Заміщені дигідро-3-гало-1н-піразол-5-карбоксилати та способи їх одержання
Номер патенту: 81104
Опубліковано: 10.12.2007
Автори: Селбі Томас Пол, Стівенсон Томас Мартін, Лам Джордж Філіп, Фройденбергер Джон Герберт







Формула / Реферат
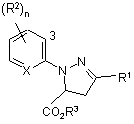
1. Сполука формули І
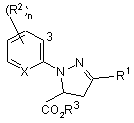 , (I)
, (I)
де R1 являє собою галоген;
кожна R2 являє собою незалежно C1-C4 алкіл, С2-С4 алкеніл, С2-С4 алкініл, С3-С6 циклоалкіл, C1-C4 галоалкіл, С2-С4 галоалкеніл, С2-С4 галоалкініл, С3-С6 галоциклоалкіл, галоген, CN, NO2, C1-C4 алкокси, С1-С4 галоалкокси, C1-C4 алкілтіо, C1-C4 алкілсульфініл, C1-C4 алкілсульфоніл, C1-C4 алкіламіно, С2-C8 діалкіламіно, C3-C6 циклоалкіламіно, C3-C6 (алкіл)циклоалкіламіно, С2-С4 алкілкарбоніл, C2-C6 алкоксикарбоніл, C2-C6 алкіламінокарбоніл, С3-C8 діалкіламінокарбоніл або C3-C6 триалкілсиліл;
R3 являє собою Η або C1-C4 алкіл;
Χ являє собою N або CR4;
R4 являє собою Η або R2; та
n дорівнює 0-3, за умови, коли Χ являє собою СН, n дорівнює принаймні 1.
2. Сполука за п. 1, яка відрізняється тим, що n дорівнює 1-3.
3. Сполука за п. 1, яка відрізняється тим, що R1 являє собою Сl або Вr; кожна R2 є незалежно Сl або Вr, і одна R2 знаходиться у 3-положенні; і Χ є N.
4. Спосіб одержання сполуки формули І, що включає обробку сполуки формули 4
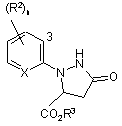 , (4)
, (4)
де R2, Х та n визначено в п. 1, а R3 являє собою C1-C4 алкіл, галогенуючим агентом;
та необов’язково перетворення утвореної сполуки у сполуку, де R3 являє собою Н.
5. Спосіб за п. 4, який відрізняється тим, що n дорівнює 1-3.
6. Спосіб за п. 4, який відрізняється тим, що R1 являє собою Сl або Вr; кожна R2 є незалежно Сl або Вr і одна R2 знаходиться у 3-положенні; R3 являє собою С1-С4 алкіл; і Χ є N.
7. Спосіб за п. 6, який відрізняється тим, що галогенуючим агентом є оксигалогенід фосфору або пентагалогенід фосфору.
8. Спосіб за п. 7, який відрізняється тим, що його проводять за відсутності основи з використанням ацетонітрилу як розчинника.
9. Сполука формули II
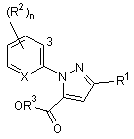 , (II)
, (II)
де
R1 являє собою галоген;
кожна R2 являє собою незалежно С1-С4 алкіл, С2-С4 алкеніл, С2-С4 алкініл, C3-C6 циклоалкіл, С1-С4 галоалкіл, С2-С4 галоалкеніл, С2-С4 галоалкініл, C3-C6 галоциклоалкіл, галоген, CN, NO2, С1-С4 алкокси, C1-C4 галоалкокси, C1-C4 алкілтіо, C1-C4 алкілсульфініл, C1-C4 алкілсульфоніл, C1-C4 алкіламіно, C2-C8 діалкіламіно, C3-C6 циклоалкіламіно, C3-C6 (алкіл)циклоалкіламіно, С2-С4 алкілкарбоніл, С2-С6 алкоксикарбоніл, C2-C6 алкіламінокарбоніл, С3-С8 діалкіламінокарбоніл або C3-C6 триалкілсиліл;
R3 являє собою Η або С1-С4 алкіл;
Χ являє собою N або CR4;
R4 являє собою Η або R2; та
n дорівнює 0-3, за умови, коли Χ являє собою СН, n дорівнює принаймні 1.
10. Сполука за п. 9, яка відрізняється тим, що n дорівнює 1-3.
11. Сполука за п. 9, яка відрізняється тим, що R1 являє собою Сl або Вr; кожна R2 є незалежно Сl або Вr, і одна R2 знаходиться у 3-положенні; і Χ є N.
12. Спосіб одержання сполуки формули II за п. 9, що включає обробку сполуки формули І, де R3 є C1-C4 алкілом,
 , (I)
, (I)
R1, R2, Х та n визначено в п. 1,
окисником, при потребі у присутності кислоти;
та необов’язково перетворення утвореної сполуки у сполуку формули II, де R3 є Н.
13. Спосіб за п. 12, який відрізняється тим, що n дорівнює 1-3.
14. Спосіб за п. 12, який відрізняється тим, що окисником є пероксид або персульфатна сіль.
15. Спосіб за п. 14, який відрізняється тим, що Χ являє собою CR4; і окисником є пероксид водню.
16. Спосіб за п. 14, який відрізняється тим, що Χ є Ν; окисником є персульфат калію; і який проводять у присутності сірчаної кислоти.
17. Спосіб за п. 12, який відрізняється тим, що у формулі І, де R1 являє собою Сl або Вr, кожна R2 є незалежно Сl або Вr, і одна R2 знаходиться у 3-положенні, R3 являє собою С1-С4 алкіл; і Χ є N.
18. Спосіб за п. 12, який відрізняється тим, що сполуку формули І одержують способом, який включає (1) обробку сполуки формули 4
, (4)
де R3 являє собою С1-С4 алкіл,
галогенуючим агентом для одержання сполуки формули І;
(2) необов’язково перетворення утвореної в (1) сполуки у сполуку, де R3 є Н.
19. Сполука формули 4
, (4)
де
кожна R2 являє собою незалежно С1-С4 алкіл, С2-С4 алкеніл, С2-С4 алкініл, C3-C6 циклоалкіл, C1-C4 галоалкіл, С2-С4 галоалкеніл, С2-С4 галоалкініл, C3-C6 галоциклоалкіл, галоген, CN, NO2, C1-C4 алкокси, С1-С4 галоалкокси, С1-С4 алкілтіо, C1-C4 алкілсульфініл, C1-C4 алкілсульфоніл, C1-C4 алкіламіно, C2-C8 діалкіламіно, C3-C6 циклоалкіламіно, C3-C6 (алкіл)циклоалкіламіно, С2-С4 алкілкарбоніл, С2-С6 алкоксикарбоніл, C2-C6 алкіламінокарбоніл, С3-C8 діалкіламінокарбоніл або C3-C6 триалкілсиліл;
Χ являє собою Ν;
R3 являє собою Н або С1-С4 алкіл; і
n дорівнює 0-3, за умови, коли Χ являє собою СН, n дорівнює принаймні 1.
20. Сполука за п. 19, яка відрізняється тим, що n дорівнює 1-3.
21. Сполука за п. 19, яка відрізняється тим, що кожна R2 являє собою незалежно Сl або Вr, і одна R2 знаходиться у 3-положенні.
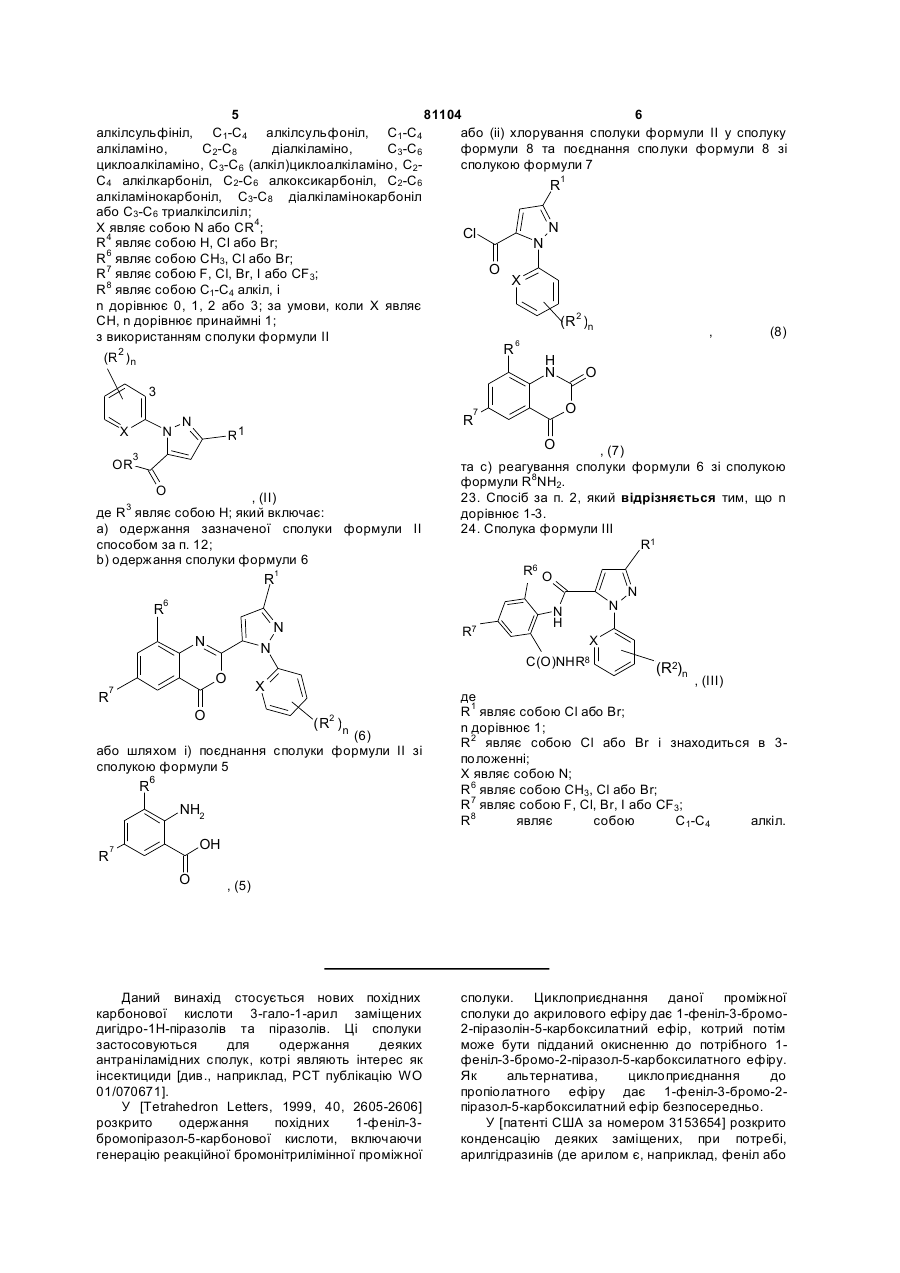
22. Спосіб одержання сполуки формули III
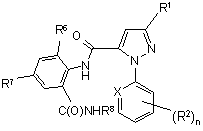 , (ІІІ)
, (ІІІ)
де
R1 являє собою галоген;
кожна R2 являє собою незалежно C1-C4 алкіл, С2-С4 алкеніл, С2-С4 алкініл, C3-C6 циклоалкіл, C1-C4 галоалкіл, С2-С4 галоалкеніл, С2-С4 галоалкініл, C3-C6 галоциклоалкіл, галоген, CN, NО2, C1-C4 алкокси, C1-C4 галоалкокси, C1-C4 алкілтіо, C1-C4 алкілсульфініл, C1-C4 алкілсульфоніл, C1-C4 алкіламіно, С2-С8 діалкіламіно, C3-C6 циклоалкіламіно, C3-C6 (алкіл)циклоалкіламіно, С2-С4 алкілкарбоніл, С2-С6 алкоксикарбоніл, С2-С6 алкіламінокарбоніл, С3-C8 діалкіламінокарбоніл або C3-C6 триалкілсиліл;
Χ являє собою N або CR4;
R4 являє собою Н, Сl або Вr;
R6 являє собою СН3, Сl або Вr;
R7 являє собою F, Сl, Вr, І або СF3;
R8 являє собою С1-С4 алкіл, і
n дорівнює 0, 1, 2 або 3; за умови, коли Χ являє СН, n дорівнює принаймні 1;
з використанням сполуки формули II
, (II)
де R3 являє собою Η; який включає:
а) одержання зазначеної сполуки формули II способом за п. 12;
b) одержання сполуки формули 6
 (6)
(6)
або шляхом і) поєднання сполуки формули ІІ зі сполукою формули 5
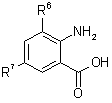 , (5)
, (5)
або (іі) хлорування сполуки формули ІІ у сполуку формули 8 та поєднання сполуки формули 8 зі сполукою формули 7
 , (8)
, (8) 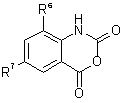 , (7)
, (7)
та с) реагування сполуки формули 6 зі сполукою формули R8NH2.
