Карбенові комплекси перехідних металів
Номер патенту: 62301
Опубліковано: 25.08.2011
Автори: Коротких Микола Іванович, Марічев Костянтин Олександрович, Швайка Олексій Павлович, Кисельов Артем Вікторович, Глиняна Наталія Валеріївна, Сабєров Вагіз Шамільович




Формула / Реферат
1. Карбенові комплекси перехідних металів загальної формули
![]()
де L означає карбеновий ліганд, вибраний з ряду (І-III):
1,3-дизаміщений імідазол-2-іліден загальної формули
 , (I)
, (I)
де R, R1 означає алкіл, арил, гетерил,
1,3-дизаміщений бензімідазол-2-іліден загальної формули
 , (II)
, (II)
де R, R1 означає алкіл, арил, гетерил,
1,3,4-тризаміщений 1,2,4-триазол-5-іліден загальної формули
 , (III)
, (III)
де R, R1, R2 означає алкіл, арил, гетерил,
М означає метал, вибраний з ряду: мідь, нікель, паладій,
X означає некарбеновий ліганд, вибраний з ряду: галогенід, феноксид, ацетоксид,
m дорівнює 1, 2, 3, 4,
n дорівнює 1, 2, 3, 4,
р дорівнює 1.
2. Карбенові комплекси за п. 1,
де L разом з Xm означає карбеновий ліганд, який містить карбеновий центр в циклічному фрагменті й атом кисню в боковому ланцюзі ряду 1,3,4-тризаміщеного 1,2,4-триазол-5-ілідену загальної формули
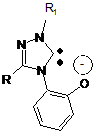 , (IV)
, (IV)
де R, R1 означає алкіл, арил, гетерил.
3. Карбенові комплекси за п. 1,
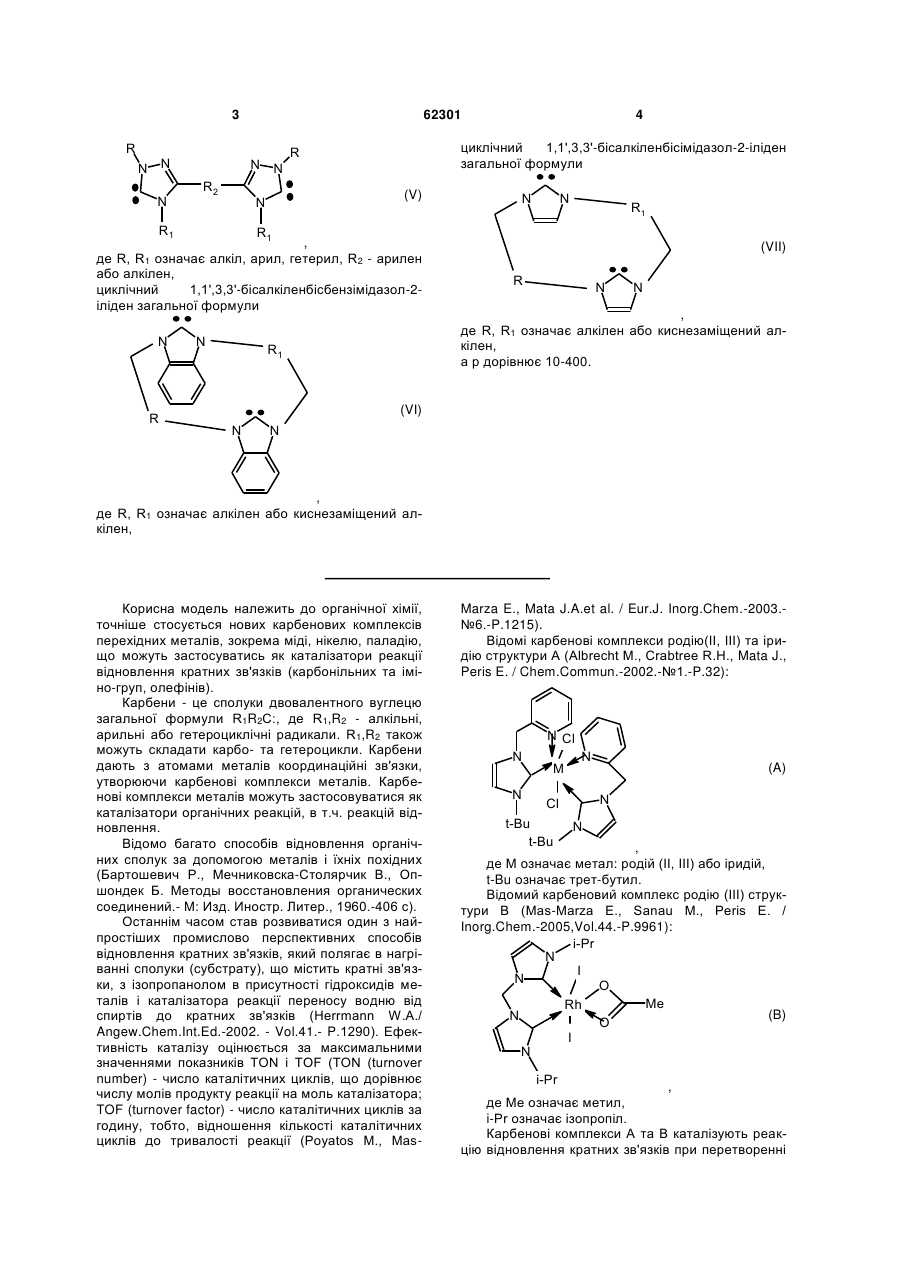
де L означає біскарбеновий ліганд, який містить один або два двовалентні радикали, вибраний з ряду (V-VII):
тетразаміщені 3,3'-місткові біс(1,2,4-триазол-5-іліден)арени або алкани загальної формули
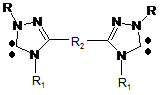 , (V)
, (V)
де R, R1 означає алкіл, арил, гетерил, R2 - арилен або алкілен,
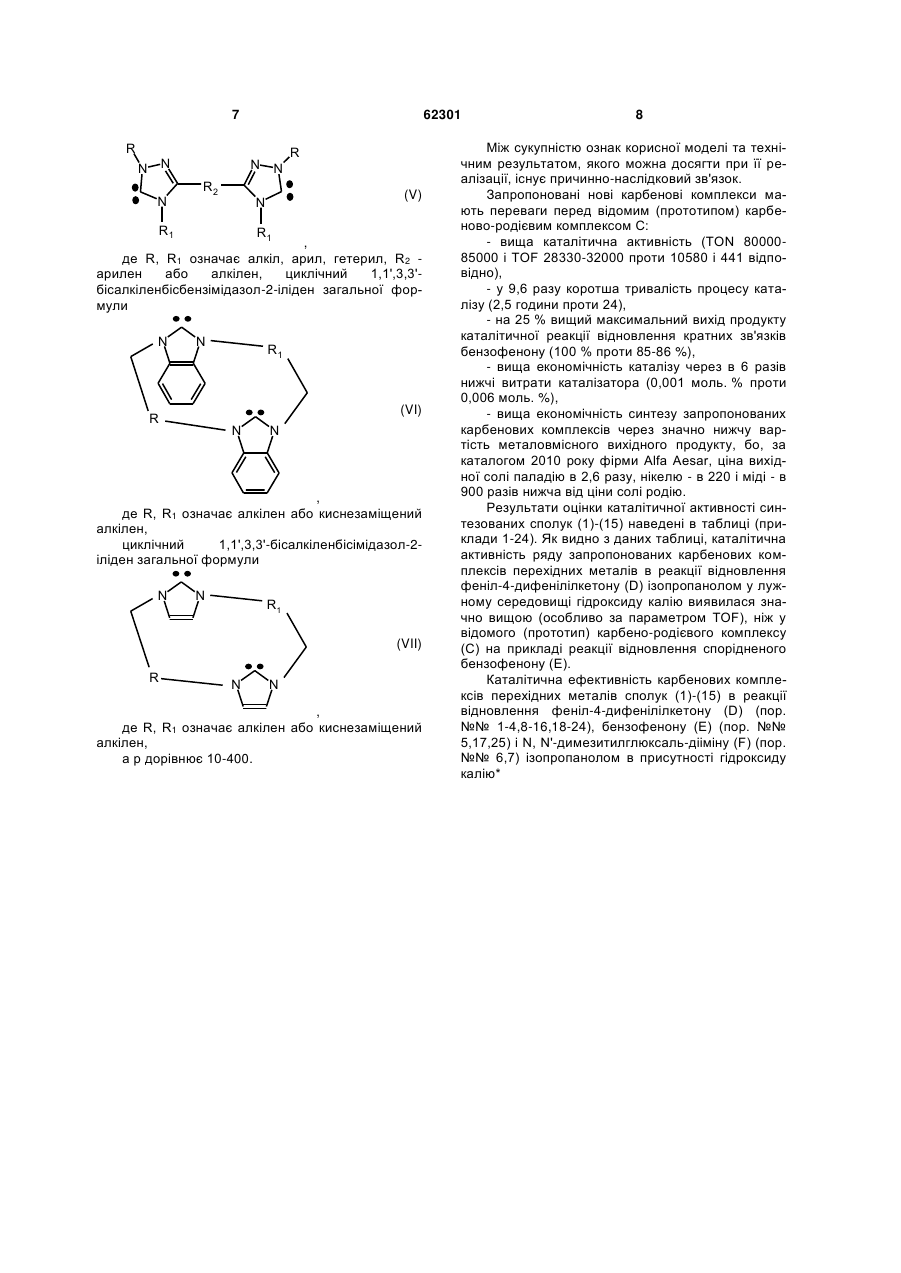
циклічний 1,1',3,3'-бісалкіленбісбензімідазол-2-іліден загальної формули
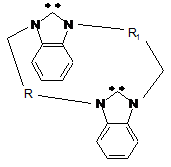 , (VI)
, (VI)
де R, R1 означає алкілен або киснезаміщений алкілен,
циклічний 1,1',3,3'-бісалкіленбісімідазол-2-іліден загальної формули
 , (VII)
, (VII)
де R, R1 означає алкілен або киснезаміщений алкілен,
а р дорівнює 10-400.
