Спосіб одержання l-ізомерних похідних пропаргіламоній хлориду
Номер патенту: 39938
Опубліковано: 16.07.2001
Автори: Тібор Монтаі, Жужанна Надь, Марія Сіладі, Тамас Каллаі







Формула / Реферат
1. Способ получения L-изомерных производных пропаргил-аммоний хлорида общей формулы (I)
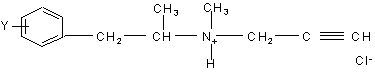 , (I)
, (I)
путем разложения D-тартрата L-изомерного амина общей формулы (II)
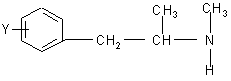 , (II)
, (II)
и взаимодействия полученного L-изомерного амина общей формулы (II) с галогенпроизводным общей формулы (V)
![]() , (V)
, (V)
где Х представляет собой атом галогена,
в присутствии основания с последующим взаимодействием полученного таким образом L-изомера общей формулы (III)
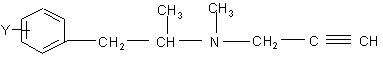 , (III)
, (III)
где Y представляет собой атом водорода или фтора, с хлористым водородом в органическом растворителе, отличающийся тем, что выделение амина в форме основания из D-тартрата L-изомерного амина общей формулы (II) производят в водной суспензии в присутствии гидроокиси аммония или основной соли щелочного металла и/или аммониевой соли, дальнейшее взаимодействие амина с 1- 1, 5 моль-эквивалентами галогенпроизводного общей формулы (V) осуществляют при температуре от 0 до 50°С в буферной системе с рН = 8-12, непосредственно образующейся в процессе выделения основания, после отделения водного слоя производят экстрагирование органического слоя, содержащего смесь L-изомерных аминов общих формул (II) и (III), водой, смесью гидроокиси аммония с водой и/или водным раствором фосфатной соли с рН =5,5 - 7,5, растворение L-изомерного амина общей формулы (II) или его солей в водном слое, селективное отделение соединений от L-изомерного амина общей формулы (III) и превращение его после перегонки в L-изомерную соль общей формулы (I).
2. Способ по п.1, отличающийся тем, что в качестве фосфатной соли используют однозамещенную соль орто-фосфорной кислоты общей формулы (IV)
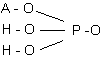 , (IV)
, (IV)
где ион А+ представляет собой ион натрия, калия или аммония. 3. Способ по п.1, отличающийся тем, что в качестве органического растворителя используют ацетон или изопропанол.
Текст
1. Способ получения L-изомерных производных пропаргиламмоний хлорида общей формулы (I): 39938 Настоящее изобретение направлено на разработку безопасного для окружающей среды способа получения 1-N-метил-N-(2-фенил-1-метил)этил-N-пропаргиламин-гидрохлорида общей формулой (Iа): CH3 CH2 CH Другой известный способ описан там же в примере 11. Под давлением проводят реакцию конденсации между 2-фенил-1-метил-этилхлоридом и N-метил-N-пропаргиламином. Реакционную смесь обрабатывают щелочью и получают требуемый продукт с вы ходом 35%. В примере 5 спецификации Венгерского патента № 151,090 описан способ, аналогичный предлагаемому нами, в части используемых исходных материалов, К 0,2 моль N-метил-N-(2-фенил-1-метил)-этиламина добавляют 0,1 моль пропаргил-бромида и нагревают реакционную смесь при 100°С в течение 2 часов. Половинное количество используемого исходного амина расходуется на связывание образующегося в результате реакции бромистого водорода. Хотя выход реакции по отношению к пропаргил-бромиду составляет 85% рацемического продукта, этот способ в целом не является подходящим с практической точки зрения, как отмечается на стр. 2 аналитического обзора спецификации Венгерского патента № 187,775, так как для регенерирования дорогоcтоящего исходного амина, используемого в качестве связующего кислоту агента, предлагается бензоилирование с последующим отделением продукта и его гидролизом, без учета выхода регенерированного амина. Способ получения оптически активных производных пропаргиламина общей формулой (la), в соответствии с данным изобретением, был впервые изложен в Примерах спецификации Венгерского патента А 154,655 без данных о выходе продукта. Согласно способу, описанному в Примере 5, N-метил-N-(2-фенил-1-метил)-этиламин реагирует вначале с пара-формальдегидом, а затем с ацетиленом в виде газа в присутствии катализатора - двухлoристой меди. В результате получают оптически активное основание, отвечающее формуле (IlIa), с вы ходом около 20%: CH3 N+ CH2 C CH , Cl H (Ia) далее именуемого "Селегилин-HCI", и 1-N-метилN-(2- (4-фтор-фенил)- 1 -метил)-этил-N-пропаргиламин-гидрохлорида общей формулой (1б): CH3 F CH2 CH CH3 N + H CH2 C CH, Cl (Iб) далее именуемого "n-фтор-Селегилин-НСl", с хорошим выходом и в свободной от посторонних примесей форме. Ссылки на способы получения производных пропаргиламина общей формулой (I) неоднократно встречаются в технической литературе. Часть известных способов следует реакционной схеме, которая существенно отличается от предлагаемой в настоящей спецификации. Исходные вещества др уги х известных способов соответствуют реагентам, используемым в данной работе. CH3 Y CH2 CH CH3 N+ H CH2 C CH (I) Cl CH3 Синтез соединения, строение которого отвечает формуле (Iа), не обладающего, однако, оптической активностью, описан в спецификации Венгерского патента № 151,090. Рацемическую форму соединения, описываемого формулой (la), получают несколькими способами. В соответствии с примерами 1 и 2 вышеназванной патентной спецификации, 1,3-дибромпропен добавляют к N-(2фенил-1-метил)-этил-N-метиламину