Похідне 3,7-діазадицикло[3,3,1] сполук, їх фармацевтично прийнятні солі, спосіб одержання та композиція на їх основі як антиаритмічні агенти
Номер патенту: 74629
Опубліковано: 16.01.2006
Автори: Геррінґ Ейдем, Кледінґбойль Дейвід, Барнвелл Ніл, Чіма Лал, Бйоре Анніка, Левквіст Карін







Формула / Реферат
1. Фармацевтично прийнятна сіль одної з таких сполук:
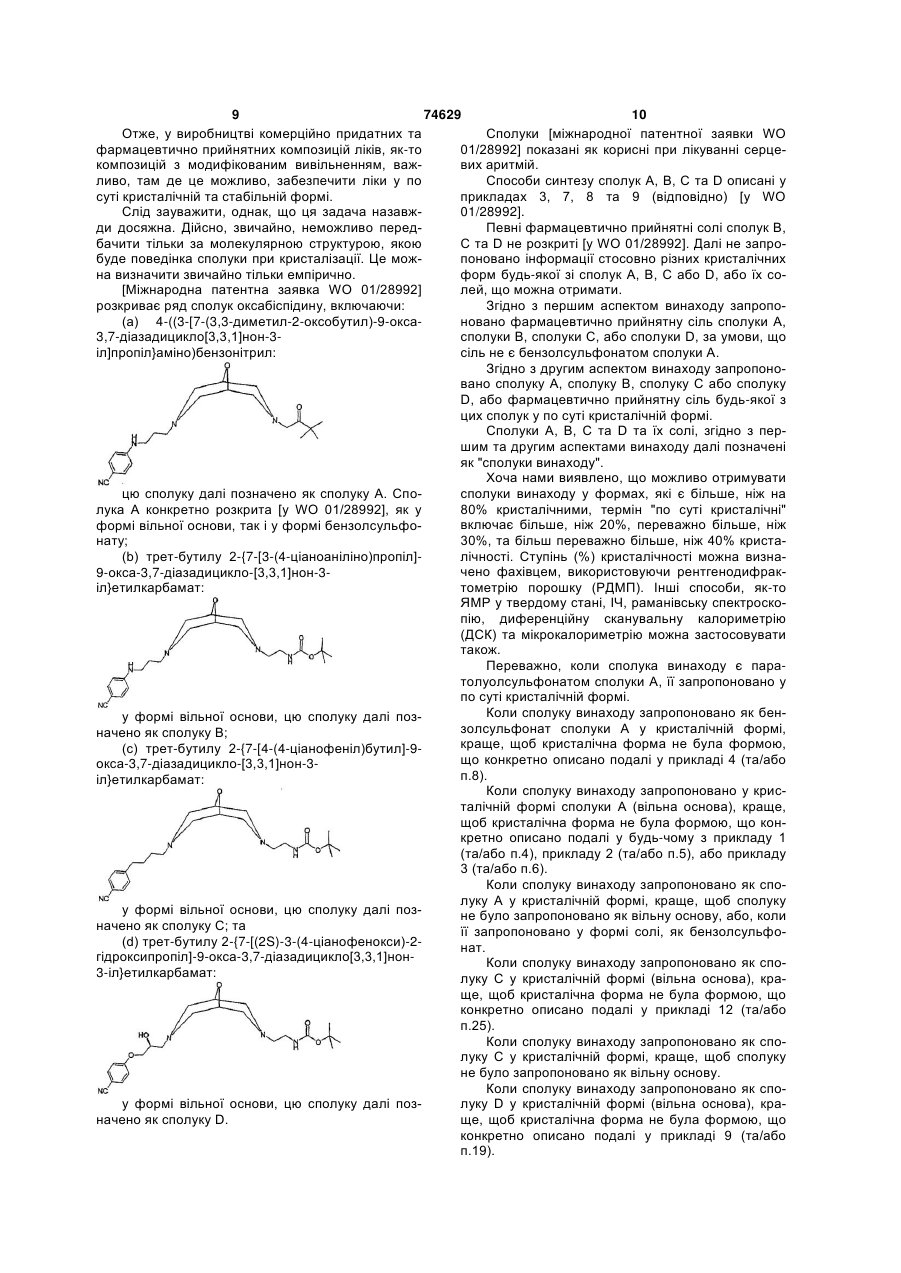
4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7-діазадицикло[3,3,1]нон-3-іл]пропіл}аміно)бензонітрил;
трет-бутилу 2-{7-[3-(4-ціаноаніліно)пропіл]-9-окса-3,7-діазадицикло[3,3,1]нон-3-іл}етилкарбамат;
трет-бутилу 2-{7-[4-(4-ціанофеніл)бутил]-9-окса-3,7-діазадицикло[3,3,1]нон-3-іл}етилкарбамат або
трет-бутилу 2-{7-1(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7-діазадицикло[3,3,1 ]нон-3-іл}етилкарбамат,
за умови, що сіль не є бензолсульфонатом 4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7-діазадицикло[3,3,1]нон-3-іл]пропіл}аміно)бензонітрилу.
2. Сполука, що вибрана з: 4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7-діазадицикло[3,3,1]нон-3-іл]пропіл}аміно)бензонітрилу;
трет-бутилу 2-{7-[3-(4-ціаноаніліно)пропіл]-9-окса-3,7-діазадицикло-[3,3,1]нон-3-іл}етилкарбамату;
трет-бутилу 1-{7-[4-(4-ціанофеніл)бутил]-9-окса-3,7-діазадицикло-[3,3,1]нон-3-іл}етилкарбамату або
трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7-діазадицикло[3,3,1]нон-3-іл}етилкарбамату,
або фармацевтично прийнятної солі будь-якої з цих сполук по суті в кристалічній формі.
3. Сполука за п. 2, якою є 4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7-діазадицикло[3,3,1 ]нон-3-іл]пропіл}аміно)бензонітрил.
4. Сполука за п. 3, що характеризується кривою диференційної сканувальної калориметрії, при швидкості нагрівання 10°С/хвилину у закритій чашці з мікроотвором під струмом азоту, виявляючи ендотермічний ефект з екстрапольованими температурними початками приблизно 121°С та 126°С; та/або рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 11,0, 7,8, 7,0, 5,7, 5,4, 5,1, 4,94, 4,71, 4,62, 4,54, 4,44, 4,34, 4,20, 3,92, 3,65, 3,51, 3,41, 3,34 та 2,89 ![]() , та/або яка по суті визначена у таблиці 1 та/або у фігурі 1.
, та/або яка по суті визначена у таблиці 1 та/або у фігурі 1.
5. Сполука за п. 3, що характеризується кривою диференційної сканувальної калориметрії, при швидкості нагрівання 10°С/хвилину у закритій чашці з мікроотвором під струмом азоту, виявляючи ендотермічний ефект з екстрапольованим температурним початком приблизно 125°С; та/або рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 10,9, 8,3, 7,8, 6,9, 6,7, 5,6 (5,64), 5,6 (5,56), 5,5, 5,4, 5,1, 5,0, 4,84, 4,78, 4,70, 4,49, 4,45, 4,36, 4,15, 4,10, 4,03, 3,97, 3,90, 3,80, 3,73, 3,47, 3,31, 2,95, 2,89, 2,85 та 2,80 ![]() , та/або яка по суті визначена у таблиці 2 та/або у фігурі 2.
, та/або яка по суті визначена у таблиці 2 та/або у фігурі 2.
6. Сполука за п. 3, що характеризується кривою диференційної сканувальної калориметрії, при швидкості нагрівання 10°С/хвилину у закритій чашці з мікроотвором під струмом азоту, виявляючи ендотермічний ефект з екстрапольованим температурним початком приблизно 122°С; та/або рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 10,4, 9,6, 5,7, 5,3, 5,2, 4,83, 4,71, 4,55, 3,83, 3,58, 3,50, 3,29, 3,22 та 3,19 ![]() , та/або яка по суті визначена у таблиці 3 та/або у фігурі 3.
, та/або яка по суті визначена у таблиці 3 та/або у фігурі 3.
7. Сіль за п. 2, якою є бензолсульфонат 4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7-діазадицикло[3,3,1 ]нон-3-іл]пропіл}аміно)бензонітрилу.
8. Сіль за п. 7, що характеризується рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 18,7, 9,4, 6,3, 4,69, 4,48, 3,76, 3,35, 3,13, 2,68, 2,35 та 1,88 ![]() , та/або яка по суті визначена у таблиці 4 та/або у фігурі 4.
, та/або яка по суті визначена у таблиці 4 та/або у фігурі 4.
9. Сіль за п. 1 або п. 2, якою є толуолсульфонат 4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7-діазадицикло[3,3,1 ]нон-3-іл]пропіл}аміно)бензонітрилу.
10. Сіль за п. 9, що характеризується кривою диференційної сканувальної калориметрії, при швидкості нагрівання 10°С/хвилину у закритій чашці з мікроотвором під струмом азоту, виявляючи ендотермічний ефект з екстрапольованим температурним початком приблизно 145°С; та/або рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 19,9, 10,0, 6,0, 4,99, 4,86, 4,38, 4,36, 4,19, 3,99 та 3,33 ![]() , та/або яка по суті визначена у таблиці 5(а) та/або у фігурі 5(а).
, та/або яка по суті визначена у таблиці 5(а) та/або у фігурі 5(а).
11. Сіль за п. 9, що характеризується кривою диференційної сканувальної калориметрії, при швидкості нагрівання 10°С/хвилину у закритій чашці з мікроотвором під струмом азоту, виявляючи ендотермічний ефект з екстрапольованим температурним початком приблизно 153°С; та/або рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 18,6, 9,3, 6,2, 4,66, 4,49, 3,73 та 3,11 ![]() , та/або яка по суті визначена у таблиці 5(b) та/або у фігурі 5(b).
, та/або яка по суті визначена у таблиці 5(b) та/або у фігурі 5(b).
12. Сіль за п. 1 або п. 2, якою є сіль гідроксинафтойної кислоти 4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7-діазадицикло[3,3,1 ]нон-3-іл]пропіл}аміно)бензонітрилу.
13. Сіль за п. 12, що характеризується рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 17,6, 12,4, 8,8, 5,7, 5,6, 5,1, 4,95, 4,88, 4,47, 4,16, 4,08, 3,84, 3,80, 3,33 та 3,03 ![]() , та/або яка по суті визначена у таблиці 6 та/або у фігурі 6.
, та/або яка по суті визначена у таблиці 6 та/або у фігурі 6.
14. Сіль за п. 1 або п. 2, якою є сіль нафталінсульфонової кислоти 4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7-діазадицикло[3,3,1]нон-3-іл]пропіл}аміно)бензонітрилу.
15. Сіль за п. 14, що характеризується рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 16,1, 8,1, 7,1, 6,3, 6,0, 5,4 (5,39), 4,96, 4,88, 4,68, 4,49, 4,34, 4,10, 4,04, 3,94, 3,44, 3,40, 3,23, 3,20 та 3,15 ![]() , та/або яка по суті визначена у таблиці 7 та/або у фігурі 7.
, та/або яка по суті визначена у таблиці 7 та/або у фігурі 7.
16. Сіль за п. 1 або п. 2, якою є сіль мезитиленсульфонової кислоти 4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7-діазадицикло[3,3,1 ]нон-3-іл]пропіл}аміно)бензонітрилу.
17. Сіль за п. 16, що характеризується рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 18,8, 9,5, 6,3 (6,33), 6,3 (6,26), 4,75, 4,50, 4,46, 3,92, 3,80 та 3,17 ![]() , та/або яка по суті визначена у таблиці 8 та/або у фігурі 8.
, та/або яка по суті визначена у таблиці 8 та/або у фігурі 8.
18. Сполука за п. 2, якою є трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-пдроксипропіл]-9-окса-3,7-діазадицикло-[3,3,1]нон-3-іл}етилкарбамат.
19. Сполука за п. 18, що характеризується кривою диференційної сканувальної калориметрії, при швидкості нагрівання 10°С/хвилину у закритій чашці з мікроотвором під струмом азоту, виявляючи ендотермічний ефект з екстрапольованим температурним початком приблизно між 100 та 102°С; та/або рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 22,3, 11,2, 8,4, 7,1, 6,4, 5,8, 5,5, 5,2, 4,91, 4,81, 4,62, 4,52, 4,32, 4,22, 4,12, 4,06, 3,91, 3,81, 3,50, 3,34 та 3,15 ![]() , та/або яка по суті визначена у таблиці 9 та/або у фігурі 9.
, та/або яка по суті визначена у таблиці 9 та/або у фігурі 9.
20. Сіль за п. 1 або п. 2, якою є метансульфонат трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7-діазадицикло[3,3,1,1]нон-3-іл}етилкарбамату.
21. Сіль за п. 20, що характеризується кривою диференційної сканувальної калориметрії, при швидкості нагрівання 10°С/хвилину у закритій чашці з мікроотвором під струмом азоту, виявляючи ендотермічний ефект з екстрапольованим температурним початком приблизно 167°С; та/або рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 12,7, 9,4, 7,3, 7,1, 6,6, 6,0, 5,4, 5,1, 4,92, 4,83, 4,27, 4,14, 4,05, 3,99, 3,87, 3,73, 3,65, 3,42, 3,37 та 3,00 ![]() , та/або яка по суті визначена у таблиці 10 та/або у фігурі 10.
, та/або яка по суті визначена у таблиці 10 та/або у фігурі 10.
22. Сіль за п. 1 або п. 2, якою є сіль гіпурової кислоти трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7-діазадицикло-[3,3,1]нон-3-іл}етилкарбамату.
23. Сіль за п. 22, що характеризується рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 16,4, 6,9, 6,2, 6,1, 5,5, 5,2, 5,1, 4,93, 4,61, 4,50, 4,28, 4,20, 4,11 та 3,68 ![]() , та/або яка по суті визначена у таблиці 11 та/або у фігурі 11.
, та/або яка по суті визначена у таблиці 11 та/або у фігурі 11.
24. Сполука за п. 2, якою є трет-бутилу 2-{7-[4-(4-ціанофеніл)бутил]-9-окса-3,7-діазадицикло[3,3,1 ]нон-3-іл}етилкарбамат.
25. Сполука за п. 24, що характеризується кривою диференційної сканувальної калориметрії, при швидкості нагрівання 10°С/хвилину у закритій чашці з мікроотвором під струмом азоту, виявляючи ендотермічний ефект з екстрапольованим температурним початком приблизно 97°С; та/або рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 19,4, 10,0, 9,1, 8,1, 6,5, 5,5, 5,2, 5,1, 4,99, 4,90, 4,46, 4,32, 4,06, 4,00, 3,85 та 3,80 ![]() , та/або яка по суті визначена у таблиці 12 та/або у фігурі 12.
, та/або яка по суті визначена у таблиці 12 та/або у фігурі 12.
26. Сіль за п. 1 або п. 2, якою є метансульфонат трет-бутилу 2-{7-[4-(4-ціанофеніл)бутил]-9-окса-3,7-діазадицикло[3,3,1]нон-3-іл}етилкарбамату.
27. Сіль за п. 26, що характеризується кривою диференційної сканувальної калориметрії, при швидкості нагрівання 10°С/хвилину у закритій чашці з мікроотвором під струмом азоту, виявляючи ендотермічний ефект з екстрапольованими температурними початками приблизно 145°С та 170°С; та/або рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 13,3, 12,3, 9,6, 7,5, 6,9, 6,7, 5,5, 5,1, 5,0, 4,89, 4,81, 4,34, 4,23, 4,20, 4,08, 3,89, 3,85 та 3,80 ![]() , та/або яка по суті визначена у таблиці 13 та/або у фігурі 13.
, та/або яка по суті визначена у таблиці 13 та/або у фігурі 13.
28. Сіль за п. 1 або п. 2, якою є толуолсульфонат трет-бутилу 2-{7-[4-(4-ціанофеніл)бутил]-9-окса-3,7-діазацикло[3,3,1 ]нон-3-іл}етилкарбамату.
29. Сіль за п. 28, що характеризується кривою диференційної сканувальної калориметрії, при швидкості нагрівання 10°С/хвилину у закритій чашці з мікроотвором під струмом азоту, виявляючи ендотермічний ефект з екстрапольованим температурним початком приблизно 138°С; та/або рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 13,2, 7,1, 5,2, 5,0, 4,67, 4,28, 4,24, 4,19, 4,08, 3,36 та 3,12 ![]() , та/або яка по суті визначена у таблиці 14 та/або у фігурі 14.
, та/або яка по суті визначена у таблиці 14 та/або у фігурі 14.
30. Сіль за п. 1 або п. 2, якою є сіль [(дифеніл-4-карбоніл)аміно]оцтової кислоти трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7-діазадицикло-[3,3,1 ]нон-3-іл)етилкарбамату.
31. Сіль за п. 30, що характеризується рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 20,4, 15,3, 11,5, 9,4, 7,7, 7,2, 5,9, 5,7, 5,4, 5,3, 5,2, 4,60, 4,54, 4,13, 3,85, 3,79, 3,64, 3,61, 3,40 та 2,94 ![]() , та/або яка по суті визначена у таблиці 15 та/або у фігурі 15.
, та/або яка по суті визначена у таблиці 15 та/або у фігурі 15.
32. Сіль за п. 1 або п. 2, якою є сіль гемібурштинової кислоти трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7-діазадицикло-[3,3,1]нон-3-іл}етилкарбамату.
33. Сіль за п. 32, що характеризується рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 18,0, 8,3, 8,0, 7,5, 7,3, 6,8, 6,1, 5,8, 5,4, 4,89, 4,79, 4,68, 4,59, 4,54, 4,43, 4,35, 4,18, 4,04, 3,99, 3,90, 3,83, 3,67, 3,58, 3,07 та 2,47 ![]() , та/або яка по суті визначена у таблиці 16 та/або у фігурі 16.
, та/або яка по суті визначена у таблиці 16 та/або у фігурі 16.
34. Сіль за п. 1 або п. 2, якою є сіль (3,4-дихлорбензоїламіно)оцтової кислоти трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7-діазадицикло-[3,3,1]нон-3-іл}етилкарбамату.
35. Сіль за п. 34, що характеризується рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 16,6, 13,8, 10,9, 6,3, 6,2, 5,5, 5,3, 5,1, 5,0, 4,60, 4,33, 4,30, 4,22, 3,85, 3,70, 3,50, 3,30, 3,16, 3,06 та 2,99 ![]() , та/або яка по суті визначена у таблиці 17 та/або у фігурі 17.
, та/або яка по суті визначена у таблиці 17 та/або у фігурі 17.
36. Сіль за п. 1 або п. 2, якою є сіль [(нафталін-2-карбоніл)аміно]оцтової кислоти трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7-діазадицикло-[3,3,1]нон-3-іл)етилкарбамату.
37. Сіль за п. 36, що характеризується рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 16,8, 6,2, 5,8, 5,6, 5,2, 5,1, 4,90, 4,76, 4,66, 4,53, 4,37, 4,31, 4,23, 4,08, 3,51, 3,25 та 3,12 ![]() , та/або яка по суті визначена у таблиці 18 та/або у фігурі 18.
, та/або яка по суті визначена у таблиці 18 та/або у фігурі 18.
38. Сіль за п. 1 або п. 2, якою є сіль 2,2,3,3,-тетраметил-1,4-дибутанової кислоти трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7-діазадицикло[3,3,1 ]нон-3-іл}етилкарбамату.
39. Сіль за п. 38, що характеризується рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 14,2, 10,3, 7,1, 6,4, 6,0, 5,6, 5,4, 5,1, 4,84, 4,77, 4,73, 4,66, 4,50, 4,22, 3,78, 3,62, 3,49, 3,35 та 3,09 ![]() , та/або яка по суті визначена у таблиці 19 та/або у фігурі 19.
, та/або яка по суті визначена у таблиці 19 та/або у фігурі 19.
40. Сіль за п. 1 або п. 2, якою є сіль 1,2-циклопентандикарбонової кислоти трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7-діазадицикло-[3,3,1]нон-3-іл}етилкарбамату.
41. Сіль за п. 40, що характеризується рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 12,9, 11,1, 10,7, 9,0, 7,7, 7,0, 6,5, 6,3, 6,1, 6,0, 5,6, 5,4, 5,3, 5,2, 4,73, 4,68, 4,31, 4,26, 4,02, 3,86, 3,61, 3,50, 3,24 та 2,90 ![]() , та/або яка по суті визначена у таблиці 20 та/або у фігурі 20.
, та/або яка по суті визначена у таблиці 20 та/або у фігурі 20.
42. Сіль за п. 1 або п. 2, якою є сіль О,О'-дибензоїлвинної кислоти трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7-діазадицикло-[3,3,1]нон-3-іл}етилкарбамату.
43. Сіль за п. 42, що характеризується рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 15,1, 13,5, 7,1, 6,6, 5,3, 5,2, 4,79, 4,50, 4,38, 4,25, 3,49 та 2,92 ![]() , та/або яка по суті визначена у таблиці 21 та/або у фігурі 21.
, та/або яка по суті визначена у таблиці 21 та/або у фігурі 21.
44. Сіль за п. 1 або п. 2, якою є сіль О,О'-ди-пара-толуоїлвинної кислоти трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7-діазадицикло-[3,3,1]нон-3-іл}етилкарбамату.
45. Сіль за п. 44, що характеризується рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 15,8, 6,8, 5,7, 5,6, 5,3 (5,25), 4,84, 4,80, 4,75, 4,39, 4,37 та 3,98 ![]() , та/або яка по суті визначена у таблиці 22 та/або у фігурі 22.
, та/або яка по суті визначена у таблиці 22 та/або у фігурі 22.
46. Спосіб отримання солі за будь-яким одним з пп. 1, 2, 7-17, 20-23 або 26-45, який включає додавання кислоти або основи (за прийнятністю), до прийнятної вільної основи сполуки, яку визначено у п. 1.
47. Спосіб за п. 46, який включає додавання кислоти до прийнятної вільної основи сполуки.
48. Спосіб отримання сполуки за будь-яким одним з пп. 1-45, який включає кристалізацію прийнятної вільної основи сполуки, яку визначено у п. 1, або підхожої фармацевтично прийнятної її солі.
49. Спосіб за п. 48, який включає кристалізацію сполуки або солі з розчинника.
50. Спосіб за п. 49, де розчинник вибирають з групи: ацетати, нижчі алкіл-спирти, аліфатичні та ароматичні вуглеводні, діалкілетери, діалкілкетони, ацетонітрил, хлоровані алкани, водні розчинники, або їх суміші.
51. Спосіб за п. 50 де розчинник вибирають з групи: С1-6 алкілацетати, лінійні або розгалужені С1-6алкілспирти, С6-12аліфатичні вуглеводні, С6-10ароматичні вуглеводні, ді-С1-6-алкілові етери, ді-С1-6алкілкетони, хлоровані метани або етани, ацетонітрил, вода, або їх суміші.
52. Спосіб за п. 51 де розчинник вибирають з групи: етилацетат, ізопропілацетат, метанол, етанол, ізопропанол, н-гептан, діетиловий етер, ацетон, дихлорметан, вода, або їх суміші.
53. Спосіб отримання солі за будь-яким одним з пп. 1, 2, 7-17, 20-23 або 26-45, який включає додавання за п. 46 або п. 47, а потім кристалізацію за будь-яким одним з пп. 48-52.
54. Спосіб отримання кристалічної форми 4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7-діазадицикло[3,3,1]нон-3-іл]пропіл}аміно)бензонітрилу; трет-бутилу 2-{7-[3-(4-ціаноаніліно)пропіл]-9-окса-3,7-діазадицикло[3,3,1]нон-3-іл}етилкарбамату; трет-бутилу 2-{7-[4-(4-ціанофеніл)бутил]-9-окса-3,7-діазадицикло[3,3,1]нон-3-іл}етилкарбамату або трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7-діазадицикло[3,3,1]нон-3-іл}етилкарбамату, або фармацевтично прийнятної солі будь-якої з цих сполук, який включає кристалізацію придатної сполуки або солі з системи розчинників, що містить комбінацію С3-7алкілового спирту та ді-С3-5алкілового етеру.
55. Спосіб за п. 54, де етером є ди-н-пропіловий етер, діізопропіловий етер або ди-н-бутиловий етер.
56. Спосіб за п. 54 або п. 55, де спиртом є н-пропанол, ізопропанол, н-бутанол, 4-метил-2-пентанол, 3-метил-1-бутанол, 2-метил-1-пропанол чи пентан-1-ол.
57. Спосіб за будь-яким одним з пп. 54 - 56, де комбінацією розчинників є н-пропанол та ди-н-пропіловий етер; ізопропанол та діізопропіловий етер; н-бутанол та ди-н-бутиловий етер; 4-метил-2-пентанол та ди-н-бутиловий етер; ізопропанол та ди-н-бутиловий етер; 4-метил-2-пентанол та діізопропіловий етер чи пентан-1-ол та діізопропіловий етер.
58. Спосіб за п. 57, де комбінацією розчинників є ізопропанол та діізопропіловий етер.
59. Спосіб за будь-яким одним з пп. 54 - 58, де сполуку нагрівають у комбінації розчинників до температури у межах 50-100°С.
60. Спосіб за будь-яким одним з пп. 54 - 59, де кристалізованою сполукою є трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7-діазадицикло[3,3,1]нон-3-іл)етилкарбамат або його фармацевтично прийнятна сіль.
61. Спосіб за п. 60, де сполукою є трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7-діазадицикло[3,3,1 ]нон-3-іл}етилкарбамат.
62. Сполука, отримана способом за будь-яким одним з пп. 46-61.
63. Сполука за будь-яким одним з пп. 1-45 або 62 для застосування як медикаменту.
64. Фармацевтична композиція, що включає сполуку за будь-яким одним з пп. 1-45 або 62 у суміші з фармацевтично прийнятним ад'ювантом, розріджувачем або носієм.
65. Сполука за будь-яким одним з пп. 1-45 або 62 для застосування при профілактиці або лікуванні аритмії.
66. Застосування сполуки за будь-яким одним з пп. 1-45 або 62 як активного інгредієнта для виробництва медикаменту для застосування при профілактиці або лікуванні аритмії.
67. Застосування за п. 66, де аритмією є передсерцева або шлуночкова аритмія.
68. Застосування за п. 66, де аритмією є передсерцева фібриляція.
69. Застосування за п. 66, де аритмією є передсерцеве миготіння.
70. Спосіб профілактики або лікування аритмії, який включає уведення сполуки за будь-яким одним з пп. 1-45 або 62 персоні, що потерпає від такого стану чи схильна до нього.
71. Спосіб за п. 70, де аритмією є передсерцева або шлуночкова аритмія.
72. Спосіб за п. 70, де аритмією є передсерцева фібриляція.
73. Спосіб за п. 70, де аритмією є передсерцеве миготіння.
