Спосіб одержання пористих координаційних полімерів на основі триядерних карбоксилатних комплексів перехідних металів і органічних лігандів, що містять залишок піридину
Номер патенту: 61912
Опубліковано: 10.08.2011
Автори: Шпаковський Ігор Валентинович, Ільїн Володимир Георгійович, Полунін Руслан Анатолійович, Гавриленко Костянтин Сергійович, Яремов Павел Степанович, Колотілов Сергій Володимирович, Литвиненко Антон Сергійович, Павліщук Віталій Валентинович, Швець Олексій Васильович





Формула / Реферат
Спосіб одержання пористих координаційних полімерів на основі триядерних карбоксилатних комплексів перехідних металів і органічних лігандів, що містять залишок піридину, який відрізняється тим, що пористий координаційний полімер являє собою полімер складу![]() ,
,
де ![]() являють собою однакові або різні метали у ступенях окиснення
являють собою однакові або різні метали у ступенях окиснення ![]() або
або ![]() ;
;
![]() - аліфатичний залишок;
- аліфатичний залишок;
![]() - ліганд, що містить щонайменше 2 піридинових фрагменти;
- ліганд, що містить щонайменше 2 піридинових фрагменти;
![]() - будь-який аніон із зарядом -1;
- будь-який аніон із зарядом -1;
![]() і інші значення в залежності від кількості піридинових груп в складі
і інші значення в залежності від кількості піридинових груп в складі ![]() ;
;
![]() або 1, в залежності від зарядів іонів металів
або 1, в залежності від зарядів іонів металів ![]() ,
,
в якому піддають взаємодії триядерні ![]() -оксоцентровані карбоксилатні комплекси перехідних металів складу
-оксоцентровані карбоксилатні комплекси перехідних металів складу ![]() ,
,
де L1 - монодентатний ліганд,
з органічними молекулами L2, що містять два або більше піридинових замісників,
причому для синтезу координаційного полімеру використовують поліядерні комплекси складу ![]() , структура яких зберігається в ході реакції, причому згадана структура входить у незмінному вигляді у склад пористого координаційного полімеру із заміщенням монодентатних лігандів
, структура яких зберігається в ході реакції, причому згадана структура входить у незмінному вигляді у склад пористого координаційного полімеру із заміщенням монодентатних лігандів ![]() у складі
у складі ![]() піридиновими фрагментами лігандів
піридиновими фрагментами лігандів ![]() .
.
Текст
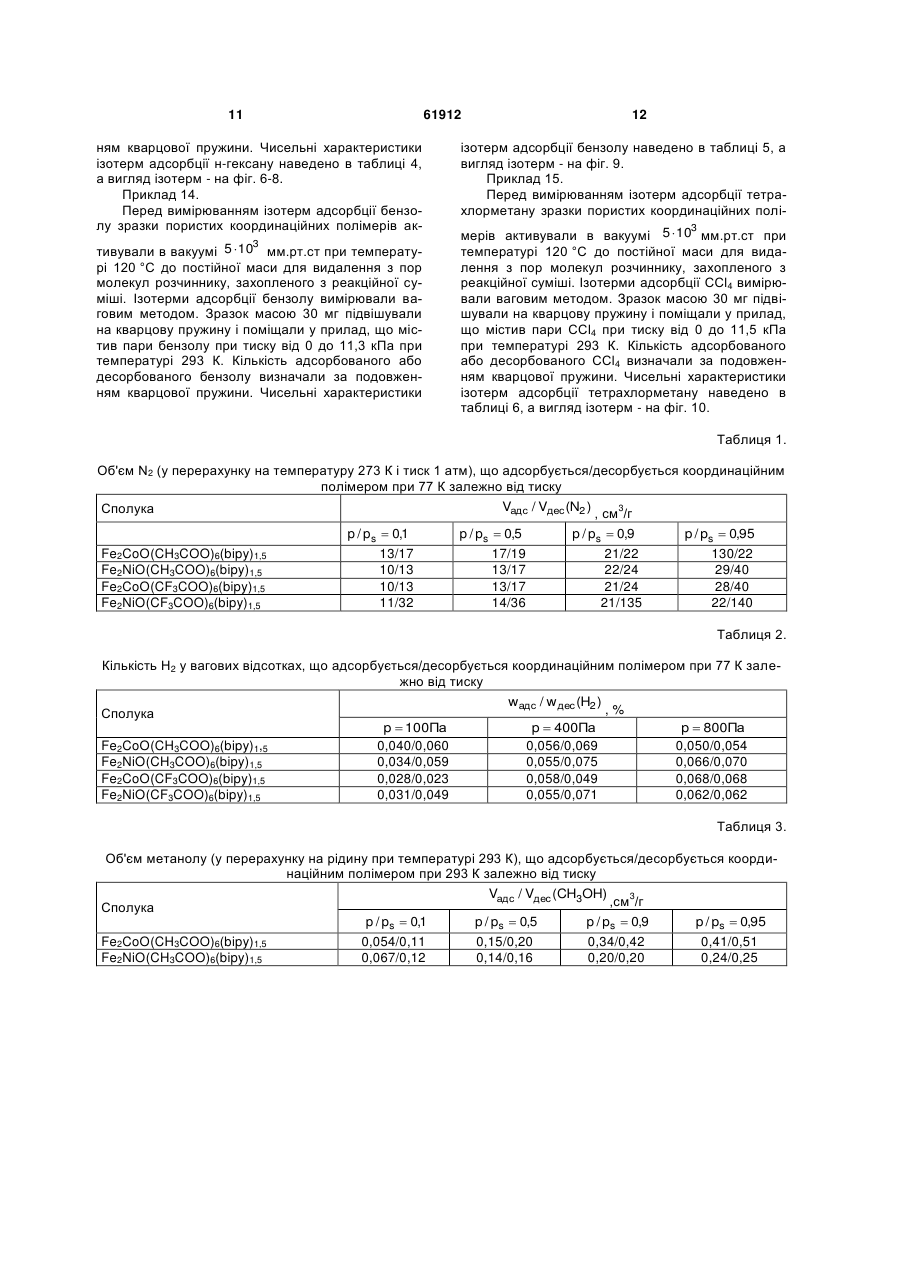
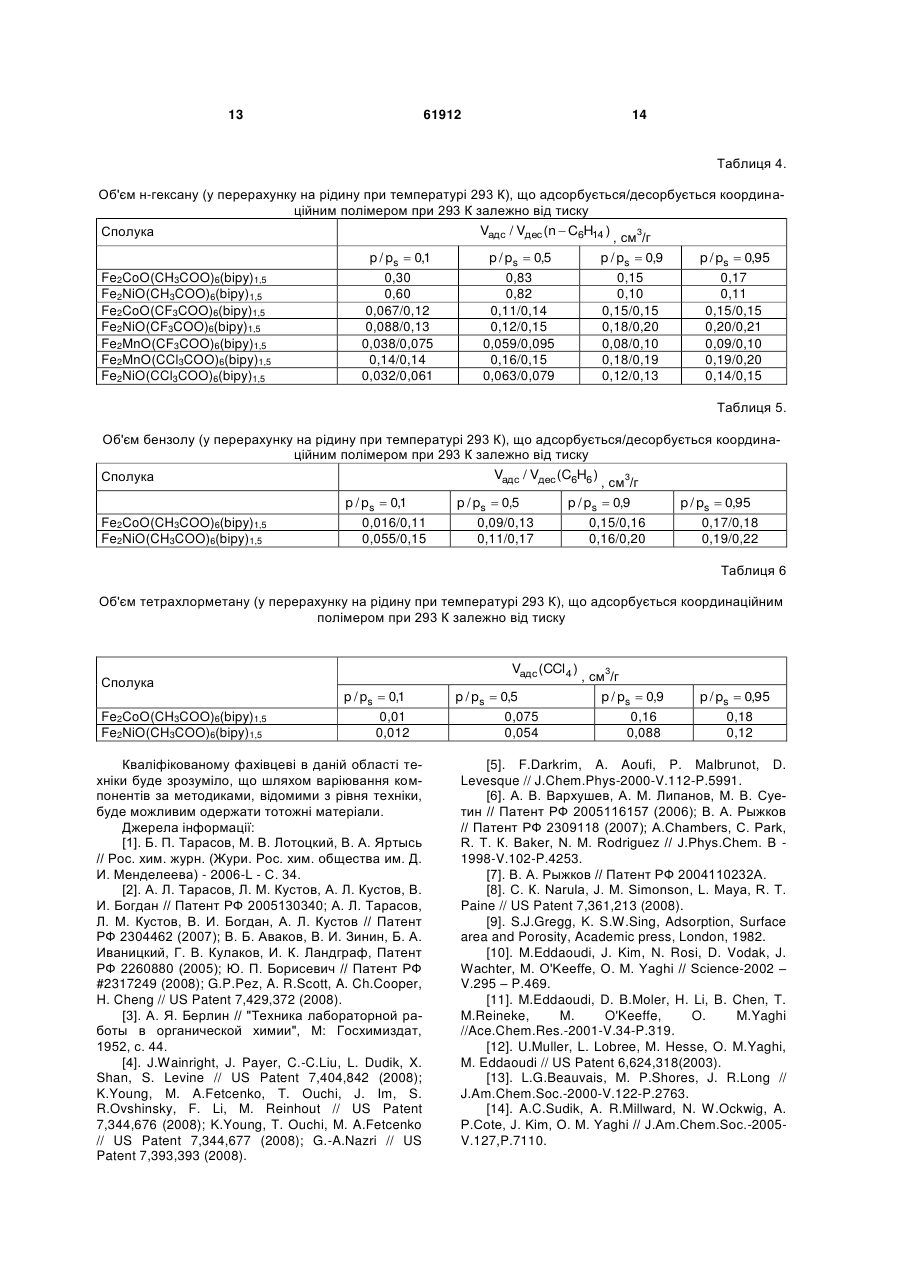
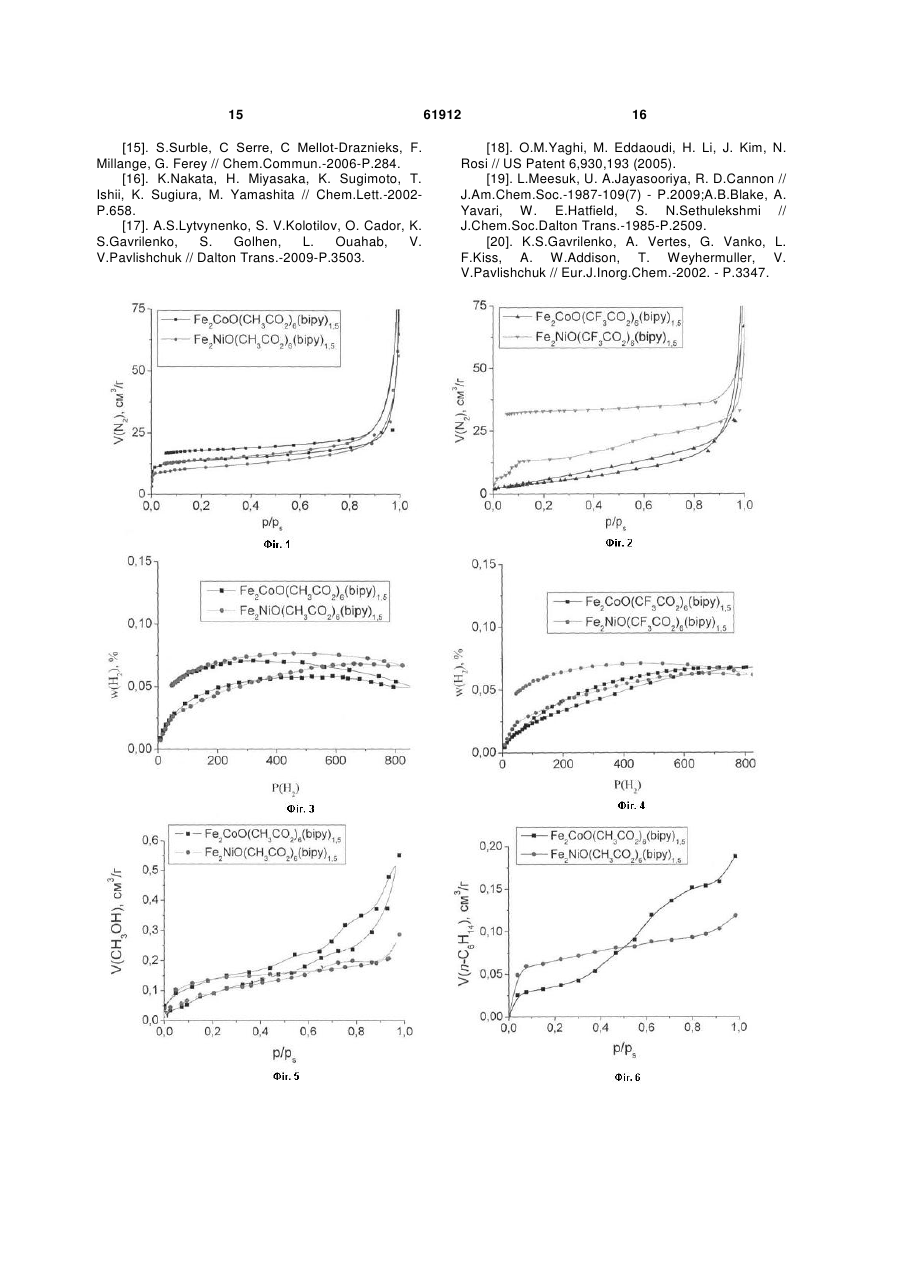
Спосіб одержання пористих координаційних полімерів на основі триядерних карбоксилатних комплексів перехідних металів і органічних лігандів, що містять залишок піридину, який відрізняється тим, що пористий координаційний полімер являє собою полімер складу MM' M' ' O(RCO 2 )6 (L2)x ( A )n , 3 на неорганічних оборотних реакціях, які йдуть з виділенням водню, таких, як розклад водяної пари під дією заліза ("вивільнення") з подальшою регенерацією заліза воднем ("зарядка", "накопичення"), або аналогічна система, робота якої основана на використанні алюмінію та ряду інших металів [2]. Проведення таких процесів пов'язане зі значними енергетичними затратами (як на першій стадії, так і для регенерації активного компоненту), а високотемпературне виробництво водню пов'язане зі значними ризиками при найдрібніших порушеннях у системі [2]. У процесах вилучення і розділення субстратів альтернативою використанню сорбентів може бути хімічне зв'язування заданих субстратів, наприклад, хімічне зв'язування води оксидом фосфору(У) для видалення води, розчиненої в органічних рідинах (наприклад, в бензолі) [3]. Недоліком такого підходу часто є необоротність використання речовини, що використовується для хімічного зв'язування, а також обмежені можливості з модифікації такої речовини для селективного зв'язування заданих субстратів. Критерії, за якими визначається ефективність сорбенту, залежать від конкретної мети його створення, і, найчастіше, включають сорбційну ємність (тобто максимальну масу субстрату, що може бути адсорбована одиницею маси або об'єму сорбенту), швидкість адсорбції і десорбції субстрату, селективність адсорбції (тобто можливість сорбції заданих речовин у присутності інших), простоту (технологічність) використання (тобто відсутність потреби у складному обладнанні і спеціальному навчанні персоналу). Сорбційна ємність сорбенту залежить від площі поверхні та об'єму пор сорбенту, а також його здатності утримувати в порах молекули субстрату (тобто від енергії адсорбції субстрату). Сорбційні характеристики пористих сорбентів можуть регулюватися шляхом варіювання структури сорбенту. Здатність утримувати в порах певні молекули може бути поліпшена (1) шляхом зміни геометричних характеристик пор, що оптимізує їх геометрію для кращої взаємодії з адсорбованими молекулами; (2) шляхом формування певних активних центрів в структурі