Спосіб одержання поліолефінових основ синтетичних мастил
Номер патенту: 84505
Опубліковано: 27.10.2008
Автори: Шамсутдінов Владімір Гарафовіч, Ханнанов Роберт Габдрахмановіч, Ільясов Габбас Лукмановіч, Демідов Міхаіл Алєксандровіч, Матковскій Пєтр Євгєньєвіч, Старцева Галіна Павловна, Троіцкій Владімір Ніколаєвіч, Яруллін Рафінат Саматовіч, Савченко Валєрій Івановіч











Формула / Реферат
1. Спосіб одержання поліолефінових основ синтетичних мастил олігомеризацією вищих олефінів, який включає стадії підготовки олефінової сировини та розчинів компонентів катіонної каталітичної системи, ізомеризації вищих лінійних альфа-олефінів, олігомеризації олефінової сировини під дією катіонної алюміній- та хлоровмісної каталітичноїсистеми - Аl(0)-НСl-(СН3)3ССl, вилучення з олігомеризату відпрацьованого каталізатора, розділення олігомеризату на фракції та гідрування виділених фракцій, причому після стадії олігомеризації та/або після стадії вилучення відпрацьованого каталізатора з олігомеризату проводять стадію дехлорування наявних в олігомеризаті монохлоровмісних олігомерів, а після стадії розділення олігомеризату на фракції проводять стадію деполімеризації високомолекулярних продуктів, виділених у вигляді кубового залишку на стадії розділення олігомеризату на фракції.
2. Спосіб за п. 1, який відрізняється тим, що олігомеризацію вищих олефінів проводять у сумішах вищих олефінів, здатних до олігомеризації, із продуктами їх олігомеризації або в сумішах вищих олефінів, здатних до олігомеризації, із продуктами їх олігомеризації та з ароматичними вуглеводнями, такими як бензол або толуол, під дією каталітичної системи Аl(0)-НСl-(СН3)3ССl при температурі від 110 до 180 °С, концентрації Аl(0) від 0,02 до 0,08 г-атом/л, мольних співвідношеннях НСl/Аl(0), змінюваних в межах від 0,002 до 0,06 та мольних співвідношеннях (СН3)3ССl/Al(0), змінюваних в межах від 1,0 до 5,0, де Аl(0) - високодисперсний порошкоподібний алюміній з розмірами частинок, які змінюються в межах від 1 до 100 мкм, наприклад, Аl(0) марки ПА-1, ПА-4, ПАП-1, АСД-4, АСД-40, АСД-Т.
3. Спосіб за пп. 1 або 2, який відрізняється тим, що як вищі олефіни використовують суміші лінійних або розгалужених альфа-олефінів з ізо-олефінами та з олефінами з внутрішньомолекулярним розташуванням подвійного зв'язку ("внутрішніми" олефінами), які містять від 4 до 14, переважно 10, атомів вуглецю, при такому співвідношенні інгредієнтів, мас. %: альфа-олефіни 0,5-99,0, ізо-олефіни 0,5-5,0, "внутрішні" олефіни - решта до 100.
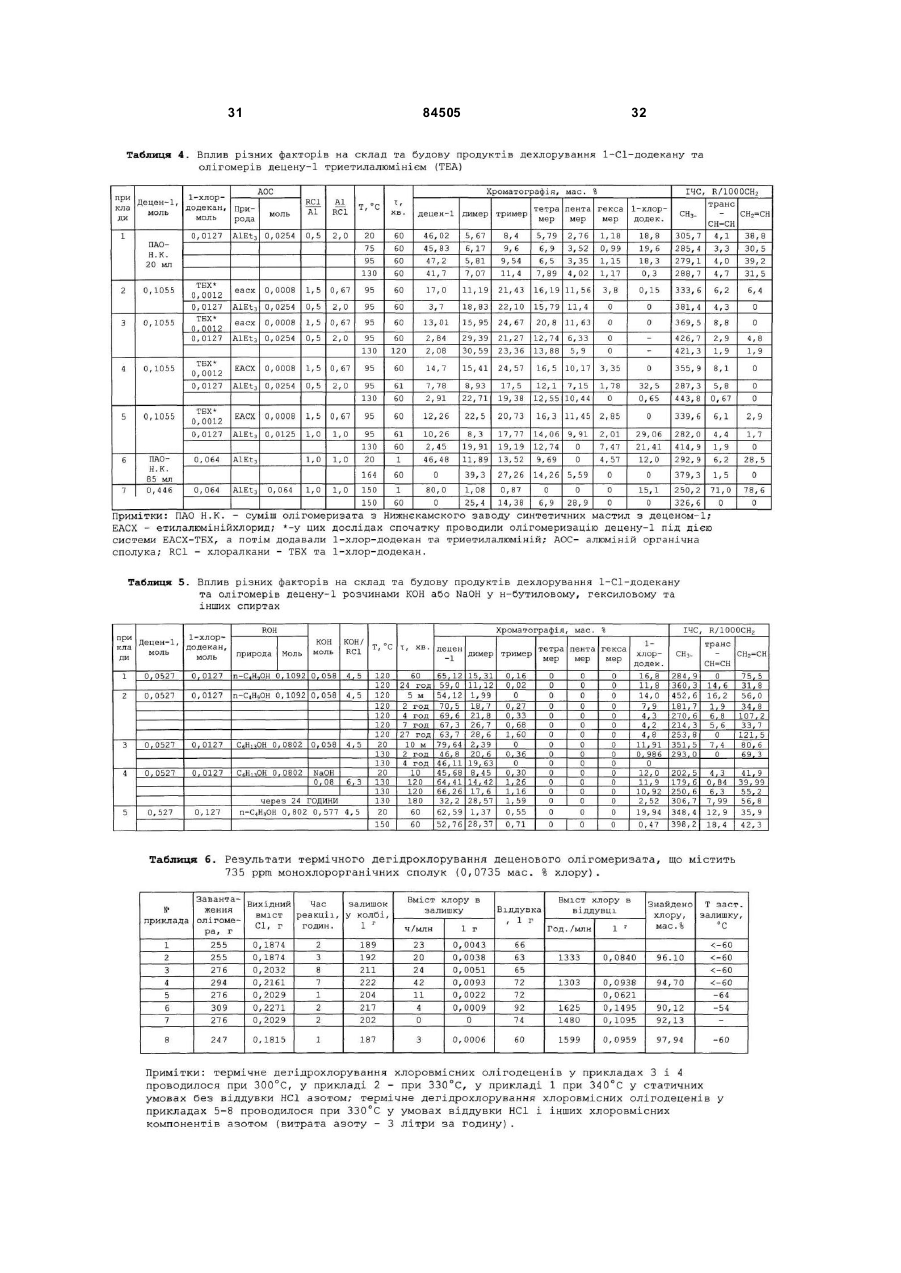
4. Спосіб за будь-яким з пп. 1-3, який відрізняється тим, що дехлорування присутніх в олігомеризаті монохлоровмісних олігомерів (RCl) здійснюють після стадії олігомеризації високодисперсним порошкоподібним металевим алюмінієм - Аl(0) з розмірами частинок, які змінюються в межах від 1 до 100 мкм, наприклад, марок ПА-1, ПА-4, АСД-4, АСД-40, АСД-Т, ПАП-1, при мольних співвідношеннях Al(0)/RCl, змінюваних в межах від 0,5 до 2,0 в інтервалі температур від 110 до 180 °С протягом від 30 до 180 хвилин.
5. Спосіб за п. 1, який відрізняється тим, що дехлорування присутніх в олігомеризаті RCl здійснюють після стадії олігомеризації триетилалюмінієм (TEA) при мольних співвідношеннях TEA/RCl, змінюваних в межах від 0,5 до 2,0, в інтервалі температур від 95 до 150 °С протягом від 30 до 180 хвилин.
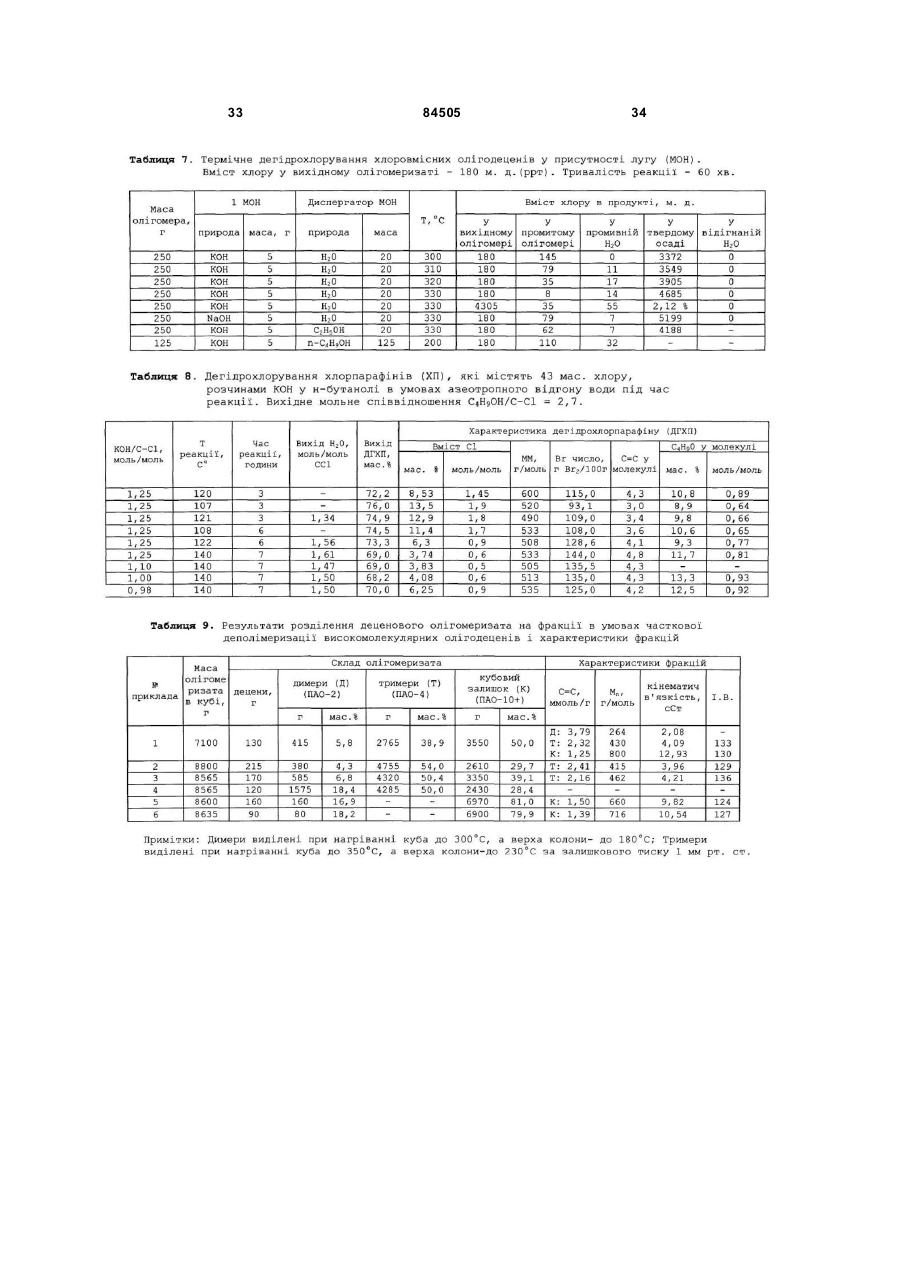
6. Спосіб за п. 1, який відрізняється тим, що дехлорування, присутніх в олігомеризаті RCl, здійснюють після стадії вилучення відпрацьованого каталізатора спиртовим розчином гідроксиду калію або натрію (МОН) при мольних співвідношеннях MOH/RCl, змінюваних в межах від 1,1 до 2,0, в інтервалі температур від 120 до 160 °С протягом від 30 до 240 хвилин.
7. Спосіб за п. 1, який відрізняється тим, що дехлорування, присутніх в олігомеризаті RCl, здійснюють після стадії вилучення відпрацьованого каталізатора шляхом термічного дегідрохлорування їх у інтервалі температур від 280 до 350 °С та тиску 1-2 бар протягом від 30 до 180 хвилин при віддуванні хлористого водню, що виділяється, азотом, діоксидом вуглецю, метаном або перегрітою водяною парою.
8. Спосіб за п. 1, який відрізняється тим, що дехлорування, присутніх в олігомеризаті RCl, здійснюють у присутності сухих гідроксидів лужних металів (МОН) при мольних співвідношеннях MOH/RCl, змінюваних в межах від 1,1 до 2,0 у інтервалі температур від 300 до 330 °С.
9. Спосіб за п. 8, який відрізняється тим, що сухі гідроксиди лужних металів одержують безпосередньо в олігомеризаті відгоном води при нагріванні суміші звільненого від відпрацьованого каталізатора олігомеризату та 5-40 %-ного водного розчину гідроксиду лужного металу при температурі, змінюваній в інтервалі від 100 до 200 °С.
10. Спосіб за п. 1, який відрізняється тим, що деполімеризацію високомолекулярних продуктів, виділених у вигляді кубового залишку на стадії розділення олігомеризату на фракції, проводять нагріванням їх при температурі, змінюваній в інтервалі від 330 до 360 °С, та тиску від 1,0 до 10,0 мм.рт.ст. протягом від 30 до 120 хвилин при безперервному вилученні продуктів з реактора деполімеризації.
