Спосіб визначення 2,4-дихлорфеноксіоцтової кислоти у водних розчинах
Номер патенту: 52180
Опубліковано: 10.08.2010
Автори: Турчин Валентина Олександрівна, Халаф Вікторія Анатоліївна, Зайцев Володимир Миколайович, Гождзінський Сергій Мартинович




Формула / Реферат
1. Спосіб визначення 2,4-дихлорфеноксіоцтової кислоти, що включає підготування проби до аналізу, відділення 2,4-дихлорфеноксіоцтової кислоти від домішок та кількісне визначення методом високоефективної рідинної хроматографії, який відрізняється тим, що підготування проби до аналізу проводять шляхом додавання до проби значного надлишку цетилтриметиламоній броміду і доведення рН до 8,5-9,0, відділення 2,4-дихлорфеноксіоцтової кислоти від домішок здійснюють пропусканням розчину через колонку з кремнеземом, модифікованим поліоксіетильованим ізооктилфенолом, промиванням колонки дистильованою водою, висушуванням з наступною десорбцією аналіту ацетонітрилом і фільтруванням елюату через мембранний фільтр, а як рухому фазу для високоефективної рідинної хроматографії беруть суміш ацетонітрилу з водою та оцтовою кислотою в об'ємному співвідношенні 40:(59-60):(0,5-1,5).
2. Спосіб за п. 1, який відрізняється тим, що доведення рН до 8,5-9,0 здійснюють розчином карбонату натрію.
3. Спосіб за п. 1, який відрізняється тим, що пропускання розчину через колонку з кремнеземом, модифікованим поліоксіетильованим ізооктилфенолом, проводять зі швидкістю 1-2 см3/хв.
4. Спосіб за п. 1, який відрізняється тим, що як рухому фазу для високоефективної рідинної хроматографії беруть суміш ацетонітрилу з водою та оцтовою кислотою в об'ємному співвідношенні 40:(59-60):(0,5-1,5), а швидкість рухомої фази становить 1,0-1,5 см3/хв.
Текст
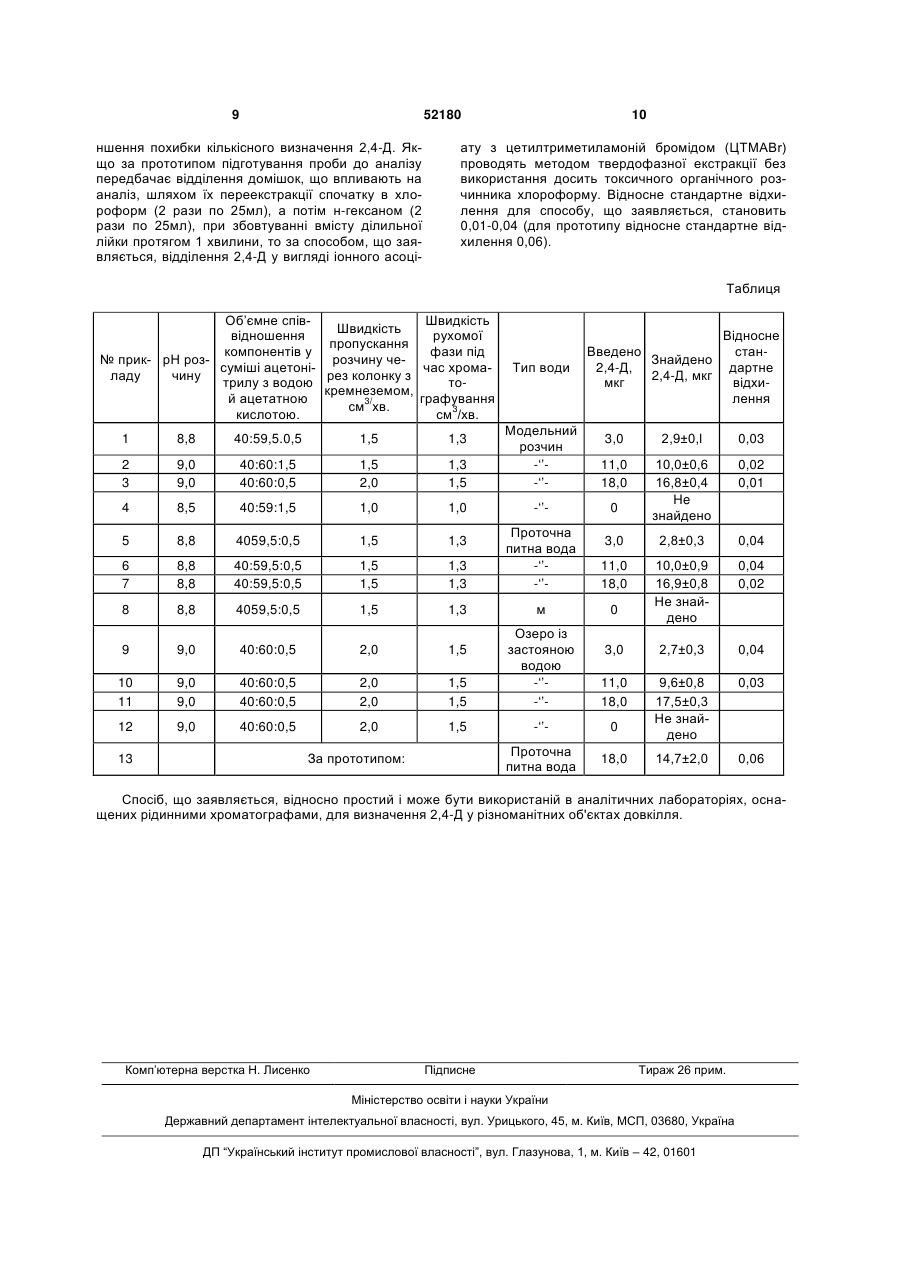
1. Спосіб визначення 2,4дихлорфеноксіоцтової кислоти, що включає підготування проби до аналізу, відділення 2,4дихлорфеноксіоцтової кислоти від домішок та кількісне визначення методом високоефективної рідинної хроматографії, який відрізняється тим, що підготування проби до аналізу проводять шляхом додавання до проби значного надлишку цетилтриметиламоній броміду і доведення рН до 8,5-9,0, відділення 2,4-дихлорфеноксіоцтової кислоти від домішок здійснюють пропусканням розчину через U 2 (19) 1 3 природних і питних водах методом капілярного зонного електрофорезу (Комарова Н.В., Карцова Л.А. II Журн. аналит. хим., 2002. Т.57. №7. С.768.). Пробу води об'ємом 1л підкисляють до рН=2,0, додають 50г NaCl і пропускають через патрон Діапак С16 зі швидкістю 15мл/хв. Патрон висушують протягом 20хв., а сорбовані речовини елюють 4мл ацетону. Елюат випарюють досуха під вакуумом, а залишок розчиняють в 1мл дистильованої води. Після центрифугування (6000 об/хв., 2хв.) отриманий розчин аналізують методом капілярного електрофорезу. Умови електрофоретичного розділення: не модифікований кварцовий капіляр з зовнішньою поліамідною плівкою довжиною 60см, внутрішнім діаметром 75мкм, ведучий електроліт 10мМ розчин тетраборату натрію (рН=9,2), робоча напруга +20кВ. Піки реєстрували в УФ діапазоні (228,8нм). Ступінь вилучення 2,4-Д після твердофазної екстракції 81,5%. Нижня межа визначення для 2,4-Д у водних зразках з врахуванням концентрування (К=250) становить 0,0005мг/л. Спільними ознаками зі способом, що заявляється, є підготування