЯМР (D2O) δ проміле: 20,4 (с); ІЧ-ФП (KBr) ν: 3406, 3055, 1581, 1456, 1399, 1341, 1096, 1009, 744, 535, 24 UA 108058 C2 -1 5 10 15 20 25 30 35 40 45 50 55 60 + 426 см ; МС (співвідношення маси до заряду) 334 [двохосновна кислота+1] ; аналітично обчислено для C16H22N4O5.H2O: C, 52,17; H, 6,57; N, 15,21; знайдено: C, 51,95; H, 6,84; N, 14,85. Речовина являє собою моноамонійну сіль D-ізоглутаміл-D-триптофану (1:1). Одержаний матеріал аморфний, що підтверджено даними РСАП. Методика 4B: Газоподібний HCl конденсують у холодному етилацетаті (133,4 г) при температурі -2 °C (температура зовнішньої льодяної бані) протягом 16 хв. Збільшення маси розчину становить 21 г. Boc-D-iGlu-D-Trp-OH (3,8 г, 8,73 ммоль) розчиняють в 50 мл вищеописаного розчину. Температуру одержаної суміші підтримують в інтервалі 0–5 °C протягом 55 хв. Реакційну суміш контролюють методом ТШХ, після чого упарюють при зниженому тиску (температура роторного випарювача: 51–52 °C) до сухого стану. Очищення флеш-хроматографією на силікагелі з використанням градієнту суміші розчинників ізопропанолу та розчину гідроксиду амонію (2830 %) (співвідношення 8:2 і 7:3, об./об.) як елюенту дає продукт (2,0 г, 62 %) у вигляді твердої 1 речовини майже білого кольору. Дані H ЯМР є подібними до даних, наведених у Прикладі 4, Методика 4A. Одержаний матеріал аморфний, що підтверджено даними РСАП. Приклад 5 Синтез моноамонійної солі D-ізоглутаміл-D-триптофану (1:1). Методика 5A: Синтез моноамонійної солі D-ізоглутаміл-D-триптофану (1:1) шляхом видалення неорганічних солей з використанням смоли AMBERLYST®15 з наступним очищенням колонковою хроматографією. Газоподібний HCl пропускають при перемішуванні крізь охолоджену льодом (0–5 °C) суспензію одержаного Boc-D-iGlu-D-Trp-OH (10,82 г, 24,96 ммоль) в етилацетаті (200 мл) протягом 2 год. Реакційну суміш далі упарюють до сухого стану. Залишок розчиняють в H 2O (30 мл) і нейтралізують до pH=6–7 за допомогою 6 н розчину NaOH. Одержаний розчин завантажують на хроматографічну колонку, заповнену смолою AMBERLYST®15, з наступним елююванням водою до pH=5–5,5, і далі 100 % ізопропанолом (pH=7) та в кінці сумішшю 25 % NH4OH/ІПА (pH=10). Фракції, що містять продукт, об'єднують та упарюють до сухого стану при зниженому тиску. Додаткове очищення залишку колонковою хроматографією на силікагелі з використанням градієнту суміші розчинника ізопропанолу та концентрованого розчину гідроксиду амонію (співвідношення 17:3, 4:1 та 7:5, об./об.) як елюент дає названий в заголовку продукт (6,68 г, 1 72,7 %) у вигляді світло-жовтої, пінистої твердої речовини. Дані H ЯМР та МС/МС підтверджують структуру (див. Методику 4A). Вміст води за даними випробування методом Карла Фішера становить 3,7 %. Методика 5B: Синтез моноамонійної солі D-ізоглутаміл-D-триптофану (1:1) шляхом видалення неорганічних солей з використанням смоли Amberlyst15 з наступним очищенням перекристалізацією. При перемішуванні крізь охолоджену на льоді (0–5 °C) суспензію Boc-D-iGlu-D-Trp-OH (10,75 г, 24,80 ммоль) в етилацетаті (200 мл) пропускають газоподібний HCl. Реакційну суміш утримують на льодяній бані (0–5 °C) протягом 2 год. Аналіз методом ТШХ (30 % аміаку в ізопропанолі) демонструє повне перетворення початкового матеріалу. Реакційну суміш