Макроциклічні пурини для лікування вірусних інфекцій
Номер патенту: 113651
Опубліковано: 27.02.2017
Автори: Бонфанті Жан-Франсуа, Мюллер Філіпп, Дубле Фредерік Марк Моріс, Рабуассон П'єр Жан-Марі Бернар, Арну Ерік П'єр Александр, Фортен Жером Мішель Клод















































































































Формула / Реферат
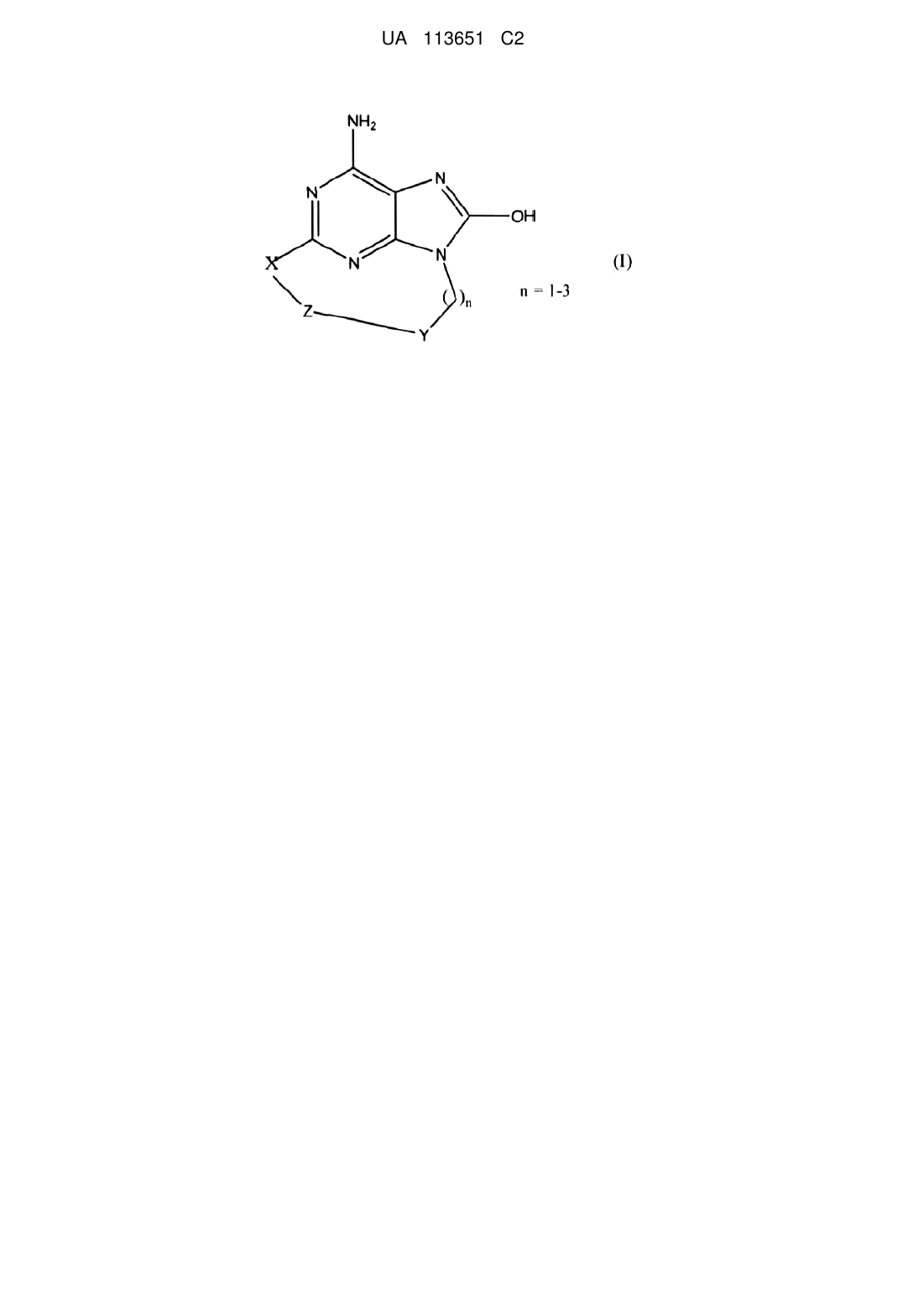
1. Сполука, що характеризується формулою (І)
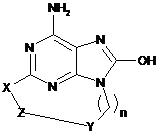 , (I)
, (I)
де n=1-3,
або її фармацевтично прийнятні солі, де X являє собою кисень, азот, сірку або
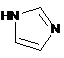 ,
,
Y являє собою ароматичне кільце або гетероциклічне кільце, що містить щонайменше азот, необов'язково заміщене одним або декількома замісниками, незалежно вибраними з С1-6алкілу, С1-4алкокси, трифторметилу або галогену,
Z являє собою насичений або ненасичений С1-10алкіл, необов'язково заміщений алкілом або алкілгідроксилом;
або Z являє собою С1-6алкіл-NН-С(О)-С1-6алкіл- або С1-6алкіл-NH-С(О)-С1-6алкіл-О-;
або Z являє собою С1-10алкіл-О-, де зазначений алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом або алкілгідроксилом,
або Z являє собою С1-6алкіл-O-С1-6алкіл-, де зазначений алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом або алкілгідроксилом,
або Z являє собою С1-6алкіл-О-С1-6алкіл-О-, де зазначений алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом або алкілгідроксилом.
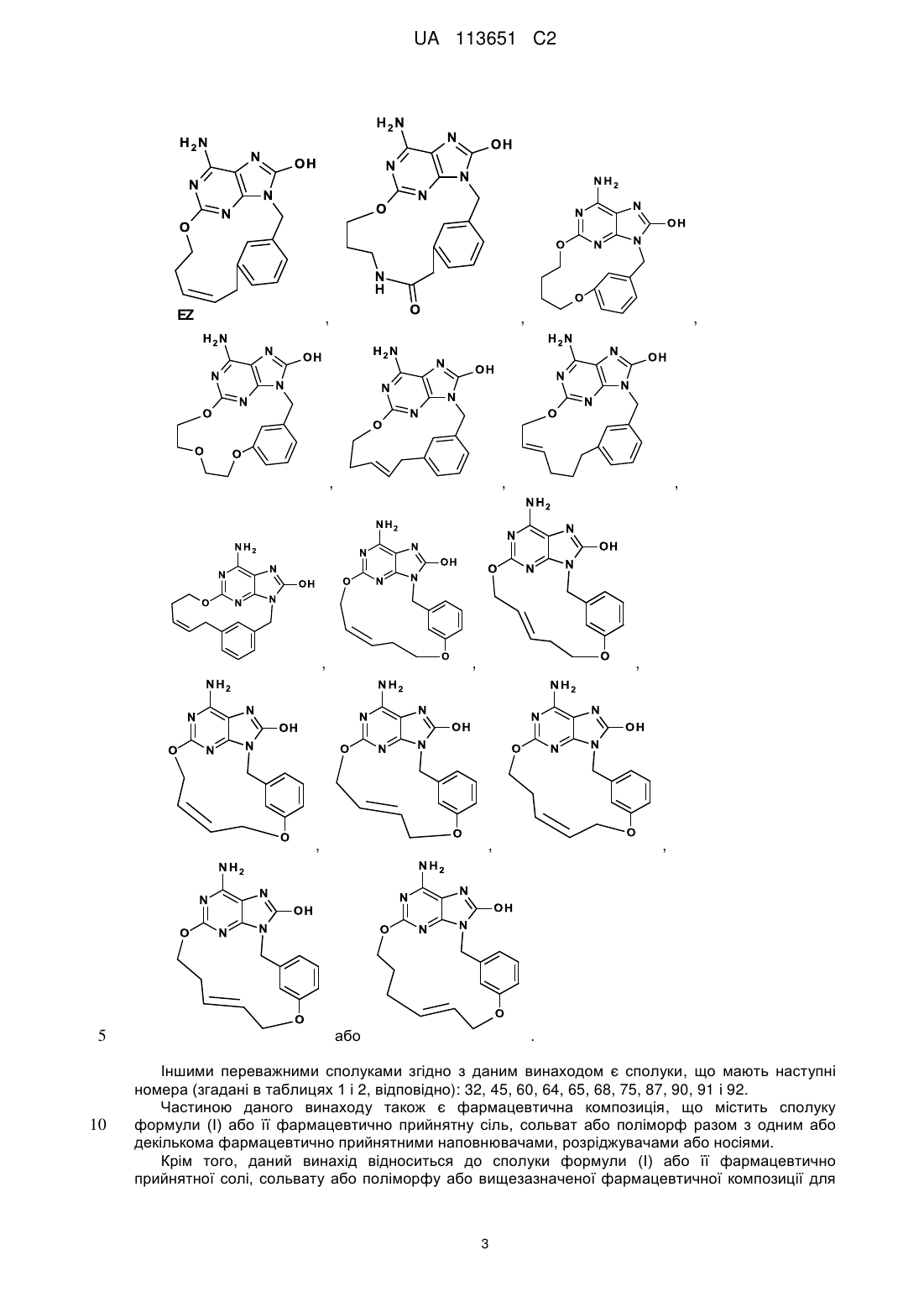
2. Сполука за п. 1, що характеризується однією з наступних формул, вибраних з групи
 ,
,  ,
, 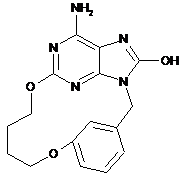 ,
,
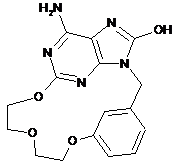 ,
, 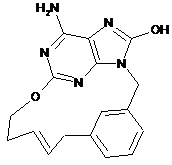 ,
, 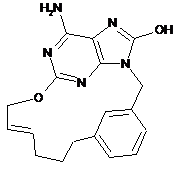 ,
,
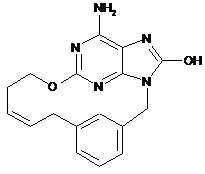 ,
, 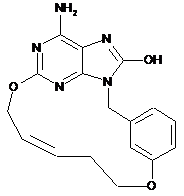 ,
, 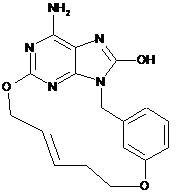 ,
,
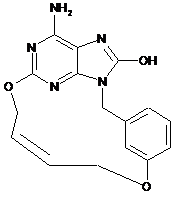 ,
, 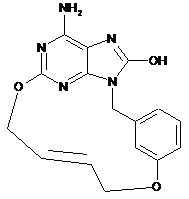 ,
, 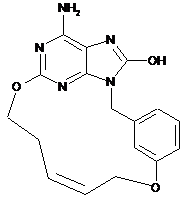 ,
,
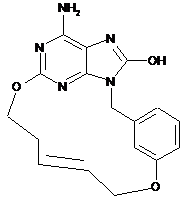 або
або 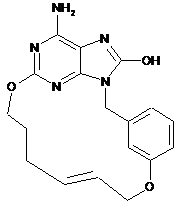 .
.
3. Фармацевтична композиція, що містить сполуку формули (І) за п. 1 або п. 2 або її фармацевтично прийнятну сіль, сольват або поліморф разом з одним або декількома фармацевтично прийнятними наповнювачами, розріджувачами або носіями.
4. Сполука формули (І) за п. 1 або п. 2 або її фармацевтично прийнятна сіль, сольват або поліморф або фармацевтична композиція за п. 3 для застосування як лікарського препарату.
5. Сполука формули (І) за п. 1 або п. 2 або її фармацевтично прийнятна сіль, сольват або поліморф або фармацевтична композиція за п. 3 для застосування у лікуванні порушення, в яке залучено модуляцію TLR7.