11. Спосіб за п. 1, який відрізняється тим, що гідрування виділених з олігомеризату вузьких фракцій олігоолефінів проводять під дією паладієвого нанесеного на оксид алюмінію каталізатора, переважно Pd(0,2 мас. %)/Аl2O3, модифікованого безводним гідроксидом калію, який беруть у кількості від 30 до 100 мас. % з розрахунку на каталізатор гідрування, при температурі, змінюваній в інтервалі від 200 до 250 °С, та тиску водню 20 ат.
Текст
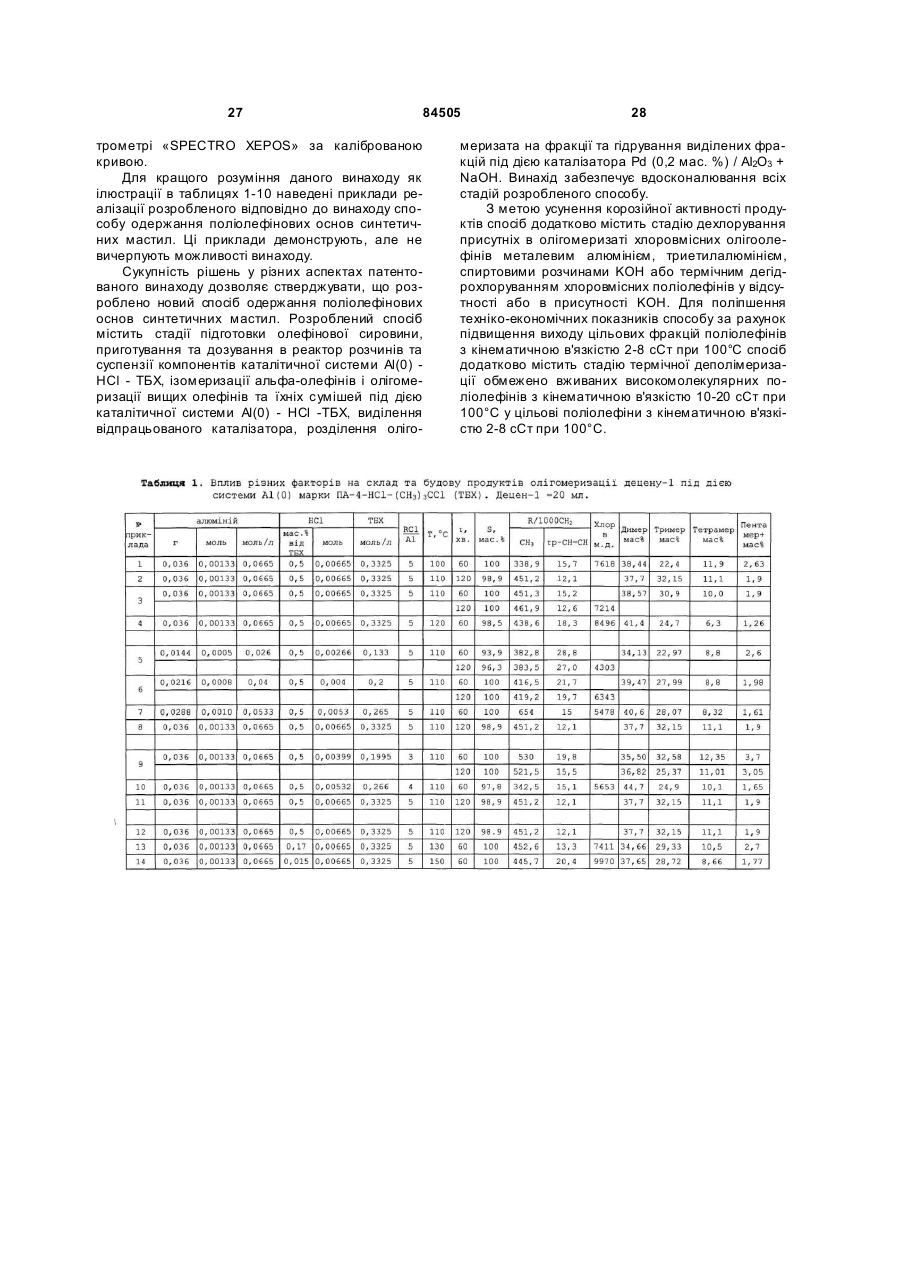
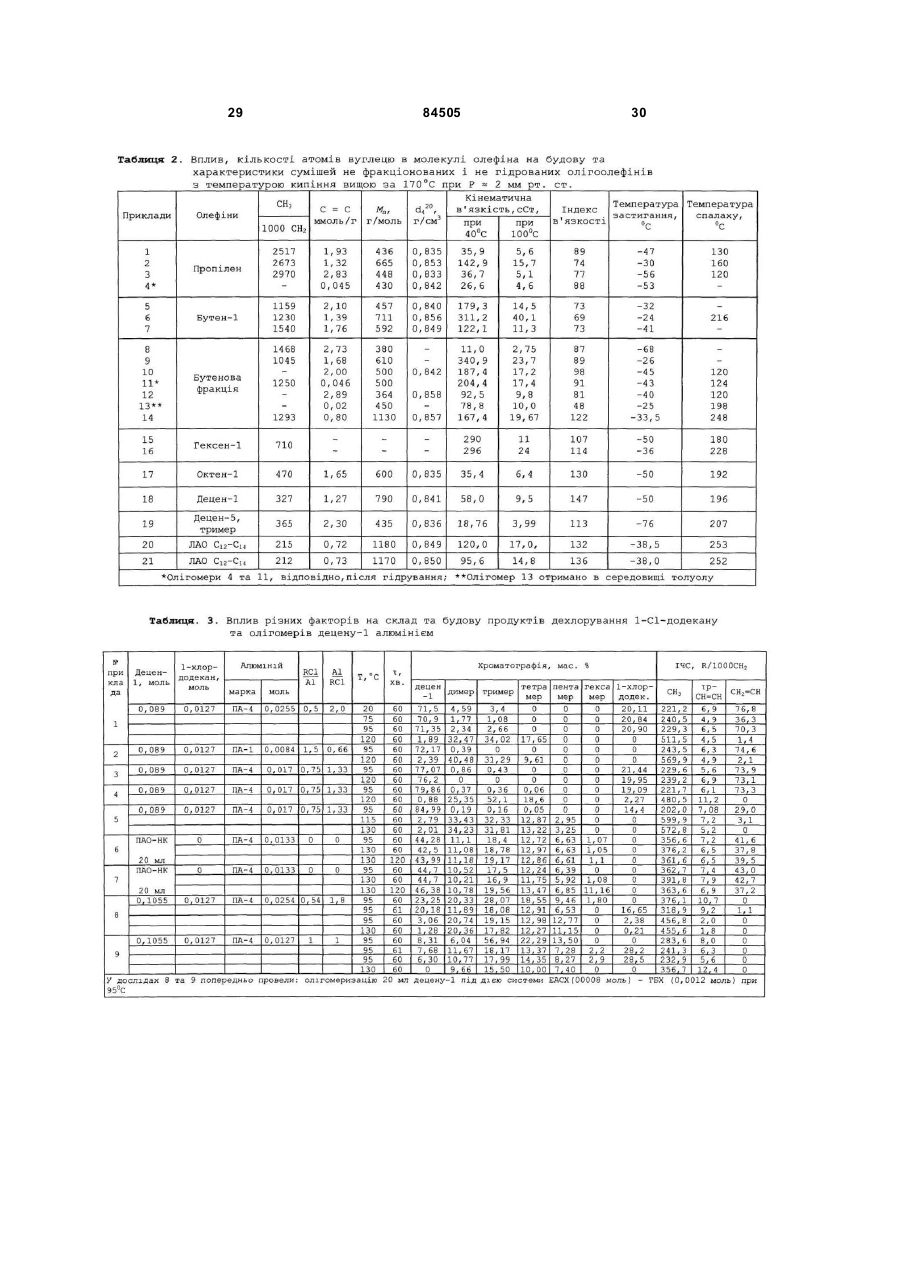
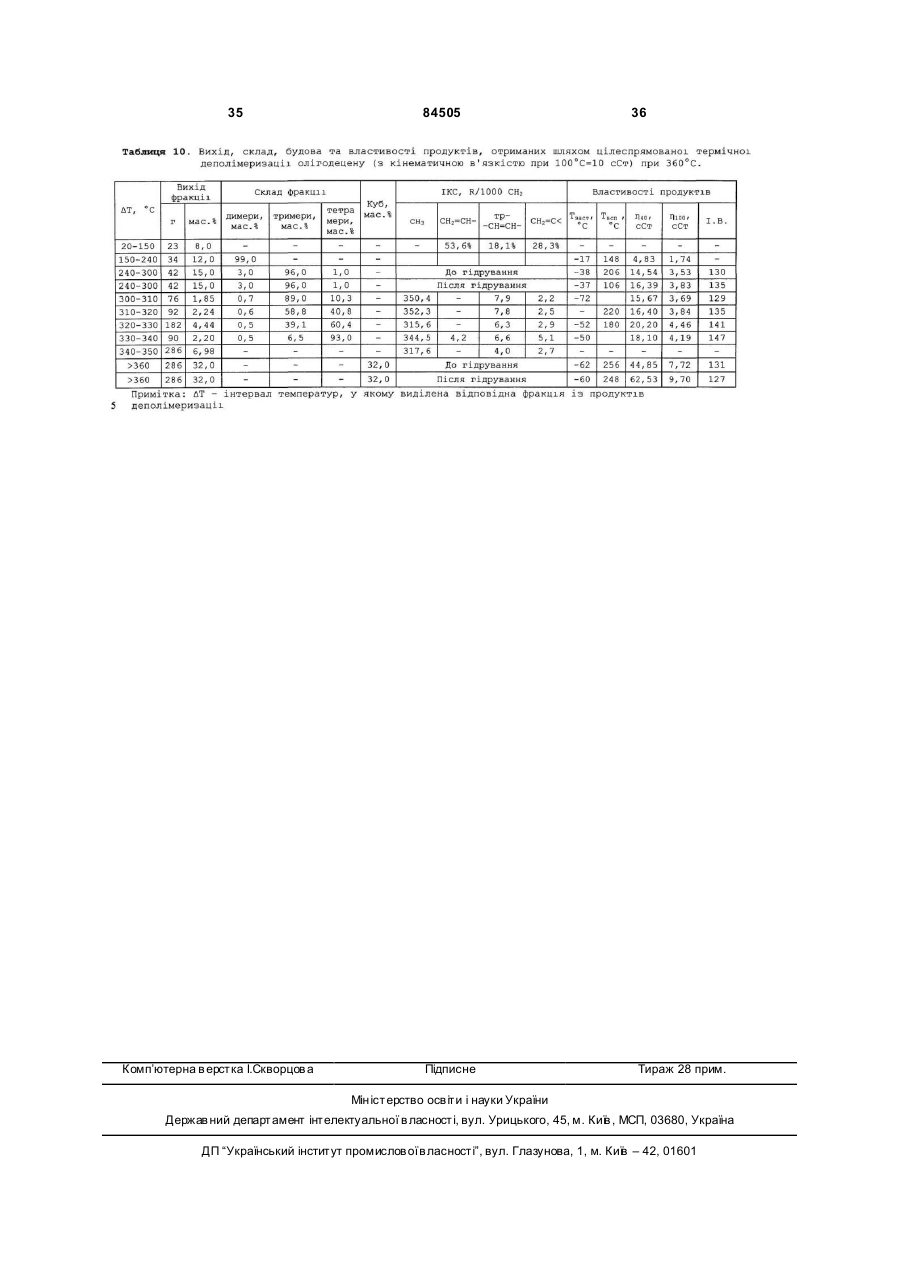
1. Спосіб одержання поліолефінових основ синтетичних мастил олігомеризацією вищих олефінів, який включає стадії підготовки олефінової сировини та розчинів компонентів катіонної каталітичної системи, ізомеризації вищих лінійних альфа-олефінів, олігомеризації олефінової сировини під дією катіонної алюміній- та хлоровмісної каталітичної системи - Аl(0)-НСl-(СН3)3ССl, вилучення з олігомеризату відпрацьованого каталізатора, розділення олігомеризату на фракції та гідрування виділених фракцій, причому після стадії олігомеризації та/або після стадії вилучення відпрацьованого каталізатора з олігомеризату проводять стадію дехлорування наявних в олігомеризаті монохлоровмісних олігомерів, а після стадії розділення олігомеризату на фракції проводять стадію деполімеризації високомолекулярних продуктів, 2 (19) 1 3 84505 4 єм (TEA) при мольних співвідношеннях TEA/RCl, змінюваних в межах від 0,5 до 2,0, в інтервалі температур від 95 до 150 °С протягом від 30 до 180 хвилин. 6. Спосіб за п. 1, який відрізняється тим, що дехлорування, присутніх в олігомеризаті RCl, здійснюють після стадії вилучення відпрацьованого каталізатора спиртовим розчином гідроксиду калію або натрію (МОН) при мольних співвідношеннях MOH/RCl, змінюваних в межах від 1,1 до 2,0, в інтервалі температур від 120 до 160 °С протягом від 30 до 240 хвилин. 7. Спосіб за п. 1, який відрізняється тим, що дехлорування, присутніх в олігомеризаті RCl, здійснюють після стадії вилучення відпрацьованого каталізатора шляхом термічного дегідрохлорування їх у інтервалі температур від 280 до 350 °С та тиску 1-2 бар протягом від 30 до 180 хвилин при віддуванні хлористого водню, що виділяється, азотом, діоксидом вуглецю, метаном або перегрітою водяною парою. 8. Спосіб за п. 1, який відрізняється тим, що дехлорування, присутніх в олігомеризаті RCl, здійснюють у присутності сухих гідроксидів лужних металів (МОН) при мольних співвідношеннях MOH/RCl, змінюваних в межах від 1,1 до 2,0 у інтервалі температур від 300 до 330 °С. 9. Спосіб за п. 8, який відрізняється тим, що сухі гідроксиди лужних металів одержують безпосередньо в олігомеризаті відгоном води при нагріванні суміші звільненого від відпрацьованого каталізатора олігомеризату та 5-40 %-ного водного розчину гідроксиду лужного металу при температурі, змінюваній в інтервалі від 100 до 200 °С. 10. Спосіб за п. 1, який відрізняється тим, що деполімеризацію високомолекулярних продуктів, виділених у вигляді кубового залишку на стадії розділення олігомеризату на фракції, проводять нагріванням їх при температурі, змінюваній в інтервалі від 330 до 360 °С, та тиску від 1,0 до 10,0 мм.рт.ст. протягом від 30 до 120 хвилин при безперервному вилученні продуктів з реактора деполімеризації. 11. Спосіб за п. 1, який відрізняється тим, що гідрування виділених з олігомеризату вузьких фракцій олігоолефінів проводять під дією паладієвого нанесеного на оксид алюмінію каталізатора, переважно Pd(0,2 мас. %)/Аl2 O3, модифікованого безводним гідроксидом калію, який беруть у кількості від 30 до 100 мас. % з розрахунку на каталізатор гідрування, при температурі, змінюваній в інтервалі від 200 до 250 °С, та тиску водню 20 ат. Винахід відноситься до технологій для нафтохімії, а саме до способу одержання поліолефінових основ синтетичних мастил шля хом катіонної олігомеризації олефінової сировини та може бути використаним в нафто хімічній промисловості. Одержані відповідно до способу запропонованого до патентування продукти можуть використовуватися як основа синтетичних поліолефінових (олігоолефінових) мастил різноманітного призначення: моторних (автомобільних, авіаційних, вертолітних, тракторних, танкових); трансмісійних, редукторних, вакуумних, компресорних, холодильних, трансформаторних, кабельних, веретенних, медичних, у складі різних мастильних матеріалів, а також як пластифікатори для пластмас, каучуків, твердих ракетних палив; сировини для одержання присадок, емульгаторів, флотореагентів, піноутворювачів, компонентів мастильно-охолоджуючих і гідравлічних рідин; високооктанових добавок до палив, тощо. Відомі способи одержання поліолефінових основ синтетичних мастил шляхом катіонної олігомеризації вищих олефінів, котрі передбачають стадію підготовки олефінової сировини та розчинів компонентів каталітичної системи, стадію олігомеризації олефінової сировини, стадію виділення з олігомеризата відпрацьованого каталізатора методом водно-лужного та наступного водного відмивання, стадію розділення очищеного олігомеризата на фракції та стадію гідрування виділених цільових фракцій. Найбільш широко відомі способи одержання поліолефінових основ синтетичних мастил відрізняються між собою тим, які сполуки катіонних ка талізаторів у них застосовуються. Відповідно до відомих способів катіонну олігомеризацію олефінів С 3-С14 (тобто олефінів, які містять від 3 до 14 атомів вуглецю) ініціюють (каталізують) за допомогою: протонних кислот (кислот Бренстеда); апротонних кислот (кислот Люїса); алкілалюміній- (або бор) галогенідів; солей стабільних карбкатіонів R+A-; природних і синтетичних алюмосилікатів, цеолітів або гетерополікислот у НФормі; різних дво- і трикомпонентних комплексів, які включають мономер; поліфункціональних каталізаторів Циглера-Натта; металоценових каталізаторів; фізичних методів стимулювання хімічних реакцій [1. Дж.Кеннеді. Катіонна полімеризація олефінів. M.: Мир, 1978. 430 C; .2. J.P.Kennedy, E.Marechal. Carbocationic Polymerization. N.-Y., 1982. 510 P.]. Найбільш широке промислове застосування як каталізатори катіонної олігомеризації олефінів та інших мономерів знайшли каталітичні системи, які включають кислоти Люїса (BF3, АІСІ 3, АІВr3, TiCl4, ZrCl4 та ін.), алкілалюміній-(або бор) галогеніди RnMX3-n (де R - алкіл С 1-С10-, арил-, алкеніл- та інші групи; M-Al або В; X-Cl, Br, І) та природні або синтетичні алюмосилікати, цеоліти та гетерополікислоти в H-Формі. При одержанні моторних ПАОМ на основі лінійних альфаолефінів (ЛАО) С6-С14 (переважно - на основі децену-1) зазвичай використовують каталітичні системи, які включають кислоти Люїса або алкілалюмінійгалогеніди. Так, відома велика кількість способів одержання поліолефінових основ синтетичних мастил, відповідно до яких як каталізатори олігомеризації ЛАО С6-С14 використовують системи які включа 5 84505 ють трифтористий бор і різні протонодонорні співкаталізатори - воду, спирти, карбонові кислоти, ангідриди карбонових кислот, кетони, поліоли та їхні суміші [1] [Пат. США 5550307 від 27,08.1996. Мкл. С07 C 2/14; Нкл. 585/525.]. Поліолефінові основи синтетичних мастил відповідно до цих способів одержують шляхом олігомеризації олефінів С6-С14 під дією згаданих борфторидних каталізаторів за температур 20-90°C у масі протягом 2-5 годин. Концентрацію трифтористого бору в реакційному середовищі варіюють у межах від 0,1 до 10 мас. %. Конверсія вихідних олефінів змінюється в межах від 80 до 99 мас. %. У результаті олігомеризації, наприклад, децену-1, утворюється суміш ди-, три-, тетра-мерів і більш високомолекулярних олігомерів. Сумарний вміст ди- і тримерів у продуктах змінюється в межах від 30 до 70 мас. %. Головним недоліком всіх способів одержання поліолефінових основ синтетичних мастил цього типу є те, що вони ґрунтуються на використанні каталізаторів, до складу яких входить дефіцитний, леткий, отруйний, корозійно-активний трифтористий бор. Крім того, через відносно низьку активність каталізаторів цього типу в олігомеризації ЛAO процес протікає протягом 2-5 годин. При промисловій реалізації цих способів використовуються дорогі великі за об'ємом і за металоємністю реактори змішування в антикорозійному виконанні. Відома [2] [патент. США 5196635 від 23,03.1993. Мкл. С07 C 2/22; Нкл. 585-532] також велика кількість способів одержання поліолефінових основ синтетичних мастил відповідно до яких олігомеризацію олефінів проводять під дією катіонних каталізаторів які включають галогеніди алюмінію та протонодонори - воду, спирти, карбонові кислоти, прості або складні ефіри, кетони (наприклад, диметиловий ефір етилен гліколя, етилен гліколь-діацетат), галоідалкіл [2] [Пат. США 5196635 від 23,03.1993. Мкл. С07 C 2/22; Нкл. 585532]. У деяких способах ці каталізатори використовують у поєднанні зі сполуками нікелю [3] [Пат. США 5 489721 від 06,02.1996. Мкл. С07 C 2/20; Нкл. 585-532]. Додавання сполук нікелю до каталізаторів, які застосовуються у згаданих способах, забезпечують можливість регулювання фракційного складу одержаних олігоолефінів. Одержання поліолефінових основ синтетичних мастил олігомеризацією альфа-(C4-C14) або внутрішніх C10-C15 олефінів (отриманих шля хом дегідрування парафінів) запропонованим способом [4] [Пат. США 4113790 від 12,09.1978. Мкл. С07 C 3/10; Нкл. 585-532] здійснюють під дією каталізаторів АІХ3 + протонодонор за температур 100140°C протягом 3-5 годин. Концентрацію АІХ3 варіюють у межах від 0,1 до 10 мол. % в розрахунку на олефіни, мольне співвідношення протонодонор/АI варіюють у межах від 0,05 до 1,25. З підвищенням цього співвідношення від 0,05 до 1,25 конверсія олефінів знижується від 99 до 12 мас. %. Способи цього типу характеризуються наступними загальними недоліками: - складною процедурою приготування каталізаторів, що включає багато операцій - сублімація та розмелювання АІСІ3 , приготування комплексу; - одержувані відповідно до цих способів ката 6 лізатори є грузлими, клейкими речовинами, погано розчинними в олефінах, через високу адгезію до охолоджуваних стінок реакторів вони погано видаляються з реакторів після завершення олігомеризації; - низькою активністю каталізаторів, які використовуються у процесі олігомеризації, що вимагає застосування більших за об'ємом металовмісних реакторів змішування; - високими видатковими коефіцієнтами за АІХ3 розраховуючи на одержані продукти; Головним загальним недоліком способів цього типу є те, що використання їх приводить до одержання в основному високомолекулярних та високов'язких продуктів, які містять до 1 мас. % хлору. Розроблено кілька способів одержання поліолефінових основ синтетичних мастил, які ґрунтуються на використанні біфункціональних комплексних каталізаторів, що включають сполуки перехідних металів (TiCl4, ZrCl4) і алкілалюмінійгалогеніди RnAlX3-n (дивися, наприклад, [5] [Пат. Великобританії 1522129. Мкл.