Композиція трамадолу з пролонгованим вивільненням з 24-годинною дією
Номер патенту: 84277
Опубліковано: 10.10.2008
Автори: Бейкон Джонатан, Оуаджі-Нджикі Патрісія Лаура, Узеруру Рашид, Рахмуні Мілу, Жерве Соня, Сміт Деймон, Ленар Венсан











Формула / Реферат
1. Тверда дозована композиція, яка містить:
ядро, що містить активний фармацевтичний інгредієнт та матрицю, і
покриття, що містить фізичну суміш полівінілацетату, полівінілпіролідону та активного фармацевтичного інгредієнта,
де активний фармацевтичний інгредієнт та матриця ядра зв’язані таким чином, що вивільнення фармацевтичного інгредієнта з матриці ядра є контрольованим, при цьому вивільнення з композиції активного інгредієнта, який знаходиться у ядрі, відбувається повільніше, ніж вивільнення активного фармацевтичного інгредієнта, що знаходиться в матриці оболонки,
причому активний фармацевтичний інгредієнт ядра та оболонки є тим самим і являє собою трамадол або його стереоізомер або фармацевтично прийнятну сіль.
2. Композиція за п. 1, яка має наступну швидкість розчинення in vitro при вимірюванні за допомогою пристрою USP типу 1 при 50-150 об./хв. у 50 мМ фосфатному буфері при рН 6,8:
від 10 до 40 мас. % трамадолу вивільняється з композиції між 0 та 2 годинами вимірювання,
від 30 до 60 мас. % трамадолу вивільняється з композиції між 2 та 7 годинами вимірювання,
від 50 до 80 мас. % трамадолу вивільняється з композиції між 7 та 12 годинами вимірювання та
від 80 до 100 мас. % трамадолу вивільняється з композиції після 20 годин вимірювання.
3. Композиція за п. 1 або 2, яка при введенні однієї дози забезпечує середню концентрацію трамадолу в плазмі, яка складає щонайменше 100 нг/мл протягом 2 години після введення та продовжує забезпечувати середню концентрацію в плазмі щонайменше 100 нг/мл протягом щонайменше 22 годин після введення.
4. Композиція за будь-яким з пп. 1-3, в якій трамадол присутній у ядрі в кількості від 30 до 50 мас. % загальної маси ядра.
5. Композиція за будь-яким з пп. 1-4, в якій трамадол присутній в оболонці в кількості від 15 до 40 мас. %.
6. Композиція за будь-яким з пп. 1-5, в якій полівінілацетат, присутній в покритті, має молекулярну масу в діапазоні від 100000 до 1000000.
7. Композиція за будь-яким з пп. 1-6, в якій полівінілпіролідон, присутній в покритті, має молекулярну масу в діапазоні від 10000 до 100000.
8. Композиція за будь-яким з пп. 1-7, в якій оболонка додатково містить ксантан.
9. Композиція за будь-яким з пп. 1-8, в якій матриця ядра являє собою зшитий високоамілозний крохмаль.
10. Композиція за будь-яким з пп. 1-9, в якій трамадол, присутній в ядрі, має розчинність, більшу ніж 500 г/л.
11. Композиція за будь-яким з пп. 1-10, в якій трамадол, присутній в оболонці, має розчинність, більшу ніж 500 г/л.
12. Композиція за будь-яким з пп. 1-11, яка являє собою пероральну фармацевтичну композицію для контрольованого вивільнення трамадолу або його стереоізомера, або його фармацевтично прийнятної солі, що вводиться один раз на день, де при початковому введенні однієї дози композиція забезпечує середню концентрацію трамадолу в плазмі щонайменше 10 нг/мл протягом 2 годин після введення та продовжує забезпечувати середню концентрацію трамадолу в плазмі щонайменше 100 нг/мл протягом щонайменше 22 годин після введення.
13. Композиція за п. 12, де середня максимальна концентрація у плазмі (Cmax) є меншою, ніж середня концентрація у плазмі, отримана через 24 години після введення (C24h), взята 2,2 рази.
14. Таблетка, що містить композицію за будь-яким з пп. 1-13.
Текст
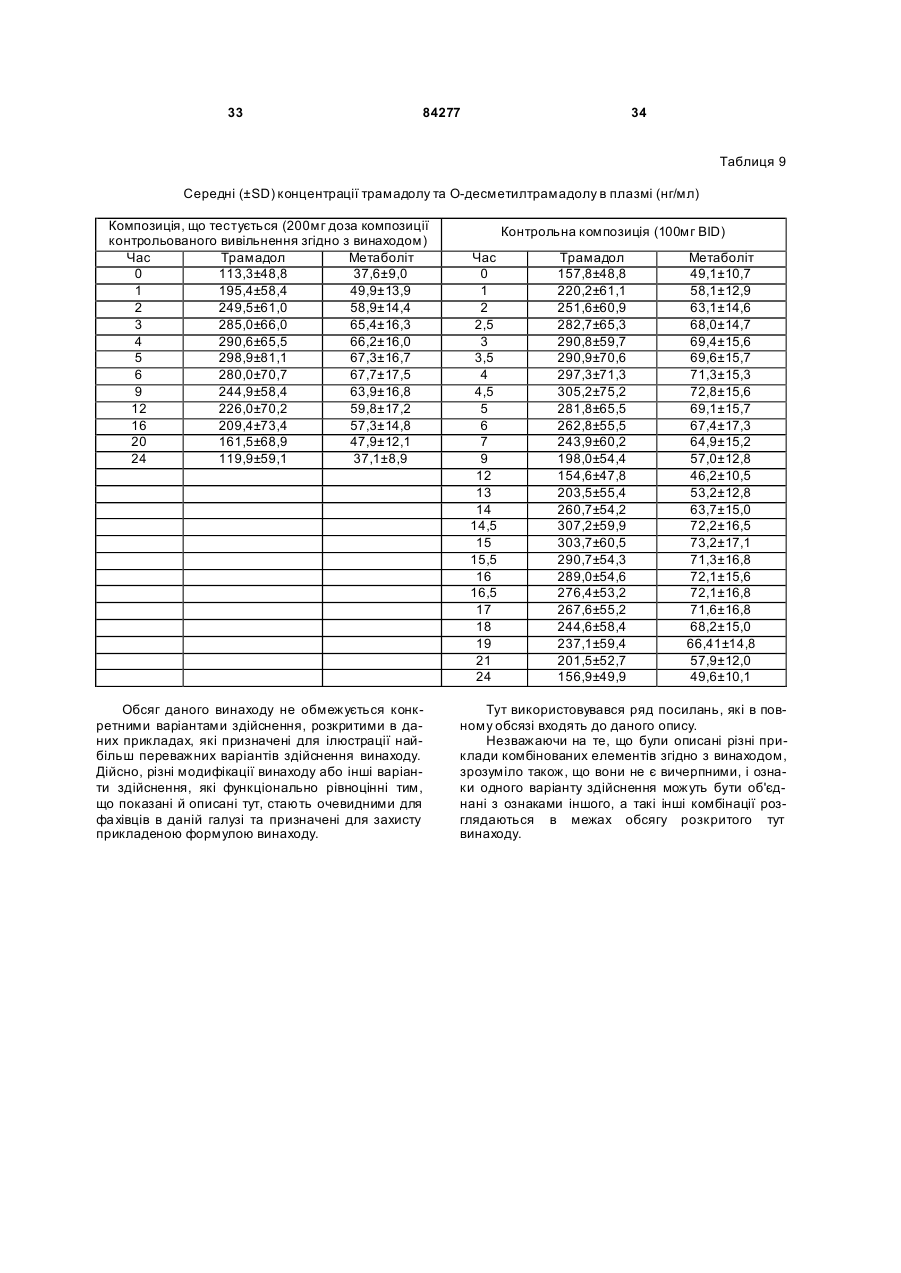
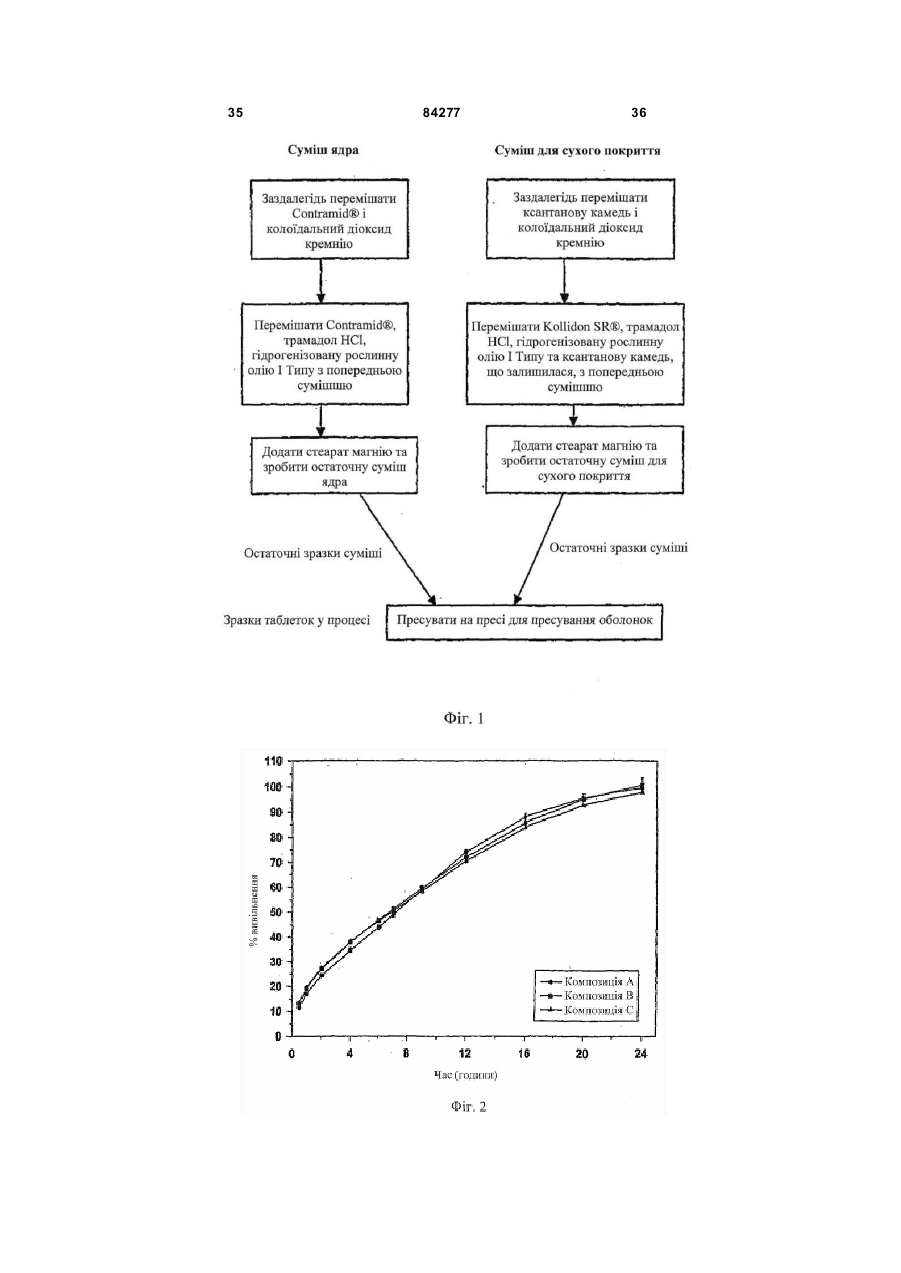
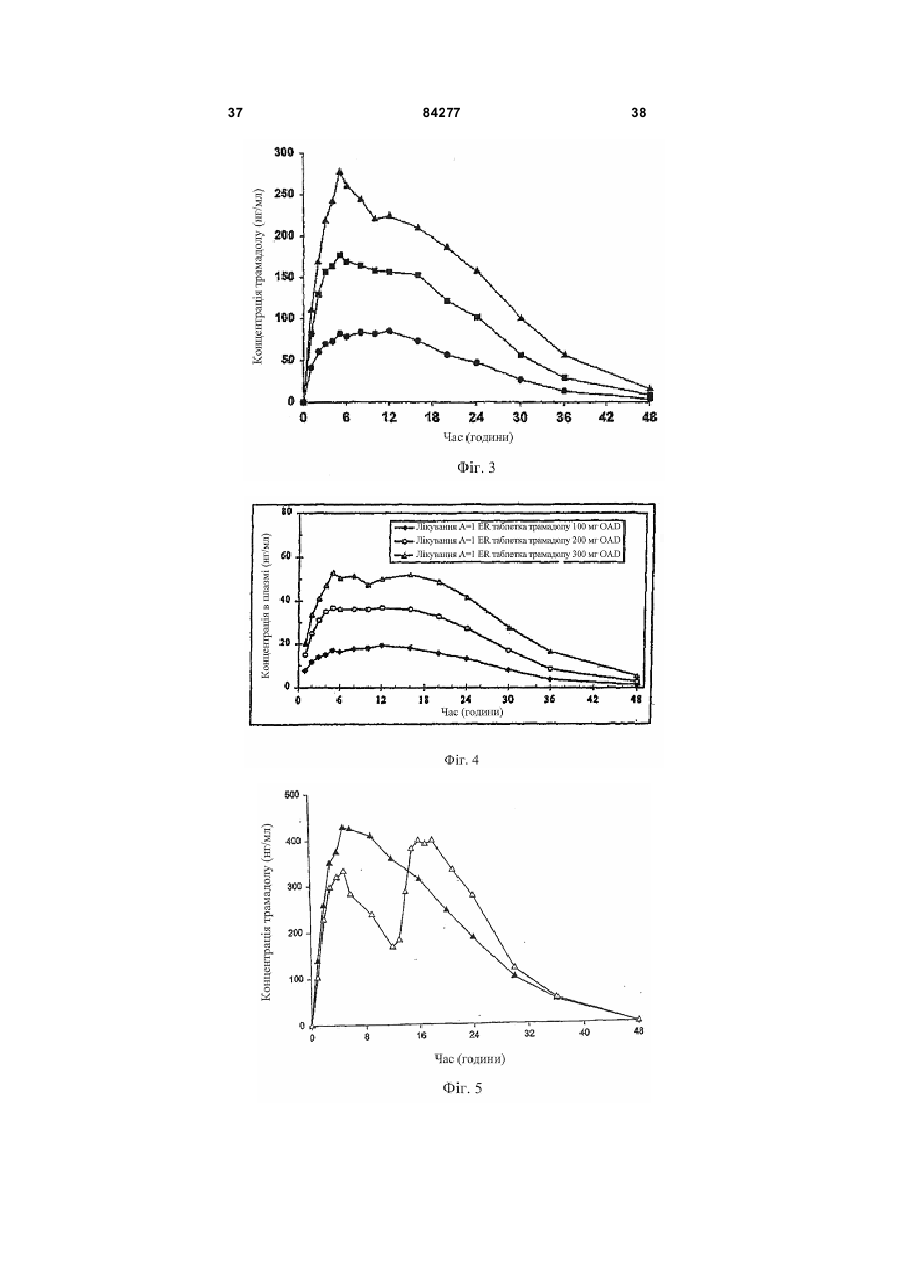
1. Тверда дозована композиція, яка містить: ядро, що містить активний фармацевтичний інгредієнт та матрицю, і покриття, що містить фізичну суміш полівінілацетату, полівінілпіролідону та активного фармацевтичного інгредієнта, де активний фармацевтичний інгредієнт та матриця ядра зв’язані таким чином, що вивільнення фармацевтичного інгредієнта з матриці ядра є контрольованим, при цьому вивільнення з композиції активного інгредієнта, який знаходиться у ядрі, відбувається повільніше, ніж вивільнення активного фармацевтичного інгредієнта, що знаходиться в матриці оболонки, причому активний фармацевтичний інгредієнт ядра та оболонки є тим самим і являє собою трама 2 (19) 1 3 84277 4 для контрольованого вивільнення трамадолу або його стереоізомера, або його фармацевтично прийнятної солі, що вводиться один раз на день, де при початковому введенні однієї дози композиція забезпечує середню концентрацію трамадолу в плазмі щонайменше 10нг/мл протягом 2 годин після введення та продовжує забезпечувати середню концентрацію трамадолу в плазмі щонайменше 100 нг/мл протягом щонайменше 22 годин після введення. 13. Композиція за п. 12, де середня максимальна концентрація у плазмі (Cmax) є меншою, ніж середня концентрація у плазмі, отримана через 24 години після введення (C24h), взята 2,2 рази. 14. Таблетка, що містить композицію за будь-яким з пп. 1-13. Галузь винаходу Даний винахід відноситься до нової, одноразового щоденного прийому, фармацевтичної композиції для контрольованого вивільнення трамадолу або його солі. Попередній рівень техніки Фармацевтичні композиції трамадолу Гідрохлорид трамадолу (НСl) був розроблений Grunental GmbH, Німеччина. Він продавався в Німеччині з 1977 (наприклад, Tramal™), у Сполучених Штата х як Ultram® з 1995. Ефективність і профіль безпеки трамадолу HCl робить його дуже придатним для пролонгованого лікування хронічного болю. Трамадол HCl є синтетичним аналгезуючим засобом центральної дії, ефективність якого була показана при різноманітних станах гострого та хронічного болю. Було показано, зокрема, що трамадол HCl, як у композиціях негайного, так і повільного вивільнення, у поєднанні з нестероїдними протизапальними лікарськими засобами (НПЗЗ) [Roth SH. "Efficacy and safety of tramadol HCl in breakthrough musculoskeletal pain attributed to osteoarthritis". J. Rheumatol 1998; 25:1358-1363. Wilder-Smith CH et al. "Treatment of severe pain from osteoarthritis with slow-release tramadol or dihydrocodeine in combination with NSAID's: a randomized study comparing analgesia, antinociception and gastrointestinal effects". Pain 2001; 91:23-31] зменшує біль, пов'язаний з остеоартритом (ОА). Після перорального введення трамадол HCl швидко і майже повністю абсорбується та ретельно метаболізується. Головними метаболічними шляхами є N- і О-деметилювання та глюкуронідування або сульфонування в печінці. Лише один метаболіт, моно-О-десметилтрамадол (М1), представляється фармакологічно активним, таким, що має приблизно в 200 разів вищу а фінність відносно m-опіоїдного рецептора, ніж рацемічний трамадол [DeJong R. "Comment on the hypoalgesic effect of tramadol in relation to CYP2D6" (comment) Pain Dig 1997; 7:245; Kogel B. et al "Involvement of metabolites in the analgesic action of tramadol" Proc. 9th World Congress on Pain, Vienna, 1999]. У здорових людей трамадол деметилюється поліморфним ензимом цитохромом Р450 2D6 (CYP2D6) до M1метаболіту. Ме ханізм дії трамадолу HCl не зрозумілий до кінця. На тваринних моделях показано, що лікарський засіб (і його активний М1-метаболіт) діє як опіоїдний агоніст, ймовірно, шляхом селективної активності відносно m-рецептора. У доповнення до активності опіоїдного агоніста, трамадол HCl інгібує зворотне захоплення деяких моноамінів (но репінефрин, серотонін), які, виявляється, сприяють аналізуючому ефекту лікарського засобу. Антиноцицептивний ефект трамадолу HCl лише частково антагонізується налоксоном у деяких тестах у тварин і людей. Крім того, внаслідок опіоїдної агоністичної активності цього лікарського засобу передбачалося, що трамадол HCl може викликати залежність; однак потенціал його зловживання виявляється низьким, і трамадол HCl не "підлягає контролю", згідно з Федеральним Актом Сполучених Штатів по Контролю Речовин 1970 як включений до списку лікарський засіб. Композиції трамадолу HCl негайного вивільнення добре відомі в даній галузі. Такі композиції, однак, вимагають частого дозування, щоб забезпечити ефективне полегшення болю. Недолік відповідності режимам високої частоти дозування може бути результатом невідповідних концентрацій лікарського препарату в плазмі і, отже, менш стійкого знеболення. Композиції щоденного дворазового прийому є більш корисними та бажаними у порівнянні з композиціями з негайним вивільненням, оскільки вони забезпечують більш тривалі періоди знеболення після введення та вимагають менш частого дозування. Композиції щоденного одноразового прийому є ще більш бажаними для підвищеної ефективності, безпеки та зручності. Вирішальним чинником, що впливає на швидкість абсорбції, і, отже, безпеку та ефективність активного лікарського інгредієнта після перорального введення в організм у таблетованій або іншій твердій лікарській формі, є швидкість вивільнення активного лікарського інгредієнта з лікарської форми після прийому всередину. Таким чином, здатність інгредієнтів лікарської форми контролювати швидкість вивільнення, яка складає основу для так званого контрольованого вивільнення, тривалого вивільнення, уповільненого вивільнення або для лікарських препаратів пролонгованої дії, які розроблені для створення повільного, рівномірного вивільнення та абсорбції активних лікарських інгредієнтів протягом періоду, що складає години, дні, тижні або місяці. Переваги таких композицій з контрольованим вивільненням включають у себе зменшення необхідної частоти введення лікарського засобу в порівнянні з загальноприйнятими лікарськими формами з негайним вивільненням, що часто призводить до поліпшення дотримання хворим режиму та схеми лікування; підтримка стабільної концентрації лікарського засобу в