Текст
1. Фармацевтично прийнятна сіль одної з таких сполук: 4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7діазадицикло[3,3,1]нон-3іл]пропіл}аміно)бензонітрил; трет-бутилу 2-{7-[3-(4-ціаноаніліно)пропіл]-9-окса3,7-діазадицикло[3,3,1]нон-3-іл}етилкарбамат; трет-бутилу 2-{7-[4-(4-ціанофеніл)бутил]-9-окса3,7-діазадицикло[3,3,1]нон-3-іл}етилкарбамат або трет-бутилу 2-{7-1(2S)-3-(4-ціанофенокси)-2гідроксипропіл]-9-окса-3,7-діазадицикло[3,3,1 ]нон3-іл}етилкарбамат, за умови, що сіль не є бензолсульфонатом 4-({3[7-(3,3-диметил-2-оксобутил)-9-окса-3,7діазадицикло[3,3,1]нон-3іл]пропіл}аміно)бензонітрилу. 2. Сполука, що вибрана з: 4-({3-[7-(3,3-диметил-2оксобутил)-9-окса-3,7-діазадицикло[3,3,1]нон-3іл]пропіл}аміно)бензонітрилу; трет-бутилу 2-{7-[3-(4-ціаноаніліно)пропіл]-9-окса3,7-діазадицикло-[3,3,1]нон-3-іл}етилкарбамату; трет-бутилу 1-{7-[4-(4-ціанофеніл)бутил]-9-окса3,7-діазадицикло-[3,3,1]нон-3-іл}етилкарбамату або трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2гідроксипропіл]-9-окса-3,7-діазадицикло[3,3,1]нон3-іл}етилкарбамату, або фармацевтично прийнятної солі будь-якої з цих сполук по суті в кристалічній формі. 2 3 74629 4 іл]пропіл}аміно)бензонітрилу. 18. Сполука за п.2, якою є трет-бутилу 2-{7-[(2S)-38. Сіль за п.7, що характеризується рентгенодиф(4-ціанофенокси)-2-пдроксипропіл]-9-окса-3,7рактограмою порошку, яка характеризується пікадіазадицикло-[3,3,1]нон-3-іл}етилкарбамат. ми з d-параметрами при 18,7, 9,4, 6,3, 4,69, 4,48, 19. Сполука за п.18, що характеризується кривою 0 диференційної сканувальної калориметрії, при 3,76, 3,35, 3,13, 2,68, 2,35 та 1,88 A , та/або яка по швидкості нагрівання 10°С/хвилину у закритій чашці з мікроотвором під струмом азоту, виявляючи суті визначена у таблиці 4 та/або у фігурі 4. ендотермічний ефект з екстрапольованим темпе9. Сіль за п.1 або п.2, якою є толуолсульфонат 4ратурним початком приблизно між 100 та 102°С; ({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7та/або рентгенодифрактограмою порошку, яка діазадицикло[3,3,1 ]нон-3характеризується піками з d-параметрами при іл]пропіл}аміно)бензонітрилу. 22,3, 11,2, 8,4, 7,1, 6,4, 5,8, 5,5, 5,2, 4,91, 4,81, 4,62, 10. Сіль за п.9, що характеризується кривою ди4,52, 4,32, 4,22, 4,12, 4,06, 3,91, 3,81, 3,50, 3,34 та ференційної сканувальної калориметрії, при швид0 кості нагрівання 10°С/хвилину у закритій чашці з 3,15 A , та/або яка по суті визначена у таблиці 9 мікроотвором під струмом азоту, виявляючи ендотермічний ефект з екстрапольованим температурта/або у фігурі 9. ним початком приблизно 145°С; та/або рентгено20. Сіль за п.1 або п.2, якою є метансульфонат дифрактограмою порошку, яка характеризується трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2піками з d-параметрами при 19,9, 10,0, 6,0, 4,99, гідроксипропіл]-9-окса-3,70 діазадицикло[3,3,1,1]нон-3-іл}етилкарбамату. 4,86, 4,38, 4,36, 4,19, 3,99 та 3,33 A , та/або яка по 21. Сіль за п.20, що характеризується кривою дисуті визначена у таблиці 5(а) та/або у фігурі 5(а). ференційної сканувальної калориметрії, при швидкості нагрівання 10°С/хвилину у закритій чашці з 11. Сіль за п.9, що характеризується кривою димікроотвором під струмом азоту, виявляючи ендоференційної сканувальної калориметрії, при швидтермічний ефект з екстрапольованим температуркості нагрівання 10°С/хвилину у закритій чашці з ним початком приблизно 167°С; та/або рентгеномікроотвором під струмом азоту, виявляючи ендодифрактограмою порошку, яка характеризується термічний ефект з екстрапольованим температурпіками з d-параметрами при 12,7, 9,4, 7,3, 7,1, 6,6, ним початком приблизно 153°С; та/або рентгено6,0, 5,4, 5,1, 4,92, 4,83, 4,27, 4,14, 4,05, 3,99, 3,87, дифрактограмою порошку, яка характеризується 0 піками з d-параметрами при 18,6, 9,3, 6,2, 4,66, 3,73, 3,65, 3,42, 3,37 та 3,00 A , та/або яка по суті 0 4,49, 3,73 та 3,11 A , та/або яка по суті визначена визначена у таблиці 10 та/або у фігурі 10. 22. Сіль за п.1 або п.2, якою є сіль гіпурової кислоу таблиці 5(b) та/або у фігурі 5(b). ти трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-212. Сіль за п.1 або п.2, якою є сіль гідроксинафтойної кислоти 4-({3-[7-(3,3-диметил-2-оксобутил)гідроксипропіл]-9-окса-3,7-діазадицикло-[3,3,1]нон3-іл}етилкарбамату. 9-окса-3,7-діазадицикло[3,3,1 ]нон-3іл]пропіл}аміно)бензонітрилу. 23. Сіль за п.22, що характеризується рентгенодифрактограмою порошку, яка характеризується 13. Сіль за п.12, що характеризується рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при 16,4, 6,9, 6,2, 6,1, 5,5, 0 піками з d-параметрами при 17,6, 12,4, 8,8, 5,7, 5,2, 5,1, 4,93, 4,61, 4,50, 4,28, 4,20, 4,11 та 3,68 A , 5,6, 5,1, 4,95, 4,88, 4,47, 4,16, 4,08, 3,84, 3,80, 3,33 0 та/або яка по суті визначена у таблиці 11 та/або у та 3,03 A , та/або яка по суті визначена у таблиці фігурі 11. 24. Сполука за п.2, якою є трет-бутилу 2-{7-[4-(46 та/або у фігурі 6. ціанофеніл)бутил]-9-окса-3,7-діазадицикло[3,3,1 14. Сіль за п.1 або п.2, якою є сіль нафталінсуль]нон-3-іл}етилкарбамат. фонової кислоти 4-({3-[7-(3,3-диметил-225. Сполука за п.24, що характеризуєтьсякривою оксобутил)-9-окса-3,7-діазадицикло[3,3,1]нон-3диференційної сканувальної калориметрії, при іл]пропіл}аміно)бензонітрилу. швидкості нагрівання 10°С/хвилину у закритій ча15. Сіль за п.14, що характеризується рентгеношці з мікроотвором під струмом азоту, виявляючи дифрактограмою порошку, яка характеризується ендотермічний ефект з екстрапольованим темпепіками з d-параметрами при 16,1, 8,1, 7,1, 6,3, 6,0, ратурним початком приблизно 97°С; та/або рент5,4 (5,39), 4,96, 4,88, 4,68, 4,49, 4,34, 4,10, 4,04, 0 генодифрактограмою порошку, яка характеризу3,94, 3,44, 3,40, 3,23, 3,20 та 3,15 A , та/або яка по ється піками з d-параметрами при 19,4, 10,0, 9,1, 8,1, 6,5, 5,5, 5,2, 5,1, 4,99, 4,90, 4,46, 4,32, 4,06, суті визначена у таблиці 7 та/або у фігурі 7. 0 16. Сіль за п.1 або п.2, якою є сіль мезитиленсу4,00, 3,85 та 3,80 A , та/або яка по суті визначена льфонової кислоти 4-({3-[7-(3,3-диметил-2у таблиці 12 та/або у фігурі 12. оксобутил)-9-окса-3,7-діазадицикло[3,3,1 ]нон-326. Сіль за п.1 або п.2, якою є метансульфонат іл]пропіл}аміно)бензонітрилу. трет-бутилу 2-{7-[4-(4-ціанофеніл)бутил]-9-окса17. Сіль за п.16, що характеризується рентгено3,7-діазадицикло[3,3,1]нон-3-іл}етилкарбамату. дифрактограмою порошку, яка характеризується 27. Сіль за п.26, що характеризується кривою дипіками з d-параметрами при 18,8, 9,5, 6,3 (6,33), 0 ференційної сканувальної калориметрії, при швид6,3 (6,26), 4,75, 4,50, 4,46, 3,92, 3,80 та 3,17 A , кості нагрівання 10°С/хвилину у закритій чашці з мікроотвором під струмом азоту, виявляючи ендота/або яка по суті визначена у таблиці 8 та/або у термічний ефект з екстрапольованими температуфігурі 8. 5 74629 6 0 рними початками приблизно 145°С та 170°С; 3,51, 3,25 та 3,12 A , та/або яка по суті визначена та/або рентгенодифрактограмою порошку, яка характеризується піками з d-параметрами при у таблиці 18 та/або у фігурі 18. 13,3, 12,3, 9,6, 7,5, 6,9, 6,7, 5,5, 5,1, 5,0, 4,89, 4,81, 38. Сіль за п.1 або п.2, якою є сіль 2,2,3,3,0 тетраметил-1,4-дибутанової кислоти трет-бутилу 4,34, 4,23, 4,20, 4,08, 3,89, 3,85 та 3,80 A , та/або 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9яка по суті визначена у таблиці 13 та/або у фігурі окса-3,7-діазадицикло[3,3,1 ]нон-313. іл}етилкарбамату. 28. Сіль за п.1 або п.2, якою є толуолсульфонат 39. Сіль за п.38, що характеризується рентгенотрет-бутилу 2-{7-[4-(4-ціанофеніл)бутил]-9-оксадифрактограмою порошку, яка характеризується 3,7-діазацикло[3,3,1 ]нон-3-іл}етилкарбамату. піками з d-параметрами при 14,2, 10,3, 7,1, 6,4, 29. Сіль за п.28, що характеризується кривою ди6,0, 5,6, 5,4, 5,1, 4,84, 4,77, 4,73, 4,66, 4,50, 4,22, 0 ференційної сканувальної калориметрії, при швид3,78, 3,62, 3,49, 3,35 та 3,09 A , та/або яка по суті кості нагрівання 10°С/хвилину у закритій чашці з мікроотвором під струмом азоту, виявляючи ендовизначена у таблиці 19 та/або у фігурі 19. термічний ефект з екстрапольованим температур40. Сіль за п.1 або п.2, якою є сіль 1,2ним початком приблизно 138°С; та/або рентгеноциклопентандикарбонової кислоти трет-бутилу 2дифрактограмою порошку, яка характеризується {7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9піками з d-параметрами при 13,2, 7,1, 5,2, 5,0, окса-3,7-діазадицикло-[3,3,1]нон-30 іл}етилкарбамату. 4,67, 4,28, 4,24, 4,19, 4,08, 3,36 та 3,12 A , та/або 41. Сіль за п.40, що характеризується рентгенояка по суті визначена у таблиці 14 та/або у фігурі дифрактограмою порошку, яка характеризується 14. піками з d-параметрами при 12,9, 11,1, 10,7, 9,0, 30. Сіль за п.1 або п.2, якою є сіль [(дифеніл-47,7, 7,0, 6,5, 6,3, 6,1, 6,0, 5,6, 5,4, 5,3, 5,2, 4,73, карбоніл)аміно]оцтової кислоти трет-бутилу 2-{74,68, 4,31, 4,26, 4,02, 3,86, 3,61, 3,50, 3,24 та 2,90 0 [(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-оксаA , та/або яка по суті визначена у таблиці 20 3,7-діазадицикло-[3,3,1 ]нон-3-іл)етилкарбамату. 31. Сіль за п.30, що характеризується рентгенота/або у фігурі 20. дифрактограмою порошку, яка характеризується 42. Сіль за п.1 або п.2, якою є сіль О,О'піками з d-параметрами при 20,4, 15,3, 11,5, 9,4, дибензоїлвинної кислоти трет-бутилу 2-{7-[(2S)-37,7, 7,2, 5,9, 5,7, 5,4, 5,3, 5,2, 4,60, 4,54, 4,13, 3,85, (4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,70 діазадицикло-[3,3,1]нон-3-іл}етилкарбамату. 3,79, 3,64, 3,61, 3,40 та 2,94 A , та/або яка по суті 43. Сіль за п.42, що характеризується рентгеновизначена у таблиці 15 та/або у фігурі 15. дифрактограмою порошку, яка характеризується 32. Сіль за п.1 або п.2, якою є сіль гемібурштинопіками з d-параметрами при 15,1, 13,5, 7,1, 6,6, 0 вої кислоти трет-бутилу 2-{7-[(2S)-3-(45,3, 5,2, 4,79, 4,50, 4,38, 4,25, 3,49 та 2,92 A , ціанофенокси)-2-гідроксипропіл]-9-окса-3,7діазадицикло-[3,3,1]нон-3-іл}етилкарбамату. та/або яка по суті визначена у таблиці 21 та/або у 33. Сіль за п.32, що характеризується рентгенофігурі 21. дифрактограмою порошку, яка характеризується 44. Сіль за п.1 або п.2, якою є сіль О,О'-ди-парапіками з d-параметрами при 18,0, 8,3, 8,0, 7,5, 7,3, толуоїлвинної кислоти трет-бутилу 2-{7-[(2S)-3-(46,8, 6,1, 5,8, 5,4, 4,89, 4,79, 4,68, 4,59, 4,54, 4,43, ціанофенокси)-2-гідроксипропіл]-9-окса-3,74,35, 4,18, 4,04, 3,99, 3,90, 3,83, 3,67, 3,58, 3,07 та діазадицикло-[3,3,1]нон-3-іл}етилкарбамату. 0 45. Сіль за п.44, що характеризується рентгено2,47 A , та/або яка по суті визначена у таблиці 16 дифрактограмою порошку, яка характеризується та/або у фігурі 16. піками з d-параметрами при 15,8, 6,8, 5,7, 5,6, 5,3 0 34. Сіль за п.1 або п.2, якою є сіль (3,4(5,25), 4,84, 4,80, 4,75, 4,39, 4,37 та 3,98 A , та/або дихлорбензоїламіно)оцтової кислоти трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9яка по суті визначена у таблиці 22 та/або у фігурі окса-3,7-діазадицикло-[3,3,1]нон-322. іл}етилкарбамату. 46. Спосіб отримання солі за будь-яким одним з 35. Сіль за п.34, що характеризується рентгенопп.1, 2, 7-17, 20-23 або 26-45, який включає додадифрактограмою порошку, яка характеризується вання кислоти або основи (за прийнятністю), до піками з d-параметрами при 16,6, 13,8, 10,9, 6,3, прийнятної вільної основи сполуки, яку визначено 6,2, 5,5, 5,3, 5,1, 5,0, 4,60, 4,33, 4,30, 4,22, 3,85, у п.1. 0 47. Спосіб за п.46, який включає додавання кисло3,70, 3,50, 3,30, 3,16, 3,06 та 2,99 A , та/або яка по ти до прийнятної вільної основи сполуки. суті визначена у таблиці 17 та/або у фігурі 17. 48. Спосіб отримання сполуки за будь-яким одним 36. Сіль за п.1 або п.2, якою є сіль [(нафталін-2з пп.1-45, який включає кристалізацію прийнятної карбоніл)аміно]оцтової кислоти трет-бутилу 2-{7вільної основи сполуки, яку визначено у п.1, або [(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-оксапідхожої фармацевтично прийнятної її солі. 3,7-діазадицикло-[3,3,1]нон-3-іл)етилкарбамату. 49. Спосіб за п.48, який включає кристалізацію 37. Сіль за п.36, що характеризується рентгеносполуки або солі з розчинника. дифрактограмою порошку, яка характеризується 50. Спосіб за п.49, де розчинник вибирають з групіками з d-параметрами при 16,8, 6,2, 5,8, 5,6, 5,2, пи: ацетати, нижчі алкіл-спирти, аліфатичні та 5,1, 4,90, 4,76, 4,66, 4,53, 4,37, 4,31, 4,23, 4,08, ароматичні вуглеводні, діалкілетери, діалкілкето 7 74629 8 ни, ацетонітрил, хлоровані алкани, водні розчинловий етер. ники, або їх суміші. 58. Спосіб за п.57, де комбінацією розчинників є 51. Спосіб за п.50 де розчинник вибирають з групи: ізопропанол та діізопропіловий етер. С1-6 алкілацетати, лінійні або розгалужені С159. Спосіб за будь-яким одним з пп.54 - 58, де С6-12аліфатичні вуглеводні, С6сполуку нагрівають у комбінації розчинників до 6алкілспирти, температури у межах 50-100°С. 10ароматичні вуглеводні, ді-С1-6-алкілові етери, діС1-6алкілкетони, хлоровані метани або етани, аце60. Спосіб за будь-яким одним з пп.54 - 59, де критонітрил, вода, або їх суміші. сталізованою сполукою є трет-бутилу 2-{7-[(2S)-352. Спосіб за п.51 де розчинник вибирають з групи: (4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7етилацетат, ізопропілацетат, метанол, етанол, діазадицикло[3,3,1]нон-3-іл)етилкарбамат або його ізопропанол, н-гептан, діетиловий етер, ацетон, фармацевтично прийнятна сіль. дихлорметан, вода, або їх суміші. 61. Спосіб за п.60, де сполукою є трет-бутилу 2-{753. Спосіб отримання солі за будь-яким одним з [(2S)-3-(4-ціанофенокси)-2-гідроксипропіл]-9-оксапп.1, 2, 7-17, 20-23 або 26-45, який включає дода3,7-діазадицикло[3,3,1 ]нон-3-іл}етилкарбамат. вання за п.46 або п.47, а потім кристалізацію за 62. Сполука, отримана способом за будь-яким одбудь-яким одним з пп.48-52. ним з пп.46-61. 54. Спосіб отримання кристалічної форми 4-({3-[763. Сполука за будь-яким одним з пп.1-45 або 62 (3,3-диметил-2-оксобутил)-9-окса-3,7для застосування як медикаменту. діазадицикло[3,3,1]нон-364. Фармацевтична композиція, що включає споіл]пропіл}аміно)бензонітрилу; трет-бутилу 2-{7-[3луку за будь-яким одним з пп.1-45 або 62 у суміші (4-ціаноаніліно)пропіл]-9-окса-3,7з фармацевтично прийнятним ад'ювантом, розрідіазадицикло[3,3,1]нон-3-іл}етилкарбамату; третджувачем або носієм. бутилу 2-{7-[4-(4-ціанофеніл)бутил]-9-окса-3,765. Сполука за будь-яким одним з пп.1-45 або 62 діазадицикло[3,3,1]нон-3-іл}етилкарбамату або для застосування при профілактиці або лікуванні трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2аритмії. гідроксипропіл]-9-окса-3,7-діазадицикло[3,3,1]нон66. Застосування сполуки за будь-яким одним з 3-іл}етилкарбамату, або фармацевтично прийнятпп.1-45 або 62 як активного інгредієнта для виробної солі будь-якої з цих сполук, який включає крисництва медикаменту для застосування при профіталізацію придатної сполуки або солі з системи лактиці або лікуванні аритмії. розчинників, що містить комбінацію С3-7алкілового 67. Застосування за п.66, де аритмією є передсерспирту та ді-С3-5алкілового етеру. цева або шлуночкова аритмія. 55. Спосіб за п.54, де етером є ди-н-пропіловий 68. Застосування за п.66, де аритмією є передсеретер, діізопропіловий етер або ди-н-бутиловий цева фібриляція. етер. 69. Застосування за п.66, де аритмією є передсер56. Спосіб за п.54 або п.55, де спиртом є нцеве миготіння. пропанол, ізопропанол, н-бутанол, 4-метил-270. Спосіб профілактики або лікування аритмії, пентанол,3-метил-1-бутанол, 2-метил-1-пропанол який включає уведення сполуки за будь-яким одчи пентан-1-ол. ним з пп.1-45 або 62 персоні, що потерпає від та57. Спосіб за будь-яким одним з пп.54 - 56, де кого стану чи схильна до нього. комбінацією розчинників є н-пропанол та ди-н71. Спосіб за п.70, де аритмією є передсерцева пропіловий етер; ізопропанол та діізопропіловий або шлуночкова аритмія. етер; н-бутанол та ди-н-бутиловий етер; 4-метил72. Спосіб за п.70, де аритмією є передсерцева 2-пентанол та ди-н-бутиловий етер; ізопропанол фібриляція. та ди-н-бутиловий етер; 4-метил-2-пентанол та 73. Спосіб за п.70, де аритмією є передсерцеве діізопропіловий етер чи пентан-1-ол та діізопропімиготіння. Представлений винахід стосується нових форм твердого стану деяких антіаритмічних ліків, фармацевтичних композицій, що їх містять, та способів їх отримання. У рецептурі композиції ліків, є важливим, щоб речовина ліків була у формі, в якій її можна зручно обробляти та поводитися з нею. Це важливо, не тільки з точки зору отримання комерційно придатного способу виробництва, але також з точки зору наступного виробництва фармацевтичних композицій, що містять активну сполуку. Крім того, у виробництві композицій ліків, важливо, щоб було забезпечено надійний, відтворюваний та постійний профіль концентрації ліків у плазмі після уведення пацієнту. Хімічна стабільність, стабільність у твердому стані та "час зберігання" активних інгредієнтів є також дуже важливими факторами. Речовина ліків та композиції, що її містять, повинні переважно ефективно зберігатися протягом суттєвого періоду часу без виявлення значної зміни фізико-хімічних характеристик активного компоненту (наприклад, його хімічного складу, густини, гігроскопічності та розчинності). Крім того, також важливо забезпечити ліки у формі, яка є такою хімічно чистою, як це можливо. Аморфні матеріали можуть представляти значні проблеми з огляду на це. Наприклад, такі матеріали звичайно важко обробляти та формувати, вони не забезпечують надійну розчинність, та як часто знаходять, є нестабільними та хімічно забрудненими. Фахівцям зрозуміло, що якщо ліки можна легко отримати у стабільній кристалічній формі, вищенаведені проблеми можна розв'язати. 9 74629 10 Отже, у виробництві комерційно придатних та Сполуки [міжнародної патентної заявки WO фармацевтично прийнятних композицій ліків, як-то 01/28992] показані як корисні при лікуванні серцекомпозицій з модифікованим вивільненням, важвих аритмій. ливо, там де це можливо, забезпечити ліки у по Способи синтезу сполук А, В, С та D описані у суті кристалічній та стабільній формі. прикладах 3, 7, 8 та 9 (відповідно) [у WO Слід зауважити, однак, що ця задача назавж01/28992]. ди досяжна. Дійсно, звичайно, неможливо передПевні фармацевтично прийнятні солі сполук В, бачити тільки за молекулярною структурою, якою С та D не розкриті [у WO 01/28992]. Далі не запробуде поведінка сполуки при кристалізації. Це можпоновано інформації стосовно різних кристалічних на визначити звичайно тільки емпірично. форм будь-якої зі сполук А, В, С або D, або їх со[Міжнародна патентна заявка WO 01/28992] лей, що можна отримати. розкриває ряд сполук оксабіспідину, включаючи: Згідно з першим аспектом винаходу запропо(а) 4-((3-[7-(3,3-диметил-2-оксобутил)-9-оксановано фармацевтично прийнятну сіль сполуки А, 3,7-діазадицикло[3,3,1]нон-3сполуки В, сполуки С, або сполуки D, за умови, що іл]пропіл}аміно)бензонітрил: сіль не є бензолсульфонатом сполуки А. Згідно з другим аспектом винаходу запропоновано сполуку А, сполуку В, сполуку С або сполуку D, або фармацевтично прийнятну сіль будь-якої з цих сполук у по суті кристалічній формі. Сполуки А, В, С та D та їх солі, згідно з першим та другим аспектами винаходу далі позначені як "сполуки винаходу". Хоча нами виявлено, що можливо отримувати сполуки винаходу у формах, які є більше, ніж на цю сполуку далі позначено як сполуку А. Спо80% кристалічними, термін "по суті кристалічні" лука А конкретно розкрита [у WO 01/28992], як у включає більше, ніж 20%, переважно більше, ніж формі вільної основи, так і у формі бензолсульфо30%, та більш переважно більше, ніж 40% кристанату; лічності. Ступінь (%) кристалічності можна визна(b) трет-бутилу 2-{7-[3-(4-ціаноаніліно)пропіл]чено фахівцем, використовуючи рентгенодифрак9-окса-3,7-діазадицикло-[3,3,1]нон-3тометрію порошку (РДМП). Інші способи, як-то іл}етилкарбамат: ЯМР у твердому стані, ІЧ, раманівську спектроскопію, диференційну сканувальну калориметрію (ДСК) та мікрокалориметрію можна застосовувати також. Переважно, коли сполука винаходу є паратолуолсульфонатом сполуки А, її запропоновано у посуті кристалічній формі. Коли сполуку винаходу запропоновано як бену формі вільної основи, цю сполуку далі поззолсульфонат сполуки А у кристалічній формі, начено як сполуку В; краще, щоб кристалічна форма не була формою, (с) трет-бутилу 2-{7-[4-(4-ціанофеніл)бутил]-9що конкретно описано подалі у прикладі 4 (та/або окса-3,7-діазадицикло-[3,3,1]нон-3п.8). іл}етилкарбамат: Коли сполуку винаходу запропоновано у кристалічній формі сполуки А (вільна основа), краще, щоб кристалічна форма не була формою, що конкретно описано подалі у будь-чому з прикладу 1 (та/або п.4), прикладу 2 (та/або п.5), або прикладу 3 (та/або п.6). Коли сполуку винаходу запропоновано як сполуку А у кристалічній формі, краще, щоб сполуку у формі вільної основи, цю сполуку далі позне було запропоновано як вільну основу, або, коли начено як сполуку С; та її запропоновано у формі солі, як бензолсульфо(d) трет-бутилу 2-{7-[(2S)-3-(4-ціанофенокси)-2нат. гідроксипропіл]-9-окса-3,7-діазадицикло[3,3,1]нонКоли сполуку винаходу запропоновано як спо3-іл}етилкарбамат: луку С у кристалічній формі (вільна основа), краще, щоб кристалічна форма не була формою, що конкретно описано подалі у прикладі 12 (та/або п.25). Коли сполуку винаходу запропоновано як сполуку С у кристалічній формі, краще, щоб сполуку не було запропоновано як вільну основу. Коли сполуку винаходу запропоновано як споу формі вільної основи, цю сполуку далі позлуку D у кристалічній формі (вільна основа), краначено як сполуку D. ще, щоб кристалічна форма не була формою, що конкретно описано подалі у прикладі 9 (та/або п.19). 