Кристалічні частинки (r)-3-(4-(2-(2-метилтетразол-5-іл)піридин-5-іл)-3-фторфеніл)-5-гідроксиметилоксазолідин-2-ондиводеньфосфату
Номер патенту: 114068
Опубліковано: 25.04.2017
Автори: Рейченбахер Катаріна, Сімсон Джаклін А., Філліпсон Дуглас, Д'югайд Роберт Дж.




































Формула / Реферат
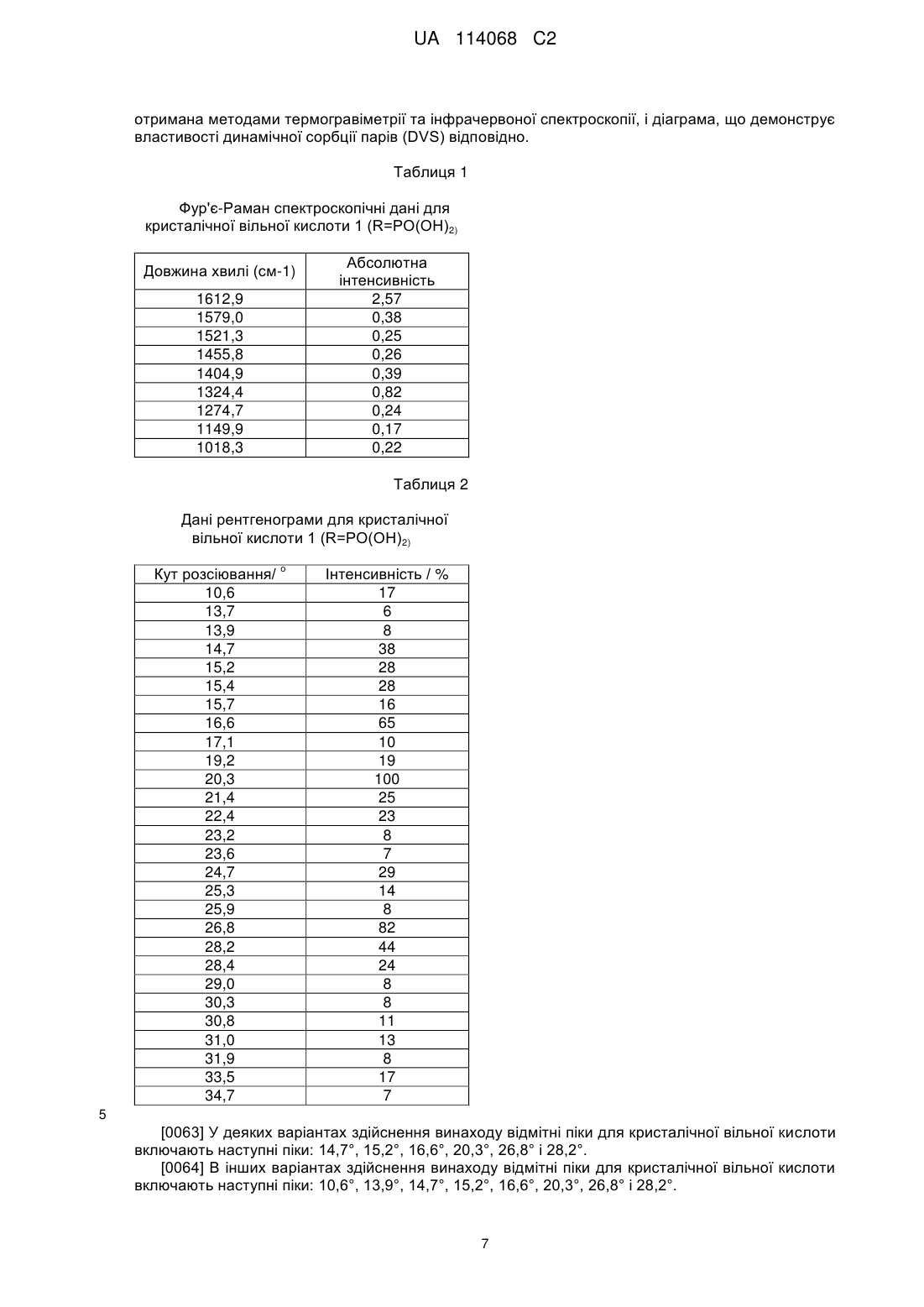
1. Кристалічні частинки, що характеризуються середнім об'ємним діаметром від 1,0 мкм до 44,0 мкм та вмістом (R)-3-(4-(2-(2-метилтетразол-5-іл)піридин-5-іл)-3-фторфеніл)-5-гідроксиметилоксазолідин-2-ондиводеньфосфату у кількості не менше ніж 96 % за вагою, а також характеризуються рентгенодифракційною картинкою порошку із піками 14,7°; 15,2°; 16,6°; 20,3°; 26,8° і 28,2°, причому залишок кристалічних частинок містить принаймні одну сполуку, вибрану з групи, що складається з:

та
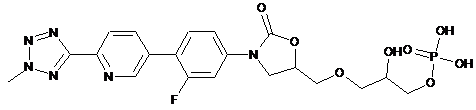 .
.
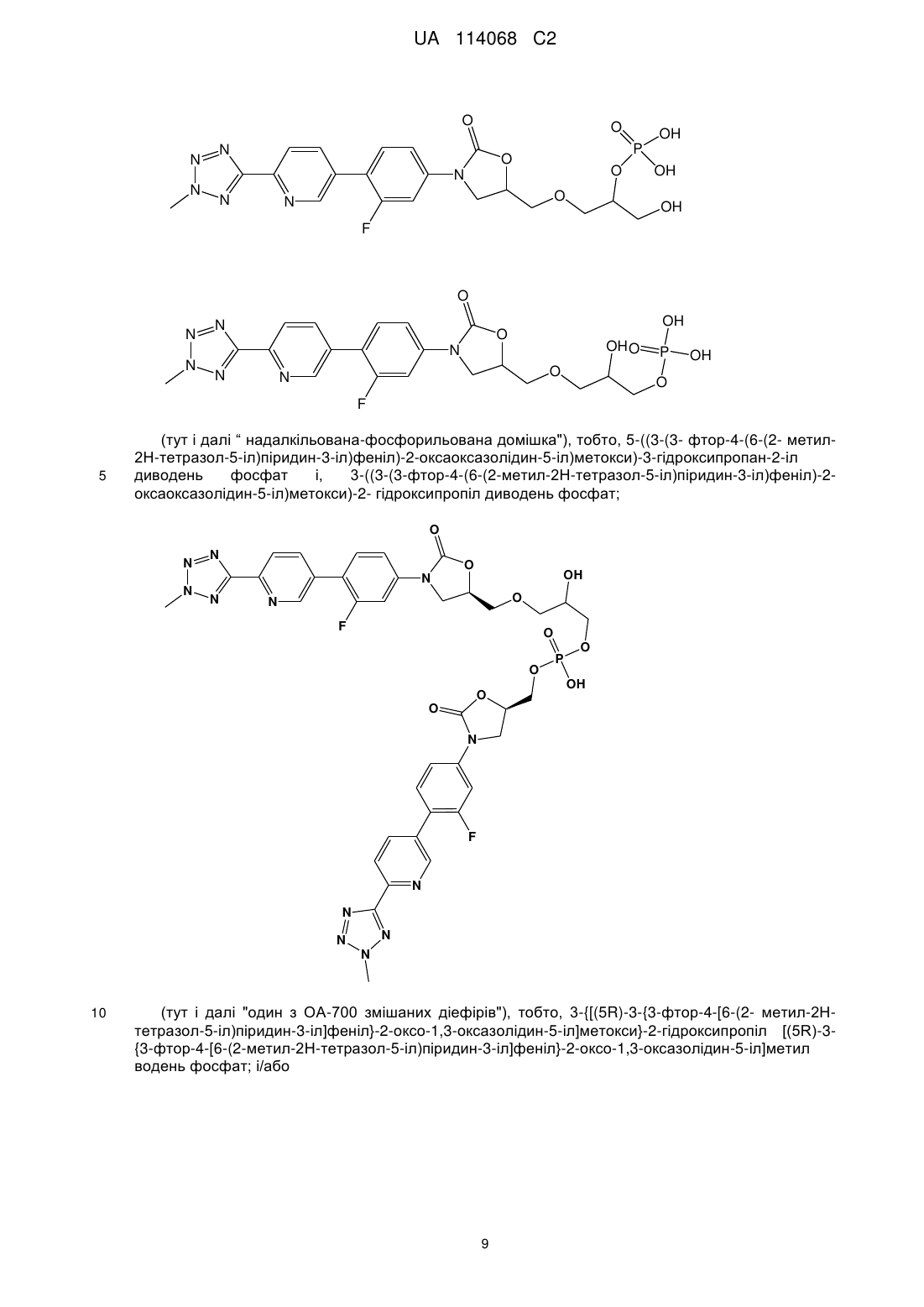
2. Кристалічні частинки за пунктом 1, причому залишок кристалічних частинок містить також принаймні одну сполуку, вибрану з групи, що складається з:

та
 .