випарюють до сухого стану під вакуумом, залишок розчиняють в H 2O (30 мл) та нейтралізують до pH=6–7 за допомогою 10 н NaOH. Одержаний гомогенний розчин завантажують на хроматографічну колонку, заповнену смолою AMBERLYST®15, з наступним елююванням водою (2450 мл) до pH=4–5,5, ізопропанолом (1000 мл) та сумішшю 25 % NH4OH/ізопропанол. Фракції, що містять продукт, об'єднують та упарюють до сухого стану. Одержують безбарвну пінисту тверду речовину, до якої додають ізопропанол (150 мл) та H 2O (30 мл). Одержану суспензію перемішують при кімнатній температурі протягом ночі. Тверду речовину відокремлюють вакуумною фільтрацією, ретельно промивають ізопропанолом (2 × 60 мл), потім EtOAc (2 × 60 мл) і в кінці сушать під вакуумом в печі при 42 °C, витримуючи при цій температурі протягом 1 ночі. Одержують практично білу тверду речовину (6,60 г, 72,2 %). Дані H ЯМР та МС/МС відповідають структурі (див. Приклад 5). Дані РСАП одержаного кристалічного матеріалу показано на фіг. 2. Вміст води за даними метода Карла Фішера становить 5,9 %. Приклад 6 Синтез моноамонійної солі D-ізоглутаміл-D-триптофану (1:1) з H-D-iGlu-D-Trp-OH. Методика 6A: Одержання моноамонійної солі D-ізоглутаміл-D-триптофану (1:1) з використанням CBz-DGlu-(γ-D-Trp-OH)-γ-OBzl як проміжної сполуки. 25 UA 108058 C2 5 10 15 20 25 30 35 40 45 50 55 ЕДК (562 мг, 2,93 ммоль) додають до розчину Z-D-Glu-OBz (990 мг, 2,67 ммоль) та Nгідроксисукциніміду (337 мг, 2,93 ммоль) в ДМФА (50 мл) на льодяній бані, та одержаний прозорий розчин перемішують протягом ночі при кімнатній температурі. H-D-Trp-OH (640 мг, 3,13 ммоль) та Et3N (1 мл) додають при кімнатній температурі. Після проходження 20 хв., матеріал змішують з водою та екстрагують етилацетатом. Об'єднані EtOAc фракції промивають 10 % розчином лимонної кислоти і далі сольовим розчином, сушать над натрію сульфатом, фільтрують, упарюють до сухого стану, сушать під вакуумом з одержанням 1,49 г CBz-D-iGlu-(γOBzl)-D-Trp-OH. Одержаний матеріал гідрогенізують з використанням 33 % (мас./мас.) 10 % Pd/C при атмосферному тиску. Після проходження 4 год., каталізатор відфільтровують за допомогою CELITE®, і фільтрат випарюють з одержанням масла. Неочищений продукт очищують флеш-хроматографією на колонці з використанням суміші ізопропанол/NH4OH (від 80:20 до 70:30, об./об.) як елюенту з одержанням названої в заголовку сполуки (813 мг). Дані 1 МС/МС та H ЯМР є подібними до сполуки, одержаної з використанням способу, показаного у Прикладі 4 вище. Методика 6B: Одержання моноамонійної солі D-ізоглутаміл-D-триптофану (1:1) з H-D-iGlu-D-Trp-OH (див. Приклад 3). H-D-iGlu-D-Trp-OH (1 г з Прикладу 3) змішують з амонію гідроксидом (0,55 M, 6 мл). Суміш перемішують та вимірюють pH, що дорівнює приблизно 4,5. Розчин гідроксиду амонію (0,55 M) додають по краплях до pH розчину 7,0–7,5. Леткі матеріали видаляють під вакуумом, і залишкове масло змішують з ізопропанолом. Утворюється білий осад. Після проходження 2 год., тверду речовину (амонійну сіль) відокремлюють вакуумною фільтрацією. Тверду речовину сушать до постійної маси (1 г) під глибоким вакуумом протягом 12 год. з одержанням амонійної солі D-ізоглутаміл-D-триптофану (1:1). Вміст води за даними метода Карла Фішера становить 4,6 %. Приклад 7 Очищення