Текст
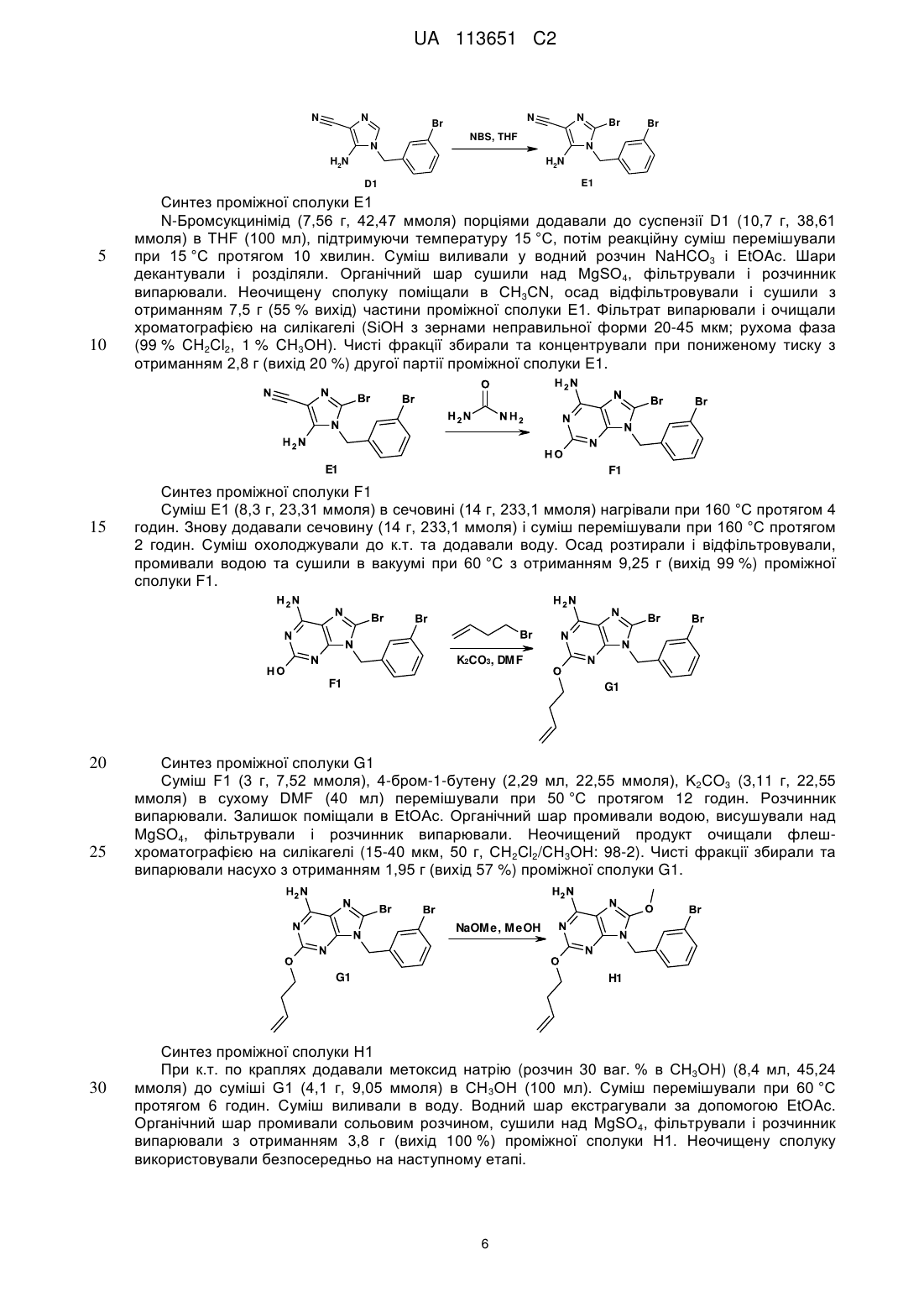
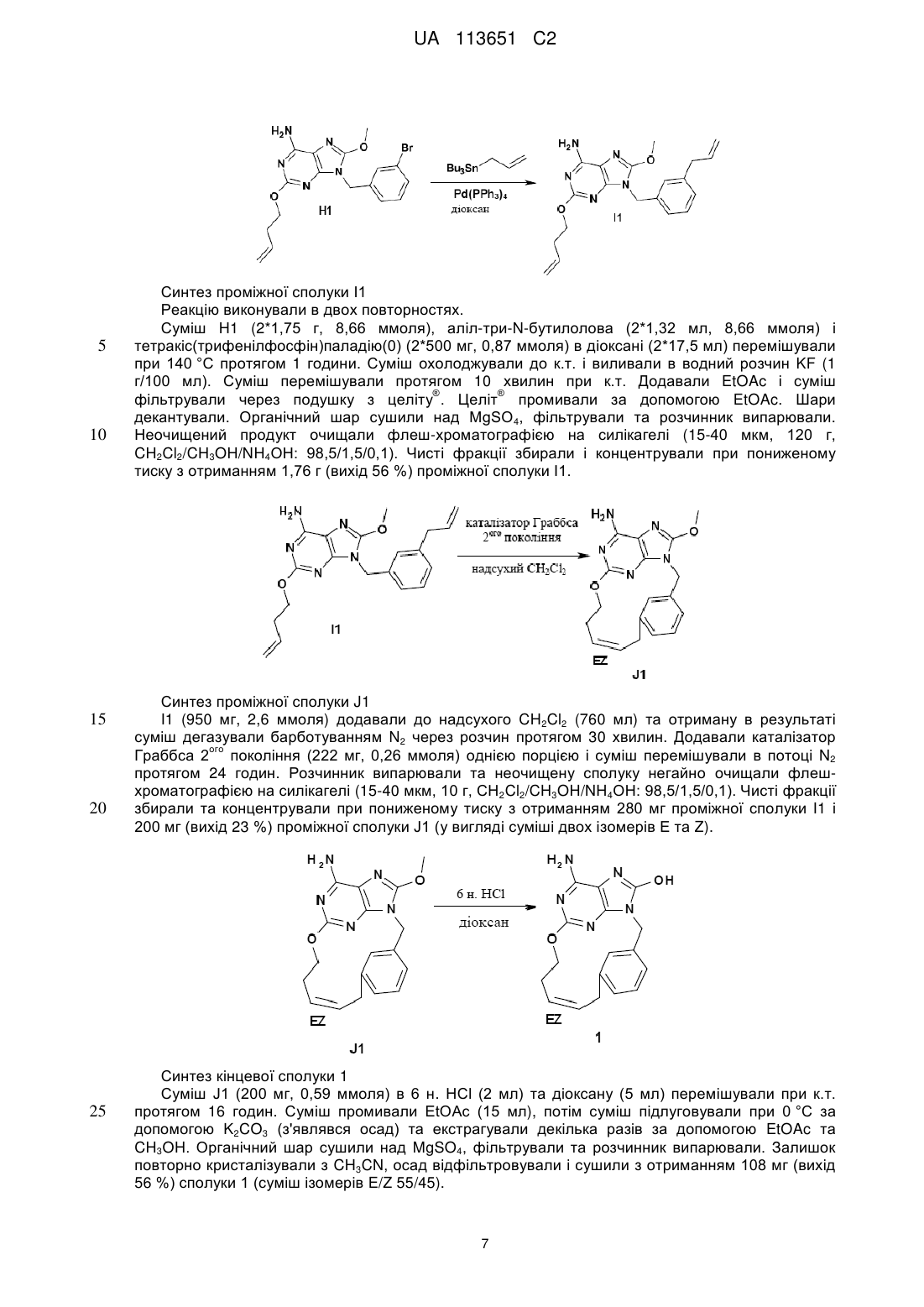
Реферат: Даний винахід стосується похідних макроциклічних пуринів, що характеризуються формулою (І), способів їхнього отримання, фармацевтичних композицій та їхнього застосування у лікуванні вірусних інфекцій. UA 113651 C2 (12) UA 113651 C2 UA 113651 C2 5 10 15 20 25 30 35 40 45 Даний винахід відноситься до похідних макроциклічних пуринів, способів їхнього отримання, фармацевтичних композицій та їхнього застосування у лікуванні вірусних інфекцій. Даний винахід відноситься до застосування похідних макроциклічних пуринів в лікуванні вірусних інфекцій, імунних або запальних порушень, за умови залучення модуляції або агонізму толл-подібних рецепторів (TLR). Толл-подібні рецептори являють собою первинні трансмембранні білки, що характеризуються позаклітинним лейцин-багатим доменом і цитоплазматичним розширенням, що містить консервативну ділянку. Врождена імунна система може розпізнавати патоген-асоційовані молекулярні паттерни за допомогою цих TLR, які експресуються на клітинній поверхні певних типів імунних клітин. Під час розпізнавання чужорідних патогенів активується утворення цитокінів та підвищення рівня експресії костимулюючих молекул на фагоцитах. Це приводить до модуляції поведінки Т-клітин. Було встановлено, що більшість видів ссавців мають від десяти до п'ятнадцяти типів толлподібних рецепторів. Загалом у людини та мишей було виявлено тринадцять TLR (під назвою TLR1-TLR13), а у інших видів ссавців було знайдено еквівалентні форми багатьох із них. Однак, еквіваленти певних TLR, виявлених у людини, присутні не у всіх ссавців. Наприклад, ген, що кодує білок, аналогічний TLR10 у людей, присутній у мишей, але, як виявляється, в певний момент в минулому був пошкоджений ретровірусом. З іншого боку, у мишей експресуються TLR 11, 12 та 13, жодний з яких не представлений у людини. У інших ссавців можуть експресуватися TLR, які не виявлені у людини. Інші види, що не є ссавцями, можуть мати TLR, що відрізняються від таких у ссавців, доказом цього служить TLR14, виявлений у риби фугу. Це може ускладнити спосіб застосування експериментальних тварин як моделей врожденого імунітету людини. Для детального огляду толл-подібних рецепторів див. наступні журнальні статті. Hoffmann, J.A., Nature, 426, p33-38, 2003; Akira, S., Takeda, K., and Kaisho, T., Annual Rev. Immunology, 21, p335-376, 2003; Ulevitch, R. J., Nature Reviews: Immunology, 4, p512-520, 2004. Раніше були описані сполуки, що проявляють активність щодо толл-подібних рецепторів, такі як похідні пурину в WO 2006/117670, похідні аденіну в WO 98/01448 і WO 99/28321 та піримідини в WO 2009/067081. Однак, існує гостра необхідність в нових модуляторах толл-подібних рецепторів, що характеризуються переважною селективністю, більш високою ефективністю, більш високою метаболічною стабільністю, а також покращеним профілем безпеки порівняно зі сполуками із відомого рівня техніки. Під час лікування певних вірусних інфекцій можуть регулярно вводитися ін'єкції інтерферону (IFN-альфа), як у випадку з вірусом гепатиту C (HCV). Більше інформації у посиланні Fried et. al. Peginterferon-alfa plus ribavirin for chronic hepatitis C virus infection, N Engl J Med 2002; 347: 97582. Доступні для перорального застосування низькомолекулярні індуктори IFN мають потенційні переваги як понижена імуногенність та зручність введення. Таким чином, нові індуктори IFN являють собою потенційно ефективний новий клас лікарських засобів для лікування вірусних інфекцій. Щодо приклада низькомолекулярного індуктора IFN, що має противірусний ефект, у літературі див. De Clercq, E.; Descamps, J.; De Somer, P. Science 1978, 200, 563-565. IFN-альфа також призначають у комбінації з іншими лікарськими засобами під час лікування певних типів раку (з посиланням на Eur. J. Cancer 46, 2849–57, та Cancer Res. 1992, 52, 1056 у якості прикладів). Агоністи TLR 7/8 також представляють інтерес як ад'юванти вакцин через їхню здатність індукувати яскраво виражену Th1 реакцію (з посиланням на Vaccines 2010, 6, 1−14, та Hum. Vaccines 2009, 5, 381−394, у якості прикладів). Згідно з даним винаходом представлена сполука формули (I), 1 UA 113651 C2 та її фармацевтично прийнятні солі, де X являє собою кисень, азот, сірку або HN N , 5 10 15 Y являє собою ароматичне кільце або гетероциклічне кільце, що містить щонайменше азот, необов'язково заміщене одним або декількома замісниками, незалежно вибраними з C1-6алкілу, C1-4алкоксі, трифторметилу або галогену, Z являє собою насичений або ненасичений C1-10алкіл, необов'язково заміщений алкілом або алкілгідроксилом; або Z являє собою C1-6алкіл-NH-C(O)-C1-6алкіл- або C1-6алкіл-NH-C(O)-C1-6алкіл-O-; або Z являє собою C1-10алкіл-O-, де зазначений алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом або алкілгідроксилом, або Z являє собою C1-6алкіл-O-C1-6алкіл-, де зазначений алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом або алкілгідроксилом, або Z являє собою C1-6алкіл-O-C1-6алкіл-О-, де зазначений алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом або алкілгідроксилом. Частину даного винаходу також складають такі сполуки формули (I), де X являє собою O, N-C1-4алкіл, NH, S або 20 HN N , 25 30 35 40 Y являє собою ароматичне кільце або гетероциклічне кільце, що містить щонайменше азот, необов'язково заміщене одним або декількома замісниками, незалежно вибраними з C1-6алкілу, C1-4алкоксі, трифторметилу, галогену, C(O)NH-C1-6алкілу, NH(CO)-C1-6алкілу, CN, NH-C1-6алкілу, N-(C1-6алкілу)2, C(O)-C1-6алкілу або OH, Z являє собою насичений або ненасичений C1-10алкіл, необов'язково заміщений алкілом, або алкілгідроксилом, або ОН; або Z являє собою C1-6алкіл-NH-C(O)-C1-6алкіл- або C1-6алкіл-NH-C(O)-C1-6алкіл-O-; або Z являє собою C1-6алкіл–NCH3-C(O)-C1-6алкіл- або C1-6алкіл–NCH3-C(O)-C1-6алкіл-O-; або Z являє собою C1-6алкіл–C(O)-NH-C1-6алкіл- або C1-6алкіл–C(O)-NH-C1-6алкіл-O-; або Z являє собою C1-6алкіл–C(O)-NCH3-C1-6алкіл- або C1-6алкіл–C(O)-NCH3-C1-6алкіл-O-; або Z являє собою C1-10алкіл-O-, де зазначений алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом, або алкілгідроксилом, або ОН, або Z являє собою C1-10алкіл-NH-, де зазначений алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом, або алкілгідроксилом, або ОН, або Z являє собою C1-6алкіл-O-C1-6алкіл-, де зазначений алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом, або алкілгідроксилом, або ОН, або Z являє собою C1-6алкіл-O-C1-6алкіл-О-, де зазначений алкіл є ненасиченим або насиченим і необов'язково може бути заміщений алкілом, або алкілгідроксилом, або ОН. Переважні сполуки, що характеризуються однією з наступних формул, згідно з даним винаходом вибрані з групи 2 UA 113651 C2 EZ , , , , , 10 , , , 5 , , , або , . Іншими переважними сполуками згідно з даним винаходом є сполуки, що мають наступні номера (згадані в таблицях 1 і 2, відповідно): 32, 45, 60, 64, 65, 68, 75, 87, 90, 91 і 92. Частиною даного винаходу також є фармацевтична композиція, що містить сполуку формули (I) або її фармацевтично прийнятну сіль, сольват або поліморф разом з одним або декількома фармацевтично прийнятними наповнювачами, розріджувачами або носіями. Крім того, даний винахід відноситься до сполуки формули (I) або її фармацевтично прийнятної солі, сольвату або поліморфу або вищезазначеної фармацевтичної композиції для 3 UA 113651 C2 5 10 15 20 25 30 35 40 45 50 55 60 застосування як лікарській препарат. Даний винахід також відноситься до сполуки формули (I) або її фармацевтично прийнятної солі, сольвату або поліморфу або вищезазначеної фармацевтичної композиції для застосування у лікуванні порушення, де залучена модуляція TLR7. Термін "алкіл" відноситься до найбільш насиченого (але який в певних сполуках згідно з даним винаходом є ненасиченим) аліфатичного вуглеводню з нерозгалуженим ланцюгом або розгалуженим ланцюгом, що містить певну кількість атомів вуглецю. Термін "галоген" відноситься до фтору, хлору, брому або йоду. Термін "алкоксі" відноситься до алкільної групи (ланцюга з вуглецю та водню), зв'язаної одинарним зв'язком з киснем, як наприклад, метоксигрупа або етоксигрупа. Фармацевтично прийнятні солі сполук формули (I) включають їхні солі приєднання кислоти та основні солі. Придатні солі приєднання кислоти утворюються з кислот, що утворюють нетоксичні солі. Придатні основні солі утворюються з основ, що утворюють нетоксичні солі. Сполуки за даним винаходом також можуть існувати в несольватованій і сольватованій формах. Термін "сольват" застосовується в даному документі для опису молекулярного комплексу, що містить сполуку за даним винаходом та одну або декілька молекул фармацевтично прийнятного розчинника, наприклад, етанолу. Термін "поліморф" відноситься до здатності сполуки за даним винаходом існувати в більш ніж одній формі або кристалічній структурі. Сполуки за даним винаходом можуть бути присутні в так званій формі "таутомеру(ів)”, якими називають ізомери органічних сполук, які легко піддаються взаємоперетворенню шляхом хімічної реакції, що має назву таутомеризація. Результатом цієї реакції є формальна міграція атома водню або протону, що супроводжується взаємною зміною одинарного зв'язку та суміжного подвійного зв'язку. Сполуки за даним винаходом можуть вводитися