.
3. Кристалічні частинки за пунктом 2, причому залишок кристалічних частинок містить також сполуку:
 .
.
4. Кристалічні частинки за пунктом 3, причому залишок кристалічних частинок містить також принаймні одну сполуку, вибрану з групи, що складається з:
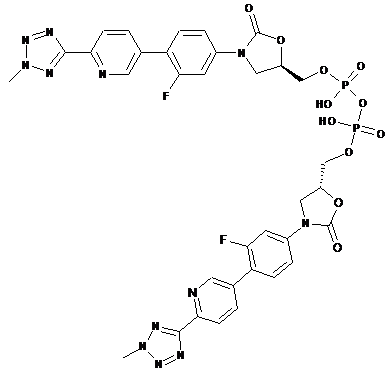
та
 .
.
5. Кристалічні частинки за пунктом 4, причому залишок кристалічних частинок містить також сполуку:
 .
.
6. Кристалічні частинки за пунктом 1, причому середній об'ємний діаметр складає від 10 мкм до 35 мкм.
7. Фармацевтична композиція, що містить кристалічні частинки за пунктом 1 і принаймні один фармацевтично прийнятний носій, наповнювач або розріджувач.
8. Фармацевтична композиція за пунктом 7, причому композиція є у формі ліків для перорального прийому.
9. Реакційна суміш, що містить кристалічні частинки за пунктом 1 та основу.
10. Реакційна суміш за пунктом 9, причому як основу беруть гідроксид натрію.
11. Фармацевтична композиція, що містить ліофілізат реакційної суміші за пунктом 9, причому ця фармацевтична композиція містить:
 ,
,
де R=PO(ONa)2; та
принаймні одну сполуку, вибрану з групи, що складається з:
та
,
та принаймні один фармацевтично прийнятний носій, наповнювач або розріджувач.
12. Фармацевтична композиція за пунктом 11, що додатково містить принаймні одну сполуку, вибрану з групи, що складається з:
та
,
та принаймні один фармацевтично прийнятний носій, наповнювач або розріджувач.
13. Фармацевтична композиція за пунктом 12, що додатково містить сполуку:
та принаймні один фармацевтично прийнятний носій, наповнювач або розріджувач.
14. Спосіб лікування бактеріальної інфекції, що включає призначення ефективної кількості фармацевтичної композиції за пунктом 7 пацієнтові, який цього потребує.
15. Спосіб за пунктом 14, причому бактеріальну інфекцію викликає грампозитивна бактерія.
16. Ліофілізований порошок для ін'єкцій, виготовлений способом, що включає змішування розчину, що містить кристалічні частинки сполуки
,
де R=РО(ОН)2,
за пунктом 1 з розчином гідроксиду натрію з утворенням динатрієвої солі вказаної сполуки та подальшою ліофілізацією одержаною розчину.
17. Спосіб лікування бактеріальної інфекції, що включає призначення ефективної кількості ліофілізованого порошку за пунктом 16 пацієнтові, який цього потребує.
18. Спосіб за пунктом 17, причому бактеріальну інфекцію викликає грампозитивна бактерія.
19. Спосіб за пунктом 18, причому ліофілізований порошок призначають у кількості від приблизно 1 мг до приблизно 500 мг в перерахунку на (R)-3-(4-(2-(2-метилтетразол-5-іл)піридин-5-іл)-3-фторфеніл)-5-гідроксиметилоксазолідин-2-ондиводеньфосфат.
20. Спосіб за пунктом 19, причому ліофілізований порошок призначають у кількості від приблизно 5 мг до приблизно 200 мг.
21. Спосіб за пунктом 19, причому ліофілізований порошок призначають у кількості приблизно 200 мг.
22. Спосіб одержання кристалічних частинок за пунктом 1, що включає додавання реакційної суміші, що містить сіль кристалічного (R)-3-(4-(2-(2-метилтетразол-5-іл)піридин-5-іл)-3-фторфеніл)-5-гідроксиметилоксазолідин-2-ондиводеньфосфату, до розчину кислоти з утворенням кристалізованого (R)-3-(4-(2-(2-метилтетразол-5-іл)піридин-5-іл)-3-фторфеніл)-5-гідроксиметилоксазолідин-2-ондиводеньфосфату;
відфільтровування кристалізованого (R)-3-(4-(2-(2-метилтетразол-5-іл)піридин-5-іл)-3-фторфеніл)-5-гідроксиметилоксазолідин-2-ондиводеньфосфату від супернатанту; та
висушування відфільтрованого кристалізованого (R)-3-(4-(2-(2-метилтетразол-5-іл)піридин-5-іл)-3-фторфеніл)-5-гідроксиметилоксазолідин-2-ондиводеньфосфату.
23. Спосіб за пунктом 22, де розчин кислоти містить НСl і етанол або НСl і THF.
Текст
Реферат: Кристалічні частинки (R)-3-(4-(2-(2-метилтетразол-5-іл)піридин-5-іл)-3-фторфеніл)-5гідроксиметилоксазолідин-2-ондиводеньфосфату, способи одержання кристалічних частинок й фармацевтичні композиції, що містять ці кристалічні форми, є корисними антибіотиками. Також похідні за даним винаходом можуть проявляти сильну антибактеріальну активність проти різних людських і тваринних патогенів, включаючи грампозитивні бактерії, такі як Staphylococi, Enterococci і Streptococi, анаеробні організми, такі як бактероїди й Clostridia, і кислотостійкі мікроорганізми, такі як Mycobacterium tuberculosis і Mycobacterium avium. Таким чином, композиції, що містять кристалічні частинки, можуть бути використані в антибіотиках. UA 114068 C2 (12) UA 114068 C2 UA 114068 C2 5 10 [0001] Ця заявка має пріоритет за Попередньою заявкою на патент США № 61/149,402, поданою 3 лютого 2009, яка наводиться тут для посилання у всій її цілісності. Область застосування винаходу. Область техніки, до якої відноситься винахід [0002] Даний опис відноситься до кристалічних частинок (R)-3-(4-(2-(2-метилтетразол-5іл)піридин-5-іл)-3-фторфеніл)-5-гідроксиметил оксазолідин-2-он диводень фосфату, способам одержання й застосування кристалічних частинок. Кристалічні частинки можуть бути використані в якості фармацевтично активної сполуки в композиціях, які є корисними для інгібування росту бактерій або для лікування пацієнтів, які страждають від бактеріальних інфекцій. Опис рівня техніки [0003] Патентна публікація US 20070155798, яка наводиться тут для посилання у всій її цілісності, розкриває ряд сильнодіючих антибактеріальних оксазолідинів, що включає O N N O N N 15 20 25 30 35 40 45 50 N OR N F де R=H, PO(OH)2 і PO(ONa)2. [0004] Хоча ця патентна заявка розкриває способи одержання сполук, таких як вільна кислота (де R=PO(OH)2) і динатрійова сіль (де R=PO(ONa)2), у ній відсутня інформація про те, що кожна із цих сполук була стабільною в кристалічній або очищеній формі. Додатково, ці способи включають використання реагентів, які є хімічно агресивними, наприклад, трифтороцтової кислоти, або вибухонебезпечними, наприклад, етилового ефіру, отже, не підходять для комерційного використання. Як описується детальніше нижче, спроби авторів даного винаходу кристалізувати динатрійову сіль призвели до одержання високо гігроскопічної, нестабільної форми солі, яка стала аморфною при висушуванні. [0005] У рівні техніки існує необхідність в утворенні стабільних, негігроскопічних кристалічних частинок вільної кислоти (де R=PO(OH)2) або її солі, які можуть бути охайно перенесені і зважені для застосування у фармацевтичних засобах. Також було б переважніше, щоб кристалічні частинки не утворювали велику кількість поліморфних модифікацій, оскільки деякі з поліморфів перешкоджають здатності відтворити ідентичні поліморфи в процесі одержання. Одержання кристалічних частинок, що мають ці властивості, є емпіричним процесом, і фахівець у даній галузі не може передбачити, які частинки вільної кислоти фармацевтичної сполуки або однієї з відповідних солей будуть кристалізуватися, чи взагалі будуть, у зазначених умовах кристалізації. Додатково фахівець у даній галузі не може передбачити, які із кристалічних частинок будуть мати кращі властивості стабільності, здатності до переносіння, відсутності гігроскопічності й відтворюваності. [0006] Додатково покращені способи