проби до аналізу, відділення 2,4-Д від домішок та кількісне визначення методом високоефективної рідинної хроматографії. Причинами, що перешкоджають одержанню потрібного технічного результату, є не досить вдалий вибір технологічних операцій способу, що призводить до зниження ефективності виділення визначуваних речовин у природних водних об'єктах з концентрацією гумінових кислот від 50 до 150мг/л. Відомий спосіб визначення феноксіоцтових кислот (зокрема 2,4-Д) та інших гербіцидів у воді. (Peruzzi М., Bartolucci G., Cioni F. // J. Chromatography А. 2000. V. 867. Р.171) Методика включає твердофазну екстракцію (ТФЕ) на макропористому кополімерному сорбенті полі(дивінілбензен-ко-вінілпіролідоні) (Oasis HLB) та визначення методом високоефективної рідинної хроматографії (ВЕРХ) з діодно-матричним детектуванням. Картриджі для ТФЕ промивали етилацетатом і метанолом. До 500мл дистильованої води, що містить 1% метанолу, додавали розчин досліджуваних речовин, щоб створити концентрацію 0,5нг/мл. Для ТФЕ додали фосфатний буфер. Зразки пропускали через картридж Oasis HLB (60мг-3мл) зі швидкістю потоку 5-10мл/хв. Після висушування картриджів, з них елюювали досліджувані речовини під вакуумом. Елюати випаровували під струменем азоту і сухі залишки розчиняли у двох порціях ацетонітрилу 1,0 і 0,5мл і аналізували методом ВЕРХ. Умови аналізу: колонка 250 4,6мм, швидкість потоку 0,8мл/хв., об'єм для інжекції 8 мкл, t=40°C, спектральний діапазон довжин хвиль 210-350 нм, водна рухома фаза 0,05 М Н3РО4 - КН2РО4 буферний розчин при рН=3, 0,01% трифтороцтова кислота, органічна частина рухомої фази ацетонітрил +0,01% трифтороцтова кислота. Процент сорбції на досліджуваному сорбенті для феноксіоцтових кислот, що аналізувалися становить 67-108% (для 2,4-Д -70%). Лінійність концентрацій зберігається в діапазоні 0,1-1,0нг/мл. Межа чутливості для досліджуваних речовин лежить в діапазоні 0,05 -0,1нг/мл. В роботі було порівняно сорбційні властивості досліджуваного сор 52180 4 бенту й інших полімерних сорбентів і показано, що полі(дивінілбензенко-вінілпіролідон) дає найкращі результати при відсутності процедури підкислення проби. Метод має такі переваги: одночасна екстракція аналітів різної полярності і кислотноосновними характеристиками, невеликий час аналізу порівняно з іншими методами твердофазного аналізу, де потрібно визначати різні класи гербіцидів, а висока сорбційна сила сорбенту дає можливість використовувати невелику масу сорбенту для картриджа, що сприяє скороченню часу аналізу і об‟єму елюату. Спільними ознаками зі способом, що заявляється, є підготування проби до аналізу, відділення 2,4-Д від домішок та кількісне визначення методом високоефективної рідинної хроматографії. Причинами, що перешкоджають досягненню потрібного технічного результату, є не досить високий відсоток виділення 2,4-Д з аналізованих об'єктів, а також те, що визначається сума гербіцидів, а визначити кількісний вміст 2,4-Д з достатньою точністю неможливо. Як прототип за найбільшим числом спільних суттєвих ознак і досягнутим результатом вибрано спосіб визначення 2,4-Д у воді (Методические указания по определению 2,4-Д в воде методом обращено-фазовой высокоэффективной жидкостной хроматографии. Утв. Мин. Здравоохранения 29.07.91, №6127-91). За цим