11 74629 12 Коли сполуку винаходу запропоновано як спододавали до кристалізаційної суміші перед провелуку D у кристалічній формі, краще, щоб сполуку денням кристалізації. не було запропоновано як вільну основу. Переважні адитивні солі включають кислотноКоли сполуку винаходу запропоновано як споадитивні солі неорганічних, а особливо, органічних луку В у кристалічній формі, краще, щоб сполуку кислот, переважно солі карбонових кислот, як-то не було запропоновано як вільну основу. гіпурової кислоти, нафтойної кислоти та гідроксиСполуки винаходу можуть бути у формі сользаміщеної нафтойної кислоти (наприклад, 1вату, гідрату або змішаного сольвату/гідрату. Согідрокси-2-нафтойної кислоти), аспарагінової кисльвати можуть бути з одним або більше органічлоти, малеїнової кислоти, бурштинової кислоти ними розчинниками, як-то нижчі алкілспирти малонової кислоти, оцтової кислоти, фумарової (наприклад, С1-4алкілспирти, наприклад, метанол, кислоти, бензойної кислоти, терефталевої кислоетанол або ізопропанол), кетони (як-то ацетон), ти, памової кислоти та гідроксибензойної кислоти; естери (як-то етилацетат) або їх суміші. солі гідрокси-кислот, як-то саліцилової кислоти, Сполуки винаходу можуть мати підвищену гліколевої кислоти, яблучної кислоти, аскорбінової стабільність у порівнянні зі сполуками/солями, що кислоти, лимонної кислоти, глюконової кислоти, розкриті [у WO 01/28992]. молочної кислоти, а також винної кислоти та її поТермін "стабільність", як визначено тут, вклюхідних, як-то Ο,Ο'-дибензоїлвинної кислоти (напричає хімічну стабільність та стабільність у твердому клад, О,O'-дибензоїл-D-винної кислоти або О,О'стані. дибензоїл-L-винної кислоти) та О,O'-ди-параТермін "хімічна стабільність", означає, що спотолуоїлвинної кислоти (наприклад, О,О'-ди-паралуки винаходу можливо зберігати у ізольованій толуоїл-D-винної кислоти або О,О'-ди-параформі, або у формі композиції, в яких їх запропотолуоїл-L-винної кислоти); солі інших дикислот, якновано у суміші з фармацевтично прийнятними то 2,2,3,3-тетраметил-1,4-дибутанова кислота та носіями, розріджувачами або ад'ювантами (напри1,2-циклопентандикарбонова кислота; та солі алклад, у пероральній формі дозування, як-то таблекіл-, арил- та алкіларилсульфонової кислоти, натка, капсула тощо), у нормальних умовах зберіганприклад, С1-8алкіл- та С6-10арил- та С1-4алкіл-С6ня, з незначним ступенем хімічної деградації або 10арил-сульфонової кислоти (ці арил- та алкіларируйнування. лсульфонові кислоти можуть бути заміщеними на Термін "стабільність у твердому стані", ознаарильній частині, наприклад, одним або більше чає, що сполуки винаходу можливо зберігати у метилом, метоксилом, гідроксилом або галогеізольованій твердій формі, або у формі твердої ном), включаючи солі бензолсульфонової кислоти, композиції, в якій її запропоновано у суміші з фартолуолсульфонової кислоти, гідрокси-заміщеної мацевтично прийнятними носіями, розріджувачами бензолсульфонової кислоти, нафталінсульфоноабо ад'ювантами (наприклад, у пероральній формі вої кислоти, нафталіндисульфонової кислоти, медозування, як-то таблетка, капсула тощо), у норзитиленсульфонової кислоти, метансульфонової мальних умовах зберігання, з незначним ступенем кислоти, етансульфонової кислоти та 2перетворення у твердому стані (наприклад, крисгідроксіетансульфонової кислоти. талізації, перекристалізації, перетворення тверКислотно-адитивні солі, особливо сполуки D3, дофазного стану, гідратування, дегідратування, що можна також згадати, включають солі, в як кисольватації або десольватації). слота є похідною гіпурової кислоти, наприклад Приклади "нормальних умов зберігання" вклюкислоти формули І, чають температури між -80 та +50°С (переважно між 0 та 40°С, а більш переважно кімнатну темпеI ратуру, як-то 15-30°С), тиски між 0,1 та 2 бар (переважно при атмосферному тиску), відносні володе гості між 5 та 95% (переважно 10-75%), та/або 460 Ar1 представляє феніл або нафтил, обидва які, люкс УФ/видимого світла, протягом подовженого як варіант, заміщені одним або більше замісникаперіоду (тобто не менше 6 місяців). У таких умовах ми, вибраними з групи: галоген (наприклад, хлор), сполуки винаходу, як можна виявити, будуть меннітрогрупа, Сгєапкіл, d-6алкоксил, гідроксил та феше, ніж на 15%, більш переважно менше, ніж на ніл; а 10%, та особливо менше, ніж на 5%, хімічно деR1, R2 та R3 незалежно представляють Η або градувати/розкладатися, або змінювати твердий С1-3алкіл. стан, за прийнятністю. Фахівцям зрозуміло, що Фахівці повинні розуміти, що коли Аr1 представищезазначені вищі та нижчі межі для температувляє феніл, a R1, R2 та R3 усі представляють Н, то ри, тиску та відносної вологості представляють кислота формули І є гіпуровою кислотою. граничні межі нормальних умов зберігання, та, що Переважні групи Аr1 включають феніл, цей деякі комбінації цих граничних меж не відбуватифеніл, як варіант, заміщено фенілом (наприклад у муться при звичайному зберіганні (наприклад, те4-позиції стосовно точки приєднання групи С(О)), мпература 50°С та тиск 0,1бар). хлором (наприклад у 3-та/або 4-позиції стосовно Переважні солі сполук А, В, С та D включають групи С(О)), нітрогрупою (наприклад у 4-позиції основно-, або переважно, кислотно-адитивні солі, стосовно С(О) груп) та/або С1-4алкілом, як-то меякі можна утворювати додаванням прийнятної кітил (наприклад у 2- та/або 4-позиції стосовно грулькості прийнятної кислоти або основи до видіпи С(О)); та нафтилом. Кращі Аr1 включають фелення (яке може включати кристалізацію). Наприніл, 4-фенілфеніл (дифеніл), 3,4-дихлорфеніл, 2клад, у випадку сполук винаходу у по суті нафтил, 4-нітрофеніл та 2,4,6-триметилфеніл. кристалічній формі, кислоту або основу можна 13 74629 14 Переважні групи R1 та R2 включають Η та меЗгідно з ще одним аспектом винаходу, запротил. Краще, щоб R1 та R2 чи обидва вони предстапоновано спосіб отримання сполуки винаходу, вляли Η або обидва представляли метил. який включає: Переважні 5,3 групи включають Н. додавання прийнятної кількості кислоти, або Коли R1 та R2 обидва представляють метил, основи до сполуки А, В, С або D для утворення 1 1 2 краще, щоб Аr представляв феніл. Коли R та R кислотно- або основно-адитивної солі; та/або кри1 обидва представляють Н, краще, щоб Аr предсталізація сполуки А, В, С або D, або солі сполуки ставляв 4-нітрофеніл, 2,4,6-триметилфеніл або, А, В, С або D. особливо, 3,4-дихлорфеніл, 2-нафтил або 4Можливо кристалізувати сполуки А, В, С та D, фенілфеніл (дифеніл). та їх фармацевтично прийнятні солі, у присутності Кислоти формули І комерційно доступні (насистеми розчинників чи без неї (наприклад, крисприклад, гіпурова кислота, 4-нітрогіпурова кислота талізувати можна з розплаву, у суперкритичних та 2-, 3- або 4-метилгіпурова кислот)а, або їх можумовах, або сублімацією). Однак, ми вважаємо за на отримати стандартними способами. краще, щоб кристалізація відбувалася з прийнятНаприклад кислоти формули І можна отриманої системи розчинників. но реакцією сполуки формули II Кристалічні кислотно- або основно-адитивні солі сполук А, В, С або D можна забезпечувати II додаванням прийнятної кількості прийнятної кис1 2 3 лоти або основи до кристалізаційної суміші (наде R , R та R визначені раніше, з хлорангідприклад, системи розчинників, включаючи сполуку ридом формули III, А, В, С або D як вільну основу) перед кристалізаціAr1C(O)CI llI 1 єю. Наприклад, органічні кислоти можна додавали де Аr визначено раніше, наприклад у присут(як варіант, у формі розчину, що містить прийнятності основи, наприклад, водного NaOH, класичний полярний розчинник (наприклад, нижчий алкіними способами Скоттена-бауманна [дивися, налспирт, як-то метанол або етанол) або ацетат, якприклад, J.Med. Chem.., 1989, 32, 1033]. то етилацетат) до сполуки А, В, С або D (як варіНейтралізація кислотою, наприклад, конц. гідрохант, у формі у розчину, де вільна основа знахолоридною кислотою, може осадити кислоту фордиться у прийнятному кристалізаційному розчинмули І, яку можна перекристалізувати, якщо необник). (Фахівцям зрозуміло, що у цьому контексті, хідно, з різних розчинників, наприклад, термін "вільна основа" означає "вільну форму" ізопропілового спирту, метанолу, етанолу, ацетону сполуки А, В, С або D (тобто форму, що не знахота води, або суміші цих розчинників. диться у формі кислотно- або основно-адитивної А Альтернативно, естерні (наприклад, естери солі)). нижчого алкілу) похідні сполук формули II, як варіФахівцям зрозуміло, що кислоту або основу ант, у формі солі, наприклад, гідрохлориду, моможна поєднати цим способом зі сполукою А, В, С жуть реагувати з хлорангідридом формули III, у або D розчиненням прийнятних матеріалів у приприсутності основи, наприклад, триетиламіну, у йнятних розчинниках, які описано вище, щонаймепридатному розчиннику, наприклад, дихлорметані, нше частково видаляючи ці розчинники, а далі з отриманням естер-аміду формули IV. знов розчиненням утвореної суміші для проведення кристалізації, як описано тут. IV Коли сполуки винаходу мають форму кислотно-адитивної солі, придатне стехіометричне спів4 де R представляє нижчий алкіл (як-то С1відношення кислоти до вільної основи знаходяться 6алкіл) або нижчий алкілфеніл (наприклад, С1у межах 0,25:1,5-1,5:1, як-то 0,45:1,25-1,25:1, 1 1 2 3 3алкілфеніл), а Аr , R , R та R визначені раніше включаючи 0,50:1-1:1. [дивися, наприклад, J. Heterocyclic Chem. 1973, 10, Система розчинників може бути гетерогенною 935, Tetrahedron 1989, 45, 1691 та J.Org. Спет., або Гомогенною та отже може включати один або 1999, 64, 8929]. Естер-аміди формули IV можуть більше органічних розчинників, як-то алкілацетати бути твердими продуктами при кімнатній темпера(наприклад, лінійні або розгалужені С1турі та можуть, отже, бути очищеними кристаліза6алкілацетати, як-то етилацетат, ізопропілацетат цією після свого утворення, за прийнятністю. Спота бутилацетат); нижчі, наприклад, лінійні або розлуки формули IV можна далі перетворити у галужені С1-6алкілспирти, як-то гексан-1-ол, 3сполуки формули І стандартним гідролізом, наприметилбутан-1-ол, пентан-1-ол, пентан-2-ол, 4клад, водним гідроксидом натрію, а потім додаметил-2-пентанол та 2-метил-1-пропанол, або С1ванням кислоти, наприклад, гідрохлоридної кисло4алкілспирт, як-то метанол, етанол, н-пропанол, ти, до осадження продукту. Перекристалізацію ізопропанол та бутанол (наприклад, н-бутанол); можна проводити далі, якщо потрібно. аліфатичні (наприклад, С6-12 як-то С7-12аліфатичні) 3 Сполуки формул І, II та IV, в яких R представвуглеводні (наприклад, ізо-гексан, ізо-октан та нляє С1-3алкіл, можна отримувати стандартним алгептан) та ароматичні вуглеводні (наприклад, токілуванням відповідної сполуки формули І, II або луол); хлоровані алкани (наприклад, хлороформ IV, в якій R3 представляє Н. та дихлорметан); діалкілкетони (наприклад, ацеСполуки формул II (та естерні похідні) та III є тон, метил-ізо-бутилкетон), ацетонітрил, діалкілекомерційно доступними або їх легко можна отритери (наприклад, діетиловий етер, димувати звичайними способами. ізопропіловий етер, ди-н-пропіловий етер, ди-нбутиловий етер та трет-бутил-метиловй етер); та/або водні розчинники, як-то вода. Суміші будь 15 74629 16 якого з вищезазначених розчинників можна застопентан-1-ол та ди-ізо-пропіловий етер. совувати. Сполуку/сіль можна додавали до суміші Різні кристалічні форми можуть мати різні розспирт/етер з системи розчинників з останньою у чинності у будь-якому даному розчиннику при попередньо змішаній формі. Альтернативно, спобудь-якій даній температурі. З огляду на це вищелуку/сіль можна розчиняти у прийнятному спирті, а зазначені розчинники можна застосовувати як "андалі до утвореного розчину можна додавати етер. тирозчинники" (тобто розчинники, в яких сполуки Солі можна також отримувати на місці, як тут равинаходу погано розчинні), які можуть тому сприяніше описано. ти кристалізації. Антирозчинники отже включають Переважно сполуку/сіль розчиняють у комбівищезазначені вуглеводні та діалкілетери. нації розчинників нагріванням до підвищеної темПридатні розчинники отже включають алкілаператури (наприклад, 50-100°С, як-то 65-90°С, цетати (як-то етилацетат або ізопропілацетат), наприклад, 75-85°С) для гарантії повного розчинижчі алкілспирти (як-то метанол, етанол та ізопнення. Далі утвореному розчину дають охолонути ропанол), хлоровані метани (як-то дихлорметан), для перебігу кристалізації. алкани (як-то н-гептан), етери (як-то діетиловий Цей спосіб кристалізації з суміші спирт/етер етер), кетони (як-то ацетон), воду тощо. переважно застосовують для забезпечення крисКристалізації сполук винаходу з прийнятної талічної форми сполуки D та її солей. Переважно системи розчинників можна досягти досягненням його можна застосовувати для забезпечення криспересичення у системі розчинників, які включає талічної сполуки D у формі вільної основи. сполуку А, В, С або D або її сіль, (наприклад, охоНами виявлено, що, коли сполуки винаходу (а лодженням, випарюванням розчиннику та/або доособливо сполуку D) отримано кристалізацією з даванням антирозчиннику (тобто розчиннику, в цієї системи розчинників, отримують високо крисякому сполуки винаходу погано розчинні (наприталічний матеріал ефективним чином з високим клад, вуглеводню, як-то ізооктан, н-гептан або ізовідтворенням кристалічного матеріалу (з гарним гексан, або діалкілетеру, як-то ди-ізопропіловий виходом),та у передбачувану кількість часу. етер, ди-н-бутиловий етер, тощо))), або зменшенСполуки винаходу можуть також бути отриманям розчинності речовини додаванням солі (як-то ними у формі сольвату (який включає форму гідNaCI), або, у випадку сполуки винаходу, що є кисрату, як-то моногідрат) або інакше (наприклад, у лотно-адитивною сіллю, додаванням надлишку формі несольватованої сполуки). прийнятної кислоти. Для гарантії, що утворюється несольватована Кристалізаційні температури та час залежать сполука, розчинник з якого відбувається кристалівід сполуки або солі, що кристалізують, концентзація, переважно слід сушити перед або протягом рації сполуки/солі у розчині та у системі розчинникристалізації для зменшення вмісту води нижче ків, які застосовують. критичного рівня, як краще не перевищувати при Кристалізацію можна також почати та/або прокристалізації. Розчинник можна сушили протягом вести стандартними способами, наприклад із закристалізації, наприклад зменшенням вмісту води травкою або без неї кристалами прийнятної крису суміші сполуки/солі, яку кристалізують та прийнталічної сполуки винаходу, та/або підгонкою pH. ятного органічного розчиннику/води з системи розКонкретний спосіб кристалізації, що можна зачинників (наприклад, збільшенням кількості органістосовувати для отримання сполуки А, В, С та, чного розчиннику, що присутній та/або особливо, D та їх солі, включає розчинення сполувидаленням води утворенням азеотропу, з настуки винаходу у системі розчинників, що містить С3пною відгонкою). Для гарантії, що утворюються гідрати (напри7алкіловий спирт та прийнятний антирозчинник диС3-7алкіловий етер. клад, моногідрати), у розчиннику, з якого відбуваЗгідно з ще одним аспектом винаходу запроється кристалізація, повинна бути присутньою вопоновано тому спосіб отримання по суті кристалічда. Вміст води при кристалізації слід переважно ної форми сполуки А, сполуки В, сполуки С або, тримати вище згаданого вище критичного рівня. особливо, сполуки D, або фармацевтично прийня"Критичний рівень" води залежить від фактотної солі будь-якої з цих сполук, спосіб включає рів, як-то температура, концентрація сполуки, яку кристалізацію доречної сполуки з системи розчинкристалізують, у розчині, профіль забруднення, та ників, що містить комбінацію С3-7алкілового спирту з системи розчинників як застосовують, але його та ди-С3-5алкілового етеру. можна визначити тривіально. Переважні етери включають ди-С3-5алкілові Отже, кристалічні гідрати можна отримати етери, як-то ди-н-пропіловий етер, дикристалізацією сполуки винаходу з системи розізопропіловий етер та ди-н-бутиловий етер. Перечинників, що містить воду, або комбінації води та важні спирти включають н-пропанол, ізо-пропанол, одного або більше органічних розчинників, вклюн-бутанол, 4-метил-2-пентанол, З-метил-1чаючи органічні розчинники, що здатні розчиняти бутанол, 2-метил-1-пропанол та пентан-1-ол. воду (наприклад, метанол, етанол, ізопропанол, Переважні комбінації розчинників включають: тощо). н-пропанол та ди-н-пропіловий етер; ізоНавпаки, кристалічні ангідрати можна отримапропанол та ди-ізопропіловий етер; ти кристалізацією сполуки винаходу з прийнятної н-бутанол та ди-н-бутиловий етер; системи органічних розчинників, які можуть бути 4-метил-2-пентанол та ди-н-бутиловий етер; висушеними, та/або їх можна сушити протягом ізо-пропанол та ди-н-бутиловий етер; кристалізації, так, щоб вміст води був нижче вище4-метил-2-пентанол та ди-ізо-пропіловий етер; зазначеного критичного рівня. Отже, несольватота 17 74629 18 вану сполуку можна отримати кристалізацією з слід проводити, оскільки кристал може втратити системи розчинників, що по суті вільна від води. воду та може відбуватися перетворення твердого Термін "по суті вільна від води ", означає, що стану, тобто вода з кристалу може втрачатися, вміст води у системі розчинників є нижче того, що якщо кристали сушать при високій температурі або призведе до утворення щонайбільше 10% гідрату під дуже зниженим тиском протягом довшого пе(наприклад, моногідрату) для будь-якої певної сисріоду. теми розчинників та умов кристалізації. Нами виявлено, що застосуванням способів Сполуки винаходу, що є безводними, містять кристалізації, які описано тут, можливо отримувати не більше 3%, переважно 2%, більш переважно сполуки винаходу з вищою хімічною чистотою, ніж 1% та більш переважно 0,5% (за масою) води, сполуки винаходу, які виділили у першому витака вода є зв'язаною (кристалогідратна вода або падку. інша) або ні. Гідрати містять не менше, ніж Подальшу очистку сполук винаходу можна 0,5моль води намоль сполуки винаходу. провести, використовуючи способи, як добре віБезводні сполуки або гідрати кристалізуються домі фахівцям. Наприклад забруднення можна залежить від кінетичних умов та до умов рівноваги видаляти перекристалізацією з прийнятної систевідповідних форм при певних умовах. ми розчинників (наприклад, етилацетат, ізопропіКоли сполуку винаходу запропоновано у крислацетат, дихлорметан, ізопропанол, н-гептан, металічній формі бензолсульфонату сполуки А, кратанол, етанол, метилетилкетон, ацетонітрил, ще, щоб ця сіль була моногідратом. пентан-2-ол, 3-метил бутан-1-ол, гексан-1-ол, вода Далі, як зрозуміло фахівцю, сполуки винаходу, або комбінації цих розчинників). Придатні темпещо мають однакову хімічну форму (вільна осноратура та час перекристалізації залежать від конва/сіль) можна також отримати у різних фізичних центрації сполуки або солі у розчині та від системи формах (наприклад, різних кристалічних формах) розчинників, яку застосовують. в різних умовах кристалізації. Кристалічна форма Коли сполуки винаходу кристалізують, або песполуки винаходу, яку отримують, залежить від рекристалізовують, як описано тут, утворена спокінетики та термодинаміки кристалізації. В деяких лука або сіль може мати форму, яка має підвищетермодинамічних умовах (система розчинників, ну хімічну стабільність та/або стабільність у температура, тиск та концентрація сполуки винатвердому стані, як згадано тут раніше. ходу), одна кристалічна форма може бути більш Фармацевтичні отримання та медичне застостабільною, ніж інша (або будь-яка інша). Однак, сування кристалічні форми, що мають відносно низьку Сполуки винаходу є корисними, оскільки вони термодинамічну стабільність можуть бути кінетичвиявляють фармакологічну активність. Вони тому но кращими. Отже, на додаток, кінетичні фактори, показані як фармацевтичні. як-то час, профіль забруднення, перемішування, Отже, згідно з ще одним аспектом винаходу присутність або відсутність затравки тощо, можуть запропоновано сполуки винаходу для застосувантакож впливати на те, які форми утворюються. ня як фармацевтичних сполук. Отже, розкриті тут способи може за прийнятністю Зокрема, сполуки винаходу виявляють міокарадаптувати фахівець для отримання різних крисдіальну електрофізіологічну активність, яку, наталічних форм. приклад, можна демонструвати у тестах, як-то Різні кристалічні форми сполук винаходу можописаних, серед іншого, [у міжнародних патентних на легко характеризувати, використовуючи спосозаявках WO 99/31100, WO 00/77000 та WO би рентгенодифрактометрії порошку (РДМП), на01/28992], доречні розкриття цих документів навеприклад як описано подалі. дене як посилання. Для гарантії, що певну кристалічну форму Сполуки винаходу, можна тому очікувати, є отримано у відсутність інших кристалічних форм, корисними при профілактиці та лікуванні аритмій, кристалізацію переважно проводять затравкою а зокрема передсерцевої та шлуночкової аритмії. ядрами та/або затравочними кристалами потрібної Певні хворобливі стани, що можна згадати, вклюкристалічної форми при по суті повній відсутності чають передсерцеву фібриляцію (наприклад, пеядер та/або затравочних кристалів інших кристаліредсерцеве мерехтіння). чних форм. Затравочні кристали прийнятної споСполуки винаходу, отже, показані при профілуки/солі можна отримати, наприклад, повільним лактиці або лікуванні хвороб серця, або при покавипарюванням розчиннику з порції розчину прийнзаннях, що стосуються хвороб серця, в яких аритятної сполуки/солі. мії, можна вважати, грають головну роль, Сполуки винаходу можна виділити, використовключаючи ішемічну хворобу серця, несподіваний вуючи способи як добре відомі фахівцям, наприсерцевий напад, інфаркт міокарду, серцеву нестаклад декантуванням, фільтруванням або центричу, серцеву хірургію та випадки тромбоемболії. фугуванням. При лікуванні аритмій, сполуки винаходу виявСполуки можна сушити, використовуючи станлені як селективні стосовно затримки серцевої дартні способи. Фахівці повинні розуміти, що темреполяризації. Хоча сполуки винаходу, як виявлепература та час сушки можуть впливати на власно, затримують серцеву реполяризацію, зокрема, тивості сполук (або солей), що мають форму при лікуванні аритмій, їх активності не обов'язково сольватів, як-то гідрати, у твердому стані, (наприобмежені цим чинником. клад, дегідратування може відбуватися при підвиЗгідно з ще одним аспектом винаходу, запрощеній температурі та/або під зниженим тиском). поновано спосіб лікування аритмії, спосіб включає Наприклад, після утворення кристалічного гідрату, уведення терапевтично ефективної кількості споможе бути критична вологість, нижче якої сушку не 19 74629 20 луки винаходу персоні, що потерпає від такого то тріпотіння-мерехтіння (torsades