у вигляді кристалічних або аморфних продуктів. Вони можуть бути отримані, наприклад, у вигляді твердої пресованої маси, порошків або плівок шляхом таких способів, як осадження, кристалізація, ліофільне сушіння, розпилювальне сушіння або сушіння випарюванням. Їх можна вводити окремо або в комбінації з однією або декількома іншими сполуками за даним винаходом або в комбінації з одним або декількома іншими лікарськими засобами. Переважно, вони будуть вводитися у вигляді складу в поєднанні з одним або декількома фармацевтично прийнятними наповнювачами. Термін "наповнювач" застосовується в даному документі для опису будь-якого інгредієнта, іншого ніж сполука(и) за даним винаходом. Вибір наповнювача великою мірою залежить від таких факторів, як конкретний спосіб введення, вплив наповнювача на розчинність та стабільність і природа лікарської форми. Сполуки за даним винаходом або будь-яка їхня підгрупа можуть бути складені в різні фармацевтичні форми для цілей введення. У якості придатних композицій можуть бути зазначені всі композиції, що зазвичай використовуються для системного введення лікарських засобів. Для отримання фармацевтичних композицій за даним винаходом ефективна кількість конкретної сполуки, необов'язково у формі солі приєднання, у вигляді активного інгредієнта об'єднується в однорідну суміш з фармацевтично прийнятним носієм, при цьому носій може бути різноманітної форми залежно від форми препарату, якій потрібний для введення. Ці фармацевтичні композиції бажано перебувають у одиничній лікарській формі, що придатна, наприклад, для перорального, ректального або черезшкірного введення. Наприклад, при отриманні композицій в пероральній лікарській формі може використовуватися будь-яке з загальноприйнятих фармацевтичних середовищ, таке як, наприклад, вода, гліколі, олії, спирти тощо у випадку пероральних рідких препаратів, таких як суспензії, сиропи, еліксири, емульсії та розчини; або твердих носіїв, таких як крохмалі, цукри, каолін, розріджувачі, змащувальні засоби, зв'язувальні засоби, засоби для покращення здатності таблеток розпадатися тощо, у випадку порошків, пілюль, капсул і таблеток. Завдяки їхній простоті введення таблетки і капсули являють собою найбільш переважні пероральні форми одиниці дозування, у цьому випадку, вочевидь, застосовують тверді фармацевтичні носії. Також включені препарати в твердій формі, які незадовго до застосування можуть бути перетворені в препарати в рідкій формі. В композиціях, придатних для черезшкірного введення, носій необов'язково включає засіб, що сприяє проникненню, та/або придатний змочувальний засіб, необов'язково в комбінації з придатними добавками будь-якої природи в мінімальних пропорціях, при цьому добавки не мають значного шкідливого впливу на шкіру. Зазначені добавки можуть полегшувати введення в шкіру та/або можуть бути корисними для отримання потрібних композицій. Ці композиції можна вводити різними шляхами, наприклад, в формі трансдермального пластиру, в формі препарату для крапкового нанесення, в формі мазі. Сполуки за даним винаходом можна також вводити 4 UA 113651 C2 5 10 15 20 25 30 шляхом інгаляції або інсуфляції за допомогою способів та складів, що використовуються в даній галузі для введення таким шляхом. Отже, переважно сполуки за даним винаходом можно вводити в легені в формі розчину, суспензії або сухого порошку. Особливо переважним є складання вищезазначених фармацевтичних композицій в одиничній лікарській формі для простоти введення і рівномірності дозування. Одиничною лікарською формою, як застосовується в даному документі, називають фізично окремі одиниці, що придатні у якості одиничних доз, при цьому кожна одиниця містить попередньо встановлену кількість активного інгредієнта, розраховану для отримання потрібного терапевтичного ефекту, в поєднанні з необхідним фармацевтичним носієм. Прикладами таких одиничних лікарських форм є таблетки (в тому числі ділені таблетки або таблетки, вкриті оболонкою), капсули, пілюлі, пакетики з порошком, пластинки, супозиторії, ін'єкційні розчини або суспензії тощо, а також їх окремі множини. Фахівці в галузі лікування інфекційних захворювань зможуть визначити ефективну кількість, виходячи з результатів тестів, представлених далі в даному документі. Загалом передбачається, що ефективна добова кількість становитиме від 0,01 мг/кг до 50 мг/кг маси тіла, більш переважно від 0,1 мг/кг до 10 мг/кг маси тіла. Може бути доцільним введення потрібної дози у вигляді двох, трьох, чотирьох або більшої кількості частин дози з відповідними інтервалами протягом дня. Зазначені частини дози можуть бути складені у вигляді одиничних лікарських форм, наприклад, таких, що містять від 1 до 1000 мг і, зокрема, від 5 до 200 мг активного інгредієнта на одиничну лікарську форму. Точне дозування і частота введення залежать від конкретної застосовуваної сполуки формули (I), конкретного стану, який лікується, тяжкості стану, лікування якого здійснюють, віку, ваги та загального фізичного стану конкретного пацієнта, а також іншої лікарської терапії, яку може отримувати індивідуум, як це добре відомо фахівцям в даній галузі техніки. Більш того, очевидно, що ефективна кількість може бути зменшена або збільшена залежно від реакції суб'єкта, який піддається лікуванню, та/або залежно від оцінки лікаря, що призначає сполуки за даним винаходом. Таким чином, вищезазначені діапазони ефективної кількості є тільки рекомендаціями і не призначені якимось чином обмежувати обсяг або застосування даного винаходу. Загальна схема отримання кінцевих продуктів: спосіб 1 , HCl EtOH A 35 B1 C D1 Синтез проміжної сполуки D1 При 10 °C 3-бромбензиламін (11,9 г, 63,96 ммоля) по краплях додавали до суміші A1 (10 г, 60,91 ммоля) і C1 (125 мг, 0,97 ммоля) в EtOH (100 мл). Суміш перемішували при к.т. протягом ночі. По краплях додавали 120 мл 1 н. NaOH і суміш перемішували при к.т. протягом 1 години. Осад відфільтровували, промивали мінімальною кількістю холодного EtOH і сушили з отриманням 12,74 г (вихід 75 %) проміжної сполуки D1. 5 UA 113651 C2 N N N NBS, THF N Синтез проміжної сполуки Е1 N-Бромсукцинімід (7,56 г, 42,47 ммоля) порціями додавали до суспензії D1 (10,7 г, 38,61 ммоля) в THF (100 мл), підтримуючи температуру 15 °C, потім реакційну суміш перемішували при 15 °C протягом 10 хвилин. Суміш виливали у водний розчин NaHCO3 і EtOAc. Шари декантували і розділяли. Органічний шар сушили над MgSO4, фільтрували і розчинник випарювали. Неочищену сполуку поміщали в CH3CN, осад відфільтровували і сушили з отриманням 7,5 г (55 % вихід) частини проміжної сполуки E1. Фільтрат випарювали і очищали хроматографією на силікагелі (SiOH з зернами неправильної форми 20-45 мкм; рухома фаза (99 % CH2Cl2, 1 % CH3OH). Чисті фракції збирали та концентрували при пониженому тиску з отриманням 2,8 г (вихід 20 %) другої партії проміжної сполуки E1. E1 15 Br E1 D1 10 Br H2N H2N 5 N N Br F1 Синтез проміжної сполуки F1 Суміш E1 (8,3 г, 23,31 ммоля) в сечовині (14 г, 233,1 ммоля) нагрівали при 160 °C протягом 4 годин. Знову додавали сечовину (14 г, 233,1 ммоля) і суміш перемішували при 160 °C протягом 2 годин. Суміш охолоджували до к.т. та додавали воду. Осад розтирали і відфільтровували, промивали водою та сушили в вакуумі при 60 °C з отриманням 9,25 г (вихід 99 %) проміжної сполуки F1. K2CO3, DMF F1 20 25 G1 Синтез проміжної сполуки G1 Суміш F1 (3 г, 7,52 ммоля), 4-бром-1-бутену (2,29 мл, 22,55 ммоля), K2CO3 (3,11 г, 22,55 ммоля) в сухому DMF (40 мл) перемішували при 50 °C протягом 12 годин. Розчинник випарювали. Залишок поміщали в EtOAc. Органічний шар промивали водою, висушували над MgSO4, фільтрували і розчинник випарювали. Неочищений продукт очищали флешхроматографією на силікагелі (15-40 мкм, 50 г, CH2Cl2/CH3OH: 98-2). Чисті фракції збирали та випарювали насухо з отриманням 1,95 г (вихід 57 %) проміжної сполуки G1. NaOMe, MeOH G1 30 H1 Синтез проміжної сполуки H1 При к.т. по краплях додавали метоксид натрію (розчин 30 ваг. % в CH3OH) (8,4 мл, 45,24 ммоля) до суміші G1 (4,1 г, 9,05 ммоля) в CH 3OH (100 мл). Суміш перемішували при 60 °C протягом 6 годин. Суміш виливали в воду. Водний шар екстрагували за допомогою EtOAc. Органічний шар промивали сольовим розчином, сушили над MgSO4, фільтрували і розчинник випарювали з отриманням 3,8 г (вихід 100 %) проміжної сполуки H1. Неочищену сполуку використовували безпосередньо на наступному етапі. 6 UA 113651 C2 5 10 15 20 25 Синтез проміжної сполуки I1 Реакцію виконували в двох повторностях. Суміш H1 (2*1,75 г, 8,66 ммоля), аліл-три-N-бутилолова (2*1,32 мл, 8,66 ммоля) і тетракіс(трифенілфосфін)паладію(0) (2*500 мг, 0,87 ммоля) в діоксані (2*17,5 мл) перемішували при 140 °C протягом 1 години. Суміш охолоджували до к.т. і виливали в водний розчин KF (1 г/100 мл). Суміш перемішували протягом 10 хвилин при к.