Спосіб кількісного визначення амінокислот
Номер патенту: 120119
Опубліковано: 25.10.2017
Автори: Мальцев Георгій Володимирович, Кашуцький Сергій Миколайович, Федосенко Ганна Олександрівна, Антонович Валерій Павлович, Єгорова Алла Володимирівна





Формула / Реферат
Спосіб кількісного визначення амінокислот, що включає приготування проби, її предколоночну дериватизацію, що включає взаємодію амінокислот з реагентом, та їх подальше хроматографічне визначення УФ детектуванням, який відрізняється тим, що як реагент використовують ди-трет-бутилдикарбонат, а хроматографічне визначення здійснюють при наступному співвідношенні компонентів рухомої фази: ацетонітрил: 0,1 % (об/об) розчин мурашиної кислоти (30:70, об/об) за довжини хвилі 200 нм.
Текст
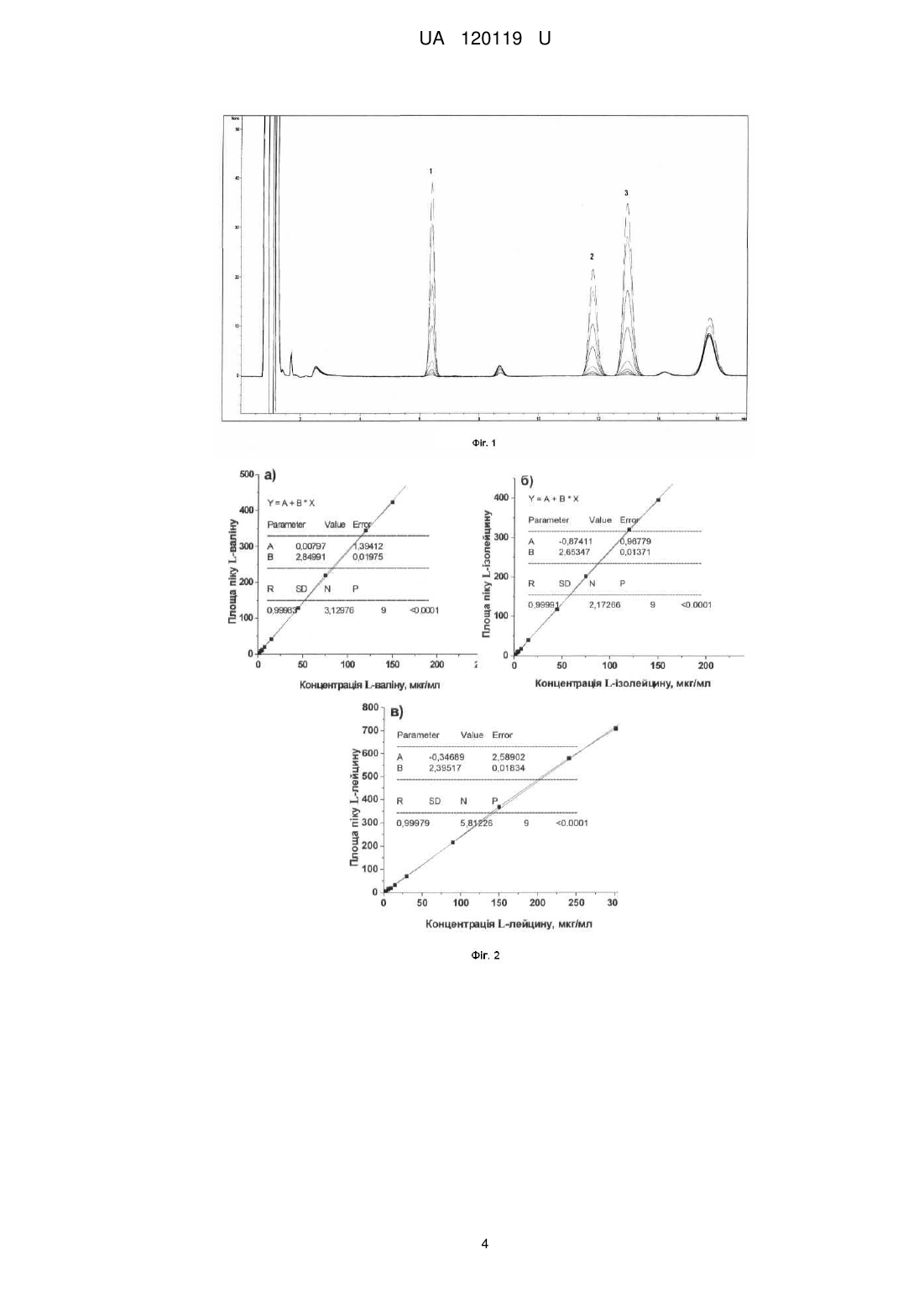
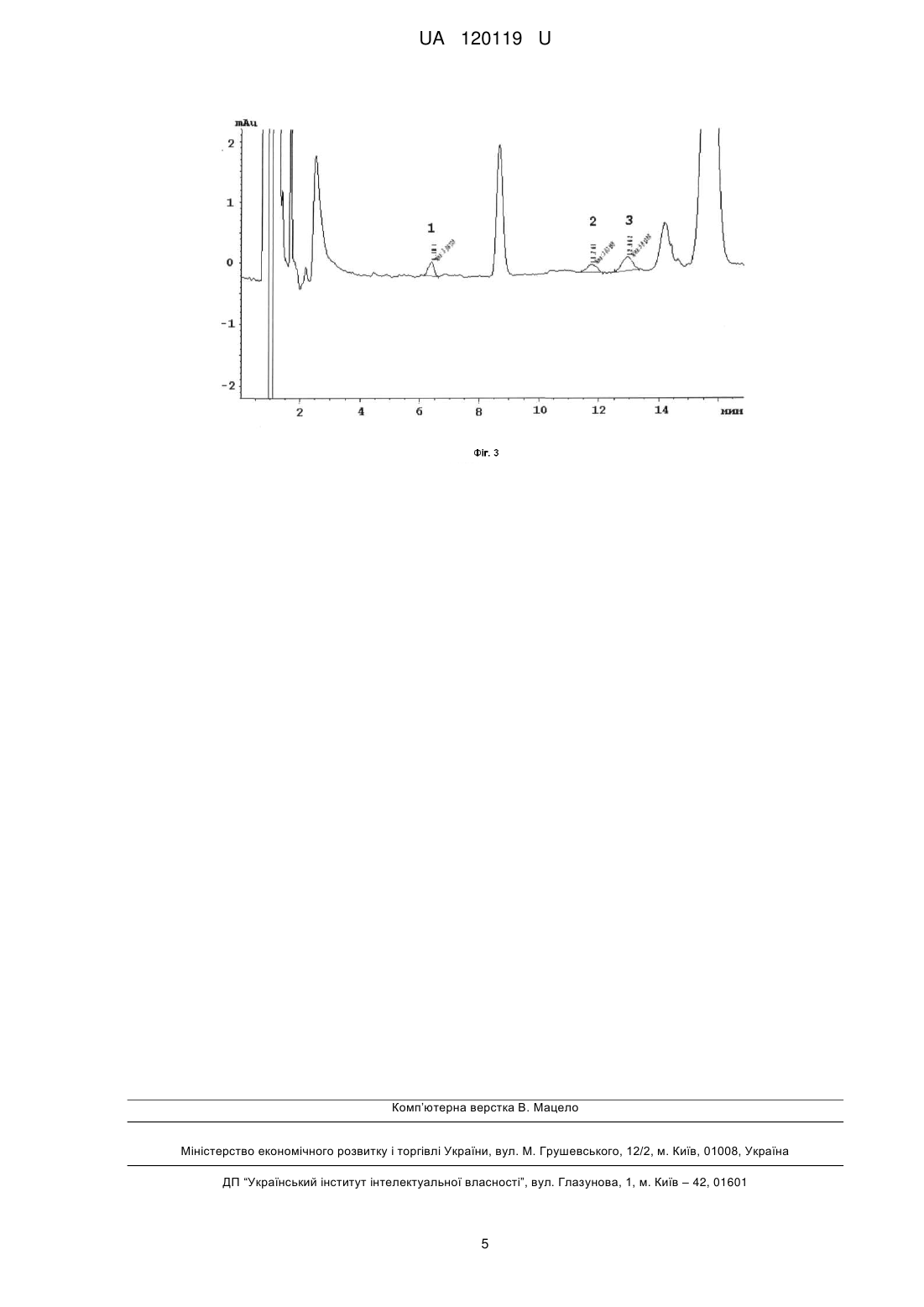
Реферат: Спосіб кількісного визначення амінокислот включає приготування проби, її предколоночну дериватизацію, що включає взаємодію амінокислот з реагентом, та їх подальше хроматографічне визначення УФ детектуванням. Як реагент використовують ди-третбутилдикарбонат, а хроматографічне визначення здійснюють при наступному співвідношенні компонентів рухомої фази: ацетонітрил: 0,1 % (об./об.) розчин мурашиної кислоти (30:70, об./об.) за довжини хвилі 200 нм. UA 120119 U (12) UA 120119 U UA 120119 U 5 10 15 20 25 30 35 40 45 Корисна модель належить до аналізу матеріалів, а саме до предколоночної дериватизації амінокислот (АК), наприклад L-валіну (В), L-лейцину (Л) та L-ізолейцину (ІЛ)) з подальшим їх визначенням методом високоефективної рідинної хроматографії (ВЕРХ). Відомий спосіб визначення амінокислот за допомогою ВЕРХ [див. Bhandare P., Madhavan P., Rao B.M., Someswar rao N. Determination of amino acid without derivatization by using HPLC-HILIC column. J. Chem. Pharm. Res. - 2010.- V. 2, № 2. - P. 372-380]. Спосіб передбачає визначення амінокислот без процедури дериватизації, а також використання прямофазної колонки Kromasil SIL 0,25 м × 4,6 мм, яка заповнена силікагелем з розміром часток 5 мкм. Визначення проводять за наступних умов: рухома фаза - 2,5 мМ розчин калію дигідрофосфату з рН 2,85: ацетонітрил (25:75); детектування за довжини хвилі 200 нм. Інтервал визначення не ароматичних амінокислот 0,4-0,65 мкг/мл. Недоліком