de pointes)), стану чи схильна до нього. легше поглинатися, або мати інші корисні фармаДля уникнення дублювання термін "лікування" кологічні властивості у порівнянні зі сполуками, включає терапевтичне лікування, а також профівідомими у рівні техніки. лактику стану. Сполуки винаходу мають перевагу в тому, що Сполуки винаходу звичайно застосовують певони мають форму, яка забезпечує підвищену легрорально, підшкірно, внутрішньовенно, інтраартекість обробки. Далі, сполуки винаходу мають періально, трансдермально, інтраназально, інгаляціревагу в тому, що їх можна отримати у формах, які ями або будь-яким іншим парентеральним мають підвищену хімічну стабільність та стабільшляхом, у формі фармацевтичних препаратів, що ність твердого стану (включаючи наприклад, нижчу містять активний інгредієнт (як вільну основу або гігроскопічність). Отже, сполуки можуть бути стабіяк сіль), у фармацевтично прийнятній формі дозульними при зберіганні протягом подовженого перівання. Залежно від розладі та пацієнта, якого ліоду. кують, а також шляху уведення, композиції можна Сполуки винаходу можуть також мати перевазастосовувати у різних дозах. гу в тому, що їх можна кристалізувати з гарними Сполуки винаходу можуть також бути поєднавиходами, з вищою чистотою, протягом меншого ними з будь-якими іншими ліками, корисними при часу, більше зручно, та з нижчою вартістю, ніж лікуванні аритмій та/або інших серцево-судинних раніше отримані форми сполук А, В, С та D та їх розладів. солі. Згідно з ще одним аспектом винаходу запроВинахід ілюстровано, але без обмеження, напоновано тому фармацевтичну композицію, що ступними прикладами, з посиланням на додані включає сполуку винаходу у суміші з фармацевтифігури, в яких: чно прийнятним ад'ювантом, розріджувачем або Фіг.1 показує рентгенодифрактограму порошку носієм. кристалічної форми сполуки А (форма В), отримаСполуки винаходу можна крім того обробляти ної способом з прикладу 1. перед формуванням у придатну фармацевтичну Фіг.2 показує рентгенодифрактограму порошку композицію. Наприклад, кристалічну форму можна кристалічної форми сполуки А (форма А), отримамолоти на менші частинки. ної способом з прикладу 2. Переважні фармацевтичні композиції включаФіг.3 показує рентгенодифрактограму порошку ють композиції у формі системи гелеутворюючої кристалічної форми сполуки А (форма С), отримаматриці, в якій сполуки винаходу запропоновані у ної способом з прикладу 3. поєднанні з полімером, що набухає у водному сеФіг.4 показує рентгенодифрактограму порошку редовищі, як-то виявленому у шлунковокристалічної форми сполуки А, бензолсульфонат, кишковому тракті. Такі системи добре відомі у рівні отриманої способом з прикладу 4. техніки [дивися, наприклад, Advanced Drugs Фіг.5(а) показує рентгенодифрактограму поDelivery Reviews, 11, 37 (1993)]. рошку кристалічної форми параКількість сполуки винаходу, яку застосовують толуолсульфонату сполуки А, (форма А), отримау такій композиції, залежить від стану та пацієнта, ної способом з прикладу 5. якого лікують, а також сполуки, яку застосовують, Фіг.5(b) показує рентгенодифрактограму поале можуть застосовувати, визначають трирошку кристалічної форми паравіально. толуолсульфонату сполуки А (форма В), отримаПридатні добові дози сполук винаходу при ліної способом з прикладу 5. куванні (наприклад, терапевтичному) людини Фіг.6 показує рентгенодифрактограму порошку складають приблизно 0,005-25,0мг/кг маси тіла кристалічної форми 1-гідрокси-2-нафтойної кислопри пероральному уведенні, та приблизно 0,005ти солі сполуки А, отриманої способом з прикла10,0мг/кг маси тіла при парентеральному застосуду 6. ванні вільної основи (тобто, у випадку солі вклюФіг.7 показує рентгенодифрактограму порошку чаючи будь-яку масу, обумовлену присутністю кристалічної форми 1,5-нафталінсульфонової киспротиіону). Переважні межі добових дозі сполуки лоти солі сполуки А, отриманої способом з приквинаходу (у формі вільної основи, як показано виладу 7. ще) при лікуванні (наприклад, терапевтичному) Фіг.8 показує рентгенодифрактограму порошку людини складають приблизно 0,005-10,0мг/кг маси кристалічної форми 2-мезитиленсульфонової кистіла при пероральному уведенні та приблизно лоти солі сполуки А, отриманої способом з прик0,005-5,0мг/кг маси тіла при парентеральному ладу 8. уведенні. Фіг.9 показує рентгенодифрактограму порошку Сполуки винаходу мають перевагу в тому, що кристалічної форми сполуки D, отриманої спосовони є ефективними проти серцевої аритмії. бом з прикладу 9. Сполуки винаходу можуть також мати переваФіг.10 показує рентгенодифрактограму порошгу в тому, що вони можуть бути більш ефективнику кристалічної форми метансульфонату сполуки ми, бути токсичними, мати ширші межі активності D, отриманої способом з прикладу 10. (включаючи виявлення будь-якої комбінації активФіг.11 показує рентгенодифрактограму порошності класу І, класу II, класу IE та/або класу IV ку кристалічної форми гіпурової кислоти солі спо(особливо активності класу І та/або класу IV на луки D, отриманої способом з прикладу 11. додаток до активності класу III)), бути більш потуФіг.12 показує рентгенодифрактограму порошжними, бути діючими довше, давати меншу побічку кристалічної форми сполуки С, отриманої спону дію (включаючи нижчий випадок про аритмії, яксобом з прикладу 12. 21 74629 22 Фіг.13 показує рентгенодифрактограму порошки коли з доречних рентгенограм та/або термограм ку кристалічної форми метансульфонату сполуки ясно (дозволяючи експериментальну похибку), що С, отриманої способом з прикладу 13. утворено по суті так ж кристалічну форму. При Фіг.14 показує рентгенодифрактограму порошпроведенні ДСК початкова температура може ваку кристалічної форми пара-толуолсульфонату ріювати у межах ±5°С (наприклад, ±2°С), а велисполуки С, отриманої способом з прикладу 14. чини інтервалів РДМП можуть варіювати у межах Фіг.15 показує рентгенодифрактограму порош±2 на останній цифрі десятинного дробу. Фахівці ку кристалічної форми [(дифеніл-4повинні розуміти, що інтенсивності РДМП можуть карбоніл)аміно]оцтової кислоти солі сполуки D, варіювати, коли їх виміряно для по суті такої ж отриманої способом з прикладу 15. кристалічної форми, з різних причин, включаючи, Фіг.16 показує рентгенодифрактограму порошнаприклад, переважну орієнтацію. ку кристалічної форми гемібурштинової кислоти Отримання А солі сполуки D, отриманої способом з прикладу 16. Отримання сполуки А та її бензолсульфонату Фіг.17 показує рентгенодифрактограму по(і) 4-[(3-гідроксипропіл)аміно]бензонітрил рошку кристалічної форми (3,4Альтернатива 1 дихлорбензоїламіно)оцтової кислоти солі сполуки Суміш 4-флуорбензонітрилу (12,0г, 99,1ммоль) D, отриманої способом з прикладу 17. та З-аміно-1-пропанолу (59,6г, 793ммоль) переміФіг.18 показує рентгенодифрактограму пошували при 80°С в інертній атмосфері протягом 3 рошку кристалічної форми [(нафталін-2годин перед додаванням води (150мл). Суміші карбоніл)аміно]оцтової кислоти солі сполуки D, давали охолонути до кімнатної температури, та отриманої способом з прикладу 18. далі екстрагували діетиловим етером. Органічний Фіг.19 показує рентгенодифрактограму пошар відділяли, сушили сульфатом натрію, фільтрошку кристалічної форми 2,2,3,3-тетраметил-1,4рували та концентрували у вакуумі, отримуючи 17г дибутанової кислоти солі сполуки D, отриманої (97%) потрібної сполуки як масло, що кристалізуспособом з прикладу 19. валося при стоянні. Фіг.20 показує рентгенодифрактограму поАльтернатива 2 рошку кристалічної форми транс-D,L-1,24-флуорбензонітрил (24,6г, 0,203моль, Aldrich циклопентандикарбонової кислоти солі сполуки D, 99%) додавали до 3-аміно-1-пропанолу (122,0г, отриманої способом з прикладу 20. 1,625моль, 8екв., Aldrich 99%) та суміш гріли до Фіг.21 показує рентгенодифрактограму по80°С протягом 5 годин під азотом. Розчину давали рошку кристалічної форми (+)-О,О'-дибензоїл-Dохолонути до 22°С та додавали воду (300мл). Мувинної кислоти солі сполуки D, отриманої спосотний розчин екстрагували двічі метиленхлоридом бом з прикладу 21. (300мл та 200мл,) та поєднані метиленхлоридні Фіг.22 показує рентгенодифрактограму поекстракти промивали водою (300мл; ГХ-аналіз рошку кристалічної форми (+)-О,О'-ди-параорганічного шару дав приблизно 1,0% залишковотолуоїл-D,L-винної кислоти солі сполуки D, отриго амінопропанолу). маної способом з прикладу 22. Альтернатива 3 Загальні способи До 4-флуорбензонітрилу (30,29г, 247,7ммоль, Рентгенодифрактометрію порошку (РДМП) 1,0екв.), додавали 3-аміно-1-пропанол (150мл, проводили, використовуючи змінну щілину на зра148,8г, 1981,5ммоль, 8,0екв.). Суміш перемішувазках, що отримано стандартними способами, [нали під азотом при кімнатній температурі (27°С), приклад описаними Giacovazzo, С. et al., (1995), доки твердий продукт не розчинився. Розчин гріли Fundamentals of Crystallography, Oxford University (масляна баня) до 77°С та тримали при цій темпеPress; Jenkins, R. та Snyder, R. л. (1996), ратурі протягом 7 годин, перед перемішуванням Introduction to X-Ray Powder Diffractometry, John при температурі довкілля протягом ночі (14 годин). Wiley & Sons, New York; Bunn, С.W. (1948), Воду (365мл) додавали та утворений мутний розChemical Crystallography, Clarendon Press, London; чин екстрагували дихлорметаном (365мл, далі або Klug, Η.P. & Alexander, L.E. (1974), X-ray 245мл). Поєднані органічні шари промивали водою Diffraction Procedures, John Wiley та Sons, New (365мл). Розчин у ДХМ продукту сушили відгонYork.] Рентгеноаналізи проводили, використовуюкою: розчинник (200мл) видаляли та замінювали чи дифрактометр Siemens D5000. свіжим ДХМ (200мл). Ще більше розчиннику Диференційну сканувальну калориметрію (250мл) видаляли для доведення загального об'є(ДСК) проводили, використовуючи прилад Mettler му розчиннику до 365мл. DSC820, стандартними способами, [наприклад (іі) 3-(4-ціаноаніліно)пропілу4описаними Hohne, Г.W.H. et al., (1996), Differential метилбензолсульфонат Scanning Calorimetry, Springer, Berlin]. Альтернатива І Термогравіметричний аналіз (ТГМА) проводиОхолоджений (0°С) розчин 4-[(3ли, використовуючи прилад Mettler Toledo TGA850. гідроксипропіл)аміно]бензонітрилу (з етапу (і) Фахівці повинні розуміти, що кристалічні фор(Альтернатива 1) вище; 17г, 96,5ммоль) у сухому ми сполук винаходу можна отримати аналогічно MeCN (195мл) обробляли триетиламіном (9,8г, способам описано тут та/або способами з прикла96,5ммоль) та далі п-толуолсульфонілхлоридом дів нижче, та можуть виявляти по суті таку ж (20,2г, 106ммоль). Суміш перемішували при 0°С РДМП рентгенограми дифракції та/або ДСК та/або протягом 90 хвилин перед концентруванням у ваТГМА термограми, як розкриті тут. Терміни "по суті куумі. Воду (200мл) додавали до залишку, та водтакі ж" РДМП-рентгенограми дифракції та/або ний розчин екстрагували ДХМ. Органічну фазу ДСК- та/або ТГМА-термограми, включають випадсушили сульфатом натрію, фільтрували та конце 23 74629 24 нтрували у вакуумі. Утворений залишок очищали отримано після витримки протягом ночі). Фази кристалізацією з ізопропанолу, отримуючи 24,6г розділяли та дихлорметановий розчин застосову(77%) потрібної сполуки. вали на наступному етапі нижче. 1 Альтернатива II Н ЯМР (400МГц, CDCl3): δ 2,55-2,65 (2Н, m), Розчин сирого 4-[(3-гідроксипропіл)аміно]2,79 (2Н, t, 74,4), 3,10-3,22 (4Н, m), 3,58-3,73 (2Н, бензонітрилу (з етапу (і) (Альтернатива 2) вище) m), 7,50-7,56 (2Н, m), 7,58-7,63 (1Н, m), 7,83-7,87 концентрували до об'єму 300мл відгонкою та до(2Н, m). давали ще 200мл·метиленхлориду і знов відганя(iv) 5-бензил-3,7-дигідрокси-1-фенілсульфонілли до 300мл (розчинена вода за способом Фішера 1,5-діазацикпооктан 0,07%). Триетиламін (20,55г, 0,203моль), а потім 4ПМС (2,5л, 10 об'ємів) додавали до дихлорме(N,N-диметиламіно)піридин (248мг, 2,0ммоль) дотанового розчину з етапу (ііі) вище. Розчин відгадавали та розчин охолоджували до 0°С. Розчин няли, доки внутрішня температура не досягне тозилхлориду (38,70г, 0,203моль) у метиленхло70°С. Приблизно 1250мл розчиннику збирали. Біриді (150мл) додавали протягом приблизно 30 льше ПМС (2,5л, 10 об'ємів) додавали, а потім хвилин з охолодженням та гарним перемішуванбензиламін (120мл, 0,7екв.) одною порцією (не ням, дозволяючи температурі досягти 5°С. Реакспостерігали нагрівання), та реакційну суміш гріли ційну суміш перемішували протягом 23 годин у при температурі кипіння під зворотним холодильмежах 3-5°С під азотом. (Через 5 годин відбувалоником протягом 6 годин (без змін від 2 годин відся осадження гідрохлориду триетиламіну. ТШХ бору зразку). Більше бензиламіну додавали (15мл) показано дуже мале, якщо це мало місце, подата розчин гріли протягом ще 2 годин. ПМС відгальше перетворення остаточного ціаноспирту на няли (приблизно 3,25л) та додавали толуол (2,5л). 20-23 годину). Воду (300мл) додавали та шари Ще відганяли розчинник (приблизно 2,4л) та далі енергійно перемішували протягом 15 хвилин. Орще додавали толуол (1л). Температура зверху ганічний розчин концентрували відгонкою при 35 була при цьому 110°С. Ще 250мл розчиннику збидо 40°С до об'єму приблизно 60-70мл. Ізопропарали при 110°С. Теоретично, це залишало продукт нол (100мл) додавали протягом 5 хвилин. (На у приблизно 2,4л толуолу при 110°С. Цей розчин цьому етапі відбувалося деяке осадження гранузастосовували на наступному етапі. 1 льованого продукту до додавання ізопропанолу. Н ЯМР (400МГц, CDCl3): δ 7,83-7,80 (4Н, m, Кристалізація відбувалася швидко після додаванАrН), 7,63-7,51 (6Н, m, ArH), 7,30-7,21 (10Н, АrН), ня ізопропанолу.) Відгонку продовжували, викори3,89-3,80 (4Н, m, СН(а)+СН(b)), 3,73 (2Н, s, стовуючи вакуум для видалення останнього метиCH2Ph(a)), 3,70 (2Н, s, CH2Ph(b)), 3,59 (2Н, dd, ленхлориду. (Подальші приблизно 30мл видаляли CHHNSO2Ar(a)), 3,54 (2H, dd, CHHNSO2Ar(b)), 3,40 та дистилят перевіряли за допомогою ГХ на відсу(2H, dd, CHHNSO2Ar(b)), 3,23 (2H, dd, тність метиленхлориду.) Кашку кристалів охолоCHHNSO2Ar(a)), 3,09-2,97 (4H, m, джували до 0 до 5°С протягом приблизно 1 годин з CHHNBn(a)+CHHNBn(b)), 2,83 (2H, dd, повільним перемішуванням та тримали протягом 1 CHHNBn(b)), 2,71 (2H, dd, CHHNBn(a)). години при 0-5°С. Кристали відфільтровували на (Дані отримували з очищеного матеріалу, що середньому спеченому фільтрі та компактний вомістить 1:1 суміш транс- (а), та цис-діолів (b)) логий шар на фільтрі обережно промивали холод(v) 3-бензил-7-(фенілсульфоніл)-9-окса-3,7ним (0°С) ізопропанолом (80мл). Шар на фільтрі діазадицикло[3,3,1]нонан сушили у вакуумі та під струмом азоту протягом Толуольний розчин з попереднього етапу (iv) ночі. Вихід: 52,6г, 78,4мол.%; ВЕРХ: 99,64%. охолоджували до 50°С. Безводну метансульфоноМікроаналіз: знайдено (теоретично): %С: 61,60 ву кислоту (0,2л) додавали. Це викликало зрос(61,67); %Н: 5,41 (5,49); %N: 8,44 (8,47); %S: тання температури від 50°С до 64°С. Через 10 9,71(9,70). хвилин додавали метансульфонову кислоту (1л) (ііі) N,N-біс(2та реакційну суміш гріли до 110°С протягом 5 гооксиранілметил)бензолсульфонамід. дин. Толуол далі відганяли з реакційної суміші; Воду (2,5л, 10 об'ємів), а потім епіхлоргідрин 1,23л збирали, (внутрішня температура не повин(500мл, 4екв.) додавали до бензолсульфонаміду на бути вище 110°С на будь-якому етапі, інакше (250г, 1екв.). Реагенти гріли до 40°С. Водний гідвихід зменшуватиметься.) Реакційну суміш далі роксид натрію (130г у 275мл води) додавали так, охолоджували до 50°С та вакуум застосовували щоб температура реакції залишалася між 40°С та для видалення залишків толуолу. Нагрівання до 43°С. Це тривало приблизно 2 години. (Швидкість 110°С та 650мбар дозволило видалити ще 0,53л. додавання гідроксиду натрію повинна бути повіль(Якщо толуол можна видалити при нижчій темпенішою на початку додавання, ніж у кінці, для підтратурі та тиску, це є кращим.) Реакційній суміші римування температури у встановлених межах.) давали охолонути до 30°С та додавали деіонізоПісля повного додавання гідроксиду натрію реаквану воду (250мл). Це викликало зростання темційну суміш перемішували при 40°С протягом 2 ператури від 30°С до 45°С. Ще додавали воду годин, далі при температурі довкілля протягом (2,15л) протягом загального часу 30 хвилин так, ночі. Надлишок епіхлоргідрину видаляли як водщоб температур була менше 54°С. Розчин охолоний азеотроп вакуумною перегонкою (приблизно джували до 30°С та далі додавали дихлорметан 40 мбар, внутрішня температура 30°С), доки біль(2л). При зовнішньому охолодженні та швидкому ше не відганялося епіхлоргідрину. Дихлорметан перемішуванні реакційну суміш підлужували дода(1л) додавали та суміш перемішували швидко ванням водного гідроксиду натрію (10М, 2л) при протягом 15 хвилин. Фазам давали розділитися швидкості, щоб тримати внутрішню температуру (це тривало 10 хвилин хоча повністю прозорі фази нижче 38°С. Це тривало 80 хвилин. Перемішуван 25 74629 26 ня припиняли та фази розділяли протягом 3 хвиВоду (500мл, 5 об'ємів), а потім 1лин. Шари розділяли. ПМС (2л) додавали до диххлорпінаколон (45,8мл, 1екв.) додавали до гідролорметанового розчину та починали відгонку. Розкарбонату натрію (114,2г, 4екв.). Розчин 3-бензилчинник (2,44л) збирали, доки температура зверху 9-окса-3,7-діазадицикло[3,3,1]нонану 2 НСl не досягне 70°С. Теоретично, це залишало про(100,0г; дивися етап (vi) вище) у воді (300мл, 3 дукт у 1,56л ПМС. Розчину далі давали охолонути об'ємів) додавали повільно, так, щоб виділення до температури довкілля протягом ночі з повільдіоксиду карбону було регульованим (20 хвилин). ним перемішуванням. Твердий осаджений продукт Реакційну суміш гріли при 65-70°С протягом 4 гофільтрували та промивали ПМС (0,5л) з отримандин. Після охолодження до температури довкілля ням жовто-коричневого продукту що, при сушці додавали дихлорметан (400мл, 4 об'ємів) та після при 50°С у вакуумі дав 50,8г (8,9% протягом 3 етаперемішування протягом 15 хвилин фази розділяпів). 20,0г цього продукту розчиняли у ацетонітрилі ли. Водну фазу промивали дихлорметаном (100мл) при нагріванні до температури кипіння під (400мл, 4 об'ємів) та органічні екстракти поєднувазворотним холодильником з отриманням блідоли. Розчин відганяли та розчинник збирали жовтого розчину. Після охолодження до темпера(550мл). Етанол (1л) додавали та відгонку продотури довкілля, кристали, що утворилися, збирали вжували. Ще збирали розчинник (600мл). Етанол фільтруванням та промивали ацетонітрилом (1л) додавали та відгонку продовжували. Ще зби(100мл). Продукт сушили у вакуумі при 40°С прорали розчинник (500мл) (температура зверху була тягом 1 годин з отриманням 17,5г (87%) потрібної при цьому 77°C). Цей розчин (з теоретичним вміссполуки. том 1150мл етанолу) застосовували безпосеред1 Н ЯМР (400МГц, CDCl3): δ 7,18-7,23 (10Н, m), ньо на наступному етапі. 1 3,86-3,84 (2H, m), 3,67 (2H, d), 3,46 (2H, s), 2,91 Н ЯМР (400МГц, CDCl3): δ 1,21 (9Н, s), 2,01(2H, d), 2,85 (2H, dd), 2,56 (2H, dd) 2,59 (2Н, m), 2,61-2,65 (2Н, m), 2,87-2,98 (4Н, m), (vi) 3-бензил-9-окса-3,73,30 (2Н, s), 3,52 (2Н, s), 3,87 (2Н, br s), 7,26 (2H, d, діазадицикло[3,3,1]нонан 2 HCl J=7,6), 7,33 (1H, dd, J=7,6, 7,6), 7,47 (2H, d, J=7,6). Концентровану гідробромідну кислоту (1,2л, 3 (viii) 3,3-диметил-1-(9-окса-3,7відн. об'ємів) додавали до твердого 3-бензил-7діазадицикло[3,3,1]нон-3-іл)-2-бутанон (фенілсульфоніл)-9-окса-3,7Паладій на вугіллі (44г, 0,4мас.екв. 61% володіазадицикло[3,3,1]нонану (400г, дивися етап (ν) гості каталізатору, Johnson Matthey Type 440L) вище) та суміш гріли при температурі кипіння під додавали до етанольного розчину з попереднього зворотним холодильником під азотом. Твердий етапу (vii) вище. Суміш гідрували при 4 бар. Реакпродукт розчиняли у кислоті при 95°С. Після нагріцію вважали завершеною через 5 годин. Каталізавання реакційної суміші протягом 8 годин, ВЕРХтор видаляли фільтруванням та промивали етааналізом показано, що реакція завершилася. Вміст нолом (200мл). Поєднані етанольні фільтрати охолоджували до кімнатної температури. Толуол були/можна застосовувати на етапі (іх) нижче. (1,2л, 3 відн. об'ємів) додавали та суміш енергійно Аналіз розчину дав 61,8г потрібного продукту в перемішували протягом 15 хвилин. Перемішуванетанолі (теоретично 1,35л; виміряно 1,65л). Порня припиняли та фази розділяли. Толуольну фазу цію продукту виділяли та очищали. Аналіз прововідкидали разом з невеликою кількістю міжфазнодили на очищеному продукті. 