Спосіб отримання білка з активністю колонієстимулювального фактора гранулоцитів та макрофагів(gm-csf) приматів
Номер патенту: 29377
Опубліковано: 15.11.2000
Автори: Кларк Стівен К., Кауфман Рандал Дж., Вонг Гордон Г., Ванг Елізабет А.


















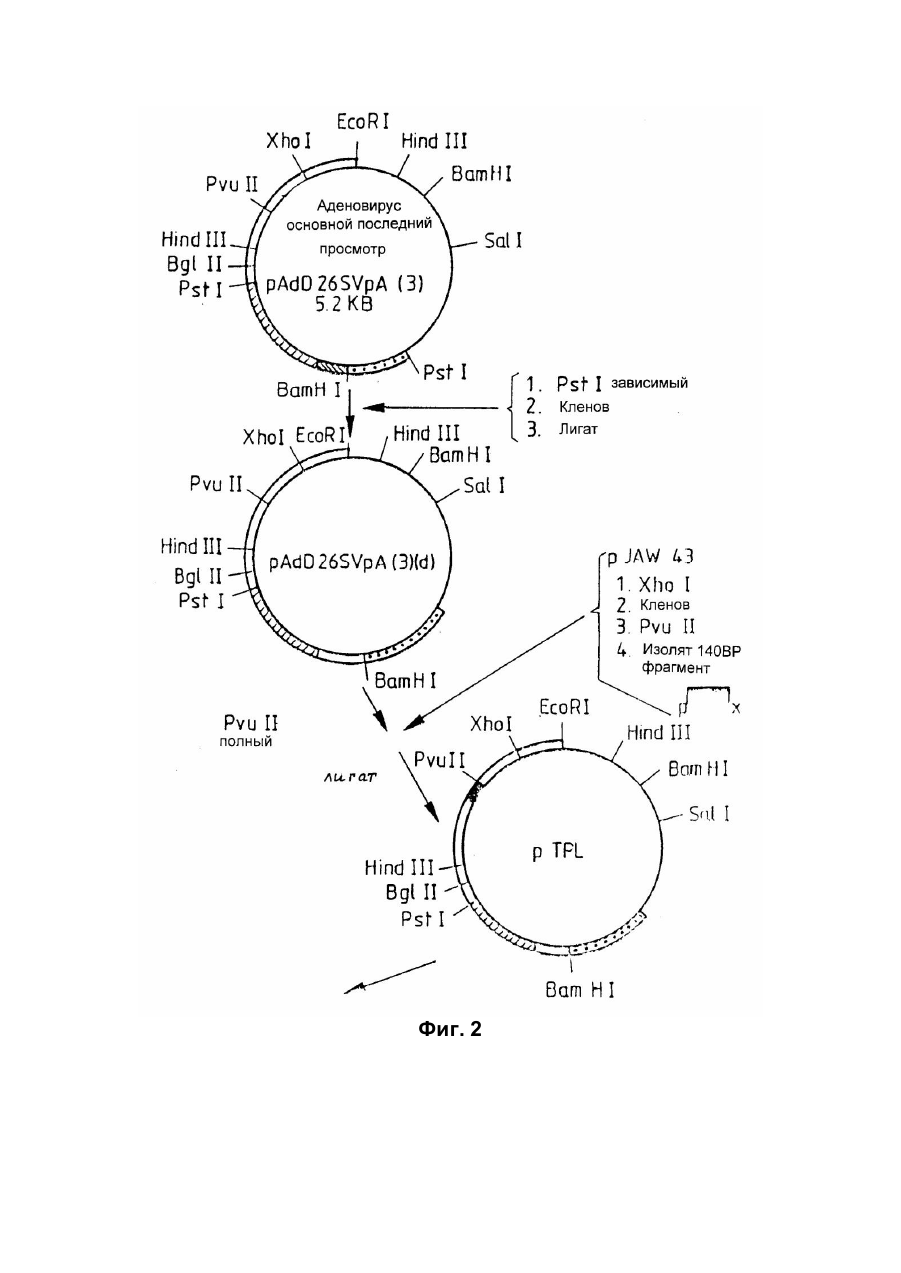
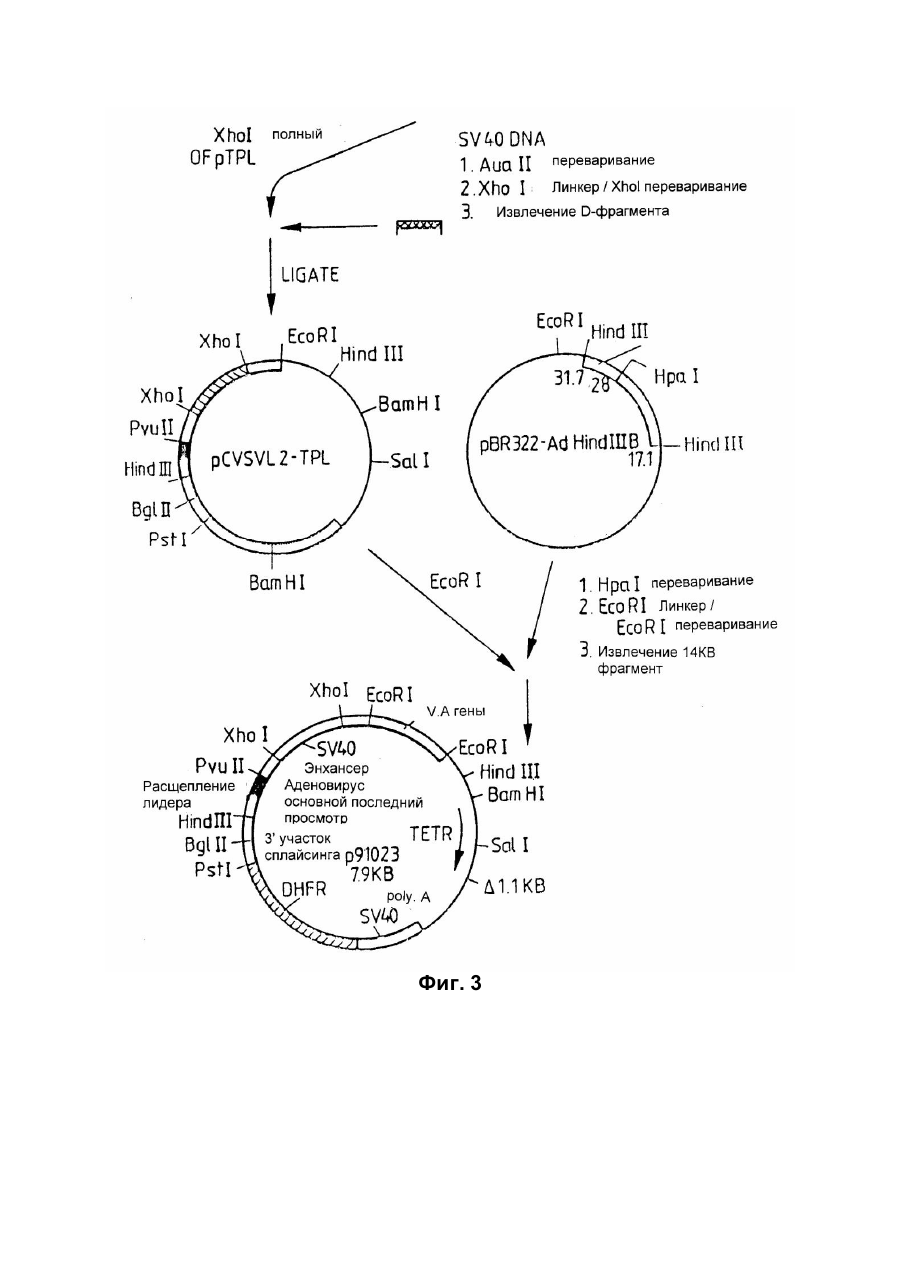
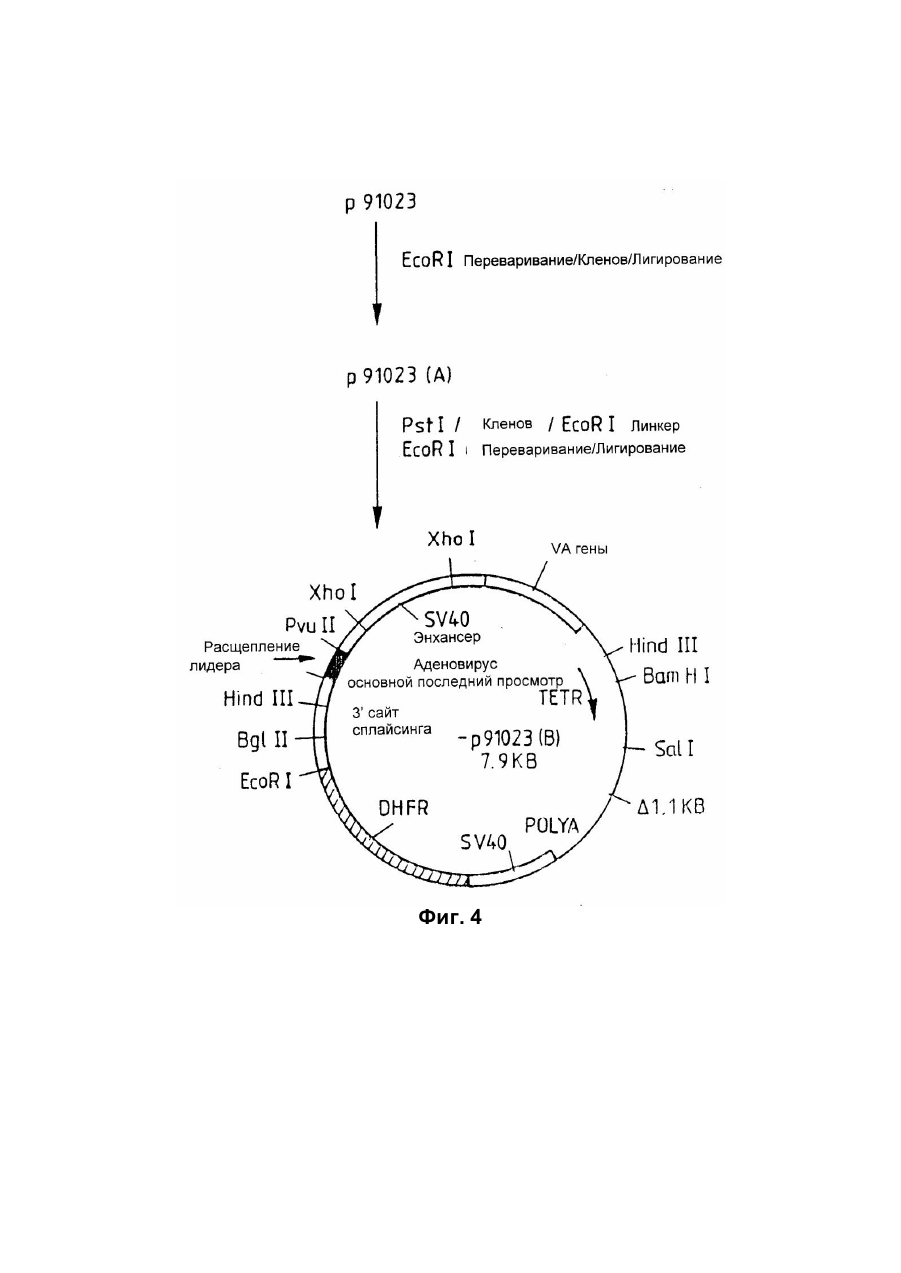
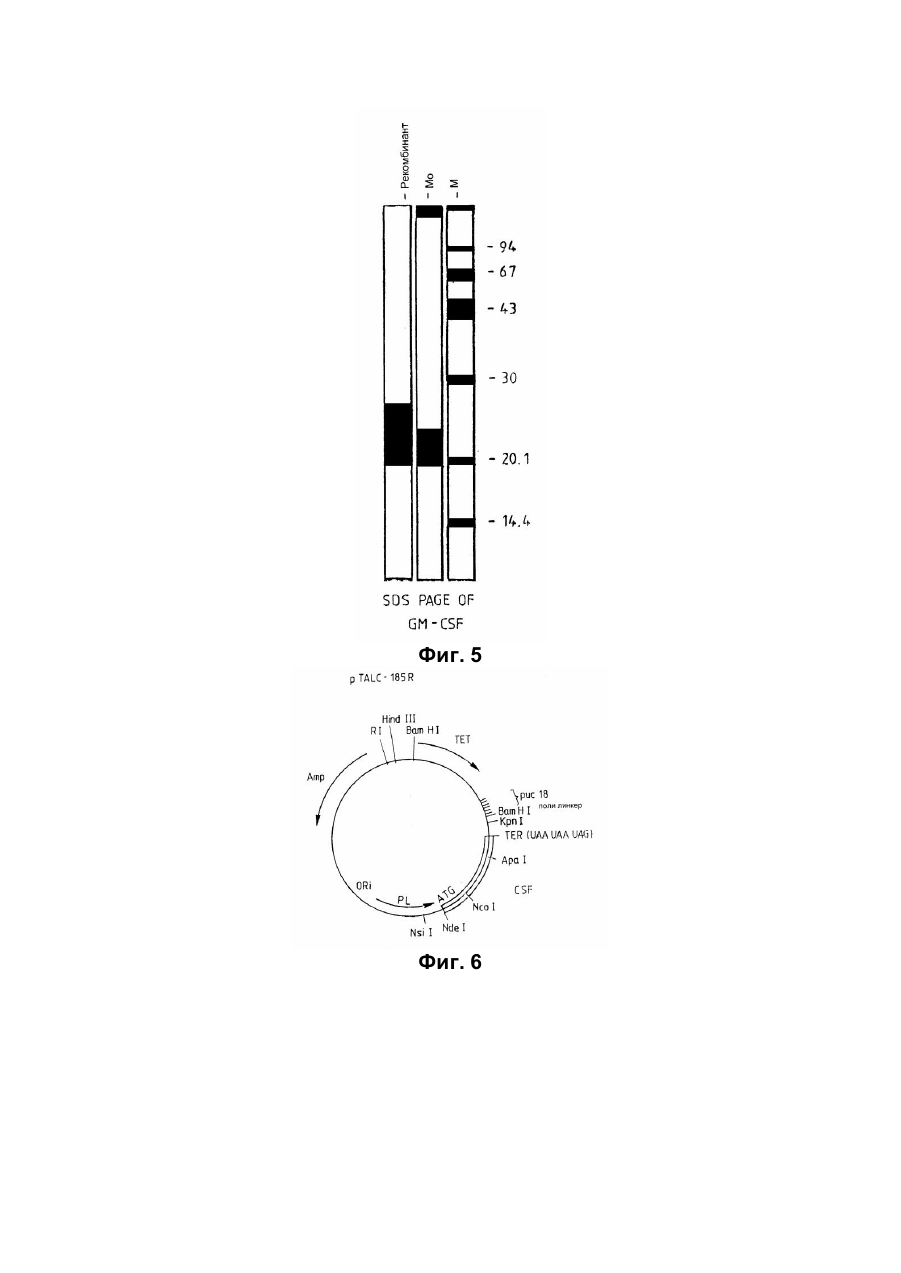

Формула / Реферат
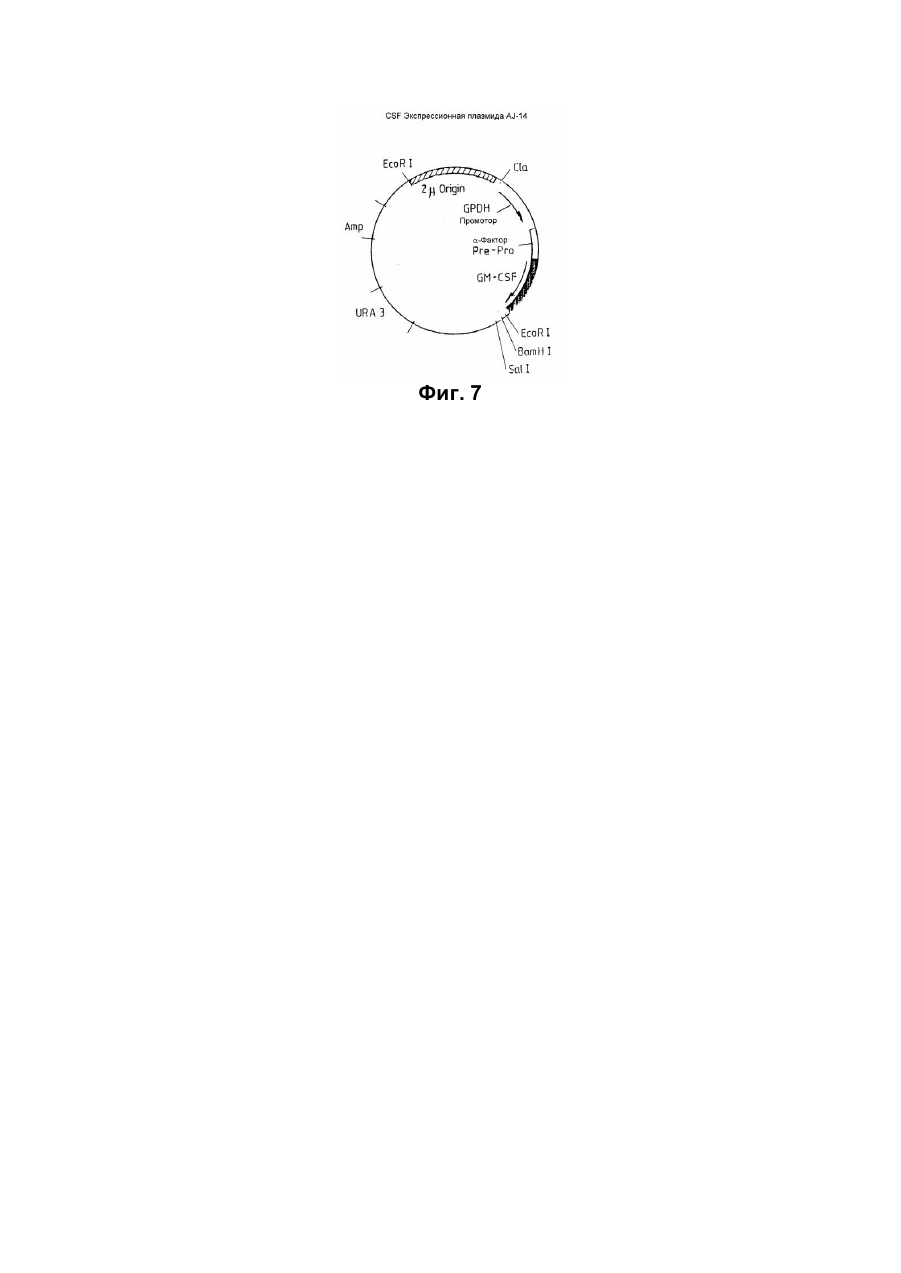
1. Способ получения белка с активностью колониестимулирующего фактора гранулоцитов и макрофагов (GM-CSF) приматов, включающий культивирование хозяйских клеток после их трансформации рекомбинантным вектором, обеспечивающим экспрессию в выбранном типе клеток встроенного в указанный вектор гена, кодирующего GM-CSF приматов, отличающийся тем, что для трансформации клеток хозяина используют рекомбинантный вектор с геном, кодирующим полипептид со следующей аминокислотной последовательностью.