и нагревают реакционную смесь, при 100°С в течение 7 часов. На первой стадии реакции получают N-(2-фенил1-метил)-этил-N-метил-N-(2- бромпропенил)-амин, который после выделения обрабатывают щелочью. После перегонки получают N-метил-N-(2фенил-1-метил)-этил-N-пропаргиламин с выходом 20% по отношению к исходному амину и 40% по отношению к используемому в реакции 1,3-дибромпропену. В соответствии с примером 7 той же патентной спецификации, N-метил-N-(2-фенил-1метил)-этиламин взаимодействует с пропаргилальдегидом в спиртовой среде в присутствии металлического алюминия. После добавления к реакционной смеси щелочи получают требуемое производное N-пропаргиламина с выходом 48,6%. CH2 CH CH3 N CH2 C CH (IIIa) Следует упомянуть также Пример 1 вышеназванной патентной спецификации, так как именно там в качестве среды впервые был использован водонерастворимый растворитель, а также была проведена реакция с пропаргил-бромидом при температуре 50-60°С. В реакции не использовался никакой активный связующий агент, и выделяющийся бромистый водород связывался с избытком амина. На основании Примеров, приведенных в вышеупомянутых венгерских патентных спецификациях, можно сделать очевидный вывод, что не существовало доступного способа, который мог бы обеспечить получение соединения, отвечающего формуле (Іа), в промышленных масштабах с хорошим выходом. Авторы венгерской патентной спецификации № 187,775 стремились устранить эти недостатки. Впервые в практике они не использовали более избыток исходного N-метил-N-(2-фенил-1метил)-этиламина для связывания кислоты, так как невозможно было эффективно решить задачу 2 39938 регенерации израсходованного с этой целью амина. В соответствии с предложенным ими способом, они выделяли оптически активное основание из L-N-метил-N-(2-фенил-1-метил)-этиламин-d-тартрата, полученного в результате раcщепления (далее именуемого в тексте L-метил-анара-D-тартрат). Этот процесс осуществляли путем добавления воды к вышеупомянутому тартрату с последующим сильным подщелачиванием водным раствором щелочи (в Примерах использовался иcключительно 40%-ый раствор гидроокиси натрия) до рН=13; выделившийся при этом в форме основания амин экстрагировали из водного раствора несмешивающимся с водой растворителем. С целью более полной экстракции амина водный слой экстрагировали повторно. В качестве несмешивающи хся с водой растворителей использовали неполярные растворители, такие как бензол, толуол, ди хлорэтан, диизопропиловый эфир. Алкилирование проводили путем взаимодействия растворенного в органическом растворителе 1-метилN-(2-фенил-1-метил)-этиламина, далее именуемого 1-метиланара, с пропаргил-бромидом при температуре 55-60°С. Реакция алкилирования, проводимая в органическом растворителе с пропаргил-бромидом при температуре 50-60°С, была упомянута впервые в Примере 1 венгерской патентной спецификации № 154,655. Из венгерской патентной спецификации № 187,775 можно далее увидеть, что новизна способа заключается в использовании водного раствора щелочи в качестве связующего кислоту агента для связывания бромистого водорода, выделяющегося при реакции алкилирования, а также в использовании в качестве реакционной среды эмульсии вода/растворитель. Реакционную смесь процесса алкилирования обрабатывают, отделяя органический слой и промывая его водой. Этот слой органического растворителя содержит непрореагировавшие исходные вещества и побочные продукты реакции, наиболее сходные по строению с основанием, соответствующим формуле (IlIa). Аминные основания, более сильные в органическом растворителе, чем амин, описываемый формулой (IlIa), можно удалить посредством экстракции водными растворами кислот. С этой целью используют неорганические кислоты с кислотной экспонентой 1,0-2,12 или органические кислоты с кислотной экспонентой 3,75-4,87, а для того, чтобы снизить растворимость основного продукта, применяют метод, аналогичный титрованию. Очищенное таким образом основание, отвечающее формуле (IlIa), растворяют в несмешивающемся с водой растворителе, обрабатывают раствором этилового спирта в соляной кислоте, и часть полученной смеси растворителей отгоняют; остаток охлаждают, в результате чего получают кристаллический гидрохлорид общей формулой (I). Максимальный выход составляет 65% (Пример 1): CH3 Y CH2 CH3 CH N+ CH2 Резюмируя все вышесказанное касательно способа, описанного в венгерской патентной спецификации № 187,775, можно сделать вывод, что указанный способ является шагом вперед по сравнению с таковым, описанным в Примере 5 венгерской патентной спецификации № 151,090, в соответствии с которым рацемическое основание "Селегилин", отвечающее формуле (IlIa), получали в очищенном виде с выходом 85% относительно пропаргил-бромида, однако по отношению к более ценному метил-анара выход составлял только 43%. В соответствии с Примерами Европейской патентной спецификации № 0,344,675 продукт, отвечающий формуле (Iа), получали в безводных растворителях в присутствии карбоната калия в качестве связующего кислоту агента путем алкилирования, причем выход конечного продукта после сложных процедур по его очистке составлял максимум 56,6%. Это является шагом назад по сравнению с известными способами. В соответствии с Европейской патентной заявкой № 0,186,680 требуемым соединением является 