Спосіб одержання цеолітовмісного мікросферичного каталізатора крекінгу
Номер патенту: 28551
Опубліковано: 16.10.2000
Автори: Яковенко Олександр Миколайович, Випирайленко Валентина Йосипівна, Роде Геральд Григорович, Назарок Володимир Іванович, Ліхньовський Руслан Володимирович, Манза Іван Андрійович, Патриляк Любов Казимирівна, Самусь Леонтій Григорович, Храновська Валентина Іванівна, Патриляк Казимир Іванович


















Формула / Реферат
1. Спосіб одержання цеолітвмісного мікросферичного каталізатора крекінгу, що передбачає проведення стадій одержання каолінової мікросфери шляхом розпилювальної сушки висококонцентрованих (55-65% мас.) водних суспензій каоліну в присутності дефлокулянта, одержання "мулітової" мікросфери шляхом прожарювання каолінової мікросфери в інтервалі температур 925-1100°С протягом не менше двох годин, одержання метакаолінової микросфери шляхом прожарювання каолінової мікроби в інтервалі температур 660-815°С протягом не менше двох годин, змішування "мулітової" мікросфери, метакаолівової мікроофери і водного розчину гідроксиду натрію, визрівання реакційної суміші, вводу до цієї суміші додаткової кількості "мулітової" мікросфери і додаткової кількості водного розчину гідроксиду натрію, кристалізації цеоліту в мікросфери, вилучення цеолітвмісної мікросфери, її промивки і сушки, перетворення цеолітвмісної мікросфери в каталізатор крекінгу шляхом іонного обміну, промивки, сушки, стабілізації і термічної активації, який відрізняється тим, що перед стадією змішування "мулітової" мікросфери, метакаолінової мікроофери і водного розчину гідроксиду натрію "мулітову" і метакаолінову мікросферу піддають роздільній "мокрій" очистці від заліза, промивці водою і сушці.
який відрізняється тим, що
2. Спосіб за п. 1, який відрізняється тим, що для очистки "мулітової" і метакаолінової мікроби використовують водні розчини кислот.
3. Спосіб за п. 1, який відрізняється тим, що очистка "мулітової" і метакаолінової мікрозфери від заліза супроводжується їх частковим деалюмінуванням.
4. Спосіб за п. 3, який відрізняється тим, що ступінь деалюмінування "мулітової" і метакаолінової мікросфери під час їхньої очистки від валіза не повинна перевищувати 0,2 і 0,6 відповідно.
5. Спосіб за п. 4, який відрізняється тим, що перед стадією визрівання завантажена в реактор суміш "мулітової" мікросфери із ступенем деалюмінування ![]() , метакаолінової мікросфери із ступенем деалюмінування в і водного розчину гідроксиду натрію з масовою часткою NaOH
, метакаолінової мікросфери із ступенем деалюмінування в і водного розчину гідроксиду натрію з масовою часткою NaOH ![]() має склад, який характеризується тим, що величина її лежить в інтервалі значень 0,16-0,20, масова частка деалюмінованої "мулітової" мікросфери в її суміші з деалюмінованою метакаоліновою мікросферою g1 визначається залежністю
має склад, який характеризується тим, що величина її лежить в інтервалі значень 0,16-0,20, масова частка деалюмінованої "мулітової" мікросфери в її суміші з деалюмінованою метакаоліновою мікросферою g1 визначається залежністю
![]() ,
,
в якій величина у1 лежить в інтервалі значень 0,8-0,9, масове співвідношення розчин гідроксиду натрію/суміш "мулітової" і метакаолінової мікросфери n1 визначається залежністю
![]() ,
,
в якій величина z1 лежить в інтервалі значень 0,6-0,7.
6. Спосіб за п.4, який відрізняється тим, що перед стадією кристалізації цеоліту до завантаженої в реактор суміші, що перебувала в ньому під час визрівання, додатково додаються "мулітова" мікросфера із ступенем деалюмінування ![]() при масовому співвідношенні додаткова кількість деалюмінованованої "мулітової" мікросфери/кількість "мулітової" і метакаолінової мікросфери при визріванні
при масовому співвідношенні додаткова кількість деалюмінованованої "мулітової" мікросфери/кількість "мулітової" і метакаолінової мікросфери при визріванні ![]() , яке визначається залежністю
, яке визначається залежністю
![]() ,
,
де величина у2 дорівнює 0,95, і водний розчин гідроксиду натрію з масовою часткою NaOH ![]() , рівною
, рівною ![]() , при масовому співвідношенні додаткова кількість розчину гідроксиду натрію/кількість "мулітової" і метакаолінової мікросферии при визріванні n2, яке визначається залежністю
, при масовому співвідношенні додаткова кількість розчину гідроксиду натрію/кількість "мулітової" і метакаолінової мікросферии при визріванні n2, яке визначається залежністю
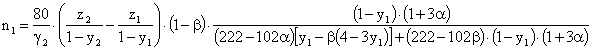
де величина z2 дорівнює величині z1.
Текст
1. Спосіб одержання цеолітвмісного мікросферичного каталізатора крекінгу, що передбачає проведення стадій одержання каолінової мікросфери шляхом розпилювальної сушки висококонцентрованих (56-65% мас.) водних суспензій каоліну в присутності дефлокулянта, одержання "мулітової" мікросфери шляхом прожарювання каолінової мікросфери в інтервалі температур 925-1100°С протягом не менше двох годин, одержання метакаолінової микросфери шляхом прожарювання каолінової мікросфери в інтервалі температур 650815°С протягом не менше двох годин, змішування "мулітової" мікросфери, метакаолінової мікросфери і водного розчину гідроксиду натрію, визрівання реакційної суміші, вводу до цієї суміші додаткової кількості "мулітової" мікросфери і додаткової кількості водного розчину гідроксиду натрію, кристалізації цеоліту в мікросфері, вилучення цеолітвмісної мікросфери, її промивки і сушки, перетворення цеолітвмісної мікросфери в каталізатор крекінгу шляхом іонного обміну, промивки, сушки, стабілізації і термічної активації, який відрізняється тим, що перед стадією змішування "мулітової" мікросфери, метакаолінової мікросфери і водного розчину гідроксиду натрію "мулітову" і метакаолінову мікросферу піддають роздільній "мокрій" очистці від заліза, промивці водою і сушці. 2. Спосіб за п. 1, що відрізняється тим, що для очистки "мулітової" і метакаолінової мікросфери від заліза використовують водні розчини кислот. 3. Спосіб за п. 1, що відрізняється тим, що очистка "мулітової" і метакаолінової мікросфери від заліза супроводжується їх частковим деалюмінуванням. 4. Спосіб за п. 3, що відрізняється тим, що ступінь деалюмінування "мулітової" і метакаолінової 28551 h2 = 80 æ z 2 z1 ö ÷ (1 - b) ´ ç g 2 ç 1 - y 2 1 - y1 ÷ ø è ´ (1 - y1)(1 + 3a) , (222 - 102a)[y1 - b( 4 - 3y1)] + (222 - 102b)(1 - y1)(1 + 3a) де величина z2 дорівнює величині z1. Винахід належить до способів одержання цеолітвмісного мікросферичного каталізатора, який застосовується в нафтопереробній промисловості для крекінгу вуглеводнів. Сучасні процеси крекінгу базуються на майже виключному використанні в них цеолітвмісних мікросферичних каталізаторів. В частинках цих каталізаторів поєднані дві компоненти - цеолітна і матрична, кожна з яких має своє призначення. Цеолітна компонента є носієм каталітичної активності і являє собою хімічну модифікацію початкової, неактивної натрійової форми цеоліту, в якій іони натрію в результаті іонного обміну заміщені на іони амонію, рідкісноземельних елементів або деякі інші. Матрична компонента відіграє роль "несучої конструкції" частинки каталізатора. Вона повинна дозволяти створювати потрібну форму частинок каталізатора, забезпечувати його необхідний гранулометричний склад і високий опір стиранню, сприяти легкому доступу до цеолітних кристалів сировини і відводу від них продуктів перетворення, бути термічне стійкою, захищати цеоліт від отруйної дії деяких сировиннх домішок. Сферичної форми частинки каталізатора діаметром до 100-160 мкм набувають під час розпилювальної сушки водної суспензії відповідного складу і практично не змінюють її в процесі реалізації подальших технологічних стадій виробництва каталізатора - прожарювання, синтезу цеолітної структури, її хімічного модифікування шляхом іонного обміну тощо. При цьому можливі два випадки: в першому випадку тверда фаза суспензії є механічною сумішшю окремо одержаних матричної і цеолітної (в активній або неактивній формі) компонент, а в другому - матеріалом, з якого за певних умов можна одночасно одержати обидві ці компоненти в уже сформованій мікросфері. У другому випадку цеолітна компонента спочатку одержується в неактивній (натрійовій) формі, яка потім може бути перетворена в активну (іонзаміщену) форму. Матеріалом, який може слугувати сировиною для одночасного одержання матричної і цеолітної компонент, а, в кінцевому рахунку, - для синтезу цеолітвмісного мікросферичного каталізатора крекінгу, є, зокрема, така природна сировина, як каолін. Спосіб одержання цеолітвмісного мікросферичного каталізатора крекінгу, який передбачає використання відповідним чином прожареної каолінової мікросфери для синтезу в ній цеоліту, одержав назву способу "in situ" ("на місці"). Загальновідомо, що іонзаміщені форми цеоліту Y виявляють високу каталітичну активність в процесах конверсії вуглеводнів, зокрема, в процесі крекінгу. Використання каоліну в способі "in situ" грунтується на можливості одержання цеоліту Y з каоліну за таких умов, які можуть бути реалізовані в промисловості. Запропоновані в патентній літературі варіанти реалізації способу "in situ" можна розділити на дві групи - групу варіантів з удосконалення технології одержання цеолітвмісного мікросферичного каталізатора крекінгу, яка базується на синтезі цеоліту Y при реагуванні каоліну, прожареного при температурі 650-815°С (метакаоліну), з розчином силікату натрію, і групу варіантів з удосконалення технології такого ж призначення, яка базується на синтезі цеоліту Y при реагуванні каоліну, прожареного при температурі 925-1100°С (умовно назвемо його "мулітом"), з розчином гідроксиду натрію. Основним джерелом алюмінію і кремнію для синтезу цеоліту Y за варіантами першої групи є метакаолін, а за варіантами другої групи - "муліт". "Муліт" це продукт перетворення метакаоліну АІ2О3×2SіO2 при вказаній вище температурі 925-1100°С в малоактивну алюмокремнійову шпінель 2АІ2O3×3SіО2 і аморфний кремнезем SiO2 з високою реакційною здатністю [1]. Пропонований винахід відноситься до другої групи варіантів реалізації способу "in situ". В цьому випадку процесу кристалізації цеоліту в мікросфері прожареного каоліну, яка здійснюється при температурі 80-100°С, передує контакт цієї мікросфери з розчином гідроксиду натрію при температурі 3050°С протягом певного часу (процес визрівання). Під час процесу визрівання кристалізація цеоліту не проходить, а відбувається просочування частинок мікросфери розчином лугу. Кристалізація цеоліту в мікросфері супроводжується значною екстракцією кремнію, внаслідок чого утворюється розчин силікату натрію. Оскільки повного перетворення в цеоліт прожареного каоліну в мікросфері не відбувається, то матричною компонентою утвореної цеолітвмісної мікросфери є пористий збагачений алюмінієм і, навпаки, збіднений кремнієм алюмосилікатний залишок від вилуговування розчином гідроксиду натрію початкової мікросфери прожареного каоліну. Серед відомих удосконалень другої групи варіантів реалізації способу "in situ" найважливішим є використання невеликих кількостей метакаоліну у вигляді порошку або мікросфери як добавки до реакційної суміші "мулітової" мікросфери і водного розчину гідроксиду натрію на стадії синтезу цеоліту Y, що дозволяє значно підвищити його вихід і збільшити опір мікросфер стиранню. При цьому необхідно, щоб "муліт" і метакаолін, які надходять на синтез цеоліту, були глибоко очищеними від сполук заліза, оскільки останні перешкоджають кристалізації цеоліту [2] і поглиблюють утворення коксу під час крекінгу [3]. Очистку каоліну від заліза (а також титану) проводять хімічними методами або шляхом поєднання цих методів з деякими фізичними методами (зокрема, з високоінтенсивною магнітною сепарацією), які їм передують [4]. Відомі дві групи методів хімічної очистки каоліну - "мокрий" і "сухий" [4, 5]. Останній метод, який базується на взаємодії присутніх в каоліні сполук заліза і титану з хлорвмісними газами (хлор, хлористий водень і ін.) при температурі 500-800°С з утворенням летких хлоридів 2 28551 заліза і титану, не дивлячись на високий ступінь очистки, не здобув широкого розповсюдження в зв'язку з тим, що в умовах високих температур відбувається втрата пластичності каоліну, що є небажаним для багатьох сфер подальшого його використання, зокрема, при формуванні мікросфери. Основним методом хімічної очистки каоліну є "мокрий" метод. В основі цього методу, який реалізується при температурах, що не перевищують 120°С [5], шляхом взаємодії сполук заліза і титану з водними розчинами кислот, відновників, сумішей кислот і відновників (з добавкою до цих розчинів інших речовин або без неї), лежить утворення добре розчинних в воді солей заліза і титану. Очистці від заліза принципово можна піддавати як вихідну фракцію каолінового порошку з суттєвим вмістом частинок субмікронного розміру, яка в подальшому використовується для одержання мікросфери з високими міцністю і насипною вагою, так і сформовану з цієї фракції мікросферу, попередньо прожарену до "муліту" чи метакаоліну. Проте, до цього часу в способі "іn situ" використовують тільки першу з названих можливостей, в тому числі і в роботах [6, 7], які є аналогами пропонованого винаходу. Спосіб одержання цеолітвміоного мікросферичного каталізатора крекінгу за технологією, описаною в роботі [6], передбачає проведення таких стадій: а) одержання каолінової мікросфери шляхом розпилювальної сушки висококонценторованих (55-65% мас.) водних суспензій очищеного каолінового поршку із значним вмістом частинок субмікронного розміру; б) одержання "мулітової" мікросфери в результаті прожарювання при температурі 925-1100°С частини каолінової мікросфери із стадії "а"; в) одержання метакаолінової мікросфери в результаті прожарювання при температурі 650815°С частини каолінової мікросфери із стадії "а"; г) змішування "мулітової" мікросфери із стадії "б", метакаолінової мікросфери із стадії "в" і водного розчину гідроксиду натрію; д) визрівання суміші із стадії "г"; е) підігрів суміші із стадії "д" і її витримування при підвищеній температурі доти, поки не пройде кристалізація цеоліту NaY в мікросферах суміші; ж) вилучення цеолітвмісної