и полученный продукт выделяют и очищают.
2. Способ по п.1, отличающийся тем, что упомянутая хозяйская клетка представляет собой дрожжевую клетку.
3. Способ по п.1, отличающийся тем, что упомянутая хозяйская клетка представляет собой прокариотическую клетку.
4. Способ по п.1, отличающийся тем, что упомянутая хозяйская клетка представляет собой клетку E.coli.
5. Способ по п.1, отличающийся тем, что включенный в рекомбинантный вектор ген кодирует GM-CSF приматов с аминокислотной последовательностью, приведенной в п.1 для CSF-Thr.
6. Способ по п.1, отличающийся тем, что включенный в рекомбинантный вектор ген кодирует GM-CSF приматов с аминокислотной последовательностью, приведенной в п.1 для CSF-lle.
7. Способ по п.1, отличающийся тем, что включенный в рекомбинантный вектор ген кодирует GM-CSF приматов с аминокислотной последовательностью, приведенной в п.1 для CSF-G.
Текст
Настоящее изобрете ние относится к получению белка, обладающего способностью стимулировать рост и диффе ренциацию первичных кроветворных прогениторных клеток, в частности факто ра стимуляции колонии (ФСК). В одном аспекте изобретение обеспечивает способ получения ФСК-белка путем технологии рекомбинантной ДНК, векторы, содержащие ген экспрессии указанного белка, микроорганизмы и клеточные линии, трансформированные указанными векто рами, и ФСК-белок, полученный таким образом. Второй аспект изобрете ния обеспечивает способ выделения и очистки ФСК-белка из природных или рекомбинантных источников и очищенный таким образом ФСК-белок, имеющий степень чистоты и уро вень активности выше те х, что бы ли описаны ранее. Найдено, что многие различные типы клеток крови происходят из многопотенциальных кроветворящи х клеток спинного мозга. Клетки спинного мозга выполняют две функции: (1) они репродуцируют са ми себя, в результате чего поддерживается популяция клеток спинного мозга в организме и (2) они обеспечивают потомство клеток, передающее различия в любых ти пах зрелых клеток крови. Клетка, которая передает различия по конкретному кроветворному пути, называется клеткой-предшественником. Клетки-предшественники для Т-лимфо цитов, В-лимфо цитов, гранулоцитов, красных кровяных клеток, тромбоцитов и эозинофи лов, так же как и более ранние предшественники, кото рые могут индивидуально приводить к увеличению некоторых ти пов зрелых клеток, были экспериментально изучены как in vivo, так и in vitro (Dexter, T.M., 1983, J. Patology 141, 415–433). Уже было определено in vitro, что пролифе рация и/или диффе ренциация предшественника каждого ти па клеток зави сит от определенных "факторов", которые происхо дили из различных источников. Например, последние предшественники красных кровяных клеток требуют фактора, называемого эритропоиэтином. Факторы, требующиеся для выживания, пролифе рации и дифференциации миелоидных предшественников, перехо дящих в форму зрелых нейтрофильных гранулоцитов, моноцитов и зрелых макрофа гов, называют факторами стимуляции колоний (ФСК). ФСК активность экстенсивно изучалась на мыша х. Большинство органов взрослых мышей продуцирует ФСК активность. Однако, композиции, содержащие ФСК активность, которые были получены из различных тканей и различными методами, различаются по своим биохи мическим характеристикам. Так, структур ное соотношение между различными факторами остается неизвестным. Кроме того, ФСК активность действует на более, чем одной стадии развития гранулоцита и макрофа га, и снова точно не известно, единственный ли фактор является ответственным за все наблюдаемые активности или же различные факторы действуют на каждой стадии. (Burgess, A. и Metcalf, Д., 1980, Blood 56, 947–957). Человеческая ФСК активность была получена из плаценты некоторых плодных тканей, макрофагов и стимулированных Т-клеток. Линия Т-клеток (Мо), которая продуцирует одну или несколько мощных ФСК активностей, была уста новлена от пациента с вариантом Т-клетки "волосатой" клетки лейкемии (лейкемический ретикулоэндотелиозис) (Golde et al., 1978, Blood, 52, 1068–1072). Способность СК активности стимулировать продуцирова ние гранулоцитов и макрофагов указывает, что фар мацевти ческие композиции, обладающие ФСК активностью, являются клинически полезными в ситуациях, когда требуется повышенное продуцирование таких типов (миелоидных) клеток. Действительно, показано, что некоторые пациенты с крайне высокими уровнями по-видимому нормальных циркулирующи х гранулоцитов имеют опухоли, которые сверх-продуцируют ФСК. В одном случае, при хирур гическом удалении опухо ли количество гранулоцитов быстро падает к нормальному уровню, четко подтверждая, что ФСК может быть полезным при регулировании количеств циркулирующих гранулоцитов. (Hocking, W. Goodinau, J. и Golde, D., Blood, 61, 600 (1983)). В частности, ФСК композиции являются клинически полезными для лечения миело-супрессии, вызванной хемотерапевтическим лечением рака или его лечением с помощью облучения. Кроме того, ФСК композиции являются полезными при лечении сильных инфекций, потому что ФСК может уве личивать и/или активировать количество гранулоцитов и/или моноцитов. Имеются различные дифференцированные типы известных ФСК активностей, включая гранулоциты. ФСК (G-ФСК), макрофаг-ФСК (М-ФСК), гранулоцит-макрофаг ФСК (GM–ФСК) и мульти-ФСК. Настоящее изобретение в частности относится к GM–ФСК. ФСК-белки являются известными из различных животных источников. Однако, настоящее изобретение в частности относится к приматному ФСК, более конкретно к человеческому ФСК и к обезьяннему ФСК. Биологическая и биохи мическая характеристи ка композиций, обладающи х ФСК активностью, и изучение этих композиций в клинических условиях было затруднено до настояще го времени недостаточным количеством и примесями ФСК-композиций человека и/или других приматов. Следует учи тывать, что должно быть желательным идентифицировать белок или белки, ответственные за ФСК активность. Кроме того, желательно иметь приматный, в частности че ловеческий источник такого ФСК, который может легко снабжать такими белками в таких количествах и такой чисто ты, кото рые достаточны для биологической и биохи мической характеристики и для использова ния их в качестве терапевти ческих агентов. Разработанные ранее методики молекулярного клонирования делают возможным клонирование нуклеотидной последовательности, которая кодирует белок, и получение такого белка в достаточном количестве, используя подхо дящую систему хо зяин-вектор, (Т. Маниатис "Мо лекулярное клонирование – Лабораторное руководство", Колд Спринг Харбор, Лаборатору, Колд Спринг, Харбор, Н.Й. 1982 г.). Затем белок может быть извлечен по известным методикам выделения и очистки. Методы клонирования, используемые до настояще го времени, могут быть сгруппированы в три основные категории: (1) способы, основанные на знании структуры белка, например, его аминокислотной последовательности; (2) способы, основанные на идентификации белка, экспрессированного клонированным геном, с использованием специфического к такому белку антитела; и (3) способы, основанные на идентифи кации видов РНК, которые могут быть транслированы для получения белка или активности, кодируемых интересующим геном. Каждый из этих классов способов ста новится трудным для применения, когда интересующий бе лок, такой как ФСК-белок, доступен в очень малом количестве. Следовательно, если трудно получить адекватное количество очищенного белка, то трудно и определить аминокислотную последовательность или даже частичные последовательности белка. Подобным образом, иденти фикацию экспрессированного белка связывающим антителом предпочти тельно проводят при использовании поликлональной антисыворотки с высоким титром. Такая анти сыворотка не может быть получена при отсутствии достаточных количеств чистого белка (антигена). Моноклональное антите ло предполагает альтернативную попытку, но требуе мое антитело также может быть получено с большим трудом при отсутствии подхо дящего антигена и такое моноклональное антите ло не может взаимодействовать с белком в том виде, в каком белок экспрессирован доступной рекомбинантной системой хозяин-вектор. Наконец, трансляция видов РНК для получения идентифи цируе мого белка или активности требует, что бы искомая РНК нахо дилась в источнике РНК в достаточном избытке, чтобы получить достоверный белок или сигнальную активность. Относительный избыток РНК, кодирующей конкретный белок, обычно параллелен избытку белка, так что редкий белок обычно кодируется редкой РНК. Мо ноклеточную ли нию использовали как в качестве исходного мате риала для очистки человеческих ФСК, так и для идентификации соответствующи х матричных РНК. Однако даже при таком относительно хорошем источнике ФСК активности оказалось очень трудно выделить белок даже для структур ных исследований. Для того, что бы решить проблемы, присущие клонированию нуклеотидной последовательности, кодирующей редкий белок, такой как ФСК, с помощью описанных выше способов, разработан новый способ. Этот способ требует только, чтобы генный продукт или его активность могли быть достоверно измерены. Подхо дящие способы определения ФСК описаны в примере 2 ниже. Во втором аспекте, разработан способ очистки, который пригоден для выделения и очистки белка ФСК как из рекомбинантных, так и природных источников при уровне очистки и активности намного более высоком, чем был ранее возможен. Краткое содержание способа с рекомбинантной ДНК согласно изобретению. В его первом аспекте настоящее изобрете ние решает проблемы известного уровня техники и обеспечивает источник белка, обладающего ФСК активностью, при использова нии технологии с рекомбинантной ДНК. В соответствии с настоящим изобретением используется новая методика клонирования, которая требует только пробу для ФСК активности, что бы клонировать кДНК, кодирующую бе лок, обладающий ФСК активностью. Итак, настоящее изобретение обеспечивает кДНК, кодирующую белок, обладающий ФСК активностью (т.е. ФСК/кДНК), микроорганизм или клеточную линию, трансформированную рекомбинантным вектором, содержащим такой ФСК/кДНК, и способ получения ФСК белка путем экспрессии указанной ФСК/кДНК при культиви ровании микроорганизма или клеточной линии. Поскольку ФСК белок продуцируется из клона в соответствии с настоящим изобрете нием, мы можем быть уверены, что это бе лок, который обладает ФСК активностью. Далее изобретение заключается в способе получения и изоляции вектора трансфор мации, содержаще го ФСК/кДНК, указанный способ состоит из: получения РНК из клетки, продуцирующей ФСК; получения полиаденилированной матричной РНК из указанной РНК; получения однониточной кДНК из указанной матричной РНК; превращения однониточной кДНК в двуниточную кДНК; вставки двуниточной кДНК в векто ры трансформации и трансформирование бактерий указанным вектором для образования колоний; отборы пулов из 200–500 колоний каждый и изоляции плазмиды ДНК из каждого пула; трансфекции плазмиды ДНК в подходящую клетку-хозяина для экспрессии ФСК белка; культивирования трансфецированных клеток и испытания надосадочного слоя на ФСК активность; отбора ФСК положительных пулов и скринирования колоний, использованных для образования пула, чтобы идентифи цировать колонии, обладающие ФСК активностью. ФСК-белки настояще го изобретения обеспечивают рост и дифференциацию гормонов для клеток миелоидной системы. Например, указывалось на их клиническое использование при лечении миелосупрессии, особенно (симпто матической) гранулоцитопении после хемотерапевтического лечения рака или после его лечения облучением. На фиг. 1 представлены последовательности ДНК, кото рые кодируют ФСК-бе лок в соответствии с настоящим изобрете нием. Последова тельность ДНК, представленная полностью, кодирует один вариант человеческого ФСК, упо минаемый как ФСК–Thr. Другой аллель кодируе т иден тичный продукт с тем исключением, что Thr в по ложении 100 заменен Ile (ФСК–Ile). Изменения, показанные выше для че ловеческой последовательности, представляют со бой различия в последовательности ДНК, кодирующей ФСК гиббона (ФСК обезьяны гиббона) (ФСК–G). Также показаны уменьшенные последова тельности аминокислот. На фиг. 2 схематически представлено получение плазмиды рТР из плазмиды pAd Д26 SV pA (3). На фиг. 3 схе матически представлено продолжение рис. 2 и иллюстрируется получение плазмиды р91023 из плазмиды рTPL. На фиг. 4 схе матически представлено продолжение рис. 3 и показана плазмида р91023 (В). На фиг. 6 схе матически представлен вектор рTALC–185P. На фиг. 7 схе матически представлен вектор AJ–14. Следующие определения приведены для облегчения понимания настоящей заявки. По мере того, как определения меняются в зависимости от значений, применяемых на данном уровне техники, приве денные ниже определения даны для контроля. Амплифи кация означает процесс, при котором клетки продуцируют копии генов внутри их хромосомальной ДНК. ФСК представляет собой биологическую активность, определенную путем анализа, описанного здесь. ФСК белок представляет собой белок из приматного источника, кото рый проявляет ФСК активность. Для целей настояще го изобретения термин ФСК-белок включает модифицированный ФСК-белок, аллальные вариации ФСК-белка и ФСК-белок, предшествующий МЕТ-остатку. Направление вниз означает направление к 3'-концу н уклеотидной последовательности. Усилитель представляет собой нуклеотидную последовательность, которая может сделать возможной транскрипцию гена независимо от положения усилителя по отношению к гену или от ориентации последовательности. Ген представляет собой деоксирибонуклеотидную последовательность, кодирующую дан ный белок. Для целей настоящей заявки ген не должен включать нетранслируе мые боковые области, та кие как сигналы начала транскрипции РНК, сайты полиаденильного присоединения, промоторы и усилители. Связывание представляет собой процесс образования фосфо ди(сложно)эфирной связи между 5',- и 3'-концами двух нитей ДНК. Оно может быть осуществлено несколькими хорошо известными ферментативными мето дами, включая связывание тупого конца Т4 лигазой. Ориентация относится к порядку нуклеотидов в последовательности ДНК. Обратная ориентация последовательности ДНК представляет собой последовательность, у кото рой порядок последовательности 5' к 3'по отношению к другой последовательности является обратным при сравнении указанного места в ДНК, из которой последовательность была получена. Такие указанные точки (места) могут вк лючать направление транскрипции других определенных последовательностей ДНК в источнике ДНК или в источнике репликации реплицируе мых векторов, со держащих последовательность. Транскрипция означает изменение генотипа клеток путем клеточного поглоще ния экзогенной ДНК. Трансфор мация может быть детектирована в некото рых случаях путем изменения фенотипа клетки. Трансформированные клетки называются трансформантами. Пре-трансформация клеток указывается в отношении родительских клеток. Трансляция означает синтез полипептида из матричной РНК. Активность фактора стимуляции колоний (ФСК) может происходить из ряда