1 го матеріалу. Кислотну фазу повертали до вихідН ЯМР (300МГц, CDCl3): δ 1,17 (9Н, s), 2,69 ної реакційної ємності та додавали одною порцією (2Н, dt, J=11,4, 2,4), 2,93 (2Н, d, J=10,8), 3,02 (2Н, гідроксид натрію (10М, 1,4л, 3,5 відн. об'ємів). Внуd, J=13,8), 3,26 (2Н, s), 3,32 (2Н, dt, J=14,1), 3,61 трішня температура зросла від 30°С до 80°С. pH (2Н, br s). перевіряли для гарантії, щоб він був більше 14. Цю реакцію можна також проводити, викорисТолуол (1,6л, 4 відн. об'ємів) додавали та темпетовуючи нижче співвідношення мас каталізатору ратура спадала від 80°С до 60°С. Після енергійностосовно бензилованого вихідного матеріалу. Цього перемішування протягом 30 хвилин фази роздіго можна досягти кількома різними способами, ляли. Водний шар відкидали разом з невеликою наприклад, використовуючи різні каталізатори (яккількістю міжфазного матеріалу. Толуольну фазу то Pd/C з відмінним від застосованого вище катаповертали до вихідної реакційної ємності, та додалізатору типу 440L завантаженням металу або вали 2-пропанол (4л, 10 відн. об'ємів). ТемператуRh/C) та/або поліпшенням властивості реакційної ру тримали між 40°С та 45°С. Концентровану гідсуміші стосовно масопереносу (фахівцям зрозумірохлоридну кислоту (200мл) додавали протягом ло, що поліпшеного масопереносу можна досягти, 45 хвилин так, щоб температура залишалася при наприклад, проведенням гідрування в більшому між 40°С та 45°С. Утворився білий осад. Суміш масштабі, ніж описано у вищенаведеній реакції), перемішували протягом 30 хвилин та далі охоловикористовуючи такі способи, співвідношення мас джували до 7°С. Продукт збирали фільтруванням, каталізатору стосовно вихідного матеріалу можна промивали 2-пропанолом (0,8л, 2 відн. об'ємів), зменшити нижче 4:10 (наприклад, між 4:10 та сушили відсмоктуванням та далі ще сушили у ва1:20.). куумній шафі при 40°С. Вихід =297г (91%). (іх) Сполука А, моногідрат бензолсульфонату 1 Н ЯМР (CD3OD + 4 краплі D2O): δ 2,70 (br d, Спосіб 1 2H), 3,09 (d, 2H), 3,47 (br s, 4H), 3,60 (s, 2H), 4,12 Карбонат калію (56,6г, 1,5екв.) та 3-(4(br.s, 2H), 7,30-7,45 (m, 5H). МС-ІАТ(іонізація при ціаноаніліно)пропіл-4-метилбензолсульфонат (диатмосферному тиску): m/2=219 [C13H18N2O+H]+. вися етап (іі) вище, 90,3г, 1екв.) додавали до ета(vii) 3,3-диметил-1-[9окса-7-(фенілметил)-3,7нольного розчину 3,3-диметил-1-(9-окса-3,7діазадицикпо[3,3,1]нон-3-іл]-2-бутанон діазадицикло[3,3,1]нон-3-іл)-2-бутанону (дивися 27 74629 28 етап (viii) вище; 61,8г з аналізу у 1,65л). Реакційну шили у вакуумі при 40°С з отриманням чистого суміш гріли при 80°С протягом 4 годин. Аналізом потрібного продукту (41,2г, 82%). показано, що реагент залишався (8,3г), тому до(Цю перекристалізацію можна проводити з бідавали ще 3-(4-ціаноаніліно)пропіл-4льшими об'ємами розчиннику якщо необхідно, у метилбензолсульфонат (12,2г), та утворену суміш підхожих реакційних ємностях, наприклад гріли при 80°С протягом 4 годин. Розчинник (1,35л) ЕtOН:вод 2:1,45 об'ємів (дало 62% відтвовідганяли, далі додавали ізопропілацетат (2,5л). рення) Розчинник (2,51л) видаляли, додавали ізопропілаЕtOН:вод 6:1,35 об'ємів (дало 70% відтвоцетат (2,5л). Розчинник (0,725л) видаляли. Внутрення)). рішня температура була при цьому 88°С. РозчинПотрібний продукт виділяли після перекристаник (0,825л) видаляли, залишаючи продукт, як лізації як моногідрат (як визначено рентгенодифізопропілацетатний розчин (теоретично у 2,04л). рактометрією одиничного кристалу). Після охолодження до 34°С, додавали воду (0,5л). Спосіб 2 Це була чорна суспензія, можливо з Pd, У суміші. (а) 3-(4-ціаноаніліно)пропілбензолсульфонат pH водної фази був 11. Гідроксид натрію (1М, До розчину 4-[(30,31л) додавали, так, щоб температура була менгідроксипропіл)аміно]бензонітрилу (з етапу (і) Альше 25°С, та суміш перемішували інтенсивно протернатива 3 вище, припустимо 43,65г, 247,7ммоль, тягом 5 хвилин. pH водної фази був 12. Фази роз1,0екв.) у дихлорметані (360мл загальний об'єм діляли та водну фазу відкидали. Ще додавали розчину) додавали послідовно триетиламін (52мл, воду (0,5л), та фази розділяли. Водну фазу відки37,60г, 371,55ммоль, 1,5екв.) та гідрохлорид тридали. Остаточний естерний розчин фільтрували метиламіну (11,89г, 123,85ммоль, 0,5екв.) одною для видалення суспендованих частинок, та фільтпорцією. Жовтий розчин охолоджували до -20°С рат далі доводили точно до 2л. Розчин далі ділили (застосовуючи баню ацетон/сухий лід або холодну на порції 2 по 1л. пластину) та обробляли розчином бензолсульфо(Для уникнення утворення потрібного продукнілхлориду (32мл, 43,74г, 247,7ммоль, 1,0екв.) у ту, що містить високий вміст паладію, можна продихлорметані (220мл, 5 об'ємів з огляду на ціаносводити таку обробку: смолу Delokcan® (12,5г, пирт) урівнюючи тиск крапельною лійкою. Розчин 25мас.%) додавали до розчину вільної основи (1л) додавали порціями так, щоб внутрішня температа суміш гріли при нагріванні до температури китура не перевищувала -14°С. Додавання проводипіння під зворотним холодильником з енергійним ли 25 хвилин до завершення. Суміш далі переміперемішуванням протягом 5 годин. Розчин далі шували протягом 35 хвилин при між -15 та -10°С. охолоджували до кімнатної температури, та переДодавали воду (365мл) та температура зростала мішували протягом 2 діб. Смолу видаляли фільтдо 10°С. Суміш охолоджували до 0°С та переміруванням.) шували, енергійно протягом 15 хвилин. Органічний Аналіз проводили для розрахунку потрібної кішар (об'єм 570мл) збирали та відганяли при атмолькості бензолсульфонової кислоти для отримансферному тиску для видалення ДХМ (450мл, темня бензолсульфонату. пература ємності 40-42°С, температур зверху 38Розчин бензолсульфонової кислоти (20,04г, 39°С). Етанол (250мл) додавали, та розчину дава1екв., у припущенні, що кислота була чистим моли охолонути до нижче 30°С перед вакуумуванногідратом) у ізопропілацетаті (200мл) додавали ням. Ще видаляли розчинник (40мл збирали, тиск протягом 5 хвилин (краще додавати повільніше, 5,2кПа (52мбар), температура ємності та зверху якщо можливо) з енергійним перемішуванням до була 21-23°С), і продукт поступово випадав з у розчину вільної основи (1л) та утворився блідорозчину. Відгонку при цьому припиняли та додажовтий осад. Температура зростала від 18°С до вали ще етанол (50мл). Суміш гріли (гаряча водя22°С. Через 10 хвилин, суміш охолоджували до на баня при 50°С) до 40°С для розчинення усього 10°С та продукт збирали фільтруванням. Продукт твердого продукту, та повільно додавали воду промивали ізопропілацетатом (250мл), відсмокту(90мл) крапельною лійкою. Розчин перемішували вали на фільтр і далі сушили у вакуумі при 40°С. повільно при кімнатній температурі (20°С) протяпротягом 2 діб з отриманням 59,0г (61% з 3гом ночі (15 годин), за цей час викристалізувалося бензил-9-окса-3,7-діазадицикло[3,3,1]нонану трохи продукту. Суміш охолоджували до -5°С (баня лід/метанол) та перемішували при цій темпера2НСl). турі протягом 20 хвилин перед збиранням блідо(Сирий бензолсульфонат альтернативно жовтого твердого продукту фільтруванням. Тверотримували додаванням 70% (за масою) водного дий продукт промивали етанолом/водою (42мл розчину бензолсульфонової кислоти до етанольEtOH, 8мл води) та сушили відсмоктуванням проного розчину вільної основи.) тягом 30 хвилин перед сушкою до постійної маси у Сирий потрібний продукт виділяють як моновакуумній шафі (40°С, 72 годин). Маса сирого гідрат. отриманого продукту була 47,42г (149,9ммоль, Етанол (500мл) та воду (250мл) додавали до 60%). Етанол (160мл, 8 об'єми) додавали до сиросирої потрібної сполуки (50,0г). Розчин гріли до го продукту (20,00 з, 63,22ммоль, 1,0екв.). Суміш 75°С. Матеріал повністю розчинявся при 55°С. перемішували під азотом та гріли до 40°С, викориРозчин тримали при 75°С протягом 5 хвилин, далі стовуючи гарячу водяну баню. Після досягнення охолоджували до 5°С протягом 1 годин. Осадженцієї температури усі тверді продукти розчинилися ня починалося при 18°С. Холодний розчин фільтз отриманням прозорого жовтого розчину. Воду рували та фільтрат промивали етанолом.водою (60мл, 3 об'єми) додавали краплями протягом пе(2:1; 150мл), відсмоктували на фільтр, та далі суріоду 10 хвилин, при цьому внутрішню температу 29 74629 30 ру підтримували у межах 38-41°С. Водяну баню (500мл, 10 об'ємів) додавали до дихлорметанововидаляли та розчину давали охолонути до 25°С го розчину та далі розчинник видаляли відгонкою протягом 40 хвилин, за цей час почалася кристалі(1,25л). Температура зверху все ще була при цьозація, суміш охолоджували до -5°С протягом 10 му 78°С. Розчину давали охолонути до темперахвилин, далі тримали при цій температурі протятури нижче температури кипіння та додавали етагом ще 10 хвилин. Блідо-жовтий твердий продукт нол (250мл, 5 об'ємів). Розчинник видаляли збирали фільтруванням, сушили відсмоктуванням (250мл). Цей гарячий розчин розбавляли етанопротягом 10 хвилин, далі сушили до постійної малом до 890мл, 17,8 об'ємів (25 об'ємів у припуси у вакуумній шафі (40°С, 15 годин). Маса отрищенні, що 100% перетворення у вільну основу). маної потрібної сполуки була 18,51г (58,51ммоль, Після нагрівання до температури кипіння під зво93% (з сирого продукту)). ротним холодильником розчин охолоджували по(b) Сполука А, моногідрат бензолсульфонату вільно. При 5°С додавали затравку потрібної споДо етанольного розчину (загальний об'єм луки. Кристалізація починалася та суміш 770мл, приблизно 20 об'ємів з огляду на амін) 3,3перемішували, при 5°С протягом 30 хвилин. Продиметил-1-(9-окса-3,7-діазадицикло[3,3,1]нон-3-іл)дукт збирали фільтруванням та промивали етано2-бутанону (припустимо 34,97г (перевірено аналілом (2 50мл, 2 1 об'ємів). Продукт далі сушили у зом), 154,5ммоль, 1,0екв.; дивися етап (viii) вище) вакуумній шафі при 40°С протягом 60 годин з додавали 3-(4-ціаноаніліно)пропілу бензол сульотриманням білуватого порошку (26,3г; 74%). 1 фонат (49,05г, 154,52ммоль, 1,0екв.; дивися етап Н ЯМР (400МГц, CDCl3): δ 7,86-7,82 (2Н, m), (а) вище) одною порцією. Утворену суміші гріли 7,39-7,32 (3Н, m), 7,30-7,26 (2Н, m), 6,47 (2Н, m), при 74°С протягом 6 годин, далі перемішували при 4,11-4,07 (4Н, m), 3,70 (2Н, s), 3,36-3,33 (4Н, m), кімнатній температурі і (20°С) протягом 65 годин 3,26 (2Н, t), 3,12 (2Н, d), 2,90 (2Н, d), 2,28-2,21 (2Н, (протягом уікенду; фахівцям зрозуміло, що реакція m), 1,06 (9H,s). 13 буде також успішною без цього пролонгованого C ЯМР (CDCl3): δ 24,07, 26,38, 41,52, 43,52, перемішування при кімнатній температурі). Етанол 56,17, 56,47, 63,17, 68,46, 96,61, 111,64, 121,03, (370мл) видаляли та додавали воду (200мл) (це 133,43. дало суміш 2:1 ЕtOН:вода, загальний об'єм MC (ІЕР-іонізація електророзпиленням): 600мл). При додаванні води, температура ємності m/z=385,1 (M+H)+ спадала від 80°С до 61°С. Розчин знов гріли до Спосіб II 70°С, далі давали охолонути природно до темпеСуміш 4-{[3-(9-окса-3,7-діазадицикло[3,3,1]нонратури довкілля протягом ночі (19 годин), при по3-іл)пропіл]аміно}-бензонітрилу (дивися отримання вільному перемішуванні. Твердий продукт спостеB(l)(vi) нижче; 5,73г, 0,02моль), карбонату калію режено на цьому етапі. Суміш охолоджували до (11,05г, 0,08моль) у MeCN (300мл) обробляли 10°С та далі перемішували при цій температурі хлорпінаколоном (4,44г, 0,032моль). Суміш перепротягом 15 хвилин перед збиранням білуватого мішували при 50°С протягом ночі перед додавантвердого продукту фільтруванням. Твердий проням ДХМ та суміш фільтрували. Шар на фільтрі дукт промивали холодною 2:1 сумішшю етадалі промивали сумішшю ДХМ та MeCN перед нол:вода (150мл), сушили відсмоктуванням протявипарюванням розчиннику з фільтрату. Утворений гом 1,25 годин, далі сушили у шафі (40°С, 20 залишок очищали хроматографією на діоксиді сигодин). Маса сирого отриманого продукту була ліцію, елюючи з градієнтом суміші етилаце57,91г (103,3ммоль, 60%). тат:метанол:метанол з аміаком (95:5:0 до 95:0:5), з Сирий продукт, як знайдено, мав чистоту отриманням потрібної сполуки (5,8г, 73,9%). 98,47% (як визначено ВЕРХ-аналізом), і його переОтримання В(І) кристалізовували (застосовуючи нижченаведений Отримання сполуки В (Спосіб І) спосіб) з отриманням потрібної сполуки з чистотою (і) Трет-бутилу 2-брометилкарбамат 99,75% (84% відтворення). Гідрокарбонат натрію (6,15г, 0,073моль) та диСпосіб перекристалізації: трет-бутилу дикарбонат (11,18г, 0,051моль) розчиЕтанол (562мл) та воду (281мл) додавали до няли у суміші води (50мл) та дихлорметану сирого отриманого вище продукту (56,2г). Розчин (150мл), далі охолоджували до 0°С. 2гріли до 75°С. Увесь матеріал розчинявся при брометиламіну гідробромід (10,0г, 0,049моль) до55°С. Розчин тримали при 75°С протягом 5 хвидавали повільно твердим, та реакційну суміші пелин, перед охолодженням до 5°С протягом 1,5 ремішували протягом ночі при 25°С. Дихлорметагодин. Осадження починалося при 35°С. Холодний новий шар розділяли, промивали водою (200мл) розчин фільтрували та зібраний осад промивали та промивали розчином гідросульфату калію етанолом: водою (2:1, 168мл). Твердий матеріал (150мл, pH=3,5). Органічний шар сушили сульфавідсмоктували на фільтрі перед сушкою у вакуумі том натрію та концентрували у вакуумі. Сире маспри 40°С з отриманням продукту (47,1г, 84%). ло хроматографували на силікагелі, елюючи дих(х) Сполука А (вільна основа) лорметаном, отримавши 7,87г (72%) потрібної Спосіб І сполуки як прозоре безбарвне масло. 1 Сирий бензолсульфонат (50,0г, 1,0екв., з етаН ЯМР (300МГц, CDCI3) δ 4,98 (bs, 1H), 3,45пу (іх) вище; Спосіб 1). додавали до водного гідро3,57 (m, 4H), 1,47 (s, 9H) ксиду натрію (1М, 500мл) промиваючи дихлормеМС-ІАТ(іонізація при атмосферному тиску): таном (1,0л, 20 об'ємів). Поєднану суміші (М+1-С5Н8О2) 126 m/z перемішували протягом 15 хвилин. Шари далі ро(іі) 3-бензил-9-окса-3,7зділяли та мала кількість міжфазного матеріалу діазадицикло[3,3,1]нонан 2НСl залишалася з верхнім водним шаром. Етанол 31 74629 32 Це отримання є альтернативним отриманню, Гідрохлорид з етапу (ііі) вище (146г, що описано в отриманні A(vi) вище. 0,411моль) та 20% Pd(OH)2-C (7,5г) завантажуваТригорлу колбу на 3л опоряджували магнітною ли у гідрогенізатор Парра. Метанол (0,5л) додавамішалкою, термометром та зворотним холодильли та ємність енергійно струшували під тиском ником. Водну гідробромідну кислоту (48%, 0,76л, водню 3,5бар. Реакцію відстежували ГХ-аналізом 4,51моль) додавали до твердого 3-бензил-7та, як знайдено, вона закінчилася через 1 годину. (фенілсульфоніл)-9-окса-3,7Каталізатор відфільтрували та фільтрат концентдіазадицикло[3,3,1]нонану (190г, 0,53моль, дивися рували, отримавши білуватий кристалічний проотримання A(v) вище) та суміш гріли при темперадукт. Сирий продукт розчиняли у гарячому ацетотурі кипіння під зворотним холодильником під азонітрилі (1,2л), та далі фільтрували гарячим. том. Твердий продукт розчиняли при 90°С. Після Фільтрат розбавляли етилацетатом (1,2л). Прозонагрівання суміші протягом 12 годин ГХ-аналізом рому розчину давали стояти протягом ночі при показано, що реакція завершилася. Вміст охолокімнатній температурі. Першу партію кристалів джували до кімнатної температури. Толуол (0,6L) збирали та сушили у вакуумі, отримавши 52г потдодавали та суміш перемішували протягом кількох рібної сполуки як білий твердий продукт. Фільтрат хвилин. Фази розділяли. Водну фазу повертали до концентрували майже досуха, далі розчиняли у вихідної реакційної ємності та водний гідроксид гарячому ацетонітрилі (0,4л) та розбавляли етинатрію (10М, 0,85л 8,5моль) додавали одною порлацетатом (0,4л). Другу партію кристалів (38г) цією. Внутрішня температура зростала до 80°С та отримали після охолодження розчину до 10°С. суміш була сильно основною. Толуол (0,8л) додаОбидві партії, як знайдено, були порівнюваними за вали коли внутрішня температур падала до 55°С. ГХ-аналізом та 1Н ЯМР-аналізом. Поєднаний виПісля енергійного перемішування протягом 30 хід: 90г (83%). хвилин, толуольну фазу відділяли та повертали до (ν) Трет-бутилу 7-[3-(4-ціаноаніліно)пропіл]1-9вихідної реакційної ємності. 2-Пропанол (1,9л) доокса-3,7-діазадицикло[3,3,1]нонан-3-карбоксилат давали та внутрішню температуру доводили до Гідрохлорид трет-бутилу 9-окса-3,7між 40°С та 50°С. Концентровану гідрохлоридну діазадицикло[3,3,1]нонан-3-карбоксилату (дивися кислоту додавали (до кислотності) при такій швидетап (iv) вище; 1,1г, 4,15ммоль) змішували з MeCN кості, щоб тримати температуру між 40 та 50°С. (46мл), вод (2,5мл) та карбонатом калію (3,5г, Утворився білий осад. Суміш перемішували протя25ммоль). Суміш перемішували протягом 4 годин гом 30 хвилин та далі охолоджували до 7°С. Білий перед додаванням хлороформу та фільтрували порошок збирали фільтруванням, промивали 2через Celite®. Фільтрат концентрували у вакуумі з пропанолом (0,4л), сушили продуванням повітря отриманням 0,933г вільної основи. Це далі змішучерез зразок протягом десяти хвилин, та далі сували з 3-(4-ціаноаніліно)пропілу 4шили у вакуумній шафі при 40°С. Вихід: 130г метилбензолсульфонатом (дивися отримання А(іі) (84%). вище; 2,1г, 6,2ммоль) та карбонатом калію (0,86г, (ііі) Трет-бутилу 7-бензил-9-окса-3,76,2ммоль) у MeCN (18мл). Утворену суміш передіазадицикло[3,3,1]нонан-3-карбоксилату гідрохмішували протягом ночі при 60°С перед концентлорид руванням у вакуумі. Залишок обробляли ДХМ Тригорлу колбу на 5л опоряджували верхньою (250мл) та 1Μ NaOH (50мл). Шари розділяли та мішалкою, термометром та патрубком для азоту. шар ДХМ промивали двічі водним NaHCO3 перед Воду (1,4л), дихлорметан (1,4 44л), гідрокарбонат сушкою сульфатом натрію та концентрували у натрію (150г, 1,79моль) та 3-бензил-9-окса-3,7вакуумі. Продукт очищали флеш-хроматографією, елюючи з градієнтом суміші толудіазадицикло[3,3,1]нонан 2НСl (130г, 0447моль, з ол:етилацетат:триетиламін (2:1:0 до 1000:1000:1), етапу (іі) вище) завантажували по порядку. Суміш з отриманням 1,47г (91%) потрібної сполуки. швидко перемішували протягом десяти хвилин та (vi) 4-{[3-(9-окса-3,7-діазадицикло[3,3,1]нон-3далі додавали повільно ди-трет-бутилу дикарбоіл)пропіл]аміно)бензонітрил нат (0,113л, 0,491моль). Суміш перемішували Потрібну сполуку отримали з виходом 96%, швидко протягом 3 годин при кімнатній температувикористовуючи спосіб, аналогічний описаному в рі. Органічний шар відділяли, сушили сульфатом отриманнях C(v) та D(iii) нижче, використовуючи магнію, фільтрували та концентрували, отримавтрет-бутилу 7-[3-(4-ціаноаніліно)пропіл]-9-окса-3,7ши 160г білуватого твердого продукту. Білуватий діазадицикло[3,3,1]нонан-3-карбоксилат (з етапу твердий продукт завантажували у тригорлу колбу (ν) вище). на 3 л, опоряджену верхньою мішалкою, термоме(vii) Сполука В тром та лійкою для додавання. Етилацетат (0,6л) До розчину трет-бутилу 2-брометилкарбамату завантажували та прозорий розчин охолоджували (4,21г, 0,019моль; дивися етап (і) вище) у DMF до -10°С. Розчин НСl у діоксані (4М) додавали (65мл) додавали 4-{[3-(9-окса-3,7краплями до pH менше 4. Гідрохлорид осаджувавдіазадицикло[3,3,1]нон-3ся та суміш перемішували протягом ще години. іл)пропіл]аміно}бензонітрил (дивися етап (vi) вище, Продукт збирали фільтруванням, промивали ети4,48г, 0,016моль) та триетиламін (3,27мл, лацетатом (0,1л), та сушили протягом ночі у ваку0,024моль). Суміш перемішували протягом ночі умній шафі. Білий кристалічний продукт важив при 35°С та далі концентрували у вакуумі. Зали146г (92% виходу). шок розчиняли у дихлорметані (80мл) та промива(iv) Трет-бутилу 9-окса-3,7ли насиченим хлоридом натрію. Водний шар екстдіазадицикло[3,3,1]нонан-3-карбоксилату гідрохлорид рагували дихлорметаном (1 150мл). Поєднані органічні екстракти сушили сульфатом натрію та 33 74629 34 концентрували у вакуумі. Сире красно-коричневе лимонну кислоту (10%, 25мл) додавали до порції маслянистого твердого продукту (5г, 138ммоль). масло хроматографували ( 2) на силікагелі, елюВодний шар відділяли та органічний шар промиючи сумішшю хлороформ:метанол:конц. NH4OH вали знов лимонною кислотою (10%, 20мл). Водні (9:1:0,02) з отриманням 3,75г (56%) потрібної спошари поєднували та обробляли твердим гідрокарлуки. 1 бонатом натрію до нейтральності. Водну фазу Н ЯМР (300МГц, CD3OD): δ 7,37-7,40 (d, екстрагували етилацетатом (2 50мл), сушили суJ=8,8Гц, 2Н), 6,64-6,67 (d, J=8,8Гц, 2Н), 3,94 (bs, 2H), 3,21-3,31 (m, 4H), 3,01 (bs, 4H), 2,47-2,59 (m, льфатом магнію та фільтрували: Фільтрат випа8H), 1,90 (bs, 2H), 1,39 (s, 9H) рювали досуха під зниженим тиском, отримуючи 13 C ЯМР (75МГц, CD3OD): δ 158,5, 134,7, потрібну сполуку як безбарвний напівтвердий про121,9, 113,2, 97,7, 80,3, 69,2, 58,8, 58,1, 57,5, 57,3, дукт, який повністю тверднув при зберіганні у хо41,9, 38,3, 28,9, 26,2. лодильнику (4,68г, 93%), темп.пл. 58-60°С. 1 MC-IAT:(M+1)=430m/z Н ЯМР (300МГц, CDCI3) δ 1,46 (9Н, s), 2,33Отримання B(ll). 2,57 (4Н, m), 2,6-2,68 (2Н, m) 2,75-2,85 (4Н, m), Отримання сполуки В (Спосіб II) 3,22 (2Н, q), 3,26 (2Н, s), 3,83 (2Н, bs), 6,17 (1Н, bs) (і) [2-(7-бензил-9-окса-3,77,2-7,4 (5Н, m). діазадицикло[3,3,1]нон-3-іл)етил]карбамової кисMC:m/z=362(MH+). лоти трет-бутиловий естер Альтернатива 2 Альтернатива 1 (a) 3-(7-бензил-9-окса-3,7(a) 2-(трет-бутилоксикарбоніламіно)етилу тодіазадицикло[3,3,1]нон-3-іл)етил)пропіонамід Тризилат етиламін (3,60г, 35,7ммоль) додавали повільно до Розчин п-толуолсульфонілхлориду (28,40г, розчину дигідрохлориду 3-бензил-9-окса-3,7148ммоль) у дихлорметані (100мл) додавали крадіазадицикло[3,3,1]нонану (дивися отримання A(vi) плями протягом 30 хвилин при 0°С до суміші третвище; 5г, 17ммоль) в етанолі (50мл). Акриламід бутилу N-(2-гідроксіетил)карбамату (20г, (1,34г, 18ммоль) додавали до цієї суміші, яку далі 120ммоль), триетиламіну (18,80г, 186ммоль) та гріли до температури кипіння під зворотним холохлориду триметиламонію (1,18г, 12,4ммоль) у дихдильником протягом 7 годин. Реакційну суміші далі лорметані (120мл). Суміш перемішували при 0°С концентрували під зниженим тиском. Воду (50мл) протягом 1 години, далі фільтрували, промиваючи та гідроксид натрію (1М, 150мл) додавали до задихлорметаном (100мл). Фільтрат промивали 10% лишку та суміш екстрагували етилацетатом лимонною кислотою (3 100мл) та розсолом (2 200мл). Поєднані органічні екстракти сушили (100мл). Органічний шар сушили сульфатом магсульфатом магнію, фільтрували та концентрували нію та далі фільтрували. Фільтрат концентрували під зниженим тиском, отримавши безбарвний твепід зниженим тиском з отриманням масла. Масло рдий продукт. Це перекристалізовували з етиларозчиняли в етилацетаті (40мл) та далі повільно цетату (50мл), отримуючи потрібну сполуку (3,80г, додавали ізогексан (160мл). Утворену кашку пе76%), темп.пл. 157-159°С. 1 ремішували при кімнатній температурі протягом 17 Н ЯМР (300МГц, CDCI3): δ 2,39 (2Н, t), 2,42годин та далі фільтрували. Зібраний твердий про2,61 (6Н, m), 2,82-2,95 (4Н, m), 3,39 (2Н, s), 3,91 дукт промивали ізогексаном (240мл,), отримуючи (2Н, bs), 5,07 (1Н, bs), 7,18-7,21 (2Н, m), 7,25- 7,39 потрібну сполуку як безбарвний порошок (25г, (ЗН, m), 9,5 (1Н, bs). 64%), темп.пл. 64-66°С. MC: m/z=290(MH+). 1 Н-ЯМР (300МГц, CDCI3,): δ 1,40 (9Н, s), 2,45 (b) [2-(7-бензил-9-окса-3,7(3Н, s), 3,38 (2Н, q),4,07 (2Н, t), 4,83 (1Н, bs) 7,34 діазадицикло[3,3,1]нон-3-іл)етил)карбамової кис(2H,d), 7,87 (2H,d). лоти трет-бутиловий естер MC: m/z=216 (MH+(316)-Boc). N-бромсукцинімід (6,0г, 33ммоль) додавали (b) [2-(7-бензил-9-окса-3,7порціями протягом 1 хвилин до розчину 3-(7діазадицикло[3,3,1]нон-3-іл)етил]карбамової кисбензил-9-окса-3,7-діазадицикло[3,3,1]нон-3лоти трет-бутиловий естер іл)етил]-пропіонамід (дивися етап (а) вище; 5г, Розчин дигідрохлориду 3-бензил-9-окса-3,712ммоль) до трет-бутоксиду калію у трет-бутанолі діазадицикло[3,3,1]нонану (дивися отримання A(vi) (1М, 81мл) та трет-бутанолу (20мл). Суміш далі вище; 10г, 34ммоль) у воді (25мл) додавали повігріли при 60-65°С протягом 30 хвилин. Реакційній льно до розчину гідрокарбонату натрію (10г, суміші давали досягти кімнатної температури та 119ммоль) у воді 10мл). Ще додавали воду (5мл) далі додавали воду (100мл). Суміш екстрагували та суміш перемішували при кімнатній температурі етилацетатом (2 50мл). Поєднані органічні екстпротягом 10 хвилин. Розчин 2-(третракти промивали розсолом (50мл), сушили сульбутилоксикарбоніламіно)етилу тозилату (дивися фатом магнію, фільтрували (промиваючи шар на етап (а) вище; 11,92г, 37ммоль) у толуол (40мл) фільтрі етилацетатом (50мл)) та далі фільтрат додавали. Цю суміш далі гріли при 65- 70°С протяконцентрували під зниженим тиском з отриманням гом 7 годин перед перемішуванням при кімнатній потрібної сполуки як коричневого масла (6,5г, температурі протягом ночі. Реакційну суміші знов >100%). 