Спосіб одержання езетимібу й проміжних продуктів, використовуваних у цьому способі
Номер патенту: 99702
Опубліковано: 25.09.2012
Автори: Кішш-Бартош Дороттья, Сьоке Каталін, Вукіч Крістіна, Боді Йожеф, Темешварі Крістіна, Гаті Тамаш, Елеш Янош





Формула / Реферат
1. Спосіб одержання езетимібу формули І
 , (І)
, (І)
в якому здійснюють наступні стадії:
a) перетворення етиленгліколевого ефіру 4-(4-фторбензоїл)масляної кислоти формули II на 4-[2-(4-фторфеніл)-[1,3]діоксалан-2-іл]масляну кислоту формули IV через невиділену проміжну сполуку формули III
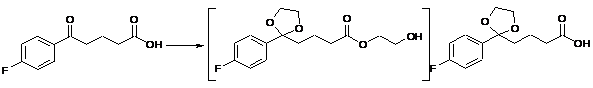
II
III
IV;
b) ацилювання хіральної сполуки формули V сполукою формули IV з одержанням ацилованої похідної оксазолідинону формули VI

IV
VI,
де сполуку формули V вибирають зі сполук формул Va, Vb, Vc або Vd
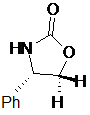
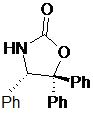

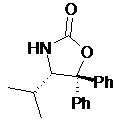
Va
Vb
Vc
Vd,
і де R1 R2 і R3 є:
у разі Va: R1=Ph, R2=R3=H,
у разі Vb: R1=R2=R3=Ph,
у разі Vc: R1=метил, R2=Ph, R3=H,
у разі Vd: R1=ізопропіл, R2=R3=Ph,
і де Ph є фенільною групою;
с) взаємодію ацилованої похідної оксазолідинону формули VI із захищеною іміносполукою формули VII і виділення сполуки формули VIII, де R4 є силільною групою

VI
VII
VIII,
циклізацію сполуки формули VIII з одержанням захищеної похідної азетидинону загальної формули IX

VIII
IX;
d) гідроліз кетальної групи сполук формули IX з одержанням сполуки формули X
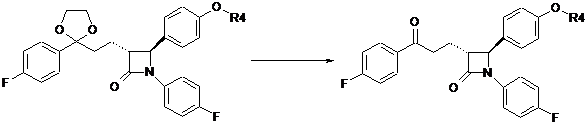
IX
X;
е) енантіоселективне відновлення сполуки загальної формули X з одержанням сполуки формули XI
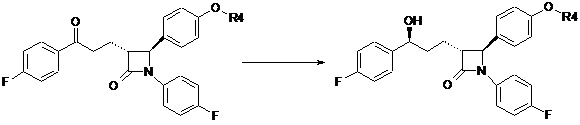
X
XI,
де одна з хіральних CBS-оксазаборолідинових сполук формул ХІІа, ХІІb, ХІІс і XIId вибрана як каталізатор
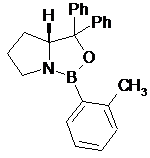
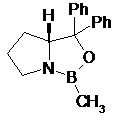
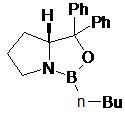
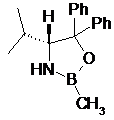
XIIa
XIIb
XIIc
XIId; і
f) видалення силільної захисної групи у сполуці загальної формули XI з одержанням кінцевого продукту езетимібу формули І
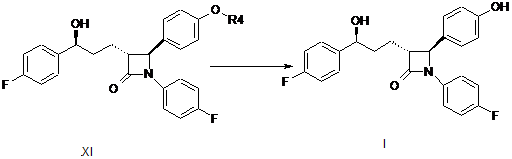 .
.
2. Спосіб одержання сполуки формули VIIIa
 , (VIIIa)
, (VIIIa)
в якому здійснюють ізомеризацію сполуки формули VIIIb у присутності похідної Ti(IV),
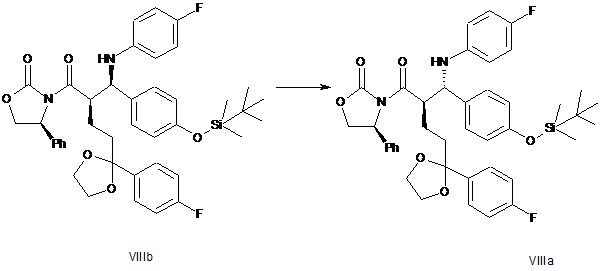 .
.
3. Сполука формули III
 , (III)
, (III)
де R є групою -О-СН2-СН2-ОН або -Н.
4. Сполука формули VI
 , (VI)
, (VI)
де R1, R2 i R3 є:
у разі Va: R1=Ph, R2=R3=H,
у разі Vb: R1=R2=R3=Ph,
у разі Vc: R1=метил, R2=Ph, R3=H,
у разі Vd: R1=ізопропіл, R2=R3=Ph,
і де Ph є фенільною групою.
5. Сполука формули VIII
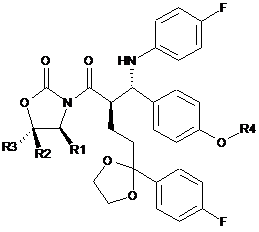 ,
,
де значення R1, R2, R3 незалежно є Va, Vb, Vc або Vd, і R4 є силільною захисною групою.
6. Сполука загальної формули IX
 ,
,
де R4 є силільною захисною групою.
7. Сполука формули Ха
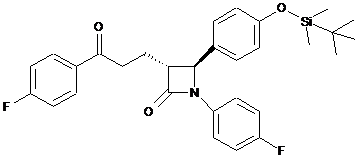 .
.
8. Сполука формули ХІа
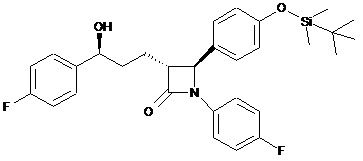 .
.