організмі, а отже, безперервний терапевтичний ефект протягом встановленого часу; і зменшення виникнення та вираженості небажаних побічних ефектів активного засобу, викликаних 5 84277 високими концентраціями в плазмі, які зустрічаються після введення лікарських форм з безперервним вивільненням. Було запропоновано та розроблено багато речовин як матрикс для контрольованого вивільнення активних лікарських інгредієнтів. До таких речовин відносяться, наприклад, полімерні матеріали, такі як полівінілхлорид, аміди поліетилену, етилцелюлоза, силікон і полі (гідроксиметилметакрилат). Див., наприклад, [Патент США №3087860 Endicott et al; Патент США №2987445 Lavesque et al.; Salomon et al. Pharm. Acta Helv., 55, 174-182 (1980); Korsmeyer, Diffusion Controlled Systems: Hydrogeis, Chap.2, pp 15-37 в Polymers for Controlled Drug Delivery, Ed Tarcha, CRC Press, Boca Raton, Fla.USA (1991); та Buri et al., Pharm.Acta Helv.55,189-197 (1980)]. Для контрольованого вивільнення застосовується також високоамілозний крохмаль, зокрема, останні досягнення були зроблені з застосуванням зшитого високоамілозного крохмалю. Наприклад, у [Патенті США №6284273 (Lenaerts et al.), виданому 4 вересня 2001, та №6419957 (Lenaerts et al.), виданому 16 липня 2002], описана тверда пероральна лікарська форма з контрольованим вивільненням у вигляді таблеток, що містять сухий порошок фармацевтичного продукту та сухий порошок поперечно зв'язаного високоамілозного крохмалю, де вказаний поперечно зв'язаний високоамілозний крохмаль являє собою матрицю, що містить суміш приблизно з 10-60% маси амілопектину та приблизно 40-90% амілози. У [Патенті США №6607748 (Lenaerts et al.), виданому 19 серпня 2003], описаний спосіб приготування зшитого високоамілозного крохмалю, який відомий під назвою Contramid®. Композиції з тривалим вивільненням, відомі в даній галузі Були запропоновані композиції з тривалим і контрольованим вивільненням, що відносяться до трамадолу HCl, і приклади таких композицій описані у [публікації Заявки на Патент США №2003/0143270, (Deboeck et al.), опублікованої 31 липня 2003; Патенті США №6254887 (Miller et al.), виданому 3 липня 2001; публікації Заявки на Патент США №2001/0036477 (Miller et al.), опублікованої 1 листопада 2001; Патенті США №6326027 (Miller et al.), виданому 4 гр удня 2001; і Патенті США №5591452 (Miller et al.), виданому 7 січня 1997; і Європейському Патенті №1190712 (Vanderbist), опублікованому 27 березня 2002]. Незважаючи на те, що на ринку існує декілька композицій трамадолу HCl з контрольованим вивільненням, під якими маються на увазі щоденні одноразові композиції, жодна з них успішно не замінила щоденні дворазові композиції трамадолу HCl. Опубліковані статті, в яких представлені порівняльні дані між передбачуваними "щоденними одноразовими" композиціями трамадолу HCl і трамадолу HCl з негайним вивільненням: Aider et al., "A Comparison of Once-Daily Tramadol with Normal Release Tramadol in the Treatment of Pain in Osteoarthritis", The Journal of Rheumatology (2002) 29(10): 2195-2199; і Bodalia et al., "A Comparison of 6 the Pharmacokinetics, Clinical Efficacy, and Tolerability of Once-Daily Tramadol Tablets with Normal Release Tramadol Capsules", Journal of Pain and Symptom Management (2003) 25(2): 142-149. Побічні ефекти від введення трамадолу HCl. До найбільш частих описаних побічних ефектів трамадолу, що спостерігаються при клінічних випробуваннях у Сполучених Штата х, відносяться запор, нудота, запаморочення/вертиго, головний біль, сонливість і блювота. Такі явища являють собою типові побічні ефекти наркотичних лікарських засобів. Епілептичні напади та анафілактичні реакції також були повідомлені, хоч оцінені випадки епілептичних нападів у пацієнтів, які отримують трамадол HCl, складають менше ніж 1% [Kazmierczak, R., and Coley, K.: Doctor letters on prescribing: evaluation of the use of tramadol HCl." Formulary 32:977-978, 1997]. Adler et al., ви ще, повідомляють про результати клінічного дослідження у порівнянні з однократною добовою композицією трамадолу з трамадолом з негайним вивільненням при лікуванні болю при остеоартриті. Автори повідомляють про схожі профілі побічних явищ у індивідів в обох гр упах лікування. Таблиця 2 у Adler et al. вказує, що більший відсоток людей, які були в гр упі однократного добового лікування, вибув через побічні ефекти, у порівнянні з тими, які вибули з другої групи лікування. У Bodalia et al., вище повідомляється про порівняльну толерантність при щоденній однократній дозі в 150мг, щоденній однократній дозі в 200мг і при трьох дозах по 50мг композиції трамадолу з нормальним вивільненням. Дана стаття, однак, не містить будь-якої інформації про те, як розробити композиції, які маються на увазі як "однократні добові", не розкриває ніяких фармакокінетичних даних після однократної дози. Цитування або посилання на будь-яке посилання в даному розділі не повинно тлумачитися як припущення, що таке посилання доступне як попередній рівень техніки по відношенню до даного винаходу. Суть винаходу Об'єкт даного винаходу повинен надавати поліпшену композицію трамадолу з пролонгованим вивільненням з 24-годинним ефективним знеболенням. Відповідно до одного об'єкта даного винаходу, надається однократна добова пероральна фармацевтична композиція для трамадолу з контрольованим вивільненням або його солі, в якому композиція, при первинному введенні, забезпечує початок знеболюючого ефекту протягом 2 годин, знеболюючий ефект якого продовжується щонайменше протягом 24 годин після введення. Відповідно до іншого об'єкта даного винаходу, надається однократна добова пероральна фармацевтична композиція для трамадолу з контрольованим вивільненням або його солі, в якому композиція при первинному введенні однієї дози забезпечує середню концентрацію у плазмі щонайменше 100нг/мл не пізніше ніж через 2 години після введення та продовжує забезпечувати сере 7 84277 дню концентрацію у плазмі щонайменше 100нг/мл щонайменше протягом 22 годин після введення. В одному з варіантів здійснення даного винаходу, надається однократна добова пероральна фармацевтична композиція для трамадолу з контрольованим вивільненням або його солі, коли композиція при первинному введенні однієї дози забезпечує середню концентрацію у плазмі щонайменше 100нг/мл не пізніше ніж через 2 години введення та продовжує забезпечувати середню концентрацію у плазмі щонайменше 100нг/мл щонайменше протягом 22 годин після введення, і де середня максимальна концентрація у плазмі (Сmax) менше взятої 2,2 рази середньої концентрації у плазмі, отриманої через 24 години після введення (С24год.). Позначення "lz" відповідає вірогідній константі заключної швидкості елімінації, що визначається нахилом лінії регресії протягом логарифмічної фази. Позначення "AUC0-max" відповідає середній площі під кривої залежності концентрації від часу в плазмі, від часу 0 до Тmax, і використовується як індикатор швидкості абсорбції лікарського засобу, або утворення метаболіту. Вона розраховується як середнє арифметичне площі під кривою залежності концентрації від часу в плазмі, від часу 0 до Тmax, розраховане для кожного індивіда, що бере участь у дослідах по вивченню біодоступності. Позначення "AUC0-¥" відповідає середній площі під кривою залежності концентрації від часу в плазмі, екстрапольованій до нескінченності. Вона розраховується як середнє арифметичне площі під кривою залежності концентрації від часу в плазмі від часу 0, екстрапольованого до нескінченності, розраховане для кожного індивіда, що бере участь у дослідах по вивченню біодоступності. Термін "знеболюючий ефект" визначається для даного винаходу як такий, що забезпечує середню концентрацію в плазмі крові, яка складає щонайменше приблизно 100нг/мл трамадолу. Позначення "С'max" означає максимум концентрації, що спостерігається в плазмі, розрахований як середнє значення індивідуальних максимальних концентрацій у плазмі крові. Вираз "контрольоване вивільнення" визначається для даного винаходу як спосіб доставки перорального лікарського засобу, де швидкість вивільнення активного фармацевтичного інгредієнта з композиції залежить не тільки від концентрації активного фармацевтичного інгредієнта, що залишається в композиції, і/або розчинності активного фармацевтичного інгредієнта в середовищі, що оточує композицію, і де проміжок часу і/або локалізація вивільнення активного інгредієнта з фармацевтичної композиції вибирається для досягнення терапевтичного ефекту або для зручності, що не пропонуються загальноприйнятими лікарськими формами. Термін "період напіввиведення" означає уявний період остаточного напіввиведення. Термін "ТСК" означає тривалість підтримки середнього рівня, тобто час, протягом якого концентрація трамадолу вище половини С'тах. Даний 8 показник являє собою індикатор форми кривої залежності концентрації від часу в плазмі. Термін "негайне вивільнення" визначається для даного винаходу як вивільнення активного інгредієнта з фармацевтичної композиції, коли швидкість вивільнення активного фармацевтичного інгредієнта з фармацевтичної композиції не гальмується матриксом контрольованого вивільнення і де інгредієнти фармацевтичної композиції розроблені так, що при прийомі всередину максимальний вплив вказаного активного фармацевтичного інгредієнта на організм відбувається за мінімальний період. Термін "первинне введення" визначається для даного винаходу як перша однократна доза композиції, що містить активний інгредієнт, який вводиться пацієнту або об'єкту, або перша доза, що вводиться пацієнту або об'єкту після відповідного періоду виведення. Термін "СЧЗ" означає середній час знаходження в організмі, який відповідає середній величині часу знаходження молекули трамадолу в організмі після перорального введення. Термін "середня