H-D-iGlu-D-Trp-OH колонковою хроматографією на силікагелі з використанням суміші ізопропанолу та води. A. Одержання Cbz-D-Glu-(γ-D-Trp-OBzl)-α-OBzl До охолодженого на льоду розчину 2,67 г Cbz-D-Glu-OBzl в 50 мл ДМФА додають 0,91 г Nгідроксисукциніміду (1,1 екв.) і 1,52 г ЕДК (1,1 екв.), та одержаний розчин перемішують на льодяній бані протягом 1 г, і потім при кімнатній температурі протягом ночі. До одержаної реакційної суміші додають 2,50 г H-D-Trp-OBzl.HCl (1,05 екв.) і 3 мл Et3N при кімнатній температурі. Реакція завершується після проходження 1 год., що підтверджують дані РХВТ. Реакційну суміш гасять за допомогою демінералізованої води на льодяній бані, після чого декілька разів екстрагують етилацетатом. Об'єднані етилацетатні екстракти промивають 10 % розчином лимонної кислоти, і далі сольовим розчином, сушать над натрію сульфатом, фільтрують та упарюють до сухого стану. Залишок наносять на силікагель в метанолі, і суміш упарюють під вакуумом. Залишок наносять на верхню частину колонки з вологим силікагелем, і цільовий продукт, Cbz-D-Glu-(γ-D-Trp-OBzl)-α-OBzl, елююють з використанням градієнту суміші розчинників (етилацетат/гексан, від 80:20 до 100:0). Цільові фракції об'єднують та упарюють під вакуумом з одержанням 4,64 г (вихід 99,6 %) названої в заголовку сполуки. Чистота за даними РХВТ (% площі піку): 93 %; умови РХВТ: колонка: Symmetry C18, 3,9 × 150 мм, 5 мкм; рухома фаза: 0,035 % HClO4 (pH=2,5) / CH3CN = градієнт (хв.-CH3CN %: 0–10, 10–100, 12–100, 14–50); швидкість потоку: 1 мл/хв.; λ: 280 нм; час утримання: 9,7 хв. B. Очищення D-ізоглутаміл-D-триптофану колонковою хроматографією з використанням суміші ізопропанолу та воду. До суспензії 4,0 г Cbz-D-Glu-(γ-D-Trp-OBzl)-α-Obzl, одержаного, як описано у Прикладі 7A вище, в 150 мл суміші MeOH/H2O (95:5, об./об.) додають 1,5 г Pd/C (37,5 % мас./мас.). Суміш 2 гідрогенізують при тиску водню 30 фунт/дюйм . Реакція завершується після проходження 75 хв., що підтверджують дані РХВТ. Каталізатор відфільтровують на шарі CELITE®, і фільтрат упарюють при зниженому тиску і температурі 45 °C. Далі залишок очищують флешхроматографією на силікагелі з використанням суміші ізопропанол/H 2O (співвідношення 80:20, об./об.). Найбільш чисті фракції (за даними РХВТ) об'єднують та упарюють під вакуумом. Таким чином, одержують названу сполуку (1,1 г, 52 %) у вигляді світло-жовтого порошку. Чистота за 1 даними РХВТ (% площі піку): 99 %. Дані МС/МС та H ЯМР відповідають цільовій структурі. Менш чисті фракції (за даними РХВТ) об'єднують, упарюють під вакуумом з одержанням ще приблизно 1,0 г (вихід 48 %) названої сполуки. Чистота за даними РХВТ (% площі піку): 96,7 %. 1 Дані МС/МС та H ЯМР відповідають цільовій структурі. 26 UA 108058 C2 5 10 15 20 25 30 Умови РХВТ є такими ж, як описано в розділі 7A вище; час утримання H-D-iGlu-D-Trp-OH становить 4,0 хв. Приклад 8 A. Одержання H-D-Pyr-D-Trp-OH (5-оксо-D-проліл-D-триптофану) з Z-D-Glu-(γ-OEt)-OH. Названа в заголовку сполука являє собою можливу синтетичну домішку H-D-iGlu-D-Trp-OH. Її синтезують незалежно та використовують як довідковий зразок при аналізі методом РХВТ продуктів, описаних у Прикладах 3–7 вище. До охолодженого на льоду розчину Z-D-Glu-(γ-OEt)-OH (1 г, 3,23 ммоль) в ДМФА (60 