т. Додавали EtOAc і суміш ® ® фільтрували через подушку з целіту . Целіт промивали за допомогою EtOAc. Шари декантували. Органічний шар сушили над MgSO4, фільтрували та розчинник випарювали. Неочищений продукт очищали флеш-хроматографією на силікагелі (15-40 мкм, 120 г, CH2Cl2/CH3OH/NH4OH: 98,5/1,5/0,1). Чисті фракції збирали і концентрували при пониженому тиску з отриманням 1,76 г (вихід 56 %) проміжної сполуки I1. Синтез проміжної сполуки J1 I1 (950 мг, 2,6 ммоля) додавали до надсухого CH2Cl2 (760 мл) та отриману в результаті суміш дегазували барботуванням N2 через розчин протягом 30 хвилин. Додавали каталізатор ого Граббса 2 покоління (222 мг, 0,26 ммоля) однією порцією і суміш перемішували в потоці N2 протягом 24 годин. Розчинник випарювали та неочищену сполуку негайно очищали флешхроматографією на силікагелі (15-40 мкм, 10 г, CH2Cl2/CH3OH/NH4OH: 98,5/1,5/0,1). Чисті фракції збирали та концентрували при пониженому тиску з отриманням 280 мг проміжної сполуки I1 і 200 мг (вихід 23 %) проміжної сполуки J1 (у вигляді суміші двох ізомерів E та Z). Синтез кінцевої сполуки 1 Суміш J1 (200 мг, 0,59 ммоля) в 6 н. HCl (2 мл) та діоксану (5 мл) перемішували при к.т. протягом 16 годин. Суміш промивали EtOAc (15 мл), потім суміш підлуговували при 0 °C за допомогою K2CO3 (з'являвся осад) та екстрагували декілька разів за допомогою EtOAc та CH3OH. Органічний шар сушили над MgSO4, фільтрували та розчинник випарювали. Залишок повторно кристалізували з CH3CN, осад відфільтровували і сушили з отриманням 108 мг (вихід 56 %) сполуки 1 (суміш ізомерів E/Z 55/45). 7 UA 113651 C2 Загальна схема отримання кінцевих продуктів: спосіб 2 5 10 Синтез проміжної сполуки K1 Суміш J1 (130 мг, 0,39 ммоля), Pd/C (10 %) (20 мг, 0,02 ммоля) в CH3OH (30 мл) гідрогенізували при атмосферному тиску H2 протягом 4 годин. Каталізатор видаляли ® ® фільтрацією через целіт . Целіт промивали за допомогою CH3OH. Фільтрат випарювали з отриманням 126 мг (вихід 96 %) проміжної сполуки K1, використовуваної як такої на наступному етапі. Синтез кінцевої сполуки 2 При к.т. суміш K1 (110 мг, 0,32 ммоля) в 6 н. HCl (1 мл) і діоксану (2 мл) перемішували протягом 6 годин. Суміш виливали на лід та нейтралізували за допомогою 3 н. NaOH. Осад відфільтровували, промивали водою, EtOH, потім діетиловим ефіром та сушили з отриманням 79 мг (вихід 75 %) сполуки 2. Загальна схема отримання кінцевих продуктів: спосіб 3 15 N N H N N K2CO3, DMF L1 20 N Br N M1 Синтез проміжної сполуки M1 L1 (3,0 г, 32,23 ммоля), 5-бром-1-пентен (4,8 г, 32,23 ммоля) і K2CO3 (5,34 г, 38,67 ммоля) в DMF (75 мл) перемішували при 60 °C протягом 12 годин. Суміш концентрували при пониженому тиску. Залишок поміщали в EtOAc. Органічний шар промивали водою, сушили над MgSO4, фільтрували та розчинник випарювали з отриманням 4,6 г (вихід 89 %) проміжної сполуки M1, використовуваної як такої на наступному етапі. 8 UA 113651 C2 Na EtOH M1 5 10 15 20 25 30 E1 N1 Синтез проміжної сполуки N1 В потоці N2 натрій (1,42 г, 62,03 ммоля) розчиняли в EtOH (80 мл) при к.т. По краплях додавали M1 (2,0 г, 12,41 ммоля), E1 (4,42 г, 12,41 ммоля) в EtOH (20 мл) і отриману в результаті суміш перемішували при 90 °C протягом 5 годин в потоці N2. Розчинник випарювали. Додавали EtOAc та воду. Суміш екстрагували за допомогою EtOAc. Органічний шар промивали сольовим розчином, сушили над MgSO4, фільтрували і розчинник випарювали. Неочищений продукт очищали хроматографією на силікагелі (SiOH з зернами неправильної форми 20-45 мкм; рухома фаза (0,5 % NH4OH, 94 % CH2Cl2, 6 % CH3OH). Чисті фракції збирали та концентрували при пониженому тиску з отриманням 3,15 г (вихід 53 %) проміжної сполуки N1. Синтез проміжної сполуки O1 Суміш N1 (0,50 г, 1,04 ммоля), аліл-три-N-бутилолова (0,32 мл, 1,04 ммоля) і тетракіс(трифенілфосфін)паладію(0) (120 мг, 0,10 ммоля) в діоксані (4 мл) перемішували при 140 °C протягом 1 години. Суміш охолоджували до к.т. та виливали у водний розчин KF (5 г/100 мл). Суміш перемішували протягом 10 хвилин при к.т. Додавали EtOAc та шари декантували. Органічний шар промивали сольовим розчином, сушили над MgSO4, фільтрували та розчинник випарювали. Неочищений продукт очищали хроматографією на силікагелі (SiOH з зернами неправильної форми 15-45 мкм; рухома фаза (0,5 % NH4OH, 95 % CH2Cl2, 5 % CH3OH). Чисті фракції збирали та концентрували при пониженому тиску з отриманням 300 мг (вихід 65 %) проміжної сполуки О1. Синтез проміжної сполуки P1 О1 (1,80 г, 4,06 ммоля) додавали до надсухого CH2Cl2 (1080 мл) і отриману в результаті суміш дегазували барботуванням N2 через розчин протягом 30 хвилин. Додавали каталізатор ого Граббса 2 покоління (346 мг, 0,41 ммоля) однією порцією і суміш перемішували при к.т. в потоці N2 протягом 24 годин. Суміш концентрували при пониженому тиску. Неочищений продукт очищали флеш-хроматографією на силікагелі (15-40 мкм, 120 г, CH2Cl2/CH3OH/NH4OH: 96/4/0,5). Чисті фракції збирали і концентрували при пониженому тиску з отриманням 1,47 г неочищеного продукту. Залишок очищали хроматографією з оберненою фазою (X-Bridge-C18 5 мкм 30*150 мм), рухома фаза (градієнт від 80 % NH4HCO3 0,5 % буфер, pH 10, 20 % CH3CN до 0 % NH4HCO3 0,5 % буфер, pH 10, 100 % CH3CN). Чисті фракції збирали і концентрували при пониженому тиску з отриманням 83 мг (вихід 5 %) проміжної сполуки P1 (у вигляді суміші двох ізомерів E та Z) і 800 мг проміжної сполуки O1. 9 UA 113651 C2 5 10 15 20 Синтез кінцевої сполуки 3 P1 (40 мг, 0,10 ммоля) в 6 н. HCl (2 мл) і діоксані (2 мл) перемішували при к.т. протягом 18 годин. При 0 °C суміш підлуговували за допомогою K2CO3 і екстрагували за допомогою EtOAc і CH3OH. Органічний шар сушили над MgSO4, фільтрували і розчинник випарювали. Неочищений продукт очищали флеш-хроматографією на силікагелі (15-40 мкм, 12 г, CH2Cl2/CH3OH/NH4OH: 90/10/0,5). Чисті фракції збирали і концентрували при пониженому тиску з отриманням 23 мг. Сполуку поміщали в діетиловий ефір, осад відфільтровували і сушили з отриманням 15 мг (вихід 40 %) сполуки 3 (у вигляді суміші двох ізомерів E/Z 70/30). Загальна схема отримання кінцевих продуктів: спосіб 4 Синтез проміжної сполуки Q1 Суміш Р1 (70 мг, 0,17 ммоля), Pd/C 10 % (18 мг, 0,02 ммоля) в CH3OH (5 мл) гідрогенізували при атмосферному тиску H2 протягом 4 годин. Каталізатор видаляли фільтрацією через ® ® подушку з целіту . Целіт промивали за допомогою CH3OH і фільтрат випарювали. Неочищений продукт очищали флеш-хроматографією на силікагелі (15-40 мкм, 10 г, CH2Cl2/CH3OH/NH4OH: 96/4/0,5). Чисті фракції збирали і концентрували при пониженому тиску з отриманням 50 мг (вихід 71 %) проміжної сполуки Q1. Синтез кінцевої сполуки 4 Q1 (50 мг, 0,12 ммоля) в 6 н. HCl (1 мл) і діоксані (1 мл) перемішували при к.т. протягом 18 годин. Осад відфільтровували, промивали діоксаном і сушили з отриманням 38 мг (вихід 71 %) сполуки 4 (1HCl, 1H2O). Загальна схема отримання кінцевих продуктів: спосіб 5 10 UA 113651 C2 , HCl EtOH R1 A1 5 10 C1 S1 Синтез проміжної сполуки S1 При 10 °C R1 (7,70 г, 34,79 ммоля) в EtOH (30 мл) по краплях додавали до суміші A1 (5,44 г, 33,14 ммоля), C1 (68 мг, 0,53 ммоля) в EtOH (25 мл). Суміш перемішували при к.т. протягом ночі. По краплях додавали 60 мл 1 н. NaOH і суміш перемішували при к.т. протягом 1 години. Суміш екстрагували за допомогою CH2Cl2 (3 рази). Об'єднані органічні шари промивали сольовим розчином, сушили над MgSO4, фільтрували і розчинник випарювали. Неочищений продукт очищали хроматографією на силікагелі (SiOH з зернами неправильної форми 20-45 мкм; рухома фаза (0,1 % NH4OH, 98 % CH2Cl2, 2 % CH3OH). Чисті фракції збирали і концентрували при пониженому тиску з отриманням 5,1 г (вихід 49 %) проміжної сполуки S1. O O O N N N N N NBS, THF N H2 N H2 N T1 S1 15 Синтез проміжної сполуки T1 При 10 °C в потоці N2 N-бромсукцинімід (2,90 г, 16,33 ммоля) порціями додавали до суміші S1 (5,1 г, 16,33 ммоля) в THF (100 мл). Суміш перемішували протягом 10 хвилин при 10 °C. Суміш виливали в 10 % розчин NaHCO3 в воді та EtOAc. Шари декантували. Органічний шар сушили над MgSO4, фільтрували і розчинник випарювали. Неочищений продукт очищали флешхроматографією на силікагелі (15-40 мкм, 80 г, CH2Cl2/CH3OH 99-1). Чисті фракції збирали і концентрували при пониженому тиску з отриманням 3,75 г (вихід 59 %) проміжної сполуки Т1. O O O O Br N N N CH3CN T1 25 30 35 O O N H 2N 20 O Br H 2N O Br N N N N O U1 Синтез проміжної сполуки U1 Бензоїлізоціанат (7,05 г, 47,92 ммоля) додавали до суміші T1 (3,75 г, 9,58 ммоля) в CH 3CN (80 мл) при перемішуванні при к.т. Суміш перемішували при к.т. протягом ночі. Розчинник випарювали, залишок розчиняли в 2-пропанолі/25 % водному NH3 1:1 (200 мл) і отриманий в результаті розчин перемішували при к.т. протягом 72 годин. Розчинник випарювали і отриману в результаті суміш виливали в воду, нейтралізували розведеною HCl і екстрагували за допомогою ® ® EtOAc. Шари фільтрували через подушку з целіту , целіт промивали за допомогою EtOAc. Фільтрат декантували. Органічний шар сушили над MgSO 4, фільтрували і розчинник випарювали. Неочищену сполуку очищали флеш-хроматографією на силікагелі (15-40 мкм, 80 г, CH2Cl2/CH3OH/NH4OH: 98/2/0,1). Чисті фракції збирали і концентрували при пониженому тиску з отриманням 1,70 г (вихід 33 %) проміжної сполуки U1. Синтез проміжної сполуки V1 Суміш U1 (1,6 г, 2,97 ммоля) в 25 % NH4OH (160 мл) та iPrOH (160 мл) перемішували при к.т. протягом 72 годин. iPrOH випарювали і додавали воду. Суміш екстрагували за допомогою EtOAc. Осад з'являвся між 2 фазами. Осад відфільтровували і сушили з отриманням 880 мг (вихід 68 %) проміжної сполуки V1. Органічний шар сушили над MgSO4, фільтрували і розчинник 11 UA 113651 C2 випарювали з отриманням 680 мг проміжної сполуки U1. K2CO3, DMF V1 5 W1 Синтез проміжної сполуки W1 Суміш V1 (520 мг, 1,20 ммоля), трет-бутил-N-(3-бромпропіл)карбамату (570 мг, 2,40 ммоля) і K2CO3 (496 мг, 3,59 ммоля) в DMF (20 мл) перемішували при 80 °C протягом 5 годин. Після охолодження до к.