Спосіб одержання і виділення глікозидів індолокарбазолу
Номер патенту: 74249
Опубліковано: 15.11.2005
Автори: Кавасакі Масасі, Хірага Соуйті, Вайссман Стівен, Ійда Такехіко, Чаєн Девід, Каматані Асаюкі






Формула / Реферат
1. Спосіб одержання кристалів сполуки І:
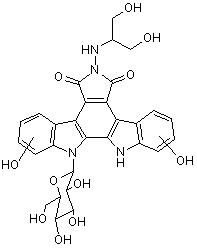 (I)
(I)
високого ступеня чистоти, при якому проводять:
(a) доведення рН кислої суміші, що складається по суті з кислоти,спирту, води і сполуки І, шляхом додання основи до одержання розчину з рН в інтервалі від приблизно 1,5 до приблизно 6,5;
(b) підтримування температури розчину зі стадії (а) в інтервалі від приблизно 50°С до приблизно 100°С; і
(c) виділення кристалів сполуки І.
2. Спосіб за пунктом 1, при якому проводять:
(a) доведення рН кислої суміші, що складається в основному з кислоти, спирту, води і сполуки І, шляхом додання основи до одержання розчину з рН в інтервалі від приблизно 1,5 до приблизно 6,5;
(b) регулювання розчину зі стадії (а) спиртом до утворення розчину, що містить від приблизно 10% (вага/об'єм) до приблизно 30% (вага/об'єм) води в спирті і з концентрацією сполуки І від приблизно 10 мл/г до 20 мл/г;
(c) доведення температури розчину зі стадії (b) до температури в інтервалі від приблизно 50°С до приблизно 100°С;
(d) додавання спирту до розчину зі стадії (с) до одержання розчину, в якому відношення (розчин:спирт) становить приблизно 3:2;
(e) витримування розчину зі стадії (d) при температурі в інтервалі від приблизно 50°С до приблизно 100°С до утворення кристалів сполуки І з утворенням суспензії; і
(f) виділення кристалів сполуки І.
3. Спосіб за пунктом 2, при якому додатково проводять видалення захисних груп з проміжної сполуки II:
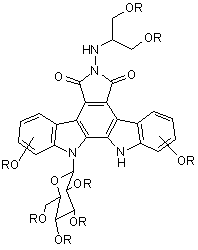 (II)
(II)
(де R незалежно один від одного означають водень або заміщену або незаміщену бензильну захисну групу; при умові, що щонайменше один R означає заміщену або незаміщену бензильну захисну групу) гідруванням в присутності каталізатора з утворенням реакційної суміші з подальшим фільтруванням реакційної суміші з одержанням суміші стадії (а).
4. Спосіб за пунктом 3, де R означає бензил.
5. Спосіб одержання сполуки III високого ступеня чистоти
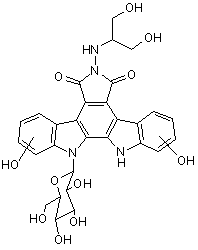 (III),
(III),
при якому проводять:
(а) доведення рН кислої суміші, що складається в основному з кислоти, спирту, води і сполуки III, шляхом додавання основи до одержання розчину з рН в інтервалі від приблизно 1,5 до приблизно 6,5;
(b) регулювання розчину зі стадії (а) спиртом до утворення розчину, що містить від приблизно 10% (вага/об'єм) до приблизно 30% (вага/об'єм) води в спирті і з концентрацією сполуки III від приблизно 10 мл/г до 20 мл/г;
(c) доведення температури розчину зі стадії (b) до температури в інтервалі від приблизно 50°С до приблизно 100°С;
(d) внесення затравки в розчин;
(e) додавання спирту до розчину до розбавлення розчину у відношенні приблизно 3:2 (розчин:спирт);
(f) витримування розчину зі стадії (е) при температурі в інтервалі від приблизно 50°С до приблизно 100°С до утворення кристалів сполуки III з утворенням суспензії; і
(g) виділення кристалів сполуки III.
6. Спосіб за пунктом 5, при якому додатково проводять видалення захисних груп з проміжної сполуки II:
(II)
(де R незалежно один від одного означають водень або заміщену або незаміщену бензильну захисну групу; при умові, що щонайменше один R означає заміщену або незаміщену бензильну захисну групу) гідруванням в присутності каталізатора з утворенням реакційної суміші з подальшим фільтруванням реакційної суміші з одержанням суміші стадії (а).
7. Спосіб за пунктом 5 або 6, де рН на стадії (а) доводять до значення в інтервалі від приблизно 1,5 до приблизно 3,5; розчин на стадії (b) доводять до розчину, що містить від приблизно 15% (вага/об'єм) до приблизно 25% (вага/об'єм) води в спирті з концентрацією сполуки III від приблизно 12 мл/г до приблизно 18 мл/г; і температуру на стадії (с) доводять до приблизно 70°С.
8. Спосіб за пунктом 7, де на стадії (а) доводять рН до приблизно 2,5; розчин на стадії (b) доводять до розчину, що містить 20% (вага/об'єм) води в спирті з концентрацією сполуки III приблизно 15 мл/г; і температуру на стадії (с) доводять до приблизно 70°С.