відомого способу є те, що використовувана колонка призначена для безводних розчинників і в рамках даного аналізу виявилася недовговічною (2 тижні). Також інтервал визначення амінокислот дуже вузький. Найбільш близьким до способу, що заявляється, є визначення амінокислот за допомогою методу ВЕРХ з попередньою процедурою предколоночної їх дериватизації [див. Bartolomeo M.P., Maisano F. Validation of a reversed-phase HPLC method for quantitative amino acid analysis J. Biomol. Techniq. - 2006. - V. 17, № 2. - P. 131-137]. Спосіб базується на використанні офталевого альдегіду (ОФА) як дериватизуючого агента. ОФА в присутності сульфовмісного нуклеофіла швидко та кількісно взаємодіє з амінокислотами при кімнатній температурі. Визначення проводять за наступних умов: рухома фаза А - 40 мМ розчин натрію дигідрофосфату (доведений до рН 7,8 за допомогою розчину натрію гідроксиду); рухома фаза В - ацетонітрил:метанол:вода (45:45:10); детектування за довжини хвилі 338 нм. Даний спосіб вибрано прототипом. Прототип та спосіб, що заявляється, мають такі спільні ознаки: - процесс предколоночної дериватизації амінокислоти, - хроматографічне визначення амінокислоти з УФ детектуванням. Але спосіб за прототипом призводить до утворення похідних ОФА, стандартизація стійкості яких в процесі хроматографічного розділення, а також вибір умов, в яких деградація похідних мінімальна, є недоліком даного методу. У способі за прототипом використовують автосамплер приладу, який дозволяє проводити дерватизацію в автоматичному режимі внесення, розведення і змішування, а також спеціальні колонки Zorbax Eclipse, AAA, що говорить про високу вартість обладнання та витратних матеріалів. Також необхідно використовувати ще другий компонент - сульфовмісний нуклеофіл (наприклад меркаптоетанол). В основу корисної моделі поставлено задачу розробити простий у виконанні спосіб кількісного визначення амінокислот, наприклад L-валіну, L-лейцину та L-ізолейцину із застосуванням предколоночної дериватизації з реагентом, який широко використовується в органічному синтезі для захисту аміногруп) - ди-трет-бутилдикарбонат (Вос2О). Перевага способу також полягає в тому, що для УФ детектування використовуються традиційні хроматографічні колонки з силікагелем октадецилсилільним для хроматографії (С18). Поставлена