мікросфери із стадії "е", її промивка і сушка; к) перетворення цеолітвмісної мікросфери із стадії "ж" в каталізатор крекінгу шляхом заміщення іонів натрію її цеолітвмісної компоненти іншими придатними для цього катіонами, промивки водою, сушки, стабілізації і термічної активаціі повітрям і (або) водяною парою. Спосіб одержання цеолітвмісного мікросферичного каталізатора крекінгу, описаний в роботі [7], відрізняється від способу, описаного в роботі [6], лише тим, що в ньому використовується не метакаолінова мікросфера, а очищений від заліза метакаоліновий порошок з розміром частинок до 510 мкм. Тому в цьому способі стадії "в" і "г" можна сформулювати таким чином: в) одержання метакаолінового порошку в результаті прожарювання при температурі 650815°С каолінового порошку; г) змішування "мулітової" мікросфери із стадії "б", метакаолінового порошку із стадії "в" і водного розчину гідроксиду натрію. Решта стадій, тобто стадії "а", "б", "д", "е", "ж", "к", є спільними для обох способів. Принагідно відзначимо, що стадії "а", "б" і "в" найбільш детально описані в роботах [6-9]. Стадії "а", "б", "в", "ж", "к" способів одержання каталізатора крекінгу, описаних в роботах [6, 7], співпадають з аналогічними стадіями способу, пропонованого у винаході. Для підвищення активності каталізатора крекінгу бажаним є збільшення виходу цеоліту під час його синтезу в мікросфері прожареного каоліну [10, 11]. Сучасні кращі промислові зразки каталізатора крекінгу вміщують до 35% цеоліту [10], а новітні розробки націлені на одержання каталізатора з вмістом цеоліту до 70% і вище [11]. Типовий вихід цеоліту, який досягається при реалізації способів одержання каталізаторів крекінгу, описаних в роботах [6, 7], становить приблизно 15-25%. При цьому час кристалізації цеоліту Y з задовільним виходом не повинен бути особливо великим (в одному з прикладів роботи [6] його оптимальна величина оцінюється приблизно в 24 години), оскільки в протилежному випадку вихід цеоліту Y знову починає знижуватися при одночасному зростанні виходу небажаної цеолітної фази (цеоліту В). З урахуванням цього обмеження на час кристалізації лише в окремих рідких випадках вдається збільшити вихід цеоліту Y до 32% за рахунок збільшення часу визрівання. Крім того, не завжди збільшення часу визрівання одночасно призводить до зростання реакційної здатності суміші під час кристалізації. Таким чином, способи одержання каталізатора крекінгу, запропоновані в роботах [6, 7], не дозволяють з достатньою ефективністю забезпечити зростання виходу цеоліту вище 20-25%. В збагаченому каоліні головним мінералом є каолініт АІ2О3×2SіO2×2Н2O. Вміст в такому каоліні домішок оксидів заліза, титану, лужних і лужноземельних елементів складає лише декілька відсотків. Тому в першому наближенні молекулярною масою як сухого каолінового порошку, який використовується для одержання каолінової мікросфери, так і самої мікросфери, можна вважати молекулярну масу каолініту Мk (Мk=258). При прожарюванні каолінової мікросфери до утворення метакаоліну і особливо до утворення "муліту" відбувається деяке ущільнення частинок каоліну всередині мікросфери і зменшення її об'єму, що якоюсь мірою компенсує повну втрату наявної в каолініті структурної води під час такого прожарювання. Молекулярною масою метакаоліну і "муліту" можна вважати молекулярну масу повністю дегідратованого каолініту Мпр (Мпр=222). Молекулярною масою Мц цеолітвмісної мікросфери, тобто мікросфери, вилученої із реакційної суміші після закінчення синтезу цеоліту, промитої і висушеної (в розрахунку на повну дегідратацію її цеолітної компоненти) буде величина Мц=Мпр- 2MSiO2 × a * +MNa2O x , (1) де MSiO2 - молекулярна маса SiO2 ( MSiO2 =60); a* - частка початкової кількості SiO2 в прожареній мікросфері, яка при екстракції кремнію під 3 28551 Виразами в правій частині рівностей (1) і (3) не передбачено вводу в пори прожареної каолінової мікросфери алюміній- і кремнійвмісних розчинів, і тому ці вирази найбільш точними є у випадку синтезу цеоліту в "мулітовій" микросфері без добавок метакаоліну. Ці вирази можуть бути перенесені і на випадок, коли в процесі синтезу цеоліту беруть участь як "мулітова", так і метакаолінова мікросфера. В цьому випадку обидва види мікросфери залишаються єдиним цілим і на стадії синтезу цеоліту, і на подальших стадіях процесу одержання каталізатора крекінгу, а тому "перетоки" оксидів алюмінію і кремнію з метакаолінової мікросфери в "мулітову" мікросферу, які мають місце в даному випадку, на розрахунок величин Мц і Мкк впливу не мають, якщо значення величини a* залишається на рівні, близькому до 0,5, а вмістом АІ2О3 в маточному розчині силікату натрію можна знехтувати. Найнижчу точність рівності (1) і (3) мають у випадку синтезу цеоліту в "мулітовій" мікросфері в присутності метакаолінового порошку (який після закінчення процесу кристалізації цеоліту відфільтровують від "мулітової" мікросфери), оскільки в цьому випадку "перетоки" оксидів алюмінію і кремнію з метакаолінового поршку в "мулітову" мікросферу при розрахунку величин Мц і Мкк за допомогою рівностей (1) і (3) не враховуються і ці величини одержуються заниженими. Якщо масову частку заліза (в розрахунку на Fe2O3) у вихідному каоліні (або в початковій каоліновій мікросфері, утвореній внаслідок розпилювальної сушки), в прожареному каоліні (для утворення метакаоліну або "муліту"), цеолітвмісній мікросфері та готовому каталізаторі позначити відповідно ак, апр, ац і акк, то є очевидним, що (4) ак×Мк=апр×Мпр=ац×Мц=акк×Мкк. За допомогою співвідношення (4) можна вести перерахунок вмісту заліза в мікросфері, яка одержується на одній стадії процесу виробництва каталізаторів крекінгу, на вміст заліза в мікросфері, яка одержується на іншій стадії цього процесу. З огляду на приблизний характер рівностей (1) і (3) бажано знаходити величину апр (або ак) не на основі перерахунку, виходячи з величини ац (або акк), а безпосередньо на основі експерименту. На жаль, значення величини ак або апр в аналогах [6, 7] не наводяться. В роботі [7] (приклад 2) наведено тільки хімічний склад готового каталізатора крекінгу, зокрема, вміст в ньому оксидів Na2O і Fе2O3, який складає відповідно 1,14 і 0,38%. Навeдено також значення величин у і М (вони дорівнюють відповідно 0,26 і 4,62). Звідси випливає, що х=0,099 (при a*=0,5). В цьому прикладі синтез цеоліту проводили в "мулітовій" мікросфері в присутності метакаолінового порошку, коли точність розрахунків, як відзначалося вище, найнижча. Для іонного обміну використовували лише розчин нітрату амонію (y=0). Із співвідношення (3) при врахуванні виразу (1 - b*) × MNa2O × x mкк = , (5) Mпр - 2MSiO2 × a * + x(1 - b*) × MNa2O час синтезу цеоліту переходить в маточний розчин силікату натрію (її в першому наближенні можна прийняти рівною 0,5 [8]); MNa2O - молекулярна маса Na2O ( MNa2O =62); x - число молів синтезованого цеоліту в розрахунку на 1 моль прожареної мікроcфери; М - мольне відношення SіO2/АІ2О3 у синтезованому цеоліті. Величина х може бути розрахована із співвідношення y(Mпр - 2МSiO2 × a*) , x= (2) MAl2O3 + MNa2O + M × MSiO2 - 63 y де у - масова частка синтезованого цеоліту в цеолітвмісній мікросфері; MAl2O3 - молекулярна маса АІ2O3 ( MAl2O3 =102). Величина Мц може бути розрахована за допомогою рівностей (1) і (2) на основі відомих (з даних рентгенофазового аналізу) величин у і М. У рівності (1) знехтувано втратою маси оксиду алюмінію з маточним розчином силікату натрію, що допустимо з огляду на результати робіт [7, 8]. Аналіз рівності (1) показує, що в інтервалі можливих значень величин у і М має місце співвідношення МцМц. В протилежному випадку має місце зворотнє співвідношення величин Мкк і Мц. Таким чином, на різних стадіях процесу одержання каталізатора крекінгу маса мікросфери змінюється (при цьому її розміри залишаються практично незмінними). яким визначається масова частка Na2О в каталізаторі m кк, випливає, що Мкк=163,9 (при mкк=0,0114). Оскільки акк=0,0038, то апр= акк×Мкк/Мпр=0,0028. Таким чином, орієнтовний вміст заліза (в перерахунку на Fе2О3) в прожареній каоліновій мікросфері 4 28551 складає приблизно 0,3%. Насправді ця величина занижена, оскільки в даному випадку заниженою є величина Мкк. Більш надійні дані відносно величини апр можна одержати з результатів, наведених в прикладі 2 роботи [8], в якому у синтезі цеоліту брала участь тільки "мулітова" мікросфера, одержана з того самого вихідного каоліну, який використовували і в аналогах [6, 7]. В цьому прикладі наведено результати хімічного аналізу як цеолітвмісної мікросфери після її синтезу, так і готового каталізатора. Величини у, М, ац і акк дорівнювали 0,226, 4,53, 0,0067 і 0,0068 відповідно. Іонний обмін проводили тільки з використанням розчину нітрату амонію (y=0). Масова частка Na2O в цеолітвмісній мікросфері m цв і каталізаторі mкк становила відповідно 0,0661 і 0,0102. З рівнянь (1) і (2) одержуємо: х=0,0868; Мц=167,4. З рівняння, яке виникає внаслідок розділення співвідношення (5) на співвідношення MNa2O × x , mц = (6) Mпр - 2MSiO2 × a * +MNa2O × x Цей спосіб передбачає проведення наступних стадій: а) одержання каолінової мікроофери шляхом розпилювальної сушки висококонцентрованих (55-65% мас.) водних суспензій очищеного каолінового порошку із значним вмістом частинок субмікронного розміру; б) одержання "мулітової" мікросфери в результаті прожарювання при температурі 925-1100°С частини або всієї кількості каолінової мікросфери із стадії "а"; в) одержання метакаоліну в результаті прожарювання при температурі 650-815°С частини каолінової мікроофери із стадії "а" або каолінового порошку; г) змішування частини "мулітової" мікросфери із стадії "б", метакаоліну із стадії "в" і водного розчину гідроксиду натрію; д) визрівання суміші із стадіїї "г"; е) вводу додаткової кількості "мулітової" мікросфери із стадії "б" і додаткової кількості водного розчину гідроксиду натрію до суміші із стадії "д"; ж) підігріву суміші із стадії "е" і її витримування при підвищеній температурі доти, поки не пройде кристалізація цеоліту в мікросферах суміші; к) вилучення цеолітвміоної мікросфери із стадії "ж", її промивки і сушки; л) перетворення цеолітвмісної мікросфери із стадії "к" в каталізатор крекінгу шляхом заміщення іонів натрію її цеолітної компоненти іншими придатними для цього катіонами, промивки водою, сушки, стабілізації і термічної активації повітрям і (або) водяною парою. Всі стадії описаного способу збігаються з аналогічними стадіями способу одержання цеолітвмісного мікросферичного каталізатора крекінгу, який пропонується у винаході. З порівняння способів, описаних в роботах [6, 7], і способу, описаного в роботі [12], випливає, що при одному і тому ж сумарному часі визрівання і кристалізації цеоліту в обидвох випадках для одержання в них приблизно одного і того ж виходу цеоліту час визрівання в другому випадку, коли у визріванні бере участь не вся необхідна для кристалізації "мулітова" мікросфера, а тільки її частина, не може бути меншим, ніж у першому. В зв'язку з тим, що кристалізацію проводять при більш високій температурі, ніж визрівання, затрати енергії на одиницю маси завантаження апаратів за одиницю часу при кристалізації вищі, ніж при визріванні. З урахуванням цих двох обставин стає очевидним, що при реалізації способу, описаного в роботі [12], сумарні енергозатрати на процес синтезу цеоліту будуть нижчими, ніж при реалізації способів, описаних в роботах [6, 7]. Крім того, за умови однакового сумарного часу визрівання і кристалізації в обидвох вищеназваних випадках і значного перевищення часу кристалізації над часом визрівання, що, як правило, завжди має місце, у другому випадку існує принципова можливість проводити визрівання в одних апаратах, а кристалізацію - в інших, що, в свою чергу, дозволяє так організувати процес, що продуктивність установки може бути суттєво підвищена [12]. Однак і в другому випадку вихід цеоліту не перевищує в середньому 20%, оскільки у прототипі [12] для одержання каолінової мікросфери використовували ту саму дрібнодис яким виражається масова частка Na2О в цеолітвмісній мікросфері m ц після синтезу цеоліту, і рівності (3) знаходимо значення Мкк (Мкк=162,8). Тепер можна одержати значення величини а пр, виходячи із значень Мц і ац, і значення цієї ж величини, виходячи із значень Мкк і акк. Позначимо перше з цих значень а'пр, а друге - а"пр. Тоді а'пр=ац×Мц/Мпр= =0,005 і а"пр=акк Мкк/Мпр=0,005. Ці значення співпадають між собою, як і слід було очікувати з теоретичних міркувань. Отже, вміст заліза (в перерахунку на Fe2O3) в прожареній каоліновій мікросфері, яку використовували в аналогах [6, 7], складає 0,5%. Існує межа, до якої можна очистити каолін від заліза "мокрим" способом [5]. За даними роботи [4] цією межею є значення величини ак на рівні 0,002-0,003 (в середньому 0,0025 або 0,25%). В перерахунку на прожарений, тобто повністю дегідратований каолін ця межа складає 0,0029 або 0,29%. Вміст заліза в прожареному каоліні, який використовували в роботах [6, 7], становить 0,50%, що значно перевищує цю межу, тобто існує можливість подальшого зниженя вмісту заліза. Механізм негативного впливу заліза на процес кристалізації цеоліту, про що згадувалося вище, пов'язаний з його адсорбцією на поверхні зародків новоутворюваної кристалічної фази цеоліту і, отже, знижує вихід цеоліту. Що вищий вміст заліза в прожарюваному каоліні, тим сильніший процес блокування росту кристалів цеоліту під час його синтезу і тим нижчий вихід цеоліту. Іншими словами, при заданому вмісті заліза в прожарюваному каоліні існує своя межа максимального виходу цеоліту і ця межа тим нижча, чим виший вміст заліза в прожарюваному каоліні. Таким чином, причиною, яка перешкоджає зростанню виходу цеоліту в аналогах [6, 7], є підвищений вміст заліза в прожарюваному каоліні, який використовували в цих роботах для синтезу цеоліту, в порівнянні з тим, якого можна досягти при поглибленні очистки прожареного каоліну від заліза. Найбільше спільних ознак з пропонованим винаходом має спосіб одержання цеолітвмісного мікрооферичного каталізатора крекінгу, описаний в роботі [12], яка є прототипом даного винаходу. 