Циклоундекадепсипептидні сполуки для лікування інфекцій, викликаних вірусом гепатиту с
Номер патенту: 102414
Опубліковано: 10.07.2013
Автори: Новаролі Цаноларі Лаура, Краббе Рафаель, Муттер Манфред, Ніколя Валері, Вуаньо Грегуар, Лисек Роберт, Гарруст Патрик, Тюрпен Олівьє, Венгер Роланд


























Формула / Реферат
1. Циклоундекадепсипептидна сполука Формули (І)
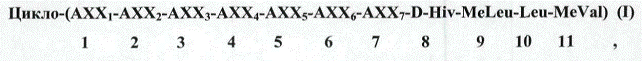
у якій:
АХХ1 являє собою MeBmt, 4-фтор-МеВmt, дигідро-MeBmt, 8-гідрокси-МеВmt;
О-ацетил-MeBmt;
АХХ2 являє собою Abu, Val, Thr, Thr(OMe), Thr(OAc), Thr(OCOCH2CH2CH2OH), Nva, 5-гідрокси-Nva;
AXX3 являє собою D-MeAla, D-3-фтор-МеАlа, D-MeSer, D-MeSer(OAc),
D-MeSer(OCH2CH2OH), D-MeSer(OCH2CH2NEt2), D-MeAsp(OMe);
AXX4 являє собою MeIle, MeMet, MeVal, MeThr, MeThr(OAc), MeAla, EtVal, EtIle, EtPhe, EtTyr, EtThr(OAc), MeThr(OAc), MeTyr, MeTyr(OAc), МеТуr(ОМе), MePhe, MeMet(Ox), де атом сірки метіоніну є сульфоксидним або сульфоновим;
АХХ5 являє собою Leu, Val, Ile;
АХХ6 являє собою MeAla, Sar, MeLeu; і
АХХ7 являє собою Gly, Ala.
2. Сполука за п. 1, яка відрізняється тим, що у Формулі (1)
АХХ1 являє собою MeBmt, 8-гідрокси-МеВmt;
АХХ2 являє собою Abu, Val, Thr, Thr(OMe), Thr(OAc), Thr(OCOCH2CH2CH2OH), 5-гідрокси-Nva;
AXX3 являє собою D-MeAla, D-3-фтор-МеАlа, D-MeSer,D-MeSer(OAc), D-MeAsp(OMe);
AXX4 являє собою MeIle, MeMet, MeMet(Ox), де Ox являє собою -SOMe, -SO2Me, MeVal, EtVal, EtIle, MeTyr; і
AXX5 являє собою Leu, Val, Ile;
AXX6 являє собою MeAla, Sar, MeLeu; і
AXX7 являє собою Gly, Ala.
3. Сполука за будь-яким з пп. 1 або 2, яка відрізняється тим, що
АХХ1 являє собою MeBmt;
АХХ2 являє собою Abu, Val, Thr;
AXX3 являє собою D-MeAla;
АХХ4 являє собою MeIle, MeVal, EtVal;
АХХ5 являє собою Leu, Val, Ile;
АХХ6 являє собою MeAla, MeLeu, Sar; і
АХХ7 являє собою Gly, Ala.
4. Сполука за будь-яким з пп. 1-3, що має наступну формулу:
цикло-(MeBmt-Abu-D-MeAla-MeVal-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal)
(Iа)
цикло-(MeBmt-Abu-D-MeAla-EtVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal)
(Ib)
цикло-(MeBmt-Val-D-MeAla-MeVal-Ile-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal)
(Іс)
цикло-(MeBmt-Abu-D-MeAla-MeVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal)
(Id)
цикло-(MeBmt-Abu-D-MeAla-MeVal-Val-MeAla-Ala-D-Hiv-MeLeu-Leu-MeVal)
(Iе)
цикло-(MeBmt-Val-D-MeAla-MeVal-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal)
(If)
цикло-(MeBmt-Abu-D-MeAla-MeVal-Ile-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal)
(Ig)
цикло-(MeBmt-Vаl-D-МеАlа-EtVal-Lеu-MeLeu-Аlа-D-Нiv-MeLeu-Lеu-MeVаl)
(Ih)
цикло-(MeBmt-Abu-D-МеАlа-МеIlе-Lеu-МеLеu-Glу-D-Ніv-МеLеu-Lеu-МеVаl)
(Iі)
цикло-(MeBmt-Val-D-MeAla-MeVal-Val-Sar-Gly-D-Hiv-MeLeu-Leu-MeVal)
(Ik)
5. Фармацевтична композиція для запобігання або лікування інфекцій, що викликаються вірусом гепатиту С (ВГС), або розладів, що викликаються ВГС, яка містить сполуку за будь-яким з пп. 1-4 разом з одним або більше фармацевтично прийнятними розріджувачами або носіями.
6. Фармацевтична комбінація, що містить щонайменше а) перший засіб, що складається із сполуки за будь-яким з пп. 1-4 або фармацевтичної композиції за п. 5, і b) другий засіб, що має здатність протидіяти реплікації ВГС.
7. Фармацевтична комбінація за п. 6 для застосування для запобігання або лікування інфекцій, що викликаються вірусом гепатиту С, або розладів, що викликаються ВГС.
8. Сполука за будь-яким з пп. 1-4 як лікарський засіб.
9. Сполука за будь-яким з пп. 1-4 як противірусний засіб.
10. Застосування сполуки за будь-яким з пп. 1-4 для одержання лікарського засобу для лікування або запобігання інфекцій, що викликаються вірусом гепатиту С, або розладів, що викликаються ВГС.
11. Спосіб запобігання або лікування інфекцій, що викликаються вірусом гепатиту С, або розладів, що викликаються ВГС, у суб'єкта, що цього потребує, який включає введення зазначеному суб'єкту терапевтично ефективної кількості сполуки за будь-яким з пп. 1-4 або фармацевтичної композиції за п. 5.
12. Спосіб пригнічення реплікації ВГС у пацієнта, який цього потребує, що включає введення зазначеному суб'єкту терапевтично ефективної кількості сполуки за будь-яким з пп. 1-4 або фармацевтичної композиції за п. 5.
13. Спосіб за будь-яким з пп. 11-12, що включає спільне введення одночасно або послідовно терапевтично ефективної кількості сполуки за будь-яким з пп. 1-4 або фармацевтичної композиції за п. 5 і щонайменше другого засобу, вибраного із засобу, що має здатність протидіяти реплікації ВГС.
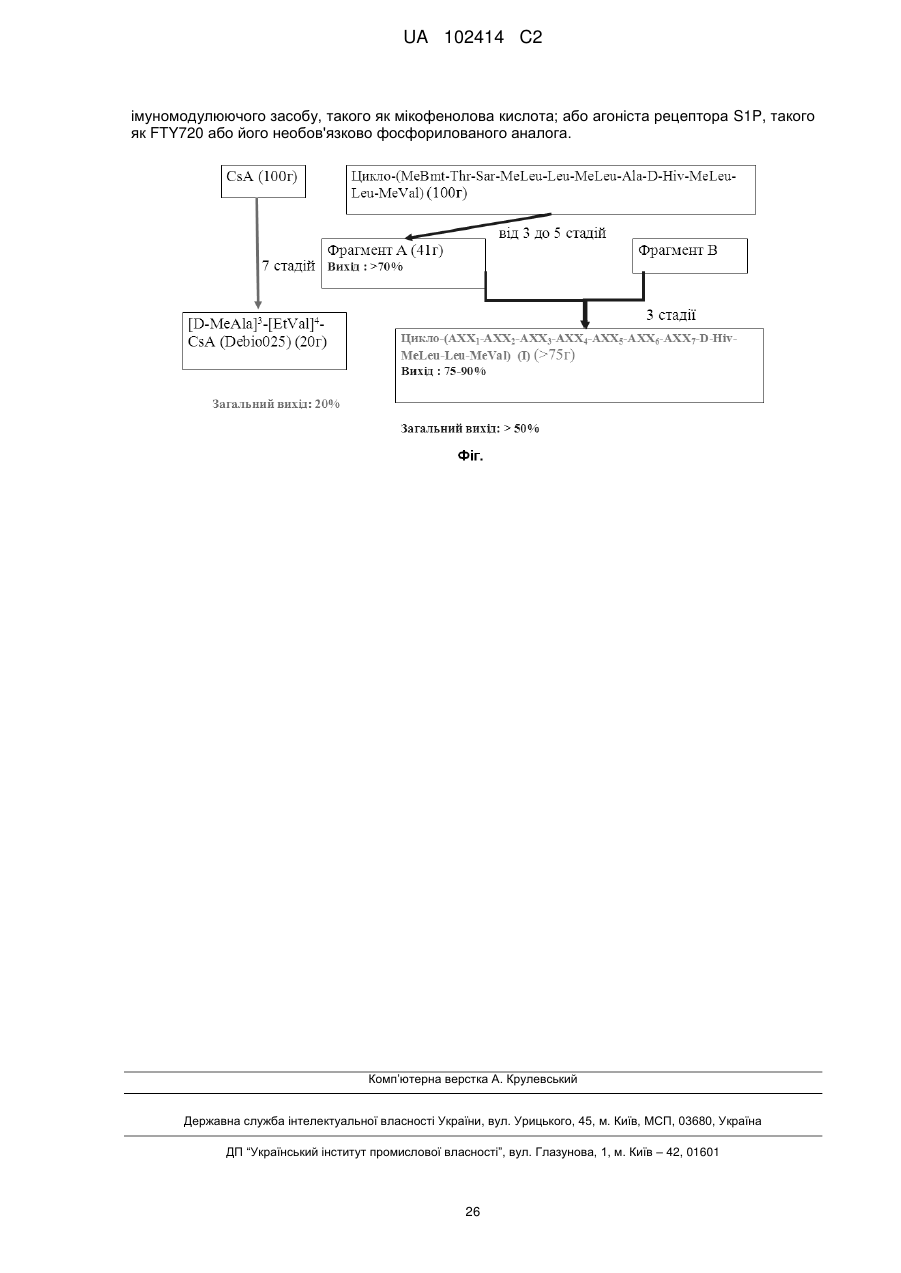
14. Спосіб за п. 13, який відрізняється тим, що зазначений засіб, який має спрямовані проти ВГС властивості, вибраний з групи, що складається з інтерферону, такого як інтерферон альфа-2а або інтерферон альфа-2b; або інтерферону, кон'югованого з водорозчинним полімером або людським альбуміном; противірусного засобу, такого як рибавірин, ламівудин, NV08 або NM283; інгібітора факторів, що кодуються ВГС, наприклад, протеази NS3-4A, хелікази або РНК-полімерази; протифіброзного засобу, такого як похідне N-феніл-2-піримідинаміну; імуномодулюючого засобу, такого як мікофенолова кислота; або агоніста рецептора S1P, такого як FTY720 або його необов'язково фосфорильованого аналога.
Текст
Реферат: Цей винахід належить до нових циклоундекадепсипептидних сполук і застосування зазначених сполук як лікарського засобу, зокрема як противірусних засобів, ще конкретніше, для UA 102414 C2 (12) UA 102414 C2 запобігання або лікування інфекцій, що викликаються вірусом гепатиту С, або розладів, що викликаються вірусом гепатиту С. UA 102414 C2 5 10 15 20 25 30 35 40 45 50 55 Цей винахід відноситься до циклоундекадепсипептидних сполук. Винахід також відноситься до застосування зазначених сполук як лікарського засобу, зокрема, як противірусних засобів, ще конкретніше, для запобігання або лікування інфекцій, що викликаються вірусом гепатиту С, або, що викликаються вірусом гепатиту С (ВГС) розладів. РІВЕНЬ ТЕХНІКИ ВГС був клонований і охарактеризований близько 15 років тому Ху (Choo) і співробітниками (див. Science 244, (1989), 359-362). ВГС належить до сімейства Flaviviridae і містить оточений оболонкою нуклеокапсид і геном у вигляді одноланцюгової РНК позитивної полярності (див. Bartenschlager et al., Antiviral Res. 60, (2003), 91-102). ВГС передається головним чином через кров, препарати крові та за допомогою вертикальної передачі під час вагітності. Введення діагностичних тестів для скринігу препаратів крові значно знизило частку нових інфекцій. Проте, ВГС залишається серйозною медичною проблемою. У цей час близько 170 мільйонів людей інфіковані ВГС. Первісний перебіг інфекції звичайно легкий. Однак імунна система найчастіше нездатна усунути вірус, і люди з хронічними інфекціями піддаються високому ризику виникнення цирозу печінки і гепатоцелюлярної карциноми (див. Poynard et al., Lancet 349, (1997), 825-832). Вакцини від ВГС не існує, а можливі способи лікування дуже обмежені (див. Manns et al., Indian J. Gastroenterol. 20 (Suppl. 1), (2001), C47-51; Tan et al., Nat. Rev. Drug Discov. 1, (2002), 867-881). Були ідентифіковані сполуки, які міцно зв'язуються з циклофіліном, але ці сполуки мали імуносупресивну дію (Nakawaga M et al., Biochemical and biophysical research communications, Academic press Inc. Orlando, FL, US, Vol. 313, №1, 2 January 2004, pages 42-47, XP004479114). У зв'язку з цим протягом останніх 20 років був проведений ряд досліджень в галузі медичної хімії з метою ідентифікації неімуносупресивних сполук, таких як NIM 811, або інших сполук, що демонструють також високу активність відносно пригнічення реплікації вірусу імунодефіциту людину 1 (ВІЛ-1) і по суті, що не мають імуносупресивної активності, див. WO 00/01715 (Wenger et al.; DEBIOPHARM SA) і Tetrahedron Lett., 41, (2000), 7193-6. Було виявлено, що неімуносупресивні сполуки, які зв'язують циклофілін, мають інгібуючу дію відносно вірусу гепатиту С (ВГС). Хронічна інфекція ВГС, яка була ідентифікована як основний збудник гепатиту, що не є гепатитом А або B, як вважають, тісно пов'язана із захворюваннями печінки, такими як хронічний гепатит, цироз печінки або гепатоцелюлярна карцинома. Розвиток цих захворювань печінки є основною проблемою суспільної охорони здоров'я. Ефективна терапія проти ВГС обмежується терапією інтерфероном або комбінацією інтерферону та рибавірину. Однак оскільки приблизно у половини пацієнтів з ВГС, яких лікують за допомогою зазначених відомих засобів, вірус не знищується, як і раніше існує більша потреба в альтернативних засобах проти ВГС. Циклоундекапептиди, що мають здатність впливати на пригнічення реплікації вірусу гепатиту С (ВГС), уже відомі з WO 2005/021028 (Novartis Pharma GMBH) і WO 2006/038088 (DEBIOPHARM SA). В Hanssons M. J. et al. Journal of Bioenergetics and biomembranes, plenum publishing, New York, NY, US, Vol. 36, №4, 1 August 2004, pages 407-413 (XP002330699) повідомляється про більш високу ефективність пригнічення переходу мітохондріальної проникності (mPT) сполуками, описаними в WO 2006/038088. Однак, незважаючи на те, що деякі з цих пептидів у цей час проходять клінічні випробування, як і раніше залишається потреба в розробці нових противірусних засобів, що мають задовільну здатність пригнічувати реплікацію, зокрема, реплікацію ВГС, поряд з поліпшеним фармакокінетичним профілем (наприклад, профіль пригнічення переносника в печінці і пов'язані з цим взаємодії лікарських засобів) і поліпшеним токсикологічним профілем (наприклад, відсутністю імуносупресивної активності та виникаючих у результаті побічних ефектів). Відповідно, завданням цього винаходу є створення додаткових неімуносупресивних сполук для застосування для запобіганні або лікування інфекцій, що викликаються вірусом гепатиту С, або розладів, що викликаються ВГС. Ці сполуки мають несподівану перевагу у вигляді зниження або можливих побічних ефектів, пов'язаних з пригніченням переносників у печінці, або можливих взаємодій лікарських засобів, пов'язаних з пригніченням переносників у печінці, або і того, й іншого. Крім того, завданням цього винаходу також є одержання сполук, які легше синтезувати, зокрема, у промисловому масштабі. КОРОТКИЙ ОПИС ВИНАХОДУ Таким чином, завданням цього винаходу було одержання нових противірусних засобів, що мають вищезгадані поліпшені властивості, і зненацька було виявлено, що цим вимогам задовольняють циклоундекадепсипептидні сполуки, визначені Формулою (I): 60 1 UA 102414 C2 5 10 15 20 25 30 35 40 45 50 у якій: AXX1 являє собою MeBmt, 4-фтор-MeBmt, дигідро-MeBmt, 8-гідрокси-MeBmt; O-ацетилMeBmt; AXX2 являє собою Abu, Val, Thr, Thr(OMe), Thr(OAc), Thr(OCOCH2CH2CH2OH), Nva, 5гідрокси-Nva; AXX3 являє собою D-MeAla, D-3-фтор-MeAla, D-MeSer, D-MeSer(OAc), D-MeSer(O-CH2CH2OH), D-MeSer(OCH2CH2NEt2), D-MeAsp(OMe); AXX4 являє собою MeIle, MeMet, MeMet(Ox), де Ox означає, що атом сірки метіоніну є сульфоксидним або сульфоновим, MeVal, MeThr, MeThr(OAc), MeThr(OtBu), MeThr(OMe), MeAla, MeIle, MeThr, EtVal, EtIle, EtPhe, EtTyr, EtThr(OAc), MeThr(OAc), MeTyr, MeTyr(OAc), MeTyr(OMe), MePhe; AXX5 являє собою Leu, Val, Ile, Ala; AXX6 являє собою MeAla, Sar, MeLeu; і AXX7 являє собою Gly, Ala. Іншим завданням винаходу є одержання фармацевтичної композиції для запобігання або лікування інфекцій, що викликаються вірусом гепатиту С, або розладів, що викликаються ВГС Формули, що містить сполуки (I) разом з одним або більше фармацевтично прийнятними розріджувачами або носіями. Додатковим завданням винаходу є одержання фармацевтичної комбінації, що містить щонайменше а) перший засіб, що складається зі сполук Формули (I) або фармацевтичної композиції, і b) другий засіб, що має властивості перешкоджати реплікації ВГС. Сполуки відповідно до Формули (I) можна застосовувати для одержання лікарського засобу для лікування або запобігання інфекцій, що викликаються вірусом гепатиту С, або розладів, що викликаються ВГС. Ще одним завданням винаходу є забезпечення способу запобігання або лікування інфекцій, що викликаються вірусом гепатиту С, або розладів, що викликаються ВГС, у суб’єкта, що цього потребує, при цьому зазначений спосіб включає введення зазначеному суб'єкту терапевтично ефективної кількості сполук Формули (I) або фармацевтичної композиції відповідно до винаходу. Альтернативно, також запропонований спосіб пригнічення реплікації ВГС у пацієнта, який цього потребує, що включає введення зазначеному суб’єкту терапевтично ефективної кількості сполук Формули (I) або фармацевтичної композиції згідно з винаходом. КОРОТКИЙ ОПИС КРЕСЛЕНЬ З метою кращого розуміння винаходу і для того, щоб показати, як цей винахід може бути реалізований, у цій заявці будуть тільки як приклад наведені конкретні варіанти реалізації, способи і процеси згідно з цим винаходом з посиланням на прикладені креслення, на яких: Фігура 1 ілюструє хімічний синтез сполук згідно з винаходом у порівнянні зі сполуками згідно 3 4 з WO 2006/038088, тобто [D-MeAla] -[EtVal] -CsA (Debio 025). ДОКЛАДНИЙ ОПИС ВИНАХОДУ Якщо не зазначено інше, усі технічні та наукові терміни, що використовуються в цій заявці, мають ті ж значення, які звичайно має на увазі фахівець в галузі техніки, до якої належить цей винахід. Незважаючи на те, що методи і матеріали, подібні з або еквівалентні тим, які описані в цій заявці, можуть бути застосовані на практиці або при дослідженні цього винаходу матеріали, придатні методи і матеріали описані нижче. Повний зміст усіх публікацій, заявок на патенти, патентів та інших посилань, згаданих у цій заявці, включено в цей опис у всій повноті за допомогою посилання. У випадку протиріччя, цей опис, включаючи визначення, є вирішальним. Крім того, матеріали, методи і приклади є лише ілюстративними та не мають на увазі обмеження. У цій заявці з метою полегшення розуміння цього винаходу дані наступні визначення. Термін “включати” звичайно застосовують у значенні “містити”, інакше кажучи, допускаючи присутність одної(го) або більше ознак або компонентів. Завданням цього винаходу є одержання циклоундекадепсипептидної сполуки Формули (I) 55 2 UA 102414 C2 5 10 15 20 25 30 35 40 45 50 55 60 у якій: AXX1 являє собою MeBmt, 4-фтор-MeBmt, дигідро-MeBmt, 8-гідрокси-MeBmt; O-ацетилMeBmt; AXX2 являє собою Abu, Val, Thr, Thr(OMe), Thr(OAc), Thr(OCOCH2CH2CH2OH), Nva, 5гідрокси-Nva; AXX3 являє собою D-MeAla, D-3-фтор-MeAla, D-MeSer, D-MeSer(OAc), D-MeSer(OCH2CH2OH), D-MeSer(OCH2CH2NEt2), D-MeAsp(OMe); AXX4 являє собою MeIle, MeMet, MeVal, MeThr, MeThr(OAc), MeAla, EtVal, EtIle, EtPhe, EtTyr, EtThr(OAc), MeThr(OAc), MeTyr, MeTyr(OAc), MeTyr(OMe), MePhe, MeMet(Ox), де атом сірки метіоніну є сульфоксидним