задача вирішується тим, що в способі визначення амінокислот, що включає приготування проби, її предколоночну дериватизацію, що включає взаємодію амінокислоти з реагентом - та її подальше хроматографічне визначення УФ детектуванням, згідно з корисною моделлю, як реагент використовують ди-трет-бутилдикарбонат, а хроматографічне визначення здійснюють при наступному співвідношенні компонентів рухомої фази: ацетонітрил: 0,1 % (об/об) розчин мурашиної кислоти (30:70, об/об) за довжини хвилі 200 нм. Новим в заявленій корисній моделі є наявність наступних ознак: предколоночна дериватизація амінокислот здійснюється дериватизуючим агентом ди-третбутилдикарбонатом (1) O CH3 O CH3 H3 C 50 55 H3 C CH3 O O O CH3 (1) - співвідношення компонентів рухомої фази: ацетонітрил: 0,1 % (об/об) розчин мурашиної кислоти (30:70, об/об); - детектування здійснюють за довжини хвилі 200 нм. Причинно-наслідковий зв'язок між сукупністю суттєвих ознак, що заявляються, і технічним результатом, що досягається, полягає в наступному: - дериватизація амінокислот з ди-трет-бутилдикарбонатом (ВОС2О) призводить до утворення стійких сполук; 1 UA 120119 U 5 визначення АК можливе на традиційних хроматографічніх колонках (С18) (не потребує спеціальних колонок); - при пробопідготовці не треба використовувати спеціальне обладнання для предколоночної дериватизації (автоматичний автосампелер). Заявлені суттєві ознаки дозволяють досягнути наступного технічного результату - спростити процес дериватизації амінокислот, а також зробити його більш доступним. В умовах розробленої методики пропонується проводити пробопідготовку в присутності 0,5 М розчину гідроксиду натрію. У лужному середовищі АК знаходяться в аніонній формі та реакція з Вос2О протікає за наступною схемою: R O H2N O 10 15 20 25 30 35 40 45 50 55 O CH3 Boc2O R H3C - CO2 - (CH3)3COH H3C O O N H O Приклад Кількісне визначення залишкових кількостей амінокислот на поверхні фармацевтичного обладнання, на якому відбувалось виробництво дієтичної добавки "ВСАА смарт" (активні фармацевтичні інгредієнти - L-валін, L-лейцин та L-ізолейцин), проводили із застосуванням ВЕРХ. Методика визначення амінокислот заснована на зміні площин їхніх піків на хроматограмах в залежності від їх концентрації. Вміст амінокислот в змивах (мкг/змив) визначають за градуювальними графіками. Градуювальний графік Розчин ди-трет-бутилдикарбонату. 