максимальна концентрація в плазмі" (Сmax) визначається для даного винаходу як максимальна середня концентрація в плазмі. Термін "середня концентрація в плазмі" визначається для даного винаходу як середнє арифметичне концентрації в плазмі крові. Позначення "tmax" відповідає проміжку часу, через який досягається Сmax. Позначення "tmax" відповідає проміжку часу, через який досягається максимальна концентрація в плазмі крові у кожного з індивідів, що беруть участь у дослідах по вивченню біодоступності. Термін "Rstart" означає час, при якому концентрація в плазмі починає знижуватися за логарифмічним законом, тобто час, при якому або завершується абсорбція лікарського засобу, або завершується утворення його метаболіту. Слово "трамадол", так, як застосовується тут, відноситься до трамадолу, його стереоізомерів і його фармацевтично прийнятних солей. Термін "стабільний стан" визначається для даного винаходу як стан, що наступає за введенням багаторазової дози, коли швидкість елімінації лікарського препарату відповідає швидкості введення, і концентрації лікарських препаратів у плазмі в даний момент у межах інтервалу дозування є приблизно одними і тими самими, від одного інтервалу дозування до іншого. Короткий опис креслень Різні ознаки та переваги даного винаходу стануть зрозумілі при більш детальному описі, наведеному нижче з посиланням на супроводжуючі креслення: Фігура 1: С хематичне представлення процесу виробництва таблеток. Фігура 2: Профілі розчинення композицій А, В і С: In vitro реалізація композицій А, В і С: в умовах USP Типу 1; натрій-фосфатний буфер, 50мМ., pH 6,8, 100об./хв. було тестовано 6 таблеток на одиницю часу. Фігура 3: Середні концентрації трамадолу в плазмі після введення однократної дози (і) дози 9 84277 100мг композиції з контрольованим вивільненням згідно з винаходом (·), (іі) дози 200мг композиції з контрольованим вивільненням згідно з винаходом ( ), і (ііі) 300мг дози композиції з контрольованим вивільненням згідно з винаходом (▲). Фігура 4: Середні концентрації Одесметилтрамадолу в плазмі після введення однократної дози, що містить або 100мг (♦), або 200мг (О), або 300мг (Δ) активних речовини композиції трамадолу (А, В і С, відповідно). Фігура 5: Середні концентрації трамадолу в плазмі після введення однократної дози (і) 2´200мг композиції з контрольованим вивільненням згідно з винаходом (▲); і (іі) Topalgic® LP 200мг BID q12h (Δ). Фігура 6: Середні концентрації Одесметилтрамадолу в плазмі після введення однократної дози (і) 2´200мг композиції з вивільненням, що контролюється згідно з винаходом (▲); і (іі) Topalgic® LP 200мг BID q12h (Δ). Фігура 7: Середня стабільна концентрація трамадолу та О-десметилтрамадолу в плазмі після введення (і) 200мг дози композиції з контрольованим вивільненням згідно з винаходом (·&ο); і (іі) Topalgic® LP 100мг BID q12h (▲&Δ). Докладний опис винаходу Ядро Ядро таблетки згідно з винаходом включає в себе щонайменше один активний інгредієнт і матрицю, дані інгредієнти взаємодіють таким чином, що вивільнення фармацевтичного інгредієнта з матриці контролюється. В особливому варіанті здійснення матриця ядра являє собою поперечно зшитий високоамілозний крохмаль, відомий під назвою Contramid® і описаний зовсім недавно в [Патенті США №6607748 (Lenaerts et al.), виданому 19 серпня 2003]. Переважна композиція, у контексті даного винаходу, представлена в [описі Патенту США №6607748]. Переважно ядро утворено шляхом перемішування інгредієнтів (у формі гранул або порошку) і потім пресування суміші для утворення ядра, поверх якого послідовно формується оболонка. Вага ядра може становити будь-який відсоток ваги всієї композиції, між 10% і 80%. Переважний відсоток залежить, окрім усього іншого, від загального дозування фармацевтичного засобу. У спеціальному варіанті здійснення, описаному далі, таблетка містить 100мг трамадолу гідрохлориду та ядро, що становить приблизно 26% від загальної ваги таблетки. В іншому варіанті здійснення таблетка містить 200мг трамадолу гідрохлориду, і ядро складає приблизно до 33% від загальної ваги таблетки. В іншому варіанті здійснення таблетка містить 300мг трамадолу гідрохлориду, і ядро становить 33% від загальної ваги таблетки. Активний засіб у ядрі В ядрі композиції згідно з винаходом присутній активний фармацевтичний інгредієнт. Придатний фармацевтичний інгредієнт згідно з винаходом являє собою будь-який такий інгредієнт, який бажано повинен бути доставлений у лікарській формі з пролонгованим вивільненням. Докладний перелік придатних фармацевтичних засобів можна знайти 10 в [The Merck Index, 12th Ed]. Переважно фармацевтичний інгредієнт є, без обмеження, гідразидом ізонікотонової кислоти, саліцилатом натрію, гідрохлоридом псевдоефедрину, сульфатом псевдоефедрину, ацетамінофеном або диклофенаком натрію, верапамілом, гліпізидом, ніфедипіном, фелодипіном, бетагістином, альбутеролом, акривастином, омепразолом, мізопростолом, трамадолом®, оксибутиніном, тримебутином, ципрофлоксацином і їх солями. У доповнення, фармацевтичний засіб може бути протигрибковим і їх солями. У доповнення, фармацевтичний засіб може бути протигрибковим засобом, як, наприклад, кетоконазол, або знеболюючим засобом, як, наприклад, ацетилсаліцилова кислота, ацетамінофен, парацетамол, ібупрофен, кетопрофен, індометацин, дифлюнісал, напроксен, кеторолак, диклофенак, толметин, суліндак, фенацетин, піроксикам, мефенамова кислота, декстрометорфан, інші нестероїдні протизапальні лікарські засоби, включаючи саліцилати, їх фармацевтично прийнятні солі або їх суміші. Проліки є частиною даного винаходу. Розчинність лікарського засобу у водному розчині може мати найрізноманітніші значення. Водорозчинність лікарського засобу може складати менше ніж 10-3г/л, більше ніж 10-3г/л, більше 10-2г/л, більше ніж 10-1г/л, більше ніж 1г/л більше ніж 10г/л, більше ніж 100г/л, більше ніж 500г/л, більше ніж 1000г/л або більше ніж 2000г/л. Переважно, розчинність складає більше ніж 100г/л. Більш переважно, щоб розчинність складала більше 500г/л. Найбільш переважно, коли розчинність складає більше 1000г/л. Лікарський засіб може задовольняти різноманітні вимоги до дозування. Наприклад, необхідне дозування лікарського засобу може складати менше 1мг/одиницю дозування, більше ніж 1мг/одиницю дозування, більше 10мг/одиницю дозування, більше 100мг/одиницю дозування, більше 200мг/одиницю дозування, більше 300мг/одиницю дозування, більше 400мг/одиницю дозування, більше 500мг/одиницю дозування або більше 1000мг/одиницю дозування. Переважно, якщо лікарський засіб складає більше 50мг/одиницю дозування. Більш переважно, коли лікарський засіб складає більше 100мг/одиницю дозування або, наприклад, більше 150мг/одиницю дозування або 200мг/одиницю дозування, або 250мг/одиницю дозування, або 300мг/одиницю дозування або більше. В особливих варіантах здійснення ядро містить трамадолгідрохлорид, причому ядро містить приблизно між 10% і 90% всього трамадолу, присутнього в таблетці, наприклад, приблизно 45мг з 100мг вмісту активної речовини таблетки (45% усієї таблетки) або приблизно 90мг з 200мг вмісту активної речовини таблетки (45% усієї таблетки) або приблизно 151мг з 300мг вмісту активної речовини таблетки (50% усієї таблетки). Матриця ядра Вивільнення з композиції активного фармацевтичного інгредієнта, що знаходиться в ядрі, відбувається повільніше, ніж вивільнення активного фармацевтичного інгредієнта, що знаходиться у 11 84277 матриці оболонки. Переважна матриця ядра являє собою поперечно зшитий високоамілозний крохмаль, відомий під назвою Contramid® і описаний в [патенті США №6607748]. У спеціальних варіантах здійснення матриця складає приблизно між 10% і приблизно 90% за вагою ядра, тобто співвідношення матриксу ядра до активного інгредієнта ядра (мас/мас.) складає приблизно між 0,1 і приблизно 10 або приблизно між 0,2 і приблизно 9, або приблизно між 0,2 і приблизно 8, або приблизно між 0,3 і приблизно 7, або приблизно між 0,4 і приблизно 6, або приблизно між 0,5 і приблизно 5, або приблизно між 0,6 і приблизно 4, або приблизно між 1 і приблизно 4, або приблизно між 1 і приблизно 3, і приблизно 1,5 і приблизно 2,5. В одному особливому варіанті здійснення ядро становить приблизно 90мг, з яких 44мг представлено Contramid® і 45мг представлено трамадолгідрохлоридом. У цьому випадку Contramid®, таким чином, становить приблизно 49 вагових відсотків ядра. Необов'язкові компоненти Ядро композиції згідно з винаходом може, за бажанням, включати в себе фармацевтично прийнятний носій або розчинник. Такі носії або розчинники відомі фахівцям в даній галузі, і можна знайти, наприклад, в [Remington's Pharmaceutical Sciences, 14 th Ed.(1970)]. Приклади таких носіїв або розчинників включають у себе лактозу, крохмаль, дикальційфосфат, сульфат кальцію, каолін, маніт і цукрову пудру. Додатково, при необхідності, можуть бути включені придатні зв'язуючі речовини, пом'якшувальні засоби та розщеплюючі засоби. За бажанням, можуть бути включені також барвники, а також підсолоджувачі або смакові домішки. Ядро композиції згідно з винаходом може, за бажанням, включати в себе додаткові інгредієнти, включаючи, без обмеження, диспергуючі засоби, такі як мікрокристалічна целюлоза, крохмаль, поперечно зшитий крохмаль, поперечно зшитий полівінілпіролідон і карбоксиметилцелюлоза натрію; смакові засоби; фарбувальні засоби; зв'язуючі засоби; консерванти; поверхнево-активні засоби та ін. Ядро може, за бажанням, також включати в себе одну або більше зв'язуючих речовин, відомих рядовому фа хівцеві в даній галузі. Придатні форми мікрокристалічної целюлози, наприклад, МКЦ-Ф101; МКЦ-102;МКЦ-105та ін. Придатні пом'якшувальні речовини, такі, наприклад, як ті, які відомі фахівцям, можуть бути також включені. Наприклад, стеарат магнію, рослинна олія, тальк, стеарилфумарат натрію, стеарат кальцію, стеаринова кислота тощо. Придатні ковзні речовини, відомі в даній галузі, можуть бути також включені. Приклади таких ковзних речовин включають в себе, без обмеження, тальк, колоїдний діоксид кремнію тощо. Кількісне співвідношення Активний засіб присутній у кількостях, що коливаються приблизно від 1 приблизно до 90мас.