Спосіб одержання n-(beta-гідроксіетил) нікотинаміду
Номер патенту: 10092
Опубліковано: 30.09.1996
Автори: Короткий Юрій Васильович, Дзвінчук Ігор Борисович, Лозинський Мирон Онуфрійович, Красавцев Іван Іванович


Формула / Реферат
Способ получения N-(![]() -гидроксиэтил)никотинамида формулы (I)
-гидроксиэтил)никотинамида формулы (I)
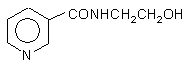
взаимодействием производных пиридинкарбоновых кислот с моноэтаноламином при нагревании с последующим выделением целевого продукта кристаллизацией из хлороформа, отличающийся тем, что в качестве производного пиридинкарбоновых кислот используют никотоновую кислоту, которую подвергают взаимодействию с моноэтаноламином при молярном соотношении, равном 1:3, соответственно, при температуре 170-180 0С в течение 8-10 часов с одновременной отгонкой реакционной воды.
Текст
Способ получения N-( pгидроксиэтил)никотинамида формулы (I) Изобретение относится к области амидов органических кислот, конкретно, к способу получения М-(Р-гидроксиэтил)никотинамида формулы: CNCH2CH2OH который используется в качестве промежуточного продукта в процессе синтеза лекарственного препарата никорандил [1]. Указанный амид упоминается в патенте [2], где описан способ его получения. Он заключается во взаимодействии никотиновой кислоты с этанолом в присутствии каталитических количеств серной кислоты. Образующийся этиловый эфир никотиновой кислоты превращают в амид путем нагревания с моноэтаноламином с отгонкой этилового спирта. Целевой продукт выделяется кристаллизацией из хлороформа с выходом 87,7%. Весь описанный процесс идет в 2 стадии по схеме соон 1. + сн.он t,H ? SO 4 CONHCH,CH,OH N взаимодействием производных пиридинкарбоновых кислот с моноэтаноламином при нагревании с последующим выделением целевого продукта кристаллизацией из хлороформа, отличающийся тем, что в качестве производного пиридинкарбоновых кислот используют никотоновую кислоту, которую подвергают взаимодействию с моноэтаноламином при молярном соотношении, равном 1:3, соответственно, при температуре 170-180 °С в течение 8-10 часов с одновременной отгонкой реакционной воды. ©1» s~~ 2 і ,CNCH,CH,OH (і) (87 7%) Описанный в патенте [2] способ получения целевого продукта имеет следующие недостатки: 1) Многостадийность технологии синтеза целевого продукта, состоящей из двух стадий. 2) Трудоемкость выделения эфира никотиновой кислоты 3) Использование серной кислоты на стадии получения эфира никотиновой кислоты Описан способ получения амида через хлорангидрид никотиновой кислоты [33] при взаимодействии последнего с моноэтаноламином ,соон COCL + SOCU HCL ONHCH,CH,OH (IV) + HjNCHXH.OH Данный способ имеет следующие недостатки: 1) использование на первой стадии токсичного реагента - хлористого тионила, 2) трудоемкость выделения целевого амида на второй стадии, 3) неустойчивость хлорангидрида никотиновой кислоты в обычных условиях. см о см а> о о 10092 Известно также, что амиды кислот получают снижается до 167°С. Через 20 - 30 минут начинает путем прямого действия на кислоты алифатичеотгоняться вода (с незначительной примесью моского ряда различных аминов, в том числе и мононоэтаноламина). Температура отгона в парах не этаноламина [4]. С ароматическими карбоновими должна превышать 120°С. В процессе отгонки кислотами эта реакция идет плохо, а с аминокисводы постепенно повышается температура кипелотами типа никотиновой - вообще не описана. ния реакционной смеси, пока не достигает через 2 Потому она может рассматриваться как отдален- 3 часа 180°С. При более высоких температурах ный прототип предлагаемого изобретения. образуются побочные продукты. Всего в опыте Задачей настоящего изобретения является отгоняется 45 - 53мл водной фракции, причем упрощение процесса получения, увеличения выосновное количество -в первые 3 часа кипячения. хода и качества оксиэтиламида (1), а также улучРеакция заканчивается через 