5 28551 персну фракцію каолінового порошку з тим же залишковим вмістом заліза, що і в аналогах [6, 7]. Таким чином, причиною, яка перешкоджує збільшенню виходу цеоліту NaY під час його синтезу в мікросферах прожареного каоліну як при реалізації способу, описаного в роботах [6, 7], так і при реалізації способу, описаного в роботі [12], є недостатньо високий ступінь очистки різних форм прожареного каоліну від заліза. В основу винаходу поставлено задачу удосконалення способу одержання цеолітвмісного мікросферичного каталізатора крекінгу шляхом введення додаткової стадії роздільної очистки від заліза прожарених форм каоліну, з тим щоб забезпечити підвищення виходу цеоліту. Більш детально задачу винаходу можна пояснити таким чином. Винахід відноситься до групи способів одержання цеолітвмісного мікросферичного каталізатора крекінгу, які базуються на утворенні цеоліту Y в частинках реагуючої з розчином гідроксиду натрію такої суміші двох форм прожареного каоліну ("муліту" і метакаоліну) з переважаючим вмістом "муліту", в якій кожна частинка цієї суміші є носієм тільки однієї форми, причому обов'язковим носієм "муліту" є мікросфера, а носієм метакаоліну може бути як мікросфера, так і порошок. Удосконалення полягає в тому, що носії кожної з названих форм прожареного каоліну піддаються роздільній очистці від заліза безпосередньо перед синтезом цеоліту. Здійснення винаходу дозволяє підвищити вихід цеоліту до 30-55% (тут і далі під виходом цеоліту розуміється його масова частка в цеолітвмісній мікросфері). Пропонований у винаході спосіб одержання цеолітвмісного мікросферичного каталізатора крекінгу передбачає проведення таких стадій. А. Одержання каолінової мікросфери Безпосередньому одержанню каолінової мікросфери шляхом розпилювальної сушки передує операція приготування водної суспензії каоліну високої концентрації (55-65% мас.). З цією метою використовують дрібнодисперсну фракцію непрожареного каолінового порошку зі значним вмістом частинок субмікронного розміру. Максимальний розмір частинок фракції не перевищує 2 мкм. Хімічна очистка від заліза вказаної фракції не є обов'язковою. Високі дисперсність, міцність і пластичністіь частинок непрожареного каоліну сприяють під час розпилювальної сушки суспензії утворенню каолінової мікросфери з високою міцністю, зокрема, з великим опором стиранню, і значною щільністю упаковки частинок каоліну в ній. Підвищенню цих якостей мікросфери сприяє також висока концентрація каоліну в частинках розпилювальної суспензії. Консистенція висококонцентрованої суспензії каоліну повинна дозволяти її транспортування до розпилюючого механізму і здійснювати сам розпил. Проте поєднати високу концентрацію каоліну в водній суспензії з її достатньою текучістю без додавання до суспензії спеціальних хімічних добавок неможливо, оскільки в воді не відбувається повного руйнування наявних у вихідному каоліні і утворюваних в ній агрегатів первинних частинок каоліну, в яких ці частинки зберігають свою форму і розміри [13]. Наявність таких агрегатів знижує текучість суспензії. Ці агрегати утворюються внаслідок адгезії ї при стиканні між собою первинних частинок каоліну. Для відвернення утворення нових агрегатів необхідно ослабити адгезійну здатність всієї поверхні первинних частинок каоліну, а для руйнування уже утворених агрегатів - знизити поверхневий натяг в місцях взаємного контакту цих частинок. Обидві цілі досягаються за рахунок адсорбції на поверхні первинних частинок каоліну спеціальних хімічних добавок - дефлокулянтів (стабілізаторів). В кінцевому рахунку використання дефлокулянтів призводить до підвищення текучості суспензії, що в свою чергу дозволяє суттєво збільшити в ній концентрацію каоліну. З відомих дефлокулянтів як добавку до водних суспензій каоліну найбільш доцільно використовувати пірофосфат натрію Na4P2О7×10H2О (або інші поліфосфати) і силікат натрію в кількостях, при яких вагове співвідношення дефлокулянт/каолін не перевищує 1/200 і 1/50 відповідно. Змішування каоліну, дефлокулянта і води для утворення суспензії можна проводити при будь-якій послідовності подачі компонентів і в будь-якій ємності, яка має пристрій для перемішування. В описуваному випадку, коли єдиним джерелом наявного в суспензії каоліну є описана вище фракція непрожареного каоліну, частина каолінової мікросфери, утвореної внаслідок наступної розпилювальної сушки суспензії, може бути перетворена в "мулітову" мікросферу, а друга її частина - в метакаолінову мікросферу. Проте каоліновою сировиною для утворення суспензії і каолінової мікросфери може бути не тільки непрожарений порошковий каолін, але і його суміш з прожареним при будь-якій температурі з інтервалу 540-1100°С каоліновим порошком з приблизно таким же розміром частинок, що і непрожарении каолін. Хімічна очистка від заліза цього прожареного каоліну перед його змішуванням з непрожареним каоліном не є обов'язковою. Така суміш придатна для одержання "мулітової" мікросфери. Якщо температура прожарювання не перевищує 800°С, то суміш такого порошку з порошком непрожареного каоліну може бути використана і для одержання метакаолінової мікросфери. Слід зауважити, що для забезпечення достатньої міцності каолінової мікросфери на основі суміші непрожареного і прожареного каоліну частка непрожареного каоліну не повинна бути нижчою від 50%. Водну суспензію каоліну з добавкою дефлокулянта піддають розпилювальній сушці для випаровування вологи із крапель суспензії і формування каолінової мікросфери із залишковою вологою, яка не перевищує 1% [14], при відсутності видалення конституційної води з наявного в мікросфері непрожареного каоліну. Для проведення цього процесу придатні розпилювальні сушарки з будьякою схемою руху теплоносія (повітря або димових газів від згоряння вуглеводневого палива) і частинок розпиленої суспензії, але перевагу слід надати прямоточним сушаркам, в яких найлегше забезпечити вказані вимоги щодо дегідратації одержаної каолінової мікросфери шляхом відповідного підбору температур теплоносія на вході в сушарку і на виході з неї, розходів теплоносія і суспензії. Розхід теплоносія і його температура на вході в сушарку в більшості випадків незмінні і відомі. В цих випадках розхід суспензії підбирають таким, щоб 6 28551 температура теплоносія на виході з прямоточної сушарки складала 120-315°С. В діапазоні цих температур теплоносія на виході із сушарки якраз і задовільняються вищенаведені показники дегідратації каолінової мікросфери, яка утворилася внаслідок розпилювальної сушки. При одержанні каолінової мікроофери в розпилювальних сушарках перевагу слід надати тим розпилюючим пристроям, які використовують відцентрові диски з окружною швидкістю на ободі диска 20-100 м/с. За цих умов забезпечується така дисперсність розпилу, яка призводить до утворення каолінової мікросфери, основну частину якої складають частинки з необхідним ефективним діаметром (40-100 мкм). З продукту, одержаного в результаті розпилювальної сушки суспензії, необхідну фракцію каолінової мікросфери можна вилучити в лабораторних умовах за допомогою сит з різними розмірами отворів, а в промислових - за допомогою аеродинамічних класифікаторів. В наступних стадіях під терміном "каолінова мікросфера" слід розуміти саме ту її фракцію, розміри частинок якої не виходять за допустимі межі. Для формування мікросфери, зокрема, на основі зразків каоліну з вмістом Fе2О3 1,12, 0,63 і 0,35% мас., використовували сушильну установку продуктивністю до 150 кг мікросфери за годину. На базі каоліну з вмістом Fе2О3 0,81% мас. мікросферу формували на лабораторній установці продуктивністю до 10 кг мікросфери за годину. Б. Одержання "мулітової" мікросфери "Мулітову" мікросферу одержують прожарюванням каолінової мікросфери в діапазоні температур 925-1100°С протягом необхідного часу. Оптимальна температура прожарювання становить приблизно 1000°С. При цій температурі повне перетворення непрожареного каоліну в "муліт" досягається за 2 години. Прожарювання каолінової мікросфери з метою її перетворення в "мулітову" мікросферу в лабораторних умовах проводили в згаданих оптимальних умовах в муфельній печі. В промислових умовах воно може здійснюватись в обертовій печі. Б. Одержання метакаоліну Метакаолін одержують прожарюванням каолінової мікросфери в діапазоні температур 650815°С протягом необхідного часу. Оптимальна температура прожарювання становить приблизно 735°С. При цій температурі повне претворения непрожареного каоліну в метакаолін досягається за 2 години. Прожарювання каолінової мікросфери з метою її перетворення в метакаолін в лабораторних умовах проводили в згаданих оптимальних умовах в муфельній печі. В промислових умовах воно може здійснюватись в обертовій печі. Стадії "А", "Б", "В" у винаході не є новими. Вони детально описані в патентній літературі, зокрема в аналогах [6, 7], які були, в свою чергу прототипами для нашого прототипу [12], де ці стадії використовували, але не описували. Єдиною відмінністю, яка існує для цієї групи стадій між винаходом і відомими патентами, є те, що на противагу відомим способам пропонований спосіб не вимагає обов'язкової хімічної очистки каолінового порошку від заліза. B1. Роздільна хімічна очистка "мулітової" і метакаолінової мікросфери від валіза У винаході пропонується роздільна хімічна очистка від заліза "мулітової" та метакаолінової мікросфери, одержаної відповідно на стадіях "Б" і "В". Стадія "В1" є новою, в аналогах [6, 7] і прототипі [12] вона відсутня. Автори цих патентів виходять з каолінового порошку, попередньо очищеного "мокрим" способом. Але використання цього способу передбачає багаторазову фільтрацію високодисперсних суспензій каолінового порошку, що становить непросту технічну проблему. Разом з тим існує можливість не проводити обов'язкову попередню хімічну очистку каолінового порошку від заліза при використанні його для одержання каолінової мікросфери, а здійснювати таку очистку "мулітової" та метакаолінової мікросфери після їх одержання (тобто після прожарювання) безпосередньо перед використанням при синтезі цеоліту. Операції фільтрації у випадку "мулітової" або метакаолінової мікросфери проходять значно швидше і легше, ніж у випадку каолінового порошку. Відсутність попередньої хімічної очистки такого порошку від заліза корисна ще й тим, що невилучене залізо під час прожарювання каолінової мікросфери при температурі 925-1100°С з метою одержання "мулітової" мікросфери каталізує процес утворення "муліту" [15]. Для проведення кислотної очистки прожареного каоліну від заліза придатним є будь-який корозійностійкий по відношенню до кислот реактор з перемішуючим пристроєм, в який завантажують певні кількості прожареного каоліну (з відомим хімічним складом, зокрема, з відомим вмістом заліза) і розчину кислоти відомої концентрації, після чого при перемішуванні суміш при заданій температурі витримують протягом певного часу. Кислотну обробку прожарених форм каоліну проводили при температурі 70-100°С протягом 16 годин з використаням розчинів кислот підвищеної концентрації. Наприклад, концентрація соляної кислоти при обробці складала 10-32%. Вагове співвідношення розчин кислоти/прожарений каолін змінювали в діапазоні 1-3. При його виборі враховували необхідність того, щоб у розчині з запасом була забезпечена така кількість кислоти, яка є стехіометрично необхідною для очистки від заліза. Рівень розчину кислоти в реакторі був вищим від висоти шару завантаженої твердої фази. Після закінчення кислотної обробки з вивантаженої з реактора суміші прожареного каоліну і розчину кислоти відфільтровували прожарений каолін, після чого його промивали водою до рН=7 і висушували при температурі 105-115°С протягом 2-4 годин. Фільтрат після очистки і промивну воду змішували і в цій суміші за допомогою тих чи інших методів аналізу визначали вміст іонів заліза і алюмінію, на основі чого розраховували відповідно глибину очистки від заліза тієї чи іншої форми прожареного каоліну і ступені її деалюмінування. Кристали каолініту - це блоки елементарних двошарових пакетів з тетраедричного кремнійкисневого шару і шару алюмокиснево-гідроксильних октаедрів [16]. Іони заліза можуть ізоморфно заміщувати іон алюміню в октаедричному шарі або проникати в міжпакетний простір. Проте, частка та 7 28551 стадії "В1" в результаті їх роздільної "мокрої" очистки від заліза, з водним розчином гідроксиду натрію. Порядок завантаження реактора реагентами може бути довільним: можна спочатку подавати тверду фазу, а потім - рідку, а можна і навпаки. У прототипі [12] на стадії "Г" рекомендований склад реакційної суміші характеризується такими показниками: масова частка "мулітової" мікросфери в твердій фазі, тобто в її суміші з метакаоліном (позначимо її у1), змінюється від 0,8 до 0,9, мольне співвідношення Nа2O/АІ2O3 (позначимо його z1) змінюється від 0,6 до 0,7 і масова частка NaOH в його водному розчині (позначимо її g1) змінюється від 0,16 до 0,20. Ці рекомендації відносяться до випадку, коли мольне співвідношення SіО2/АІ2О3 в "муліті" і метакаоліні дорівнює 2, тобто до випадку використання недеалюмінованих "мулітової" мікросфери і метакаоліну. Саме цей випадок найчастіше зустрічається на практиці. В зв'язку з тим, що із стадії "В1" на стадію "Г" у винаході надходять деалюміновані "мулітова" мікросфера і метакаолін, ступінь деалюмінування яких становить відповідно a і b, склад реакційної суміші повинен бути відкоректований з врахуванням величин a і b, тобто з врахуванням того, що в кожній з двох форм прожареного каоліну мольне співвідношення SіО2/АІ2О3 дорівнює не 2, а 2/(1-a) для "муліту" і 2/(1-b) для метакаоліну. При цьому будемо виходити з того, що, на відміну від оксидів Na2O і Н2O, не вся наявна в суміші кількість оксидів SiО2 і АІ2О3 є достатньо реакційноздатною (активною) по відношенню до розчину гідроксиду натрію. Тому при змішуванні деалюмінованої "мулітової" мікросфери, деалюмінованого метакаоліну і розчину NaOH рекомендовані в прототипі [12] межі концентрації водного розчину гідроксиду натрію g1 залишаються без зміни, а замість мольних співвідношень SіО2/АІ2О3 і Nа2O/АІ2O3, де під SiO2 і АІ2O3 розуміється вся наявна в