1-n-фтор-Селегилин-НСl. При алкилировании исходят из рацемического и оптически-активного амина, соответственно; конечный продукт получают из 1-n-фтор-метиланара-d-тартрата и пропаргил-бромида, причем реакция алкилирования в каждом Примере осуществляется в присутствии безводного растворителя и карбоната калия в качестве связующего кислоту агента или в эмульсионной среде вода/растворитель с использованием гидроокиси натрия в качестве кислотосвязующего агента. Максимальный выход конечного продукта составляет 47,1% (Пример 5). После обзора раскрытых в патентных описаниях способов можно сделать вывод, что хотя технологическая сторона получения соединений общей формулой (I) развивалась, в настоящее время не существуе т доступного способа, посредством которого эти соединения можно было бы получать с хорошим выходом, простым и не загрязняющим окружающую среду методом, в очень чистом виде. Настоящее изобретение предлагает способ, позволяющий устранить перечисленные недостатки. Неожиданно было обнаружено, что, если в процессе выделения основания общей формулой (II) и в ходе реакции пропаргилирования в качестве реакционной среды использовать не органический растворитель, а водный буферный раствор, включающий винную кислоту, а также гидроокись аммония либо основные соли щелочных металлов и/или соли аммония в качестве связывающего кислоту агента, с рН=8-12, то реакция пропаргилирования может быть осуществлена с высокой степенью превращения исходных веществ без стадии отделения основания общей формулой (II). Используемые умеренные значения рН и низкая температура реакции (в интервале от 0 до 50°С) приводят к значительному сокращению нежелательных побочных реакций, благодаря чему небольшие количества непрореагировавших исходных веществ и примесей могут быть удалены из сферы реакции путем экстрагирования гидроокисью аммония и водой или при помощи метода селективного солеобразования. Данный метод по H C CH Cl (I) 3 39938 зволяет также получать соли соединений общей формулой (I) с превосходным выходом из такого растворителя, применение которого в известных ранее способах было невозможно из-за низкого качества полученного материала. CH3 Y CH 3 CH CH 2 торых приводит к растворению основного продукта общей формулой (III). Еще одно преимущество способа, в соответствии с настоящим изобретением, состоит в том, что отпадает необходимость использования несмешивающихся с водой растворителей, таких, в основном, как бензол, толуол, с целью регенерации конечного продукта общей формулой (I). Применение таких растворителей является большим недостатком, с точки зрения возможности промышленного использования, регенерации, а также с точки зрения здоровья людей и охраны окружающей среды, В промышленно развитых странах, где существует контроль качества, благодаря строгому тесту на наличие остатков растворителей, нельзя использовать исходные материалы для фармацевтической промышленности, полученные из растворителей, не смешивающихся с водой. Применяя данный способ, можно использовать вместо смеси растворителей, обладающих перечисленными недостатками, водорастворимый растворитель, предпочтительнее, ацетон или изопропанол, с целью получения соединений общей формулой (I). Использование ацетона в качестве реакционной среды обладает еще одним преимуществом, благодаря избирательной растворяющей способности этого растворителя; удерживая в растворенном состоянии загрязняющие вещества, присутствующие в минимальном количестве, ацетон позволяет получить конечный продукт такой степени чистоты, которая намного превышает требования, предъявляемые к качеству данного продукта,. Чистота, определяемая методом ХПЛС (HPLC), составляет минимум 99,9%, а содержание известных и неизвестных примесей не превышает 0,1%. Селегилин-гидрохлорид в соответствии со способом, предлагаемым настоящим изобретением, получают с очень хорошим выходом, составляющим около 91%. Наилучший, из до сих пор полученных, выход составлял 85% (Пример 5 описания венгерского патента № 151,090), но это величина, рассчитанная по отношению к алкилирующему агенту и полученная при использовании в реакции двойного избытка основания метиланара относительно основания Селегилина, отвечающего формуле (III), неустановленной степени чистоты. Cсылка на стр. 2 описания венгерского патента № 187,775, согласно которому расчетный уровень потерь при регенерации избытка исходного амина составляет 40-50%, что делает данный способ весьма неэкономичным. В соответствии с описанием венгерского патента № 187,775, его авторы не использовали более избыточного количества L-метил-анара, но при этом значительно снизился выход конечного продукта. Наилучший выход по отношению к исходному L-метил-анара-Dтартрату составил 65% (Пример 1). Из маточника и промывных вод было выделено еще 7.6-19% конечного продукта, но компонентный состав полученного материала приведен не был, и поэтому не представляется также возможным определить, какая дополнительная обработка требуется для получения продукта, по чистоте соответствующего первоначально выделенному. Данный опыт, свидетельствует о том, что в таком случае вы ход составляет только 30%. N H (II) Преимущества данного способа можно подытожить следующим образом. Выделение оптически активного вторичного амина осуществляется в щелочной