Текст
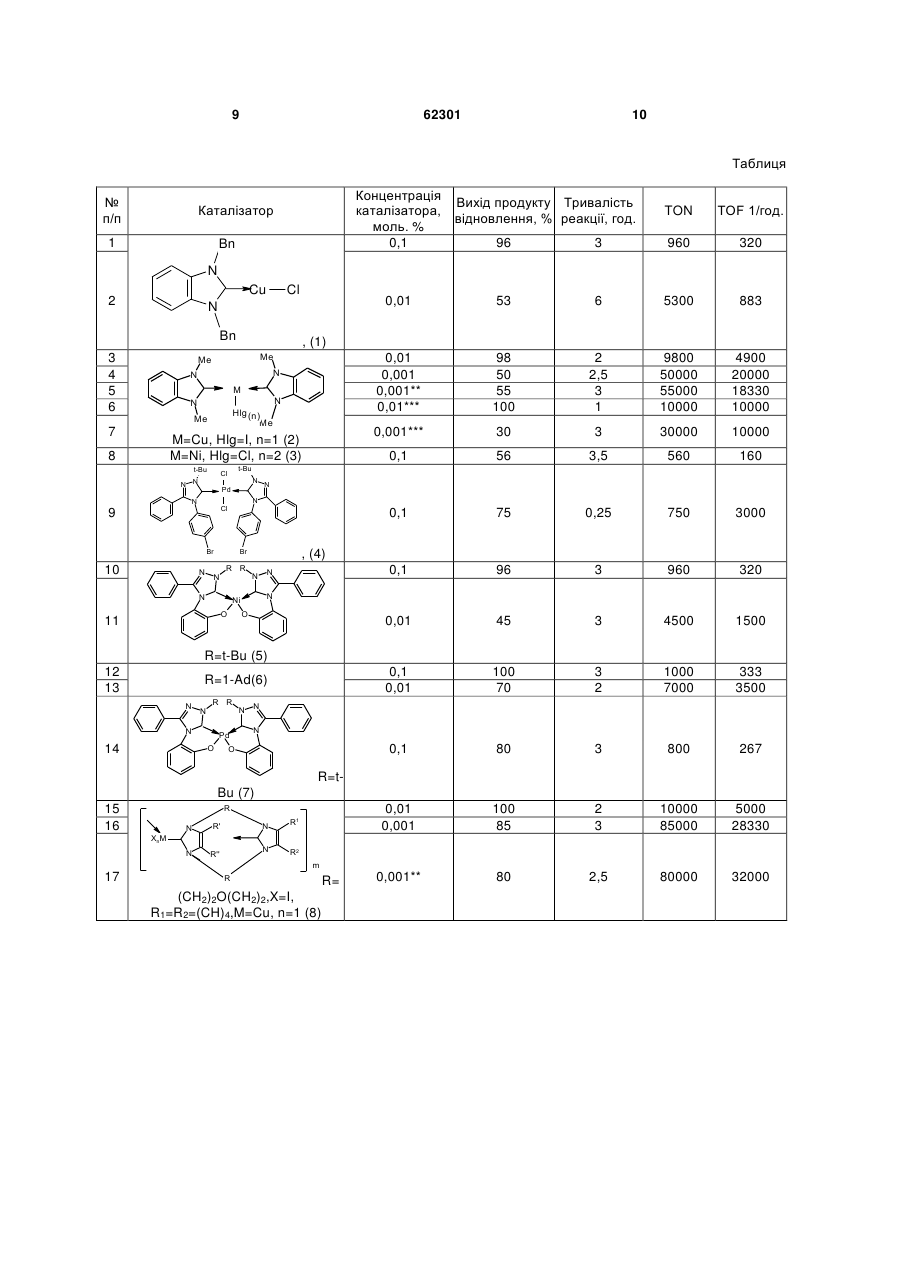
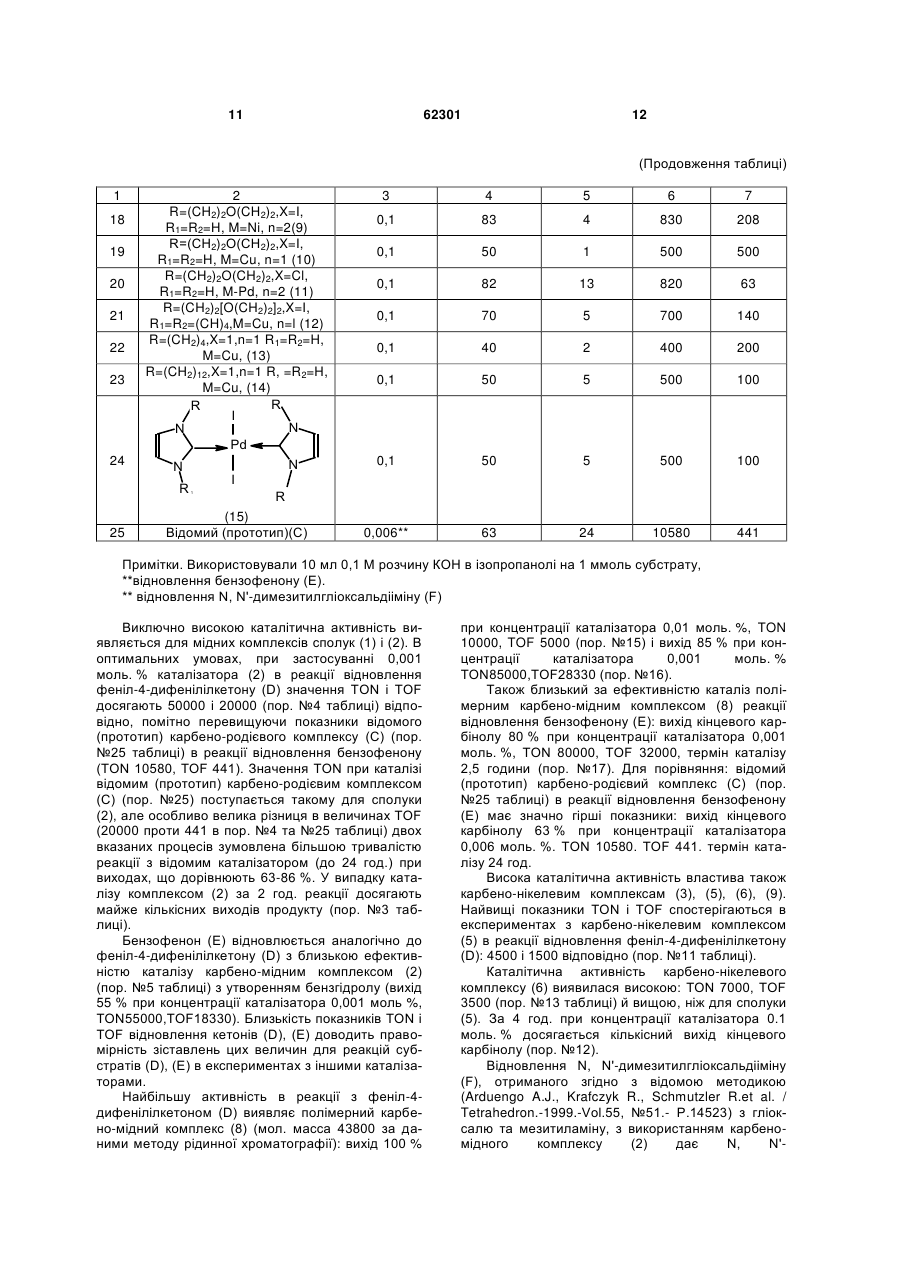
1. Карбенові комплекси перехідних металів загальної формули (LnMXm )p , 3 62301 R N N N N N R2 R1 циклічний 1,1',3,3'-бісалкіленбісімідазол-2-іліден загальної формули R (V) N R N N R1 R1 , де R, R1 означає алкіл, арил, гетерил, R2 - арилен або алкілен, циклічний 1,1',3,3'-бісалкіленбісбензімідазол-2іліден загальної формули N 4 N R1 (VII) R N N , де R, R1 означає алкілен або киснезаміщений алкілен, а р дорівнює 10-400. (VI) N N , де R, R1 означає алкілен або киснезаміщений алкілен, Корисна модель належить до органічної хімії, точніше стосується нових карбенових комплексів перехідних металів, зокрема міді, нікелю, паладію, що можуть застосуватись як каталізатори реакції відновлення кратних зв'язків (карбонільних та іміно-груп, олефінів). Карбени - це сполуки двовалентного вуглецю загальної формули R1R2C:, де R1,R2 - алкільні, арильні або гетероциклічні радикали. R1,R2 також можуть складати карбо- та гетероцикли. Карбени дають з атомами металів координаційні зв'язки, утворюючи карбенові комплекси металів. Карбенові комплекси металів можуть застосовуватися як каталізатори органічних реакцій, в т.ч. реакцій відновлення. Відомо багато способів відновлення органічних сполук за допомогою металів і їхніх похідних (Бартошевич Р., Мечниковска-Столярчик В., Опшондек Б. Методы восстановления органических соединений.- М: Изд. Иностр. Литер., 1960.-406 с). Останнім часом став розвиватися один з найпростіших промислово перспективних способів відновлення кратних зв'язків, який полягає в нагріванні сполуки (субстрату), що містить кратні зв'язки, з ізопропанолом в присутності гідроксидів металів і каталізатора реакції переносу водню від спиртів до кратних зв'язків (Herrmann W.A./ Angew.Chem.Int.Ed.-2002. - Vol.41.- P.1290). Ефективність каталізу оцінюється за максимальними значеннями показників TON і TOF (TON (turnover number) - число каталітичних циклів, що дорівнює числу молів продукту реакції на моль каталізатора; TOF (turnover factor) - число каталітичних циклів за годину, тобто, відношення кількості каталітичних циклів до тривалості реакції (Poyatos M., Mas Marza E., Mata J.A.et al. / Eur.J. Inorg.Chem.-2003.№6.-P.1215). Відомі карбенові комплекси родію(ІІ, III) та іридію структури A (Albrecht М., Crabtree R.H., Mata J., Peris E. / Chem.Commun.-2002.-№1.-P.32): N Cl N N M N N Cl t-Bu t-Bu (A) N , де М означає метал: родій (II, III) або іридій, t-Bu означає трет-бутил. Відомий карбеновий комплекс родію (III) структури В (Mas-Marza E., Sanau М., Peris Е. / Inorg.Chem.-2005,Vol.44.-P.9961): i-Pr N I N O Me Rh (B) N O I N i-Pr , де Me означає метил, i-Pr означає ізопропіл. Карбенові комплекси А та В каталізують реакцію відновлення кратних зв'язків при перетворенні 5 62301 бензофенону до бензгідролу з близькою ефективністю: виходи карбінолу дорівнюють 90-99 %, TON890-900,TOF-50-72. Недоліком відомих карбенових комплексів А та В є неекономічність їх синтезу через дуже високу вартість вихідних сполук, що містять родій чи іридій, та низька каталітична активність в реакції відновлення кратних зв'язків на прикладі бензофенону. Відомий вибраний за прототип карбеновий комплекс родію(ІІІ), який є найближчим до комплексів, що заявляються, по суті та досягнутому технічному результату та має структуру С (Poyatos М., Mas-Marza Е., Mata J.A.et al. / Eur.J. Inorg.Chem.-2003.-№6.-P.1215): Br N Rh (С) Br Br R N R1 , де R, R1 означає алкіл, арил, гетерил, 1,3-дизаміщений бензімідазол-2-іліден загальної формули R N , де R, R1 означає алкіл, арил, гетерил, 1,3,4-тризаміщений 1,2,4-триазол-5-іліден загальної формули R1 N Bu (II) N N N (I) N R1 N Bu N 6 , де Вu означає бутил. Каталітична активність відомого (прототипу) карбенового комплексу С щодо реакції відновлення кратних зв'язків бензофенону виявилася вищою за відомі аналоги А та В: показники TON і TOF для цього каталізатора досягають значень 10580 і 441 відповідно. Недоліками відомого (прототипу) карбенового комплексу С є: недостатньо висока каталітична активність та неекономічність каталізу (довготривалість процесу, недостатньо високий вихід продукту каталітичної реакції та висока вартість синтезу цього каталізатора). В основу корисної моделі поставлено задачу запропонувати нові карбенові комплекси з включенням міді, нікелю чи паладію, які синтезують з низькими економічними затратами, які проявляють високу каталітичну активність при короткій тривалості процесу, низьких витратах каталізатора та високому виході продукту каталітичної реакції з відновлення кратних зв'язків (карбонільних та іміногруп). Запропоновані нові карбенові комплекси дешеві, виходи продуктів каталітичної реакції сягають 100 %, показники TON і TOF для найкращих варіантів нових каталізаторів досягають значень 80000-85000 і 28330-32000 відповідно (за прототипом 10580 і 441), витрати каталізатора в 6 разів нижчі, ніж за прототипом (0,001 моль. % проти 0,006 моль. %), тривалість процесу каталізу скорочується у 9,6 разу (2.5 год. проти 24). Поставлена задача вирішується тим, що створено карбенові комплекси перехідних металів загальної формули (LnMXm)p, де L означає карбеновий ліганд, вибраний з ряду (І-ІІІ): 1,3-дизаміщений імідазол-2-іліден загальної формули N (III) N R R2 , де R, R1,R2 означає алкіл, арил, гетерил, М означає метал, вибраний з ряду: мідь, нікель, паладій, X означає некарбеновий ліганд, вибраний з ряду: галогенід, феноксид, ацетоксид, m дорівнює 1,2, 3,4, n дорівнює 1,2, 3,4, р дорівнює 1. 2. Карбенові комплекси за п. 1, де L разом з Хm означає карбеновий ліганд, який містить карбеновий центр в циклічному фрагменті й атом кисню в боковому ланцюзі ряду 1,3,4-тризаміщеного 1,2,4-триазол-5-ілідену загальної формули R1 N R N N O (IV) , де R, R1 означає алкіл, арил, гетерил. 3. Карбенові комплекси за п. 1, де L означає біскарбеновий ліганд, який містить один або два двовалентні радикали, вибраний з ряду (V-VIII): тетразаміщені 3,3'-місткові біс(1,2,4-триазол-5іліден)арени або алкани загальної формули 7 62301 R N N N N N R2 (V) N R1 R R1 , де R, R1 означає алкіл, арил, гетерил, R2 арилен або алкілен, циклічний 1,1',3,3'бісалкіленбісбензімідазол-2-іліден загальної формули N N R1 (VI) R N N , де R, R1 означає алкілен або киснезаміщений алкілен, циклічний 1,1',3,3'-бісалкіленбісімідазол-2іліден загальної формули N N R1 (VII) R N N , де R, R1 означає алкілен або киснезаміщений алкілен, а р дорівнює 10-400. 