Одинична дозована фармацевтична форма
Номер патенту: 108594
Опубліковано: 25.05.2015
Автори: Ліппер Роберт А., Меннінг Марк М., Оліяй Реза, Янг Тайінь, Хассейн Мунір А., Єжевскі Роберт Л., Дахл Терренце К.










Формула / Реферат
1. Одинична дозована форма, яка включає тенофовір DF та емтрицитабін у першому окремому відділенні, одержаному шляхом сухого гранулювання, та ефавіренц з лаурилсульфатом натрію у другому окремому відділенні, і зазначені відділення є фізично сполученими та знаходяться у контакті один з одним.
2. Одинична дозована форма за п. 1, у якій перше та друге окремі відділення розташовані у вигляді шарів у контакті один з одним, причому тенофовір DF та емтрицитабін містяться у першому шарі, а ефавіренц та лаурилсульфат натрію містяться у другому шарі.
3. Одинична дозована форма за п. 2, у якій шари орієнтовані горизонтально уздовж осі дозованої форми.
4. Одинична дозована форма за п. 2, яка придатна для перорального введення.
5. Одинична дозована форма за п. 1, у якій друге окреме відділення одержане шляхом мокрого гранулювання з високим зсувним зусиллям.
6. Одинична дозована форма за п. 5, у якій загальна кількість ефавіренцу, емтрицитабіну та тенофовіру DF є більшою, ніж приблизно 60 % за масою композиції.
7. Одинична дозована форма за п. 6, яка додатково включає стеарат магнію, кроскармелозу натрію, мікрокристалічну целюлозу та гідроксипропілцелюлозу.
8. Одинична дозована форма за п. 7, у якій приблизний відсоток за масою ефавіренцу, тенофовіру DF, емтрицитабіну, стеарату магнію, кpocкapмeлози натрію, мікрокристалічної целюлози, лаурилсульфату натрію та гідроксипропілцелюлози становить, приблизно 39, приблизно 19, приблизно 13, приблизно 2, приблизно 7, приблизно 17, приблизно 1 та приблизно 2, відповідно.
9. Одинична дозована форма за п. 8, яка при введенні пацієнтові пероральним шляхом забезпечує для ефавіренцу, емтрицитабіну та тенофовіру DF практично такі самі AUC та Сmах, що й для затверджених FDA продуктів Truvada та Sustiva.
10. Одинична дозована форма за п. 9, яка має масу приблизно від 1200 мг до 2300 мг, включаючи будь-яку оболонку, що необов'язково може бути нанесена.
11. Контейнер, який включає одиничну дозовану форму за п. 1 та десикант.
12. Спосіб приготування дозованої форми за будь-яким з пп. 1-3, у якому: готують перше окреме відділення, що включає тенофовір DF та емтрицитабін, шляхом сухого гранулювання,
готують друге окреме відділення, що включає ефавіренц та лаурилсульфат натрію, шляхом мокрого гранулювання, та фізично сполучують обидва відділення одне з одним.
13. Спосіб за п. 12, у якому зазначені відділення розміщують у шарах у контакті один з одним.
14. Спосіб за п. 12, у якому зі стеаратом магнію комбінують підданий сухому гранулюванню тенофовір DF та емтрицитабін, та зі стеаратом магнію комбінують підданий мокрому гранулюванню ефавіренц та лаурилсульфат натрію, а потім пресують у двошарову таблетку.
15. Спосіб антивірусної терапії, у якому пацієнтові, що потребує антивірусної терапії, перорально вводять одиничну дозовану форму за будь-яким з пп. 1-3.
16. Спосіб за п. 15, у якому одиничну дозовану форму вводять тільки один раз на добу.
17. Спосіб за п. 16, у якому антивірусна терапія являє собою спрямовану проти ВІЛ терапію.
18. Двошарова таблетка, яка містить емтрицитабін та тенофовір DF у першому шарі та ефавіренц і лаурилсульфат натрію у другому шарі, причому зазначені шари знаходяться у контакті один з одним.
19. Двошарова таблетка за п. 18, яка має масу менше ніж 2,5 грама.
Текст
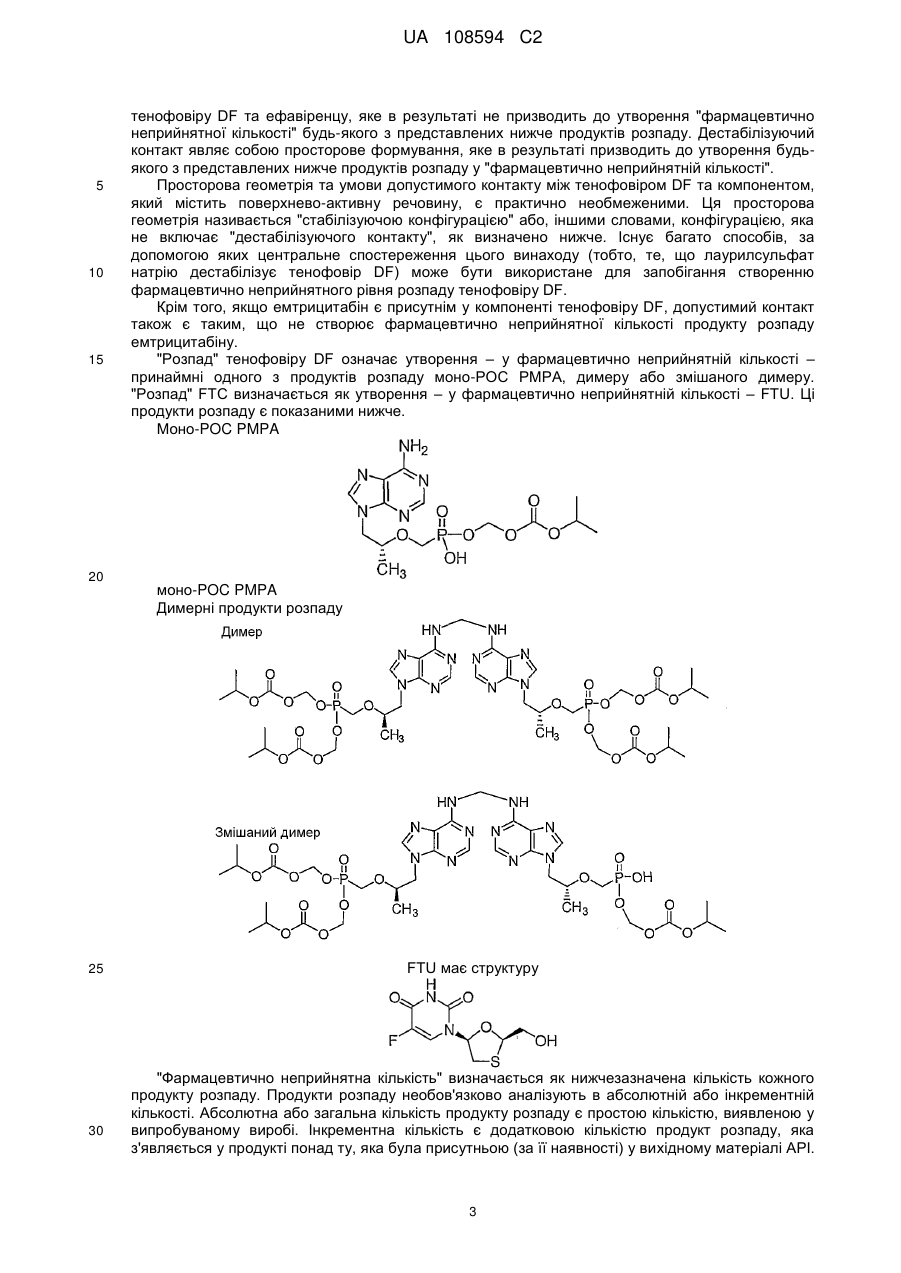
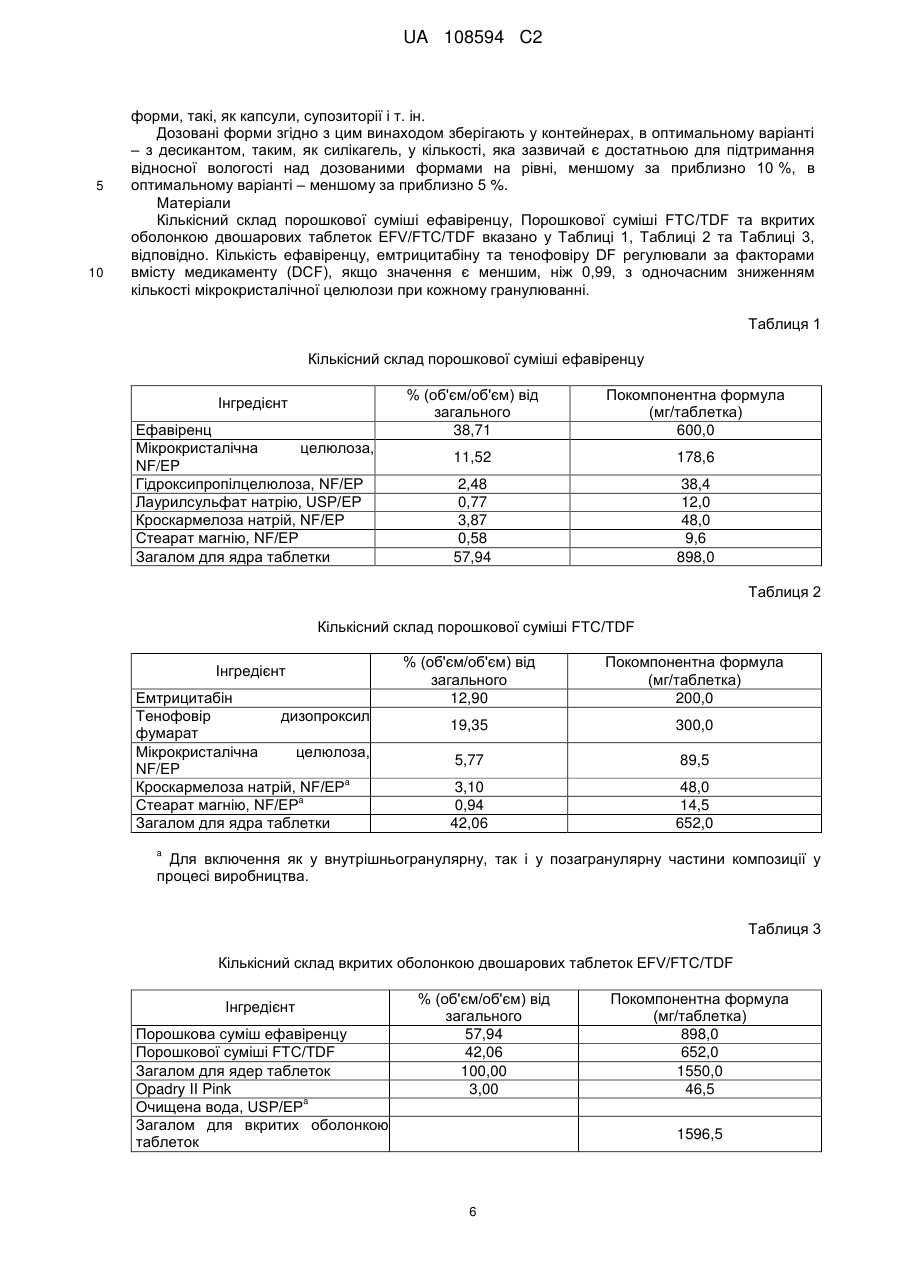
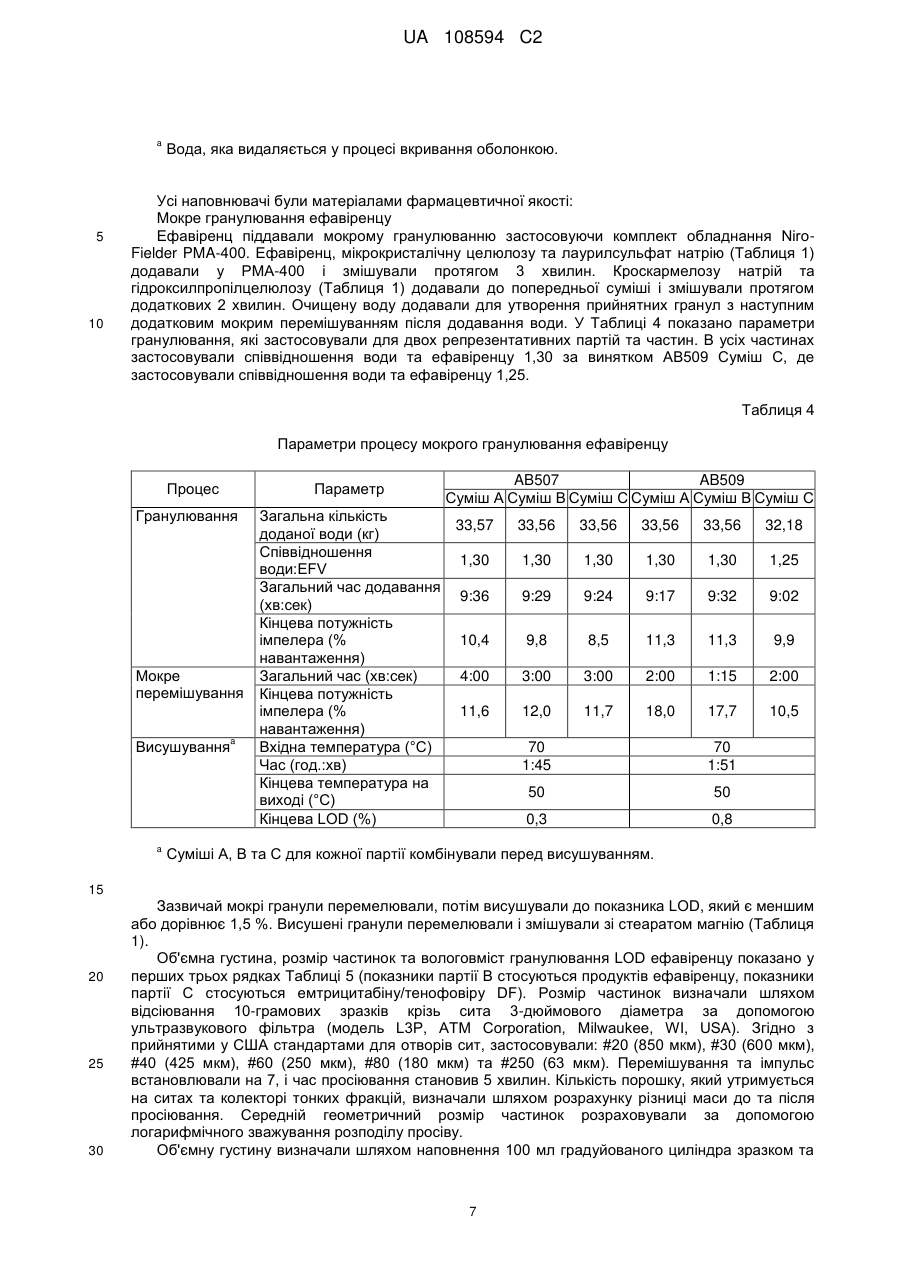
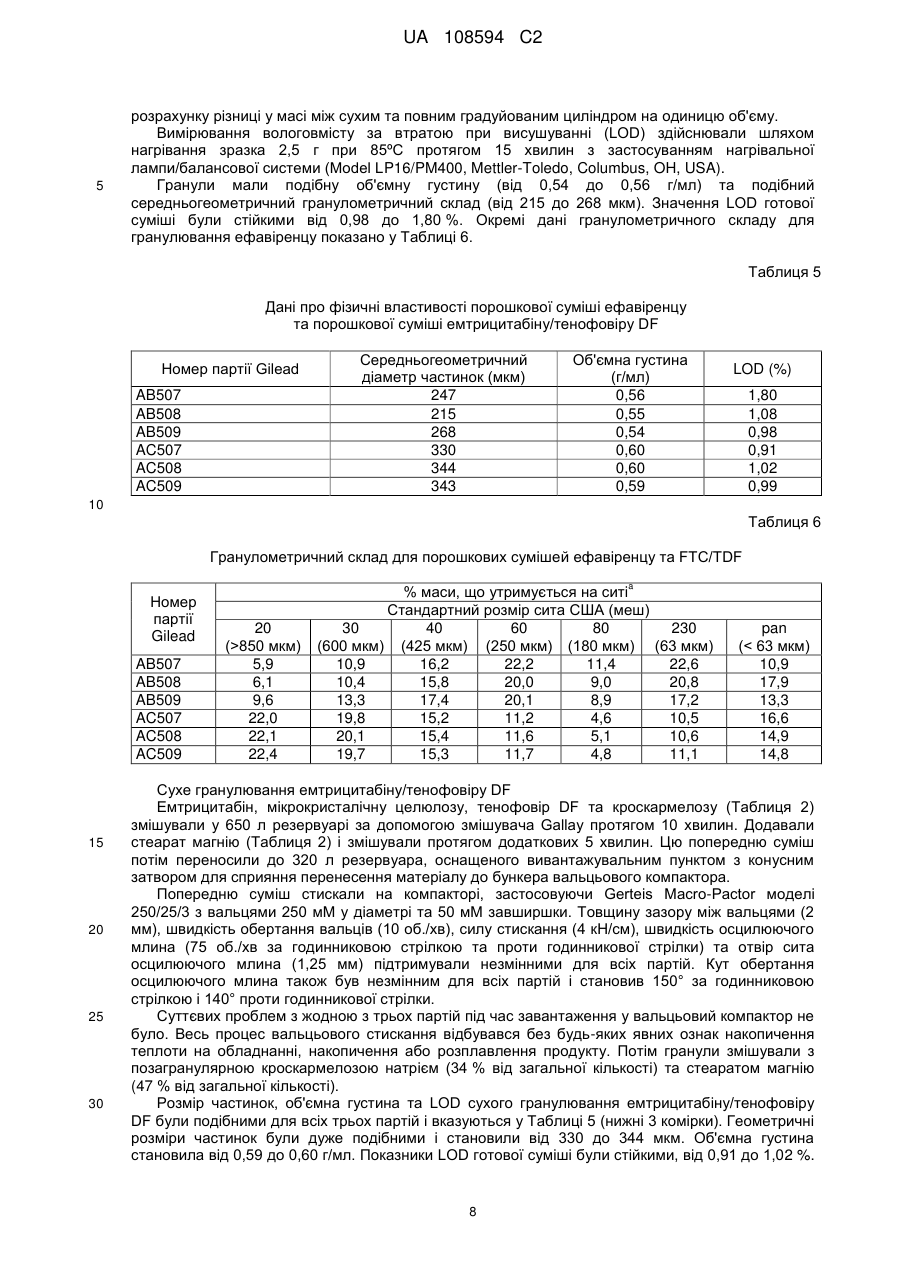
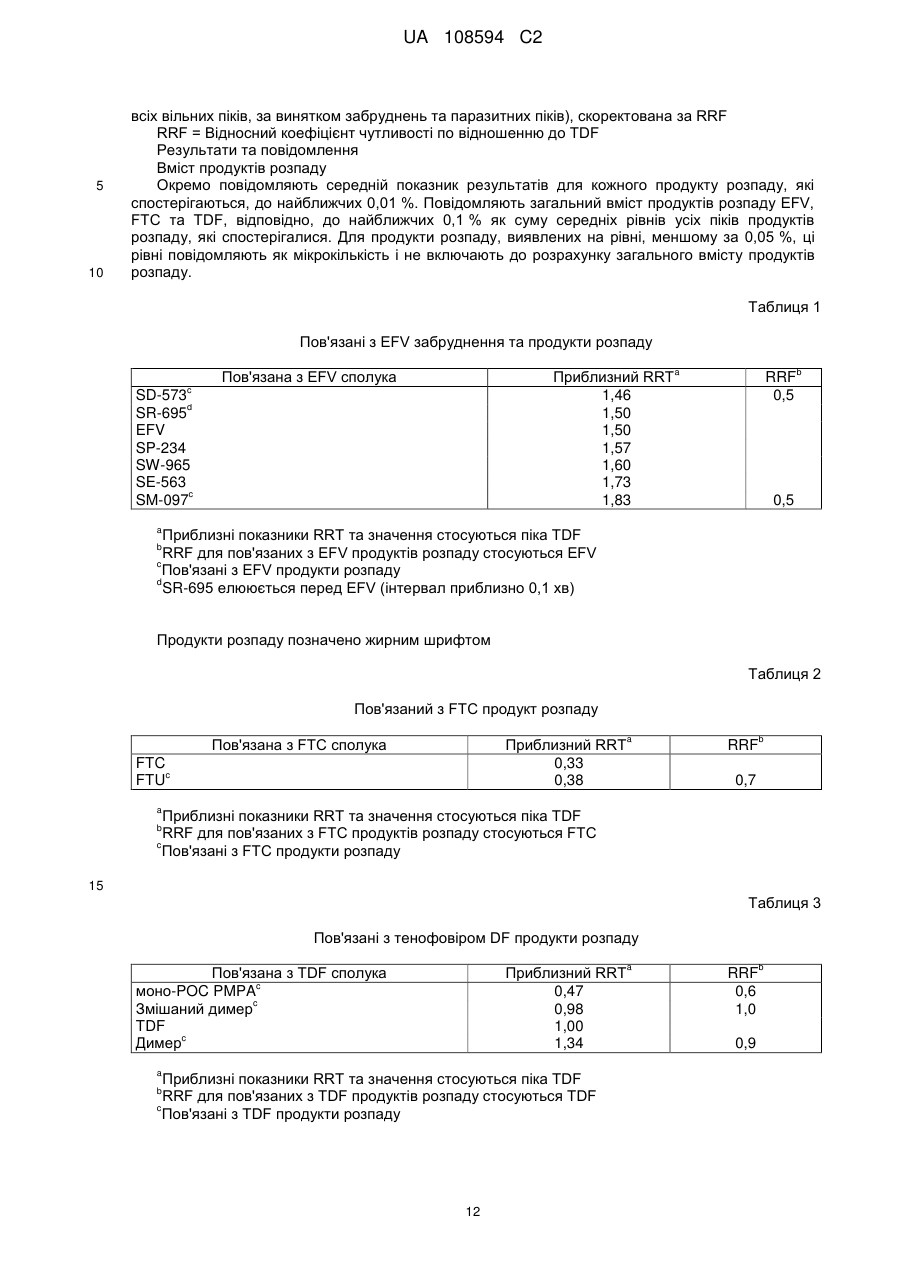
Реферат: Винахід стосується фармацевтичного продукту, який містить емтрицитабін та тенофовір DF у першому окремому відділенні, одержаному шляхом сухого гранулювання, та ефавіренц та лаурилсульфат натрію у другому окремому відділенні, де зазначені відділення є фізично сполученими та знаходяться у контакті один з одним. UA 108594 C2 (12) UA 108594 C2 UA 108594 C2 5 10 15 20 25 30 35 40 45 50 55 60 Рівень техніки Ця заявка стосується продуктів для лікування вірусних інфекцій, зокрема, ВІЛ- інфекцій, з застосуванням відомих антивірусних сполук ефавіренцу (торгова назва Sustiva, також відомий як EFV), емтрицитабіну (торгова назва Emtriva, також відомий як FTC) та тенофовіру DF (дизопроксил фумарат, також відомий як TDF) (торгова назва Viread, продається у комбінації з емтрицитабіном під торговою назвою Truvada). Truvada виробляють шляхом мокрого гранулювання емтрицитабіну та тенофовіру DF (WO 04/64845), що за відповідних обставин утворює хімічно стійку дозовану форму. Цей продукт не містить ефавіренцу. Бажаною вважають спрямовану проти ВІЛ терапію з застосуванням ефавіренцу, а також емтрицитабіну та тенофовіру DF (далі – "потрійна комбінація"; див. WO 04/64845). Однак виробництво комерційно ефективного продукту потрійної комбінації вимагає відповідності жорстким вимогам Управління з контролю за продуктами та ліками щодо біоеквівалентності, які висуваються до комерційних продуктів, Viread (тенофовір дизопроксил фумарату), Emtriva (емтрицитабіну) та Sustiva (ефавіренцу) і прийнятного для пацієнтів розміру таблетки для полегшення ковтання. Початкові спроби простого комбінування трьох медикаментів (активних фармацевтичних проміжних сполук або API) у єдину, практично гомогенну композицію, вироблену шляхом мокрого гранулювання, не дозволяли одержати хімічно стійку таблетку. Тенофовір DF у цій комбінованій таблетці був дуже нестійким і швидко розпадався при випробуваннях стійкості. Композиція ефавіренцу несподівано виявилася несумісною з тенофовіром DF, що, очевидно, зумовлюється поверхнево-активною речовиною (лаурилсульфатом натрію), яка виявляється в ефавіренці цієї композиції. Було здійснено ще одну спробу одержання потрійної комбінації, на цей раз із застосуванням сухого гранулювання трикомпонентної комбінації без поверхнево-активної речовини. В результаті одержали таблетку, яка не забезпечувала біоеквівалентності щодо ефавіренцу у клінічних випробуваннях на людях. Пікова концентрація ефавіренцу в потоці крові та залежність концентрації від часу (Cmax та AUC) були нижчими за параметри, визначені для таблеток комерційного компаратора, Sustiva (ефавіренцу). Автори винаходу дійшли висновку, що принаймні поверхнево-активна речовина у таблетках потрійної комбінації (ефавіренц/емтрицитабін/тенофовір дизопроксил фумарату) є необхідною для досягнення біоеквівалентності з Sustiva. Крім того, комбіновані таблетки виготовляли шляхом мокрого гранулювання ефавіренцу з поверхнево-активною речовиною та іншим наповнювачем, окремого виготовлення Truvada шляхом сухого гранулювання, змішування гранулятів, пресування суміші у таблетки з наступним вкриванням таблеток оболонкою. Несподівано цей підхід також виявився невдалим у досягненні потрібної біоеквівалентності між комерційним