Текст
Реферат: Даний винахід описує новий, легко реалізований у промисловості і рентабельний спосіб, що включає лише невелику кількість стадій і оснований на використанні нових проміжних продуктів, для одержання 1-(4-3(R)-[3(S)-(4-фторфеніл)-3-гідроксипропіл]-4(3)-(4-гідроксифеніл)2-азетидинону (езетимібу) згідно з наступною реакційною схемою: (І), (II), (III), (IV), (V), (VI), (VII), (VIII), (IX), (X), (XI), де сполуки загальних формул II, IV, VI, VIII, IX, X і XI є новими речовинами, формула III означає невиділений проміжний продукт, R1, R2 і R3 представлені сполуками формул Va-Vd, (Va), (Vb), (Vc), (Vd) і R4 є силільною, наприклад, трет-бутилдиметилсилільною, трет-бутилдифенілсилільною групою. O O O O OH O O OH F F II O O OH F O III IV UA 99702 C2 (12) UA 99702 C2 F O R4 O O HN O O O V O N N R3 R1 O F R2 O R3 F R3 R1 O O HN N R2 R1 O O R2 R4 O F VI VII O O R4 O O VIII O N F N F O O F F IX X O OH R4 O OH N F N F O O F F X XI O HN Rh O HN O H O HN O Ph Rh H Vb O O O O Vd O OH O F O F II IV III O O F R4 O O R3 O O O V O R2 N VII F O R1 R3 O O F VIII O O O R4 O N F O R4 R1 R2 R3 VI O HN O N O N F O OH F R2 Ph O O R4 N F O O F F IX X O OH R4 OH OH N F O N F O F F XI I HN Ph HN HN O O Ph H H Va Ph Vb Ph O O O O H 3C Ph Vc HN O H O Ph H Ph Vc OH R1 HN O C3H Ph Va HN R4 Ph Vd O Ph UA 99702 C2 5 10 Галузь техніки, до якої належить винахід Даний винахід стосується нового способу одержання езетимібу, тобто, 1-(4)-3(R)-[3(S)-(4фторфеніл)-3-гідроксипропіл]-4(S)-(4-гідроксифеніл)-2-азетидинону формули I. Крім того, даний винахід стосується нових проміжних продуктів, використовуваних у даному способі. Рівень техніки У розвинених країнах значна частина смертей викликана серцево-судинними порушеннями. Ці захворювання, головним чином, ініціюються атеросклерозними змінами коронарних артерій. Серед чинників ризику, що приводять до розвитку хвороби, як-от високий кров'яний тиск, діабет, паління тощо, найбільш важливою є висока концентрація холестерину в сироватці крові. Активні інгредієнти й композиції, що зменшують концентрацію сироваткового холестерину, є корисними агентами в лікуванні і запобіганні атеросклерозу. OH OH F 15 20 25 30 35 40 45 50 N O F Езетиміб, тобто 1-(4)-3(R)-[3(S)-(4-фторфеніл)-3-гідроксипропіл]-4(S)-(4-гідроксифеніл)-2азетидинон формули I є активним інгредієнтом деяких сучасних фармацевтичних препаратів, що продаються на ринку і виявляють значний гіпохолестеринемічний ефект, використовуваних для лікування й запобігання атеросклерозу, та описаних у патенті США № 5 767 115 (Schering Co. U.S.A.) і Європейському патенті № 720 599. Перші синтетичні способи здобуття езетимібу і його похідних опубліковані в цих описах. Згідно з одним із описаних у них способів відповідну транс-похідну азетидинону отримують в одній стадії з реакції основи [4-(бензилокси)бензиліден]-(4-фторфеніл)аміну з метил-4(хлорформіл)бутиратом, і після гідролізу й утворення хлорангідриду даним 3-[2-(4бензилoксифеніл)-1-(4-фторфеніл)-4-оксоaзетидин-3-іл]пропіонілхлоридом ацилують (4фторфеніл)цинкхлорид у присутності тетракис(трифенілфосфін)паладію. Чистий 1-(4фторфеніл)-3(R)-[3-оксо-3-(4-фторфеніл)пропіл)]-4(S)-(4-бензилоксифеніл)-2-азетидинон отримують розділенням з використанням хіральної ВЕРХ, і потім кінцевий продукт езетиміб отримують подальшим енантіоселективним відновленням і каталітичним гідруванням. У цьому способі кільце заміщеного азетидинону не утворюється енантіоселективним способом, тому передостанній проміжний продукт очищають способом хіральної колонкової хроматографії. Таким чином, щонайменше 50% останнього проміжного продукту втрачається, що значно збільшує вартість процедури. Щоб уникнути використання дорогої хіральної хроматографії, в патенті США 5919672 (Schering Co.) використовували мікробіологічний і ферментативний способи розділення. Хоча мікробіологічний спосіб знижує вартість розділення рацемату, навіть у цьому випадку вихід на стадії розділення не може бути збільшений більше 50%. У Європейському патенті № 720 599 (Schering Co.) розкриті способи одержання деяких тризаміщених похідних азетидинону, що мають гіпохолестеринемічну активність. Для утворення β-лактамного кільця описані одностадійний і двохстадійний способи, і формування арилгідроксіалкільного бічного ланцюга проводять декількома способами. Для синтезу езетимібу запропонований енантіоселективний спосіб. Cпочатку азетидинове кільце утворюється в результаті двохстадійного синтезу з метилового ефіру 5-оксо-5-((S)-2-оксо-4фенілоксазолідин-3-іл)пентанової кислоти і [4-(бензилокси)бензиліден]-(4-фторфеніл)аміну. Ацилування проводять за допомогою одержаного 3-[(2S,3R)-2-(4-бензилоксифеніл)-1-(4фторфеніл-4-оксоазетидин-3-іл]пропіонілхлориду у присутності (4-фторфеніл)-цинкхлоридутетракис(трифенілфосфін)паладію. Даний проміжний продукт 1-(4-фторфеніл)-3(R)-[3-оксо-3-(4фторфеніл)пропіл)]-4(S)-(4-бензилоксифеніл)-2-азетидинон очищають колонковою хроматографією, потім після енантіоселективного відновлення оксогрупи і видалення захисної групи отримують активний інгредієнт. З попередніх процедур спосіб, що стратегічно відрізняється, був опублікований у міжнародній заявці на патент WO 97/45 406 і в патенті США № 5739321 (Schering Co). Згідно з цими публікаціями енантіоселективне утворення транс-заміщеного проміжного продукту азетидинону виконують взаємодією 4(S)-гідроксибутиролактону із захищеним іміном у присутності основи, потім 3-(4-фторфеніл)-3-гідроксипропільну бічну групу формують синтезом, 1 UA 99702 C2 5 10 15 20 25 30 35 40 45 50 55 60 що складається з декількох стадій, з використанням як вихідної речовини вищезазначеного проміжного продукту 1-(4-фторфеніл)-3(R)-[3-оксо-3-(4-фторфеніл)пропіл)]-4(S)-(4бензилоксифеніл)-2-азетидинону. Бензольну захисну групу видаляють каталітичним гідруванням. Інший реакційний шлях розкрито в патенті США № 5 856 473 (Schering Co.). Згідно з описом (3R,4S)-4-(4-бензилоксифеніл)-1-(4-фторфеніл)-3-[(E)-3-(4-фторфеніл)аліл]азетидин-2-oн, що містить подвійний зв'язок у бічному ланцюзі, отримують алкілуванням 1-4-(фторфеніл)-4(S)-(4бензилоксифеніл)-2-азетидинон-4-фторцинамоїлбромідом, або енантіоселективним синтезом з використанням як вихідної речовини (S)-3-[5-(4-фторфеніл)-пент-3-eноїл]-4-фенілоксазолідин-2oну. Проміжний продукт 1-(4-фторфеніл)-3(R)-[3-оксо-3-(4-фторфеніл)пропіл)]-4(S)-(4бензилоксифеніл)-2-азетидинон отримують окисленням бічного ланцюга, після чого кінцевий продукт езетиміб отримують видаленням захисної групи вищезазначеним енантіоселективним відновленням. Ці енантіоселективні процедури зазвичай застосовують згодом при використанні багатостадійних способів синтезу з оптично чистими похідними азетидинону, отриманими відносно дорогими енантіоселективними синтетичними способами. Ключовий проміжний продукт, 1-(4-фторфеніл)-3(R)-[3-оксо-3-(4-фторфеніл)пропіл)]-4(S)-(4-бензилоксифеніл)-2азетидинон, очищають лише хроматографічно, що значно збільшує вартість промислових способів. У заявках на патент WO 2000/34 240 (Schering Co.) і WO 1995/08 532 і в європейському патенті 0 720 599 (Schering Co.) описаний покращений і ефективніший енантіоселективний спосіб синтезу езетимібу. Згідно з даною процедурою спочатку 3-[(S)-5-(4-фторфеніл)-5гідроксипентаноїл]оксазолідин-2-oн отримують з 98%-ною чистотою (de, діастереомерний надлишок) з відповідної оксосполуки енантіоселективним відновленням. 