способом 100мл відфільтрованої проби поміщають у плоскодонну конічну колбу місткістю 250мл, додають 4г натрій гідрокарбонату (зваженого на технічних терезах) і розчиняють при перемішуванні. Після цього розчин переносять у ділильну лійку на 250мл. Для видалення з проби домішок, що впливають на аналіз, проводять їх переекстракція спочатку в хлороформ (2 рази по 25мл), а потім н-гексаном (2 рази по 25мл), при збовтуванні вмісту ділильної лійки протягом 1хв. Розчинники, що містять супутні домішки проби, відкидають, а водний розчин зливають в колбу місткістю 250мл і поступово приливають туди 1мл хлороводневої кислоти, розчин перемішують при легкому збовтуванні. Після припинення виділення пухирців вуглекислого газу розчин переносять у ділильну лійку і проводять переекстракцію 2,4-Д у діетиловий ефір (2 рази по 20мл) протягом 1хв. Водний розчин відкидають, а об'єднаний ефірний екстракт відмивають водою до нейтрального значення рН. Фільтрують через прожарений натрій сульфат в круглодонну колбу зі шліфом місткістю 100мл, випарюють на ротаційному упарювачі при 30-35°С до невеликого об'єму, переносять у мірну пробірку і випарюють в струмені азоту особливої чистоти. Сухий залишок проби розчиняють в 0,2мл дистильованої води і проводять хроматографічне визначення 2,4-Д методом ВЕРХ (високоефективної рідинної хроматографії). Нерухома фаза - "Силасорб C18", зернистість 5 мкм. Рухома фаза - вода з метанолом в об'ємному співвідношенні 7:3, швидкість потоку 200мкл/хв., довжина хвилі світлового потоку 280нм. Кількість 2,4-Д в аналізованій пробі визначають шляхом порівняння площі піків хроматограми аналізованого зразка і стандартних розчинів з відомим вмістом 2,4-Д. 5 Спільними ознаками зі способом, що заявляється, є: підготування проби до аналізу, відділення 2,4-Д від домішок та кількісне визначення методом високоефективної рідинної хроматографії. Причинами, що перешкоджають одержанню потрібного технічного результату, є не досить вдалий вибір параметрів способу-прототипу, а також надмірна складність процесу проведення вилучення 2,4-Д із об'єктів аналізу. В основу корисної моделі поставлена задача у способі визначення 2,4-Д шляхом зміни параметрів та введення нових операцій розробити відносно просту та ефективну методику визначення 2,4Д у водних розчинах, спростити процедуру підготовки проби до аналізу та зменшити похибку кількісного визначення 2,4-Д. Поставлена задача вирішується тим, що у способі визначення 2,4-дихлорфеноксіоцтової кислоти, який включає підготування проби води до аналізу, відділення 2,4-дихлорфеноксіоцтової кислоти від домішок та кількісне визначення методом високоефективної рідинної хроматографії, згідно з корисною моделлю, підготування проби до аналізу проводять шляхом додавання до проби значного надлишку цетилтриметиламоній броміду і доведення рН до 8,5-9,0, відділення 2,4дихлорфеноксіоцтової кислоти від домішок здійснюють пропусканням розчину через колонку з кремнеземом, модифікованим поліоксіетильованим ізооктилфенолом, промиванням колонки дистильованою водою, висушуванням з наступною десорбцією аналіту ацетонітрилом і фільтруванням елюату через мембранний фільтр, а рухомою фазою для високоефективної рідинної хроматографії беруть суміш ацетонітрилу з водою й оцтовою кислотою в об'ємному співвідношенні 