продуктом, Sustiva (ефавіренц) та клінічно досліджуваним матеріалом (тобто, запропонованим комерційним продуктом потрійної комбінації). Вимагався новий рівень винаходу для подолання недоліків більш прямолінійних підходів до потрійної комбінованої дозованої форми. Паралельно перебуваюча на розгляді заявка U.S.S.N. 60/771,353 (з однаковою датою подання, вказана авторами шляхом посилання) стосується подолання іншої перешкоди, яка трапляється при одержанні потрійної комбінованої дозованої форми, тобто, зменшення розміру комбінованого продукту. Хоча в існуючому рівні техніки повідомляється про успішне виробництво хімічно стійких композицій Truvada (WO04/64845), ці композиції містять відносно низькі пропорції наповнювача відносно API. В результаті збільшення пропорції наповнювачів та мокрого гранулювання комбінації з трьох API несподівано було одержано композицію, в якій тенофовір DF був дуже нестійким. Як повідомляється в U.S.S.N. 60/771,353, вважалося, що застосування достатньої кількості води для здійснення мокрого гранулювання ефавіренцу (який має відносно низьку розчинність порівняно з емтрицитабіном та тенофовіром DF) викликає розчинення двох останніх API в евтектичну суміш. Евтектичну суміш висушують під час гранулювання для утворення склоподібного або аморфного продукту, в якому тенофовір DF є хімічно нестійким порівняно з кристалічним API. Забезпечення достатньої кількості наповнювача для виправлення впливу надлишку води не відповідало меті одержання потрійної комбіновано пероральної дозованої форми з контрольованими пропорціями. Як описано в U.S.S.N. 60/771,353, ця перешкода долалася шляхом сухого гранулювання композиції емтрицитабіну та тенофовіру DF, тобто, гранулювання композиції без її контакту дестабілізуючою кількістю рідкої води. Невключення води (зокрема, рідкої води) або зменшення присутності води до несуттєвої кількості усуває небажане утворення евтектичної суміші й підвищує стійкість одержаного в результаті фармацевтичного продукту. 1 UA 108594 C2 5 10 15 20 25 30 35 40 45 50 55 60 Незважаючи на переваги, які дає сухе гранулювання емтрицитабіну/тенофовіру DF, все одно існує потреба у подоланні несподіваної несумісності тенофовіру DF та поверхневоактивної речовини, яку застосовують у композиції Sustiva. Короткий опис винаходу Згідно з цим винаходом, стійкість та біоеквівалентність для продукту потрійної комбінації досягається шляхом забезпечення багатокомпонентної дозованої форми, в якій один компонент включає тенофовір DF та, необов'язково, емтрицитабін, а інша включає принаймні ефавіренц. Інший варіант втілення винаходу стосується дозованої форми, яка включає компонент тенофовіру DF та компонент поверхнево-активної речовини, які не перебувають у дестабілізуючому контакті з компонентом тенофовіру DF. Детальний опис винаходу Дозована форма згідно з цим винаходом включає ефавіренц, емтрицитабін та тенофовір DF. Як було зазначено, тенофовір DF та ефавіренц належать до окремих компонентів. Емтрицитабін зазвичай включається до компонента тенофовіру DF, але в інших варіантах втілення емтрицитабін може бути присутній як окремий компонент або змішаний з компонентом ефавіренцу. Його місце не є критичним для практичного втілення цього винаходу. Необхідно лише, щоб емтрицитабін був присутній у дозованій формі, і щоб компонент тенофовіру DF був практично відокремлений від поверхнево-активної речовини у компоненті ефавіренцу. Будьякий спосіб, домішка, особливість або конфігурація процесу, які відповідно мінімізують контакт поверхнево-активної речовини з тенофовіром DF, є придатними для практичного втілення цього винаходу. Термін "компонент" означає фізично відокремлену одиницю або відділення, яке перебуває у фізичному зв'язку і в контакті з іншими компонентами. Це не означає, що одиниці або відділення не перебувають у фізичному контакті. Фактично, перевагу віддають варіантові, коли вони перебувають у фізичному контакті й утворюють єдиний засіб, виріб або композицію. Ступінь зв'язку є лише таким, що вимагається для полегшення перорального споживання композиції як єдиної дозованої форми. Цей винахід не охоплює, наприклад, пакети для пацієнтів з продуктами Sustiva та Truvada в окремих комірках або вмістищах, або в інших поєднаннях, які здебільшого передбачають окреме фасування розчинів (хоча, звичайно, композиції згідно з цим винаходом необов'язково можуть бути упаковані або розфасовані у будь-який традиційний спосіб, прийнятний за даних обставин). Зазвичай компоненти дозованої форми згідно з цим винаходом формують у кілька шарів, як правило, у двошарову структуру, як показано у наведеному прикладі втілення. Однак, якщо емтрицитабін є присутнім як окремий компонент, дозована форма включає щонайменше тришарову структуру. Окремий компонент не обов'язково відповідає кожному медикаментові (наприклад, дозовані форми необов'язково включають 2 шари для кожного з компонентів, загалом 6). Таким чином, одинична доза включає шаруваті структури з багатьох компонентів. Не обов'язково має бути однакова кількість кожного компонента, наприклад, шарів, для кожного медикаменту або комбінації медикаментів, якщо загальна доза всіх компонентів у сумі складає потрібну кількість. Можуть бути прийнятними інші засоби просторового формування компонентів, якщо забезпечується потрібний ступінь відокремлення тенофовіру DF та поверхнево-активної речовини. Наприклад, замість формування плоских шарів уздовж осі таблетки компоненти необов'язково можуть розташовуватися кільцями, причому кожне кільце або циліндр містить окремий компонент. Інший альтернативний варіант передбачає застосування процесу нанесення покриття пресуванням для зв'язування компонентів. Компоненти зазвичай перебувають у прямому контакті один з одним, тобто, без бар'єра або захисного шару між ними. В інших варіантах втілення передбачено бар'єр між несумісними компонентами. Відповідним прикладом цього варіанта втілення винаходу може бути багатокамерна капсула, в якій несумісні компоненти є розподіленими по окремих камерах. В альтернативному варіанті необов'язково пропонується таблетка, яка містить один інкапсульований компонент, диспергований або розподілений у несумісному компоненті. Зазвичай ретельне пряме змішування несумісних компонентів є небажаним, якщо не передбачаються засоби захисту компонента тенофовіру DF від поверхнево-активної речовини. У типових варіантах втілення компоненти дозованої форми згідно з цим винаходом є просторово сформованими таким чином, щоб компонент тенофовіру DF не вступав у дестабілізуючий контакт з поверхнево-активною речовиною у компонент ефавіренцу. "Дестабілізуючий" означає будь-який контакт між тенофовіром DF та поверхнево-активною речовиною, який може викликати фармацевтично неприйнятний розпад тенофовіру DF. Стабілізуюча конфігурація являє собою будь-яке просторове формування компонентів 2 UA 108594 C2 5 10 15 20 25 30 тенофовіру DF та ефавіренцу, яке в результаті не призводить до утворення "фармацевтично неприйнятної