3-[(S)-5-(4-фторфеніл)5-гідроксипентаноїл]-оксазолідин-2-oн і N-(4-гідроксибензиліден)-4-фторанілін силілують in situ хлортриметилсиланом у одній судині. Відповідний продукт β-аміноамід отримують обробкою суміші реагентом TiCl4 у присутності основи з використанням процедури, добре відомої в даній галузі. Автори виявили несподівану стабільність триметилсилільної групи, що є захисною групою для фенольної ОН-групи. Попри стабільність силільної групи, проміжний продукт вдалося виділити лише з 65% виходом після обробки й додаткової стадії силілування. Езетиміб отримують після циклізації β-аміноаміду й подальшого видалення захисних груп. У цій процедурі утворення 3(S)-гідроксигрупи здійснюють на початку синтезу відносно дорогим енантіоселективним способом, потім продукт виділяють після додаткових стадій реакції й процедури очищення. Стереоселективне утворення 3(S)-OH групи є однією з ключових стадій одержання езетимібу. В кожній із вищезазначених процедур використовують один із варіантів способів енантіоселективного відновлення, каналізованого СBS-оксазаборолідином, добре відомим з літератури (E.J. Corey et al., J.Am.Chem.Soc. 1987, 109, 5551-5553). Величина de (діастереомерний надлишок), що досягається, складає 88-98%, як і має бути в типових випадках. Патенти США 5886171 і 5856473 (Schering Co.) описують спосіб енантіоселективного відновлення з використанням CBS-оксазаборолідинового каталізатора, в якому захищений 1-(4фторфеніл)-3(R)-[3-оксо-3-(4-фторфеніл)-пропіл)]-4(S)-(4-гідроксифеніл)-2-азетидинон перетворюється на захищений 1-(4-фторфеніл)-3(R)-[3-гідрокси-3-(4-фторфеніл)пропіл)]-4(S)-(4гідроксифеніл)-2-азетидинон. Патенти США 6207822 і 6627757 (Schering Co.) описують вживання подібних відновних агентів і хіральних каталізаторів для перетворення 3-[5-(4-фторфеніл)-5-оксопентаноїл]-4(S)-4феніл-1,3-оксазолідин-2-ону на 3-[5(S)-5-(4-фторфеніл)-5-гідроксипентаноїл]-4(S)-4-феніл-1,3оксазолідин-2-он. У патенті США 5618707 і в заявці на патент WO 1997/12 053 (Schering Co.) описана інша можливість енантіоселективного відновлення, коли попередній проміжний продукт 3-[5-(4фторфеніл)-5-оксопентаноїл]-4(S)-4-феніл-1,3-оксазолідин-2-он перетворюють стереоселективним мікробіологічним відновленням на 3-[5(S)-5-(4-фторфеніл)-5гідроксипентаноїл]-4(S)-4-феніл-1,3-оксазолідин-2-он. Величина de >95% (діастереомерний надлишок), що досягається, схожа з величиною, отриманою з використанням CBSоксазаборолідинового каталізатора. У патенті США 6133001 стереоселективне мікробіологічне відновлення описано для перетворення 1-(4-фторфеніл)-3(R)-[3-оксо-3-(4-фторфеніл)пропіл)]-4(S)-(4-гідроксифеніл)-2азетидинону на 1-(4-фторфеніл)-3(R)-[3-гідрокси-3-(4-фторфеніл)пропіл)]-4(S)-(4-гідроксифеніл)2-азетидинон (езетиміб). Кінцевий продукт отримують у малих кількостях і очищають 2 UA 99702 C2 5 10 препаративною тонкошаровою хроматографією. У міжнародній заявці на патент WO 2005/066 120 (Ranbaxy Ltd.) енантіоселективне відновлення оксогрупи 3-[5-(4-фторфеніл)-5-оксопентаноїл]-4(S)-4-феніл-1,3-оксазолідин-2-ону й 1-(4-фторфеніл)-3(R)-[3-оксо-3-(4-фторфеніл) пропіл)]-4(S)-(4-гідроксифеніл)-2-азетидинону виконують за допомогою (-)-B-хлордіізопінокамфеїлборану, досягаючи селективності, подібної отриманій при CBS-відновленні. Проте цікаво, що при відновленні 20 г 1-(4-фторфеніл)-3(R)-[3оксо-3-(4-фторфеніл)пропіл)]-4(S)-(4-гідроксифеніл)-2-азетидинону в такий спосіб після очищення колонковою хроматографією отримують тільки 3 г кінцевого продукту езетимібу. Суть винаходу Даний винахід забезпечує новий, легко реалізований у промисловості й економічний спосіб, що складається тільки з невеликої кількості стадій, і заснований на використанні нових проміжних продуктів для одержання 1-(4-3(R)-[3(S)-(4-фторфеніл)-3-гідроксипропіл)]-4(S)-(4гідроксифеніл)-2-азетидинону (езетимібу) відповідно до наступної реакційної схеми: O O O O O OH O F O OH F IV III O O F R4 O R1 O R3 R2 O O O V O N VII F O R1 R3 O O F VI VIII O O O R4 O N F O R4 R1 R2 R3 R2 O HN O N O N F O OH F II HN O R4 N F O O F F IX X O OH R4 OH OH N F O N F O F F XI 15 Ph HN HN HN O O Ph H H Va Ph Vb Ph O O O O 20 I де • сполуки загальних формул II, IV, VI, VIII, IX, X і XI є новими • формула III є невиділеним проміжним продуктом • R1, R2 і R3 представлені сполуками формул Va-Vd H 3C Ph HN O H Ph O Ph Vd Vc і R4 є силільною, наприклад трет-бутилдиметилсилільною, трет-бутилдифенілсилільною групою. Детальний опис винаходу 3 UA 99702 C2 5 10 15 20 Зважаючи на недоліки відомих синтетичних процедур одержання езетимібу, автори спробували розробити промисловий безпечний спосіб одержання, що включає рентабельні технологічні стадії, що забезпечують активний інгредієнт з чистотою, що задовольняє вимогам фармакології. Автори поставили завдання розробити таку стратегію синтезу, яка не передбачає утомливих технологічних стадій або стадій, що вимагають екстремальних умов, і в якому проміжні продукти можуть бути отримані простими процедурами з високою ефективністю й можуть бути виділені з високою чистотою. Для захисту функціональних груп автори прагнули використовувати такі захисні групи, які є стабільними під час синтезу, можуть бути введені простими й дешевими способами й селективно видалені. Автори мали намір здійснювати дорожчі технологічні стадії в кінці синтезу і регенерувати дорогі допоміжні матеріали (наприклад, оптично активні). Під час експериментів автори несподівано виявили, що в наступному реакційному шляху синтезу з використанням комбінації спеціальних захисних груп у більшості випадків виходять такі чудові проміжні продукти, які можуть бути легко очищені простими способами і з високими ефективностями, завдяки їхній відмінній здатності кристалізуватися. Проміжні продукти, що не кристалізуються, могли бути використані на наступних стадіях без очищення. Була відкрита нова реакція, не опублікована раніше в літературі, де стереоізомерна сполука, що утворюється як побічний продукт у реакції типу реакції Манніха, каталізованої Ti(IV), могла бути перетворена на бажаний проміжний продукт. Процедура даного винаходу, що використовує нові проміжні продукти, включає сім стадій, перерахованих нижче. Стадія 1: 4-(4-фторбензоїл)масляну кислоту (II) перетворюють на 4-[2-(4-фторфеніл)-[1,3]діоксолан-2іл]масляну кислоту (IV) через невиділену проміжну сполуку (III). O O O O OH O F 25 O O OH O O OH F F II IV III Стадія 2: Хіральний оксазолідинон (V) ацилують 4-[2-(4-фторфеніл)-[1,3]діоксолан-2-іл]масляною кислотою (IV) з одержанням похідної