або сульфоновим; AXX5 являє собою Leu, Val, Ile; AXX6 являє собою MeAla, Sar, MeLeu; і AXX7 являє собою Gly, Ala. Згідно з визначенням, прийнятим Міжнародними союзом теоретичної і прикладної хімії (IUPAС), циклодепсипептиди являють собою природні або синтетичні сполуки, що містять послідовності залишків аміно- і гідроксилкарбонових кислот (звичайно -аміно- і гідроксикислоти), і залишки замкнені в кільце (див. IUPAC Compendium of Chemical Terminology, nd 2 Edition (1997). Такі циклодепсипептиди представляють як гетеродетні пептиди, у яких щонайменше один амідний зв'язок замінений на складноефірний зв'язок (див. J. Peptide Sci. 10:115-8 (2004)). Сполуки згідно з цим винаходом містять одинадцять залишків, причому десять з них являють собою -амінокислоти, а один – -гідроксикислоту. Ця -гідроксикислота являє собою (2R)-2-гідрокси-3-метилбутанову кислоту, також відому як D--гідроксиізовалеріанова кислота і позначувану скорочено як H-D-HIV-OH. У Формулі (I) ця гідроксикислота перебуває в положенні 8. По кінцевий карбоксильній групі вона утворює амідний зв'язок з аміногрупою -амінокислоти в положенні 9, а саме, N-метиллейцину, і за рахунок гідроксильної групи – складноефірний зв'язок з карбоксильною групою -амінокислоти в положенні 7, а саме аланіну або гліцину. Зазначені -амінокислоти Формули (I) названі з використанням трилітерного коду, що звичайно використовується для позначення амінокислот, і їх конфігурація є L-конфігурацією, якщо не зазначено інше. Нумерація залишків починається з AXX1, що представляє собою Nметил-(4R)-4-[(E)-2-бутеніл]-4-метил-L-треонін або MeBmt і його структурні похідні, як визначено вище. Коли алкільна група, така як метильна група Me або етильна група Et, з'являється перед абревіатурою амінокислоти, це означає, що така алкільна група приєднана до аміногрупи зазначеного амінокислотного залишку. Перевага сполук згідно з цим винаходом може полягати у введенні складноефірного зв’язку всередину макроциклічного кістяка у порівнянні з регулярним циклічним амідним кістяком відповідних циклоундекапептидних сполук, описаних у відомому рівні техніки. Не заглиблюючись у теорію, вважають, що заміна амідного зв’язку на складноефірний між амінокислотою AXX7 у положенні 7 і D-Hiv у положенні 8 дозволяє значно впливати на конформаційні та фізико-хімічні властивості, такі як підвищена конформаційна гнучкість (в P.J. Flory, Statistical Mechanics of Chain Molecules, Hanser Publishers, NY, 1988) і ліпофільність, а також відсутність донорного водневого зв'язку. Вважають, що ці структурні особливості трансформуються у виражені відмінності у фізикохімічних, фармакокінетичних і біологічних властивостях сполук згідно з цим винаходом у порівнянні з класом аналогів, отриманих з циклоспорину А (CsA). Можливе розумне пояснення спостережуваної більш високої стійкості до замін амінокислот у систематичних дослідженнях взаємозв'язку структура-активність (SAR) може полягати у збільшенні конформаційного простору відносно біоактивної конформації (V. Mikol et al., J. Mol. Biol. (1998) 283, 451-461). На противагу циклоундекапептидним аналогам, відомим з рівня техніки, єдина або множинні заміни в положеннях у фрагменті між амінокислотою у положенні 2 і амінокислотою у положенні 7 природного циклоундекадепсипептиду, такого як отриманого згідно або патенту США № 5116816, Приклад 2, або WO 02/092033, Приклад 4, Стадія 4-1, приводять до збереження високої здатності зв'язуватися з Циклофіліном, що очевидно зі списку сполук, які задовольняють вимогам цього винаходу. Насамперед, спостерігали підвищення здатностей зв'язуватися з циклофіліном A (Сyp A) до 2-4 разів для сполук згідно з винаходом у порівнянні зі 3 4 сполуками згідно з WO 2006/038088, тобто [D-MeAla] -[EtVal] -CsA (Debio 025). Крім поліпшеного активного профілю (див. Приклади 5 і 6), новий клас сполук також дозволяє запропонувати поліпшений спосіб одержання, особливо в промисловому масштабі. Винахід також включає хімічні модифікації сполук Формули (I) з метою продовження їх часу 3 UA 102414 C2 5 10 15 20 25 30 35 40 45 50 55 60 перебування в кровотоці. Приклади придатних похідних поліетиленгліколю, що мають цю властивість, описані в, наприклад, у заявці на патент США 2005171328 (NEKTAR THERAPEUTICS AL CORP) або патенті США № 6713454 (NOBEX CORP). Ще краще, сполуки згідно з цим винаходом визначені Формулою (I), у якій: AXX1 являє собою MeBmt, дигідро-MeBmt, 8-гідрокси-MeBmt; AXX2 являє собою Abu, Val, Thr, Thr(OMe), Thr(OAc), Thr(OCOCH2CH2CH2OH), 5-гідроксиNva; AXX3 являє собою D-MeAla, D-3-фтор-MeAla, D-MeSer, D-MeSer(OAc), D-MeAsp(OMe); AXX4 являє собою MeIle, MeMet, MeMet(Ox), де Ox означає, що атом сірки метіоніну є сульфоксидним або сульфоновим, MeVal, EtVal, EtIle, MeTyr, MeTyr(OAc), MeTyr(OMe), MeThr, MeThr(OtBu), MeThr(OAc), MeThr(OMe), MePhe; AXX5 являє собою Leu, Val, Ile, Ala; AXX6 являє собою MeAla, Sar, MeLeu; і AXX7 являє собою Gly, Ala. Ще краще, сполуки Формули (I) визначені наступними значеннями: AXX1 являє собою MeBmt, 8-гідрокси-MeBmt; AXX2 являє собою Abu, Val, Thr, Thr(OMe), Thr(OAc), Thr(OCOCH2CH2CH2OH), 5-гідроксиNva; AXX3 являє собою D-MeAla, D-3-фтор-MeAla, D-MeSer, D-MeSer(OAc), D-MeAsp(OMe); AXX4 являє собою MeIle, MeMet, MeMet(Ox), где Ox являє собою -SOMe, -SO2Me, MeVal, EtVal, EtIle, MeTyr; і AXX5 являє собою Leu, Val, Ile; AXX6 являє собою MeAla, Sar, MeLeu; і AXX7 являє собою Gly, Ala. В окремому варіанті реалізації винаходу сполуки Формули (I)визначені наступними значеннями: AXX1 являє собою MeBmt; AXX2 являє собою Abu, Val, Thr; AXX3 являє собою D-MeAla; AXX4 являє собою MeIle, MeVal, EtVal; AXX5 являє собою Leu, Val, Ile; AXX6 являє собою MeAla, MeLeu, Sar; і AXX7 являє собою Gly, Ala. Відповідно до одного з найкращих варіантів реалізації винаходу, сполуки Формули (I) представлені наступними формулами: цикло-(MeBmt-Abu-D-MeAla-MeVal-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-EtVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Val-D-MeAla-MeVal-Ile-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeVal-Val-MeAla-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Val-D-MeAla-MeVal-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeVal-Ile-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Val-D-MeAla-EtVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeIle-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Val-D-MeAla-MeVal-Val-Sar-Gly-D-Hiv-MeLeu-Leu-MeVal) (Ia) (Ib) (Ic) (Id) (Ie) (If) (Ig) (Ih) (Ii) (Ik) Перераховані вище сполуки відповідають повній оптимізації найбільш важливих критеріїв цього винаходу, а саме вдосконаленню хімічного синтезу, фармакологічних властивостей, фармакокінетичних і токсикологічних профілів. Примітно, що зазначені сполуки показують набагато меншу ефективність, у порівнянні з Debio 025, пригнічення переносників у печінці, таких як MRP2 або OATP1B1. Таким чином, ці сполуки, як очікують, зменшать або можливі несприятливі ефекти (наприклад, гіпербілірубінемію), або можливі взаємодії лікарських засобів (наприклад, з інгібіторами гідроксиметилглутарил-кофермент А (HMG-CoA) редуктази, такими як аторвастатин), пов'язані з пригніченням переносників у печінці, або навіть і те, й інше. Додаткові сполуки згідно з винаходом являють собою наступні: цикло-(MeBmt-Val-D-MeAla-MeVal-Val-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeThr(OtBu)-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) 4 UA 102414 C2 5 10 15 цикло-(MeBmt-Val-D-MeAla-EtVal-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeVal-Leu-Sar-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Val-D-MeAla-MeVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Thr-D-MeAla-MeVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Val-D-MeAla-MeVal-Ile-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Val-D-MeAla-MeIle-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeIle-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Val-D-MeAla-MeVal-Val-MeAla-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Val-D-MeAla-MeVal-Leu-MeAla-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeThr-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Thr(OMe)-D-MeAla-MeVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-EtVal-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeTyr-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Val-D-MeAla-MeVal-Leu-Sar-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Thr-D-MeAla-EtVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Val-D-MeAla-MeVal-Val-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeVal-Ala-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeVal-Ile-MeAla-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Val-D-MeAla-MeVal-Val-Sar-Gly-D-Hiv-MeLeu-Leu-MeVal) 20 Інші сполуки, що представляють інтерес, визначені наступними формулами: 25 30 35 40 45 50 55 60 цикло-(MeBmt-Thr(OMe)-D-MeAla-EtVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-((8-гідрокси)MeBmt-Abu-D-MeAla-MeVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-(5-OH)Nva-D-MeAla-MeVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Thr(OR) (R = -COCH3, -COCH2CH2CH2OH)-D-MeAla-MeVal-Leu-MeLeu-Ala-DHiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeVal-Val-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Val-D-MeSer-MeVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeSer-MeVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeSer(OAc)-MeVal-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAsp(OMe)-MeVal-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Val-D-(3-фтор)MeAla-MeVal-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-(3-фтор)MeAla-MeIle-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeMet-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeMet(Ox)(Ox = -SOMe, -SO2Me)-Leu-MeLeu-Gly-D-Hiv-MeLeuLeu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MePhe-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeTyr(OR) (R = Ac, Me)-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-MeThr(OR) (R = Me, Ac)-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-(3-фтор)MeAla-EtVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-(MeBmt-Abu-D-MeAla-EtIle-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-((дигідро)MeBmt-Abu-D-MeAla-MeVal-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) цикло-((8-гідрокси)MeBmt-Abu-D-MeAla-EtVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) цикло-((8-гідрокси)MeBmt-Val-D-MeAla-EtVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) Сполуки цього винаходу можуть бути отримані за допомогою класичних пептидних способів (пептидний синтез у розчині або твердофазний синтез; в Houben-Weyl, Methods of Organic Chemistry, Vol. E 22d, Ed.-in-chief: M. Goodman, Thieme Verlag, Stuttgart, 2003) і органічній хімії або біотехнології, наприклад, застосовуючи хімічні методи, як описав Wenger в Helv. Chim. Acta, 67, 502-25, 1984 або в Helv. Chim. Acta, 66, 2672-702 (1983), і використовуючи HATU як реагент для реакції сполучення, описаного в Rich, D.H. et al, Comparative studies of the coupling of Nmethylated, sterically hindered amino acids during SPPS, Tetr. Letters. 35, 5981-5984 (1994). Наприклад, одна з можливих загальних схем складається з одержання двох фрагментів, а саме, Фрагмента А і Фрагмента B, що містять придатні залишки з, якщо необхідно, придатними захисними групами і групами, що активують, і, на останніх стадіях одержання, зв'язування їх разом з одержанням ундекапептиду, який потім циклізують у циклоундекадепсипептид. Наприклад, Фрагмент (Ax) може бути наступним: H-D-Hiv-MeLeu-Leu-MeVal-AXX1-OH (Ax) 5 UA 102414 C2 і фрагмент (Bx) наступним: 5 10 15 20 25 30 35 40 45 50 55 60 H-AXX2-AXX3-AXX4-AXX5-AXX6-AXX7-OR, (R являє собою алкільну групу) (Bx), тоді Ax і Bx поєднують в ундекадепсипептид Ax-Bx,H-D-Hiv-MeLeu-Leu-MeVal-AXX1-AXX2AXX3-AXX4-AXX5-AXX6-AXX7-OR (R = алкіл, Н), і фінальна стадія являє собою макролактонізацію. Фрагмент А, який містить залишок -гідроксикислоти D-Hiv, може бути отриманий розкладанням природного циклоундекадепсипептиду (а саме Цикло-(MeBmt-Thr-Sar-MeLeu-LeuMeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal), одержання зазначеного циклоундекадепсипептиду описане або в патенті США № 5116816, Приклад 2, або в WO 02/092033, Приклад 4, Стадія 4-1. Перевага сполук згідно з винаходом перед відомим рівнем техніки полягає не тільки в його активному профілі (див. Приклади 5 і 6), але також і в поліпшеному способі одержання, особливо в промисловому масштабі. 1. При застосуванні як вихідних речовин природних сполук (CsA і Цикло-(MeBmt-Thr-SarMeLeu-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal), див. Фігуру 1, загальні виходи синтезу циклоундекадепсипептидних аналогів перевищують 50 % у порівнянні з 20 % для DEBIO 025 3 4 ([D-MeAla] -[EtVal] -CsA, як описано в WO 2006/038088, DEBIOPHARM SA). 2. Синтез дорогого дипептидного похідного Boc-D-MeAla-EtVal-OH (що включає 4 хімічні стадії) не потрібен. Крім того, деякі зі сполук містять С-кінцевий гліцин (Ia, Ic, If, Ig, Ii, Ik), який полегшує останню стадію синтезу (макролактонізація, немає епімеризації). 3. Вартості реагентів і вихідних сполук значно нижче у випадку сполук згідно з винаходом. Слід зазначити, що “реагент Меервейна”, що застосовується для реакції відкриття кільця CsA, не потрібен. 4. Фрагмент B (гексапептид AXX2-AXX7) може бути ефективно отриманий за допомогою стандартного твердофазного пептидного синтезу із застосуванням комерційних вихідних сполук (див. Фігуру 1). 5. При оптимізації одержання Фрагмента А з вихідної природної сполуки, також як і конденсації (фрагмента А с фрагментом B), макроциклізації і кінцевих стадій очищення, загальний вихід для одержання фармакологічно цікавих сполук може доходити до 80%. 