т. суміш виливали в воду і екстрагували за допомогою EtOAc. Органічний шар промивали водою, сольовим розчином, сушили над MgSO4, фільтрували і розчинник випарювали з отриманням 800 мг (вихід >100 %) проміжної сполуки W1. Неочищену сполуку використовували безпосередньо на наступному етапі. NaOH 30% EtOH W1 10 15 20 X1 Синтез проміжної сполуки X1 Суміш W1 (0,60 г, 1,01 ммоля), бензилтриетиламонію хлориду (11,5 мг, 0,05 ммоля) в NaOH 30 % (15 мл) і EtOH (15 мл) перемішували при 60 °C протягом 4 годин. Суміш концентрували наполовину. Регулювали pH до 5 за допомогою 3 н. HCl. Суміш екстрагували за допомогою EtOAc (двічі). Об'єднані органічні шари сушили над MgSO4, фільтрували і розчинник випарювали з отриманням 500 мг (вихід 98 %) проміжної сполуки X1. Синтез проміжної сполуки Y1 При 0 °C 4 н. HCl в діоксані (1,25 мл, 4,99 ммоля) по краплях додавали до суміші X1 (0,50 г, 1 ммоль)в діоксані (2,5 мл). Суміш перемішували при к.т. протягом 12 годин. Суміш випарювали насухо з отриманням 450 мг (вихід >100 %) проміжної сполуки Y1. Неочищену сполуку використовували без будь-якого додаткового очищення на наступному етапі. EDCI HOBT DIPEA DMF Y1 5 Синтез кінцевої сполуки 5 12 UA 113651 C2 5 10 15 1-(3-Диметиламінопропіл)-3-етилкарбодіїміду гідрохлорид (520 мг, 2,72 ммоля) і 1гідроксибензотриазол (367 мг, 2,72 ммоля) повільно додавали до суміші Y1 (370 мг, 0,91 ммоля), діїзопропілетиламіну (0,78 мл, 4,53 ммоля) в DMF (270 мл). Суміш перемішували при к.т. протягом 24 годин. Розчинник випарювали насухо. Залишок поміщали в CH2Cl2-CH3OH (9010) і промивали водою. Осад з'являвся в лійці для декантації. Осад відфільтровували з отриманням 103 мг. Цей осад поміщали в гарячий EtOH, перемішували зі зворотним холодильником протягом 1 години, охолоджували до к.т. і відфільтровували. Неочищений продукт очищали із застосуванням хроматографії з оберненою фазою (X-Bridge-C18 5 мкм 30*150 мм), рухома фаза (градієнт від 75 % NH4HCO3 0,5 % буфер, pH 10, 25 % CH3OH до 0 % NH4HCO3 0,5 % буфер, pH 10, 100 % CH3OH). Чисті фракції збирали і концентрували при пониженому тиску з отриманням 8 мг (чистого продукту) і 50 мг (неочищеного продукту). Неочищений продукт очищали із застосуванням хроматографії з оберненою фазою (X-BridgeC18 5 мкм 30*150 мм), рухома фаза (градієнт від 90 % трифтороцтової кислоти 0,05 %, 10 % CH3OH до 0 % трифтороцтової кислоти 0,05 %, 100 % CH3OH). Чисті фракції збирали і концентрували при пониженому тиску з отриманням 20 мг. 2 фракції (8 мг і 20 мг) об'єднували, поміщали в діоксан і CH3CN і додавали 0,50 мл 4 н. HCl в діоксані. Суміш перемішували при к.т. протягом 2 годин. Осад відфільтровували і сушили з отриманням 28 мг (вихід 7 %) сполуки 5 (сіль HCl). Загальна схема отримання кінцевих продуктів: спосіб 6 20 OH O + HN O O Z1 N HO A2 B2 Синтез проміжної сполуки B2 Розчин етилгліколяту (10,0 г, 96,06 ммоля) в диметиламіні (40 % розчин у воді) (100 мл) 13 UA 113651 C2 перемішували при к.т. протягом 16 годин і концентрували в вакуумі. Залишок поміщали в EtOH і знову концентрували. Цикл виконували 3 рази з отриманням 9,75 г (вихід 98 %) проміжної сполуки B2. Cl + N HO N NaH DMF N O N N O O N C2 B2 5 10 D2 Синтез проміжної сполуки D2 При 0 °C в потоці N2 додавали NaH (2,16 г, 54,13 ммоля) до розчину B2 (4,09 г, 39,69 ммоля) в DMF (36 мл) при к.т. Суміш перемішували при к.т. протягом 30 хвилин і додавали 6хлорніконітрил C2 (5,0 г, 36,09 ммоля) (з виділенням тепла) і суміш перемішували при к.т. протягом 16 годин. Додавали 10 % водний розчин NaHCO3 (150 мл), потім додавали сольовий розчин. Водний шар екстрагували за допомогою EtOAc (двічі). Органічний шар сушили над MgSO4, фільтрували і розчинник випарювали з отриманням 7,0 г (вихід 95 %) проміжної сполуки D2. N O N H2 Pd/C 10% MeOH H2 N N O N N O O D2 15 20 E2 Синтез проміжної сполуки E2 Додавали Pd/C 10 % (2,0 г) до розчину D2 (7,0 г, 34,11 ммоля) в MeOH (140 мл). Реакційну суміш перемішували протягом 16 годин при к.т. в атмосфері H2 (1 атм.). Додавали Pd/C 10 % (1,5 г, 0,04 ммоля) і реакційну суміш перемішували за тих самих умовах протягом 4 годин. ® ® Каталізатор фільтрували через подушку з целіту . Целіт промивали за допомогою CH3OH і фільтрат концентрували в вакуумі. Перед очищенням цю фракцію об'єднували з іншою партією. Залишок очищали флеш-хроматографією на силікагелі (15-40 мкм, 120 г, CH2Cl2/CH3OH/NH4OH: 92/8/0,5). Чисті фракції збирали і концентрували при пониженому тиску з отриманням 2,2 г (вихід 27 %) проміжної сполуки Е2. C1 EtOH E2 25 30 G2 F2 Синтез проміжної сполуки G2 E2 (2,2 г, 10,51 ммоля) в EtOH (10 мл) по краплях додавали до розчину F2 (1,64 г, 10,01 ммоля) і аніліну гідрохлориду (20 мг, 0,16 ммоля) в EtOH (10 мл) при 10 °C. Реакційну суміш перемішували при к.т. протягом 20 годин. Водний розчин 1 M NaOH (25 мл) по краплях додавали до розчину при 10 °C і отриману в результаті суміш перемішували при к.т. протягом 1 години. Осад відфільтровували, промивали мінімумом холодного EtOH і сушили в вакуумі з отриманням 2,25 г (вихід 75 %) проміжної сполуки G2. G2 використовували безпосередньо на наступному етапі без додаткового очищення. N N N N Br NBS N N H 2N O N THF H 2N O G2 35 N N N O H2 O Синтез проміжної сполуки H2 Розчин N-бромсукциніміду (1,47 г, 8,24 ммоля) в THF (50 мл) по краплях додавали упродовж 25 хвилин до розчину G2 (2,25 г, 7,49 ммоля) в THF (80 мл) при 0 °C. Суміш перемішували при 0 °C протягом 30 хвилин, а потім при к.т. протягом 45 хвилин. Суміш поміщали в CH2Cl2, промивали насиченим водним розчином NaHCO3, потім сольовим розчином, сушили над MgSO4, фільтрували і концентрували. Неочищену сполуку кристалізували з CH3CN, осад 14 UA 113651 C2 відфільтровували і сушили з отриманням 0,79 г (вихід 27 %) проміжної сполуки H2. Фільтрат випарювали і залишок очищали флеш-хроматографією на силікагелі (15-40 мкм, 50 г, CH2Cl2/CH3OH/NH4OH: 97/3/0,1). Чисті фракції збирали і концентрували з отриманням 0,71 г (25 % вихід) проміжної сполуки Н2. H2N O N N N Br H2N NH2 Br N N N H2N O N HO N O N 5 10 O H2 N N O I2 Синтез проміжної сполуки I2 Суміш Н2 (1,4 г, 3,69 ммоля) в сечовині (13,3 г, 221,51 ммоля) нагрівали при 160 °C протягом 6 годин. Суміш охолоджували до к.т. і додавали воду. Осад розтирали і відфільтровували, промивали водою і сушили в вакуумі при 60 °C з отриманням 1,05 г (вихід 67 %) проміжної сполуки I2. K2CO3 DMF J2 I2 15 K2 Синтез проміжної сполуки K2 Суміш I2 (1,23 г, 2,91 ммоля), трет-бутил-N-(3-бромпропіл)карбамату J2 (1,04 г, 4,37 ммоля), K2CO3 (604 мг, 4,37 ммоля) в DMF (20 мл) перемішували при 50 °C протягом 12 годин. Розчинник випарювали. Залишок поміщали в EtOAc. Органічний шар промивали водою, сушили над MgSO4, фільтрували і розчинник випарювали. Залишок очищали флеш-хроматографією на силікагелі (15-40 мкм, 80 г, CH2Cl2/CH3OH/NH4OH: 95/5/0,5). Чисті фракції збирали і концентрували з використанням 0,97 г (вихід 57 %) проміжної сполуки К2. Na MeOH K2 20 25 30 L2 Синтез проміжної сполуки L2 При к.т. натрій (446 мг, 19,42 ммоля) додавали до MeOH (30 мл). Суміш перемішували до розчинення натрію (з виділенням тепла). Додавали K2 (750 мг, 1,29 ммоля) і суміш перемішували при 50 °C протягом 16 годин в потоці N2. Додавали воду і pH регулювали до 5-6. Водний шар екстрагували за допомогою EtOAc. Органічний шар сушили над MgSO4, фільтрували і розчинник випарювали з отриманням 0,54 г (вихід 83 %) проміжної сполуки L2. Синтез проміжної сполуки M2 При 0 °C 4 М HCl в діоксані (2,68 мл, 10,73 ммоля) по краплях додавали до суміші L2 (0,54 г, 1,07 ммоля) в діоксані (20 мл). Суміш перемішували при к.т. протягом 12 годин. Суміш випарювали насухо з отриманням 0,74 г (вихід >100 %). Неочищену сполуку використовували без будь-якого додаткового очищення на наступному етапі. 15 UA 113651 C2 H2N H2N N OH N N O N EDCI HOBT N O N N DIPEA DMF OH OH N O N H2 N N O M2 HN 6 O O 5 10 Синтез кінцевої сполуки 6 1-(3-Диметиламінопропіл)-3-етилкарбодіїміду гідрохлорид (675 мг, 3,52 ммоля) і гідроксибензотриазол (476 мг, 3,52 ммоля) повільно додавали до суміші M2 (500 мг, 1,17 ммоля), діїзопропілетиламіну (1,01 мл, 5,87 ммоля) в DMF (360 мл). Суміш перемішували при к.т. протягом 24 годин. Розчинник випарювали насухо. Залишок поміщали в воду. Осад відфільтровували, промивали водою і сушили. Залишок очищали хроматографією із оберненою фазою (X-Bridge-C18 5 мкм 30*150 мм), рухома фаза (градієнт від 90 % трифтороцтової кислоти 0,05 %, 10 % MeOH до 0 % трифтороцтової кислоти 0,05 %, 100 % MeOH). Чисті фракції збирали і концентрували з отриманням 75 мг сполуки 6 і 100 мг (неочищеного осаду). Чисту фракцію (75 мг) поміщали в діоксан і CH3CN і додавали 1 мл 4 н. HCl в діоксані. Суміш перемішували при к.т. протягом 2 годин. Осад відфільтровували і сушили з отриманням 57 мг (вихід 12 %) сполуки 6 (сіль HCl). Загальна схема отримання кінцевих продуктів: спосіб 7 15 Br O N O K2CO3 O N TBABr CH3CN N2 O O2 Синтез проміжної сполуки O2 Додавали 4-бром-1-бутен (8,6 мл, 84,17 ммоля) до суміші N2 (15 г, 56,11 ммоля), K2CO3 16 UA 113651 C2 5 10 15 (23,3 г, 168,33 ммоля), тетрабутиламонію броміду (1,81 г, 5,61 ммоля) в CH3CN (75 мл). Отриману в результаті суміш перемішували зі зворотним холодильником протягом 18 годин. Суміш охолоджували до к.