кількості" будь-якого з представлених нижче продуктів розпаду. Дестабілізуючий контакт являє собою просторове формування, яке в результаті призводить до утворення будьякого з представлених нижче продуктів розпаду у "фармацевтично неприйнятній кількості". Просторова геометрія та умови допустимого контакту між тенофовіром DF та компонентом, який містить поверхнево-активну речовину, є практично необмеженими. Ця просторова геометрія називається "стабілізуючою конфігурацією" або, іншими словами, конфігурацією, яка не включає "дестабілізуючого контакту", як визначено нижче. Існує багато способів, за допомогою яких центральне спостереження цього винаходу (тобто, те, що лаурилсульфат натрію дестабілізує тенофовір DF) може бути використане для запобігання створенню фармацевтично неприйнятного рівня розпаду тенофовіру DF. Крім того, якщо емтрицитабін є присутнім у компоненті тенофовіру DF, допустимий контакт також є таким, що не створює фармацевтично неприйнятної кількості продукту розпаду емтрицитабіну. "Розпад" тенофовіру DF означає утворення – у фармацевтично неприйнятній кількості – принаймні одного з продуктів розпаду моно-POC PMPA, димеру або змішаного димеру. "Розпад" FTC визначається як утворення – у фармацевтично неприйнятній кількості – FTU. Ці продукти розпаду є показаними нижче. Моно-POC PMPA моно-POC PMPA Димерні продукти розпаду FTU має структуру "Фармацевтично неприйнятна кількість" визначається як нижчезазначена кількість кожного продукту розпаду. Продукти розпаду необов'язково аналізують в абсолютній або інкрементній кількості. Абсолютна або загальна кількість продукту розпаду є простою кількістю, виявленою у випробуваному виробі. Інкрементна кількість є додатковою кількістю продукт розпаду, яка з'являється у продукті понад ту, яка була присутньою (за її наявності) у вихідному матеріалі API. 3 UA 108594 C2 5 10 15 20 25 30 35 40 45 50 55 60 Крім того, кількість продукту розпаду необов'язково вимірюють у два моменти часу. Одним є час випуску на ринок. Іншим є момент після поміщення в умови зберігання з нижчеописаних умов, тобто, під час терміну придатності, як вказується нижче. Загальна кількість під час випуску (перший комерційний продаж) Не більше, ніж приблизно 3 %, зазвичай приблизно 1,5 %, моно-POC PMPA, Не більше, ніж приблизно 1 %, зазвичай приблизно 0,5 % димеру, Не більше, ніж приблизно 0,5 %, зазвичай приблизно 0,25 % змішаного димеру. Менше, ніж приблизно 0,5 %, зазвичай приблизно 0,2 % FTU Загальна кількість протягом терміну придатності (зберігання при 25 °C/60 % відносної вологості протягом 24 міс.) Не більше, ніж приблизно 10 %, зазвичай приблизно 5 % моно-POC PMPA, Не більше, ніж приблизно 2 %, зазвичай приблизно 1 % димеру, Не більше, ніж приблизно 2 %, зазвичай приблизно 1 % змішаного димеру. Не більше, ніж приблизно 4 %, зазвичай приблизно 2 % FTU Інкрементна кількість під час випуску (перший комерційний продаж) Не більше, ніж приблизно 2 %, зазвичай приблизно 0,5 %, моно-POC PMPA, Не більше, ніж приблизно 0,6 %, зазвичай приблизно 0,1 % димеру, Не більше, ніж приблизно 0,3 %, зазвичай приблизно 0,05 % змішаного димеру. Менше, ніж приблизно 0,4 %, зазвичай приблизно 0,1 % FTU Інкрементна кількість протягом терміну придатності (зберігання при 25 °C/60 % відносної вологості протягом 24 міс.) Не більше, ніж приблизно 9 %, зазвичай приблизно 4 % моно-POC PMPA, Не більше, ніж приблизно 1,6 %, зазвичай приблизно 0,6 % димеру, Не більше, ніж приблизно 1,8 %, зазвичай приблизно 0,8 % змішаного димеру. Не більше, ніж приблизно 3,9 %, зазвичай приблизно 1,9 % FTU. Відсоток продуктів розпаду є кількістю продукту розпаду, виміряна шляхом порівняння часу утримання при HPLC. У порівнянні часу утримання при HPLC час утримання головних піків, який спостерігають у таблетках, має бути у межах 2 % часу утримання головних піків у контрольній стандартній композиції, яка містить ефавіренц, емтрицитабін та тенофовір DF під час аналізу, який виявився характерним для ефавіренцу, емтрицитабіну та тенофовіру DF. Відсоток визначають шляхом ділення загальної кількості тенофовіру DF та трьох продуктів розпаду на кількість окремого продукту розпаду, згідно з визначенням шляхом HPLC. Ці параметри застосовують для того, щоб визначити, чи відповідає випробувана композиція вимогам стабілізуючого контакту. Наприклад, потрійна комбінована дозована форма необов'язково передбачається як формовий виріб, який включає порції пресованих гранул компонента тенофовіру DF, диспергованого у матриксі компонента ефавіренцу. Можливим є варіювання розмірів порцій при утворенні композиції. Цю сукупність потенційних продуктів після цього піддають випробуванню або зберігають за вищевказаних умов, а потім випробують, для аналізу утворення продуктів розпаду тенофовіру DF та/або FTC. Якщо одержаний в результаті продукт після випуску не містив більше за вказану граничну кількість будь-яких одного або кількох з 4 забруднювачів, перелічених для будь-якого з 4 прикладів аналізу, то контакт вважають стабілізуючим. Звичайно, спеціаліст у даній галузі може застосовувати жорсткіші стандарти, але це є справою вибору і не обмежує обсяг цього винаходу. В оптимальних варіантах втілення емтрицитабін та тенофовір DF комбінують і цей компонент приготовляють шляхом сухого гранулювання (U.S.S.N. 60/771,353). В оптимальних варіантах втілення, композицію, яка включає підданий сухому гранулюванню тенофовір DF та емтрицитабін, застосовують в одному компоненті дозованих форм згідно з цим винаходом. Сухе гранулювання саме по собі є загальновідомим способом фармацевтичного виробництва. Зазвичай, API комбінують з наповнювачами і змащувальним наповнювачем, а потім стискають для утворення маси. Як правило, цю масу потім подрібнюють або перемелюють, потім просіюють для одержання потрібного розміру частинок. Гранульований продукт пресують у таблетки, поміщають у капсули або іншим традиційним способом чином формують у єдину дозовану форму. Стискання у масу здійснюють за допомогою традиційного обладнання. Зазвичай API та наповнювач пропускають через вальцьовий пристрій для стискання. Однак необов'язково можуть застосовуватись інші засоби стискання суміші API, наприклад, стискання у порції (або "злежування"). Процес сухого гранулювання є способом, згідно з яким суху композицію API та вибраного(их) наповнювача(ів) стискають для утворення маси, яку у разі необхідності подрібнюють або перемелюють, а потім необов'язково просіюють для одержання гранул 4 UA 108594 C2 5 10 15 20 25 30 35 40 45 50 55 60 потрібного розміру. Стискання у масу здійснюють за допомогою традиційного обладнання. Зазвичай API та наповнювач пропускають через вальцьовий пристрій для стискання. Однак можуть застосовуватися й інші засоби стискання суміші API, наприклад, стискання у порції (або "злежування"). Композиція, яка включає підданий сухому гранулюванню емтрицитабін та тенофовір DF, є продуктом процесу сухого гранулювання. Ця композиція суттєвою мірою утримує кристалічні API і є практично