9. Спосіб за пунктом 8, при якому додатково проводять стадію регулювання суспензії після стадії (f) таким чином, щоб перед виділенням кристалів сполуки III на стадії (g) вміст води в суспензії був знижений до інтервалу від приблизно 1% (вага/об'єм) до приблизно 10% (вага/об'єм).
10. Спосіб за пунктом 5, при якому проводять:
(a) доведення рН кислої суміші, що складається в основному з кислоти, спирту, води і сполуки III, шляхом додання основи до одержання розчину з рН приблизно 2,5;
(b) регулювання розчину зі стадії (а) ізопропіловим спиртом до утворення розчину, що містить від приблизно 20% (вага/об'єм) води в ізопропіловому спирті і з концентрацією сполуки І приблизно 15 мл/г;
(c) доведення температури розчину зі стадії (b) до температури приблизно 70°С;
(d) внесення затравки в розчин;
(e) додавання ізопропілового спирту до розчину до одержання розчину, в якому співвідношення (розчин:спирт) становить приблизно 3:2;
(f) витримування розчину зі стадії (е) при температурі приблизно 70°С до утворення кристалів сполуки І з утворенням суспензії;
(g) регулювання суспензії таким чином, щоб вміст води в ній становив приблизно 3% (вага/об'єм);
(h) витримування суспензії при приблизно 70°С перед охолоджуванням до приблизно 22°С; і
(і) виділення кристалів сполуки III.
11. Спосіб за пунктом 10, при якому додатково проводять видалення захисних груп з проміжної сполуки II:
(II)
(де R незалежно один від одного означають водень або заміщену або незаміщену бензильну захисну групу; при умові, що щонайменше один R означає заміщену або незаміщену бензильну захисну групу) гідруванням в присутності каталізатора з утворенням реакційної суміші з подальшим фільтруванням реакційної суміші з одержанням суміші стадії (а).
12. Спосіб за пунктом 11, де нижчим алкіламіном на стадії (а) є триетиламін.
Текст
Даний винахід відноситься до нового способу одержання глікозидів індолокарбазолу високого ступеня чистоти, які інгібують ріст пухлинних клітин і внаслідок цього застосовні при лікуванні раку у ссавців і подібних (захворювань). В області хіміотерапії раку вже застосовується велике число сполук для практичного застосування як протипухлинних агентів. Однак залишається необхідність в розробці більш ефективних сполук, які ефективні проти різних пухлин [дивись Proceedings of the 47th General Meeting of the Japan Cancer Society, стор. 12-15 (1988)]. Це приводить до необхідності розробки похідних індолокарбазолу. [Дивись патенти US №№ 4487925; 4552842; 4785085; 5591842 і 5922860; патент Японії 20277/91; Journal of Antibiotics, том 44, стор. 723-728 (1991); патенти WO91/18003; WO98/07433; і ЕРО545195 ΑΙ]. Показано, що вказані сполуки діють як інгібітори топоізомерази і тому застосовні при лікуванні раку [Cancer Chemother. Pharmacol. 34 (suppl): S41-S45 (1994)]. Успіх вказаних сполук в лікуванні численних ракових захворювань примушує розробляти вдосконалені способи їх синтезу, [дивись Bioorg. & Med. Chem. Letters 2000, 10, 419; Tetrachedron 1997, 53, 5937; Tetrahedron 1997, 53, 585 і Synthesis 1976, 414]. Однак раніше відомі способи мають численні проблеми, такі як використання небажаних розчинників, солей ртуті або срібла, низький вихід і утворення небажаних побічних продуктів, що приводять до обов'язкових стомлюючих або затяжних стадій очищення. Наприклад, раніше відомі способи одержання глікозидів індолокарбазолу III високого ступеня чистоти вимагають процедур очищення, таких як комбінації обробки вугіллям, хроматографії і/або перекристалізації сирого матеріалу, які є трудомісткими, що вимагають багато часу і небезпечними, особливо якщо здійснюються в промисловому масштабі, через високу цитотоксичну природу продукту [дивись Bioorg & Med Chem Letters 1999, 3307; і Tetrahedron 1997, 585 (описаний синтез структурно подібної сполуки, що вимагає повторного розчинення сирого продукту для одержання чистого матеріалу)]. Отже, метою даного винаходу є розробка нового шляху до вказаних протипухлинних речовин, похідних індолопіролокарбазолу, який дозволив би подолати проблеми, властиві раніше відомим синтезам, особливо шляху, що дає продукт достатнього ступеня чистоти, щоб можна було використовувати його в подальших композиціях «як такий». Даний винахід відноситься до нового способу одержання глікозидів індолокарбазолу формули І високого ступеня чистоти, які інгібують ріст пухлинних клітин і внаслідок цього застосовні при лікуванні ракових захворювань у ссавців і подібних (захворювань). Один з варіантів здійснення даного винаходу ілюструється способом одержання сполуки формули І високого