реакционной среде, вследствие чего отпадает необходимость таких операций, как экстракция растворителем и разделение слоев, что повышает выход и не наносит ущерба окружающей среде (описание венгерского патента 187,775). В ходе реакции пропаргилирования не используется ни разведение растворителем, ни щелочная гидроокись в качестве связующего кислоту агента; алкилирование проводится в виннокислой буферной смеси, обеспечивающей рН, равное 8-1-, в интервале температур от 0 до 50°С. Условия реакции таковы, что полученное аминное основание общей формулой (III) почти не содержит загрязнений или исходных ве ществ. CH3 Y CH2 CH3 CH N CH2 C CH (III) Степень чистоты аминного основания общей формулой (III), полученного способом, предлагаемым настоящим изобретением, позволяет удалять небольшие количества не прореагировавших исходных ве ществ и побочных продуктов путем водной или водно-аммиачной экстракции или при помощи простой и весьма избирательной методики образования двойных солей. Для этого используют кислые соли общей формулой (IV) в виде водных растворов. A H H O O O P O (IV) В этой формуле А+ представляет собой ион натрия, калия или аммония. Кислые соли общей формулой (IV) обладают очень низкой кислотностью, рКа=7,21 (рКа - показатель степени кислотной диссоциации или кислотная экспонента), следовательно, при их использовании для очистки от примесей в предложенном способе отсутствует опасность растворения аминного основания общей формулой (III), так как основность таких соединений также слишком низка для образования солей. Неорганические кислоты с рКа=1,0-2,12 и органические кислоты с рКа=3,75-4,87, применяемые для очистки в соответствии с описанием венгерского патента № 187,775, являются сильными или средней силы кислотами, использование ко 4 39938 Максимальный выход при получении n-фторСелегилин-гидрохлорида, указанный в Примере 5 Европейской патентной заявки № 0,186,680, составляет только 47,1% в противоположность 85%ному выходу, достигаемому при использовании способа, являющегося предметом настоящего изобретения. Настоящее изобретение относится к способу получения L-изомерных производных пропаргиламмоний-хлорида общей формулой (I) путем разложения D-тартрата L-изомера амина общей формулой (II) под действием основания с последующим взаимодействием L-изомерного амина общей формулой (II) с галогенпроизводным общей формулой (V) в присутствии основания; полученный таким образом L-изомер общей формулой (III) реагирует с хлористым водородом в органическом растворителе, причем в приведенных общи х формулах X представляет собой атом галогена, а Y атом водорода или фтора. Данный способ характеризуется выделением амина в форме основания из D-тартрата L-изомерного амина общей формулой (II), где Y - как указано выше, в водной суспензии с гидроокисью аммония или основной солыо щелочного металла и/или аммония; далее, каждое соединение взаимодействует с галогенпроизводным общей формулой (V), где X - как указано выше, в буферной системе, непосредственно образующейся в процессе выделения основания и имеющей значение рН от 8 до 12, при температурах от 0 до 50°С; водный слой отделяют; органический слой, содержащий смесь L-изомерных аминов общими формулами (II) и (III), экстрагируют водным раствором гидроокиси аммония и/или водным раствором, содержащим смесь гидроокиси аммония и фосфатной соли, при рН=5,5-7,5; при этом L-изомерный амин общей формулой (II) или его соль растворяется в водном слое, и его избирательно отделяют от L-изомерного амина общей формулой (III); после перегонки L-изомерный амин общей формулой (III) переводят в Lизомерную соль общей формулой (I) общеизвестным методом: X - CH2 - C CH вывают, промывают ацетоном и высушивают. Получают 101,8 г L-N-метил-N-(2-фенил-1-метил)этил-N-пропаргиламин-гидрохлорида; выход составляет 91%. Чистота, по данным анализа методом ХПЛС (НРLС), составляет 99,9%. Известные и неизвестные общие загрязнители составляют менее, чем 0,1%. Пример 2. 149,7 г (0,5 моль) L-метил-анара-D-тартрата, 210 мл воды и 210 г концентрированной гидроокиси аммония перемешивают в течение 10 минут, после чего при температуре 20-25°C добавляют 65,5 г (0,55 моль) пропаргил-бромида. Реакционную смесь перемешивают при 30-35°С в течение часа, а затем при 40-45°C - в течение еще одного часа. После охлаждения до 20-25°С и разделения слоев процедуру продолжают, как описано в Примере 1. Выход Селегилин-гидрохлорида составляет 100,7 г (90%). Чистота полученного продукта соответствуе т таковой, указанной в Примере 1. Пример 3. Реакцию алкилирования проводят, как описано в Примере 2, и осуществляют разделение слоев при температуре 20-25°С. Верхний слой дважды встряхивают с водой (по 25 мл), затем дважды - с 10%-ным (по весу) раствором дигидрофосфата натрия (по 30 г ), а затем опять с 25 мл воды. Смесь разделяют. Верхний слой перегоняют, как описано в Примере 1, и обрабатывают, как описано там же. Выход конечного продукта составляет 98,5 г (88%). Чистота, на основании данных анализа методом ХПЛС(НРLС), составляет минимум 99,8%, количество известных и неизвестных примесей не превышает 0,05%. Пример 4. Смешивают 149,7 г (0,5 моль) L-метил-анараD-тартрата, 175 г концентрированной гидроокиси аммония и 175 мл воды. После 10-минутного перемешивания при 20-25°С