1 гріли до 50°С та фази розділяли. Водний шар ексН ЯМР (300МГц, CDCI3): δ 1,46 (9Н, s), 2,4трагували толуолом (40мл) при 50°С. Поєднані 2,58 (4Н, m), 2,58-2,7 (2Н, m) 2,75-2,91 (4Н, m), органічні шари промивали насиченим гідрокарбо3,22 (2Н, q), 3,28 (2Н, s), 3,83 (2Н, bs), 6,19 (1Н, bs) натом натрію (25мл). Розчинники випарювали під 7,2-7,42 (5Н, m). зниженим тиском, отримуючи суміш масла та твеMC: m/z=316(MH+). рдого продукту (13г, >100%). Етилацетат (50мл) та Альтернатива 3 35 74629 36 1 (а) 3-бензил-9-окса-3,7Н ЯМР (300МГц, CDCI3): δ 1,42 (9Н, s), 2,31 діазадицикло[3,3,1]нонан (3Н, s), 2,62 (6Н, s) 3,40 (2Н, q), 4,01 (2Н, t), 4,83 Усі об'єми та еквіваленти виміряні з огляду на (1H,bs), 6,98 (2H,s) кількість дигідрохлориду 3-бензил-9-окса-3,7(с) [2-(7-бензил-9-окса-3,7діазадицикло[3,3,1]нонану (дивися отримання A(vi) діазадицикло[3,3,1]нон-3-іл)етил]карбамової кисвище), що застосовували. Толуол (420мл, 7 об'єлоти трет-бутиловий естер, 2,4,6мів) та водний розчин гідроксиду натрію (2М, триметилбензолсульфонат 420мл, 7 об'ємів, 4,0екв.) додавали до дигідрохлоГарячий (28°С) розчин 2-(третриду 3-бензил-9-окса-3,7бутилоксикарбоніламіно)етилу 2,4,6діазадицикло[3,3,1]нонану (60,07г, 206,03ммоль, триметилбензолсульфонату (70,93г, 206,03ммоль, 1,0екв., дивися отримання A(vi) вище). Суміш пе1,0екв., дивися етап (b) вище) у толуолі (240мл, 4 ремішували під азотом, гріли до 60°С та тримали об'єми) додавали до розчину 3-бензил-9-окса-3,7при цій температурі протягом 30 хвилин, за цей діазадицикло[3,3,1]нонану (44,98г, 206,03ммоль, час утворилися два прозорі шари. Нижчий водний 1,0екв.) у толуолі (240мл, 4 об'єми) (дивися етап шар видаляли, та толуольний розчин потрібної (а) вище). Утворений розчин швидко перемішувасполуки (вільна основа) сушили азеотропною відли під азотом з нагрівання при 68°С протягом 8 гонкою при атмосферному тиску (загальний об'єм годин. Реакційну суміші перемішували при темпевидаленого розчиннику =430мл; загальний об'єм ратурі довкілля протягом 84 годин. Тонкий білий доданого толуолу =430мл), далі концентрували до твердий осад утворився у блідо-жовтому розчині. об'єму 240мл (4 об'єми). Аналіз за Фішером на Суміш охолоджували до +9°С, та потрібну сполуку цьому етапі показав 0,06% води у розчині. Висузбирали фільтруванням. Реакційну ємність промишений розчин потрібної сполуки (теоретично вали толуолом (100мл) та додавали до фільтру. 44,98г, 206,03ммоль, 1,0екв.) застосовували як Шар на фільтрі промивали толуолом (150мл). Бітакий на наступному етапі. лий твердий продукт сушили відсмоктуванням (b) 2-(трет-бутилоксикарбоніламіно)етилу2,4,6протягом 15 хвилин, далі сушили до постійної матриметилбензолсульфонат си у вакуумі при 40°С протягом 23 годин. Вихід Триетиламін (65мл, 465,3ммоль, 1,5екв.) доотриманої потрібної сполуки був 79,61г, давали одною порцією до розчину трет-бутилу N141,7ммоль, 69%. Поєднані фільтрат та промивки (2-гідроксіетил)карбамату (50,11г, 310,2ммоль, (670мл.) промивали водним розчином гідроксиду 1,0екв.) у дихлорметані (250мл, 5 об'єми). Розчин натрію (2М, 200мл, 3,3 об'єми). Суміш гріли до охолоджували до -10°С та гідрохлорид тримети60°С та тримали при цій температурі протягом 20 ламіну (14,84г, 155,1ммоль, 0,5екв.) додавали одхвилин з швидким перемішуванням. Два шари далі ною порцією. Утворену суміш охолоджували подарозділяли. Толуольний розчин концентрували до льше до -15°С, перемішували протягом 5 хвилин, 200мл вакуумною перегонкою (темп.кип.50-54°С далі обробляли розчином мезитиленсульфонілхпри 650-700мбар; темп.кип.46°С при 120мбар у лориду (74,74г, 341,2ммоль., 1,1екв.) у дихлормекінці). В процесі відгонки розчин став мутним внатані (250мл, 5 об'єми), протягом 28 хвилин так, слідок утворення потрібної сполуки, було припущоб внутрішня температура залишалася нижче щено, що 20% початкової кількості 3-бензил-910°С. Після повного додавання утворився осад і окса-3,7-діазадицикло[3,3,1]нонану залишалося у суміш перемішували при -10°С протягом ще 30 фільтраті, відтак ще 2-(третхвилин. Додавали воду (400мл, 8 об'єми) та увесь бутилоксикарбоніламіно)етилу 2,4,6осад розчинявся. Суміш перемішували швидко триметилбензолсульфонат (14,20г, 41,21ммоль, протягом 5 хвилин, а далі два шари розділяли. 0,2екв.) додавали одною порцією (завантажувати Розчинник замінювали з дихлорметану на ізопрояк твердий краще, ніж як розчин у толуолі). Мутний панол відгонкою під зниженим тиском. Розчинник розчин гріли при 67°С протягом 8 годин з швидким видаляли (450мл) та замінювали ізопропанолом перемішуванням, та далі перемішували при тем(450мл) (початковий тиск був 450мбар, пературі довкілля протягом 11 годин. Суміш охотемп.кип.24°С; кінцевий тиск був 110мбар, лоджували до +8°С, та потрібну сполуку збирали темп.кип.36°С). У кінці відгонки, розчинник (150мл) фільтруванням. Реакційну ємність промивали ще видаляли для доведення об'єму до 350мл, (7 толуолом (2 30мл), та додавали до фільтру. Білий об'ємів з огляду на кількість трет-бутилу N-(2твердий продукт сушили відсмоктуванням протягідроксіетил)карбамату, що застосовували). Розгом 15 хвилин, далі сушили до постійної маси у чин охолоджували до 25°С, далі повільно додававакуумі при 40°С протягом 7 годин, вихід потрібної ли воду (175мл) з перемішуванням, внаслідок чого сполуки був 23,25г, 41,39ммоль, 20%. Поєднаний розчин поступово мутнів. Жодний твердий продукт вихід потрібної сполуки (білий твердий продукт) не осаджувався на цьому етапі. Ще додавали воду був 102,86г, 183,11ммоль, 89%, темп.пл 190(125мл), та твердий осад починав утворюватися 190,5°С. 1 після додавання приблизно 75мл. Внутрішня темН ЯМР (300МГц, CDCI3): δ 1,43 (9H, s), 2,17 пература зросла від 25°С до 31°С. Суміш перемі(3H, s), 2,51 (6H, s), 2,73-2,80 (2H, m), 2,90-2,94 шували повільно та охолоджували до 7°С. Твер(4Н, m), 3,14-3,22 (4Н, m), 3,37 (2Н, bm), 3,89 (2Н, дий продукт збирали фільтруванням, промивали bs), 4,13 (2Н, bs), 6,74 (2Н, s), 7,12 (1H,bt), 7,42сумішшю ізопропанол:вода (1:1, 150мл) та сушили 7,46 (5H,m) у вакуумі при 40°С протягом 21 годин з отриман(ii) [2-(9-окса-3,7-діазадицикло[3,3,1]нон-3ням потрібної сполуки як білого кристалічного твеіл)етил]карбамовоі кислоти трет-бутиловий естер рдого продукту (92,54г, 87%),. темп.пл.73,5°С. Спосіб 1 Гідрокарбонат натрію (0,058г, 0,069ммоль) та 5% Pd/C (0,250г, Johnson Matthey 37 74629 38 Type 440) додавали до розчину [2-(7-бензил-9Matthey type 440L). Поєднану суміш далі гідрували окса-3,7-діазадицикло[3,3,1]нон-3під тиском водню 4бар протягом 24 годин. Реакцію іл)етил]карбамовоі кислоти трет-бутилового естевідстежували за допомогою ТШХ, використовуючи ру (дивися етап (i), Альтернатива 1 вище, 1г, пластину з діоксидом силіцію з рухомою фазою 2,77ммоль) в етанолі (10мл). Суміш далі гідрували Х:ДХМ 1:1 за об'ємом, (X представляє хлоропри 500кПа (5бар) протягом 18 годин. Реакційну форм:метанол:концентрований аміак 80:18:2 за суміш фільтрували через Cehte® та далі промиваоб'ємом). Візуалізацію здійснювали УФ-світлом ли етанолом (20мл). Розчин концентрували під (254нм) та фарбуванням водним перманганатом зниженим тиском з отриманням масла. Це розчикалію. Цим показано повну витрату вихідного маняли у дихлорметані (20мл) та промивали гідроктеріалу та появу потрібної сполуки. Реакційну сусидом натрію (1М, 10мл). Органічну фазу відділяміші підлужували водним гідроксидом натрію (10М, ли, сушили сульфатом магнію та далі 8мл, 0,9мольекв.), далі фільтрували через кізельфільтрували. Фільтрат концентрували під знижегур. Шар на фільтрі промивали етанолом (100мл, ним тиском з отриманням потрібної сполуки як 2,0 об'ємів). Утворений розчин потрібної сполуки жовтого твердого продукту (0,67г, 87%), темп.пл (припустимо 24,15г, 100%) застосовували безпо91-93°С. середньо у наступній реакції. 1 Н ЯМР (300МГц, CDCI3): δ 1,46 (9Н, S), 2,25 (ііі) Сполука В (2Н, t), 2,53-2,65 (2Н, m) 2,95-3,06 (4Н, m), 3,2-3,38 Спосіб І (4Н, m), 3,64 (2Н, bs), 4,65 (1Н, bs) MC: 3-(4-ціаноаніліно)пропіл-4m/z=272(MH+). метилбензолсульфонат (дивися отримання А(іі) Спосіб 2: [2-(7-бензил-9-окса-3,7вище; 0,30г, 0,92ммоль) та карбонат калію (0,2г, діазадицикло[3,3,1]нон-3-іл)етил]-карбамової кис1,38ммоль) додавали до розчину [2-(9-окса-3,7лоти трет-бутилового естеру 2,4,6діазадицикло[3,3,1]нон-3-іл)етил]карбамової кистриметилбензолсульфонат (320г, 1,0моль екв., лоти трет-бутилового естеру (дивися етап (іі), 1,0від. об'ємів/масу, дивися етап (і), Альтернатива Спосіб 1 вище; 0,250г, 0,92ммоль) в етанолі (5мл). 3 вище), толуол (640мл, 2,0 об'ємів) та водний Реакційну суміші гріли до 70°С протягом 10 годин гідроксид натрію (1М, 1,6л, 5,0 об'ємів) перемішуперед концентрацією суміші під зниженим тиском. вали разом протягом 15 хвилин та шари далі розЗалишок розподіляли між етилацетатом (20мл) та діляли. Органічний шар з вмістом [2-(7-бензил-9гідроксидом натрію (1М, 10мл). Водну фазу знов окса-3,7-діазадицикло[3,3,1]нон-3екстрагували етилацетатом (20мл). Поєднані оріл)етил]карбамовоі кислоти трет-бутилового естеганічні фази концентрували під зниженим тиском з ру розбавляли етанолом (690мл, 2,16 об'ємів) та отриманням жовтого твердого продукту (0,290г). водою (130мл, 0,4 об'єми). Лимонну кислоту Твердий продукт розчиняли в етилацетаті (10мл) (32,83г, 0,3мольекв.) та 5% Pd/C (20,8г, 0,065маста цей розчин промивали розчином лимонної кисекв. 61% води вологий каталізатор, Johnson лоти (0,250г) у воді (10мл). Водну фазу відділяли, Matthey type 440L) додавали. Поєднану суміш далі підлужували гідроксидом натрію (1М, 10мл) та ексгідрували під тиском водню 4бар протягом 24 готрагували етилацетатом (2 10мл). Усі органічні дин. Реакцію відстежували за допомогою ТШХ, фази поєднували, сушили сульфатом магнію та використовуючи пластину з дюксидом силіцію з далі фільтрували (промиваючи фільтровані тверді рухомою фазою Х:ДХМ (1:1 за об'ємом, X предпродукти етилацетатом (10мл)). Фільтрат концентставляє хлороформ метанол концентрований амірували під зниженим тиском з отриманням жовтоак 80:18:2 за об'ємом) Візуалізацію здійснювали го твердого продукту (0,160г). Це суспендували в УФ-світлом (254нм) та фарбуванням водним перетилацетаті (0,2мл) та далі фільтрували з отриманганатом калію. Цим показано повну витрату манням потрібної сполуки (0,050г, 12%), темп.пл. вихідного матеріалу та появу потрібної сполуки. 113-115°С. 1 Реакційну суміші фільтрували через кізельгур та Н-ЯМР (400МГц, ДМСО-d6): δ 1,32 (9Н, s), 1,7 промивали етанолом (590мл, 1,84 об'єми). Утво(2Н, qt), 2,20 (2Н, t), 2,22-2,3 (4Н, m), 2,38-3,1 (2Н, рений розчин потрібної сполуки (припустимо m) 2,8-2,85 (4Н, m), 3,05 (2Н, q), 3,19 (2Н, q), 3,79 154,85г, 100%) застосовували безпосередньо у (2Н, bs), 6,47 (1Н, t), 6,66 (2Н, d), 6,69 (1Н, t), 7,41 наступній реакції. (2Н, d). MC: m/z=430 (МН+). Спосіб 3: [2-(7-бензил-9-окса-3,7Спосіб II діазадицикло[3,3,1]нон-3-іл)етил]-карбамової кисДо розчину [2-(9-окса-3,7лоти трет-бутилового естеру 2,4,6діазадицикло[3,3,1]нон-3-іл)етил]карбамової кистриметилбензолсульфонат (50г, 1,0моль екв., лоти трет-бутилового естеру з етапу (іі) (Спосіб 3) 1,0відн. об'єм/маса, дивися етап (і), Альтернатива вище (припустимо 24,15г, 1,0моль екв. 3 вище), толуол (100мл, 2,0 об'ємів) та водний 1,0маса/об'єм) у суміші толуолу (приблизно гідроксид натрію (1М, 100л, 2,0 об'ємів) перемішу100мл), етанолу (приблизно 200мл) та води (прибвали разом протягом 20 хвилин, далі при 30°С лизно 14мл), додавали безводний карбонат калію протягом 10 хвилин та шари далі розділяли. Орга(18,58г, 1,5мольекв.). Твердий 3-(4нічний шар з вмістом [2-(7-бензил-9-окса-3,7ціаноаніліно)пропілу бензолсульфонат (28,17г, діазадицикло[3,3,1]нон-3-іл)етил]карбамової кис1,0мольекв., дивися отримання А(іх), Спосіб 2, лоти трет-бутилового естеру розбавляли етаноетап (а) вище) додавали та поєднану суміш гріли лом (100мл, 2,0 об'ємів). До цього додавали роздо 70°С протягом 6 годин. Реакцію відстежували чин лимонної кислоти (5,14г, 0,3моль екв.) у воді за допомогою ТШХ, використовуючи пластину з (5мл, 0,1 об'єм), а потім 5% Pd/C (1,50г, 0,03масдіоксидом силіцію з рухомою фазою Х:ДХМ 1:1 за екв. 61% води вологий каталізатор, Johnson об'ємом (в якій X представляє хлоро 39 74629 40 форм:метанол:концентрований аміак 80:18:2 за Потрібну сполуку отримували додаванням тооб'ємом). Візуалізацію здійснювали УФ-світлом луолусульфонілхлориду до 4-(4(254нм) та фарбуванням водним перманганатом гідроксибутил)бензонітрилу (дивися етап (іі) вище). калію. Цим показано повну витрату вихідного ма(iv) трет-бутил у 7-[4-(4-ціанофеніл)бутил]-9теріалу та появу потрібної сполуки. Реакційну суокса-3,7-діазадицикло[3,3,1]нонан-3-карбоксилат міш охолоджували, та розчинник концентрували у Тригорлу колбу на 2л опоряджували магнітною вакуумі. Залишок розподіляли між толуолом мішалкою, термометром та зворотним холодиль(200мл) та водою (200мл). Шари розділяли, та ником. Колбу завантажували розчином 4-(4органічну фазу концентрували у вакуумі з отриціанофеніл)бутилу толуолсульфонату (72г, манням жовтого твердого продукту (38,6г). 0,218моль, дивися етап (ііі) вище) у диметилфорОтримання С маміді (0,55л). Додавали гідрохлорид трет-бутилу Отримання сполуки С 9-окса-3,7-діазадицикло[3,3,1]нонан-3(і) 4-(4-ціанофеніл)бут-3-ин-1-ол карбоксилату (48,2, 0,182моль, дивися отримання Карбонат калію (376,7г, 2,5моль екв.) розчиняB(l)(iv) вище), а потім карбонат калію (62,9г, ли у суміші 1,2-диметоксіетану (ДМЕ, 1,2л, 6 об'є0,455моль). Гетерогенну суміш перемішували промів) та вод (1,2л, 6 об'ємів). Паладій на вугіллі тягом 22 годин при 85°С. ТШХ-аналіз показав пов(20г, 0,01моль екв., 10% Johnson Matthey type 87L, ну витрату вихідного матеріалу. Реакційну суміші 60% води), трифенілфосфін (11,5г, 0,04моль екв.) охолоджували до кімнатної температури та розбата додавали йодид купруму(І) (4,2г, 0,02моль екв.). вляли водою (0,5л). Суміш екстрагували етилаце4-бромбензонітрил (200г, 1моль екв.) далі додавататом (3 0,4л) та органічні фракції поєднували. ли, промиваючи сумішшю ДМЕ (200мл, 1 об'єм) та Після промивки водою (2 200мл) та розсолом води (200мл, 1 об'єм). Цю суміш перемішували (200мл) органічний шар сушили сульфатом магшвидко під азотом протягом мінімум тридцяти нію, фільтрували та концентрували у вакуумі. Сихвилин. Розчин бут-3-ин-1-олу (92,1мл, 1,1моль ре коричневе масло очищали хроматографією на екв.) у ДМЕ (200мл, 1 об'єм) та води (200мл, 1 силікагелі, елюючи сумішшю 3:2 гекоб'єм) додавали краплями протягом 5 хвилин. Посан/етилацетат, отримавши 34г (48% виходу) потєднану суміш далі гріли до 80сС протягом 3 годин. рібної сполуки як білуватий твердий продукт. Реакцію відстежували за допомогою ВЕРХ стосов(v) 4-[4-(9-окса-3,7-діазадицикпо[3,3,1]нон-3но зникнення арилброміду та утворення потрібної іл)бутил]бензонітрил сполуки. Після витрати усього вихідного матеріалу Тригорлу колбу на 2л опоряджували магнітною реакційну суміші охолоджували до 25°С та фільтмішалкою, термометром та лійкою для додавання. рували через кізельгур. Шар на фільтрі промивали Колбу завантажували трет-бутилу 7-[4-(4окремо толуолом (1,6л, 8 об'ємів). Суміш ціанофеніл)бутил]-9-окса-3,7ДМЕ:вода частково концентрували у вакуумі для діазадицикло[3,3,1]нонан-3-карбоксилатом (34г, видалення більшої частини ДМЕ. Це далі об'єдну88ммоль, з етапу (iv) вище) та дихлорметаном вали з промивкою толуолом. Толуольний шар кон(440мл). Трифлуороцтову кислоту (132мл) додацентрували у вакуумі, отримуючи потрібний алкін вали повільно при кімнатній температурі. Розчин як жовтий твердий продукт, який сушили у вакуумперемішували протягом 3 годин, в цей час ТШХній шафі протягом ночі при 40°С. аналізом показано повну витрату вихідного матеВихід 182,88г, 97%. ріалу. Вміст переносили у одногорлу колбу та кон1 Н ЯМР (300МГц, CDCl3): δ 7,599-7,575 (d, центрували у вакуумі. Залишок розчиняли у дихJ=7,2Гц, 2H, CH), 7,501-7,476 (d, J=7,5Гц, 2Н, СН), лорметані (500мл) та промивали насиченим 3,880-3,813 (q, 2H, СН2), 2,751-2,705 (t, 2H, СН2), розчином гідрокарбонату натрію. Водний шар від1,791-1,746 (t, 1H, ОН), темп.пл,79,6-80,5°С. діляли та екстрагували дихлорметаном (2 200мл). (іі) 4-(4-гідроксибутил)бензонітрил Поєднані органічні шари промивали розсолом 4-(4-ціанофеніл)бут-3-ин-1-ол (40г, 1мас-екв., (200мл), сушили сульфатом магнію та концентрудивися етап (і) вище) в етанолі (200мл, 5 об'ємів) вали у вакуумі, отримавши 25,8г (100% виходу) та паладій на вугіллі (20г, 0,5мас-екв., 10% потрібної сполуки як білуватий твердий продукт. Johnson Matthey type 487, 60% води) перемішуваСирий матеріал застосовували на наступному ли швидко під тиском водню 5 бар протягом 5 гоетапі без подальшої очистки. дин. Реакцію відстежували за допомогою ВЕРХ (vi) Сполука С стосовно зникнення вихідного матеріалу, та утвоТригорлу колбу на 3л опоряджували магнітною рення потрібної сполуки. Реакційну суміші фільтмішалкою, термометром та зворотним холодильрували через кізельгур та промивали етанолом ником. Колбу завантажували неочищеним 4-[4-(9(80мл, 1 об'єми). Етанольний розчин концентруваокса-3,7-діазадицикло[3,3,1]нон-3ли у вакуумі, отримуючи потрібний спирт як жовтоіл)бутил]бензонітрилом (25,8г, 88ммоль, з етапу (ν) коричневе 34 масло. Вихід 36,2г, 88,5%. вище), дихлорметаном (0,88л) та трет-бутилу 21 Н ЯМР (300МГц, CDCI3): δ 7,550-7,578 (d, брометилкарбаматом (дивися отримання В(І)(і) J=8,4Гц, 2Н), 7,271-7,298 (d, J=8,1Гц, 2H), 3,646вище, 27,7г, 123ммоль). Триетиламін (0,0197л, 3,688 (t, 2H), 2,683-2,733 (t, 2H), 1,553-1,752 (m, 0,141моль) додавали далі. Прозорий розчин гріли 4H). при температурі кипіння під зворотним холодиль13 С ЯМР (300МГц, CDCI3): δ 148,04 (С), 132,16 ником протягом 12 годин під азотом та далі охоло(С), 119,1 (С), 109,64 (С), 62,46 (С), 35,77(С),32,08 джували до кімнатної температури. Хід реакції (С), 27,12 (С). відстежували за допомогою ТШХ-аналізу, як знай(ііі) 4-(4-ціанофеніл)бутилу толуолсульфонат дено, вона закінчилася у цей час. Реакційну суміші переносили у ділильну лійку та промивали послі 41 74629 42 довно водою (200мл), 15% водним гідроксидом (ііі) 4-{[(2S)-2-гідрокси-3-(9-окса-3,7натрію (200мл), водою (200мл) та розсолом діазадицикло[3,3,1]інон-3-ілпропіл](200мл). Органічний шар сушили сульфатом магокси}бензонітрил нію та концентрували у вакуумі. Утворене жовте Тригорлу колбу на 3л опоряджували магнітною в'язке масло хроматографували на силікагелі, мішалкою, термометром та лійкою для додавання і елюючи спершу сумішшю 9:1 дихлормезавантажували неочищеним трет-бутилу 7-[(2S)-3тан/метанол, далі сумішшю 9:1:0,02 дихлорме(4-ціанофенокси)-2-гідроксипропіл]-9-окса-3,7тан/метанол/28% водний гідроксид амонію, отридіазадицикло[3,3,1]нонан-3-карбоксилатом (100г, з муючи потрібну сполуку (25,1г, 66% виходу), як етапу (іі) вище) та дихлорметаном (1,15л). білуватий твердий продукт. Ранішні фракції (5,1г) з Трифлуороцтову кислоту (0,352л) додавали хроматографії, як знайдено, містили малу кількість повільно при кімнатній температурі та утворений менш полярного забруднення (за ТШХ-аналізом) розчин перемішували протягом 3 годин, в цей час елюючи сумішшю 9:1:0,05 дихлормеТШХ-аналізом показано завершення реакції. Вміст тан/метанол/28% водний гідроксид амонію), а пізпереносили в одногорлу колбу та концентрували у ніші фракції (20г) давали одну пляму за ТШХвакуумі. Залишок розчиняли у дихлорметані (1,2л) аналізом. Ранішні фракції (5,1г) поєднували з інта промивали насиченим гідрокарбонатом натрію. шою партією потрібної сполуки (7,1г, містить трохи Водний шар відділяли та екстрагували дихлормезабруднень) та хроматографували на силікагелі, таном (2 0,2л). Поєднані органічні шари промиваелюючи спершу сумішшю 19:1 дихлормели розсолом (0,25л), сушили сульфатом магнію та тан/метанол, та далі сумішшю 9:1 дихлормеконцентрували у вакуумі, отримавши 73г (100% тан/метанол, отримавши блідо-жовтий порошок виходу) потрібної сполуки як білуватого твердого (5,5г). Порошок розчиняли у дихлорметані (200мл). продукту. Неочищений матеріал застосовували на Утворений розчин промивали послідовно 25% вонаступному етапі. дним гідроксидом натрію (50мл), вод (50мл), та (iv) Сполука D розсолом (40мл). Матеріал далі сушили сульфаСпосіб І том магнію та концентрували у вакуумі, отримуючи Тригорлу колбу на 2л опоряджували магнітною потрібну сполуку як білуватий порошок (5г). Фракмішалкою, термометром та зворотним холодильцію 20г розчиняли у дихлорметані (500мл). Органіником. Колбу завантажували неочищеним 4-{[(2S)чний шар промивали послідовно 25% водним гід2-гідрокси-3-(9-окса-3,7-діазадицикло[3,3,1]нон-3роксидом натрію (100мл), водою (100мл), та іл)пропіл]-окси}бензонітрилом (73г, з етапу (ііі) вирозсолом (100мл). Матеріал далі сушили сульфаще), дихлорметаном (0,7л) та трет-бутилу 2том магнію та концентрували у вакуумі, отримуючи брометилкарбаматом (дивися отримання В(І)(і) потрібну сполуку як білуватий порошок (19г). Парвище, 74г, 0,330моль). Триетиламін (52мл, тії змішували разом. 