оксазолідинону (VI) O HN O R1 O O O O R2 R3 O O N V OH O F F R1 R2 R3 VI IV 30 де R1, R2 і R3 представлені наступними структурами (Va-Vd): Ph HN HN HN O O Ph H H Va Ph Vb Ph O O O O H 3C Ph HN O H Ph O Ph Vd Vc Стадія 3: Наступний ацилований оксазолідинон (VI) реагує з іміном (VII), і виділяють сполуку формули (VIII) 4 O UA 99702 C2 F O O O O O N F O R1 O + O HN N R3 N R1 R2 O R4 O R3 R2 O F VI 5 O R4 F VII де R4 означає силільну, наприклад, трет-бутилдиметилсилільну бутилдифенілсилільну групу. Стадія 4: Захищений азетидинон (IX) отримують циклізацією сполуки формули (VIII). VIII (TBDMS), трет F O O R3 O HN O O R4 O N R2 O R4 R1 N F O O F O IX F VIII Стадія 5: Сполуку формули (X) одержують гідролізом кетальної групи сполуки формули (IX). O O O O R4 O R4 N F O N F O F IX F X Стадія 6: Енантіоселективним відновленням сполуки формули X одержують сполуку формули XI. 10 R4 R4 N F N F O OH O O O O F 15 F X XI Стадія 7: Видаленням силільної захисної групи у сполуці формули XI отримують кінцевий продукт, езетиміб (I). O OH F OH OH R4 N F O N O I F F XI 20 У наступній частині всі стадії описані детальніше: Стадія 1: У інертному, безводному розчиннику, наприклад, у дихлорметані, у присутності сильної кислоти, наприклад, концентрованої сірчаної кислоти або п-толуолсульфонової кислоти, переважно концентрованої сірчаної кислоти, і допоміжного матеріалу, що зв'язує воду, 5 UA 99702 C2 5 10 15 20 25 30 35 40 45 наприклад, триметил-орто-форміату, 4-(4-фторбензоїл) масляна кислота (II) взаємодіє в одній стадії з етилгліколем при температурі 20-25°C. Реакцію зупиняють додаванням основи, наприклад NaHCO3. Розчинник замінюють на спиртний розчинник, переважно, на метанол, і утворений складноефірний проміжний продукт (III) гідролізують розчином основи, переважно, розчином гідроксиду калію. Утворену 4-[2-(4-фторфеніл)-[1,3]діоксалан-2-іл] масляну кислоту (IV) виділяють після концентрації реакційної суміші, потім після підкислення слабкою кислотою, наприклад, винною кислотою, лимонною кислотою, переважно лимонною кислотою, з подальшою екстракцією відповідним розчинником, наприклад етилацетатом. Продукт очищають кристалізацією з неполярного розчинника, наприклад, з н-гексану або н-гептану. Стадія 2: Продукт із стадії 1 перетворюють на змішаний ангідрид у інертному, безводному розчиннику, наприклад тетрагідрофурані або дихлорметані, переважно в тетрагідрофурані, з використанням 1-1,7-кратної молярної кількості, переважно, 1,05-1,10 еквіваленту хлорангідриду, наприклад, півалоїлхлориду в присутності триетиламіну при температурі в діапазоні від -20 до -10C. Оксазолідінон формули V, переважно, S-(+)-4-феніл-2-оксазолідинон (Va) додають у розчин отриманого змішаного ангідриду у присутності відповідного активуючого агента, наприклад, хлориду літію (LiCl) або 4-диметиламінопіридину, переважно хлориду літію, потім розчин перемішують протягом 4-8 годин при температурі в діапазоні від -20 до 25°C. Продукт виділяють екстракцією й очищають кристалізацією. Стадія 3: Спосіб A: Продукт стадії 2 взаємодіє з іміном формули VII (де R4 є, переважно, третбутилдиметилсилільною групою) в інертному, безводному розчиннику, наприклад дихлорметані, і в атмосфері N2 при температурі в діапазоні від -40 до -25°C у присутності TiCl4 і Ti(IV)ізопропоксиду й у присутності третинної основи, наприклад, діізопропілетиламіну протягом 1-2 годин Реакцію зупиняють додаванням спирту, переважно ізопропілового спирту, і продукт (VIII) виділяють екстракцією й після упарювання очищають при його перемішуванні з метанолом. Для захисту фенольного гідроксилу використовують силільну захисну групу, переважно трет-бутилдиметилсилільну групу, яка особливо переважно порівняно з іншими групами силільного типу, здатними відщеплюватися навіть у м'якших умовах, і в поєднанні з іншими захисними групами алкільного й ацильного типу. Оскільки трет-бутилдиметилсилільна захисна група стабільна в умовах синтезу, немає необхідності повторно силілувати перед виділенням проміжний продукт, який під час обробки частково втрачає свою захисну групу. Крім того, третбутилдиметилсилільна захисна група може бути легко видалена при обробці кислотою за відсутності побічних реакцій. З іншого боку, видалення захисних груп бензильного типу вимагає або технологічно більш трудомісткого і небезпечнішого каталітичного гідрування, або способу видалення сильнішою кислотою. На жаль, карбенієвий катіон, що утворюється під час відщеплення захисних груп бензильного й алкільного типу в кислому середовищі, приводить до утворення значної кількості побічних продуктів алкілування фенільного кільця. Як добре відомо з літератури, видалення захисних груп ацильного типу основою супроводжується значною кількістю побічних реакцій (наприклад, розкриттям лактамного циклу). Експерименти авторів довели, що в рівноважній реакції типу реакції Манніха, каталізованої Ti(IV), окрім очікуваної R4=трет-бутилдиметилсилільної похідної (VIIIa), також утворюється ізомерний побічний продукт (VIIIb) у значних кількостях. Автори показали, що з використанням як вихідних речовин з (VIIIa) і (VIIIb) в описаних вище умовах реакції утворюються ті самі продукти, що і при проведенні реакції з використанням як вихідних речовин (VIa) та (VIIa). При відповідному виборі параметрів експерименту рівновага може бути зрушена в напрямі, що сприяє утворенню (VIIIa), і продукт може бути виділений з виходом 73-78%. 6 UA 99702 C2 F O O O O O O N O Si Ph Si O O N F HN O O O + Ph N Ti(OiPr)4/TiCl4 DIPEA, DKM F VIIIa + F F VIIa VIa O O HN O N Ph O Si O O F 5 10 VIIIb Ізомер (VIIIb), який може бути присутнім у матковому розчині, отриманому після фільтрування продукту з метанольної суспензії, може бути перетворений на (VIIIa) в умовах Ti(IV)-каталізованої реакції типу реакції Манніха. Таким чином, вихід реакції може бути значно підвищений. З цією метою метанольний матковий розчин упарюють, розчинник замінюють на відповідний розчинник, наприклад толуол, розчин обезбарвлюють силікагелем і потім після фільтрування упарюють. При використанні цієї процедури з цієї суміші може бути отримана додаткова кількість продукту формули VIII таким чином: Осад після упарювання розчиняють у дихлорметані й у присутності Ti(IV)-ізопропоксиду й третинної основи, наприклад діізопропілетиламіну, розчин перемішують у інертній атмосфері, наприклад N2, при температурі в діапазоні від -40 до -25C протягом 1-2 годин. Чистий продукт формули (VIIIa) виділяють способом, описаним вище. Спосіб B: OH O TBDMS-Cl/DIPEA/DKM N N F F VIIb 15 20 25 30 Si VIIa У альтернативній процедурі спочатку одержують сполуку формули (VIIa) in-situ в дихлорметані у присутності діізопропілетиламіну (DIPEA) реакцією (E)-(4-гідроксибензиліден)(4-фторфеніл)аміну (VIIb) з трет-бутилдиметилсилілхлоридом (TBDMS-Cl), потім розчин отриманого продукту формули VIIa використовують, як описано у Способі A. Стадія 4: Продукт із стадії 4 формули VIII силілують у відповідному розчиннику, наприклад у тетрагідрофурані, толуолі, метил-трет-бутиловому ефірі або ацетонітрилі, переважно, в ацетонітрилі, придатним силілуючим агентом, наприклад, біс(триметилсиліл)ацетамідом, при температурі в діапазоні від 20 до 25ºC протягом 1-3 годин. Фторовмісну сполуку, переважно тригідрат тетрабутиламонійфториду, додають до суміші в каталітичній кількості (0,1-10 мол.