40:(59-60):(0,51,5). Згідно з корисною моделлю, доведення рН до 8,5-9,0 здійснюють розчином карбонату натрію. Згідно з корисною моделлю, пропускання розчину через колонку з кремнеземом, модифікованим поліоксіетильованим ізооктилфенолом проводять зі швидкістю 1-2см3/хв. Згідно з корисною моделлю, рухомою фазою для високоефективної рідинної хроматографії беруть суміш ацетонітрилу з водою й оцтовою кислотою в об'ємному співвідношенні 40:(59-60):(0,51,5), а швидкість рухомої фази 1,0-1,5см3/хв. Технічним результатом способу, що заявляється, є спрощення процедури підготування проби до аналізу та зменшення похибки кількісного визначення 2,4-Д. Відносне стандартне відхилення для способу, що заявляється, становить 0,01-0,04 (для прототипу відносне стандартне відхилення 0,06). Для здійснення способу, що заявляється, використовували такі пристрої та реагенти: - рідинний хроматограф Shimadzu SPD-6A з УФ детектором (№28A7224LP); - колонку Kromasil C18 (150 3,2мм, розмір часточок 5мкм); - перистальтичний насос; - іономер «И-160 М»; - мембранні фільтри (пористість 0,45мкм, d=25мм); 52180 6 - магнітну мішалку; - стакани на 100мл; - пробірки на 10мл; - мірні колби на 50мл, на 100мл; - 2,4-дихлорфеноксіоцтову кислоту фірми Dow Elanco, чистота 99,5%; - кремнезем, модифікований поліоксиетильованим ізооктилфенолом (носій - силохром С-120 (Ставрополь) з питомою площею поверхні 120м2/г); - дистильовану воду; - етиловий спирт кваліфікації х.ч.; - ацетонітрил для ВЕРХ фірми Merck без додаткового очищення; - оцтову кислоту кваліфікації х.ч.; - натрію карбонат кваліфікації х.ч.; - натрію гідроксид кваліфікації х.ч.; - цетилтриметиламоній бромід фірми «Меrk» з чистотою 99,0%; - хлороводневу кислоту із фіксаналу; - кальцію хлорид кваліфікації х.ч.; - натрію хлорид кваліфікації х.ч.; - очищене повітря. Спосіб, що заявляється, здійснювали таким чином. До проби досліджуваного водного розчину чи зразка води з певного природного об'єкта, яку в разі необхідності концентрують і фільтрують, додають у значному надлишку цетилтриметиламоній бромід (ЦТМАВr). Доводять рН розчину до 8,5-9,0 за допомогою розчину Na2СО3, або іншого лужного 3 розчину. Приготовлений розчин (50см ) пропускають із швидкістю 1-2см3/хв. через колонку (d=10мм, h=10мм), заповнену модифікованим поліоксіетильованим ізооктилфенолом кремнеземом (SiO2-TX). Після цього сорбент промивають водою, висушують і проводять десорбцію 2,4-Д ацетонітрилом. В усіх випадках отриманий елюат фільтрують через мембранний фільтр (пористість 0,45мкм, d=25мм) і визначають у ньому вміст 2,4-Д методом ВЕРХ. Для хроматографування використовують рідинний хроматограф, наприклад, Shimadzu SPD-6A з УФ детектором (№28A7224LP). Умови хроматографування: колонка, заповнена обернено-фазовим сорбентом Kromasil C18 (150x3,2 мм, розмір частинок 5 мкм); температура термостата колонки 35°С. Рухома фаза: суміш ацетонітрилу з водою та ацетатною кислотою в об'ємному співвідношенні 40:(5960):(0,5-1,5). Швидкість потоку рухомої фази 1,01,5см3/хв. Умови детектування: X=220нм. На одержаній хроматограмі вимірюють час утримування, а також висоту піка. За градуювальним графіком визначають концентрацію 2,4-Д. Побудова градуювального графіка: Для приготування серії стандартних розчинів у стакани на 100мл вносили по 1мл ацетонітрильного розчину, що містив 2, 4, 8, 16, 20мкг стандартного розчину 2,4-Д. Ацетонітрил у кожному стакані випаровували в струмені очищеного повітря. До сухого залишку приливали по 20мл водного розчину цетилтриметиламоній броміду (ЦТМАВr) з молярною концентрацією 9 10-4моль/дм3, доводили рН до 8,80 розчином Nа2СО3 і переносили в мірну колбу ємністю 50мл. Загальний об'єм доводили до 50мл 7 дистильованою водою. 