0,359моль) далі додавали. Прозорий розчин гріли Отримання D при температурі кипіння під зворотним холодильОтримання сполуки D ником протягом 16 годин та далі охолоджували до (і) 4-[(2S)-оксиранілметокси]бензонітрил кімнатної температури. Реакційну суміш переноКарбонат калію (414г) та (R)-(-)-епіхлоргідрин сили у ділильну лійку та промивали послідовно (800мл) додавали до перемішуваного розчину пводою (100мл) та розсолом (100мл). Органічний ціанофенолу (238г) у 2,0л MeCN та реакційну сушар сушили сульфатом магнію, фільтрували та міш гріли при температурі кипіння під зворотним концентрували у вакуумі. Утворене жовте в'язке холодильником в інертній атмосфері протягом 2 масло очищали хроматографією на силікагелі, годин. Гарячий розчин фільтрували та фільтрат елюючи спершу сумішшю 9:1 дихлормеконцентрували, отримуючи прозоре масло, яке тан/метанол, далі сумішшю 9:1:0,02 дихлормекристалізували з ди-ізопропілового етеру, отримутан/метанол/28% водний гідроксид амонію, отриючи продукт з виходом 90%. мавши білуватий пінистий твердий продукт (40г). (іі) Трет-бутилу 7-[(2S)-3-(4-ціанофенокси)-2Твердий продукт розчиняли у дихлорметані гідроксипропіл]-9-окса-3,7(200мл) та промивали послідовно 20% водним діазадицикло[3,3,1]нонан-3-карбоксилат гідроксидом натрію (100мл) та водою (100мл). ОрТригорлу колбу на 3л опоряджували магнітною ганічний шар сушили сульфатом магнію та концемішалкою та термометром і завантажували третнтрували у вакуумі, отримуючи потрібну сполуку як бутилу 9-окса-3,7-діазадицикло[3,3,1]нонан-3білуватий твердий продукт (35,4г, 67% виходу у карбоксилатом як вільною основою (53,7г, три етапи). 0,235моль, отримано з гідрохлориду, дивися Спосіб II ізопропанол (5мл) та воду (0,5мл) доотримання B(l) (iv) вище), 4-[(2S)давали до [2-(9-окса-3,7-діазадицикло[3,3,1]нон-3оксиранілметокси]бензонітрилом (41,2г, іл)етил]карбамової кислоти трет-бутилового есте0,235моль, дивися етап (і) вище), та 10:1 (за об'єру (дивися отримання В(ІІ)(іі), Спосіб І вище; 0,43г, мом) розчином 2-пропанол/вода (0,94л). Суміш 1,6ммоль) та додавали 4-[(2S)перемішували при 60°С протягом 20 годин, протяоксиранілметокси]бензонітрил (0,280г, 1,6ммоль, гом цього часу вихідні матеріали поступово витрадивися етап (і) вище). Суміш гріли при 66°С протячалися (за ТШХ-аналізом). Суміш охолоджували гом 19 годин (реакція завершилася за 2 години). та концентрували у вакуумі, отримавши 100г Розчинник випарювали досуха під зниженим тис(>100% виходу) потрібної сполуки як білого тверком з отриманням потрібної сполуки як білуватого дого продукту. Неочищений матеріал застосовутвердого продукту (0,71г, 100%). 1 вали на наступному етапі. Н ЯМР (300МГц, CDCI3): δ 1,41 (1Н, s), 2,32,75 (6Н, m), 2,75-3,0 (3Н, m), 3,1-3,38 (3Н, m), 3,88 43 74629 44 (2Н, s), 3,95-4,19 (3Н, m), 5,85 (1Н, bs), 6,99 (2Н, Приклад 1 d),7,6(2H, d). Кристалізація сполуки А (Спосіб (І); форма В) 1 H -ЯМР (300МГц, ДМСО-d6) δ 1,35 (9Н, s), Сполуку А (246,58г; отримано аналогічно спо2,12-2,59 (7Н, m), 2,63-2,78 (1Н, m), 2,78-2,9 (4Н, собам, що описано в отриманні А вище, хоча в m), 3,2 (2Н, q), 3,78 (2Н, m), 4-4,1 (2Н, m), 4,12-4,19 цьому випадку матеріал знов поєднано з маточни(1Н, m), 5,3 (1Н, bs), 6,61 (1Н, t), 7,15 (2Н, d), 7,76 ком, основу промивали NaOH, а далі випарювали (2Н, d). MC: m/z=447 (MH+). з отриманням твердого продукту) суспендували в Спосіб III: Розчин [2-(9-окса-3,7етилацетаті (500мл) та далі гріли для розчинення діазадицикло[3,3,1]нон-3-іл)етил]карбамової киствердого продукту. Отримали прозорий розчин. лоти трет-бутилового естеру з отримання В(Н)(іі), Етилацетат (80мл) видаляли відгонкою для азеотСпосіб 2 вище (припустимо 154,85г, 1,0моль екв., ропної сушки сполуки (залишаючи 1,7 об'ємів ети1,0маса/об'єм) у суміші толуолу (приблизно лацетату). Розчин далі переносили гарячим у по640мл), етанолу (приблизно 1280мл) та води (припередньо нагріту колбу на 1 л з фланцем близно 130мл), підлужували водним гідроксидом (температура при переносі була 68°С). Розчину натрію (10М, 51мл, 0,9моль-екв.). Твердий 4-[(2,S)давали охолонути природно (повністю видаляли оксиранілметокси]бензонітрил (99,80г, 1,0моль джерело теплоти) під азотом з перемішуванням екв.; дивися етап (і) вище) додавали та поєднану верхньою мішалкою. Після 40 хвилин охолодженсуміш гріли до 70°С протягом 4 годин. Реакцію ня починалася кристалізація (внутрішня темперавідстежували за допомогою ТШХ, використовуючи тура була 38°С). Температура розчину була між пластину з діоксидом силіцію з рухомою фазою 38-40°С протягом наступних 1,5 годин. Розчин далі Х:ДХМ 1:1 за об'ємом (в якій X представляє хлоохолоджували до 20°С на бані вода/лід та залироформ:метанол:концентрований аміак 80:18:2 за шали протягом 1 годин. Продукт фільтрували, об'ємом). Візуалізацію здійснювали УФ-світлом промивали холодним (0°С) етилацетатом (330мл), (254нм) та фарбуванням водним перманганатом а далі сушили протягом ночі у вакуумі при 40°С з калію. Цим показано повну витрату вихідного маотриманням 126,83г (вихід 51%, у припущенні, що теріалу та появу потрібної сполуки. Реакційну сувихідний матеріал був сухим). міш охолоджували, фільтрували через кізельгур та Кристали аналізували РДМП, результати попромивали етанолом (620мл, 4,0 об'єми). Це дало казані нижче (таблиця 1) і у Фіг.1. розчин потрібної сполуки (припустимо 254,38г, Таблиця 1 100% th, 2,4л, 1,0маса/об'єм для реакції). Цей розчин завантажували у колбу, що була пристосована d-параметр (Å) Інтенсивність (%) для перегонки під зниженим тиском. Градуювальну 11,0 81 помітку наносили на стінку цієї колби. Розчинник 8,3 2 7,8 5 (1250мл) видаляли при між 50°С та 35°С, 320мбар 7,0 24 та 100мбар. Далі 4-метилпентан-2-ол (1500мл) 6,7 2 додавали для досягнення градуювальної помітки. 5,9 3 Розчинник (1250мл) видаляли при між 35°С та 5,7 16 80°С, 220мбар та 40мбар. Більше 4-метилпентан5,5 4 5,4 8 2-ол (1500мл) додавали для досягнення градую5,1 44 вальної помітки. Розчинник (1250мл) видаляли при 4,94 7 між 62°С та 76°С, 100мбар та 90мбар. Поєднану 4,71 4 суміш охолоджували до менше 25°С та додавали 4,62 5 4,54 33 водний гідроксид натрію (2М, 1,27л, 5,0 об'ємів). 4,44 100 Шари розділяли та органічний шар фільтрували 4,34 12 через кізельгур з отриманням прозорого розчину 4,20 7 (1,2л). Цей розчин завантажували у чисту колбу, 4,12 2 яка була пристосована для перегонки під зниже3,92 5 3,65 15 ним тиском. Розчинник (450мл,) видаляли при між 3,51 9 52°С та 55°С, 90мбар та 35мбар. Теоретично, 3,47 2 продукт при цьому залишався у 2 об'ємах 43,41 4 метилпентан-2-олу. Ди-н-бутиловий етер (1,27л, 5 3,34 5 об'ємів) додавали та розчину давали охолонути 3,31 0,8 3,26 2 повільно до кімнатної температури, що викликало 3,04 2 утворення осаду. Суміш охолоджували від кімнат2,89 4 ної температури до приблизно 10°С. Продукт зби2,82 3 рали фільтруванням та промивали попередньо 2,77 2 змішаним розчином ди-н-бутилового етеру (320мл, 2,70 2 2,58 2 1,25 об'ємів) та 4-метилпентан-2-олу (130мл, 0,50 2,44 1 об'ємів). Вологий продукт сушили у вакуумі при 2,34 4 55°С до постійної маси з отриманням потрібної 2,18 3 сполуки як білого твердого продукту (193,6г, 76%), 2,06 3 темп.пл,99-101°С. 1 Н-ЯМР (300МГц, CDCl3):δ 1,41 (9Н, s), 2,3Кристалічну комірку було визначено на одини2,75 (6Н, m), 2,75-3,0 (5Н, m), 3,1-3,38(3H, m), 3,88 чному кристалі за результатом рентгеноаналізу. (2Н, s), 3,95-4,19 (3Н, m), 5,85 (1Н, bsj, 6,99 (2Н, d), Вона була орторомбічною, з просторовою групою 7,6 (2Н, d). 45 74629 46 2,80 6 Р212121 та такими параметрами: а=9,096Å, 2,74 3 b=11,077Å, с=22,136Å, α=β=γ=90°, та V=2230,3Å3. 2,69 2 ДСК показано два ендотерми з екстрапольо2,63 2 ваними температурними початками приблизно 2,56 2 121°С та 126°С. ТГМА показано зменшення маси приблизно 0,1% (за масою) між 110 та 130°С. ДСК показано ендотерм з екстрапольованим Приклад 2 температурним початком приблизно 125°С. ТГМА Кристалізація сполуки А (Спосіб (II); форма А) показано зменшення маси приблизно 0,1% (за Етилацетат (250мл) додавали до сполуки А масою) між 120 та 130°С. (188,10г, отримано аналогічно способам, що опиПриклад 3 сано в отриманні А вище) та суміш гріли. Гарячий Кристалізація сполуки А (Спосіб (III); форма С) отриманий розчин фільтрували (залишку не було). Сполуку А як моногідрат бензолсульфонату При фільтруванні з фільтрованого розчину оса(4,5кг, отримано аналогічно способам, описаним в джувалося трохи твердого продукту. Це знов розотриманні А вище) додавали до дихлорметану чиняли в етилацетаті (50мл) та фільтрували. По(89,7кг). Водний гідроксид натрію (1М, 46,9кг) доєднані фільтрати концентрували видаленням давали та реакційну суміш перемішували протягом етилацетат (120мл). Остаточному розчину давали 45 хвилин. Кінцева температура вмісту була охолонути до кімнатної температури (кристалізація 16,9°С. Дві фази залишали протягом 68 хвилин починалася при 40°С). Кашку далі охолоджували для розділення та водний шар (48,8кг) відкидали. до 10°С протягом 1 годин. Твердий продукт фільтОрганічний шар фільтрували у чистий резервуар рували, промивали холодним етилацетатом та фільтрували. Етанол (72кг) додавали. Розчин(100мл) та далі сушили у вакуумі при 40°С з отриник (101,1кг) відганяли (3 години 20 хвилин.). Кінманням 126,7г (68%). цева температура вмісту була 76,0°С. Етанол Кристали аналізували РДМП, результати по(25,4кг) додавали та ще відганяли 11,6кг розчинказані нижче (таблиця 2) та у Фіг.2. нику (1 година 30 хвилин.), кінцева температура вмісту була 79,3°С. Розчин охолоджували до -17°С Таблиця 2 протягом ночі але не спостерігали кристалізації. Ще 32,7кг розчиннику відганяли та реакційну суміш d-параметр (Å) Інтенсивність (%) охолоджували до 15°С протягом ночі, протягом 10,9 65 8,3 54 цього часу продукт кристалізувався. Реакційну 7,8 9 суміш охолоджували до 0°С та перемішували про7,5 3 тягом 30 хвилин. Продукт відфільтровували та 7,0 3 промивали етанолом (7,3кг) з отриманням волого6,9 6 го твердого продукту (2,7кг). Продукт сушили при 6,8 2 6,7 12 кімнатній температурі протягом 2 годин 16 хвилин 6,4 2 (33мм Hg), далі при 40-45°С протягом 15 годин 24 5,9 2 хвилин (29мм Hg) з отриманням 2,18кг продукту, 5,6 (5,64) 26 який мав 99,76% чистоти, 0,3% за масою етанолу. 5,6 (5,56) 8 5,5 7 Кристали аналізували РДМП, результати по5,4 20 казані нижче (таблиця 3) та у Фіг.3. 5,3 5,1 5,0 4,90 4,84 4,78 4,70 4,49 4,45 4,36 4,25 4,15 4,10 4,03 3,97 3,90 3,80 3,73 3,60 3,52 3,47 3,44 3,39 3,35 3,31 3,23 3,13 3,01 2,95 2,93 2,89 2,85 3 27 14 3 8 8 16 64 100 26 4 6 15 9 6 11 5 15 2 4 8 3 2 2 6 1 2 1 5 3 5 5 Таблиця 3 d-параметр (Å) 10,4 9,6 8,5 7,0 5,7 5,3 5,2 4,83 4,71 4,55 4,39 3,96 3,83 3,58 3,50 3,43 3,29 3,22 3,19 3,12 3,08 2,99 2,86 2,66 2,63 2,55 2,37 Інтенсивність (%) 4 100 0,6 0,9 9 6 20 22 6 6 0,6 0,5 1 2 1 0,4 1 6 2 0,3 0,5 0,3 0,3 0,6 0,8 0,9 0,7 47 2,31 2,19 2,13 2,10 74629 0,4 0,6 0,4 0,3 Кристалічну комірку було визначено на одиничному кристалі за результатом рентгеноаналізу. Вона була орторомбічною, з просторовою групою C2cb та такими параметрами: а=10,685Å, b=19,391Å, с=21,071Å, α=β=γ=90°, та V=4365,8Å3. ДСК показано ендотерм з екстрапольованим температурним початком приблизно 122°С. ТГМА показано зменшення маси приблизно 0,3% (за масою) між 25 та 140°С. Приклад 4 Кристалізація сполуки А, моногідрату бензолсульфонату 1,88г (4,9ммоль) сполуки А (отримано аналогічно способам, що описано в отриманні А вище) розчиняли у 23мл етилацетату. 0,807г (5,1ммоль) бензолсульфонової кислоти розчиняли у 4,5мл метанолу. Розчин бензолсульфонової кислоти додавали до перемішуваного розчину сполуки А. Бензолсульфонат осаджувався після приблизно 10 хвилин. Осад тримали при 4°С протягом ночі. Кристали відфільтровували, промивали етилацетатом та сушили у вакуумі протягом ночі. Вихід 1,6г (60%). Продукт перекристалізовували з суміші ЕtOН:вода (1:1). Кристали аналізували РДМП, результати показані нижче (таблиця 4) та у Фіг.4. Таблиця 4 d-параметр (Å) 18,7 9,4 7,5 6,6 6,4 6,3 5,1 4,85 4,77 4,69 4,63 4,48 4,40 4,19 4,13 4,05 3,92 3,89 3,76 3,61 3,35 3,31 3,21 3,13 3,07 3,00 2,68 2,35 2,31 2,09 2,00 1,88 Інтенсивність (%) 100 13 0,3 0,2 0,2 2 0,3 0,3 0,3 37 0,7 1 0,2 0,6 0,2 0,3 0,2 0,3 28 0,2 1 0,3 0,2 14 0,2 0,4 2 1 0,3 0,6 0,4 1 Кристалічну комірку було визначено на одиничному кристалі за результатом рентгеноаналізу. Вона була моноклінічною, з просторовою групою Р21/с та такими параметрами:=18.833Å, b=9,293Å, с=16,271Å, α=90°, β=94,94°, γ=90°, та V=2837,1Å3. 48 Продукт є моногідратом без добре визначеної температури плавлення. ДСК показано ендотерм з екстрапольованим температурним початком у межах приблизно 85-110°С. ТГМА показано зменшення маси приблизно 3% (за масою) між 60 та 160°С. Приклад 5 Кристалізація сполуки А, паратолуолсульфонату (форми А та В) Сполуку А (1,14г, 2,96ммоль, отримано аналогічно способам, що описано в отриманні А вище (наприклад, до додавання бензолсульфонової кислоти в описаному тут способі для утворення бензолсульфонату сполуки А)) розчиняли в етилацетаті (20мл). Розчин пара-толуолсульфонової кислоти (PTSA) (0,57г, 2,99ммоль) у метанолі (0,3мл) додавали краплями. Додавали промивку пара-толуолсульфонової кислоти у 0,1мл метанолу, та ще 5мл етилацетату. Біла кашка утворилася протягом кількох хвилин. Це перемішували протягом 1 години. Твердий продукт відфільтровували та промивали 5мл етилацетату. Вихід 1,4г (84,8%). Твердий продукт (1,40г) толуолсульфонату змішували з 15мл етилацетату та гріли при температурі кипіння під зворотним холодильником. До кашки додавали 0,85мл метанолу. Протягом кількох секунд твердий продукт перейшов у розчин. Йому давали охолонути та через кілька хвилин спостерігали кристалізацію. Скоро це затверділо. До цього додавали ще етилацетат (10мл) та метанол (0,15мл). Густу кашку далі перемішували при 0°С протягом 30 хвилин. Тверді продукти відфільтровували та промивали етилацетатом (5мл). Цьому давали висохнути при відсмоктуванні. Вихід 1,10г (66,6%). Порцію цього (500мг) сушили при 50°С під зниженим тиском, отримуючи 0,440г. Порцію солі толуолсульфонової кислоти перекристалізовували з ацетону. Кристали (форма А) аналізували РДМП, результати показані нижче (таблиця 5(а)) та у Фіг.5(а). Порцію солі толуолсульфонової кислоти суспендували у фосфатному буфері (іонна сила 0,1Μ при pH3), а потім декантували. Кристали (форма В) аналізували РДМП, результати показані нижче (таблиця 5(b)) та у Фіг.5(b). Таблиця 5(а) d-параметр (Å) 19,9 10,0 8,7 7,6 7,0 6,6 6,4 6,0 5,7 5,5 5,2 4,99 4,86 4,49 4,38 4,36 4,19 3,99 3,93 3,77 3,59 Інтенсивність (%) 100 30 0,5 1 0,9 0,7 1 4 1 0,8 1 33 4 2 4 8 5 5 1 1 2 49 3,40 3,33 3,29 3,19 3,08 2,86 2,22 2,11 2,09 2,00 74629 1 5 1 0,7 1 0,7 0,7 0,6 0,7 1 Для форми А ДСК показано ендотерм з екстрапольованим температурним початком приблизно 145°С. ТГМА показано зменшення маси приблизно 3,3% (за масою) між 20 та 120°С. Таблиця 5(b) d-параметр (Å) 18,6 9,7 9,3 7,6 6,2 5,0 4,66 4,49 3,73 3,11 2,33 Інтенсивність (%) 100 0,6 43 0,4 8 0,9 20 2 8 4 0,6 Для форми В ДСК показано ендотерм з екстрапольованим температурним початком приблизно 153°С. ТГМА показано зменшення маси приблизно 4,4% (за масою) між 25 та 120°С. Приклад 6 Кристалізація солі 1-гідрокси-2-нафтойної кислоти сполуки А До сполуки А (0,60г, 1,56ммоль, отримано аналогічно способам, що описано в отриманні А вище (наприклад, до додавання бензолсульфонової кислоти в описаному тут способі для утворення бензолсульфонату сполуки А)) в етилацетаті (10мл) додавали 1-гідрокси-2-нафтойну кислоту (0,323г, 1,7ммоль) у метанолі (1мл). Розчин не утворився. Суміш перемішували при кімнатній температурі протягом 30 хвилин. Не було помічено кристалізації. Розчин далі охолоджували протягом 30 хвилин. Спостерігали невелику кількість кристалів. Розчинники випарювали досуха, тверді продукти переносили в етилацетат (5мл) та гріли при температурі кипіння під зворотним холодильником. До часткового розчину додавали метанол (0,2мл}. Утворену кашку охолоджували сумішшю лід/вода, фільтрували та промивали етилацетатом (2мл). Твердий продукт далі сушили у шафі при 50°С під зниженим тиском протягом 24 годин. Вихід 0,510г (57%). Кристали аналізували РДМП, результати показані нижче (таблиця 6) та у Фіг.6. Таблиця 6 d-параметр (Å) 17,6 12,4 8,8 7,9 7,5 6,6 6,4 6,2 Інтенсивність (%) 100 16 44 4 5 2 2 5 50 5,7 5,6 5,2 5,1 4,95 4,88 4,47 4,40 4,21 4,16 4,08 3,95 3,84 3,80 3,64 3,55 3,33 3,03 2,96 2,63 5 29 4 6 7 9 20 4 4 8 6 5 5 8 3 3 6 7 2 4 Приклад 7 Кристалізація солі 1,5-нафталінсульфонової кислоти сполуки А Сполуку А (0,490г, 1,27ммоль, отримано аналогічно способам, що описано в отриманні А вище (наприклад, до додавання бензолсульфонової кислоти в описаному тут способі для утворення сполуки А, бензолсульфонат)) розчиняли в етилацетаті (10мл). Розчин 1,5-нафталінсульфонової кислоти у метанолі (0,5мл) додавали. Протягом кількох хвилин з'явився білий твердий продукт. Це перемішували при кімнатній температурі протягом 30 хвилин та далі охолоджували сумішшю лід/вода. Кілька порцій білого твердого продукту були помічені у кашці. Тверді продукти відфільтровували з отриманням 0,34г білого твердого продукту. Це переносили у метанол (50мл) та воду (100мл) та гріли при температурі кипіння під зворотним холодильником, до отримання прозорого розчину. Розчину давали досягти кімнатної температури температур, а далі охолоджували сумішшю лід/вода протягом 30 хвилин. Тверді продукти відфільтровували з отриманням білого порошку (0,150г). Твердий продукт сушили у шафі при 50°С під зниженим тиском протягом 5 годин. 1 Н-ЯМР демонструє співвідношення кислоти до основи 1:2. Кристали аналізували РДМП, результати показані нижче (таблиця 7) та у Фіг.7. Таблиця 7 d-параметр (Å) 16,1 8,6 8,1 7,8 7,1 6,8 6,3 6,0 5,5 5,4 (5,43) 5,4 (5,39) 5,3 4,96 4,88 4,68 4,62 4,49 4,34 4,10 Інтенсивність (%) 100 4 19 2 7 3 10 6 3 3 4 2 17 22 18 4 9 8 12 51 4,04 3,94 3,55 3,44 3,40 3,23 3,20 3,15. 2,87 2,72 2,19 2,18 74629 6 13 2 5 15 11 5 4 2 3 3 3 Приклад 8 Кристалізація солі 2-мезитиленсульфонової кислоти сполуки А: Розчин 2-мезитиленсульфонової кислоти (0,276г) у метанолі (0,3мл) додавали до перемішуваного розчину сполуки А (0,45г, отримано аналогічно способам, що описано в отриманні А вище (наприклад, до додавання бензолсульфонової кислоти в описаному тут способі для утворення бензолсульфонату сполуки А)) в етилацетаті (10мл). Осадження відбувалася негайно. Кашку перемішували при кімнатній температурі протягом 30 хвилин та далі охолоджували сумішшю лід/вода. Продукт відфільтровували та промивали етилацетатом (3мл) з отриманням потрібної сполуки (0,60г, 88%). Отриману сіль розчиняли в етилацетаті (10мл) та гріли при температурі кипіння під зворотним холодильником. Метанол (3мл) додавали з отриманням прозорого розчину. Розчину давали охолонути до кімнатної температури та далі перемішували при 0-5°С протягом 1 годин. Кашку фільтрували, отримавши безбарвний твердий продукт (0,370г, 54% для двох етапів). Кристали аналізували РДМП, результати показані нижче (таблиця 8) та у Фіг.8. Таблиця 8 d-параметр (Å) 18,9 9,5 8,8 7,8 6,3 (6,33) 6,3 (6,26) 5,7 5,3 5,1 4,98 4,75 4,62 4,50 4,46 4,41 4,29 4,24 4,13 3,92 3,80 3,69 3,54 3,36 3,17 3,04 2,38 2,11 Приклад 9 Кристалізація сполуки D Інтенсивність (%) 100 25 1 1 10 9 3 2 3 2 20 2 4 5 2 1 1 2 5 12 1 2 1 6 1 2 1 52 Спосіб І: Сполуку D (отримано аналогічно способам, що описано тут раніше) спершу очищали хроматографією на колонці з силікагелем, використовуючи суміш 9:1:0,05 дихлорметан/метанол/28% водний аміак як елюент. Утворений світло-жовтий порошок (203г) розчиняли у дихлорметані (400мл) та далі жовтий розчин розбавляли н-гептаном (2л) до помутніння суміші. Суміш перемішували енергійно при кімнатній температурі та затравлювали кристалами сполуки D (отримано повільним випарюванням розчиннику з малої порції розчину потрібної сполуки, отриманої аналогічно цьому способу). Велика порція продукту осадилася після перемішування протягом 2 годин. Утворену кашку фільтрували та далі сушили у вакуумі при 40°С, отримавши 179г потрібної сполуки як білуватий порошок. Друга порція (44г) очищеного матеріалу з колонки дала ще 25г потрібної сполуки після подібної перекристалізації. Спосіб II: Суміш сполуки D (отримано аналогічно способам, що описано тут раніше, (дивися особливо Отримання D(iv), Спосіб III вище); 14,29г), ізопропанолу (28мл) та ди-ізопропілового етеру (140мл) гріли до 80°С. Розчин фільтрували гарячим для його очистки та далі знов гріли до 80°С. Розчину далі давали охолонути до кімнатної температури, при цьому починав утворюватися осад. Після перемішування протягом 2 годин осад збирали фільтруванням, промивали сумішшю ізопропанол:ізопропіловий етер (1:6, 70мл) та далі відсмоктували на фільтр. Вологий продукт сушили у вакуумі при 70°С протягом ночі з отриманням кристалічної сполуки D як білого твердого продукту (10,1г, 70%). 1 Н ЯМР (300МГц, CDCI3): δ 1,41 (9Н, s), 2,32,75 (6Н, m), 2,75-3,0 (5Н, m), 3,1-3,38(3Н, m), 3,88 (2Н, s), 3,95-4,19 (ЗН, m), 5,85 (1Н, bs), 6,99 (2Н, d), 7,6 (2Н, d) Спосіб III: Сполуку D (отримано аналогічно способам, що описано тут раніше; 1,0г) розчиняли у гарячому метилізобутилкетоні (10мл). Додавали ізо-гексан (30мл) та розчин декантували від невеликої кількості нерозчиненого матеріалу у чисту колбу. Розчин охолоджували протягом ночі у холодильнику, що викликало утворення гранульованого осаду. Розчину давали нагрітися до кімнатної температури та далі твердий продукт збирали фільтруванням. Твердий продукт відсмоктували на фільтр, а далі сушили у вакуумі при 40°С з отриманням білуватого твердого продукту (0,73г). Спосіб IV: Сполуку D (отримано аналогічно способам, що описано тут раніше; 1,0г) розчиняли у гарячому ізопропілацетаті (10мл). Додавали ізооктан (20мл) та розчин декантували від невеликої кількості нерозчиненого матеріалу у чисту колбу. Розчин охолоджували протягом ночі у холодильнику, що викликало утворення гранульованого осаду. Розчину давали нагрітися до кімнатної температури та далі твердий продукт збирали фільтруванням. Твердий продукт відсмоктували на фільтр, далі сушили у вакуумі при 40°С з отриманням білуватого твердого продукту (0,82г). Спосіб V: Сполуку D (отримано аналогічно способам, що описано тут раніше; 1,0г) розчиняли у гарячому водному етанолі (50%, 20мл). Розчин охолоджували протягом уікенду у холодильнику, 53 74629 54 що викликало утворення голчатих кристалів. Криссмолу знов розчиняли у можливо мінімальній кільтали збирали фільтруванням та відсмоктували на кості гарячого етилацетату. Розчин затравлювали фільтр. Сушка у вакуумі при 40°С дала безбарвпопередньо отриманими кристалами солі (отриманий твердий продукт (0,48 г). но повільним випарюванням розчиннику з невелиСпосіб VI: Сполуку D (отримано аналогічно кої порції розчину потрібної сполуки, отриманої способам, що описано тут раніше; 100мг) розчианалогічно цьому способу) та залишали у холодиняли у толуолі (1мл). Додавали ізо-октан (2мл) та льнику протягом кількох діб для кристалізації. мутний розчин охолоджували протягом уікенду у Утворені кристали збирали фільтруванням та холодильнику, що викликало утворення дрібного промивали невеликою кількістю холодного етилагранульованого осаду. Розчину давали нагрітися цетату (отримано 280мг, 91%). до кімнатної температури та далі твердий продукт Спосіб II: Сполуку D (4,00г, отримано аналогізбирали фільтруванням. Твердий продукт відсмокчно способам, що описано в отриманні D вище) тували на фільтр, далі сушили у вакуумі при 40°С, розчиняли в етилацетаті (40мл). Розчин метансуотримавши білуватий твердий продукт (62мг). льфонової кислоти (0,87г) в етилацетаті (40мл) Кристали (зі способів І-VI) аналізували РДМП, додавали. Після 3 хвилин розчин затравлювали результати показані нижче (таблиця 9) та у Фіг.9. попередньо отриманою сіллю (14мг, зі способу І вище), що викликало негайне утворення осаду. Таблиця 9 Після 5 хвилин твердий продукт збирали фільтруванням та промивали етилацетатом перед відсмоd-параметр (Å) Інтенсивність (%) ктуванням на фільтр. Сушка у вакуумі при 40°С 22,3 45 протягом 3,5 годин дала білий кристалічний твер11,2 16 8,4 41 дий продукт (4,71г, 97%), темп.