С07 C 2/22. Нкл. СЗР;В] При застосуванні, як передбачено у цих способах, бі функціональних каталітичних систем типу TiCl4, - Rn Al X3-n утворюється два типи активних центрів - катіонні та аніонно-координаційні. Через це олігомеризація олефінів C3-C14 під дією катіонних активних центрів практично у всіх випадках супроводжується полімеризацією олефінів C3C14 під дією аніонно-координаційних активних центрів у нерозчинні високомолекулярні поліолефіни, які важко видаляються із реактора. При цьому під дією біфункціональних комплексних каталізаторів у всіх випадках утворюються високомолекулярні високов'язкі олігоолефіни, які не можуть використовуватися як основи найбільш широко вживаних моторних мастил. Це є головним недоліком способів такого типу. Відповідно до деяких способів у катіонних процесах полімеризації, олігомеризації та алкілювання широко застосовуються також двокомпонентні розчинні монофункціональні каталітичні системи, які включають алкілалюмінійгалогенід RnAl X3-n і галоідорганічну сполуку R'X при мольному співвідношенні R'X/RnAl X3- n = 1,0-5,0 (де R CH3, С2Н5, С3Н7 або ізо-С 4Н9; X - хлор, бром або йод; n = 1,0; 1,5 або 2,0; R’ - H [6] [ Пат. США 4952739. 28,08.1990, Мкл. С07 С07С/18; Нкл. 585/18; 585/511], первинний, вторинний або третинний алкіл, аліл або бензил [7] [ Пат. ФРН 2304314. 1980, Мкл. С08 F 110/20]. У каталітичних системах цього типу Rn AlX3-n є основою каталізатора, a RX -співкаталізатором. Відповідно до цих способів каталітичні системи RnAlX3-n – R’ X використовуються для ініціювання катіонної олігомеризації окремих лінійних альфа-олефінів або їх сумішей, від пропілену до тетрадецену включно в поліальфа-олефінові основи синтетичних мастил у середовищі вихідних олефінів або сумішей їх із продуктами олігомеризації та парафіновими, ароматичними або галоідовмісними вуглеводнями за температур до 250°C. Катіонні активні центри ([R’+ (RnAlX4-n)-] і R’+) у каталітичних системах RnAl X3-n -R' утворюються у відповідності до наступної спрощеної схеми: 7 84505 RnAl X3-n + R’X « [R’+ (RnAlX4-n)-] « R’+ + (RnAl X4-n)Утворення катіонних активних центрів у розглянути х каталітичних системах відбувається з дуже високою швидкістю. Завдяки цьому відразу ж після змішування розчинів компонентів розглянутих каталітичних систем досягається висока концентрація катіонних активних центрів, і процес олігомеризації протікає без індукційного періоду з дуже високою початковою швидкістю. При цьому 95-98 відсоткова конверсія вихідних олефінів в олігомерні продукти за температур 20-200°C досягається протягом шести-одинієї хвилини, відповідно. Такий характер кінетики олігомеризації лінійних альфа-олефінів (ЛАО) під дією розглянутих каталітичних систем забезпечує можливість проведення процесу олігомеризації у форсованому ізотермічному режимі в реакторах витиснення трубчастого типу при термінах перебування від 1 до 10 хвилин [8] [ Пат. РФ 2201799. 29,09.2000, Мкл. 7 У 01 J 8/06, C 08 F 10/10; Бюл. Зобр. 2003. № 10]. При одержанні оліго-олефінових основ синтетичних мастил відповідно до цих способів у ході олігомеризації ЛAO в масі або в середовищі парафінових вуглеводнів під дією каталітичних систем RnAlX3-n- R’X утворюються високорозгалужені олігомери, які застигають за низьких температур, та містять один ди-, три- або тетраалкілзаміщений подвійний зв'язок і до 0,2 мас.% монохлоролігоолефінів, а при олігомеризації в середовищі або в присутності ароматичних вуглеводнів (бензол, толуол, нафталін) утворюються олігоалкілароматичні олієподібні продукти (теломери), які не містять подвійних зв'язків [9] [Пат. РФ 2199516 від 18,04.2001 p. MKH 7 С07С 2/22. Бюл. Зобр. №6 від 27,02.2003р.]. Головним недоліком способів одержання оліго-олефінових основ синтетичних мастил шляхом олігомеризації олефінів під дією каталітичних систем RnAlX3-n – R’ є те, що застосування їх у процесі олігомеризації ЛAO (зокрема - децену-1) приводить до утворення переважно високомолекулярних продуктів із широким молекулярно-масовим розподілом і з низьким (менше 20 мас. %) вмістом цільових низькомолекулярних фракцій (димерів і тримерів децену-1). Іншим недоліком способів, які грунтуються на використанні каталітичних систем цього типу є те, що одержані у такий спосіб димери децену-1 є лінійними та після гідрування мають температуру застигання, яка перевищує мінус 20°C. Для усунення цього недоліку, тобто для підвищення селективності та поліпшення технікоекономічних показників способу, розроблено спосіб одержання оліго-олефінових основ синтетичних мастил відповідно до якого, не гідровані димери децену-1 направляють у рецикл на співолігомеризацію їх з деценом-1 у широко вживані три- і тетрамери децену [10] [Пат. США 4263467. 02,04.1981]. Співолігомеризацію димерів децену-1 (43,8 мас.% у шихті) з деценом-1 (40мас.% децену-1 і 15,4 мас. % декану в шихті) за цим способом здійснюють під дією системи BF3/SiO 2 (D = 0,8-2,0 мм) + Н2О (65 м.д. у шихті) за температур 15-30°C, тиску 1-6,9 ат при витраті 8 шихти 2,5 л за годину на 1 л каталізатора. Вміст димерів децену-1 на виході з реактора зменшується від 43,8 до 20,7 мас. %, а вміст тримерів децену-1 зростає до 41,8 мас. %. Це рішення дозволяє кваліфіковано використовувати не утилізовані ( такі, що застигають за високих температур ) лінійні димери децену-1. Недоліком цього рішення є різке зниження продуктивності процесу, який грунтується на цьому способі. Третім загальним недоліком всіх способів у яких використовуються каталітичні системи RnAlX3n – R’X є те, що каталітичні системи, які у них застосовуються, включають горючу самозаймисту на повітрі, небезпечну при виробництві, при транспортуванні та при використанні алюмінійорганічну сполуку Rn Al X3-n. І, нарешті, четвертим загальним недоліком всіх способів заснованих на використанні каталітичних систем Rn AlX3-n – R’ X є те, що під дією цих каталітичних систем утворюються продукти, які містять до 1,0 мас. % хлору у вигляді монохлоролігоолефінів. Найбільш близькими до запропонованого даним винаходом способу одержання олігоолефінових основ синтетичних мастил є способи катіонної полімеризації, олігомеризації олефінів і алкілювання ароматичних вуглеводнів олефінами під дією каталітичних систем, які включають металевий алюміній. Власне сам металевий алюміній не є каталізатором згаданих процесів. Для забезпечення цим системам каталітичної активності алюміній звичайно використовують у комбінації із співкаталізатором. Так, наприклад, відомі способи полімеризації, олігомеризації та теломеризації олефінів, а також алкілювання ароматичних вуглеводнів олефінами, які містять металевий алюміній і галоідорганічну сполуку [11] [Пат. США 3343911. Нкл. 260-683.15. 1969]. Найбільш близьким за технічною сутністю і за тим результатом, який досягається, до запропонованого даним винаходом способу одержання поліолефінових основ синтетичних мастил є спосіб олігомеризації та полімеризації олефінів під дією каталітичної системи, яка включає металевий алюміній і чотирихлористий вуглець [12] [A.C. CPCP 803200, Нкл. B01 J 31/14. 1979]. Каталізатор для олігомеризації та полімеризації олефінів при цьому способі одержують шля хом взаємодії металевого алюмінію із чотирихлористим вуглецем за температур 40-80°С і співвідношенні мас алюмінію до чотирихлористого вуглецю рівному 1 : (20-80) у середовищі чотирихлористого вуглецю у відсутності олефінів в інертній атмосфері [12] [A.C. CPCP 803200, Нкл. B01 J 31/14. 1979]. Відповідно до цього способу спочатку, у відсутність олефінів, в інертній атмосфері одержують твердий чорний продукт невідомої сполуки, який надалі використовується як каталізатор олігомеризації альфа-олефінів і полімеризації ізобутилену. Ми розглядаємо цей спосіб як прототип для розробленого нами способу одержання поліолефінових основ синтетичних мастил. Недоліком способу - прототипу є використання в цьому способі чотирихлористого вуглецю в складі застосованої каталітичної системи при високому 9 84505 співвідношенні ССI4/АI (0). Це приводить до входження до складу продуктів великої кількості (до 3,0 мас. %) хлору, який важко з них видаляється. Іншим недоліком способу - прототипу [12] [A.C. CPCP 803200, Нкл. B01 J 31/14. 1979] є низька активність, низька продуктивність і низька селективність щодо цільових продуктів каталітичної системи Al(0)-CCl 4, яка застосовується при цьому способі. Недоліком способу-прототипу є також багатостадійність і висока трудомісткість приготування та використання каталізатора олігомеризації олефінів і полімеризації ізобутилену з алюмінію та CCl4. Загальним завданням даного технічного рішення є усунення всіх вищевказаних недоліків відомих способів. Основним конкретним завданням даного винаходу була розробка способу одержання поліолефінових основ синтетичних мастил з використанням удосконаленої каталітичної системи для катіонної олігомеризації лінійних альфа-олефінів (ЛAO) С 3¸С14, яка характеризувалася б підвищеною активністю та підвищеною продуктивністю, забезпечувала б підвищення керованості процесом олігомеризації, зокрема, дозволила б регулювати швидкість олігомеризації, підвищувати ви хід цільових низькомолекулярних фракцій олігомерів (наприклад, димерів і тримерів децену-1), підвищити розгалуженість ланцюга продуктів олігомеризації та знизити температуру їх застигання, а також підвищила б безпеку використання її в процесі олігомеризації олефінів. Другим завданням даного винаходу було спрощення способу приготування та використання каталітичної системи олігомеризації олефінів, яка включає металевий алюміній. Сформульовані завдання вирішені в даному винаході в результаті вдосконалення всіх основних стадій способу одержання поліолефінових основ синтетичних мастил. Розроблений спосіб одержання поліолефінових основ синтетичних мастил, згідно із даним винаходом, так само як і будь-який інший аналогічний спосіб, містить стадію підготовки олефінової сировини та розчинів компонентів катіонної каталітичної системи, стадію ізомеризації вищи х лінійних альфа-олефінів, стадію олігомеризації олефінової сировини під дією катіонної алюмінієвмісної каталітичної системи, стадію виділення з олігомеризата відпрацьованого каталізатора, стадію розділення олігомеризата на фракції та стадію гідрування виділених фракцій. Крім того, він після стадії олігомеризації та/або після стадії виділення відпрацьованого каталізатора з олігомеризата, додатково містить стадію дехлорування наявних в олігомеризаті монохлоровмісних олігомерів, а після стадії розділення олігомеризата на фракції він передбачає стадію деполімеризації високомолекулярних продуктів, виділених з олігомеризата у вигляді кубового залишку на стадії розділення олігомеризата на фракції (пункт 1 формули винаходу). Ці стадії мають на меті поліпшення технікоекономічних показників способу, для вирішення специфічних хімічних проблем і для підвищення гнучкості розробленого способу щодо продуктів. 