%), переважно, в кількості 0,5-1 мол.%. Таку реакційну суміш, отриману при циклізації, додатково перемішують протягом 0,5-3 годин, переважно, протягом 0,5 години, потім реакцію зупиняють водою, і продукт формули IX виділяють за допомогою розчинника алканового типу, наприклад н-гексану. Хіральний допоміжний матеріал S-(+)-4-феніл-2-оксазолідинон (Va), після витягання з ацетонітрильної фази і його подальшої концентрації екстрагують дихлорметаном і очищають 7 UA 99702 C2 5 10 15 20 кристалізацією. Стадія 5: Сполуку формули IX, отриману на стадії 4, обробляють в інертному розчиннику, наприклад дихлорметані, мінеральною глиною кислотного типу, переважно, з монтморилонітом при температурі в діапазоні від 20 до 25ºC протягом 3-6 годин. У цих умовах третбутилдиметилсилільна захисна група стабільна, і кетальна захисна група може бути селективно видалена. Отриманий таким чином продукт формули X відокремлюють простим фільтруванням і після упарювання очищають кристалізацією. Стадія 6: У процедурі авторів енантіоселективне відновлення для утворення 3-(S)-гідроксильної групи проводять наприкінці синтезу. В цьому випадку особливі витрати на дорогий хіральний каталізатор є нижчими. Оскільки асиметричний центр входить до складу оптично чистого індивідуального ізомеру, очищення кінцевого продукту спрощується до розділення двух діастереомерів. Відповідно, одержану таким чином сполуку формули X на стадії 5 відновлюють відновним агентом боранового типу, таким як боран-диметилсульфід, боран-тетрагідрофуран, боран-діетиланілін або катехін-боран, переважно, сумішшю боран-диметилсульфіду і борантетрагідрофурану у присутності хірального каталізатора CBS-оксазаборолідинового типу, добре відомого для досягнення даної мети, в інертному розчиннику, наприклад дихлорметані, в інертній атмосфері, наприклад, у N2, при температурі в діапазоні від -20 до 20ºC, переважно, від -5 до +5C. Хіральний CBS-оксазаборолідин (сполуку XIIa-XIId), переважно, оксазаборолідин (сполуку XIIa), використовують як каталізатор. H Ph N B XIIa 25 30 35 40 45 50 H Ph H Ph Ph Ph O O N B n-Bu O CH3 Ph N B CH3 XIIb XIIc Ph Ph O HN B CH3 XIId Продукт виділяють екстракцією і потім вводять в наступну реакцію без очищення. Стадія 7: Отриманий таким чином продукт формули XI нагрівають у суміші з розбавленим водним розчином хлористоводневої або сірчаної кислоти, переважно, з розчином сірчаної кислоти і спиртним розчинником, наприклад, метанолом або ізопропіловим спиртом, переважно ізопропіловим спиртом, при температурі в діапазоні від 50 до 70ºC протягом 1-3 годин. Кінцевий продукт кристалізують з реакційної суміші з додаванням води, потім очищають перекристалізацією. Переваги даного винаходу підсумовані таким чином: a) у способі авторів на новому шляху синтезу, заснованому на використанні нових сполук, ключові проміжні продукти, завдяки їхній чудовій здатності кристалізуватися можуть бути ефективно очищені простими операціями кристалізації; b) для захисту фенольної ОН-групи використовують групу силільного типу, переважно третбутилдиметилсилільну групу, яка є переважнішою, ніж ті, що відщеплюються у м'якших умовах, наприклад, порівняно з групами алкільного і ацильного типу; c) у енантіоселективній каталізованій Ti(IV) реакції типу реакції Манніха, відповідний проміжний продукт (VIIIa) утворюється з високим виходом (85-90%), проте стереоізомерний побічний продукт не втрачається в рівноважній реакції, а головним чином перетворюється на бажаний проміжний продукт; d) таким чином, велика частина хіральної допоміжної речовини, S-(+)4-феніл-2оксазолідинону (>70% введеної кількості) регенерується простим способом у процесі синтезу; e) у цій процедурі енантіоселективне відновлення з утворенням 3-(S)-гідроксильної групи проводять наприкінці синтезу. Завдяки цьому спеціальні витрати на дорогий хіральний каталізатор є меншими. Оскільки асиметричний центр входить до складу оптично чистого індивідуального ізомеру, очищення кінцевого продукту спрощується до розділення двох діастереомерів. Підводячи підсумки, можна сказати, що в даному винаході розкрита така нова процедура, яка може бути використана для рентабельного одержання езетимібу в промисловому масштабі. Чистота активного інгредієнта, що одержується з використанням цієї процедури, може відповідати сучасним дедалі зростаючим вимогам до якості фармацевтично активних інгредієнтів. 8 UA 99702 C2 5 10 15 20 25 30 35 40 45 50 55 60 Приклади Наступні приклади є ілюстративними, і не призначені для обмеження обсягу заявленого винаходу. Приклад 1 Одержання 4-[2-(4-фторфеніл) -[1,3]діоксалан-2-іл]масляної кислоти (IV) 21,0г (0,1 моль) 4-(4-фторбензоїл)масляної кислоти (II) зважують у круглодонній колбі на 500 мл і суспендують у 210 мл дихлорметану. При безперервному перемішуванні до суспензії по краплях додають 28мл (31,2г, 0,5моль) етилгліколю, 32 мл (31,04 г, 0,3 моль) триметил-оформіату і 0,5 мл концентрованої сірчаної кислоти. Реакційну суміш перемішують при 20-25ºC протягом 3-6 годин. Реакцію контролюють аналітично тонкошаровою хроматографією. Коли кетон закінчується, про що судять за даними тонкошарової хроматографії, які показують зникнення його плями, реакцію зупиняють додаванням 5 г твердого NaHCO 3. Суспензію перемішують протягом 05 хв, потім розчинник видаляють випаровуванням і осад розчиняють у 150 мл метанолу. Цей розчин охолоджують на водяній бані з льодом і додають при охолоджуванні 100 мл 10%-ного розчину NAOH. Колбу закривають і каламутну суміш перемішують при 20-25ºC протягом 1 год. Гідроліз контролюють аналітично тонкошаровою хроматографією. Коли складний ефір закінчується, про що судять за даними тонкошарової хроматографії, які показують зникнення його плями, метанол видаляють випаровуванням у вакуумі, і при інтенсивному охолоджуванні на водяній бані з льодом додають розчин 350 мл 10% лимонної кислоти для досягнення значення кислого pH у діапазоні від 3 до 4. Осаджений продукт екстрагують 200 мл етилацетату. Водну фазу двічі екстрагують 50-50 мл етилацетату і потім об'єднану органічну фазу промивають до нейтральної реакції 5x50 мл води. Етилацетатний розчин сушать над безводним Na2SO4, дегідратуючий агент відфільтровують, і фільтрат упарюють у вакуумі. Осад після упарювання кристалізують додаванням 50 мл нгексану при 0C. Кристалічний матеріал сполуки (IV) виділяють фільтруванням і сушать. Вихід: 23 г (90%). Точка плавлення: 65-67C. 1 ° Дані H ЯМР: (500 МГц, DMSO-d6, 25 C) δ 1,41-1,52 (м, 2H), 1,79-1,87 (м, 2H), 2,18 (т, J = 7.5 Hz, 2H), 3,63-3,73 (м, 2H), 3,91-4,01 (м, 2H), 7,13-7,22 (м, 2H), 7,37-7,45 (м, 2H), 11,97 (шир с, 1H) м.