одержання вільної кислоти розкриті в патентній заявці US 12/577,089, переданої Траюс Терап'ютікс, Інк., яка наводиться тут для посилання у всій її цілісності. Існують складності у фільтрації кристалічного матеріалу й переробці кристалічного матеріалу в дозовані форми, такі як таблетки, тому що вільна кислота утворює дрібні частинки, які подовжують термін процесу. Таким чином, у рівні техніки існує необхідність в утворенні кристалічних частинок сполуки й відповідних способах, які долають ці складності. [0007] Додатковою перевагою було б мати очищену сполуку, яка б була прийнятною для застосування у фармацевтичних композиціях. Короткий опис сутності винаходу. [0008] Неочікувано (R)-3-(4-(2-(2-метилтетразол-5-іл) піридин-5-іл)-3-фторфеніл)- 5гідроксиметил оксазолідин-2-он диводень фосфат 1 (R=PO(OH)2) виявився більш стабільним і негігроскопічним, ніж вже перевірені сольові форми. Додатково, на відміну від звичайної кристалізації, де умови кристалізації, наприклад, розчинник і температура, визначають конкретну кристалічну форму, одн й т сам кристалічна сполука 1 (R=PO(OH) 2), була отримана з використанням різних розчинників й умов кристалізації. Таким чином, ц кристалічна сполука були дуже стабільною, відтворюваною й ідеальною для комерційного виробництва, оскільки вона зменшила ймовірність того, що інші поліморфи будуть утворювати забруднюючі домішки під час виробництва. Однак, у всіх попередніх випробуваннях вільна кислота кристалізувалася у вигляді дрібних частинок, ускладнюючи фільтрування й переробку. 1 UA 114068 C2 5 10 15 [0009] Для подолання складностей з фільтруванням і переробкою кристалічного (R)-3-(4-(2(2-метилтетразол-5-іл)піридин-5-іл)-3-фторфеніл)-5-гідроксиметил оксазолідин-2-он диводень фосфату 1 (R=PO(OH)2) описані тут процеси призводять до значного зменшення терміну фільтрування, допомагають уникнути використання токсичних розчинників і значно полегшують одержання дозованих форм, таких як таблетки. Було виявлено, що здійснення різних процесів можна контролювати за допомогою розподілу за розмірами частинок отриманого матеріалу, що є корисним для їх одержання та для комерційного й фармацевтичного використання. Неочікувано було встановлено, що процес збільшення розміру частинок зменшує кількість домішки димеру в порівнянні із процесом одержання вільної кислоти, описаної в патентній заявці США 12/577,089. Таким чином, також представлені різні способи одержання й використання кристалічних частинок. [0010] Додатково, використовуючи способи одержання вільної кислоти, описані в патентній заявці США 12/577,089, права на яку передані тому ж патентовласникові, що й по цій заявці, і застосовуючи описані тут способи кристалізації, кристалічна вільна кислота, що має, щонайменше, 96 % чистоти за вагою, може бути отримана у формі, яка містить сполуку, що має наступну формулу: O N N O N N N Cl N F 20 (тут і далі "домішки, що містять хлор", тобто (R)-5-(хлорметил)-3-(3-фтор-4-(6-(2- метил-2Нтетразол-5-іл) піридин-3-іл)феніл) оксазолідин-2-он у кількості менше 1 %. [0011] Подібним чином, використовуючи способи одержання вільної кислоти, розкритої в патентній заявці США 12/577,089, права на яку передані тому ж правовласникові, що й ця заявка, і використовуючи описані тут способи кристалізації, кристалічна вільна кислота, що має, щонайменше, 96 % чистоту за вагою може бути отримана у формі, яка містить сполуку, що має наступну формулу: O N N O N N 25 30 35 40 45 N OH N F (тут і далі "TR-700"), тобто, 5R)-3-{3-фтор-4-[6-(2-метил-2H-1,2,3,4-тетразол-5-іл)- піридин-3іл]-феніл}- 5-гідроксиметил-1,3-оксазолідин-2-он, у кількості, менше 1 %. [0012] Кристалічна вільна кислота може мати один або більш атрибутів, описаних тут. [0013] У деяких аспектах очищений кристалічний (R)-3-(4-(2-(2-метилтеразол- 5-іл) піридин5-іл)-3-фторфеніл)-5-гідроксиметил оксазолідин-2-он диводень фосфат, тобто вільна кислота має чистоту, щонайменше, близько 96 % за вагою. У деяких варіантах здійснення винаходу кристалічна вільна кислота має середній діаметр, щонайменше, близько 1,0 мкм. [0014] У деяких варіантах здійснення винаходу фармацевтична композиція містить вільну кислоту або сіль і, щонайменше, один фармацевтично прийнятний носій, екципієнт або дилюєнт. [0015] У деяких варіантах здійснення винаходу спосіб лікування бактеріальної інфекції включає введення ефективної кількості кристалічної вільної кислоти або її солі суб'єктові, який цього потребує. Способи можуть також включати лікування бактеріальної інфекції, що включає введення вільної кислоти, фармацевтичної композиції або солі суб'єктові, який цього потребує. [0016] У деяких аспектах способи одержання вільної кислоти включають висушування кристалічного (R)-3-(4-(2-(2-метилтетразол-5-іл) піридин-5-іл)-3-фторфеніл)-5-гідроксиметил оксазолідин-2-он диводень фосфату або фармацевтичної композиції, що містить його сіль. [0017] Ці та інші варіанти здійснення винаходу детальніше описані нижче. Короткий опис фігур [0018] Фігура 1 являє собою Фур'є-Раман спектр кристалічної сполуки 1 (R=PO(OH) 2). [0019] Фігура 2 являє собою рентгенограму кристалічної сполуки 1 (R=PO(OH) 2). [0020] Фігура 3 являє собою термограму диференціальної скануючої калориметрії (DSC) кристалічної сполуки 1 (R=PO(OH)2). 1 [0021] Фігура 4 демонструє H NMR спектр сполуки 1 (R=PO(OH)2). 2 UA 114068 C2 5 10 15 20 25 30 35 40 45 50 55 60 [0022] На Фігурі 5 зображена діаграма кристалічної сполуки 1 (R=PO(OH) 2) методами термогравіметрії й інфрачервоної спектроскопії. [0023] Фігура 6 являє собою діаграму, що демонструє властивості динамічної сорбції парів (DVS) кристалічної сполуки 1 (R=PO(OH)2). [0024] Фігура 7 являє собою схематичний процес одержання сполуки 1 (R=PO(OH) 2) (TR-701 FA) у формі таблеток. [0025] Фігура 8 являє собою схематичний процес одержання сполуки 1 (R=PO(OH) 2) (TR-701 FA) у вигляді розчину для ліофілізації. [0026] Фігура 9 являє собою схематичний процес одержання сполуки 1 (R=PO(OH) 2) (TR-701 FA) для ін'єкції, 200 мг/ампула: стерильне фільтрування, наповнення й ліофілізація. [0027] Фігура 10 являє собою розподіл частинок за розмірами кристалічної вільної кислоти без контролювання розміру частинок, як описано тут. [0028] Фігура 11 являє собою розподіл частинок за розмірами кристалічної вільної кислоти, виконаний з використанням лабораторних процесів для контролю розміру частинок, представленого тут. [0029] Фігура 12 розподіл частинок за розмірами кристалічної вільної кислоти з використанням збільшеного у масштабі виробничого процесу для контролю розміру частинок, описаного тут. Детальний опис кращих варіантів здійснення винаходу [0030] R)-3-(4-(2-(2-метилтетразол-5-іл)піридин-5-іл)-3-фторфеніл)-5-гідроксиметил оксазолідин-2-он диводень фосфат 1 (R=PO(OH) 2), який іноді називається "вільна кислота" або "TR-701 FA" і деякі його солі були отримані в різних умовах кристалізації для визначення матеріалу, який утворює найстабільнішу й найменш гігроскопічну кристалічну сполуку. Емпіричний процес одержання кристалічних частинок вільної кислоти та її