системі кількість молів відповідного оксиду, необхідно використовувати мольні співвідношення SіО2*/АІ2О3* і Na2O/Al2O3*, де під SiO2* і АІ2O3* слід розуміти кількість активних молів оксидів кремнію і алюмінію відповідно. При цьому межі значень величин співвідношень SіО2*/АІ2О3* і Nа2O/АІ2O3* повинні бути розраховані на основі рекомендованих у прототипі [12] значення співвідношення SіО2/АІ2О3=2 і меж значень величини z1 з врахуванням критерію поділу оксидів кремнію і алюмінію на активні і неактивні. Будемо вважати, що в "мулітовій" мікросфері оксиди кремнію і алюмінію незруйнованої частини фази алюмокремнійової шпінелі 2АІ2O3×3SіО2 є неактивними, а мольне співвідношення названих оксидів в цій частині становить 3/2. Вся остання кількість SiO2 і АІ2О3 є активною. Неактивніcть оксидів кремнію і алюмінію в алюмокремнійовій шпінелі має відносний характер, її слід розуміти не як хімічну індиферентність цих оксидів, а як відзначення того факту, що здатність до взаємодії з NaOH для них значно нижча, ніж для відповідних активних оксидів. Таким чином, поділ наявних в реакційній суміші оксидів кремнію і алюмінію на активні і неактивні є лише засобом, який дозволяє звести процес синтезу цеоліту при використанні як недеалюмінованих, так і деалюмінованих "мулітової" і метакаолінової мікросфери, до спільної основи, що означає підтримання в обидвох випадках мольних кого "внутрішнього" заліза в його сумарній кількості, яка знаходиться в каоліні, невелика. Основна частина присутнього в каоліні заліза припадає на залізовмісні мінерали з різною розчинністю в кислотах [5, 16], більшість яких представлена оксидами і гідроксидами заліза, такими як гематит aFе2О3, гідрогематит Fе(ОН)3×nН2О, гетит НFеО2, лепідокрит FеООН, лимонит Fе2О3×nН2O тощо. Частково ці мінерали (з колоїдними розмірами частинок) каолініту перебувають у вигляді адсорбційної плівки на поверхні частинок каолініту, а частково (мінерали з більш крупними розмірами частинок) у вигляді окремих частинок. Найлегше кислоти розчиняють колоїдні частинки. З оксидів заліза найповільніше розчиняється в кислотах Fе2О3 [17]. В умовах прожарювання каоліну при температурах 700-1100°С в окислювальному середовищі саме цей оксид заліза є основним. Він, як і інші оксиди заліза та інших металів, при прожарюванні знижує свою активність [18-20]. Незважаючи на це, в умовах кислотної обробки при використанні концентрованих розчинів кислот досягається значна глибина очистки прожарених форм каоліну від заліза (40-60%). При цьому залишкова концентрація заліза (в перерахунку на Fe2О3) в прожареній мікросфері знижується до 0,28-0,36%. Зниження вмісту заліза в прожареній мікросфері в порівнянні з прототипом [12], для якого ця величина становить 0,5%, призводить до зменшення адсорбції заліза на поверхні утворюваних під час синтезу кристалів цеоліту і послаблює процес блокування їх росту, що, в свою чергу, сприяє підвищенню виходу цеоліту. З іншого боку, відомо, що алюміній в каоліні, прожареному при температурі 700-1100°С, значно активніший, ніж в непрожареному каоліні, не тільки по відношенню до розчину гідроксиду натрію, але і по відношенню до розчинів кислот [21-23]. Це використовують, зокрема, для екстракції оксиду алюмінію концентрованими розчинами кислот з метакаоліну. Він деалюмінується значно швидше, ніж "муліт". Отже очистка прожарених форм каоліну від заліза концентрованими розчинами кислот неминуче буде супроводжуватися розчиненням оксиду алюмінію. Якщо очистку проводити так, щоб ступінь деалюмінування цих форм не перевищував певного допустимого рівня, який вибирається з умови забезпечення необхідного вмісту оксиду алюмінію в каталізаторі, то можна сподіватись на те, що глибока очистка "мулітової" та метакаолінової мікросфери від шкідливих домішок заліза, яка досягається у винаході, дозволить підвищити вихід цеоліту під час його наступного синтезу. Виходячи з вказаної умови, допустимі ступені деалюмінування "мулітової" і метакаолінової мікросфери було оцінено відповідно в 20 і 60%. В необхідності контролю за ступенем деалюмінування як "мулітової", так і метакаолінової мікросфери, випливає необхідність їх роздільної кислотної обробки. Суміш таких частково деалюмінованих "мулітової" і метакаолінової мікросфер далі використовували для синтезу цеоліту. Г. Змішування "мулітової" та метакаолінової мікросфери і водного розчину гідроксиду натрію На стадії "Г" відбувається змішування "мулітової" та метакаолінової мікросфери, одержаних на 8 28551 h1 - масове співвідношення поданого в реактор водного розчину NaOH концентрації g1 до суміші "мулітової" і метакаолінової мікросфери. Мають місце очевидні співвідношення: р1=x[2a+(1-a)/2]+(1-x1) [2b+2(1-b)]= = (9) 2-3(1-a)х1/2, (10) q1=(1-b)(1-х1), k1=p1/q1, (11) Мд.мул.=(1-a) MAl2O3 + 2MSiO2 (12) співвідношень SіO2*/АІ2O3* і На2O/АІ2О3* (позначимо їх відповідно k1 і m1) на одному і тому ж рівні. З рекомендацій прототипу [12] і прийнятого поділу оксидів SiO2 і АІ2O3 на активні і неактивні випливає, що для стадії "Г" k1=2+y1/2(1-у1) (7) і m1=z1/(1-y1). (8) На основі вибраних конкретних значень у1 і z1 можна розрахувати конкретні значення величин k1 і m1. Введемо такі позначення: х1 - мольна частка "мулітової" мікросфери в її суміші з метакаоліновою; p1 - число активних молів SiO2, які припадають на 1 моль суміші "мулітової" і метакаолінової мікросфери; q1 - число активних молів АІ2O3, які припадають на 1 моль названої суміші; n1 - число молів Na2O, які подають в реактор в перерахунку на 1 моль суміші "мулітової" і метакаолінової мікросфери; Mд.мул. - молекулярна маса деалюмінованої "мулітової" мікросфери; Мд.мет. - молекулярна маса деалюмінованої метакаолінової мікросфери; Мсм. - молекулярна маса суміші деалюмінованих "мулітової" і метакаолінової мікросфери, яка подається в реактор; g1 - масова частка "мулітової" мікросфери в цій суміші; Мд.мет.=(1-b) MAl2O3 + 2MSiO2 , (13) (14) Мсм.=x1×Mд.мул.+(1-x1)×Мд.мeт., (15) n1=m 1×q1 Із співвідношень (9)-(11) випливає, що 2k 1( -b) - 4 x1 = . (16) 2k 1(1 - b) - 3(1 - a ) Основними характеристиками складу реакційної суміші на стадії "Г" є величини g1 і h1. Перша з них з врахуванням співвідношень (7), (12), (13), (14) і (16) може бути розрахована на основі рівності x 1M д.мул. , g1 = (17) Мсм. а друга - з врахуванням співвідношень (7), (8), (10), (12), (13), (14), (15) і (16) - на основі рівності 2 × MNaOH h1 = × h1, (18) g1 × Mсм. де MNaOH - молекулярна маса NaOH (МNaOH=40). Рівності (17) і (18) мають відповідно такий остаточний вигляд: ( 222 - 102a )[y 1 - b( 4 - 3y 1 )] , g1 = (17а) (222 - 102a )[ y 1 - b( 4 - 3 y 1 )] + (222 - 102b) × (1 - y 1 ) × (1 + 3a ) h1 = 80 1 + 3a z 1 ( -b) , g1 ( 222 - 102a )[ y 1 - b( 4 - 3 y 1 )] + (222 - 102b) × (1 - y 1 ) × (1 + 3a ) (18а) стю розчину NaOH з масовою часткою гідроксиду натрію g2. Необхідне масове співвідношення названих додаткових кількостей до сумарної кількості твердої фази (тобто суми "мулітової" і метакао-лінової мікросфери), введеної в реактор на стадії "Г", позначимо відповідно q і h2. У прототипі [12] остаточний рекомендований склад реакційної суміші на стадії "Е" після вводу в реактор додаткових кількостей "мулітової" мікросфери і розчину гідроксиду натрію харктеризуються такими показниками: масова частка "мулітової" мікросфери в твердій фазі, тобто в її суміші з метакаоліновою (позначимо її у2), становить 0,95, мольне співвідношення На2О/АІ2О3 (позначимо його z2) змінюється від 0,6 до 0,7 і масова частка NaOH в його водному розчині (позначимо її g) змінюється від 0,16 до 0,20. Ці рекомендації відносяться до випадку використання недеалюмінованих "мулітової" і метакаолінової мікросфери, тобто до випадку, коли кожна з величин a і b дорівнює нулю. В зв'язку з тим, що на стадії "Е" використовують деалюміновані "мулітову" і метакаолінову мікросферу, ступені деалюмінування яких дорівнюють відповідно a і b, остаточний склад реакційної суміші на цій стадії мусить бути відкоректований з врахуванням істиних значень величин a і b. Для здійснення такого коректування будемо вважати, Таким чином, склад реакційної суміші на стадії "Г" визначається на основі рекомендацій прототипу [12] (щодо величин у1, z1 і g1) і експериментальне визначених ступенів деалюмінування "мулітової" і метакаолінової мікросфери (a і b відповідно) на стадії "В1". Наведені формули використовували для розрахунку складу реакційної суміші на стадії "Г". Д. Визрівання реакційної суміші із стадії "Г" Визрівання реакційної суміші із стадії "Г" проводили при температурі 40±1°С протягом 4-8 годин. З цією метою реакційну суміш, яку завантажували в круглодонну колбу, оснащену мішалкою, термометром і зворотним холодильником, підігрівали за допомогою водяної або пісочної бані. Суміш при цьому перемішували за допомогою мішалки. В деяких випадках визрівання проводили в сушильній шафі при вищезгаданій температурі і тривалості процесу. В цих випадках реакційну суміш завантажували в запаяні пробірки. Пробірки періодично струшували. Е. Ввід додаткових кількостей "мулітової" мікросфери із стадії "Б" і водного розчину гідроксиду натрію до суміші із стадії "Д" На стадії "Е" відбувається завантаження реактора, що містить в собі реакційну суміш із стадії "Д", додатковою кількістю "мулітової" мікросфери зі ступенем деалюмінування a і додатковою кількі 9 28551 як і для стадії "Г", що в "мулітовій" мікросфері окконцентрації g2 (в розрахунку на 1 моль суміші сиди кремнію і алюмінію незруйнованої частини "мулітової" і метакаолінової мікросфери, які поалюмокремнійової шпінелі є неактивними, а решта дають в реактор на стадії "Г"). цих оксидів, а також Na2О і Н2O є активними. З Мають місце очевидні співвідношення: цього випливає, що величина g2 корекції не потреp2=(1-x2){x1[2a+(1-a)/2]+2(1-x1]}+x2[2a+ бує, а також те, що замість величин у2 і z2 склад (21) +(1-a)/2]=(1-x2)A0+x2B0 твердої реакційної суміші повинен характеризува(22) A0=[4=3x1(1-a)]/2, тися мольними співвідношеннями SіO2*/АІ2O3* і (23) B0=(3a+1)/2, Nа2O/АІ2O3* (позначимо їх відповідно k2 і m2), де (24) q2=(1-x2)(1-x1)(1-b)=(1-x2)q1, під SiO2* і АІ2O3* розуміється активна частина окk2=p2/q2, (25) сидів кремнію і алюмінію відповідно. Враховуючи n2=(m2-m1)q1. (26) рекомендації прототипу [12] і принятий поділ оксиІз співвідношень (21), (24) і (25) випливає, що дів SiO2 і АІ2O3 на активні і неактивні, доходимо x2=(q1k2-A0)/(q1k2+B0-A0). (27) висновку, що на стадії "Е" Основними характеристиками складу реакційk2=2+у2/2(1-у2) (19) ної суміші на стадії "Е" є величини q і h2. Перша з i них з врахуванням співвідношень (10), (16), (19), m2=z2/(1-у2). (20) (22), (23) і (27) може бути розрахована на основі На основі вибраних конкретних значень y2 і z2 рівності можна розрахувати конкретні значення величин k2 x2 g і m2. q= × 1, (28) 1 - x 2 x1 Введемо такі додаткові позначення: а друга з врахуванням співвідношень (10), (12)-(16) x2 - мольна частка додатково поданої в реактор і (26) - на основі рівності на стадії "Е" "мулітової" мікросфери в її суміші з твердою фазою продукту із стадії "Д"; 2MNaOH h2 = × n2 . (29) р2 - число активних молів SіО2 в одному молі g 2Mсм. такої суміші; Рівності (28) і (29) мають відповідно такий осq2 - число активних молів АІ2О3 в одному молі таточний вигляд: цієї ж суміші; n2 - число молів Na2O, які додатково подають в реактор на стадії "Е" у вигляді розчину NaOH y - y1 (1 - b)( 222 - 102a ) × , q= 2 (28а) 1 - y 2 ( 222 - 102a )[ y 1 - b( 4 - 3 y 1 )] + ( 222 - 102b)(1 - y 1 )(1 + 3a) h2 = z1 ö (1 - y1)(1 + 3a ) 80 æ z 2 ÷ (1 - b ) × ç , g 2 ç 1 - y 2 1 - y1 ÷ ( 222 - 102a )[ y1 - b( 4 - 3 y1 )] + (222 - 102b)(1 - y1)(1 + 3 a ) ø è При цьому слід добиватися [12] виконання рівностей (30) g2=g1 i z2=z1. (31) Таким чином, склад реакційної суміші на стадії "Е" визначається на основі рекомендацій прототипу [12] (щодо величин у2, z2 і g2) і експериментально визначених ступенів деалюмінування "мулітової" і метакаолінової мікроофери (a і b відповідно) на стадії "В1". Наведені формули використовували для розрахунку складу реакційної суміші на стадії "Е". Ж. Кристалізація цеоліту Після вводу в реактор додаткової кількості "мулітової" мікросфери і розчину гідроксиду натрію температуру піднімали до 83±1°С і витримували реакційну суміш при цій температурі протягом 2048 годин. Реатором слугувала круглодонна колба, оснащена мішалкою, термометром і зворотним холодильником, яку підігрівали за допомогою водяної або пісочної бані. Реакційну суміш при цьому перемішували за допомогою мішалки. В дяких випадках кристалізацію цеоліту проводили в сушильній шафі при вказаних температурі і тривалості процесу, використовуючи запаяні пробірки. Пробірки періодично струшували. Стадії "Г", "Д", "Е", "Ж" є спільними для винаходу і прототипу [12]. Відмінність між винаходом і прототипом для цих стадій полягає в тому, що у (29а) винаході рекомендований у прототипі склад реакційних сумішей на стадіях "Г" і "Е" коректують з врахуванням ступенів деалюмінування "мулітової" мікросфери a і метакаолінової мікросфери b на стадії "В1". К. Вилучення цеолітвміоної мікросфери із стадії "Ж", її промивка і сушка Після закінчення кристалізації і охолодження реактора цеолітвмісну мікросферу відділяли від маточного розчину силікату натрію і пилу, одержаного в результаті часткового руйнування мікоросфери під час синтезу цеоліту, шляхом фільтрування. Для цієї мети можна використовувати центрифугування [7, 9]. Потім цеолітвмісну мікросферу промивали водою для відмивання розчину силікату натрію до рН=7 в промивній воді і висушували в сушильній шафі при температурі 105-115°С протягом 2-4 годин. Принагідно зазначимо, що операції промивки і сушки цеолітвмісної мікросфери не є обов'язковими [6-9], якщо цеолітвмісна мікросфера не піддається рентгенофазовому аналізу. За даними рентгенофазового аналізу в одержаній це-олітвмісній мікросфері вміст цеоліту Y з мольним співвідношенням SіO2/АІ2О3 4,6-5,0 лежить в межах 30-55% мас., що перевищує аналогічний показник прототипу (~20%). Дифрактограми зразків цеолітвмісної мікросфери були зняті на напівавтоматичному дифрактометрі ДРОН-УМ1. Рентгенофазовий аналіз про 10 28551 термообробці 