10 Зокрема, стадія дехлорування монохлоролігодеценів, які містяться в олігомеризаті та утворюються в ході олігомеризації, призначена для перетворення ковалентно пов'язаного з вуглецем у монохлоролігодеценах, так званого, «органічного» хлору в іонно зв'язанний з металами, так званий, «іонний» хлор. Отриманий із хлоровмісних олігодеценів «іонний» хлор разом з відпрацьованим катіонним каталізатором, як і в інших способах катіонної олігомеризації олефінів або алкілювання, далі видаляється з олігомеризата методом воднолужного відмивання. Відповідно до даного винаходу поліолефінові основи синтетичних мастил одержують шляхом олігомеризації вищих оле фінів у сумішах олігомеризованих вищи х олефінів із продуктами їх олігомеризації або в сумішах олігомеризованих ви щих олефінів із продуктами їх олігомеризації та з ароматичними вуглеводнями під дією трикомпонентної, безпечної при транспортуванні, зберіганні та використанні, стійкої на повітрі, доступної катіонної каталітичної системи Al(0) - HCl - (СН3)3ССІ за температур від 110 до 180°C, концентраціях Al(0) від 0,02 до 0,08 г-атом/л, мольних співвідношеннях НСI/АI (0), що змінюються в межах від 0,002 до 0,06 і мольних співвідношеннях RCl/Al(0), що змінюються в межах від 1,0 до 5,0, де Al (0) - високо дисперсний порошкоподібний алюміній з розмірами часток, які коливаються в межах від 1 до 100 мкм, наприклад, Al (0) марки ПА-1, ПА-4, АСД-4, АСД-40, АСД-Т (пункт 2 формули винаходу). Індивідуальні компоненти системи Al(0) - HCl (СН3)3ССІ не є каталізаторами олігомеризації вищи х олефінів. Попередники катіонних активних центрів і самі катіонні активні центри олігомеризації вищих олефінів у цій системі утворюються в послідовності багатьох хімічних реакцій між компонентами системи. Застосований у складі каталітичної системи, що розглядається металевий високодисперсний алюміній складається із часток алюмінію, покритих щільною не реакційноздатною алюмінійоксидною оболонкою. Через це він стійкий на повітрі та за температур 20-110°Cпрактично не реагує із HCl і з (CH3)3ССI. Реакція Al(0) із HCl і з (СН3)3ССІ починається тільки за температур вищи х за 110°C. Відповідно до розробленого способу одержання поліолефінових основ синтетичних мастил у розробленій каталітичній системі алюміній у першу чергу реагує із хлористим воднем. HCl, принаймні, частково руйнує алюмінійоксидну плівку на поверхні часток алюмінію та забезпечує можливість протікання реакції алюмінію із третбутилхлоридом. Інакше кажучи, хлористий водень у розглянутій системі є активатором металевого алюмінію. Реакція алюмінію із третбутилхлоридом протікає за наступною спрощеною схемою: 2Al(0) + 3(СН3)зССI ®2 [(CH 3)3С]1, 5 АIСI1,5 (1) Сесквітретбутилалюмінійхлорид (1), який утворюється, таким же чином як і HCl, реагує з алюмінійоксидною оболонкою на поверхні часток алюмінію. Це призводить до прискорення процесу утворення (1) і до повного розчинення металевого алюмінію. Активація алюмінію забезпечується дуже малою кількістю хлористого водню, який розчи 11 84505 няють у третбутилхлориді (ТБХ) у процесі його одержання за реакцією ізобутилену із хлористим воднем. Мольне співвідношення HCl/Al(0) у каталітичній системі Al(0) - HCl - (CH 3)3ССI варіюють в інтервалі від 0,002 до 0,06 шляхом зміни концентрації HCl у ТБХ від 0,015 до 0,5 мас. %. Концентрацію алюмінію в реакційному середовищі в процесі олігомеризації олефінової сировини змінюють в інтервалі від 0,02 до 0,08 гатом/л. При концентраціях алюмінію нижче 0,02 гатом/л олігомеризація не протікає через інгібуючу дію присутніх в олефінах домішок, а при концентраціях алюмінію вище 0,08 г-атом/л різко зростає питома витрата компонентів каталізатора. Оптимальна концентрація алюмінію в реакційному середовищі змінюється в межах від 0,03 до 0,04 гатома/л. Мольне співвідношення ТБХ/Аl(0) варіюють у межах від 1,0 до 5,0, оптимальне мольне співвідношення ТБХ/Аl(0) дорівнює 3,5. При мольних співвідношеннях ТБХ/АІ(0) менших за 3,5 розчиняється тільки частина металічного алюмінію, що міститься в системі Al(0) - HCl - (CH 3) 3ССІ а при мольних співвідношеннях ТБХ/Аl(0) ви щих за 4,0 різко зростає вміст хлору в олігомерах. Первинні катіонні активні центри в розглянутій каталітичній системі утворюються за схемою: [(CH3)3C]1,5 AlCl1,5 + (CH3) 3ССl® - (CH3) 3С+ {[ (CH 3) 3С] 1,5 АlСl2,5}- (2) ТБХ при цьому виконує функції співкаталізатора. Олігомеризація олефінів під дією системи Al(0) - HCl -(СН3)3ССІ з високою швидкістю та з високою конверсією олефінів у продукти (вище 95 мол. %) протікає за температур від 110 до 180°C. У ході олігомеризації альфа-олефінів під дією системи Al(0) - HCl - (CH3) 3ССІ відбувається часткова ізомеризація альфа-олефінів у суміш позиційних і геометричних ізомерів олефінів із внутрішнім розташуванням подвійних зв'язків, які співполімеризуються з вихідним альфа-олефіном. Це приводить до підвищення розгалуженості молекул одержаних продуктів і до зниження температури їх застигання. У випадку олігомеризації децену-1 застосування системи Al(0) - HCl -(СН3)3ССІ забезпечує зниження відсотку високомолекулярних олігодеценів Сбо+ від 50 (прототип) до 8 мас. %. Олігодецени, які утворюються під дією цієї системи містять від 4300 до 9970м. д. хлору (таблиця 1). Використання системи Al(0)-HCl-(CH 3) 3ССІ в процесі олігомеризації забезпечує рішення проблеми регулювання фракційного складу та розгалуженості продуктів олігомеризації деценів. Витрати компонентів цієї каталітичної системи за масою не перевищують відповідних показників кращих відомих (у тому числі борфторидних) каталізаторів. Важливою, з практичного погляду, особливістю розробленого способу є те, що взаємодію алюмінію з активатором (HCl) і співкаталізатором (ТБХ) проводять безпосередньо в процесі олігомеризації в середовищі сумішей олігомеризованих олефінів із продуктами олігомеризації та додатково ароматичними вуглеводнями, які додаються (бензолом, толуолом, нафталіном). Саме це рішення виключає роботу з концентрованими високореакційними попередниками та продуктами реа 12 кцій утворення активних центрів і підвищує безпеку способу. Відповідно до даного винаходу для олігомеризації вищих олефінів беруть суміші лінійних або розгалужених альфа-олефінів із ізо-олефінами та із олефінами з внутрішньомолекулярним розташуванням подвійного зв'язку (з "внутрішніми" олефінами), які містять від 3 до 14 (переважно - 10) атомів вуглецю, при наступному співвідношенні інгредієнтів, мас. %: альфа-олефіни 0,5-99,0; ізоолефіни 0,5-5,0; "внутрішні" олефіни - решта до 100 мас. %. З наведених у таблиці 2 даних видно, що за інших однакових умов збільшення числа атомів вуглецю в молекулах олефінів приводить до зменшення розгалуженості молекул олігоолефінів і до підвищення їхнього індексу в'язкості. Найпоширеніші способи одержання поліолефінових основ синтетичних мастил, у яких як олефінову сировину використовують децен-1. Це обумовлено тим, що тримери децену-1, які виділені з олігомерів децену-1 після гідрування характеризуються унікальним комплексом фізичних властивостей (кінематична в'язкість при 100°C дорівнює 3,9 сСт, індекс в'язкості = 130, температура застигання = мінус 60°С, температура спалаху = 215-220°С). Таке поєднання властивостей забезпечує можливість використання їх як основи широко вживаних синтетичних і напівсинтетичних моторних (автомобільних, авіаційних, вертолітних, тракторних, танкових) та інших олив. Тому, найкращою сировиною для одержання поліолефінових основ синтетичних мастил є децен-1. Відповідно до даного способу наявність у вихідному децену-1 домішок ізоолефінів (від 0,5 до 11,0 мас. %) і олефінів із внутрішньомолекулярним розташуванням подвійного зв'язку (від 0,5 до 5,0 мас. %) практично не впливає на фізико-хімічні характеристики виділених із продуктів не гідрованих і гідрованих фракцій. У ході катіонної олігомеризації децен-1 перед входженням до олігомерного продукта під дією каталітичної системи Al(0)-HCl-(СН3) 3ССІ та інших каталітичних систем, які містять сполуки алюмінію ізомеризується в суміш позиційних і геометричних ізомерів децену із внутрішньомолекулярним розташуванням подвійних зв'язків. Децени із внутрішньомолекулярним розташуванням подвійних зв'язків (включаючи індивідуальний децен5) під дією каталітичної системи Al (0)-HCl-(CH 3) 3ССІ так само легко (але більш повільно) олігомеризуються, як і децен-1. У ході олігомеризації деценів із внутрішньомолекулярним розташуванням подвійних зв'язків утворюються більш розгалужені, ніж у випадку децену-1 ( і тому вони застигають за більш низьких температур ) молекули олігодеценів. Із класичного стадійного механізму процесів катіонної олігомеризації олефінів під дією хлоровмісних (у тому числі -і алюмінійорганічних) катіонних каталізаторів випливає [1. Дж.Кеннеді. Катіонна полімеризація олефінів. M: Мир, 1978. 430 C; 2. J.P.Kennedy, E.Marechal. Carbocationic Polymerization. N.-Y., 1982. 510 P.], що олігомери, які утворюються в цих процесах можуть містити «органічно» зв'язаний хлор (тобто хлор, пов'язаний з атомом вуглецю молекул олігодеценів у 13 84505 складі олігомеризата). Теоретичні розрахунки та експериментальні дані показують, що від 2 до 10% молекул олігодеценів, отриманих під дією застосованих катіонних алюмінієвмісних каталізаторів, містять у своєму складі по одному атому хлору, а 90-98% молекул олігодеценів містять по одному подвійному зв'язку. Після дезактивації та видалення з олігомеризата методом водно-лужного відмивання за температури +95°C відпрацьованого катіонного каталізатора, олігомеризат містить, приблизно, від 1000 до 10000 м. д. (0,1-1,0 мас. %) хлору, зв'язаного з атомами вуглецю молекул олігодецену. Хлор в олігодецени потрапляє при (практично незворотному) обриві ланцюга в результаті захоплення зростаючим карбкатіоном аніона хлору з аніонного фрагмента катіонного активного центра (АЦ). Схема цього процесу на прикладі найпростішого каталізатора RCl + АІСІ3 (® R+ AlCl-) має наступний вигляд (3): R-(C10H20)nC10H20+ AlCl- (АЦ) ® R-(С10Н20) nС10Н20Сl + AlCl3 Хлор, який міститься в олігомеризаті та у цільових фракціях викликає корозію відповідного устаткування не тільки на всіх стадіях процесу одержання олігодеценів, але та у процесі використання олігодеценових основ синтетичних мастил. Через це хлор необхідно видаляти не тільки з основних фракцій олігодеценів, але та з олігомеризата на найбільш ранніх стадіях їх одержання. Відповідно до даного винаходу дехлор ування наявних в олігомеризаті монохлоровмісних молекул олігомерів (RCl) здійснюють як після стадії олігомеризації, так і після стадії виділення з