вільною від висушеного евтектичного емтрицитабіну/тенофовіру DF. Вона зазвичай містить менше, ніж приблизно 15 % за масою висушеної евтектичної суміші, як правило, менше, ніж приблизно 10 % і зазвичай менше, ніж приблизно 5 %. Процес сухого гранулювання здійснюють за відсутності дестабілізуючої кількості води, причому "дестабілізуюча" означає кількість рідкої води, яка може викликати фармацевтично неприйнятний розпад тенофовіру DF та/або FTC, як визначено авторами. Якщо дозована форма згідно з цим винаходом включає підданий сухому гранулюванню компонент емтрицитабіну/тенофовіру DF, то кількість допустимого продукту розпаду у кінцевій дозованій формі все одно є такою самою, як вказана вище, тобто, кількість води, яка має вплив через контакт, разом або окремо, не є такою, щоб в результаті утворювалися продукти розпаду, які не відповідають описаним вище стандартам. Звичайно, на вибір можна спочатку випробувати сухі грануляти на їх рівень продуктів розпаду і, у разі їх придатності, потім рецептувати їх у дозовану форму згідно з цим винаходом, а потім визначати, чи призводить контакт до збільшення продуктів розпаду, що виключає одержану в результаті дозовану форму з числа тих, що відповідають визначеним параметрам. Зв'язана, захоплена або абсорбована вода зазвичай є присутньою у наповнювачі. Ця вода не має суттєвого негативного впливу на стійкість тенофовіру DF і, таким чином, не виключається з сухих гранулятів, які необов'язково застосовують у дозованій формі згідно з цим винаходом. Зазвичай рідка вода (яка додається або утворюється in situ) з будь-якого джерела, наприклад, через хімічні реакції, конденсацію, з захопленого льоду і т. ін., має бути виключена з гранулювання. Однак незначну кількість рідкої води необов'язково додають під час гранулювання. Ця кількість зазвичай має бути меншою, ніж приблизно 5 % за масою, як правило, меншою, ніж приблизно 1 % за масою, однак вода може утворюватися або подаватися. Вода є присутньою в кінцевому продукті гранулювання у кількості до приблизно 10 % за масою (Karl Fischer), але в оптимальному варіанті – меншій, ніж 0,1 % за масою. Однак допустима кількість води може змінюватися, залежно від інших чинників у гранулюванні, наприклад, типу наповнювача, температури і т. ін. Наприклад, якщо включається гігроскопічний наповнювач, він перетворює додану воду на зв'язану форму. Необхідно лише, щоб вода не призводила до розпаду тенофовіру DF та/або емтрицитабіну в кінцевому продукті. Як правило, воду виключають з етапу попереднього гранулювання (приготування композиції, яка має бути застосована безпосередньо у гранульованій формі), а також під час самого процесу гранулювання. Відсутність води або визначення "сухий" не означає відсутності рідини. Гранулювання з органічними розчинниками також є можливим за умови виключення дестабілізуючої кількості води. Сухе гранулювання в результаті забезпечує продукт, який містить мінімальну кількість води. Кількість води у грануляті продукту або виготовлених з нього дозованих формах, вимірюють за втратою при висушуванні (LOD) або способом Карла Фішера (Karl Fischer). Показники LOD композицій згідно з цим винаходом становлять приблизно 15 %, приблизно 10 %, приблизно 5 % або зазвичай менше, ніж приблизно 3 % за масою. Вода за Карлом Фішером складає приблизно від 0,1 до 10 % за масою, як правило, менше, ніж приблизно 5 % за масою або менше, ніж приблизно 2 %. Кількість води у готових композиціях, на відміну від гранулятів, залежить від води у грануляті, а також незначної кількості води, яку застосовують під час наступних етапів процесу, таких, як нанесення покриття. Ця кількість води, яку додають на більш пізніх етапах, ніж гранулювання, зазвичай не впливає на стійкість API емтрицитабіну/тенофовіру DF, а отже може змінюватись у значних допустимих межах. Описаний нижче спосіб виробництва стосується одержання потрійної комбінованої таблетки, яка містить ефавіренц, емтрицитабін та тенофовір DF. У цьому конкретному варіанті втілення останні два медикаменти поміщають у частину таблетки, яка є окремою, але контактує з частиною таблетки, яка містить ефавіренц. Однак слід розуміти, що компонент емтрицитабіну та тенофовіру DF таблетки, який є варіантом втілення цього винаходу, необов'язково виготовляють як самостійний продукт і не обов'язково у поєднанні з компонентом ефавіренцу. У цьому варіанті описану нижче суху гранульовану проміжну сполуку емтрицитабіну/тенофовіру DF просто пресують у таблетки або традиційно переробляють на інші традиційні єдині дозовані 5 UA 108594 C2 5 10 форми, такі, як капсули, супозиторії і т. ін. Дозовані форми згідно з цим винаходом зберігають у контейнерах, в оптимальному варіанті – з десикантом, таким, як силікагель, у кількості, яка зазвичай є достатньою для підтримання відносної вологості над дозованими формами на рівні, меншому за приблизно 10 %, в оптимальному варіанті – меншому за приблизно 5 %. Матеріали Кількісний склад порошкової суміші ефавіренцу, Порошкової суміші FTC/TDF та вкритих оболонкою двошарових таблеток EFV/FTC/TDF вказано у Таблиці 1, Таблиці 2 та Таблиці 3, відповідно. Кількість ефавіренцу, емтрицитабіну та тенофовіру DF регулювали за факторами вмісту медикаменту (DCF), якщо значення є меншим, ніж 0,99, з одночасним зниженням кількості мікрокристалічної целюлози при кожному гранулюванні. Таблиця 1 Кількісний склад порошкової суміші ефавіренцу Інгредієнт Ефавіренц Мікрокристалічна целюлоза, NF/EP Гідроксипропілцелюлоза, NF/EP Лаурилсульфат натрію, USP/EP Кроскармелоза натрій, NF/EP Стеарат магнію, NF/EP Загалом для ядра таблетки % (об'єм/об'єм) від загального 38,71 Покомпонентна формула (мг/таблетка) 600,0 11,52 178,6 2,48 0,77 3,87 0,58 57,94 38,4 12,0 48,0 9,6 898,0 Таблиця 2 Кількісний склад порошкової суміші FTC/TDF Інгредієнт Емтрицитабін Тенофовір дизопроксил фумарат Мікрокристалічна целюлоза, NF/EP a Кроскармелоза натрій, NF/EP a Стеарат магнію, NF/EP Загалом для ядра таблетки % (об'єм/об'єм) від загального 12,90 Покомпонентна формула (мг/таблетка) 200,0 19,35 300,0 5,77 89,5 3,10 0,94 42,06 48,0 14,5 652,0 a Для включення як у внутрішньогранулярну, так і у позагранулярну частини композиції у процесі виробництва. Таблиця 3 Кількісний склад вкритих оболонкою двошарових таблеток EFV/FTC/TDF Інгредієнт Порошкова суміш ефавіренцу Порошкової суміші FTC/TDF Загалом для ядер таблеток Opadry II Pink a Очищена вода, USP/EP Загалом для вкритих оболонкою таблеток % (об'єм/об'єм) від загального 57,94 42,06 100,00 3,00 Покомпонентна формула (мг/таблетка) 898,0 652,0 1550,0 46,5 1596,5 6 UA 108594 C2 a 5 10 Вода, яка видаляється у процесі вкривання оболонкою. Усі наповнювачі були матеріалами фармацевтичної якості: Мокре гранулювання ефавіренцу Ефавіренц піддавали мокрому гранулюванню застосовуючи комплект обладнання NiroFielder PMA-400. Ефавіренц, мікрокристалічну целюлозу та лаурилсульфат натрію (Таблиця 1) додавали у PMA-400 і змішували протягом 3 хвилин. Кроскармелозу натрій та