Спосіб одержання нітрату бутоконазолу
Номер патенту: 87674
Опубліковано: 10.08.2009
Автори: Веркне Папп Ева, Шебок Ференц, Цібула Ласло, Надьне Багдь Юдіт, Добай Ласло





Формула / Реферат
1. Спосіб одержання нітрату 1-[4-(4-хлорфеніл)-2-(2,6-дихлорфенілтіо)-н-бутил]імідазолу формули (І), при якому здійснюють такі стадії:
a) взаємодію 1-хлор-4-хлорфеніл-2-бутанолу з імідазолом з утворенням 1-[4-(4-хлорфеніл)-2-гідрокси-н-бутил]імідазолу формули (IV)
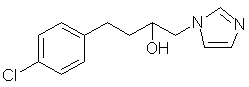 , (IV)
, (IV)
b) взаємодію сполуки формули (IV), одержаної на стадії а), з хлористим тіонілом з утворенням 1-[4-(4-хлорфеніл)-2-хлор-н-бутил]імідазолу формули (V)
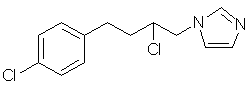 , (V)
, (V)
та
c) взаємодію сполуки формули (V), одержаної на стадії b), з 2,6-дихлортіофенолом,
який відрізняється тим, що стадію а) проводять у суміші розчинника, який не змішується з водою, і водного розчину гідроксиду або карбонату лужного металу у присутності каталізатора міжфазового перенесення;
стадію b) здійснюють у розчині 1,2-дихлоретану у присутності диметилформаміду, де також використовують 1-1,2 моль хлористого тіонілу у розрахунку на кількість сполуки формули (IV); і стадію с) проводять без виділення 1-[4-(4-хлорфеніл)-2-(2,6-дихлорфенілтіо)-н-бутил]імідазолу формули (VI)
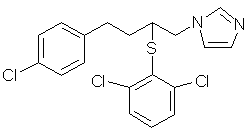 , (VI)
, (VI)
і до бутоконазолу (VI), що знаходиться у розчині, одержаному на стадії с), додають азотну кислоту, і продукт виділяють у вигляді нітратної солі.
2. Спосіб за п. 1, в якому як розчинник, який не змішується з водою, переважно використовують ароматичний вуглеводень.
3. Спосіб за п. 2, в якому як ароматичний вуглеводень переважно використовують толуол.
4. Спосіб за п. 1, в якому як гідроксид або карбонат лужного металу краще використовують гідроксид натрію або карбонат натрію.
5. Спосіб за п. 1, в якому на стадії b) переважно використовують хлористий тіоніл у кількості 1,1 моль.
6. Спосіб одержання нітрату бутоконазолу, в якому принаймні 95 % частинок речовини мають діаметр менше 75 мкм і принаймні 99 % частинок мають діаметр менше 250 мкм, який відрізняється тим, що нітрат бутоконазолу високої чистоти розчиняють у суміші метанолу і метилізобутилкетону у співвідношенні 1-1,5:1 (за об'ємом), потім цей розчин додають до метилізобутилкетону, охолодженого до температури від 5 °С до -15 °С, і виділяють одержаний продукт.
7. Спосіб за п. 6, в якому температура охолодження складає від -5 °С до -10 °С.
8. Спосіб за п. 6, в якому у розчині вихідного матеріалу об'ємне співвідношення метанолу/метилізобутилкетону переважно становить 1,25:1.