добавляют 41,0 г (0,55 моль) пропаргилхлорида. Далее следуют процедуре, описанной в Примере 1 или в Примере 3. Выход конечного продукта составляет 95 г (85%). Чистота полученного продукта идентична таковой, указанной в Примере 1. Пример 5. Смешивают 178,6 г (0,5 моль) n-фтор-L-метиланара-D-тартрат-дигидрата, 210 г концентрированной гидроокиси аммония и 210 мл воды, и полученную смесь перемешивают в течение 10 минут при 20-25°С. Затем смесь охлаждают до 0°С и добавляют 65,5 г (0,55 моль) пропаргил-бромида. Смесь перемешивают при температуре 0-5°С, а затем полтора часа - при 20-25°С. Слои разделяют. Вер хний маслянистый слой встряхивают с насыщенным раствором хлористого натрия (2 ра- за по 30 г) и с раствором дигидрофосфата натрия (10% по весу, 2 раза по 30 г), затем опять с насыщенным раствором хлористого натрия (2 раза по 30 г), после чего слои разделяют. Верхний слой, содержащий n-фтор-Селегилин в форме основания, перегоняют в вакууме при давлении 0,10,2 кПа. Отогнанный материал растворяют в 300 мл ацетона и доводят рН до значения 2,5 путем пропускания через раствор газообразного хлористого водорода при 15-25°С. Суспензию (V) Детально способ, являющийся предметом настоящего изобретения, иллюстрируется следующими Примерами. Пример 1. Смешивают 149,7 г (0,5 моль) L-метил-анараD-тартрата и 210 г концентрированной гидроокиси аммония и перемешивают при 20-25°С в течение 10 минут. Добавляют 65,5 г (0,55 моль) пропаргилбромида и перемешивают реакционную смесь в течение 3 часов при 30-35°С. Затем добавляют 210 мл воды и разделяют слои при 20-25°С. Маслянистый слой встряхивают со смесью 25 мл воды и 25 г концентрированной гидроокиси аммония, а затем с 50 мл воды, и разделяют слои. Верхний слой (Селегилин в форме основания) перегоняют в вакууме при давлении 0,1-0,2 кПа. Отгон растворяют в 300 мл ацетона и доводят раствор до рН=2-2,5 посредством пропускания газообразного хлористого водорода при 20-30°С. Суспензию подвергают кристаллизации в течение 2 часов при температуре -10°С, осадок отфильтро 5 39938 подвергают кристаллизации в течение 2 часов при температуре -10°С, осадок отфильтровывают, промывают ацетоном и высушивают. В результате получают 102,3 г n-фтор-Селегилин-гидрохлорида с выходом 8 5%. Чистота продукта, согласно данным анализа методом HPLC, составляет 99,9%. Содержание известных и неизвестных примесей не превышает 0,1%. Пример 6. Смешивают 149,7 г (0,5 моль) L-метил-анараD-тартрата, 750 мл воды и 414,6 г (3,0 моль) карбоната калия и перемешивают в течение 10 минут при 30-35°С. Затем добавляют 65,5 г (0,55 моль) пропаргил-бромида и перемешивают в течение часа при 35-40°С, и еще один час - при 40-45°С. Смесь вновь охлаждают до температуры 20-25°С и отделяют нижний слой. Верхний маслянистый слой встряхивают с водой (5 раз по 50 мл, вновь разделяют слои, после чего верхний слой перегоняют и обрабатывают, как описано в Примере 1. Выход Селегилин-гидрохлорида составляет 101,8 г (91%). Чистота, согласно данным анализа методом ХПЛС (HPLC), составляет 99.9%. Содержание известных и неизвестных примесей не превышает 0,1%. Пример 7. Смешивают 149,7 г (0,5 моль) L-метил-анараD-тартрата, 750 мл воды и 318,0 г (3 моль) карбоната натрия и перемешивают в течение 10 минут при 30-35°С. Затем добавляют 65,5 г (0,55 моль) пропаргил-бромида и перемешивают сначала в течение часа при 35-40°С, а затем еще полчаса при 45-50°С. После охлаждения смеси до 20-25°С отделяют нижний слой. Верхний маслянистый слой встряхивают с водой (5 раз по 50 мл) и вновь разделяют слои. Верхний слой перегоняют и обрабатывают, как описано в Примере 1. Выход конечного продукта составляет 101,8 г (93%). Чистота, согласно данным анализа методом ХПЛС (НРLС), составляет 99,9%. Содержание известных и неизвестных примесей не превышает 0,1%. Пример 8. Реакционную смесь, полученную в результате реакции алкилирования, проведенной в соответствии с Примером 6 или 7, разделяют при 2025°С. Верхний слой встряхивают с водой (дважды по 50 мл), затем с 10%-ным (по весу) раствором дигидрофосфата натрия (дважды по 30 г), после чего вновь разделяют слои. Верхний слой перегоняют и обрабатывают, как описано в Примере 1. Выход конечного продукта составляет 99,5 г (89%). Чистота, согласно данным анализа методом ХПЛС (HPLС), составляет 99,9%. Содержание известных и неизвестных примесей не превышает 0.05%. Пример 9. Смешивают 88,3 г (0,25 моль) n-фтор-L-метиланара-D-тартрат-дигидрата, 375 мл воды и 138,2 г (1 моль) карбоната калия и перемешивают в течение 10 минут при 30-35°С. Смесь охлаждают до 10-15°С, добавляют 35,7 г (0,3 моль) пропаргилбромида и сначала перемешивают полчаса при 15-20°С, а потом еще 2,5 часа при 20-25°С. Слои разделяют. Верхний маслянистый слой встряхивают с 25 мл воды, 25 г насыщенного раствора хлористого натрия, 10%-ным (по весу) раствором дигидрофосфата натрия (дважды по 15 г) и вновь с насыщенным раствором хлористого натрия (дважды по 25 г), после чего разделяют слои. Верхний маслянистый слой, содержащий n-фторСелегилин в виде основания, перегоняют в вакууме при давлении 0,1-0,2 кПа. Отогнанное вещество растворяют в 150 мл ацетона и доводят рН раствора до 2,5-3,5 путем пропускания газообразного хлористого водорода при 15-25°С. Суспензию оставляют кристаллизоваться на 2 часа при температуре -10°С, осадок отфильтровывают, промывают ацетоном и высушивают. В результате получают 45,3 г n-фтор-Селегилин-гидрохлорида с выходом 75%. Чистота, в соответствии с данными анализа методом ХПЛС (НРLС), составляет 99,9%. Содержание известных и неизвестных примесей не превышает 0,1%. Пример 10. Смешивают 88,3 г (0,25 моль) n-фтор-L-метиланара-D-тартрат-дигидрата, 375 мл воды и 159,0 г (1,5 моль) карбоната натрия и перемешивают в течение 10 минут при 30-35°С. Далее процедуру проводят в соответствии с Примером 9. Выход конечного продукта составил 45,3 г (75%). Чистота, в соотве тствии с данными анализа методом ХПЛС (HPLС), идентична таковой, указанной в Примере 9. Пример 11. 