21,90 г ди-трет-бутилдикарбонату (CAS 24424-99-5) поміщають у мірну колбу місткістю 100 мл, доводять об'єм розчину ацетонітрилом до позначки та перемішують. Розчин РСЗ L-лейцину, РСЗ L-валіну, РСЗ L-ізолейцину. У мірну колбу місткістю 100,0 мл вносять 300,0 мг L-лейцину (Л), 150,0 мг L-валіну (В), 150,0 мг L-ізолейцину (ІЛ), розчиняють в 70 мл води та доводять до позначки тим же розчинником. У мірні колби місткістю 100,0 мл вносять 0,1; 0,2; 0,3; 0,5; 1,0; 3,0; 5,0; 8,0 та 10,0 мл отриманого розчину, додають 2,0 мл 0,5 М розчину натрію гідроксиду, 30 мл метанолу та 1,0 мл розчину ди-трет-бутилдикарбонату, перемішують на магнітній мішалці протягом 30 хв. (часу перемішування дотримуватися обов'язково). Доводять об'єм розчину метанолом до позначки та перемішують. Розчини фільтрують крізь мембранний фільтр (0,20 мкм; Minisart RC 15, "Sartorius", Німеччина). Хроматографування проводять на рідинному хроматографі з УФ-детектором за таких умов: - колонка з нержавіючої сталі розміром 0,10 м × 4,6 мм, заповнена силікагелем октадецилсилільним для хроматографії з розміром часток 3,5 мкм; - рухома фаза: суміш ацетонітрил: 0,1 % (об/об) розчин мурашиної кислоти (30:70 об/об); - швидкість рухомої фази 1,0 мл/хв.; - об'єм проби, що вводиться, 10 мкл; - детектування за довжини хвилі 200 нм; - температура колонки 30 °C; - час хроматографування 17 хв. Хроматографування проводять в ізократичному режимі (на Фіг. 1 наведено хроматограми розчинів РСЗ АК для градуювальних графіків, де 1-L-валін, 2-L-ізолейцин, 3-L-лейцин). За отриманими результатами будують градуювальні графіки, відкладаючи на осі абсцис концентрації амінокислот (Л / В / ІЛ), а по осі ординат - значення інтенсивності абсорбції за довжини хвилі 200 нм (площі піків, AU) Градуювальні залежності площ піків від концентрації амінокислот (Л / В / ІЛ) наведено на Фіг. 2. Межі виявлення (MB) L-лейцину, РСЗ L-валіну, РСЗ L-ізолейцину. складають - 3,57 мкг/мл, 1,61 мкг/мл и 1,20 мкг/мл, відповідно. Методика визначення Випробовуваний розчин Сваб (аплікатор Alpha® Sampling Swab марки ТХ 71) зі змивом з поверхні фармацевтичного обладнання (площа змиву -100,0 см) поміщають в лабораторній стакан місткістю 25 мл, додають 2,0 мл 