23. Спосіб за п. 2, який відрізняється тим, що n дорівнює 1-3.
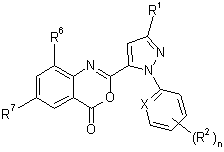
24. Сполука формули ІІІ
, (ІІІ)
де
R1 являє собою Сl або Вr;
n дорівнює 1;
R2 являє собою Сl або Вr і знаходиться в 3-положенні;
Χ являє собою N;
R6 являє собою СН3, Сl або Вr;
R7 являє собою F, Сl, Вr, І або СF3;
R8 являє собою С1-С4 алкіл.
Текст
1. Сполука формули І (R2 )n 2 (13) 1 3 9. Сполука формули II 2 (R ) n 3 X N N 81104 4 18. Спосіб за п. 12, який відрізняється тим, що сполуку формули І одержують способом, який включає (1) обробку сполуки формули 4 (R2)n 3 R1 X 3 OR O , (II) де R1 являє собою галоген; кожна R2 являє собою незалежно С1-С4 алкіл, С2С4 алкеніл, С2-С4 алкініл, C3-C6 циклоалкіл, С1-С4 галоалкіл, С2-С4 галоалкеніл, С2-С4 галоалкініл, C3C6 галоциклоалкіл, галоген, CN, NO2, С1-С4 алкокси, C1-C4 галоалкокси, C1-C4 алкілтіо, C1-C4 алкілсульфініл, C1-C4 алкілсульфоніл, C1-C4 алкіламіно, C2-C8 діалкіламіно, C3-C6 циклоалкіламіно, C3-C6 (алкіл)циклоалкіламіно, С2С4 алкілкарбоніл, С2-С6 алкоксикарбоніл, C2-C6 алкіламінокарбоніл, С3-С8 діалкіламінокарбоніл або C3-C6 триалкілсиліл; R3 являє собою Η або С1-С4 алкіл; Χ являє собою N або CR4; R4 являє собою Η або R2; та n дорівнює 0-3, за умови, коли Χ являє собою СН, n дорівнює принаймні 1. 10. Сполука за п. 9, яка відрізняється тим, що n дорівнює 1-3. 11. Сполука за п. 9, яка відрізняється тим, що R1 являє собою Сl або Вr; кожна R2 є незалежно Сl або Вr, і одна R2 знаходиться у 3-положенні; і Χ є N. 12. Спосіб одержання сполуки формули II за п. 9, що включає обробку сполуки формули І, де R3 є C1-C4 алкілом, (R2)n 3 X N N CO2R3 R1 , (I) R1, R2, Х та n визначено в п. 1, окисником, при потребі у присутності кислоти; та необов’язково перетворення утвореної сполуки у сполуку формули II, де R3 є Н. 13. Спосіб за п. 12, який відрізняється тим, що n дорівнює 1-3. 14. Спосіб за п. 12, який відрізняється тим, що окисником є пероксид або персульфатна сіль. 15. Спосіб за п. 14, який відрізняється тим, що Χ являє собою CR4; і окисником є пероксид водню. 16. Спосіб за п. 14, який відрізняється тим, що Χ є Ν; окисником є персульфат калію; і який проводять у присутності сірчаної кислоти. 17. Спосіб за п. 12, який відрізняється тим, що у формулі І, де R1 являє собою Сl або Вr, кожна R2 є незалежно Сl або Вr, і одна R2 знаходиться у 3положенні, R3 являє собою С1-С4 алкіл; і Χ є N. N H N O CO2R3 , (4) де R3 являє собою С1-С4 алкіл, галогенуючим агентом для одержання сполуки формули І; (2) необов’язково перетворення утвореної в (1) сполуки у сполуку, де R3 є Н. 19. Сполука формули 4 (R2)n 3 X N H N O CO2R3 , (4) де кожна R2 являє собою незалежно С1-С4 алкіл, С2С4 алкеніл, С2-С4 алкініл, C3-C6 циклоалкіл, C1-C4 галоалкіл, С2-С4 галоалкеніл, С2-С4 галоалкініл, C3C6 галоциклоалкіл, галоген, CN, NO2, C1-C4 алкокси, С1-С4 галоалкокси, С1-С4 алкілтіо, C1-C4 алкілсульфініл, C1-C4 алкілсульфоніл, C1-C4 алкіламіно, C2-C8 діалкіламіно, C3-C6 циклоалкіламіно, C3-C6 (алкіл)циклоалкіламіно, С2С4 алкілкарбоніл, С2-С6 алкоксикарбоніл, C2-C6 алкіламінокарбоніл, С3-C8 діалкіламінокарбоніл або C3-C6 триалкілсиліл; Χ являє собою Ν; R3 являє собою Н або С1-С4 алкіл; і n дорівнює 0-3, за умови, коли Χ являє собою СН, n дорівнює принаймні 1. 20. Сполука за п. 19, яка відрізняється тим, що n дорівнює 1-3. 21. Сполука за п. 19, яка відрізняється тим, що кожна R2 являє собою незалежно Сl або Вr, і одна R2 знаходиться у 3-положенні. 22. Спосіб одержання сполуки формули III R1 R6 O R7 N N H N X C(O)NHR8 (R2)n , (ІІІ) де R1 являє собою галоген; кожна R2 являє собою незалежно C1-C4 алкіл, С2С4 алкеніл, С2-С4 алкініл, C3-C6 циклоалкіл, C1-C4 галоалкіл, С2-С4 галоалкеніл, С2-С4 галоалкініл, C3C6 галоциклоалкіл, галоген, CN, NО2, C1-C4 алкокси, C1-C4 галоалкокси, C1-C4 алкілтіо, C1-C4 5 81104 6 алкілсульфініл, C1-C4 алкілсульфоніл, C1-C4 або (іі) хлорування сполуки формули ІІ у сполуку алкіламіно, С2-С8 діалкіламіно, C3-C6 формули 8 та поєднання сполуки формули 8 зі циклоалкіламіно, C3-C6 (алкіл)циклоалкіламіно, С2сполукою формули 7 1 С4 алкілкарбоніл, С2-С6 алкоксикарбоніл, С2-С6 R алкіламінокарбоніл, С3-C8 діалкіламінокарбоніл або C3-C6 триалкілсиліл; Χ являє собою N або CR4; N Cl R4 являє собою Н, Сl або Вr; N R6 являє собою СН3, Сl або Вr; O R7 являє собою F, Сl, Вr, І або СF3; X R8 являє собою С1-С4 алкіл, і n дорівнює 0, 1, 2 або 3; за умови, коли Χ являє СН, n дорівнює принаймні 1; (R2 )n , (8) з використанням сполуки формули II 6 R 2 (R ) n H N O 3 O 7 R N X N R1 O , (7) 3 OR та с) реагування сполуки формули 6 зі сполукою формули R8NH2. O , (II) 23. Спосіб за п. 2, який відрізняється тим, що n де R3 являє собою Η; який включає: дорівнює 1-3. а) одержання зазначеної сполуки формули II 24. Сполука формули ІІІ способом за п. 12; R1 b) одержання сполуки формули 6 R6 O 1 R N 6 N R N H N R7 X N N O 7 C(O)NHR8 X R O (R2 )n (6) або шляхом і) поєднання сполуки формули ІІ зі сполукою формули 5 6 R NH2 R (R2)n , (ІІІ) де R1 являє собою Сl або Вr; n дорівнює 1; R2 являє собою Сl або Вr і знаходиться в 3положенні; Χ являє собою N; R6 являє собою СН3, Сl або Вr; R7 являє собою F, Сl, Вr, І або СF3; R8 являє собою С1-С4 алкіл. OH 7 O , (5) Даний винахід стосується нових похідних карбонової кислоти 3-гало-1-арил заміщених дигідро-1Н-піразолів та піразолів. Ці сполуки застосовуються для одержання деяких антраніламідних сполук, котрі являють інтерес якінсектициди [див., наприклад, РСТ публікацію WO 01/070671]. У [Tetrahedron Letters, 1999, 40, 2605-2606] розкрито одержання похідних 1-феніл-3бромопіразол-5-карбонової кислоти, включаючи генерацію реакційної бромонітрилімінної проміжної сполуки. Циклоприєднання даної проміжної сполуки до акрилового ефіру дає 1-феніл-3-бромо2-піразолін-5-карбоксилатний ефір, котрий потім може бути підданий окисненню до потрібного 1феніл-3-бромо-2-піразол-5-карбоксилатного ефіру. Як альтернатива, циклоприєднання до пропіолатного ефіру дає 1-феніл-3-бромо-2піразол-5-карбоксилатний ефір безпосередньо. У [патенті США за номером 3153654] розкрито конденсацію деяких заміщених, при потребі, арилгідразинів (де арилом є, наприклад, феніл або 7 81104 8 нафтил, котрі заміщені, при потребі, нижчим Даний винахід також стосується сполуки алкілом, нижчим алкокси або галогеном) з деякими Формули II ефірами фумарової або малеїнової кислоти з одержанням похідних 3-піразолідинон карбонової кислоти. У неперевірених [японських патентних публікаціях за номерами 9-316055 та 9-176124] розкрито одержання похідних ефіру піразолкарбонової кислоти та піразолінових похідних, відповідно, коїрі заміщені алкілом у 1-му положенні. [J. Med. Chem. 2001, 44, 566-578] розкриває одержання 1-(3-ціанофеніл)-3-метил-1Н-піразол-5де R1 являє собою галоген (і X, R2, R3 та n є карбонової кислоти та її застосування в одержанні такими, як визначено вище для Формули І), і інгібіторів фактора зсілості крові Х а. способу одержання сполуки Формули II. Даний Даний винахід запроваджує технологію для спосіб включає (3) обробку сполуки Формули І зручного одержання 3-гало-5-карбоксилат-1-арилокисником, при потребі, у присутності кислоти з заміщених дигідро-1Н-піразолів та піразолів. утворенням сполуки Формули II; і коли сполука Даний винахід стосується сполуки Формули І Формули І, де R3 являє собою С1-С4 алкіл, використовується для приготування сполуки Формули II, де R3 є Н, (4) перетворення сполуки, утвореної в (2), в сполуку Формули ll, де R3 є Н. Даний винахід також запроваджує сполуки Формули 4, де X являє собою Ν, та їх використання у приготуванні сполук Формул І та II, де X являє собою N (і R2, R3 та n є такими, як визначено вище для Формули І). Даний винахід також включає спосіб одержання сполуки Формули III, де R1 являє собою галоген; кожна R2 являє собою, незалежно, С1-С4 алкіл, С2-С4 алкеніл, С2-С4 алкініл, С3-С6 циклоалкіл, С1С4 галоалкіл, С2-С4 галоалкеніл, С2-С4 галоалкініл, С3-С6 галоциклоалкіл, галоген, CN, NO2, С1-С4 алкокси, С1-С4 галоалкокси, С1-С4 алкілтіо, С1-С4 алкілсульфініл, С1-С4 алкілсульфоніл, С1-С4 алкіламіно, С2-C8 діалкіламіно, С3-С6 циклоалкіламіно, С3-С6 (алкіл)циклоалкіламіно, С2С4 алкілкарбоніл, С2-C6 алкоксикарбоніл, С2-С6 де X, R1, R2 та n є такими, як визначено вище алкіламінокарбоніл, С3-C8 діалкіламінокарбоніл для Формули II; R6 являє собою СН3, СІ або Br; R7 абo С3-С6 триалкілсиліл; являє собою F, СІ, Br, І або СF3, і R8 являє собою R3 являє собою N або С1-С4 алкіл; 4 С1-С4 алкіл, з використанням сполуки Формули II, X являє собою N або CR ; де R6 є Н. Цей спосіб характеризується R4 являє собою N або R2; та приготуванням сполуки Формули II за способом, як n дорівнює 0-3, за умови, коли X являє СН, n зазначено вище. дорівнює принаймні 1. У лищенаведеному викладі вираз "алкіл", що Даний винахід стосується також способу використовується окремо або у складних словах, одержання сполуки Формули І, що включає (1) таких як "алкілтіо" або "галоалкіл", включає алкіли обробку сполуки Формули 4 з прямими або розгалуженими ланцюгами, такі як метил, етил, n-пропіл, і-пропіл, або різні бутилові, пентилові чи гексилові ізомери. "Алкеніл" може включати алкени з прямими або розгалуженими ланцюгами, такі як 1-пропеніл, 2-пропеніл, та інші бутенілові, пентенілові та гексенілові ізомери. "Алкеніл" також включає полієни, такі як 1,2пропадієніл та 2,4-гексадієніл. "Алкініл" включає Г алкіни з прямими та розгалуженими ланцюгами, 2 такі як 1-пропініл, 2-пропініл та різні бутинілові, (де X, R та n є такими, як описано вище для пентинілові та гексинілові ізомери. "Алкініл" може Формули І, і R3 являє собою С1-С4 алкіл) також включати складові, що мають множину галогенуючим агентом з утворенням сполуки потрійних зв'язків, такі як 2,5-гексадіініл. "Алкокси" Формули І; і при одержанні сполук Формули І, де включає, наприклад, метокси, етокси, пR3 являє собою Н, (2) перетворення сполуки, 3 пропілокси. ізопропілокси та різні бутокси, утвореної в (1), у сполуку, де R є Н. пентокси і гексилокси ізомери. "Алкоксиалкіл" 9 81104 10 означає алкокси заміщення по алкілу. Приклади "алкоксиалкілу" включають СН3ОСН2, СН3ОСН2СН2, СН3СН2 ОСН2, СН3СН2СН2СН2ОСН2 та СН3СН2ОСН2СН2. "Алкілтіо" включає алкілтіо складові з розгалуженими або прямими ланцюгами, такі як метилтіо, етилтіо, і різні де R2 і n є такими, як визначено вище, і "3" пропілтіо, бутилтіо, пентилтіо та гексилтіо ізомери. вказує на 3-положення для замісників на даній "Циклоалкіл" включає, наприклад, циклопропіл, складовій. циїлобутил, циклопентил та