100%-ною водяною парою при температурі 800-820°С протягом 2-3 годин. Зазначимо, що наявні у винаході стадії "К" і "Л" використовувались як в аналогах [6, 7], так і в прототипі [12]. На початку роботи було вивчено динаміку формування активності синтезованих каталізаторів в модельній реакції крекінгу кумолу в залежності від кількості циклів іонний обмін-промивка мікросфери водою-сушка-ультрастабілізація. Це дослідження показало доцільність обмеження кількості вказаних циклів до чотирьох. В подальшій роботі автори при синтезі каталізаторів проводили тільки чотири обміни. Зразки цеолітвміоної мікросфери (ЦВМС), одержані в даній роботі, відрізнялися між собою не тільки умовами синтезу цеоліту (температурним режимом і тривалістю стадій визрівання та кристалізації, складом реакційної суміші на цих стадіях), але і вмістом в них заліза, який залежав від того, чи проводили очистку вихідного каоліну, чи його прожарених форм ("муліту" і метакаоліну) і в яких умовах таку очистку здійснювали (в умовах деалюмінування чи в умовах його відсутності). Частину ЦВМС було одержано з використанням зразків вихідного каоліну (з вмістом Fe2О3 1,12 і 0,63% мас.), які не були піддані очистці від заліза ні перед формуванням мікросфери, ні після її прожарювання. Перший з цих зразків назвемо каоліном 1, другий - каоліном 2. Ряд зразків ЦВМС було одержано на основі каоліну з початковим вмістом в ньому Fе2О3, який становив 0,81% мас. При цьому частину з них було одержано з цього каоліну, очищеного від заліза до залишкового вмісту останнього, рівного 0,35% мас. (в перерахунку на Fе2О3). Вказану очистку проводили в умовах практичної відсутності деалюмінування (її здійснювали в шість прийомів з використанням на кожному з них свіжої 7,5%-ної соляної кислоти при температурі 95°С і ваговому співвідношенні розчин кислоти/каолін, рівному 2; тривалість очистки при кожному прийомі складала 2 години; після кожного прийому проводили промивку каоліну водою до нейтральної реакції і сушку протягом 4 годин при 115-120°С). Назвеvj цей очищений каолін каоліном 3. Одержану з такого каоліну мікросферу після її прожарювання у вказаних вище умовах використовували для синтезу цеоліту без додаткової очистки. Таким чином, при використанні каолінів 1, 2 і 3 як сировини для одержання каталізатора крекінгу на синтез цеоліту надходили недеалюміновані "мулітова" і метакаолінова мікроcфера. Друга частина каоліну з початковим вмістом в ньому Fе2О3 0,81% мас. була використана для формування мікросфери і одержання її прожарених форм - "муліту" і метакаоліну, які перед синтезом цеоліту піддавали роздільній очистці від заліза концентрованою соляною кислотою (умови цієї очистки наведено при описі стадії "В1"). Така очистка супроводжувалася суттєвим деалюмінуванням "муліту" і метакаоліну. Деякі зразки ЦВМС (незалежно від наявності або відсутності очистки каоліну від заліза і місця цієї очистки в технологічному ланцюгу одержання каталізатора) були використані для її перетворення в каталізатор крекінгу, як це описано вище. Ко водили з дотриманням відомих рекомендацій [24, 25]. Л. Перетворення цеолітвмісної мікросфери в каталізатор крекінгу В одержаній на стадії "К" цеолітвмісній мікросфері цеоліт знаходиться в неактивній, натрійовій формі. Для переведення його в форму, яка б виявляла каталітичну активність в процесі крекінгу вуглеводнів, іони натрію необхідно замінити іншими. Найчастіше з цією метою використовують іони амонію і рідкісноземельних елементів. Після іонного обміну необхідно проводити промивку іонзаміщеної цеолітвмісної мікросфери від залишків іонообмінного розчину і її сушку. Часто проводять також прожарювання промитої мікросфери при температурі 500-600°С протягом 1-4 годин, що сприяє стабілізації цеоліту. Сукупність операцій іонний обмін-промивка-сушка або іонний обмін-промивкасушка-стабілізація може повторюватись декілька разів. Завершальною операцією перетворення цеолітвмісної мікросфери із стадії "К" в готовий каталізатор крекінгу є термоактивація іонзаміщеної цеолітвмісної мікросфери повітрям і (або) водяною парою при температурі 750-850°С протягом 1-4 годин. Цю операцію можна проводити як на установці одержання каталізатора крекінгу, так і на установці його використання, тобто на установці каталітичного крекінгу. Іонний обмін проводили при температурі 8085°С протягом 1,5-2,5 годин в обертовому автоклаві з нержавіючої сталі об'ємом біля 400 мл, куди завантажували наважку цеолітвмісної мікросфери (10-50 г) із стадії "К" і наважку іонообмінного розчину нітрату амонію (або лантану). Масове співвідношення розчин нітрату/цеолітвмісна мікросфера лежало в межах 1,5-2,0. Концентрацію розчину нітрату при цьому вибирали такою, щоб співвідношення кількість грам - еквівалентів катіону в розчині кількість грам - еквівалентів початкового натрію в цеолітній фазі знаходилось в межах 1-2. Перебіг іонного обміну контролювали за концентраціями витісненого натрію і відповідного обмінного катіону в розчині. При цьому концентрацію натрію визначали методом полум'яної спектрофотометрії [26], а концентрацію лантану - трилонометричним методом [27]. Розбіжність результатів по лантану і витісненому натрію не перевищувала 5% відносних. Катіонний склад зразків, зокрема, вміст залишкового натрію, визначали також через їх розчинення в фтористоводневій кислоті, опираючись на стандартні аналітичні методики [28]. Процес заміщення іонів натрію проводили таким чином, що цикл операцій іонний обмінпромивка мікросфери водою до нейтральної реакції на воронці Бюхнера або фільтрі Шотта-сушка (при температурі 110-120°С протягом 2-3 годин)стабілізація (ультрастабілізація) цеоліту (при температурі 550-570°С протягом 2-3 годин) повторювали 2-5 разів, причому так, що розчин нітрату амонію використовували або на всіх циклах або на перших циклах (в цьому випадку на останніх циклах використовували розчин нітрату лантану). Після завершення останнього циклу в деяких випадках висушену і стабілізовану іонзаміщену цеолітвмісну мікросферу (свіжий каталізатор) піддавали 11 28551 жний такий зразок ЦВМС породжував серію каталізаторів, які відрізнялися між собою катіонним складом цеолітної компоненти. Оскільки параметри всіх операцій перетворення ЦВМС в каталізатор крекінгу, як згадувалося вище, коливалися в дуже вузькому діапазоні значень, то цей склад визначався, головним чином, кількістю циклів операцій іонний обмін-промивка-сушка-ультрастабілізація і природою іонообмінного розчину (нітрат амонію чи нітрат лантану), який використовували під час іонного обміну в кожному з цих циклів. Кожна серія мала свій номер. Всередині серії зразки свіжого каталізатора шифрували таким чином, щоб відбити кількість циклів, на яких при іонному обміні використовували нітрат амонію, і кількість циклів, на яких використовували нітрат лантану. Наприклад, шифр 25-4УС означає, що на всіх чотирьох циклах одержання каталізатора серії 25, кожен з яких починався з іонного обміну і закінчувався ультрастабілізацією, при іонному обміні використовували нітрат амонію, а шифр 26-3УС-ЛУС означає, що каталізатор серії 26 одержано за 4 цикли, в перших трьох з яких під час іонного обміну використовували нітрат амонію, а в останньому циклі – нітрат лантану. Термопарооброблені зразки каталізатора шифрували аналогічним чином з добавкою скорочення "ТПО". Основними методами оцінки якості одержаних зразків каталізаторів було визначення їх кислотності методом термопрограмованої десорбції (ТПД) аміаку і активності в модельній реакції крекінгу кумолу. Причому для одержання зіставимих результатів ці ж характеристики час від часу знімали на "еталонних" зразках - деяких промислових каталізаторах (фірми Енгельгард, фірми Грейс і каталізаторі виробництва Уфимської каталізаторної фабрики). Кислотність і активність - одні з найважливіших показників ефективності каталізатора крекінгу. Вибір даних методів визначення цих показників зумовлювався швидкістю їх реалізації, простотою (наприклад, невеликим набором основних продуктів крекінгу - пропілену, бензолу, толуолу, етилбензолу і залишкового кумолу - при використанні модельної реакції корекінгу кумолу), можливістю використання малих наважок каталізатора, високою точністю та інформативністю одержуваних результатів. Завдяки цьому в сучасних дослідженнях широко використовують як визначення кислотності каталізатора крекінгу методом ТПД аміаку [29-32], так і визначення його активності в модельній реакції крекінгу кумолу [33-36]. Обидві методики описано нижче. Для більш повного вивчення зразків синтезованих каталізаторів при необхідності використовували методи хімічного і рентгенофазового аналізів, адсорбції, інфрачервоної спектроскопії, електронного парамагнітного резонансу тощо. Для кращих зразків проводили оцінку тих показників їх якості, які регламентуються чинною нормативно-технічною документацією [37], з використанням діючих стандартних методик випробування каталізаторів крекінгу [28]. Фіг. 1. Фрагменти дифрактограм деяких синтезованих зразків ЦВМС з різним вмістом цеоліту NaY. 2q - кут дифракції (град.), а -ЦВМС-7; б ЦВМС-8; в - ЦВМС-9. Зліва від пунктирних ліній вказаний кінцевий кут попереднього фрагмента, справа - початковий кут даного фрагмента. Фіг. 2. Спектри ТПД аміаку для свіжих зразків каталізаторів. І - інтенсивність сигналу детектора (мВ), Т температура (°С). 1 - фірми Енгельгард; 2 - фірми Грейс; 3 – Уфимської каталізаторної фабрики; 4 - 25-4УС; 5 25-3УС-ЛУС; 6 - 26-4УС; 7 - 26-3УС-ЛУС; 8 - 293УС; 9 - 29-4УС; 10 - 29-3УС-ЛУС. Фіг. 3. Залежність конверсії кумолу с (%. мас.) від числа імпульсів і для свіжих каталізаторів. а: 1 - 25-4УС; 2 -26-4УС; 3 - 29-4УС; 4 - 29-4УС; 5 – фірми Енгельгард. б: 1 - 25-3УС-ЛУС; 2 - 26-3УС-ЛУС; 3 - 29-3УСЛУС; 4 - фірми Енгельгард. Фіг. 4. Термостабілізатор активності мікросферичного каталізатора крекінгу. 1 - корпус; 2 - каталізатор; 3 - стакан для каталізатора; 4 - втулка; 5 - термопарна кишеня; 6 трійник; 7 - штуцер пароперегрівача; 8 - прокладка; 9, 12 - накидна гайка; 10 - сальникове ущільнення; 11 - гранд-букса; 13 - термопари; 14 - пічка; 15 - теплоізоляція; 16 - пробка; 17 - контрольне дзеркало. Фіг. 5. Спектри ТПД аміаку для стабілізованих зразків каталізаторів. І - інтенсивність сигналу детектора (мВ), Т температура (°С). Промислові зразки (а); 1 - фірми Енгельгард; 2 - фірми Грейс; 3 - Уфимської каталізаторної фабрики; 4 - фірми Енгельгард (свіжий). Декатіоновані зразки (б): 1 - 25-4УС-ТПО; 2 26-4УО-ТПО; 3 - 29-4УС-ТПО. Лантан-декатіоновані зразки (в): 1 - 25-3УСЛУС-ТПО; 2 - 26-3УС-ЛУС-ТПО; 3 - 29-3УС-ЛУСТПО. Фіг. 6. Залежність конверсії кумоду с (% мас.) від числа імпульсів І для стабілізованих каталізаторів. Декатіоновані зразким (а): 1 - 25-4УС-ТПО; 2 26-4УС-ТПО; 3 - 29-4УС-ТПО. Лантан-декатіоновані зразки (б): 1 - 25-3УСЛУС-ТПО; 2 - 26-3УС-ЛУС-ТПО; 3 - 29-3УС-ЛУСТПО. Залежність 4 відноситься до стабілізованого зразка фірми Енгельгард. Приклад 1 Цим прикладом ілюструється одержання каолінової мікросфери і її прожарених форм. Вихідною сировиною для одержання каолінової мікросфери слугувала фракція збагаченого каоліну Просянівського каолінового родовища (Дніпропетровська область), 82% мас. якої складали частинки з розміром до 5 мкм. Вміст заліза (в перерахунку на Fе2О3) в цій фракції складав 0,81% мас. З цієї вихідної фракції каоліну одержували більш дрібнодисперсну фракцію з суттєвим вмістом частинок субмікронного розміру. Для цього у високих скляних циліндрах ємністю 20 літрів готовили суспензію вихідної фракції каоліну в дистильованій воді концентрацією 10% мас. В циліндр завантажували 2 кг вихідної фракції каоліну, 18 л дистильованої води, а також 10 г дефлокулянта 12 28551 лювальній сушарці продуктивністю по випарюваній воді 10 кг/год за допомогою відцентрового розпилюючого механізму. Швидкість обертання розпилюючого диска при цьому складала 18000 обертів за хвилину, а його діаметр - 120 мм. Робочий об'єм сушильної камери складав 0,9 м3, а її діаметр - 1200 мм. Теплоносієм в сушарці слугувало повітря, підігріте в електрокалорифері. Температура теплоносія на вході в сушильну камеру становила 280-300°С, а на виході з неї - 140-150°С. В процесі розпилювальної сушки відбувається випаровування води із крапель розпиленої суспензії і утворення каолінової мікроофери. Як показала експериментальна перевірка, залишкова вологість каолінової мікросфери не перевищує 1% мас. Від одержаної мікросфери відсіювали за допомогою сит частинки, розмір яких перевищує 160 мкм. За описаною процедурою було одержано декілька партій каолінової мікросфери масою 3-5 кг кожна. Якщо в процесі подальших досліджень виникала потреба в метакаоліновій або "мулітовій" мікросфері, то їх одержували шляхом прожарювання частини каолінової мікросфери в муфельній печі протягом двох годин при температурі 735 і 1000°С відповідно. В необхідних випадках проводили вилучення з "мулітової" і метакаолінової мікросфери дрібних частинок, менших 20 мкм, водою шляхом декантації. В деяких наступних прикладах "мулітову" мікросферу будемо називати мікросферою А, а метакаолінову мікросферу - мікросферою В. Приклад 2 Даним прикладом ілюструється вплив умов роздільної "мокрої" очистки "мулітової" і метакаолінової мікросфери від заліза на глибину цієї очистки, а також на ступінь деалюмінування згаданих форм прожареного каоліну. Як реагент використовували соляну кислоту концентрацією 30% мас. Очистці від заліза і деалюмінуванню піддавали наважки масою 125 г "мулітової" або метакаолінової мікросфери (мікросфери А і В відповідно), одержаних у прикладі 1. Оскільки в розрахунку на повністю дегідратований каолін сумарний вміст оксидів заліза, титану, лужних і лужноземельних елементів становив лише 4,0% мас. (решту складали оксиди алюмінію і кремнію), то при розрахунку вмісту Fе2О3 і АІ2О3 в початковій (неочищеній) "мулітовій" і метакаоліновій мікросфері апр і впр відповідно першу з цих величин розраховували за допомогою рівняння (4), а другу визначали як вміст оксиду алюмінію в повністю дегідратованому каолініті. Таким чином, апр=акМк/Мпр=0,0094 і M Al2O2 102 впр = = = 0,46. MAl2O3 + 2MSiO2 222 (пірофосфату натрію) для запобігання