ступеня чистоти, який включає стадії: (a) доведення рН кислої суміші, що складається в основному з спирту, кислоти, води і сполуки І, шляхом додання основи до утворення розчину з рН в інтервалі від приблизно 1,5 до приблизно 6,5; (b) підтримання температури розчину стадії (а) в інтервалі від приблизно 50°С до приблизно 100°С; і (c) виділення кристалів сполуки І. У відповідності до другого варіанту здійснення даного винаходу пропонується спосіб, що включає стадії: (a) доведення рН кислої суміші, що складається в основному з кислоти, спирту, води і сполуки І, шляхом додавання основи до утворення розчину з рН в інтервалі від приблизно 1,5 до приблизно 6,5; (b) регулювання розчину стадії (а) спиртом до одержання розчину, що містить від приблизно 10% (вага/об'єм) до приблизно 30% (вага/об'єм) води в спирті з концентрацією сполуки І приблизно від 10мл/г до 20мл/г; (c) доведення температури розчину стадії (b) до температури в інтервалі від приблизно 50°С до приблизно 100°С; (d) додання спирту до розчину стадії (с) таким чином, щоб розбавити розчин в співвідношенні 3:2 (розчин:cпирт); (є) витримування розчину стадії (d) при температурі в інтервалі від приблизно 50°С до приблизно 100°С до появи кристалів сполуки І з утворенням суспензії; і (f) виділення кристалів сполуки І. Додатковий варіант здійснення винаходу представляє спосіб, що додатково включає видалення захисних груп проміжної сполуки II: (де R незалежно один від одного означають водень або заміщену або незаміщену бензильну захисну групу; причому щонайменше один R означає незаміщену або заміщену бензильну захисну групу) гідруванням в присутності каталізатора з утворенням реакційної суміші з подальшим фільтруванням реакційної суміші, для одержання суміші стадії (а). В альтернативному варіанті здійснення винаходу R означає бензил в способі, описаному вище. У третьому варіанті здійснення даного винаходу пропонується спосіб одержання сполуки III високого ступеня чистоти що включає стадії: (а) доведення рН кислої суміші, що складається в основному з кислоти, спирту, води і сполуки III, шляхом додання основи до утворення розчину з рН в інтервалі від приблизно 1,5 до приблизно 6,5; (b) регулювання розчину стадії (а) спиртом до одержання розчину, в якому міститься від приблизно 10% (вага/об'єм) до приблизно 30% (вага/об'єм) води в спирті з концентрацією сполуки III приблизно від 10 мл/г до 20 мл/г; (c) доведення температури розчину стадії (b) до температури в інтервалі від приблизно 50°С до приблизно 100°С; (d) внесення в розчин затравки; (є) додання спирту до одержаного розчину таким чином, щоб розбавити розчин у (відношенні) 3:2 (розчин:cпирт); (f) витримування розчину стадії (є) при температурі в інтервалі від приблизно 50°С до приблизно 100°С до утворення кристалів сполуки III з одержанням суспензії; і (g) виділення кристалів сполуки III. Відповідно до третього варіанту здійснення винаходу спосіб додатково передбачає стадію видалення захисних груп з проміжної сполуки II: (де R незалежно один від одного означають водень або заміщену або незаміщену бензильну захисну групу; причому щонайменше один R означає заміщену або незаміщену бензильну захисну групу) шляхом гідрування в присутності каталізатора з утворенням реакційної суміші, з подальшим фільтруванням реакційної суміші з одержанням суміші стадії (а). Ще одним варіантом здійснення винаходу є спосіб, описаний вище, де рН на стадії (а) доводять до рН в інтервалі від приблизно 1,5 до приблизно 3,5; розчин на стадії (b) регулюють таким чином, щоб він мав від приблизно 15% (вага/об'єм) до приблизно 25% (вага/об'єм) води в спирті і концентрація сполуки III була від приблизно 12 мл/г до приблизно 18мл/г; і температуру на стадії (с) доводять до приблизно 70°С. Додатковим виконанням є спосіб, вказаний вище, де рН на стадії (а) доводять до рН приблизно 2,5; розчин на стадії (b) регулюють таким чином, щоб розчин мав приблизно 20% (вага/об'єм) води в спирті і концентрація сполуки III була приблизно 15 мл/г; і температуру на стадії (с) доводять до приблизно 70°С. Даним винаходом охоплюється також спосіб, описаний вище, що додатково передбачає стадію регулювання суспензії стадії (f) таким чином, щоб вміст води меншав до від приблизно 1% (вага/об'єм) до приблизно 10% (вага/об'єм) перед виділенням кристалів сполуки III на стадії (g). Переважним варіантом здійснення винаходу є спосіб одержання сполуки III високого ступеня чистоти, який включає стадії: (a) доведення рН кислої суміші, що складається в основному з кислоти, спирту, води і сполуки III, шляхом додання нижчої