циклогексил. Вираз "галоген", як окремо, так і у складних "Циклоалкілалкіл" вказує на алкільну групу, що словах, таких як "галоалкіл", включає фтор, хлор, заміщена циклоалкільною групою, і включає, бром або йод. Крім того, при застосуванні у наприклад, циклопропілметил, циклобутилетил. складних словах, таких як "галоалкіл", зазначений циклопентилпропіл та циклогексилметил. алкіл може бути частково або цілком заміщений "Циклоалкіламіно" означає, що амінний азотний атомами галогену, котрі можуть бути тими самими атом приєднаний до циклоалкільного радикалу та або різними. Приклади "галоалкілу" включають атому водню, і включає такі групи як F3C, СІСН2, CF3CH2 та CF3CCI 2. Вирази циклопропіламіно, циклобутиламіно, "галоалкеніл", "галоалкініл", галоалкокси" і подібні циклопентиламіно та циклогексиламіно. визначаються аналогічно до виразу "галоалкіл". "(Алкіл)циклоалкіламіно" означає Приклади "галоалкенілу" включають (Сl)2С=СНСН2 циклоалкіламінну групу, де атом водню заміщений та СF3СН2СН= СНСН2. Приклади "галоалкокси" алкілрадикалом; приклади включають групи, такі включають CF3O, CCI 3CH2O, HCF2CH2CH2O та як (алкіл)циклопропіламіно, CF3CH2O. (алкіл)циклобутиламіно, (алкіл)циклопентиламіно Приклади "алкілкарбонілу" включають та (алкіл)циклогексиламіно. Краще, коли алкілом у С(О)СН3, С(О)СН2СН2СН3 та С(О)СН(СН3)2 (алкіл)циклоалкіламіно є С 1-С4 алкіл, тоді як Приклади "алкоксикарбонілу" включають циклоалкілом у циклоалкіламіно та СН3ОС(=О), СН3СН2ОС(=О), СН3СН2СН2ОС(=О), (алкіл)циклоалкіламіно є С 3-С6 циклоалкіл. (СН3)2СНОС(=О) та різні бутоксиабо Вираз "арил" у даній заявці стосується пентоксикарбонільні ізомери. Вирази ароматичного кільця або кільцевої системи, або "алкіламінокарбоніл" та "діалкіламінокарбоніл" гетероароматичного кільця чи кільцевої системи, включають, наприклад, СН3 NНС(=О), що, при потребі, заміщена. Вираз "ароматична CH3CH2NHC(=O) та (CH3)2NC(=O). кільцева система" означає цілком ненасичені Загальна кількість вуглецевих атомів у групі карбоцикли та гетероцикли, в яких принаймні одне замісника вказується префіксом- "Сi-Сj", де і та j кільце поліциклічної кільцевої системи є дорівнюють 1-8. Наприклад, С1-С3 алкілсульфоніл ароматичним. Ароматичний вказує на те. що відповідає сполукам від метилсульфонілу до кожен із кільцевих атомів знаходиться суттєво в пропілсульфонілу. У вищезазначеному викладі, одній і тій же площині, і має р-орбіталь, коли сполука Формули І містить гетгроароматичне перпендикулярну до площини даного кільця, і де кільце, всі замісники приєднані до даного кільця (4n+2) p електронів, коли n є 0 або цілим через наявний вуглець або азот шляхом позитивним числом, зв'язані з даним кільцем заміщення водню на зазначених атомах вуглецю згідно з правилом Гюккеля. Вираз "ароматична чи азоту. карбоциклічна кільцева система" включає цілком Коли група містить замісника, який може бути ароматичні карбоцикли та карбоцикли, у яких воднем, наприклад, R4, тоді, якщо цей замісник принаймні одне кільце поліциклічної кільцевої береться як водень, вважається, що це системи є ароматичним (наприклад, феніл та еквівалентно тому, що зазначена група нафтил). Вираз "гетероароматичне кільце або незаміщена. кільцева система" включає цілком ароматичні Деякі сполуки даного винаходу можуть гетероцикли та гетероцикли, у яких принаймні існувати як один або більше стереоізомерів. Різні одне кільце поліциклічної кільцевої системи є стереоізомери включають енантіомери, ароматичним, і у яких принаймні один кільцевий діастереомери, атропоізомери та геометричні атом не є вуглецем, і може містити 1-4 ізомери. Фахівцеві у даній галузі зрозуміло, що гетероатоми, що вибираються незалежно із групи, один стереоізомер може бути більш активним котра складається із азоту, кисню та сірки, за та/або може виявляти сприятливі ефекти, коли умови, що кожне гетероароматичне кільце містить його відносна кількість перевищує кількість інших не більше 4 азотів, не більше 2 кленів та не стереоізомерів, або коли він відокремлений від більше 2 сірок (де ароматичний вказує на те, що інших стереоізомерів. Крім того, фахівцеві відомі правило Гюккеля задовольняється). Зазначені способи відокремлення, збагачення та/або гетероциклічні кільцеві системи можуть бути селективного одержання зазначених приєднані через наявний вуглець або азот шляхом стереоізомерів. заміщення водню на зазначених атомах вуглецю Відповідно, сполуки даного винаходу можуть або азоту. У більш конкретному викладі, вираз бути присутніми як суміш стереоізомерів, окремі "арил" стосується складової стереоізомери або як оптично активна форма. Кращими у плані вартості, легкості синтезу та/або найбільшої придатності є: Перевага 1. Сполуки Формули І, де 11 81104 12 R1 являє собою СІ або Br; кожна R2 є, незалежно, СІ або Br, і одна R2 знаходиться у третьому положенні; і X є N. Перевага 2. Сполуки Формули І, де R1 являє собою СІ або Br; де R3 являє собою С1-С4 алкіл, X є Ν; та у присутності основи, з одержанням сполуки n дорівнює 0. Формули 4 Заслуговують на увагу сполуки Формули І (включаючи, але не обмежуючись Перевагою 1), де n дорівнює 1-3. Перевага 3. Сполуки Формули 2, де X є N. Перевага 4. Сполуки Формули II, де R1 являє собою СІ або Br; кожна R2 є, незалежно, СІ або Br, і одна R2 знаходиться у третьому положенні; і X є N. Перевага 5. Сполуки Формули II, де R1 являє собою СІ або Br; X є N; та n дорівнює 0. де X, R2 та n є такими, як визначено вище, і R3 Заслуговують на увагу сполуки Формули II є N або С1-С4 алкілом. (включаючи, але не обмежуючись Перевагою 3 та Потім сполука Формули 4, де R3 є С1-С4 Перевагою 4), де n дорівнює 1-3. 3 алкілом, може бути (1) оброблена галогенуючим Перевага 6. Сполуки Формули 4 (де R являє агентом з одержанням сполуки Формули І; і коли собою С1-С4 алкіл), де кожна R2 є, незалежно, СІ одержують сполуки Формули І, де R3 є Н, (2) або Br, і одна R2 знаходиться у 3-положенні. 3 перетворюють сполуку, утворену в (1), у сполуку, Перевага 7. Сполуки Формули 4 (де R являє де R3 є Н. собою С1-С4 алкіл), де X є N;i n дорівнює 0. Заслуговують на увагу сполуки Формули 4 (де R3 являє собою С1-С4 алкіл), включаючи, але не обмежуючись Перевагою 6, де n дорівнює 1-3. 3-положення ідентифіковане як цифра "3", що зображена на арильній складовій, включеній у Формулу І, Формулу ll та Формулу 4 вище. Заслуговують на увагу сполуки Формули ll, у яких, коли R1 являє собою СІ або Br, n дорівнює 1, і R2, що вибрана із СІ або Br, знаходиться у 3Потім сполука Формули І, що одержана у (1) положенні; тоді X є N. Включені сполуки, де n або (2), може бути (3) оброблена окисником, при дорівнює 1-3. потребі, у присутності кислоти з одержанням Заслуговують на увагу сполуки Формули II, у сполуки Формули II; і коли сполуки Формули І, де яких, коли R1 являє собою СІ або Br, n дорівнює 1, R3 є С1-С4 алкілом, використовують для і R2, що вибрана із СІ або Br, знаходиться у 34 одержання сполук Формули II, де R3 є Н, (4) положенні; тоді X є CR . Включені сполуки, де n перетворюють сполуку, утворену в (3), у сполуку дорівнює 1-3. Формули II, де R3 є Н. Способами, яким віддається перевага, є ті, що включають розглянуті вище сполуки, яким віддається перевага. Способи, які заслуговують на увагу, включають розглянуті вище сполуки, котрі заслуговують на увагу. Особливої уваги заслуговує спосіб одержання сполуки Формули І, де n дорівнює 1-3; і спосіб одержання сполуки Формули II, де n дорівнює 1-3. Ступеневий процес одержання сполук Формули І та Формули II, що запроваджений у даному винаході, включає (а) обробку сполуки Формули 2 Схема І ілюструє стадію (а). сполукою Формули 3 13 81104 14 засобів етерифікації. Перевага віддається сполукам Формули 4, де R3 є С1-С4 алкілом. Потрібний продукт, сполука Формули 4, може бути виділений за допомогою способів, що добре відомі фахівцям у даній галузі, таких як кристалізація, екстракція або дистиляція. На стадії (1), як показано на Схемі 2, сполуку Формули 4 обробляють галогенуючим агентом звичайно у присутності розчинника. Галогенуючі реагенти, що можуть використовуватись, На стадії (а) сполуку Формули (2) обробляють включають оксигалогеніди фосфору, сполукою Формули (3), де R3 є С1-С4 алкілом тригалогеніди фосфору, пентагалогеніди (можуть бути використані ефіри фумарової або фосфору, тіонілхлорид, малеїнової кислот, або їх суміш) у присутності дигалотриалкілфосфорани, основи та розчинника. Основою є, типово, дигалодифенілфосфорани, оксалілхлорид та металалкоксидна сіль, така як натрій метоксид, фосген. Перевага віддається оксигалогенідам кглій метоксид, натрій етоксид, калій етоксид, калій фосфору та пентагалогенідам фосфору. Для трет-бутоксид, літій трет-бутоксид і таке подібне. досягнення повного перетворення слід Відносно сполуки Формули 2 слід використовувати використовувати принаймні 0,33 еквівалента більше 0,5 еквівалента основи, краще, від 0,9 до оксигалогеніду фосфору відносно сполуки 1,3 еквівалента. Слід використовувати більше 1,0 Формули 4, краще, від 0,33 до 1,2 еквівалента. Для еквівалента сполуки Формули 3, краще, від 1,0 до досягнення повного перетворення слід 1,3 еквівалента. Можуть застосовуватись полярні використовувати принаймні 0,20 еквівалентів протонні та полярні апротонні органічні пентагалогеніду фосфору відносно сполуки розчинники, такі як спирти, ацетонітрил, Формули 4, краще, від приблизно 0,20 до 1,0 тетрагідрофуран, Ν,Ν-диметилформамід, еквівалента. Для даної реакції перевага диметилсульфоксид і таке подібне. Перевага віддається сполукам Формули 4, де R3 є С1-С4 віддається спиртам, таким як метанол та етанол. алкілом. Найкраще, коли спирт береться той самий, що утворює ефір фумарової або малеїнової кислот та алкоксидну основу. Дану реакцію звичайно проводять шляхом змішування сполуки Формули 2 та основи у даному розчиннику. Потім дану суміш нагрівають або охолоджують до потрібної температури, і сполуку Формули 3 додають протягом деякого часу. Звичайно температури реакції складають від 0°С до точки кипіння використаного розчинника. Дана реакція може проводитись під тиском, що перевищує атмосферний, з метою підвищення точки кипіння Типові розчинники для зазначеного даного розчинника. Загалом, перевага віддається галогенування включають галогеновані алкани, температурам від приблизно 30 до 90°С. такі як дихлорометан, хлороформ, хлоробутан і Швидкість додавання може бути настільки таке подібне, ароматичні розчинники, такі як великою, наскільки дозволяє теплопередача. бензол, ксилол, хлорбензол і таке