2,4-Д, що знаходився в розчині у вигляді іонного асоціату з цетилтриметиламоній бромідом (ЦТМАВr), вилучали пропусканням через колонку з кремнеземом, модифікованим поліоксіетильованим ізооктилфенолом (SiO2-TX). Адсорбційну колонку (h=10мм, d=10мм) заповнювали водною суспензією SiO2-TX, який є ефективним адсорбентом для твердофазної екстракції (ТФЕ) фенольних сполук у формі їх іонних асоціатів (ІА) з ЦТМАВr. Перед використанням сорбент SiO2-VTX кондиціювали (переводили в активну форму) послідовним пропусканням через колонку 1 мл ацетонітрилу, 1мл етилового спирту, 5мл суміші етилового спирту з дистильованою водою у співвідношенні 1:1, а потім - 20 мл дистильованої води. Через колонку, заповнену SiO2-TX, пропускали кожний розчин 2,4-Д зі швидкістю 1,5 мл/хв. Потім кожну колонку промивали 20 мл дистильованої води і проводили десорбцію 2,4-Д порціями по 1мл ацетонітрилу. Елюат фільтрували через мембранний фільтр (пористість 0,45мкм, d=25мм) і використовували для наступного кількісного визначення вмісту 2,4-Д методом високоефективної рідинної хроматографії. Хроматографування проводили на рідинному хроматографі Shimadzu SPD6A з УФ детектором (№28A7224LP). Умови хроматографування: колонка, заповнена оберненофазовим сорбентом Kromasil C18 (150x3,2 мм, розмір частинок 5 мкм); температура термостата колонки 35°С. Рухома фаза: суміш ацетонітрилу з водою й ацетатною кислотою в об'ємному співвідношенні 40:59,5:0,5. Швидкість потоку рухомої фази 1,3мл/хв. Умови детектування: =220нм. В інжектор хроматографа вводили порцію (3мкл) виготовленого стандартного розчину 2,4-Д з певною концентрацією. На одержаній хроматограмі визначали час утримування, а також висоту піка. За одержаними результатами серії стандартних розчинів будували градуювальний графік залежності висоти піка від концентрації. Одержаний графік використовували для кількісного визначення 2,4-Д у модельних розчинах. Для перевірки можливості способу, що заявляється, визначати 2,4-Д у різних об'єктах, готували модельні розчини що містили солі кальцію, натрію з концентраціями близькими до природних об'єктів. В такі модельні розчини вводили певну кількість 2,4-Д. Крім того, певну кількість 2,4-Д вводили у зразки води з різних природних джерел (проточна питна вода з водопровідної мережі, озеро із застояною водою). Після цього проводили кількісне визначення 2,4-Д так, як описано вище. Можливість здійснення способу, що заявляється, підтверджується наступними прикладами конкретної реалізації. Приклад 1. До 30 мл модельного розчину, що містив 3мкг 2,4-Д, додавали значний надлишок ЦТМАВr - цетилтриметиламоній броміду (20мл водного розчину ЦТМАВr з молярною концентрацією 9 104 моль/дм3), доводили рН до 8,80 розчином Na2CO3. Приготовлений розчин (50см3) пропускали із швидкістю 1,5см3/хв. через колонку (d=10мм), заповнену модифікованим поліоксіетильованим