алкіламінової основи для утворення розчину з рН приблизно 2,5; (b) регулювання розчину стадії (а) ізопропіловим спиртом до одержання розчину, в якому приблизно 20% (вага/об'єм) води в ізопропіловому спирті з концентрацією сполуки І приблизно 15мл/г; (c) доведення температури розчину стадії (b) до температури приблизно 70°С; (d) внесення затравки в розчин; (e) додання до розчину ізопропілового спирту для одержання розчину, в якому співвідношення (розчин:ізопропіловий спирт) становить приблизно 3:2. (f) витримування розчину стадії (є) при температурі приблизно 70°С до утворення кристалів сполуки І з утворенням суспензії; (g) регулювання суспензії таким чином, щоб вміст води в ній становив приблизно 3% (вага/об'єм); (h) витримування суспензії при приблизно 70°С перед охолоджуванням до приблизно 22°С; і (і) виділення кристалів сполуки III. У додатковому варіанті переважного здійснення винаходу спосіб, що пропонується додатково включає видалення захисних груп з проміжної сполуки II: (де R незалежно один від одного означають водень або заміщену або незаміщену бензильну захисну групу; причому щонайменше один R означає заміщену або незаміщену бензильну захисну групу) шляхом гідрування в присутності каталізатора з подальшим фільтруванням реакційної суміші, надаючи суміш стадії (а). Крім того, в об'ємі даного винаходу розкривається спосіб, описаний вище, де нижчим алкіламіном на стадії (с) є триетиламін. Даний винахід надає продукт, який може безпосередньо кристалізуватися з реакційного середовища без додаткових стадій очищення. Крім того, запропонований спосіб дає кристали з більш стабільною кристалічною геометрією, 3-D-трапецоїди на відміну від раніше відомих способів, (що дають) 2-D-голки, що доведено кінетикою розчинності. Сполука І і сполука III можуть бути синтезовані згідно з способом, описаним в патенті US №5591842, опублікованому 7 січня 1997, приведеному тут як посилання. Порошок сполуки III, одержаний за способом, описаним в патенті WO 95/30682 і в патенті US №5591842, кристалізують за способом даного винаходу. Використаний в даному описі термін «високого ступеня чистоти» відноситься до продукту, що містить 1% або менше загальних домішок, як виміряно за допомогою ВЕРХ (високоефективної рідинної хроматографії). «Кислотна суміш» відноситься до суміші, яка має рН менше приблизно 7,0. Більш переважно рН кислотної суміші менше приблизно 2,5. Для даного опису види кислот, які можуть бути використані, включають, але не обмежують, безводні або водні HF, HCl, НВr, НІ, HNO3, НсlО4, сірчану, фосфорну, пропіонову кислоти, MsOH, TsOH, монофосфатну сіль, дифосфатну сіль, фосфатну змішану сіль, карбонові кислоти або галогеніди амонію. Фосфатна змішана сіль може бути представлена як М1 М2НРО4, де М1 і М2 незалежно вибрані з Н, Na, Κ, ΝΗ4ΟΗ, калій-натрію і подібних. Більш переважно як кислота вибрана НСl. Вибір каталізатора в описаних реакціях гідрогенолізу може бути очевидний для фахівців в даній області. Відповідними каталізаторами є паладій на вугіллі, Pd(OH)2, нікель Ренея, вольфрамові каталізатори, Rh/AI 2O3 і подібні. Переважними є паладієві каталізатори, такі як паладій на вугіллі. «Внесення затравки» відноситься до введення в розчин кристалів (затравкових кристалів), щоб каталізувати осадження кристалів з розчину. Внесення затравки може бути здійснене доданням затравкових кристалів в твердому сухому вигляді, або затравкові кристали можуть бути додані у вигляді суспензії в рідині. «Затравковими кристалами» можуть бути кристали тієї ж сполуки, які індукують осадження, або можуть бути іншою сполукою. У цьому випадку переважно внесення затравки у вигляді суспензії тієї ж сполуки. «Витримування» означає витримку розчину при постійній температурі і об'ємі протягом певного періоду часу. Час реакцій в даному винаході не є критичним, якщо спеціально не вказано інакше, і може бути легко встановлено фахівцями в даній області. «Фільтрування» означає пропущення розчину через деяке середовище, так що частки речовини видаляються. Вибір середовища не є критичним і може бути легко вибраний на практиці. Фільтрування може здійснюватися через целіт, солка-флок, пісок, склуватий шлак, діатомову землю і подібні. «Спирт» означає органічну молекулу з лінійним або розгалуженим ланцюгом з 1-5 атомами вуглецю з щонайменше однією гідроксильною групою, виступаючою як активна група. Спиртами є метанол, пропанол, ізопропанол, бутанол, втор-бутанол і т.