% загальної ваги ядра, переважно, приблизно від 10 приблизно до 70мас.% від загальної композиції ядра, більш переважно - приблизно від 20 прибли 12 зно до 60мас.% від загальної композиції ядра і, ймовірно, найчастіше - приблизно від 30 приблизно до 50мас.% загальної композиції ядра. Звичайно, загальна кількість усіх інгредієнтів становить 100мас.%, і рядові фахівці в даній галузі, можуть змінювати кількості в межах встановлених діапазонів для досягнення корисних композицій. Покриття (оболонка) Покриття лікарської форми включає в себе фізичну суміш полівнілацетату та полівінілпіролідону та активного фармацевтичного інгредієнта(ів) оболонки. Оболонка може також включати в себе поперечно зшитий високоамілозний крохмаль, наприклад, Contramid® та інші можливі інгредієнти. У переважному варіанті здійснення оболонка формується шляхом сухого пресування. Вага оболонки може становити будь-який відсоток ваги загальної композиції, приблизно між 10% і 90%, але переважно в більш високій частині даного діапазону. Оболонка, таким чином, зазвичай складає приблизно між 20% і 90% (мас/мас.) таблетки згідно з винаходом або приблизно 25% і приблизно 90% або приблизно 30% і приблизно 85%, або приблизно 35% і приблизно 85%, або приблизно 40% і приблизно 85%, або приблизно 45% і приблизно 85%, або приблизно 25% і приблизно 90%, або приблизно 45% і приблизно 90%, або приблизно 50% і приблизно 90%, або приблизно 50% і приблизно 85%, або приблизно 55% і приблизно 90%, або приблизно 55% і приблизно 85%, або приблизно 55% і приблизно 80%, або приблизно 60% і приблизно 90%, або приблизно 60% і приблизно 85%, або приблизно 60% і приблизно 80%, або приблизно 60% і приблизно 75%, або приблизно 65% і приблизно 90%, або приблизно 65% і приблизно 85%, або приблизно 65% і приблизно 80%, або приблизно 65% і приблизно 75%, або приблизно 65% і приблизно 70% або приблизно 75%. Оболонка часто включає в себе необов'язковий зв'язуючий засіб. Полівінілацетат і полівінілпіролідон покриття (оболонки) Відсоткова вага суміші полівінілацетат/полівінілпіролідону в оболонці може бути в широкому діапазоні значень. В залежності від розчинності у воді активного інгредієнта в оболонці, кількість суміші полівінілацетат/полівінілпіролідону в оболонці може бути підібрана. У [публікації Патенту США №2001/0038852] описані шляхи підбору. Наприклад, для активних інгредієнтів, які у більшій мірі розчинні у воді, суміш полівінілацетат/полівінілпіролідону може складати приблизно від 20% приблизно до 80мас.% оболонки, переважно приблизно від 30 приблизно до 65мас.% або приблизно від 40 приблизно до 55мас.%. В особливому варіанті здійснення, описаному нижче, Kollidon™ SR складає приблизно 45% за вагою оболонки, яка становить приблизно 31% за вагою трамадолгідрохлориду та приблизно 23% ксантанової камеді. Для активних інгредієнтів, від помірно розчинних до слабо розчинних у воді, кількість суміші полівінілацетат/полівінілпіролідону часто менша, як описано в [Публікації Патенту США №2001/0038852]. 13 84277 Співвідношення ваги полівінілацетату до полівінілпіролідону в суміші полівінілацетат/полівінілпіролідон може складати широкий діапазон значень. Переважно таке співвідношення складає приблизно між 6:4 та 9:1; більш ймовірно приблизно між 7:3 і 6:1, ще більш переважно приблизно 8:2. Молекулярна вага полівінілацетатного інгредієнта в суміші полівінілацетат/полівінілпіролідон може складати широкий діапазон значень. Переважно середня молекулярна вага полівінілацетату складає приблизно від 100 приблизно до 1000000; або, приблизно від 1000 приблизно до 1000000; або приблизно від 10000 приблизно до 1000000; або приблизно від 100000 приблизно до 100000, або приблизно 450000. Молекулярна вага полівінілпіролідонового інгредієнта в суміші полівінілацетат/полі вінілпіролідон може складати широкий діапазон значень. Середня молекулярна вага полівінілпіролідону може складати приблизно від 100 приблизно до 10000000; або приблизно 1000 приблизно до 1000000; або приблизно 5000 приблизно до 500000; або приблизно 10000 приблизно до 100000; або приблизно 50000. Суміш полівінілацетату та полівінілпіролідону може бути приготована різноманітними способами, включаючи просте перемішування порошків полівінілпіролідону та полівінілацетату. У переважному варіанті здійснення така суміш являє собою висушений шляхом розпилення порошок колоїдної дисперсії розчину полівінілацетату та полівінілпіролідону. За бажанням, застосовується лаурилсульфат натрію як стабілізатор для запобігання агломерації під час процесу сушіння розпиленням і/або застосовується колоїдний кремній для поліпшення властивостей текучості суміші полівінілацетат/полівінілпіролідон. За бажанням, полівінілацетат і полівінілпіролідон можуть бути формовані довільно або у блоксополімері. Необов'язкові компоненти Придатні зв'язуючі засоби згідно з винаходом включають у себе, без обмеження, рослинні екстракти, смоли, синтетичні або натуральні полісахариди, поліпептиди, алгінати, синтетичні полімери або їх суміш. Придатні рослинні екстракти, які повинні використовуватися як желатинізуючі засоби, включають у себе, без обмеження, агар, ispaghula, подорожник, червець айвовий, вогнівку або їх суміш. Придатні смоли, які повинні використовуватися як желатинізуючі засоби, включають у себе, без обмеження, ксантанову камедь, гуарову камедь, аравійську камедь, ghatti камедь, камедь карайі, трагакантову камедь або їх суміш. Придатні синтетичні або натуральні гідрофільні полісахариди, що використовуються як желатинізуючі засоби, включають у себе, без обмеження, гідроксіалкілцелюлозу, простий ефір целюлози, складний ефір целюлози, нітроцелюлози, декстрин, агар, карагенан, пектин, фурцеларан, крохмаль або похідні крохмалю, поперечно зшитий високоамілозний крохмаль або їх суміш. Придатні поліпептиди, що застосовуються як желатинізуючі засоби, включають у себе, без об 14 меження, желатин, колаген, поліжелін або їх суміш. Придатні альгінати, що застосовуються як желатинізуючі засоби, включають у себе, без обмеження, альгінову кислоту, пропіленглікольальгінат, альгінат натрію або їх суміш. Придатні синтетичні полімери, що застосовуються як желатинізуючі засоби, включають у себе, без обмеження, карбоксивініловий полімер, полівініловий спирт, полівінілпіролідон, поліетиленоксид, поліетиленгліколі, співполімери етиленоксиду та пропіленоксиду та їх співполімери або їх суміш. У переважному варіанті здійснення, желатинізуючий засіб являє собою камедь, таку як ксантанова камедь, гуарова камедь, аравійська камедь, ghatti камедь, камедь карайі, трагакантова камедь або їх суміш, РЕО 7000000 та НРМС К100М. У найбільш переважному варіанті здійснення, желатинізуючий засіб являє собою ксантанову камедь. Активний засіб у покритті (оболонці) Придатний активний фармацевтичний інгредієнт згідно з винаходом являє собою будь-який активний засіб, який бажано наданий у лікарській формі з пролонгованим вивільненням. Докладний перелік придатних фармацевтичних засобів можна знайти в [The Merck Index, 12th Ed]. Переважно фармацевтичний інгредієнт являє собою, без обмеження, гідразид ізонікотинової кислоти, саліцилат натрію, гідрохлорид псевдоефедрину, сульфат псевдоефедрину, ацетомінофен або диклофенак натрію, верапаміл, гліпізид, ніфедипін, фелодипін, бетагістин, альбутерол, акривастин, омепразол, мізопростол, трамадол®, оксибутинін, тримебутин, ципрофлоксацин і їх солі. У доповнення, фармацевтичний засіб може бути протигрибковим засобом, як наприклад, кетоконазол або аналгезуючим засобом, як наприклад, ацетилсаліцилова кислота, ацетамінофен, парацетамол, ібупрофен, кетопрофен, індометацин, дифлюнісал, напроксен, кеторолак, диклофенак, толметин, суліндак, фенацетин, піроксикам, мефенамова кислота, декстрометорфан, інші нестероїдні протизапальні лікарські засоби, включаючи саліцилати, їх фармацевтично прийнятні солі або їх суміші. Розчинність лікарського засобу у водному розчині може мати значення в широкому діапазоні. Водорозчинність фармацевтичного засобу може бути менше ніж 10-3г/л, більше ніж 10-3г/л, більше ніж 10-2г/л, більше ніж 10-1г/л, більше ніж 1г/л, більше ніж 10г/л, більше ніж 100г/л, більше ніж 500г/л, більше ніж 1000г/л або більше ніж 2000г/л. Переважно розчинність складає більше ніж 100г/л. Більш переважно, якщо розчинність складає більше ніж 500г/л або навіть 1000г/л. Лікарський засіб може задовольняти різні вимоги до дозування. Наприклад, необхідне дозування лікарського засобу може бути менше ніж 1мг/одиницю дозування, більше ніж 1мг/одиницю дозування, більше ніж 10мг/одиницю дозування, більше ніж 100мг/одиницю дозування, більше ніж 200мг/одиницю дозування, більше ніж 300мг/одиницю дозування, більше ніж 400мг/одиницю дозування, більше ніж 500мг/одиницю дозування або більше ніж 15 84277 1000мг/одиницю дозування. Переважно лікарський засіб складає більше ніж 50мг/одиницю дозування. Більш переважно, якщо лікарський засіб складає більше ніж 100мг/одиницю дозування. Найбільш переважно, лікарський засіб складає більше ніж 200мг/одиницю дозування. Оболонка може складати приблизно між 5% і приблизно 90% маси фармацевтично активного інгредієнта, або приблизно між 5% і приблизно 80% мас. арі, або, приблизно між 10% і приблизно 70% мас. арі, або, приблизно між 10% і приблизно 60% мас. арі, або, приблизно між 15% і приблизно 50% за мас. арі, або, приблизно між 15% і приблизно 45% мас. арі, або, приблизно між 15% і приблизно 40% мас. арі, або, приблизно