8 Між сукупністю ознак корисної моделі та технічним результатом, якого можна досягти при її реалізації, існує причинно-наслідковий зв'язок. Запропоновані нові карбенові комплекси мають переваги перед відомим (прототипом) карбеново-родієвим комплексом С: - вища каталітична активність (TON 8000085000 і TOF 28330-32000 проти 10580 і 441 відповідно), - у 9,6 разу коротша тривалість процесу каталізу (2,5 години проти 24), - на 25 % вищий максимальний вихід продукту каталітичної реакції відновлення кратних зв'язків бензофенону (100 % проти 85-86 %), - вища економічність каталізу через в 6 разів нижчі витрати каталізатора (0,001 моль. % проти 0,006 моль. %), - вища економічність синтезу запропонованих карбенових комплексів через значно нижчу вартість металовмісного вихідного продукту, бо, за каталогом 2010 року фірми Alfa Aesar, ціна вихідної солі паладію в 2,6 разу, нікелю - в 220 і міді - в 900 разів нижча від ціни солі родію. Результати оцінки каталітичної активності синтезованих сполук (1)-(15) наведені в таблиці (приклади 1-24). Як видно з даних таблиці, каталітична активність ряду запропонованих карбенових комплексів перехідних металів в реакції відновлення феніл-4-дифенілілкетону (D) ізопропанолом у лужному середовищі гідроксиду калію виявилася значно вищою (особливо за параметром TOF), ніж у відомого (прототип) карбено-родієвого комплексу (С) на прикладі реакції відновлення спорідненого бензофенону (Е). Каталітична ефективність карбенових комплексів перехідних металів сполук (1)-(15) в реакції відновлення феніл-4-дифенілілкетону (D) (пор. №№ 1-4,8-16,18-24), бензофенону (Е) (пор. №№ 5,17,25) і N, N'-димезитилглюксаль-дііміну (F) (пор. №№ 6,7) ізопропанолом в присутності гідроксиду калію* 9 62301 10 Таблиця № п/п Концентрація Вихід продукту Тривалість каталізатора, відновлення, % реакції, год. моль. % 0,1 96 3 Каталізатор 1 Bn TON TOF 1/год. 960 320 N Cu 2 Cl 0,01 t-Bu N Pd 4900 20000 18330 10000 30 3 30000 10000 56 3,5 560 160 75 0,25 750 3000 0,1 96 3 960 320 45 3 4500 1500 0,1 0,01 100 70 3 2 1000 7000 333 3500 0,1 80 3 800 267 0,01 0,001 100 85 2 3 10000 85000 5000 28330 0,001** 80 2,5 80000 32000 N N N Cl 9 Br Br 10 9800 50000 55000 10000 t-Bu Cl N N 2 2,5 3 1 0,1 N Hlg (n) Me M=Cu, Hlg=I, n=1 (2) M=Ni, Hlg=Cl, n=2 (3) 8 98 50 55 100 0,01 M 7 883 0,001*** N N Me 5300 , (1) Me Me N 6 0,1 Bn 3 4 5 6 53 0,01 0,001 0,001** 0,01*** N N N R R N , (4) N N Ni O 11 N O R=t-Bu (5) 12 13 R=1-Ad(6) N N R R N Pd O 14 N N N O R=tBu (7) 15 16 R N N N R'' R1 N R' R2 XnM m 17 R (CH2)2O(CH2)2,X=I, R1=R2=(CH)4,M=Cu, n=1 (8) R= 11 62301 12 (Продовження таблиці) 1 18 19 20 21 22 23 24 2 R=(CH2)2O(CH2)2,X=I, R1=R2=H, M=Ni, n=2(9) R=(СН2)2О(СН2)2,X=I, R1=R2=H, M=Cu, n=1 (10) R=(CH2)2O(CH2)2,X=Cl, R1=R2=H, M-Pd, n=2 (11) R=(CH2)2[O(CH2)2]2,X=I, R1=R2=(CH)4,M=Cu, n=l (12) R=(CH2)4,X=1,n=1 R1=R2=H, M=Cu, (13) R=(CH2)12,X=1,n=1 R, =R2=H, M=Cu, (14) R R I N N Pd N N I R R (15) Відомий (прототип)(С) 3 4 5 6 7 0,1 83 4 830 208 0,1 50 1 500 500 0,1 82 13 820 63 0,1 70 5 700 140 0,1 40 2 400 200 0,1 50 5 500 100 0,1 50 5 500 100 0,006** 63 24 10580 441 1 25 Примітки. Використовували 10 мл 0,1 М розчину КОН в ізопропанолі на 1 ммоль субстрату, **відновлення бензофенону (Е). ** відновлення N, N'-димезитилгліоксальдііміну (F) Виключно високою каталітична активність виявляється для мідних комплексів сполук (1) і (2). В оптимальних умовах, при застосуванні 0,001 моль. % каталізатора (2) в реакції відновлення феніл-4-дифенілілкетону (D) значення TON і TOF досягають 50000 і 20000 (пор. №4 таблиці) відповідно, помітно перевищуючи показники відомого (прототип) карбено-родієвого комплексу (С) (пор. №25 таблиці) в реакції відновлення бензофенону (TON 10580, TOF 441). Значення TON при каталізі відомим (прототип) карбено-родієвим комплексом (С) (пор. №25) поступається такому для сполуки (2), але особливо велика різниця в величинах TOF (20000 проти 441 в пор. №4 та №25 таблиці) двох вказаних процесів зумовлена більшою тривалістю реакції з відомим каталізатором (до 24 год.) при виходах, що дорівнюють 63-86 %. У випадку каталізу комплексом (2) за 2 год. реакції досягають майже кількісних виходів продукту (пор. №3 таблиці). Бензофенон (Е) відновлюється аналогічно до феніл-4-дифенілілкетону (D) з близькою ефективністю каталізу карбено-мідним комплексом (2) (пор. №5 таблиці) з утворенням бензгідролу (вихід 55 % при концентрації каталізатора 0,001 моль %, TON55000,TOF18330). Близькість показників TON і TOF відновлення кетонів (D), (Е) доводить правомірність зіставлень цих величин для реакцій субстратів (D), (Е) в експериментах з іншими каталізаторами. Найбільшу активність в реакції з феніл-4дифенілілкетоном (D) виявляє полімерний карбено-мідний комплекс (8) (мол. масса 43800 за даними методу рідинної хроматографії): вихід 100 % при концентрації каталізатора 0,01 моль. %, TON 10000, TOF 5000 (пор. №15) і вихід 85 % при концентрації каталізатора 0,001 моль. % TON85000,TOF28330 (пор. №16). Також близький за ефективністю каталіз полімерним карбено-мідним комплексом (8) реакції відновлення бензофенону (Е): вихід кінцевого карбінолу 80 % при концентрації каталізатора 0,001 моль. %, TON 80000, TOF 32000, термін каталізу 2,5 години (пор. №17). Для порівняння: відомий (прототип) карбено-родієвий комплекс (С) (пор. №25 таблиці) в реакції відновлення бензофенону (Е) має значно гірші показники: вихід кінцевого карбінолу 63 % при концентрації каталізатора 0,006 моль. %. TON 10580. TOF 441. термін каталізу 24 год. Висока каталітична активність властива також карбено-нікелевим комплексам (3), (5), (6), (9). Найвищі показники TON і TOF спостерігаються в експериментах з карбено-нікелевим комплексом (5) в реакції відновлення феніл-4-дифенілілкетону (D): 4500 і 1500 відповідно (пор. №11 таблиці). Каталітична активність карбено-нікелевого комплексу (6) виявилася високою: TON 7000, TOF 3500 (пор. №13 таблиці) й вищою, ніж для сполуки (5). За 4 год. при концентрації каталізатора 0.1 моль. % досягається кількісний вихід кінцевого карбінолу (пор. №12). Відновлення N, N'-димезитилгліоксальдііміну (F), отриманого згідно з відомою методикою (Arduengo A.J., Krafczyk R., Schmutzler R.et al. / Tetrahedron.-1999.-Vol.55, №51.