агрегації частинок каоліну. Після ретельного перемішування з метою створення однакової концентрації суспензії в усіх точках об'єму циліндра суспензію відстоювали протягом 70-90 годин. Після цього верхній шар суспензії висотою Н (см) відсифонювали в спеціальну ємність (кристалізатор із термостійкого скла). Необхідну висоту розраховували із співвідношення Н=tvоc, де t - час відстоювання суспензії (години), voc=0,333 см/год - швидкість осідання сферичних твердих частинок діаметром 1 мкм в сильно розбавленій водній суспензії, в якій на процес осідання даної частинки не впливають інші частинки суспензії, тобто коли осідання частинок відбувається в умовах виконання закону Стокса. Якби закон Стокса при осіданні частинок каоліну справді виконувався, то у верхньому шарі суспензії висотою Н сантиметрів через t годин відстоювання залишились би тільки частинки каоліну, розмір яких не перевищує 1 мкм. Насправді ж, частинки каоліну не мають сферичної форми, а суспензія є досить концентрованою і незалежного від інших частинок суспензії осідання даної частинки не відбувається, тобто закон Стокса, строго кажучи, при цьому не виконується, а у верхньому шарі суспензії висотою Н можуть бути в наявності і частинки каоліну, розміри яких перевищують 1 мкм. Проте, безсумнівно, що каолін в відсифонованому верхньому шарі суспензії є більш дрібнодисперсним, ніж у вихідній фракції каоліну, і що він відзначається значним вмістом каолінових частинок субмікронного розміру. Відсифонований шар суспензії ретельно перемішували в кристалізаторі, після чого у фарфорову чашку відбирали наважку суспензії об'ємом 50-100 мл, в якій шляхом випаровування води в сушильній шафі при температурі 110130°С протягом часу, достатнього для досягнення постійної ваги чашки з наважкою (8-12 годин), визначали концентрацію суспензії в кристалізаторі (вона складала біля 5% мас.). Далі проводили часткове випаровування води із суспензії, нагріваючи при перемішуванні кристалізатор із суспензією на електричній плитці і періодично зважуючи його на технічній вазі з метою контролю за наближенням маси суспензії до необхідної кінцевої маси, концентрація каоліну в якій становила б приблизно 60% мас., як це необхідно для проведення розпилювальної сушки суспензії при одержанні каолінової мікросфери. Ця кінцева маса суспензії може бути легко розрахована на основі відомих початкової маси суспензії в кристалізаторі і концентрації каоліну в ньому. Наприклад, якщо початкова маса суспензії в кристалізаторі становить 8,4 кг, а її концентрація - 5% мас., то кінцева маса суспензії 60%-ної концентрації повинна становити (8,4´0,05):0,6=0,7 кг. Коли кінцева маса суспензії досягала необхідного значення, випаровування води із суспензії припиняли. Оскільки під час випаровування води пірофосфат натрію втрачає свої дефлокулюючі властивості внаслідок його гідролізу з утворенням фосфату, який дефлокулюючими властивостями не відзначається, то в одержану каолінову суспензію знову додавали пірофосфат натрію з розрахунку 0,3-0,5% від маси наявного в суспензії каоліну і суспензію добре перемішували. Зразки каолінової суспензії, одержані описаним способом, розпилювали в прямоточній розпи Масове співвідношення розчин соляної кислоти/наважка мікросфери ("мулітової" або метакаолінової) підтримували на рівні 1,8. В дослідах змінювали температуру і тривалість очистки. Саму очистку здійснювали в термостатованій колбі ємністю 500 мл, оснащеній мішалкою, термометром і зворотним холодильником, за методикою, наведеною при описі технологічної стадії "В1". Ця методика передбачає таку послідовність операцій: очистку, відфідьтрування мікросфери, її промивку водою до нейтральної реакції, сушку мі 13 28551 кросфери при температурі 115°С протягом 3 годин, хімічний аналіз суміші фільтрату і промивної води на загальний вміст в ній іонів заліза, а також на вміст іонів алюмінію. Визначення вмісту в суміші названих іонів проводили відповідно сульфосаліциловим [38] і тригонометричним [39] методами. Результати хімічного аналізу суміші використовували для розрахунку ступеню деалюмінування взятої наважки прожареної каолінової мікросфери, глибини її очистки від заліза і залишкової концентрації Fе2О3 в очищеній прожареній мікросфері. Введемо позначення: V - об'єм суміші фільтрату і промивної води, л; с1 і с2 - загальна концентрація іонів заліза і концентрація іонів АІ+3 в цій суміші відповідно, г/л; d1 і d2 - глибина очистки "мулітової" і метакаолінової мікросфери від заліза; d1 і d2 - масова частка залишкового заліза (в перерахунку на Fе2О3) в очищеній "мулітовій" і метакаоліновій мікроcфері відповідно, % мас. Якщо кислотній очистці піддавали "мулітову" мікросферу масою m, то ступінь деалюмінування цієї мікросфери a, глибину її очистки від заліза d1 і масову частку залишкового заліза (в перерахунку на Fе2О3) в очищеній мікросфері d1 визначали за допомогою виразів a=(102/54)(Vc2/впрm, d1=(160/112)(Vc1/aпрm) і a прm(1 - d1 )100 a пр (1 - d1 )100 , d1 = = m[(1 - a )впр + (1 - впр )] (1 - aвпр ) і метакаоліновій мікросфері на вихід цеоліту Y при його синтезі в них у присутності розчину гідроксиду натрію. Деалюмінованими "мулітовою" і метакаоліновою мікросферою слугували зразки очищеної від заліза мікросфери, одержані в прикладі 2, а недеалюмінованими - зразки відповідним чином прожареної мікросфери, утвореної з каолінів 1, 2 і 3 (див. опис стадії "Л"). Синтез цеоліту проводили в круглодонній колбі так, як це наведено при описі стадій "Д" і "Ж". Умови ситензу: тривалість визрівання tвизр=6 год.; температура визрівання tвизр=40±1°С; тривалість кристалізації tкр=48 год.; температура кристалізації tкр=83±1°С; масова частка гідроксиду натрію в розчині, який подають в реактор на стадіях "Г" і "Е", g=0,19; спосіб подачі реагентів в реактор - всю метакаолінову мікросферу вводять на стадії "Г", а "мулітову" мікросферу і розчин NaOH вводять частково на стадії "Г", частково - на стадії "Е"; наважка метакаолінової мікросфериGм=20 г. При цьому згідно з рекомендаціями прототипу [12] було вибрано наступні значення необхідних для розрахунку складу реакційної суміші на стадіях "Г" і "Е" величини у1, z1, у2 і z2 (їх реально потрібно було б забезпечувати, якби на синтез цеоліту надходили недеалюміновані "мулітова" і метакаолінова мікросфера): у1=0,9 - масова частка "мулітової" мікросфери в її суміші з метакаоліновою на стадії "Г"; z1=0,65 - мольне співвідношення Na2O/Al2O3 на стадії "Г"; у2=0,95 - масова частка "мулітової" мікросфери в її суміші з метакаоліновою на стадії "Е"; z2=0,65 - мольне співвідношення Na2O/Al2O3 на стадії "Е". Результати досліджень наведено в табл. 2, в якій (крім ступеню деалюмінування "мулітової" і метакаолінової мікросфери a і b та величин g1, h1, q і h2, які відповідно визначаються за допомогою формул (17а), (18а), (28а) і (29а)) використано додаткові позначення: d1 і d2 - вміст залишкового Fe2O3 відповідно в "мулітовій" і метакаоліновій мікросфері перед синтезом цеоліту, % мас.; G1 і Q1 відповідно наважка "мулітової" мікроcфери і розчину NaOH, які завантажують в реактор на стадії "Г", г; G2 і Q2 - відповідно наважка "мулітової" мікросфери і розчину NaOH, які додатково завантажують в реактор на стадії "Е", г; ур - вміст цеоліту Y в цеолітвмісній мікросфері (після її синтезу, вилучення, промивки водою до нейтральної реакції і сушки при температурі 115°С протягом трьох годин) за даними рентгенофазового аналізу, % мас. На фіг. 1 представлено фрагменти рентгенівських дифрактограм ЦВМС-7, ЦВМС-8 і ЦВМС-9. В дослідах 7, 8 і 9, в яких для синтезу цеоліту використовували недеалюміновану "мулітову" і метакаолінову мікросферу, одержану на основі каолінів 1, 2 і 3 з початковим вмістом Fе2О3 1,12, 0,63 і 0,35% мас. відповідно, величини d1 і d2 розраховували за допомогою рівняння (4). Величини G1, Q1, G2 i Q2 розраховували відповідно за допомогою залежностей G1=Gm[g1/(1-g1)], Q1=h1Gm[1/(1-g1)], G2=qGm [1/(1-g1)], Q2=h2Gm[1/(1-g1)]. Кожний дослід повторювали 4 рази. В табл. 2 наведено усереднені дані (щодо вмісту цеоліту Y в ЦВМС). Наважки цеолімтвмісної мікросфери, одержані в паралельних дослідах, змішували. в яких числа 102, 54, 160 і 112 означають відповідно молекуклярну масу оксиду алюмінію, подвоєну атомну масу алюмінію, молекулярну масу Fе2О3 і подвоєну атомну масу заліза. При заміні в наведених виразах a на b, d1 на d2 і d1 на d2 за їх допомогою розраховували cтупінь деалюмінування, глибину очистки від заліза і масову частку залишкового заліза в очищеній мікросфері у випадку, коли кислотній очистці піддавали метакаолінову мікросферу масою m. Вплив температури і тривалості солянокислотної обробки "мулітової" і метакаолінової мікроcфери на ефективність їх очистки від заліза і cтупінь деалюмінування демонструється табл. 1. Умови очистки: масове співвідношення розчин соляної кислоти/наважка "мулітової" або метакаолінової мікросфери j=1,8; концентрація соляної кислоти ск=30% мас.; наважка "мулітової" або метакаолінової мікросфери m=125 г. Для накопичення значних кількостей очищеної від заліза мікросфери кожний дослід повторювали 8 разів. В табл. 1 наведено усереднені дані. Наважки очищеної мікросфери, одержані в паралельних дослідах, змішували. Як видно з цієї таблиці, підвищення температури і збільшення тривалості солянокислотної обробки призводить до зростання як ефективності очистки прожарених форм каоліну від заліза, так і ступеню їх деалюмінування. За одних і тих же умов глибини очистки від заліза "мулітової" і метакаолінової мікросфери близькі між собою, тоді як деалюмінування метакаоліну проходить значно швидше, ніж деалюмінування "муліту". Приклад 3 Цим прикладом ілюструється вплив концентрації залишкового заліза (в перерахунку на Fе2О3) в деалюмінованих і недеалюмінованих "мулітовій" 14 28551 Як видно з цієї таблиці, існує чітка тенденція зростання виходу цеоліту із зменшенням концентрації залишкового заліза в мікроcфері. Приклад 4 Даним прикладом ілюструється вплив тривалості кристалізації цеоліту Y на його вміст в цеолітвмісній мікросфері. З цією метою було одержано зразки цеолітвмісної мікросфери ЦВМС-3а, ЦВМС-3б і ЦВМСЗв, умови синтезу яких повністю збігалися з умовами синтезу зразка ЦВМС-3 в прикладі 3 за винятком часу кристалізації. Вихід цеоліту в одержаних зразках визначали за допомогою рентгенофазового аналізу. Одержані результати наведено в табл. 3. Як видно з цієї таблиці, із збільшенням часу кристалізації вихід цеоліту Y зростає, але темп цього зростання поступово знижується. Приклад 5 Цим прикладом ілюструється процес перетворення зразків ЦВМС в зразки каталізатора крекінгу, методика якого детально описана в описі стадії "Л"). Такому перетворенню піддавались ЦВМС-7, ЦВМС-8 і ЦВМС-9 з прикладу 3. Одержані з них каталізатори складали відповідно серії 25, 26 і 29. Було синтезовано такі каталізатори: 25-4УС, 253УС-ЛУС, 26-4УС, 26-3УС-ЛУС, 29-3УС, 29-4УС, 29-3УС-ЛУС. Процес перетворення кожної цеолітвмісної мікросфери в той чи інший зразок каталізатора крекінгу повторювали три рази, причому кожен раз наважка свіжої ЦВМС складала 50 г. Всі три одержані зразки каталізатора змішували. Умови синтезу каталізатора: температура іонного обміну – 85°С; тривалість іонного обміну в одному циклі іонний обмін-промивка-сушка-ультрастабілізація - 2 год.; наважка розчину нітрату амонію - 100 г; концентрація цього розчину - 8% мас.; промивка каталізатора водою - до нейтральної реакції; температура сушки – 120°С; тривалість сушки - 2 год.; температура ультраотабілізації –560°С; тривалість ультрастабілізації - 2 год.; кількість циклів іонний обмін-промивка-сушка-ультраотабілізація - 3 або 4. В табл. 4 наведено дані з гранулометричного і хімічного складу каолінів 1, 2 і 3, які були сировиною для одержання ЦВМС-7, ЦВМС-8 і ЦВМС-9 відповідно. З даних, наведених в цій таблиці, випливає, що за своїм гранулометричним і хімічним складом каоліни 1, 2 і 3 близькі між собою. Суттєво вони відрізняються лише вмістом Fe2O3. Цеолітвмісні мікросфери ЦВМС-7, ЦВМС-8 і ЦВМС-9 було одержано за одних і тих же умов (див. приклад 3). При їх синтезі використовували недеалюміновану "мулітову" і метакаолінову мікросферу, що можна розглядати як позитивний момент, оскільки на синтез цеоліту ніяким чином не впливав різний ступінь деалюмінування "мулітової" і метакаолінової мікросфери. Усі ці фактори дозволяють стверджувати, що можливі відмінності в якості зразків каталізаторів, одержаних за одних і тих же умов і за допомогою одних і тих же процедур, але із зразків різної сировини (ЦВМС), які відрізняються, по суті, лише одним показником - вмістом Fe2O3, зумовлені саме цим показником. Вплив вмісту Fe2O3 y вихідному каоліні (а, отже, і в ЦВМС і каталізаторі) на кислотність і активність одержаних зразків каталізаторів буде продемонстровано в наступних прикладах. У цьому прикладі ми обмежимося лише демонстрацією катіонного складу цеолітної компоненти каталізаторів (табл. 5), визначеного за даними хімічного аналізу. Як видно з цієї таблиці, за ступенем декатіонування і вмістом лантану одержані через однакові операції і однакову їх кількість зразки каталізаторів різних серій близькі між собою. Приклад 6 Проведемо оцінку кислотності наших свіжих зразків каталізаторів методом ТПД аміаку, базуючись на зразках, одержаних у прикладі 5, в зіставленні їх із свіжими зразками промислових каталізаторів крекінгу. Методика вивчення кислотності названим методом зводилась до наступного. Спочатку проводили термопрограмовану контрольовану дегідратацію наважки каталізатора (0,1 г) в потоці гелію в температурному діапазоні від 50 до 500°С в спеціальному мікрореакторі, вмонтованому в хроматографічну схему з детектором по теплопровідності - катарометром. Після охолодження дегідратованого зразка до температури 200°С на нього за допомогою відкаліброваної петлі контрольовано подавали до проскоку певне число доз аміаку. Надлишковий аміак віддували при 200°С, після чого починали його термопрограмовану десорбцію в температурному інтервалі від 200 до 500°С. При досягненні кінцевої температури програма автоматично відключалась, після чого реактор витримували в ізотермічному (500°С) режимі до виходу