т. Розчинник випарювали. Додавали воду та EtOAc. Шари декантували. Органічний шар сушили над MgSO4, фільтрували і розчинник випарювали. Залишок очищали флеш-хроматографією на силікагелі (15-40 мкм, 120 г, гептан/EtOAc: 93-7). Чисті фракції збирали і концентрували з отриманням 14,8 г (вихід 82 %) проміжної сполуки O2. Синтез проміжної сполуки P2 При 0 °C по краплях додавали 1 н. HCl (120 мл, 119,47 ммоля) до суміші O2 (19,2 г, 59,74 ммоля) в Et2O (250 мл). Суміш перемішували при 0 °C протягом 30 хвилин, потім енергійно перемішували при к.т. протягом 12 годин. Отримані в результаті шари декантували. Водний шар підлуговували до pH 8 за допомогою K2CO3 (порошок), потім екстрагували за допомогою Et2O (3 рази). Водний шар насичували K2CO3, потім знову екстрагували за допомогою CH2Cl2 (2 рази). Органічні шари об'єднували, сушили над MgSO4, фільтрували і розчинник випарювали з отриманням 7,9 г (вихід 84 %) проміжної сполуки P2. O H2N LiAlH4 O H2N THF Q2 P2 20 Синтез проміжної сполуки Q2 В потоці N2 LiAlH4 (4,4 г, 114,50 ммоля) суспендували в THF (150 мл) при 10 °C. По краплях додавали P2 (9 г, 57,25 ммоля) в THF (150 мл). Забезпечували нагрівання реакційної суміші до к.т. і перемішували при к.т. протягом 30 хвилин. Реакційну суміш охолоджували до -10 °C і гасили додаванням води (5 мл), 3 н. NaOH (5 мл) і знову води (14 мл). Суспензію фільтрували ® ® через подушку з целіту . Целіт промивали за допомогою THF і фільтрат концентрували в вакуумі. Залишок поміщали в EtOAc, сушили над MgSO4, фільтрували і розчинник випарювали з отриманням 5,3 г (вихід 80 %) проміжної сполуки Q2. Q2 25 30 35 OH Et3N DMAP CH2Cl2 R Синтез проміжної сполуки R2 При 0 °C трет-бутилдиметилсилілу хлорид (1,31 г, 8,68 ммоля) додавали до суміші Q2 (1,0 г, 8,68 ммоля), Et3N (1,33 мл, 9,55 ммоля), 4-диметиламінопіридину (106 мг, 0,87 ммоля) в CH2Cl2 (30 мл). Суміш перемішували при к.т. протягом 24 годин. Додавали воду і шари декантували. Органічний шар сушили над MgSO4, фільтрували і розчинник випарювали з отриманням 1,70 г (вихід 85 %) проміжної сполуки R2. Синтез проміжної сполуки S2 Розчин 4,6-дигідрокси-2-метилтіопіримідину (50 г, 316,09 ммоля) в трифтор-оцтовій кислоті (210 мл) перемішували при к.т. протягом 30 хвилин. Суміш охолоджували до 5 °C, потім по краплях додавали димлячу HNO3 (19,5 мл, 426,73 ммоля) при 5 °C. Упродовж додавання температуру підтримували на рівні 10-15 °C. Видаляли льодяну баню і, коли температура 17 UA 113651 C2 5 досягала 20 °C, відбувалося бурхливе виділення тепла (від 20 °C до 45 °C за 5 секунд). Суміш перемішували при к.т. протягом 16 годин. Суміш виливали в суміш води і льоду. Осад відфільтровували і промивали водою. Осад сушили в вакуумі при 50 °C з отриманням 42 г (вихід 65 %) проміжної сполуки S2. Цю проміжну сполуку використовували безпосередньо на наступному етапі без будь-якого додаткового очищення. POCl3 T2 S2 10 15 20 Синтез проміжної сполуки T2 N, N-диметиланілін (76,7 мл, 0,61 моля) по краплях додавали до POCl3 (93,7 мл, 1,01 моля) при 0 °C. S2 (41 г, 201,79 ммоля) додавали порціями при 0 °C, а потім суміш нагрівали до 100 °C протягом 2 годин. Розчин концентрували в вакуумі і видаляли залишковий POCl3 азеотропним випарюванням з толуолом (3 рази). Отримане в результаті масло поміщали в розчин CH2Cl2гептану (70-30) і фільтрували через скляний фільтр з SiO2. Фільтрат концентрували і залишок очищали препаративною LC (SiOH з зернами неправильної форми 20-45 мкм, 1000 г DAVISIL), рухома фаза (80 % гептан, 20 % CH2Cl2). Чисті фракції збирали і концентрували з отриманням 37,8 г (вихід 78 %) проміжної сполуки Т2. Синтез проміжної сполуки U2 Розчин 2M NH3 в iPrOH (115 мл, 229,31 ммоля) по краплях додавали до розчину Т2 (36,7 г, 152,87 ммоля) і Et3N (23,4 мл, 168,16 ммоля) в THF (360 мл) (упродовж додавання температуру підтримували на рівні к.т. за допомогою бані з льодяною водою). Реакційну суміш перемішували при к.т. протягом 5 годин. Суміш випарювали насухо. Воду і EtOAc додавали до залишку. Шари розділяли і водний шар екстрагували за допомогою EtOAc (двічі). Об'єднані органічні шари сушили над MgSO4, фільтрували і видаляли розчинник при пониженому тиску з отриманням 34,5 г (вихід 100 %) проміжної сполуки U2. Et3N THF 25 30 U2 V2 Синтез проміжної сполуки V2 Етилхлорформіат (13,5 мл, 138,90 ммоля) додавали до розчину U2 (39,8 г, 126,27 ммоля) і Et3N (26,5 мл, 189,40 ммоля) в THF (1300 мл). Суміш перемішували при к.т. протягом 6 годин і розчинник частково випарювали при пониженому тиску. Залишок поглинали CH2Cl2 і водою. Шари розділяли, водний шар екстрагували за допомогою CH2Cl2 (двічі). Об'єднані органічні шари висушували над MgSO4, фільтрували і видаляли розчинник при пониженому тиску. Залишок очищали за допомогою LC (SiOH з зернами неправильної форми 20-45 мкм, 1000 г DAVISIL), рухома фаза (градієнт від 85 % гептану, 15 % AcOEt до 80 % гептану, 20 % AcOEt). Чисті фракції збирали і концентрували з отриманням 35 г (вихід 95 %) проміжної сполуки V2. 18 UA 113651 C2 5 Синтез проміжної сполуки X2 Суміш V2 (5,0 г, 17,08 ммоля), W2 (2,85 г, 17,08 ммоля), K2CO3 (3,54 г, 25,6 ммоля) і NaI (2,56 г, 17,08 ммоля) в ацетоні (200 мл) перемішували при к.т. протягом 48 годин. Суміш відфільтровували і фільтрат випарювали насухо. Залишок очищали флеш-хроматографією на силікагелі (15-40 мкм, 220 г, CH2Cl2/гептан 50-50). Чисті фракції збирали і концентрували з отриманням 7,4 г (вихід 100 %) проміжної сполуки Х2. Cl NH2 NO 2 N N S N O S 15 N O N O X2 10 NO 2 N NH3 THF O Y2 Синтез проміжної сполуки Y2 Розчин X2 (7,20 г, 17,03 ммоля) і NH3 30 % в воді (100 мл) в THF (100 мл) перемішували при к.т. протягом 2 годин. Розчинник видаляли при пониженому тиску. Залишок суспендували в воді і екстрагували за допомогою CH2Cl2. Органічний шар промивали водою і сушили над MgSO4, фільтрували і розчинник випарювали в вакуумі з отриманням 7,1 г (вихід 100 %) проміжної сполуки Y2 (жовте масло). Цю проміжну сполуку використовували безпосередньо на наступному етапі. m CPBA CH2Cl2 Z2 Y2 20 Синтез проміжної сполуки Z2 3-Хлорпероксибензойну кислоту (2,44 г, 9,91 ммоля) в CH 2Cl2 (20 мл) по краплях додавали до розчину Y2 (2,0 г, 4,96 ммоля) в CH2Cl2 (100 мл) при к.т. Суміш перемішували при к.т. протягом 20 годин. До суміші додавали водний розчин Na2S2O3 (5 екв.). Шари розділяли і водний шар екстрагували за допомогою CH2Cl2 (двічі). Об'єднані органічні шари промивали насиченим водним розчином NaHCO3, сушили над MgSO4, фільтрували і розчинник видаляли при пониженому тиску з отриманням 2,70 г (вихід >100 %) проміжної сполуки Z2. Проміжну сполуку використовували безпосередньо на наступному етапі без додаткового очищення. Et3N CH3CN Z2 R2 A3 25 Синтез проміжної сполуки A3 Суміш Z2 (2,16 г, 4,96 ммоля), R2 (1,70 г, 7,44 ммоля) і Et3N (1,04 мл, 7,44 ммоля) в CH3CN (70 мл) перемішували при к.т. протягом 2 годин. Додавали воду і суміш екстрагували за 19 UA 113651 C2 допомогою EtOAc (двічі). Органічний шар промивали сольовим розчином, сушили над MgSO4, фільтрували і розчинник випарювали. Залишок очищали флеш-хроматографією на силікагелі (15-40 мкм, 90 г, CH2Cl2/CH3OH 99,5-0,5). Чисті фракції збирали і концентрували з отриманням 1,10 г (вихід 38 %) проміжної сполуки A3. 5 10 15 20 25 30 Синтез проміжної сполуки B3 A3 (1,05 г, 1,80 ммоля) додавали до надсухого CH2Cl2 (230 мл) і отриману в результаті суміш ого дегазували барботуванням N2 через розчин протягом 30 хвилин. Каталізатор Граббса 2 покоління (153 мг, 0,18 ммоля) додавали однією порцією і суміш перемішували при к.т. в потоці N2 протягом 24 годин. Суміш концентрували. Залишок очищали препаративною LC (SiOH з зернами неправильної форми 15-40 мкм, 300 г, MERCK), рухома фаза (80 % гептан, 20 % AcOEt). Чисті фракції збирали і концентрували з отриманням 0,70 г (вихід 70 %) проміжної сполуки B3. Синтез кінцевої сполуки 7 Fe (385 мг, 6,90 ммоля) додавали до суміші B3 (640 мг, 1,15 ммоля) в AcOH (6,8 мл) і воді (1,36 мл). Суміш нагрівали при 100 °C із застосуванням однорежимного мікрохвильового пристрою (Biotage Initiator) з виходом потужності, що варіює від 0 до 400 Вт, протягом 40 ® хвилин. Суміш фільтрували через подушку з целіту і промивали за допомогою AcOH. Фільтрат концентрували в вакуумі і насухо випарювали разом з толуолом (двічі). Залишок поміщали в CH2Cl2/MeOH/NH4OH 90-10-0,5. Осад фільтрували (осад (1,0 г) містив очікувану сполуку) і фільтрат випарювали для очищення хроматографією. Залишок (фільтрату) очищали флеш-хроматографією на силікагелі (15-40 мкм, 80 г, CH2Cl2/CH3OH/NH4OH: 90-10-0,5). Чисті фракції збирали і випарювали з отриманням 52 мг фракції 1. Попередньо отриманий осад очищали хроматографією (сполуку і SiO2 змішували перед елююванням). Залишок очищали флеш-хроматографією на силікагелі (15-40 мкм, 25 г, CH2Cl2/CH3OH/NH4OH: 90-10-0,5). Чисті фракції збирали і концентрували з отриманням 80 мг фракції 2. Фракцію 1 та фракцію 2 об'єднували, а потім здійснювали тверднення з CH3CN з отриманням 95 мг (вихід 23 %) сполуки 7 (E-ізомер з 3,5 % Z-ізомеру). Загальна схема отримання кінцевих продуктів: спосіб 8 20 UA 113651 C2 5 10 Синтез проміжної сполуки C3 V2 (1,7 г, 5,8 ммоля), 3-метоксибензилхлорид (0,93 мл, 6,4 ммоля), K2CO3 (2 г, 14,5 ммоля) і йодид натрію (0,87 г, 5,8 ммоля) в ацетоні (60 мл) перемішували при к.