д. Переважним спиртом є ізопропіловий спирт. Термін «заміщена бензильна захисна група» означає, але не обмежує, пара-МеО-бензил, ортонітробензил, пара-нітробензил, пара-галогенбензил (де галогеном може бути хлор, бром і йод), 2,6дихлорбензил, дифенілметил, трифенілметил і подібні. Додаткові відповідні захисні групи можуть бути знайдені в Protective Groups in Organic Chemustry Peter G. Wuts і Theodora W. Green, John Wiley & Sons, 3-е вид. (1999). Даний винахід передбачає стадію, де рН фільтрату доводять до певного рівня. Значення рН може бути встановлене за допомогою будь-якої відповідної основи, такої як триетиламін, діізопропілетиламін, трибутиламін, піридин, 2,6-лутидин, 2,4,6-лутидин, ДБУ (діазабіцикло[5.4.0]ундец-7-ен), DBN (1,5діазабіцикло[4.3.0]но-5-ен), діізопропіламін, Ν,Ν-диметиланілін, DABCO (1,4-діазабіцикло[2.2.2]октан), Nалкілморфолін і подібні. Переважні нижчі алкіламінові основи. Найбільш переважний триетиламін. «Суспензія» відноситься до суспензії твердої речовини або кристалів в рідині. Тверда речовина може бути частково, неповністю або повністю нерозчинна в рідині. Схема А ілюструє узагальнений підхід до одержання біологічно активних глікозидів індолокарбазолу шляхом видалення захисних груп/кристалізації з одержанням продукту з високим виходом і високого ступеня чистоти, запобігаючи необхідності в подальших стадіях очищення перед складанням рецептури. ПРИКЛАДИ Приклади сприяють подальшому розумінню винаходу. Використані конкретні матеріали, види і умови мають на увазі подальшу ілюстрацію винаходу і не обмежують області його застосування. ПРИКЛАД 1 Спосіб одержання сполуки 1-1 описаний в патенті WO95/30682 (еквівалент: патент US 5804564), приведеному тут як посилання. Етанол (14,0 л) додавали по краплях до двофазної суміші сполуки 1-1 (1,55кг, 1,44 моль), толуолу (5,6л) і 48%-ного водного КОН (4,15кг) в 50-літровій скляній посудині протягом 0,5 години при кімнатній температурі, підтримуючи внутрішню температуру нижче 30°С. Одержану темно-червону суміш перемішували при 20-30°С 12 годин, причому суміш за цей час перетворювалася в гомогенний червоний розчин. Потім суміш додатково витримували при -5°С протягом години, після чого повільно додавали 10%ну водну лимонну кислоту (23,5кг) до одержання суміші з рН 7,7-8,0, підтримуючи внутрішню температуру нижче 5°С. Одержану суміш нагрівали і перемішували при 25-30°С 7 годин, періодично додаючи за цей час додатково 10%-ну водну лимонну кислоту (1,77кг), зберігаючи значення рН при 7,5-8,0. Суміш потім екстрагували МТВЕ (трет-бутилметиловим ефіром) (15,5л) і відділений органічний шар промивали 3%-ним водним NaCl (2x3,1л) і 25%-ним водним NaCl (3,1л), сушили (Na2SO4) і обробляли вугіллям (Darco G-60, 155г, кімнатна температура, 1 година). Профільтрований розчин концентрували у вакуумі до об'єму 6 л і діяли струменем MeCN (2x15л), кожний раз упарюючи у вакуумі до об'єму 6л (залишковий толуол: 9%). Суміш потім розбавляли MeCN до одержання 23,3л розчину і повільно додавали МеОН (3,0л) за 0,5 години при 22-25°С з подальшим внесенням затравки продукту (1-2), яка ініціювала кристалізацію. Одержану суміш потім витримували при вказаній температурі 1 годину з подальшим повільним доданням МеОН (17,6л) протягом 1 години. Одержану жовту суспензію витримували при 22-25°С 1 годину і потім витримували при 0-5°С 3 години. Кристали відділяли фільтруванням, промивали сумішшю (9:1 по об'єму) MeCN/MeOH (15,5л) і сушили у вакуумі, одержуючи 1-2. 1 Н-ЯМР (270 МГц, CDCl3, м.д.): 10,79 (1Н, шир.с,), 9,04 (1Н, д, J=9,2Гц), 8,95 (1Н, д, J=9,6Гц), 7,26 (32Н, м), 6,17 (2Н, д, J=7,3Гц), 5,85 (1Н, д, J=8,2Гц), 4,89 (10Н, м), 4,32 (1Н, т, J=8,9Гц), 3,96 (6Н, м), 3,13 (1Н, д, J=10,2Гц). ПРИКЛАД 2 Додавали 12,1%-ний водний NaCIO (попередньо відтитрований водним Na2S2O3, 4,06кг, 6,61 моль) до суміші, що перемішується, 1,3-дибензилокси-2-пропанолу (сполука 2-1, 1,50кг, 5,51моль), вільного радикала 2,2,6,6-тетраметил-1-піперидинілокси (TEMPO, 86,0г, 0,550моль), MeCN (20,6л) і 3%-ного водного NaHCO3 (15,5кг, 5,51моль) в 50-літровій скляній посудині при 0°С за 1,5 години, підтримуючи внутрішню температуру 0-5°С. Одержану суміш перемішували ще протягом години при 0-5°С і екстрагували МТВЕ, метил-третбутиловим ефіром (41л) при температурі нижче 10°С. Відділений органічний шар промивали 10%-ним водним Na2SO3 (5,0кг) при температурі нижче 10°С і потім 5%-ним водним NaCl (3,0кг) і 1%-ним NaCl (3,0кг) при кімнатній температурі. Ясно-червоний органічний шар потім піддавали ВЕРХ (високоефективній рідинній хроматографії) і підраховували вміст 1,48кг (5,49 моль) бажаного кетону (сполука 2-2). Потім розчин вміщували в 50-літрову посудину і концентрували у вакуумі (баня 