Текст
1. Спосіб одержання нітрату 1-[4-(4хлорфеніл)-2-(2,6-дихлорфенілтіо)-нбутил]імідазолу формули (І), при якому здійснюють такі стадії: a) взаємодію 1-хлор-4-хлорфеніл-2-бутанолу з імідазолом з утворенням 1-[4-(4-хлорфеніл)-2гідрокси-н-бутил]імідазолу формули (IV) 3 Одним з об'єктів цього винаходу є нітрат бутоконазолу з частками заданого розміру та спосіб його одержання. Іншим об'єктом цього винаходу є фармацевтична композиція, яка містить нітрат бутоконазолу високого ступеня чистоти з частками заданого розміру у суміші з добавками, відомими з рівня техніки. Нітрат бутоконазолу (хімічна назва: нітрат 1-[4(4-хлорфеніл)-2-(2,6-дихлорофенілтіо)-нбутил]імідазолу) являє собою сполуку формули (І) він відноситься до арилетилімідазолових сполук, має фунгіцидну активність і може бути використаний для лікування піхвових інфекцій, викликаних головним чином грибком Candida albicans. Азоли мають протигрибковий вплив шляхом модифікації синтезу ергостеролу у грибкових клітинах; зокрема, імідазоли інгібують фермент 14aдиметилази і таким чином призводять до підвищення рівня 14a-метилстеролів, які, в свою чергу, викликають зміну проникності клітинної мембрани, що призводить до деструкції грибкових клітин [Tetrahedron: Asymmetry Vol 4, No. 7, pp. 15211526, 1993]. Першим способом одержання нітрату бутоконазолу є багатостадійний синтез, описаний у патенті США 4078071. Для одержання ключового інтермедіата (проміжної речовини) формули (IV) (1[4-(4-хлорфеніл)-2-гідрокси-н-бутил]-імідазол) використовують дві схеми: 87674 4 фоксиду, і епоксид, що утворюється у цій реакції, виділяють після складної обробки. Одержаний таким чином епоксид перетворюють на похідну імідазолу у перебігу досить тривалої реакції у присутності диметилформаміду, після чого інтермедіат формули (IV) (1-[4-(4-хлорфеніл)-2-гідрокси-нбутил]імідазол) виділяють і очищують на додатковій стадії способу. Зі сполук, що мають кінцевий подвійний зв'язок (VII), на стадії епоксидування надкислотою (речовина підвищеної вибухонебезпечності) одержують епоксид, який потім перетворюють на (1-[4-(4хлорфеніл)-2-гідрокси-н-бутил]-імідазол) (IV) способом, описаним вище. Згідно з іншою схемою як вихідний матеріал використовують ядовиту ароматичну a-галоїдкетосполуку, яка вступає у реакцію з імідазолом з утворенням відповідного кетоімідазолу, який, у свою чергу, відновлюють металогідридним комплексом (потенційно небезпечним реагентом) з утворенням ключового інтермедіата (IV). Реакційну суміш обробляють відомим способом. Послідовність синтезу, описана у [J. Med. Chem., 1978, Vol. 21, No. 8, pp. 840-843], виглядає таким чином: 1-хлор-4-хлорфеніл-2-бутанол (II) обробляють імідазолом (III) Згідно з першою з них спочатку одержують епоксидну сполуку з ароматичного альдегіду або з олефінової сполуки, яка має подвійний зв'язок на кінці; потім епоксидна сполука взаємодіє з імідазолом з одержанням ключового інтермедіата. Ароматичний альдегід (VIII) обробляють дорогими та небезпечними реагентами (йодидом триметилсульфоксонію та гідридом натрію) у присутності осушеного диметисуль у присутності гідриду натрію у диметилформаміді, який використовують як розчинник. Ця реакція заміщення відбувається впродовж тривалого часу і призводить до утворення (1-[4-(4хлорфеніл)-2-гідрокси-н-бутил]імідазолу) (IV) з низьким виходом (51,7%). На наступній стадії синтезу нітрату бутоконазолу (1-[4-(4-хлорфеніл)-2гідрокси-н-бутил]імідазол) (IV) обробляють хлористим тіонілом (який одночасно є реагентом і розчинником) при температурі 65-70°С з утворенням 1-[4-(4-хлорфеніл)-2-хлор-н-бутил]імідазолу формули (V). 