6. У цілому, зазначені аспекти приводять до гарної ефективності і, отже, вартості продуктів, особливо відносно синтезу сполук згідно з винаходом у промисловому масштабі. Здатність сполук згідно з цим винаходом пригнічувати реплікацію ВГС була показана в аналізі із застосуванням реплікону, а істотну відсутність імуносупресивної активності оцінили в аналізі проліферативної активності Т-клітин (див. Приклади 5 і 6). “Неімуносупресивна” сполука згідно з цим винаходом слід розуміти як сполука, яке щонайменше в 30 разів менш імуносупресивна, ніж циклоспорин А (CsA) (визначають як відношення IC50 (половина концентрації, що викликає максимальне пригнічення) сполуки до IC 50 Циклоспорину А (IC50 сполуки/IC50 CsA)), краще менше ніж в 150 разів, менше навіть ніж в 200 разів і краще навіть менше ніж в 300 разів. В ідеалі, сполуки винаходу мають неімуносупресивну активністю in vitro, що складає щонайменше дві логарифмічні різниці з CsA в аналізі викликаної Конканаваліном А проліферативної активності Т-клітин. Відповідно, сполуки згідно з цим винаходом запропоновано застосовувати як лікарський засіб, зокрема, як противірусний засіб, а ще конкретніше, для запобігання або лікування інфекцій, що викликаються вірусом гепатиту С, або розладів, що викликаються ВГС. Інфекції гепатиту С або розладу, що викликаються ВГС, являють собою, наприклад, хронічний гепатит, цироз печінки або рак печінки, наприклад, гепатоцелюлярну карциному. Сполуки згідно з цим винаходом можуть також бути застосовані, наприклад, як профілактичне лікування немовлят, народжених від інфікованих ВГС матерів, або працівників охорони здоров'я, підданих дії вірусу, або реципієнтів трансплантатів, наприклад, реципієнтів трансплантатів органу або тканини, наприклад, трансплантата печінки, з метою усунення можливої повторної інфекції ВГС після трансплантації. “Лікування” відноситься і до терапевтичного лікування, і до профілактичних або попереджувальних заходів. Ті, хто потребує лікування включають тих, хто вже страждає розладом, також як і тих, у кого захворювання потрібно попередити. Отже, ссавці, яких потрібно лікувати згідно з цим винаходом, можуть бути продіагностовані як такі, що мають розлад або можуть бути схильні або сприйнятливі до розладу. “Ссавець” для цілей лікування відноситься до будь-якої тварини, що класифікується як ссавець, включаючи людей, домашніх і сільськогосподарських тварин або домашніх вихованців, таких як собаки, коні, кішки, корови, мавпи і т.д. Краще, ссавець являє собою людину. 6 UA 102414 C2 5 10 15 20 25 30 35 40 45 50 55 60 Термін “терапевтично ефективна кількість” відноситься до кількості лікарського засобу, ефективної для лікування захворювання або розладу у ссавця. Сполуки згідно з цим винаходом можуть бути застосовані, наприклад, перорально пацієнтам, що потребують цього, наприклад, включені в попередньо концентровану мікроемульсію. Сполуки згідно з цим винаходом, їх фармацевтично прийнятні солі та їх проліки, у відповідних випадках, можуть бути застосовані у формі фармацевтичної композиції, у якій вони об'єднані з фармацевтично прийнятним ад’ювантом, розріджувачем або носієм, з метою запобігання або лікування інфекцій, що викликаються вірусом гепатиту С, або розладів, що викликаються ВГС. Згідно з цим винаходом також запропонована фармацевтична композиція, що містить сполуки згідно з цим винаходом або його фармацевтично прийнятну сіль, як визначено вище в цій заявці, разом з фармацевтично прийнятним ад’ювантом, розріджувачем або носієм. Що стосується придатних наповнювачів, розріджувачів і ад’ювантов, можна зробити посилання на стандартну літературу, що їх описує, наприклад, на главу 25.2 тому 5 джерела “Comprehensive Medicinal Chemistry”, Pergamon Press 1990 і на “Lexikon der Hilfsstoffe für Pharmazie, Kosmetik und angrenzende Gebiete”, автор H.P. Fiedler, Editio Cantor, 2002. Сполуки згідно з цим винаходом можуть також бути поміщені в мікрокапсули, отримані, наприклад, за допомогою методів коацервації або міжфазної полімеризації, наприклад, у мікрокапсули на основі гідроксиметилцелюлози або желатину і поліметилметацилатні мікрокапсули, відповідно, у колоїдних системах доставки лікарських засобів (наприклад, ліпосомах, альбумінових мікросферах, мікроемульсіях, наночастинках і нанокапсулах) або в макроемульсіях. Такі методи запропоновані в Remington’s Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980). Можуть бути отримані препарати з уповільненим вивільненням. Придатні приклади препаратів з уповільненим вивільненням включають напівпроникні матриці з твердих гідрофобних полімерів, що містять сполуки згідно з цим винаходом, матриці яких перебувають у вигляді виробів певної форми, наприклад, плівок або мікрокапсул. Приклад матриць з уповільненим вивільненням включають поліефіри, гідрогелі (наприклад, полі(2гідроксиетилметакрилат) або полівініловий спирт), полілактиди (патент США № 3773919), співполімер L-глутамінової кислоти і [гамма]-етил-L-глутамату, етилен-вінілацетат, що не розкладається, сополімери молочної та гліколевої кислот, що розкладаються, як такі, що застосовуються для одержання лікарського засобу LUPRON DEPOT(TM) (ін’єкційні мікросфери, що складені з сополімера молочної та гліколевої кислот і леупролида ацетату), і поліD-(-)-3гідроксиолійну кислоту. Добова доза згідно з цим винаходом буде за необхідністю варіюватися залежно від організму, що підлягає лікуванню, конкретного способу застосування і тяжкості та виду захворювання, що зазнає лікування. Відповідно, оптимальне дозування може бути визначено практикуючим лікарем, що проводить лікування конкретного пацієнта. Фармацевтичні композиції згідно з цим винаходом можуть бути отримані у вигляді кремів, гелів, розчинів, мазей, суспензій або пластирів і.т.д. у випадку місцевого застосування; для застосування шляхом інгаляції, наприклад, у вигляді аерозолів або сухих порошків; для перорального введення, наприклад, у формі таблеток, капсул, гелів, сиропів, суспензій, розчинів, порошків або гранул; для ректального або вагінального введення, наприклад, у вигляді супозиторій; або для парентеральної ін'єкції (включаючи внутрішньовенну, підшкірну, внутрішньом'язову, внутрішньосудинну або вливання) у вигляді стерильного розчину, суспензії або емульсії. Активне сполука згідно з винаходом може бути введена будь-яким традиційним способом. Вона може бути введена парентерально, наприклад, у формі розчинів для ін'єкцій або суспензій, або у формі складів-депо для ін'єкцій. Краще, вона може бути введена перорально у формі питних розчинів або суспензій, таблеток або капсул. Фармацевтичні композиції для перорального введення, що містять циколундекадепсипептидну сполуку згідно з винаходом, описані в Прикладах. Як презентовано в прикладах, зазначені фармацевтичні композиції, як правило, містять циколундекадепсипептидну сполуку згідно з винаходом і одну або більше фармацевтично прийнятних речовин-носіїв. Як правило, ці композиції є концентрованими і їх необхідно об'єднати з придатним розріджувачем, наприклад, водою перед застосуванням. Фармацевтичні композиції для парентерального введення, як правило, також включають один або більше наповнювачів. Можливі наповнювачі включають ізотонічний агент, буфер або інший контролюючий рН агент і консервант. Ці наповнювачі можуть бути додані для збереження композиції і для досягнення кращих діапазонів рН (приблизно 6,5-7,5) і осмолярності (приблизно 7 UA 102414 C2 5 10 15 20 25 30 35 40 45 50 55 60 300 мосмоль/л). Додаткові приклади складів для перорального введення можна знайти в патентах США №№ 5525590 і 5639724 та заявці на патент США 2003/0104992. При пероральному способі введення зазначено дозування циклоундекадепсипептидної сполуки згідно з винаходом для введення від рази 3 на добу до разу на тиждень може становити від приблизно 1 мг/кг до приблизно 100 мг/кг, краще від приблизно 1 мг/кг до приблизно 20 мг/кг. При внутрішньовенному способі введення зазначене відповідне дозування може становити від приблизно 1 мг/кг до приблизно 50 мг/кг, краще від приблизно 1 мг/кг до приблизно 25 мг/кг. Під ефективною кількістю циклоундекадепсипептидної сполуки згідно з винаходом розуміють кількість, яка при багаторазовому застосуванні в курсі програми лікування до пацієнта, що потребує лікування інфекції ВГС, приводить до об'єктивної клінічної відповіді, такої як статистично значиме зниження титру ВГС у сироватці або значиме зниження активності сироваткової аланінамінотрансферази (ALT) у пацієнта. Численні фактори будуть прийняті до уваги практикуючим лікарем при визначенні пробних доз для перевірки ефективності фармацевтичної композиції, що містить сполуки згідно з цим винаходом, проти інфекції ВГС. Першорядними серед них є токсичність і період напіввиведення обраного циклоундекадепсипептидної сполуки згідно з винаходом. Додаткові фактори включають вагу пацієнта, вік пацієнта, загальний стан пацієнта (включаючи значимі системні або великі захворювання, включаючи декомпенсовану хворобу печінки, важке існуюче порушення функції кісткового мозку та інші вірусні інфекції), стадію інфекції ВГС (гостра у порівнянні з хронічною) обумовлену, наприклад, рівнями сироваткової аланінамінотрансферази (ALT), специфічний генотип ВГС, попередню терапію інфекції ВГС, присутність інших лікарських засобів в організмі пацієнта і т.п. Курс лікування потребує багаторазового застосування фармацевтичної композиції згідно з винаходом. Як правило, відповідна доза лікарського засобу буде застосована 3-7 разів на тиждень, і тривалість лікування може становити від приблизно 4 тижнів до 6 місяців, краще від приблизно 4 тижнів до приблизно 12 місяців. Лікування може супроводжуватися визначеннями ВГС у сироватці і вимірюванням рівнів сироваткової ALT. Кінцевою точкою лікування є вірусологічна відповідь, тобто відсутність ВГС наприкінці курсу лікування, через кілька місяців після початку лікування або через кілька місяців після завершення лікування. ВГС у сироватці може бути виміряний на рівні РНК такими методами як кількісна полімеразна ланцюгова реакція в режимі реального часу (RT-PCR) або нозернблоттінг або на рівні білка за допомогою імуноферментного аналізу або імуноаналізу з посиленою хемілюмінесценцією вірусних білків. Кінцева точка може також включати визначення рівня сироваткової ALT у нормальному діапазоні. Фармацевтична композиція згідно з цим винаходом може містити один або більше інших інгредієнтів, активних проти інфекції ВГС, на додаток до сполук згідно з цим винаходом, таких як, наприклад, інший противірусний лікарський засіб, наприклад, рибавірин або інтерферон альфа. Сполуки згідно з винаходом і подібні інші активні складові можуть бути застосовані спільно як частина однієї й тієї ж фармацевтичної композиції або можуть бути застосовані окремо як частина придатного режиму дозування, розробленого з метою одержання користі від комбінованого лікування. Придатний режим дозування, кількість кожної дози, що застосовується, і специфічні інтервали між дозами кожного активного засобу будуть залежати від специфічної комбінації активних засобів, що застосовуються, стану пацієнта, що підлягає лікуванню, та інших факторів, обговорених у попередній частині. Зазначені додаткові активні складові можуть, загалом, бути застосовані в кількостях, менших ніж або рівних тим, у яких вони є ефективними як окремі лікарські засоби. Схвалені Федеральним агентством США по контролю над лікарськими засобами (FDA) дозування для даних активних засобів, які одержали схвалення FDA для застосування у людей, привселюдно доступні. Сполуки згідно з винаходом можуть бути застосовані як єдина складова або разом з іншими лікарськими засобами, наприклад, з лікарським засобом, що мають активність проти ВГС, наприклад, інтерфероном, наприклад, інтерфероном альфа-2а або інтерфероном альфа-2b, наприклад, Інтроном А (Intron A®), Рофероном (Roferon®), Авонексом (Avonex®), Ребіфом (Rebif®) або Бетафероном (Betaferon®), або інтерфероном, з'єднаним з водорозчинним полімером або людським альбуміном, наприклад, Альбуфероном (Albuferon®) (Human Genome Science), противірусним засобом, наприклад, рибавірином, ламівудином, NV08 або NM283, інгібітором факторів, що кодуються ВГС, таких як серинова протеаза NS3-4A, хеліказа або РНКполімераза, або проліками цього інгібітора, протифіброзним засобом, наприклад, похідним Nфеніл-2-піримідинаміну, наприклад, іматинібом, імуномодулюючим засобом, наприклад, мікофеноловою кислотою, її сіллю або проліками, наприклад, мікофенолятом натрію або мікофеноляту мофетилом, або агоністом рецептора S1P, наприклад, FTY720 або його факультативно фосфорильованим аналогом, наприклад, як описано в EP 627406А1, ЕР 8 UA 102414 C2 5 10 15 20 25 30 35 40 45 50 55 60 778263А1, ЕР 1002792А1, WO 02/18395, WO 02/76995, WO 02/06268, JP2002316985, WO 03/29184, WO 03/29205, WO 03/62252 і WO 03/62248, повний зміст яких включено в цю заявку за допомогою посилання. У конкретних варіантах реалізації типовий інтерферон, що застосовується у цьому винаході, обраний з групи, що складається з Інтрону А (Intron A®); Пегінтрону (Peg-intron®); Роферону (Roferon®); Пегасису (Pegasys®); Берофору (Berofor®); Sumiferon®; Велферону (Wellferon®); Інфергену (Infergen®); Альферону (Alferon®); Віраферону (Viraferon®); Альбуферону (Albuferon®) (Human Genome Science); Ребіфу; Omniferon; Омеги і їх комбінацій. Мається на увазі, що кон’югати інтерферону з водорозчинним полімером включають, головним чином, кон’югати з гомополімерами на основі поліалкілен-оксиду, такі як поліетиленгліколь (ПЕГ) або поліпропіленгліколі, поліоксиетиленування поліолі, їхні співполімери і їх блокові співполімери. Як альтернативи полімерам на основі поліалкілен-оксиду можуть бути ефективно застосовані неантигенні матеріали, такі як декстран, полівінілпіролідони, поліакриламіди, полівінілові спирти, полімери на основі вуглеводів і т.