мл), додають N-гідроксисукцинімід (409 мг, 3,56 ммоль), ЕДКI (682 мг, 3,56 ммоль), та одержаний розчин перемішують на льодяній бані протягом 1 год., після чого перемішують при кімнатній температурі протягом ночі. До одержаної реакційної суміші додають H-D-Trp-OH (792 мг, 3,88 ммоль) та Et3N (1 мл) при кімнатній температурі. Додають воду після проходження 1,5 год. при температурі льодяної бані. Суміш екстрагують декілька разів етилацетатом. Об'єднані етилацетатні фракції промивають 10 % розчином лимонної кислоти, і далі сольовим розчином, сушать над натрію сульфатом, фільтрують, упарюють до сухого стану та сушать під вакуумом з одержанням 410 мг фракції неочищеного продукту A. Водну фракцію упарюють під вакуумом на бані з температурою 55 °C. Залишок розчиняють в CH2Cl2, і органічну фракцію промивають 10 % розчином лимонної кислоти (1 × 20 мл) і сольовим розчином, сушать над натрію сульфатом, фільтрують, упарюють практично до сухого стану при температурі 50 °C з одержанням фракції неочищеного продукту B. Обидві фракції неочищеного продукту A та B об'єднують та гідрогенізують над 10 % Pd/C (40 % мас./мас. Pd/C) при атмосферному тиску з використанням балону з воднем при кімнатній температурі протягом 2,75 год. Реакційну суміш фільтрують крізь CELITE®. Фільтрат, що містить продукт H-D-iGlu(γ-OEt)-D-Trp-OH, аналізують за допомогою РХВТ (такий же спосіб, як описано в 7A вище). Час утримання для піку продукту становить 4,54 хв. Фільтрат упарюють до сухого стану при зниженому тиску на бані з температурою приблизно 45 °C. Аналіз методом РХВТ демонструє часткове перетворення продукту з Rt 4,54 хв. на інший продукт з піком R t 4,45 хв. Неочищений продукт очищують флешхроматографією на колонці з використанням ізопропанолу та концентрованого розчину NH 4OH (елюювання з градієнтом від 90:10 до 80:20, об./об.). Аналіз методом РХВТ одержаного 1 матеріалу (670 мг) демонструє пік із значенням R t 4,45 хв. Прояснення структури H ЯМР спектроскопією показало, що продукт після обробки являє собою циклічну сполуку, показану нижче: O HO O N H NH 1 35 40 45 50 HN O H ЯМР (CD3OD) δ проміле: 7,60 (д, J=7,8 Гц, 1H), 7,31 (д, J=8,0 Гц, 1H), 7,10 (с, 1H), 7,07 (т, J=7,8 Гц, 1H), 7,00 (т, J=7,4 Гц, 1H), 4,68-4,65 (м, 1H), 4,05-4,02 (м, 1H), 3,44 (дд, J=14,6 Гц, J=4,6 Гц, 1H), 3,23-3,18 (м, 1H), 2,34-2,24 (м, 1H), 2,13-2,05 (м, 1H), 2,00-1,92 (м, 1H) та 1,76-1,68 (м, + 1H). MS (співвідношення маси до заряду): 316 [M+1] , 188 (100 %). 13 C ЯМР (CD3OD) δ проміле: 181,6, 177,5, 174,2, 138,0, 129,3, 124,5, 122,4, 119,8, 119,5, 112,3, 111,7, 58,3, 56,3, 30,2, 28,8 та 26,5. Метод РХВТ: колонка Symmetry C18, 5 мкм, 3,9 × 150 мм, WAT 046980; рухома фаза 0,035 % HClO4 /CH3CN, градієнт; спосіб: хв.-CH3CN %: 0–10 %, 10–100 %, 12–100 %, 14–50 %; швидкість потоку: 1,0 мл/хв.; детектор λ: 254 нм. B. Незалежний синтез 5-оксо-D-проліл-D-триптофану з (R) (+)-2-піролідин-2-5-карбонової кислоти (H-D-Pyr-D-Trp-OH). (R) (+)-2-Піролідин-2,5-карбонову кислоту (2,09 г, 0,016 моль) додають до 250 мл дихлорметану. Діізопропілетиламін (3,12 мл, 0,018 моль) додають, і розчин стає прозорим. Гідроксибензотриазолу гідрат (HOBt.H2O, 2,48 г, 0,016 моль) додають при температурі 0 °C. Додають ЕДКI (4,64 г, 0,025 моль). Після проходження 