подібне, ефіри, Типовий час додавання складає від 1 хвилини до 2 такі як тетрагідрофуран, р-діоксан, діетиловий годин. Оптимальна температура реакції та час ефір і таке подібне, та полярні апротонні додавання варіюють у залежності від ідентичності розчинники, такі як ацетонітрил, Ν,Νсполук Формули 2 та Формули 3. Після додавання диметилформамід і таке подібне. При потребі, реакційну суміш можна витримувати протягом можуть додаватись такі органічні основи як деякого часу при температурі реакції. У залежності триетиламін, піридин, Ν,Ν-диметилаланін або від температури реакції потрібний час витримки подібні. Може також додаватись каталізатор, такий може варіювати від 0 до 2 годин. Типові терміни як Ν,Ν-диметилформамід. Перевага віддається витримки складають від 10 до 60 хвилин. Потім процесу, де розчинником виступає ацетонітрил, і дана реакційна маса може бути підкислена основа відсутня. Типовим є те, що коли як шляхом додавання органічної кислоти, такої як розчинник використовується ацетонітрил, не оцтова кислота і таке подібне, або неорганічної потрібні ані основа ані каталізатор. Перевага кислоти, такої як соляна, сірчана кислота і таке віддається процесу, де сполуку Формули 4 подібне. У залежності від умов реакції та засобів перемішують в ацетонітрилі. Потім протягом виділення можна одержати сполуки Формули 4, де 3 3 зручного часу додають галогенуючий агент, і дану R є Н, або сполуки Формули 4, де R є С1-С4 суміш витримують при потрібній температурі до алкілом. Наприклад, сполука Формули 4, де R3 є завершення реакції. Типова температура реакції С1-С4 алкілом, може бути гідролізована in situ у 3 від 20°С до температури кипіння ацетонітрилу, і сполуку Формули 4, де R є Н, коли у даній тривалість реакції типово складає менше 2 годин. реакційній суміші присутня вода. Сполуки Формули 3 Потім дану реакційну масу нейтралізують 4, де R є Н, можуть бути легко трансформовані у неорганічною основою, такою як бікарбонат сполуки Формули 4, де R3 є С1-С4 алкілом, з натрію, гідроксид натрію і таке подібне, або використанням добре відомих у даній галузі 15 81104 16 продукт, сполука Формули II, де R3 є С1-С4 органічною основою, такою як ацетат натрію. алкілом, може бути виділений з використанням Потрібний продукт, сполуку Формули І, можна способів, що відомі фахівцям у даній галузі, виділяти з використанням способів, що відомі включаючи кристалізацію, екстракцію та фахівцям у даній галузі, включаючи кристалізацію, дистиляцію. екстракцію та дистиляцію. На стадії (2) сполука Формули І, де R3 є С1-С4 алкілом, ефір, може бути гідролізована у сполуку Формули І, де R3 є Н, карбонову кислоту. Гідроліз може бути каталізований кислотами, металічними іонами та ферментами. Прикладом кислоти, що може бути застосована для каталізування гідролізу, зазначають йодотриметилсилан [див. Advanced Organic Chemistry, Third Ed., Jerry March, John Wiley & Sons, Inc. New York, 1985, p.p.334338, де подано огляд способів]. Гідролітичні способи, що каталізуються основами, не рекомендуються для гідролізу сполук Формули І і можуть призвести до розкладу. Карбонова кислота На стадії (4), як показано на Схемі 4, сполука може бути виділена з використанням способів, що Формули II, де R3 є С1-С4 алкілом, ефір, може бути відомі фахівцям у даній галузі, включаючи перетворена у сполуку Формули II, де R3 є Н, кристалізацію, екстракцію та дистиляцію. карбонову кислоту. Способи для перетворення На стадії (3), як показано на Схемі 3, сполуку ефірів у карбонові кислоти добре відомі фахівцям Формули І обробляють окисником, при потребі, у у даній галузі. Сполуки Формули II (R3 є С1-С4 присутності кислоти. Як вихідний матеріал для алкілом) можуть бути перетворені у сполуки стадії (3) перевага віддається сполуці Формули І, Формули II (R3 є Н) з використанням множини де R3 є С1-С4 алкілом (тобто, продукт стадії (1), способів, включаючи нуклеофільне розщеплення якому віддається перевага, є кращим як вихідний за безводних умов, або з використанням матеріал для стадії (3)).3азначеним окисником гідролітичних методів, включаючи використання як можуть слугувати пероксид водню, органічні кислот, так і основ [див. T.W. Greene and P. G. M. пероксиди, персульфат калію, персульфат натрію, Wuts, Protective Groups in Organic Synthesis, 2 nd персульфат амонію, моноперсульфат калію ed., John Wiley & Sons, Inc., New York, 1991, (наприклад, ОхоnеÒ) або перманганат калію. Для pp.224-269, де подано огляд способів]. Для досягнення повного перетворення слід способу Схеми 4 перевага віддається застосовувати принаймні один еквівалент каталізованим основою гідролітичним способам. окисника відносно сполуки Формули І, краще, від Придатні основи включають лужні метали (такі як приблизно одного до двох еквівалентів. Це гідроксиди літію, натрію або калію). Наприклад, окиснення типово прoводиться у присутності даний eфір може бути розчинений у суміші води та спирту, такого як етанол. Після обробки розчинника. Таким розчинником може слугувати ефір, такий як тетрагідрофуран, р-діоксан і таке гідроксидом натрію або гідроксидом калію даний подібне, органічний ефір, такий як етилацетат, ефір піддається омиленню з утворенням натрієвої диметилкарбонат і таке подібне, або полярний або калієвої солі даної карбонової кислоти. апротонний органічний розчинник, такий як Ν,ΝПідкислення сильною кислотою, такою як соляна диметилформамід, ацетонітрил і таке подібне. киcлота або сірчана кислота, дає карбонову Кислоти, що придатні для використання на стадії кислоту. Карбонова кислота може бути виділена з використанням методів, що відомі фахівцям у окиснення, включають неорганічні кислоти, такі як сірчана кислота, фосфорна кислота і таке подібне, даній галузі, включаючи кристалізацію, екстракцію органічні кислоти, такі як оцтова кислота, бензойна та дистиляцію. кислота і таке подібне. При використанні кислоти її кількість має бути більше 0,1 еквівалента відносно сполуки Формули І. Для досягнення повного перетворення може використовуватись від одного до п'яти еквівалентів кислоти. Для сполук Формули І, де X є CR2, перевага віддається такому окиснику як пероксид водню, і краще, коли окиснення проводиться за умов відсутності кислоти. Для сполук Формули І, де X є N, перевага віддається такому окиснику як персульфат калію, і краще, коли окиснення проводиться у присутності сірчаної кислоти. Дана реакція може проводитись шляхом Слід зазначити, що деякі сполуки Формули І, перемішування сполуки Формули І у потрібному де R1 являє собою галоген, можуть бути одержані розчиннику та, коли застосовується, кислоті. Потім з інших сполук Формули І, де R1 є іншим галогеном при зручній швидкості може додаватись окисник. або являє собою сульфонатну групу, таку як pТемпература реакції звичайно варіює від низької, толуолсульфонат, бензолсульфонат та близько 0°С, до точки кипіння даного розчинника, метансульфонат. Наприклад, сполука Формули І, щоб досягти прийнятної тривалості реакції до її де R1 являє собою Br, може бути одержана завершення, краще, менше 8 годин. Потрібний 17 81104 18 етанолу, 80,0мл (0,214моль) 21% етоксиду натрію шляхом обробки бромистим воднем відповідної в етанолі та 20,0мл (0,203моль) фенілгідразину. сполуки Формули І, де R1 являє собою СІ або рОранжевий розчин обробляли по краплях 40,0мл толуолсульфонат. Дана реакція проводиться у (0,247моль) діетилмалеату протягом приблизно 18 придатному розчиннику, такому як дибромометан, хвилин. За перші п'ять хвилин уведення дихлорометан або ацетонітрил. Дана реакція температура реакційної маси зросла від 25 до може бути проведена при атмосферному або 38°С. Для обмеження температури реакції у межах близькому до нього тиску, або при тиску, що 38-42°С подальше введення проводилось з перевищує атмосферний, в автоклаві. Коли R1 у періодичним використанням водяної бані. вихідній сполуці Формули І являє собою галоген, Утворений в результаті оранжево-червоний такий як СІ, краще, коли дана реакція проводиться розчин витримували за умов навколишнього у такий спосіб, що галогеноводень, утворений в середовища протягом 30 хвилин. Потім він був результаті реакції, вилучається шляхом уведений у ділильну лійку, що містила 20,0мл барботування або за допомогою інших придатних (0,349моль) льодяної оцтової кислоти та 700мл методів. Дана реакція може проводитись при води. Дану суміш екстрагували 250мл температурах від приблизно 0°С до 100°С, дихлорометану. Даний екстракт осушували над найбільш зручно при температурі навколишнього сульфатом магнію, фільтрували і потім середовища (наприклад, від 10 до 40°С), і ще концентрували на роторному випарнику. Утворене краще, при температурах від приблизно 20 до в результаті жовто-чорне масло (52,7г) розводили 30°С. Добавка кислотного каталізатора Льюїса 100мл ефіру, після чого кристалізація даного (наприклад, триброміду алюмінію для одержання продукту була досить швидкою, щоб зумовити сполуки Формули І, де R1 є Br) може полегшити м'яке кипіння. Суспензію витримували протягом 2 протікання даної реакції. Продукт Формули І годин за умов навколишнього середовища. Потім виділяють за допомогою звичайних методів, що її охолоджували до приблизно 0°С. Продукт відомі фахівцям у даній галузі, включаючи виділяли методом фільтрування, промивали екстракцію, дистиляцію та кристалізацію. Вихідні сполуки Формули І, де R1 являє собою 2´20мл холодного ефіру і потім висушували на галоген, можуть бути одержані як вже показано на фільтрі у повітряному середовищі. Отриманий Схемі 2. Вихідні сполуки Формули І, де R1 являє продукт складався із 29,1г (61%) собою сульфонатну групу, можуть бути одержані у висококристалічного білого порошку. 1Н ЯМР не схожий спосіб із відповідних сполук Формули 4 з виявив наявності значних домішок. Фільтрат використанням стандартних методів, таких як концентрували до 20,8г коричневого масла. Аналіз обробка сульфонілхлоридом (наприклад, рданого масла виявив присутність додаткових 6,4г толуолсульфонілхлоридом) та основою, такою як (13%) потрібного продукту. Отже, загальний вихід третинний амін (наприклад, триетиламін), у реакції складав 74%. 