5 Потім реакційну суміш випарюють до сухого залишку. Для видалення надлишку хлористого тіонілу, сильної корозійно-активної речовини, потрібне спеціальне обладнання; це також відноситься до обробки відходів, тобто цей спосіб відрізняється високим ступенем екологічного ризику. Залишок розчиняють у дихлорметані, розчин підлужують шляхом додавання водного розчину карбонату калію. Фази розділяють, органічний шар промивають водою, висушують над сульфатом магнію і випарюють, щоб одержати 1-[4-(4хлорфеніл)-2-хлор-н-бутил]-імідазол (V) у вигляді смоли. Згадана смола, розчинена в ацетоні, взаємодіє з 2,6-дихлортіофенолом у присутності карбонату калію впродовж тривалого часу. По завершенні реакції неорганічні солі видаляють при фільтруванні, розчинник випарюють, і залишок розподіляють між водою та ефіром. Нітрат бутоконазолу осаджують азотною кислотою з ефірного шару. Кінцевий продукт кристалізується у вигляді пластинок білого кольору із суміші ацетону та етилацетату (вихід: 84%). Метою винаходу є розробка способу, в якому є можливим одержання активної речовини високого ступеня чистоти і з високим виходом, і, крім того, в якому не потрібне використання розчинників, які є вогненебезпечними або вибухонебезпечними (ефір), канцерогенними (диметилформамід) або корозійними (хлористий тіоніл) рідинами, та реагентів (наприклад, гідрид натрію), які є вогненебезпечними або вибухонебезпечними речовинами. Несподівано було виявлено, що у перебігу взаємодії вихідного матеріалу I-хлор-4-хлорфеніл2-бутанолу (II) з імідазолом (III) у суміші толуолу та водного розчину гідроксиду натрію у присутності каталізатору міжфазового перенесення, ключовий інтермедіат (1-[4-(4-хлорфеніл)-2-гідрокси-нбутил]імідазол) (IV) утворюється з високим виходом (95%) впродовж меншого часу. Далі була вивчена можливість застосування альтернативних розчинників для заміни хлористого тіонілу, який виконує функцію розчинника на стадії перетворення (1-[4-(4-хлорфеніл)-2-гідроксин-бутил]імідазолу) (IV) на (1-[4-(4-хлорфеніл)-2хлор-н-бутил]імідазол) (V). При використанні інертних розчинників, таких як дихлорметан, толуол, хлорбензол і диметилформамід, у результаті реакції хлорування одержували в'язку реакційну суміш, обробити яку не було можливо. Однак, несподівано було виявлено, що при розчиненні (1-[4(4-хлорфеніл)-2-гідрокси-н-бутил]імідазолу) (IV) у 1,2-дихлоретані у процесі взаємодії з приблизно еквімолярною кількістю хлористого тіонілу у присутності каталітичної кількості диметилформаміду при температурі 30-35°С утворюється кристалічна суспензія, яка легко перемішується впродовж усього часу реакції, в результаті чого процес хлорування добігає кінця з кількісним виходом 1-[4-(4хлорфеніл)-2-хлор-н-бутил]імідазолу (V). Оскільки 87674 6 сполука є досить чистою, її не ізолюють, але відокремлюють шляхом екстракції і піддають взаємодії безпосередньо з 2,6-дихлортіофенолом у метилізобутилкетоні з утворенням 1-[4-(4-хлорфеніл)-2(2,6-дихолорфенілтіо)-н-бутил]імідазолу (VI) (бутоконазолу). Бутоконазол виділяють з розчину метилізобутилкетону у формі нітратної солі високого ступеня чистоти (індивідуальні домішки, максимум 0,10%) за рахунок додавання концентрованої азотної кислоти. Активна речовина - нітрат бутоконазолу звичайно входить до складу мазі і у такому вигляді надходить у продаж. Для одержання сполуки відповідної якості принаймні 95% часток активної речовини повинні мати діаметр менше 75мкм і принаймні 99% з них повинні бути менше 250мкм. Метою винаходу є розробка способу, який забезпечує одержання продукту з вищезгаданими частками заданого розміру. Несподівано було встановлено, що частки у бажаному діапазоні розмірів можуть бути одержані без способу подрібнення (стадії, що потребує використання спеціального обладнання), таким способом, при якому нітрат бутоконазолу розчиняють у суміші метанолу та метилізобутилкетону, потім цей гарячий розчин додають при перемішуванні до метилізобутилкетону, попередньо охолодженому до температури 5°С або нижче. При таких умовах одержують кінцевий продукт з частками бажаного розміру. Таким чином, одним з об'єктів цього винаходу є нітрат бутоконазолу високого ступеня чистоти формули (І) (хімічна назва: нітрат 1-[4-(4хлорфеніл)-2-(2,6-дихлорфенілтіо)-нбутил]імідазолу) із вмістом хімічних домішок максимум 0,1мас.