гідроксилпропілцелюлозу (Таблиця 1) додавали до попередньої суміші і змішували протягом додаткових 2 хвилин. Очищену воду додавали для утворення прийнятних гранул з наступним додатковим мокрим перемішуванням після додавання води. У Таблиці 4 показано параметри гранулювання, які застосовували для двох репрезентативних партій та частин. В усіх частинах застосовували співвідношення води та ефавіренцу 1,30 за винятком AB509 Суміш C, де застосовували співвідношення води та ефавіренцу 1,25. Таблиця 4 Параметри процесу мокрого гранулювання ефавіренцу Процес Параметр Гранулювання Мокре перемішування Висушування a a Загальна кількість доданої води (кг) Співвідношення води:EFV Загальний час додавання (хв:сек) Кінцева потужність імпелера (% навантаження) Загальний час (хв:сек) Кінцева потужність імпелера (% навантаження) Вхідна температура (°C) Час (год.:хв) Кінцева температура на виході (°C) Кінцева LOD (%) AB507 AB509 Суміш A Суміш B Суміш C Суміш A Суміш B Суміш C 33,57 33,56 33,56 33,56 33,56 32,18 1,30 1,30 1,30 1,30 1,30 1,25 9:36 9:29 9:24 9:17 9:32 9:02 10,4 9,8 8,5 11,3 11,3 9,9 4:00 3:00 3:00 2:00 1:15 2:00 11,6 12,0 11,7 18,0 17,7 10,5 70 1:45 70 1:51 50 50 0,3 0,8 Суміші A, B та C для кожної партії комбінували перед висушуванням. 15 20 25 30 Зазвичай мокрі гранули перемелювали, потім висушували до показника LOD, який є меншим або дорівнює 1,5 %. Висушені гранули перемелювали і змішували зі стеаратом магнію (Таблиця 1). Об'ємна густина, розмір частинок та вологовміст гранулювання LOD ефавіренцу показано у перших трьох рядках Таблиці 5 (показники партії B стосуються продуктів ефавіренцу, показники партії C стосуються емтрицитабіну/тенофовіру DF). Розмір частинок визначали шляхом відсіювання 10-грамових зразків крізь сита 3-дюймового діаметра за допомогою ультразвукового фільтра (модель L3P, ATM Corporation, Milwaukee, WI, USA). Згідно з прийнятими у США стандартами для отворів сит, застосовували: #20 (850 мкм), #30 (600 мкм), #40 (425 мкм), #60 (250 мкм), #80 (180 мкм) та #250 (63 мкм). Перемішування та імпульс встановлювали на 7, і час просіювання становив 5 хвилин. Кількість порошку, який утримується на ситах та колекторі тонких фракцій, визначали шляхом розрахунку різниці маси до та після просіювання. Середній геометричний розмір частинок розраховували за допомогою логарифмічного зважування розподілу просіву. Об'ємну густину визначали шляхом наповнення 100 мл градуйованого циліндра зразком та 7 UA 108594 C2 5 розрахунку різниці у масі між сухим та повним градуйованим циліндром на одиницю об'єму. Вимірювання вологовмісту за втратою при висушуванні (LOD) здійснювали шляхом нагрівання зразка 2,5 г при 85ºC протягом 15 хвилин з застосуванням нагрівальної лампи/балансової системи (Model LP16/PM400, Mettler-Toledo, Columbus, OH, USA). Гранули мали подібну об'ємну густину (від 0,54 до 0,56 г/мл) та подібний середньогеометричний гранулометричний склад (від 215 до 268 мкм). Значення LOD готової суміші були стійкими від 0,98 до 1,80 %. Окремі дані гранулометричного складу для гранулювання ефавіренцу показано у Таблиці 6. Таблиця 5 Дані про фізичні властивості порошкової суміші ефавіренцу та порошкової суміші емтрицитабіну/тенофовіру DF Номер партії Gilead AB507 AB508 AB509 AC507 AC508 AC509 Середньогеометричний діаметр частинок (мкм) 247 215 268 330 344 343 Об'ємна густина (г/мл) 0,56 0,55 0,54 0,60 0,60 0,59 LOD (%) 1,80 1,08 0,98 0,91 1,02 0,99 10 Таблиця 6 Гранулометричний склад для порошкових сумішей ефавіренцу та FTC/TDF a Номер партії Gilead AB507 AB508 AB509 AC507 AC508 AC509 15 20 25 30 20 (>850 мкм) 5,9 6,1 9,6 22,0 22,1 22,4 % маси, що утримується на ситі Стандартний розмір сита США (меш) 30 40 60 80 230 (600 мкм) (425 мкм) (250 мкм) (180 мкм) (63 мкм) 10,9 16,2 22,2 11,4 22,6 10,4 15,8 20,0 9,0 20,8 13,3 17,4 20,1 8,9 17,2 19,8 15,2 11,2 4,6 10,5 20,1 15,4 11,6 5,1 10,6 19,7 15,3 11,7 4,8 11,1 pan (< 63 мкм) 10,9 17,9 13,3 16,6 14,9 14,8 Сухе гранулювання емтрицитабіну/тенофовіру DF Емтрицитабін, мікрокристалічну целюлозу, тенофовір DF та кроскармелозу (Таблиця 2) змішували у 650 л резервуарі за допомогою змішувача Gallay протягом 10 хвилин. Додавали стеарат магнію (Таблиця 2) і змішували протягом додаткових 5 хвилин. Цю попередню суміш потім переносили до 320 л резервуара, оснащеного вивантажувальним пунктом з конусним затвором для сприяння перенесення матеріалу до бункера вальцьового компактора. Попередню суміш стискали на компакторі, застосовуючи Gerteis Macro-Pactor моделі 250/25/3 з вальцями 250 мM у діаметрі та 50 мM завширшки. Товщину зазору між вальцями (2 мм), швидкість обертання вальців (10 об./хв), силу стискання (4 кН/см), швидкість осцилюючого млина (75 об./хв за годинниковою стрілкою та проти годинникової стрілки) та отвір сита осцилюючого млина (1,25 мм) підтримували незмінними для всіх партій. Кут обертання осцилюючого млина також був незмінним для всіх партій і становив 150° за годинниковою стрілкою і 140° проти годинникової стрілки. Суттєвих проблем з жодною з трьох партій під час завантаження у вальцьовий компактор не було. Весь процес вальцьового стискання відбувався без будь-яких явних ознак накопичення теплоти на обладнанні, накопичення або розплавлення продукту. Потім гранули змішували з позагранулярною кроскармелозою натрієм (34 % від загальної кількості) та стеаратом магнію (47 % від загальної кількості). Розмір частинок, об'ємна густина та LOD сухого гранулювання емтрицитабіну/тенофовіру DF були подібними для всіх трьох партій і вказуються у Таблиці 5 (нижні 3 комірки). Геометричні розміри частинок були дуже подібними і становили від 330 до 344 мкм. Об'ємна густина становила від 0,59 до 0,60 г/мл. Показники LOD готової суміші були стійкими, від 0,91 до 1,02 %. 8 UA 108594 C2 5 10 15 20 25 30 35 40 45 50 55 60 Готові порошкові суміші мали дуже стійкі фізичні властивості. Гранули ефавіренцу та тенофовір DF мають середньогеометричний розмір частинок необов'язково у межах приблизно від 100 до 600 мкм, об'ємну густину необов'язково у межах приблизно від 0,1 до 1 г/мл і показники LOD необов'язково у межах приблизно від 0,1 до 10 % за масою. Готові суміші Масу гранул ефавіренцу та позагранулярного стеарату магнію відповідним чином регулювали на основі вихідної кількості сухих гранул емтрицитабіну/тенофовіру DF. Гранули ефавіренцу та сухі гранули емтрицитабіну/тенофовіру DF змішували у V-блендері об'ємом 3 кубічні фути протягом 10 хвилин. Додавали стеарат магнію і змішували протягом додаткових 5 хвилин. Зразки готової порошкової суміші брали з 10 різних місць після перемішування і аналізували на однорідність суміші. Готові порошкові суміші ефавіренцу та емтрицитабіну/тенофовіру DF демонстрували прийнятну однорідність і гомогенність суміші для всіх трьох активних інгредієнтів, що вказувало на стійкість композиції незалежно від розміру частинок або об'ємної густини сухих гранул емтрицитабіну/тенофовіру DF та гранул ефавіренцу. Процедура гранулювання та змішування має бути задовільною