пл. 170-2°С (розкл.) 7,5 5 Спосіб III: Метансульфонат сполуки D (0,8г; 7,1 14 отримано способом описаним у способі II вище) 6,4 17 гріли у ацетонітрилі (8мл, 10 об'ємів). При 70°С 5,8 60 розчин утворився. Йому давали охолонути до тем5,5 13 5,2 7 ператури довкілля. Продукт збирали фільтруван4,91 31 ням, та промивали невеликим об'ємом холодного 4,81 28 ацетонітрилу. Це спочатку сушили у вакуумі при 4,69 14 40°С (температура плавлення 169,4-69,9°С 4,62 10 4,52 100 (розкл.), остаточного розчиннику ацетонітрилу 4,32 77 0,08% за масою (ГХ)). Це далі сушили у вакуумі 4,22 14 при 80°С, остаточного розчиннику ацетонітрилу 4,12 35 менше, ніж 0,02% за масою (ГХ). Чистота за ВЕРХ 4,06 7 99,92%. 3,91 9 3,72 4 Спосіб IV: Суміш метансульфонату сполуки D 3,53 5 (2,0г,моль екв., отримано способом, описаним у 3,50 9 способі II вище) та ацетонітрил (20мл, 10 відн. 3,42 5 об'ємів) гріли до 80°С. При цій температурі розчин 3,34 12 утворився. Розчину давали охолонути до 3,15 8 3,03 5 25°С±5°С та далі охолоджували до 5°С±5°С. Про2,98 3 дукт збирали фільтруванням та промивали холод2,92 3 ним ацетонітрилом (4мл, 2 відн. об'ємів). Волог 2,83. 4 твердий продукт сушили у вакуумі при 80°С. Це 2,77 4 дало потрібну сполуку (0,15г, 7,5%) як білуватий 2,75 5 2,67 6 твердий продукт. 99,97% за ВЕРХ. Остаточного 2,37 3 розчиннику ацетонітрилу менше, ніж 0,02% за ма2,11 5 сою (ГХ). Спосіб V: Суміш метансульфонату сполуки D Кристалічну комірку було визначено на одини(10,0г,моль екв.; отримано аналогічно способу, що чному кристалі за результатом рентгеноаналізу. описано у способі II вище, за винятком того, що у Вона була орторомбічною, з просторовою групою цьому випадку бутилацетат замість етилацетату P212121 та такими параметрами: а=5,870Å, застосовували як розчинник) та пентан-2-олу 3 b=9,098Å, с=45,101Å, α=β=γ= 90°, а V=2408,6Å . (100мл, 10 відн. об'ємів) гріли до 85°С з отриманДСК показано ендотерм з екстрапольованим ням розчину. Розчину давали охолонути до температурним початком приблизно 100-102°С. 25°С±5°С та далі охолоджували до 5°С±5°С. ПроТГМА показано зменшення маси приблизно 0,4% дукт збирали фільтруванням та промивали холод(за масою) між 90 та 110°С. ним пентан-2-олом (20мл, 2 відн. об'ємів). Вологий Приклад 10 твердий продукт сушили у вакуумі при 40°С протяКристалізація солі метансульфонової кислоти гом 12 годин. Це дало потрібну сполуку (6,72г, сполуки D 67,2%) як білуватий твердий продукт. 99,47% за Спосіб І:. Сполуку D (250мг, отримано аналогіВЕРХ. чно способам, що описано в отриманні D вище) Спосіб VI; Суміш метансульфонату сполуки D розчиняли у метанолі (10мл). Метансульфонову (3,0г, 1,0моль екв.; отримано способом, описаним кислоту (56мг, 38мкл) додавали до розчину. Метау способі V вище) та З-метилбутан-1-олу (15мл, 5 нол видаляли під зниженим тиском та остаточну відн. об'ємів) гріли до 90°С з отриманням розчину. 55 74629 56 Розчину давали охолонути до 25°С±5°С та далі Розчин гіпурової кислоти (0,8г) у метанолі охолоджували до 5°С±5°С. Продукт збирали філь(20мл) додавали до розчину сполуки D (2,00г, труванням та промивали холодним З-метилбутанотримано аналогічно способам, що описано у при1-олом (6мл, 2 відн. об'єми). Волог твердий прокладі 9 вище) у метанолі (5мл). Далі, розчин кондукт сушили у вакуумі при 40°С. Це дало потрібну центрували у вакуумі, з отриманням масла. Діетисполуку як білуватий твердий продукт (2,43г, 81%). ловий етер (20мл) додавали та суміш знов Спосіб VII: Суміш метансульфонату сполуки D концентрували з отриманням піни. Перемішування (2,0г, 1,0моль екв.; отримано способом, описаним у етері (100мл) протягом ночі та фільтрування дау способі V вище) та гексан-1-ол (15мл, 5 відн. ли вихід 2,22г потрібної сполуки (79%). Сіль сушиоб'ємів) гріли до 80°С з отриманням розчину. Сули у вакуумі при 34°С, отримуючи 2,19г (78%). міші давали охолонути до 25°С±5°С та далі охоПрисутність солі гіпурової кислоти підтверлоджували до 5°С±5°С. Продукт збирали фільтруджено ЯМР. ванням та промивали холодним гексан-1-олом Кристали аналізували РДМП, результати по(6мл, 2 відн. об'єми). Вологий твердий продукт казані нижче (таблиця 11) та у Фіг.11. Таблиця 11 сушили у вакуумі при 40°С. Це дало потрібну сполуку як білуватий твердий продукт (2,43г, 81%). d-параметр (Å) Інтенсивність (%) Кристали (Способи І -VII) аналізували РДМП, 16,4 100 результати показані нижче (таблиця 10) та у 13,8 3 6,9 8 Фіг.10. Таблиця 10 d-параметр (Å) 12,7 9,4 7,3 7,1 6,8 6,6 6,4 6,0 5,4 5,1 4,92 4,83 4,74 4,66 4,45 4,27 4,14 4,05 3,99 3,87 3,73 3,65 3,42 3,37 3,31 3,22 3,12 3,00 2,96 2,92 2,89 2,81 2,71 2,63 2,57 2,50 2,41 Інтенсивність (%) 100 6 20 11 3 11 3 17 18 21 38 22 5 4 4 11 14 30 38 33 9 7 19 6 3 5 4 6 5 5 4 3 2 5 2 3 3 Кристалічну комірку було визначено на одиничному кристалі за результатом рентгеноаналізу. Вона була орторомбічною, з просторовою групою Р212121 та такими параметрами: а=7,772Å, b=14,250Å, с=25,750Å, α=β=γ=90°, та V=1717,2Å3. ДСК показано ендотерм з екстрапольованим температурним початком приблизно 167°С. ТГМА показано зменшення маси приблизно 1,2% (за масою) між 25 та 100°С. Приклад 11 Кристалізація солі гіпурової кислоти сполуки D 6,2 6,1 5,6 5,5 5,2 5,1 4,93 4,82 4,61 4,50 4,28 4,20 4,11 3,68 3,54 3,27 6 25 5 7 17 15 13 4 18 26 30 6 8 8 5 5 Приклад 12 Кристалізація сполуки С Сполуку С отримували аналогічно способу, що описано в отриманні С вище, за винятком того, що після кінцевий етап способу проводили так: тригорлу колбу на 3л опоряджували магнітною мішалкою, термометром та зворотним холодильником. Колбу завантажували неочищеним 4-[4-(9-окса3,7-діазадицикло[3,3,1]нон-3іл)бутил]бензонітрилом (дивися отримання C(v) вище, 25,8г, 88ммоль), дихлорметаном (0,88л) та трет-бутилу 2-брометилкарбаматом (дивися отримання В(І)(і) вище, 27,7г, 123ммоль). Триетиламін (0,0197л, 0,141моль) додавали далі. Прозорий розчин гріли при температурі кипіння під зворотним холодильником протягом 12 годин під азотом та далі охолоджували до кімнатної температури. Хід реакції відстежували за допомогою ТШХаналізу, як знайдено, вона закінчилася у цей час. (іі) Реакційну суміш переносили у ділильну лійку та промивали послідовно водою (200мл), 15% водним гідроксидом натрію (200мл), водою (200мл), та розсолом (200мл). Органічний шар сушили сульфатом магнію та концентрували у вакуумі. Утворене жовте в'язке масло хроматографували на силікагелі, елюючи спершу сумішшю 9:1 дихлорметан/метанол, далі сумішшю 9:1:0,02 дихлорметан/метанол/28% водний гідроксид амонію, отримавши сиру сполуку С (25,1г, 66% виходу) як білуватий твердий продукт. Ранішні фракції (5,1г) з хроматографії, як знайдено, містили невелику кількість менш полярного забруднення (за ТШХ-аналізом, елюючи сумішшю 9:1:0,05 дихлор 57 74629 58 метан/метанол/28% водний гідроксид амонію), а кількох діб для кристалізації. Утворені кристали пізніші фракції (20г) дали одну плями за ТШХзбирали фільтруванням та промивали невеликою аналізом. Ранішні фракції (5,1г) поєднували з покількістю холодного ізопропанолу (отримано передньо отриманими порціями сполуки С (7,1г, 220мг, 72%). містить трохи забруднень) та хроматографували Кристали аналізували РДМП, результати пона силікагелі, елюючи спершу сумішшю 19:1 дихказані нижче 5 (таблиця 13) та у Фіг.13. лорметан/метанол, а далі сумішшю 9:1 дихлормеТаблиця 13 тан/метанол, отримавши блідо-жовтий порошок (5,5г). Порошок розчиняли у дихлорметані (200мл). d-параметр (Å) Інтенсивність (%) Утворений розчин промивали послідовно 20% во13,3 100 дним гідроксидом натрію (50мл), водою (50мл), та 12,3 5 9,6 5 розсолом (40мл). Матеріал далі сушили сульфа7,5 6 том магнію та концентрували у вакуумі, отримав6,9 6 ши сиру потрібну сполуку як білуватий порошок 6,8 3 (5г). 20г фракції розчиняли у дихлорметані 6,7 12 (500мл). Органічний шар промивали послідовно 6,4 2 6,2 2 20% водним гідроксидом натрію (100мл), водою 6,0 2 (100мл), та розсолом (100мл). Матеріал далі су5,5 12 шили сульфатом магнію та концентрували у ваку5,3 2 умі, отримавши як білуватий порошок (19г). Партії 5,1 13 5,0 5 змішували разом. 4,89 5 Кристали аналізували РДМП, результати по4,81 4 казані нижче (таблиця 12) та у Фіг.12. Таблиця 12 d-параметр (Å) 19,4 10,0 9,1 8,1 6,5 5,5 5,2 5,1 4,99 4,90 4,60 4,46 4,32 4,06 4,00 3,85 3,80 3,66 3,56 3,44 3,33 3,16 2,94 2,82 2,69 2,44 Інтенсивність (%) 24 16 64 100 84 19 41 21 20 21 9 30 25 78 33 15 23 10 5 17 4 5 8 4 4 5 ДСК показано ендотермічний пік з екстрапольованим температурним початком приблизно 97°С. ТГМА показано зменшення маси приблизно 0,6% (за масою) між 25 та 120°С. Приклад 13 Кристалізація метансульфонату сполуки С Сполуку С (250мг, отримано аналогічно способу, що описано у прикладі 12 вище) розчиняли у метанолі (10мл). Метансульфонову кислоту (56мг, 38мкл) додавали до розчину. Метанол видаляли під зниженим тиском та остаточну смолу знов розчиняли у можливо мінімальній кількості гарячого ізопропанолу. Розчин затравлювали попередньо отриманою сіллю (отримано повільним випарюванням розчиннику з невеликої порції розчину потрібної сполуки, отриманої аналогічно цьому способу) та залишали у холодильнику протягом 4,34 4,23 4,20 4,08 3,99 3,89 3,85 3,80 3,68 3,52 3,49 3,43 3,39 3,33 3,25 3,01 2,94 2,90 2,80 2,49 2,40 4 4 6 5 2 8 5 4 2 2 2 2 2 2 3 2 2 1 1 1 2 ДСК показано ендотермічні піки з екстрапольованим температурним початком приблизно 145°С (другорядний) та 170°С (головний). ТГМА показано зменшення маси приблизно 0,9% (за масою) між 25 та 105°С. Приклад 14 Кристалізація пара-толуолсульфонату сполуки С Сполуку С (250мг, отримано аналогічно способу, що описано у прикладі 12 вище) розчиняли у метанолі (10мл). Толуолсульфонову кислоту (110мг) додавали до розчину. Метанол видаляли під зниженим тиском та остаточну смолу знов розчиняли у можливо мінімальній кількості гарячого етилацетату. Розчин затравлювали попередньо отриманою сіллю (отримано повільним випарюванням розчиннику з невеликої порції розчину потрібної сполуки, отриманої аналогічно цьому способу) та залишали у холодильнику протягом кількох діб для кристалізації. Утворені кристали збирали фільтруванням та промивали невеликою кількістю холодного етилацетату (отримано 280мг, 78%). Кристали аналізували РДМП, результати показані нижче (таблиця 14) та у Фіг.14. 59 74629 Таблиця 14 d-параметр (Å) 13,2 8,1 7,5 7,1 6,6 6,4 6,3 6,0 5,6 5,4 5,2 5,0 4,97 4,86 4,67 4,42 4,28 4,24 4,19 4,12 4,08 4,03 4,01 3,92 3,82 3,78 3,66 3,57 3,46 3,36 3,32 3,12 Інтенсивність (%) 100 0,5 0,7 3 0,7 0,9 0,7 0,8 1 2 3 4 2 0,8 2 0,7 3 7 2 1 2 2 1 1 0,7 0,8 0,8 0,5 1 2 1 2 ДСК показано ендотермічний пік з екстрапольованим температурним початком приблизно 138°С. ТГМА показано зменшення маси приблизно 0,2% (за масою) між 25 та 150°С. Приклад 15 Сполука D, сіль [(дифеніл-4карбоніл)аміно1оцтової кислоти (а) [(Дифеніл-4-карбоніл)аміно]оцтової кислоти метиловий естер Дихлорметан (50мл) та далі триетиламін (11,2мл, 79,6ммоль, 2,0екв.) додавали до гідрохлориду метилового естеру гліцину (5,0г, 39,8ммоль, 1,0екв.). Суміш перемішували та охолоджували до -5°С, використовуючи баню метанол/лід. Суспензію дифеніл-4-карбонілхлориду (8,26г, 39,8ммоль, 1,0екв.) у дихлорметані (25мл) додавали протягом 22 хвилин. Суміш перемішували протягом 3 годин при -5°С, та далі перемішували при кімнатній температурі протягом ночі (16 годин). Воду (75мл) додавали та суміш перемішували швидко протягом 30 хвилин при кімнатній температурі. Шари розділяли. Органічний шар промивали водою (75мл), далі випарювали досуха, використовуючи роторний випарник з отриманням білуватого твердого продукту (6,58г, 62%). 1Н-ЯМР (300МГц, CDCI3) δ 3,82 (s, 3Н), 4,29 (d, J=5,1Гц, 2H), 6,68 (s, 1H), 7,3-7,5 (m, 3Н), 7,62 (d, J=4,8Гц, 2H), 7,68 (d, J=8,1Гц, 2H), 7,90(d, J=8,4Гц, 2Н), темп.пл. 127-128°С (b) [(Дифеніл-4-карбоніл)аміно]оцтова кислота [(Дифеніл-4-карбоніл)аміно]оцтової кислоти метиловий естер (6,58г, 25ммоль, 1,0екв., з етапу (а) вище) додавали у колбу, а потім додавали водний гідроксид натрію (1М, 84мл, 50ммоль, 2,0екв.). Суміш гріли до 50°С, використовуючи ма 60 сляну баню протягом 5 годин. Розчин далі перемішували протягом ночі (16 годин) при кімнатній температурі. При охолодженні утворився білий осад. Суміш охолоджували до 5°С, використовуючи баню лід/вода. Концентровану гідрохлоридну кислоту (8мл) додавали дуже повільно до охолодженого розчину, щоб температур не зросла вище 10°С. Суміш перемішували протягом 15 хвилин та далі фільтрували. Білий твердий продукт сушили на повітрі протягом 30 хвилин та далі сушили у вакуумі при 40°С протягом 16 годин з отриманням білуватого твердого продукту (5,75г, 93%). 1 Н-ЯМР (300МГц, ДМСО-d6): δ 3,95 (d, J=5,7Гц, 2H), 7,35-7,5 (m, 3Н), 7,7-7,8 (m, 4Н), 7,97 (d, J=6,9Гц, 2H), 8,89 (t, J=6,0Гц, 1Н), 12,58 (s, 1Н), темп.пл. 217-217,5°C. (c) Перекристалізація [(дифеніл-4карбоніл)аміно]оцтової кислоти Метанол (100мл, 20 об'ємів) додавали до [(дифеніл-4-карбоніл)аміно]оцтової кислоти (5,0г, з етапу (b) вище). Суміш гріли до 62°С, використовуючи масляну баню при перемішуванні. Утворений блідо-оранжевий розчин тримали при цій температурі протягом 10 хвилин. Розчину давали охолонути до кімнатної температури, та далі охолоджували до 5°С, використовуючи баню лід/вода. Кристалізація починалася приблизно при 30°С. Осад збирали фільтруванням, сушили на повітрі протягом 15 хвилин, далі сушили у вакуумі при 40°С протягом 26 годин з отриманням безбарвних кристалів (2,9г, 58%). 1 Н-ЯМР (300МГц, ДМСО-d6) δ 3,95 (d, J=5,7Гц, 2H), 7,35-7,5 (m, 3Н), 7,7-7,8 (m, 4Н), 7,97 (d, J=6,9Гц, 2Н), 8,89 (t, J=6,0Гц, 2Н), 12,58 (s, 1H). (d) Сполук D, сіль [(дифеніл-4карбоніл)аміно]оцтової кислоти [(Дифеніл-4-карбоніл)аміно]оцтову кислоту (1,14г, дивися етапи (b) або (с) вище) та сполуку D (2г, отримано аналогічно способам, що описано тут раніше) розчиняли у гарячому ізопропанолі (40мл). При охолодженні до кімнатної температури утворився кристалічний осад, який фільтрували, промивали ізопропанолом (2 20мл) та відсмоктували на фільтр. Сушка протягом 6 годин у вакуумі при 40°С дала сіль як безбарвний кристалічний твердий продукт (2,50г, 80%). 1 Н-ЯМР (300МГц, ДМСО-d6): δ 1,34 (9Н, s), 2,25 (2Н, t), 2,3-2,5 (4Н, m), 2,6-2,7 (1Н, m), 2,7-2,8 (1Н, m), 2,85-3,0 (4Н, m), 3,0-3,1 (2Н, m), 3,82 (2Н, s), 3,88 (2Н, d), 3,95-4,05 (2Н, m), 4,1-4,2 (1Н, m), 6,65 (1Н, t), 7,14 (2Н, d), 7,35-7,55 (3Н, m), 7,7-7,85 (6Н, m), 7,96 (2Н, d), 8,75 (1Н, t), темп.пл. 143143,5°С. Кристали аналізували РДМП, результати показані нижче (таблиця 15) та у фігурі 15. Таблиця 15 d-параметр (Å) 20,4 15,3 11,5 10,3 10,0 9,4 7,7 7,2 Інтенсивність (%) 45 100 24 4 6 11 34 23
ДивитисяДодаткова інформація
Назва патенту англійськоюA derivative of 3,7-diazabicyclo [3,3,1] compounds , pharmaceutically acceptable salts thereof, a method for producing and composition based thereon as antiarrhythmic agents
Автори англійськоюBjoere Annika
Назва патенту російськоюПроизводное 3,7-диазадицикло[3,3,1] соединений, их фармацевтически приемлемые соли, способ получения и композиция на их основании как антиаритмические агенты
Автори російськоюБйоре Анника
МПК / Мітки
МПК: C07D 498/08, A61P 9/06, A61K 31/5386
Мітки: солі, похідне, фармацевтично, агенти, прийнятні, 3,7-діазадицикло[3,3,1, композиція, антиаритмічні, основі, одержання, сполук, спосіб
Код посилання
<a href="https://ua.patents.su/37-74629-pokhidne-37-diazadiciklo331-spoluk-kh-farmacevtichno-prijjnyatni-soli-sposib-oderzhannya-ta-kompoziciya-na-kh-osnovi-yak-antiaritmichni-agenti.html" target="_blank" rel="follow" title="База патентів України">Похідне 3,7-діазадицикло[3,3,1] сполук, їх фармацевтично прийнятні солі, спосіб одержання та композиція на їх основі як антиаритмічні агенти</a>
Похідне триазолу, його фармацевтично прийнятні солі або проліки, що є агоністами 5-нt1-подібних рецепторів, спосіб його отримання та фармацевтична композиція
Номер патенту: 27672
Опубліковано: 15.09.2000
Автори: Стріт Леслі Дж., Бейкер Реймонд, Матасса Віктор Г.
МПК: C07D 401/14, C07D 403/06, C07D 409/04
Мітки: солі, проліки, фармацевтична, отримання, агоністами, похідне, спосіб, рецепторів, прийнятні, фармацевтично, триазолу, 5-нt1-подібних, композиція
Текст:
...призначення або призначення шля хом ін'єкцій , включають водні розчини , ароматизо вані сиропи, во дні або масляні емульсії, ароматизовані емульсі ї, з такими маслами, як бавовняне масло, сезамове масло, кокосове масло, а також еліксири та подібні засоби . Відпо відні диспер гуючі та суспензуючі аген ти для водни х суспензій включають синте ти чні і натуральні смоли, такі як трагант, акація, альгінат, декстран, натрієва сіль...
2,3,4,5-тетрагідро-1,4-бензотіазепіни, їх стереоізомери або їх фармацевтично прийнятні солі, спосіб їх одержання та фармацевтична композиція на їх основі

Номер патенту: 41342
Опубліковано: 17.09.2001
Автори: Ніколь Кеннет Джон, Хоуслі Джон Росіндейл, Сарджент Брюс Джеремі, Джеффері Джеймс Едвард
МПК: A61P 25/08, A61K 31/554, A61K 31/55, C07D 281/00
Мітки: композиція, фармацевтична, основі, фармацевтично, солі, прийнятні, 2,3,4,5-тетрагідро-1,4-бензотіазепіни, спосіб, одержання, стереоізомери
Формула / Реферат:
1. 2,3,4,5-тетрагидро-1,4-бензотиазепины общей формулы II:их стереоизомеры и их фармацевтически приемлемые соли, где n=0 или 1,R1, R2, R1 и R7 каждый представляет собой Н,R3 и R3 независимо представляют Н или метил, или вместе представляют имино, метилимино, фенилимино, гидроксиимино или метоксиимино группу,R5 представляет Н или метил, или, когда R3 и R4 независимо представляют Н или метил, то R5...
(1r,2s,4r)-(-)-2-[(2′-{n,n-диметиламіно}-етокси)]-2-[феніл]-1,7,7-три-[метил]-біцикло[2,2,1]гептан високого ступеня чистоти, його фармацевтично прийнятні кислотні адитивні солі, спосіб одержання цих сполук та лікарські засоби

Номер патенту: 61122
Опубліковано: 17.11.2003
Автори: ШІМІГ Дьюла, Суладьі Янош, Лукач Дьюла, Надь Кальман, Краснаі Дьйордь, САБО Тібор, НЕМЕТ Норберт, Порч-Маккаі Марта, Будаі Золтан, Мезеі Тібор, ДОНАТ ВЕРЕЦКЕІ Дьйордьі
МПК: C07C 217/12
Мітки: фармацевтично, засоби, 1r,2s,4r)-(-)-2-[(2'-{n,n-диметиламіно}-етокси)]-2-[феніл]-1,7,7-три-[метил]-біцикло[2,2,1]гептан, адитивні, лікарські, кислотні, сполук, високого, одержання, солі, ступеня, прийнятні, чистоти, цих, спосіб
Формула / Реферат:
1. (1R,2S,4R)-(-)-2-[(2'-{N,N-диметиламіно}-етокси)]-2-[феніл]-1,7,7-три-[метил]-біцикло[2,2,1]гептан формули (І)і його фармацевтично прийнятні кислотно-адитивні солі, який відрізняється тим, що він містить не більше 0,2% (1R,3S,4R)-3-[(2'-{N,N-диметиламіно}-етил)]-1,7,7-три-[метил]-біцикло[2,2,1]гептан-2-ону формули V
Похідні піролзаміщених уреїдів і їх фармацевтично прийнятні солі, спосіб їх одержання і фармацевтична композиція, яка має інгібуючу ангіогенез активність

Номер патенту: 32513
Опубліковано: 15.02.2001
Автори: Монгеллі Нікола, Гранді Маріа, Чомеі Маріна, Біазолі Джованні, Пайо Альфредо
МПК: C07D 215/02, A61K 31/40, C07D 407/14, C07D 319/00
Мітки: композиція, прийнятні, уреїдів, фармацевтична, яка, солі, ангіогенез, одержання, піролзаміщених, має, активність, похідні, інгібуючу, фармацевтично, спосіб
Формула / Реферат:
(57) 1. Производные пирролзамещенных уреидов общей формулы (I)где m и n - одинаковые, каждый - целое число от 1 до 3,W - кислород;В - группы одинаковые, каждая из них - нафталиновое или хинолиновое кольцо, замещенное 1-3 группами сульфокислоты, или 2-дезокси-D-глюкозо-6-сульфат, или 2-дезокси-D-гпюкозо-6-фосфат, и их фармацевтически приемлемые соли.2. Соединения по п. 1 формулы (I), отличающиеся тем, что в...
Похідні n-заміщеного 4-феніл-4-піперидинкарбоксаміду та їх фармацевтично прийнятні солі, що проявляють анестезувальну та анальгезувальну дію, спосіб їх одержання та фармацевтична композиція

Номер патенту: 26403
Опубліковано: 30.08.1999
Автори: Аск Анна-Лена, Сандберг Руне
МПК: C07D 211/64, C07D 211/52, A61P 25/04, A61P 29/00, A61K 31/451, A61K 31/445
Мітки: проявляють, солі, 4-феніл-4-піперидинкарбоксаміду, похідні, n-заміщеного, анальгезувальну, спосіб, фармацевтично, фармацевтична, композиція, одержання, дію, прийнятні, анестезувальну
Формула / Реферат:
1. Производные N-замещенного 4-фенил-4-пиперидинкарбоксамида общей формулыгде R1 представляет собой алкильную группу с 2 - 6 атомами углерода или алкоксиалкильную группу R4O(CH2)m, в которой R4 представляет собой алкильную группу с 1 - 4 атомами углерода и m равно 2 - 4;R2 и R3 являются одинаковыми или различными и каждый представляет собой алкильную группу с числом атомов углерода до 6, или R2 и R3 образуют вместе цепь...
Попередній патент: Пристрій для відокремлення листового матеріалу зі стосу
Наступний патент: Спосіб захисту посівів сільськогосподарських культур від бур`янів
Випадковий патент: Пристрій для орієнтації виробів