% . Іншим об'єктом цього винаходу є спосіб одержання згаданого вище продукту, при цьому згаданий спосіб має у своєму складі стадії: a) взаємодії І-хлор-4-хлорфеніл-2-бутанолу з імідазолом формули (III) з утворенням сполуки формули (IV); b) взаємодії сполуки формули (IV), одержаної на стадії а), з хлористим тіонілом з утворенням сполуки формули (V); і c) взаємодії сполуки формули (V), одержаної на стадії b), з 2,6-дихлортіофенолом, який відрізняється тим, що: стадію а) проводять у суміші розчинника, який не змішується з водою, і водного розчину гідроксиду або карбонату лужного металу у присутності каталізатора міжфазового перенесення; стадію b) здійснюють у розчині 1,2дихлоретану у присутності диметилформаміду, в якій також використовують 1-1,2моль хлористого 7 87674 8 тіонілу у розрахунку на кількість сполуки формули Одержання нітрату 1-[4-(4-хлорФеніл-2-(2,6(IV); і дихлорфенілтіо-н-бутил]імідазолу (І) стадію с) проводять без виділення продукту Суспендують у 125мл 1,2-дихлоретану 25г (VI); і (0,1моль) 1-[4-(4-хлорфеніл)-2-гідрокси-2-ндо бутоконазолу (VI), що перебуває у розчині, бутил]імідазолу (IV), до цієї суспензії додають диодержаному на стадії с), додають азотну кислоту, і метилформамід (1мл) і хлористий тіоніл (13,6г; продукт виділяють у вигляді нітратної солі. 0,11моль) при температурі 30-32°С, і реакційну У кращому варіанті здійснення винаходу як суміш витримують при температурі 35-38°С впророзчинник, що не змішується з водою, використодовж 1,5 годин при перемішуванні. По завершенні вують ароматичний вуглеводень, як гідроксид або реакції хлорування гомогенний розчин охолоджукарбонат лужного металу використовують гідроють до температури 15-18°С, надлишок хлористоксид натрію або карбонат натрію, і на стадії с) виго тіонілу розкладають водою (10мл), потім знову користовують 1,1моль хлористого тіонілу. до розчину додають воду (80мл). Після перемішуВ іншому варіанті здійснення винаходу аромавання при температурі 20-22°С впродовж 0,5 годитичним вуглеводнем є толуол. ни фази відокремлюють, і органічний шар екстраДодатковим об'єктом винаходу є нітрат бутогують водою (30мл). До водного розчину додають коназолу високого ступеня чистоти, який містить метилізобутилкетон (250мл), і значення рН суміші максимум 0,1мас.% хімічних домішок, в якому прирегулюють у діапазоні 8,5-9, додаючи 15г наймні 95% часток речовини мають діаметр мен(0,14моль) карбонату натрію, розчиненого у воді ше 75мкм і принаймні 99% часток мають діаметр (70мл). Суміш перемішують при температурі 22менше 250мкм. 25°C впродовж 0,5 години, фази відокремлюють, з Ще одним об'єктом винаходу є спосіб одерорганічного шару відганяють фракцію (50мл) для жання згаданого вище продукту, виходячи з нітравидалення води, і до розчину, що залишився, доту бутоконазолу високого ступеня чистоти, таким дають 26,8г (0,15моль) 2,6-дихлортіофенолу і 40г чином, що вихідний матеріал розчиняють у суміші (0,29моль) сухого карбонату калію. Суспензію пеметанолу і метилізобутилкетону у співвідношенні ремішують при температурі 105-108°С в атмосфе1-1,25:1 (за об'ємом), охолодженій до температури рі азоту впродовж 3-4 годин. По завершенні реакції від 5°С до -15°С, потім виділяють одержаний пронеорганічні солі видаляють фільтрацією при темдукт. У кращому варіанті здійснення способу температурі 22-25°С, фільтрат промивають і освітлюпература охолодження складає від -5°С до -10°С, ють, додаючи активоване вугілля, і підкислюють а об'ємне співвідношення метапрозорий розчин до рН 3-3,5 шляхом додавання нол/метилізобутилкетон дорівнює 1,25:1. приблизно 8-9мл 65%-ної азотної кислоти. Розчин До обсягу винаходу також входить фармацевперемішують при тій самій температурі впродовж тична композиція, яка містить згаданий вище про1 години, потім температуру знижують до 8-12°С. дукт, одержувана у перебігу згаданого вище споОдержані кристали фільтрують і промивають, щоб собу у вигляді суміші з добавками, відомими з одержати 48г вологого нітрату 1-[4-(4-хлорфеніл)рівня техніки. 