солей призвів до відбору кристалічної вільної кислоти, яка додатково до кращої стабільності й відсутності гігроскопічності, могла бути відтворена в різних умовах кристалізації, потім ці частинки були очищені й висушені. [0031] Більшість із перевірених солей було важко одержати в кристалічній формі або вони були нестабільними в очищеній або висушеній формі. Наприклад, що стосується моно-натрієвої солі, одержання стабільного гідрату виявлено не було. Також матеріал містив понад 10 % за вагою води й у такий спосіб матеріал був дуже гігроскопічним, і при цьому нестабільним для бажаного використання. [0032] Був отриманий кристалічний гідрат динатрійової солі, але він виявився нестабільним і містив 19,6 % води за вагою. Однак динатрійова сіль виявилася дуже добре розчинною. У результаті висушування гідрату були отримані аморфні зразки. Зміст води в аморфному зразку склав 6,2 % за вагою. [0033 Кристалічний твердий матеріал не був виділений для дикалійової солі. [0034] Була отримана напів-кальційова сіль у вигляді кристалу, однак, вона була занадто гігроскопічною. [0035] Був отриманий кристалічний матеріал напів-магнійової солі, але як виявилося, він містив різні гідрати солі, а присутність різних поліморфів є небажаним для застосування в лікарських формах. В одному експерименті магнієва сіль мала температуру плавлення 152,8° C, що вказує на меншу стабільність цього матеріалу в порівнянні з вільною кислотою. [0036] Отримані кристали вільної кислоти, які не були гігроскопічними при фільтруванні й висушуванні, продемонстрували розчинність в 0,1 мг/мл (pН = 3,2 насиченого розчину). Температура плавлення кристалічного матеріалу склала 255-258 °C, і в такий спосіб матеріал був дуже стабільним при відносно високій температурі. [0037] У цілому умови кристалізації звичайно є критичними для формування конкретного поліморфа; однак, неочікувано виявилося, що один і той самий поліморф вільної кислоти був отриманий у різних умовах, у яких утворювалася кристалічна вільна кислота. [0038] У деяких варіантах здійснення винаходу кристалічний матеріал є негігроскопічним, тому він не поглинає й утримує воду з атмосферного повітря. У деяких варіантах здійснення винаходу "негігроскопічний" матеріал має вміст води менше приблизно 5 %, 4 %, 3 %, 2 %, 1 %, 0,9 %, 0,8 %, 0,7 %, 0,6 %, 0,5 %, 0,4 %, 0,3 %, 0,2 % або 0,1 % води за вагою. [0039] Переважно, вільна кислота може бути використана як для одержання твердої лікарської форми, так і форми для внутрішньовенного (IV) введення. При аналізі було виявлено, що динатрійова сіль, хоча і є нестабільною для твердих форм, таких як таблетки, була дуже добре розчинною й прийнятною для форм IV введення. При цьому в інших варіантах здійснення винаходу, стерильний ліофілізований порошок для ін'єкцій був отриманий з динатрійової солі in situ з гідроксидом натрію й наступною ліофілізацією отриманого розчину. Динатрійова сіль є 3 UA 114068 C2 5 10 15 20 25 30 35 40 45 50 55 60 добре розчинною, і тому краще відновлювати її стерильною водою з одержанням розчину. У деяких варіантах здійснення винаходу отриманий розчин може бути доданий у ємність для внутрішньовенного введення. Ємність може містити ізотонічний розчин, такий як 0,9 % хлорид натрію або 5 % декстрозу. [0040] У деяких варіантах здійснення винаходу, сольовий розчин, такий як динатрійова або мононатрійова сіль, може бути ліофілізований шляхом заморожування розчину в ліофілізаторі до приблизно -50 –30 °C зі швидкістю приблизно 0,1-1 градуси/хвилину й витримування протягом 200-700 хвилин, у цій точці в камері ліофілізатора створюється тиск приблизно 200250 міліторр і температура збільшується до -30-10 °C зі швидкістю 0,5-3 градуси/хвилину. Продукт витримують при -30-10 °C протягом приблизно 1000-2500 хвилин і потім температуру збільшують до приблизно 21-35 °C зі швидкістю 0,1 – 1 градус/хвилину й витримують протягом 1000-2500 хвилин з одержанням бажаного продукту. [0041] У варіантах деяких способів одержання кристалічна вільна кислота (R)-3-(4-(2-(2метилтетразол- 5-іл) піридин-5-іл)- 3-фторфеніл)- 5-гідроксиметил оксазолідин-2-он диводень фосфат 1 (R=PO(OH)2) може бути отримана підкисленням водяного розчину відповідної солі, такої як динатрійова сіль 1 (R=PO(ONa)2). [0042] Будь-яка сіль вільної кислоти 1 (R=PO(OH) 2) може бути використана для відновлення вільної кислоти за допомогою підкислення. У деяких варіантах здійснення сіль являє собою сіль лужного або лужноземельного металу. В інших варіантах здійснення винаходу сіль являє собою сіль лужного металу, таку як динатрійова сіль 1 (R=PO(OH)2). [0043] Було виявлено, що вибір кислоти не є критичним. Будь-яка кислота є у достатній мірі кислотоутворюючою для подвійного протонування динатрійового фосфату 1 (R=PO(ONa) 2) або іншої солі з одержанням вільної кислоти 1 (R=PO(OH) 2). У деяких варіантах здійснення винаходу кислота являє собою HСl, HBr або H2SO4. [0044] Після розчинення солі (R)-3-(4-(2-(2-метилтетразол- 5-іл)- піридин-5-іл)- фторфеніл)-5гідроксиметил оксазолідин-2-он диводень фосфату, і потім підкислення сольового розчину з утворенням кристалів, кристали можуть бути відфільтровані з супернатанту. У деяких варіантах здійснення винаходу вологі кристали можуть бути висушені, наприклад, з використанням вакууму або ліофілізації кристалів. [0045] У деяких варіантах здійснення винаходу термін "кристалічний" відноситься до однорідного кристалічного матеріалу кристалічного (R)-3-(4-(2-(2-метилтетразол-5-іл)піридин-5іл)-3-фторфеніл)-5-гідроксиметил оксазолідин-2-он диводень фосфату, таким як по суті чисті кристали. [0046] Терміни "приблизно, "близько" і "по суті" використані тут для зазначення кількості, приблизної до тієї, що заявляється, при якій усе ще виконується бажана функція або досягається бажаний результат. Наприклад, терміни "приблизно", "близько" і “ по суті" можуть відноситися до кількості, яка відрізняється від значення, що заявляється, у межах менше 10 %, менше 5 %, менше 1 %, менше 0,1 % і менше 0,01 %. [0047] Наприклад, у фармацевтичній промисловості, стандартною практикою є забезпечення по суті чистого матеріалу при виготовленні фармацевтичних композицій. Таким чином, у деяких варіантах здійснення винаходу "по суті чистий" відноситься до ступеня чистоти, необхідного для одержання лікарських засобів, які можуть включати, наприклад, невелику кількість аморфного або іншого матеріалу, при цьому матеріал усе ще має достатню здатність до переносіння, не є гігроскопічним і має чистоту, прийнятну для фармацевтичного застосування. У деяких варіантах здійснення винаходу кристалічна вільна кислота, яка є по суті чистою, містить, щонайменше, приблизно 96 % кристалічної вільної кислоти за вагою, наприклад, щонайменше, приблизно 96,1 %, 96,2 %, 96,3 %, 96,4 %, 96,5 %, 96,6 %, 96,7 %, 96,8 %, 96,9 %, 97 %, 97,1 %, 97,2 %, 97,3 %, 97,4 %, 97,5 %, 97,6 %, 97,7 %, 97,8 %, 97,9 %, 98 %, 98,1 %, 98,2 %, 98,3 %, 98,4 %, 98,5 %, 98,6 %, 98,7 %, 98,8 %, 98,9 %, 99 %, 99.1 %, 99,2 %, 99,3 %, 99,4 %, 99,5 %, 99,6 %, 99,7 %, 99,8 %, 99,9 % або 100 % кристалічної вільної кислоти за вагою. У деяких варіантах здійснення винаходу ди- або мононатрійова сіль у фармацевтичних сполуках, описаних тут, містить, щонайменше, 96 %, 96,1 %, 96,2 %, 96,3 %, 96,4 %, 96,5 %, 96,6 %, 96,7 %, 96,8 %, 96,9 %, 97 %, 97,1 %, 97,2 %, 97,3 %, 97,4 %, 97,5 %, 97,6 %, 97,7 %, 97,8 %, 97,9 %, 98 %, 98,1 %, 98,2 %, 98,3 %, 98,4 %, 98,5 %, 98,6 %, 98,7 %, 98,8 %, 98,9 %, 99 %, 99,1 %, 99,2 %, 99,3 %, 99,4 %, 99,5 %, 99,6 %, 99,7 %, 99,8 %, 99,9 % або 100 % кристалічної солі за вагою. В отриманих лікарських засобах корисним є забезпечення нев'язкого твердого кристалічного (R)-3-(4-(2-(2-метилтетразол- 5-іл) піридин-5-іл)- 3фторфеніл)- 5-гідроксиметил оксазолідин-2-он диводень фосфату, який може бути перенесений і охайно зважений для застосування, наприклад, у таблетках і капсулах. Таким чином, у деяких варіантах здійснення винаходу кристалічний матеріал перебуває в придатній до переносіння 4 UA 114068 C2 5 10 15 20 25 30 35 40 45 50 55 60 формі, так, що частинки не щільно прилипають одна до одної або до посудини, у якій вони містяться так, що можливо однорідно перенести матеріал з посудини. [0048] Одержання вільної кислоти (R)-3-(4-(2-(2-метилтетразол- 5-іл) піридин-5-іл)- 3фторфеніл)- 5-гідроксиметил оксазолідин-2-он диводень фосфату 1 (R=PO(OH)2) і його динатрійової солі 1 (R=PO(ONa)2) описане в патентній публікації № 2007/0155798 і патентній заявці США №12/577,089, права на останню із зазначених заявок передані тому ж правовласникові, що зазначений у цій заявці. [0049] У варіантах здійснення винаходу в деяких способах одержання кристалічна вільна кислота (R)-3-(4-(2-(2-метилтетразол-5-іл) піридин-5-іл)- 3-фторфеніл)- 5-гідроксиметил оксазолідин-2-он диводень фосфату 1 (R=PO(OH) 2) може бути отримана шляхом підкислення водяного розчину відповідної солі, наприклад, динатрійової солі 1 (R=PO(ONa) 2). [0050] Для відновлення вільної кислоти підкисленням може бути використана будь-яка сіль вільної кислоти 1 (R=PO(OH)2). У деяких варіантах здійснення винаходу сіль являє собою сіль лужного або лужноземельного металу. В інших варіантах здійснення винаходу сіль являє собою сіль лужного металу, такого як динатрійова сіль 1 (R=PO(OH) 2). [0051] У додаткових варіантах здійснення винаходу вільна кислота як така може бути використана для одержання кристалічних частинок наприклад, шляхом розчинення у диполярному апротонному розчиннику: диметил сульфоксиді (DMSO) або 2-піролідоні (NMP); а потім додавання розчинника, що викликає кристалізацію, такого як етанол, ацетон, ацетонітрил, діоксан, гептан, ізопропиловий спирт, метанол, тетрагідрофуран, толуол, вода, дихлорметан, метил ізобутил кетон і етилацетат. У деяких варіантах здійснення винаходу розчинники, що застосовуються для розчинення або для кристалізації, можуть являти собою чистий розчинник або суміш чистих розчинників або можуть бути у формі рідини, пари або другого шару. У деяких варіантах здійснення винаходу з останніх двох випадків розчинник, що викликає кристалізацію, може бути використаний згідно зі способом вирощування кристалів дифузією з парової фази, або способом розшарування розчинників, обидва з яких добре відомі з рівня техніки. [0052] У додаткових способах одержання вільна кислота може бути розчинена, щонайменше, в одному диполярному апротонному розчиннику, такому як DMSO або NMP при підвищеній температурі, і кристалічна вільна кислота 1 (R=PO(OH) 2) одержується шляхом охолодження розчину згідно з добре відомими способами з рівня техніки. Розчинник може бути чистим сам по собі або являти собою суміш чистих розчинників. [0053] При одержанні лікарських засобів корисно забезпечити тверду кристалічну сполуку, яка може бути легко переведена в дозовані форми, наприклад, таблетки. Також бажано скоротити термін, необхідний для одержання сполуки. У деяких варіантах здійснення винаходу для того, щоб задовольнити ці потреби, описаний спосіб одержання кристалічної сполуки 1 (R=PO(OH)2), який призводить до одержання великого розміру частинок, що значно скорочує термін фільтрування, оскільки, як відомо, дрібні частинки сповільнюють процес фільтрування. В інших варіантах здійснення винаходу кристалічна речовина 1 (R=PO(OH) 2) має визначений розподіл частинок за розмірами, що прямо випливає зі способу одержання незалежно від фільтрування матеріалу для одержання такого розподілу. [0054] У деяких варіантах здійснення винаходу більший розмір частинок кристалів сполуки 1 (R=PO(OH)2) може бути отриманий шляхом використання процедури осадження при високій температурі. Додатково, у варіантах, де для одержання вільної кислоти із солі використовується кислота, було виявлено, що збільшення швидкості, при якій реакційну суміш додають до кислоти, впливає на розмір частинок і робить частинки більшими. При цьому реакційна суміш може взаємодіяти з розчином кислоти так швидко, як це можливо, так, що по суті відбувається негайний контакт із розчином кислоти. У звичайних способах реакційна суміш взаємодіє з розчином кислоти повільніше, тому що розчин кислоти додають до реакційної суміші й у такий спосіб реакційна суміш не може взаємодіяти з розчином кислоти деякий час після додавання розчину кислоти, що призводить до значно меншого розміру частинок. Було виявлено, що дія зворотна до цієї стадії, тобто додавання реакційної суміші до розчину кислоти дозволяє реакційній суміші практично негайно взаємодіяти протягом терміну введення реакційної суміші в розчин кислоти, що призводить до одержання матеріалу з більшим розміром частинок. У деяких варіантах здійснення винаходу негайний контакт здійснюють додаванням реакційної суміші до розчину кислоти. Реакційна суміш може бути прокачана через розчин кислоти протягом терміну, наприклад, протягом кількох годин, наприклад 1-4 години. [0055] У деяких варіантах здійснення винаходу водний етанол- або розчин TR-701FA, що містить ТГФ, може бути отриманий додаванням розчину бікарбонату натрію, наприклад, 2-10 % розчину за вагою, такого як 5 % розчин. У деяких варіантах здійснення винаходу розчин може бути доданий до водяного розчину кислоти й етанолу або ТГФ із утворенням вільної кислоти. У 5 UA 114068 C2 5 10 15 20 25 30 35 40 45 50 55 деяких варіантах здійснення винаходу може бути використано приблизно від 0,5-10, приблизно 1,5-3,0 або приблизно 2,2 еквівалента 1 M HСl. Додатково в деяких варіантах здійснення винаходу можуть бути використані приблизно 1-10 об'ємів, 2-6 об'ємів або 4 об'єми етанола. Також може бути використаний ТГФ. У деяких варіантах здійснення винаходу розчин, що містить соляну кислоту й етанол, може підтримуватися при температурі приблизно 40-100° C, приблизно 60-70 °C або приблизно 65-70 °C. Вміст кислоти й спирту можуть бути приведені у відповідність. Під час цього додавання TR-701FA закристалізувався з одержанням меншої кількості невеликих кристалів у продукті в порівнянні з раніше розкритими способами. [0056] У деяких варіантах здійснення винаходу етанол або ТГФ перешкоджають гелеутворенню вільної кислоти під час процесу. [0057] Звичайний