матеріалу, які завдяки своїй електронній будові матимуть значну енергію взаємодії з адсорбатом. Перевагами сорбентів на основі пористих матеріалів, у порівнянні з сорбентами на основі дисперсних частинок [4], є велика площа поверхні поряд зі стабільністю матеріалу (немає проблеми агрегування частинок сорбенту), а також можливість модифікації сорбційних характеристик пор шляхом вибору певного способу створення пористої структури сорбенту. Наразі використовуються сорбенти на основі речовин, що належать до різних класів, зокрема: кремнеземні сорбенти і цеоліти [5], активоване вугілля [6], вуглецеві нанотрубки [7], нанотрубки невуглецевої природи [8] тощо. Промислові зразки цеолітів мають площу по2 верхні порядку 400-700 м /г, а площа поверхні деяких зразків кремнеземних сорбентів може досягати 1300 м /г (наприклад, МСМ-48). Вуглецеві 2 матеріали мають площу поверхні до 3000 м /г (ти 61912 4 2 пові зразки – до 1000 м /г), але поряд зі значною масовою сорбційною ємністю (відношення кількості речовини або маси адсорбату до одиниці маси сорбенту), об'ємна сорбційна ємність таких матеріалів (відношення кількості речовини або маси адсорбату до одиниці об'єму сорбенту) є досить невисокою через низьку густину вуглецевого матеріалу (оскільки густина найщільнішого вуглецевого матеріалу з шаруватою структурою - графіту 3 знаходиться в межах 2,09-2,23 г/см , а насипна густина вуглецевих пористих матеріалів є набагато меншою) [9]. Пористі координаційні полімери (металоорганічні каркасні сполуки) є речовинами, які побудовано шляхом зв'язування металвмісних структурних елементів (комплексних сполук) органічними містковими лігандами [10]. У якості структурних елементів такі сполуки можуть містити моно- або поліядерні комплекси перехідних металів [11]. У якості місткових лігандів можуть використовуватися аніони органічних ароматичних дикарбонових кислот (наприклад, 1,4-бензолдикарбонової кислоти), діаміни з конформаційно-жорсткою структурою (наприклад, 4,4'-біпіридин) тощо [11]. Перевагами пористих координаційних полімерів перед сорбентами інших класів є можливість безпосереднього введення іона перехідного металу в склад сполуки (що надає можливості використання таких сполук як каталізаторів [12], активних елементів сенсорів [13] тощо), а також можливості варіювання сорбційних характеристик пористого матеріалу шляхом вибору певної комбінації іонів металів і органічних місткових лігандів. Серед пористих координаційних полімерів, що містять поліядерні структурні блоки, переважна кількість таких сполук утворюється з неорганічних вихідних речовин, будова яких не відповідає будові поліядерного структурного елементу у складі координаційного полімеру. Наприклад, координаційні полімери, що містять у своєму складі триядерні карбоксилатні комплекси заліза(III) і описуються формулою [NH2(CH3)2]8[Fe12O4(SO4)12(O2C-R-CO2)6 x(py)] або [NH2(CH3)2]8[Fe12O4(SO4)12(R(CO2)3)4 x(ру)], де R - ароматичний радикал, утворюються з сульфату заліза(III) [14]. Недоліком такого підходу є неможливість передбачення складу поліядерного структурного елементу в координаційному полімері, який утворюється в результаті реакції (цей недолік не стосується відтворення вже досліджених процесів і синтезу вже досліджених пористих координаційних полімерів за відомими методиками). В свою чергу, неможливість передбачення складу і будови поліядерного "будівельного блоку" у складі координаційного полімеру, що утворюється в реакційній суміші, обмежує можливості створення сполук з наперед заданими властивостями, що залежать від будови таких поліядерних "будівельних блоків". На відміну від наведеного вище підходу, отримання пористих координаційних полімерів з поліядерних комплексів зі збереженням їх будови може дозволити певною мірою передбачувати топологію кристалічної ґратки і властивості таких полімерів, оскільки вони певною мірою визначати 5 муться топологією і властивостями поліядерних структурних елементів. Єдиним прикладом збереження триядерного остову при утворенні пористого координаційного полімеру є реакція утворення сполук складу [Fe3O(O2C-R-CO2)3 n(H2O)]m, де R- аліфатичний або ароматичний радикал, виходячи з триядерних вихідних комплексів [Fе3О(СН3СО2)6(Н2О)3](СН3СО2) [15]. Проте і в цьому разі, беручи до уваги, що утворення координаційного полімеру неминуче має пройти через стадію (або ряд послідовних реакцій) заміни усіх шести карбоксильних груп у складі + [Fе3О(СН3СО2)6(Н2О)3] , а саме такі карбоксильні 3+ групи зв'язують іони Fe в триядерний комплекс, не можна вважати доведеним припущення про збереження триядерного остову в ході реакції на відміну від його розкладу до моноядерних сполук з подальшою самозборкою ("відновленням"). Відомі приклади утворення координаційних полімерів з поліядерних комплексів зі збереженням будови поліядерного