т. протягом 16 годин. Розчин відфільтровували і фільтрат випарювали при пониженому тиску. Неочищений продукт очищали препаративною LC (SiOH з зернами неправильної форми 15-40 мкм, 80 г, Merck, рухома фаза гептан/CH2Cl2 70/30). Чисті фракції збирали і концентрували з отриманням 1,4 г (вихід 58 %) проміжної сполуки C3. Синтез проміжної сполуки D3 C3 (1,4 г, 3,4 ммоля) перемішували в NH 3 30 % в воді (30 мл) і THF (30 мл) при к.т. протягом 16 годин. Суміш концентрували і залишок сушили азеотропним випарюванням з EtOH (двічі) з отриманням 1,3 г (вихід 97 %). Неочищений продукт використовували без додаткового очищення на наступному етапі. m CPBA CH2Cl2 D3 15 E3 Синтез проміжної сполуки E3 3-Хлорпероксибензойну кислоту (2,04 г, 8,3 ммоля) додавали до розчину D3 (1,3 г, 3,3 ммоля) в CH2Cl2 (80 мл) при к.т. Суміш перемішували при к.т. протягом 20 годин. До суміші додавали водний розчин Na2S2O3 (2,61 г, 16,52 ммоля). Шари розділяли і водний шар 21 UA 113651 C2 екстрагували за допомогою CH2Cl2 (двічі). Об'єднані органічні шари промивали насиченим водним розчином NaHCO3, сушили над MgSO4, фільтрували і розчинник випарювали з отриманням 1,4 г (вихід 100 %) проміжної сполуки E3. K2CO3 CH3CN E3 5 10 15 20 F3 Синтез проміжної сполуки F3 Суміш E3 (1,4 г, 3,3 ммоля), 4-аміно-1-бутанолу (0,45 мл, 3 ммоля) і K2CO3 (414 мг, 4,9 ммоля) в CH3CN (65 мл) перемішували при 80 °C протягом 1 години 30 хвилин. Солі фільтрували і до фільтрату додавали воду. Суміш екстрагували за допомогою CH2Cl2 (двічі). Органічний шар сушили над MgSO4, фільтрували і розчинник випарювали. Неочищений продукт очищали препаративною LC (SiOH з зернами неправильної форми 15-40 мкм, 80 г, Merck, рухома фаза CH2Cl2/MeOH/NH4OH 98/2/0,1). Чисті фракції збирали і концентрували з отриманням 1,2 г (вихід 84 %) проміжної сполуки F3. Синтез проміжної сполуки G3 Fe (1,54 г, 27,6 ммоля) додавали до суміші F3 (1,2 г, 2,76 ммоля) в AcOH (24 мл) і воді (8,6 мл). Суміш енергійно перемішували при к.т. протягом 24 годин. Реакційну суміш концентрували ® в вакуумі і залишок розводили EtOAc і водою. Суміш фільтрували через подушку із целіту і промивали за допомогою EtOAc. Шари розділяли і органічний шар промивали насиченим водним розчином NaHCO3 (двічі), потім сольовим розчином, сушили над MgSO4, фільтрували і концентрували в вакуумі. Залишок очищали хроматографією на колонці з силікагелем (15-40 мкм, 40 г) в CH2Cl2/MeOH/NH4OH (90/10/0,5). Чисті фракції збирали і концентрували. Здійснювали твердіння цієї фракцію з CH3CN/діїзопропілового етеру з отриманням 0,70 г (вихід 71 %) проміжної сполуки G3. BBr 3 CH2Cl2 G3 25 30 H3 Синтез проміжної сполуки H3 При -60 °C в потоці N2 BBr3 (5,6 мл, 5,6 ммоля) по краплях додавали до суміші G3 (400 мг, 1,1 ммоля) в CH2Cl2 (40 мл). Суміш перемішували при -60 °C протягом 1 години, а потім при к.т. протягом 12 годин в потоці N2. По краплях додавали 1 мл CH3OH при 0 °C. Потім суміш виливали в насичений розчин K2CO3 в воді. Суміш екстрагували розчином CH2Cl2/MeOH. Органічний шар сушили над MgSO4, фільтрували і розчинник випарювали. Неочищену сполуку очищали хроматографією на колонці з силікагелем (15-40 мкм, 40 г) в CH2Cl2/MeOH/NH4OH (90/10/0,5). Чисті фракції збирали і концентрували. Здійснювали твердіння залишку з CH3CN/діїзопропілового етеру з отриманням 265 мг (вихід 69 %) проміжної сполуки H3. 22 UA 113651 C2 PPh3 DIAD THF 8 H3 5 10 Синтез кінцевої сполуки 8 При к.т. в потоці N2 повільно по краплях додавали розчин діїзопропілазодикарбоксилату (0,27 мл, 1,36 ммоля) в THF (5 мл) до суміші H3 (235 мг, 0,68 ммоля), PPh3 (358 мг, 1,36 ммоля) в THF (50 мл). Суміш перемішували при к.т. протягом 6 годин. Реакційну суміш виливали в воду з льодом і додавали EtOAc. Суміш підлуговували водним розчином NaHCO3 10 % в воді, потім органічний шар розділяли, сушили над MgSO4, фільтрували і розчинники випарювали насухо. Неочищену сполуку очищали хроматографією на колонці з силікагелем (15-40 мкм, 40 г) в CH2Cl2/MeOH/NH4OH (95/5/0,1). Чисті фракції збирали і випарювали. Здійснювали твердіння залишку з CH3CN/діїзопропілового ефіру з отриманням 75 мг (вихід 34 %) сполуки 8. Загальна схема отримання кінцевих продуктів: спосіб 9 NaH DMF J3 B2 I3 15 Синтез проміжної сполуки J3 При 0 °C в потоці N2 додавали NaH (3,28 г, 82 ммоля) до розчину B2 (7,32 г, 71 ммоль) в DMF (80 мл). Суміш перемішували при к.т. протягом 30 хвилин і додавали I3 (10 г, 54,6 ммоля) (з виділенням тепла) і суміш перемішували при к.т. протягом 4 годин. Додавали 10 % водний 23 UA 113651 C2 розчин NaHCO3 (150 мл), а потім сольовий розчин (150 мл). Отриману в результаті суміш екстрагували за допомогою EtOAc (двічі). Органічний шар сушили над MgSO4, фільтрували і розчинник випарювали. Залишок поміщали в мінімум AcOEt, осад відфільтровували і сушили з отриманням проміжної сполуки J3 (9,04 г, вихід 81 %). BH3/THF K3 J3 5 10 Синтез проміжної сполуки K3 При 0 °C в потоці N2 по краплях додавали BH3/THF (110 мл, 39 ммолей) до розчину J3 (9,0 г, 43,9 ммоля) в THF (60 мл). Суміш перемішували при к.т. протягом 2 годин, потім гасили за допомогою 2M HCl і перемішували при к.т. протягом 12 годин. Реакційну суміш випарювали насухо. Залишок поміщали в CH2Cl2-CH3OH-NH4OH 90-10-1. Осад відфільтровували (мінеральні речовини), а фільтрат концентрували. Виконували очищення флеш-хроматографією на силікагелі (15-40 мкм, 330 г, CH2Cl2/CH3OH/NH4OH: 96/4/0,5-90/10/0,5). Чисті фракції збирали і випарювали насухо з отриманням проміжної сполуки K3 (6,2 г, вихід 73 %). C1 EtOH K3 L3 F2 15 20 Синтез проміжної сполуки L3 К3 (6,2 г, 29,5 ммоля) в EtOH (30 мл) по краплях додавали до розчину F2 (4,6 г, 28 ммоль) і аніліну гідрохлориду (56 мг, 0,43 ммоля) в EtOH (25 мл) при 10 °C. Реакційну суміш перемішували при к.т. протягом 20 годин. Водний розчин 1M NaOH (25 мл) по краплях додавали до розчину при 10 °C і отриману в результаті суміш перемішували при к.т. протягом 1 години. Осад відфільтровували, промивали мінімумом холодного EtOH і сушили в вакуумі. Маточні шари концентрували, отримували другий осад в CH2Cl2, фільтрували та сушили в вакуумі. Дві партії об'єднували з отриманням проміжної сполуки L3 (2,44 г, вихід 29 %). NBS THF L3 25 30 M3 Синтез проміжної сполуки M3 Розчин NBS (0,326 г, 1,83 ммоля) в THF (15 мл) по краплях додавали упродовж 25 хвилин до розчину L3 (0,5 г, 1,67 ммоля) в THF (15 мл) при 0 °C. Суміш перемішували при 0 °C протягом 30 хвилин, а потім при к.т. протягом 45 хвилин. Суміш поміщали в CH2Cl2, промивали насиченим водним розчином NaHCO3, сушили над MgSO4, фільтрували і випарювали в вакуумі. Здійснювали твердіння цієї неочищеної сполуки з CH3CN. Осад відфільтровували і сушили з отриманням проміжної сполуки M3 (216 мг, вихід 34 %). M3 N3 Синтез проміжної сполуки N3 Суміш М3 (1,04 г, 2,74 ммоля) в сечовині (4,9 г, 82,28 ммоля) нагрівали при 160 °C протягом 24 UA 113651 C2 4 годин. Знову додавали сечовину (3 г, 2,64 ммоля) і суміш перемішували при 160 °C протягом 12 годин. Суміш охолоджували до к.т. і додавали воду. Осад розтирали і відфільтровували, промивали водою і сушили в вакуумі при 60 °C з отриманням проміжної сполуки N3. Неочищену сполуку використовували безпосередньо на наступному етапі. K2CO3 DMF J2 O3 N3 5 10 Синтез проміжної сполуки O3 Суміш N3 (неочищеного), J2 (1,557 г, 6,54 ммоля), K2CO3 (904 мг, 6,54 ммоля) в DMF (30 мл) перемішували при 50 °C протягом 12 годин. Розчинник випарювали. Залишок поміщали в EtOAc. Органічний шар промивали водою, сушили над MgSO4, фільтрували і розчинник випарювали. Неочищену сполуку очищали препаративною LC (SiOH з зернами неправильної форми 15-40 мкм 300 г, Merck), рухома фаза: 0,3 % NH4OH, 97 % CH2Cl2, 3 % MeOH з отриманням проміжної сполуки O3 (280 мг, вихід 11 %). Na MeOH O3 P3 15 20 25 Синтез проміжної сполуки P3 При к.т. Na (167 мг, 7,25 ммоля) додавали до MeOH (11 мл). Суміш перемішували до розчинення Na (з виділенням тепла). Додавали O3 (280 мг, 0,48 ммоля) і суміш перемішували при 50 °C протягом 16 годин в потоці N2. Додавали воду і pH регулювали (за допомогою 1 н. HCl) до 5-6. Водний шар екстрагували за допомогою EtOAc. Водну фазу насичували порошком K2CO3 та екстрагували за допомогою AcOEt. Об'єднані органічні фази сушили над MgSO4, фільтрували і розчинник випарювали з отриманням проміжної сполуки P3 (140 мг, вихід 58 %). Синтез проміжної сполуки Q3 При 0 °C HCl (4М в діоксані) (0,7 мл, 2,78 ммоля) по краплях додавали до суміші Р3 (140 мг, 2,78 ммоля) в діоксані (5 мл). Суміш перемішували при к.т. протягом 12 годин. Розчинник випарювали насухо з отриманням проміжної сполуки Q3. Неочищену сполуку використовували на наступному етапі без будь-якого додаткового очищення. EDCI HOBT DIPEA DMF Q3 30 9 Синтез кінцевої сполуки 9 1-(3-Диметиламінопропіл)-3-етилкарбодіїміду гідрохлорид (318 мг, 1,66 ммоля) і гідроксибензотриазол (224 мг, 1,66 ммоля) повільно додавали до суміші Q3 (неочищена), діїзопропілетиламіну (0,476 мл, 2,76 ммоля) в DMF (170 мл). Суміш перемішували при к.