8 - 1 0 часов. Реакшение экологических параметров. ционной смеси дают остыть до 90"С и отгоняют из Поставленная задача решается осуществленее избыток моноетаноламина при 10 - 12мм рт. нием предлагаемого способа, сущность которого ст., отбирая фракцию в интервале 80 - 100°С. Гозаключается в нагревании смеси моноэтаноламирячий остаток переливают из реактора в стакан на с никотиновой кислотой без катализатора при емкостью 0,8 - 1,0л, дают ему остыть до 70 - 80"С температуре 170 - 160"С с одновременной отгони разбавляют при перемешивании 0,5мл хлорокой реакционной воды при атмосферном давлеформа. Раствор охлаждают до 40°С и вызывают нии кристаллизацию. Раствор оставляют на 3 - 4 часа /\.ООСН /^/ХХЖ^СНрН при 20 - 25% а затем выдерживают 3 - 4ч. при 5°С. Выкристаллизовавшийся продукт отфильтровырґ +шп\р\р\ 17f>180°» Ю вают, промывают хлороформом (330мл) и сушат V на воздухе. Выход 229 - 250г (55 - 60%). (О Для более полного извлечения продукта фильтрат встряхивают в делительной воронке с Существенными признаками заявляемого спо50мл воды. Отслоившийся водный раствор отдесоба являются нагревание смеси монозтаноламиляют и экстрагируют кипящим хлороформом в на и никотиновой кислоты в соотношении 3 : 1 экстракторе непрерывного действия. Дополни(мольн.) при температуре 170 - 180°С с одновретельно получают 79г продукта. Общий выход 308 менной отгонкой выделяющейся реакционной во329г (74 - 79%). ды. Найдено %: С 57,95; Н 10,12; 16,78, Т. пл. 91 Техническим результатом осуществленного 92°С способа являются: сведение процесса получения Вычислено %: С 57,82;Н 10,07; Н 16,85. амида к одной стадии и экологическая чистота ИК спектр (КВг, см"1): 1640 (С = 0), 3260 (NH, технологии при высоком выходе и качестве целе3330 (ОН). вого продукта, экономия сырья и сбережение ПМР спектр (ацетон 6 ) . м 3,48 - 3,74 (4Н, энергоресурсов СН2СН2), м. 4,50 (ОН), м. 7,34 (1Н), м. 7,51 (NH), м. Пример 1 Получение N-(P8,01 (1Н), м. 8,51 (1Н), м. 8,84 (1Н). гидроксиэтил)никотинамида Контроль протекания реакции и чистоту проСмесь 308г (2,5моля) никотиновой кислоты и дукта осуществляли, с помощью высокоэффек447мл (7,5моля моноэтаноламина в трехгорлом тивной жидкостной хроматографии. реакторе (емкостью 1,0л), снабженном термометСписок использованной литературы ром, а также дефлегматором (высотой 25см) с 1 Патент США 424/266 №4200640. РЖ хим 1982 2 насадкой Вюрца с термометром, алонжем и при86П емником, кипятят при нагревании на масляной 2 Chem. Abstr. 1984, V 100 138906g, Pharm. Ind. бане 10 часов. Температура в нижней части удер1983 №11.2-3 живается при 200 - 205°С. Реактор погружают в 3 Span. ES 536,328 (C07D 211/60). C.A. 1986 баню так, чтобы уровень реакционной смеси был 190955X ниже уровня масла в бане на 3 - 5см. Вначале 4 К.Бюлер, Д.Пирсон,-Органические синтезы, TOW реакционная смесь прогревается до 170 - 173"С, 2, "Мир", Л, 1978, с. 384 (прототип) затем, по мере образования воды, температура ДП «Український інститут промислової власності» (Укрпатент) вуя Сім'ї Хохлових, 15, м. Київ, 04119, Україна (044)456-20- 90 ТОВ "Міжнародний науковий комітеТ' вул Артема, 77, м Київ, 04050, Україна (044)216-32-71
ДивитисяДодаткова інформація
Автори англійськоюKorotkyi Yurii Vasyliovych, Krasavtsev Ivan Ivanovych, Dzvinchuk Ihor Borysovych, Lozynskyi Myron Onufriovych, Lozynskyi Myron Onufriiovych
Автори російськоюКороткий Юрий Васильевич, Красавцев Иван Иванович, Дзвинчук Игорь Борисович, Лозинский Мирон Онуфриевич
МПК / Мітки
МПК: C07C 231/00, C07C 7/00
Мітки: n-(beta-гідроксіетил, спосіб, одержання, нікотинаміду
Код посилання
<a href="https://ua.patents.su/2-10092-sposib-oderzhannya-n-beta-gidroksietil-nikotinamidu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання n-(beta-гідроксіетил) нікотинаміду</a>
Beta-[beta’-(2,4-ді-трет-амілфенокси)-етокси] етилхлорид як напівпродукт для одержання речовин, які мають антимікробні властивості