остову, наприклад, утворення полімеру складу [Мn6O2((СН3)3ССO2)10((СН3)3ССO2Н)2(bipy)] (bіру = 4,4'-біпіридин) виходячи з шестиядерного комплексу [Мn6O2((СН3)3ССO2)10((СН3)зCCO2Н)4] [16], але такі сполуки не є пористими і нездатні сорбувати субстрати. Варто відзначити, що використання поліядерних комплексів як вихідних речовин в синтезі координаційних полімерів не завжди веде до утворення сполук, що містять бажані поліядерні структурні елементи, і реакції часто супроводжуються розпадом або перегрупуванням вихідних сполук. Так, наприклад, реакція триядерного ацетату заліза(III)марганцю(II) складу Fe2MnO(CH3CO2)6 з мурашиною кислотою призвела до утворення координаційного полімеру складу Fе3О(СН3СО2)6Мn(НСО2)3, в якому є триядерний + фрагмент Fe3O(CH3CO2)6 , що утворився внаслідок деструкції Fe2MnO(CH3CO2)6 з подальшою + самозборкою іншого катіону - Fe3O(CH3CO2)6 [17]. Таким чином, отримання координаційного полімеру виходячи з певного поліядерного комплексу зі збереженням будови поліядерного остову не є тривіальною задачею. Прототипом даної корисної моделі є спосіб синтезу ряду пористих координаційних полімерів складу M4O(L)3 (M- метал, зокрема, перехідний 2+ метал, наприклад, Zn , L - діаніон дикарбонової кислоти, наприклад, 1,4-бензолдикарбонової), здатних сорбувати різні субстрати, зокрема, метан [18]. Запропонований спосіб синтезу базується на утворенні ізоретикулярних пористих металоорганічних каркасів, побудованих шляхом з'єднання октаедричних кластерів М4О(СО2)6 карбоксилатними містками з утворенням тривимірних кристалічних структур. Означені сполуки можуть бути функціоналізовані шляхом використання місткових молекул, що містять замісники (такі, як Br, NH2, ОС3Н7, ОС5Н11 -С2Н4- і -С4Н4- тощо); розмір пор можна варіювати шляхом заміни залишку С6Н4 на біфенільні, тетрагідропіренові, піренові та терфенільні фрагменти тощо. Синтез таких матеріалів полягає у розчиненні солі цинку (Zn(NO3)2) і ліганду (дика 61912 6 рбонової кислоти) у відповідному розчиннику (наприклад, диметилформаміді), після чого у деяких випадках додається основа. Густина отриманих 3 сполук лежить в межах від 1 до 0,2 г/см . Запропонований спосіб передбачає самозбірку тетраядерних комплексів М4О(СО2)6 (М - метал, CO2 - карбоксильна група зі складу карбонової кислоти) у реакційній суміші. Недоліком прототипу є придатність описаного способу для синтезу лише координаціних поліме6+ рів, що містять структурний блок Zn4O , який утворюється в реакційній суміші шляхом самозбо2+ рки з іонів Zn . Описаний в прототипі спосіб є непридатним для одержання координаційних полімерів, побудованих на базі інших металвмісних структурних елементів, зокрема, але не обмежуючись цим, комплекси парамагнітних іонів 3d металів. Запропонована нами корисна модель описує спосіб синтезу пористих координаційних полімерів шляхом взаємодії триядерних 3-оксоцентрованих карбоксилатних комплексів перехідних металів (Fe, Co, Ni, Mn, тощо) складу MM'M''O(RCO2)6(L1)3(A)n, де М, М", М" - однакові або різні метали у ступенях окиснення +2 або +3 (Fe, Co, Ni, Mn, тощо), R - аліфатичний залишок (Н, СН3,CF3, ССl3 тощо), L1 - монодентатний ліганд (Н2О, CH3CN, піридин тощо), А - будь-який аніон з зарядом -1,n=0 або 1, з молекулами L2, що містять два або більше піридинових замісників (4,4'діпіридин, 1,2-біс(4-піридил)етилен тощо). Запропонований нами спосіб ґрунтується на заміщенні лігандів L1 в MM'M'O(RCO2)6(L1)3(A)n атомами азоту піридинвмісного ліганду, внаслідок чого кожен піридинвмісний ліганд зв'язує як мінімум два n+ триядерних блоки MM'M'O(RCO2)6 , а кожен триядерний блок зв'язується трьома піридинвмісними лігандами, атоми азоту яких займають певні позиції в координаційних сферах іонів М, М', М", які у вихідних сполуках було зайнято донорними атомами молекул L1. Одержані координаційні полімери мають склад MM'M'O(RCO2)6(L2)x(A)n, (якщо L2 містить два піридинових фрагменти, тоді х = 1,5, а п залежить від зарядів іонів М, М', М"), мають полімерну будову і здатні сорбувати субстрати різної природи (азот, водень, спирти, алкани, ароматичні молекули, галогенвмісні молекули тощо). Запропонований нами спосіб відрізняється від способу, описаного у прототипі, тим, що для синтезу координаційного полімеру використовуються готові поліядерні комплекси, структура яких зберігається в ході реакції і які входять у незмінному вигляді у склад пористого координаційного полімеру. В даному контексті вираз "у незмінному вигляді" означає, що в поліядерній частці n+ MM'M'O(RCO2)6(L1)3 зберігається загальна кількість іонів металів, зберігаються усі місткові групи, зберігається топологія поліядерної частинки (спосіб зв'язування іонів металів і їх взаємне розташування), але при цьому монодентатні ліганди L1 заміщуються піридиновими фрагментами лігандів L2. Перевагою запропонованого