для рецептування у більших масштабах. Пресування ядра таблетки Готову порошкову суміш ефавіренцу/емтрицитабіну/тенофовіру DF пресували у ядра таблеток за допомогою преса для штампування двошарових таблеток, Stokes Genesis Model 757, з 41 позицією, оснащеного плоскими верхніми/ рельєфними "123" нижніми пуансонами у формі капсули (20,0 мM x 10,4 мм). Задана маса ядер таблеток становила 1550 мг. Зразки ядер таблеток брали щонайменше з 20 розташованих на однаковій відстані одне від одного місць під час циклу штампування і аналізували на однорідність вмісту. В цілому порошкові суміші задовільно піддавалися пресуванню на ротаційному пресі для таблетування щодо твердості, крихкості, товщини зовнішнього вигляду таблеток та мінливості маси таблетки. Операцію пресування здійснювали на швидкості приблизно 500 таблеток/хвилину (швидкість преса 12 об./хв) або приблизно 0,8 кг/хвилину для забезпечення задовільної однакової маси таблеток. Вкривання таблеток оболонкою Придатні оболонки для покриття вибирають шляхом традиційного відбору серед композицій серійного виробництва. Ці заходи є загальновідомими серед спеціалістів у даній галузі. Кожну партію ядер таблеток розділяли на дві підгрупи для нанесення оболонок і вкривали у 48дюймовому котлі Thomas Engineering COMPU-LAB, застосовуючи систему розпилення з подвійною насадкою. Усі ядра таблеток вкривали оболонками, застосовуючи 15 % (об'єм/об'єм) водну суспензію для покриття Opadry II Pink, яку застосовували протягом 24 годин з моменту приготування. Усі ядра таблеток вкривали до заданого збільшення маси у 3,0 % при заданій швидкості напилення 180 г/хв, що відповідає нормалізованій швидкості напилення від 1,5 до 2,3 г/хв/кг таблеток. Аналіз продуктів розпаду шляхом HPLC Таблетки ефавіренцу/емтрицитабіну/тенофовіру DF (таблетки EFV/FTC/TDF) аналізують за допомогою HPLC на EFV, FTC та TDF, застосовуючи зовнішні еталонні стандарти. Продукти розпаду EFV, FTC та TDF визначають за нормалізацією площі з відповідним застосуванням відносних коефіцієнтів чутливості. Ідентичність EFV, FTC та TDF підтверджується шляхом порівняння їх часу утримання з показниками еталонних стандартів. Приготування стандартних та випробуваних розчинів Стандартний та випробуваний розчинник 25 мM фосфатний буфер, pH 3 Зважують і переносять 3,4 г одноосновного фосфату калію, безводного, в 1 л мірну колбу. Додають приблизно 800 мл води і змішують до розчинення. Рівень pH доводять до 3,0±0,1 фосфорною кислотою, а потім розводять до потрібного об'єму водою. Випробуваний розчинник (40:30:30 25 мM фосфатний буфер, pH 3:ацетонітрил:метанол) Комбінують 400 мл 25 мM фосфатного буфера, pH 3, 300 мл ацетонітрилу та 300 мл метанолу і змішують. Дають врівноважитися до навколишньої температури. 50:50 ацетонітрилу:метанолу Комбінують 500 мл ацетонітрилу та 500 мл метанолу і змішують. Дають врівноважитися до навколишньої температури. Стандартний розчин Точно зважують приблизно 60 мг еталонного стандарту EFV, 20 мг еталонного стандарту FTC та 30 мг еталонного стандарту TDF і переносять у 100 мл мірну колбу. У колбу додають приблизно 80 мл випробуваного розчинника (40:30:30) і змішують або обробляють 9 UA 108594 C2 5 10 15 20 25 30 35 40 45 50 55 60 ультразвуком до розчинення. Розводять до потрібного об'єму випробуваним розчинником (40:30:30) і ретельно змішують. Кінцева концентрація кожного компонента становить приблизно 0,6 мг/мл EFV, 0,2 мг/мл FTC і 0,3 мг/мл TDF. Розчини для випробування придатності системи Стандарт для перевірки чутливості Приготовляють 10 мкг/мл вихідного розчину FTU шляхом точного зважування приблизно 10 мг автентичної речовини FTU з поміщенням у 100 мл мірну колбу. Додають випробуваний розчинник (40:30:30) до приблизно 80 % об'єму і змішують або обробляють ультразвуком до розчинення. Розводять до потрібного об'єму випробуваним розчинником (40:30:30) і ретельно змішують. За допомогою піпетки 10 мл цього розчину поміщають у 100 мл мірну колбу. Розводять до потрібного об'єму випробуваним розчинником (40:30:30) і ретельно змішують. Приготовляють стандарт для перевірки чутливості, який містить 0,2 мг/мл FTC та 0,2 мкг/мл FTU (0,10 % відносно FTC). Точно відважують 20 мг FTC у 100 мл мірну колбу. За допомогою піпетки Class A переносять 2,0 мл вихідного розчину FTU у ту ж саму колбу. У колбу ще додають випробуваний розчинник (40:30:30) і змішують або обробляють ультразвуком до розчинення. Розводять до потрібного об'єму випробуваним розчинником (40:30:30) і ретельно змішують. В альтернативному варіанті перед розведенням до потрібного об'єму додають 2,0 мл 10 мкг/мл вихідного розчину FTU до стандартного розчину. Приготування зразка для таблеток EFV/FTC/TDF Міцність та вміст продуктів розпаду в таблетках EFV/FTC/TDF визначають шляхом аналізу складеного розчину, приготовленого з десяти таблеток. Кінцева концентрація кожного компонента у випробуваному розчині становить приблизно 0,6 мг/мл EFV, 0,2 мг/мл FTC і 0,3 мг/мл TDF. a) Поміщають десять таблеток в 1 л мірну колбу і додають 400 мл 25 мM фосфатного буфера, pH 3, у цю мірну колбу. б) Змішують шляхом інтенсивного перемішування протягом приблизно 75 хвилин. в) У колбу додають 50:50 ацетонітрил:метанол до рівня приблизно на 2 см нижче мітки об'єму. г) Врівноважують розчин до навколишньої температури шляхом перемішування протягом години. Розводять до потрібного об'єму 50:50 ацетонітрилом:метанолом. Ретельно змішують шляхом перевертання колби або перемішування за допомогою магнітної мішалки. д) За допомогою шприцевого фільтра 0,45 мкм зі шприцом фільтрують приблизно 10 мл з етапу (г) для наступного розведення. Зливають перші 2 мл фільтрату. е) За допомогою піпетки Class A переносять 5,0 мл фільтрату з етапу (д) у 50 мл мірну колбу і розводять до потрібного об'єму випробуваним розчинником (40:30:30). Ретельно змішують. ХРОМАТОГРАФІЯ 1. Застосовують установку для HPLC, оснащену УФ-детектором та електронною системою збору даних. 2. Застосовують колонку для HPLC, вн. д. 4,6 мM на довжину 250 мM, набивка C12 з оберненням фаз, розмір частинок 4 мкм, розмір пор 80 Å. 3. Буфер мобільної фази: приготовляють 20 мM буфера ацетату амонію, pH 4,6; рівень pH у разі потреби регулюють оцтовою кислотою. 4. Градієнт мобільної фази: елююють з буфером мобільної фази:ацетонітрилом від 99:1 до 1:99 протягом 67 хвилин. 