клеточных источников, включая кондиционированную среду из перифе рических моноядерных клеток крови, ткани легких и плаценты, и костного мозга, мочи от анемичных пациентов, сыворотки, и нормальных и неопластичных клеток Т-лимфо цита и моноядерного фагоцита. Одна клеточная линия, которая продуцирует ФСК, представляет собой Мо клеточную линию, депонированную и доступную публике из АТСС под кодовым номером СР 8066. ФСК, продуцированный этой клеточной линией, известен как гранулоцитный макрофаг ФСК (или GM–ФСК), и, разумеется, как человеческий ФСК. Одним источником ФСК гиббона является линия Т-клеток, обозначенных ИСД М А– 144 и депонированных и доступ ных для публики из АТСС под кодовым номером НВ 9370, депонированы 29 сентября 1983 г. Для то го, что бы изолировать ФСК клон в соо тветствии с настоящим изобрете нием, была использована новая процедура, кото рая требуе т толь ко техники определе ния ФСК-активности. Снача ла иденти фи цируют клетку, ко торая продуцируе т ФС К ак тивность, та кую как Т-лим фо цитные клетки (или другие исто чники, та кие как представлены вы ше). За тем со бирают мРНК клетки. Предпочти тельно используют Т-лимфо цитные клетки. В та ком случае отделяют связанную с мембраной мРНК, кото рая содержит мРНК для лим фо кина, о т сво бодной мРНК в кле тках. Э то о тделение, как полагают, обо га щае т собранную мРНК в 5–10 раз последова тельностя ми для лимфо кина и таким образом сокращает усилия, вк ладывае мые в иденти фи кацию це лево го ФСК кло на. За тем получают полиа денилированн ую матричную РНК хро мато графией на оли го-dT-целлюло зе. Готовят банк кДНК из мРНК, используя вектор, подхо дящий для трансфекции в хозяина для экспрессии целевого белка, обладающе го ФСК активностью. Сначала получают нить кДНК с использованием стандартных методик, применяя полученную вы ше мРНК. Затем превращают гибрид РНК/кДНК в двуниточную форму кДНК. Затем кДНК может быть вставлена в подхо дящий вектор. Предпочти тельная система хозяин-вектор для изоляции клона ФСК основана на экспрессии ФСК кДНК в подхо дящем векторе трансформации. Подхо дящий вектор трансформации может обеспечить случайное введение ДНК в клетки млекопитающи х. (Mellou P., V. Parker, Y. Glu zman Т. Maniatis, 1981, Cell, 27, 279–288). Для изоляции целевых ФСК-трансформантов не требуется, что бы все клетки популяции стабильно содержали экзогенные гены, которые экспрессируют це левой ФСК-продукт. Возможно временное вхождение экзогенных генов в субпопуляцию клеток, так что субпопуляция будет экспрессировать целевой продукт в течение нескольких дней. Поскольку нет нужды в отбираемом маркере в векторе трансфор мации для трансфекции ДНК и системы экспрессии в соответствии с настоящим изобретением, экзогенная ДНК может быть потеряна при росте клеток в течение 1–2 недель. Однако через 2–3 дня после трансфекции подхо дящи х клеток млекопитающи х, как было обнаружено, целевые продук ты могут быть синте зированы и могут быть детектированы. Выбор системы хозяин-вектор основан на развитии клеточных линий CV–1 обезьян, трансфор мированных молекулой ДНК репликация-происхождение-дефектных SV 40 (Gluzman, Y., Cell, 23, 175–182, 1981). Трансфор мированные CV–1 клетки обезьян, содержащие дефектную SV 40 ДНК, обозначенные COS (CV– 1, дефектное происхождение, SV 40), не содержат полной копии генома SV 40, но продуцируют вы сокие уровни широкого Т антигена и разрешают репликацию ДНК 40. Они также эффективно поддерживают репликацию SV 40, содержащи х делеции в ранней области, и бактериальных плазмид, которые содержат 40 источников репликации (Myors, R.M. and Tjian, R. 1980, PNA 77, 6491–6495). Следовательно, такая система обеспечивает средство амплифи кации трансфе цированной экзогенной ДНК через репликацию ДНК медиированных SV 40, чтобы повысить уровень мРНК и белка, экспрессированного из экзогенной ДНК. Однако также являются полезными другие подобные системы. Векторы, использованные для ФСК экспрессии, обычно содержат различные элементы, такие как уси лите ли, промоторы, интроны, сайты полиаденилирования, 3' некодирующие области и активато ры трансляции, как будет описано ниже. Здесь векторы могут включать уси лители. Усилители функционально отличаются от промоторов, но очевидно работают совместно с промоторами. Их функция на клеточном уровне еще недостаточно понятна, но их уни кальной характерной чертой является способность активировать или делать возможной транскрипцию независимо от положения или ориентации. Промоторы должны быть расположены вверх от гена, тогда как усилители могут нахо диться вверх или 5' от промотора, вн утри гена как интрона, или вниз от гена между ге ном и сайтом полиаденилирования. Вставленные промото ры не являются функциональными, а вставленные усилите ли являются. Усилители являются цис-активными, а именно они действуют на промоторы, только если они нахо дятся в одной и той же нити ДНК. Общую дискуссию об усилителях смотри у Khoury et al., Cell, 33, 313–314 (1983). Предпочти тельные усилители для использования в клетках млекопитающи х получают из ви русов животных, таких как обезьяний вирус 40, вирус полиомы, вирус бычьей папилломы, ретровирус и аденовирус. Идеально, уси литель должен быть из вируса, для которого клетка-хозяин является рекомендованной, т.е. который обычно заражает клетки хозяина. Вирусные усилители могут быть легко получены из публично доступ ных вирусов. Усиливающие области для некоторых ви русов, а именно вируса саркомы Рауса и обезьяньего вируса 40, хорошо известны. Смотри Luein et al., Cell 33, 705–716 (1983). Из традиционной молекулярной биологии известно расположение этих областей на основе опубликованных карт рестрикции для указанных вирусов и, в случае необходимости, можно модифицировать сайты, чтобы сделать возможной сращи вание усилителя с вектором, как желательно. Например, смотри Кауфман и др., J. Mol. Biol., 159, 601–621 (1982) и Mol. Cell Biol. 2 (11), 1304–1319 (1982). Альтернативно усилитель может быть синтезирован из данных последовательностей; размеры вирусных уси лите лей (обычно менее примерно 150 пар оснований) являются достаточно маленькими, чтобы это могло быть осуществлено на практике. Другим элементом, который должен присутствовать в векторе, является сайт полиаденилированного сращи вания (или присоединения). Он представляет собой последовательность ДНК, расположенную вниз от транслируе мых областей гена, вскоре вниз от кото рого в свою очередь добавляются ринонуклеотиды остановки транскрипции и аденина для образования полиаденинового нуклеотидного хвоста на 3' конце матричной РНК. Полиаденилирование является важным при стабилизации матричной РНК против разложения в клетке, ситуа ция, при кото рой снижается уровень матричной РНК и, следовательно, уровень белкового продукта. Эукариотные сайты полиаденилирования хорошо известны. Consensus последовательность существуе т среди эукариотных генов: гексануклеотид 5'–AAUAAA–3' найден 11–30 нуклеотидов от точки, в которой начинается полиаденилирова ние. Последовательности ДНК, содержащие сайты полиаденилирования, могут быть получены из вирусов в соответствии с опубликованными сообщениями. Типичные последовательности полиаденилирования могут быть получены из бета-глобина мыши и поздней или ранней областей генов обезьяньего ви руса 40, но предпочтительными являются вирусные сайты полиаденилирования. Поскольку эти последовательности являются известными, они могут быть синтезированы in vitro и связаны в вектор традиционным способом. Последовательность, которая отделяет сайт полиаденилирования от трансляционного стоп-кодона, является предпочтительно нетранслируемой последовательностью ДНК, та кой как непромотированный эукариотический ген. Поскольку та кие последовательности и гены не обеспечены промото ром, они не будут экспрессироваться. Последова тельность должна простираться на значительное расстояние, порядка примерно 1000 оснований, от стоп-кодона до сайта полиаденилирования. Такая 3'-нетранслируемая последовательность обычно в результате приводит к повышению выхо да продукта. Вектор может заканчиваться примерно за 30 пар оснований вниз от consensus последовательности полиаденилирования, но предпочтительно сохранить 3'-последовательности, найденные вниз по ходу от сайта полиаденилирования, в окружении их дикого типа. Эти последовательности обычно протягиваются примерно на 200–600 пар оснований вниз от сайта полиаденилирования. Наличие интронов в нетранслируемом транскрибируемом участке вектора может уве личить выход продукта. Такие интроны могут быть получены из других источников, чем клетки хозяина или источники генов. Например, гибридный интрон, состоящий из 5' сайта сращивания от второго интрона трехчастного лидера аденовируса и 3' сайта сращивания от гена иммуноглобулина, вставленного вниз от сайта старта транскрипции в основной последний промотор аденовируса, приводит в результате к увеличению выхода продукта. В предпочтительном варианте вектора клонирования ФСК и экспрессии имеется ген активатора трансляции. Активаторы трансляции являются генами, которые кодируют или белок или продукты РНК, которые влияют на трансляцию целевой мРНК. Лучшим примером является аденовирус-ассоциированный (VA) ген (VAI), который считывается в коротких типах РНК, которые взаимодействуют с последовательностями в 5' нетранслируе мой области основных поздних мРНК аденовируса (Thimmappaya et al., 1982, Ce, 3, 543). Необходимые последовательности трансляционной активации с помощью VA РНК ле жат внутри трехчастного лидера последней мРНК аденовируса. Трехчастный лидер аденовируса сращи вают вместе с несоприкасающи мися областями генома аденовируса и нахо дится на 5'-конце основных последних транскриптов аденовируса. VA РНК может взаимодействовать для активации трансляции мРНК, которые содер жат последовательность трехчастного лидера. Итак, предпочтительный вектор клонирования кДНК и экспрессии содержит соединенную форму трехчастного лидера и гены VA аденовируса. Эти векторы могут быть синтезированы по методикам, хорошо известным специалистам в данной области. Компоненты векто ров, такие как уси лители, промоторы и т.п., могут быть получены из природных источников или синтезированы, как описано выше. В основном, если найдено, что компоненты в ДНК доступны в большом количестве, а именно, такие компоненты, как вирусные функции, или если они могут быть синте зированы, например, сайты полиаденилирования, тогда при соответствующем использовании рестрикционных ферментов могут быть получены большие количества вектора при простом культивировании организма-источника, переваривании его ДНК соответствующий эндонуклеазой, отделении фрагментов ДНК, идентифи кации ДНК, содержащей интересующий элемент, и его извлечении. Обычно вектор трансформации должен быть введен в малом количестве и затем связан в подхо дящий автономно реплицирующийся синтетический вектор, та кой как прокариотная плазмида или фаг. В большинстве случаев может быть использована плазмида рВР322. Смотри Кауфман и др., выше. Синтетический вектор используют для клонирования связанных векто ров трансформации обычным образом, а именно путем трансфекции возможного прокариотного организма, репликации синтетического вектора с большим числом копий и извлечения синтетического вектора при лизисе клеток и отделения синтетического вектора от клеточного дебриса. Векторы, содержащие кДНК, приготовленную из клетки, которая продуцирует ФСК активность, затем трансфе цируют в E.coli и помещают на чашки Петри приблизительно 2000 колоний на чашку. Колонии вынимают на нитроцеллюлозный фильтр и фильтр переносят на новую пластину, которую хранят как первый оригинал. После выращи вания этих колоний делают реплики и сравнивают с оригиналом, тщательно отмечая, что бы секции фильтров реплик могли быть идентифи цированы с соответствующим участком пласти ны оригинала. Каждый фильтр реприку разрезают на секции, содержащие заранее определенное число колоний на секцию, предпочтительно 200–500 колоний на секцию. Колонии с каждой секции соскабливают в среду, такую как L-бульон, бакте рии собирают центрифугированием и отделяют плазмиду ДНК. Плазмиду ДНК каждой секции трансфецируют в подхо дяще го хо зяина для экспрессии белка. Предпочти тельный синтетический вектор здесь является мутантом E.coli плазмиды рВР 322, в кото рой были делецированы последовательности, которые являются вредными для эукариотных клеток. Смотри Кауфман и др., выше. Использование этого мутанта устраняет необходимость в делении остатка плазмиды перед трансфекцией. После выращи вания трансфе цированных клеток среды проверяют на ФСК активность. Положительная проба показывает, что колония, содержащая ФСК/кДНК, нахо дится на конкретной секции на фильтре. Для определения какой из клонов на секции исходного первого оригинала фильтра содержит ФСК/кДНК, каждый клон на секции фильтра пересаживают и выращивают. За тем культуры помещают на матрицу П улы получают от каждого горизонтального ряда и вертикальной колонки матрицы. Гото вят образцы ДНК из каждого пула культуры и трансфе цируют в клетку-хозяина для экспрессии. Надосадочные слои для каждого пула проверяют на ФСК активность. Одна вертикальная колонка пула и горизонтальный ряд пула должны продуцировать ФСК активность. Клон, общий для этих пулов, будет содержать ФСК/кДНК. Если матрица содержит больше одного положительного клона, больше чем одна колонка и ряд должны быть положительными. В таком случае может быть необходимым дальнейший скрининг малого количества клонов. Вырезают ФСК/кДНК из клонов рестрикционными ферментами и можно провести определение аминокислотной последова тельности обычными методами. Можно легко оценить что описанная здесь процедура может быть использована для получения ФСК/кДНК из любого источника. Полная последовательность ФСК/кДНК в соответствии с изобрете нием представлена на рис. 1 вместе с предсказанной аминокислотной последовательностью транслированного ФСК белка. Последовательность ДНК, кодирующая белок, проявляющий ФСК активность, в соответствии с настоящим изобретением, такая как представлена на рис. 1, может быть модифицирована с помощью традиционных методик для получения вариантов в конечном ФСК белке, который еще обладает ФСК активностью в описанных здесь ниже пробах. Так, например, одна, две, три, че тыре или пять аминокислот могут быть заменены другими аминокислотами. В бельгийском патенте № 898016, приведенном здесь в качестве уровня техники, описана одна такая типичная методика для замены цистеина, например, серином. В соответствии с настоящим изобретением ФСК/кДНК включает природный ФСК/кДНК ген, предшествующий АТС кодону, и ФСК/кДНК, кодирующей аллельные вариации ФСК-белка. Один аллель представлен на рис. 1. Другой аллель, который мы обнаружили, имеет тимидиновый остаток в положении 365 вместо цитозинового остатка, показанного на рис. 1. ФСК-белок настоящего изобрете ния включает 1-метиониновое производное ФСК-белка (Met–ФСК) и аллельные вариации ФСК-белка. Зрелый ФСК-белок, представленный последовательностью на