Номер патенту: 10153
Опубліковано: 30.09.1996
Автори: Свертілова Ізабела Олександрівна, Ютілов Юрій Михайлович
МПК: A61P 31/04, C07C 39/00, A61K 31/02
Мітки: антимікробні, мають, властивості, речовин, напівпродукт, одержання, beta-[beta'-(2,4-ді-трет-амілфенокси)-етокси, етилхлорид
Формула / Реферат:
b-b'-(2,4-Ди-трет-амилфенокси)-этокси]-этилхлорид формулы в качестве полупродукта для получения веществ, обладающих антимикробными свойствами.
Спосіб одержання сірчанокислого ефіру 4-beta-оксиетилсульфоніланіліну

Номер патенту: 5795
Опубліковано: 29.12.1994
Автори: Вугеншмідт Валерій Григорович, Беляєв Володимир Львович, Красюк Іван Іванович, Дєєв Олександр Сергійович, Доценко Лариса Сергіївна, Золова Тамара Андріївна, Коротенко Таміла Олександрівна
МПК: C07C 303/00, C07C 211/46
Мітки: 4-beta-оксиетилсульфоніланіліну, ефіру, одержання, сірчанокислого, спосіб
Формула / Реферат:
Способ получения сернокислого эфира 4-b-оксиэтилсульфониланилина восстановлением N-замещенного n-аминобензолсульфохлорида сульфитом натрия при 30-35°С в щелочной среде с последующим оксиэтилированием этиленхлор-гидрином соответствующей сульфиновой кислоты при нагревании и рН 6,9-7 с дальнейшим использованием стадий гидролиза в присутствии минеральной кислоты при нагревании и этерификации концентрированной серной кислотой при...
П-толуолсульфонат 5-метокси-6-метилтіо-2-(2, 5, 5триметил-3-етокси-2-циклогексеніліден)-метил-3етилбензотіазолія як проміжний продукт для синтезу барвників, спосіб його одержання та способи одержання барвників

Номер патенту: 2560
Опубліковано: 26.12.1994
Автори: Толмачов Олексій Іванович, Вальшин Галі Карімович, Сломінський Юрій Леонідович, Романов Микола Миколайович, Чепелева Лариса Василівна
Мітки: спосіб, синтезу, п-толуолсульфонат, барвників, 5триметил-3-етокси-2-циклогексеніліден)-метил-3етилбензотіазолія, способи, проміжний, одержання, 5-метокси-6-метилтіо-2-(2, продукт
Формула / Реферат:
(57) - Толуолсульфонат 5-метокси-6-метклтио-2-(2,5,5-триметил-3-этокси-2-циклогексенилиден)-метил-3-этилбензотиазолия формулы 12. Способ получения n-толуолсульфоната 5-метокси-6-метилтио-2-(2,5,5-триметил-3-этокси-2-циклогексенилиден)-метил-3-этилбензотиазолия формулы Іотличающийся тем, что n-толуолсульфонат 2-метил-5-метокси -6-метилтио-3 -этилбензотиазолия формулы ІІподвергают взаимодействию с...
Спосіб одержання beta-галактозидази

Номер патенту: 2463
Опубліковано: 26.12.1994
Автори: Черних Світлана Ігорівна, Коробко Вячеслав Григорович, Кордюм Віталій Арнольдович, Славченко Ірина Юріївна
МПК: C12N 15/56
Мітки: beta-галактозидази, одержання, спосіб
Формула / Реферат:
Способ получения b-галактозидазы путем заражения штамма бактерий ЕксЬепсЫа соіі фагом с геном b-галактозидазы и с мутациями, задерживающими лизис клеток, l рlаsС1857Qаm117Ram54 с последующим культивированием штамма, отличающийся тем, что, с целью повышения вы-хода b-галактозидазы, фагом заражаютштамм, содержащий плазмиду рZ56 с геном b-галактозидазы, а культивирование проводят при 37 °С в течение 7—12 ч.
Спосіб одержання діоксіду свинцю beta-модифікації

Номер патенту: 6188
Опубліковано: 29.12.1994
Автори: Зіньков Валерій Миколайович, Гордєєв Валерій Іванович, Фельдман Володимир Давидович, Кисіль Микола Григорович, Корнишева Галина Олександрівна, Крапівіна Валентина Яківна, Бобир Наталія Федорівна, Абакумова-Бойченко Ірина Миколаївна
МПК: C01G 21/00
Мітки: спосіб, свинцю, beta-модифікації, діоксиду, одержання
Формула / Реферат:
(57) Способ получения диоксида свинца /J-модификации, включающий окисление соли свинца персульфатом аммония или натрия, или калия в щелочной среде при нагревании, отделение, промывку и сушку осадка, отличающийся тем, что в качестве соля свинца используют сульфат или нитрат свинца, перед окислением осуществляют взаимодействие соли свинца со щелочью при температуре 55-65°С, а окисление полученной смеси проводят при температуре 85-95°С при...
Попередній патент: Полімерна композиція для еластичних матеріалів
Наступний патент: Спосіб лікування захворювань товстої кишки
Випадковий патент: Біологічний спосіб очищення води і грунту на дні водоймищ від нафти та нафтопродуктів