40°С) до приблизно 8л з промиванням н-гептаном (2x7,5л) з утворенням гептанової суміші (залишки МТВЕ і MeCN 0,005% і 0,90% відповідно) з подальшим розбавленням н-гептаном до одержання об'єму 37,5л. Потім суміш нагрівали до 70°С і додавали розчин Boc-NHNH2 (801г, 6,06моль) в толуолі (1,5л). Одержану суміш перемішували при температурі вище 70°С 3 години з подальшим охолоджуванням до 60°С. Вносили затравку продукту (речовина 2-3) і одержану суміш витримували при 59-61°С 1 годину для ініціювання кристалізації. Суміш потім залишали охолоджуватися до кімнатної температури і витримували протягом ночі. Кристали відділяли фільтруванням при 20°С, промивали н-гептаном (7,5л), потім сумішшю (7:3 по об'єму) н-гептан/ізо-РгОН (4,5л) і н-гептаном (4,5л) і сушили у вакуумі з одержанням сполуки 2-3 у вигляді безбарвних голок. По краплях потім додавали розчин сполуки 2-3 (1,64кг, 4,27моль) і ТГФ (5,9л) до суспензії, що перемішується, NaBH4 (364г, 9,62моль) і ТГФ (7,2л) в 50-літровій скляній посудині при 0°С, підтримуючи внутрішню температуру нижче 5°С. До одержаної суміші потім додавали по краплях BF3OEt2 (920г, 6,48моль), підтримуючи внутрішню температуру нижче 10°С. Одержану безбарвну суспензію перемішували при 0-5°С протягом 1 години, після чого додавали по краплях 6н водний NaCl (4,29кг, 23,5моль) за 1 годину, підтримуючи внутрішню температуру нижче 20°С (Обережно: бурхливе виділення газу). Одержану безбарвну суспензію нагрівали і перемішували при 60-65°С 2 години, за які виділення газу припинялося. До суміші при 3°С повільно додавали дегазований 2н водний NaOH (12,9л, 25,8моль), підтримуючи внутрішню температуру нижче 20°С, з подальшим нагріванням одержаної суміші до кімнатної температури і екстрагуванням із застосуванням дегазованого МТВЕ (40л). Відділений органічний шар промивали дегазованою водою (6,6л), потім дегазованим розсолом (6,5л) і дегазованою водою (3,3л). Органічний шар потім розбавляли дегазованим МТВЕ до утворення 57-літрового розчину і додавали затравку продукту (сполука 2-4) з подальшим додаванням по краплях розчину щавлевої кислоти (177г, 1,97моль) і дегазованого МТВЕ (1,97л) за 15 хвилин, які кристалізували продукт. Одержану безбарвну суспензію витримували при кімнатній температурі протягом ночі і відділяли кристали фільтруванням, промивали МТВЕ (12,3л) і сушили у вакуумі, одержуючи сполуку 2-4 (1,25кг, 88%, 99,9%-на чистота по ВЕРХ) у вигляді безбарвних пластинок. 1 H-ЯМР (270МГц, ДМСО-d6, ч/млн): 7,41-7,26 (м, 10Н), 5,91-5,62 (шир.м, 4Н), 4,50 (с, 4Н), 3,56 (шир.д, J=4,9Гц, 4Н), 3,34 (м, 1Н). 13С-ЯМР (68МГц, ДМСО-d6, ч/млн): 164,7, 138,2, 128,2, 127,5, 127,4, 72,3, 68,3, 59,8. ПРИКЛАД 3 У 22-літрову посудину, що продувається азотом, при 22°С завантажували DMA (8,3 л), сполуку 1-2 (1,00кг; 0,94 моль) і сполуку 2-4 (350г; 1,06 моль). Одержану суспензію дегазували при перемішуванні прикладенням до посудини вакууму (40-80 тор) циклами по 5 хвилин і заповненням азотом (три цикли). Вміст нагрівали до 65°С за 30 хвилин, під час чого розчин ставав гомогенним. Швидко додавали триетиламін (146мл; 1,05моль) і розчин витримували при 65°С 3 години. Вміст охолоджували до 45°С і переносили в 50-літрову циліндричну посудину, що містить продутий азотом МТВЕ (17,0л), охолоджену до 10°С. Вміст знов охолоджували до 10°С і за 10 хвилин додавали продуту азотом воду (4,7л), підтримуючи внутрішню температуру нижче 30°С. До двофазної суміші при 22°С додавали 2М соляну кислоту (440мл). Після збовтування при 22°С протягом 10 хвилин шари розділяли і органічний шар промивали водою (3х3,8л). Органічний шар концентрували у вакуумі до 5л (20-25°С) і для видалення МТВЕ обробляли декількома потоками ТГФ. Видалення розчинника у вакуумі дало бажану сполуку 3-1. 1 H-ЯМР (270МГц, CDCl3, ч/млн): 10,63 (1Н, шир.с,), 9,24 (1Н, шир.д, J=9,6Гц), 9,16 (1Н, шир.д, J=9,6Гц), 7,50-6,84 (42Н, м), 6,20 (2Н, шир.д, J=7,6Гц), 5,84 (1Н, д, J=8,6Гц), 5,33 (1Н, шир.д, J=3,0Гц), 5,21 (1Н, д, J=12,2Гц), 5,19 (1Н, д, J=11,9Гц), 5,16 (1Н, д, J=12,2Гц), 5,08 (1Н, д, J=11,9Гц), 5,08 (1Н, д, J=10,9Гц), 4,96 (1Н, д, J=10,9Гц), 4,89 (1Н, д, J=10,9Гц), 4,85 (1Н, д, J=10,9Гц), 4,72 (1Н, д, J=12,9Гц), 4,68 (1Н, д, J=12,9Гц), 4,62-4,48 (4Н, м), 4,33 (1Н, дд, J=9,6, 9,6Гц), 4,06-3,77 (7Н, м), 3,72 (4Н, д, J=5,6Гц), 3,04 (1Н, д, J=9,9Гц). 