розподіл частинок за розмірами вимірюється аналізатором лазерної дифракції розміру частинок, а саме Malvern Mastersizer. D10 (мкм) являє собою діаметр, нижче якого лежить 10 % загального об'єму частинок. D50 (мкм) являє собою медіанний діаметр. D90 (мкм) являє собою діаметр, нижче якого лежить 90 % загального об'єму частинок. [0058] У деяких варіантах здійснення винаходу, коли розмір частинок не контролюється, 10 % загального об'єму частинок може мати діаметр менше приблизно 0,28 мкм, медіанний діаметр може бути приблизно 0,79 мкм, і 90 % загального об'єму частинок може мати діаметр менше приблизно 0,44 мкм. Контроль (за збільшенням) розміру частинок з використанням описаних тут способів призводить до того, що частинки стають значно більше за розміром в цілому. [0059] У деяких варіантах здійснення винаходу, коли розмір частинок контролюється з використанням описаних тут способів для збільшення розміру частинок, 10 % загального об'єму частинок може мати середній діаметр, щонайменше, приблизно 0,5 мкм, і/або медіанний діаметр може бути, щонайменше, приблизно 1,0 мкм, і/або 90 % загального об'єму частинок можуть мати середній діаметр, щонайменше, приблизно 45 мкм. У деяких варіантах здійснення винаходу, коли розмір частинок контролюється (для збільшення розміру частинок), 10 % загального об'єму може мати середній діаметр приблизно 0,5-10 мкм, наприклад, приблизно 1-5 мкм. Наприклад, коли розмір частинок контролюється (для збільшення розміру частинок), 10 % загального об'єму може мати середній діаметр приблизно 0,5, 0,6, 0,7, 0,8, 0,9, 1,0, 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2,0, 2,1, 2,2, 2,3, 2,4, 2,5, 2,6, 2,7, 2,8, 2,9, 3,0, 3,1, 3,2, 3,3, 3,4, 3,5, 3,6, 3,7, 3,8, 3,9, 4,0, 4,1, 4,2, 4,3, 4,4, 4,5, 4,6, 4,7, 4,8, 4,9, 5,0, 5,1, 5,2, 5,3, 5,4, 5,5, 5,6, 5,7, 5,8, 5,9, 6,0, 6,1, 6,2, 6,3, 6,4, 6,5, 6,6, 6,7, 6,8, 6,9, 7,0, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8, 7,9, 8,0, 8,1, 8,2, 8,3, 8,4, 8,5, 8,6, 8,7, 8,8, 8,9, 9,0, 9,1, 9,2, 9,3, 9,4, 9,5, 9,6, 9,7, 9,8, 9,9 або 10,0 мкм. [0060] У деяких варіантах здійснення винаходу, коли розмір частинок контролюється (для збільшення розміру частинок), медіанний діаметр може бути більше, ніж приблизно 1,0 мкм і мати середній медіанний діаметр приблизно 1-44 мкм, приблизно 1-40 мкм, приблизно 10-35 мкм, приблизно 20-30 мкм або приблизно 25-29, наприклад, приблизно 27 мкм. У деяких варіантах здійснення винаходу, коли розмір частинок контролюється для збільшення розміру частинок, середній медіанний діаметр може приблизно дорівнювати 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 38, 39, 40, 41, 42, 43 або 44 мкм. Наприклад, середній медіанний діаметр може приблизно дорівнювати 25, 25,1, 25,2, 25,3, 25,4, 25,5, 25,6, 25,7, 25,8, 25,9, 26, 26,1, 26,2, 26,3, 26,4, 26,5, 26,6, 26,7, 26,8, 26,9, 27, 27,1, 27,2, 27,3, 27,4, 27,5, 27,6, 27,7, 27,8, 27,9, 28, 28,1, 28,2, 28,3, 28,4, 28,5, 28,6, 28,7, 28,8, 28,9 або 29 мкм. [0061] У деяких варіантах здійснення винаходу, коли розмір частинок контролюється, 90 % загального об'єму частинок може мати середній діаметр, щонайменше, приблизно 45 мкм, такий як приблизно 45-100, приблизно 45-80, приблизно 55-75 або приблизно 64-68, наприклад, приблизно 66. У деяких варіантах здійснення винаходу, коли розмір частинок контролюється, 90 % загального об'єму частинок може мати середній діаметр приблизно 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 або 100 мкм. Наприклад, 90 % загального об'єму частинок може мати середній діаметр приблизно 64, 64,1, 64,2, 64,3, 64,4, 64,5, 64,6, 64,7, 64,8, 64,9, 65, 65,1, 65,2, 65,3, 65,4, 65,5, 65,6, 65,7, 65,8, 65,9, 66, 66,1, 66,2, 66,3, 66,4, 66,5, 66,6, 66,7, 66,8, 66,9, 67, 67,1, 67,2, 67,3, 67,4, 67,5, 67,6, 67,7, 67,8, 67,9 або 68 мкм. [0062] Кристалічна вільна кислота 1 (R=PO(OH) 2) може бути охарактеризована Фур'є-Раман спектром, як показано на Фігурі 1 і рентгенограмою, як показано на Фігурі 2, з відповідними даними, представленими в Таблиці 1 і 2 відповідно. На Фігурі 3, Фігурі 4, Фігурі 5 і Фігурі 6 1 показані термограма диференціальної скануючої калориметрії (DSC), H NMR спектр, діаграма, 6 UA 114068 C2 отримана методами термогравіметрії та інфрачервоної спектроскопії, і діаграма, що демонструє властивості динамічної сорбції парів (DVS) відповідно. Таблиця 1 Фур'є-Раман спектроскопічні дані для кристалічної вільної кислоти 1 (R=PO(OH)2) Довжина хвилі (см-1) 1612,9 1579,0 1521,3 1455,8 1404,9 1324,4 1274,7 1149,9 1018,3 Абсолютна інтенсивність 2,57 0,38 0,25 0,26 0,39 0,82 0,24 0,17 0,22 Таблиця 2 Дані рентгенограми для кристалічної вільної кислоти 1 (R=PO(OH)2) Кут розсіювання/ 10,6 13,7 13,9 14,7 15,2 15,4 15,7 16,6 17,1 19,2 20,3 21,4 22,4 23,2 23,6 24,7 25,3 25,9 26,8 28,2 28,4 29,0 30,3 30,8 31,0 31,9 33,5 34,7 о Інтенсивність / % 17 6 8 38 28 28 16 65 10 19 100 25 23 8 7 29 14 8 82 44 24 8 8 11 13 8 17 7 5 [0063] У деяких варіантах здійснення винаходу відмітні піки для кристалічної вільної кислоти включають наступні піки: 14,7°, 15,2°, 16,6°, 20,3°, 26,8° і 28,2°. [0064] В інших варіантах здійснення винаходу відмітні піки для кристалічної вільної кислоти включають наступні піки: 10,6°, 13,9°, 14,7°, 15,2°, 16,6°, 20,3°, 26,8° і 28,2°. 7 UA 114068 C2 [0065] У деяких варіантах здійснення винаходу кристалічна вільна кислота містить домішки, які присутні в кількості менше 1 % очищеної кристалічної вільної кислоти. Ці домішки включають O N N O N N N OH N F 5 тобто 5(R)-3-{3-фтор-4-[6-(2-метил-2H-1,2,3,4-тетразол-5-іл)-піридин-3-іл]-феніл}-5гідроксиметил-1,3-оксазолідин-2-он ("TR-700") і/або O N N O N N N Cl N F 10 15 20 25 30 35 тобто (R)-5-(хлорметил)-3-(3-фтор-4-(6-(2-метил-2Н-тетразол-5-іл)піридин-3-іл)феніл) оксазолідин-2-он ("домішка, що містить хлор"). [0066] Із звичайно одержуваного матеріалу, що має домішки, які були ідентифіковані з використанням ВЕРХ у Прикладі 15, наявно щонайменше, 2 % за вагою домішки, що містить хлор. В очищеній кристалічній вільній кислоті, одержаної з використанням способу одержання вільної кислоти, описаної в патентній заявці США 12/577,089, права на яку були передані тому ж правовласникові, що й за цією заявкою, і способів кристалізації, описаних тут, домішка, що містить хлор, наявна в кількості приблизно меншее 1 % за вагою, наприклад, меншее приблизно 0,9 %, 0,8 %, 0,7 %, 0,6 %, 0,5 %, 0,4 %, 0,3 %, 0,2 % або 0,1 % за вагою кристалічної вільної кислоти. У деяких варіантах здійснення винаходу кількість домішки, що містить хлор, може бути знижена до 0,1 % за вагою, наприклад, менше ніж приблизно 0,09 %, 0,08 %, 0,07 %, 0,06 %, 0,05 %, 0,04 %, 0,03 %, 0,02 % або 0,01 % за вагою кристалічної вільної кислоти. У деяких варіантах здійснення винаходу очищена кристалічна вільна кислота взагалі не містить цієї домішки. [0067] Зі звичайно одержуваного матеріалу, що має домішки, які були ідентифіковані з використанням ВЕРХ у Прикладі 15, наявно щонайменше, 1 % за вагою TR-700 домішки. В очищеній кристалічній вільній кислоті, отриманої з використанням способу