олігомеризата відпрацьованого каталізатора Al(0)-НСІ(СН3)3ССl (пункт 1 формули винаходу). При розробці даного способу одержання поліолефінових основ синтетичних мастил розроблено 5 варіантів вирішення проблеми дехлорування. У першому варіанті даного способу дехлорування RCl проводять високодисперсним порошкоподібним металевим алюмінієм - Al(0) з розмірами часток, які коливаються в межах від 1 до 100 мкм (наприклад, марок ПА-1, ПА-4, ПАП-1, АСД-4, АСД-40, АСД-Т) при мольних співвідношеннях Al(0)/RCl, які коливаються в межах від 0,5 до 2,0, в інтервалі температур від 110 до 180°C протягом від 30 до 180 хвилин (пункт 4 формули винаходу) (таблиця 3). Через високу енергію зв'язку C - Cl, хлор із хлоралканів, які містять фрагменти -CH2Cl за допомогою хімічних реагентів видаляється з великими труднощами. Найлегше із хлоралканів видаляється хлор, який міститься у фрагментах R3ССІ. Тому як тест-індикатор швидкості та глибини дехлорування хлоралканів використовували 1хлордодекан, який містить фрагмент –СН2СІ. З таблиці 3 видно, що за температур, які не перевищують 95°C під дією алюмінію марки ПА-4 дехлорування 1-хлордодекану та хлоровміснкх олігодеценів не відбувається. Підвищення температури до 120°C і вище приводить до повного дехлорування згаданих хлоралканів. Реакція дехлорування 1-хлордодекану алюмінієм у середовищі децену-1 приводить до майже 100%-ної олігомеризації децену-1. Це свідчить про те, що реакція алюмінію з 1-хлордодеканом протікає в такий же спосіб, як і 14 реакція алюмінію із ТБХ (тобто із проміжним утворенням катіонних активних центрів олігомеризації). При цьому реакція алюмінію з RCl приводить до перетворення ковалентно -зв'язаного з вуглецем хлору в іонно- зв'язаний з металом хлор, який на стадії водно-лужного відмивання видаляється з олігомеризата. Відповідно до другого варіанту дехлор ування, запропонованого патентованим способом, наявні в олігомеризаті монохлоровмісні олігодецени (RCl) дехлорують після стадії олігомеризації триетилалюмінієм (TEA) при мольних співвідношеннях TEA/RCl, які змінюються в межах від 0,5 до 2,0, в інтервалі температур від 95 до 150°C протягом від 30 до 180 хвилин (пункт 5 формули винаходу). Дехлорування RCl триетилалюмінієм за вказаних вище умов протікає відповідно до наступної схеми (4): RCl + (C2H5)3Al® {R+ [ (C2H5) 3АІС1]-}® RH + C2H4 + (C2H5) 3 АlСl (4) З таблиць (3, 4) видно, що де хлорування 1хлордодекану алюмінієм і триетилалюмінієм відбувається тільки за температур 130-164°С, як при відсутності відпрацьованого катіонного каталізатора, так і в присутності продуктів його еволюції в ході олігомеризації. Видно також, що конверсія децену-1 у продукти олігомеризації в ході реакції дехлорування RCl алюмінієм і триетилалюмінієм зростає. Це узгоджується зі схемами (3, 4), відповідно до яких реакції RCl з алюмінієм і триетилалюмінієм протікають через стадію утворення катіонного активного центра (R+ [(C2H5) зАІСІ]-}, що ініціює олігомеризацію децену-1. Загальною перевагою першого та другого варіантів дехлорування RCl за допомогою Al(0) і TEA, відповідно до даного винаходу, є те, що реакції RCl зі згаданими дехлоруючими реагентами проводять безпосередньо після олігомеризації в присутності відпрацьованих, але не видалених з олігомеризата, продуктів перетворення застосованої катіонної каталітичної системи Al(0) - HCl ТБХ. Алкіл-алюміній хлориди, які утворюються при дехлоруванні видаляються з олігомеризата одночасно з відпрацьованим каталізатором на стадії водно-лужного відмивання. Це спрощує технологічне оформлення цієї стадії, але вимагає підвищеної витрати дисперсного алюмінію або використання TEA. Первинні та вторинні хлоралкани за звичайних умов (тобто за температур, які не перевищують 100°C) не гідролізуються водою та водними розчинами гідрооксидів натрію або калію. Відповідно до третього варіанта даного способу одержання поліолефінових основ синтетичних мастил, дехлорування монохлоровмісних олігомерів (RCl), що утворюються в ході олігомеризації і присутні в олігомеризаті, здійснюють до або після стадії виділення відпрацьованого каталізатора спиртовим (бутанольним або гексанольним - ROH) розчином гідрооксида калію або натрію (MOH) при мольних співвідношеннях MOH/RCl, які змінюються в межах від 1,1 до 2,0, в інтервалі температур від 120 до 160°C протягом від 30 до 240 хвилин (таблиця 5) (пункт б формули винаходу). Замість згаданих ROH як розчинник KOH можна використовувати 15 84505 моноетиловий ефір етиленгліколю. Концентрацію MOH в ROH варіюють у межах від 1 до 5 мас. %. З таблиці 5 видно, що за інших однакових умов швидкість реакції та конверсія дехлорування різко знижуються при заміні KOH на NaOH. Із цієї причини KOH є кращим. Реакція дехлорування RCl спиртовими розчинами MOH навіть за 120°C протікає повільно. Підвищення температури від 120 до 150°C приводить до різкого підвищення швидкості реакції. При 150°C реакція завершується протягом 60 хвилин. Дехлорування RCl відповідно до цього варіанта способу протікає за схемою, яка включає проміжне утворення алкоксидів лужних металів, які реагують далі із хлоралканами RCl: KOH + C 4H9OH ® C 4H9OK + H2O C4H9OK + RCl ®-» KCl + C 4H9OR Хлориди натрію та калію в згаданих спиртах в умовах реакції дехлорування хлоралканів є не розчинними, що дозволяє виділяти їх з реакційної маси осадженням з наступним розчиненням осаду солі MCl водою. Найбільш простим з розроблених варіантів дехлорування хлоровмісних олігодеценів є варіант, відповідно до якого дехлорування присутніх в олігомеризаті монохлоровмісних олігомерів (RCl), здійснюють після стадії виділення відпрацьованого каталізатора шляхом термічного дегідрохлорування RCl в інтервалі температур від 280 до 350°C і тисках 1-2 бар протягом від 30 до 180 хвилин при віддувці хлористого водню, який виділяється, азотом, діоксидом вуглецю, метаном (природним газом) або перегрітою водяною парою (таблиця 6) (пункт 7 формули винаходу). Термічне дегідрохлорування при цьому варіанті способу здійснюють у нагрівачі-випарювачі атмосферної колонки при інтенсивному перемішуванні олігомеризата віддувочним газом або перегрітою водяною парою. Цей варіант дехлорування хлоровмісних олігомерів децену має і переваги і недоліки: він забезпечує глибокий ступінь дегідрохлорування олігомеризата (на 97-98%), не вимагає застосування нових реагентів, але приводить до ускладнення оформлення атмосферної колонки. Термічне дегідрохлорування хлоровмісних сполук починається за температур вищи х за 250°C. Швидкість термічного дегідрохлорування істотно залежить від будови хлоровмісної сполуки. У випадку хлоровмісних сполук, які мають бета-СН зв'язки відносно зв'язків C- Cl, найбільш термостійкими є первинні галоідалкіл, найменш термостійкими є третинні галоідалкіл. Термічне дегідрохлорування R3CCI з помітною швидкістю протікає навіть при 100°C. Виходячи зі складу ви хідних реагентів і стадійного механізму олігомеризації децену-1 можна зробити висновок, що в олігомеризаті можуть бути присутніми всі теоретично можливі для кожного конкретного випадку типи хлоровмісних сполук (ті, які містять та ті, які не містять бетаС-Н зв'язку первинні, вторинні та третинні галоідалкіли). Швидкості, і за інших однакових умов, ступені дегідрохлорування первинних, вторинних і третинних галоідалкілів, які містять бета- C-H зв'язки зазвичай зростають із підвищенням температури. За температур 250-300°C дегідрохлорування 16 хлоровмісного олігомеризата протікає відносно повільно і є, ймовірно, зворотнім протягом 6-10 годин. Хлористий водень, який виділяється в процесі дегідрохлорування відразу ж може приєднуватися до молекул олігодеценів, які містять подвійні зв'язки. Цьому сприяють відносно висока концентрація подвійних зв'язків різних типів у молекулах олігодеценів. За інших однакових статичних умов швидкість і ступінь дегідрохлорування олігомеризата зростають із підвищенням температури від 300 до 330°C. Деполімеризація олігодеценів при цьому не відбувається. При 330°C і часі перебування - дві години, майже весь органічно зв'язаний хлор, який міститься в олігомерах видаляється з олігомеризата у вигляді HCl. В олігомеризаті після термообробки залишається близько 20 м. д. органічно зв'язаного хлору (тобто близько 0,002 мас. %). В умовах віддувки хлористого водню та пароподібних компонентів з олігомеризата азотом при 330°C і часі перебування одна година в олігомеризаті залишається тільки 3 ч/млн (ррт) органічно зв'язаного хлору. З о триманих даних витікає, що ступінь видалення хлоровмісних вуглеводнів з олігомеризата = 99,94 мас. %. Вміст хлорорганічних сполук у рецикльованій децен-декановій фракції не перевищує 160 ч/млн (що відповідає 0,016 мас. %). Ця фракція може бути змішана зі свіжим деценом, який надходить на установку, а о тримана суміш може використовуватися як вихідна сировина для одержання олігодеценів. Термічне дегідрохлорування хлоровмісних молекул олігодеценів за температур 300-3300C і загальному часі перебування дві години протікає майже кількісно за наступною схемою: R- (C10H20) х-1-С10Н 21Сl®HCl + R-(C10H20) x-1C10H20 ( = ) Хлористий водень, який виділяється при цьому, віддувається азотом, або перегрітою водяною парою та разом з парами води та вуглеводнів надходить спочатку до конденсатора, а з нього направляється в скрубер для нейтралізації HCl водним розчином гідрооксиду натрію. З метою виключення виділення хлористого водню у вільному стані в паро-газову фазу, дегідрохлорування присутніх в олігомеризаті монохлоровмісних олігомерів (RCl), відповідно до даного винаходу (пункт 8 формули винаходу) здійснюють в V інтервалі температур від 300 до 330°C у присутності сухи х гідрооксидів лужних металів (MOH) при мольних співвідношеннях MOH/RCl що змінюються в межах від 1,1 до 2,0 (таблиця 7). Для забезпечення максимально можливого ступеня диспергування часток не розчинних в олігомеризаті сухи х гідрооксидів лужних металів їх одержують відповідно до даного винаходу (пункт 9 формули винаходу) безпосередньо в олігомеризаті шляхом відгону води при нагріванні суміші звільненого від відпрацьованого каталізатора олігомеризата та 540%-ного водного розчину гідрооксиду лужного металу за температур, які змінюються в інтервалі температур від 100 до 200°C (таблиця 7). При підвищенні температури від 100 до 200°C відбувається випарювання і повне видалення води з реакційної маси. Наступне дегідрохлорування 17 84505 хлоровмісних оліго-деценів проводять протягом однієї години за температур 300, 310, 320 і 330°C. З таблиці 7 видно, що за інших однакових умов залишковий вміст хлору в олігомері з підвищенням температури від 300 до 330°C різко знижується та при 330°C досягає 8 ч/млн. (8 ррт) при ступені дегідрохлорування 95.56 мас. %. Хлористий водень у газову фазу не виходить - він повністю реагує з