5. Виявлення піка: УФ при 262 нM 6. Об'єм ін'єкції: 10 мкл. За вказаних умов хроматографії піки часу утримання FTC, TDF та EFV становлять 11, 33 та 50 хвилин, відповідно. ПОСЛІДОВНІСТЬ ІН'ЄКЦІЙ Вводять випробуваний розчинник принаймні двічі як контроль для забезпечення врівноваження колонки і для розпізнавання будь-яких можливих паразитних піків. Вводять стандарт для перевірки чутливості або стандартний розчин, який містить приблизно 0,10 % FTU, для вимірювання чутливості виявлення. Здійснюють п'ять повторів введення стандартного розчину 1 (R1) з наступною однією ін'єкцією стандартного розчину 2 (R2). Розраховують теоретичні тарілки та коефіцієнта асиметрії піків для ін'єкцій стандартного розчину. Для визначення ідентичності, міцності та продуктів розпаду ін'єкції випробуваного розчину здійснюють двічі. Усі випробувані розчини повинні розмежовуватися ін'єкціями стандартного розчину. Як правило, рекомендується не більше десяти ін'єкцій випробуваного розчину між 10 UA 108594 C2 5 10 15 20 25 30 35 40 45 50 розмежовувальними ін'єкціями стандартного розчину. ПРИДАТНІСТЬ СИСТЕМИ Теоретичні тарілки та коефіцієнт асиметрії піків Розраховують кількість теоретичних тарілок (N) та коефіцієнти асиметрії (T) для піків EFV, FTC та TDF хроматограми стандартного розчину. Формули для визначення N та T вказуються у діючій Фармакопеї США. Значення цих параметрів повинні відповідати критеріям: N < 40 000, і 0,8 ≤ T ≥ 2,0. Перевірка чутливості Для перевірки чутливості використовують пік FTU у стандарті для перевірки чутливості, присутній при приблизно 0,10 %. Розраховують відсоток площі піка FTU за допомогою відповідного RRF (вказаного у Таблиці 2), який застосовують для стандарту для перевірки чутливості, за допомогою розрахунку відсотка окремого продукту розпаду. Порівнюють цей результат з теоретичним відсотком FTU для стандарту для перевірки чутливості таким чином: FTU Визначений Чутливість FTU Теоретични й Де: FTUВизначений = відсоток площі FTU, визначений для стандарту для перевірки чутливості або стандартного розчину FTUТеоретичний = теоретичний відсоток площі FTU для стандарту для перевірки чутливості або стандартного розчину Чутливість повинна становити 0,70-1,30. ОЦІНКА ТА РОЗРАХУНКИ Ідентифікація продуктів розпаду Застосовують відповідні параметри виявлення (такі, як пороговий пік, мінімальна площа піка і т. ін.), що дозволяє виявляти піки, присутні при 0,05 % або менше. Ідентифікують забруднення та продукти розпаду EFV, FTC та TDF, присутні у хроматограмах ін'єкцій випробуваного розчину, шляхом фіксації відносного часу утримання (RRT) вторинних піків, які спостерігалися, з відкиданням будь-яких піків, які не стосуються зразка. Визначають кількість лише продуктів розпаду. Розраховують середній показник результатів усіх ін'єкцій випробуваного розчину до найближчих 0,01 %. У випадках, коли продукт розпаду не виявляється або є нижчим за поріг включення до однієї ін'єкції та/або зразка, застосовують лише визначені результати розрахунку (тобто, не розглядають як нульове значення). час утримання вторинного піка RRT час утримання піка тенофовіру дизопрокси лу Показники RRT та відносного коефіцієнта чутливості (RRF) можливих забруднень та продуктів розпаду для EFV є показаними у Таблиці 1, і продукти показано жирним шрифтом. Забруднення та продукти розпаду для FTC є показаними у Таблиці 2, і продукти розпаду показано жирним шрифтом. Забруднення та продукти розпаду для TDF є показаними у Таблиця 3, і продукти розпаду показано жирним шрифтом. Оскільки RRT може бути різним, ідентичність забруднень та продуктів розпаду у разі потреби може бути підтверджена шляхом порівняння з автентичними речовинами (або з піками забруднень та продуктів розпаду в еталонному стандарті). Визначення вмісту продуктів розпаду Кількісне визначення продуктів розпаду FTC Визначають рівень кожного продукту розпаду FTC, який спостерігають у хроматограмах ін'єкцій випробуваного розчину за допомогою такої формули: І Продукт розпаду (%) х RRF х 100 ТРА Де: I = Площа піка продукту розпаду TPA = Загальна площа піка (площа FTC та всіх відповідних продуктів розпаду, за винятком забруднень та паразитних піків), скоректована за RRF RRF = Відносний коефіцієнт чутливості по відношенню до FTC 8.4.3 Визначення кількості продуктів розпаду TDF Визначають рівень кожного продукту розпаду TDF, який спостерігають у хроматограмах ін'єкцій випробуваного розчину за допомогою такої формули: І Продукт розпаду (%) х RRF х 100 ТРА Де: I = Площа піка продукту розпаду або вільного піка TPA = Загальна площа піка (площа головного піка TDF, усіх відповідних продуктів розпаду та 11 UA 108594 C2 5 10 всіх вільних піків, за винятком забруднень та паразитних піків), скоректована за RRF RRF = Відносний коефіцієнт чутливості по відношенню до TDF Результати та повідомлення Вміст продуктів розпаду Окремо повідомляють середній показник результатів для кожного продукту розпаду, які спостерігаються, до найближчих 0,01 %. Повідомляють загальний вміст продуктів розпаду EFV, FTC та TDF, відповідно, до найближчих 0,1 % як суму середніх рівнів усіх піків продуктів розпаду, які спостерігалися. Для продукти розпаду, виявлених на рівні, меншому за 0,05 %, ці рівні повідомляють як мікрокількість і не включають до розрахунку загального вмісту продуктів розпаду. Таблиця 1 Пов'язані з EFV забруднення та продукти розпаду Пов'язана з EFV сполука Приблизний RRT 1,46 1,50 1,50 1,57 1,60 1,73 1,83 c SD-573 d SR-695 EFV SP-234 SW-965 SE-563 c SM-097 a RRF 0,5 b 0,5 a Приблизні показники RRT та значення стосуються піка TDF RRF для пов'язаних з EFV продуктів розпаду стосуються EFV c Пов'язані з EFV продукти розпаду d SR-695 елююється перед EFV (інтервал приблизно 0,1 хв) b Продукти розпаду позначено жирним шрифтом Таблиця 2 Пов'язаний з FTC продукт розпаду Пов'язана з FTC сполука Приблизний RRT 0,33 0,38 FTC c FTU a RRF b 0,7 a Приблизні показники RRT та значення стосуються піка TDF RRF для пов'язаних з FTC продуктів розпаду стосуються FTC c Пов'язані з FTC продукти розпаду b 15 Таблиця 3 Пов'язані з тенофовіром DF продукти розпаду Пов'язана з TDF сполука c моно-POC PMPA c Змішаний димер TDF c Димер Приблизний RRT 0,47 0,98 1,00 1,34 a Приблизні показники RRT та значення стосуються піка TDF RRF для пов'язаних з TDF продуктів розпаду стосуються TDF c Пов'язані з TDF продукти розпаду b 12 a RRF 0,6 1,0 0,9 b UA 108594 C2 Джерела United States Pharmacopeia Pharmacopeial Forum 26(4) 2000. 5 10 15 20 25 30 35 40 45 ФОРМУЛА ВИНАХОДУ 1. Одинична дозована форма, яка включає тенофовір DF та емтрицитабін у першому окремому відділенні, одержаному шляхом сухого гранулювання, та ефавіренц з лаурилсульфатом натрію у другому окремому відділенні, і зазначені відділення є фізично сполученими та знаходяться у контакті один з одним. 2. Одинична дозована форма за п. 1, у якій перше та друге окремі відділення розташовані у вигляді шарів у контакті один з одним, причому тенофовір DF та емтрицитабін містяться у першому шарі, а ефавіренц та лаурилсульфат натрію містяться у другому шарі. 3. Одинична дозована форма за п. 2, у якій шари орієнтовані горизонтально уздовж осі дозованої форми. 4. Одинична дозована форма за п. 2, яка придатна для перорального введення. 5. Одинична дозована форма за п. 1, у якій друге окреме відділення одержане шляхом мокрого гранулювання з високим зсувним зусиллям. 6. Одинична дозована форма за п. 5, у якій загальна кількість ефавіренцу, емтрицитабіну та тенофовіру DF є більшою, ніж приблизно 60 % за масою композиції. 7. Одинична дозована форма за п. 6, яка додатково включає стеарат магнію, кроскармелозу натрію, мікрокристалічну целюлозу та гідроксипропілцелюлозу. 