1 придатному розчиннику, такому як дихлорометан. Н ЯМР (DMSO-d6) d 10,25 (s, 1Н), 7,32 (t, 2H), Без додаткових уточнень, можна вважати, що 7,15 (d, 2H), 7,00 (t, 1H), 4,61 (dd, 1H), 4,21 (q, 2H), фахівець у даній галузі, використовуючи 2,95 (dd, 1H), 1,25 (t, 3H). попередній опис, може застосувати даний винахід Приклад 2 у самому повному обсязі. Тому наступні Приклади Одержання етил 5-оксо-2-феніл-3мають лише ілюстративний характер і не піразолідинкарбоксилату (альтернативна назва обмежують ніяким чином даний винахід. етил 1-феніл-3-піразолідинон-5-карбоксилат) з Необов'язково, щоб вихідний матеріал для використанням діетилфумарату наступних Прикладів одержувався з У 500-мл чотиригорлу колбу, обладнану використанням препаративних методів, що описані механічною мішалкою, термометром, крапельною в інших Прикладах. Під відсотками маються на лійкою, зворотним холодильником та трубкою для думці вагові відсотки, за виключенням сумішей введення азоту, завантажували 150мл розчинників для хроматографії або коли є абсолютного етанолу, 15,0г (0,212моль) 96% спеціальне застереження. Якщо нема етоксиду натрію в етанолі та 20,0мл (0,203моль) спеціального застереження, частини та відсотки фенілгідразину. Дану оранжеву суміш обробляли для сумішей розчинників для хроматографії подані по краплях 40,0мл (0,247моль) діетилфумарату в об'ємних відсотках. 1Н ЯМР спектри подані у протягом 75 хвилин. Підчас уведення температура млн. -1 відносно нижньої енергетичної границі реакційної маси зросла від 28 до 37°С, і кінцева тетраметилсилану; "s" означає синглет, "d" температура складала 32°С. Утворений в означає дублет, "t" означає триплет, "q" означає результаті дещо каламутний оранжевий розчин квартет, "т" означає мультиплет, "dd" означає витримували за умов навколишнього середовища дублет дублетів, "dt" означає дублет триплетів, та протягом 135 хвилин. Потім реакційну суміш "br s" означає широкий синглет. виливали у ділильну лійку, що містила 15,0мл Приклад 1 (0,262моль) льодяної оцтової кислоти та 700мл Одержання етил 5-оксо-2-феніл-3води. Дану суміш екстрагували 150мл піразолідинкарбоксилату (альтернативна назва дихлорометану. Даний екстракт осушували над етил 1-феніл-3-піразолідинон-5-карбоксилат) з сульфатом магнію, фільтрували і потім використанням діетилмалеату концентрували на роторному випарнику. Утворене У 300-мл чотиригорлу колбу, обладнану в результаті коричнево-жовто масло (41,3г) механічною мішалкою, термометром, краплинною розводили 100мл ефіру. Додавали кілька лійкою, зворотним холодильником та трубкою для затравочних кристалів. Дану суміш витримували введення азоту, завантажували 80мл абсолютного протягом 30 хвилин за умов навколишнього 19 81104 20 хлорофеніл)гідразину. Даний пурпуровий розчин середовища. Потім її охолоджували до 0°С. нагрівали до 35°С. Потім його обробляли по Продукт виділяли методом фільтрування, краплях 19,0мл (0,117моль) діетилмалеату промивали 2´20мл холодного ефіру і потім протягом приблизно 23 хвилин. Для обмеження висушували на фільтрі у повітряному середовищі температури реакції у межах 35-40°С введення протягом приблизно 15 хвилин. Даний продукт проводилось з періодичним використанням складався із 9,5г (20%) висококристалічного білого водяної/льодяної бані. Дану реакційну суміш порошку. 1 Н ЯМР не виявив наявності значних витримували при цій температурі протягом 30 домішок. Фільтрат концентрували до 31г хвилин. Потім вона була введена у ділильну лійку, коричневого масла. Аналіз даного масла виявив що містила 10,0мл (0,175моль) льодяної оцтової присутність додаткових 7,8г (16%) потрібного кислоти та 400мл води. Дану суміш екстрагували продукту. Отже, загальний вихід реакції складав 2´100мл дихлорометану. Даний екстракт 36%. осушували над сульфатом магнію, фільтрували і Приклад 3 потім концентрували на роторному випарнику. Одержання етил 5-оксо-2-(2-піридиніл)-3Утворене в результаті темно-коричневе масло піразолідинкарбоксилату (альтернативна назва (31,0г) кристалізувалось при витримці. Даний етил 1-(2-піридиніл)-3-піразолідинон-5матеріал суспендували у 100мл ефіру, і суспензію карбоксилат) перемішували протягом приблизно 1 години. У 200-мл чотиригорлу колбу, обладнану Продукт виділяли фільтрацією, промивали 50мл механічною мішалкою, термометром, крапельною ефіру і потім висушували протягом ночі при лійкою, зворотним холодильником та трубкою для кімнатній температурі під вакуумом. Отриманий введення азоту, завантажували 18мл абсолютного продукт складався із 12,5г (46%) кристалічного етанолу, 18,0мл (0,0482моль) 21% етоксиду порошку. 1 Н ЯМР не виявив наявності значних натрію в етанолі, та 5,00г (0,0458моль) 2домішок. Фільтрат концентрували до 16,3г гідразинопіридину. Даний розчин нагрівали до коричневого масла. Аналіз даного масла виявив 34°С. Потім його обробляли по краплях 9,0мл присутність додаткових 6,7г (25%) потрібного (0,056моль) діетилмалеату протягом 20 хвилин. продукту. Отже, загальний вихід реакції складав Температура даної реакційної маси підчас 71%. уведення зросла максимально до 48°С. Утворений 1 в результаті оранжевий розчин витримували за Н ЯМР (DMSO-d6) d 10,14 (s, 1Н), 7,47 (6, 1H), умов навколишнього середовища протягом 85 7,32 (m, 2H), 7,14 (t, 1H), 4,39 (d, 1H), 4,19 (q, 2H), хвилин. Потім його виливали у ділильну лійку, що 3,07 (dd, 1H), 2,29 (d, 1H), 1,22 (t, 3H). містила 4,0мл (0,070моль) льодяної оцтової Приклад 5 кислоти та 300мл води. Дану суміш екстрагували Одержання етил 2-(3-хлоро-2-піридиніл)-52´50мл дихлорометану. Даний екстракт оксо-3-піразолідинкарбоксилату (альтернативна назва етил 1-(3-хлоро-2піридиніл)-3-піразолідиноносушували над сульфатом магнію, фільтрували і 5-карбоксилат) потім концентрували на роторному випарнику. У 2-л чотиригорлу колбу, обладнану Утворене в результаті оранжеве масло (10,7г) механічною мішалкою, термометром, крапельною піддавали флешхроматографії на колонці із 200г лійкою, зворотним холодильником та трубкою для силікагелю з використанням 4% метанолу у введення азоту, завантажували 250мл хлороформі як елюенту (50мл фракції). Фракції 9абсолютного етанолу та 190мл (0,504моль) 21% 12 випарювали на роторному випарнику з етоксиду натрію в етанолі. Дану суміш нагрівали зі одержанням 3,00г оранжевого масла, котре зворотним холодильником при температурі містило 77% потрібного продукту, 15% приблизно 83°С. Потім її обробляли 68,0г хлороформу та 8% діетил 2-етоксибутандіоату. (0,474моль) 3-хлоро-2(1Н)-піридинон гідразоном Фракції 13-17 концентрували з одержанням 4,75г (альтернативна назва 3-хлоро-2оранжево-жовтого масла, що містило 94% гідразинопіридин). Дану суміш піддавали потрібного продукту та 6% хлороформу. Фракції повторному нагріванню зі зворотним 18-21 концентрували з одержанням 1,51г холодильником протягом 5 хвилин. Потім жовту оливково-зеленого масла, котре містило 80% суспензію обробляли по краплях 88,0мл потрібного продукту та 20% хлороформу. (0,544моль) діетилмалеату протягом 5 хвилин. Загальний вихід потрібного продукту складав 8,0г Підчас уведення швидкість кипіння помітно (74%). 1 зростала. По завершенню додавання весь Н ЯМР (DMSO-d6) d 10,68 (br, 1Н), 8,22 (d, вихідний матеріал розчинився. Утворений в 1H), 7,70 (t, 1H), 6,90 (m, 2H), 5,33 (dd, 1H), 4.17 (q. результаті оранжево-червоний розчин нагрівали зі 2H), 3,05 (dd, 1H), 2,48 (dd, 1H), 1,21 (t, 3H). зворотним холодильником протягом 10 хвилин. Приклад 4 Після охолодження до 65°С реакційну суміш Одержання етил 2-(2-хлорофеніл)-5-оксо-3обробляли 50,0мл (0,873моль) льодяної оцтової піразолідинкарбоксилату (альтернативна назва кислоти. Утворився осад. Дану суміш розводили етил 1-(2-хлорофеніл)-3-піразолідинон-5650мл води, після чого зазначений осад карбоксилат) розчинився. Оранжевий розчин охолоджували на У 250-мл чотиригорлу колбу, обладнану льодяній бані. Продукт почав осаджуватись при механічною мішалкою, термометром, крапельною 28°С. Суспензію витримували при температурі лійкою, зворотним холодильником та трубкою для приблизно 2°С протягом 2 годин. Даний продукт введення азоту, завантажували 40мл абсолютного виділяли шляхом фільтрації, промивали 3´50мл етанолу, 40,0мл (0,107моль) 21% етоксиду натрію 40% водного етанолу і потім висушували на в етанолі та 14,5г (0,102моль) (2 21 81104 22 водного розчину бікарбонату натрію. Органічний фільтрі у повітряному середовищі протягом шар осушували над сульфатом магнію, приблизно 1 години. Даний продукт складався із фільтрували, потім концентрували на роторному 70,3г (55%) висококристалічного світловипарнику. Неочищений продукт складався із 1,50г оранжевого порошку. 1Н ЯМР не виявив наявності оранжевого масла, яке кристалізувалось при значних домішок. 1 витримці. Аналіз даного неочищеного продукту Н ЯМР (DMSO-d6) d 10,18 (s, 1Н), 8,27 (d, 1Н), методом 1Н ЯМР показав, що він складається із 7,92 (d, 1H), 7,20 (dd, 1H), 4,84 (d, 1H), 4,20 (q, 2H), 65% потрібного продукту та 35% вихідного 2,91 (dd, 1H), 2,35 (d, 1H), 1,22 (t, 3H). матеріалу. Тому вихід потрібного продукту складав Приклад 6 приблизно 18%. Одержання етил 3-хлоро-4,5-дихлоро-1-фенілПриклад 6С 1Н-піразол-5-карбоксилату (альтернативна назва Застосування оксихлориду фосфору у етил 1-феніл-3-хлоро-2-піразолін-5-карбоксилат) хлороформі у присутності триетиламіну Приклад 6А У 100-мл двогорлу колбу, обладнану Застосування оксихлориду фосфору в магнітною мішалкою, термометром, зворотним ацетонітрилі за відсутності основи холодильником та трубкою для введення азоту, У 500-мл чотиригорлу колбу, обладнану завантажували 20мл хлороформу, 2,00г механічною мішалкою, термометром, крапельною (0,00854моль) етил 5-оксо-2-феніл-3лійкою, зворотним холодильником та трубкою для піразолідинкарбоксилату, 1,30мл (0,00933моль) введення азоту, завантажували 150мл триетиламіну, 2 краплі Ν,Ν-диметилформаміду та ацетонітрилу, 25,0г (0,107моль) етил 5-оксо-20,0850мл (0,00912моль) 21% оксихлорлду феніл-3-піразолідинкарбоксилату та 11,0мл фосфору. При додаванні оксихлориду фосфору (0,18моль) оксихлориду фосфору. Світло-жовтий зразу ж розпочиналась енергійна реакція. Дану розчин нагрівали до 78-80°С протягом 45 хвилин. суміш нагрівали зі зворотним холодильником при Після охолодження до 54°С утворену в результаті 64°С протягом 25 хвилин. Утворений в результаті суміш глибокого синьо-зеленого кольору жовтий розчин розводили 50мл води і потім обробляли по краплях розчином 25,0г (0,298моль) обробляли 3,0г (0,036моль) твердого бікарбонату бікарбонату натрію у 250мл води. Підчас уведення натрію. Двофазову суміш перемішували протягом протягом 15 хвилин відокремлювалось оранжеве 50 хвилин за умов навколишнього середовища. масло. Після перемішування протягом 5 хвилин Потім її переносили до ділильної лійки та величина рН даної суміші складала близько 1. розводили 100мл дихлорометану. Органічний шар Протягом приблизно 3 хвилин додавали ще 10,0г відокремлювали і потім промивали послідовно (0,119моль) бікарбонату натрію у вигляді твердої 50мл 5,5% водним розчином соляної кислоти та речовини, в результаті чого кінцева величина рН 50мл 3,8% водним розчином карбонату натрію. складала близько 6. Дану суміш розводили 400мл Промитий органічний шар осушували над води, в результаті чого оранжеве масло сульфатом магнію, фільтрували і потім кристалізувалось. Утворену кристалічну масу концентрували на роторному випарнику. подрібнювали за допомогою шпателя. Продукт Неочищений продукт складався із 1,90г жовтого виділяли фільтрацією, промивали 4´100мл води і масла, котре кристалізувалось при витримці. потім висушували на фільтрі у повітряному Аналіз зазначеного неочищеного продукту середовищі протягом приблизно 2 годин. методом 1Н ЯМР показав, що він містить близько Отриманий продукт складався із 24,5г (91%) 94% потрібного продукту, 2% вихідного матеріалу пушистого кристалічного порошку світло-жовтого та 2% неідентифікованих домішок. Тому вихід кольору. Будь-яких суттєвих домішок 1Н ЯМР не потрібного продукту складав близько 83%. виявив. 