самописця на нульову або горизонтальну лінії, що свідчило про повну десорбцію аміаку, і чого, на жаль, не завжди вдавалось досягти за прийнятні відрізки часу. На фіг. 2 представлено ТПД-спектрограми одержаних в прикладі 5 семи зразків свіжих каталізаторів в порівнянні із спектрограмами промислових зразків. ТПД-спектр каталізатора фірми Енгельгард (крива 1) свідчить про більш-менш рівномірний розподіл кислотних центрів різної сили по температурному діапазону. Спектр зразка фірми Грейс (крива 2) виявляє перекос в бік сильних кислотних центрів, тоді як спектр зразка виробництва Уфимської каталізаторної фабрики (крива 3), навпаки, свідчить про перевагу в ньому слабких кислотних центрів, хоча при цьому за числом сильних кислотних центрів він не поступається ні зразку фірми Енгельгард, ні зразку фірми Грейс. Крім того, уфимський зразок характеризується також найбільшим числом кислотних центрів в цілому серед досліджуваних трьох промислових каталізаторів. Одержані нами зразки характеризуються більш-менш рівним розподілом кислотних центрів за силою (криві 4-10) з деяким в цілому перекосом в бік слабких кислотних центрів. Число активних центрів помітно зростає при переході від зразків серії 25 до зразків серії 26 і 29. Цікава особливість проглядається між зразками 4УС і 3УС-ЛУС в кожній серії щодо загального числа кислотних центрів. Так, якщо в серії 25 лантанвмісний зразок відзначався більшим числом кислотних центрів (крива 5), ніж декатіонований (крива 4), а в серії 26 15 28551 обидва зразки за цим показником (особливо щодо слабких кислотних центрів) близькі (криві 6 і 7), хоча декатіонований зразок (крива 6) має деяку перевагу над лантанвмісним (крива 7), то в серії 29 лантанвмісний зразок (крива 10) за числом кислотних центрів однозначно поступається декатіонованому (крива 9). Із зіставлення ТПД-спектрів зразків 29-3УС (крива 8) і 29-4УС (крива 9) видно, що декатіонування і ультрастабілізація приводять до зменшення числа кислотних центрів головним чином - слабких. В цілому можна зробити висновок, що одержані нами кращі зразки свіжих каталізаторів (серії 29) за своєю кислотністю зіставимі з промисловими каталізаторами крекінгу і що за інших рівних умов підвищення вмісту цеоліту в каталізаторі (при переході від серії 25 до серії 26 і 29) призводить до зростання його кислотності. Приклад 7 Проведемо оцінку активності наших свіжих каталізаторів в модельній реакції крекінгу кумолу, базуючись на зразках, одержаних у прикладі 5, в зіставленні їх із свіжим каталізатором крекінгу фірми Енгельгард. Визначення активності зразків в модельній реакції крекінгу кумолу проводили в імпульсному режимі на установці, створеній на базі хроматографа з полум'яно-іонізаційним детектором. Тестування зразків зводилось до попередньої дегідратації наважки (0,1 г) в потоці гелію при 500°С протягом 1 години, охолодження до певної температури (як правило, 320±2°С) і послідовної подачі імпульсів кумолу об'ємом 2 мкл кожний. В залежності від активності досліджуваних зразків число імпульсів коливалось від 3-4 до 10-20. Як видно з фіг. 3, активність декатіонованих і лантандекатіонованих зразків при переході від серії 25 до серії 29 помітно зростає, перевищуючи активність зразка фірми Енгельгард у випадку каталізатора 29-4УС (фіг. 3а, крива 4) і дещо поступаючись активності цього зразка в початковий період роботи у випадку каталізатора 29-3УС-ЛУС (фіг. 3б, крива 3). Таким чином, каталітичні дані для синтезованих нами зразків дуже непогано узгоджуються з даними по кислотності: збільшення вмісту цеоліту в зразках має наслідком більш високу, порівняно рівномірно розподілену за силою кислотність і, відповідно, більш високу активність і стабільність в реакції крекінгу. За своєю каталітичною активністю в реакції крекінгу кумолу синтезовані нами зразки каталізаторів знаходяться на рівні кращих свіжих промислових каталізаторів (зокрема, каталізатора фірми Енгельгард). Приклад 8 В цьому прикладі описані пристрій і процедура термопаростабілізації мікросферичного цеолітвмісного каталізатора крекінгу, які були використані авторами. Свіжий зразок каталізатора шляхом термопарообробки за певних жорстких умов можна перевести в стан, ідентичний рівноважному каталізатору. Для моделювання умов стабілізації свіжосинтезованих зразків було створено спеціальну установку, яка дозволяла піддавати зразки контрольованій термопарообробці в умовах чистої водяної пари при температурах до 1000°С протягом необхідного часу. Основним вузлом установки (фіг. 4) є термопаростабілізатор (далі - стабілізатор), корпус якого 1 виконаний із товстостінної трубки з нержавіючої сталі внутрішнім діаметром 21 мм. Каталізатор 2 розмішується в тонкостінному стакані 3 і утримується втулкою 4, запресованою в стакані 3 на певній висоті. Зазор між зовнішньою стінкою стакану і внутрішньою стінкою корпуса складає 0,2 мм. Крізь втулку з зазором, який (для запобігання просипанню каталізатора) не перевищує 0,01 мм, по осі реактора проходить термопарна кишеня 5. Стакан 3 опирається на глухо загвинчений в нижню частину корпусу стабілізатора трійник 6, який слугує як для подачі перегрітої до 150200°С пари через штуцер 7, так і для утримання термопарної кишені 5 всередині стабілізатора. Штуцер 7 з'єднаний з трійником 6 через тефлонову прокладку 8 за допомогою накидної гайки 9, а термопарна кишеня ущільнена з використанням звичайного сальникового ущільнення, яке включає тефлонову набивку 10, гранд-буксу 11 і накидну гайку 12. Для регулювання і контролю температурного режиму в зоні каталізатора служать дві хромель-алюмелеві термопари 13, гарячі спаї яких розміщуються відповідно посередині шару каталізатора і безпосередньо за шаром, як показано на фігурі точками по осі стабілізатора. По всій висоті корпусу стабілізатора розміщена пічка 14 із ніхромового дроту з теплоізоляцією 15. Верхня частина корпусу стабілізатора оснащена загвинчуваною пробкою 16 з отвором для виходу пари. Як і сам корпус, всі металеві деталі стабілізатора виконані з нержавіючої сталі. Стабілізатор розміщено у витяжній шафі на окремому стенді, який дозволяє здійснювати його легкий зйом і повторну установку при перезавантаженні. Крім показаного на фіг. 4 вузла термопаростабілізації, установка в цілому включає ще колбу з водою, паропровід з пароперегрівачем для підводу утворюваної пари через штуцер 7 до шару каталізатора, регулятори напруги на пічку 14 і пароперегрівач паропроводу, потенціометри для регулювання і вимірювання температури, а також відповідні електричні та термопарні роз'єми. Термопаростабілізацію синтезованих зразків здійснювали таким чином. В згаданій колбі підігрівали воду до рівномірного кипіння (вносили декілька цеолітних гранул) і (при від'єднаному від стабілізатора штуцері 7) встановлювали через паропровід з пароперегрівачем рівномірний потік перегрітої до 150-200°С пари. При вивернутій пробці 16 в знятий зі стенду стабілізатора стакан 3 з використанням лійки, носик якої опирався на втулку 4, вносили зважену на технічній вазі певну кількість повітряно-сухого каталізатора, як правило, 5 г. Завертали пробку 16, стабілізатор закріплювали на стенді, з'єднували електричні і термопарні роз'єми, після чого каталізатор підігрівали до температури 700-750°С із швидкістю 20-25°С за хвилину. При досягненні цієї температури паропровід з перегрітою парою через штуцер 7 і прокладку 8 під'єднували до трійника 6 і ущільнювали підкруткою накидної гайки 9. Злегка перегріта водяна пара починала омивати стакан 3, витісняючи повітря з корпусу стабілізатора. Крім того, пара попадала на 16 28551 шар каталізатора через зазор між втулкою 4 і термопарною кишенею 5. Встановлення режиму потоку пари якісно контролювали періодичним розташуванням дзеркала 17 на виході пари через отвір пробки 16. Через 5-10 хвилин від початку подачі пари досягалась задана температура 750-900°С. В сталому режимі з точністю витримування температури ±5°С термопарообробку проводили протягом необхідного часу - від 1 до 6 годин (найчастіше протягом 2-3 годин). При завершенні цього часу припиняли подачу напруги на пічку 14, зразу ж від'єднували штуцер 7 від трійника 6 і давали можливість системі охолонути до температури, яка не перевищувала 100°С. Потім, роз'єднавши електричні і термопарні роз'єми, стабілізатор знімали зі стенду і висипали термопарооброблений каталізатор. Для адекватності моделювання стабілізації каталізатора в описаній установці його натурній стабілізації в промисловій установці каталітичного крекінгу автори відібрали рівноважний каталізатор фірми Енгельгард із працюючої установки AT "Укртатнафта", визначили його активність в модельній реакції крекінгу кумолу, а після здійснили пошук умов термопарообробки свіжого каталізатора цієї ж фірми в описаній системі, які дозволили одержати термопарооброблений каталізатор, ідентичний за своєю активністю рівноважному. Ідентичність досягнута при температурі і тривалості термопарообробки, які становили 800°С і 2 години відповідно. В знайдених умовах проводили термопаростабілізацію власних зразків. Приклад 9 Проведемо оцінку кислотності наших (стабілізованих каталізаторів методом ТПД аміаку, базуючись на зразках, синтезованих в прикладі 5, в зіставленні їх із стабілізованими (рівноважними) промисловими каталізаторами. Вивчення кислотності стабілізованих зразків каталізаторів проводили за методикою, описаною в прикладі 6. На фіг. 5 представлено ТПД-спектрограми шести стабілізованих зразків каталізаторів, одержаних в прикладі 5, а також трьох промислових каталізаторів (фірми Енгельгард, фірми Грейс і виробництва Уфимської каталізаторної фабрики). Для порівняння також наведено ТПД-спектрограму свіжого каталізатора фірми Енгельгард. Термопарообробка свіжих зразків в жорстких умовах, описаних в прикладі 8, призводить до їх глибоких змін, що проявляється в дуже значному зменшенні числа кислотних центрів. Це наочно можна бачити з порівняння кислотності свіжого і стабілізованого зразків фірми Енгельгард (фіг. 5а, криві 1 і 4). Але певна залишкова кислотність в стабілізованих зразках зберігається. Найбільшим числом кислотних центрів характеризується стабілізований зразок каталізатора виробництва Уфимської каталізаторної фабрики (фіг. 5а, крива 3) - його ТПД-спектр "поглинає" спектри зразків фірм Грейс і Ентельгард, а найменшим - стабілізірований зразок фірми Грейс (фіг. 5а, крива 2). Стабілізований зразок фірми Енгельгард займає проміжне становище. Характерно, що, як і для відповідно свіжих зразків, ТПДспектри для уфимського зразка мають перекос в сторону слабких кислотних центрів, для зразка фірми Грейс - навпаки, в сторону сильних кислотних центрів, а для зразка фірми Енгельгард вони відзначаються більш-менш рівномірним розподілом кислотних центрів за силою. На фіг. 5б і 5в наведено ТПД-спектри власних каталізаторів. Всі спектри декатіонованих зразків (4УС-ТПО, фіг. 5б) дуже близькі як за своїм зовнішнім виглядом, так і за площами під ними. Серед спектрів лантандекатіонованих зразків (3УС-ЛУСТПО, фіг. 5в) помітно видається спектр зразка серії 26, в якому переважають сильні кислотні центри. Зразки каталізаторів серій 25 і 29 відзначаються рівним спектром у всьому програмованому температурному діапазоні. В цілому можна зробити висновок, що за своєю залишковою кислотністю наші стабілізовані зразки каталізаторів зіставимі із промисловими каталізаторами. Приклад 10 Проведемо оцінку активності наших стабілізованих каталізаторів в модельній реакції крекінгу кумолу, базуючись на зразках, одержаних у прикладі 5, в зіставленні їх з промисловим каталізатором фірми Енгельгард, який, як було з'ясовано в результаті спеціального дослідження, є найбільш активним і селективним серед випробуваних промислових каталізаторів. Визначення активності стабілізованих каталізаторів в модельній реакції крекінгу кумолу проводили за методикою, описаною в прикладі 7, за винятком того, що випробування зразків здійснювали не при температурі 320±2°С (як у випадку свіжих каталізаторів), а при температурі 400±2°С. На фіг. 6 наведено залежності конверсії кумолу від числа імпульсів для наших каталізаторів. Для ряду 4УС-ТПО із збільшенням вмісту цеолітної фази при переході від зразка серії 25 до зразків серії 26 і 29 (фіг. 6а, криві 1, 2, 3) активність зростає, причому відповідні залежності розташовані еквідистантно по відношенню одна до одної. Залежність для зразка фірми Енгельгард (фіг. 6а, крива 4) розташована дещо нижче від залежності 3, але більш полого, і для 8-10 імпульсів обидві криві зближуються. Суттєво по-іншому виглядають закономірності для каталізатор-ного ряду 3УС-ЛУС-ТПО (фіг. 6б): попередня одноманітність зникає. Залежності для зразків серії 25 і 26 - доволі круті (криві 1 і 2) за явно переважаючої активності зразка серії 26. Але найбільш високою і стабільною для імпульсів 7-10 активністю відзначається зразок серії 29 (крива 3). Вона помітно вища за активність промислового зразка фірми Енгельгард. Таким чином, стабілізовані зразки синтезованих каталізаторів кращої серії 29 мають таку активність в модельній реації крекінгу кумолу, яка не поступається активності кращих промислових зразків каталізаторів. Приклад 11 Цим прикладом ілюструється відповідність показників якості синтезованих каталізаторів вимогам діючих технічних умов ТУ 38.1011366-91 [37]. Для випробування було взято зразок каталізатора 4УС серії 19 (це серія каталізаторів, одержаних за методикою, детально описаною в описі стадії "Л"), на основі ЦВМС-5 з прикладу 3). 17 28551 Процес перетворення ЦВМС-5 в каталізатор крекінгу 19-4УС повторювався три рази, причому кожен раз наважка свіжої ЦВМС-5 складала 50 г. Інші умови синтезу каталізатора: температура іонного обміну – 85°С; тривалість іонного обміну в одному циклі іонний обмін-промивка-сушка-ультрастабілізація - 2 год.; наважка розчину нітрату амонію - 100 г; концентрація цього розчину - 8% мас.; промивка каталізатора водою - до нейтральної реакції; температура сушки – 120°С; тривалість сушки - 2 год.; температура ультрастабілізації – 560°С; тривалість ультрастабілізації - 2 год.; кількість циклів іонний обмін-промивка-сушка-ультрастабілізація - 4. Всі три одержані зразки каталізатора змішували. В табл. 6 наведено значення всіх передбачених ТУ 38.1011366-91 показників якості цього каталізатора в їх порівнянні з регламентними нормами вказаних технічних умов. Визначення цих показників здійснювали у відповідності із стандартними методиками випробування каталізатора крекінгу [28]. Як видно з цієї таблиці, за своєю якістю випробуваний каталізатор перевищує вимоги ТУ 38.1011366-91 по всіх показниках. Джерела інформації 1. Уорелл У. Глины и керамическое сырье. М.: Мир, 1978. С. 178-179. 