рис. 1 начинается с последовательности Ala, Pro, Ala, Arg..., начало которой обозначено стрелкой после нуклеотида номер 59 на рис. 1. Met–ФСК– будет начинаться с последовательности Met, Ala, Pro, Ala, Arg... Аллельная вариация, представленная на рис. 1, имеет Thr у аминокислотного остатка номер 100 (начиная от Ala после стрелки) и может быть упомянута как ФСК(Thr). Другая вариация имеет Ile остаток у положения 100 и может быть упомянута как ФСК(Ile). Очищен ный ФСК-белок настояще го изобретения проявляет удельную активность по крайней мере 107 единиц/мг белка и предпочтительно по крайней мере 4 × 107 единиц/мг при пробе с человеческими клетками костного мозга. Систе мы вектор-хозяин для экспрессии ФСК могут быть прокариотическими или эукариоти ческими, но сложность ФСК может сделать предпочтительной систему экспрессии млекопитающи х. Экспрессия легко осуществляется при трансформации прокариоти ческих или эукариотических клеток подхо дящим векто ром ФСК. Последовательность ДНК, полученная по описанной выше процедуре, может быть экспрессирована непосредственно в клетках млекопитающи х под контролем подходящего промотора. Гетерологические промоторы, хо рошо известные специалистам в данной области, могут быть использованы. Для экспрессии ФСК в прокариотических или дрожжевых клетках должна быть удалена лидерная последовательность (или секреторная последовательность). Положение кодона для N-терминала зрелого ФСК белка показано на рис. 1. Это может быть сделано с использованием стандартных методик, известных специалистам в данной области. Как только получают целевой ФСК/кДНК клон, используют известные и подхо дящие средства для экспрессии ФСК белка, а именно, вставку в подхо дящий вектор и трансфекцию вектора в подхо дящую клетку-хо зяин, отбор трансформированных клеток и культивирование этих трансфор мантов для экспрессии ФСК активности. Подхо дящи ми клетками-хозяевами являются бактерии, например, E.coli, дрожжи, клетки млекопитающих, например СНО, и насекомых. Полученный таким образом, ФСК белок может иметь метиониновую груп пу у N-терминала белка (здесь называемого Met–ФСК). Зрелый белок, полученный из прокариоти ческих или эукариоти ческих клеток, иначе, должен быть идентичным по аминокислотной последовательности, но эукариоти ческий продукт может быть гликозилированным в той же самой степени или в другой, чем природный продукт. Различные методы получения ФСК-белка в соответствии с соглашением показаны в приведенных ниже примерах. Другие способы или мате риалы, например, векто ры, будут легко оценены специалистами в данной области на основе примеров и последующе го описания. ФСК-белок, эспрессированный в соответствующи х прокариотных или эукариотных клетках, может быть извлечен с помощью методик очистки и разделения, известных специалистам в данной области. Однако, как указано, настоящее изобретение также обеспечивает способ очистки, пригодный для ФСК белка как из рекомбинантного, так и природного источников, с помощью которого ФСК-белок может быть получен с высокой степенью чистоты и активности. Краткое описание заявленного способа очистки. Настоящее изобретение разрешает проблемы известного уровня техники и обеспечивает способ очистки белка, обладающе го ФСК-активностью. ФСК-белок в соответствии с настоящим изобретением обладает удельной активностью по крайней мере 1 × 107 единиц/мг белка, предпочти тельно по крайней мере 2 × 107 единиц/мг белка и более предпочтительно по крайней мере примерно 4 × 107 единиц/мг белка при пробе с человеческим костным мозгом. В соотве тствии с настоящим изоб ретением способ очистки ФСК-белка состоит из: осаждения белка суль фа том аммония при 80% насыщении для образования ша рика, содержаще го ФСК-белок; повтор ного суспендирова ния шарика в буфер ном растворе при рН в ин терва ле от при мерно 6 до примерно 8; нанесения буфер ного раство ра, содержаще го ФСК, на хро мато графи ческую ко лонку, элюи рова ния буферным раство ром, содержащим хло ристый натрий, и сбора фракций, обладающи х ФСК активностью; объединения активных фрак ций, нанесения их на колонку с С4 обрати мой фазой и элюирова ния 0–90% гра диентом аце тонитрила для сбо ра активной фракции. Краткое описание фигур, относящи хся к процессу очистки. Фиг. 5 иллюстрирует СДГ–ПАГЭ анализ очищенного ФСК белка. Детальное описание способа очистки по изобретению. ФСК-белок, очищаемый в соответствии со способом изобретения, может происхо дить из любого природного источника, описанного выше в качестве исходного источника для рекомбинантного способа ДНК, например, Мо клеточной линии или UCD MZA–144 гиббоновой клеточной линии. Альтернативно ФСК-белок может быть получен с использованием методик рекомбинантной ДНК изобретения. ФСК из любого источника может быть очищен по способу настояще го изобретения. Кондиционированную среду из любого источника ФСК белка предпочтительно концентрируют ультрафильтрацией до концентрации белка по крайней мере около 0,1 мг белка/мл. Затем белок осаждают при добавлении сульфа та аммония до 80% насыще ния. Полученный шарик повторно суспендируют в водном растворе, забуферированном при рН в интерва ле от примерно 6 до примерно 8. Примерами подхо дящи х буфе ров являются Трис-HCl, HEPES, цитрат натрия и т.п. Буферный раствор фракционируют хро матографией на колонке. Подходящи ми материалами для использования при колоночной хроматографии являются октилсефароза, ДЕАЕ-ультрогель, АсА44-ультрогель, АсА-54 ультрогель и т.п. Один или несколько из этих мате риалов могут быть использованы последовательно для получения более высокой чистоты. Фракции от каждой колонки собирают и проверяют на ФСК активность. Активные фракции объединяют и разбавляют трифторуксусной кислотой (ТФУК), гептафтормасляной кислотой (ГФМК) или т.п. и наносят на колонку с С4 обратимой фазой. ФСК активность затем элюируют, используя 0–90% ацетонитрильный градиент в ТФУК или ГФМК, предпочтительно при концентрации от 0,10% или 0,15% (объем/объем), соответственно, в зави симости от того, какая кислота была использована при нанесении объединенных фракций на колонку. Фракции, имеющие ФСК активность, анализируют СДС-электрофорезом на полиакриламидном геле (13,5% гель, как описано Лэммли, wature 227, 6810 (1970)). Дополнительные обработки с использованием указанных выше материалов для хроматографических колонок могут до полнительно очистить ФСК белок для гомогенности. Очищенный ФСК белок, фракционированный на СДС–ПАГЭ, показывает гете рогенный ФСК белок, имеющий кажущуюся молекулярную массу в ин тервале от примерно 15000 до примерно 26000 дальтон. Такой кажущийся размер гетерогенности имеется благодаря экстенсивному гликозилированию белка и представляет собой обычную ха рактеристику гликопротеинов. Фракционирование менее очищенных образ цов из Мо клеток кондиционированной среды с помощью СДС–ПАГЭ (в невосстановительных условиях) и проверка белка, элюированного из геля, показывает наличие второго белка, обладающе го ФСК активностью, имеющего кажущуюся молекулярную массу от примерно 28000 до 30000. ФСК активность связывается и элюируется из октилсефа розы, ДЕАЕ ультрогеля и колонки с С4 обратимой фазы. Примерно 60% ФСК активности связывается Кон-А сефа розой (40% протекает через) и может быть элюировано альфа метилманнозидом. Мо лекулярно-массовый анализ рекомбинантного ФСК гельфильтрацией при низком содержании соли показывает, что примерно 30% активности элюируется с определенной молекулярной массой около 19000, но 70% материала нахо дится в качестве димера, элюируется в положении, соответствующем молекулярной массе примерно 38000. Если 1 м хлористо го натрия включают в эту колонку, вся активность элюируется в виде ши рокого пика примерно при 19000. Очищенный ФСК является стабильным в течение по крайней мере 16 часов при инкубировании при 4оС (рН 7,4) в 4 М гуанидин гидрохлориде; в 10 мМ ЭДТА; 10 мМ 2-меркаптоэтанола; и в 30% (объем/объем)этаноле. ФСК активность также является стабильной в 0,1% трифторуксусной кислоте (ТФУК) (рН 2,0) и 0,1% ТФУК плюс 25% (объем/объем)ацетонитрила. Как указывалось ранее, ФСК белок в соответствии с настоящим изобрете нием используется при лечении миелосуп рессии, такой как (симптоматическая) гранулоципения, например, вызываемая химиотерапевтическим лечением рака или его лечением при облучении. В дополнение к этому, ФСК белки изобретения, как указывалось, используются при лечении серьезной инфекции. Для такой цели обычно показана дозировка примерно 200–1000 мкг/пациента. ФСК-белок предпочтительно вводят пациенту вн утривенно с подхо дящим фармакологическим носителем. Примерами таких носителей являются фармакологический фи зиологический раствор и человеческий сывороточный альбумин в фи зиологическом растворе. Кроме того, ФСК белки изобретения обладают други ми активностями и применениями. Например, было показано, что мышиные ФСК акти вируют нейтрофи лы. Следовательно, следует ожидать, что приматные ФСК настоящего изобретения также будут активировать нейтрофи лы. Поэтому физиологические функции ФСК могут быть разнообразными (?). В костном мозге этот лимфокин может стимулировать пролиферацию и дифференциацию эффекторных клеток для защи ты хо зяина, тогда как в периферии могут быть активированы новые и существующие клетки. В локализованном иммунологическом ответе ФСК может сохранять циркулирующие нейтрофи лы внутри или вне площа дей воспаления. Несоответствующая локализация и/или активация нейтрофи лов может быть включена в патофи зиологию различных иммуно-медиированных болезней, таких как равматоидный артрит. Изобретение будет более понятно при ссылке на следующие иллюстративные варианты, кото рые являются чисто примерными и не должны рассматриваться как ограничивающие область настоящего изобретения, описанную в фор муле изобретения. В примерах, если нет других указаний, температура дана в оС. Рестрикционные эндонуклеазы используются в таких условиях и по методике, рекомендуемой их коммерческими поставщиками. Реакции связывания проводят, как описано Маниатисом и др., выше, на стр. 245–6, описание которых приведено здесь в качестве уровня техники, используется буферный раствор, описанный на стр. 246, и концентрации ДНК 1–100 мкг/мл при температуре 23оС для ДНК с тупыми концами и 16оС для ДНК с липкими концами. Проводят электрофо рез в 0,5–1,5% агалозных ге лях, содержащих 90 мМ Трис-бората, 10 мМ ЭДТА. Всю радиомеченную ДНК метят 32Р, какая бы методика метки не использовалась. "Быстрый преп" означает быстрое, маломасштабное продуцирова ние бактериофага или плазмиды ДНК, например, как описано Маниати сом и др., вы ше, стр. 365–373. Пример А. Стадия 1. Культивирование Мо клеточной линии. Мо клетки (АТСС CRZ 8066) выращи вают тра диционным образом на Альфа (6% CO2) сре де или на среде Искова (10% СО2), содержащей 20% сыворотки зародыша теленка (СЗТ), 2 мМ глютамина, 100 u/мл стрепто мицина и 100 мкг/мл пенициллина. Клетки должны быть субкультивированы каждые 4–5 дней. Клетки подсчитывают и вы севают в колбы Фалькона Т–175 в 100–150 мл среды при плотности 3–4 × 105 клеток/мл. Клетки должны удваиваться в 20% СЗТ каждые 4–7 дней. Скорость роста не является постоянной и клетки могут иногда казаться остановившимися в росте, затем прохо дят через вспышки роста. Мо клетки могут быть выращены на среде, не содержащей сыво ротки. Выживание немного лучше, когда клетки не промывают при переносе из СЗТ на среду, не содержащую сы воротки. Оптимальная плотность на среде, не содержащей сыворотки (СНС) составляет 5 × 105 клеток/мл. Клетки будут расти медленно (или по крайней мере сохранять постоянное число) в те чение 3 дней в среде, не содержащей сыворотки, а затем должны быть подпитаны 20% СЗТ в те чение по крайней мере 4 дней. Рост по такому гра фику (3 дня СНС, 4 дня 20% СЗТ) может быть повторен еженедельно, если требуется СНС среда, без какого-либо ущерба для клеток в те чение нескольких месяцев. Стадия 2. Проба на ФСК активность. А. Проба с костным мозгом. Получают свободный костный мозг. Отделяют спикулы вытягиванием через иглу 20, 22, а затем 25 калибра. Разбавляют 1:1 сте рильным забуфе рированным фосфатом фи зиологическим раствором (ФБР) (комнатная температура) и наслаивают Фиколл-Пак (примерно 30 мл ВМ–ФБР на 6 мл Фиколла). Перемешивают при 1500 об/мин в течение 30 минут при комнатной температуре. Удаляют жир и ФБР слой и отбрасывают. Отбирают пипеткой слой малой плотности. Промывают 2 раза ФБР и считают. Располагают клетки на пластине в RPMI (получен от ГИБКО как RPMI 1640) плюс 10% HTFCS (инактивированная теплом СЗТ) в течение 3 часов для уда ления приклеивши хся клеток. Среда для пластины (готовится свежая): 20% СЗТ 0,3% агаpа, растворенного в во де, охлажденного до 40о 2х Искова (1:1 объем/объем с агаром) 1% P/S конечная концентрация 100 u/мл стрепто мицина, 100 мкг/мл пенициллина 10-4 М альфа-тиоглицерина в 2х Искова из 10-2 М исходного Агар, о хлажденный до 40о. Смесь с другими ингредиентами. Охлаждение на водяной бане до 37–38о и хранение при этой температуре. Через 3 часа отбирают пипеткой неприклеившиеся клетки. Перемешивают и подсчитывают. До бавляют 2 × 105 клеток/мл посевной среды и хра нят при контролируемой температуре водяной бани 37–38оС. Прибавляют образцы (например, среды от трансфецированных клеток, обычно 10 мкл образца) в первый ряд углублений пластины микротитратора в двойной повторности. Прибавляют 100 мкл суспензии клеток в каждое углубление. Прибавляют дополнительные 50 мкл суспензии клеток в каждое углубление в первом ряду. Тща тельно смеши вают и переносят 50 мкл раствора из первого ряда в следующий ряд и т.д. и продолжают разбавления 1:3 поперек пластины. Обертывают пласти ну парафильмом. Инкубируют 10–14 дней при 10% СО2, 37оС в полностью влажной атмосфе ре и подсчитывают колонии. Для подсчета колоний считают общее число колоний, кото рое выросло в каждом углублении. В каждой пробе несколько углублений засевают без включения образца (хо лостой) для получе ния основы для отсче та колоний. Среднее число колоний, кото рые выросли в контрольных углублениях, вычи тают из числа колоний, найденных в каждом углублении, содержащем образцы. Одна единица СЗТ представляет собой количество, ко торое будет сти мулировать образование одной колонии больше контрольного уровня на 105 клеток человеческого костного мозга (засеивается 105 клеток/мл), когда концентрация СЗТ является суб-насыщен ной. Суб-насыщен ная концентрация определяется разбавлением и сравнением числа колоний при различных разбавлениях для нахож дения концентрации чуть ниже уровня насыщения. Для этой пробы подсчитывают колонии, содержащие гранулоциты, моноциты или оба типа клеток. Типы клеток в колониях определяются отбором колоний и окрашиванием индивидуальных клеток. Б. Проба с KG–1 клетками. KG–1 клетки (Blod, 56, № 3 (1980)) выращи вают в среде Искова +10% СЗТ, переносят 2 раза в неделю и вы севают каждый пассаж 2 × 105 клеток/мл. Клетки используют для пробы только между пассажем 30– 35. Проба является такой же, как описанная выше для костного мозга, с тем исключением, что KG1 клетки высевают в ага ровую смесь при 4 × 103 клеток/мл. Число колоний, выросши х в каждом углублении, определяют и вычитают контрольный счет хо лостого опыта, как и в описанном выше опыте с костным мозгом. Одна KG–1 СЗТ единица/мл означает, такую концентрацию СЗТ, которая будет сти мулировать половину максимального числа (насыщение) KG–1 колоний для роста. Максимальное число получают при включении уровня насыще ния СЗТ в нескольких углублениях. Стадия 3. Конструирование вектора р91023(В). Вектор трансформации представляет собой pAdD26SV pA(3), описанную Кауфманом и др., Мol. Cell Biol. 2(11): 1304–1319 (1982). Он имеет структуру, представленную на рис. 2. Короче, эта плазмида содержит мышиный ген кДНК дигидрофолат редук тазы (ДГФР), который является основным последним промотором аденовируса 2 (Ad2) при транскрипционном контроле. 