нами способу у порівнянні з прототипом є можливість одержання координаційних полімерів із заданих поліядерних 7 комплексів, що певною мірою може дозволити регулювати топологію координаційного полімеру і, в більшому ступені, його властивості, обумовлені наявністю такого поліядерного комплексу в якості структурного елементу. Запропонований спосіб дозволяє використовувати будь-які поліядерні комплекси, що мають вакантні координаційні місця в координаційних сферах іонів металів або містять ліганди, що можуть бути заміщені іншими (містковими) лігандами. Єдиною додатковою вимогою до поліядерних комплексів, які використовуються для синтезу пористих координаційних полімерів, є стабільність в розчині протягом часу, потрібного для кристалізації пористого координаційного полімеру. Характеристики пористої структури (розмір пор, площа поверхні, об'єм пор) залежать від топології поліядерного комплексу і просторової будови місткового ліганду. Необхідною умовою утворення двовимірних структур є наявність щонайменше трьох центрів зв'язування в поліядерному комплексі або у ліганді (три вакантних координаційних місця або три донорних атоми, відповідно). Координаційні полімери, одержані за запропонованим нами способом, можуть розглядатися не лише як сорбенти, а і як основа для створення мультифункціональних матеріалів, що поєднують здатність сорбувати певні субстрати з іншими властивостями (магнітними, спектральними тощо), існування яких обумовлене наявністю металвмісних структурних елементів. Корисну модель було реалізовано на прикладі координаційних полімерів, що відрізняються складом триядерного карбоксилатного комплексу (іони металів, замісники в карбоксилатних лігандах, будова піридинвмісних молекул L2), а також на прикладі сорбції різних субстратів (азот, водень, метанол, н-гексан, бензол і ССl4). Перелік фігур та креслень: На фіг. 1 наведено ізотерми сорбції азоту координаційними полімерами Fе2СоО(СН3СО2)6(bіру)1,5 і Fe2NiO(CH3CO2)6(bipy)1,5 (bipy=4,4'-біпіридин) при 77 К. На фіг. 2 наведено ізотерми сорбції азоту координаційними полімерами Fe2CoO(CF3CO2)6(bipy)1,5 і Fe2NiO(CF3CO2)6(bipy)1,5 (bipy=4,4'-біпіридин) при 77 К. На фіг. З наведено ізотерми сорбції водню координаційними полімерами Fe2CoO(CH3CO2)6(bipy)1,5 і Fe2NiO(CH3CO2)6(bipy)1,5 при 77 К. На фіг. 4 наведено ізотерми сорбції водню координаційними полімерами Fe2CoO(CF3CO2)6(bipy)1,5 і Fe2NiO(CF3CO2)6(bipy)1,5 при 77 К. На фіг. 5 наведено ізотерми сорбції метанолу координаційними полімерами Fe2Co(CH3CO2)6(bipy)1,5 Fe2Ni(CH3CO2)6(bipy)1,5 при 293 К. На фіг. 6 наведено ізотерми сорбції гексану координаційними полімерами Fe2Co(CH3CO2)6(bipy)1,5 Fe2Ni(CH3CO2)6(bipy)1,5 при 293 К. 61912 8 На фіг. 7 наведено ізотерми сорбції гексану координаційними полімерами Fe2CoO(CF3CO2)6(bipy)1,5,Fe2NiO(CF3CO2)6(bipy )1,5,Fe2MnO(CF3CO2)6(bipy)1,5 при 293 К. На фіг. 8 наведено ізотерми сорбції гексану координаційними полімерами Fe2NiO(CCl3CO2)6(bipy)1,5,Fe2MnO(CCl3CO2)6(bi py)1,5 при 293 К. На фіг. 9 наведено ізотерми сорбції бензолу координаційними полімерами Fe2Co(CH3CO2)6(bipy)1,5,Fe2Ni(CH3CO2)6(bipy)1,5 при 293 К. На фіг. 10 наведено ізотерми сорбції ССІ4 координаційними полімерами Fe2Co(CH3CO2)6(bipy)1,5,Fe2Ni(CH3CO2)6(bipy)1,5 при 293 К. Дана корисна модель підтверджується, але не обмежується наведеними нижче прикладами. Приклади ілюструють одержання вихідних триядерних комплексів (приклади 1-3), пористих координаційних полімерів з використанням 4,4'біпіридину, Мру (приклади 4-9), ізотерми сорбції азоту (приклад 11), ізотерми сорбції водню (приклад 12), ізотерми сорбції метанолу (приклад 13), ізотерми сорбції н-гексану (приклад 14), ізотерми сорбції бензолу (приклад 15), ізотерми сорбції тетрахлорметану (приклад 16). Триядерні комплекси II ІІІ [М Ре 2О(СН3COO)6(Н2О)3]-2Н2О (М = Ni, Co, Mn) були синтезовані за описаними методиками [19]. II III Триядерні комплекси [M Fe 2O(CF3COO)6(H2O)3]2H2O (М = Ni, Co, Mn) було синтезовано за описаними методиками [20] з урахуванням їх модифікування [17]. Приклад 1. Синтез Fe2CoO(CCl3COO)6(H2O)3.9,9 г (61 ммоль) ССl3СООН розчинили в 6,6 мл води. 1 г (1,7 ммоль) Fе2СоО(СН3СОО)6(Н2О)3 розчинили в одержаному розчині. Суміш, що утворилася, упарили при нагріванні до видалення приблизно половини об'єму розчинника. При охолодженні випали чорні кристали Fe2CoO(CCl3COO)6(H2O)3, які було відфільтровано та висушено на повітрі. Вихід 1,45 г (70 %). Дані аналізу: теор. для Fe2MnO(CCl3COO)6(H2O)3H2O/eкcп.: С, 11,7/12,3; Н, 0,65/0,52. Приклад 2. Синтез Fe2NiO(CCl3COO)6(H2O)3.9,9 г (61 ммоль) ССl3СООН розчинили в 6,6 мл води. 1 г (1,7 ммоль) Fe2NiO(CH3COO)6(H2O)3 розчинили в одержаному розчині. Суміш, що утворилася, упарили при нагріванні до видалення приблизно половини об'єму розчинника. При охолодженні випали чорні кристали