- P.14523) з гліоксалю та мезитиламіну, з використанням карбеномідного комплексу (2) дає N, N' 13 62301 димезитилетилендіамін з виходом 100 % при концентрації каталізатора 0,01 моль %, TON 10000, TOF 10000 (пор. №6 таблиці), а при концентрації цього ж каталізатора 0,001 моль % TON 30000, a TOF 10000 (пор. №7). Значну, хоча й дещо меншу, ніж сполуки (1)(9), каталітичну активність показують сполуки (10)(15), дозволяючи досягати хороших виходів продуктів реакції (40-82 %) при незначних витратах часу (1-13 год., TON 400-820, TOF 63-500). Механізм каталізу карбеновими комплексами перехідних металів реакції відновлення кратних Ar' (LnMXm-1 O-Pr-i)p p O Ar' Ar p зв'язків (карбонільних й іміногруп) точно не встановлено. Однак, на основі спостережень обміну лігандів в комплексах можно передбачити, що він схожий на дію ізопропоксиду алюмінію в реакції Меєрвейна-Понндорфа-Верлея, з тією різницею, що як каталітичний центр, виконуючого донорноакцепторну функцію в шестичленному перехідому комплексі, виступає комплексоутворюючий атом перехідного металу (схема каталізу, рівняння 1 і 2): (LnMXm-1 O-Pr-i)p+p KK (LnMXm)p + p i - PrOK Ar 14 Ar H O M p i-PrOH, X O (1) H3C Ar' p + (LnMXm)p+ OH -p i - PrO p CH3 O (2) LnXm-1 де L - карбеновий ліганд; X - інший ліганд (галогенід, феноксид, ацетоксид) Схема каталізу У випадку карбенових комплексів перехідних металів спочатку, ймовірно, відбувається обмін галогенід-йона на алкоксид-аніон з утворенням комплексного алкоксиду металу (рівняння 1). Далі, по аналогії з відновленням за МеєрвейномПонндорфом-Верлеєм, здійснюється взаємодія карбонільної сполуки з вакантною d-орбіталлю перехідного металу та гідридний перенос з алкоголят-йона на карбонільний атом вуглецю в циклічному перехідному стані. Аналогічно в хелатних комплексах може відбуватися витіснення фенолятнона з координаційної сфери металу з наступним відновленням структури хелату після виділення ацетону. Таким чином, пропоновані карбенові комплекси перехідних металів загальної формули (LnMXm)p, з лігандами I-VII є каталізаторами реакції відновлення кратних зв'язків (карбонільних, імінових). В оптимальних варіантах використання карбеномідних комплексів (2), (8) концентрація каталізатора може вибиратися на рівні тисячних мольного процента (TON до 50000-85000, TOF до 18000-32000) при високих виходах продуктів (спиртів), що свідчить про високу ефективність каталізу та дає можливість застосування каталізаторів в промисловості. CH3 N Cl CH3 N N Cl H3C (3) Bn Bn N N N Cl CuCl, NEt3 CH3CN N Cu NiCl22PPh3 CH3CN Cl Bn Bn (1a) (1) Схема 1 Біскарбеновий комплекс 1,3диметилбензімідазол-2-ілідену (ліганд загальної структури II) і йодиду міді(І) (2) одержували при взаємодії 2-ціанометил-1,3-диметил-2Hбензімідазоліну (2а) з йодидом міді(І) в органічних розчинниках (ацетонітрил, диметилформамід) відповідно до схеми 2 (приклад 2). CH3 H3C Ni N Способи одержання сполук відповідно до пропонованої корисної моделі. Звичайно сполуки відповідно до цього винаходу одержують за відомими або новими методиками з використанням вихідних речовин, що наявні у продажу, які отримують відповідно до звичайних хімічних способів. Застосовувана методика одержання сполуки за корисною моделлю залежить від конкретної необхідної сполуки. Так, монокарбеновий комплекс 1,3-диметил-бензімідазол-2-ілідену (ліганд загальної структури II) і хлориду міді(І) (1) отримували шляхом взаємодії 1,3дибензилбензімідазолій хлориду (1а) з хлоридом міді(І) в присутності триетиламіну згідно зі схемою 1 (див. методики одержання сполук відповідно до корисної моделі, приклад 1). CH3 Cul N H3C N N Cu N CN CH3 (2a) CH3CN N CH3 N I H3C (2) 15 62301 Схема 2 Лінійний біскарбеновий комплекс 1,3диметилбензімідазол-2-ілідену (ліганд загальної структури II) і хлориду нікелю (3) одержували шляхом взаємодії 2-ціанометил-1,3-диметил-2Hбензімідазоліну (2а) з бістрифенілфосфіновим комплексом нікель хлориду в ацетонітрилі згідно зі схемою 2 (приклад 3). Лінійний біскарбеновий комплекс 1-трет-бутил3-феніл-4-n-бромфеніл-1,2,4-триазол-5-ілідену (ліганд загальної структури III) і хлориду паладію (4) одержували шляхом взаємодії 1-трет-бутил-3феніл-4-n-бромфеніл-1,2,4-триазол-5-ілідену (4а) з паладій хлоридом в органічному розчиннику (ацетонітрилі або суміші ацетонітрилу та тетрагідрофурану) (схема 3, приклад 4). t-Bu t-Bu N N N N PdCl2 N 2 t-Bu Cl N Pd N N N Cl CH3CN Br Br Br (4a) (4) Схема 3 Хелатні карбенові комплекси 1-алкіл-3-феніл4-(феніл-2-оксид)-1,2,4-триазол-5-ілідену (ліганд загальної структури IV) і нікелю (5),(6) або паладію (7) отримували шляхом взаємодії 1-заміщених 3феніл-4-(феніл-2-оксид)-1,2,4-триазолієвих солей (5а), (6а) з нікель хлоридом або паладій хлоридом в присутності трет-бутоксидів металів в диметилсульфоксиді (схема 4, приклади 5-7). 16 нікелю та паладію відповідно до схеми 6 (приклади 9-14). R' N R'' N N R'' N N MCl2 N OH N R R N KOBu-t DMSO N N N M O O ClO4 (5a), (6a) (5)-(7) (5), (5a), (7) R=t-Bu; (6), (6a) R=1-Ad; (5), (6)M=Ni; (7)M=Pd Схема 4 Полімерний карбеновий комплекс бісалкіленбісбензімідазол-2-ілідену (ліганд загальної структури VI) (8) синтезовано з відповідного ацетилацетонату біскарбенієвої солі (8а) з йодидом міді(І) в ацетонітрилі (схема 5, приклад 8). R R R' R'' N N R' N N R'' R (8a) 2W MXn N N N R'' R' N R' R'' XnM n R (8) R = (СН2)2О(СН2)2,R1,R2 = (СН)4 (8), (8а), М = Сu, X=1,n=1 (8), W = ОС(СН3)СНССН3 (8а) Схема 5 Полімерні комплекси бісалкіленбісбензімідазол-2-ілідену та бісалкіленбісімідазол-2-ілідену (ліганди загальних структур VI, VII) (9)-(14) одержані дією гідриду натрію на ацетонітрильні або диметилсульфоксидні розчини біскарбенієвих солей (9а), (12а)-(14а) в присутності солей міді(І), R' N N R'' R' N N MXn, NaH R'' XnM DMSO m R 2W R (9)-(14) (9a), (11a)-(14a) R = (CH2)2 (CH2)2, R1,R2=H(9), (9a), M=Ni, ,X=1,n=2 (9), W=CIO4 (9а); R = (CH2)2O(CH2)2,R1,R2=H, M=Cu, X=I, n-1 (10); R = (CH2)2O(CH2)2,R1,R2=H, M=Pb, X=Cl, n=2(11); R = (CH2)2[O(CH2)2]2,R1,R2=(CH)4 (12), (12a), M=Cu, X=I, n-1(12),W=ClO4 (12a); R = (CH2)4,R1,R2=H (13, (13a), M=Cu, X=I, n=1 (13,W=CIO4 (13a) (13a); R = (CH2)12,R1,R2=H (14), (14a), M=Cu, X=I, n=1 (14), W=CIO4 (14a) Схема 6 Карбеновий комплекс імідазол-2-ілідену (ліганд загальної структури І) і паладій йодиду (15) отримано шляхом реакції імідазолієвої солі (15а) з трет-бутоксидом калію в присутності йодиду паладію (схема 7). R R N t-BuOK, Pdl2 N R N N Pd DMSO X R I N 1 R N I R 1 (15) (15a) R N R R N R' R=Me Схема 7 Біскарбеновий комплекс 3,3'-ариленбіс-1,2,4триазол-5-ілідену (ліганд загальної структури V) і хлориду міді (16) отримано шляхом прямої реакції біскарбену (16а) з хлоридом міді(І) в суміші ацетонітрилу і тетрагідрофурану (схема 8). R N N N N N R2 R1 N R1 (16a) R R N CuCl THF/CH3CN Cl Cu N N N R2 R1 N R1 N R Cu Cl (16) R=1-Ar, R1=Ph, R2=м-фенілін Схема 8 Приклади одержання сполук відповідно до корисної моделі. Спектри протонного ядерного магнітного резо1 нансу ( Н ЯМР) знімали на спектрометрі Bruker Avance II400 (400 МГц) з використанням стандарту 13 Me4Si ( 0,00). Вуглецеві спектри С ЯМР (100 МГц) знімали на спектрометрі Bruker Avance II400 (400 МГц) з використанням стандарту Me4Si ( 0,00). Позначення в спектрах ЯМР: с - синглет, д дублет, м - мультиплет. Приклад 1. Одержання (1,3дибензилбензімідазол-2-іліден)міді(І) хлориду (1). Суміш 1 г (3 ммоль) 1,3-дибензилбензімідазолій хлориду (2а) і 0,15 г (1,5 ммоль) хлориду міді розчиняли в 15 мл сухого ацетонітрилу, додавали 0,50 мл (3,6 ммоль) триетиламіну и нагрівали до 17 кипіння зі зворотним холодильником. Після 2 год. кип'ятіння додавали ще 0,50 мл триетиламіну. По завершенні реакції продукт висаджували водою, відфільтровували, промивали сумішшю ізопропіловий/спирт-петролейний етер у співвідношенні 1:3 і сушили над КОН. Вихід неочищеної сполуки (1) - 1 г (95 %), після перекристалізації з ацетонітрилу - 0,7 г (66 %). Т. пл. 175-177 °C. Знайдено, %: С 63,6, Н 4,6, Сl 8,6, Сu16,1,N 7,2.C21H19ClCuN2. 1 Обчислено. %: С 63,5, Н 4,6. СІ 8,9, Сu16,0,N7,1. Н ЯМР (AMSO-d6), , м.ч.: 5,76 с (4Н, CH2N), 7,43 м 13 (14Н, Аr). С ЯМР (AMSO-d6), , м.д.: 51,2(CH2N), 111,8,123,4,127,3,127,7,128,5 (Аr), 133,4,136,3 2 (ipso-C, Аr), 188,6 (С ). Приклад 2. Одержання біс(1,3диметилбензімідазол-2-іліден) міді(І) йодиду (2). До суспензії 1,3 г (6,8 ммоль) йодиду міді(І) в 4 мл ацетонітрилу додавали 2,55 г (13,6 ммоль) 1,3диметил-2-ціанометил-2Я-бензімідазоліну та кип'ятили суміш протягом 2 год. Отриманий осад комплексу (3) відфільтровували, промивали діетиловим етером і сушили. Вихід комплексу (2) 2,2 г (67 %). Т. пл. 220-221 °C (ацетонітрил). Знайдено, %: С 44,5, Н 4,1, Сu13,4, І 26,5,N11,4. С18Н20СuIN4. Обчислено, %: С 44,8, Н 4,2, 1 Сu13,2,126,3,N11,6. Н ЯМР (DMSO-d6), , м.ч.: 4,06 с (6Н) (CH3N), 7,40 д (2Н), 7,65 д (2H)(J 2,6 Гц) (Аr). 13 С ЯМР (CDCl3), м.ч.: 34,3 (CH3N), 110,9 (С4.7), 123,0 (С5,6), 134,0 (ipso-C) (Аr), 190,7 (С2). Приклад 3. Одержання біс-(1,3диметилбензімідазол-2-іліден)нікелю хлориду (3). Отримували аналогічно комплексу (2) з 1,66 г (2,54 ммоль) комплексу хлориду нікелю з двома молекулами трифенілфосфіну та 0,95 г (5,08 ммоль) 1,3-диметил-2-ціанометил-2H-бензімідазоліну в 6 мл ацетонітрилу. Вихід 1,07 г (100 %). Т. пл. 275278 °C (ацетонітрил). Знайдено, %: С 51,5, Н 4,9, Сl16,7,N13,2,Ni14,1.C18H20Cl2N4Ni. Обчислено, %: 1 С 51,2, Н 4,8, Сl16,8,N13,3,Ni13,9. Н ЯМР (DMSOd6), , м.ч.: 4,47 с (12Н, CH3N), 7,30-7,78 м (8Н, Аr). 1З С ЯМР (DMSO-d6+Py-d6), , м.ч.: 35,7 (CH3N), 2 110,4,123,8 (Аr), 134.9.144,2 (ipso-C, Аr), 180,6 (С ). Приклад 4. Одержання біс(1-трет-бутил-3феніл-4-n-бромфеніл-1,2,4-триазол-5іліден)паладій хлориду (4). До суспензії 0,05 г (0,28 ммоль) паладій хлориду в 1 мл ацетонітрилу додавали 0,2 г (0,56 ммоль) 1-трет-бутил-3-феніл-4n-бромфеніл-1,2,4-триазол -5-ілідену (4а) в 1 мл толуену та перемішували суміш протягом 0,5 год. Осад сполуки (4) (0,15 г) відфільтровували і сушили, а з маточного розчину випаровуванням розчинника виділяли ще 0,1 г комплексу (4). Загальний вихід 0,25 г (100 %). Т. пл. 160-163 °C (субл.). Знайдено, %: С 48,7, Н 4,0, Вr18,3, СІ 8,1,N 9,4,Pd11,9.C36H36Br2Cl2N6Pd. Обчислено, %: С 1 48,6, Н 4,1, Вr18.0, Сl 8,0,N 9,4,Pd12,0. Н ЯМР (CDCl3), , м.ч.: 1,64 с (9Н, СН3С), 7,26-7,67 м (18Н. 13 Аr). С ЯМР (CDC13). , м.ч.: 30,1 (СН3С), 62,9 (СН3С), 127,8,128,6,128,7.129,1,130,4,131,6,132,4.132,9 1N 3 2 (Аr), 143,2 (C ), 151,4 (С ), 171,0 (C N). Приклад 5. Одержання біс-[1-(1-трет-бутил)-3феніл-4-(феніл-2-оксид)-1,2,4-триазол-5іліден]нікелю (5). 62301 18 Стадія 1. Одержання 1-трт-бутил-3-феніл-4-(2гідроксифеніл)-1,2,4-три-азолій перхлорату (5а). Суміш 1,38 мл (12,65 ммоль) трет-бутилхлориду й 1,9 г (12,65 ммоль) йодиду натрію кип'ятили в 7 мл оцтової кислоти протягом 2 год. По охолодженні осад хлориду натрію відфільтровували, додавали 1 г (4,22 ммоль) 3-феніл-4-(2-гідроксифеніл)-1,2.4триазолу та кип'ятили протягом двох діб. Оцтову кислоту відмивали розведеннями петролейним етером (2 × 20 мл). Олієподібний залишок розчиняли в 50 мл киплячої води, додавали невеличку кількість сульфіту натрію до зникнення темного забарвлення, перефільтровували з активованим вугіллям і до нього додавали гарячий розчин 1,22 г (10 ммоль) перхлорату натрію. Осад відфільтровували й сушили. Вихід 1,37 г (83 %). Т. пл. 194195 °C (50 % водний розчин оцтової кислоти). Знайдено, %: С 54,8, Н 5,0,N10,8, Сl 8,9.C18H20Cl2N3O5. Обчислено, %: С 54,9, Н 1 5,1,N10,7, Сl 9,0. НЯМР спектр (DMSO-d6), , м.ч.: 1,75 с (9Н, СН3), 7,07 м (2Н, Аr), 7,05-7,69 м (7Н, Аr), 10,61 (с, 1H, CHN), 10,81 (с, 1Н, ОН). Стадія 2. Одержання біс-[1-(1-трет-бутил)-3феніл-4-(феніл-2-оксид)-1,2,4-триазол-5іліден]нікелю (5). Суміш 0,4 г (1,07 ммоль) 1-(1трет-бутил)-3-феніл-4-(2-гідроксифеніл)-1,2,4триазолій перхлорату (5а), 0,07 г (0,54 ммоль) хлориду нікелю розчиняли в 5 мл диметилсульфоксиду та при перемішуванні додавали 0.24 г (2,14 ммоль) трет-бутоксиду калію. Перемішували 1 год. Реакційну масу виливали в воду й осад відфільтровували. Далі осад розчиняли в дихлорметані, фільтрували через тонкий шар (1 см) силікагелю (70/230 мкм), розчинник випаровували. Вихід комплексу (5) 0,29 г (42 %). Т. пл. 272-274 °C (диметилсульфоксид). Знайдено, %: С 67,4, Н 5,6,N13,0, № 9,0.C36H36N6O2Ni. Обчислено, %: С 67,2, Н 1 5,6,N13,1,Ni 9,1. НЯМР (та.), , м.ч.: 1,87 (с, 18Н, 13 СН3С), 6,68 (м, 5Н), 7,41 (м, 25Н) (Аr). СЯМР (тв.), , м.ч.: 31,0 (СН3С), 62,3 (СН3С), 113,9,122,9,126,4,127,9,128,7,129,3. (Аr), 130,5 1N 3 20 2 (C ), 148,8 (С ) 160.5 (С ). 163,8 (C N). Приклад 6. Одержання біс-[1-(1-адамантил)-3феніл-4-(феніл-2-оксид)-1,2,4-триазол-5-іліден] нікелю (6). Суміш 0,15 г (0,32 ммоль) 1-(1адамантил)-3-феніл-4-(2-гідроксифеніл)-1,2,4триазолій перхлорату (6а), отриманого згідно з методикою роботи (Короткіх М.І., Кисельов А.В., Пехтерева Т.М., Швайка О.П., Каулі А.Г., Джонс Дж. Н. / Доповіді НАН України.-2005.-№6.-С. 150155) шляхом кватернізації відповідного триазолу 1-бромадамантаном в оцтовій кислоті, 0,02 г (0,16 ммоль) хлориду нікелю й 0,068 (0,64 ммоль) карбонату натрію нагрівали при 80 °C в 2 мл диметилсульфоксиду впродовж 2 год. По охолодженні реакційну масу виливали у воду й осад відфільтровували. Осад розчиняли в дихлорметані, фільтрували через тонкий шар силікагелю (70/230 мкм), розчинник випаровували. Вихід 0,1 г (77 %). Т. пл. 288-290 °C (ацетонітрил). Знайдено, %: С 72,3, Н 6,2,N10,5,Ni 7,1.C48H48N6NiO2. Обчислено, %: С 1 72,1, Н 6,1,N10,5,Ni 7,3. Спектр Н ЯМР (CDCl3), , м.