ізооктилфенолом кремнеземом (SiO2-TX), який 52180 8 попередньо кондиціювали (переводили в активну форму), пропускаючи послідовно 1мл ацетонітрилу, 1мл етилового спирту, 5мл суміші етилового спирту з дистильованою водою у співвідношенні 1:1, а потім - 20мл дистильованої води. Потім колонку промивали водою, висушували і проводили десорбцію 2,4-Д ацетонітрилом. Отриманий елюат фільтрували через мембранний фільтр (пористість 0,45мкм, d=25мм). В інжектор хроматографа вводили порцію (3мкл) профільтрованого елюату і визначали у ньому вміст 2,4-Д методом ВЕРХ. Для хроматографування використовували рідинний хроматограф Shimadzu SPD-6A з УФ детектором (№28A7224LP). Умови хроматографування: колонка, заповнена обернено-фазовим сорбентом Khromasil C18 (150 3,2мм, розмір частинок 5мкм); температура термостата колонки 35°С. Рухома фаза: суміш ацетонітрилу з водою та ацетатною кислотою в об'ємному співвідношенні 40:59,5:0,5. Швидкість потоку рухомої фази 1,3см3/хвил. Умови детектування: =220нм. На одержаній хроматограмі вимірювали час утримування, а також висоту піка. За градуювальним графіком визначали вміст 2,4-Д. Кількісне визначення 2,4-Д проводили 3 рази і розраховували відносне стандартне відхилення. Результати кількісного визначення 2,4-Д: уведено 3,0мкг 2,4-Д; знайдено 2,9±0,1мкг; відносне стандартне відхилення 0,03. Приклади 2, 3. Кількісне визначення 2,4-Д проводили так, як описано у прикладі 1, за винятком того, що брали модельні розчини із різним умістом 2,4-Д, та змінювали параметри способу в межах заявлених інтервалів. Параметри способу та результати кількісного визначення наведено у прикладах 2, 3 таблиці. Приклад 4. Для дослідження взяли модельний розчин, що не містив 2,4-Д. Результати наведено у прикладі 4 таблиці. Приклади 5-8. Кількісне визначення 2,4-Д проводили так, як описано у прикладі 1, за винятком того, що брали зразки проточної питної води з водопровідної мережі із різним умістом 2,4-Д, та змінювали параметри способу в межах заявлених інтервалів. Параметри способу та результати кількісного визначення наведено у прикладах 5-8 таблиці. Приклади 9-12. Для кількісного визначення 2,4-Д брали зразки води з озера із застояною водою, що містили різні кількості 2,4-Д. Кількісне визначення проводили так, як описано у прикладі 1, за винятком того, що зразки фільтрували та змінювали параметри способу в межах заявлених інтервалів. Параметри способу та результати кількісного визначення наведено у прикладах 9-12 таблиці. Приклад 13. Для порівняння кількісне визначення 2,4-Д у модельному розчині провели за методикою, що наведена у прототипі. Результати наведено у прикладі 13 таблиці. Наведені приклади переконливо підтверджують досягнення технічного результату: спрощення процедури підготування проби до аналізу та зме 9 52180 ншення похибки кількісного визначення 2,4-Д. Якщо за прототипом підготування проби до аналізу передбачає відділення домішок, що впливають на аналіз, шляхом їх переекстракції спочатку в хлороформ (2 рази по 25мл), а потім н-гексаном (2 рази по 25мл), при збовтуванні вмісту ділильної лійки протягом 1 хвилини, то за способом, що заявляється, відділення 2,4-Д у вигляді іонного асоці 10 ату з цетилтриметиламоній бромідом (ЦТМАВr) проводять методом твердофазної екстракції без використання