0,5 М розчину натрію гідроксиду, 3,0 мл метанолу та 1,0 мл розчину ди-трет-бутилдикарбонату, перемішують на магнітній мішалці протягом 30 хв. (часу перемішування дотримуватися обов'язково). Отриманий розчин кількісно переносять у мірну колбу місткістю10,0 мл, доводять об'єм розчину метанолом до позначки та перемішують. Розчин фільтрують крізь мембранний фільтр (0,20 мкм; Minisart RC 25, "Sartorius", Німеччина). Далі роблять як при побудові градуювального графіку. 2 UA 120119 U 5 10 15 Вміст амінокислот (X), в мкг/змив розраховують за формулою: X=С10 де: С - концентрація амінокислоти (Л / В / ІЛ) на хроматограмі випробовуваного розчину, отримана за градуювальним графіком, мкг/мл. На Фіг. 3 наведена хроматограма розчину після обробки свабу, яким робили змиви з поверхні фармацевтичного обладнання (таблетпрес Korsch EKO, де 1-L-валін, 2-L-изолейцин, 3-L-лейцин). Встановлено залишкові кількості АК: L-валін - 10,8 мкг/сваб, L-ізолейцин - 14,7 мкг/сваб, L-лейцин -24,9 мкг/сваб. Результати визначення залишкових кількостей амінокислот на поверхні таблетпресу Korsch ECO не вище за практичне гранично допустиме значення їх залишків в змиві (250 мкг/сваб), розраховане по фактору безпеки 0,1 %, що свідчить про задовільну якість очищення даного обладнання. Визначення ступеня вилучення У модельних дослідах в ході валідації методики робили змиви свабом, змоченим водой, з 2 поверхні (100,0 см ), на яку штучно наносили (0,1 мл розчину з концентрацією 1500,0 мкг/мл Lлейцину, 750,0 мкг/мл L-валіну; 750,0 мкг/мл L-ізолейцину наносили та висушували). Далі готували випробуваний розчин як у розділі "Методика визначення" (отримували концентрації: лейцину - 15,0 мкг/мл; валіну - 7,5 мкг/мл; ізолейцину - 7,5 мкг/мл). Ступінь вилучення амінокислот з поверхні фармацевтичного обладнання наведений у таблиці. 20 Таблиця Номер змиву Ступінь вилучення L-лейцину, % Ступінь вилучення L-валіну, % Ступінь вилучення L-ізолейцину, % 25 1 90,0 91,7 91,2 2 92,6 91,9 93,2 3 90,3 92,1 91,7 4 91,5 91,4 93,5 5 90,8 93,1 92,8 Встановлено, що ступінь вилучення амінокислот з поверхні фармацевтичного обладнання становить 90,0 % - 93,5 % (див. таблиця). Таким чином, спосіб дозволяє отримати більш стійкі сполуки амінокислот (наприклад, Lваліну, L-лейцину, L-ізолейцину) з тільки одним дереватизуючим