13 С-ЯМР (68МГц, CDCl3, ч/млн): 168,8, 168,7, 159,4, 159,3, 143,2, 142,9, 138,0, 137,9, 137,6, 136,9, 136,8, 136,6, 136,0, 130,2, 128,7, 128,6, 128,5, 128,4, 128,3, 128,2, 128,2, 128,1, 128,0, 127,9, 127,8, 127,7, 127,6, 127,5, 127,4, 127,3, 126,9, 126,6, 119,4, 119,1, 118,0, 116,9, 116,7, 116,1, 110,4, 96,7, 96,3, 85,8, 84,7, 80,9, 77,4, 77,2, 76,0, 75,9, 75,4, 74,9, 73,9, 73,3, 73,2, 70,7, 70,4, 69,9, 69,8, 66,7, 58,7, 49,4, 30,9, 27,0. ПРИКЛАД 4 Обережно: продукт даної реакції, сполука 4-1 є цитотоксичною сполукою: У 5-галонний автоклав завантажували 10%-ний Pd на вугіллі (вологість 50%; 112г), потім розчин 12-b-D(2,3,4,6-тетра-О-бензилглюкопіранозил)-12,13-дигідро-2,10-дибензилокси-6-[(2-бензилокси-1(бензилоксиметил)етил]аміно]-5Н-індоло[2,3-а]піроло[3,4-с]карбазол-5,7(6Н)-діону (3-1) в ТГФ (тетрагідрофурані) (розчин 175г/л; 6,4л; зразок 1,12кг), ізопропіловий спирт (IPА) (7,9л) і 3н НСl (224мл). Вміст гідрували при 40740 фунтів на кв.дюйм при швидкому перемішуванні 4-14 годин, при цьому вбиралося 110% від теоретичної кількості водню. Вміст охолоджували до 25°С і реакційну суміш фільтрували через шар солка-флок, який промивали 3/2 ІРА/ТГФ (1x3л). Фільтрат доводили до рН 2,5 (інтервал 1,5-6,5), використовуючи 1М триетиламіну в ІРА (приблизно 600 мл), з подальшим додаванням води (4,0л). Завантаження концентрували при атмосферному тиску до об'єму 7,5л. Перегонку продовжували при постійному об'ємі завантаження, додаючи суміш (4/1) IP А/води (6,5л). Вміст води знижувався до 20% (вага/об'єм) (інтервал 10-30% води), доданням в посудину ІРА (приблизно 9л), підтримуючи об'єм завантаження 7,5л. Вміст охолоджували до 70°С і вносили затравку (5,0г) у вигляді суспензії в ІРА (50мл). Завантаження витримували при 70°С 1 годину з подальшим додаванням ІРА (5,0л) за 90 хвилин. Завантаження витримували при 70°С 9-24 годин, протягом яких маса продукту кристалізувалася. Постійний об'єм перегонки, що живиться ІРА, (17л) приводив до зменшення вмісту води до 3% (вага/об'єм) (інтервал 1-10%). Суспензію витримували при 70°С протягом 3-6 годин і потім охолоджували до 22°С і витримували 1 годину. Суспензію фільтрували і коржик на фільтрі промивали ІРА (2,5л) і потім метанолом (1,5л), після чого сушили 6 годин у вакуумі при 38°С, одержуючи продукт, 4-1, у вигляді оранжевої твердої речовини з чистотою більше 99 А% і виходом більше 80%. Дані ЯМР (константи поєднання (J) приведені в герцах): 1 Н ЯМР (400,13МГц, ДМСО-d6) дані для головного ротамера d 11,23 (с, 1Н), 9,80 (с, 1Н), 9,77 (с, 1Н), 8,90 (д, J=8,4, 1Н), 8,82 (д, J=8,4, 1Н), 7,21 (шир.с, 1Н), 7,01 (шир.с, 1Н), 6,84 (перекривання м, 6Н), 6,00 (д, J=8,0, 1H), 5,88 (т, J=3,6, 1Н), 5,57 (д, J=2,4, 1Н), 5,34 (д, J=4,4, 1Н), 5,13 (д, J=4,4, 1Н), 4,94 (д, J=4,4, 1Н), 4,56 (т, J=5,6,2Н), 4,04 (дд, J=11,2, 3,2, 1Н), 3,95 (перекривання м, 2Н), 3,81 (дд, J=10,4, 4,0, 1Н), 3,53 (перекривання м, 6Н); 13 С ЯМР (100,64МГц, ДМСО-d6) дані для головного ротамера d 169,03, 168,94, 157,79, 157,63, 144,38, 143,12, 129,46, 127,92, 125,19 (2С), 118,91, 117,57, 115,94, 114,32, 114,23, 113,92, 110,30, 110,24, 97,54, 97,49, 84,49, 78,39, 76,77, 72,88, 67,53, 62,59, 60,47 (2С), 58,33,21. Аналіз ВЕРХ Параметри ВЕРХ: Колонка: YMC ODS-AQ (250x4,6мм) Швидкість потоку: 1,5мл/хв. Визначення: 228нм Рухома фаза: А=0,1% Н3РО4 водний В=ацетонітрил Градієнт: Хв. 0 40 60 61 65 А(%) 85 74 30 85 85 В(%) 15 26 70 15 15 Об'єм вприскування: 10мкл Температура: 25°С
ДивитисяДодаткова інформація
Назва патенту англійськоюA process for making and isolation of indolocarbazole glycosides
Назва патенту російськоюСпособ получения и выделения гликозидов индолокарбазола
МПК / Мітки
МПК: C07H 19/23, C07H 19/044
Мітки: спосіб, індолокарбазолу, одержання, виділення, глікозидів
Код посилання
<a href="https://ua.patents.su/6-74249-sposib-oderzhannya-i-vidilennya-glikozidiv-indolokarbazolu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання і виділення глікозидів індолокарбазолу</a>
Спосіб вилучення та виділення хлориду натрію з потоку відходів процесу одержання n-фосфонометилімінодіоцтової кислоти

Номер патенту: 73350
Опубліковано: 15.07.2005
Автор: Філліпс Скотт Гордон
МПК: C02F 1/74, B01D 9/02, C02F 1/02, C07F 9/38, C02F 1/00, C02F 1/04, C01D 3/00
Мітки: виділення, кислоти, спосіб, n-фосфонометилімінодіоцтової, одержання, процесу, вилучення, хлориду, відходів, потоку, натрію
Формула / Реферат:
1. Спосіб вилучення та виділення хлориду натрію з потоку відходів процесу одержання N-фосфонометилімінодіоцтової кислоти, який відрізняється тим, що потік відходів нейтралізують NaOH до рН приблизно 7, випаровують з потоку нейтралізованих відходів воду при атмосферному або більш низькому тиску при температурі від 40 до 130°С доти, поки не випадає осад NaCl, фільтрують осад при температурі від 35 до 110°С для виділення NaCl з фільтрату і...