5' сайт сращи вания включен в ДНК аде новируса, а 3' сайт сращи вания, происхо дящий из ге на иммуноглобулина, нахо дится между Ad2 основным последним промотором и последовательностью, кодирующей ДГФР. SV40 ранний сайт полиаденилирования нахо дится вниз от последовательности, кодирующей ДГФР. Секция прокариотного происхождения pAd26SVpA(3) взята из pSVOd (Mellon P., Parker V., Gluzman J. and Maniatis T., 1981, Cell 27: 279–288) и не содержит рВР322 последовательностей, известных для ингибирования репликации в клетках млекопитающих (Lusky M. and Botchan M., 1981, Nature (London) 293:79–81). pAdD26SVpA(3) превращают в плазмиду pCVSV2, как показано на рис. 2. pAdD26 pA(3) превращают в плазмиду p AdD26 pA(3) путем делеции одного или двух Pst сайтов в p AdD26 pA(3). Это осуществляется при частичном переваривании PstI (используя недостаточность ферментной активности, что бы таким образом могла быть получена субпопуляция линеаризированных плазмид, в кото рых отщеплен только один Pst сайт), затем проводят обработку по Клейноу, свя зывание для рециркуляризации плазмиды, трансформацию E.coli и скринирова ние для делеции P I сайта, локализованного у 3' SV 40 последовательности полиаденилирования. Трехчастный лидер аденовируса и гены ассоциированных ви русов (VA ге ны) вставляют в pAdD26SVpA(3) (d), как показано на рис. 2. Сначала pAdD26SVpA(3) (d) расщепляют Pvu II, чтобы сделать линейную молекулу открытой внутри 3' участка первого из трех элементов, составляющи х трехчастный лидер. Затем p JAW 43 (Zain et al., 1979, Cell, 16, 851) переваривают Xho I, обрабатывают по Клейноу, переваривают P II и изолируют фрагмент 140 пар оснований, содержащий вто рой и часть третьего лидеров, с помощью электрофо реза на полиакриламидном геле (6% в Трис-боратном буфере; Ма ниатис и др. (1982) выше). Фрагмент 140 пар основа ний затем связывают с Pvu II переваренной pAdD26SVpA(3) (d). Продукт связывания используют для трансформации E.coli для резистентности к тетрациклину и скринируют колонии, используя процедуру Грюнштейна-Хогнесса с применением 32Р-меченного зонда гибридизации для фрагмента 140 пар оснований. Получают ДНК из положительно гибридизованных колоний, чтобы прове рить, нахо дится ли реконструи рованный Pvu II сайт в 5' или в 3' концах вставленной определенной ДНК 140 пар оснований во 2-м или 3-м лидерах последних аденови руса. При правильной ориентации Pvu II сайт нахо дится на 5' стороне вставки 140 пар оснований. Эту плазмиду обозначают pTPL на рис. 2. Ava II D фрагмент SV 40, содержащий SV 40 уси ливающую последовательность, получают при переваривании SV 40 ДНК ферментом Ava II, затуплении концов фрагментом Клейноу Pol I, связывании Xh I лингеров с фрагментами, переваривании Xho I для открытия Xho I сайта и изоляции четвертого самого большого (D) фрагмента гельэлектрофо резом. Затем этот фрагмент связывают с разрезанной Xho I pTPL, получая плазмиду pCVSVL2–TPL. Ориентация SV 40 D фрагмента в pC VSVl2–TPL будет такой, что SV 40 последний промотор находится в той же ориентации, как основной последний промотор аденовируса. Для вве дения генов ассоциированного вируса (VA) аденовируса в pCVSVL2–TPL сначала конструируют плазмиду pBR322, которая содержит фрагмент аденовируса ти па 2 Hind III B. ДНК аденовируса ти па 2 переваривают Hind III и изолируют фрагмент В после гельэлектрофо реза. Затем этот фрагмент вставляют в pBR322, которую заранее переварили Hind III. После трансформации E.coli для резистенции к ампициллину скринируют рекомбинаты для вставки фрагмента Hind III и определяют ориентацию вставки при переваривании рестракционным ферментом. pB322–Ad Hind III B содержит фрагмент Hind III B аденовируса типа 2 в ориентации, представленной на рис. 3. Как показано на рис. 3, VA ге ны обычно получают из плазмиды pBR322–Ad Hind III B при переваривании Hpa I, добавлении EcoRI линкеров и переваривании EcoRI и извлечении фрагмента 1,4 кв. Фрагмент, имеющий EcoRI липкие концы, затем связывают по EcoRI сайту pTPL (которая была ранее переварена EcoRI). После трансформации E. coli НВ101 и отбора на резистентность к тетрациклину, колонии скринируют п утем гибридизации на фильтре с специфи ческим зондом ДНК для VA ге нов. ДНК получают из положительно гибридизированных клонов и характеризуют переварива нием рестрикционными эндонуклеазами. Полученную плазмиду обозначают р91023. Удаляют оба EcoRI сайта в р91023. р91023 разрезают полностью EcoRI, генерируют два фрагмента ДНК, один примерно 7 кв, а другой – пример 1,3 кв, содержащие VA ге ны. Концы обоих фрагментов являются заполненными при использовании фрагмента Клейноу Pol I, а затем оба фрагмента, а именно 1,3 кв и 7 кв связывают вместе. Плазмиду р91023(А), содержащую VA ге ны, и подобную р91023, но делецированную на 2 сайта EcoRI, иденти фицируют с помощью скрининга Грунштейна-Хогнесса с фрагментом VA гена и с помощью традиционного анализа сайтов рестрикции. Затем удаляют единственный сайт Pst I в р91023(А) и заменяют сайтом EcoRI. р91023(А) разрезают для завершения Pst I, а за тем обрабатывают фрагментом Клейноу Pol I для ге нерации полных концов. EcoRI линкеры связывают с затупленным Pst I сайтом р91023(А). Линейную р91023(А) с EcoRI линкерами, присоединенными к тупому Pst I сайту, отделяют от неприсоединенных линкеров и переваривают для завершения EcoRI, а затем повторно связывают. Извлекают плазмиду р91023(В) и иденти фицируют, чтобы иметь структуру, подобную р91023(А), но с EcoRI сайтом, расположенным на месте предыдуще го Pst I сайта. Стадия 4. Получение кДНК банка. Мо клетки индуцируют в те чение 16–20 часов с помощью РНА и РМА для уси ления продуцирования ими лимфокина. Эти клетки высевают при 5 × 105 клеток/мл в среду Искова с 20% СЗТ, 0,3% (объем/объем) РНФА и 5 нг/мл ТРА. Клетки собирают центрифугированием. Шарик клеток снова суспендируют в 20 мл охлажденного на льду ги потонического буфера (RSB буфер: 0,01 М Трис–HCl, рН 7,4, 0,01 М KCl, 0,0015 M MgCl2, 1 мкг/мл циклогексимида, 50 единиц/мл РНК-син и 5 мМ дитиотрейнола). Клеткам дают набухнуть на льду в течение 5 минут, затем механически разрывают 10 ударами плотной гарнитуры стеклянного гомогенизатора. Гомогенат центрифугируют при низкой скорости (2000 об/мин в центрифуге Бекман J 6) для удаления ядер и нелизированных клеток. Надосадочный слой хранят на льду до те х пор, пока шарик из ядер не суспендируют повторно в 10 мл RSB и снова не отцентрифугируют с низкой скоростью. Этот второй надосадочный слой объединяют с первым и центрифугируют объединенные надосадочные слои с низкой скоростью для удаления остаточного загрязнения ядрами и нелизированными клетками. Надосадочный слой из этой операции доводят до 0,15 М KCl добавлением 2М KCl, затем центрифугируют с вы сокой скоростью (25000 об/мин, Бекман SW ротор в течение 30 минут), чтобы получить ша рик из мембран. Мембранный шарик тщательно промывают холодным RSB, затем повторно суспендируют в 12 мл RSB, содержащем 2 М саха розы и 0,15 М KCl. Гото вят два прерывистых гра диента в центрифужных тр убках Бекман SW 41 путем наслаивания 6 мл мембранного раствора в 2 М сахарозы на 2 мл RSB с 2,5 М саха розы и 0,15 М KCl. Трубки заполняют доверху путем наслаивания сверху 2,5 мл PSB, содержащих 1,3 М са харозы и 0,15 М KCl. Эти градиенты перемешивают в те чение 4 часов при 27000 об/мин (Бекман, ротор SV/41) при 4оС. Мембранный слой (при границе фаз между 2,0 М и 1,3 М саха розы) тща тельно уда ляют сбоку, используя иглу 18 калибра и шприц. Объединяют мембранные фракции из двух гра диентов и разбавляют 1 объемом дистиллированной воды, затем смешивают с 0,5% Тритона Х–100 и 0,5% деоксихо лата натрия, затем экстрагируют равным объемом фенола. Водный слой повторно экстрагируют смесью 1:1 фенола и хлороформа и, наконец, равным объемом хлороформа. Наконец, осаждают соединенную с мембраной РНК добавлением хлористого натрия до 0,25 М и 2,5 объемов холодного этанола и инкубируют всю ночь при -20оС. Осажденную РНК собирают центрифугированием (4000 об/мин в течение 10 минут в центрифуге Бекман J–6) и повторно суспендируют в 1 мл дистиллированной воды. Из 2 × 109 клеток получают приблизительно 1 мг РНК. Матричную РНК (мРНК) изолируют из всей РНК хроматографией на колонке с 0,5 мл олиго-dT-целлюлозой. Кратко, РНК нагревают в те чение 5 минут, быстро охлаждают на льду, затем разбавляют в 5 раз при комнатной температуре связывающим буфе ром (0,5 M LiCl 0,01 М Трис-HCl, рН 7,4, 0,002 М ЭДТА, и 0,1% СДС). РНК в связывающем буфе ре переносят на колонку в олиго-dt-целлюлозой, уравно вешенную связывающим буфе ром, при комнатной температуре. Колонку промывают 5 мл связывающе го буфера, затем 5 мкл 0,15 M LiCl, 0,01 М Трис-HCl, рН 7,4, 0,002 М ЭДТА и 0,1% СДС. На-конец, элюируют мРНК 2 мл 0,01 М Трис-HCl, рН 7,4, 0,002 М ЭДТА и 0,1% СДС мРНК осаждают добавлением хлористого натрия до 0,25 М и 2,5 объемов этанола и инкубируют всю ночь при -20оС. Осажденную мРНК собирают центрифугированием (30000 об/мин в течение 30 минут в роторе SW55 Бекмана). Трубку тща тельно сливают и ша рик мРНК повторно суспендируют в 50 мл воды. Повторно суспендированную мРНК доводят до 0,25 М NaCl; затем экстрагируют 1 раз смесью 1:1 фенола и хло роформа; затем 3 раза хло роформом. Осаждают мРНК добавлением 2,5 объемом этанола. Смесь замораживают и оттаивают несколько раз на бане сухой лед/эта нол, затем центрифугируют 15 минут в центрифуге Эпнендорфа. Трубку тща тельно сливают и шарик мРНК повторно суспендируют в 20 мкл дистиллированной воды. Конечный выход составляет примерно 30 мкг мРНК. Готовят первую нить кДНК, используя стандартные методики. Кратко, 10 мкг мембранной мРНК разбавляют в 100 мкл реакционной смеси синтеза кДНК, содержащей 300 мМ Трис, рН 8,4, 140 мМ KCl, 10 мМ MgCl2, 10 мМ b-меркаптоэтанола, 500 мМ каждой из dATF, dGTP, dCTP и dTTP, 5 мкг олиго-dT-(фосфорилированного и со средним размером 12–18) в качестве праймера, 150 m Кu32P dCTP (400 К/ммоль) и 20 единиц ингибито ра рибонуклеазы РНК-син. Реакцию инициируют до бавлением 100 единиц обратной транскриптазы и инкубируют 30 минут при 42 оС. Реакцию останавливают добавлением ЭДТА до 40 мМ и разлагают РНК инкубированием в течение 20 минут при 65оС в 0,2 М NaOH. Основание нейтрализуют добавлением 20 мкл Трис, рН 7,4. Реакционную смесь затем экстрагируют фенолом/хлороформом, снова экстрагируют 50 мкл 10 мМ Трис, рН 7,5 мМ ЭДТА (ТЕ) и объединяют водные фазы. Однониточную кДНК превращают в двуниточную ин кубированием в те чение 12 часов при 16оС с 40 единицами фрагмента Клейноу ДНК полимеразы 1 в 100 мкл реакционной смеси, содержащей 50 мМ фосфата калия, рН 7,4, 2,3 мМ ДТТ, 2-меркаптоэтанол, 10 мМ MgCl2, 150 Молей каждой из четырех деоксинуклеотидтрифосфа тов и 25 mKu32P dCTP. Реакцию прекращают экстракцией фенолом/хлороформом и удаляют невошедшие трифосфа ты пропусканием водной фазы через колонку с 1 мл сефадексом G-50. Выведенные фракции объединяют и осаждают этанолом. Шарик кДНК промывают хо лодным этанолом, затем повторно суспендируют в 200 мкл 20 мМ Трис, рН 8,0, 1 мМ ЭДТА 80 m Молярном 5-аденозил-метионине и 300 единицах EcoRI метилазы в те чение 60 минут при 37оС. Реакцию останавливают экстрацией фенолом/хлороформом и собирают метилированную кДНК осаждением этанолом. Шарик кДНК промывают 70% этанола, затем повторно суспендируют в 200 мкл Sl буфе ра (Маниатис и др.) и инкубируют с 200 единицами Sl-нуклеазы при 30оС в течение 30 минут. Реакцию останавливают экстракцией фенолом/хлороформом и собирают кДНК осаждением этанолом. Двуниточную кДНК притуп ляют инкубирова нием в 100 мкл 20 мМ Трис, рН 7,4 50 мМ NaCl, 10 мМ 2меркаптоэтанола и 500 m Молярных всех че тырех деоксинуклеотидтрифосфатов с 25 единицами Клейноу при комнатной температуре в течение 30 минут. Реакцию останавливают экстракцией фенолом/хлороформом и собирают кДНК осаждением этанолом. Связывают кДНК в 50 млк Т4 лигазного буфе ра (Маниатис и др.) с 500 рМолей RI линкеров, полученных от Нью Инглэнд Биолабс (последовательность: pCGGAATTCCG), используя 2000 единиц Т4 лигазы в течение ночи при 16оС. Реакцию оста навливают инкубированием при 70о в течение 20 минут, затем разбавляют до 300 мкл так, чтобы конечная концентрация соли составляла 0,1 М NaCl, 10 мМ MgCl2, 50 мМ Трис-Cl, рН 7,4. Затем кДНК переваривают 2 минуты при 37оС 700 единицами КсoRI. Реакцию останавливают экстракцией фенолом/хлороформом и собирают кДНК осаждением этанолом. Шарик повторно суспендируют в 50 мкл ТЕ и пропускают через колонку 5 мл СI–4B. Выведенные фракции собирают, объединяют и осаждают этанолом. Осажденную кДНК подвергают электрофо резу через 1% агарозный гель в Трис-ацетатном буфе ре в присутствии 1 мкг/мл этидинийбромида. Изолируют из гелия кДНК с размером в интервале 500–4000 пар оснований, используя стандартную методику со стеклянным порошком. Элюированную кДНК экстрагируют фенолом/хлорофор мом, осаждают эта нолом и шарик (после промывки этанолом) повторно суспендируют в 60 мкл ТЕ. Конечный выход составляет 100–500 нг кДНК. Получение вектора экспрессии р91023(В) описано выше. Связывают переваренный EcoRI и обработанный фосфата зой вектор (500 нг) со 100 нг кДНК в 100 мкл реакционной смеси (стандартная Т4 лигазная реакционная смесь) в течение ночи при 16оС. Реакцию останавливают экстракцией фенолом/хлороформом, затем связанную кДНК собирают осаждением этанолом после добавления 5 мкг т-РНК в качестве носителя. Осажденную этанолом ДНК промывают 70% этанола, затем повторно суспендируют в 100 мкл ТЕ. Эту ДНК используют в 4 мкл аликвотов для трансформации E.coli МС 1061 (4 мкл в 100 мкл трансформационной среды). Каждую из 25 стансформаций переносят в 150 мм чашки Петри с 1% агара, L-бульоном и 10 мкг/мл тетрациклина (Tet-пластины) и инкубируют всю ночь при 37оС. На каждой пластине вырастает приблизительно 2000 колоний, в результате получают около 50000 колоний. После достижения примерно 0,5 мм в диаметре колонии переносят на нитроцеллюлозные диски (137 мм), осто рожно размещая сухой фильтр на поверхности пластины, затем мягко отщепляют фильтр. Все колонии на пластине переносят на фильтр, который затем помещают (колонии сбоку Side up) на свежую Tet-пластину. После выращи вания колоний в течение нескольких