досить токсичного органічного розчинника хлороформу. Відносне стандартне відхилення для способу, що заявляється, становить 0,01-0,04 (для прототипу відносне стандартне відхилення 0,06). Таблиця Об‟ємне співШвидкість Швидкість відношення рухомої пропускання компонентів у фази під № прик- рН розрозчину чесуміші ацетонічас хромаладу чину рез колонку з трилу з водою токремнеземом, й ацетатною графування 3/ см хв. кислотою. см3/хв. Тип води Введено Знайдено 2,4-Д, 2,4-Д, мкг мкг Відносне стандартне відхилення 3,0 2,9±0,l 0,03 1,3 1,5 Модельний розчин -„‟-„‟ 11,0 18,0 0,02 0,01 1,0 1,0 -„‟ 0 10,0±0,6 16,8±0,4 He знайдено 4059,5:0,5 1,5 1,3 8,8 8,8 40:59,5:0,5 40:59,5:0,5 1,5 1,5 8 8,8 4059,5:0,5 9 9,0 10 11 12 1 8,8 40:59,5.0,5 1,5 1,3 2 3 9,0 9,0 40:60:1,5 40:60:0,5 1,5 2,0 4 8,5 40:59:1,5 5 8,8 6 7 13 3,0 2,8±0,3 0,04 1,3 1,3 Проточна питна вода -„‟-„‟ 11,0 18,0 0,04 0,02 1,5 1,3 м 0 10,0±0,9 16,9±0,8 He знайдено 40:60:0,5 2,0 1,5 9,0 9,0 40:60:0,5 40:60:0,5 2,0 2,0 9,0 40:60:0,5 2,0 3,0 2,7±0,3 0,04 1,5 1,5 Озеро із застояною водою -„‟-„‟ 11,0 18,0 0,03 1,5 -„‟ 0 9,6±0,8 17,5±0,3 He знайдено Проточна питна вода 18,0 14,7±2,0 0,06 За прототипом: Спосіб, що заявляється, відносно простий і може бути використаній в аналітичних лабораторіях, оснащених рідинними хроматографами, для визначення 2,4-Д у різноманітних об'єктах довкілля. Комп‟ютерна верстка Н. Лиcенко Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod of determining 2,4-dichlorphenoxyacetic acid in water solutions
Автори англійськоюTurchyn Valentyna Oleksandrivna, Zaitsev Volodymyr Mykolaiovych, Khalaf Viktoria Anatoliivna, Hozhdzinskyi Serhii Martynovych
Назва патенту російськоюСпособ определения 2,4-дихлорфеноксиуксусной кислоты в водных растворах
Автори російськоюТурчин Валентина Александровна, Зайцев Владимир Николаевич, Халаф Виктория Анатольевна, Гождзинський Сергей Мартынович
МПК / Мітки
МПК: A01N 37/10, B01D 15/08
Мітки: спосіб, розчинах, визначення, кислоти, 2,4-дихлорфеноксіоцтової, водних
Код посилання
<a href="https://ua.patents.su/5-52180-sposib-viznachennya-24-dikhlorfenoksioctovo-kisloti-u-vodnikh-rozchinakh.html" target="_blank" rel="follow" title="База патентів України">Спосіб визначення 2,4-дихлорфеноксіоцтової кислоти у водних розчинах</a>
Спосіб визначення тринітротолуолу та нітрату амонію в водних розчинах, що утворилися під час витягнення амотолу із боєприпасів, які утилізуються

Номер патенту: 27149
Опубліковано: 25.10.2007
Автори: Закотей Валентина Григорівна, Маренець Марина Олександрівна, Белова Людмила Андріївна, Межевич Геннадій Васильович, Буллер Михайло Фридрихович, Ярманова Світлана Павлівна
МПК: G01N 30/02, C06B 25/00
Мітки: нітрату, боєприпасів, визначення, витягнення, амонію, водних, спосіб, розчинах, утворилися, амотолу, тринітротолуолу, утилізуються
Формула / Реферат:
Спосіб визначення концентрації тринітротолуолу та нітрату амонію в водних розчинах, що утворилися під час витягнення амотолу із боєприпасів, що утилізуються, який включає пряме ареометричне визначення щільності нітрату амонію та фотоколориметрування пофарбованого розчину аддукту тринітротолуолу з сульфітом натрію, який відрізняється тим, що попередньо вибухову речовину витягають з розчину нітрату амонію методом екстракції, екстрагування...