п. Подібні кон’югати інтерферону з полімером описані в патентах США №№ 4766106; 4917888, заявці на європейський патент № 0236987, заявці на європейський патент № 0510356 і міжнародної публікації заявки № WO 95/13090. Оскільки полімерна модифікація достатньо зменшує антигенні відповіді, немає потреби в тому, щоб чужорідний інтерферон був повністю аутогенним. Інтерферон, що застосовується для одержання полімерних кон’югатів, може бути отриманий з екстракту ссавців, наприклад, інтерферон людини, жуйної тварини або бика, або зроблений рекомбінантним способом. Інші форми інтерферонів включають інтерферон бета, гама, тау і омега, такі як Ребіф (Інтерферон бета-1а) придбаний в Serono, Omniferon (природний інтерферон) придбаний в Viragen або Омега Інтерферон (Omega Interferon) придбаний в Boehringer Ingelheim. Пероральні інтерферони такі як пероральний інтерферон альфа, придбаний в Amarillo Biosciences. Кращими є кон’югати інтерферону з поліетиленгліколем, також відомі як пегільовані інтерферони. Особливо кращі кон’югати інтерферону являють собою пегільовані альфа-інтерферони, наприклад, пегільований інтерферон альфа-2а, пегільований інтерферон альфа-2b; пегільований консенсус інтерферон або пегільований очищений продукт а. Пегільований інтерферон альфа-2а описаний, наприклад, у європейському патенті № 593868 і комерційно доступний, наприклад, під торговельної маркою Пегасис (PEGASYS®) (Hoffmann-La Roche). Пегільований інтерферон альфа-2b описаний, наприклад, у європейському патенті № 975369 і комерційно доступний, наприклад, під торговельної маркою Пегінтрон (PEG-INTRON A®) (Schering Plough). Пегільований консенсус інтерферон описаний в WO 96/11953 (повністю включений у цю заявку за допомогою посилання). Кращі пегільовані альфа-інтерферони являють собою пегільований інтерферон альфа-2а і пегільований інтерферон альфа-2b. Також кращим є пегільований консенсус інтерферон. Добові дозування щодо застосовуваного супутнього засобу будуть варіюватися залежно від застосовуваної сполуки, виду організму, способу застосування і тяжкості стану, що підлягає лікуванню. Наприклад, ламівудин може бути застосований в добовому дозуванні 100 мг. Пегільований інтерферон може бути введений парентерально від одного до трьох разів на тиждень, краще один раз на тиждень, у загальній тижневій дозі, що становить від 2 до 10 мільйонів міжнародних одиниць (IU), ще краще від 5 до 10 мільйонів IU, найкраще від 8 до 10 мільйонів IU. Дані значення можуть відповідати від 0,5 до 2,0 мікрограмам/кілограм на тиждень розраховуючи на щотижневе приймання, приймання три рази на тиждень, приймання через день або щоденне приймання. В інших варіантах реалізації інтерферон альфа являє собою пегільований інтерферон альфа-2а, і кількість застосовуваного пегільованого інтерферону альфа-2а становить від 20 до 250 мікрограмів/кілограм на тиждень розраховуючи на щотижневе приймання, приймання три рази на тиждень, приймання через день або щоденне приймання. Краще, пегільований інтерферон альфа-2а (peg-IFNa2a) застосовують у кількості 180 мікрограмів один раз на тиждень. Противірусні засоби, що звичайно застосовуються для лікування ВГС, також включені в комбінацію згідно з цим винаходом. Подібні засоби включають сполуки або біопрепарати, які ефективно пригнічують утворення і/або реплікацію вірусу в організмі ссавця. Зазначені засоби включають такі засоби як рибавірин (1-бета-D-рибофуранозил-1Н-1,2,4-триазол-3-карбоксамід), придбаний в Valeant Pharmaceuticals, Inc., Costa Mesa, CA; Ребетол (Rebetol®), придбаний в Schering-Plough Corporation, Kenilworth, NJ і Копегус (Copegus®), придбаний в Hoffmann-La Roche, Nutley, NJ, аналоги рибавірину в стадії розробки, такі як Левовірин і Вірамідин, придбані в Valeant, і Мізорибіну монофосфат. 9 UA 102414 C2 5 10 15 20 25 30 35 40 45 50 55 60 Крім того, комбінація згідно з цим винаходом може додатково включати застосування інгібіторів протеаз, дія яких заснована на субстратному інгібуванні серинової протеази NS3-4A ВГС, інгібіторів протеази NS3, дія яких не заснована на субстратному інгібуванні; фенантренхінонів, тіазолідинів і бензанілідів і.т.п., які, як показали, мають активність проти інфекції, що викликається ВГС. Інгібітори протеази для лікування ВГС запропоновані, наприклад, у патенті США № 6004933 (Spruce et al); патенті США № 5990276 (Zhang et al); патенті США № 5538865 (Reyes et al.); WO 02/008251 (Corvas International, Inc.), WO 02/08187 і WO 02/008256 (Schering Corporation); патентах США №№ 6534523, 6410531 і 6420380 (Boehringer Ingelheim) і WO 02/060926 (Bristol Myers Squibb); WO 02/48172 і WO 02/18198 (Schering Corporation); WO 02/48157 і WO 02/48116 (Bristol Myers Squibb); WO 98/17679 (Vertex Pharmaceuticals), повний зміст яких включено в цю заявку за допомогою посилання. Як приклад, можуть бути застосовані інгібітори протеази NS3, дія яких не заснована на субстратному інгібуванні, такі як похідні 2,4,6-тригідрокси-3-нітробензаміду (Sudo K. et al., Biochemical and Biophysical Research Communications, 1997, 238 643-647; Sudo K. et al. Antiviral Chemistry and Chemotherapy, 1998, 9, 186), включаючи RD3-4082 і RD3-4078. Додаткові експериментальні засоби, які, як було показано, є ефективними в терапії ВГС, включають інгібітори серинової протеази NS3-4A ВГС BILN 2061, придбаний в Boehringer Ingelheim, VX-950, придбаний в Vertex, SCH-6, SCH-7 і SCH-351633, придбані в Schering-Plough, та інші інгібітори протеази ВГС у доклінічній і клінічній стадії розробки, придбані в Glaxosmithkline, Bristol Myers Squibb, Abbot, Roche, Merck, Pfizer і Gilead. Додаткові засоби, які перебувають у цій час у клінічній стадії розробки для лікування ВГС і які можуть бути застосовані в комбінації з винаходом, описаним у цій заявці, включають TMC 435350 (Tibotec) і ITM-191 (Intermune). Також можуть бути застосовані аналоги нуклеозидів. Прикладом одного з таких аналогів є телбівудин, придбаний в Idenix (US 6444652, US 6596700 і WO 0196353). Нуклеозидні або ненуклеозидні інгібітори РНК-залежної РНК-полімерази NS5B ВГС, такі як 2’-C-метил-3’-O-Lваліновий ефір рибофуранозилцитидину (NM283, Idenix), запропонований в WO 2004/002422, можуть також бути застосовані. Розгалужені нуклеозиди, запропоновані в WO 01/90121 і WO 01/92282 (Idenix Pharmaceuticals) можуть також бути застосовані. Повний зміст цих міжнародних заявок на патент включено в цю заявку за допомогою посилання. Додаткові терапевтичні сполуки можуть включати антисмислові молекули, спрямовані проти генома ВГС, або антисмислову послідовність, комплементарну кожній з частин генома ВГС, яка підвищує ефективність терапії. Також передбачені інгібітори інших мішеней у життєвому циклі ВГС, такі як Celgosivir (MBI 3253), інгібітор процесінга глікопротеїну, придбаний в Migenix, інгібітор злиття, придбаний в Trimeris, ACH-0137171, придбаний в Achillion. Агоністи рецептора, такі як агоністи Toll-подібного рецептора (“TLR”), включають ANA245, ANA971, ANA975 (US 5041426, 4880784) від Anadys; агоніст TLR-9 Cpg-10101 (Coley Pharmaceuticals); інгібітор інозинмонофосфатдегідрогенази (IMPDH), мікофенолову кислоту, її сіль або проліки мікофенолят натрію або мікофеноляту мофетил, або Merimebodib (VX-497, придбаний в Vertex); тімозин альфа-1 (Задаксин або його комбінація, придбаний в SciClone); SCV-07 (SciClone), Belerofon (поліпшений інтерферон альфа (IFN-), придбаний в Nautilus); CIVACIR (імуноглобулін проти гепатиту С), придбаний в NABI, або агоніст рецептора S1P, наприклад, FTY720 або його факультативно фосфорильований аналог, наприклад, запропонований в EP 627406A1, EP 778263A1, EP 1002792A1, WO 02/18395, WO 02/76995, WO 02/06268, JP2002316985, WO 03/29184, WO 03/29205, WO 03/62252 і WO 03/62248, повні описи яких включені в цю заявку за допомогою посилання; Резиквімод [VML 600], придбаний в 3M Pharmaceuticals, аналог іміквімоду, що є потужним індуктором інтерферону альфа та інших цитокінів. Крім того, сполуки згідно з цим винаходом можуть бути застосовані в комбінації з Інтерлейкіном-10 (Shering-Plough), амантадином (Symmetrel), придбаним в Endo Labs Solvay, інгібітором каспази IDN-6556, придбаним в Idun Pharma, ВГС/MF59, придбаним в Chiron, CEPLENE (гістаміну дихлорид), придбаним в Maxim, IDN-6556, придбаним в Idun PHARM, T67, інгібітором бета-тубуліну, придбаним в Tularik, FK788, придбаним в Fujisawa Healthcare, Idbi 016 (Siliphos, пероральна фітосома, що містить комплекс сілібін-фосфатидилхолін), Dication, придбаним в Immtech, очисником крові, придбаним в Aethlon Medical, UT 231 B, придбаним в United Therapeutics; Hepex-c SM1, Ppvo-bay55-8800 (овечий парапоксвірус), придбаними в Bayer; Refanalin (міметик фактора росту гепатоцитів (HGF)), придбаним в Angion, R803 (Rigel), JTK-003, JTK-002 і JTK-109 (усі придбані в Japan Tobacco), ВГС-086 (Viropharma/Wyeth), ISIS-14803 (ISIS Pharmaceuticals), GS-9132 (інгібітор полімерази, придбаний в Achillion pharmaceuticals), ВГС-793 (Pharmasset and Roche), R1626 (Roche). 10 UA 102414 C2 5 10 15 20 25 30 35 40 45 50 55 60 Інші сполуки, що представляють інтерес, включають Ursodiol (EP 00269516, Axcan Pharma), HE-2000 (аналог дегідроепіандростерону (DHEA), Colthurst Ltd), EHC-18 (імуномодулятор, Enzo biochem), гістаміну дигідрохлорид (агоніст гістамінового рецептора H2, WO 09104037, EsteroAnstalt), нітазоксанід (Romark), 1-аміноалкілциклогексани (патент США № 6034134), алкілліпіди (патент США № 5922757), вітамін Е та інші антиоксиданти (патент США № 5922757), жовчні кислоти (патент США № 5846964), N-(фосфоноацетил)-L-аспарагінову кислоту (патент США № 5830905), бензолдикарбоксаміди (патент США № 5633388), похідні поліаденілової кислоти (патент США № 5496546), 2’,3’-дидезоксиінозин (патент США № 5026687), бензімідазоли (патент США № 5891874), екстракти рослин (патент США № 5837257; патент США № 5725859 і патент США № 6056961) і піперидини (патент США № 5830905); N-(фосфоноацетил)-Lаспарагінову кислоту, бензолдикарбоксаміди, похідні поліаденілової кислоти, інгібітори глікозилювання та неспецифічні цитопротективні засоби, які перешкоджають ушкодженню клітини, що викликається вірусною інфекцією. Сполуки згідно з винаходом можуть також бути застосовані в комбінації з вакциною або заснованими на застосуванні антитіл підходами до лікування ВГС. Терапевтичні вакцини включають вакцини, придбані в Intercell (Терапевтична пептидна вакцина IC41 ВГС), Epimmune/Genecor, Merix, Tripep (Chiron-VacC), імунотерапію (Therapore), придбану в Avant, Тклітинну терапію, придбану в CellExSys, моноклональні антитіла XTL-002, придбані в STL, ANA 246 і ANA 246, придбані в Anadys, терапевтичну вакцину, спрямовану на білок оболонки E2, придбану в Innogenetics, моноклональні антитіла (mAb) проти білка оболонки Е2, придбані в XTL Bio, GI-5005 (GlobeImmune Inc), InnoVac-C (WO 9967285, Innogenetics), IC-41 (Intercell), інтерферон альфа-n3 (Interferon Sciences). Відповідно до вищесказаного, згідно з цим винаходом запропонований інший додатковий аспект, фармацевтична комбінація, що містить щонайменше а) перший засіб, що складається зі сполуки згідно з винаходом, або фармацевтичну композицію згідно з цим винаходом і b) другий засіб, що має здатність протидіяти реплікації ВГС. Зокрема, ця фармацевтична комбінація призначена для застосування в запобіганні або лікуванні інфекцій, що викликаються вірусом гепатиту С, або розладів, що викликаються ВГС. Також цей винахід включає спосіб запобігання або лікування інфекцій, що викликаються вірусом гепатиту С, або розладів, що викликаються ВГС, у суб’єкта, що цього потребує, що включає введення зазначеному суб’єкту терапевтично ефективної кількості сполуки згідно з винаходом або фармацевтичною композицією згідно з винаходом. Крім того, запропонований спосіб пригнічення реплікації ВГС у пацієнта, що цього потребує, який включає введення зазначеному суб’єкту терапевтично ефективної кількості сполуки згідно з винаходом або фармацевтичною композицією згідно з винаходом. Іншим завданням винаходу є спосіб, що включає спільне введення, одночасно або послідовно, терапевтично ефективної кількості сполуки згідно з винаходом або фармацевтичною композицією згідно з винаходом і супутнього засобу, обраного із засобів, що мають властивості діяти проти ВГС. Терміни “спільне введення” або “комбіноване введення” або подібні їм, що використовуються в цій заявці, позначають застосування обраних терапевтичних засобів до окремого пацієнта і включають схему лікування, у якій засоби не обов'язково застосовують тим самим способом застосування або в один і той же час. Введення фармацевтичної комбінації згідно з винаходом приводить до корисного ефекту, наприклад, синергетичного терапевтичного ефекту, у порівнянні з монотерапією, у випадку якої застосовують тільки одну з її фармацевтично активних складових. Кращою синергічною комбінацією є комбінація циклоундекадепсипептидної сполуки згідно з винаходом з інтерфероном, факультативно з'єднана з полімером. Додаткова краща комбінація являє собою комбінацію сполук Формули (I) згідно з винаходом з мікофеноловою кислотою, її сіллю або проліками, або з агоністом рецептора S1P, наприклад, FTY720. Повний зміст усіх патентів, заявок на патент і публікацій, наведених у цій заявці, включено в цю заявку за допомогою посилання. Фахівці в цій галузі техніки візьмуть до уваги, що винахід, описаний в цій заявці, підлягає змінам і модифікаціям, відмінним від тих, які конкретно описані. Слід розуміти, що винахід включає всі подібні зміни і модифікації без відхилення від його сутності або основних характеристик. Винахід також включає всі стадії, особливості, композиції та сполуки, названі або позначені в цьому описі, індивідуально або спільно, і будь-яку і усі комбінації або будь-які дві або більше зазначених стадій або особливостей. Таким чином, слід вважати, що цей опис у всіх його аспектах є ілюстративним і не обмежуючим, причому, обсяг винаходу визначений 11 UA 102414 C2 5 10 15 20 25 30 35 40 45 50 55 60 прикладеною формулою винаходу, і будь-які зміни, які можуть бути внесені в межах сутності і які є еквівалентними, вважаються включеними в цей винахід. У цьому описі наведені різні посилальні матеріали, повний зміст кожного з яких включено в цю заявку за допомогою посилання. Для більш повного розуміння вищенаведений опис посилається на наступні Приклади. Ці Приклади, однак, наведені для пояснення способів застосування цього винаходу і не обмежують обсяг винаходу. ПРИКЛАДИ Приклад 1a: Одержання циклоундекадепсипептиду (Iа): Цикло-(MeBmt-Abu-D-MeAla-MeVal-Leu-MeLeuGly-D-Hiv-MeLeu-Leu-MeVal) (Iа) 1. Одержання Фрагмента (Аа), починаючи з природного циклоундекадепсипептиду Цикло(MeBmt-Thr-Sar-MeLeu-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) (див. US 5116816, Приклад 2, або в WO 02/092033), Приклад 4, Стадія 4-1): H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-OH (Aa) 1.1 Варіант А Одержання Цикло-(MeBmt-Thr-(O-(N-імідазоліл)карбоніл)Sar-MeLeu-Leu-MeLeu-Ala-D-HivMeLeu-Leu-MeVal) (1A). Розчин Цикло-(MeBmt-Thr-Sar-MeLeu-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) (US 5116816, Приклад 2, або в WO 02/092033, Приклад 4, Стадія 4-1) (3,00 г, 2,40 ммоль, 1,0 екв.) і 1,1’карбонілдиімідазолу (1,17 г, 7,21 ммоль, 3,0 екв.) в 15 мл безводного CH 2Cl2 перемішували при кімнатній температурі протягом 2,5 годин. Протікання реакції контролювали за допомогою аналітичної надпродуктивної рідинної хроматографії (НПРХ, UPLC). Ступінь перетворення становив 81,1 %. Додавали додаткову кількість 1,1’-карбонілдиімідазолу (0,39 г, 2,40 ммоль, 1,0 екв.) у реакційну суміш. Після додаткового перемішування протягом 16 годин ступінь перетворення становив 98,8 %. Розчин упарювали при зниженому тиску. Залишок розчиняли в AcOEt (45 мл) і промивали послідовно 10%-вою лимонною кислотою (45 мл) і розчином солі (45 мл). Органічну фазу сушили над Na 2SO4, фільтрували і упарювали при зниженому тиску з одержанням суміші двох сполук (1А) і (2А) у вигляді білого порошку. + + НПРХ-ІЕР-MS (m/z): (1A): 1342,75 [M + H] ([C68H117N12O15] ; розр. 1342,73), + + (2A): 1274,77 [M + H] ([C65H113N10O15] ; розр. 1274,65) Одержання Цикло-(MeBmt-Thr(O,N-карбоніл)-Sar-MeLeu-Leu-MeLeu-Ala-D-Hiv-MeLeu-LeuMeVal) (2А). Розчин сполуки (1А), отриманої за допомогою попередньої реакції (3,41 