часов го товят реплику каждого из фильтров, помещая све жий влажный фильтр точно на первоначальный фильтр, прижимая их друг к другу, отщепляя их, затем возвращая каждый фильтр на свежую Tet-пластину и инкубируя пластины всю ночь при 37оС. Каждую реплику тща тельно маркируют, чтобы ее можно было сравнить с ориги нальным фильтром. Стадия 5. Приготовление пула плазмид ДНК. Каждый из 25 фильтров-реплик осторожно разрезают на восемь частей, используя скальпель и отмечая ориентацию каждой восьмой части относительно оригинального фильтра. Соскабливают колонии с каждой секции в 10 мл L-бульона. Бакте рии собирают центрифуги рованием (3000 об/мин, 10 минут, центрифуга Бекман J–6), повторно суспендируют в 0,6 мл 25% саха розы, 50 М Трис-HCl, рН 8,0, и превращают в протопласты добавлением 0,12 мл 5 мг/мл лизоцима и инкубируют на льду в течение 5–10 минут. Протопласты снова инкубируют при комнатной температуре 10 минут, затем добавляют 0,125 мл 0,5 М ЭДТА, затем лизируют при добавлении 0,12 мл 10% СДС в 50 мМ Трис–HCl, рН 8,0. Лизат осторожно смешивают, инкубируют при комнатной температуре 15 минут, затем осаждают белок и хромосомальную ДНК добавлением 0,3 мл 5 М NaCl. После инкубирования на льду в течение 15 минут лизат центрифугуют на центрифуге Эппендорфа в течение 30 минут на хо лоду. Осторожно удаляют недосадочный слой, оставляя внизу вязкий шарик ДНК/белок и разбавляют, добавляя 2,5 мл воды. Смесь экстрагируют 1 мл фенола, разделяют слои центрифугированием (10К в течение 10 минут в роторе Серволл SS–34) и удаляют водный слой в свежую трубку. Осаждают ДНК добавлением 0,5 мл 5 М NaCl и 7,5 мл холодного этанола, замораживают смесь несколько раз на бане сухой лед/эта нол. Осадок собирают центрифугированием (10К, 15 минут в Сорволл SS–34), повторно суспендируют в 0,3 мл 0,3 М ацетата натрия и снова осаждают (в трубке Эппендорфа) добавлением 1 мл этанола. После 10–15 минут на бане сухой лед-эта нол собирают осажденную ДНК центрифуги рованием (5 минут в центрифуге Эппендорфа) и готовый шарик снова суспендируют в 100 мкл стерильного ТЕ (10 мМ Трис, рН 8,0, 1 мМ ЭДТА). Из типичной операции получают 5–10 мкг плазмиды ДНК. Каждое приготовление содержит ДНК из 200–500 колоний на первоначальном фильтре. Всего приготовлено 200 образцов ДНК из 25 фильтров. Стадия 6. Изолирование ФСК клона. Каждым из образцов ДНК со стадии 5 отдельно стансфе цируют М6 COS клетки обезьяны, как описано ниже. М6 клетки выращи вают рутинно в среде Игла, модифи цированной Дальбекко "ДМЕ, доступная из Джибко", содержащей 10% инактивированной теплом сыворотки зародыша теленка (HIFCS), разделяют дважды в неделю при разбавлении 1:6. Через 24 часа после разделения М6 клетки гото вы для трансфекции. За 24 часа до трансфекции высевают 1,2 × 108 М6 клеток (деление 1:6) в Клеточный Фактор (доступный от Нунк) в 1,5 л ДМЕ + +10% HIFCS. Непосредственно перед трансфекцией пластины отсасывают и дважды промывают 7 мл не содержащей сыворотки ДМЕ. ДНК растворяют в 0,1 М Трис (рН 7,3) и прибавляют в ДМЕ среде, содержащей 2 мМ глютамина, 100 мкг/мл стрептомицина, 100 U/мл пенициллина и 0,25 мг/мл ДЕАЕ Декстрана, доводя до 4 мл раствором Трис-ДНК. Прибавляют 4 мл среды, содержащей растворенную ДНК, на пластину, содержащую М6 COS клетки, и инкубируют в те чение 12 часов. После инкубации клетки промывают один или два раза 7 мл SFDME. Затем прибавляют 5 мл ДМЕ с 10% HIFCS, 100 U/мл пенициллина, 100 мкг/мл стрептомицина, 2 мМ глютамина и 0,1 мМ хло рокина и инкубируют клетки 2,5 часа. Через 2,5 часа промывают один раз SF ДМЕ и прибавляют 10 мл ДМЕ + 10% HIFCS /пластину. Через 30 часов отсасывают среду и подают 4 мл/пластину ДМЕ + 10% HIFCS. Собирают урожай, удаляя кондиционированную сре ду после 24–26 часов дополнительной инкубации. Кондиционированную среду после каждой трансфекции проверяют на ФСК активность, используя KG–1 пробу. Для каждого образца, положительного на ФСК активность, должен быть иденти фицирован клон на первоначальном основном фильтре, ответственный за ФСК активность. Например, для одной трансфекции, положительной на ФСК активность, пересаживают все колонии с секции первоначального основного фильтра, с которого происхо дит образец трансфекции ДНК. Некоторые из этих 320 колоний пересаживают в 3 мл L-бульона плюс 10 мкг/мл тетрациклина. Культуры выращи вают в течение ночи. 320 колоний помещают на матрицу 18 х 18. Готовят пулы из каждого горизонтального ряда и верти кальной колонки матрицы (всего 36 пулов) (примечание: последний горизонтальный ряд имеет только 14 клонов). Готовят образцы ДНК из каждой объединенной в пул культуры, затем используют для трансфекции COS-клеток. Проверяют надосадочные слои этих трансфекций, используя пробу KG–1 колонии. Из этой группы трансфекций получают два положительных: один – в вертикальной колонке, другой – в горизонтальном ряду. Общая культура этих пулов со держит ФСК-клон. Изолируют 12 индивидуальных клонов из этой культуры и гото вят минипрер ДНК из 10 мл культуры в L-бульоне, как описано выше. Образцы 10 мкг ДНК из этих приготовлений переваривают EcoRI и анализируют полученные в результате фрагменты ДНК с помощью электрофореза на агарозном геле. Девять из двенадцати клонов имеют общую вставку приблизительно 750 пар оснований. ДНК из четырех из этих клонов и остальные три клона вводят в М6 COS клетки, как описано выше. Надосадочные слои от этих трансфекций прове ряют, используя KG–1 пробу, а также пробу на ФСК с костным мозгом. Четы ре клона, каждый из которых содержит фрагмент 750 пар оснований, все направляют экспрессию М6 COS клеток с высоким уровнем ФСК активности, как определено в любой пробе, тогда как другие три клона нет. Следовательно, кодирующая область для ФСК должна быть локализова на внутри вставки 750 пар оснований. Последова те льность ДН К, коди рующая ФСК, бы ла уда лена из векто ра трансфор мации в по ложитель ном клоне п утем пе рева рива ния EcoRI и бы ла определена ее ами нокислотная последо ватель ность с использова нием стан дартны х ме то дов ди деокси секвен сирова ния после субк лониро вания фра гменто в в М1 3 век то ры для получе ния последо ва тель ности , по казанной на рис. 1. П лаз мида, p91023 /В/– ФСК, кото рая сна чала показала направлен ную ФСК-эксп рессию в COS кле тках, бы ла обозначе на pCSF–1. Эта плаз мида была де пони рова на в Коллекции к уль тур американского типа в штамме E.coli–MC1061 под номером хранения АТСС 39754 2 июля 1984 года. Стадия 7. Экспрессия ФСК-белка. М6 COS клетки обезьяны, трансформированные вектором р91023/В/ содержащим ФСК/кДНК, как выделено на стадии 6, вы ращивают, как описано на стадии 6, для получения ФСК-белка в культуральной среде. А именно 1 мг этой ДНК (рС5F) растворяют в 1 мл 0,1 М Трис рН 7,3 и прибавляют к 600 мл ДМЕ, содержащим 2 мМ глютамина, 100 11/мл стрепто мицина, 100 мкг/мл пенициллина (P/S) и 0,25 мг/мл ДЕАЕ Декстрана (молекулярная масса 500000, получено из Фармациа). 600 мл раствора ДНК ДЕАЕ Декстрана прибавляют к М6 COS клеткам в уста новку для культивирования клеток и инкубируют при 37 оС в те чение 12 часов. После инкубирования клетки промывают один раз 900 мл SF ДМЕ, затем инкубируют 2,5 ча са с 600 мл ДМЕ, содержащими 0,1 мМ хло рокина, 10% HIFCS, 2 мМ глютамина и P/S. После отсасывания среды, содержащей хлорокин, клетки промывают SF ДМЕ и подают 1500 мл ДМЕ с 10% HIFCS. Че рез 30 часов клетки промывают SF ДМЕ, среду заменяют 800 мл SF ДМЕ и помещают трансфе цированные клетки в условия среды при 37оС на 24 часа. Отработанную кондиционированную среду отсасывают и заменяют другими 800 мл SF ДМЕ. Клетки оставляют на 24 часа в этой среде, затем собирают отработанную среду. По возможности быстро после сбора концентрируют образец отработанной среды в 20 раз ультрафильтрацией под давлением, используя камеру Амикон на 2,5 л с УМ5 мембраной (5000 MW отрезок). Стадия 8. Очистка рекомбинантного ФСК. 200 мл концентрированной отработанной среды (из 4 л исходного материала – стадия 7) доводят до 30% насыщения сульфатом аммония при добавлении твердого сульфа та аммония и удаляют осажденный белок центрифугированием. Надосадочный слой доводят до 80% насыще ния сульфатом аммония добавлением более твердого сульфата аммония и осажденный белок собирают центрифугированием. Шарик повторно суспендируют в 5 мл 20 мМ цитрата натрия, рН 6,1, содержащем 1 М NaCl. Растворенный белок наносят на колонку 1,6 х 100 см Ультрогель АсА54, уравнове шенную тем же самым буфе ром, ВСК активность, элюированная с колонки, имеет кажущийся молекулярный вес 19 к дальтон или после примерно 90 мл. Наблюдали, что если проводить гель-фильтрацию с низкой ионной силой, ФСК активность элюируют с колонки в двух положениях с кажущи мися молекулярными массами примерно 19 к дальтон и 38 к дальтон, подтверждая, что СМ–ФСК может легко образовать димеры. Объединяют активные фракции и доводят до 0,15% ТФУК (добавлением 10% ТФУК) и наносят на колонку Ви дак С4 (0,46 х 25 см), уравновешенную 0,1% ТФА. Колонку проявляют при линейном градиенте 0–90% ацето нитрила (1 мл/мин, всего 340 мл) в 0,1% ТФУК. ФСК-активность элюируется между 39% и 43% ацетонитрила (фракции 16-активность элюируется между 39% и 43% ацетонитрила) (фракции 16–20). Анализируют образец 20 мкл фракции 19 с помощью электрофореза на СДС-полиакриламидном геле (13,5% гель, как описано Леммли, Nature 227, 680 (1970)). Наблюдалась одна широкая полоса белка с кажущей ся молекулярной массой 18–26 к дальтон. Более широкий размер полосы для ФСК является общим признаком гликопротеинов и утончается, отражая экстенсивное, но переменное добавление углевода. Белок из фракции 19 подвергают разложению на Эдману, используя га зофазный микросеквенатор Апплайд Биосистемс. Из приблизительно 20 мкг нанесенного белка получают последова тельность из первых 16 аминокислот (A–P–A–R–S–P–S–P–S–T–Q–P–W–E–H). Высокий выход этой единственной белковой последовательности хорошо подтверждает, что ФСК-белок во фракции 19 был очищен до гомогенности. Биопроба показывает, что фракция 19 имеет 3 × 107 единиц на А280 единиц поглощения. Поскольку ти пичные белки в водном растворе дают коэффициенты экстинкции в интервале от 0,8 до 1,2 А280 единиц поглоще ния на миллиграмм белка, удельная активность очищенного ФСК находится между примерно 1 × 107 и примерно 4 × 107 единиц/мг при испытании с использованием пробы с человеческим костным мозгом. Пример В. Клонирование ФСК гиббона. Стадия 1. Получение мРНК из Т-клеток гиббона. Образец линии Т-клеток гиббона, обозначенной ИСД–MLA 144, культивируют в течение нескольких недель в RPMI 1640 (полученной от Гибко) и 20% сыворотки зародыша теленка (CЗТ) до тех пор, пока не будет получено 1 × 109 клеток всего. Клетки индуцируют для продуцирования высоких уровней ФСК путем активации в течение 24 часов в присутствии 10 нг/мл 12-0-тетрадеканоилфорбол-13-ацетата (ТФА) в RPMI 1640 плюс 1% СЗТ. Собирают выращенные клетки центрифугированием (1000 об/мин, 5 минут), промывают один раз фосфатным буферным солевым раствором (ФБР) и снова собирают центрифугированием. Из этих клеток готовят связанную с мембраной полисомальную (МВР) мРНК, используя ту же самую процедуру, что описана в примере А для получения РНК Мо клеток. Стадия 2. Реакция однонитрочной кДНК. Разбавляют 6 мкг МВР мРНК (из стадии 1) в 50 мкл реакционной смеси для синтеза кДНК (смотри пример А – ста дия 4) и инициируют реакцию добавлением обратной транскриптазы. После 30 минут ин кубирования при 42оС реакцию останавливают добыванием ЭДТА до 50 мМ и разбавляют водой до 100 мкл. Смесь экстрагируют фенолом/хлороформом и снова экстрагируют хлороформом. Отделяют гибриды кДНК/РНК от непрореагировавших трифосфатов хроматографией на колонке с Сефа розой CL–4B. Выведенные фракции объединяют и собирают гибриды осаждением этанолом. Конечный выход составляет 570 нг. Стадия 3. Реакция двуниточной кДНК. Снова суспендируют ша рик однониточной кДНК (стадия 2) в 50 мл воды и проводят синтез второй нити в стандартной реакционной смеси с E.coli. Полимеразой 1, E.coli лигазой и РНКазой Н. Реакционную смесь инкубируют в те чение ночи при 16оС и затем инкубируют 1 час при 37оС. Реакцию останавливают добавлением ЭДТА и экстрагируют фенолом/хлороформом. Отделяют кДНК от непрореаги ровавши х трифос фа тов хроматографией на колонке с Сефарозой CL–4B, выведенные фракции объединяют и собирают кДНК осаждением этанолом. Стадия 4. Получение рекомбинантной кДНК. Снова суспендируют шарик кДНК (стадия 3) в 75 мкл воды. Гомополимерные С "хвосты" присоединяют к концам кДНК при добавлении 10 мкл раствора кДНК к 25 мкл стандартной реакционной смеси с концевой трансферазой и инкубируют при 30оС в течение 5 минут. Реакцию останавливают добавлением ЭДТА до 40 мМ и тепловой инактивацией при 68оС в течение 10 минут. Закаливают 10 нг этой кДНК с хвостами с 50 нг G-хвостовой рBR322 (получена от НЕН) в 10 мкл 10 мМ Трис, рН 7,5, 1 мМ ЭДТА и 100 мМ NaCl. Закаленную реакционную смесь инкубируют в те чение 10 минут при 68оС, а затем 2 часа при 57оС. Стадия 5. Трансформация бакте рий. Выращи вают E.coli штамм МС1061 в L-бульоне, охлаждают на льду, со бирают центрифугирова нием и обрабатывают CaCl2 для приготовления их для трансформации. Затем инкубируют 5 мкл закаленной реакционной смеси кДНК с 200 мкл обработанных CaCl2 бактерий. Проводят 50 таких трансформаций, используя всю закаленную кДНК, и наносят на пластины с 1% агаром L-бульоном, размером 15 см, содержащие 10 мкг/мл тетрациклина. На каждой пластине вырастает приблизительно 1000 колоний. Стадия 6. Посев реплик. Пересаживают 10000 колоний из трансфор мации каждую с помощью зубочистки, переносят на свежие пласти ны (500 на пластину в решетке) и выращи вают всю ночь при 37оС. Затем колонии снимают с каждой пластины, прижимая сухой нитроцеллюлозный фильтр плотно к поверхности пластины. Для каждого из этих основных исходных фильтров готовят два фильтра-реплики. Основные исходные фильтры хранят при 4оС, а фильтры-реплики обрабатывают основанием и сушат, чтобы подгото вить их к гибридизации. Стадия 7. Приготовление 32Р-меченых зондов гибридизации. Изолируют вставку кДНК из pCSF–1 перевариванием рестрикционным ферментом EcoRI и электрофо резом на агарозном геле с Трис-ацетатом и этидинийбромидов. Полосу, содержащую фрагмент кДНК, вырезают из геля и очищают по мето дике со стеклянным порошком. Затем прибавляют 300 нг фрагмента кДНК к 1 мкл 10 х Т4 ДНК Полимеразного буфера (0,33 М Трисацетата, рН 7,9, 0,66 М ацетата калия, 0,1 М аце тата магния и 10 мМ дитиотрейтола), и 3 единицам Т4 ДНК Полимеразы "Нью Ингленд Биолабс" и разбавляют водой до 10 мкл. После инкулирования 5–10 минут при 37оС, эту смесь объединяют с 1 мкл 10 х Т4 ДНК Полимеразного буфера; 1 мкл 2 мМ раствора каждого из dCTP, dTTP, dGTP, 10 мкл 32PdATP /10 m Кu/мкл 3000 Ku/ммоль" и 3 единицами Тг ДНК Полимеразы. Реакционную смесь инкубируют в течение 20 минут при 37 оС. Затем прибавляют 1 мкл 2 мМ dATP и реакционную смесь инкубируют е ще дополнительные 10 минут при 37оС. Отделяют не включенные трифосфаты от меченной кДНК хроматографией