2. Murat М., Amokrane A., Bastide J.-P. // Comptes rendus Acad. sci. Ser. 2. 1990. V. 310. № 13. P. 1725-1730. 3. Нефедов Б.К., Радченко Е.Д., Алиев P.P. Катализаторы процессов углубленной переработки нефти. М.: Химия, 1992. С. 72. 4. Каолины Украины. Справочник. Ред. Ф.Д. Овчаренко. Киев: Наукова думка, 1982. С. 216-224. 5. Лапин В. В. Автореферат дисс. ... канд. хим. наук. Ленинградская лесотехническая академия им. С.М. Кирова. Л.: 1969. 15 с. 6. Патент США 3647718, опубл. 07.03.1972, МПК B01j11/40; C01b33/28. 7. Патент США 3633165, опубл. 16.05.1972, МПК C01b33/23; B01j11/40. 8. Патент США 3657154, опубл. 18.04.1972, МПК B01j11/40. 9. Патент США 3932268, опубл. 13.01.1976, МПК C01g11/04. 10. Avidan A.A. Akzo Catalysts Symposium 1991. Fluid Catalytic Cracking / Ed. B. van Keulen. Amersfort, The Netherlands: Akzo Chemicals Division, 1991, p. 47. 11. Патент США 5023220, опубл. 11.06.1991, МПК B01j29/08; B01j21/16. 12. Патент США 4581341, опубл. 08.04.1986, МПК B01j29/08. Прототип. 13. Химическая энциклопедия. М.: Советская энциклопедия, т. 2, 1990. С. 816-818. 14. Порубанский А.Е. Каолиновая промышленность в СССР и за рубежом (обзор). М.: Стройиздат, 1969. С. 19. 15. Permelee C.W., Rodriguez A.R. // J. Am. Ceram. Soc. 1942. V. 25. № 1. P. 1. 16. Грум-Гржимайло О.С., Макаров В.В., Гелвановский В.П. Труды института "НИИстройкерамика". М.: Стройиздат, 1990. Вып. 66. С. 26-30. 17. Нестеренко А.П., Сенин Е.В. Кинетика растворения оксидов железа и расчет ионных равновесий в дезактивирующих растворах. М.: ЦНИИатоминформ, 1987. С. 2. 18. Краткая химическая энциклопедия. М.: Советская энциклопедия, т. 2, 1963. С. 18. 19. Химическая энциклопедия. М.: Советская энциклопедия, т. 2, 1990. С. 131. 20. Неницеску К. Общая химия. М.: Мир, 1968. С. 663-665. 21. Лайнер А.И. Производство глинозема. М.: Металлургия, 1978. С. 320. 22. Olsen R.S., Bullard S.J, Grusensky W.G., Mrazek R.V., Henry J.L. // Rept. Invest. Bur. Mines US. Dep. Inter. 1983. № 8744. 11 p. 23. Пат. США 4388280, опубл. 14.06.1983, МПК C01f7/20. 24. Хейкер Д.М., Зевин Л. С. Рентгеновская дифрактометрия. М.: Физматгиз, 1963. С. 201-227. 25. Васильев Е.К., Нахмансон М.С. Качественный рентгенофазовый анализ. Новосибирск: Наука, 1986. С. 7-33, 36-48. 26. Полуэктов Н.С. Методы анализа по фотометрии пламени. М.: Госхимиздат, 1959. С.137139. 27. Несмеянова Т.О., Твердохлебова Т.Г. // Тр. ГрозНИИ. 1974. Вып. 27. Ч. 2. С.12-15. 28. Отраслевой стандарт "Катализаторы крекинга микросферические и молотые. Медоды испытаний" (ОСТ 38.01161-78). Издание официальное. Министерство нефтеперерабатывающей и нефтехимической промышленности СССР. М.: 1978. С. 37-45, 63-84. 29. Karge H.G,, Dondur V., Wetkamp J. // J. Phys. Chem. 1991. V. 95. № 1. P. 283-288. 30. Восмериков А.В., Барабашин Я.E., Ерофеев В.И. // Журн. прикл. химии. 1995. Т. 68. № 5. С. 789-792. 31. Osako К., Ono Y. // Bull. Chem. Soc. Jpn. 1993. V. 66. № 3. P. 755-759. 32. Ono Y. // Catal. Rev. - Sсi. eng. 1992. V. 34. № 3. P. 176-226. 33. Arata K., Hino M., Yamagata N. // Bull. Chem. Soc. Jpn. 1990. V. 63. № 1. P.244-246. 34. Vijayakumar S., Vijaya C., Rengara J.K., Sivasankar B. // Bull. Chem. Soc. Jpn. 1994. V. 67. № 11. P. 3107-3111. 35. Sedran U.A. // Catal. Rev. - Sci. Eng. 1994. V. 36. № 3. P. 405-431. 36. Bielanski A., Malecka A. Proceedings of the International Symposium on Zeolite Catalysis / Siofok, Hungаrу. 1985. P.389-396. 37. Технические условия "Катализаторы крекинга микросферические" (ТУ 38.1011366-91). Чечено-Ингушский центр стандартизации и метрологии. Грозный, 1991. С. 2-3. 38. Шарло Г. Методы аналитической химии. М.: Химия, 1969. С.775-776. 39. Несмеянова Т.С., Муляева Е.Н. // Тр. ГрозНИИ. 1974. Вып. 27. Ч. 2. С. 9-12. 18 28551 Таблиця 1 Залежність ефективності очистки від заліза і ступеню деалюмінування "мулітової" та метакаолінової мікросфери від температури і тривалості їх обробки соляною кислотою Номер досліду Температура, °С Тривалість очистки, год. 1 2 3 4 5 6 7 8 9 10 11 12 70 70 70 90 90 90 70 70 70 90 90 90 1 2 3 1 2 3 1 2 3 1 2 3 Масова частка залишкового заліза (в перахунку на Fе2О3) в очищеній прожареній мікросфері, % мас. d1 d2 0,69 0,50 0,36 0,67 0,46 0,28 0,77 0,60 0,44 0,77 0,55 0,37 Глибина очистки d1 0,273 0,481 0,632 0,305 0,533 0,728 d2 0,302 0,497 0,653 0,316 0,551 0,713 Ступінь деалюмінування a 0,039 0,067 0,094 0,053 0,098 0,142 b 0,317 0,452 0,559 0,323 0,481 0,594 Таблиця 2 Залежність виходу цеоліту Y при його синтезі в прожарених формах каолінової мікросфери від концентрації в них залишкового заліза (в перерахунку на Fе2О3) Номер досліду 1 2 3 4 5 6 7 8 9 d1 a d2 b g1 h1 G1 0,69 0,50 0,36 0,67 0,46 0,28 1,30 0,73 0,41 0,039 0,067 0,094 0,053 0,098 0,142 0 0 0 0,77 0,60 0,44 0,77 0,55 0,37 1,30 0,73 0,41 0,317 0,452 0,559 0,323 0,481 0,594 0 0 0 0,834 0,761 0,635 0,826 0,722 0,535 0,900 0,900 0,900 1,637 2,039 2,670 1,706 2,280 3,199 1,233 1,233 1,233 100,5 63,7 34,8 94,9 51,9 23,0 180,0 180,0 180,0 Продовження табл. 2 Номер досліду 1 2 3 4 5 6 7 8 9 Q1 q h2 G2 Q2 ур 197,2 170,6 146,3 196,1 164,0 137,6 246,6 246,6 246,6 0,570 0,417 0,280 0,559 0,375 0,217 0,900 0,900 0,900 1,637 2,039 2,670 1,706 2,280 3,199 1,233 1,233 1,233 68,7 34,9 15,3 64,3 27,0 9,3 180,0 180,0 180,0 197,2 170,6 146,3 196,1 164,0 137,6 246,6 246,6 246,6 27 33 44 29 37 53 21 25 39 Шифр ЦВМС ЦВМС-1 ЦВМС-2 ЦВМС-3 ЦВМС-4 ЦВМС-5 ЦВМС-6 ЦВМС-7 ЦВМС-8 ЦВМС-9 Таблиця 3 Залежність вмісту цеоліту Y в зразках ЦВМС від тривалості його кристалізації Шифр ЦВМС ЦВМС-3а ЦВМС-3б ЦВМС-3в ЦВМС-3 Тривалість кристалізації цеоліту Y, год. 20 29 39 43 19 Вихід цеоліту, % мас. 18 30 39 44 28551 Таблиця 4 Гранулометричний і хімічний склад каолінів Назва показника 1. Вміст фракції з розміром частинок, більших 10 мкм, % мас. 2. Вміст фракції з розміром частинок, менших 2 мкм, % мас. 6. Хімічний склад, % мас. SiО2 Al2O3 Fе2O3 ТiO2 СаО MgO К2O Na2O втрати при прожарюванні Каолін 1 Каолін 2 Каолін 3 6,5 2,6 8,0 73,0 76,0 67,0 46 38 1,12 1,16 0,12 0,28 0,62 0,11 12,4 47 38 0,63 1,17 0,10 0,25 0,63 0,12 12,1 46 38 0,35 1,23 0,35 0,39 0,92 0,16 12,6 Таблиця 5 Катіонний склад синтезованих каталізаторів крекінгу, (% від обмінної ємності) Зразок 25-4УС 25-3УС-ЛУС 26-4УС 26-3УС-ЛУС 29-3УС 29-4УС 29-3УС-ЛУС Вміст La+3 5,87 6,90 2,15 Ступінь декатіонування 99,61 93,36 99,74 87,14 96,71 99,70 97,79 Вміст залишкового Na+ 0,39 0,77 0,26 5,96 3,29 0,30 0,06 Таблиця 6 Відповідність показників якості каталізатора 19-4УС вимогам ТУ 38.1011366-91 Значення показника для каталізатора 19-4УС Назва показника якості і одиниця його виміру 1. Гранулометричний склад, % мас крупніше 0,160 мм, дрібніше 0,100 мм, дрібніше 0,040 мм, дрібніше 0,020 мм, 2. Насипна вага, кг/м3 3. Масова частка вологи, що видаляється при 800°С, % мас. 4. Зносостійкість, % мас. 5. Стабільна активність (після стабілізації при 775°С), вагова швидкість подачі сировини 20 год-1, % 6. Масова частка оксиду натрію, % мас. 7. Масова частка оксиду алюмінію, % мас. 20 Норма ТУ 38.1011366-091 95,0 5,0 1,0 900 не більше 2,0 не менше 93,0 не більше 25,0 не більше 4,0 не менше 750 3,0 не більше 5,0 96,0 не менше 95,0 51,0 не менше 42,0 0,16 37,0 не більше 0,5 не менше 19,0 28551 Фіг. 1 Фіг. 2 Фіг. 3 21 28551 Фіг. 4 Фіг. 5 Фіг. 6 __________________________________________________________ ДП "Український інститут промислової власності" (Укрпатент) Україна, 01133, Київ-133, бульв. Лесі Українки, 26 (044) 295-81-42, 295-61-97 __________________________________________________________ Підписано до друку ________ 2002 р. Формат 60х84 1/8. Обсяг ______ обл.-вид. арк. Тираж 35 прим. Зам._______ ____________________________________________________________ УкрІНТЕІ, 03680, Київ-39 МСП, вул. Горького, 180. (044) 268-25-22 ___________________________________________________________ 22
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for obtaining zeolite-containing micro-spherical catalyst of cracking
Автори англійськоюPatryliak Kazymyr Ivanovych, Nazarok Volodymyr Ivanovych, Vypyrailenko Valentyna Yosypivna, Khranovska Valentyna Ivanivna, Patryliak Liubov Kazymyrivna, Manza Ivan Andriiovych, Rode Herald Hryhorovych, Likhniovskyi Ruslan Yakovych, Yakovenko Oleksandr Mykolaiovych, Samus' Leontii Hryhorovych
Назва патенту російськоюСпособ получения цеолитсодержащего микросферического катализатора крекинга
Автори російськоюПатриляк Казимир Иванович, Назарок Владимир Иванович, Випирайленко Валентина Иосифовна, Храновская Валентина Ивановна, Патриляк Любовь Казимировна, Манза Иван Андреевич, Роде Геральд Григорьевич, Лихневський Руслан Владимирович, Яковенко Александр Николаевич, Самусь Леонтий Григорьевич
МПК / Мітки
МПК: B01J 29/00
Мітки: каталізатора, мікросферичного, крекінгу, цеолітовмісного, одержання, спосіб
Код посилання
<a href="https://ua.patents.su/22-28551-sposib-oderzhannya-ceolitovmisnogo-mikrosferichnogo-katalizatora-krekingu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання цеолітовмісного мікросферичного каталізатора крекінгу</a>
Спосіб одержання цеолітного каталізатора ізомеризації нормальних парафінових вуглеводнів

Номер патенту: 24297
Опубліковано: 07.07.1998
Автори: Ільїн Володимир Георгійович, Соломаха Володимир Миколайович, Цуприк Іванна Миколаївна, Левчук Микола Миколайович, Манза Іван Андрійович, Патриляк Казимир Іванович, Бобонич Федір Михайлович, Волошина Юлія Геннадіївна
МПК: B01J 29/00, B01J 37/08, C07C 5/00, B01J 37/02
Мітки: каталізатора, одержання, ізомеризації, парафінових, цеолітного, вуглеводнів, спосіб, нормальних
Формула / Реферат:
Спосіб одержання цеолітного каталізатора ізомеризації нормальних парафінових вуглеводнів, що включає обробку вихідного матеріалу амонійвмісними сполуками, прожарювання, обробку кислотою, нанесення паладія і/або платини та активацію в потоці водню, який відрізняється тим, що оброблену амонійвмісними сполуками каталітичну композицію перед кислотною обробкою попередньо прожарюють при 623 - 973К.
Компонент каталізатора (спів) полімеризації альфа-олефінів, спосіб його одержання та спосіб одержання поліолефінів

Номер патенту: 27689
Опубліковано: 16.10.2000
Автори: Саккеті Маріо, Говоні Габріелє, Чароккі Антоніо
МПК: C08F 10/00, C08F 4/60, C08F 10/06, C08F 4/654, C08F 4/02, C08F 110/00, C08F 4/6592, C08F 4/64
Мітки: спів, поліолефінів, альфа-олефінів, одержання, спосіб, каталізатора, компонент, полімерізації
Формула / Реферат:
(57) 1. Компонент катализатора (со) полимеризации альфа-олефинов, представляющий собой твердый продукт взаимодействия аддукта дихлорида магния со спиртом с тетрахлоридом титана и диизобутилфталатом, отличающийся тем, что он представляет собой твердый продукт взаимодей ствия, полученный с использованием аддукта дихлорида магния со спиртом, содержащего 0,4 -1,7моль спирта на 1моль дихлорида магния, и имеющий мольное...
Спосіб приготування каталізатора для одержання метанолу

Номер патенту: 3443
Опубліковано: 27.12.1994
Автори: Рижак Ігор Олександрович, Калінченко Федір Володимирович, Ілько Едуард Григорович, Хруцький Олег Павлович, Фірсов Олег Петрович, Калякін Сергій Володимирович, Павелко Віктор Захарович
МПК: B01J 37/04, C07C 29/154, B01J 23/80, B01J 23/76, C07C 29/153
Мітки: одержання, каталізатора, спосіб, приготування, метанолу
Формула / Реферат:
1. Способ приготовления катализатора для получения метанола, включающий смешение аммиачно-карбонатного раствора солей меди и цинка с водной суспензией гидроксида алюминия, подачу суспендированной реакционной массы в реактор с жидкой средой при постоянном барботаже диоксида углерода и повышенной температуре с последующим отделением осадка, сушкой и прокаливанием, отличающийся тем, что водную суспензию гидроксида алюминия или суспензию...
Спосіб приготування каталізатора для одержання ароматичних вуглеводнів

Номер патенту: 18598
Опубліковано: 25.12.1997
Автор: Нільс Йорген Блом
МПК: B01J 37/08, B01J 21/04, B01J 27/04, B01J 23/08, B01J 23/04, B01J 21/08
Мітки: спосіб, приготування, каталізатора, ароматичних, вуглеводнів, одержання
Формула / Реферат:
1. Способ приготовления катализатора для получения ароматических углеводородов, включающий нагревание смеси, содержащей источник оксида кремния, источник оксида алюминия, галлия или их смеси, источник оксида натрия и/или калия или любого другого катиона, способного к замене натрия или калия, и воду, при температуре 95 - 190°C до образования кристаллов цеолита с последующими выделением и сушкой полученных кристаллов, отличающийся тем, что...
Спосіб одержання каталізатора відновлення оксидів азоту на основі неблагородних металів

Номер патенту: 23824
Опубліковано: 16.06.1998
Автори: Степанова Ірина Ігорівна, Ведь Марина Віталіївна, Сахненко Микола Дмитрович, Богоявленська Олена Володимирівна
МПК: B01J 23/70, B01J 37/34, B01J 37/02
Мітки: каталізатора, металів, азоту, неблагородних, спосіб, оксидів, основі, одержання, відновлення
Формула / Реферат:
Способ получения катализатора восстановления оксидов азота на основе неблагородных металлов, включающий катодное выделение металлоксидных сплавов неблагородных металлов (Сu, Ni, Fe), отличающийся тем, что каталитически активные пленки на основе этих сплавов получают на неэлектропроводном носителе переменного состава типа nАІ2О3 × xSiO2 электрохимическим способом, причем носитель. пропитывают лакосажевой суспензией, вязкость которой...
Попередній патент: Піднімально-перекидний або перекидний пристрій для випорожнення сміттєвих баків
Наступний патент: Пристрій супутникового телевізійного приймання /варіанти/
Випадковий патент: Спосіб видобування високопарафінистої нафти