2-(2,6-дихлорфенілтіо)-н-бутил]імідазолу, що відКрім того, винахід ілюструють подальші приповідає 42,6г (90%) сухого продукту. клади, які не обмежують приклади здійснення виПриклад 3. находу. Одержання нітрату 1-[4-(4-хлорфеніл)-2-(2,6Прикладі. дихлорфенілтіо)-н-бутилімідазолу з частками заОдержання (1-[4-(4-хлорфеніл)-2-гідрокси-нданого розміру бутил]імідазолу) (IV) Розчиняють 48г вологого нітрату 1-[4-(4До розчину, який містить 56,7г (0,26моль) Іхлорфеніл)-2-(2,6-дихлорфенілтіо)-нхлор-4-хлорфеніл-2-бутанолу [J. of Medicinal бутилімідазолу, одержаного як описано у прикладі Chemistry, 1978. Vol. 21. No. 8. p. 842] у 200мл то2, у суміші метанолу (120мл) і метилізобутилкетолуолу, додають 36,2г (0,9моль) гідроксиду натрію, ну (96мл) при температурі 65-70°С, і цей розчин розчиненого у 100мл води, 6,4г (0,028моль) хловводять тонким струменем у 96мл метилізобутилриду бензилтриетиламонію і 35,2г (0,51моль) імікетону, охолодженого до -5°С. Одержану кристалідазолу (III). Реакційну суміш нагрівають до темпечну суспензію витримують при температурі 0-10°С ратури 93-95°С впродовж однієї години, потім при перемішуванні впродовж 1 години, потім фільтемпературу знижують до 60°С, фази розділяють, і трують, промивають метилізобутилкетоном (2 рази до органічного шару додають воду (100мл). Потім по 15мл) при температурі 0-5°С і висушують при суміш перемішують спочатку при температурі 2250°С у вакуумі до постійної ваги. 25°С впродовж 1 години, потім при температурі 0Суха вага: 40,5г (95%) 5°С впродовж двох годин. Кристали відокремлюЗагальна кількість домішок: 0,05% (визначено ють фільтрацією, промивають водою (2 рази по методом високоефективної рідинної хроматографії 35мл) з температурою 0-5°С, одержуючи 74г воло(ВЕРХ)) гого (1-[4-(4-хлорфеніл)-2-гідрокси-нРозміри часток:
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparation of butoconazole nitrate
Автори англійськоюCzibula Laszlo, Dobay Laszlo, Werkne Papp Eva, Nagyne Bagdy Judit, Sebok Ferenc
Назва патенту російськоюСпособ получения нитрата бутоконазола
Автори російськоюЦибула Ласло, Добай Ласло, Веркне Папп Эва, Надьне Багдь Юдит, Шебок Ференц
МПК / Мітки
МПК: C07D 233/60, A61P 31/10, A61K 31/4174
Мітки: нітрату, одержання, бутоконазолу, спосіб
Код посилання
<a href="https://ua.patents.su/5-87674-sposib-oderzhannya-nitratu-butokonazolu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання нітрату бутоконазолу</a>
Спосіб спільного одержання розчину нітрату кальцію та нітрату амонію

Номер патенту: 59598
Опубліковано: 15.09.2003
Автори: Кіященко Дмитро Володимирович, Криштопа Павло Петрович, Барабаш Іван Іванович, Сергієнко Іван Данилович, Пояркова Ізольда Федорівна, Купрін Віталій Павлович
МПК: C05C 5/00
Мітки: спільного, одержання, амонію, спосіб, кальцію, розчину, нітрату
Формула / Реферат:
Спосіб спільного одержання розчину нітрату кальцію та нітрату амонію, що включає спочатку одержання нітрату кальцію прямим розкладенням вапняку з надлишком азотної кислоти, подальшою його амонізацією, який відрізняється тим, що в реактор, обладнаний механічним піногасником та мішалкою, вводять спочатку азотну кислоту концентрації не вище 58 %, а потім вапняк, при цьому надлишок азотної кислоти становить не менш ніж 80 % порівняно із...