одержання вільної кислоти, описаного у патентній заявці США 12/577,089, права на яку були передані тому ж правовласникові, що й за цією заявкою, і способів кристалізації, описаних тут, TR-700 домішка наявна в кількості приблизно меншее 1 % за вагою. У деяких варіантах здійснення винаходу кристалічна вільна кислота містить менше 0,9 %, 0,8 %, 0,7 %, 0,6 %, 0,5 %, 0,4 %, 0,3 %, 0,2 % або 0,1 % за вагою TR-700 домішки. У деяких варіантах здійснення винаходу очищена кристалічна вільна кислота взагалі не містить TR-700 домішки. [0068] Додатково, очищена кристалічна вільна кислота, отримана з використанням способу одержання вільної кислоти, описаного у патентній заявці США 12/577,089, права на яку були передані тому ж правовласникові, що й за цією заявкою, і способів кристалізації, описаних тут, може також відрізнятися від кристалічної вільної кислоти, отриманої звичайним способом, наявністю наступних сполук. Наприклад, наступні домішки не були виявлені в зразку кристалічної вільної кислоти, отриманої звичайним способом, як показано в Прикладі 15: O N N O N HN N P F 40 O O N HO OH (тут і далі " дез-метил TR-701"), тобто диводень ((5R)-3-{3-фтор-4-[6-(2H-1,2,3,4-тетразол- 5іл)- 3-піридиніл]феніл}-2- оксо-1,3-оксозолан- 5-іл)метил фосфат; 8 UA 114068 C2 O O N N O O N N N OH P O N OH OH F O N OH N O OH O N N N O N P OH O F 5 (тут і далі “ надалкільована-фосфорильована домішка"), тобто, 5-((3-(3- фтор-4-(6-(2- метил2Н-тетразол-5-іл)піридин-3-іл)феніл)-2-оксаоксазолідин-5-іл)метокси)-3-гідроксипропан-2-іл диводень фосфат і, 3-((3-(3-фтор-4-(6-(2-метил-2Н-тетразол-5-іл)піридин-3-іл)феніл)-2оксаоксазолідин-5-іл)метокси)-2- гідроксипропіл диводень фосфат; O N N O OH N N N O N F O O O P O OH O N F N N N N N 10 (тут і далі "один з OA-700 змішаних діефірів"), тобто, 3-{[(5R)-3-{3-фтор-4-[6-(2- метил-2Нтетразол-5-іл)піридин-3-іл]феніл}-2-оксо-1,3-оксазолідин-5-іл]метокси}-2-гідроксипропіл [(5R)-3{3-фтор-4-[6-(2-метил-2Н-тетразол-5-іл)піридин-3-іл]феніл}-2-оксо-1,3-оксазолідин-5-іл]метил водень фосфат; і/або 9 UA 114068 C2 N N N N N O F HO N O O HO O P O O F N N O N N 5 10 15 20 25 30 35 40 O N N (тут і далі "інший з OA-700 змішаних діефірів"), тобто 2-{[(5R)-3-{3-фтор-4-[6-(2- метил-2Нтетразол-5-іл) піридин-3-іл]феніл}-2-оксо-1,3-оксазолідин-5-іл]метоксі}-1-гідроксіетил [(5R)-3-{3фтор-4-[6-(2-метил-2Н-тетразол-5-іл)піридин-3-іл]феніл}-2-оксо-1,3-оксазолідин-5-іл]метил водень фосфат. [0069] Фахівці в даній галузі визначать, що також можуть бути легко отримані різні заміщені 13 15 ізотопами варіанти (наприклад, через заміщення водню дейтерієм, вуглецю - C, азоту - N, 32 або фосфору - P). Усі такі варіанти включені в обсяг домагань за винаходом. [0070] У різних варіантах здійснення винаходу очищена кристалічна вільна кислота, описана тут, може бути використана окремо, у комбінації з іншими описаними тут сполуками або в комбінації з одним або більше іншими агентами, активними в терапевтичній галузі, описаній тут. [0071] В іншому аспекті цей опис стосується фармацевтичної композиції, що містить один або кілька фізіологічно прийнятних поверхнево-активних агентів, додаткових носіїв, дилюєнтів, ексципієнтів, розгладжуючих компонентів, суспензійних агентів, агентів, що утворюють плівку й допоміжних речовин, для утворення оболонки або їх комбінації й композиції, описані тут. Додаткові прийнятні носії або дилюєнти для терапевтичного використання добре відомі у фармацевтичній галузі і описані, наприклад, в Remington's Pharmaceutical Sciences, 18th Ed., Mack Publishing Co., Easton, PA (1990), який наводиться тут для посилання у всій його цілісності. У фармацевтичній композиції можуть бути представлені консерванти, стабілізатори, барвники, підсолоджувачі, віддушки, ароматизатори й таке інше. Наприклад, в якості консервантів можуть бути додані бензоат натрію, аскорбінова кислота й ефіри п-гідроксибензойної кислоти. Додатково можуть бути використані антіоксиданти й суспендуючі агенти. У різних варіантах здійснення винаходу спирти, ефіри, сульфурировані аліфатичні спирти й таке інше можуть бути використані в якості поверхнево-активних агентів; сахароза, глюкоза, лактоза, крохмаль, мікрокристалічна целюлоза, кристалічна целюлоза, манітол, світлий безводний силікат, алюмінат магнію, метасилікат алюмінат магнію, синтетичний силікат алюмінію, карбонат кальцію, гідрокарбнат натрію, гідрофосфат кальцію, кальцій карбоксиметил целюлоза, і таке інше можуть бути використані в якості екципієнтів; стеарат магнію, тальк, загартоване масло й таке інше можуть бути використані в якості розгладжуючих агентів; кокосове масло, маслинове масло, кунжутне масло, арахісове масло, соєве масло можуть бути використані в якості суспензійних агентів або лубрикантів; фталат ацетату целюлози у вигляді похідного вуглеводу, такого як целюлоза або сахароза або метилацетат-метакрилат співполімер у вигляді похідного полівінілу може бути використаний у якості суспензійного агента; і пластифікатори, такі як ефір фталати й таке інше можуть бути в якості суспензійних агентів. [0072] Термін "фармацевтична композиція" відноситься до суміші сполуки, описаної тут, з іншими хімічними компонентами, такими як дилюєнти або додаткові носії. Фармацевтична композиція полегшує введення сполуки в організм. В галузі техніки існують численні способи введення фармацевтичної композиції, включаючи, без обмеження, пероральний, ін'єкційний, аерозольний, парентеральний і локальний. Фармацевтичні композиції можуть також бути отримані шляхом взаємодії вільної кислоти з неорганічною або органічною основою, такою як гідроксид натрію або гідроксид магнію. У деяких варіантах здійснення винаходу представлені фармацевтично прийнятні солі описаних тут сполук (наприклад, отриманих in situ під час 10 UA 114068 C2 5 10 15 20 25 30 35 40 45 50 55 60 виготовлення внутрішньовенної фармацевтичної форми). У деяких варіантах здійснення винаходу гідроксид натрію використовується для одержання ліофілізованого порошку, який містить сіль вільної кислоти, одержуваної in situ. [0073] Термін "носій" відноситься до хімічної сполуки, яка полегшує введення сполуки в клітини або тканини. [0074] Термін "дилюєнт" відноситься до хімічних сполук, розведених водою, які розчиняють цільову композицію, а також стабілізують біологічно активну форму сполуки. Солі, розчинені в буферних розчинах, використовуються в якості дилюєнтів у рівні техніки. Одним із широко використовуваних буферних розчинів є сольовий розчин фосфатного буфера, тому що він імітує склад солей у людській крові. Оскільки буферні солі можуть контролювати pН розчину при низькій концентрації, буферний дилюєнт рідко змінює біологічну активність сполуки. Як використовується тут, термін "екципієнт" відноситься до інертної речовини, яка надає композиції, без обмеження, об'єм, стійкість, стабільність, зв'язуючу здатність, зволоження, здатність до розпаду. "Дилюєнт" являє собою вид екципієнту. [0075] Термін "фізіологічно прийнятний" відноситься до носія або дилюєнту, який не змінює біологічну активність або властивості