1 Приклад 7 Н ЯМР (DMSO-d6) d 2,74 (t, 2Н), 6,88 (d, 2Н), Одержання етил 3-хлоро-4,5-дигідро-1-(26,83 (t, 1Н), 5,02 (dd, 1H), 4,14 (q, 2H), 3,68 (dd, піридиніл)-1Н-піразол-5-карбоксилату 1HJ, 3,34 (d, 1H), 1,16 (t, 3H). (альтернативна назва етил 1-(2-піридиніл)-3Приклад 6В хлоро-2-піразолін-5-карбоксилат) Застосування оксихлориду фосфору у У 250-мл чотиригорлу колбу, обладнану хлороформі за відсутності основи механічною мішалкою, термометром, зворотним У 100-мл двогорлу колбу, обладнану холодильником та трубкою для введення азоту, механічною мішалкою, термометром, крапельною завантажували 50мл ацетонітрилу, 4,70г лійкою, зворотним холодильником та трубкою для (0,0188моль) 5-оксо-2-(2-піридиніл)-3введення азоту, завантажували 50мл хлороформу. піразолідинкарбоксилату та 2,00мл (0,0215моль) 5,00г (0,0213моль) етил-5-оксо-2-феніл-3оксихлориду фосфору. Дана суміш піразолідинкарбоксилату, 2,10мл (0,0225моль) саморозігрівалась від 22 до 33°С. Після витримки оксихлориду фосфору та 2 краплі Ν,Νпротягом 60 хвилин за умов навколишнього диметилформаміду. Червоно-оранжевий розчин середовища відбирали пробу. Аналіз методом 1Н нагрівали зі зворотним холодильником при 64°С ЯМР виявив 70% перетворення вихідного протягом 60 хвилин. Утворену в результаті суміш, матеріалу у потрібний продукт. Дану суміш жовто-коричневу рідину та тверду смолисту нагрівали зі зворотним холодильником при 85°С речовину глибокого зеленого кольору, піддавали протягом 80 хвилин. Нагрівальну сітку вилучали. нагріванню зі зворотним холодильником протягом Утворений в результаті жовто-оранжевий розчин 140 хвилин. Потім її розводили 100мл розводили 50мл води. Потім його обробляли по дихлорометану і переносили у ділильну лійку. краплях 3,9г (0,049моль) 50% водної каустичної Даний розчин промивали два рази 50мл 6% 23 81104 24 сульфату магнію та 12г силікагелю, і дану соди, що дало рН близько 7,5. Після суспензію перемішували з допомогою магнітної перемішування протягом 20 хвилин величина рН мішалки протягом 30 хвилин. Суспензію, яка даної суміші упала до 3. Додавали ще 3,0г набула глибокого синьо-зеленого кольору, (0,038моль) 50% водної каустичної соди, після фільтрували для вилучення сульфату магнію та чого величина рН підвищилась до приблизно 9,0. силікагелю. Осад на фільтрі промивали 100мл Для встановлення рН на рівні близько 7,5 дихлорометану. Фільтрат концентрували на додавали невелику кількість концентрованої роторному випарнику. Одержаний продукт соляної кислоти. Дану нейтралізовану суміш складався із 92,0г (93%) темно-бурштинового переносили до ділильної лійки, що містила 300мл масла. Єдиними домішками, що були виявлені води та 100мл дихлорометану. Органічний шар методом 1Н ЯМР, були 1,0% вихідного матеріалу відокремлювали, осушували над сульфатом та 0,7% ацетонітрилу. магнію, фільтрували і потім концентрували на 1 роторному випарнику. Одержаний продукт Н ЯМР (DMSO-d6) d 8,12 (d, 1Н), 7,84 (d, 1H), складався із 4,10г (84%) блідо-жовтого масла, 7,00 (dd, 1H), 5,25 (dd, 1H), 4,11 (q, 2H), 3,58 (dd, котре кристалізувалось при витримці. Єдиними 1H, 3,26 (dd, 1H), 1,15 (t, 3H). домішками, що були виявлені методом 1Н ЯМР, Приклад 9 були 1,0% вихідного матеріалу та 0,6% Одержання етил 3-бромо-1-(3-хлоро-2ацетонітрилу. піридиніл)-4,5-дигідро-1Н-піразол-5-карбоксилату 1 Н ЯМР (DMSO-d6) d 8,18 (d, 1Н), 8,63 (t, 1H), (альтернативна назва етил 1-(3-хлоро-28,13 (d, 1H), 7,80 (t, 1H), 5,08 (dd, 1H), 4,11 (m, 2H), піридиніл)-3-бромо-2-піразолін-5-карбоксилат) 3,65 (dd, 1H), 3,27 (dd, 1H), 1,14 (t, 3H). Приклад 9а Приклад 8 Використання оксиброміду фосфору Одержання етил 3-хлоро-1-(3-хлоро-2У 1-л чотиригорлу колбу, обладнану піридиніл)-4,5-дигідро-1Н-піразол-5-карбоксилату механічною мішалкою, термометром, зворотним (альтернативна назва етил 1-(3-хлоро-2холодильником та трубкою для введення азоту, піридиніл)-3-хлоро-2-піразолін-5-карбоксилат) завантажували 400мл ацетонітрилу, 50,0г У 2-л чотиригорлу колбу, обладнану (0,185моль) етил 2-(3-хлоро-2-піридиніл)-5-оксо-3механічною мішалкою, термометром, зворотним піразолідинкарбоксилату та 34,0г (0,119моль) холодильником та трубкою для введення азоту, оксиброміду фосфору. Оранжеву суспензію завантажували 1000мл ацетонітрилу, 91,0г нагрівали зі зворотним холодильником при 83°С (0,337моль) етил 2-(3-хлоро-2-піридиніл)-5-оксо-3протягом 20 хвилин. Утворений в результаті піразолідинкарбоксилату та 35,0мл (0,375моль) каламутний оранжевий розчин нагрівали зі оксихлориду фосфору. Після додавання зворотним холодильником протягом 75 хвилин, і оксихлориду фосфору дана суміш за цей час утворився щільний рудуватосаморозігрівалась від 22 до 25°С, і утворювався коричневий кристалічний осад. Зворотний осад. Світло-жовту суспензію нагрівали зі холодильник був замінений на дистиляційну зворотним холодильником при 83°С протягом 35 насадку, і було зібрано 300мл каламутного хвилин, після чого даний осад розчинявся. безбарвного дистиляту. Друга 1-л чотиригорла Утворений в результаті оранжевий розчин колба, обладнана механічною мішалкою, була нагрівали зі зворотним холодильником протягом завантажена 45г (0,54моль) -бікарбонату натрію та 45 хвилин, після чого він став чорно-зеленим. 200мл води. Протягом 5 хвилин до суспензії Зворотний холодильник був замінений на бікарбонату натрію додавали концентровану дистиляційну насадку, і 650мл розчинника було реакційну суміш. Утворена в результаті двофазова вилучено шляхом дистиляції. Друга 2-л суміш енергійно перемішувалась протягом 5 чотиригорла колба, обладнана механічною хвилин, і за цей час виділення газу припинилось. мішалкою, завантажувалась 130г (1,55моль) Дану суміш розводили 200мл дихлорометану і бікарбонату натрію та 400мл води. Протягом 15 потім перемішували протягом 75 хвилин. Суміш хвилин до суспензії бікарбонату натрію додавали обробляли 5г Celite 545Ò і потім фільтрували для концентровану реакційну суміш. Утворену в вилучення коричневої смолистої речовини. результаті двофазову суміш енергійно Фільтрат переносили до ділильної лійки. перемішували протягом 20 хвилин, і за цей час Коричневий органічний шар (400 мл) виділення газу припинилось. Дану суміш відокремлювали і потім обробляли 15г сульфату розводили 250мл дихлорометану і потім магнію та 2,0г активованого вугілля Darco G60. перемішували протягом 50 хвилин. Суміш Утворену в результаті суспензію перемішувади з обробляли 11г діатомової землі Celite 545Ò і потім допомогою магнітної мішалки протягом 15 хвилин і фільтрували для вилучення чорної, смолистої потім фільтрували для вилучення сульфату магнію речовини, що інгібувала розділення фаз. Оскільки та активованого вугілля. Фільтрат зеленого даний фільтрат повільно розділявся на дві чіткі кольору обробляли 3г силікагелю та перемішували фази, його розводили 200мл дихлорометану та протягом кількох хвилин. Силікагель глибокого синьо-зеленого кольору вилучали шляхом 200мл води і ще раз обробляли 15г Celite 545Ò. фільтрації, і фільтрат концентрували на роторному Дану суміш фільтрували, і фільтрат переносили у випарнику. Одержаний продукт складався із 58,6г ділильну лійку. Був відокремлений більш важкий (95%) світло-бурштинового масла, котре органічний шар глибокого зеленого кольору. 50 кристалізувалось після витримки. Єдиною міліметровий забруднений шар повторно домішкою, що була виявлена методом 1Н ЯМР, фільтрували і додавали до даного органічного був ацетонітрил у кількості 0,3%. шару. Органічний розчин (800мл) обробляли 30г 25 81104 26 промивали 15мл води. Вологий осад розчиняли у Н ЯМР (DMSO-d6) d 8,12 (d, 1Н), 7,84 (d, 1H), 100мл дихлорометану. Даний розчин осушували 6,99 (dd, 1H), 5,20 (dd, 1H), 4,11 (q, 2H), 3,60 (dd, над сульфатом магнію, фільтрували і потім 1H), 3,29 (dd, 1H), 1,15 (t, 3H). концентрували на роторному випарнику. Продукт Приклад 9В складався із 1,24г (приблизно 79%) оранжевого Використання пентаброміду фосфору масла, котре кристалізувалось після витримки. У 1-л чотиригорлу колбу, обладнану Виходячи із 1Н ЯМР даних, чистота продукту механічною мішалкою, термометром, зворотним складала близько 95%. xoлодильником та трубкою для введення азоту, 1 Н ЯМР (DMSO-d6) d 7,50 (s, 5Н), 7,20 (s, 1Н), завантажували 330мл ацетонітрилу, 52,0г 7,92 (d, 1H), 4,18 (q, 2H), 1,14 (t, 3H). (0,193моль) етил 2-(3-хлоро-2-піридиніл)-5-оксо-3Приклад 10В піразолідинкарбоксилату та 41,0г (0,0952моль) Використання діоксиду марганцю пентаброміду фосфору. Оранжеву суспензію У 100-мл двогорлу колбу, обладнану нагрівали зі зворотним холодильником при 84°С магнітною мішалкою, термометром, зворотним протягом 20 хвилин. Утворена в результаті холодильником та трубкою для введення азоту, цегляно-червона суміш нагрівалась зі зворотним завантажували 3,00г (0,0119моль) етил 3-хлорохолодильником протягом 90 хвилин, і за цей час 4,5-дигідро-1-феніл-1Н-піразол-5-карбоксилату. утворився щільний рудувато-коричневий 25мл хлороформу та 2,50г (0,0245моль) кристалічний осад. Зворотний холодильник був активованого діоксиду марганцю. Дану суміш замінений на дистиляційну насадку, і було зібрано нагрівали зі зворотним .