30 хв., додають H-D-Trp-OBzl.HCl (4,76 г, 0,016 моль), з наступним додаванням діізопропілетиламіну (3,12 мл, 0,018 моль). Розчин залишається прозорим в точці часу 5 хв., і його перемішують протягом 16 год. Розчинник випарюють до сухого стану, і матеріал розподіляють між етилацетатом та 10 % кислотою хлористоводневою. Етилацетатну фракцію промивають 10 % розчином натрію бікарбонату, після чого сольовим розчином. Органічну фракцію сушать над натрію сульфатом та упарюють з одержанням пінистої твердої речовини. Одержана тверда речовина являє собою бензил-5-оксо 27 UA 108058 C2 5 10 D-проліл-D-триптофанат. Без очищення, зразок неочищеного бензил-5-оксо-D-проліл-Dтриптофанату (1,55 г) розчиняють в метанолі (30 мл) та гідрогенізують над 10 % Pd/C при тиску 2 водню 45 фунт/дюйм протягом 2 год. Каталізатор фільтрують крізь броунмілерит, і фільтрат випарюють з одержанням червоної піни. Одержаний матеріал очищують колонковою хроматографією (градієнт елюювання: 10 % метанолу в дихлорметані, і далі 50 % ізопропанолу в дихлорметані) з одержанням твердої речовини (419 мг). Чистота матеріалу може бути перевірена методом ТШХ (30 % водний розчин аміаку/ізопропанол). Одержаний матеріал являє собою 5-оксо-D-проліл-D-триптофан з такими ж даними ЯМР, як і для матеріалу з розділу A. Приклад 9 Синтез моноамонійної солі L-ізоглутаміл–L-триптофану (1:1). HN CO2+ H3N 15 20 25 30 35 40 45 50 L H L N O CO2 NH4+ A: Синтез Boc-L-Glu-(γ-L-Trp-O-трет-Bu)-α-O-трет-Bu Розчин Boc-L-Glu-O-трет-Bu (1,50 г, 4,9 ммоль) в CH2Cl2 (50 мл) охолоджують до -3 °C та перемішують протягом 15 хв. Далі послідовно додають 1-гідроксибензотриазол (HOBt, 1,00 г, 7,4 ммоль), N-(3-диметиламінопропіл)-N’-етилкарбодііміду гідрохлорид (ЕДК, 1,42 г, 7,4 ммоль) та діізопропілетиламін (ДІПЕА, 1,30 мл, 7,4 ммоль). Після цього, розчин H-L-Trp-O-трет-Bu.HCl (2,20 г, 7,4 ммоль) та ДІПЕА (1,30 мл, 7,4 ммоль) в CH2Cl2 (20 мл) додають по краплях. Одержану суміш перемішують при температурі льоду (від –3 °C до 0 °C) протягом 1 год., і далі дозволяють нагрітися до кімнатної температури та перемішують протягом ночі в атмосфері азоту. Реакційну суміш випарюють до сухого стану. Залишок розподіляють між EtOAc (40 мл) та насиченим розчином NaHCO3 (100 мл). Органічну фракцію відокремлюють, промивають 10 % розчином лимонної кислоти, і далі сольовим розчином (30 мл). Органічну фракцію сушать над натрію сульфатом, фільтрують та упарюють до одержання густого масла. Очищення залишку колонковою хроматографією на силікагелі з використанням градієнту суміші розчинників гексан/EtOAc (співвідношення 85:15, 80:20 та 60:40, об./об.) як елюенту дає названий в 1 заголовку продукт (2,55 г, 95 %) у вигляді білої твердої речовини. H ЯМР (ДМСО-d6) δ проміле: 10,84 (с, 1H), 8,18 (д, J=7,5 Гц, 1H), 7,52 (д, J=7,7 Гц, 1H), 7,35 (д, J=8,0 Гц, 1H), 7,12-7,15 (м, 2H), 7,06 (т, J=7,6 Гц, 1H), 6,98 (т, J=6,8 Гц, 1H), 4,41 (к, J=6,7 Гц, 1H), 3,73-3,80 (м, 1H), 2,94-3,12 (м, 13 2H), 2,13-2,21 (м, 2H), 1,60-1,85 (м, 2H), 1,28-1,38 (м, 27H); C ЯМР (ДМСО-d6) δ проміле: 171,5 (C), 171,4 (C), 171,1 (C), 155,5 (C), 136,1 (C), 127,2 (C), 123,5 (CH), 120,9 (CH), 118,3 (CH), 118,1 (CH), 111,3 (CH), 109,7, 80,2, 78,0, 53,9 (CH), 53,6 (CH), 31,5 (CH 2), 28,2 (CH3), 27,9 (CH3), 27,6 + (CH3), 27,5 (CH3), 27,2 (CH2), 26,6 (CH2); МС (співвідношення маси до заряду) 546 [M+1] ; аналітично обчислено для C29H43N3O7,0,5H2O: C, 62,80; H, 8,00; N, 7,58; знайдено: C, 62,69; H, 8,56; N, 7,57. B: Синтез моноамонійної солі L-ізоглутаміл-L-триптофану (1:1) Газоподібний HCl пропускають при перемішуванні крізь охолоджений на льоду (0–5 °C) розчин Boc-L-Glu-(-L-Trp-O-трет-Bu)-γ-O-трет-Bu, одержаний, як описано вище (2,38 г, 4,4 ммоль), в CH2Cl2 (40 мл) протягом 5 год. Реакційна суміш стає мутною в ході пропускання газоподібного HCl. Температуру реакції підтримують на рівні нижче 30 °C шляхом охолодження на льоду. Реакційну суміш далі упарюють з одержанням білої твердої речовини (маса неочищеного матеріалу = 2,80 г). Очищення неочищеного зразка (482 мг) з використанням зворотно-фазної високоефективної флеш-хроматографії (HPFC™, Biotage) з використанням колонки C18HS M+40 та градієнту суміші розчинників 15 мМ розчину NH4OAc/CH3CN як елюенту дає названу сполуку після 1 випарювання розчинника та сушіння матеріалу виморожуванням (150 мг). H ЯМР (ДМСО-d6) δ проміле: 7,57 (д, J=7,8 Гц, 1H), 7,39 (д, J=8,1 Гц, 1H), 7,11-7,14 (м, 2H), 7,05 (т, J=7,2 Гц, 1H), 4,44-4,48 (м, 1H), 3,42 (т, J=5,7 Гц, 1H), 3,25 (дд, J=14,7, 4,7 Гц, 1H), 2,97-3,03 (м, 1H), 2,14-2,19 -1 (м, 2H), 1,72-1,85 (м, 2H); ІЧ-ФП (KBr) ν: 3057, 1581, 1400, 745 см ; МС (співвідношення маси до + заряду) 334 [двохосновна кислота+1] ; аналітично обчислено для C16H22N4O5.H2O: C, 52,17; H, 6,57; N, 15,21; знайдено: C, 51,92; H, 6,80; N, 14,94. Речовина являє собою моноамонійну сіль Lізоглутаміл-L-триптофану (1:1). Приклад 10 Синтез моноамонійної солі H-D-ізоглутаміл-L-триптофану (1:1). 28
ДивитисяДодаткова інформація
Автори англійськоюTam, Tim, Fat, N'zemba, Blaise, Leung-Toung, Regis, Wang, Yingsheng, Zhao, Yanqing, Yu, Lily
Автори російськоюТам Тим Фет
МПК / Мітки
МПК: C07K 1/107, A61K 38/05, C07K 1/30, A61P 17/06, A61P 37/00, C07K 1/16, C07K 5/037, C30B 7/00, C07K 5/02
Мітки: d-ізоглутаміл-d-триптофан, d-ізоглутаміл-d-триптофану, сіль, моноамонійна, кристалічний
Код посилання
<a href="https://ua.patents.su/44-108058-kristalichnijj-d-izoglutamil-d-triptofan-i-monoamonijjna-sil-d-izoglutamil-d-triptofanu.html" target="_blank" rel="follow" title="База патентів України">Кристалічний d-ізоглутаміл-d-триптофан і моноамонійна сіль d-ізоглутаміл-d-триптофану</a>
Дикалієва сіль n-сукцин – dl-триптофану, що має антиангінальну та антигіпоксантну дію

Номер патенту: 33865
Опубліковано: 15.02.2001
Автори: Гриневич Олександр Йосипович, Горчакова Надія Олександрівна, Чекман Іван Сергійович, Ткаченко Олександр Петрович, Ніженківська Ірина Володимирівна
МПК: C07C 209/20, A61K 31/405, A61P 9/02
Мітки: має, антигіпоксантну, антиангінальну, сіль, дію, dl-триптофану, n-сукцин, дикалієва
Текст:
...об'єму дихання (ХОД) на 3-й хвилині після введення. Такий рівень легеневої вентиляції утримувався протягом всього наступного періоду спостереження (30 хв), коливаючись у межах 88-92% вихідного рівня ХОД. Описані зміни рівня вентиляції легень в умовах нормоксії під впливом сполуки були викликані зменшенням частоти дихальних рухів (ЧД), виражених в тій же мірі, як і зниження ХОД. Інший компонент показника ХОД, що має значно більше значення для...