т. 25 UA 113651 C2 5 протягом 24 годин. Розчинник випарювали насухо. Залишок поміщали в воду. Осад відфільтровували, промивали водою і сушили. Неочищену сполуку очищали за допомогою хроматографії із оберненою фазою (X-Bridge-C18 5 мкм 30*150 мм), рухома фаза (градієнт від 90 % NH4HCO3 0,5 %, 10 % CH3CN до 0 % NH4HCO3 0,5 %, 100 % CH3CN) з отриманням кінцевої сполуки 9 (37 мг, вихід 18 %). Загальна схема отримання кінцевих продуктів: спосіб 10 H2, Pd/C (10%) CH3OH/THF 7 10 15 20 10 Синтез кінцевої сполуки 10 Суміш сполуки 7 (100 мг, 0,27 ммоля), Pd/C (10 %) (14,5 мг, 0,014 ммоля) в CH3OH/THF 50/50 (10 мл) гідрогенізували при атмосферному тиску H2 протягом 4 годин. Каталізатор видаляли фільтрацією через фільтр (Сhromafil Xtra 0,45 мкм). Фільтрат концентрували. Здійснювали твердіння цієї фракції з CH3CN, осад відфільтровували і сушили з отриманням кінцевої сполуки 10 (81 мг, вихід 81 %). Загальна схема отримання кінцевих продуктів: спосіб 11 Синтез проміжної сполуки R3 При 0 °C в потоці N2 додавали NaH (705 мг, 17,6 ммоля) до розчину 1,4-бутандіолу (3,2 г, 35,26 ммоля) в DMF (30 мл). Суміш перемішували протягом 30 хвилин при к.т., потім додавали E3 (2,5 г, 5,87 ммоля). Суміш перемішували при кімнатній температурі протягом 1 години. Додавали лід і суміш екстрагували за допомогою EtOAc. Органічний шар сушили над MgSO4, фільтрували і розчинник випарювали. Виконували очищення флеш-хроматографією на силікагелі (15-40 мкм, 80 г, CH2Cl2/CH3OH/NH4OH: 97/3/0,1). Чисті фракції збирали і випарювали насухо з отриманням проміжної сполуки R3 (1,78 г, вихід 70 %). 26 UA 113651 C2 5 Синтез проміжної сполуки S3 Порошок заліза (2,27 г, 40,65 ммоля) додавали до суміші R3 (1,77 г, 4,07 ммоля) в AcOH (35 мл) і воді (11 мл). Суміш перемішували при 50 °C протягом 8 годин. Реакційну суміш розводили водою і підлуговували за допомогою K2CO3 10 % в воді. Додавали EtOAc і CH3OH, і отриману в ® ® результаті суміш фільтрували через подушку з целіту . Целіт промивали за допомогою CH2Cl2/CH3OH (80/20). Фільтрат декантували. Органічний шар сушили над MgSO4, фільтрували і розчинник випарювали. Фракцію поміщали в CH3CN, осад відфільтровували і сушили з отриманням проміжної сполуки S3 (1,2 г, вихід 82 %). BBr 3 CH2Cl2 S3 T3 10 15 Синтез проміжної сполуки T3 При -60 °C в потоці N2 BBr3 (13,6 мл, 13,6 ммоля) по краплях додавали до суміші S3 (980 мг, 2,727 ммоля) в CH2Cl2 (40 мл). Суміш перемішували при -60 °C протягом 1 години в потоці N2. Суміш перемішували 5 годин при 0 °C. По краплях додавали 5 мл CH3OH при -60 °C. Потім суміш виливали в насичений розчин K2CO3. Суміш екстрагували за допомогою CH2Cl2/CH3OH. Органічний шар сушили над MgSO4, фільтрували і розчинник випарювали з отриманням проміжної сполуки T3 (0,55 г, вихід 49 %), який використовували безпосередньо на наступному етапі без додаткового очищення. K2CO3 DMF 11 T3 20 25 Синтез кінцевої сполуки 11 Суміш T3 (537 мг, 1,32 ммоля), K2CO3 (182 мг, 1,32 ммоля) в DMF (71 мл) перемішували при 80 °C протягом 12 годин. Неочищену суміш відфільтровували і фільтрат концентрували при пониженому тиску. Залишок поміщали в мінімум DMF і додавали 5 г SiO2 35-70 мкм. Отриману в результаті суспензію випарювали насухо і поміщали в верхню частину 50 г хроматографічної колонки і елюювали з градієнтом CH2Cl2-CH3OH-NH4OH від 95-5-0,5 до 90-10-0,5. Фракції, що містять очікувані сполуки, об'єднували і концентрували при пониженому тиску. Тверду речовину кристалізували з CH3CN, осад відфільтровували і сушили з отриманням кінцевої сполуки 11 (33 мг, вихід 8 %). Загальна схема отримання кінцевих продуктів: спосіб 12 27 UA 113651 C2 5 10 15 Синтез проміжної сполуки V3 V2 (1,4 г, 4,8 ммоля), U3 (1,44 г, 4,8 ммоля), K2CO3 (1,65 г, 12 ммоль) і NaI (0,72 г, 4,8 ммоля) в ацетоні (60 мл) перемішували при к.т. протягом 16 годин. Розчин відфільтровували і фільтрат випарювали при пониженому тиску. Неочищений продукт очищали препаративною LC (SiOH з зернами неправильної форми 15-40 мкм, 80 г, Merck, рухома фаза гептан/CH2Cl2 85/15) з отриманням проміжної сполуки V3 (2,3 г, вихід 94 %). Синтез проміжної сполуки W3 V3 (2,3 г, 4,5 ммоля) перемішували в NH3 (30 % в воді) (40 мл) і THF (40 мл) при к.т. протягом 16 годин. Суміш концентрували в вакуумі і залишок сушили азеотропним випарюванням EtOH (двічі). Неочищений продукт очищали препаративною LC (SiOH з зернами неправильної форми 15-40 мкм, 40 г, Merck, рухома фаза гептан/AcOEt 85/15) з отриманням проміжної сполуки W3 (1,25 г, вихід 56 %). m CPBA CH2Cl2 W3 20 X3 Синтез проміжної сполуки X3 Додавали 3-хлорпероксибензойну кислоту (2,04 г, 8,2 ммоля) до розчину W3 (1,3 г, 3,3 ммоля) в CH2Cl2 (70 мл) при к.т. Суміш перемішували при к.т. протягом 20 годин. До суміші додавали водний розчин Na2S2O3 (5 екв.). Два шари розділяли і водний шар екстрагували за допомогою CH2Cl2 (двічі). Об'єднані органічні шари промивали насиченим водним розчином 28
ДивитисяДодаткова інформація
Назва патенту англійськоюMacrocyclic purines for the treatment of viral infections
Автори англійськоюBonfanti, Jean-Francois, Fortin, Jerome Michel Claude, Muller, Philippe, Doublet, Frederic Marc Maurice, Raboisson, Pierre Jean-Marie Bernard, Arnoult, Eric Pierre Alexandre
Автори російськоюБонфанти Жан-Франсуа, Фортен Жэром Мишель Клод, Мюллер Филипп, Дубле Фрэдэрик Марк Морис, Рабуассон Пьер Жан-Мари Бернар, Арну Эрик Пьер Александр
МПК / Мітки
МПК: C07D 498/16, A61K 31/52, C07D 487/16
Мітки: вірусних, макроциклічні, лікування, інфекцій, пурини
Код посилання
<a href="https://ua.patents.su/118-113651-makrociklichni-purini-dlya-likuvannya-virusnikh-infekcijj.html" target="_blank" rel="follow" title="База патентів України">Макроциклічні пурини для лікування вірусних інфекцій</a>
Похідні піперидинопіримідину для лікування вірусних інфекцій

Номер патенту: 112668
Опубліковано: 10.10.2016
Автори: МакГован Девід Крейг, Даубі Хамлічі Мурад, Рабуассон П'єр Жан-Марі Бернар, Йонкерс Тім Хьюго Марія
МПК: C07D 471/04, A61K 31/519
Мітки: інфекцій, піперидинопіримідину, похідні, вірусних, лікування
Формула / Реферат:
1. Сполука формули (І) (I)або її фармацевтично прийнятна сіль, деА вибраний з групи, що складається з СН2 і NCOR2 в будь-якій стереохімічній конфігурації,В вибраний з групи, що складається з СН2 і NCOR4 в будь-якій стереохімічній конфігурації,за умови, що, якщо А являє собою NCOR2, тоді В не являє собою NCOR4,X вибраний з СН2 в...
Дієтична добавка – природна сила 6 – для лікування запальних захворювань лор-органів та інфекцій дихальних шляхів (ларинготрахеїти, бронхіти, пневмонії, астматичний компонент), респіраторних вірусних інфекцій

Номер патенту: 88547
Опубліковано: 25.03.2014
Автор: Матюшенко Роман Анатолійович
МПК: A61K 35/00
Мітки: інфекцій, респіраторних, шляхів, захворювань, природна, астматичний, пневмонії, компонент, лікування, запальних, сила, ларинготрахеїти, дієтична, бронхіті, дихальних, вірусних, лор-органів, добавка
Формула / Реферат:
Дієтична добавка - природна сила 6 - для лікування запальних захворювань ЛОР-органів та інфекцій дихальних шляхів (ларинготрахеїти, бронхіти, пневмонії, астматичний компонент), респіраторних вірусних інфекцій, що містить квітки бузини чорної, траву череди, яка відрізняється тим, що додатково містить листя підбілу (мати й мачуха), листя подорожника великого, листя м'яти перцевої, траву деревію, траву звіробою, квітки нагідок при наступному...
Спосіб лікування гострих респіраторних вірусних інфекцій у дорослих хворих

Номер патенту: 65097
Опубліковано: 15.03.2004
Автори: Фролов Аркадій Федорович, Шаповалова Ірина Олександрівна, Фролов Валерій Мітрофанович, Пустовий Юрій Григорович
МПК: A61K 31/18
Мітки: хворих, гострих, вірусних, інфекцій, спосіб, респіраторних, дорослих, лікування
Формула / Реферат:
Спосіб лікування гострих респіраторних інфекцій у дорослих хворих, що включає введення препаратів, що одночасно мають інтерфероногенні, протизапальні, жарознижувальні та анальгезуючі властивості, який відрізняється тим, що хворим вводять амізон усередину по 0,25 г 3-4 рази на добу після вживання їжі, протягом 5-7 діб поспіль.
Спосіб лікування гострих респіраторних вірусних інфекцій у дорослих

Номер патенту: 52236
Опубліковано: 16.12.2002
Автори: Шаповалова Ірина Олександрівна, Пустовий Юрій Григорович, Фролов Валерій Мітрофанович
МПК: A61P 11/00
Мітки: гострих, респіраторних, лікування, дорослих, спосіб, вірусних, інфекцій
Формула / Реферат:
Спосіб лікування гострих респіраторних вірусних інфекцій у дорослих, що включає введення індукторів ендогенного інтерферону, який відрізняється тим, що як індуктор інтерферону та протизапальний препарат хворим вводять усередину мефенамову кислоту по 0,25-0,5 г 3-4 рази на день після вживання їжі протягом 5-7 днів поспіль.
Способи й сполуки для лікування вірусних інфекцій paramyxoviridae

Номер патенту: 111163
Опубліковано: 11.04.2016
Автори: Перріш Джей П., Рей Едріан С., Макман Річард Л., Теодор Дороті Агнес
МПК: C07D 309/10, C07H 19/00
Мітки: інфекцій, способи, paramyxoviridae, лікування, сполуки, вірусних
Формула / Реферат:
1. Застосування сполуки Формули II в лікуванні захворювання, викликаного інфекцією Раrаmуxoviridae, де сполука Формули II має структуру:, Формула IIабо її фармацевтично прийнятної солі; де:R1 є Н або галогеном;R2 є ORa або галогеном;R3 є Н або ORa;R5 є H;R6 є CN, (С1-С8)алкілом, (С2-С8)алкенілом або...
Попередній патент: Гербіцидна композиція та спосіб її одержання
Наступний патент: Електроліт для пасивації срібла та срібних покриттів
Випадковий патент: Спосіб профілактики та лікування постлапароскопічного больового плечолопаткового синдрому