г, 2,40 ммоль) у сухому ДМСО (15 мл) перемішували і нагрівали при 100 °С протягом 2 годин в атмосфері аргону. Протікання реакції контролювали за допомогою аналітичної НПРХ. Досягали повного перетворення. Далі суміш розчиняли в AcOEt (45 мл) і промивали послідовно 1М HCl (30 мл) і 23,2%-вим водним розчином NaCl (15 мл), потім 11,6%-вим водним розчином NaCl (45 мл). Органічну фазу сушили над Na2SO4, фільтрували і упарювали при зниженому тиску з одержанням циклоундекадепсипептиду Цикло-(MeBmt-Thr(O,N-карбоніл)-Sar-MeLeu-Leu-MeLeuAla-D-Hiv-MeLeu-Leu-MeVal) (2A) у вигляді білого порошку. + + НПРХ-ІЕР-МС (m/z): 1274,65 [M + H] ([C65H113N10O15] ; розр. 1274,65). Одержання H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-OMe (метиловий ефір фрагменту Аа) (3A). До розчину циклодепсипептиду, отриманого за допомогою попередньої реакції, (1,00 г, 0,78 ммоль) в 30 мл MeOH (сухого), охолодженому до 0°С, додавали однією порцією Ca(OMe) 2 (0,24 г, 2,35 ммоль) в атмосфері аргону. Контроль повноти протікання реакції за допомогою НПРХ через 5 годин при 0°С і потім через 1 годину при кімнатній температурі показала 98,6 % перетворення. Розчин охолоджували до 0°С і потім нейтралізували водним розчином 10%-вої лимонної кислоти. Метанол упарювали. Водну суміш доливали до 100 мл суміші AcOEt/NaCl 23,2 % (1:1 об./об.). Органічну фазу сушили над Na 2SO4, фільтрували і упарювали при зниженому тиску. Неочищену суміш розділяли за допомогою колонкової хроматографії з одержанням пентапептиду Н-D-Hiv-MeLeu-Leu-MeVal-MeBmt-OMe (3A). + + НПРХ-ІЕР-МС (m/z): 669,16 [M + H] ([C35H65N4O8] ; розр. 669,48). Одержання фрагменту (Аа) H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-OH (4). Розчин пентапептиду, отриманого за допомогою попередньої реакції (3А) (1,15 г, 1,72 ммоль, 1,0 екв.) у ТГФ (8,6 мл) і воді (1,1 мл) охолоджували до 0°С у водно-крижаній бані, потім додавали протягом 20 секунд 2M LiOH (1,72 мл, 3,44 ммоль, 2,0 екв.). Охолоджуючу баню потім видаляли і суміш (pH = 12-13) перемішували при кімнатній температурі протягом приблизно 3 годин. Контроль повноти протікання реакції за допомогою НПРХ показав повне перетворення. Далі розчин охолоджували до 0°С і нейтралізували 10%-вою лимонною кислотою. 12 UA 102414 C2 5 10 15 20 25 30 35 40 45 50 55 60 Тетрагідрофуран упарювали при зниженому тиску. Залишок розчиняли в AcOEt (50 мл) і промивали 10%-вою лимонною кислотою (3 мл) і 23,2%-вим водним розчином NaCl (50 мл) (рН 3). Органічну фазу сушили над Na2SO4, фільтрували і упарювали при зниженому тиску з одержанням Фрагменту (Аа) H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-OH (4) у вигляді білого порошку. + + НПРХ-ІЕР-МС (m/z) 655,21 [M + H] ([C34H63N4O8] ; розр. 655,46). 1.2 Варіант B Одержання H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-Thr-Sar-MeLeu-Leu-MeLeu-Ala-OMe До розчину природного Цикло-(MeBmt-Thr-Sar-MeLeu-Leu-MeLeu-Ala-D-Hiv-MeLeu-LeuMeVal) (отриманого згідно або US 5116816, Приклад 2, або WO 02/092033, Приклад 4, Стадія 41) (3,0 г, 2,40 ммоль) у сухому МеОН (90 мл), охолодженому до 0°С, додавали в атмосфері аргону MeONa (0,519 г, 9,6 ммоль, 4,0 екв.). Контроль повноти протікання реакції за допомогою ВЕРХ через 1 годину при 0°С і потім через 2 години при кімнатній температурі показує її завершення. Розчин охолоджували до 0°С, потім нейтралізували (рН 5-6) водним розчином 10%-вої лимонної кислоти. Метанол упарювали. Водну суміш виливали в 150 мл етилацетату. Органічний шар промивали водою (20 мл) і розчином солі (20 мл) і сушили над MgSO 4, фільтрували. Розпарювання розчину під вакуумом і флеш-хроматографія на силікагелі привели до одержання чистого H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-Thr-Sar-MeLeu-Leu-MeLeu-Ala-OMe. Одержання H-D-Hiv(O-(N-імідазоліл)карбоніл)-MeLeu-Leu-MeVal-MeBmt-Thr(O-(N-імідазоліл) карбоніл)-Sar-MeLeu-Leu-MeLeu-Ala-OMe (1B) Розчин ундекапептиду, отриманого за допомогою попередньої реакції, (2,96 г, 2,31 ммоль, 1,0 екв.) і 1,1’-карбонілдиімідазолу (1,50 г, 9,25 ммоль, 4,0 екв.) в 53 мл безводного CH 2Cl2 перемішували при кімнатній температурі протягом 22 годин. Протікання реакції контролювали за допомогою аналітичної ВЕРХ і ТШХ. Розчин упарювали при зниженому тиску. Залишок розчиняли в 150 мл етилацетату і промивали послідовно 10%-вою лимонною кислотою (30 мл), водою (30 мл) і розчином солі (30 мл). Органічну фазу сушили над Na 2SO4, фільтрували і упарювали при зниженому тиску. Неочищений продукт очищали за допомогою флешхроматографії з одержанням H-D-Hiv(O-(N-імідазоліл)карбоніл)-MeLeu-Leu-MeVal-MeBmt-Thr(O(N-імідазоліл)карбоніл)-Sar-MeLeu-Leu-MeLeu-Ala-OMe (1В) у вигляді білого порошку. Одержання H-D-Hiv(O-(N-імідазоліл)карбоніл)-MeLeu-Leu-MeVal-MeBmt-Thr(O,N-карбоніл)Sar-MeLeu-Leu-MeLeu-Ala-OMe (2В) Розчин ундекапептиду, отриманого за допомогою попередньої реакції, (3,26 г, 2,22 ммоль) у сухому ДМСО (40 мл) перемішували і нагрівали при 100 °С протягом 3,5 годин в атмосфері аргону. Протікання реакції контролювали за допомогою ТШХ і аналітичної ВЕРХ. Далі суміш розчиняли в AcOEt (200 мл) і промивали послідовно водним розчином 10%-вої лимонної кислоти (2 рази, 30 мл), H2O (1х30 мл) і насиченим водним розчином NaCl (1x30 мл). Органічну фазу сушили над Na2SO4, фільтрували і упарювали при зниженому тиску. Неочищений продукт очищали за допомогою флеш-хроматографії. Залишок після розпарювання розчинників сушили у високому вакуумі з одержанням ундекапептиду H-D-Hiv(O-(N-імідазоліл)карбоніл)-MeLeu-LeuMeVal-MeBmt-Thr(O,N-карбоніл)-Sar-MeLeu-Leu-MeLeu-Ala-OMe (2В) у вигляді білого порошку. Одержання H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-OMe (3В) До розчину ундекапептиду (2В), отриманого за допомогою попередньої реакції, (3,05 г, 2,17 ммоль) в 100 мл МеОН (сухого), охолодженому до 0°С, додавали однією порцією МеОК (0,456 г, 6,53 ммоль) в атмосфері аргону. Контроль повноти протікання реакції за допомогою ВЕРХ через 1 годину при 0°С і потім 1 годину при кімнатній температурі показав її завершення. Розчин охолоджували до 0°С і потім нейтралізували водним розчином 10%-вої лимонної кислоти. Метанол упарювали. Водну суміш виливали в 100 мл суміші AcOEt/H 2O (1:1 об./об.). Органічний шар промивали H2O (1х10 мл) і розчином солі (1х10 мл). Органічну фазу сушили над Na 2SO4, фільтрували і упарювали при зниженому тиску. Неочищену суміш розділяли за допомогою препаративної ВЕРХ з одержанням пентапептиду H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-OMe (3В). Одержання H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-OH (4, Фрагмент Aa) Розчин пентапептиду, отриманого за допомогою попередньої реакції, (1,265 г, 1,88 ммоль, 1,0 екв.) у ТГФ (18,8 мл) охолоджували до 0°С у водно-крижаній бані, потім додавали по краплях протягом 20 хвилин 0,2 М LiOH (18,8 мл, 3,66 ммоль, 2,0 екв.). Охолоджуючу баню потім видаляли і суміш (рН = 12-13) перемішували при кімнатній температурі протягом приблизно 2,5 годин (контроль ТШХ). Далі розчин охолоджували до 0°С і нейтралізували 0,1 М НCl (рН 2-3). Тетрагідрофуран упарювали при зниженому тиску. Залишок розчиняли в AcOEt (100 мл) і промивали 10%-вою лимонною кислотою (1х20 мл), Н2О (1х20 мл) і розчином солі (1х20 мл). Органічну фазу сушили над Na2SO4, фільтрували і упарювали при зниженому тиску. Неочищену суміш розділяли за допомогою колонкової хроматографії (СС) з одержанням пентапептиду (натрієвої солі) H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-ONa у вигляді білого порошку. 13 UA 102414 C2 + 5 10 15 20 25 30 35 40 45 50 55 60 + HR-MALDI-МС: 677,46 [M + Na] ([C34H62N4NaO8] ; розр. 677,4465). 2. Одержання Фрагменту (Ва): H-Abu-D-MeAla-MeVal-Leu-MeLeu-Gly-OMe (Ва) 2.1. Одержання Boc-MeLeu-Gly-OMe До розчину комерційно доступного Н-Gly-OMe•HCl (4,0 г, 31,8 ммоль, 1,0 екв.) у сухому ДХМ (240 мл), охолодженому до 0°С, додавали в атмосфері аргону DIPEA (32,88 мл, 189,0 ммоль, 6,0 екв.). Далі, через 15 хвилин, додавали однією порцією HATU (15,72 г, 41,34 ммоль, 1,3 екв.) і Boc-MeLeu-OH (7,8 г, 31,8 ммоль, 1,0 екв.). Контроль повноти протікання реакції за допомогою ТШХ через 15 хвилин при 0°С і потім 22,45 години при кімнатній температурі показав її завершення. Реакцію зупиняли додаванням 10%-го NaHCO3 (40 мл) і перемішували протягом 15 хвилин. Реакційну суміш розбавляли ДХМ (400 мл) і промивали 10%-вою лимонною кислотою (1х80 мл), H2O (1x80 мл) і розчином солі (1х80 мл). Органічну фазу сушили над Na 2SO4, фільтрували і упарювали при зниженому тиску. Неочищену суміш розділяли з одержанням складного ефіру Boc-MeLeu-Gly-OMe у вигляді білої олійної піни. + + НПРХ-ІЕР-МС (m/z) 339,0234 [M + Na] ([C15H28N2NaO5] ; розр. 339,1896). 2.2. Одержання H-MeLeu-Gly-OMe Розчин дипептиду, отриманого за допомогою попередньої реакції, (9,9 г, 31,29 ммоль, 1,0 екв.) у ТФО/ДХМ (30 мл, 2:3 об./об.) витримували протягом 45 хвилин при 0°С і 2 годин при кімнатній температурі, і розчинники видаляли при зниженому тиску. Неочищений продукт сушили у високому вакуумі (20 хвилин). Далі додавали ДХМ (60 мл) і суміш розтирали при 0°С з DIPEA (2,0 мл) до рН 7-8 для нейтралізації надлишку ТФО. Після розпарювання і висушування неочищену амінну сіль дипептиду H-MeLeu-Gly-OMe застосовували для наступної стадії комбінації без додаткового очищення. + + НПРХ-ІЕР-МС (m/z) 217,0546 [M + H] ([C10H21N2O3] ; розр. 217,1552). 2.3. Одержання Boc-Leu-MeLeu-Gly-OMe До розчину дипептиду, отриманого за допомогою попередньої реакції, (3,38 г, 15,63 ммоль, 1,0 екв.) у сухому ДХМ (160 мл), охолодженому до 0°С, додавали в атмосфері аргону DIPEA (8,08 мл, 46,89 ммоль, 3,0 екв.). Далі, через 10 хвилин, додавали однією порцією HATU (7,72 г, 20,14 ммоль, 1,3 екв.) і Boc-Leu-OH (3,98 г, 17,19 ммоль, 1,1 екв.). Контроль повноти протікання реакції за допомогою ТШХ через 15 хвилин при 0°С і потім 15,45 години при кімнатній температурі показав її завершення. Реакцію зупиняли додаванням 10%-го NaHCO3 (36 мл) і перемішували протягом 15 хвилин. Реакційну суміш розбавляли ДХМ (240 мл) і промивали 10%-вою лимонною кислотою (1х60 мл), H2O (1x60 мл) і розчином солі (1х60 мл). Органічну фазу сушили над Na2SO4, фільтрували і упарювали при зниженому тиску. Неочищену суміш розділяли c допомогою СС з одержанням складного ефіру Boc-Leu-MeLeu-Gly-OMe у вигляді жовтуватої олії. + + НПРХ-ІЕР-МС (m/z) 430,0120 [M + H] ([C21H40N3O6] ; розр. 430,2917). 2.4. Одержання H-Leu-MeLeu-Gly-OMe Розчин трипептиду, отриманого за допомогою попередньої реакції, (3,0 г, 6,98 ммоль, 1,0 екв.) у ТФО/ДХМ (12 мл, 1:1 об./об.) витримували протягом 1 години при 0°С і 2 годин при кімнатній температурі і розчинники видаляли при зниженому тиску. Неочищений продукт сушили у високому вакуумі (15 хвилин). Далі додавали ДХМ (20 мл) і суміш розтирали при 0°С з DIPEA до рН 7-8 для нейтралізації надлишку ТФО. Після розпарювання і висушування неочищену амінну сіль H-Leu-MeLeu-Gly-Ome застосовували для наступної стадії комбінації без додаткового очищення. + + + НПРХ-ІЕР-МС (m/z) 330,0227 [M + H] ([C16H32N3O4] ; розр. 330,2393); 352,0016 [M + Na] + + + ([C16H31N3NaO4] ; розр. 352,4248); 659,1626 [2M + H] ([C32H63N6O8] ; розр. 659,1626). 2.5. Одержання Boc-MeVal-Leu-MeLeu-Gly-OMe До розчину трипептиду, отриманого за допомогою попередньої реакції, (2,3 г, 6,98 ммоль, 1,0 екв.) у сухому ДХМ (105 мл), охолодженому до 0°С, додавали в атмосфері аргону DIPEA (3,61 мл, 20,34 ммоль, 3,0 екв.). Далі, через 10 хвилин, додавали однією порцією HATU (3,45 г, 9,07 ммоль, 1,3 екв.) і Boc-MeVal-OH (1,78 г, 7,68 ммоль, 1,1 екв.). Контроль повноти протікання реакції за допомогою ТШХ через 15 хвилин при 0°С і потім 15,45 години при кімнатній температурі показав її завершення. Реакцію зупиняли додаванням 10%-го NaHCO3 (25 мл) і перемішували протягом 15 хвилин. Реакційну суміш розбавляли ДХМ (150 мл) і промивали 10%-вою лимонною кислотою (1х50 мл), H2O (1x50 мл) і розчином солі (1х50 мл). Органічну фазу сушили над Na2SO4, фільтрували і упарювали при зниженому тиску. Неочищену суміш розділяли з одержанням складного ефіру Boc-MeVal-Leu-MeLeu-Gly-OMe у вигляді жовтуватої олії. + + + НПРХ-ІЕР-МС (m/z) 543,0484 [M + H] ([C27H51N4O7] ; розр. 543,3758); 565,0467 [M + Na] + ([C27H50N4NaO7] ; розр. 565,3577). 14 UA 102414 C2 5 10 15 20 25 30 35 40 45 50 55 60 2.6. Одержання H-MeVal-Leu-MeLeu-Gly-OMe Розчин тетрапептиду, отриманого за допомогою попередньої реакції, (3,56 г, 6,56 ммоль, 1,0 екв.) у ТФО/ДХМ (12 мл, 1:1 об./об.) витримували протягом 1 години при 0°С і 2 годин при кімнатній температурі та видаляли розчинники при зниженому тиску. Неочищений продукт сушили у високому вакуумі (приблизно 15 хвилин). Далі додавали ДХМ (45 мл) і суміш розтирали при 0°С з DIPEA до рН 7-8 для нейтралізації надлишку ТФО. Після розпарювання і висушування неочищену амінну сіль тетрапептиду H-MeVal-Leu-MeLeu-Gly-OMe застосовували для наступної стадії комбінації без додаткового очищення. + + + НПРХ-ІЕР-МС (m/z) 443,0694 [M + H] ([C22H43N4O5] ; розр. 443,3233); 465,0063 [M + Na] + ([C22H42N4NaO5] ; розр. 465,3053). 2.7. Одержання Boc-D-MeAla-MeVal-Leu-MeLeu-Gly-OMe До розчину тетрапептиду, отриманого за допомогою попередньої реакції, (2,90 г, 6,56 ммоль, 1,0 екв.) у сухому ДХМ (110 мл), охолодженому до 0°С, додавали в атмосфері аргону DIPEA (3,39 мл, 19,68 ммоль, 3,0 екв.). Далі, через 10 хвилин, додавали однією порцією HATU (3,24 г, 8,53 ммоль, 1,3 екв.) і Boc-D-MeAla-OH (1,46 г, 7,21 ммоль, 1,1 екв.). Контроль повноти протікання реакції за допомогою ТШХ через 15 хвилин при 0°С і потім 17,45 години при кімнатній температурі показав її завершення. Реакцію зупиняли додаванням 10%-го NaHCO3 (25 мл) і перемішували протягом 15 хвилин. Реакційну суміш розбавляли ДХМ (150 мл) і промивали 10%-вою лимонною кислотою (1х50 мл), H2O (1x50 мл) і розчином солі (1х50 мл). Органічну фазу сушили над Na2SO4, фільтрували і упарювали при зниженому тиску. Неочищену суміш розділяли за допомогою СС з одержанням складного ефіру Boc-D-MeAla-MeVal-Leu-MeLeu-GlyOMe у вигляді жовтуватої піни. + + + НПРХ-ІЕР-МС (m/z) 628,56 [M + H] ([C31H58N5O8] ; розр. 628,4285); 650,53 [M + Na] + ([C31H57N5NaO8] ; розр. 650,4105). 2.8. Одержання H-D-MeAla-MeVal-Leu-MeLeu-Gly-OMe Розчин пентапептиду, отриманого за допомогою попередньої реакції, (1,6 г, 2,55 ммоль, 1,0 екв.) у ТФО/ДХМ (6 мл, 1:1 об./об.) витримували протягом 1 години при 0°С і 2 годин при кімнатній температурі і розчинники видаляли при зниженому тиску. Неочищений продукт сушили у високому вакуумі (приблизно 15 хвилин). Далі додавали ДХМ (20 мл) і суміш розтирали при 0°С з DIPEA до рН 7-8 для нейтралізації надлишку ТФО. Після розпарювання і висушування неочищену амінну сіль пентапептиду H-D-MeAla-MeVal-Leu-MeLeu-Gly-OMe застосовували для наступної стадії комбінації без додаткового очищення. + + НПРХ-ІЕР-МС (m/z) 528,49 [M + H] ([C26H50N5O6] ; розр. 528,3761). 