на колонке с Сефа дексом G100. Готовят вто рой зонд из синтетического олигонуклеоти да, имеющего последовательность: ATC TGG CTG C AC AG которая комплементарна аминоконцу области, кодирующей ФСК. Этот оли гонуклеотид мечен по 32P dАТР на его 5'-конце с использованием реакции полинуклеотидкиназы. Стадия 8. Изоляция клонов ФСК кДНК. В стандартной процедуре скрининга гибридизацией гибридизуют некоторые 45 клонов с Т4 меченной pCSF–1 кДНК. Из этих приблизительно 20 также гибридизуется с меченным олигонуклеотидным зондом. Кодирующую область одного из них секвенсировали и данные последовательности показывают число замещений основа ния, некоторые из которых являются результатом в различии аминокислот в экспрессированном белке. Эти различия представлены на рис. 1 выше последовательности ДНК для ге на человеческого ФСК, клонированного в примере А. Пример С. Клонирование ФСК из мРНК лимфоцита периферического кровотока. Стадия 1. Получение мРНК из лимфо цитов периферического кровотока. Лимфоциты перифе рического кровотока были получены из четырех побочных продук тов плазмафореза "получены от Ред Кромм" при фракционировании на градиенте Фиколл-Гипак. Малая плотность в RPMI 1640 в присутствии 5% сыворотки зародыша теленка, 0,17% фито геммаглюти нина и 10 нг/мл форбал миристат ацета та (РМА) при плотности 2 × 106 клеток (мл) (всего получено 6 × 109 клеток). Собирают клетки центрифугированием (1000 об/мин, 5 минут), один раз промывают солевым фосфатным буфе ром (PBS) и снова собирают центрифугированием. Гото вят цитоплазматическую РНК при осторожном лизисе, при котором клетки снова суспендируют в 50 мл холодного Тритонового буфе ра для лизиса (140 мМ NaCl, 1,5 мМ MgCl2, 10 мМ Трис, рН 8,6, 0,5% Тритон Х–100) с 10 мМ дитиотрейтола (ДТТ) и 50 единиц/мл РНКсин (получена от Биотек). Этот ли зат делят на 2 равные части и каждую часть наслаивают на 10 мл подушки лизисного буфера, содержаще го 20% саха розы. Удаляют ядра клеток центрифугированием на холоду (4оС, 400 об/мин в течение 5 минут). Осторожно удаляют верхний слой (цитоплазматический экстракт) и прибавляют додецилсульфат натрия (СДС) до конечной концентрации 1%. Этот раствор дважды экстрагируют равным объемом фенола/хлорофор ма (смесь 1:1) и осаждают РНК добавлением 2,5 объемов этанола. Осажденную РНК собирают центрифуги рованием (15 минут при 4000 об/мин.) и снова суспендируют в 0,01 М Трис, рН 7,5 1 мМ ЭДТА, 0,25 М NaCl (ТЕ буфер плюс 0,25 М NaCl) и снова осаждают добавлением 2,5 объемов холодного эта нола. Наконец собирают РНК центрифугированием и снова суспендируют в 5 мл воды. Конечный выход 7,5 мг. Изолируют матричную РНК из всей цитоплазматической РНК селекцией на олиго-dT-целлюлозе. Нагревают 2,5 мг всей РНК до 65о в течение 5 минут. Прибавляют NaCl до 0,5 М и дают РНК остыть до комнатной температуры. Эту РНК пропускают через колонку с 1 мл олиго-dT-целлюлозы, уравновешенную в ТЕ + 0,5 М NaCl (связующий буфер). Удаляют несвязанную РНК экстенсивным промыванием колонки связующим буфером. Связанную матричную РНК элюируют 3 мл воды и осаждают добавлением 0,2 мл 4 М NaCl и 2,5 объемов холодного этанола. Осажденную мРНК собирают центрифуги рованием (30 минут при 25000 об/мин). Конечный шарик (приблизительно 100 мкг) повторно суспендируют в 50 мкл воды. Стадия 2. Реакция однониточной кДНК. Разбавляют 20 мкг PBL мРНК в 50 мкл реакции синтеза кДНК, содержащей 100 мМ Трис, рН 8,4, 140 мМ KCl, 10 мМ MgCl2, 10 мМ 2-меркаптоэтанола, 400 мкмМ каждого из dATP, dGTP, dCTP и dTTP, 5 мкг олиго-dT (средний размер 12–18) в качестве праймера, 25 mKu 32PdCTP (400 m Ku/моль) и 20 единиц ингибитора рибонуклеазы РНК син. Реакцию инициируют добавлением 60 единиц обратной транскриптазы при 37оС и инкубируют 30 минут при 42оС. Реакцию останавливают добавлением ЭДТА до 40 мМ и экстрагируют равным объемом насыщенного водой фенола. Фенольную фа зу снова экстрагируют 50 мкл ТУ буфера. Водные фазы объединяют. Отделяют гибриды кДНК/РНК от непрореагировавших трифосфа тов, пропуская объединенные водные фазы через колонку с 5 мл Сефарозы СL–4B, (получена от Сигмы), уравновешенную ТЕ. Фракции, которые отводят из колонки, объединяют, доводят до 2500 мМ NaCl и осаждают нуклеиновые кислоты добавлением 2,5 объемов холодного этанола. Гибриды собирают центрифугированием в течение 30 минут при 4000 об/м. Конечный шарик (2,5 мкг кДНК) снова суспендируют в 50 мкл воды. Стадия 3. Реакция двуниточной кДНК. Синтезируют двуниточную кДНК при совместном действии ферментов E.coli ДНК Полимеразы 1, E.coli ДНК лигазы и E.coli РНКазы Н. Реакционная смесь (50 мкл) содержит 20 мМ Трис, рН 8,0, 4 м М MgCl 2, 1,2 мМ ЭДТА, 25 М Н АД, 100 mМ каждого из dATP, dGTP, dCTP и dTTP, и 50 mKu 32PdCTP (3000 Ku/ммоль). Проводят реакцию при добавлении 3 единиц ДНК Полимеразы 1,05 единиц ДНК лигазы и 0,75 единиц РНКазы и инкубировании при 16 о в течение 18 часов, затем 1 час при 37оС, а затем останавливают добавлением ЭТА до 40 мМ и экстрагируют равным объемом фенола. Фенольную фазу снова экстрагируют 50 мкл ТЕ, объединяют водные фазы и отделяют к ДНК от непрореагировавших трифосфатов хро мографией на колонке с Сефарозой CL–4B, как описано выше для однониточной кДНК. Основываясь на включенном 32Р, однониточная кДНК количественно превращается в двуниточную форму. Стадия 4. Получение рекомбинантной кДНК. Прибавляют гомополимерные С "хвосты" к конца кДНК при осторожном нагревании 400 нг кДНК в 50 мкл реакционной смеси, содержащей 1 мМ 2-меркаптоэтанола, 1 мМ СoCl2 и 9 единиц концевой деоксинуклеоти дилтрансферазы, при 30оС в течение 5 минут. Реакцию прекращают добавлением ЭДТА до 40 мМ и нагревают до 68оС в течение 10 минут. Закаливают 200 нг этой хвостовой кДНК с 500 нг G-хвостового рАТ153 (получена от Амершем) в 100 мкл 10 мМ Трис, рН 7,5, 1 мМ ЭДТА и 100 мМ NaCl. Реакцию закаливания проводят при 57оС в течение 2 часов после 3 минут предварительной инкубации при 68оС. Стадия 5. Трансформация бакте рий. Продукт реакции закаливания кДНК используют непосредственно для трансформации E.coli штамм МС1061. Используют све жую колонию бактериальных клеток для инокуляции 50 мл L-бульона и выращивают несколько часов до тех пор, пока оптическая плотность при 550 нм не достигнет 0,25. Клетки замораживают на льду и со бирают центрифугированием (2000 об/мин в течение 10 минут). Ша рик повторно суспендируют в 10 мл холодного 0,1 м CaCl2 и оставляют на льду на 10 минут. Кле тки собирают центрифугированием (2000 об/мин в течение 5 мин) и повторно суспендируют в 2,5 мл 0,1 М CaCl2. Затем 10 мкл реакционной смеси закаливания кДНК инкубируют с 200 мкл обработанных CaCl2 бактерий в течение 30 минут на льду, а затем 2 минуты при 37оС, затем добавляют 0,8 мл L-бульона и наконец инкубируют 30 минут при 37оС. Осуществляют 20 из этих трансформаций, используя всю закаленную кДНК. Каждую трансфор мированную смесь наливают на пластины с 1% агаром L-бульоном (15 см диаметр), содержащее 10 мкг/мл тетрациклина. Из 20 трансформаций все 20 таких пластин были залиты и инкубированы всю ночь при 37 оС. В среднем приблизительно 1500 бактериальных колоний выросло на каждой пластине, всего 30000 клонов. Стадия 6. Посев реплик. Первоначальные колонии, выросшие на каждой пластине, переносят на 137 мм нитроцеллюлозные фильтры прижиманием сухо го фильтра к верхушкам колоний и снятием их с пласти ны. Готовят две идентичных реплики для каждого исходного фильтра по стандартной методике посева реплик, в которой каждый ориги нальный фильтр осторожно помещают сбоку колонии на стерильный квадрат фильтровальной бумаги (Ватман ЗММ), оставляя на квадратном кусочке стекла. Новый предварительно увлажненный нитроцеллюлозный фильтр осторожно накладывают поверх основного фильтра, покрывают вто рым стерильным квадратом фильтровальной бумаги и полный сэндвич затем зажимают вместе с первым и вторым куском стекла. Нумеруют сэндвичевые фильтры и 3 отверстия малого диаметра пробивают через них асимметрично, чтобы можно было точно выровнять в будущем. Затем уда ляют реплики с основы и помещают колонии сбоку на новые пластины с L-бульоном агаром, содержащие те трациклин. Немедленно готовят вто рую реплику идентичным образом. Каждый основной фильтр возвращают на пласти ну и все пласти ны инкубируют при 37оС в те чение нескольких часов до тех пор, пока бактериальные колонии не достигнут примерно 1 мм в диаметре. Основные исходные фильтры хранят при 4оС и готовят реплики для гибридизации, как описано ниже. Стадия 7. Приготовление фильтров для гибридизации. Каждый фильтр-реплику (стадия 6) помещают сбоку колонии на фильтровальную бумагу (Ватман 3 ММ), смоченную в 0,5 М NaOH, 1,5 М NaCl в течение нескольких минут. Фильтры переносят для нейтрализации на фильтровальную бумагу, смоченную в 1 м Трис, рН 7,5 1,5 М NaCl в течение 2 минут, а затем переносят на вторую груп пу фильтров для нейтрализации в те чение 5–10 минут. Наконец фильтры поме щают на фильтры, смоченные SS C-буфе ром (0,015 М цитрата натрия, 0,15 М NaCl, рН 7,4) в течение 5 минут, сушат на воздухе и прогревают в вакууме при 80оС в течение 1–2 часов. Стадия 8. Изоляция клонов ФСК к ДНК. Дубликатные фильтры зондируют с ра диоактивно меченной вставкой рФСК–1 кДНК, приготовленной как описано выше в примере В. Гибридизуют в кДНК 20 колоний. Пересаживают 20 из них с основного фильтра и выращи вают всю ночь в L-бульоне для дальнейше го анализа. Рестрикционный фермент переваривает (PttI) образцы ДНК (быстрый преп) из этих клонов, показывая что 3 близки к полной длине. Один из них был секвенсирован. Последовательность области, кодирующей ФСК, этого клона идентична соответствующей последовательности pCSF–1 (а именно, имеет Т в положении 365–CSF(Ile)). Пример Д. Очистка ФСК из линии Мо клеток. Инкубируют отработанную кондиционированную МО среду, не содержащую сыво ротки (40 л), при 55оС в те чение 30 минут для инакти вации HTLV–11 вируса, ассоциированного с клеточной линией. Эту среду концентрируют ультрафильтрацией по давлениям, используя Пелликон Газетт с мембраной РТGC (1,5 кв.фута), которая имеет отрезок с молекулярной массой 10000. Затем белок концентрируют осаждением сульфа том аммония (80% насыще ния). Повторно суспендируют конечный шарик белка (800 мг) в 100 мл 20 мМ трис/оксиметил/ аминомета на гидрохлорида /Трис-HCl/, рН 7,4, и диализуют против то го же самого буфе ра (3 раза с 4 л загрузки каждый раз). Диализованный белок наносят на колонку 2,5 х 10 см с ДЕАЕ (диэтиламиноэтил) – ультрагелем, уравновешенным в том же буфе ре. Колонку промывают 800 мл 20 мМ Трис-НСl, рН, 7,4, затем элюируют ФСК-активность 800 мл 20 мМ Трис-HCl, рН 7,4, содержаще го 0,12 М NaCl. Собирают фракции 10 мл и проверяют на ФСК. Объединяют активные фракции (3) и концентрируют в 6 раз (до 5 мл) ультрафильтрацией под давлением (мембрана Амикон УМ5, отрезок с молекулярной массой 5000). Концентрированный образец из ДЕАЕ колонки наносят на колонку 1,6 х 100 см с АсА44 ультрагелем (акриламид-агарозный ультрагель, имеющий фракционирование 10–130 к дальтон), уравновешенным 20 мМ N-2-оксиэтилпиперазин-N-2- этансульфо новой кислотой (HEPES), рН 7,4, 50 мМ NaCl и 0,01% полиэтиленгликоля (ПЭГ–8000). ФСК активность элюируют с колонки с кажущейся молекулярной массой 30 к дальтон. Объединяют активные фракции и доводят до 0,15% (объем/объем) трифторуксусной кислоты (ТФУК) добавлением 10% ТФУК и наносят на колонку 1 х 25 см с Ви дак С4 обратной фазой. Колонку проявляют с линейным градиентом 0–90% ацетонитрила в 0,1% ТФУК (объем/объем) при 4 мл/мин (всего 1000 мл). ФСК активность элюируется приблизительно при 47% (объем/объем) ацетонитрила. Объединенные активные фракции доводят до 0,05% (объем/объем) гептафтормасляной кислоты (ГФМК) при добавлении половины объема 0,15% (объем/объем) ГМФК и наносят на колонку (0,46 х 25 см) с Видак С4, уравновешенной 0,15% (объем/объем) ГФМК. Колонку проявляют с линейным градиентом 0–90% (объем/объем) ацетонитрила в 0,15% (объем/объем) ГФМК при 1 мл/мин. (Всего 340 мл). ФСК активность элюируется примерно при 53% (объем/объем) ацетонитрила. Было найдено, что активными являются фракции 37–44 (1 мл каждая). Концентрируют 0,15 мл фракции 40 в 4 раза (используя САВАНТ Спид Вак Концентратор) и прибавляют 40 мкл 2 х СДС гель образца буфе ра (0,125 М Трис-НСl, рН 6,8, 4% СДС, 20% глицерина и 0,004% бромфе нола голубого). Эти образцы кипят 2 минуты и наносят на 13,5% СДС гель (Лэммли U., Nature 227, 680 (1970)) (см. рис. 2). Было определено, что фракция 40 имеет 110000 единиц ФСК костного мозга мл. Это соответствует примерно 3,0 × 107 единиц на А280 единиц поглоще ния. Поскольку типичные белки имеют коэффи циенты экстинкции в интервале между 0,8 и 1,2 А280 единиц/мг, очищен ный ФСК имеет удельную активность в интервале от примерно 1 × 107 до примерно 4 × 107 единиц/мг в пробе с костным мозгом. Подвергают разложению по Эдману 1 мкг образца очищенного GM–ФСК, используя га зофазный микросексенсатор нанесенный биосистем. Была определена последовательность остатков 3–5, являющаяся Ala Arg Ser. Пример Е. Сотрансформация и амплифи кация ФСК последовательности в СНО клетках. Плазмиду р91023(В)– CSF, вво дят в СНО ДГФР-дефицитные клетки ДИКХ–В11 (Chasin and Urlaub PVAS 77: 4216, 1980) путем слияния протопластов, как описано (Sandri – goldin et al., Mel. Cell Biol 1, 743–752, 1981). Рост и поддержание СНО клеток описаны (Кауфман и Шарп, J. Mol. Bio, 150, 501–621 (1981)). Для слияния протопластов вводят р91023(В)–CSF–1 в E.coli НВ101 и выращи вают бактерии в 50 мл м 9 солей, содержащих 0,5% казаининовых кислот, 0,5% глюкозы, 0,012% MgSO4, 5 мкг/мл тиамина, и 10 мкг/мл тетрациклина, до поглоще ния 0,6 при 600 нм. Добавляют хлорамфеникол до 250 мкг/мл и инкубируют культуру при 37оС дополнительно 16 часов для того, что бы амплифицировать число копий плазмиды. Клетки центрифугируют при 3000 g в течение 10 минут при 4 оС и суспендируют в 2,5 мл охлажденной 20% сахарозы в 50 мМ Трис-Cl, рН 8,0. Прибавляют лизоцим (0,5 мл 5 мл/мл раствора в 0,25 М Трис-Cl, рН 8,0) и смесь выдерживают на льду в течение 5 минут. Прибавляют ЭДТА (1 мл 0,025 М ЭДТА, рН 8,0) и выдерживают на льду еще 5 минут, затем медленно прибавляют 1,0 мл 0,05 М Трис-Cl, рН 8,0. Инкубируют суспензию в течение15 минут при 37оС до тех пор, пока бактерии не превратятся в протопласты. Затем суспензию медленно разбавляют 20 мл предварительно подогретой среды, содержащей 10% саха розы и 10 мМ MgCl2, и выдерживают 15 минут при 37оС. Раствор протопластов (приблизительно 109/мл) прибавляют к СНО, ДГФР де фицитным ДИКХ–В11 клеткам в пласти не с 6 углублениями (приблизительно 1 × 106) клеток (углубление) при соотношении примерно 1–2 × 104 протопластов, клетку и протопласты собирают в шарик в клетки при центрифугировании при 2000 об/мин в течение 8 минут в дисковом роторе вращающе гося микротитратора центрифуги IEC Мо дель К. После центрифугирования удаляют надосадочный слой отсасыванием. Прибавляют 2 мл раствора полиэтиленгликоля 50 г РЕО – 1450 (Baler Chem Co) в 50 мл среды в каждое углубление пластины с 6 углублениями. Клетки снова