ч.: 1,57 (м, 12Н), 2,02 (м, 12Н), 2,74 (м, 6Н) (Ad); 6,33 (м, 2Н), 6,56 (м, 2Н), 7,06 (м, 4Н), 7,22 (м, 4Н). 13 7,48 (м, 6Н) (Аr). Спектр С ЯМР (CDCl3), , м.ч.: 19 62301 5 29,3,35.1,42,8,62,3 (ipso-C) (Ad), 112,6 (C N), 1 1 126,1,126,5 (C N, C C), 3 121,6,121,9,128,6,128,9,130,7 (Ar), 148,8 (C ), 160,3 20 5 (C ), 162,9 (C ). Приклад 7. Одержання біс-[1-(1-трет-бутил)-3фенил-4-(феніл-2-оксид)-1,2,4-триазол-5-іліден] паладію (7). Суміш 0,45 г (1,12 ммоль) 1-(1адаман-тил)-3-феніл-4-(2-гідроксифеніл)-1,2,4триазолій перхлорату (6а), 0,1 г (0,56 ммоль) хлориду паладію й 0,24 г (2,24 ммоль) карбонату натрію нагрівали при 80 °C в 2 мл диметилсульфоксиду протягом 2 год. По охолодженні реакційну масу виливали у воду й осад відфільтровували. Осад розчиняли в хлористому метилені, фільтрували через тонкий шар силікагелю (70/230 мкм), розчинник випаровували. Вихід 0,25 г (64 %). Т. пл. 140 °C. Знайдено, %: С 62,7, Н 5,1,N12,4,Pd15,3.C36H36N6O2Pd. Обчислено, %: С 1 62,6, Н 5,3,N12.2,Pd15,4. Н ЯМР, , м.ч.: 1,60 (с, 13 9Н, СН3С), 7,39 (м, 9Н, Аr). С ЯМР, , м.ч.: 30,8 (СН3С), 62,6 (СН3С), 114,6,121,4,124,4,128,5,128,6,129,4,130,2 (Аr), 1С 1N 3 20 126,9 (С ), 148,2 (C ), 153,0 (С ) 161,7 (С ), 2 171,1 (C N). Приклад 8. Одержання полімерного йодиду 1,1',3,3'-біс(3-оксапенти-лен)бісбензімідазол-2іліденміді (І) (8). Стадія 1. Одержання ацетилацетонату 1,1',3,3'-біс(3-оксапентилен)бісбензімідазолію (8а). До розчину 0,22 г (1,96 ммоль) трет-бутоксиду калію в 5 мл абсолютного метанолу одноразово додавали розчин 0,196 г (1,96 ммоль) ацетилацетону у 2 мл абсолютного метанолу. Отриманий розчин ацетилацетонату калію додавали до розчину 0,44 г (0,98 ммоль) хлориду 1,1',3,3'-біс(3оксапентилен)бісбензімідазолію, що одержували згідно з методикою роботи (Короткіх М.І., Марічев К.О., Кисельов А.В., Швайка О.П./ Ukrainica bioorganica acta.-2008.-T.6, №2.-С. 22-27) з бензімідазолу і 2,2'-дихлордіетилового етеру в 20 мл суміші абсолютних ізопропанолу і метанолу (2:1) і перемішували. Одразу випадав осад хлориду калію, який відфільтровували; маточний розчин випаровували, а залишок розтирали з діетиловим етером. Продукт висушували в вакуумі для повного видалення розчинників і залишків ацетилацетону. Всі операції виконувались в атмосфері азоту. Вихід 0,56 г (99 %). Т. пл. 158-161 °C. Знайдено, %: С 66,2, Н 6,8,N 9,7.C32H40N4О6. Обчислено, %: С 1 66,7, Н 7,0,N 9,7. Н ЯМР (DMSO-d6), , м.ч.: 1,74 с (12Н, СН3); 2,50 с (2Н, СНСО); 3,78,3,79,3,80,3,82,3,84,3,85,3,96 м (8Н, СН2О); 4,72 с (8Н, CH2N); 7,58 м, 8,05 м (8Н, Аr); 10,56 с (2Н, CHN). Стадія 2. Одержання полімерного йодиду 1,1",3,3'-біс(3-оксапентилен)біс-бензімідазол-2іліден міді(І) (8). До розчину 2,0 г (3,4 ммоль) ацетилацетонату 1,1",3,3'-біс(3оксапентилен)бісбензімідазолію (8а) в 30 мл безводного ацетонітрилу при кімнатній температурі додавали крапельно розчин 0,66 г (3,4 ммоль) йодиду міді(1) в 15 мл абсолютного ацетонітрилу в атмосфері азоту при постійному перемішуванні. Продукт одразу ж починав кристалізуватися з розчину. Після додавання всієї кількості йодиду міді(І) 20 суміш витримували протягом 40 хв. при кімнатній температурі. Осад відфільтровували та промивали на фільтрі діетиловим етером в атмосфері азоту. Отримували кристали зеленуватого кольору (8). Вихід 1,6 г (82 %). Т. пл. 164-165 °C. Знайдено, %: С 46,9, Н 4,3,N 9,8, І 22,4, Си 11,3.C22H24IN4O2CU. 1 Обчислено, %: С 46,6, Н 4,3,N 9,9,122,4, Сu11,2. Н ЯМР (DMSO-d6), , м.ч.: 3,36 с (8Н, СН2О), 3,92,4,6 м (8Н, CH2N), 6,60,7,30,7,95 м (8Н, Аr). РХ: Mw43800, Мn42400 (Mw/Mn 1,0402). Приклад 9. Одержання полімерного йодиду 1,1",3,3'-біс[(3-оксапенти-лен)бісімідазол-2іліден]нікелю (9). До розчину 0,2 г (0,42 ммоль) перхлорату 1,1",3,3'-біс(3оксапентилен)бісімідазолію (9а), який одержували (Короткіх М.І., Марічев К.О., Кисельов А.В., Швайка О.П./ Ukrainica bioorganica acta.-2008.-Т.6, №2.С.22-7) з імідазолу і 2,2'-дихлордіетилового етеру, в 1 мл диметилсульфоксиду додавали 0,036 г (0,84 ммоль) 55 %-ного гідриду натрію в мінеральній олії. Реакційну суміш премішували при кімнатній температурі протягом 10 хв., потім додавали 0,13 г (0,42 ммоль) йодиду нікелю(ІІ) та перемішували 20 хв. Осад, що випав, відфільтровували, промивали 0,5 мл диметилсульфоксиду та 15 мл діетилового етеру, сушили при 100 °C. Вихід 0,23 г (92 %). Т, пл. вище 300 °C. Знайдено, %: С 28,0, Н 3,7,I43,8,N 9,4, № 9,6. С14Н20I4NiO2. Обчислено, %: С 28,6, Н 3,4,143,1,N 9,5,Ni10.0. Приклад 10. Одержання полімерного 1,1",3,3'біс(3-оксапентилен)бісіміда-зол-2-іліденміді(І) йодиду (10). До розчину 1 г (2,1 ммоль) перхлорату 1,1",3,3'-біс(3-оксапентилен)бісімідазолію (9а) в 8 мл диметилсульфоксиду додавали 0,18 г (4,2 ммоль) 55 %-ного гідриду натрію в мінеральній олії. Після виділення теоретичної кількості водню додавали 0,4 г (2,1 ммоль) йодиду міді(І). Перемішували протягом 2 год. Розчин продукту фільтрували, продукт висаджували 30 мл води. Осад комплексу відфільтровували, промивали 15 мл води, сушили та переосаджували водою з диметилформаміду. Вихід 0,6 г (62 %). Т. пл. 180-182 °C (розкл.). Знайдено, %: С 35,9, Н 4,4, І 27,0.N12,1, Сu13,4.C14H20CuIN4O2. Обчислено, %: С 36,0, Н 1 4,3,127,2,N12,0, Сu13,6. Н ЯМР (DMSO-d6), , м.ч.: 3,45 м, 3,80 м, (8Н, СН2О), 4,41 м (8Н, CH2N), 6,60,7,30,7,76 (4Н, CHN). РХ: Mw164800, Мn100000 (Mw/Mn 1,65). Приклад 11. Одержання полімерного 1,1",3,3'біс(3-оксапентилен)бісіміда-зол-2-іліденпаладію хлориду (11). До суспензії 0,40 г (0,84 ммоль) перхлоратної солі (9а) в 6 мл піридину додавали 0,19 г (1,68 ммоль) трет-бутоксиду калію і перемішували протягом 2 год. Осад перхлорату калію відфільтровували. До розчину додавали 0,15 г (0,84 ммоль) хлориду паладію (II) та перемішували реакційну суміш протягом 3 год. Продукт, нерозчинний в піридині, відфільтровували та промивали 5 мл гарячого диметилформаміду та 5 мл ацетонітрилу. Вихід 0,26 г (68 %). Т. пл. більше 300 °C. Знайдено. %: С 37,3, Н 4,3, Сl15,7,N15,5,Pd12,3.C14H20Cl2N4O2Pd. Обчислено, %: С 37,1, Н 4,4, Сl15,6,N15,6,Pd12,4. Приклад 12. Одержання полімерного 1,1",3,3'біс(3,6-діоксаоктилен)біс-бензімідазол-2 21 іліденміді(І) йодиду (12). До розчину 0,2 г (0,30 ммоль) перхлоратної солі 1,1",3,3'-біс(3,6діоксаоктилен)бісбензімідазолію (12а), що одержували згідно з методикою роботи (Короткіх М.