2.9. Одержання Boc-Abu-D-MeAla-MeVal-Leu-MeLeu-Gly-OMe До розчину пентапептиду, отриманого за допомогою попередньої реакції, (1,33 г, 2,52 ммоль, 1,0 екв.) у сухому ДХМ (60 мл), охолодженому до 0°С, додавали в атмосфері аргону DIPEA (1,30 мл, 7,56 ммоль, 3,0 екв.). Далі, через 10 хвилин, додавали однією порцією HATU (1,25 г, 3,28 ммоль, 1,3 екв.) і Boc-Abu-OH (0,56 г, 2,77 ммоль, 1,1 екв.). Контроль повноти протікання реакції за допомогою ТШХ через 15 хвилин при 0°С і потім ночі при кімнатній температурі показав її завершення. Реакцію зупиняли додаванням 10%-го NaHCO3 (15 мл) і перемішували протягом 15 хвилин. Реакційну суміш розбавляли ДХМ (100 мл) і промивали 10%-вою лимонною кислотою (1х40 мл), H2O (1x40 мл) і розчином солі (1х40 мл). Органічну фазу сушили над Na2SO4, фільтрували і упарювали при зниженому тиску. Неочищену суміш розділяли за допомогою СС з одержанням складного ефіру Boc-Abu-D-MeAla-MeVal-Leu-MeLeuGly-OMe у вигляді жовтуватої піни. + + + НПРХ-ІЕР-МС (m/z) 713,1513 [M + H] ([C35H65N6O9] ; розр. 713,4813); 735,1010 [M + Na] + ([C35H64N6NaO9] ; розр. 735,4632). 2.10. Одержання H-Abu-D-MeAla-MeVal-Leu-MeLeu-Gly-OMe (Вa) Розчин гексапептиду, отриманого за допомогою попередньої реакції, (0,82 г, 1,15 ммоль, 1,0 екв.) у ТФО/ДХМ (6 мл, 1:1 об./об.) витримували протягом 1 години при 0°С і 1 години при кімнатній температурі і розчинники видаляли при зниженому тиску. Неочищений продукт сушили у високому вакуумі (приблизно 15 хвилин). Далі додавали ДХМ (25 мл) і суміш розтирали при 0°С з DIPEA до рН 7-8 для нейтралізації надлишку ТФО. Після розпарювання та висушування неочищену амінну сіль гексапептиду H-Abu-D-MeAla-MeVal-Leu-MeLeu-Gly-OMe застосовували для наступної стадії комбінації без додаткового очищення. + + НПРХ-ІЕР-МС (m/z) 613,1925 [M + H] ([C30H57N6O7] ; розр. 613,4289). 3. Комбінація Фрагменту (Aa) і Фрагменту (Ва) 3.1. Одержання D-Hiv-MeLeu-Leu-MeVal-MeBmt-Abu-D-MeAla-MeVal-Leu-MeLeu-Gly-OMe До розчину гексапептиду Н-Abu-D-MeAla-MeVal-Leu-MeLeu-Gly-OMe, отриманого за допомогою попередньої реакції, (0,7 г, 1,14 ммоль, 1,0 екв.) у сухому ДХМ (33 мл), 15 UA 102414 C2 5 10 15 20 25 30 35 40 45 50 55 60 охолодженому до 0°С, додавали в атмосфері аргону DIPEA (0,59 мл, 3,42 ммоль, 3,0 екв.). Далі, через 10 хвилин, додавали однією порцією HATU (0,56 г, 1,48 ммоль, 1,3 екв.) і пентапептид НD-Hiv-MeLeu-Leu-MeVal-MeBmt-ОН (0,75 г, 1,14 ммоль, 1,0 екв.). Контроль повноти протікання реакції за допомогою ТШХ через 15 хвилин при 0°С і потім 16,45 години при кімнатній температурі показав її завершення. Реакцію зупиняли додаванням 10%-го NaHCO3 (10 мл) і перемішували протягом 15 хвилин. Реакційну суміш розбавляли ДХМ (130 мл) і промивали 10%-вою лимонною кислотою (1х35 мл), H2O (1x35 мл) і розчином солі (1х35 мл). Органічну фазу сушили над Na2SO4, фільтрували і упарювали при зниженому тиску. Неочищений продукт очищали за допомогою СС з одержанням ундекапептиду D-Hiv-MeLeu-Leu-MeVal-MeBmt-Abu-DMeAla-MeVal-Leu-MeLeu-Gly-OMe у вигляді білої піни. + + + НПРХ-ІЕР-МС (m/z) 625,6903 [M/2 + H] ([C32H59N5O7] ; розр. 625,4415); 1249,7793 [M + H] + ([C64H117N10O14] ; розр. 1249,8751). 3.2. Одержання D-Hiv-MeLeu-Leu-MeVal-MeBmt-Abu-D-MeAla-MeVal-Leu-MeLeu-Gly-OH Розчин ундекадепсипептиду, отриманого за допомогою попередньої реакції, (1,30 г, 1,04 ммоль, 1,0 екв.) у ТГФ (10,4 мл) охолоджували до 0°С у водно-крижаній бані, потім додавали по краплях протягом 10 хвилин 0,2 М LiOH (10,4 мл, 2,08 ммоль, 2,0 екв.). Охолоджуючу баню залишали і суміш (рН = 12-13) перемішували при 0°С протягом приблизно 20 хвилин і 30 хвилин при кімнатній температурі (контроль ТШХ). Далі розчин охолоджували до 0°С і нейтралізували водним розчином 10%-вої лимонної кислоти (рН 3-4). Розчин упарювали при зниженому тиску. Залишок розчиняли в AcOEt (140 мл) і промивали Н2О (1х30 мл) і розчином солі (1х30 мл). Органічну фазу сушили над Na2SO4, фільтрували та упарювали при зниженому тиску. Неочищений продукт очищали за допомогою СС з одержанням ундекапептиду D-Hiv-MeLeuLeu-MeVal-MeBmt-Abu-D-MeAla-MeVal-Leu-MeLeu-Gly-OH у вигляді білого порошку. + + НПРХ-ІЕР-МС (m/z) 1235,7690 [M + H] ([C63H115N10O14] ; розр. 1235,8594). 4. Макролактонізація: одержання цикло-(MeBmt-Abu-D-MeAla-MeVal-Leu-MeLeu-Gly-D-HivMeLeu-Leu-MeVal) (Ia) До розчину DMAP (0,507 г, 4,144 ммоль, 4,0 екв.) і бензотриазол-1ілокситрипіролідинфосфонію гексафторфосфату (PyBOP) (1,078 г, 2,071 ммоль, 2,0 екв.) у ДХМ (128 мл) додавали по краплях протягом 2 годин розчин у ДХМ (20 мл) ундекадепсипептидної кислоти, отриманої за допомогою попередньої реакції (1,28 г, 1,036 ммоль, 1,0 екв.). Суміш перемішували протягом ночі (22 годин) після завершення додавання кислоти; потім суміш переносили в ділильну лійку. Загальний час реакції = 24 години. Розчин промивали 0,1 М Hcl (20 мл) і органічний шар відокремлювали та сушили над MgSO 4, фільтрували і концентрували при зниженому тиску. Неочищений продукт очищали за допомогою СС, у такий спосіб одержуючи циклоундекадепсипептид цикло-(MeBmt-Abu-D-MeAla-MeVal-Leu-MeLeu-Gly-D-HivMeLeu-Leu-MeVal) (Ia) у вигляді білого твердого залишку. Далі отриману сполуку очищали повторно за допомогою препаративної ВЕРХ (з одержанням цикло-(MeBmt-Abu-D-MeAla-MeValLeu-MeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal) у вигляді білого порошку. Чистота 99,38 % (80 °С, 30 хвилин). (C63H112N10O13, MW = 1217,6226 г/моль). НПРХ (40 ˚C, ACQUITY НПРХ BEH C18 1,7 мкм, 214 нм 50 95 %, 7 хв) tR = 2,923 хв. НПРХ Загальний вихід при одержанні ундекадепсипептиду цикло-(MeBmt-Abu-D-MeAla-MeVal-LeuMeLeu-Gly-D-Hiv-MeLeu-Leu-MeVal), починаючи з природного ундекадепсипептиду Цикло(MeBmt-Thr-Sar-MeLeu-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal), становить більше 50 %. Приклад 1b: Сполуки (Ic), (If), (Ig), (Ii) і (Ik) можуть бути отримані за допомогою послідовності реакцій, аналогічної наведеній в Прикладі 1а. Приклад 2: Одержання циклоундекадепсипептиду (Ib): цикло-(MeBmt-Abu-D-MeAla-EtVal-Leu-MeLeu-AlaD-Hiv-MeLeu-Leu-MeVal) (Ib) 2. Одержання Фрагменту (Bb): H-Abu-D-MeAla-EtVal-Leu-MeLeu-Ala-OMe (Bb) 2.1. Одержання Boc-MeLeu-Ala-OMe До розчину комерційно доступного Н-Ala-OMe•HCl (2,00 г, 14,32 ммоль, 1,0 екв.) у сухому ДХМ (130 мл), охолодженому до 0°С, додавали в атмосфері аргону DIPEA (14,68 мл, 86,0 ммоль, 6,0 екв.). Далі, через 15 хвилин, додавали однією порцією HATU (6,52 г, 17,18 ммоль, 1,2 екв.) і Boc-MeLeu-ОН (3,512 г, 14,32 ммоль, 1,0 екв.). Контроль повноти протікання реакції за допомогою ТШХ через 15 хвилин при 0°С і потім 48 годин при кімнатній температурі (RT = 2 дні 15 хвилин) показав її завершення. Реакцію зупиняли додаванням 10%-го NaHCO3 (20 мл) і перемішували протягом 15 хвилин. Реакційну суміш розбавляли ДХМ (200 мл) і промивали 16 UA 102414 C2 5 10 15 20 25 30 35 40 45 50 55 60 10%-вою лимонною кислотою (1х40 мл), H2O (1x40 мл) і розчином солі (1х40 мл). Органічну фазу сушили над Na2SO4, фільтрували і упарювали при зниженому тиску. Неочищену суміш розділяли за допомогою СС з одержанням складного ефіру Boc-MeLeu-Ala-OMe у вигляді блідої жовтуватої олії. Зразок (30 мг) очищали повторно за допомогою напівпрепаративної зверненофазової ВЕРХ (ЗФ-ВЕРХ, RP-HPLC) і ліофілізовували пептид з одержанням складного ефіру Boc-MeLeu-Ala-OMe у вигляді білого порошку. 2.2. Одержання H-MeLeu-Ala-OMe Розчин дипептиду, отриманого за допомогою попередньої реакції, (1,70 г, 5,14 ммоль, 1,0 екв.) у ТФО/ДХМ (10 мл, 2:3 об./об.) витримували при 0°С протягом 1,5 години і розчинники видаляли при зниженому тиску. Неочищений продукт сушили у високому вакуумі (20 хвилин). Далі додавали ДХМ (10 мл) і суміш розтирали при 0°С з DIPEA до рН 7-8 для нейтралізації надлишку ТФО. Далі реакційну суміш розбавляли ДХМ (60 мл) і промивали H 2O (1x10 мл) і розчином солі (1х10 мл). Органічну фазу фільтрували, сушили (Na2SO4), упарювали і сушили у високому вакуумі. Неочищену амінну сіль H-MeLeu-Ala-OMe застосовували для наступної стадії комбінації без додаткового очищення. 2.3. Одержання Boc-Leu-MeLeu-Ala-OMe До розчину дипептиду, отриманого за допомогою попередньої реакції (7), (2,17 г, 9,42 ммоль, 1,0 екв.) у сухому ДХМ (108 мл), охолодженому до 0°С, додавали в атмосфері аргону DIPEA (6,45 мл, 37,68 ммоль, 4,0 екв.). Далі, через 10 хвилин, додавали однією порцією HATU (4,65 г, 12,25 ммоль, 1,3 екв.) і Boc-Leu-ОН (2,39 г, 10,63 ммоль, 1,1 екв.). Контроль повноти протікання реакції за допомогою ТШХ через 15 хвилин при 0°С і потім 15 годин при кімнатній температурі (15 годин 15 хвилин) показав її завершення. Реакцію зупиняли додаванням 10%-го NaHCO3 (10 мл) і перемішували протягом 15 хвилин. Реакційну суміш розбавляли ДХМ (50 мл) і промивали 10%-вою лимонною кислотою (1х20 мл), H2O (1x20 мл) і розчином солі (1х20 мл). Органічну фазу сушили над Na2SO4, фільтрували і упарювали при зниженому тиску. Неочищену суміш розділяли за допомогою СС з одержанням трипептиду Boc-Leu-MeLeu-Ala-OMe у вигляді жовтуватої олії. Зразок очищали повторно за допомогою напівпрепаративної ЗФ-ВЕРХ і ліофілізовували пептид з одержанням трипептиду Boc-Leu-MeLeu-Ala-OMe у вигляді білого порошку. 2.4. Одержання H-Leu-MeLeu-Ala-OMe Розчин трипептиду, отриманого за допомогою попередньої реакції, (2,00 г, 4,50 ммоль, 1,0 екв.) у ТФО/ДХМ (10 мл, 2:3 об./об.) витримували при 0°С протягом 2 годин і розчинники видаляли при зниженому тиску. Продукт сушили у високому вакуумі (20 хвилин) з одержанням неочищеної речовини. Далі додавали ДХМ (20 мл) і суміш розтирали при 0°С з DIPEA до рН 7-8 (по лакмусовому паперу) для нейтралізації надлишку ТФО. Після розпарювання і висушування неочищений амін H-Leu-MeLeu-Ala-OMe застосовували для наступної стадії комбінації без додаткового очищення. Зразок (25 мг) очищали повторно за допомогою напівпрепаративної ЗФВЕРХ і ліофілізовували продукт з одержанням трипептиду H-Leu-MeLeu-Ala-OMe у вигляді твердого залишку. 2.5. Одержання Boc-D-MeAla-EtVal-OH Boc-D-MeAla-EtVal-OH одержували, починаючи з Boc-D-MeAla-Val-OМе, застосовуючи BuLi і Триетилоксонійфторборат з наступним гідролізом метилового ефіру згідно з джерелом [Jean François. Guichou, PhD thesis entitled “De nouveaux analogues de Cyclosporine A comme agent anti-VIH-1”, Faculté des Sciences, Université De Lausanne, 2001, p. 121-122]. 2.6. Одержання Boc-D-MeAla-EtVal-Leu-MeLeu-Ala-OMe До розчину трипептиду H-Leu-MeLeu-Ala-OMe (0,096 г, 0,291 ммоль, 1,0 екв.) у сухому ДХМ (8 мл), охолодженому до -8°С, додавали в атмосфері аргону DIPEA (0,15 мл, 0,87 ммоль, 3,0 екв.). Далі, через 10 хвилин, додавали однією порцією HATU (0,13 г, 0,35 ммоль, 1,2 екв.) і дипептид Boc-D-MeAla-EtVal-OH (0,1 г, 0,29 ммоль, 1,0 екв.). Контроль повноти протікання реакції за допомогою ВЕРХ через 10 хвилин при -8°С і потім 40 хвилин при кімнатній температурі показав її завершення. Реакцію зупиняли додаванням 10%-го NaHCO3 (1 мл) і перемішували протягом 15 хвилин. Реакційну суміш розбавляли ДХМ (20 мл) і промивали 10%вою лимонною кислотою (1х5 мл), H2O (1x5 мл) і розчином солі (1х5 мл). Органічну фазу сушили над Na2SO4, фільтрували і упарювали при зниженому тиску. Неочищену суміш розділяли за допомогою СС з одержанням пентапептиду Boc-D-MeAla-EtVal-Leu-MeLeu-Ala-OMe у вигляді олії. Зразок очищали повторно за допомогою напівпрепаративної ЗФ-ВЕРХ і ліофілізовували продукт з одержанням пентапептиду Boc-D-MeAla-EtVal-Leu-MeLeu-Ala-OMe у вигляді білого порошку. 2.7. Одержання H-D-MeAla-EtVal-Leu-MeLeu-Ala-OMe Розчин пентапептиду, отриманого за допомогою попередньої реакції, (0,45 г, 0,69 ммоль, 1,0 17 UA 102414 C2 5 10 15 20 25 30 35 40 45 50 55 60 екв.) у ТФО/ДХМ (4,0 мл, 2:3 об./об.) витримували при 0°С протягом 1,5 години і розчинники видаляли при зниженому тиску. Залишок сушили у високому вакуумі (0,5 години). Далі додавали ДХМ (10 мл) і суміш розтирали при 0°С з DIPEA до рН 7-8 (по лакмусовому паперу) для нейтралізації надлишку ТФО. Після розпарювання і висушування неочищену амінну сіль пентапептиду H-D-MeAla-EtVal-Leu-MeLeu-Ala-OMe застосовували для наступної стадії комбінації без додаткового очищення. Зразок (30 мг) очищали за допомогою напівпрепаративної ЗФ-ВЕРХ і ліофілізовували пептид з одержанням вільного аміну пентапептиду H-D-MeAla-EtVal-Leu-MeLeu-Ala-OMe у вигляді білого порошку. 2.8. Одержання Boc-Abu-D-MeAla-EtVal-Leu-MeLeu-Ala-OMe До розчину неочищеного пентапептиду, отриманого за допомогою попередньої реакції, (0,2 г, 0,36 ммоль, 1,0 екв.) у сухому ДХМ (10 мл), охолодженому до 0°С, додавали в атмосфері аргону DIPEA (0,18 мл, 1,08 ммоль, 3,0 екв.). Далі, через 5 хвилин, додавали однією порцією HATU (0,19 г, 0,5 ммоль, 1,4 екв.) і Boc-Abu-OH (0,088 г, 0,42 ммоль, 1,1 екв.). Контроль повноти протікання реакції за допомогою ТШХ через 15 хвилин при 0°С і потім 1,15 години при кімнатній температурі (RT = 1,5 години) показав її завершення. Реакцію зупиняли додаванням 10%-го NaHCO3 (2 мл) і перемішували протягом 15 хвилин. Реакційну суміш розбавляли ДХМ (50 мл) і промивали 10%-вою лимонною кислотою (1х10 мл), H2O (1x10 мл) і розчином солі (1х10 мл). Органічну фазу сушили над Na2SO4, фільтрували і упарювали при зниженому тиску. Неочищену суміш розділяли за допомогою СС з одержанням гексапептиду Boc-Abu-D-MeAla-EtVal-LeuMeLeu-Ala-OMe у вигляді жовтуватої олії. Зразок (30 мг) очищали повторно за допомогою напівпрепаративної ЗФ-ВЕРХ і ліофілізовували продукт з одержанням гексапептиду Boc-Abu-DMeAla-EtVal-Leu-MeLeu-Ala-OMe у вигляді білого порошку. 2.9. Одержання H-Abu-D-MeAla-EtVal-Leu-MeLeu-Ala-OMe Фрагмент (Bb) Розчин гексапептиду, отриманого за допомогою попередньої реакції, (0,187 г, 0,25 ммоль, 1,0 екв.) у ТФО/ДХМ (3,0 мл, 2:3 об./об.) витримували при 0°С протягом 1,5 години і розчинники видаляли при зниженому тиску. Неочищений продукт сушили у високому вакуумі (0,5 години). Далі додавали ДХМ (3 мл) і суміш розтирали при 0°С з DIPEA до рН 7-8 (по лакмусовому паперу) для видалення ТФО. Після розпарювання та висушування неочищений амін H-Abu-DMeAla-EtVal-Leu-MeLeu-Ala-OMe застосовували для наступної стадії комбінації без додаткового очищення. Зразок неочищеного продукту (23 мг) очищали повторно за допомогою напівпрепаративної ЗФ-ВЕРХ і ліофілізовували пептид з одержанням Фрагменту (Ва) H-Abu-DMeAla-EtVal-Leu-MeLeu-Ala-OMe у вигляді білого порошку. + + НПРХ-ІЕР-МС (m/z) 641,259 [M + H] ([C32H61N6O7] ; розр.641,4602). 