д. Приклад 2 Одержання (S)-3-{4-[2-(4-фторфеніл)-[1,3]діоксалан-2-іл]бутирил}-4-фенілоксазолідин-2-ону (VIа) 42 г (165 ммоль) сполуки формули IV, продукту прикладу 1, розчиняють у 340мл безводного тетрагідрофурану й судину продувають сухим газоподібним N 2. Розчин охолоджують до -20ºC і додають 55 мл (390 ммоль) триетиламіну. Через краплинну воронку додають суміш 40 мл тетрагідрофурану і 20,2 мл півалоїлхлориду (19,8 г, 164 ммоль) протягом приблизний 30 хв при температурі в діапазоні від -10C до -20ºC. Суміш, що містить осад, перемішують протягом 2 годин при температурі в діапазоні від -10C до -20C, і потім до неї послідовно додають 24,45 г (150 ммоль) твердого S(+)-4-феніл-2-оксазолідинону (Va) і 7,5 г (177 ммоль) безводного хлориду літію. Потім суспензію перемішують протягом 4 годин при нагріванні до 20-25ºC. Реакцію контролюють аналітично тонкошаровою хроматографією. Коли пляма S(+)-4-феніл2-оксазолідинону зменшується до 3%, реакцію зупиняють додаванням 300 мл толуолу і 150 мл насиченого розчину хлориду амонію. Фази розділяють, потім водну фазу екстрагують 50 мл толуолу. Об'єднаний толуольний розчин промивають 2x150 мл розчином 10% лимонної кислоти, 2x150 мл розчином 1M NAOH і, нарешті, 3x150 мл води. Органічну фазу сушать над безводним Na2SO4, дегідратуючий агент відфільтровують і фільтрат упарюють у вакуумі. Осад кристалізують при 0ºC в 150 мл ізопропілового спирту. Продукт (VIa) сушать у вакуумі у присутності P2O5. Вихід: 55,7г (93%). Точка плавлення: 100-102°C 25 [α] D =+54,3, (c=l, дихлорметан) 1 Дані H ЯМР: (500 МГц, DMSO-d6, 25ºС) δ 1,42-1,56 (м, 2H), 1,76-1,85 (м, 2H), 2.80 (дт, J = 17,2, 7,5 Hz, 1H), 2,90 (дт, J = 17,2, 7,5 Hz, 1H), 3,61-3,71 (м, 2H), 3,89-3,99 (м, 2H), 4,13 (дд, J = 8,7, 3,6 Hz, 1H), 4,71 (т, J = 8,7 Hz, 1H), 5,43 (дд, J – 8,7, 3,6Hz, 1H), 7,12-7,19 (м, 2H), 7,23-7,28 (м, 2H), 7,29-7,34 (м, 1H), 7,34-7,42 (м, 4H) м.д. Приклад 3 Одержання (S)-3-{(R)-2-[(S)-[4-(трет-бутилдиметилсиланілокси)фенілу]-(4фторфеніламін)метил]-4-[2-(4-фторфеніл)-[1,3]діоксалан-2-іл]бутирил}-4-фенілоксазолідин-2ону (VIIIà) Одержання титантрихлоридізопропоксидного реагенту 0,95 мл (0,9 г, 3,2 ммоль) ізопропоксиду Ti(IV) додавали до розчину 0,99 мл (1,71 г, 9 ммоль) 9 UA 99702 C2 5 10 15 20 25 30 35 40 45 50 55 60 TiCl4, приготованому в 34 мл дихлорметану при температурі 0ºC і в атмосфері N 2. Суміш перемішують протягом 15 хв при 0ºС. Даний розчин використовують на наступній стадії конденсації. Конденсація (спосіб A) 4,0 г (10 ммоль) сполуки формули VIa і 6,6 г (20 ммоль) іміносполуки формули VIla зважують у судині на 250 мл, забезпеченій магнітною мішалкою, термометром, краплинною воронкою і вхідним отвором для N2, і розчиняють у 50 мл дихлорметану. Суміш охолоджують до -40C і додають 3,6 мл (20,7 ммоль) DIPEA. Розчин титантрихлоридізопропоксидного реагенту поступово додають з краплинної воронки протягом приблизно 30 хв. Суміш перемішують протягом 1 години при температурі в діапазоні від -30 до -40C, потім реакцію зупиняють додаванням 25 мл ізопропілового спирту і 50 мл дихлорметану при температурі між -30 і -40C і після цього реакцію перемішують додатково протягом 30 хв при тій самій температурі. Отриману таким чином помаранчеву суспензію повільно виливають у 100 мл тартратного буферу при pH=7, потім після 15-хвилинного перемішування фази розділяють. Водну фазу екстрагують додатковою кількістю дихлорметану (3x30 мл), потім об'єднаний дихлорметановий розчин промивають 30 мл води, сушать безводними Na 2SO4, дегідратуючий агент відфільтровують і фільтрат упарюють у вакуумі. До осаду додають 50мл метанолу, отриману таким чином суспензію перемішують при 20-25ºC протягом 10 хв і потім продукт виділяють фільтруванням. Білу кристалічну сполуку (Vllla) сушать у вакуумі у присутності P2О5. Вихід: 5,5 г (76%). Конденсація (спосіб B) 25,8 г (120 ммоль) (E)-(4-гідроксибензиліден)-(4-фторфеніл) аміну зважують у судині на 2 л, забезпеченій магнітною мішалкою, термометром, краплинною воронкою і вхідним отвором для N2, його розчиняють у 500 мл дихлорметану, потім додають 57,8 мл (332 ммоль) діізопропілетиламіну (DIPEA) при 20-25ºC. Додають 19,9 г (132 ммоль) третбутилдиметилсилілхлориду і розчин перемішують при 20-25ºC протягом 1-2 годин. Гідроліз контролюють аналітично тонкошаровою хроматографією. Коли на хроматограмі зникає пляма вихідної речовини, (E)-(4-гідроксибензиліден)-(4-фторфеніл)аміну, додають 40 г (100 ммоль) сполуки (Vla) і суміш охолоджують до температури в діапазоні від -25 до 30ºC. Приблизно через 30-хвилинний період через краплинну воронку поступово додають розчин 9,5 мл (9 г, 32 ммоль) тетраізопропоксиду титану й 9,9 мл (17,1 г, 90 ммоль) тетрахлориду титану(TiCl4) в 340 мл дихлорметану при 0ºC. Суміш перемішують протягом 0,5 години при температурі від -25 до -30C, реакцію в суміші зупиняють додаванням 250 мл ізопропілового спирту і 500 мл дихлорметану при температурі в діапазоні від -30 до 40ºC і після цього перемішують протягом ще 30 хв при тій самій температурі. Отриману таким чином суміш повільно виливають у 1000 мл тартратного буфера при pH=7, потім після 15-хвилинного перемішування фази розділяють. Водну фазу екстрагують додатковою кількістю дихлорметану (3x250 мл), потім об'єднаний дихлорметановий розчин промивають 300 мл води, сушать безводними Na2SO4, дегідратуючий агент фільтрують і фільтрат упарюють у вакуумі. До осаду додають 500 мл метанолу, отриману таким чином суспензію перемішують при 20-25ºC протягом 10 хв, і потім продукт виділяють фільтруванням. Білу кристалічну сполуку (Vllla) сушать у вакуумі у присутності P2О5. Вихід: 57 г (78%). Точка плавлення: 211-213°C 25 [α] D =-0,9, (c=l, дихлорметан) 1 Дані H ЯМР: (500 МГц, CDCl3, 25°C) δ 0,17 (с, 6H), 0,97 (с, 9H), 1,22-1,35 (м, 1H), 1,66-1,90 (м, 3H), 3,58-3,77 (м, 2H), 3,84-3,96 (м, 2H), 4,21 (дд, J = 8,7, 2,9 Hz, 1H), 4,26 (д, J = 9,1 Hz, 1H), 4,46-4,57 (м, 1H), 4,66 (т, J = 8,7 Hz, 1H), 5,06 (шир, IH), 5,44 (дд, J = 8,7, 2,9 Hz, IH), 6,33-6,41 (м, 2H), 6,65-6,78 (м, 4H), 6,91-6,98 (м, 2H), 7,02-7,13 (м, 6H), 7,13-7,19 (м, 1H), 7,25-7,31 (ср, 2H) м.д. Обробка маткового розчину Отриманий метанольний матковий розчин упарюють, розчинник замінюють на 200 мл толуолу, до толуольного розчину додають 10 г силікагелю Si 60, суспензію перемішують при 2025ºC протягом 15 хв. Силікагель відфільтровують, промивають толуолом і фільтрат упарюють. Залишок після упарювання розчиняють у 100 мл дихлорметану, суміш охолоджують до -30C і додають 7 мл (40 ммоль) DIPEA в атмосфері N2. 