новим реагентом - ди-третбутилдикарбонатом, проводити хроматографування на традиційних хроматографічних колонках (С18), що робить аналіз дешевшим та значно його спрощує у порівнянні з прототипом. ФОРМУЛА КОРИСНОЇ МОДЕЛІ 30 35 Спосіб кількісного визначення амінокислот, що включає приготування проби, її предколоночну дериватизацію, що включає взаємодію амінокислот з реагентом, та їх подальше хроматографічне визначення УФ детектуванням, який відрізняється тим, що як реагент використовують ди-трет-бутилдикарбонат, а хроматографічне визначення здійснюють при наступному співвідношенні компонентів рухомої фази: ацетонітрил: 0,1 % (об./об.) розчин мурашиної кислоти (30:70, об./об.) за довжини хвилі 200 нм. 3 UA 120119 U 4 UA 120119 U Комп’ютерна верстка В. Мацело Міністерство економічного розвитку і торгівлі України, вул. М. Грушевського, 12/2, м. Київ, 01008, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 5
ДивитисяДодаткова інформація
МПК / Мітки
МПК: G01N 33/15, G01N 30/02, G01N 30/00
Мітки: спосіб, кількісного, амінокислот, визначення
Код посилання
<a href="https://ua.patents.su/7-120119-sposib-kilkisnogo-viznachennya-aminokislot.html" target="_blank" rel="follow" title="База патентів України">Спосіб кількісного визначення амінокислот</a>
Спосіб визначення кількісного складу рухомого продукту, апаратура для визначення кількісного вмісту інсуліну в рухомому продукті, застосування апаратури і установка для розфасовки розчинів і дисперсій, що включ

Номер патенту: 90847
Опубліковано: 10.06.2010
Автори: Прінц Хайно, Крістіансен Крістіан-Петер, Плосс Ханс-Йоахім, Мертенс Ріхард
МПК: B07C 5/342, A61B 5/00, G01N 21/31
Мітки: вмісту, дисперсій, спосіб, інсуліну, застосування, розчинів, установка, апаратури, складу, включ, продукту, розфасовки, кількісного, продукти, рухомому, апаратура, визначення, рухомого
Формула / Реферат:
1. Спосіб визначення кількісного складу рухомого продукту з наступними етапами: опромінення продукту джерелом випромінювання у діапазоні ближнього інфрачервоного випромінювання;приймання випромінювання, яке передається через рухомий продукт в первинній упаковці, і отримання вихідного сигналу відповідно до інтенсивності прийнятого випромінювання при декількох різних довжинах хвиль;визначення на основі вихідного сигналу за...