3. Зв'язування фрагменту (Аа) і фрагменту (Bb) 3.1. Одержання H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-Abu-D-MeAla-EtVal-Leu-MeLeu-Ala-OMe До розчину гексапептиду H-Abu-D-MeAla-EtVal-Leu-MeLeu-Ala-OMe, отриманого за допомогою попередньої реакції, (0,16 г, 0,25 ммоль, 1,0 екв.) у сухому ДХМ (10 мл), охолодженому до 0°С, додавали в атмосфері аргону DIPEA (0,214 мл, 1,26 ммоль, 5,0 екв.). Далі, через 5 хвилин, додавали однією порцією HATU (0,12 г, 0,31 ммоль, 1,25 екв.) і пентадепсипептид H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-OH (4), отриманий так, як зазначено вище (0,16 г, 0,25 ммоль, 1,0 екв.). Контроль повноти протікання реакції за допомогою ТШХ через 15 хвилин при 0°С і потім 1,15 години при кімнатній температурі (RT = 1,5 години) показав її завершення. Реакцію зупиняли додаванням 10%-го NaHCO3 (3 мл) і перемішували протягом 15 хвилин. Реакційну суміш розбавляли ДХМ (40 мл) і промивали 10%-вою лимонною кислотою (1х8 мл), H2O (1x8 мл) і розчином солі (1х8 мл). Органічну фазу сушили над Na 2SO4, фільтрували і упарювали при зниженому тиску. Неочищену суміш розділяли за допомогою СС з одержанням ундекапептиду H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-Abu-D-MeAla-EtVal-Leu-MeLeuAla-OMe у вигляді білої піни. Аналітичний зразок (18 мг) очищали повторно за допомогою напівпрепаративної ЗФ-ВЕРХ і ліофілізовували продукт з одержанням ундекапептиду H-D-HivMeLeu-Leu-MeVal-MeBmt-Abu-D-MeAla-EtVal-Leu-MeLeu-Ala-OMe у вигляді білого порошку. 3.2. Одержання H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-Abu-D-MeAla-EtVal-Leu-MeLeu-Ala-OH Розчин метилового ефіру ундекапептиду, отриманого за допомогою попередньої реакції, (0,27 г, 0,21 ммоль, 1,0 екв.) у ТГФ (2,5 мл) охолоджували до 0°С у водно-крижаній бані, потім додавали по краплях протягом 10 хвилин 0,2 М LiOH (2,11 мл, 0,42 ммоль, 2,0 екв.). Охолоджуючу баню потім видаляли і суміш (рН = 12-13, по лакмусовому паперу) перемішували при кімнатній температурі протягом 1 години 20 хвилин (контроль ТШХ). Далі розчин охолоджували до 0°С и підкисляли 1,0 M HCl до рН 3-4. Розчин упарювали при зниженому тиску. Залишок розчиняли в AcOEt (40 мл) і промивали 10%-вою лимонною кислотою (1х8 мл), Н2 ПРО (1х8 мл) і розчином солі (1х8 мл). Органічну фазу сушили над Na 2SO4, фільтрували і упарювали при зниженому тиску. Неочищену суміш розділяли за допомогою СС з одержанням 18 UA 102414 C2 5 10 15 20 25 30 35 40 45 50 55 60 кислоти H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-Abu-D-MeAla-EtVal-Leu-MeLeu-Ala-OH у вигляді білого порошку. Зразок (40 мг) очищали повторно за допомогою напівпрепаративної ЗФ-ВЕРХ і ліофілізовували продукт з одержанням ундекапептиду H-D-Hiv-MeLeu-Leu-MeVal-MeBmt-Abu-DMeAla-EtVal-Leu-MeLeu-Ala-OН у вигляді білого порошку. 4. Макролактонізація: Одержання цикло-(MeBmt-Abu-D-MeAla-EtVal-Leu-MeLeu-Ala-D-HivMeLeu-Leu-MeVal) (Ib) До розчину DMAP (0,048 г, 0,39 ммоль, 4,0 екв.) і бензотриазол-1ілокситрипіролідинфосфонію гексафторфосфату (PyBOP) (0,102 г, 0,196 ммоль, 2,0 екв.) у ДХМ (12 мл) додавали по краплях протягом 1,5 години розчин у ДХМ (4 мл) ундекапептиду, отриманого за допомогою попередньої реакції (0,124 г, 0,098 ммоль, 1,0 екв.). Після завершення додавання кислоти суміш перемішували (RT = 24 години), потім переносили в ділильну лійку. Розчин промивали 1 М HCl (2 мл) і органічний шар відокремлювали та сушили над MgSO 4, фільтрували і концентрували при зниженому тиску. Неочищений продукт очищали за допомогою колонкової хроматографії, у такий спосіб одержуючи циклоундекадепсипептид цикло-(MeBmt-Abu-D-MeAla-EtVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) (Ia) у вигляді білого твердого залишку. Зразок (77 мг) очищали за допомогою напівпрепаративної ЗФ-ВЕРХ і ліофілізовували продукт з одержанням циклоундекадепсипептиду цикло-(MeBmt-Abu-D-MeAlaEtVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) (Ib) у вигляді білого порошку. + + + НПРХ-ІЕР-МС (m/z) 1245,787 [M + H] ([C65H117N10O13] ; розр. 1245,8802), 623,465 [M/2 + H] (розр. 623,444). Приклад 3: Одержання цикло-(MeBmt-Val-D-MeAla-EtVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) (Ih) Одержання (Ih) проводили аналогічно одержанню сполуки, описаної в Прикладі 2. Фрагмент (Bh) одержували, застосовуючи умови реакцій, аналогічні описаним у Прикладі 2 для одержання Фрагменту (Bb). Потім фрагмент (Bh) поєднували з Фрагментом (Аа), застосовуючи умови реакцій, подібні описаним у Прикладі 2. Потім проводили макролактонізацію, застосовуючи умови, аналогічні описаним у Прикладі 2а, з одержанням циклоундекадепсипептиду цикло-(MeBmt-Val-D-MeAla-EtVal-Leu-MeLeu-Ala-DHiv-MeLeu-Leu-MeVal) (Ih). + + 2+ НПРХ-ІЕР-МС (m/z) 1259,888 [M + H] ([C66H119N10O13] ; розр. 1259,8958); 1260,788 [M + 2H] 2+ ([C66H120N10O13] ; розр. 1260,9037). Приклад 4: Одержання цикло-(MeBmt-Abu-D-MeAla-MeVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) (Id) Фрагмент H-Abu-D-MeAla-MeVal-Leu-MeLeu-Ala-OMe (Bd) одержували, застосовуючи умови реакцій, подібні описаним у Прикладі 1 для одержання Фрагменту (Bа). Потім проводили комбінацію з Фрагментом (Аа) і макролактонізацію, застосовуючи умови реакцій, аналогічні описаним у Прикладі 1, з одержанням циклоундекадепсипептиду цикло-(MeBmt-Abu-D-MeAlaMeVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu-MeVal) (Id). + + НПРХ-ІЕР-МС (m/z) 1261,888 [M + H] ([C66H117N10O14] ; розр. 1261,8751) Приклад 5: Інгібіторні ефекти в системі реплікону ВГС З метою визначення, чи мають циклоундекадепсипептидні сполуки згідно з цим винаходом активність проти ВГС, були проведені експерименти, у яких порівнювали інгібуючу дію цих сполук у системі реплікону ВГС. Аналізи проводили на клітинах Huh 9-13, що містять реплікон Con1 вірусу гепатиту С генотипу 1b (Lohmann et al. 2001. J.Virol. 75: 1437-1449; і Lohmann et al. 1999. Science. 285: 110113). Клітини пересівали в середовище для росту клітин DMEM з додаванням 10 % сиворотки ембріонів великої рогатої худоби (FCS), 1 % амінокислот, що не є незамінними, 1 % пеніциліну/стрептоміцину і 2 % Geneticin (Invitrogen) у співвідношенні 1:4-1:5 і культивували 2 протягом 3-4 днів у культуральних матрацах для тканин площею 75 см (Techno Plastic Products), клітини збирали і висівали в досліджуване середовище (DMEM, 10 % FCS, 1 % амінокислот, що не є незамінними, 1 % пеніциліну/стрептоміцину) у концентрації 5000 клітин/лунка (100 мкл/лунка) в 96-лункові титраційні мікропланшети для тканинних культур (Falcon, Beckton Dickinson) для оцінки цитостатичного/цитотоксичного і противірусного ефекту. Мікропланшет для титруванн інкубували протягом ночі (37 °С, 5 % СО 2, відносн вологіст 95-99 %) з одержанням моношару клітин, що не злився. Щільність клітин і цитостатичні ефекти оцінювали в паралельних культурах у звичайних 96ямкових планшетах (Beckton-Dickinson), застосовуючи MTT-тест (CellTiter 96 AQueous NonRadioactive Cell Proliferation Assay, Promega). У даному тесті 3-(4,5-диметилтіазол-2-іл)-5-(3карбоксиметоксифеніл)-2-(4-сульфофеніл)-2Н-тетразолій (MTS) зазнає біологічного 19 UA 102414 C2 5 10 15 20 25 30 35 відновлення до формазану, який визначають кількісно при 498 нм за допомогою спектрофотометра для прочитання планшетів. Вироблення формазану прямо пов'язана з 2 кількістю живих клітин. Вимірювали поглинання на довжині хвилі 498 нм (Safire , Tecan) і оптичні щільності (значення OD) переводили в процентне відношення до неопрацьованих контролів. Сполуки розчиняли в ДМСО в концентрації 4 ммоль/л. Стокові розчини негайно застосовували для постановки експериментів. Сполуки були перевірені в 24 різних концентраціях у діапазоні від 0,0001 мкмоль/л до 100 мкмоль/л. Найвища концентрація ДМСО в аналізі становила 2,5 % (при кінцевій концентрації сполуки, рівній 100 мкм). Під час постановки аналізу процентний вміст ДМСО зменшували одночасно зі зменшенням концентрації сполуки. Для оцінки противірусних ефектів середовище для проведення аналізу видаляли і планшети з сухим моношаром зберігали при -80°С до екстракції. Після відтавання планшетів при кімнатній температурі клітинний моношар лізували за допомогою 100 мкл лізуючого буфера для виділення кДНК з клітин (Invitrogen). Лізис клітин продовжували протягом 10 хвилин при кімнатній температурі, після чого всю рідину переносили в планшет для полімеразної ланцюгової реакції (ПЦР) (Axygen). Планшет для ПЦР інкубували протягом 15 хвилин при 75 °С (Т3, Biometra). Лізат розбавляли 1:2 водою, що не містить рибонуклеаз/дезоксирибонуклеаз, після чого 5 мкл переносили в планшет для ПЦР у режимі реального часу (Applied Biosystems). Вміст РНК реплікону визначали кількісно, застосовуючи метод кількісної одностадійної ПЦР у режимі реального часу (RT-qПЦР): Low Rox One-Step RT-qPCR master mix, Abgene). Як прямий і зворотний праймер використовували 5’-CCA GAT CAT CCT GAT CGA CCA G-3’ і 5’CCG GCT ACC TGC CCA TTC-3’, відповідно. Флюорогенним зондом був 5'ACATCGCATCGAGCGAGCACGTAC-3'. Аналізи зразків виконували, застосовуючи SDS7500F (Applied Biosystems, стандартний термоциклічний профіль: 30 хвилин при 48 °С, 10 хвилин при 95 °С, 40 циклів по 15 секунд при 95 °С і 1 хвилину при 60 °С), після чого кількості РНК реплікону переводили в процентне відношення до неопрацьованих контролів. EC50 (значення, отримане з кривої доза-ефект) представляє концентрацію, при якій спостерігають 50%-ве пригнічення реплікації вірусу. CC 50 (значення, отримане з кривої дозаефект) представляє концентрацію, при якій метаболічна активність клітин знижена на 50 % у порівнянні з метаболічною активністю неопрацьованих клітин. Результати цих експериментів дозволили виконати розрахунки EC 50 ± SD (стандартне відхилення або Std. Dev.), яка являє собою ефективну концентрацію, необхідну для пригнічення реплікації реплікону ВГС на 50 %, і СC50 ± SD (у тих випадках, коли можливо), яка являє собою концентрацію, необхідну для пригнічення проліферації експоненціально зростаючих клітин на 50 %. Значення EC50 і СC50 ± SD розраховували відповідно як середнє значення всіх значень EC50 і СC50, отриманих з 3 індивідуальних кривих доза-ефект. Показник селективності (SI), що показує терапевтичне вікно сполуки, обчислювали як відношення СC 50 і EC50. У таблиці 1 показані результати порівняння значень EC50, СC50 (мкмоль/л) і SI обраних сполук у клітинах Huh 9-13. Таблиця 1 Сполука Debio 025 (Ia) (Ib) (Ic) (Id) (Ie) (If) (Ig) (Ih) (Ii) (Ik) EC50 (мкмоль/л) середнє 0,034 0,034 0,037 0,022 0,025 0,07 0,025 0,033 0,022 0,041 0,075 SD 0,02 0,019 0,023 0,006 0,013 0,02 0,02 0,016 0,02 0,023 0,033 CC50 (мкмоль/л) середнє 2,5 60,1 52,3 86,9 44,4 8,3 81,8 67 20,5 25,4 9,8 SI SD 0,6 3,3 0,9 73,5 1791,8 1398 4024,1 1745,9 117,9 3236,8 2045,2 950 623,8 132 40 Вважається, що концентрація сполуки викликає справжній противірусний ефект у системі реплікону ВГС, коли при цій конкретній концентрації ефективність проти реплікону значно перевищує 70%-вий поріг, і спостерігають зниження метаболічної активності не більше ніж на 30 20 UA 102414 C2 %. 5 10 15 Як показано в таблиці 1, дані результати, отримані в клітинах Huh 9-13, показують, що зазначені сполуки селективно інгібують реплікацію реплікону вірусу гепатиту С у клітинній культурі та демонструють ефективність від такої ж до більш високої, ніж в Debio 025, зі значеннями EC50 у двозначному наномолярному діапазоні, і показники селективності від таких же до більш високих. Приклад 6: Неімуносупресивна активність З метою визначення, чи мають циклоундекадепсипептидні сполуки згідно з цим винаходом неімуносупресивну активність, були проведені експерименти, які порівнювали дозозалежне пригнічення викликаної Конканаваліном А проліферації Т-клітин (див. Таблицю 2). Аналізи проводили згідно з методикою, описаною Дайтоном зі співавторами (Dayton et al.) (1992. Mol. Pharmacol. 41. 671-676) и Mishell et al. (1980. Cell proliferation in selected methods in cellular immunology, V, XXIX, W.H. Freeman Co., San Francisco, CA. 153-160). IC50 (половина максимальної інгібуючої концентрації) сполук одержували з кривої доза-ефект і порівнювали з IC50 CsA (відношення IC50). Таблиця 2 Сполука Циклоспорин A (CsA) (Ia) (Ib) (Ic) (Id) (Ie) (If) (Ig) (Ih) (Ii) (Ik) 20 25 30 35 40 45 IC50 (мікромоль/л) 0,006 2 9,44 0,984 1,435 2 7,725 2,69 1,74 2,69 3,44 Відношення IC50 1 333 1573 164 239 333 1288 448 290 448 573 У цьому дослідженні Сполуки (Ia), (Ib) і (Id) щонайменше мають приблизно в 300 разів меншу імуносупресивну дію, ніж циклоспорин А (CsA) (у показнику відношення IC 50). Приклад 7: Взаємодії з переносниками в печінці З метою визначення, чи взаємодіють циклоундекадепсипептидні сполуки згідно з цим винаходом з печіночними переносниками, проводили експерименти, які порівнювали залежне від концентрації пригнічення відносно білка 2, пов'язаного з множинною лікарською стійкістю (MRP2), поліпептиду 1В1, що переносить органічні аніони (OATP1B1), насосу, що експортує солі жовчних кислот (BSEP), і поліпептиду, котранспортуючому натрієву сіль таурохолевої кислоти (NTCP). Зазначені переносники залучені в гепатобіліарну циркуляцію ендогенних сполук, таких як солі жовчних кислот або білірубін (для розгляду див. C. Pauli-Magnus et al, J. Hepatology, 2005, 43: 342-357). Деякі з них також беруть участь в усуненні ксенобіотиків. MRP2 і BSEP експресували за допомогою SOLVO Biotechnology у клітинах яєчників Spodoptera frugiperda (Sf9), інфікуючи клітини комах рекомбінантним бакуловірусом. Пригнічення транспорту субстратів, що містять радіоактивний зонд (естрадіол-17бетаглюкуронід і таурохолат для MRP2 і BSEP відповідно), у перехідні зсередини назовні мембранні пухирці, що містять переносник, вимірювали підрахунком сцинтиляцій. Аналізи OATP1B1 і NTCP виконували за допомогою клітин яєчників китайського хом'яка (СНО), що експресують дані людські захоплюючі переносники. Кількість стерпного субстрату, що містить зонд (естрон-3-сульфат і таурохолат для OATP1B1 та NTCP відповідно) визначали за допомогою рідинної сцинтиляції або флюоресцентної фотометрії після лізису клітин. IC50 (що визначають як концентрацію, необхідну для пригнічення транспорту субстрату, що містить зонд, на 50 %, половина максимальної інгібіторної концентрації) сполук одержували з кривих концентрація-ефект і порівнювали з IC50 Debio 025 (відношення IC50 виражене як “кількість разів Debio 025” у таблицях). MRP2 являє собою головну рушійну силу для потоку солей жовчних кислот незалежно від виділення жовчі і переносить широкий спектр органічних аніонів, включаючи диглюкуронід білірубіну, кон’югати солей жовчних кислот, так як і деякі лікарські засоби. У таблиці 3 показані 21 UA 102414 C2 результати взаємодії обраних сполук стосовно цього переносника. Таблиця 3 MRP2 Debio 025 (Ib) (Ih) (Id) (Ie) (Ic) (Ia) (If) 5 Кратність Debio 025 1 n.a. n.a. n.a. 0,73 6,6 n.a. n.a. IC50 (мкM) 15 n.i. n.i. n.i. 20 2,2 n.i. n.i. n.i. немає пригнічення n.a. не застосовне OATP1B1 переносить солі жовчних кислот незалежним від натрію способом. Крім того, численні лікарські засоби є субстратами OATP. У таблиці 4 показані результати взаємодії обраних сполук стосовно OATP1B1. 