гідрооксидом калію та виявляється у вигляді KCl у твердому осаді, який повністю розчиняється у воді. Промивання дегідрохлорованого олігомеризата водою після вивантаження його з реактора та аналіз промивної води на хлор свідчать про те, що деяка частина (менше 0,5 мас. %) хлористого калію, що утворився, міститься у завислому стані в дегідрохлорованому олігомере. Розроблені варіанти дехлорування молекул олігодеценів, які містять хлор (тобто - хлоралканів) є універсальними та можуть використовуватися самостійно для вирішення аналогічних завдань в інших хімічних процесах, у нафтопереробці, а також при дехлоруванні моно-, ди- і поліхлоровмісних аліфатичних і ароматичних вуглеводнів, олігомерів, полімерів, нафтови х фракцій і різних рідких і твердих хлоровмісних органічних відходів (пункт 10 формули винаходу). Для приклада в таблиці 8 наведені дані про дегідрохлорування рідких поліхлорпарафінів, які містять 44 мас. % хлору, бутанольним розчином KOH. Це рішення використовується, зокрема, при одержанні синтетичної оліфи та олігомерів ацетилену. Спосіб одержання поліолефінових основ синтетичних мастил, запропонований в патенті, передбачає утилізацію тих фракцій високомолекулярного олігодецену, які не знаходять застосування, шляхом їх деполімеризації. Стадія деполімеризації високомолекулярних олігодеценів призначена для корегування молекулярно-масового розподілу та фракційного складу олігодеценів, одержаних на стадії олігомеризації. Деполімеризацію високомолекулярних продуктів, виділених у вигляді кубового залишку на стадії розділення олігомеризата на фракції, відповідно до розробленого способу (пункт 11 формули винаходу), проводять нагріванням їх до температур від 330 до 360°C і тискові від 1,0 до 10,0 мм рт. ст. протягом від 30 до 120 хвилин з безперервним видаленням продуктів з реактора деполімеризації в систему атмосферної та двох вакуумних колон для розділення олігомеризата на фракції. Розділення деценового олігомеризата на фракції та деполімеризацію виділеного кубового залишку, який містить високомолекулярні олігодецени, проводили на вакуумній установці французької компанії «GECIL». Комп'ютеризована автоматизована установка (model «minidist С»), яка тут використовувалася, включає дві колони, два куби об'ємом 10 і 22 літра, з електрообігрівом, скляний збірник, близько 10 приймачів для фракцій, що виділяються, вакуумний пост, вакуумний насос, вакуумметр і два пристрої для вимірювання температури в кубі та у верхній частині колони. Збірник був оснащений автоматичною системою контролю швидкості відбору фракцій. Вакуумна 18 система цієї установки забезпечує можливість фракціонування олігомеризата за чітко заданого залишкового тиску. Перша колона з регулярною сітковою насадкою та зі зрошенням може функціонувати за атмосферного або зниженого тиску (аж до 2,0 мм рт. ст.). Др уга - вакуумна колона (без насадки та без зрошення) може функціонувати при глибокому вакуумі (навіть коли залишковий тиск дорівнює 1,0-0,01 мм рт. ст.) за температури до 370°C. Розглянута дистиляційна установка оснащена також автоматичним вузлом визначення маси фракцій. Вся інформація про деталі розділення олігомеризата надходить у комп'ютер, де вона накопичується та обробляється. Зокрема, комп'ютер за спеціальною програмою, виходячи з реальних значень температури куба та верхньої частини колони та залишкового тиску в колоні визначає віртуальну температуру процесу за атмосферного тиску (Тв), яка збігається з умовною температурою фракціонування (Ту) , знайденою за допомогою відомої номограми T-P. Розділення олігомеризата на фракції проводили на першій колоні з регулярною насадкою (15 теоретичних тарілок) з кубом близько 20 літрів. Деполімеризацію високомолекулярних олігодеценів проводили на другій (вакуумній) колоні без регулярної насадки та без зрошення з кубом близько 10 літрів. В куб з електрообігрівом першої колони завантажували від 5 до 10 кг очищеного від каталізатора та від органічно зв'язаного хлору олігомеризата. Розділення олігомеризата на фракції проводили в режимі повільного підвищення температури від 20 до 300°C. Легко киплячі компоненти, а також ПАО-2 і П АО-4 з олігомеризата виділяли на першій колоні з насадкою та зі зрошенням. ПАО-6 і ПАО-8 виділяли на другій вакуумній системі. Було встановлено, що в процесі розділення деценового олігомеризата на вузькі фракції за температур, що перевищують 330°C спостерігається термічна деполімеризація олігодеценів. В інтервалі температур 300-330°C за залишкового тиску меншому за 5 мм рт. ст. з олігомеризата виділяються тримери, які там залишилися та тетрамери децену. При 360°C протягом трьох годин за залишкового тиску в колоні меншому за 3 мм рт. ст. у кубі відбувається практично повна деполімеризація високомолекулярних олігомерів децену. Отримані при цьому продукти були розділені на фракції. Установлено, що найбільш легка фракція, виділена за температур 20-150°C, із продуктів деполімеризації високомолекулярних олігодеценів складається із суміші олефінів (переважно деценів) з вінільними (53,6 %), транс-вініленовими (18,1 %) і вініліденовими (28,3%) подвійними зв'язками. Виділені в інтервалах температур 150-240 і 240300°C продукти є димерами та тримерами децену, відповідно. Це підтверджено методом газової хроматографії та збігом основних властивостей цих фракцій із властивостями стандартних зразків димерів і тримерів деценів. У кубовому залишку після розділення продуктів деструкції на фракції містилися не деполімеризовані високомолекулярні олігомери деценів. Описаний склад продуктів свідчить про те, що в процесі термообробки високомолекулярних олі 19 84505 годеценів відбувається (певно, статистична) їх деполімеризація. Виявлений ефект має велике практичне значення, тому що дозволяє, якщо буде потреба, простим способом переробляти високомолекулярні олігодецени в ди-, три- і тетрамери деценів. Результати розділення деценового олігомеризата на фракції в умовах часткової деполімеризації високомолекулярних компонентів наведені в таблиці 9. Деполімеризація високомолекулярних олігомерів децену (ПАО-10¸ПАО-20) з кінематичною в'язкістю при 100°C цілеспрямовано проводилася за температур 330-360°C за залишкового тиску на верху колони деполімеризатора 1 мм рт. ст., а в Природа фракції Газоподібні BB Рідкі BB ПАО-2 ПАО-4 ПАО-6 Газоподібні BB Рідкі BB ПАО-2 ПАО-4 ПАО-6 кубі колони 3-5 мм рт. ст. Для кращого розуміння роботи деполімеризатора приводимо опис одного з типових дослідів. Приклад. У куб деполімеризатора було завантажено 3000 г олігомера децену з температурою кипіння при залишковому тиску на верху колони 1 мм рт. ст. вище 360°C. У результаті деполімеризації цього олігодецену за температури 350°C протягом трьох годин було відігнано 2258 г (75,27 мас. %) продуктів деполімеризації. Виділені через верх реактора-колони продукти деполімеризації олігодецену-1 у загальній кількості 2258 г були повторно розділені на наступні фракції: Інтервал T викіпання фракції, °C 20-30 30-150 150-240 240-330 330-360 При деполімеризації олігодеценів з температурою кипіння більшою за 300°C за температури куба 360°C, температури стінок деполімеризатора Природа фракції 20 Вихід фракції Мас. % г 60 154 156 830 1058 3500C і залишковому тиску в конденсаторі 1 мм рт. ст. були отримані наступні результати: Інтервал T викіпання фракції, °C 20-30 30-150 150-240 240-330 330-360 2,6 6,8 6,9 36,8 46,9 Вихід фракції мас. % г 62,8 188,4 376,8 471,0 2041,0 å=3148,0 2,0 6,0 12 15,0 65,0 100,0 Примітка: BB - вугле водні, T - температура. Для виявлення оптимальних умов функціонування деполімеризатора вивчений вплив температури на кінетику термічної деполімеризації високомолекулярного олігодецену з температурою кипіння вищою за 330°C. З отриманих даних можна зробити висновок, що: 1. Деполімеризатор виходить на заданий стаціонарний температурний режим протягом 10-20 хвилин; 2. Після виходу деполімеризатора на заданий температурний режим термічна деполімеризація високомолекулярного олігодецену протікає з постійною швидкістю; 3. Швидкість деполімеризації, вихід продуктів деполімеризації та конверсія високомолекулярного олігодецену в цільові продукти з підвищенням температури від 34 0 до 360°C монотонно зростають; 4. Енергія активації деполімеризації, яка при цьому спостерігається дорівнює 27,5 ккал/моль, Ig А = 9,9073; 5. При 360°C протягом години досягається більш ніж семидесяти відсоткова конверсія високомолекулярного олігодецену в продукти деполімеризації, які за вказаної температури та при залишковому тиску в кубі не більше 3 мм рт. ст. відганяються з деполімеризатора та направляються в колону для повторного розділення; 6. Продукти деполімеризації відрізняються від продуктів олігомеризації децену-1 не тільки тим, що поряд з молекулами із внутрішніми (вініленовими) і вініліденовими подвійними зв'язками вони містять значну кількість (близько 30 мол. %) молекул з вінільними подвійними зв'язками, але й фізико-хімічними властивостями (Таблиці 9, 10). 7. Термічна деполімеризація високомолекулярних оліго-олефінів є хімічним ендотермічним процесом. Для забезпечення деполімеризації 1000 кг високо-молекулярних олігодеценів за годину 21 84505 повинен бути передбачений нагрівач загальною потужністю 116 кВт. Тепловий баланс деполімеризатора такий: Надходження тепла - 116 кВт/годину. Витрата тепла: -обігрів високомолекулярних олігодеценів до 360 °С - 30 кВт/годину; ендотермічна реакція деполімеризації при 360°С 33 кВт/годину; -випарювання продуктів, які утворилися, - 28 кВт/годину; -тепловий резерв - 11 кВт/годину; -втрати тепла -14 кВт/годину. Важливо відзначити, що умови процесу (температура, тиск) впливають на склад продуктів деполімеризації та на їхні характеристики (таблиці 9, 10). Особливо великий вплив на склад та характеристики продуктів деполімеризації має швидкість видалення їх з реактора. При підвищенні температури та тиску, а також при зменшенні часу відгону продуктів з деполімеризатора глибина деполімеризації олігодеценів різко підвищується. Вид хроматограмм димерів і тримерів, а також Час утрим., хв 10,9 13,8 15,9 17,7 24,2 Час утрим., хв 1,09 3,83 7,33 10,81 13,71 15,93 17,75 25,20 високі значення температур їх застигання, свідчать про-те, що в ході термічної деполімеризації високомолекулярних олігодеценів за високого залишкового тиску (більшому за 10 мм рт. ст.) на колоні з регулярною насадкою та зі зрошенням відбувається їх парафінізація. Зазначені умови термічної деполімеризації, коли продукти первинної деполімеризації не видаляються із зони реакції, а багаторазово вертаються в куб, певно, сприяють протіканню ланцюгового розпаду олігодеценів. Цей висновок підтверджується прикладом, який проводився на колоні без регулярної насадки та без зрошення за залишкового тиску 1,0-0,6 мм рт. ст. У цьому