2 мл розчину титантрихлоридізопропоксидного реагенту, приготованого з (1,9 г, 6,74 ммоль) тетраізопропоксид титану і 1,81 мл (3,12 г, 16,3ммоль) TiCl4, додають через краплинну воронку протягом 30-хвилинного періоду. Реакційну суміш перемішують при температурі між -30 і -40°C, потім чистий продукт формули VIIIa виділяють таким самим способом, як у разі реакції конденсації. Вихід: 8,0 г. Об'єднаний вихід: 65 г (89%) 10 UA 99702 C2 5 10 15 20 25 30 35 40 45 50 55 60 Приклад 4 Одержання (3R,4S)-4-[4-(трет-бутилдиметилсиланілокси)феніл]-1-(4-фторфеніл)-3-(2-[2-(4фторфеніл)-[1,3]діоксалан-2-іл]етилу}азетидин-2-ону (IX, R4=TBDMS) 20,25 г (28 ммоль) сполуки формули Vllla суспендують у 556 мл безводного ацетонітрилу при 20-25ºC, потім додають 13,6 мл (56 ммоль) N,O-біс-(триметилсиліл) ацетаміду. Реакційну суміш перемішують при 20-25°C протягом 2 годин, потім додають 0,1 г (0,28 ммоль) тригідрату тетрабутиламонійфториду і додатково перемішують при тій самій температурі. В кінці реакції (0,5-1 год.) суспензія стає прозорим розчином. Реакцію контролюють аналітично тонкошаровою хроматографією. Коли зникає пляма вихідної речовини, аміносполуки з лінійним ланцюгом (Vllla), реакційну суміш розбавляють 556 мл води і 556 мл н-гексану. Після розділення фаз водну ацетонітрильну фазу екстрагують 556 мл н-гексану. Об'єднану н-гексанову фазу сушать безводними Na2SO4, дегідратуючий агент відфільтровують і фільтрат упарюють у вакуумі. Отриманою таким чином сполуку (IXa) є масло, яке використовують без подальшого очищення на наступній стадії реакції. 1 Дані H ЯМР: (500 МГц, DMSO-d6, 25ºC) δ (м.д.) 0,16 (с, 3H), 0,16 (с, 3H), 0,92 (с, 9H), 1,701,82 (м, 2H), 1,89-2,09 (м, 2H), 3,07 (тд, J= 7,7, 2,3 Hz, IH), 3,62-3,72 (м, 2H), 3,91-4,01 (м, 2H), 4,85 (д, J = 2,3 Hz, IH), 6,80-6,86 (м, 2H), 7,07-7,22 (м, 6H), 7,24-7,29 (м, 2H), 7,38-7,43 (м, 2H) м.д. Регенерація S(+)-4-феніл-2-оксазолідинону, поверненого як побічний продукт з водної ацетонітрильної фази: Ацетонітрільну водну фазу, отриману, як описано вище, концентрують до об'єму приблизно в 500 мл і продукт, осаджений з розчину, що залишився, екстрагують дихлорметаном (2x100 мл). Об'єднаний дихлорметановий розчин упарюють, осад кристалізують із суміші етилацетату і н-гексану. Регенерований S(+)-4-феніл-2-оксазолідинон виділяють фільтруванням. Вихід: приблизно 3,9 г (приблизно 85%, з розрахунку на введений Vllla). Приклад 5 Одержання (3R,4S)-4-[4-(трет-бутилдиметилсиланілокси)фенілу]-1-(4-фторфеніл)-3-[3-(4фторфеніл)-3-оксопропіл]азетидин-2-ону (X, R4=TBDMS) Приблизно 17 г сполуки, одержаної за прикладом 4 (IX, R4=TBDMS) (з умістом, щонайменше: 15,8 г, 28 ммоль), розчиняють у 330 мл дихлорметану і додають при 20-25°C 42 г монтморилоніту K10. Гетерогенну суміш перемішують при 20-25ºC протягом 2-4 годин. Реакцію контролюють аналітично тонкошаровою хроматографією. Після зникнення плями вихідного матеріалу на хроматограмі реакційну суміш фільтрують, відфільтрований монтморилоніт K10A спочатку промивають 50 мл дихлорметану, а потім 3x50 мл сумішшю дихлорметану й метанолу (2:1 об./об.). Об'єднаний фільтрат упарюють, осад кристалізують з суміші етанолу і води при 0ºC. Вихід: 11,6 г висушеного продукту (80%, об'єднані стадії 4 і 5.) Точка плавлення: 110-112°C 25 [α] D =+4,0°, (c=l, дихлорметан) 1 Дані H ЯМР: (500 МГц, DMSO-d6, 25°C) δ 0,16 (с, 3H), 0,17 (с, 3H), 0,93 (с, 9H), 2,12-2,23 (м, 2H), 3,14-3,30 (м, 3H), 4,99 (д, J = 2,3 Hz, 1H), 6,81-6,88 (м, 2H), 7,10-7,18 (м, 2H), 7,20-7,27 (м, 2H), 7,29-7,38 (м, 4H), 7,99-8,07 (м, 2H) м.д. Приклад 6 Одержання (3R,4S)-4-[4-(трет-бутилдиметилсиланілокси)феніл]-1-(4-фторфеніл)-3-[(S)-3-(4фторфеніл)-3-гідроксипропіл]азетидин-2-ону (XIa) 5,00 г (9,6 ммоль) (3R,4S)-4-[4-(трет-бутилдиметилсиланілокси)фенілу]-1-(4-фторфеніл)-3-[3(4-фторфеніл)-3-оксопропіл]азетидин-2-ону розчиняють у 9,6 мл дихлорметану, що не містить воду, і потім додають 0,5 М толуольний розчин 1,92 мл (0,96 ммоль) (R)-o-толіл-CBSoксазаборолідину. Суміш охолоджують до температури в діапазоні від 0 до -5C і при цій температурі протягом 6 годин додають 1,9 мл 1,0 M дихлорметанового розчину борандиметилсульфіду. Реакційну суміш перемішують при цій температурі, поки за даними тонкошарової хроматографії не зникне пляма вихідного кетону. Потім додають 10 мл метанолу, 0,5 мл 5%-ного розчину пероксиду водню і 10 мл 2M сірчаної кислоти. Після перемішування цієї суміші протягом 0,5 години фази розділяють. Органічну фазу промивають 50 мл 2N сірчаної кислоти і потім 50 мл 5%-ного розчину сульфіту. Розчин сушать над безводним сульфатом натрію, фільтрують і упарюють. Вихід: 5,05 г безбарвного масла. Діастереомерний надлишок: >98% de (хіральна ВЕРХ) 1 Дані H ЯМР: (500 МГц, DMSO-d6, 25ºC) δ 0,17 (с, 3Н), 0,18 (с, 3Н), 0,93 (с, 9Н), 1,65-1,94 (м, 4H), 3,07-3,15 (м, 1H), 4,46-4,54 (м, 1H), 4,88 (д, J = 2,3 Hz, 1H), 5,29 (д, J = 4,5 Hz, 1H), 6,83-6,89 (м, 2H), 7,07-7,17 (м, 4H), 7,19-7,25 (м, 2H), 7,27-7,34 (м, 4H) м.д. Приклад 7 11 UA 99702 C2 5 10 15 20 Одержання (3R,4S)-1-(4-фторфеніл)-3-[(S)-3-(4-фторфеніл)-3-гідроксипропіл]-4-(4гідроксифенілу)азетидин-2-ону (I, езетимібу) 5,0 г (9,6 ммоль) (3R,4S)-4-[4-(трет-бутилдиметилсиланілокси)феніл]-1-(4-фторфеніл)-3-[(S)3-(4-фторфеніл)-3-гідроксипропіл)азетидин-2-ону (XI, R4=TBDMS) розчиняють у 35 мл 2пропанолу й додають 10 мл 2M сірчаної кислоти. Розчин нагрівають при 60-70ºC протягом 1-2 годин і потім йому дають охолодитися. Продукт кристалізують додаванням деіонізованої води. Кристалічний продукт відфільтровують і промивають водою до нейтральної реакції. Вихід: 3,2 г (81%, об'єднані стадії 7 і 8.) 1 Дані H ЯМР: (500 МГц, DMSO-d6, 25ºC) δ 1,65-1,92 (м, 4H), 3,05-3,13 (м, 1H), 4,46-4,55 (м, 1H), 4,81 (д, J= 2,3 Hz, 1H), 5,29 (д, J = 3,7 Hz, 1H), 6,74-6,80 (м, 2H), 7,08-7,17 (м, 4H), 7,19-7,26 (м, 4H), 7,28-7,35 (м, 2H), 9,54 (м, 1H) м.д. Приклад 8 Одержання (E)-[4-(трет-бутилдиметилсиланілоксибензиліден]-(4-фторфеніл)аміну (Vlla) 21,5г (0,1 моль) (E)-(4-гідроксибензиліден]-(4-фторфеніл)аміну (VIIb) розчиняють у 125 мл безводного тетрагідрофурану, до розчину додають 10,2 г (0,15моль) імідазолу, і потім до нього додають по краплях 40 мл тетрагідрофуранового розчину 18,8г (0,125 моль) третбутилдиметилсилілхлориду при 20-25ºC. Реакційну суміш перемішують при цій температурі до зникнення в реакційній суміші, за даними тонкошарової хроматографії, вихідного матеріалу. Очікуваний час реакції складає 1-2 години. Реакційну суміш розбавляють 50 мл толуолу і виливають її в 100 мл води. Водну фазу екстрагують 50 мл толуолу і потім об'єднану органічну фазу промивають 3 x 50 мл води до нейтральної реакції. Розчин упарюють і продукт кристалізують з холодного н-гексану. Вихід: 28 г (85%). ФОРМУЛА ВИНАХОДУ 25 1. Спосіб одержання езетимібу формули І OH OH N F O F 30 , (І) в якому здійснюють наступні стадії: a) перетворення етиленгліколевого ефіру 4-(4-фторбензоїл)масляної кислоти формули II на 4[2-(4-фторфеніл)-[1,3]діоксалан-2-іл]масляну кислоту формули IV через невиділену проміжну сполуку формули III O O O O O O OH II 35 O O OH F F F O OH III IV; b) ацилювання хіральної сполуки формули V сполукою формули IV з одержанням ацилованої похідної оксазолідинону формули VI 12 UA 99702 C2 O HN O R3 R1 O O O O O O R2 N V OH O F F O R3 R1 IV R2 VI, де сполуку формули V вибирають зі сполук формул Va, Vb, Vc або Vd O O O HN HN