88,3 г (0,25 моль) n-фтор-L-метил-анара-Dтартрат-дигидрата, 105 г концентрированной гидроокиси аммония и 105 г воды смешивают и перемешивают в течение 10 минут при 20-25°С. Затем смесь охлаждают до 5-10°С и добавляют 32,8 г (0,275 моль) пропаргил-бромида. Смесь перемешивают в течение часа при 5-10°С и еще один час - при 20-25°С, а затем нагревают до 40-45° и перемешивают еще один час. Слои разделяют, верхний маслянистый слой встряхивают со смесью 2 х 25 мл воды, и 25 мл концентрированной гидроокиси аммония, и с 2 х 25 г насыщенного раствора хлористого натрия и разделяют слои. Далее смесь обрабатывают, как описано в Примере 9. Выход конечного продукта составляет 48,3 г (80%). Чистота, согласно данным анализа методом ХПЛС (HPLC), соответствует таковой, указанной в Примере 9. Пример 12. 88,3 г (0,25 моль) n-фтор-L-метил-анара-Dтартрат-дигидрата перемешивают в течение 10 минут при 20-25°С в 105 г концентрированной гидроокиси аммония. Затем смесь охлаждают до 510°С и добавляют 32,8 г (0,275 моль) пропаргилбромида. Смесь перемешивают в течение часа при 5-10°С, затем час при 25-30°С и разделяют слои. Верхний маслянистый слой встряхивают с 25 мл воды, 25 г насыщенного раствора соли, 2 х 15 г 9%-ного (по весу) раствора дигидрофосфата натрия и 1%-ного (по весу) динатрий-гидрофосфа та, а также с 25 г насыщенного раствора хлористого натрия, после чего слои разделяют. Далее обработку проводят, как описано в Примере 9. 6 39938 Выход конечного продукта составляет 48,3 г (80%). Чистота соответствует таковой, указанной в Примере 9. Пример 13. Смешивают 88,3 г (0,25 моль) n-фтор-L-метиланара-D-тартрат-дигидрата, 105 г концентрированной гидроокиси аммония и 105 г воды и перемешивают в течение 10 минут при 20-25°С. Затем смесь охлаждают до 5°С и добавляют 32,8 г (0,275 моль) пропаргил-бромида. Смесь перемешивают в течение получаса при 5-10°С и в течение полутора часов - при 25-30°С. Смесь охлаждают до 20-25°С и разделяют слои. Верхний маслянистый слой встряхивают с 25 мл воды, 25 г насыщенного раствора хлористого натрия, 2 х 15 г водного раствора, содержащего 2% по весу ди натрий-гидрофосфата и 8% по весу дигидрофосфата натрия, после чего опять с 25 г насыщенного раствора хлористого натрия. Слои разделяют. Верхний слой, содержащий n-фтор-Селегилин в форме основания, перегоняют в вакууме под давлением 0,1-0,2 кПа. Отогнанный материал растворяют в 150 мл изопропилового спирта и доводят рН до 3,0-3,5 путем добавления хлористоводородной (соляной) кислоты при 15-25°С. Суспензию оставляют кристаллизоваться на 2 часа, осадок отфильтровывают, промывают изопропиловым спиртом и высушивают. В результате получают 48,3 г (80%) n-фторСелегилин-гидрохлорида. Качество продукта соответствует таковому, указанному в Примере 9. __________________________________________________________ ДП "Український інститут промислової власності" (Укрпатент) Україна, 01133, Київ-133, бульв. Лесі Українки, 26 (044) 295-81-42, 295-61-97 __________________________________________________________ Підписано до друку ________ 2001 р. Формат 60х84 1/8. Обсяг ______ обл.-вид. арк. Тираж 50 прим. Зам._______ ____________________________________________________________ УкрІНТЕІ, 03680, Київ-39 МСП, вул. Горького, 180. (044) 268-25-22 ___________________________________________________________ 7
ДивитисяДодаткова інформація
МПК / Мітки
МПК: C07C 211/29, C07C 209/08, C07C 211/27
Мітки: спосіб, пропаргіламоній, l-ізомерних, одержання, хлориду, похідних
Код посилання
<a href="https://ua.patents.su/7-39938-sposib-oderzhannya-l-izomernikh-pokhidnikh-propargilamonijj-khloridu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання l-ізомерних похідних пропаргіламоній хлориду</a>
Спосіб одержання похідних 1-ціклогексіл-4-аріл4-піперідінкарбонових кислот або їх кислотноадитивних солей, або їх стереохімічних ізомерних форм (його варіанти)

Номер патенту: 2700
Опубліковано: 26.12.1994
Автори: Джоан Уільямс, Марсель Геребернус Марія Льюікс, Раймон Стокброекс
Мітки: спосіб, кислот, солей, форм, стереохімічних, його, кислотноадітівних, похідних, варіанти, ізомерних, 1-ціклогексіл-4-аріл4-піперідінкарбонових, одержання
Формула / Реферат:
(57) 1. Способ получения производных 1-циклогексил-4-арил-4-пиперидинкар-боновой кислоты общей формулыгде R1 - водород или низший алкил; R2 - гидроксил, низший алкоксил, низший алкокси - низший алкоксил, фенокси - низший алкил, фенокси - низший алкоксил, в котором фенил может быть замещен низшим алкоксилом или низшим алкилом, амино, ди(низший алкил)амино, ди (низший алкил)амино-низший алкоксил, 4-морфолинил, 1-пирролидинил,...