Спосіб одержання гранульованого амонію сульфат- нітрату

Номер патенту: 39625
Опубліковано: 17.05.2004
Автори: Криштопа Павло Петрович, Пояркова Ізольда Федорівна, Кирилюк Олександр Федорович, Білик Андрій Михайлович, Березюк Зеновій Миколайович, Галечко Богдан Миронович, Барабаш Іван Іванович, Сергієнко Іван Данилович, Хижніченко Леонід Пилипович
МПК: C05C 1/00, C05C 1/02, B01J 2/00, C05C 13/00, C05G 5/00, C05G 1/00, C05C 3/00
Мітки: спосіб, сульфат, нітрату, амонію, гранульованого, одержання
Формула / Реферат:
Спосіб одержання гранульованого амонію сульфат-нітрату (АSN), що включає грануляцію, сушіння та охолодження готового продукту, який відрізняється тим, що плав нітрату амонію вмістом не менше 88 % мас. напилюють на суміш сульфату амонію та ретуру, після чого масу гранулюють в барабані-грануляторі при температурі 70-90 °С, підтримуючи вологість в межах 2,5-4,0 %, причому розчин зі стадії очистки "хвостових" газів, який містить...
Спосіб одержання нітрату графіту, що терморозширюється

Номер патенту: 61363
Опубліковано: 17.11.2003
Автори: Ярошенко Олександр Павлович, Магазинський Олександр Миколайович, Галушко Леонід Якович, Мисик Роман Дмитрович, Шологон Віктор Іванович, Савоськін Михайло Віталійович
МПК: C01B 31/04
Мітки: спосіб, одержання, нітрату, терморозширюється, графіту
Формула / Реферат:
Спосіб одержання нітрату графіту, що терморозширюється, який містить обробку природного лускатого графіту димучою азотною кислотою при вилученні газуватих продуктів з реакційної маси та його подальше сушіння до постійної ваги при температурі навколишнього середовища, який відрізняється тим, що графіт після його обробки димучою азотною кислотою перед сушінням додатково витримують у насиченому водяною парою повітрі протягом 18-24 годин.
Спосіб одержання нітрату графіту, що терморозширюється

Номер патенту: 79387
Опубліковано: 11.06.2007
Автори: Савоськін Михайло Віталійович, Ярошенко Олександр Павлович, Шологон Віктор Іванович, Хрипунов Сергій Васильович
МПК: C01B 31/04
Мітки: одержання, графіту, нітрату, терморозширюється, спосіб
Формула / Реферат:
Спосіб одержання нітрату графіту, що терморозширюється, який включає обробку природного лускатого графіту димлячою азотною кислотою при вилученні газуватих продуктів з реакційної маси, який відрізняється тим, що обробку графіту димлячою азотною кислотою здійснюють при температурі від 65 °С до 90 °С.
Пристрій для одержання розчину нітрату амонію та спосіб одержання розчину нітрату амонію

Номер патенту: 76139
Опубліковано: 17.07.2006
Автори: Савчук Микола Петрович, Білик Руслан Володимирович, Мазніченко Сергій Васильович, Малишко Микола Семенович
Мітки: розчину, одержання, пристрій, нітрату, амонію, спосіб
Формула / Реферат:
1. Пристрій для одержання розчину нітрату амонію, що містить циліндричний корпус із днищем і штуцерами для відводу розчину, виміру рівня і тиску, кришку зі штуцером для відводу сокової пари, який відрізняється тим, що усередині корпуса на деякій відстані від днища змонтований вузол нейтралізації, що містить два концентрично встановлені стакани, сполучені між собою двома рядами отворів, що виконані у внутрішньому стакані внизу біля днища...
Попередній патент: Рейкова транспортна система
Наступний патент: Аспартати, придатні як компоненти композицій для покриттів, та їх одержання
Випадковий патент: Спосіб одержання суміші d-пантотенової кислоти і її солей