прикладі як вихідний олігомеризат використовувався кубовий залишок, виділений з деценового олігомеризата після водно-лужного дегідрохлорування. Він містив близько 30 м. д. хлор у та мав наступний склад: Компонент Тример Тетрамер Пентамер Гексамер Гептамер У куб колони-деполімеризатора було завантажено 1484.6 г охарактеризованого вище дегідрохлорованого високо-молекулярного олігодецену. Спочатку в кубі при перемішуванні олігомеризата потужною електромагнітною мішалкою проводився поступовий розігрів олігомеризата до 360°C. Деполімеризація високомолекулярного олігодецену починалася за температури 330°C і здійснювалася в основному при 360°C. Реальна і віртуальна температура куба та верху колони визначалися втратами тепла, витратою тепла на розігрів олігомеризата та продуктів деполімеризації, на термічну деполімеризацію олігодеценів і на випарювання продуктів деполімеризації. На термограмі протікання всіх цих ендотермічних процесів проявилося у вигляді декількох ендо-ефектів. Швидкість деполімеризації (точніше - швидкість відгону продуктів термічної деполімеризації) у часі змінювалася складним чином. Спочатку швидкість процесу в міру підйому температури від 330 до 360°C монотонно зростала до приблизно 13 мл продуктів за хвилину. Однак на 170 хвилині цілком раптово відбулося стрибкоподібне зростання (майже в 3 Вміст, мас. % 19,93 32,91 21,75 11,48 13,93 рази) швидкості відгону продуктів термічної деполімеризації. Оскільки температура та залишковий тиск при цьому залишалися незмінними, то можна припустити, що відзначене прискорення процесу визначається його вироджено-ланцюговим характером. У результаті деполімеризації високомолекулярних олігодеценів за зазначених вище умов в уловлювачі зібраний конденсат, виділено дві фракції продуктів та отриманий кубовий залишок. Конденсат складався в основному (98,3 мас. %) з вуглеводнів C4-C12. Частка вуглеводнів (парафінів та олефінів) у конденсаті монотонно знижувалася від C4 до С12. Такий склад конденсату, певно, відображає співвідношення різних відгалужень у молекулах олігодеценів. Якщо це припущення відповідає дійсності, то можна зробити висновок про те, що в молекулах олігодеценів найбільше міститься бутильних відгалужень. Перша фракція продуктів термічної деполімеризації, виділена в кількості 657,7 г при віртуальних температурах від 209 до 575°С має дуже складний склад: Компонент До C12 C12-C18 Димер Тример Тетрамер Пентамер Гексамер Гептамер З наведеної таблиці видно, що ця фракція є сумішшю 1,43 мас. % низькомолекулярних продуктів термічної деполімеризації, а також вихідних і 22 Вміст, мас. % 0,34 1,09 2,39 3,42 3,88 21,42 46,96 20,49 отриманих у результаті деполімеризації ди-, три-, тетра- і більше високомолекулярних олігодеценів. Температура застигання цієї фракції = -39°С. Вона 23 84505 знижується до -49°С після виділення з неї низькомолекулярних C4-C18 вуглеводнів. Друга фракція продуктів термічної деполімеризації високомолекулярних олігодеценів, виділена в кількості 295,1 г при віртуальних температурах 575-534 °С (за залишкового тиску 0,6 мм рт. ст.), також як і перша фракція, є складною сумішшю низькомолекулярних продуктів термічної деполімеризації, а також вихідних і отриманих у результаті деполімеризації ди-, три-, тетра- і більше високомолекулярних олігодеценів. Температура застигання цієї фракції = -33°С, але після виділення з неї низькомолекулярних C4-C18 вуглеводнів 24 знижується до -45°C. Вміст низькомолекулярних продуктів деполімеризації (3,0 мас. %) у другій фракції більше ніж у два рази перевищує вміст низькомолекулярних продуктів деполімеризації (1,43 мас. %) у складі першої фракції. Це вказує на підвищення глибини деполімеризації. Загальна конверсія вихідних олігодеценів у продукти деполімеризації перевищувала 70 мас. %. Сумарний вихід другої та третьої фракцій (952,8 г) = 64,18 мас. %. Кубовий залишок (458,8 г = 30,9 мас. %) складається із суміші вихідних та деполімеризованних високомолекулярних олігодеценів: Час утрим., хв Компонент 16,10 17,98 25,41 Пентамер Гексамер Гептамер+ Вміст, мас. % 0,45 2,43 97,11 Він має температуру застигання =-30°C. Сукупність отриманих даних дозволяє зробити наступні загальні висновки: високомолекулярні олігодецени методом термічної деполімеризації за температур 330-360°C та залишковому тиску 1,0 мм рт. ст. можна переробити з високою конверсією в суміш низькомолекулярних продуктів; із суміші термічної деполімеризації високомолекулярних продуктів методом вакуумної дистиляції можна виділити фракції продуктів, які за фракційним складом та за в'язкістними властивостями наближаються до ПАО-2, ПАО-4 та П АО-6; температури застигання виділених фракцій перевищують температури застигання ПАО-2, ПАО-4 та П АО-6, виділених з ви хідного олігомеризата, але є істотно нижчими, ніж температури застигання відповідних за в'язкістними властивостями не компаундованих фракцій мінеральних мастил. Якщо буде необхідно це дозволяє рекомендувати їх після гідрування до використання в суміші з відповідними основними продуктами як основи синтетичних та напівсинтетичних мастил. Наявність стадії деполімеризації забезпечує можливість переробки всіх обмежено вживаних високомолекулярних олігодеценів з кінематичною в'язкістю при 100°C, яка змінюється в межах від 10 до 20 сСт у широко вживані поліолефіни з кінематичною в'язкістю при 100°C, яка змінюється в межах від 2 до 8 сСт (тобто в ПАО-2, ПАО-4, ПАО-6 та ПАО-8, відповідно). Розділення олігомеризата на вузькі фракції відповідно до способу запропонованого патентом, як ми вже відзначали, відбувається так само, як і в інших відомих способах, за винятком того, що глибокий вакуум у колонах розділення забезпечується оригінальною вакуумною паро-ежекторною системою. Продукти, які виділяються на стадії розділення олігомеризата на фракції, у всі х випадках є сумішами ненасичених і хлоровмісних молекул олігодеценів. Залишковий вміст хлору у фракціях, які виділяються не перевищує 10 ч/мдн. (10 ррт). Для підвищення термоокислювальної стабільності одержаних продуктів у всі х відомих способах їх гідрують. Гідрування виділених з олігомеризата вузьких фракцій олігоолефінів відповідно до даного винаходу проводять під дією паладієвого нанесеного на оксид алюмінію каталізатора (переважно Pd(0,2 мас. %)/Аl2О 3), модифікованого безводним NaOH, який беруть у кількості від 30 до 100 мас. % розраховуючи на каталізатор гідрування, за температур, що змінюються в інтервалі температур від 200 до 250°C при тиску водню 20 ат. Температура та тиск на стадії гідрування відповідно до способу запропонованого патентом є значно нижчими, ніж у випадку інших відомих способів. Гідрування вузьких фракцій олігодеценів за зазначених вище умов дозволяє гідрувати не тільки C=C, але й C-Cl зв'язки олігодеценів. Проведення гідрування олігодеценових фракцій у присутності безводного NaOH забезпечує підвищення активності та продуктивності каталізатора гідрування в результаті нейтралізації хлористого водню, який утворюється при гідруванні C-Cl зв'язків, які залишилися у фракціях хлоровмісних олігодеценів, що гідр уються. Це рішення, крім того, усуває корозію реакторів гідрування та допоміжного устаткування та запобігає прискореній дезактивації хлористим воднем паладієвого каталізатора гідрування. При розробці способу одержання поліолефінових основ синтетичних мастил використовувалися наступні реактиви: децен-1, виділений із продуктів олігомеризації етилена під дією триетилалюмінію на ВАТ «Нижнєкамскнєфтєхім» (ТУ 2411-057-05766801-96) , а також децен-1, виділений із продуктів олігомеризації етилена під дією триетилалюмінію в м. Нератовіце (Чехія) компанією «Сполана». Використаний децен-1 виробництва ВАТ «НКНХ» мав наступну груповий склад: CH2=CH-83,4 мол. %; транс-CH=CH- 5,4 4 мол. %; СН2=С < 11,2 мол. %. Перед застосуванням децен-1 сушили над прожареними при 600°C молекулярними ситами NaX; н-гептан, марки "ета 25 84505 лонний", який застосовується як розчинник для приготування розчинів компонентів каталізаторів, сушили перегонкою над натрієвим дротом; AOC загальної формули RnAlCl3-n, (R - C2H5; n = 1,5, 3,0) очищали перегонкою за зниженого тиску. Підготовлені олефіни, розчинники та AOC зберігали в інертній атмосфері в герметичних посудинах. AOC використовували у вигляді розведених нгептанових розчинів. Олігомеризацію децену-1 здійснювали під дією систем Al (0) - HCl - (CH 3) 3ССІ (ТБХ) та Al(0) (C2H5) 1.5 АІСІ 1.5 (EACX) -(CH 3) зССІ, у яких HCl та EAC X виконували функції активаторів алюмінію марок АСД-4, АСД-40, ПА-1 та ПА-4. При розробці способу, запропонованого патентом в основному використовували алюміній марки ПА-4. Швидкість олігомеризації децену-1 під дією згаданих каталітичних систем лімітується швидкістю розчинення алюмінію третбутилхлоридом. Цей процес передує утворенню активних центрів олігомеризації децену-1. Включення в згадані каталітичні системи активаторів алюмінію скорочує або усуває індукційний період. Як співкаталізатор в обох згаданих системах використовували трет-бутилхлорид - (СН3)3ССІ, який був отриманий за реакцією ізобутилену із сухим HCl. ТБХ містив 0,5 мас. % HCl (ТУ 6-09-071338-83). У попередніх дослідженнях було встановлено, що за сукупністю характеристик кращим активатором алюмінію є HCl, а кращим співкаталізатором у процесі олігомеризації децену-1 є ТБХ. Тому основні дослідження виконані із застосуванням каталітичної системи Al(0) марки ПА-4 - HCl - ТБХ Олігомеризацію децену-1 проводили в термостатованому висушеному скляному або металевому реакторах змішування в атмосфері сухого аргону при безперервному інтенсивному перемішуванні реакційної маси за допомогою електромагнітної мішалки. Тестування каталітичних систем у процесі катіонної олігомеризації децену-1 та інших олефінів проводили в такий спосіб: у термостатований до заданої температури реактор послідовно завантажували алюміній, децен-1 або інший олефін, а потім розчин HCl у ТБХ. Al завантажували в реактор у вигляді заздалегідь підготовленої в інертній атмосфері навііски, яка зберігалася в скляній запаяній ампулі. Слідом за завантаженням у реактор всіх згаданих реагентів (зазвичай відразу ж) починалася олігомеризація олефіна, яка супроводжувалася помітним підвищенням температури реакційного середовища (олігомеризата). Реакцію продовжували протягом запланованого часу, а потім зупиняли шляхом введення в реактор водного розчину лугу (NaOH) або етанола. Отриманий олігомеризат з реактора вивантажували далі в проміжну ємність. Відпрацьований (дезактивований) каталізатор з олігомеризата видаляли методом водно-лужного та водного відмивання. Вміст децену-1 (або іншого вихідного олефіна) у реакційній масі (олігомеризаті) у ході та після олігомеризації (тобто поточну та загальну конверсію) визначали методом газової хроматографії на приладах ЛХМ-8-МД, ЛХМ-2000 і «Hewlett-Packard 5880A» (внутрішній стандарт -пентадекан) і мето 26 дом ІЧ-спектроскопії на приладі "Specord M-80". Для цього в заданий момент часу з реактора під аргоном відбирали частину реакційної маси, яку відразу ж змішували з етанолом або 5 %-ним водним розчином NaOH в умовах інтенсивного перемішування. Реакційну суміш після припинення олігомеризації багаторазово промивали дистильованою водою в закритій лійці. Децен-1, який не прореагував, а також ди-, три- та тетрадецени з олігомеризата виділяли на вакуумній колоні з кубом з електрообігрівом до 360°C за залишкового тиску від 1 до 10 мм рт.