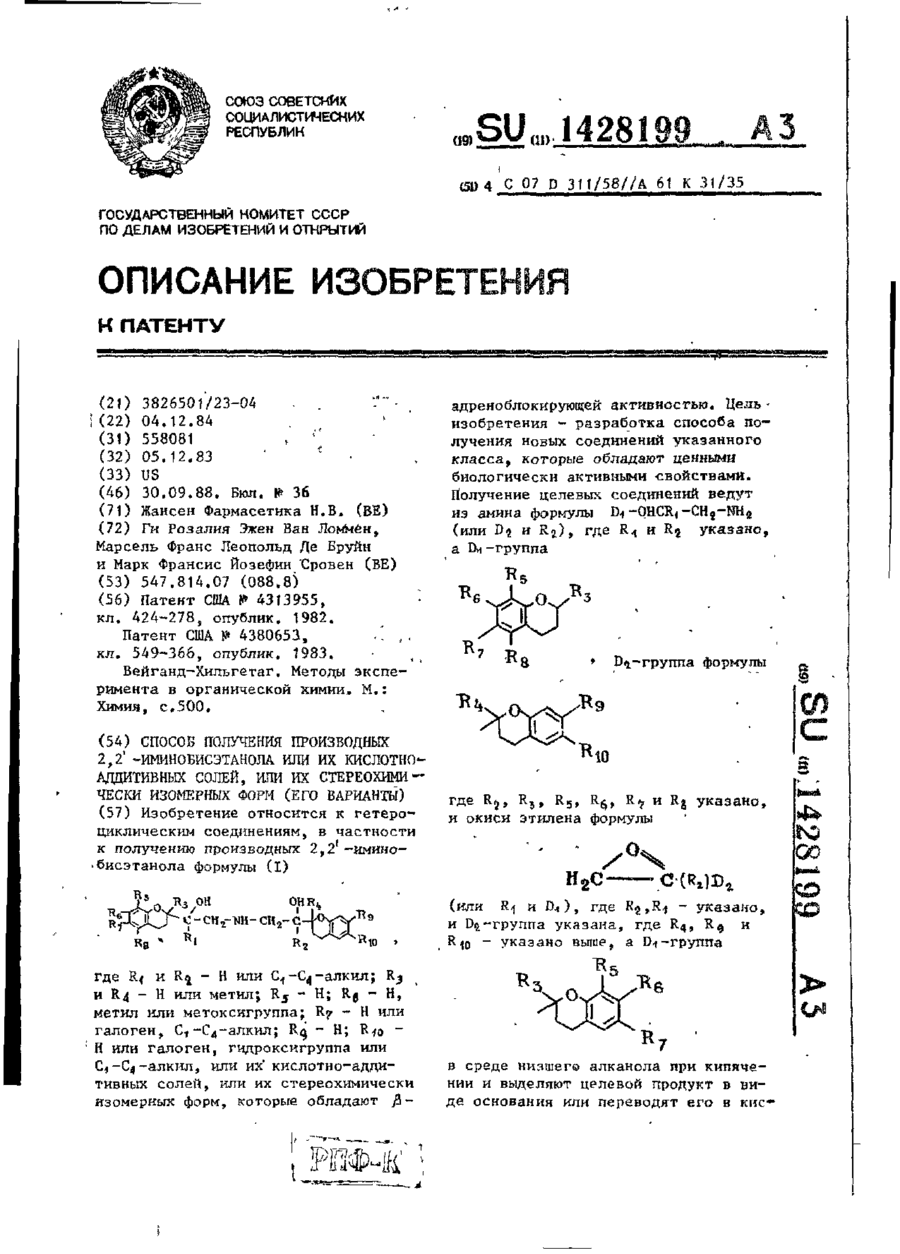
Спосіб одержання похідних 2,2′-імінобіоетанола або їх кислотно-адітівних солей, або їх стереохімічно ізомерних форм (його варіанти)
Номер патенту: 2707
Опубліковано: 26.12.1994
Автори: Марсель Франс Леопольд Де Бруйн, Гі Розалія Ежен Ван Ломмен, Марк Франсіс Йозефін Сровен
МПК: C07D 307/81, A61P 25/02, C07D 311/58, C07D 311/66, C07D 311/70, C07D 311/22, C07D 407/04, C07D 307/79, C07D 311/92, A61K 31/35, A61K 31/352
Мітки: варіанти, солей, кислотно-адітивних, форм, його, ізомерних, похідних, стереохімічно, спосіб, одержання, 2,2'-імінобіоетанола
Формула / Реферат:
1. Способ получения производных 2,2'-иминобисэтанола формулы Ігде R1 и R2— водород или С1 — С2 алкил; R3 и R4— водород или метил; R5 — водород; R6— водород, метил или метоксигруппа; R7 —водород или галоген; С1 — С4-алкид, ацетиламиногруппа, цианогруппа гидроксигруппа или метил-сульфониламиногруппа; R8 — водород или С1 — С4-алкил; R9 — водород; R10 — водород или галоген, гидроксигруппа или С1—С4-алкил, или их кислотно-аддитивных...