Fe2NiO(CCl3COO)6(H2O)3, які було відфільтровано та висушено на повітрі. Вихід 1,45 г (70 %). Дані аналізу: теор. для Fе2NiО(СС13СОО)6(Н2О)2(ССl3СООН)/експ.: С, 12,3/12,4; Н, 0,37/0,67. Приклад 3. Синтез Fe2MnO(CCl3COO)6(H2O)3.9,9 г (61 ммоль) СС13СООН розчинили в 6,6 мл води. 1 г (1,7 ммоль) Ре2МnО(СН3COO)6(Н2О)3 розчинили в одержаному розчині. Результуючу суміш упарили при нагріванні до видалення приблизно половини об'єму розчинника. При охолодженні випали чорні 9 кристали Fе2МnО(ССl3COO)6(Н2О)з, які було відфільтровано та висушено на повітрі. Вихід 1,45 г (70 %). Дані аналізу: теор. дляFе2МnО(ССl3COO)6(Н2О)2(ССl3СООН)/експ.: С, 12,4/12,7; Н, 0,37/0,67. Приклад 4. Синтез Fe2NiO(CH3COO)6(bipy)1.5:300 мг (0,5 ммоль) Fе2NiО(СН3COO)6(Н2О)3 розчинили в 5 мл ДМФ, до розчину одразу ж після розчинення додали 10 мл CH3CN та 0,5 мл СН3СООН. До одержаного розчину при перемішуванні додали розчин bіру (118 мг, 0,75 ммоль) в 20 мл CH3CN. Через кілька годин осад відфільтрували, промили ацетонітрилом та висушили на повітрі. Вихід 230 мг (60 %). Дані аналізу, теор. для Fe2NiO(CH3COO)6(bipy)1.5-2H2O-CH3CN /експ.: С, 40,90/40,70; Н, 4,35/4,39;N, 6,58/6,92. Приклад 5. Синтез Fе2СоО(CH3COO)6(bіру)1.5:300 мг (0,5 ммоль) Ре2СоО(СН3СОО)6(Н2О)3 розчинили в 5 мл ДМФ, до розчину одразу ж після розчинення додали 10 мл CH3CN та 0,5 мл СН3СООН. До одержаного розчину при перемішуванні додали розчин bіру (118 мг, 0,75 ммоль) в 20 мл CH3CN. Через кілька годин осад відфільтрували, промили ацетонітрилом та висушили на повітрі. Вихід 230 мг (60 %). Дані аналізу, теор. для Fe2CoO(CH3COO)6(bipy),.5-2H2O-l, 5CH3CN /експ.: С, 41,30/41,40; Н, 4,41/4,43;N, 7,22/7,09. Приклад 6. Синтез Fe2CoO(CF3COO)6(bipy)1.5:460 мг (0,5 ммоль) Fe2CoO(CF3COO)6(H2O)3 розчинили в 10 мл ДМФ та 5 мл CH3CN. До одержаного розчину при перемішуванні додали розчин bіру (118 мг, 0,75 ммоль) в 20 мл CH3CN. Через кілька годин осад відфільтрували, промили ацетонітрилом та висушили на повітрі. Вихід 186 мг (34 %). Дані аналізу, теор. для Fe2CoO(CF3COO)6(bipy)1.52H2O /експ.: С, 28,56/29,08; Н, 1,41/1,65;N, 3,70/3,57. Приклад 7. Синтез Fe2NiO(CF3COO)6(bipy)1.5:460 мг (0,5 ммоль) Fe2CoO(CF3COO)6(H2O)3 розчинили в 10 мл ДМФ та 5 мл CH3CN. До одержаного розчину при перемішуванні додали розчин bipy (118 мг, 0,75 ммоль) в 20 мл CH3CN. Через кілька годин осад відфільтрували, промили ацетонітрилом та висушили на повітрі. Вихід 312 мг (57 %). Дані аналізу, теор. для Fe2NiO(CF3COO)6(bipy)1.52H2O /експ.: С, 28,56/28,60; Н, 1,41/1,64;N, 3,70/3,62. Приклад 8. Синтез Fe2NiO(CCl3COO)6(bipy)1,53H2O CH3CN:500 мг (0,6 ммоль) Fе2NiО(ССl3СОО)6(Н2О)3 та 100 мг (0,6 ммоль) ССl3СООН було розчинено в 10 мл ацетонітрилу та додано 200 мг (1,3 ммоль) 4,4'-біпіридилу. Одразу після додавання випав зелено-коричневий аморфний осад, який було відфільтровано, промито ацетонітрилом та висушено на повітрі. Вихід 540 мг (60 %). Дані аналізу: теор. для Fe2NiO(CCl3COO)6(bipy)1,5(H2O)3(CH3CN)/ експ.: С, 23,3/22,9; Н, 1,41/1,65;N, 3,8/4,1. Приклад 9. Синтез Fe2MnO(CCl3COO)6(bipy)1,5 CH3CN:500 мг (0,6 ммоль) Fe2MnO(CCl3COO)6(H2O)3 та 100 мг (0,6 ммоль) ССl3СООН було розчинено в 10 мл 61912 10 ацетонітрилу та додано 200 мг (1,3 ммоль) 4,4'біпіридилу. Одразу після додавання випав зеленокоричневий аморфний осад, який було відфільтровано, промито ацетонітрилом та висушено на повітрі. Вихід 520 мг (60 %). Дані аналізу: теор. для Fe2MnO(CCl3COO)6(bipy)1,5 CH3CN/ експ.: С, 24,3/24,1; Н, 1,05/1,42;N, 3,9/3,7. Приклад 10. Перед вимірюванням ізотерми адсорбції азоту зразки пористих координаційних полімерів активу3 вали в вакуумі 5-10- мм.рт.ст. при температурі 120 °C до постійної маси для видалення з пор молекул розчинника, захопленого з реакційної суміші. Ізотерми адсорбції азоту вимірювали з використанням приладу Sorptomatic1990 при температурі 77 К. Кількість азоту, адсорбованого при певному тиску, вимірювали за зменшенням тиску у вимірювальній посудині після введення фіксованої кількості цього газу. Вимірювання проводили в діапазоні тисків від 0 до 101,3 кПа. Чисельні характеристики ізотерм адсорбції азоту наведено в таблиці 1, а вигляд ізотерм - на фіг. 1 і 2. Приклад 11. Перед вимірюванням ізотерми адсорбції водню зразки пористих координаційних полімерів ак3 тивували в вакуумі 5-10- мм.рт.ст при температурі 120 °C до постійної маси для видалення з пор молекул розчинника, захопленого з реакційної суміші. Ізотерми адсорбції водню вимірювали з використанням приладу Sorptomatic1990 при температурі 77 К. Кількість водню, адсорбованого при певному тиску, вимірювали за зменшенням тиску у вимірювальній посудині після введення фіксованої кількості цього газу. Вимірювання проводили в діапазоні тисків від 0 до 101,3 кПа. Чисельні характеристики ізотерм адсорбції водню наведено в таблиці 2, а вигляд ізотерм - на фіг. 3 і 4. Приклад 12. Перед вимірюванням ізотерм адсорбції метанолу зразки пористих координаційних полімерів 3 активували в вакуумі 5 10 мм.