Спосіб одержання каталізатора реакції виділення кисню

Номер патенту: 68287
Опубліковано: 15.07.2004
Автори: Іванова Наталія Дмитрівна, Болдирєв Євген Іванович, Стадник Ольга Олександрівна
МПК: C25B 11/00, B01J 23/75, B01J 27/06
Мітки: одержання, спосіб, кисню, каталізатора, реакції, виділення
Формула / Реферат:
Спосіб одержання каталізатора реакції виділення кисню шляхом синтезу оксидної сполуки кобальту, який відрізняється тим, що каталізатор одержують електролізом водного розчину, внаслідок чого каталізатор містить ОН- -групу, електроліз проводять з розчину, що містить, г/л: сульфат кобальту CoSO4.7H2O 10-30 фторид амонію NH4F 20-50 вода H2O ...
Стимулятор росту збудника туберкульозу “рідин” (варіанти), живильне середовище для виділення збудника туберкульозу (варіанти), спосіб одержання живильного середовища (варіанти), спосіб виділення збудника туберк

Номер патенту: 43467
Опубліковано: 17.12.2001
Автори: Багрій Петро Іванович, Власенко Володимир Васильович
МПК: C12N 1/38, C12N 1/20, C12R 1/32, C12Q 1/04, C12N 1/02
Мітки: стимулятор, спосіб, рідин, росту, збудника, середовища, одержання, варіанти, живильне, туберк, середовище, туберкульозу, виділення, живильного
Формула / Реферат:
1. Стимулятор росту збудника туберкульозу, що містить хімічні сполуки, до складу яких входять елементи: натрій, кисень, водень, вуглець, який відрізняється тим, що характеризується наступним складом інгредієнтів, мас. %: сахароза х/ч 2,0-5,0 спирт ректифікований 0,1-0,4 кальцій хлористий 0,1-0,15 натрій двовуглекислий ...
Спосіб виділення та очищення таксанових сполук, зокрема паклітакселу (варіанти)

Номер патенту: 68355
Опубліковано: 16.08.2004
Автор: Ліу Джиан
МПК: C07D 305/00
Мітки: сполук, спосіб, очищення, таксанових, паклітакселу, виділення, зокрема, варіанти
Формула / Реферат:
1. Спосіб виділення та очищення таксанових аналогів з джерела, що містить таксани, який відрізняється тим, що включає: екстрагування джерела таксанів в органічному екстрагенті; покривання слабкого абсорбційного середовища вказаним екстрагентом та завантаження вказаного середовища у колонку, що містить абсорбент; елюювання на першій стадії сумішшю органічних розчинників при тиску 68,95-137,90 кПа (10-20 psi) для одержання фракцій, що містять...
Композиція для одержання діоксиду хлору та спосіб його одержання (варіанти)

Номер патенту: 71973
Опубліковано: 17.01.2005
Автори: Бойруп-Андресен Сесілія, Тенні Джоел Д., Ядеше Гунілла
МПК: D21C 9/10, C01B 11/00
Мітки: варіанти, композиція, спосіб, одержання, діоксиду, хлору
Формула / Реферат:
1. Стійка при зберіганні композиція, придатна як сировина для одержання діоксиду хлору, яка є водним розчином, що містить від близько 1 до близько 6,5 моль/л хлорату лужного металу, від близько 1 до близько 7 моль/л пероксиду водню і щонайменше один із наступних компонентів: станат лужного металу, піридинкарбонова кислота або комплексоутворюючий агент на основі фосфонової кислоти, де рН водного розчину складає від близько 1 до близько...
Попередній патент: Пристрій для оснащення в технології утворення ливарної форми (варіанти)
Наступний патент: Фармакологічно активні уридин-естери
Випадковий патент: Система протипожежної сигналізації