між 20% і приблизно 35% мас. арі, або, приблизно між 20% і приблизно 30% мас. арі. В особливих варіантах здійснення, описаних нижче, вага трамадолу в 100мг таблетці трамадолу становить приблизно 21% мас. оболонки. Вага трамадолу в 200мг таблетці трамадолу становить приблизно 31% мас. оболонки. Вага трамадолу в 300мг таблетці трамадолу становить приблизно 30% мас. оболонки. Шляхи введення Таблетована композиція згідно з винаходом може бути введена, без обмеження, через ряд шляхів, таких, наприклад, як пероральний, сублінгвальний і ректальний. Переважним шляхом введення композицій згідно з винаходом є пероральний. Композиції згідно з винаходом, які підходять для перорального введення, можуть бути представлені як дискретні одиниці, такі як таблетки та гранули. Переважно композиції згідно з винаходом представлені в таблетованій формі. Такі таблетки можуть бути традиційно сформовані шляхом пресування або формування. Пресовані таблетки можуть бути приготовані шляхом пресування суміші одного або більше компонентів, описаних вище, у відповідному пристрої. Формовані таблетки можуть бути отримані шляхом компресії вищезгаданих інгредієнтів у відповідному пристрої, при цьому вищезгадані інгредієнти можуть бути, якщо потрібно, зволожені інертним рідким розчинником. Таблетки можуть бути, якщо потрібно, вкриті (оболонкою) і/або мати розпізнавальні знаки, видимі споживачеві. Таблетка може також бути отримана у різних видах, наприклад, не вкрита оболонкою, вкрита сухою оболонкою або з плівковим покриттям тощо. Таблетка може також мати різноманітні форми (наприклад, овальну, сферичну тощо) і розміри. Докладне обговорення таблеток може бути знайдене у посиланнях, наприклад, в [The Theory and Practice of Industrial Pharmacy by Lachman et al., 3 thEd. (Lea&Fibiger, 1986)]. Профіль розчинення композиції з пролонгованим вивільненням Активний засіб композиції має наступний in vitro профіль розчинення, при вимірюванні за допомогою пристрою USP Типу І в 50мМ. фосфату, pH 6,8 і при перемішуванні між 50 і 150об./хв.: середня швидкість між 10% і 30% за годину засобу вивільняється між 0 і 2 годинами, при тестуванні in vitro із застосуванням пристрою USP 16 Типу І в 50мМ фосфа ту, pH 6,8 і при перемішуванні між 50 і 150об./хв.; або між 10% і 40% засобу вивільняється з композиції між 0 і приблизно 2 годинами вимірювання, приблизно між 30% і 60% засобу вивільняється з композиції між 2 і приблизно 7 годинами вимірювання, приблизно між 50% і 80% засобу вивільняється з композиції між 7 і приблизно 12 годинами вимірювання, і приблизно між 80% і 100% засобу вивільняється з композиції після приблизно 20 годин вимірювання; або, більш переважно, приблизно між 15% і 35% засобу, вивільняється з композиції через 2 години, приблизно між 40% і 60% засобу що вивільняться з композиції через 7 годин, приблизно між 60% і 80% засобу вивільняється з композиції за 12 годин і приблизно між 85% і 100% засобу вивільняється з композиції при вимірюванні через 20 годин, або між 20% і 40% засобу вивільняється з композиції через 2 години, приблизно між 40% і 60% засобу вивільняється з композиції через 7 годин, приблизно між 60% і 80% засобу вивільняється з композиції за 12 годин, і приблизно між 85% і 100% засобу вивільняється з композиції після 20 годин вимірювання. Даний винахід буде легше зрозумілий шляхом посилання на наступні приклади, які надані швидше для ілюстрації винаходу, ніж для обмеження його обсягу. Приклади Поперечно зшитий високоамілозний крохмаль, що застосовується в даних прикладах, створений способом, що включає в себе етапи зв'язування поперечними зв'язками та хімічного модифікування, з подальшим желатинізуванням і висушуванням. Даний спосіб описаний докладно в деталях у [Патенті США №6607748 (Lenaerts et al.), виданому 19 серпня, 2003], і відомий на ринку під назвою Contramid®; він описаний у Прикладах 1 та 2. Приклад 1 A. Перехресне зв'язування Високоамілозний крохмаль (30,0кг), що містить приблизно 70% мас/мас. амілози (СІ АмілоГель 03003) вміщується до реактора. У даний реактор додається вода (55,0л), що містить гідроксид натрію (30,0г) і сульфат натрію (2,40кг). Результуюча суспензія нагрівається до температури 30°С. Оксихлорид фосфору (22,5г) додається в реакційну суміш, яка вступає в реакцію протягом 1 години. B. Хімічна модифікація, гідроксипропілювання Неочищена реакційна суміш з Частини А переноситься до реактора гідроксипропілювання. Реакційна суміш нагрівається до 40°С більше 30 хвилин, і реакція очищується азотом. Після повного очищення додається пропіленоксид (1,80кг). Реакційна суміш зберігається при 40°С протягом 20 годин. Реакційна суміш нейтралізується 0,1Н H2SO4 (1:2об./об.) до pH 5,5. Крохмальний осад промивають за допомогою кошикової центрифуги зі швидкістю 1200об./хв. Отриманий крохмальний осад повторно емульгують у 35л води та центрифугують другий раз. Отриманий крохмальний осад висушують у сушарці миттєвої дії при температурі на вході 160°С та температурі на виході 60°С. С. Желатинізування. 17 84277 Модифікований гранулярний крохмальний осад розчиняють у демінералізованій воді, з утворенням осаду, у концентрації приблизно 8% у розрахунку на суху речовину. Результуюча суспензія має відносну щільність 1,032кг/л по відношенню до води. pH модифікованої крохмальної суспензії доводиться до 6,0. Потім розчин нагрівається до 160°С прямим введенням пари (Schlick Model 825). Коливання температури складають не більше ніж ±1°С. Суспензія тримається у колонці для зберігання протягом 4 хвилин при температурі 160°С і тиску приблизно 5,5 бар. Потім тиск зменшується. до атмосферного шляхом флеш-пропускання. Потім розчин тримають при 95°С в резервуарі для зберігання. Д. Сушіння розпиленням Сушіння суспензії з Частини С здійснюється шляхом застосування баштової розпилювальної сушарки Niro FSD 4, обладнаної 0,8мм клапаном і подачею 10л/годину. Температура на вході фіксується як 300°С, а температура на виході -120°С. Отриманий порошок являє собою допоміжну речовину з контрольованим вивільненням з наступними властивостями: Властивості Вміст вологи 4,5% Об'ємна щільність 150г/л Уявна щільність 210г/л pH 5,4 Пікове значення розміру частинки (Лазерний вимірювач частинок Sympatec) 50мкм Приклад 2 А. Перехресне зв'язування Високоамілозний крохмаль (30,0кг), що містить приблизно 70% мас/мас, амілози (СІ АмілоГель 03003) вміщується до реактора. У даний реактор додається вода (55,01), що містить гідроксид натрію (30,0г) і сульфат натрію (2,40кг). Результуюча суспензія нагрівається до температури 30°С. Триметофосфат натрію (45г) додається в реакційну суміш, яка вступає в реакцію протягом однієї години. В. Хімічна модифікація, гідроксипропілювання Неочищена реакційна суміш з частини А переноситься до реактора гідроксипропілювання. Реакційна суміш нагрівається до 40°С більше 30 хвилин, і реакція очищується азотом. Після повного очищення додається пропіленоксид (1,80кг). Реакційна суміш зберігається при 40°С протягом 20 годин. Реакційну суміш нейтралізують 0,1Н H2SO4 (1:2об./об.) до pH 5,5. Крохмальний осад промивають за допомогою кошикової центрифуги при швидкості 1200об./хв. Отриманий крохмальний осад повторно емульгується в 35л води та центрифугується другий раз. Отриманий крохмальний осад висушується в сушарці миттєвої дії при температурі на вході 160°С і температурі на виході 60°С. С. Желатинізування Модифікований гранулярний крохмальний осад розчиняють у демінералізованій воді, з утворенням осаду, у концентрації приблизно 8% у розрахунку на суху речовину. Результуючий осад має відносну щільність 1,032кг/л по відношенню до 18 води. pH модифікованого крохмального осаду доводять до 6,0. Потім розчин нагрівають до 160°С прямим введенням пари (Schlick Model 825). Коливання температури складають не більше ніж ±1°С. Суспензія тримається в колонці для зберігання протягом 4 хвилин при температурі 160°С і тиску приблизно 5,5 бар. Потім тиск зменшують до атмосферного шляхом флеш-пропускання. Потім розчин тримають при 95°С в резервуарі для зберігання. Д. Сушіння розпиленням Сушіння суспензії з Частини С здійснюється шляхом застосування баштової розпилювальної сушарки Niro FSD 4, обладнаної 0,8мм. клапаном і подачею 10л/годину. Температура на вході становить 300°С, і температура на виході - 120°С. Отриманий порошок представляє допоміжну речовину з контрольованим вивільненням з наступними властивостями: Властивості Вміст вологи 5,2% Об'ємна щільність 103г/л Уявна щільність 155г/л pH 5,3 Пікове значення розміру частинки (Лазерний вимірювач частинок - Sympatec) 70мкм Lubritab® являє собою продукт, що продається Penwest Pharmaceuticals Co. (Cedar Rapids, ΙΑ, USA). Kollidon™ SR представляє продукт, що виготовляється BASF (Germany). Encompress™ являє собою дикальційфосфатдигідрат, який може бути придбаний у Mendell (Patterson, NY). Трамадолгідрохлорид може бути отриманий з Chemagis Ltd., з Hashlosha Street, P.O.Bo x 9091, 61090, Tel Avi v, Israel. Способи синтезу та очищення трамадолу описані, наприклад, у [Патентах США №3652589, 5414129, 5672755, 5874620, 5877351 і 6169205]. Спосіб виготовлення Таблетки згідно з винаходом можуть бути виготовлені згідно зі способом, загалом викладеним у технологічній схемі способу на Фігурі 1 та описаному детально нижче. Дозування за масою: Початкові речовини розподіляються по чітко маркованим контейнерам. Попередня суміш ядра: Перемішати частину Contramid® та колоїдального діоксиду кремнію та пропустити крізь #30-меш-сито у відповідний контейнер. Суміш ядра: Помістити частину Contramid® до змішувача. Пропустити трамадолгідрохлорид крізь #30-меш-сито і додати до змішувача. Промити контейнер частиною Contramid® і додати до змішувача. Просіяти гідрогенізовану рослинну олію типу І крізь #30-меш-сито і додати до змішувача. Додати