рт.ст при температурі 120 °C до постійної маси для видалення з пор молекул розчинника, захопленого з реакційної суміші. Ізотерми адсорбції метанолу вимірювали ваговим методом. Зразок масою 30 мг підвішували на кварцову пружину і поміщали у прилад, що містив пари метанолу при тиску від 0 до 12,5 кПа при температурі 293 К. Кількість адсорбованого або десорбованого метанолу визначали за подовженням кварцової пружини. Чисельні характеристики ізотерм адсорбції метанолу наведено в таблиці 3, а вигляд ізотерм - на фіг. 5. Приклад 13. Перед вимірюванням ізотерм адсорбції нгексану зразки пористих координаційних полімерів 3 активували в вакуумі 5 10 мм.рт.ст при температурі 120 °C до постійної маси для видалення з пор молекул розчинника, захопленого з реакційної суміші. Ізотерми адсорбції н-гексану вимірювали ваговим методом. Зразок масою 30 мг підвішували на кварцову пружину і поміщали у прилад, що містив пари н-гексану при тиску від 0 до 16 кПа при температурі 293 К. Кількість адсорбованого або десорбованого н-гексану визначали за подовжен 11 ням кварцової пружини. Чисельні характеристики ізотерм адсорбції н-гексану наведено в таблиці 4, а вигляд ізотерм - на фіг. 6-8. Приклад 14. Перед вимірюванням ізотерм адсорбції бензолу зразки пористих координаційних полімерів ак3 тивували в вакуумі 5 10 мм.рт.ст при температурі 120 °C до постійної маси для видалення з пор молекул розчиннику, захопленого з реакційної суміші. Ізотерми адсорбції бензолу вимірювали ваговим методом. Зразок масою 30 мг підвішували на кварцову пружину і поміщали у прилад, що містив пари бензолу при тиску від 0 до 11,3 кПа при температурі 293 К. Кількість адсорбованого або десорбованого бензолу визначали за подовженням кварцової пружини. Чисельні характеристики 61912 12 ізотерм адсорбції бензолу наведено в таблиці 5, а вигляд ізотерм - на фіг. 9. Приклад 15. Перед вимірюванням ізотерм адсорбції тетрахлорметану зразки пористих координаційних полі3 мерів активували в вакуумі 5 10 мм.рт.ст при температурі 120 °C до постійної маси для видалення з пор молекул розчиннику, захопленого з реакційної суміші. Ізотерми адсорбції ССl4 вимірювали ваговим методом. Зразок масою 30 мг підвішували на кварцову пружину і поміщали у прилад, що містив пари ССl4 при тиску від 0 до 11,5 кПа при температурі 293 К. Кількість адсорбованого або десорбованого ССl4 визначали за подовженням кварцової пружини. Чисельні характеристики ізотерм адсорбції тетрахлорметану наведено в таблиці 6, а вигляд ізотерм - на фіг. 10. Таблиця 1. Об'єм N2 (у перерахунку на температуру 273 К і тиск 1 атм), що адсорбується/десорбується координаційним полімером при 77 К залежно від тиску Vадс / Vдес (N2 ) 3 Сполука , см /г p / ps 0,1 p / ps 0,5 p / ps 0,9 p / ps 0,95 Fe2CoO(CH3COO)6(bipy)1,5 13/17 17/19 21/22 130/22 Fe2NiO(CH3COO)6(bipy)1,5 10/13 13/17 22/24 29/40 Fe2CoO(CF3COO)6(bipy)1,5 10/13 13/17 21/24 28/40 Fe2NiO(CF3COO)6(bipy)1,5 11/32 14/36 21/135 22/140 Таблиця 2. Кількість Н2 у вагових відсотках, що адсорбується/десорбується координаційним полімером при 77 К залежно від тиску wадс / w дес (H2 ) ,% Сполука p 100Па p 400Па p 800Па Fe2CoO(CH3COO)6(bipy)1,5 0,040/0,060 0,056/0,069 0,050/0,054 Fe2NiO(CH3COO)6(bipy)1,5 0,034/0,059 0,055/0,075 0,066/0,070 Fe2CoO(CF3COO)6(bipy)1,5 0,028/0,023 0,058/0,049 0,068/0,068 Fe2NiO(CF3COO)6(bipy)1,5 0,031/0,049 0,055/0,071 0,062/0,062 Таблиця 3. Об'єм метанолу (у перерахунку на рідину при температурі 293 К), що адсорбується/десорбується координаційним полімером при 293 К залежно від тиску Vадс / Vдес (CH3OH) 3 ,см /г Сполука p / ps 0,1 p / ps 0,5 p / ps 0,9 p / ps 0,95 Fe2CoO(CH3COO)6(bipy)1,5 0,054/0,11 0,15/0,20 0,34/0,42 0,41/0,51 Fe2NiO(CH3COO)6(bipy)1,5 0,067/0,12 0,14/0,16 0,20/0,20 0,24/0,25 13 61912 14 Таблиця 4. Об'єм н-гексану (у перерахунку на рідину при температурі 293 К), що адсорбується/десорбується координаційним полімером при 293 К залежно від тиску Vадс / Vдес (n C6H14 ) 3 Сполука , см /г p / ps 0,1 p / ps 0,5 p / ps 0,9 p / ps 0,95 Fe2CoO(CH3COO)6(bipy)1,5 0,30 0,83 0,15 0,17 Fe2NiO(CH3COO)6(bipy)1,5 0,60 0,82 0,10 0,11 Fe2CoO(CF3COO)6(bipy)1,5 0,067/0,12 0,11/0,14 0,15/0,15 0,15/0,15 Fe2NiO(CF3COO)6(bipy)1,5 0,088/0,13 0,12/0,15 0,18/0,20 0,20/0,21 Fe2MnO(CF3COO)6(bipy)1,5 0,038/0,075 0,059/0,095 0,08/0,10 0,09/0,10 Fe2MnO(CCl3COO)6(bipy)1,5 0,14/0,14 0,16/0,15 0,18/0,19 0,19/0,20 Fe2NiO(CCl3COO)6(bipy)1,5 0,032/0,061 0,063/0,079 0,12/0,13 0,14/0,15 Таблиця 5. Об'єм бензолу (у перерахунку на рідину при температурі 293 К), що адсорбується/десорбується координаційним полімером при 293 К залежно від тиску Vадс / Vдес (C6H6 ) 3 Сполука , см /г Fe2CoO(CH3COO)6(bipy)1,5 