холодильником при 62°С 220мл каламутного безбарвного дистиляту. Друга протягом 1 години. Аналіз проби реакційної маси, 1-л чотиригорла колба, обладнана механічною виконаний методом 1Н ЯМР, показав приблизно мішалкою, була завантажена 40г (0,48моль) 6% перетворення вихідного матеріалу у потрібний бікарбонату натрію та 200мл води. Протягом 5 етил 1-феніл-3-хлоропіразол-5-карбоксилат. Дану хвилин до суспензії бікарбонату натрію додавали суміш витримували ще 5 годин при нагріванні зі концентровану реакційну суміш. Утворена в зворотним холодильником. Аналіз другої проби результаті двофазова суміш енергійно дав близько 9% перетворення. перемішувалась протягом 10 хвилин, і за цей час Приклад 10С виділення газу припинилось. Дану суміш Використання гіпохлориту натрію розводили 200мл дихлорометану і потім У 100-мл двогорлу колбу, обладнану перемішували протягом 10 хвилин. Суміш магнітною мішалкою, термометром, зворотним обробляли 5г Celite 545Ò і потім фільтрували для холодильником та трубкою для введення азоту, вилучення пурпурової смолистої речовини. Осад завантажували 1,00г (0,00396моль) етил 3-хлорона фільтрі промивали 50мл дихлорометану. 4,5-дигідро-1-феніл-1Н-аіразол-5-карбоксилату, Фільтрат переносили до ділильної лійки. 10мл ацетонітрилу, 0,55г (0,0040моль) первинного Пурпурово-червоний органічний шар (400мл) кислого фосфату натрію моногідрату та 5,65г відділяли і потім обробляли 15г сульфату магнію (0,00398моль) 5,25% водного гіпохлориту натрію. та 2,2г активованого вугілля Darco G60. Утворену Оранжевий розчин витримували за умов в результаті суспензію перемішували з допомогою навколишнього середовища протягом 85 хвилин. магнітної мішалки протягом 40 хвилин. Суспензію Аналіз проби реакційної маси, виконаний методом фільтрували для вилучення сульфату магнію та 1 Н ЯМР, показав приблизно 71% перетворення активованого вугілля. Фільтрат концентрували на вихідного матеріалу у два основних продукти. роторному випарнику. Одержаний продукт Даний розчин нагрівали до 60°С протягом 60 складався із 61,2г (95%) темно-бурштинового хвилин. Аналіз другої проби не виявив підвищення масла, котре кристалізувалось після витримки. ступеня перетворення у порівнянні з першою Єдиною домішкою, що була виявлена методом 1Н пробою. Дану реакційну суміш обробляли ЯМР, був ацетонітрил у кількості 0,7%. 1 додатковими 3,00г (0,00211моль) 5,25% водного Н ЯМР (DMSO-d6) d 8,12 (d, 1Н), 7,84 (d, 1H), гіпохлориту натрію. Після витримки протягом 60 6,99 (dd, 1H), 5,20 (dd, 1H), 4,11 (q, 2H), 3,60 (dd, хвилин при 60°С реакційну масу додавали до 1H), 3,29 (dd, 1H), 1,15 (t, 3H). 100мл води. Дану суміш екстрагували 100мл Приклад 10 дихлорометану. Екстракт відокремлювали, Одержання етил 3-хлоро-1-феніл-1Н-піразолосушували над сульфатом магнію, фільтрували і 5-карбоксилату (альтернативна назва етил 1потім концентрували на роторному випарнику. феніл-3-хлоропіразол-5-карбоксилат) Неочищений продукт складався із 0,92г червоноПриклад 10А оранжевого масла. 1Н ЯМР показав, що Використання пероксиду водню неочищений продукт складався, головним чином, У 100-мл двогорлу колбу, обладнану із етил 3-хлоро-1-(4-хлорофеніл)-4,5-дигідро-1Нмеханічною мішалкою, термометром, зворотним піразол-5-карбоксилату (альтернативна назва холодильником та трубкою для введення азоту, етил 1-(4-хлорофеніл)-3-хлоро-2-піразолін-5завантажували 1,50г (0,00594моль) етил 3-хлорокарбоксилат) та етил 3-хлоро-1-(2-хлорофеніл)4,5-дигідро-1-феніл-1Н-піразол-5-карбоксилату та 4,5-дигідро-1Н-піразол-5-карбоксилату 15мл ацетонітрилу. Дану суміш нагрівали до 80°С. (альтернативна назва етил 1-(2-хлорофеніл)-3Потім її обробляли 0,700мл (0,00685моль) 30% хлоро-2-піразолін-5-карбоксилат) у відношенні 2:1. водним розчином пероксиду водню. Дану суміш Даний ізомер може бути відокремлений методом витримували при 78-80°С протягом 5 годин. Потім хроматографії на силікагелі з використанням 10% реакційну масу додавали до 70мл води. етилацетату у гексанах як елюенту. Осаджений продукт виділяли фільтрацією і потім 1 27 81104 28 відфільтровували ще в теплому стані (50-65°С) Н ЯМР для етил 3-хлоро-1-(4-хлорофеніл)для вилучення білого осаду. Осад на фільтрі 4,5-дигідро-1Н-піразол-5-карбоксилату (DMSO-d6) промивали 2´50мл ацетонітрилу. Даний фільтрат d 7,28 (d, 2Н), 6,89 (d, 2Н), 5,08 (dd, 1Н), 4,14 (q, концентрували до приблизно 200мл на роторному 2H), 3,71 (dd, 1H), 3,37 (dd, 1H), 1,16 (t, 3H). 1Н випарнику. Другу 1-л чотиригорлу колбу, ЯМР для етил 3-хлоро-1-(2-хлорофеніл)-4,5обладнану механічною мішалкою, завантажували дигідро-1Н-піразол-5-карбоксилату (DMSO-d6) d 400мл води. Дану концентровану реакційну масу 7,41 (d, 1Н), 7,30 (m, 2H), 7,14 (m, 1H), 5,22 (dd, додавали до води протягом приблизно 5 хвилин. 1H), 3,90 (q, 2H), 3,68 (dd, 1H), 3,38 (dd, 1H), 0,91 (t, Отриманий продукт виділяли фільтрацією, 3H). промивали 100мл 20% водного розчину Приклад 11 ацетонітрилу, 75мл води і потім висушували на Одержання етил 3-хлоро-1(3-хлоро-2фільтрі у повітряному середовищі протягом 1 піридиніл)-1Н-піразол-5-карбоксилату години. Одержаний продукт складався із 36,6г (альтернативна назва етил 1-(3-хлоро-2(90%) кристалічного оранжевого порошку. піридиніл)-3-хлоропіразол-5-карбоксилат) Єдиними домішками, що були виявлені з У 2-л чотиригорлу колбу, обладнану допомогою 1Н ЯМР, були приблизно 1% невідомих механічною мішалкою, термометром, зворотним речовин та 0,5% ацетонітрилу. холодильником та трубкою для введення азоту, 1 Н ЯМР (DMSO-d6) d 8,59 (d, 1Н), 8,39 (d, 1H), завантажували 99,5г (0,328моль) 95% чистого 7,22 (dd, 1Н), 7,35 (s, 1H), 4,16 (q, 2H), 1,09 (t, 3H). етил 3-хлоро-1-(3-хлоро-2-тридиніл)-4,5-дигідроПриклад 13 1Н-піразол-5-карбоксилату, 1000мл ацетонітрилу Одержання 3-хлоро-1-(3-хлоро-2-піридиніл)та 35,0мл (0,661моль) 98% сірчаної кислоти. Після 1Н-піразол-5карбонової кислоти (альтернативна уведення сірчаної кислоти дана суміш назва 1-(3-хлоро-2-піридиніл)-3-хлоропіразол-5саморозігрівалась від 22 до 35°С. Після карбонова кислота) перемішування протягом кількох хвилин дану У 1-л чотиригорлу колбу, обладнану суміш обробляли 140г (0,518моль) персульфату механічною мішалкою, термометром та трубкою калію. Дану суспензію піддавали нагріванню зі для введення азоту, завантажували 79,3г зворотним холодильником при 84°С протягом 4,5 (0,270моль) 97,5% етил 3-хлоро-1-(3-хлоро-2годин. Утворену в результаті оранжеву суспензію піридиніл)-1Н-піразол-5-карбоксилату, 260мл відфільтровували ще в теплому стані (50-65°С) метанолу, 140мл води та 13,0г (0,325моль) для вилучення тонкого білого осаду. Осад на таблеток гідроксиду натрію. Дана суміш фільтрі промивали 50мл ацетонітрилу. Даний саморозігрівалась від 22 до 35°С, і вихідний фільтрат концентрували до приблизно 500мл на матеріал починав розчинятись після додавання роторному випарнику. Другу 2-л чотиригорлу гідроксиду натрію. Після перемішування протягом колбу, обладнану механічною мішалкою, 45 хвилин за умов навколишнього середовища завантажували 1250мл води. Дану концентровану весь вихідний матеріал розчинився. Утворений в реакційну масу додавали до води протягом результаті розчин глибокого оранжевоприблизно 5 хвилин. Отриманий продукт виділяли коричневого кольору концентрували до приблизно фільтрацією, промивали 3´125мл 25% водним 250мл на роторному випарнику. Потім дану розчином ацетонітрилу, один раз 100мл води і концентровану реакційну суміш розводили 400мл потім висушували протягом ночі у вакуумі при води. Водний розчин екстрагували 200мл ефіру. кімнатній температурі. Одержаний продукт Водний шар переносили у 1-л колбу Ерленмеєра, складався із 79,3г (82%) кристалічного оранжевого обладнану магнітною мішалкою. Потім розчин порошку. Єдиними домішками, що були виявлені з обробляли по краплях 36,0г (0,355моль) допомогою 1Н ЯМР, були приблизно 1,9% води та концентрованої соляної кислоти протягом 0,6% ацетонітрилу. 1 приблизно 10 хвилин. Даний продукт виділяли Н ЯМР (DMSO-d6) d 8,59 (d, 1Н), 8,38 (d, 1H), шляхом фільтрації, повторно суспендували 7,71 (dd, 1H), 7,31 (s, 1H), 4,16 (q, 2H), 1,09 (t, 3H). 2´200мл води, промивали один раз 100 мл води і Приклад 12 потім висушували на фільтрі у повітряному Одержання етил 3-бромо-1-(3-хлоро-2середовищі протягом 1,5 години. Одержаний піридиніл)-1Н-піразол-5-карбоксилату продукт складався із 58,1г (83%) кристалічного (альтернативна назва етил 1-(3-хлоро-2світло-коричневого порошку. Єдиною домішкою, піридиніл)-3-бромопіразол-5-карбоксилат) що була виявлена методом 1Н ЯМР, було У 1-л чотиригорлу колбу, обладнану приблизно 0,7% ефіру. механічною мішалкою, термометром, зворотним 1 Н ЯМР (DMSO-d6) d 13,95 (brs, 1Н), 8,56 (d, холодильником та трубкою для введення азоту, 1H), 8,25 (d, 1H), 7,68 (dd, 1H), 7,20 (s, 1Н). завантажували 40,2г (0,121моль) етил 3-бромо-1Приклад 14 (3-хлоро-2-піридиніл)-4,5-дигідро-1Н-піразол-5Одержання 3-бромо-1-(3-хлоро-2-піридиніл)карбоксилату, 300мл ацетонітрилу та 13,0мл 1Н-піразол-5-карбонової кислоти (альтернативна (0,245моль) 98% сірчаної кислоти. Після назва 1-(3-хлоро-2-піридиніл)-3-бромопіразол-5додавання сірчаної кислоти дана суміш карбонова кислота) саморозігрівалась від 22 до 36°С. Після У 300-мл чотиригорлу колбу, обладнану перемішування протягом кількох хвилин дану механічною мішалкою, термометром та трубкою суміш обробляли 48,0г (0,178моль) персульфату для введення азоту, завантажували 25,0г калію. Дану суспензію піддавали нагріванню зі (0,0756моль) 98,5% чистого етил 3-бромо-1-(3зворотним холодильником при 84°С протягом 2 хлоро-2-піридиніл)-1Н-піразол-5-карбоксилату, годин. Утворену в результаті оранжеву суспензію 1 29 81104 30 1,83ммоль) та триетиламіну (0,19г, 1,88ммоль). 75мл метанолу, 50мл води та 3,30г (0,0825моль) Потім реакційну суміш підігрівали до кімнатної таблеток гідроксиду натрію. Дана суміш температури та перемішували протягом ночі. саморозігрівалась від 29 до 34°С, і вихідний Потім дану суміш розводили дихлорометаном матеріал починав розчинятись після додавання гідроксиду натрію. Після перемішування протягом (200мл) та промивали водою (3´70мл). Органічну 90 хвилин за умов навколишнього середовища фазу висушували та випарювали з одержанням весь вихідний матеріал розчинився. Утворений в титульного продукту у вигляді масла (13,7г, 87% результаті розчин темно-оранжевого кольору вихід), котрий повільно утворював кристали. концентрували до приблизно 90мл на роторному Продукт, що піддавали рекристалізації із випарнику. Потім дану концентровану реакційну етилацетат/гексани, плавився при 99,5-100°С. суміш розводили 160мл води. Водний розчин ІК (нуйол (nujol)): 1740, 1638, 1576, 1446, 1343, екстрагували 100мл ефіру. Водний шар 1296, 1228, 1191, 1178, 1084, 1027, 948, 969, 868, переносили у 500-мл колбу Ерленмеєра, 845 cм-1. 