Спосіб кількісного визначення флуконазолу у капсулах

Номер патенту: 48381
Опубліковано: 10.03.2010
Автори: Бурлака Юлія Віталіївна, Васюк Світлана Олександрівна, Тарханова Ольга Олександрівна
МПК: G01N 21/78
Мітки: флуконазолу, спосіб, визначення, капсулах, кількісного
Формула / Реферат:
Спосіб кількісного визначення флуконазолу в капсулах, який полягає у розчиненні проби, фільтруванні отриманого розчину та вимірюванні абсорбції, який відрізняється тим, що розчиняють пробу в хлороформі, застосовують кольорореагент - розчин бромтимолового синього в хлороформі, та вимірюють абсорбцію у видимій області спектра при довжині хвилі 422 нм.
Спосіб кількісного спектрофотометричного визначення фенілефрину гідрохлориду або тимололу малеату у присутності бензалконію хлориду

Номер патенту: 95908
Опубліковано: 12.01.2015
Автори: Бевз Наталія Юріївна, Віслоус Ольга Олександрівна, Георгіянц Вікторія Акопівна, Криванич Олександр Валерійович
МПК: G01J 3/42
Мітки: спектрофотометричного, гідрохлориду, спосіб, тимололу, кількісного, визначення, фенілефрину, малеату, хлориду, бензалконію, присутності
Формула / Реферат:
Спосіб кількісного спектрофотометричного визначення фенілефрину гідрохлориду або тимололу малеату в присутності бензалконію хлориду, який відрізняється тим, що проводять зв'язування бензалконію хлориду 5 % розчином калію дихромату, вимірювання оптичної густини розчинів проводять відповідно за довжин хвиль 296 нм та 298 нм.
Спосіб кількісного визначення мезатону в розчині для ін’єкцій

Номер патенту: 76557
Опубліковано: 10.01.2013
Автори: Васюк Світлана Олександрівна, Жук Юлія Миколаївна
МПК: G01N 21/78
Мітки: ін'єкцій, кількісного, розчині, мезатону, спосіб, визначення
Формула / Реферат:
Спосіб кількісного визначення мезатону в розчині для ін'єкцій, який включає розчинення проби, додавання реагенту та вимірювання абсорбції, який відрізняється тим, що пробу розводять ацетоном, застосовують кольорореагент - розчин бромкрезолового зеленого в ацетоні, та вимірюють абсорбцію у видимій області спектра при довжині хвилі 410 нм.
Спосіб кількісного визначення хромонів

Номер патенту: 30471
Опубліковано: 15.11.2000
Автори: Підпружников Юрій Васильович, Лисоченко Лілія Михайлівна, Калоєв Василь Васильович
МПК: G01N 21/25, A61K 36/23
Мітки: кількісного, хромонів, визначення, спосіб
Формула / Реферат:
Спосіб кількісного визначення хромонів, шляхом екстракції їх з аналізуємого зразку екстрагентом, фільтрування, установлювання потрібної концентрації фільтрату і спектрофотометрування, який відрізняється тим, що аналізуємий зразок екстрагують хлороформом з такою кратністю екстракції і до такого розведення екстракту, щоб припустима концентрація суми хромонів у перерахунку на келлін дорівнювала 0,02-0,04 мг на 1 мл готового екстракту, а...
Попередній патент: Спосіб консервування силосу із кукурудзи
Наступний патент: Спосіб підвищення надійності робочого обладнання малогабаритного навантажувача пмтс 1200 на базі вихідної 3d моделі
Випадковий патент: Спосіб одержання феромагнітного порошку