Fe2NiO(CH3COO)6(bipy)1,5 p / ps 0,1 0,016/0,11 0,055/0,15 p / ps 0,5 0,09/0,13 0,11/0,17 p / ps 0,9 0,15/0,16 0,16/0,20 p / ps 0,95 0,17/0,18 0,19/0,22 Таблиця 6 Об'єм тетрахлорметану (у перерахунку на рідину при температурі 293 К), що адсорбується координаційним полімером при 293 К залежно від тиску Сполука Fe2CoO(CH3COO)6(bipy)1,5 Fe2NiO(CH3COO)6(bipy)1,5 Vадс (CCl 4 ) p / ps 0,1 0,01 0,012 Кваліфікованому фахівцеві в даній області техніки буде зрозуміло, що шляхом варіювання компонентів за методиками, відомими з рівня техніки, буде можливим одержати тотожні матеріали. Джерела інформації: [1]. Б. П. Тарасов, М. В. Лотоцкий, В. А. Яртысь // Рос. хим. журн. (Жури. Рос. хим. общества им. Д. И. Менделеева) - 2006-L - С. 34. [2]. А. Л. Тарасов, Л. М. Кустов, А. Л. Кустов, В. И. Богдан // Патент РФ 2005130340; А. Л. Тарасов, Л. М. Кустов, В. И. Богдан, А. Л. Кустов // Патент РФ 2304462 (2007); В. Б. Аваков, В. И. Зинин, Б. А. Иваницкий, Г. В. Кулаков, И. К. Ландграф, Патент РФ 2260880 (2005); Ю. П. Борисевич // Патент РФ #2317249 (2008); G.P.Pez, A. R.Scott, A. Ch.Cooper, H. Cheng // US Patent 7,429,372 (2008). [3]. А. Я. Берлин // "Техника лабораторной работы в органической химии", М: Госхимиздат, 1952, с. 44. [4]. J.Wainright, J. Payer, C.-C.Liu, L. Dudik, X. Shan, S. Levine // US Patent 7,404,842 (2008); K.Young, M. A.Fetcenko, T. Ouchi, J. Im, S. R.Ovshinsky, F. Li, M. Reinhout // US Patent 7,344,676 (2008); K.Young, T. Ouchi, M. A.Fetcenko // US Patent 7,344,677 (2008); G.-A.Nazri // US Patent 7,393,393 (2008). p / ps 0,5 0,075 0,054 3 , см /г p / ps 0,9 0,16 0,088 p / ps 0,95 0,18 0,12 [5]. F.Darkrim, A. Aoufi, P. Malbrunot, D. Levesque // J.Chem.Phys-2000-V.112-P.5991. [6]. А. В. Вархушев, А. М. Липанов, М. В. Суетин // Патент РФ 2005116157 (2006); В. А. Рыжков // Патент РФ 2309118 (2007); A.Chambers, С. Park, R. Т. К. Baker, N. М. Rodriguez // J.Phys.Chem. В 1998-V.102-P.4253. [7]. В. А. Рыжков // Патент РФ 2004110232А. [8]. С. К. Narula, J. М. Simonson, L. Maya, R. Т. Paine // US Patent 7,361,213 (2008). [9]. S.J.Gregg, K. S.W.Sing, Adsorption, Surface area and Porosity, Academic press, London, 1982. [10]. M.Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe, О. М. Yaghi // Science-2002 – V.295 – P.469. [11]. M.Eddaoudi, D. B.Moler, H. Li, B. Chen, T. M.Reineke, M. O'Keeffe, O. M.Yaghi //Ace.Chem.Res.-2001-V.34-P.319. [12]. U.Muller, L. Lobree, M. Hesse, O. M.Yaghi, M. Eddaoudi // US Patent 6,624,318(2003). [13]. L.G.Beauvais, M. P.Shores, J. R.Long // J.Am.Chem.Soc.-2000-V.122-P.2763. [14]. A.C.Sudik, A. R.Millward, N. W.Ockwig, A. P.Cote, J. Kim, О. М. Yaghi // J.Am.Chem.Soc.-2005V.127,P.7110. 15 [15]. S.Surble, С Serre, С Mellot-Draznieks, F. Millange, G. Ferey // Chem.Commun.-2006-P.284. [16]. K.Nakata, H. Miyasaka, K. Sugimoto, T. Ishii, K. Sugiura, M. Yamashita // Chem.Lett.-2002P.658. [17]. A.S.Lytvynenko, S. V.Kolotilov, O. Cador, K. S.Gavrilenko, S. Golhen, L. Ouahab, V. V.Pavlishchuk // Dalton Trans.-2009-P.3503. 61912 16 [18]. O.M.Yaghi, M. Eddaoudi, H. Li, J. Kim, N. Rosi // US Patent 6,930,193 (2005). [19]. L.Meesuk, U. A.Jayasooriya, R. D.Cannon // J.Am.Chem.Soc.-1987-109(7) - P.2009;A.B.Blake, A. Yavari, W. E.Hatfield, S. N.Sethulekshmi // J.Chem.Soc.Dalton Trans.-1985-P.2509. [20]. K.S.Gavrilenko, A. Vertes, G. Vanko, L. F.Kiss, A. W.Addison, T. Weyhermuller, V. V.Pavlishchuk // Eur.J.Inorg.Chem.-2002. - P.3347. 17 Комп’ютерна верстка І.Скворцова 61912 Підписне 18 Тираж 23 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod of producing porous coordination polymers based on trinuclear carboxylate complexes of transition metals and organic ligands containing residue of pyridine
Автори англійськоюKolotilov Serhii Volodymyrovych, Havrylenko Kostiantyn Serhiiovych, Lytvynenko Anton Serhiiovych, Polunin Ruslan Anatoliiovych, Shpakovskyi Ihor Valentynovych, Shvets Oleksii Vasyliovych, Yaremov Pavlo Stepanovych, Ilin Volodymyr Heorhiiovych, Pavlischuk Vitalii Valentynovych
Назва патенту російськоюСпособ получения пористых координационных полимеров на основе трехъядерных карбоксилатных комплексов переходных металлов и органических лигандов, содержащих остаток пиридина
Автори російськоюКолотилов Сергей Владимирович, Гавриленко Константин Сергеевич, Литвиненко Антон Сергеевич, Полунин Руслан Анатольевич, Шпаковский Игорь Валентинович, Швец Алексей Васильевич, Яремов Павел Степанович, Ильин Владимир Георгиевич, Павлищук Виталий Валентинович
МПК / Мітки
МПК: B01D 53/44
Мітки: містять, органічних, металів, основі, пористих, координаційних, триядерних, залишок, полімерів, комплексів, карбоксилатних, одержання, перехідних, лігандів, спосіб, піридину
Код посилання