8. Одинична дозована форма за п. 7, у якій приблизний відсоток за масою ефавіренцу, тенофовіру DF, емтрицитабіну, стеарату магнію, кpocкapмeлози натрію, мікрокристалічної целюлози, лаурилсульфату натрію та гідроксипропілцелюлози становить, приблизно 39, приблизно 19, приблизно 13, приблизно 2, приблизно 7, приблизно 17, приблизно 1 та приблизно 2, відповідно. 9. Одинична дозована форма за п. 8, яка при введенні пацієнтові пероральним шляхом забезпечує для ефавіренцу, емтрицитабіну та тенофовіру DF практично такі самі AUC та Сmах, що й для затверджених FDA продуктів Truvada та Sustiva. 10. Одинична дозована форма за п. 9, яка має масу приблизно від 1200 мг до 2300 мг, включаючи будь-яку оболонку, що необов'язково може бути нанесена. 11. Контейнер, який включає одиничну дозовану форму за п. 1 та десикант. 12. Спосіб приготування дозованої форми за будь-яким з пп. 1-3, у якому: готують перше окреме відділення, що включає тенофовір DF та емтрицитабін, шляхом сухого гранулювання, готують друге окреме відділення, що включає ефавіренц та лаурилсульфат натрію, шляхом мокрого гранулювання, та фізично сполучують обидва відділення одне з одним. 13. Спосіб за п. 12, у якому зазначені відділення розміщують у шарах у контакті один з одним. 14. Спосіб за п. 12, у якому зі стеаратом магнію комбінують підданий сухому гранулюванню тенофовір DF та емтрицитабін, та зі стеаратом магнію комбінують підданий мокрому гранулюванню ефавіренц та лаурилсульфат натрію, а потім пресують у двошарову таблетку. 15. Спосіб антивірусної терапії, у якому пацієнтові, щопотребує антивірусної терапії, перорально вводять одиничну дозовану форму за будь-яким з пп. 1-3. 16. Спосіб за п. 15, у якому одиничну дозовану форму вводять тільки один раз на добу. 17. Спосіб за п. 16, у якому антивірусна терапія являє собою спрямовану проти ВІЛ терапію. 18. Двошарова таблетка, яка містить емтрицитабін та тенофовір DF у першому шарі та ефавіренц і лаурилсульфат натрію у другому шарі, причому зазначені шари знаходяться у контакті один з одним. 19. Двошарова таблетка за п. 18, яка має масу менше ніж 2,5 грама. 50 Комп’ютерна верстка В. Мацело Державна служба інтелектуальної власності України, вул. Василя Липківського, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 13
ДивитисяДодаткова інформація
Автори англійськоюDahl Terrence C., Menning, Mark, M., Oliyai, Reza
Автори російськоюДахл Терренце К., Олияй Реза
МПК / Мітки
МПК: A61K 31/535, A61K 31/675, A61K 31/513, A61K 9/26, A61K 9/24, A61P 31/18
Мітки: одинична, дозована, форма, фармацевтична
Код посилання
<a href="https://ua.patents.su/15-108594-odinichna-dozovana-farmacevtichna-forma.html" target="_blank" rel="follow" title="База патентів України">Одинична дозована фармацевтична форма</a>
Фармацевтична одинична дозована форма опіоїдного анальгетика та інгібітора циклооксигенази-2

Номер патенту: 72193
Опубліковано: 15.02.2005
Автори: Берч Рональд М., Голденхейм Пол Д., Саклер Річард С.
МПК: A61K 9/20, A61K 9/28, A61K 9/48, A61F 2/02
Мітки: форма, дозована, опіоїдного, циклооксигенази-2, одинична, інгібітора, фармацевтична, анальгетика
Формула / Реферат:
1. Фармацевтична одинична дозована форма, що містить аналгезивну комбінацію, яка містить (а) інгібітор СОХ-2 та/або щонайменше одну його фармацевтично прийнятну сіль і (б) оксикодон та/або щонайменше одну його фармацевтично прийнятну сіль, причому зазначений інгібітор СОХ-2 має щонайменше в 9 разів більшу специфічність по відношенню до СОХ-2, ніж до СОХ-1, як in vivo (що визначається шляхом вимірювання ED50), так і/або in vitro (що...
Оральна фармацевтична складова одинична дозована форма у вигляді таблетки, спосіб її одержання, упаковка у вигляді блістера та спосіб інгібування секреції шлункової кислоти і/або лікування шлунково-кишкових зап

Номер патенту: 41946
Опубліковано: 15.10.2001
Автори: ЛЕВГРЕН Курт Інгмар, БЕРГСТРАНД Понтус Йохн Арвід
МПК: A61K 9/26, A61K 31/444, A61P 1/04
Мітки: форма, шлункової, фармацевтична, одинична, оральна, зап, шлунково-кишкових, одержання, складова, кислоти, вигляді, дозована, інгібування, спосіб, упаковка, секреції, лікування, блістера, таблетки
Формула / Реферат:
1. Оральная фармацевтическая составная единичная дозированная форма в виде таблетки, включающая наполнители для таблеток и индивидуально покрытые слоями энтеросолюбильного покрытия единицы материала сердцевины, содержащие активное вещество в виде омепразола или одного из его индивидуальных энантиомеров или щелочной соли омепразола или щелочной соли одного из его индивидуальных энантиомеров, покрытые одним или более слоем(ями), по крайней...
Одинична дозована форма, що включає прищеплений співполімер полівінілового спирту-поліетиленгліколю і стероїдний естроген

Номер патенту: 102680
Опубліковано: 12.08.2013
Автори: Функе Адріан, Генераль Саша, Теребезі Ільдіко
МПК: A61K 9/00, A61K 47/30, A61K 31/56, A61P 15/00
Мітки: полівінілового, стероїдний, включає, прищеплений, естроген, спирту-поліетиленгліколю, форма, дозована, одинична, спів)полімер
Формула / Реферат:
1. Одинична дозована форма, що включає матрикс тонкої водорозчинної плівки, де зазначений матрикс плівки включаєa) прищеплений співполімер полівінілового спирту-поліетиленгліколю (PVA-PEG прищеплений співполімер) як водорозчинний матриксний співполімер;b) активний інгредієнт, що являє собою стероїдний естроген, вибраний з...
Тверда фармацевтична дозована форма

Номер патенту: 106634
Опубліковано: 25.09.2014
Автори: Пернчоб Нантарат, Шнайдер Петер, Айзенрайх Вольфрам
МПК: A61P 3/10, A61K 31/351, A61K 31/155, A61K 9/20
Мітки: тверда, фармацевтична, дозована, форма
Формула / Реферат:
1. Тверда фармацевтична дозована форма, що включає інгібітор SGLT-2-1-хлор-4-(β-D-глюкопіраноз-1-ил)-2-[4-((S)-тетрагідрофуран-3-ілокси)бензил]бензол, інший лікарський засіб - гідрохлорид метформіну і один або більше фармацевтичних ексципієнтів,де діапазон доз інгібітора SGLT-2 становить від приблизно 1 мг до приблизно 25 мг, іде діапазон доз гідрохлориду метформіну становить від приблизно 100 мг до приблизно 1500 мг,...
Фармацевтична дозована форма

Номер патенту: 81279
Опубліковано: 25.12.2007
Автори: Довгий Віктор Петрович, Вараксін Максім Ігорович, Сєдова Наталія Олександрівна, Вараксін Ігор Вікторович
МПК: A61K 9/44, A61K 9/22, A61K 9/52, A61K 9/32, A61K 9/68
Мітки: форма, дозована, фармацевтична
Формула / Реферат:
1. Фармацевтична дозована форма з регульованим вивільненням і перемінними швидкостями вивільнення, що містить полімерну оболонку й один або більше фармацевтично активних інгредієнтів, яка відрізняється тим, що фармацевтично активний інгредієнт у рідкому або желеподібному стані знаходиться в герметичній еластичній капсулі з полімерної нерозчинної у воді, слині і шлунковому соці оболонці, що має концентратори напруг.2. Фармацевтична...
Попередній патент: Спосіб пакування та зберігання свіжої зелені
Наступний патент: Шлагбаум
Випадковий патент: Карбюраторний електроагрегат