ст. Фракційний склад олігомеризата визначали на хроматографах ЛХМ-8-МД, ЛХМ-2000 і "HewlettPackard 5880A" з іонізаційно полум'яними детекторами в режимі програмування температури від 20 до 350°C зі швидкістю підвищення температури 810 град/хв. При хроматографуванні олігодеценів використовували колонки з нержавіючої сталі (0,4 x 70 - 0,4 х 200 см), заповнені хроматоном NAWD MCS з 3,0 % силікону OV-17, хромосорбом W-AW з 3 % Dexil - 300 або Паропаком-Q. Середньоеквівалентний діаметр часток вказаних носіїв 0,200 -0,250 мм. Швидкість подачі попередньо очищеного газу-носія (гелій, азот) становила ~ 40, водню ~ 30, повітря ~ 300 мл/хв. Пробу у випарник хроматографа вводили за допомогою мікрошприца (0,2 - 1,0 мкл) після того як температура випарника досягала 350°C. Тривалість аналізу 50 хв. Ідентифікацію хроматографічних піків, проводили методом додавання реперних вуглеводнів (пентадекана) і шляхом порівняння із хроматограмами сумішей вуглеводнів відомого складу. Кількісну обробку хроматограм здійснювали за інтегральними площами піків, які визначали за допомогою комп'ютерного інтегратора або методом тріангуляції. Фракційний склад олігомеризата визначали також методом фракціонування його на вакуумній ректифікаційній колоні. Результати цих кількісних аналізів збігалися з точністю ± 3 мас. %. Ненасиченість олігомерів (тобто вміст у молекулах олігомерів подвійних зв'язків) визначали методом озонолізу на аналізаторі подвійних зв'язків АДС-4М. Будову не перетворених деценів і продуктів олігомеризації кількісно визначали методом ІЧспектороскопії на приладі «Specord М-80», а також методами спектроскопії ПМР і ЯМР 13C. Спектри ПМР і ЯМР 13C реєстр ували за кімнатної температури на імпульсному спектрометрі ЯМР АС-200Р (200 МГц) фірми «Bruker». Для зняття спектрів ПМР і ЯМР 13C готували 10-20 %-ні розчини продуктів у дейтерохлороформі. Як стандарт використовували тетраметилсилан. Домішки іонного хлору в олігомерах визначали аргентометричним методом за Фольгардом шляхом титрування водного екстракту. Ковалентнопов'язаний з молекулами олігомерів хлор спочатку переводили в іонну форму шля хом мокрого спалювання проби у кварцовому реакторі або за допомогою біфеніл натрію за методом UOP 395-66 з наступним аргентометричним титруванням його за Фольгардом. У деяких випадках вміст ковалентнопов'язаного з молекулами олігомерів хлору визначали рентгенофлюоресцентним методом на спек 27 84505 трометрі «SPECTRO XEPOS» за каліброваною кривою. Для кращого розуміння даного винаходу як ілюстрації в таблицях 1-10 наведені приклади реалізації розробленого відповідно до винаходу способу одержання поліолефінових основ синтетичних мастил. Ці приклади демонструють, але не вичерпують можливості винаходу. Сукупність рішень у різних аспектах патентованого винаходу дозволяє стверджувати, що розроблено новий спосіб одержання поліолефінових основ синтетичних мастил. Розроблений спосіб містить стадії підготовки олефінової сировини, приготування та дозування в реактор розчинів та суспензії компонентів каталітичної системи Al(0) HCl - ТБХ, ізомеризації альфа-олефінів і олігомеризації вищих олефінів та їхніх сумішей під дією каталітичної системи Al(0) - HCl -ТБХ, виділення відпрацьованого каталізатора, розділення оліго 28 меризата на фракції та гідрування виділених фракцій під дією каталізатора Pd (0,2 мас. %) / АІ2О3 + NaOH. Винахід забезпечує вдосконалювання всіх стадій розробленого способу. З метою усунення корозійної активності продуктів спосіб додатково містить стадію дехлорування присутніх в олігомеризаті хлоровмісних олігоолефінів металевим алюмінієм, триетилалюмінієм, спиртовими розчинами KOH або термічним дегідрохлоруванням хлоровмісних поліолефінів у відсутності або в присутності KOH. Для поліпшення техніко-економічних показників способу за рахунок підвищення виходу цільових фракцій поліолефінів з кінематичною в'язкістю 2-8 сСт при 100°C спосіб додатково містить стадію термічної деполімеризації обмежено вживаних високомолекулярних поліолефінів з кінематичною в'язкістю 10-20 сСт при 100°C у цільові поліолефіни з кінематичною в'язкістю 2-8 сСт при 100°C. 29 84505 30 31 84505 32 33 84505 34 35 Комп’ютерна в ерстка І.Скворцов а 84505 Підписне 36 Тираж 28 прим. Міністерство осв іт и і науки України Держав ний департамент інтелектуальної в ласності, вул. Урицького, 45, м. Київ , МСП, 03680, Україна ДП “Український інститут промислов ої в ласності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparation of polyolefinic bases of synthetic oils
Автори англійськоюMatkovskii Pietr Yevhienievich, Troitskii Vladimir Nikolaievich, Startseva Haliva Pavlovna, Savchenko Valierii Ivanovich, Demidov Mikhail Alieksandrovich, Shamstudinov Vladimir Harafovich, Iliasov Habbas Lukmanovich, Lkannanov Robert Habdrakhmanovich, Yarullin Rafinat Samatovich
Назва патенту російськоюСпособ получения полиолефиновых основ синтетических масел
Автори російськоюМатковский Петр Евгєньевич, Троицкий Владимир Николаевич, Старцева Галина Павловна, Савченко Валерий Иванович, Демидов Михаил Александрович, Шамсутдинов Владимир Гарафович, Ильясов Габбас Лукманович, Ханнанов Роберт Габдрахманович, Яруллин Рафинат Саматович
МПК / Мітки
МПК: C07C 2/00, C10G 19/00, C10G 50/00, C10G 29/00, C10G 31/00
Мітки: поліолефінових, основ, спосіб, одержання, синтетичних, мастил
Код посилання
<a href="https://ua.patents.su/18-84505-sposib-oderzhannya-poliolefinovikh-osnov-sintetichnikh-mastil.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання поліолефінових основ синтетичних мастил</a>
Спосіб одержання синтетичних спиртів

Номер патенту: 74732
Опубліковано: 16.01.2006
Автори: Кухар Валерій Павлович, Євдокименко Віталій Олександрович, Бортишевський Валерій Анатолійович, Мельникова Світлана Львівна, Ткаченко Тетяна Вікторівна, Моторний Валентин Григорович, Бойко Володимир Васильович
МПК: C07B 41/00, C07C 37/00, C07C 29/04
Мітки: одержання, спосіб, синтетичних, спиртів
Формула / Реферат:
Спосіб одержання синтетичних спиртів загальної формули СxH2x+1OH (x=2-16) шляхом гідратації відповідних олефінів CxH2x при температурах 100-180 °С та тисках 0,2-10,0 МПа, який відрізняється тим, що процес проводять в реакційній зоні, утвореній двома каталітичними протонопровідними сульфованими або фторсульфованими полімерорганічними мембранами, крізь які створюють протікання різних за величиною і односпрямованих протонних струмів у...
Спосіб одержання рідких синтетичних речовин з вуглецевмісних твердих відходів

Номер патенту: 78628
Опубліковано: 10.04.2007
Автори: Кульчицький Юрій Ігорович, Лотоцький Ігор Михайлович, Стефаник Юрій Васильович, Гвоздевич Олег Васильович, Подольський Мирослав Романович
МПК: C10G 1/00, C10L 1/00, C10J 1/00
Мітки: спосіб, речовин, одержання, синтетичних, твердих, вуглецевмісних, рідких, відходів
Формула / Реферат:
Спосіб одержання рідких синтетичних речовин з вуглецевмісних твердих відходів, при якому в газифікаторі проводять газифікацію подрібненої вуглецевмісної сировини високоенергетичною водяною парою, яку виробляють у парогенераторі, з одержанням оксиду вуглецю та водню як синтез-газу, очищають від золи синтез-газ, який направляють в золоуловлювач, і охолоджують його потоком води з наступним синтезом вуглеводнів у реакторі синтезу - синтезаторі, в...
Спосіб одержання рідких синтетичних речовин з вугільного пласта

Номер патенту: 79893
Опубліковано: 25.07.2007
Автори: Стефаник Юрій Васильович, Павлюк Мирослав Іванович, Храмов Володимир Миколайович, Подольський Мирослав Романович, Гвоздевич Олег Васильович
МПК: C10J 3/02, C10G 1/00, E21B 43/295, E21C 41/18
Мітки: спосіб, синтетичних, речовин, пласта, одержання, вугільного, рідких
Формула / Реферат:
Спосіб одержання рідких синтетичних речовин із вугільного пласта, який включає буріння дуттьової та газовідвідної свердловин на вугільний пласт, з'єднання свердловин по пласту каналом, розпал пласта навколо вибою дуттьової свердловини, почергову подачу нагрітого повітря для підігріву пласта до температури 1500 К і води в нагрітий пласт через дуттьову свердловину, почерговий відвід продуктів горіння та синтез-газу, що утворюються, через...
Спосіб одержання депресорної присадки до мастил

Номер патенту: 18670
Опубліковано: 25.12.1997
Автори: Літовченко Микола Романович, Клименко Петро Лукич, Цапенко Юрій Тимофійович, Марусяк Оксана Володимирівна, Євтушенко Ніна Олексіївна, Сопкіна Алля Костянтинівна
МПК: C10M 129/10, C10N 30/02
Мітки: депресорної, присадки, одержання, мастил, спосіб
Формула / Реферат:
Способ получения депрессорной присадки к смазочным маслам путем алкилирования фенола хлорпарафином при температуре 135 - 160°C в присутствии хлорида алюминия с последующим разбавлением продукта алкилирования маслом, отличающийся тем, что разбавленный маслом продукт алкилирования обрабатывают газообразным аммиаком при температуре 80 - 130°C до достижения pH отходящих газов, равного 8 - 9, или подвергают последовательной обработке газообразным...
Спосіб одержання високолужних алкілсаліцилатних присадок до мастил

Номер патенту: 46969
Опубліковано: 17.06.2002
Автори: Загородній Ігорь Васільєвіч, Гарист Павло Григорович, Пальшін Міхаіл Васільєвіч, Пилат Ярослава Іванівна, Лабуза Ігор Володимирович, Павлік Роман Любомирович, Павлів Богдан Омелянович, Іваськевич Іван Васильович, Угрин Ярослава Антонівна, Косінов Лєонід Алєксандровіч, Баженов Владіслав Пантєлєймоновіч
МПК: C10M 159/20
Мітки: присадок, мастил, спосіб, алкілсаліцилатних, одержання, високолужних
Формула / Реферат:
Спосіб одержання високолужних алкілсаліцилатних присадок до мастил, що включає взаємодію алкілсаліцилових кислот з числом атомів вуглецю в алкільному ланцюзі 16-18 з гідроксидом кальцію і діоксидом вуглецю при нагріванні в присутності вуглеводного розчину з наступним відокремленням промотору і механічних домішок, який відрізняється тим, що обробку розчину алкілсаліцилових кислот в мінеральній оливі проводять в присутності промотору води в...
Попередній патент: Спосіб дослідження біокорозії металів
Наступний патент: Мата дезінфекційна
Випадковий патент: Спосіб прогнозування технічного стану об'єктів