<a href="https://ua.patents.su/9-61912-sposib-oderzhannya-poristikh-koordinacijjnikh-polimeriv-na-osnovi-triyadernikh-karboksilatnikh-kompleksiv-perekhidnikh-metaliv-i-organichnikh-ligandiv-shho-mistyat-zalishok-piridin.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання пористих координаційних полімерів на основі триядерних карбоксилатних комплексів перехідних металів і органічних лігандів, що містять залишок піридину</a>
Метод формування пористих магнітних композитів, що містять гомо- та гетерометалічні наночастинки оксидів 3d металів в пористих матрицях, шляхом термічного або мікрохвильового розкладу поліядерних комплексів в р

Номер патенту: 32259
Опубліковано: 12.05.2008
Автори: Касьян Наталія Володимирівна, Колотілов Сергій Володимирович, Павліщук Віталій Валентинович, Василенко Інна Вілоріївна, Швець Олексій Васильович, Котенко Ігор Євгенович
МПК: B01J 20/00
Мітки: гомо, оксидів, розкладу, формування, мікрохвильового, комплексів, шляхом, поліядерних, наночастинки, містять, гетерометалічні, метод, матрицях, композитів, пористих, термічного, магнітних, металів
Формула / Реферат:
1. Метод формування пористих магнітних композитів, що містять гомо- та гетерометалічні наночастинки оксидів 3d металів в пористих матрицях, шляхом термічного або мікрохвильового розкладу поліядерних комплексів в розчинах з високою температурою кипіння для різних за природою субстратів, зокрема органічних та неорганічних речовин, газів, сполук біологічного походження, що полягає в інкорпоруванні магнітних наночастинок оксидів 3d металів в...
Спосіб одержання комплексів n-оксидів похідних піридину з хлористим марганцем

Номер патенту: 87789
Опубліковано: 10.08.2009
Автор: Дульнєв Петро Георгійович
МПК: C07F 19/00, A01P 21/00, C07D 213/89, A01N 43/00, C07D 213/00
Мітки: піридину, похідних, n-оксидів, хлористим, комплексів, марганцем, одержання, спосіб
Формула / Реферат:
Спосіб одержання комплексів N-оксидів похідних піридину з хлористим марганцем в середовищі розчинника, який відрізняється тим, що як розчинник використовують воду, а процес проводять при температурі до 50 °С і мольному співвідношенні реагентів 1,01-1,02 : 1.
Спосіб одержання сорбенту для вилучення перехідних металів з розчинів

Номер патенту: 51520
Опубліковано: 26.07.2010
Автори: Трохимчук Анатолій Костянтинович, Ульберг Зоя Рудольфівна, Андріанова Олена Борисівна, Лещенко Віталій Миколайович
МПК: B01J 20/10, B01J 45/00
Мітки: металів, розчинів, вилучення, спосіб, сорбенту, перехідних, одержання
Формула / Реферат:
1. Спосіб одержання сорбенту для вилучення іонів перехідних металів з розчинів взаємодією силікагелю і зшивального агента з наступною обробкою отриманого модифікованого силікагелю комплексоутворюючим агентом, який відрізняється тим, що як зшивальний агент використовують полігексаметиленгуанідин (ПГМГ) загальної формули:,де Х - аніон неорганічної кислоти, n =...
Спосіб нанесення покриттів на основі сполук перехідних металів у вакуумі на металеві поверхні

Номер патенту: 40387
Опубліковано: 15.03.2004
Автори: Борисова Ніна Миколаївна, Дабіжа Євген Вікторович, Бондар Іван Васильович, Новіков Микола Васильович, Золотухін Олександр Віталійович
МПК: C23C 14/02, C23C 14/32
Мітки: нанесення, покриттів, металеві, перехідних, поверхні, сполук, спосіб, металів, основі, вакуумі
Формула / Реферат:
Спосіб нанесення покриттів на основі сполук перехідних металів у вакуумі на металеві поверхні, що включає попередню очистку поверхні підкладки активним тліючим розрядом, кінцеву очистку шляхом конденсації на неї металу з іонним бомбардуванням, формування потоку металевої плазми за допомогою вакуумно-дугового розряду у розрідженій атмосфері реакційного газу, розташування виробу у плазмовому потоці і конденсацію потоку плазми, який...
Екструдер для виготовлення виробів з композицій на основі термопластичних полімерів і органічних наповнювачів

Номер патенту: 42573
Опубліковано: 15.10.2001
Автори: Суханов Владімір Пєтровіч, Сезонов Максим Вікторович, Жидов Ніколай Вікторовіч, Біденко Василь Дмитрович, Бородін Ігорь Владіміровіч, Мікульонок Ігор Олегович
МПК: B27N 3/08, B29C 47/38
Мітки: виробів, виготовлення, основі, полімерів, екструдер, органічних, наповнювачів, композицій, термопластичних
Формула / Реферат:
1. Екструдер для виготовлення виробів з композицій на основі термопластичних полімерів і органічних наповнювачів, що містить порожнистий корпус з вікнами для завантаження органічного наповнювача й термопластичного полімеру, розташований у корпусі з можливістю обертання щонайменше один шнек з ущільнювальним елементом, що розділяє порожнину корпуса на дві частини, сполучені одна з одною за допомогою масопроводу, який відрізняється...
Попередній патент: Ущільнюючий пристрій обертової печі
Наступний патент: Спосіб керування трифазним фільтрокомпенсуючим перетворювачем з використанням нейронечіткого регулятора
Випадковий патент: Фундамент будівлі, споруди