1 обладнану магнітною мішалкою. Потім розчин Н ЯМР (CDCІ 3) d 8,01 (dd, J=1,4, 4,68Гц, 1H), обробляли по краплях 8,50г (0,0389моль) 7,95 (d, J=8,4Гц, 1H), 7,56 (dd, J=1,6, 7,8Гц. 1Н). концентрованої соляної кислоти протягом 7,36 (d, J=8,4Гц, 2H), 6,79 (dd, J=4,6, 7,7Гц, 1H), приблизно 10 хвилин. Даний продукт виділяли 5,72 (X із ABX, J=9, 11,8Гц, 1H), 4,16 (q, 2H). 3,33 шляхом фільтрації, повторно суспендували (1/2 AB у зразку ABX, J=17,5, 11,8Гц, 1Н), 3,12 (1/2 2´40мл води, промивали один раз 25мл води і AB у зразку ABX, J=17,3, 9Гц, 1Н), 2,45 (s, 3H), 1,19 потім висушували на фільтрі у повітряному (t, 3H). середовищі протягом 2 годин. Одержаний продукт З використанням способів, що описані у складався із 20,9г (91%) кристалічного рудуватоданому тексті та відомі у даній галузі, можуть бути коричневого порошку. Єдиними домішками, що одержані наступні сполуки Таблиць 1-3. У були виявлені методом 1Н ЯМР, були приблизно зазначених Таблицях використані наступні 0,8% невідомих речовин та 0,7% ефіру. скорочення: t - третинний, s - вторинний, n 1 нормальний, і - ізо, Me - метил, Et - етил. Рr Н ЯМР (DMSO-d6) d 13,95 (br s, 1H), 8,56 (d, пропіл, і-Рr - ізопропіл та t-Bu - третинний бутил. 1H), 8,25 (d, 1H), 7,68 (dd, 1H), 7,25 (s, 1H). Приклад 15 Одержання етил 3-бромо-1-(3-хлоро-2пірвдиніл)-4,5-дигідро-1Н-піразол-5-карбоксилату із етил 3-хлоро-1-(3-хлоро-2-піридиніл)-4,5дигідро-1Н-піразол-5-карбоксилату з використанням бромистого водню Бромистий водень перепускали через розчин етил 3-хлоро-1-(3-хлоро-2-піридиніл)-4,5-дигідро1Н піразол-5-карбоксилату (8,45г, 29,3ммоль) у дибромометані (85мл). За 90 хвилин потік газу припиняли, і реакційну суміш промивали водним розчином бікарбонату натрію (100мл). Органічну фазу висушували та випарювали під зниженим тиском з одержанням титульного продукту у вигляді масла (9,7г, 99% вихід), що кристалізувалось при витримці. 1 Н ЯМР (CDCІ 3) d 8,07 (dd, J=1,6, 4,8Гц, 1Н), 7,65 (dd, J=1,6, 7,8Гц, 1Н), 6,85 (dd, J=4,7, 7,7Гц, 1Н), 5,25 (X із ABX, 1Н, J=9,3, 11,9Гц), 4,18 (q, 2H), 3,44 (1/2 AB у зразку АВХ, J=11,7, 17,3Гц, 1H), 3,24 (1/2 AB у зразку ABX, J=9,3, 17,3Гц, 1 N), 1,19 (t, 3H). Наступний Приклад 16 ілюструє одержання етил 1-(3-хлоро-2-піридиніл)-4,5-дигідро-3-[[(4метилфеніл)сульфонал]окси]-1Н-піразол-5карбоксилату, котрий може бути використаний для одержання етил 3-бромо-1-(3-хлоро-2-тридиніл)4,5-дигідро-1Н-піразол-5-карбоксилату за способом, подібним до описаного у Прикладі 15. Приклад 16 Одержання етил 1-(3-хлоро-2-піридиніл)-4,5дигідро-3-[[(4-метилфеніл)сульфонал]окси]-1Нпіразол-5-карбоксилату Триетиламін (3,75г, 37,1ммоль) додавали по краплях до суміші етил 2-(3-хлоро-2-піридиніл)-5оксо-3-піразолідинкарбоксилату (10,0г, 37,1ммоль) та р-толуолсульфонілхлориду (7,07г, 37,1ммоль) в дихлорометані (100мл) при 0°С. Додавали додаткові порції р-толуолсульфонілхлориду (0,35г, 31 81104 32 де X, R1, R2 та n є такими, як визначено вище; R6 являє собою СН3, СІ або Br; R7 являє собою F, СІ, Br, І або CF3; і R8 являє собою С1-С4 алкіл. Сполуки Формули III корисні як інсектициди. Сполуки Формули III можуть бути одержані із сполук Формули II (та, у свою чергу, із сполук Формули 4 та І) з використанням процесів, що показані на Схемах 5-7. Сполучення піразолкарбонової кислоти Формули llа (сполука Формули ll, де R3 є Н) з антраніловою кислотою Формули 5 дає бензоксазинон Формули 6. На Схемі 5 бензоксазичон Формули 6 одержують безпосередньо через послідовне додавання метансульфонілхлориду у присутності третинного аміну, такого як триетиламін або піридин, до піразолкарбонової кислота Формули llа з наступним додаванням антранілової кислоти Формули 5 з наступним повторним додаванням третинного аміну та метансульфонілхлориду. Цей процес дає, загалом, добрі виходи бензоксазинону. Схема 6 зображує альтернативний процес одержання бензоксазинонів Формули 6, включаючи сполучення хлориду піразолової кислоти Формули 8 з ізатиновим ангідридом Формули 7 для прямого одержання бензоксазинону Формули 6. Застосування Сполуки Формул І, II та 4 використовуються як синтетичні проміжні сполуки для одержання сполуки Формули III Для цієї реакції придатні такі розчинники як піридин або піридин/ацетонітрил. Кислотні хлориди Формули 8 можуть бути одержані із відповідних кислот Формули llа за допомогою відомих способів, таких як хлорування тіонілхлоридом або оксалілхлоридом. Сполуки Формули III можуть бути одержані шляхом реакції бензоксазинонів Формули 6 з С1-С4 алкіламінами як зазначено на Схемі 7. Дана реакція може проводитись у чистому вигляді або в різновиді придатних розчинників, включаючи тетрагідрофуран, діетиловий ефір, дихлорометан або хлороформ, при оптимальних температурах, що варіюють від кімнатної температури до температури перегонки даного розчинника. Загальна реакція бензоксазинонів з амінами для одержання антраніламідів добре задокументована 33 81104 у хімічній літературі. Огляд по хімії беноксазинону [див. у Jakobsen et al., Biorganic and Medicinal Chemistry 2000, 8, 2095-2103 та наведених там посиланнях. Див. також Coppola, J. Heterocyclic Chemistry 1999, 36, 563-588]. 34
ДивитисяДодаткова інформація
Назва патенту англійськоюSubstituted dihydro 3-halo-1h-pyrazole-5-carboxylates and their preparation
Автори англійськоюFreudenberger John Herbert, Lahm George Philip, Selby Thomas Paul, Stevenson Thomas Martin
Назва патенту російськоюЗамещенные дигидро-3-гало-1н-пиразол-5-карбоксилаты и способы их получения
Автори російськоюФройденбергер Джон Герберт, Лам Джордж Филип, Сэлби Томас Пол, Стивенсон Томас Мартин
МПК / Мітки
МПК: C07D 401/04, C07D 231/06, C07D 231/14, C07D 231/16, C07D 231/08, C07D 231/22, A01N 43/56
Мітки: одержання, заміщені, дигідро-3-гало-1н-піразол-5-карбоксилати, способи
Код посилання
<a href="https://ua.patents.su/17-81104-zamishheni-digidro-3-galo-1n-pirazol-5-karboksilati-ta-sposobi-kh-oderzhannya.html" target="_blank" rel="follow" title="База патентів України">Заміщені дигідро-3-гало-1н-піразол-5-карбоксилати та способи їх одержання</a>
Спосіб одержання 5-[2-етокси-5-(4-метилпіперазин-1-іл-сульфоніл)-феніл]-1-метил-3-н-пропіл-1,6-дигідро-7н-піразол-[4,3-d]-піримідин-7-ону та способи одержання проміжних продуктів

Номер патенту: 78692
Опубліковано: 25.04.2007
Автори: Доші Мадхукант Мансукхлал, Шріканде Атул Анант, Моді Шіріш Бхагванлал
МПК: C07D 237/00
Мітки: проміжних, одержання, спосіб, 5-[2-етокси-5-(4-метилпіперазин-1-іл-сульфоніл)-феніл]-1-метил-3-н-пропіл-1,6-дигідро-7н-піразол-[4,3-d]-піримідин-7-ону, способи, продуктів
Формула / Реферат:
1. Cпосіб одержання 5-[2-етокси-5-(4-метилпіперазин-1-ілсульфоніл)-феніл]-1-метил-3-н-пропіл-1,6-дигідро-7Н-піразол-[4,3-d]-піримідин-7-ону (V) та його фармацевтично прийнятних солей, який включає такі стадії:a) взаємодія 1-метил-4-аміно-3-н-пропілпіразол-5-карбоксаміду (І) із хлористим воднем у розчиннику, такому як ізопропанол, з одержанням 1-метил-4-аміно-3-н-пропілпіразол-5-карбоксамідгідрохлориду (II);b) взаємодія сполуки...
Спосіб одержання 3-гало-4,5-дигідро-1н-піразолів

Номер патенту: 79799
Опубліковано: 25.07.2007
Автор: Енніс Гері Девід
МПК: C07D 231/06, C07D 401/04
Мітки: спосіб, 3-гало-4,5-дигідро-1н-піразолів, одержання
Формула / Реферат:
1. Спосіб одержання 3-гало-4,5-дигідро-1Н-піразолової сполуки формули Іа, (Ia)деХ1 є галогеном;кожний R3 незалежно є C1-С4 алкілом, С2-С4 алкенілом, C2-C4 алкінілом, C3-C6 циклоалкілом, C1-С4 галоалкілом, С2-С4 галоалкенілом, C3-C6 галоалкінілом, C3-C6 галоциклоалкілом, галогеном, CN, NO2, C1-С4 алкокси групою, C1-С4 галоалкокси групою, C1-С4...
Способи одержання проміжних продуктів пестицидів

Номер патенту: 69451
Опубліковано: 15.09.2004
Автори: Ванжелісті Манюель, Ансель Жан-Ерік, Верспрумі П'єр, Перрен-Жане Жилль
МПК: C07B 61/00, C07C 211/45, C07D 213/73, C07D 231/44, C07C 209/42, C07C 211/52
Мітки: способи, одержання, проміжних, продуктів, пестицидів
Формула / Реферат:
1. Спосіб одержання сполуки формули (І) , (I)в якій R1 являє собою галогеналкіл, галогеналкокси або -SF5; W являє собою N або CR3; і R2 і R3 кожний незалежно являє собою водень або хлор; або її кислотно-адитивної солі; який передбачає гідрогеноліз сполуки формули (II) (II)або...
b-арил-a-оксизаміщені алкілкарбонові кислоти, способи їх одержання, проміжні сполуки, способи їх одержання, фармацевтична композиція та спосіб лікування за допомогою цих сполук

Номер патенту: 73917
Опубліковано: 17.10.2005
Автори: Лохрей Відіа Бхушан, Лохрей Брадж Бхушан, Рамануджам Раджагопалан, Баджі Ашок Чаннавеераппа, Калчар Шіварамаййа, Чакрабарті Ранджан
МПК: C07D 265/38, C07D 413/06, C07D 265/28, C07D 417/06, C07D 279/00, C07C 69/734, C07C 59/00
Мітки: лікування, b-арил-a-оксизаміщені, фармацевтична, сполук, цих, сполуки, одержання, кислоти, проміжні, композиція, спосіб, допомогою, способи, алкілкарбонові
Формула / Реферат:
1. Сполука формули (І) , (І)де R1, R2, R3 і R4 являють собою водень, гідрокси, (С1-С3)-алкіл; цикл А являє собою феніленове кільце; Х являє собою гетероатом, вибраний серед атомів кисню або сірки; Аr являє собою фенілен або бензофураніл; R5 являє собою водень, алкіл або утворює зв'язок разом з R6; R6 являє собою водень, алкіл або утворює зв'язок разом з...
Біциклічна сполука, способи її одержання, фармацевтичні композиції, що її містять,способи лікування та попередження різних захворювань, проміжні сполуки та способи їх одержання

Номер патенту: 72883
Опубліковано: 16.05.2005
Автори: Баджі Ашок Чаннавеераппа, Рамануджам Раджагопалан, Калчар Шіварамаййа, Лохрей Відіа Бхушан, Чакрабарті Ранджан, Лохрей Брадж Бхушан
МПК: A61K 9/14, A61K 9/20, A61P 3/10, C07D 265/36, A61P 19/10, A61K 9/10, A61P 17/06, C07D 413/06, A61K 31/538, A61P 25/28, A61P 13/12, C07D 279/00, A61P 3/04, A61K 9/48, A61P 3/06, A61P 9/10, A61K 9/08, A61K 31/5415
Мітки: одержання, сполука, лікування, сполуки, композиції, попередження, способи, захворювань, біциклічна, проміжні, різних, містять,способи, фармацевтичні
Формула / Реферат:
1. Сполука формули (І), (I)де групи R1, R2, R3, R4 і групи R5 і R6, коли вони приєднані до атома вуглецю, можуть бути однаковими або різними і означають водень, галоген, гідрокси або необов'язково заміщену групу, вибрану з алкілу, алкокси, фенілу, карбонової кислоти або сульфонової кислоти ; один або обидва замісники R5 і R6 можуть також означати оксогрупу,...
Попередній патент: Різальний верстат настільного типу
Наступний патент: Друкарська фарба для трафаретного друку на водній основі
Випадковий патент: Таблетка кветіапіну з пролонгованим вивільненням і спосіб її отримання