O H H Ph Ph Ph , 10 HN O H3C , Vb Ph O Ph H Ph Va 5 HN O O Ph , , Vc Vd і де R1, R2 і R3 є: у разі Va: R1=Ph, R2=R3=H, у разі Vb: R1=R2=R3=Ph, у разі Vc: R1=метил, R2=Ph, R3=H, у разі Vd: R1=ізопропіл, R2=R3=Ph, і де Ph є фенільною групою; с) взаємодію ацилованої похідної оксазолідинону формули VI із захищеною іміносполукою формули VII і виділення сполуки формули VIII, де R4 є силільною групою F O O O O O N F R1 R4 O + O O N R3 R3 R2 O HN N R2 R1 F O O R4 O F VI 15 VII VIII, циклізацію сполуки формули VIII з одержанням захищеної похідної азетидинону загальної формули IX F O O O R3 O HN O O N R2 R1 O O R4 F N O F O F 13 R4 UA 99702 C2 VIII IX; d) гідроліз кетальної групи сполук формули IX з одержанням сполуки формули X O O O O R4 N F N F O O F F IX 5 X; е) енантіоселективне відновлення сполуки загальної формули X з одержанням сполуки формули XI O O R4 O OH N F R4 N F O O F F X 10 R4 O XI, де одна з хіральних CBS-оксазаборолідинових сполук формул ХІІа, ХІІb, ХІІс і XIId вибрана як каталізатор N O B H Ph Ph H Ph Ph H Ph Ph N CH3 O CH3 O N B H Ph Ph HN B n , Bu , O B CH3 ,і , XIIa XIIb XIIc XIId f) видалення силільної захисної групи у сполуці загальної формули XI з одержанням кінцевого продукту езетимібу формули І 15 O OH N F OH R4 OH N F O O F F I XI 2. Спосіб одержання сполуки формули VIIIa 14 . UA 99702 C2 F O O O HN N Si O Ph O O F , (VIIIa) в якому здійснюють ізомеризацію сполуки формули VIIIb у присутності похідної Ti(IV), F O O O F HN O N O O HN N Si Si O Ph Ph O O F VIIIb VIIIa . 3. Сполука формули III O O O OR F , (III) де R є групою -О-СН2-СН2-ОН або -Н. 4. Сполука формули VI O O O O N F 10 15 O O F 5 O R1 O R2 R3 , (VI) де R1, R2 i R3 є: у разі Va: R1=Ph, R2=R3=H, у разі Vb: R1=R2=R3=Ph, у разі Vc: R1=метил, R2=Ph, R3=H, у разі Vd: R1=ізопропіл, R2=R3=Ph, і де Ph є фенільною групою. 5. Сполука формули VIII 15 UA 99702 C2 F O O O HN N R4 R3 R2 O R1 O O F , де значення R1, R2, R3 незалежно є Va, Vb, Vc або Vd, і R4 є силільною захисною групою. 6. Сполука загальної формули IX O R4 O O N F O F , де R4 є силільною захисною групою. 7. Сполука формули Ха 5 O Si O N F O F . 8. Сполука формули ХІа O Si OH N F O F . 10 Комп’ютерна верстка Г. Паяльніков Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 16
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for the production of ezetimibe and intermediates used in this process
Автори англійськоюBodi, Jozsef, Eles, Janos, Szoke Katalin, Vukics Krisztina, Gati Tamas, Temesvari Krisztina, KISS-BARTOS DOROTTYA
Назва патенту російськоюСпособ получения эзетимиба и промежуточных продуктов, используемых в этом способе
Автори російськоюБоди Йожеф, Элеш Янош, Секе Каталин, Вукич Кристина, Гати Тамаш, Темешвари Кристина, Кишш-Бартош Дороттья
МПК / Мітки
МПК: C07F 7/02, C07D 317/30, C07D 413/06, C07D 405/06, C07D 205/08
Мітки: продуктів, одержання, використовуваних, способи, проміжних, цьому, спосіб, езетимібу
Код посилання
<a href="https://ua.patents.su/18-99702-sposib-oderzhannya-ezetimibu-jj-promizhnikh-produktiv-vikoristovuvanikh-u-comu-sposobi.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання езетимібу й проміжних продуктів, використовуваних у цьому способі</a>
Спосіб одержання анілінів, застосування аміаку в цьому способі та спосіб амінування

Номер патенту: 86284
Опубліковано: 10.04.2009
Автори: Вальтер Харальд, Ламберт Клеменс, Тоблер Ханс, Корсі Камілла, Еренфройнд Йозеф
МПК: C07C 211/45, C07C 209/10
Мітки: цьому, застосування, аміаку, одержання, амінування, способи, спосіб, анілінів
Формула / Реферат:
1. Спосіб одержання сполуки формули І, (I)у якій R1, R2 і R3 всі незалежно один від іншого означають водень або метил, який відрізняється тим, що сполуки формули II, (II)у якій R1, R2 і R3 є такими, як визначено для формули І, і Х означає бром або хлор, вводять у реакцію з...
Спосіб одержання біологічно активного похідного 1,2,4-триазолу і проміжні сполуки, які використовуються у цьому способі

Номер патенту: 64712
Опубліковано: 15.03.2004
Автор: Паломо Колл Альберто
МПК: C07C 241/00, C07C 243/00, C07D 249/08
Мітки: спосіб, сполуки, 1,2,4-триазолу, біологічно, одержання, похідного, проміжні, способи, активного, використовуються, цьому
Спосіб одержання 5-[2-етокси-5-(4-метилпіперазин-1-іл-сульфоніл)-феніл]-1-метил-3-н-пропіл-1,6-дигідро-7н-піразол-[4,3-d]-піримідин-7-ону та способи одержання проміжних продуктів

Номер патенту: 78692
Опубліковано: 25.04.2007
Автори: Шріканде Атул Анант, Моді Шіріш Бхагванлал, Доші Мадхукант Мансукхлал
МПК: C07D 237/00
Мітки: 5-[2-етокси-5-(4-метилпіперазин-1-іл-сульфоніл)-феніл]-1-метил-3-н-пропіл-1,6-дигідро-7н-піразол-[4,3-d]-піримідин-7-ону, проміжних, способи, одержання, продуктів, спосіб
Формула / Реферат:
1. Cпосіб одержання 5-[2-етокси-5-(4-метилпіперазин-1-ілсульфоніл)-феніл]-1-метил-3-н-пропіл-1,6-дигідро-7Н-піразол-[4,3-d]-піримідин-7-ону (V) та його фармацевтично прийнятних солей, який включає такі стадії:a) взаємодія 1-метил-4-аміно-3-н-пропілпіразол-5-карбоксаміду (І) із хлористим воднем у розчиннику, такому як ізопропанол, з одержанням 1-метил-4-аміно-3-н-пропілпіразол-5-карбоксамідгідрохлориду (II);b) взаємодія сполуки...
Способи одержання проміжних продуктів пестицидів

Номер патенту: 69451
Опубліковано: 15.09.2004
Автори: Перрен-Жане Жилль, Ансель Жан-Ерік, Верспрумі П'єр, Ванжелісті Манюель
МПК: C07D 231/44, C07C 211/45, C07C 211/52, C07D 213/73, C07C 209/42, C07B 61/00
Мітки: пестицидів, одержання, продуктів, способи, проміжних
Формула / Реферат:
1. Спосіб одержання сполуки формули (І) , (I)в якій R1 являє собою галогеналкіл, галогеналкокси або -SF5; W являє собою N або CR3; і R2 і R3 кожний незалежно являє собою водень або хлор; або її кислотно-адитивної солі; який передбачає гідрогеноліз сполуки формули (II) (II)або...
Спосіб одержання оцтової кислоти та застосування індію, кадмію, ртуті, галію, цинку як стабілізатора в цьому способі

Номер патенту: 79316
Опубліковано: 11.06.2007
Автори: Кі Леслі Енн, Пул Ендрю Девід, Пейн Марк Джон
МПК: C07C 53/08, C07C 51/12
Мітки: кадмію, індію, стабілізатора, способи, цьому, оцтової, кислоти, цинку, застосування, ртуті, одержання, галію, спосіб
Формула / Реферат:
1. Спосіб одержання оцтової кислоти карбонілюванням метанолу і/або його реакційноздатної похідної монооксидом вуглецю, який відрізняється тим, що його здійснюють в щонайменше одній реакційній зоні карбонілювання, що містить рідку реакційну композицію, яка включає іридієвий каталізатор карбонілювання, метилйодидний співкаталізатор, воду в обмеженій концентрації, оцтову кислоту, метилацетат, щонайменше один промотор, вибраний з рутенію, осмію...
Попередній патент: Людське моноклональне антитіло, яке специфічно зв’язується з лігандом-1 запрограмованої загибелі клітин (pd-l1)
Наступний патент: Застосування імуногенної композиції streptococcus pneumoniae
Випадковий патент: Спосіб одержання біциклічних складних ефірів або їх фармацевтично прийнятних солей