попередню суміш ядра до змішувача. Додати Contramid®, що залишився, до змішувача і перемішати всі інгредієнти. Просіяти стеарат магнію крізь #30-меш-сито і додати суміш з іншими інгредієнтами. Розподілити суміш у відповідний контейнер і визначити як суміш ядра. Попередня суміш сухого покриття: Змішати частину ксантанової камеді та весь колоїдний діоксид кремнію та пропустити крізь #30-меш-сито. 19 84277 Суміш сухого покриття: Помістити частину Kollidon™ SR у змішувач. Пропустити трамадолугідрохлорид крізь сепаратор Kason з #30-меш-сито у відповідний контейнер і додати до змішувача. Промити контейнер з ксантановою камеддю, що залишилася, і додати до змішувача. Просіяти гідрогенізовану рослинну олію Типу І крізь #30-мешсито і додати до змішувача. Помістити вкриту сухою оболонкою попередню суміш і залишки Kollidon™ SR до змішувача та перемішати з усіма інгредієнтами. Просіяти стеарат магнію крізь #30 20 меш-сито та перемішати з іншими інгредієнтами. Розподілити грануляцію у відповідний контейнер і визначити як суміш сухого покриття. Пресування: Застосувати прес Manesty DryCota для виробництва спресованих вкритих таблеток. Приклад 3 Композиції А, В і С, як показано в Таблиці 1, були виготовлені у відповідності до способів, викладених вище. Таблиця 1 Рецептури композицій А, В і С для трамадолу з контрольованим вивільненням 1) Інгредієнт Ядро Трамадолгідрохлорид Contramid® Гідрогенізована рослинна олія Кремній Стеарат магнію Загальна вага ядра 2) Оболонка Трамадолгідрохлорид Кремній Kollidon SR® Ксантанова камедь Гідрогенізована рослинна олія Стеарат магнію Загальна вага оболонки 3) Вкрита оболонкою таблетка Трамадолгідрохлорид Contramid® Гідрогенізована рослиннаолія Кремній Стеарат магнію Kollidon SR® Ксантанова камедь Загальна вага вкритої оболонкою таблетки: Композиція А Композиція В Композиція С % мг/ таблетку % мг/таблетку % мг/таблетку 50 48,3 0,75 0,2 0,75 100 45 43,47 0,675 0,18 0,675 90 50 48,3 0,75 0,2 0,75 100 90 86,94 1,35 0,36 1,35 180 63,25 35,05 0,75 0,20 0,75 100 151,8 84,1 1,8 0,5 1,8 240 21,15 0,20 51,42 25,72 1,00 0,50 100 55 0,52 133,7 66,86 2,6 1,3 260 30,56 0,20 45,16 22,58 1,00 0,50 100,00 110 0,72 162,58 81,3 3,6 1,8 360 30,56 0,20 45,16 22,58 1,00 0,50 100 148,5 1,0 219 109,5 4,9 2,4 485 28,57 12,42 0,94 0,20 0,56 38,20 19,11 100 43,47 3,275 0,7 1,975 133,7 66,86 37,04 16,10 0,92 0,20 0,58 30,11 15,06 200 86,94 4,95 1,08 3,15 162,58 81,3 41,38 11,60 0,92 0,20 0,58 30,21 15,10 300 84,1 6,7 1,5 4,2 219 109,5 100 350 100 540 100 725 Профілі розчинення композицій А, В і С показані на Фігурі 2. Композиція трамадолу щоденного одноразового прийому Даний винахід відноситься до таблетованої композиції з контрольованим вивільненням, яка забезпечує знеболюючий ефект не пізніше ніж через 2 години після перорального введення, який продовжується щонайменше протягом 24 годин після введення. 200мг доза композиції з контрольованим вивільненням згідно з винаходом несподівано забезпечує швидкий початок знеболюючого ефекту, не пізніше ніж через 2 години після перорального введення, та середню концентрацію трамадолу в плазмі між 100нг/мл і 200нг/мл щонайменше протягом 24 годин після однократної дози. Крім того, при стабільному стані середня концентрація трамадолу в плазмі зберігається між 100нг/мл і 350нг/мл. Несподівано було показано, що композиції з контрольованим вивільненням згідно з винаходом забезпечують повний клінічний ефект щонайменше протягом 24 годин після перорального введення. Дослідження біодоступності Об'єкт даного винаходу дає можливість гнучкого вибору дозування для пацієнтів з різними потребами у знеболенні, за допомогою композиції для однократного добового прийому. Один з варіантів здійснення даного винаходу пов'язаний з композицією для однократного добового прийому, яка при первинному прийомі всередину дози в 200мг забезпечує бажаний ранній початок дії, але при якій середні концентрації трамадолу в плазмі щонайменше 45нг/мл досягаються між 2 і 24 годинами. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного 21 84277 добового прийому, яка при первинному прийомі дози в 200мг забезпечує необхідний ранній початок дії, але при якій середні концентрації трамадолу в плазмі, щонайменше 100нг/мл досягаються між 2 і 24 годинами. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози в 300мг забезпечує необхідний ранній початок дії, але при якій середні концентрації трамадолу в плазмі щонайменше 150нг/мл досягаються між 2 і 24 годинами. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози в 400мг забезпечує необхідний ранній початок дії, але при якій середні концентрації трамадолу в плазмі щонайменше 180нг/мл досягаються між 2 і 24 годинами. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує С' max для співвідношення доз приблизно від 0,9 приблизно до 1,0. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує концентрацію трамадолу в плазмі, яка неухильно підвищується до досягнення піка концентрацій трамадолу при Тmax, що складає приблизно від 4 годин приблизно до 6 годин. Переважно Тmax складає приблизно від 5 годин приблизно до 5,5 години. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує концентрацію трамадолу в плазмі, що після Тmax знижується повільно та рівномірно, відображуючи безперервну абсорбцію, у доповнення до процесів елімінації. Переважно зниження концентрації трамадолу в плазмі після Тmax відбувається згідно з логарифмічним законом, з середнім уявним періодом напіввиведення приблизно між 5,5 години і приблизно 6,5 години. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, що при первинному прийомі дози забезпечує концентрацію трамадолу в плазмі, яка, після Тmax знижується повільно, але рівномірно, відображуючи безперервну абсорбцію, у доповнення до процесів елімінації, та абсорбція якої продовжується щонайменше протягом 20 годин з часу початку абсорбції прийнятої дози. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, що при первинному прийомі дози забезпечує концентрацію трамадолу в плазмі, яка, після Тmax знижується згідно з логарифмічним законом, з уявною константою швидкості остаточної елімінації (lz), що становить приблизно 0,12год.-1. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує середній час знаходження (СЧЗ) 22 трамадолу, в межах приблизно від 15 годин приблизно до 18 годин. Додатковий варіант здійснення згідно з винаходом забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує тривалість середньої величини (ТСК) трамадолу, в межах приблизно від 22,5 години приблизно до 25,4 години. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує відношення С'max до AUC0-¥, що складає приблизно від 0,04год.-1 приблизно до 0,06год.-1. Переважно відношення С'max до AUC0-¥ складає приблизно від 0,04год.-1 приблизно до 0,05год.-1. Відношення С'max / AUC0-¥, застосовується для оцінки швидкості абсорбції лікарського засобу. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує середнє значення AUC0-24 концентрації трамадолу в плазмі, яка зростає пропорційно дозі, в діапазоні доз від 100мг до 300мг композиції контрольованого вивільнення. Додатковий варіант здійснення згідно з винаходом забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози в 100мг забезпечує середнє AUC0-Tmax приблизно від 610нг×годину/мл приблизно до 630нг×годину/мл. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози 200мг забезпечує середнє AUC0-Tmax приблизно від 910нг×годину/мл приблизно до 920нг×годину/мл. Додатковий варіант здійснення згідно з винаходом забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози 300мг забезпечує середнє AUC0-Tmax приблизно від 1570нг×годину/мл, приблизно до 1590нг×годину/мл. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує середнє співвідношення AUC0-24 /AUC0-¥ концентрації трамадолу в плазмі, яка коливається в межах приблизно від 70% і приблизно до 85%. Переважно середнє співвідношення AUC0-24/AUC0-¥ концентрації трамадолу в плазмі коливається в межах приблизно від 74% і приблизно до 80%. У результаті приблизно від 15% приблизно до 30% введеної дози продовжує циркулювати в плазмі 24 години після введення, в залежності від введеної дози. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує відношення С'max до дози, вивільненої у плазму крові в перші 24 години (AUC0-24 /AUC0-¥, помножене на дозу) приблизно від 1,10 приблизно до 1,35. Переважно співвідно 23 84277 шення складає приблизно від 1,15 приблизно до 1,31. Додатковий варіант здійснення згідно з винаходом забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує відношення C'max/T max для введеної дози приблизно від 0,10 приблизно до 0,20. Переважно це відношення складає приблизно від 0,12 приблизно до 0,19. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного 24 добового прийому, яка при первинному прийомі дози забезпечує нахил у нг/мл-годину, після пікового рівня концентрації у плазмі крові, який не перевищує коефіцієнт приблизно 0,035 загальної дози, введеної у мг. Переважно коефіцієнт становить приблизно 0,03. Параметри фармакокінетики трамадолу композиції з контрольованим вивільненням представлені у Таблиці 2. Таблиця 2 Узагальнені результати фармакокінетичних параметрів трамадолу Композиція. Вміст Кількісн. актив - Доза показник (мг) розпоної речоділу в ини (мг) 100 100 Середнє арифметичне SD 200 200 Середнє арифметичне SD 300 300 Середнє арифметичне SD 200 400 Середнє арифметичне SD С'max AUC0-¥ AUC0-T max (нг/мл) (нг/мл) (нг/мл) Період С'max/ Rstart напівСЧЗ (го- ТСК (го- AUC0-24 AUC0-¥ lz (го(гов иве-1 дина) дина) (нг/мл) (годи- дина ) дина) дення -1 на ) (година) AUC0-24/ AUC0-¥ 91,3 2108 625 0,0442 0,118 21,2 6,11 16,03 22,5 1635 78,9 26,83 196,55 731 4416 0,0052 915 471 0,024 0,0455 0,118 4,3 22,9 1,31 6,11 2,13 16,46 3,4 23,5 465 3374 6,60 77,2 58,33 290,08 1192 6741 567 1578 0,0108 0,025 0,0432 0,115 5,0 24,8 1,26 6,30 2,28 17,60 4,5 25,4 860 4900 8,1 73,9 147,16 487,35 2156 9332 1338 HO 0,0126 0,023 0,0544 0,120 4,4 21,1 1,52 6,11 3,03 15,33 6,6 HO 1544 7471 10,1 80,0 210,43 3767 но 0,0198 0,027 6,5 1,53 2,83 HO 2887 10,1 HO - не обчислений Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує відношення С'max, обчислене відносно концентрації у плазмі крові Одесметилтрамадолу, для дози трамадолу приблизно від 0,19 приблизно до 0,22. Переважно відношення складає приблизно від 0,20 до 0,21. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує концентрацію Одесметилтрамадолу в плазмі, яка підвищується рівномірно, доки не досягне піка концентрацій трамадолу при Тmax приблизно від 8 годин приблизно до 16 годин. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує концентрацію Одесметилтрамадолу в плазмі, яка після Тmax зменшується повільно, але рівномірно, відображуючи безперервну абсорбцію трамадолу та утворення послідовних метаболітів, у доповнення до проце сів елімінації. Переважно зменшення концентрації О-десметилтрамадолу в плазмі відбувається згідно з логарифмічним законом, з уявним середнім періодом напіввиведення приблизно від 6,7 години і приблизно до 8,1 години. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує утворення метаболіту щонайменше протягом 18 годин. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози, після Тmax забезпечує зниження концентрації О-десметилтрамадолу в плазмі згідно з логарифмічним законом, з константою швидкості уявної остаточної елімінації (lz), що становить приблизно 0,1 години-1. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує тривалість середньої величини (ТСК) О-десметилтрамадолу в діапазоні приблизно від 25,6 години приблизно до 28,1 години. 25 84277 Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує відношення С'max до AUC0-¥, обчислене відносно концентрації Одесметилтрамадолу в плазмі, приблизно 0,04 години-1. Відношення С'max/AUC0-¥ застосовується для оцінки швидкості утворення метаболіту. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує середнє AUC0-24, обчислене відносно концентрації О-десметилтрамадолу в плазмі, яка зростає пропорційно дозі, в діапазоні дозування від 100мг до 300мг композиції контрольованого вивільнення. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози 100мг забезпечує середнє AUC0-Tmax відносно концентрації О-десметилтрамадолу в плазмі приблизно від 175нг×годину/мл приблизно до 180мг×годину/мл. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози 200мг забезпечує середнє AUC0-Tmax відносно концентрації О-десметилтрамадолу в плазмі приблизно від 530нг×год./мл приблизно до 550нг×год./мл. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози 300мг забезпечує середнє AUC0-Tmax відносно 26 концентрації О-десметилтрамадолу в плазмі приблизно від 580нг×годину/мл приблизно до 590нг×годину/мл. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка при первинному прийомі дози забезпечує середнє відношення AUC0-24 /AUC0-¥, концентрації Одесметилтрамадолу в плазмі, що коливається в межах приблизно від 65% і приблизно до 80%. Переважно середнє відношення AUC0-24/AUC0-¥, концентрації О-десметилтрамадолу в плазмі коливається в межах приблизно від 68% і приблизно до 75%. У результаті приблизно від 25% приблизно до 32% активного метаболіту продовжує циркулювати в плазмі 24 години після введення дози. Додатковий варіант здійснення даного винаходу забезпечує композицію для однократного добового прийому, яка, при первинному прийомі дози забезпечує відношення С'max, розрахованого відносно концентрації О-десметилтрамадолу в плазмі, до концентрації О-десметилтрамадолу в плазмі крові у перші 24 години (AUC0-24/AUC0-¥, помножене на дозу трамадолу) приблизно від 0,0025 приблизно до 0,0035. Переважно відношення складає приблизно від 0,0027 приблизно до 0,0031. Параметри фармакокінетики Одесметилтрамадолу композиції з контрольованим вивільненням представлені в Таблиці 3. Таблиця 3 Узагальнені результати фармакокінетичних параметрів трамадолу Композиція. Вміст Кількісн. актив - Доза показник (мг) розпоної ділу речов ини (мг) 100 100 Середнє арифметичне SD 200 200 Середнє арифметичне SD 300 300 Середнє арифметичне SD 200 400 Середнє арифметичне SD HO - не обчислений С'max AUC0-¥ AUC0-T max (нг/мл) (нг/мл (нг/мл) С'max/ AUC0-¥ (година-1) Rstart Період ТСК lz (годи- (годи-на) напів- (година) в ивена-1) дення (година) AUC0-24 (нг/мл) AUC0-24/ AUC0-¥ (%) 20,38 520 179 0,0394 0,106 23,1 6,96 25,6 380 72,5 6,67 43,13 170 1080 92 540 0,0054 0,0395 0,256 0,111 4,2 25,1 1,91 6,69 2,9 26,3 123 782 7,69 71,3 16,53 59,88 328 1641 164 587 0,0079 0,0374 0,029 0,102 4,0 25,8 1,84 7,36 5,0 28,1 259 1107 8,8 67,9 19,19 538 114,34 2866 312 HO 0,0092 0,0457 0,029 0,094 3,6 18,7 2,21 8,14 6,6 HO 346 1909 11,0 74,6 46,39 HO 0,0147 0,028 5,5 2,98 HO 651 10,9 773 27 84277 Приклад 4 (і)Пропорційність дози - однократна доза Вивчення біодоступності проводилося для оцінки пропорційності дози між трьома дозуваннями активної речовини (100мг, 200мг і 300мг). Дане вивчення проводилося з відповідним періодом вимивання між кожним введенням. Дози брали у 27 здорових добровольців в умовах голодування. 28 На фігурі 3 зображені залежності середньої концентрації у плазмі трамадолу від часу, отримані у суб'єктів після введення композиції згідно з винаходом із контрольованим вивільненням (дози 100мг, 200мг і 300мг трамадолу HCl). Дані, використані для створення Фігури 3, включені до Таблиці 4. Таблиця 4 Середнє (±SD) концентрації трамадолу в плазмі (нг/мл) Час 0 1 2 3 4 5 6 8 10 12 16 20 24 30 36 48 100мг доза композиції згідно з 200мг доза композиції згідно з 300мг доза композиції згідно з винаходом контрольованого винаходом контрольованого винаходом контрольованого вивільнення вивільнення вивільнення 0 0 0 41,8±14,1 82,5±24,1 110,2±36,7 60,0±14,6 129,2±25,7 168,6±52,1 69,2±20,2 156,5±37,0 218,1±82,3 72,5±21,8 164,0±44,9 242,0±96,2 81,7±24,2 177,2±61,8 277,1±153,8 77,9±24,7 169,2±58,1 260,3±134,8 83,0±25,6 164,1±52,7 243,6±127,1 81,0±24,7 157,8±57,8 219,8±101,6 84,4±25,3 156,4±55,9 223,4±85,1 73,0±24,1 152,8±42,0 209,9±70,2 56,4±19,4 121,0±34,4 185,7±62,7 47,2±20,9 101,6±38,2 157,0±60,4 26,8±15,0 56,4±28,3 99,9±50,3 13,2±9,4 29,1±18,7 55,9±37,9 3,7±3,5 8,5±6,7 15,7±13,1 Результати даного дослідження показали, що дози в 100мг, 200мг і 300мг композиції контрольованого вивільнення згідно з винаходом є пропорційними відносно швидкості та міри абсорбції трамадолу та швидкості і міри утворення Одесметилтрамадолу. Було проведено вивчення біодоступності для характеристики фармакокінетичних властивостей композиції згідно з винаходом з контрольованим вивільненням і демонстрацією схожої дії лікарського засобу і/або його активного метаболіту в порівнянні з контрольним продуктом. Приклад 5 (іі) Порівняння двократної добової композиції з однократною дозою 2´200 дозування композиції контрольованого вивільнення згідно з винаходом порівнювали з композицією щоденного дворазового прийому, Topalgic® LP (200мг) таблетки, виготовлені Laboratoires Hoechst Houde, у порівняльному ви вченні біодоступності після введення в умовах голодування 24 здоровим добровольцям. Фармакокінетичні результати введення композиції з контрольованим вивільненням згідно з винаходом порівнювали з результатами дворазового введення (через 12-годинні інтервали) контрольної композиції, щоб оцінити біоеквівалентність між тестовим і контрольним продуктом. На основі розрахунку 90% довірчого інтервалу відношень тестового до контрольних середніх геометричних значень, міра впливу (така, що визначається оцінкою AUC0-t і AUC0-¥ трамадолу після коректування дози) була в межах загальноприйнятого біоеквівалентного інтервалу 80-125% для логарифмічно перетворених параметрів. Таким чином, було виявлено, композиція з контрольованим вивільненням згідно з винаходом і щоденна дворазова композиція є біоеквівалентними у плані загального впливу трамадолу. Результати для трамадолу AUC0-¥ представлені в Таблиці 5. 29 84277 30 Таблиця 5 Порівняння AUC0-¥ (Однократна доза в порівнянні зі щоденною дворазовою композицією) Лікування Середнє арифметичне±SD (нг×годину/мл) Середнє геометричне співвідношення (90% довірчого інтервалу) 2´200мг доза композиції згідно з винаходом з контрольованим вивільненням 1´200 Topalgic® LP BID 9332±3767 103 (98-109) 8897±3124 На фігурі 5 представлені графіки тимчасової залежності середнього арифметичного концентрації трамадолу в плазмі, отриманій після введення однократної добової композиції згідно з винаходом з контрольованим вивільненням і контрольним продуктом, що вводиться в день через 12 годинні інтервали у 24 здорових добровольців. Дані, використані для отримання фігури 5, включені до Таблиці 6. Таблиця 6 Середні концентрації трамадолу в плазмі (нг/мл)Час 0 1 2 3 4 5 6 9 12 16 20 24 30 36 48 Тестова композиція Конц. 2´200мг доза композиції згідно з винаходом 0 138,49±58,62 257,56±81,20 350,21±166,42 373,93±124,33 427,66±166,90 424,72±176,20 408,61±196,28 357,88±162,48 312,70±153,34 243,94±117,93 184,96±102,9 99,78±61,60 51,01±43,33 0 на фігурі 6 представлені графіки тимчасової залежності середнього арифметичного концентрації у плазмі О-десметилтрамадолу, отриманого після однократного добового введення композиції контрольованого вивільнення згідно з винаходом і Контрольна композиція Час Конц. 200мг BID 0 1 2 3 4 5 6 9 12 13 14 15 16 17 18 21 24 30 36 48 0 101,93±43,72 226,89±72,90 296,35±99,46 318,22±91,27 330,88±98,68 281,67±85,95 236,39±87,89 167,41±65,49 181,96±70,51 284,67±126,76 378,82±136,23 396,87±146,56 388,83±142,32 396,38±140,65 331,81±121,52 275,00±110,61 118,69±64,92 54,04±39,07 0 контрольного продукту протягом одного дня з 12 годинними інтервалами 24 здоровим добровольцям. Дані, використані для отримання фігури 6, включені до Таблиці 7. 