Калієва сіль n-сукцин-d,l-триптофану, яка має кардіотонічну дію

Номер патенту: 11771
Опубліковано: 25.12.1996
Автори: Натаров Валерій Володимирович, Антоненко Людмила Іванівна, Чекман Іван Сергійович, Нікішина Людмила Євгеніївна, Воловельський Лев Натанович, Горчакова Надія Олександрівна, Корюкіна Віра Миколаївна, Фролькис Раіса Аронівна, Мудрая Ірина Сергіївна, Братусь Віктор Васильович, Ткачук Валерій Віталійович, Французова Стелла Борисівна, Растрепіна Інна Олександрівна
МПК: C07D 209/20, A61P 9/00, A61K 31/405
Мітки: n-сукцин-d,l-триптофану, яка, кардіотонічну, дію, калієва, має, сіль
Виділений і очищений кристалічний білок bacillus thuringiensis, композиція кристалічного білка, очищений сегмент нуклеїнової кислоти, що кодує кристалічний білок, спосіб застосування сегмента нуклеїнової кислот

Номер патенту: 75317
Опубліковано: 17.04.2006
Автори: Донован Вілльям П., Слейні Аннетт С., Донован Джудіт С.
МПК: C12Q 1/68, C12N 1/21, C07K 14/325, C07K 16/12, C12R 1/38, G01N 33/68, C12N 15/32, C12N 15/82, C12R 1/07, C12R 1/19, A01N 63/00, C07K 14/195
Мітки: кодує, нуклеїнової, bacillus, білка, білок, кристалічний, спосіб, кислот, виділений, сегмента, thuringiensis, кислоти, очищений, сегмент, композиція, кристалічного, застосування
Формула / Реферат:
1. Виділений і очищений кристалічний білок Bacillus thuringiensis, що містить амінокислотну послідовність SEQ ID № 3 або SEQ ID № 4.2. Білок за п. 1, який відрізняється тим, що вказаний кристалічний білок виділяють із Bacillus thuringiensis EG10327 (NRRL B-21365), EG11402 (NRRL B-21366) або EG11403 (NRRL B-21367).3. Білок за п. 2, який відрізняється тим, що вказаний кристалічний білок має розмір приблизно 29 kДа або приблизно 14...
Мононатрій глутаміл-триптофан, який має імуностимулюючу дію, та спосіб його одержання

Номер патенту: 32271
Опубліковано: 15.12.2000
Автори: Височин Ігор Олександрович, Семеній Валерій Якович, Винокуров Юрій Віталійович, Головченко Володимир Іванович, Гершкович Олександр Абрамович
МПК: A61K 38/05
Мітки: мононатрій, спосіб, має, дію, імуностимулюючу, глутаміл-триптофан, одержання
Текст:
...Розчин охолоджують і залишають в холодильнику при температурі -2 °С на ніч. Осад, який випав, відфільтровують, промивають 20 - 30 мл холодної води, 2 - 30 мл холодного ізопропанолу. Осад піддають тонкошарової хроматографії в тій же системі. При цьому присутні дві плями: з Rf=0,5 (глутаміл-триптофан) і з Rf=0,2 (глутамінова кислота). Для очистки від глутамінової кислоти осад розчиняють у 300 мл гарячої води, додають ї л ізопропанолу і...
Спосіб одержання дикалієвої солі n-сукциноїл-d, l-триптофану

Номер патенту: 25680
Опубліковано: 15.08.2002
Автори: Кустова Світлана Петрівна, Карпова Тетяна Олексіївна, Масловська Людмила Валентинівна, Скачек Ірина Борисівна, Бодрих Олена Андріївна, Нікішина Людмила Євгеніївна
МПК: A61K 31/395, C07D 209/20
Мітки: дикалієвої, n-сукциноїл-d, одержання, спосіб, l-триптофану, солі
Формула / Реферат:
Спосіб одержання дикалієвої соді N-сукциноіл-Д,-триптофану формулишляхом взаємодії Д, -триптофану формулиз янтарним ангідридом, який відрізняється тим, що реакцію проводять у середовищі сухого діоксану при температурі кипіння реакційної маси протягом 1,5 години, суміш охолоджують, виливають у хлороформ і утворений осадок обробляють спиртовим розчином гідроокису калія з послідуючим виділенням цільового продукту...