Спосіб кількісного визначення іонів важких металів у водних розчинах паперовою хроматографією

Номер патенту: 31106
Опубліковано: 15.12.2000
Автори: Борисенко Юлія Володимирівна, Голубєв Анатолій Васильович, Чеховська Людмила Михайлівна
МПК: G01N 30/90, G01N 33/44
Мітки: важких, металів, спосіб, кількісного, паперовою, водних, визначення, хроматографією, розчинах, іонів
Текст:
...розчином осадника - 10% розчином карбамата амонію. На підсушений просочений папір калібровочним капіляром наносять стандартні розчини об'ємом 5мкл. -4Одержують чіткі кольорові круги різних діаметрів, які зменшуються зі зменшенням концентрації хрому таким чином отримують тести , круговоі хроматографії. У таблиці 1 наведені величини діаметрів кругів, що утворилися на папері, просоченому 10% розчином карбамату амонію, 0.04М розчином реактиву...
Спосіб визначення золота і/або паладію у водних розчинах

Номер патенту: 41777
Опубліковано: 10.06.2009
Автори: Трохимчук Анатолій Костянтинович, Ульберг Зоя Рудольфівна, Гудима Наталія Валеріївна
МПК: G01N 31/20, G01N 31/22
Мітки: золота, визначення, спосіб, розчинах, водних, паладію
Формула / Реферат:
1. Спосіб визначення золота і/або паладію у водних розчинах, що включає підкислення розчину і обробку його сорбентом на основі хімічно модифікованого силікагелю, який відрізняється тим, що як сорбент використовують силікагель з хімічно прищепленими групами N-(2-меркаптофеніл)- або N-(4-меркаптофеніл)-N'-пропілсечовини та про наявність у розчині шуканого металу судять за забарвленням використаного сорбенту.2. Спосіб за п. 1, який...
Спосіб визначення токсичності речовин у водних розчинах

Номер патенту: 24287
Опубліковано: 25.06.2007
Автори: Абдураманова Ельвіра Рустамівна, Кацев Андрій Моисейович, Стародуб Микола Федорович
МПК: C12Q 1/00, G01N 33/18
Мітки: розчинах, водних, речовин, визначення, токсичності, спосіб
Формула / Реферат:
Спосіб визначення токсичності речовин у водних розчинах, що включає змішування розведення морських світляних бактерій з аналізованими розчинами, інкубацію їх при температурі 20-25 °С, вимірювання інтенсивності біолюмінесценції і порівняння її з контролем, який відрізняється тим, що використовують для розведення бактерій середовище з рН=5,5-5,8, що містить неінгібуючі концентрації катіонних поверхнево-активних речовин від 0,4 до...
Безекстракційний спосіб спектрофотометричного визначення вмісту алкілсульфатів у водних розчинах

Номер патенту: 55250
Опубліковано: 17.03.2003
Автори: Шаповалов Сергій Андрійович, Чорна Тетяна Олександрівна
МПК: G01N 21/78
Мітки: розчинах, безекстракційний, спосіб, вмісту, спектрофотометричного, визначення, алкілсульфатів, водних
Формула / Реферат:
Безекстракційний спосіб спектрофотометричного визначення вмісту алкілсульфатів у водних розчинах, за яким до аналізованого розчину додається барвник у суміші з безколірною речовиною та вимірюється оптична густина розчину при певній довжині хвилі, який відрізняється тим, що до аналізованого розчину додають барвник тетрабромфенілфлуорон концентрацією моль/дм3 -
Попередній патент: Спосіб оцінки перебігу генералізованого пародонтиту
Наступний патент: Спосіб визначення термінів початку ентерального харчування в ранньому післяопераційному періоді
Випадковий патент: Пристрій для утилізації тепла з складських одиниць твердого палива