31 84277 32 Таблиця 7 Середні (±SD) концентрації О-десметилтрамадолу в плазмі (нг/мл) Час 0 1 2 3 4 5 6 9 12 16 20 24 30 36 48 Тестова композиція Конц. 2´200мг доза композиції згідно з винаходом 0 29,82±17,0 57,8±17,0 76,3±31,6 84,9±30,9 98,0±41,4 100,6±41,7 99,9±41,7 96, 52±38,8 83,9±32,6 68,2±28,8 57,6±28,0 33,2±20,0 0 0 Приклад 6 (ііі) Порівняння двократної добової композиції та композиції безперервного вивільнення Дозування в 200мг композиції з контрольованого вивільнення згідно з винаходом порівнювали з двократною добовою композицією, таблетки Topalgic® LP (100мг), виготовлені Laboratoires Hoechst Houde, при порівняльному вивченні біодоступності після багаторазового введення в умовах голодування у 26 здорових добровольців. Результати даного дослідження показали, що композиція контрольованого вивільнення згідно з Контрольна композиція Час Конц. 200мг BID 0 1 2 3 4 5 6 9 12 13 14 15 16 17 18 21 24 30 36 48 0 17,7±14,6 48,3±17,5 66,2±25,9 74,3±26,2 80,64±29,2 74,3±26,1 68,1±24,6 56,6±22,1 59,1±23,8 75,1±32,6 92,6±38,0 96,7±37,0 97,0±34,5 100,4±33,6 93,0±32,4 83,3±37,8 44,4±21,6 18,1±16,8 0 винаходом є еквівалентною контрольному продукту за швидкістю та мірою абсорбції трамадолу та швидкістю та мірою утворення Одесметилтрамадолу. Порівняльна біодоступність двох продуктів оцінювалася на основі довірчого інтервалу для основної змінної AUCSS для трамадолу та О-десметилтрамадолу відносно загальноприйнятого діапазону біоеквівалентності від 80% до 125%. Результати для трамадолу AUCSS представлені в Таблиці 8. Таблиця 8 Порівняння AUCSS (Композиція один раз на день у порівнянні з щоденною дворазовою) Лікування Середнє арифметичне± SD (нг×годину/мл) 2´200мг доза композиції контрольованого вивільнення згідно з винаходом Topalgic® LP 100мг BID На фігурі 7 представлені графіки тимчасових кривих середнього арифметичного концентрації трамадолу в плазмі та О-десметилтрамадолу, після введення 200мг дози однократної добової композиції контрольованого вивільнення згідно з ви 5185±460 Відношення середніх геометричних (90% довірчий інтервал) 92,4 (87,5-97,5) 5538±214 находом та контрольного продукту (Topalgic® LP 100мг BID) протягом одного дня з 12-годинними інтервалами. Дані, використані для отримання фігури 7, включені до Таблиці 9. 33 84277 34 Таблиця 9 Середні (±SD) концентрації трамадолу та О-десметилтрамадолу в плазмі (нг/мл) Композиція, що тестується (200мг доза композиції контрольованого вивільнення згідно з винаходом) Час Трамадол Метаболіт 0 113,3±48,8 37,6±9,0 1 195,4±58,4 49,9±13,9 2 249,5±61,0 58,9±14,4 3 285,0±66,0 65,4±16,3 4 290,6±65,5 66,2±16,0 5 298,9±81,1 67,3±16,7 6 280,0±70,7 67,7±17,5 9 244,9±58,4 63,9±16,8 12 226,0±70,2 59,8±17,2 16 209,4±73,4 57,3±14,8 20 161,5±68,9 47,9±12,1 24 119,9±59,1 37,1±8,9 Обсяг даного винаходу не обмежується конкретними варіантами здійснення, розкритими в даних прикладах, які призначені для ілюстрації найбільш переважних варіантів здійснення винаходу. Дійсно, різні модифікації винаходу або інші варіанти здійснення, які функціонально рівноцінні тим, що показані й описані тут, стають очевидними для фа хівців в даній галузі та призначені для захисту прикладеною формулою винаходу. Контрольна композиція (100мг BID) Час 0 1 2 2,5 3 3,5 4 4,5 5 6 7 9 12 13 14 14,5 15 15,5 16 16,5 17 18 19 21 24 Трамадол 157,8±48,8 220,2±61,1 251,6±60,9 282,7±65,3 290,8±59,7 290,9±70,6 297,3±71,3 305,2±75,2 281,8±65,5 262,8±55,5 243,9±60,2 198,0±54,4 154,6±47,8 203,5±55,4 260,7±54,2 307,2±59,9 303,7±60,5 290,7±54,3 289,0±54,6 276,4±53,2 267,6±55,2 244,6±58,4 237,1±59,4 201,5±52,7 156,9±49,9 Метаболіт 49,1±10,7 58,1±12,9 63,1±14,6 68,0±14,7 69,4±15,6 69,6±15,7 71,3±15,3 72,8±15,6 69,1±15,7 67,4±17,3 64,9±15,2 57,0±12,8 46,2±10,5 53,2±12,8 63,7±15,0 72,2±16,5 73,2±17,1 71,3±16,8 72,1±15,6 72,1±16,8 71,6±16,8 68,2±15,0 66,41±14,8 57,9±12,0 49,6±10,1 Тут використовувався ряд посилань, які в повному обсязі входять до даного опису. Незважаючи на те, що були описані різні приклади комбінованих елементів згідно з винаходом, зрозуміло також, що вони не є вичерпними, і ознаки одного варіанту здійснення можуть бути об'єднані з ознаками іншого, а такі інші комбінації розглядаються в межах обсягу розкритого тут винаходу. 35 84277 36 37 84277 38 39 Комп’ютерна в ерстка Л.Литв иненко84277 Підписне 40 Тираж 28 прим. Міністерство осв іт и і науки України Держав ний департамент інтелектуальної в ласності, вул. Урицького, 45, м. Київ , МСП, 03680, Україна ДП “Український інститут промислов ої в ласності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюSustained-release tramadol formulations with 24-hour efficacy
Автори англійськоюLenaerts Vincent, Ouadji-Njiki Patricia Laure, Bacon Jonathan, Ouzerourou Rachid, Gervais Sonia, Rahmouni Milloud, Smith Damon
Назва патенту російськоюКомпозиция трамадола с пролонгированным высвобождением с 24-часовым действием
Автори російськоюЛенар Венсан, Оуаджи-Нджики Патрисия Лаура, Бейкон Джонатан, Узеруру Рашид, Жерве Соня, Рахмуни Милу, Смит Деймон
МПК / Мітки
МПК: A61K 31/135, A61P 25/04, A61K 9/22
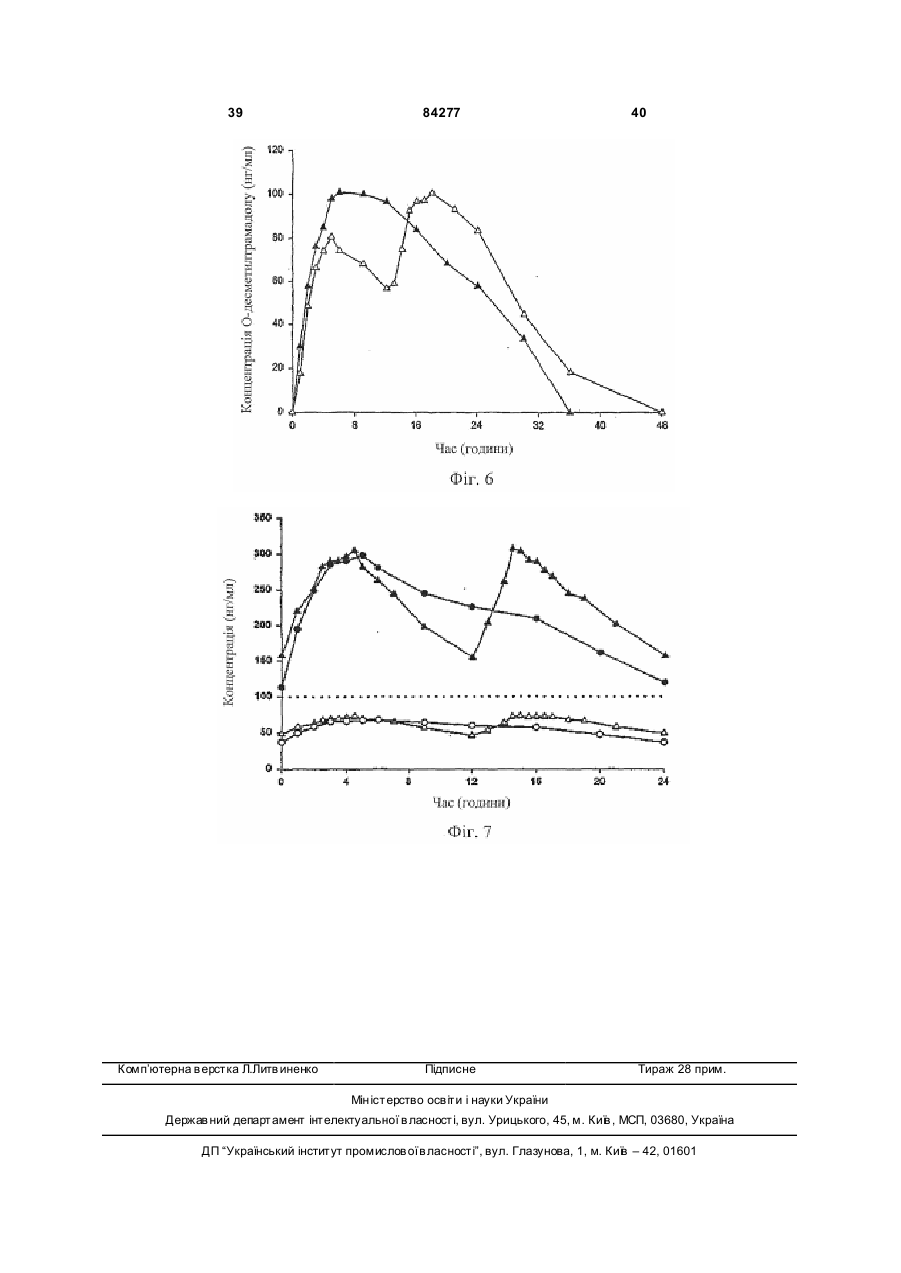
Мітки: композиція, дією, 24-годинною, вивільненням, пролонгованим, трамадолу
Код посилання
<a href="https://ua.patents.su/20-84277-kompoziciya-tramadolu-z-prolongovanim-vivilnennyam-z-24-godinnoyu-diehyu.html" target="_blank" rel="follow" title="База патентів України">Композиція трамадолу з пролонгованим вивільненням з 24-годинною дією</a>
Фармацевтична композиція з контрольованим вивільненням, що містить гідрохлорид трамадолу, і спосіб її одержання

Номер патенту: 76411
Опубліковано: 15.08.2006
Автори: Седларова Гелена, Земанек Марьян, Гаттнар Ондрей, Разус Любослав, Варга Іван
МПК: A61P 29/00, A61K 9/22, A61K 31/135
Мітки: гідрохлорид, спосіб, трамадолу, фармацевтична, містить, композиція, одержання, контрольованим, вивільненням
Формула / Реферат:
1. Фармацевтична композиція в таблетованій формі з контрольованим вивільненням, що містить гідрохлорид трамадолу, яка відрізняється тим, що вона містить від 100 до 200 мг активного інгредієнта в суміші з тонкоподрібненими складними ефірами гліцерину з вищими жирними кислотами в кількості від 10 до 53 % мас., солями фосфорної кислоти і лугів в кількості від 20 до 41 % мас., неіонними вінілпіролідоновими полімерами в кількості від 1,15 до 1,75...
Композиція з пролонгованим вивільненням антибіотика(ів) та спосіб виготовлення імплантанта за її допомогою

Номер патенту: 71030
Опубліковано: 15.11.2004
Автори: Фогт Себастіан, Кюн Клаус-Дітер, ШНАБЕЛЬРАУХ Маттіас
МПК: A61P 31/00, A61K 31/65, A61K 9/22, A61L 27/00, A61K 31/726, A61K 47/20
Мітки: композиція, імплантанта, вивільненням, спосіб, допомогою, антибіотика(ів, виготовлення, пролонгованим
Формула / Реферат:
1. Композиція антибіотика/антибіотиків, яка відрізняється тим, що містить суміш, яка складається із принаймні одного амфіфільного компонента як представника алкілсульфатів, арилсульфатів, алкіларилсульфатів, циклоалкілсульфатів, алкілциклоалкілсульфатів, алкілсульфаматів, циклоалкілсульфаматів, алкілциклоалкілсульфаматів, арилсульфаматів, алкіларилсульфаматів, алкілсульфонатів, сульфонатів-2-жирних кислот, арилсульфонатів,...
Галенова форма препарату з пролонгованим вивільненням молсидоміну

Номер патенту: 73972
Опубліковано: 17.10.2005
Автор: Жезі Джозеф-Мішель
МПК: A61K 31/535, A61K 9/20, A61P 9/00
Мітки: пролонгованим, галенова, препарату, форма, молсидоміну, вивільненням
Формула / Реферат:
1. Тверда пероральна форма галенового препарату молсидоміну з пролонгованим вивільненням вказаної речовини, яка відрізняється тим, що містить від 14 до 24 мг молсидоміну або одного з його активних метаболітів на одиницю дози і тим, що швидкість її розчинення in vitro [вимірювана спектрофотометрично при довжині хвилі 286 або 311 нм відповідно до засобу, що викладений в Pharmacopee Europeenne, 3-е вид. (або U.S.P. XXIV) при 50 об/хв. у 500 мл...
Таблетка з миттєвим і в подальшому пролонгованим вивільненням діючих речовин та спосіб одержання таблетки

Номер патенту: 55503
Опубліковано: 15.04.2003
Автори: Орландо Лоренс, Саславскі Олівер
МПК: A61K 9/22
Мітки: вивільненням, діючих, таблетка, одержання, таблетки, подальшому, спосіб, пролонгованим, миттєвим, речовин
Формула / Реферат:
1. Багатошарова таблетка з миттєвим і в подальшому пролонгованим вивільненням діючих речовин, що містить щонайменше два накладених один на одний шари, яка відрізняється тим, що:- перший зовнішній шар складається з суміші ексципієнтів і першої діючої речовини, цей перший шар дозволяє здійснювати миттєве вивільнення першої діючої речовини;- другий шар, що контактує з першим шаром, складається з біологічно нерозкладного інертного...
Фармацевтична композиція, що містить велику кількість одиниць трамадолу, і спосіб її приготування

Номер патенту: 52679
Опубліковано: 15.01.2003
Автори: Рабер Марк, Момбергер Гельмут, Шмід Вольфганг, Кухн Дітер
МПК: A61P 29/00, A61K 9/52, A61K 31/135
Мітки: композиція, трамадолу, фармацевтична, містить, приготування, одиниць, спосіб, велику, кількість
Формула / Реферат:
1. Складна фармацевтична композиція, що включає активну речовину, носій та допоміжні фармацевтично прийнятні речовини, яка відрізняється тим, що окремі гранули з однаковим або різним уповільненням дії складаються з вкритого трамадолом або його фізіологічно прийнятними солями як активною речовиною носія, вкритого оболонкою з одного або декількох шарів мембрани для регульованого вивільнення активної речовини, які складаються з речовини,...
Попередній патент: Лікарський засіб проти туберкульозу
Наступний патент: Автоматичний запит на повтор по зворотній лінії зв’язку
Випадковий патент: Спосіб переробки сталеплавильного пилу