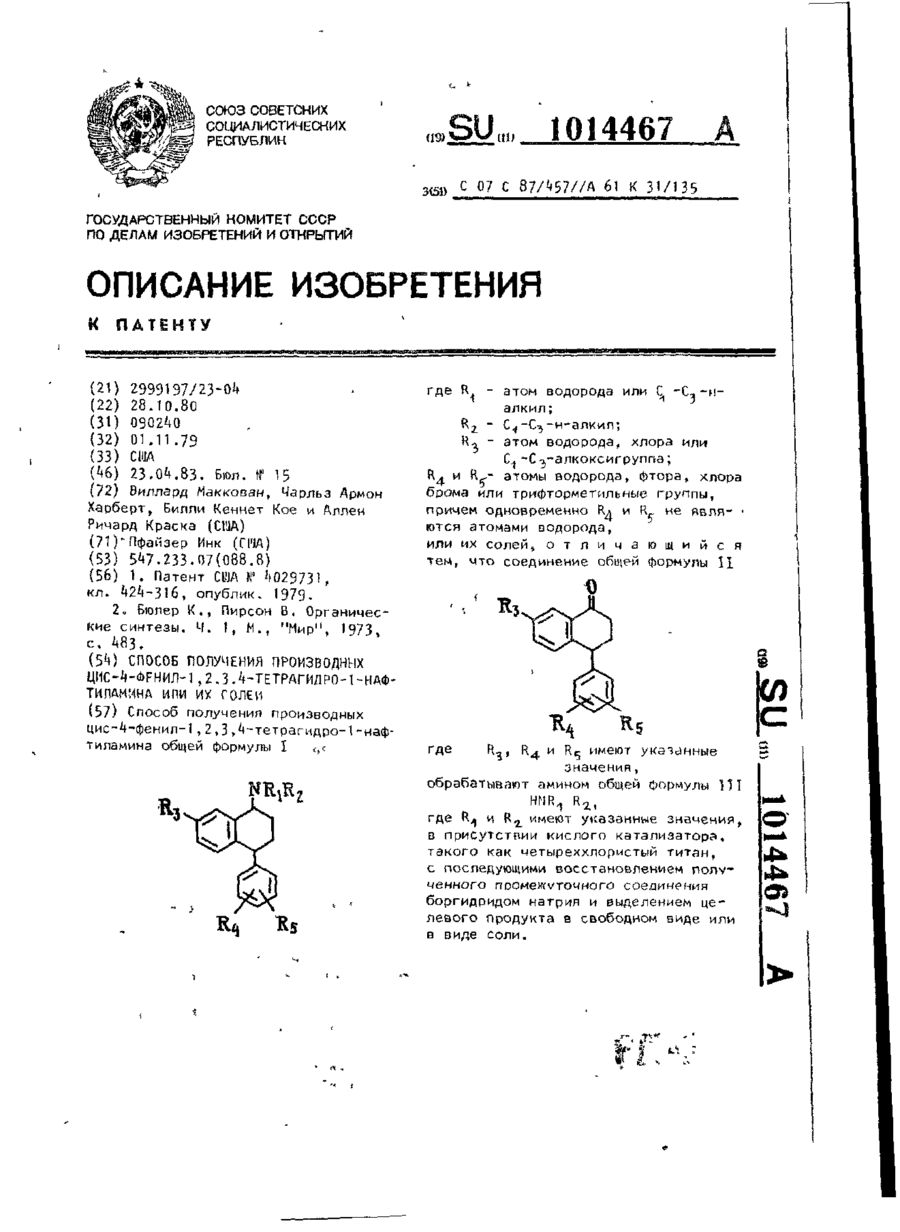
Спосіб одержання похідних ціс-4-феніл-1,2,3,4-тетрагідра-1-нафтіламіна або його солей
Номер патенту: 6301
Опубліковано: 29.12.1994
Автори: Аллен Річард Краска, Чарльз Армон Херберт, Віллард Маккован, Біллі Кеннет Кое
МПК: C07C 213/00, A61K 31/137, C07C 67/00, A61P 25/26, C07C 49/697, C07C 209/00, C07C 255/58, C07C 45/00, C07C 211/42, C07C 45/46, C07C 45/28, A61K 31/135, A61K 31/13, C07C 211/41, A61K 31/165, C07C 57/00, C07C 43/21, A61P 25/24, C07C 217/74
Мітки: похідних, ціс-4-феніл-1,2,3,4-тетрагідра-1-нафтіламіна, одержання, спосіб, солей
Формула / Реферат:
Способ получения производных цис-4-фенил-1,2,3,4-тетрагидро-1-нафтиламина общей формулы Ігде R1 - атом водорода или С1-С3-н-алкил; R2-С1-С3-н-алкил; R3 - атом водорода, хлора или С1-С3 - алкоксигруппа; R4 и R5 - атомы водорода, фтора, хлора, брома или трифторметильные группы, причем одновременно R4 и R5 не являются атомами водорода, или их солей, отличающийся тем, что соединение общей формулы IIгде R3, R4 и R5...
Спосіб одержання похідних n-(2-оксо-3-оксазолідініл)ацетамідів

Номер патенту: 7033
Опубліковано: 31.03.1995
Автори: Рудольф Зандмайєр, Ханспетер Шеллінг, Йост Харр
МПК: C07D 413/12, A01N 43/76, C07D 263/26, C07D 263/22
Мітки: спосіб, похідних, n-(2-оксо-3-оксазолідініл)ацетамідів, одержання
Формула / Реферат:
(57) Способ получения производных N-{2-оксо-3-оксазолидинил)ацетамидов, общей формулыгдеотличающийся тем, что проводят внутримолекулярную конденсацию соединения общей формулы
Спосіб одержання похідних пірімідінтріона

Номер патенту: 7040
Опубліковано: 31.03.1995
Автори: Бернард Зігфрід, Карел Вальтер(без Громадянства), Філіпп Гольд-Оберт, Діран Мелконіан, Стефан Хугентоблер, Жіндріх Фахта(без Громадянства)
МПК: C07D 239/62
Мітки: похідних, спосіб, пірімідінтріона, одержання
Формула / Реферат:
Формула изобретенияСпособ получения производных пиримидинтриона общей формулыгде R1 и R2 одинаковы или различны, фенил или низший С1-С4-алкил;R3-CH2CH(OCONH2)CH2OR,где R – С1-С5-алкил,путем взаимодействия натриевой соли малонилмочевины с галоидалкоксипро-панолкарбаматом в эквимолярных количествах при нагревании, отличающийся тем, что, с целью повышения выхода, к смеси производного малонилмочевины...
Попередній патент: Спосіб десорбції галію
Наступний патент: Гірка настойка “старка кіровоградська”
Випадковий патент: Спосіб діагностики розвитку атеросклерозу