І., Марічев К.О., Кисельов А.В., Швайка О.П. Синтез похідних краункарбеноїдів // Ukrainica bioorganicaacta.-2008, Т.6, №2.- С.22-27) з бензімідазолу і 8,8'-дихлор-5-оксаоктану, в 5 мл ацетонітрилу та додавали 0,026 г (0,60 ммоль) 55 %-ного гідриду натрію в мінеральній олії. Реакційну суміш нагрівали при 60 °C протягом 20 хв. Кінець реакції контролювали за об'ємом водню, що виділився. Суспензію 0,057 г (0,30 ммоль) йодиду міді(І) в 6 мл ацетонітрилу додавали до реакційної суміші й кип'ятили протягом 30 хв. Олієподібний продукт відділяли від ацетонітрилу декантацією, промивали 2 рази по 10 мл ацетонітрилу, при охолоджені продукт кристалізувався. Розтирали з діетиловим етером. Продукт розчиняли в 10 мл диметилформаміду при кип'ятінні з активованим вугіллям та фільтрували. Продукт як світло-зелений порошок висаджували 100 мл води, фільтрували й сушили при 60 °C. Вихід 0,12 г (61 %). Т. пл. 175-178 °C (розкл.). Знайдено, %: С 47,5, Н 5,0, Сu 9,6, І 19,2,N 8,7. С26Н32СuIN4О4. Обчислено, %: С 47,7, Н 1 4,9, Сu 9,7, І 19,4,N 8,6. Н ЯМР (DMSO-d6), , м.ч.: 3,40 с, 3,79, (16Н, СН2О), 4,59 м (8Н. CH2N). 6,98,7.31,7,62 м (8Н, Аr). РХ: Mw47100.Mn35500 (Mw/Mn 1.32). Приклад 13. Одержання 1,Г, 3,3'біс(тетраметилен)бісімідазол-2-іліден міді(I) йодиду (13). Стадія 1. Одержання 1,1",3,3'-біс-(1,4тетраметилен)бісімідазолій перхлорату (13а). Суміш 13,0 г (0,068 моль) 1,4тетраметиленбісімідазолу (136) і 7,9 мл (0,068 моль) 1-хлор-4-бромбутану та нагрівали протягом 3 год. при 120 °C до завершення утворення бісімідазолієвої солі (13а). До реакційної суміші додавали 2 мл диметилформаміду і продовжували нагрівання ще 6 год. Сіль в хлоридній формі виділяли висадженням надлишком етеру (50 мл). Олієподібний продукт переводили у перхлорат додаванням надлишку перхлорату натрію (9.8 г, 0,08 моль) осад відфільтровували, сушили. Перекристалізували з води. Вихід чистої солі 22,3 г (49 %). Т. пл. 78-82 °C (вода). Знайдено, %: С 38,0, Н 5,1, Сl15,7,N12,7.C14H22Cl2N4O8. Обчислено, %: С 37,8, Н 5,0, Сl15,9,N12,7. Вихідний бісімідазол 1 (136): Н ЯМР (DMSO-d6), , м.ч.: 1,60 с (СН2С), 5 4 3,94 с (CH2N), 6,87 с (C HN), 7,12 с (C HN), 7,60 с 2 1 (C HN). Бісімідазолієва сіль (13а): Н ЯМР (DMSO4d6), , м.ч.: 1,81 с (СН2С), 4,19 с (CH2N), 7,75 с (C 5 2 HN), 9,09 с (C HN). Стадія 2. Одержання 1,1",3,3'біс(тетраметилен)бісімідазол-2-іліден міді(І) йодиду (13). До розчину 0,2 г (0,45 ммоль) перхлорату 1,1",3,3'-біс(тетраметилен)бісімідазолію (13а) в 6 мл ацетонітрилу додавали 0,04 г (0,90 ммоль) 55 %-ного гідриду натрію в мінеральній олії. Реакційну суміш нагрівали при 60 °C протягом 20 хв. Кінець реакції контролювали за об'ємом водню, що виділився. Суспензію 0,086 г (0,45 ммоль) йодиду міді(І) в 8 мл ацетонітрилу додавали до реакційної суміші, перемішували при 60 °C протягом 1 62301 22 год. Осад продукту відфільтровували та промивали 20 мл ацетонітрилу. Пересаджували діетиловим етером з диметилформаміду. Вихід 0,16 г (82 %). Т. пл. 230-235 °C (розкл.). Знайдено, %: С 38,9, Н 4,5, Сu14,5, І 29,1,N12,8.C14H20CuIN4. Об1 числено, %: С 38,7, Н 4,6, Сu14,6, І 29,2,N12,9. Н ЯМР (DMSO-d6), 8, м.ч.: 1,49 м (4Н, СН2С), 1,75 м, (4Н, СН2С), 3,38 м, (4Н, CH2N), 4,18 м (8Н, CH2N), 6.47 (2Н, CHN), 7.35 (1H, CHN), 7.78 (1Н, CHN). PX:Mw225000, Мn76800 (Mw/Mn 2,93). Приклад 14. Одержання полімерного 1,1",3,3'біс-(1,12-додекаметилен)біс-імідазол-2іліден)міді(ї) йодиду (14). Стадія 1. Одержання 1,1",3,3'-біс-(1,12додекаметилен)бісімідазолій перхлорату (14а). Отримували аналогічно солі (12а) з 0,4 г (1,32 ммоль) 1,12-додекаметиленбісімідазолу (146) і 0,3 мл (1,46 ммоль) 1,12-дихлор до декану при 120 °C протягом 4 год. Продукт виділено у вигляді майже безбарвної олії. Вихід 0,5 г (56 %). Знайдено, %: С 53,9, Н 8,3, Сl10,4,N 8,5.C30H54CI2N4O8. Обчислено, %: С 53,8, Н 8,1, Сl10,6,N 8,4. Вихідний бісімідазол 1 (146): Н ЯМР (DMSO-d6), , м.ч.: 1,26 с (СН2С), 5 1,75 с (CH2CN), 3,91 с (CH2N), 6,89 с (C HN), 7,04 с 4 2 (C HN), 7,45 с (C HN). Бісімідазолієва сіль (14а): 1 Н ЯМР (DMSO-d6), , м.ч.: 1,23 с (СН2С), 1,77 с 45 (CH2CN), 4,15 с (CH2N), 7,76 с (C ' HN), 2 9,14c(C HN). Стадія 2. Одержання полімерного 1,Г, 3,3'-біс(1,12-додекаметилен)бісімідазол-2-іліден)міді(І) йодиду (14). До розчину 0,23 г (0,49 ммоль) солі (14а) в 15 мл ацетонітрилу додавали 0,064 г (0,98 ммоль) 37 %-ного гідриду натрію в мінеральній олії. Реакційну суміш нагрівали при 50 °C протягом 20 хв., додавали 0,094 г (0,49 ммоль) йодиду міді(І) й перемішували при 50 °C протягом 2 год. Утворений олієподібний продукт відділяли від ацетонітрилу та розтирали з 30 мл води. Осад фільтрували, промивали 20 мл води, сушили при 60 °C. Переосаджували діетиловим етером із диметилсульфоксиду. Вихід 0,18 г (80 %). Т. пл. 75-78 °C. Знайдено, %: С 55.2. Н 8,4, Сu 9,3, І 18,7,N 8,5.C31H56CuIN4. Обчислено. %: С 55,1, Н 8,4, Сu 1 9,4,118,8,N 8,4. Н ЯМР (DMSO-d6), , м.ч.: 1,22 м (36Н, СН2С), 1,52 м, (4Н, СН2С), 1,78 м, (8Н, СН2С), 4,11 м (8Н, CH2N), 6,46,7,40,7,82 (4Н, CHN). Приклад 15. Біс(1,3-диметилімідазол-2іліден)паладію(ІІ) йодид (15). До розчину 0,1 г (0,277 ммоль) йодиду паладію та 0,77 г (3,91 ммоль) 1,3-диметилімідазолій перхлорату в 0,5 мл диметилсульфоксиду додавали 0,062 г (0,554 ммоль) трет-бутоксиду калію. Перемішували протягом 8 год. Диметилсульфоксид екстрагували 30 мл діетилового етеру. Залишок обробляли 150 мл води. Осад, що випав, фільтрували та сушили. Порошок розчиняли в 20 мл хлороформу та фільтрували через силікагель (70/230 мкм). Розчинник випаровували, а жовтий порошок залишку перекристалізовували з суміші хлороформ/гексан, 1:3.R, 0,90. Вихід 0,1 г (65 %). Т. пл. 299-300 °C. Знайдено, %: С 21,8, Н 2,9,I46,0,N10,1.C10H16I2N4Pd. Об1 числено, %: С 21,7, Н 2,9,I 45,9, N 10,1. Спектр Н ЯМР (CDC13), , м.ч.: 3,83 (с, 6Н), 7,31 (с, 2Н, ім.) (транс-ізомер); 3,75 (с, 3Н), 3,86 (с, 3Н), 7,13 (с, 13 1H, ім.), 7,23 (с, 2H, ім.) (транс-ізомер). Спектр С 23 62301 ЯМР (CDC13), , м.ч.: 37,9 (СН3), 123,2 (С -ім.), 2 167,1 (С ) (транс-ізомер); 33,7,37,3 (СН3), 4,5 2 120,6,122,4 (С -ім.), 167,1 (С ) (транс-ізомер). Приклад 16. Комплекс 1.3-біс(1-адамантил-4феніл-1.2,4-триазол-5-іліден-3-іл)бензену з хлоридом міді(І) (16). Суспензію 0.28 г (0,44 ммоль) біскарбену (16а) і 0,088 г (0,88 ммоль) хлориду міді(1) в 4 мл суміші тетрагідрофуран-ацетонітрил 1:1 перемішували протягом 2 год. Розчин випаровували у вакуумі та фільтрували через тонкий шар силікагелю в хлороформі. Після відгонки хлороформу вихід комплексу (16) 0,33 г (90 %). Т. пл. 268-270 °C. Знайдено, %: С 60,3, Н 5,0, С1 8,7, Сu15,3,N 9,9.C42H44N6Cl2Cu2. Обчислено, %: С 1 60,7, Н 5,3, С1 8,5, Сu 15,3, N 10,1. Спектр Н ЯМР (CDCl3), , м.