10 Таблиця 4 OATP1B Debio 025 (Ib) (Ih) (Id) (Ie) (Ic) (Ia) (If) Кратність Debio 025 1 0,07 0,04 0,05 0,72 0,18 0,02 0,03 IC50 (мкM) 0,42 6,2 12 7,9 0,58 2,3 18 15 BSEP являє собою основну систему виведення солей жовчних кислот, залучену в секрецію холефільних сполук з клітини печінки в жовчний каналець. У таблиці 5 показані результати взаємодії обраних сполук стосовно BSEP. 15 Таблиця 5 BSEP Debio 025 (Ib) (Ih) (Id) (Ie) (Ic) (Ia) (If) Кратність Debio 025 1 1,6 0,80 0,42 0,40 1,5 0,60 0,50 IC50 (мкM) 0,24 0,15 0,30 0,57 0,60 0,16 0,40 0,48 NTCP опосередковує залежне від натрію поглинання солей жовчних кислот з кровообігу в системі ворітної вени, яке необхідне для утворення жовчі. Таблиця 6 показує результати взаємодії обраних сполук стосовно NTCP. 20 22 UA 102414 C2 Таблиця 6 NTCP Кратність Debio 025 1 0,05 n.a. 1,2 0,12 0,11 n.a. n.a. IC50 (мкM) 3,2 65 n.i. 2,5 26 30 n.i. n.i. Debio 025 (Ib) (Ih) (Id) (Ie) (Ic) (Ia) (If) n.i. немає пригнічення n.a. не застосовне 5 10 15 Крім зазначених досліджень in vitro за участю печіночних переносників, циклоундекадепсипептидні сполуки згідно з цим винаходом також оцінювали відносно пригнічення in vitro ферментів метаболізму, також як і фармакокінетичних властивостей у тварин in vivo. Як показано в таблицях 3-6, результати експериментів по переносу in vitro показують, що ці сполуки демонструють набагато меншу здатність інгібувати деякі печіночні переносники у порівнянні з Debio 025. Зокрема, деякі сполуки демонструють відсутність пригнічення відносно MRP2 і/або до 50 разів меншу активність стосовно OATP1B1. Таким чином, ці сполуки, як очікують, зменшать або можливі несприятливі ефекти (наприклад, гіпербілірубінемію), пов'язані з пригніченням переносників у печінці, або можливі взаємодії між лікарськими засобами (наприклад, з інгібіторами редуктази HMG-CoA, такими як аторвастатин), пов'язані з пригніченням переносників у печінці, або навіть і те, й інше. Список скорочень D-Hiv г HCOOH ВЕРХ HR-MALDI L--аміно-n-олійна кислота ацетил етилацетат оцтова кислота L-аланін трет-бутилоксикарбоніл насос, що експортує солі жовчних кислот трет-бутил метоксид кальцію колонкова хроматографія дихлорметан N,N-диізопропілетиламін 4-(N,N-диметиламіно)піридин диметилсульфоксид еквівалент мас-спектрометрія з іонізацією електророзпиленням N-етил-L-валін 9-флуоренілметоксикарбоніл N-[(диметиламіно)-1H-1,2,3-триазол[4,5-b]піридин-1-ілметилен]- Nметилметанамінію гексафторфосфат N-оксид D-гідроксиізовалеріанова кислота грам мурашина кислота високоефективна рідинна хроматографія мас-спектрометрія MALDI-TOF високої роздільної здатності HR-Q-TOF мас-спектрометрія Q-TOF високої роздільної здатності Im2CO N,N-карбонілдиімідазол (CDI) Abu Ac AcOEt AcOH Ala Boc BSEP t-Bu Ca(MeO)2 CC ДХМ DIPEA DMAP ДМСО Екв. ІЕР-МС EtVal Fmoc HATU MS MS 23 UA 102414 C2 кг KOMe MALDI MALDI-TOF Me D-MeAla MeBmt MeLeu MeCN MeOH MeVal мг мл MRP2 МС MtBE NMR NTCP OATP1B1 PyBOP ЗФ ЗФ-ВЕРХ ЧР Thr ТФО tR ТГФ Tj TШХ Tr НПРХ об. кілограм метоксид калію лазерна десорбція/іонізація зразка в матриці часопролітна мас-спектрометрія метил N-метил-D-аланін N-метил-(4R)-4-[(E)-2-бутеніл]-4,4-диметил-L-треонін N-метил-L-лейцин ацетонітрил метанол N-метил-L-валін міліграм мілілітр білок 2, зв’язаний з множинною лікарською стійкістю мас-спектрометрія метил-трет-бутиловий ефір ядерний магнітний резонанс поліпептид, котранспортуючий натрієву сіль таурохолевої кислоти поліпептид 1В1, що переносить органічні аніони (бензотриазол-1ілокси)-трис(піролідино)фосфонію гексафторфосфат зверненофазовий зверненофазова високоефективна рідинна хроматографія час реакції L-треонін трифтороцтова кислота час утримання тетрагідрофуран температура сорочки тонкошарова хроматографія температура в реакторі надпродуктивна рідинна хроматографія об’єми (1 г цільової необробленої речовини означає 1 об’єм) ФОРМУЛА ВИНАХОДУ 1. Циклоундекадепсипептидна сполука Формули (І) 5 10 15 20 25 у якій: АХХ1 являє собою MeBmt, 4-фтор-МеВmt, дигідро-MeBmt, 8-гідрокси-МеВmt; О-ацетил-MeBmt; АХХ2 являє собою Abu, Val, Thr, Thr(OMe), Thr(OAc), Thr(OCOCH2CH2CH2OH), Nva, 5-гідроксиNva; AXX3 являє собою D-MeAla, D-3-фтор-МеАlа, D-MeSer, D-MeSer(OAc), D-MeSer(OCH2CH2OH), D-MeSer(OCH2CH2NEt2), D-MeAsp(OMe); AXX4 являє собою MeIle, MeMet, MeVal, MeThr, MeThr(OAc), MeAla, EtVal, EtIle, EtPhe, EtTyr, EtThr(OAc), MeThr(OAc), MeTyr, MeTyr(OAc), МеТуr(ОМе), MePhe, MeMet(Ox), де атом сірки метіоніну є сульфоксидним або сульфоновим; АХХ5 являє собою Leu, Val, Ile; АХХ6 являє собою MeAla, Sar, MeLeu; і АХХ7 являє собою Gly, Ala. 2. Сполука за п. 1, яка відрізняється тим, що у Формулі (1) АХХ1 являє собою MeBmt, 8-гідрокси-МеВmt; АХХ2 являє собою Abu, Val, Thr, Thr(OMe), Thr(OAc), Thr(OCOCH2CH2CH2OH), 5-гідрокси-Nva; AXX3 являє собою D-MeAla, D-3-фтор-МеАlа, D-MeSer, D-MeSer(OAc), D-MeAsp(OMe); AXX4 являє собою MeIle, MeMet, MeMet(Ox), де Ox являє собою -SOMe, -SO2Me, MeVal, EtVal, EtIle, MeTyr; і AXX5 являє собою Leu, Val, Ile; AXX6 являє собою MeAla, Sar, MeLeu; і 24 UA 102414 C2 5 10 15 20 25 30 35 40 AXX7 являє собою Gly, Ala. 3. Сполука за будь-яким з пп. 1 або 2, яка відрізняється тим, що АХХ1 являє собою MeBmt; АХХ2 являє собою Abu, Val, Thr; AXX3 являє собою D-MeAla; АХХ4 являє собою MeIle, MeVal, EtVal; АХХ5 являє собою Leu, Val, Ile; АХХ6 являє собою MeAla, MeLeu, Sar; і АХХ7 являє собою Gly, Ala. 4. Сполука за будь-яким з пп. 1-3, що має наступну формулу: цикло-(MeBmt-Abu-D-MeAla-MeVal-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu(Iа) MeVal) цикло-(MeBmt-Abu-D-MeAla-EtVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu(Ib) MeVal) цикло-(MeBmt-Val-D-MeAla-MeVal-Ile-MeLeu-Gly-D-Hiv-MeLeu-Leu(Іс) MeVal) цикло-(MeBmt-Abu-D-MeAla-MeVal-Leu-MeLeu-Ala-D-Hiv-MeLeu-Leu(Id) MeVal) цикло-(MeBmt-Abu-D-MeAla-MeVal-Val-MeAla-Ala-D-Hiv-MeLeu-Leu(Iе) MeVal) цикло-(MeBmt-Val-D-MeAla-MeVal-Leu-MeLeu-Gly-D-Hiv-MeLeu-Leu(If) MeVal) цикло-(MeBmt-Abu-D-MeAla-MeVal-Ile-MeLeu-Gly-D-Hiv-MeLeu-Leu(Ig) MeVal) цикло-(MeBmt-Vаl-D-МеАlа-EtVal-Lеu-MeLeu-Аlа-D-Нiv-MeLeu-Lеu(Ih) MeVаl) цикло-(MeBmt-Abu-D-МеАlа-МеIlе-Lеu-МеLеu-Glу-D-Ніv-МеLеu-Lеu(Iі) МеVаl) цикло-(MeBmt-Val-D-MeAla-MeVal-Val-Sar-Gly-D-Hiv-MeLeu-Leu-MeVal) (Ik). 5. Фармацевтична композиція для запобігання або лікування інфекцій, що викликаються вірусом гепатиту С (ВГС), або розладів, що викликаються ВГС, яка містить сполуку за будь-яким з пп. 14 разом з одним або більше фармацевтично прийнятними розріджувачами або носіями. 6. Фармацевтична комбінація, що містить щонайменше а) перший засіб, що складається із сполуки за будь-яким з пп. 1-4 або фармацевтичної композиції за п. 5, і b) другий засіб, що має здатність протидіяти реплікації ВГС. 7. Фармацевтична комбінація за п. 6 для застосування для запобігання або лікування інфекцій, що викликаються вірусом гепатиту С, або розладів, що викликаються ВГС. 8. Сполука за будь-яким з пп. 1-4 як лікарський засіб. 9. Сполука за будь-яким з пп. 1-4 як противірусний засіб. 10. Застосування сполуки за будь-яким з пп. 1-4 для одержання лікарського засобу для лікування або запобігання інфекцій, що викликаються вірусом гепатиту С, або розладів, що викликаються ВГС. 11. Спосіб запобігання або лікування інфекцій, що викликаються вірусом гепатиту С, або розладів, що викликаються ВГС, у суб'єкта, що цього потребує, який включає введення зазначеному суб'єкту терапевтично ефективної кількості сполуки за будь-яким з пп. 1-4 або фармацевтичної композиції за п. 5. 12. Спосіб пригнічення реплікації ВГС у пацієнта, який цього потребує, що включає введення зазначеному суб'єкту терапевтично ефективної кількості сполуки за будь-яким з пп. 1-4 або фармацевтичної композиції за п. 5. 13. Спосіб за будь-яким з пп. 11-12, що включає спільне введення одночасно або послідовно терапевтично ефективної кількості сполуки за будь-яким з пп. 1-4 або фармацевтичної композиції за п. 5 і щонайменше другого засобу, вибраного із засобу, що має здатність протидіяти реплікації ВГС. 14. Спосіб за п. 13, який відрізняється тим, що зазначений засіб, який має спрямовані проти ВГС властивості, вибраний з групи, що складається з інтерферону, такого як інтерферон альфа2а або інтерферон альфа-2b; або інтерферону, кон'югованого з водорозчинним полімером або людським альбуміном; противірусного засобу, такого як рибавірин, ламівудин, NV08 або NM283; інгібітора факторів, що кодуються ВГС, наприклад, протеази NS3-4A, хелікази або РНКполімерази; протифіброзного засобу, такого як похідне N-феніл-2-піримідинаміну; 25 UA 102414 C2 імуномодулюючого засобу, такого як мікофенолова кислота; або агоніста рецептора S1P, такого як FTY720 або його необов'язково фосфорилованого аналога. Комп’ютерна верстка А. Крулевський Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 26
ДивитисяДодаткова інформація
Назва патенту англійськоюCycloundecadepsipeptide compounds for treating hepatitis c infections
Автори англійськоюWenger, Roland, Mutter, Manfred, Garrouste, Patrick, Lyser, Robert, Turpin, Olivier, Vuagniaux, Gregoire, Nicolas, Valerie, Novaroli Zanolari, Laura, Crabbe, Rafael
Назва патенту російськоюЦиклоундекадепсипептидные соединения для лечения инфекций, вызванных вирусом гепатита с
Автори російськоюВенгер Роланд, Муттер Манфред, Гарруст Патрик, Лисек Роберт, Тюрпен Оливье, Вуаньо Грегуар, Николя Валери, Новароли Цанолари Лаура, Краббе Рафаэль
МПК / Мітки
МПК: C07K 11/00, A61P 31/12, A61K 38/13, C07K 7/64
Мітки: гепатиту, циклоундекадепсипептидні, викликаних, лікування, вірусом, сполуки, інфекцій
Код посилання
<a href="https://ua.patents.su/28-102414-cikloundekadepsipeptidni-spoluki-dlya-likuvannya-infekcijj-viklikanikh-virusom-gepatitu-s.html" target="_blank" rel="follow" title="База патентів України">Циклоундекадепсипептидні сполуки для лікування інфекцій, викликаних вірусом гепатиту с</a>
Сполуки бензофурану, композиції та способи лікування та профілактики інфекцій, викликаних вірусом гепатиту с, і пов’язаних з ним захворювань

Номер патенту: 79834
Опубліковано: 25.07.2007
Автори: Кулкарні Бхеемашанкар А., Ріс Девід Дж., Блек-Ледж Чарльз В., Райнхардт Джейсон А., Фенг Хао, Ніц Теодор Дж., Саха Ашіз К., Бейлі Томас Р., Дель Веччіо Альфред М., Бернс Крістофер Дж., Лессен Томас А., Суесток Джон, Шерк Сюзан Р., Фе Томас Х., Денг Юіджун
МПК: C07D 405/06, C07D 407/12, C07D 417/14, C07D 413/14, C07D 407/04, C07D 409/04, C07D 417/04, C07D 405/04, C07D 413/12, C07D 417/12, C07D 413/04, C07D 405/12, C07D 307/84
Мітки: вірусом, сполуки, пов'язаних, інфекцій, гепатиту, композиції, бензофурану, ним, викликаних, захворювань, профілактики, способи, лікування
Формула / Реферат:
1. Сполука формули , Iде:R1 означає радикал, який вибирають із групи, яка включає водень, алкіл, галоген і ціаногрупу,R2 означає радикал, який вибирають із групи, яка включає водень, заміщений або незаміщений алкіл, заміщену або незаміщену алкоксигрупу, гідрокси, циклоалкіл, циклоалкілокси, поліфторалкіл, поліфторалкокси, галоген, аміногрупу,...
Застосування індольної сполуки для лікування або попередження інфікування вірусом гепатиту с

Номер патенту: 101465
Опубліковано: 10.04.2013
Автори: Фазал Гулрез, Пупар Марк-Андре, Гіллард Джеймс, Жолікьор Ерік, Гуле Сільві, Ранкур Жан, Цантрізос Йола С., Кюколь Жорж, Больо П'ер Луі, Пуар'є Мартен
Мітки: гепатиту, вірусом, індольної, попередження, лікування, інфікування, сполуки, застосування
Формула / Реферат:
1. Застосування сполуки, представленої формулою IIc, або її енантіомера, діастереомеру або таутомера:, (IIс) у якій: R1 вибирають з групи, яка складається з: Η та (С1-6)алкілу; R2 вибирають з: 6- або 10-членного арилу або Het, кожний з яких необов'язково монозаміщений або дизаміщений...
Вакцинний препарат та спосіб лікування хворого з інфекцією вірусу гепатиту або схильного до інфікування вірусом гепатиту

Номер патенту: 44688
Опубліковано: 15.03.2002
Автори: Воє П'єр, Гарсон-Джонсон Наталі Марі-Жозеф Клод, Хозер П'єр, Тіріар Клотільда
МПК: A61K 39/29, A61K 39/295
Мітки: хворого, схильного, гепатиту, інфікування, вірусу, препарат, інфекцією, вакцинний, вірусом, спосіб, лікування
Формула / Реферат:
1. Вакцинный препарат, содержащий антиген вируса гепатита и фармацевтически приемлемый носитель, отличающийся тем, что он содержит антиген вируса гепатита в комбинации с 3-0-дезацилированным монофосфорил-липидом А в количестве 10-100 мкг на дозу.2. Вакцинный препарат по п. 1, отличающийся тем, что в качестве носителя он содержит алюминат.3. Вакцинный препарат по п. 1, отличающийся тем, что в качестве носителя он содержит...
1-гідрокси-2-піридони як засіб для лікування інфекцій шкіри, викликаних резистентними до антибіотиків бактеріями

Номер патенту: 56192
Опубліковано: 15.05.2003
Автори: Маркус Астрид, Бон Манфред, Кремер Карл Теодор
МПК: A61P 17/00, A61K 31/44
Мітки: антибіотиків, інфекцій, викликаних, 1-гідрокси-2-піридони, бактеріями, резистентними, лікування, засіб, шкіри
Формула / Реферат:
1. Застосування 1-гідрокси-2-піридону формули І: ,(I)в якій R1, R2, R3 однакові або різні означають атом водню або алкіл з 1-4 атомами вуглецю, іR4 означає насичений вуглеводневий радикал з 6-9 атомами вуглецю або радикал формули II ,(II)причомуХ означає S або О,У означає атом водню або від 1 до 2 атомів галогену,...
Композиція та спосіб лікування захворювань, викликаних вірусом герпесу, та інших інфекційних захворювань

Номер патенту: 65537
Опубліковано: 15.04.2004
Автор: Сквайєрз Меріл
МПК: A61K 39/385, A61K 36/00, A61K 45/00, A01N 33/12, A01N 65/00, A61K 36/28, A61P 31/22, A61K 31/14, A61P 31/12, A61K 31/13, A61K 31/19
Мітки: інших, вірусом, інфекційних, спосіб, викликаних, герпесу, лікування, композиція, захворювань
Формула / Реферат:
1. Композиція, що містить рослинний компонент та поверхнево-активну речовину, яка відрізняється тим, що рослинний компонент являє собою трав’янисту рослину роду Echinacea, а поверхнево-активна речовина являє собою галогенід бензалконію.2. Композиція за п. 1, яка відрізняється тим, що композиція містить приблизно від 2% мас. до 90% мас. трав’янистої рослини роду Echinacea та приблизно від 0,005% мас. до 0,8% мас. галогеніду...
Попередній патент: Спосіб отримання електропровідного волокнистого матеріалу
Наступний патент: Багатофункціональний підсилювач шинкаренка
Випадковий патент: Вихровий енергетизатор