центрифуги руют при 2000 об/мин в течение 90 секунд, уда ляют раствор полиэтиленгликоля и пластины три раза промывают 4 мл среды/углубление. Затем клетки трипсинизируют, суспендируют в 10 мл среды, содержащей 10% сыворотки зародыша те ленка, и центрифугируют в конической трубке при 500 об/мин в клинической центрифуге. Ша рики клеток из 3 углублений объединяют и высевают в чашку 10 см для культуры ткани. К каждой пластине добавляют свежую среду, со держащую 100 мкг/мл канамицина, тимидина, аденоксина, деоксиаденозина, пенициллина и стрептомицина и 10% диализованной сыворотки зародыша теленка. Канамицин вводят для предотвраще ния роста любых бакте рий, которые имеют способность превращаться в протопласты. Через 2 дня клетки подкультивируют 1:15 в альфа-среде с 10% диализованной сыворотки теленка, пенициллином и стрепто мицином, но без нуклеозидов. Затем клетки снова подают в ту же самую селективную среду (лишенную н уклеозидов) через 4–5 дней. Колонии появляются через 10–12 дней после субкульти вирования в селективной среде. Следовали двум схемам отбора на метотрексат (МТХ) и амплифи кации. В первой схе ме изолируют еди ные независимо клонированные трансформанты на основе экспрессии ДГФР и выращи вают последовательно каждый клон в условиях для амплификации числа копий выше указанной ДНК, а именно рост при увеличивающейся концентрации метотрексата. По второй схеме изолируют п ул из множественных независимых трансформантов на основе ДГФР экспрессии и размножают вместе в условиях амплифи кации указанной выше ДНК, а именно рост при повы шающихся концентрациях метотрексата. Затем изолируют индивидуальные клоны из массы отобранной популяции и анализируют на GM–ФСК экспрессию. Эти клоны, показывающие самые высокие уровни экспрессии, снова выращивают в условиях дальнейшей амплифи кации вышеуказанной ДНК (а именно рост в условиях повышающейся концентрации метотрексата в культуральной среде). В одном эксперименте семь ДГФР+ трансфор мантов объединяют в а льфа-сре ду, ли шен ную н уклеозидов. Эти кле тки последова тельно вы ращи вают при посте пенном уве личе нии концентраций МТХ, начиная с 0,02 mМ, затем 0,1, 0 ,5 и 2,0 m М МТХ. При пробе на GM–ФСК-ак тивность в KG–1 пробе клеток, эти клетки продуци руют от 3000 до 12000 единиц/мл. Отобранную популяцию клонируют при 0,5 mМ.МТХ и в 2,0 М.МТХ. Кло ны, получен ные в 0,5 mМ.МТХ (010, Д2 и В6) после дователь но отбирает для роста в 2,0 mМ.МТХ. При испыта нии на GM–ФСК акти вность в пробе на КG–1 клетках, клонированные клето чные линии продуцир уют от 15000 до 300000 единиц/мл GM– ФСК акти вности. GM–ФСК, продуцированный согласно этому примеру, имеет аминокислотную последовательность, приведенную для ФСК–Thr на рис. 1. Пример Г. Экспрессия GM–ФСК в E.coli. GM–ФСК экспрессируется в E.coli из вектора pTALC–185R, схематическое описание которого приведено на рис. 6. Последовательность, кодирующая GM–ФСК, начинается с синтетической последовательности ATG.CCA.CCA.CCT.CCT.TCT. CCA. TCT.CC A.CTC. ACT, которая определяет начальные 11 аминокислотных остатков зрелого GM–ФСК. Остальная последовательность, кодирующая GM–ФСК в pTALC–185R является идентичной последовательности pCSF–1, нуклеотиды 97–447 вслед за последова тельностью TAR TAP TAG. Сра зу после трип летного тер минато ра имеется рUМ–18 полилинкер. Ген резистен тности к тетрациклину из pBR322 был вставлен в проти во положной ориента ции к ФСК ген у, 100 основа ний вниз от рUC-18 поли линкера. Ген резистентности к тетрациклину не сет свой собствен ный просмотр. Продолжая движение проти в ча совой стрелки имеется следующий ген для b-лакта мазы вслед за pUC18/CoL EI/ источником репликации. Конечным структурным признаком плазмиды перед возвраще нием к ФСК последовательностям является PL промотор. Этот промотор по существу описан А. Скатцманом и М. Ро зенбергом (в "Лабораторном руководстве по молекулярному клонированию" (1982)), Колд Спринг Харбeр Лаборатори, с. 419). ФСК экспрессия обеспечивается PL промотором после термической индукции в подхо дящем штамме-хозяине E.coli. Родительский штамм, используемый для конструирова ния всех штаммов, представляет собой W 3110 IacIoL8 (P. Brent and M. Plashne, PNAS 78 (1981) 4204–4208). Фрагмент l ДНК (l нуклеотиды 34399–38214) интегрируют в хро мосому W 3110 IacIоL8 у Iac Z локуса. Интеграцию осуществляют с использованием вектора интеграции, состоящего из рBR325 последовательностей, несущей гены для резистентности к хлорамфениколу и ампициллину, а также pBR322 источника репликации (F.Bolivar, Gene, 4:(1978), с. 121-136). Встpаивают l ДНК в lac L ген, котоpый сам находится в плазмиде в качестве фpагмента, пpостиpающегося от сайта Bsteil в Lacl до сайта Tilli вниз от lac L. Интегpация ДНК в хpомосомальную копию lac L достигается гомологической рекомбинацией и нахо дят Iac+, чувствительные к ампициллину, резистентные к хлорамфе николу колонии. Второй рекомбинационный случай приводит к уда лению всех экстра-плазмидных последовательностей, но приводит к тому, что интегрированный l ДНК фрагмент скринируется на лактозных пласти нах МакКонкея. Во втором рекомбинантном случае исходный Iac+, amp, cam P фенотип изменяется на Iac-, ampS, cam S фенотип. Полученный в результате штамм назван GL400 и является lP при 30о и lS при 42оС. Этот фенотип демонстрирует существование функциональной хромосомальной копии Сl857 аллеля. GL400 дает lon- при PL трансдук ции из лизата, вы росшего на штамме SG20252 (Iac D и 1169, ага 139 гр. Sl. lon D 100, Th 10) Th 10 заготавливают скринированием на TetS на селективной среде. (S. Maloy, W. Nunn, J. Bacterial 145 (1981) 1110–1112). Готовый штамм-хозяин называется G1413 (IacIoL8, LacZ D (lCl, PEX, № 1, lon D 100). pTALC–185R транспормируют в G1413. Культуру этого штамма выращивают всю ночь при 30 оС в 5 мл индукционной среды, содержащей 7 мкг/мл тетрациклина. Индукционная среда содержит, на 1 л: 20 г. казаининовых кислот, 6 г. Na2HPO4 × 17H2O 3 г. KH2PO4 0,5 г. NaCl 1 г. NH4Cl 1% глицерина 2 мг витамина В1 2 мг CaCl2 × 2H2O 0,2 г MgCl2 × 6H2O Инокулируют 25 мл этой среды, содержащей 7 мкг/мл тетрациклина, 125 мкл ночной культуры и встряхи вают при 30оС на водяной бане до тех пор, пока культура не достигнет плотности при А550 0,5. Затем быстро перехо дит на 40о во дяную ба ню и встряхи вают еще 2 ча са, что бы дать возможность пройти синтезу GM–ФСК. Собирают урожай клеток и проверяют их на со держание ФСК электрофорезом на СДС-полиакриламидном геле. В эти х условиях GM–ФСК аккумулируется приблизительно до 5% от клеточного белка. Пpимеp С. Экспрессия GM–ФСК в Saсcharomyces Cerevisial. А. Вектор конструирования. Конструируют плазмиду, которая содержит ген для фермента в ура цил биосинтети ческом пути (ИРАЗ) в качестве ге на селекции и 2 источника репликации. Эта плазмида происходит из YIp5 (Botstein et al., Gene, 8, c.c. 17–24 (1979)) с добавлением фрагмента, содержаще го источник репликации из 2 микрон плазмиды дрожжей. В. Изоляция гена для Глицеральдегид фосфат. Дегидрогеназы (SPDH). Изолировали из дрожжей два гена для GГДН (Holland and Hollan Journal of Biological Chemistry 255, c.c. 2596–2605 (1980)). Олигонуклеотидный зонд, синте зированный из опубликованной последовательности, используют для изоляции GГДН гена из плазмидной библиотеки дрожжевой геномной ДНК по стандартным методикам. Плазмида, содержащая целый GAP491 ген, бы ла депонирована ранее (АТСС № 39777). С. Получение промотора глицеральдегид фосфат де гидрогеназы для экспрессии гетерологического гена. Конструируют плазмиду, которая позволяет природное распределение GРДН промотора с начала целевого гетерологи ческого структурального гена. Это осуществляется при введении Кр I сайта в непосредственной близости к инициирующему метиониновому кодону GРДН структурального гена. Затем вставляют промотор "cas sette" в дрожжевой вектор экспресcии YOp1. Д. Изоляция гена для L фактора. Из дрожжей изолировали ген для фактора L, зреюще го фе ромона (Kurjan and Herskavitz, Cell 30, c.c. 933–943 (1982)). Олигонуклеотидный зонд, синтезированный из этой последовательности, используют для изоляции гена из плазмидной библиотеки дрожжевой геномной ДНК по стандартным методикам. Е. Получение плазмиды экспрессии ФСК. Конструируют по стандартным методикам из опис анных выше эле ментов и гена человеческого ФСК вектор экспрессии (AJ14, рис. 7). В этом векторе удалили природную лидерную последовательность ФСК и встави ли последовательнос ть, кодирующую зре лый ФСК, вб лизи пре-про l факто ра. Сое динения между СРДН промото ром, пре-про последова тельностью l фак тора и последова тельностью зрелого ФСК уто чнены (ниже) и были подтверждены деокси нуклеотидным секвенсированием. AAATAAAC AAAATG. CGTTTTCCTTCA....... AAA AGA GAG GCG GAA GCT.GC A CCC GCC CGC TCG... Г. Экспрессия GM–ФСК. Плазмиду AJ14 трансформируют в штамм Saccharourgces Cerevisial. Культивируют клетки для продуцирования ФСК. последовательности Фиг. 1 Фиг. 2 Фиг. 3 Фиг. 4 Фиг. 5 Фиг. 6 Фиг. 7 Тираж 50 екз. Відкрите акціонерне товариство «Патент» Україна, 88000, м. Ужгород, вул. Гагаріна, 101 (03122) 3 – 72 – 89 (03122) 2 – 57 – 03
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for preparing protein with activity of granulocyte macrophage-colony stimulating factor (gm-csf) of primates
Автори англійськоюCLARK Steven K., Kaufman Randal G., Wong Gordon G., Wong Elizabeth A.
Назва патенту російськоюСпособ получения белка с активностью колониестимулирующего фактора гранулоцитов и макрофагов (gm-csf) приматов
Автори російськоюКларк Стивен К., Кауфман Рандал Дж., Вонг Гордон Г., Ванг Элизабет А.
МПК / Мітки
МПК: C12N 15/81, C07K 14/535, C12N 1/21, A61K 38/16, C07K 14/00, C07K 14/435, A61K 35/74, C12P 21/02, C12N 15/00, C12N 1/19, C07K 14/52, C12N 15/70, C07K 14/53, C12N 1/20, C12N 5/00, C12N 5/10, C07K 1/16, C12N 15/85, A61K 38/00, C12N 15/09, C07H 21/04, C07K 1/20, A61K 35/12, C12P 21/00
Мітки: фактора, активністю, спосіб, приматів, отримання, колонієстимулювального, гранулоцитів, білка, макрофагів(gm-csf
Код посилання
<a href="https://ua.patents.su/24-29377-sposib-otrimannya-bilka-z-aktivnistyu-koloniehstimulyuvalnogo-faktora-granulocitiv-ta-makrofagivgm-csf-primativ.html" target="_blank" rel="follow" title="База патентів України">Спосіб отримання білка з активністю колонієстимулювального фактора гранулоцитів та макрофагів(gm-csf) приматів</a>
Кристал білка, зшитого багатофункціональним зшивальним агентом (варіанти), пристрій, що містить кристал білка та спосіб отримання аспартаму

Номер патенту: 27035
Опубліковано: 28.02.2000
Автори: Нейвіа Мен'юел А., Ст.Клер Ненсі Л.
МПК: C12M 1/34, C12N 9/14, C12Q 1/00, C07K 5/072, G01N 33/531, C12P 21/06, G01N 33/66, C12M 1/40, C12N 9/36, A61K 38/46, C12P 21/02, A61K 38/43, C12N 11/00, C12N 9/18, C12N 9/78, C12N 9/52, C12Q 1/37, C12N 9/66
Мітки: містить, отримання, білка, зшитого, пристрій, зшивальним, кристал, багатофункціональним, спосіб, варіанти, агентом, аспартаму
Формула / Реферат:
1. Кристалл белка, сшитого многофункциональным сшивающим агентом, отличающийся тем, что он обладает устойчивостью к экзогенному протеолизу, такой, что указанный белковый кристалл сохраняет, по меньшей мере, 91% его исходной активности после инкубирования в течение, по меньшей мере, трех часов в присутствии проназы (Pronase™) в концентрации, вызывающей у растворимой формы белка, кристаллизующейся с образованием этого белкового кристалла,...
Спосіб отримання лікарського препарату на основі білка із мікроводоростей

Номер патенту: 22431
Опубліковано: 03.03.1998
Автори: Кірпенко Юрій Олексійович, Бадзюк Валентин Вікторович, Сіренко Лідія Якимівна, Кірпенко Наталія Іванівна, Паршиков Олександр Вікторович
МПК: A61K 38/01
Мітки: мікроводоростей, отримання, препарату, основі, білка, спосіб, лікарського
Формула / Реферат:
1. Способ получения лекарственного препарата на основе белка из водорослей, предусматривающий изготовление белкового гидролизата, воздействие на него реагентом, термическую обработку и сушку, отличающийся тем, что в качестве реагента, воздействующего на белковый гидролизат, используют этиловый спирт с содержанием алкоголя 30-70%, который вводят в гидролизат при достижении им pH 4,5-6,5, после чего определяют массу выпавшего в осадок белка,...
Спосіб отримання гранулоцитарного колонієстимулюючого фактора

Номер патенту: 27263
Опубліковано: 15.09.2000
Автори: Юічі Хірата, Тацумі Ямазакі, Ясуо Секіморі, Сігеказу Нагата, Масаюкі Цутія, Осамі Ямамото
МПК: C12N 15/27
Мітки: спосіб, фактора, колонієстимулюючого, отримання, гранулоцитарного
Формула / Реферат:
(57) Способ получения гранулоцитарного коло-ниестимулирующего фактора, предусматривающий конструирование рекомбинантной плазмид-ной ЦНК, содержащей фрагмент ДНК, кодирующий гранулоцитарный колониестимулирующий фактор, трансформацию штаммов Eschenchia coli или культивируемых клеток животных рекомбинантной плазмидной ДНК, культивирование трансформантов и выделение целевого продукта, отличающийся тем, что конструируют...
Спосіб отримання білка, який має опосередковану фактором vii активність у згортанні крові

Номер патенту: 27262
Опубліковано: 15.09.2000
Автори: Вудбері Річард Г., Хейджен Фредерік С., Грей Чарлз Л., Муррей Марк Дж., Басбі Шарон Дж., Інслі Маргарет І., Беркнер Кетлін Л.
МПК: C12P 21/02, C12N 15/12, C12N 15/70, C12N 9/64
Мітки: спосіб, білка, отримання, фактором, крові, активність, згортанні, опосередковану, має
Текст:
...со структурой, кодируе мой 5'-окончанием к ДНК клона puCVII2115. Для получения дополнительных данных по последовательности, два на номолякарбоксиметилированной малой цепи расщепляли 12 час бычьим химотрипсином (1:100 вес/вес, фер мент:субстрат) в 0,1 М бикарбонате аммония при рН = 7,8 и 37 оС. Полученные фрагменты очищают ЖХВД на колонке с обращенной фазой Micro Pack С18 с использованием указанных выше растворителей в градиенте 0–30...
Спосіб отримання речовини з протипухлинною активністю

Номер патенту: 19910
Опубліковано: 25.12.1997
Автори: Завальнюк Андрій Кіндратович, Ситенко Валентина Казимирівна, Танасієнко Ольга Андріївна, Загадарчук Надія Леонідівна, Хом'як Ольга Григорівна, Лісовенко Галина Степанівна, Затула Дмитро Григорович, Сядро Таміла Андріївна
МПК: A61P 35/00, C12Q 1/02, A61K 35/68, C12P 21/00
Мітки: активністю, отримання, протипухлинною, речовини, спосіб
Текст:
...1 '12991 44 Изобретение относится к медицине, роторном испарителе и очищают. Актива именно к онкологии, и касается леность полученного вещества в конценткарственных веществ, обладающих прорации 0,06 мг/мл составляет 60% гибетивоопухолевой активностью. ли асцитных опухолевых клеток. Целью изобретения является повы5 П р и м е р 2. Бульонную культуру шение специфической активности за Вас. mesentericus АБ-56 культивируют счет использования в...
Попередній патент: Портативний індикатор стану серцево-судинної діяльності людини та моніторна система стану серцево-судинної діяльності людей
Наступний патент: Похідне n-(1,2-цис-2-фторциклопропіл)піридонкарбонової кислоти
Випадковий патент: Запірно-пломбувальний пристрій