ч.: 1,75 м (12Н, СН2,Ad), 2,25 м (СН, Ad), 2,38 м (12Н, СН2,Ad), 7,33 м, 7,52 м (СН 13 аром.). Спектр С ЯМР (CDCl3), , м.ч.: 29,0,35,2,43,0,60,8 (Ad), 125,2 (ipso4,5 Комп’ютерна верстка Л. Купенко 24 СN127,0,128,9,129,1,129,7,129,8,130,5,136,7 (С 3 аром.) 149,8 (триаз. C N), 176,5 (С:). Приклад 17. Методика проведення каталітичної реакції. До 1 ммоль субстрату додавали 10 мл 0.1 М розчину КОН в ізопропанолі та необхідну кількість каталізатора (див. табл.). Реакційну суміш кип'ятили, контролюючи хід реакції методом тонкошарової хроматографії (ТШХ). Аналіз вихідної сполуки та продукту відновлення під час реакції проводили наступним чином. Ізопропанол випаровували до початку кристалізації залишку вихідної сполуки (об'єм приблизно 3 мл), охолоджували й осад відфільтровували. Через вельми низьку розчинність кетону в ізопропанольному розчині лугу в маточному розчині не залишається більше кетону (контроль ТШХ). Маточний розчин розводили 12 мл води й утворений осад карбінолу відфільтровували, сушили, виміряли вихід продукту. Для очистки до спектральної чистоти карбінол перекристалізовували з ізопропанолу. Підписне Тираж 23 прим. Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюCarbene complexes of transition metals
Автори англійськоюKorotkikh Mykola Ivanovych, Saberov Vahiz Shamilovych, Kyselov Artem Viktorovych, Marichev Kostiantyn Oleksandrovych, Hlyniana Natalia Valeriivna, Shvaika Oleksii Pavlovych
Назва патенту російськоюКарбеновые комплексы переходных металлов
Автори російськоюКоротких Николай Иванович, Саберов Вагиз Шамильевич, Киселев Артем Викторович, Маричев Константин Александрович, Глиняна Наталия Валериевна, Швайка Алексей Павлович
МПК / Мітки
МПК: C07D 233/00, C07D 277/00, C07D 239/00, C07D 249/00, C07C 209/00, C07D 235/00
Мітки: металів, карбенові, перехідних, комплекси
Код посилання
<a href="https://ua.patents.su/12-62301-karbenovi-kompleksi-perekhidnikh-metaliv.html" target="_blank" rel="follow" title="База патентів України">Карбенові комплекси перехідних металів</a>
Спосіб одержання сорбенту для вилучення перехідних металів з розчинів

Номер патенту: 51520
Опубліковано: 26.07.2010
Автори: Андріанова Олена Борисівна, Трохимчук Анатолій Костянтинович, Ульберг Зоя Рудольфівна, Лещенко Віталій Миколайович
МПК: B01J 45/00, B01J 20/10
Мітки: перехідних, спосіб, розчинів, вилучення, одержання, металів, сорбенту
Формула / Реферат:
1. Спосіб одержання сорбенту для вилучення іонів перехідних металів з розчинів взаємодією силікагелю і зшивального агента з наступною обробкою отриманого модифікованого силікагелю комплексоутворюючим агентом, який відрізняється тим, що як зшивальний агент використовують полігексаметиленгуанідин (ПГМГ) загальної формули:,де Х - аніон неорганічної кислоти, n =...
Хелати перехідних металів з олігопероксидними лігандами для низькотемпературного ініціювання радикальних реакцій

Номер патенту: 17450
Опубліковано: 06.05.1997
Автори: Мітіна Наталія Євгенівна, Воронов Станіслав Андрійович, Заіченко Олександр Сергійович
МПК: C08F 4/50, C08F 4/42, C08F 4/80, C08F 4/36, C08F 4/78
Мітки: перехідних, лігандами, низькотемпературного, хелати, ініціювання, олігопероксидними, реакцій, радикальних, металів
Формула / Реферат:
Хелати перехідних металів з олігопероксидними лігандами для низькотемпературного ініціювання реакцій загальної формулиL' - гетерофункціональні олігопероксидні ліганди структури:
Спосіб виготовлення порошків феромагнітних сплавів рідкісноземельних металів, перехідних металів та бору і пристрій для здійснення способу

Номер патенту: 51229
Опубліковано: 15.11.2002
Автори: Басараба Юрій Борисович, Булик Ігор Іванович, Путілов Юрій Григорович, Панасюк Володимир Васильович
МПК: B22F 9/00, H01F 7/02, H01F 7/00
Мітки: пристрій, здійснення, бору, феромагнітних, виготовлення, металів, спосіб, способу, порошків, сплавів, перехідних, рідкісноземельних
Формула / Реферат:
1. Пристрій очищення питної води, розміщений у водонапірній башті, що включає бак з дахом і дном та ствол, в якому знаходиться контактне завантаження, розташоване у приймальній обичайці з перфорацією у нижній частині, перегородку, крізь яку проходить трубопровід подачі вихідної води, оснащений аератором, і під якою знаходиться плаваюче фільтруюче завантаження з дренажно-розподільчою системою, який відрізняється тим, що плаваюче фільтруюче...
Спосіб одержання хелатів перехідних металів з олігопероксидними лігандами

Номер патенту: 85344
Опубліковано: 12.01.2009
Автори: Заіченко Олександр Сергійович, Зелікова Ольга Володимирівна, Мітіна Наталія Євгенівна, Волошиновський Анатолій Степанович
МПК: C08F 8/42, C09K 11/77
Мітки: олігопероксидними, перехідних, спосіб, лігандами, металів, хелатів, одержання
Формула / Реферат:
Спосіб одержання хелатів з олігопероксидними лігандами, що включає взаємодію гетерофункціонального олігопероксиду з сіллю перехідного металу, який відрізняється тим, що як гетерофункціональний олігопероксид використовують ліганд формули: , (I)де n = 22,16-54,09 %, m = 12,78-21,50 %, l = 11,93-32,50 %,або
Спосіб одержання пористих координаційних полімерів на основі триядерних карбоксилатних комплексів перехідних металів і органічних лігандів, що містять залишок піридину

Номер патенту: 61912
Опубліковано: 10.08.2011
Автори: Литвиненко Антон Сергійович, Павліщук Віталій Валентинович, Яремов Павел Степанович, Колотілов Сергій Володимирович, Ільїн Володимир Георгійович, Шпаковський Ігор Валентинович, Полунін Руслан Анатолійович, Гавриленко Костянтин Сергійович, Швець Олексій Васильович
МПК: B01D 53/44
Мітки: пористих, лігандів, перехідних, піридину, металів, триядерних, полімерів, координаційних, комплексів, спосіб, основі, містять, карбоксилатних, залишок, одержання, органічних
Формула / Реферат:
Спосіб одержання пористих координаційних полімерів на основі триядерних карбоксилатних комплексів перехідних металів і органічних лігандів, що містять залишок піридину, який відрізняється тим, що пористий координаційний полімер являє собою полімер складу,де являють собою однакові або різні метали у...
Попередній патент: Спосіб дії на призабійну зону свердловини
Наступний патент: Сполука з антигіпоксичною й антиоксидантною дією
Випадковий патент: Електронагрівач