сполуки. [0076] Фармацевтичні сполуки, описані тут, можуть бути введені людині (пацієнтові) самостійно або в складі фармацевтичної композиції, де вони змішуються з іншими активними інгредієнтами, у якості комбінованої терапії або прийнятними носіями або екципієнтом (екципієнтами). Техніки виготовлення й введення сполук за цією заявкою можуть бути виявлені в "Remington's Pharmaceutical Sciences, ” Mack Publishing Co., Easton, PA, 18th edition, 1990. [0077] Прийнятні способи введення можуть включати, наприклад, пероральний, ректальний, введення через слизову оболонку, локальний або інтестинальний спосіб; парентеральну доставку, включаючи внутрішнм'язову, внутрішньовенну, інтрамедулярну ін'єкцію, також інтратекальну, пряму інтравентрикулярну, інтраперитонеальну, інтраназальну або внутрішньоочну ін'єкцію. Сполука може бути введена у вигляді дозованої форми із тривалим або контрольованим вивільненням, включаючи, ін'єкцію речовин уповільненого всмоктування, осмотичні насоси, таблетки, трансдермальні (включаючи електротранспорт) ділянки й таке інше, для пролонгованого й/або тимчасового, імпульсного введення із заздалегідь визначеною швидкістю. [0078] Фармацевтичні композиції за даним винаходом можуть бути виготовлені відомим способом, наприклад за допомогою звичайного перемішування, розчинення, гранулювання, виготовлення драже, розтирання на порошок, емульсифікації, капсулювання або таблетирування. [0079] Фармацевтичні композиції можуть бути отримані звичайним способом з використанням одного або кількох фізіологічно прийнятних носіїв, що включають екципієнти й допоміжні речовини, які полегшують підготовку активних сполук для включення у форму, яка може бути використана фармацевтично. Сполука залежить від обраного способу введення. Кожна з добре відомих технік, дилюєнтів, носіїв і ексципієнтів можуть бути використані в якості прийнятних і зрозумілих, наприклад, зазначені в Remington's Pharmaceutical Sciences, дивитися вище. [0080] Речовини для ін'єкцій можуть бути отримані у звичайних формах або у вигляді рідких розчинів або суспензій, твердих формах, що прийнятні для розчинення або суспендування в рідині до ін'єкції або у вигляді емульсій. Прийнятні ексципієнти являють собою, наприклад, воду, сольовий розчин, декстрозу, манітол, лактозу, лецитин, альбумін, глютамат натрію, гідрохлорид цистеїну й таке інше. Додатково, якщо бажано, фармацевтичні композиції для ін'єкцій можуть містити невелику кількість нетоксичних допоміжних речовин, таких як зволожуючі агенти, рН буферні агенти й таке інше. Фізіологічно сумісні буфери включають, без обмеження, розчин Хенкса (Hanks), розчин Ринджера (Ringer) або фізіологічного сольового розчину. Якщо бажано, може бути використана абсорбція, що поліпшує виготовлення. [0081] Для введення через слизову оболонку у формах можуть бути використані агенти, необхідні для проникнення через бар'єр. [0082] Фармацевтичні форми для парентерального введення, наприклад, за допомогою болюсної ін'єкції або безперервним уливанням, включають водяні розчини активних сполук у водорозчинній формі. Додатково, суспензії активних сполук можуть бути отримані у вигляді прийнятних масляних ін'єкційних суспензій. Водні ін'єкційні суспензії можуть містити речовини, які збільшують в'язкість суспензії, такі як натрій карбоксиметилцелюлоза, сорбітол або декстран. Необов'язково суспензія може також містити прийнятні стабілізатори або агенти, які збільшують розчинність сполук, що дозволяють одержати висококонцентровані розчини. Сполуки для ін'єкцій можуть бути представлені в одиничній дозованій формі, наприклад, в 11 UA 114068 C2 5 10 15 20 25 30 35 40 45 50 55 60 ампулах або контейнерах, що містять кілька доз, з додаванням консерванту. Композиції можуть містити такі форми у вигляді суспензій, розчинів або емульсій у масляному або водному наповнювачі, і можуть містити агенти для одержання сполуки, наприклад, суспендуючі, стабілізуючі і/або диспергуючі агенти. Альтернативно, перед застосуванням активний інгредієнт може мати вигляд порошку для одержання лікарської форми з прийнятним наповнювачем, наприклад, стерильною апірогенною водою. [0083] Для перорального введення композиція може бути складена комбінуванням композиції, що складає інтерес, і фармацевтично прийнятних носіїв, добре відомих з рівня техніки. Такі носії можуть бути використані додатково до катіонного полімерного носія, що дає можливість приготувати композиції за винаходом у вигляді таблеток, пігулок, драже, рідин, гелів, сиропів, суспензій і таке інше для перорального застосування пацієнтами, що потребують лікування. Фармацевтичні форми для перорального використання можуть бути отримані комбінуванням активних сполук із твердим екципієнтом, необов'язковим подрібненням отриманої суміші, і обробкою суміші гранул після додавання прийнятних допоміжних речовин, якщо бажано, одержання таблеток або ядерець драже. Прийнятні ексципієнти являють собою, зокрема, наповнювачі, такі як цукор, включаючи лактозу, сахарозу, манітол або сорбітол; целюлозні форми, такі як кукурудзяний крохмаль, пшеничний крохмаль, рисовий крохмаль, картопляний крохмаль, желатин, трагакантова камедь, метилцелюлоза, гідроксипропілметилцелюлоза, натрій карбоксиметилцелюлоза й/або полівінілпіролідон (PVP), наприклад, Povidone. Якщо бажано, можуть бути додані дезінтегруючі агенти, такі як крос-зшитий полівінілпіролідон (наприклад, Crospovidone), агар або альгінова кислота або її сіль, така як альгінат натрію. Ядерця драже покриваються прийнятною оболонкою. Для цієї мети можуть бути використані концентровані розчини цукрів, які необов'язково містять гуміарабік, тальк, полівінілпіролідон, карбополь гель, поліетиленгліколь, і/або діоксид титану, лакові розчини й прийнятні органічні розчинники або суміші розчинників. Барвники або пігменти можуть бути додані до оболонки таблеток або драже для ідентифікації або для характеристики різних комбінацій доз активних сполук. З цією метою можуть бути використані концентровані розчини цукрів, які можуть необов'язково містити гуміарабік, тальк, полівінілпіролідон, карбополь гель, поліетиленгліколь, і/або диоксид титану, лакові розчини й прийнятні органічні розчинники або суміші розчинників. Барвники або пігменти можуть бути додані до оболонки таблеток або драже для ідентифікації або для характеристики різних комбінацій доз активних сполук. [0084] Фармацевтичні форми, які можуть бути використані перорально, включають push-fit капсули (тверді капсули із двох частин), вироблені з желатину, а також м'які, герметичні капсули з желатину й пластифікатору, такого як гліцерол або сорбітол. Тверді капсули можуть містити активні інгредієнти в суміші з наповнювачем, таким як лактоза, зв'язувальною речовиною, таким як крохмалі й/або лубриканти, такі як тальк або стеарат магнію й необов'язково стабілізатори. У м'яких капсулах активні сполуки можуть бути розчинені або суспендовані в прийнятних рідинах, таких як жирні масла, рідкі парафіни або рідкі поліетиленгліколі. Додатково можуть бути додані стабілізатори. Усі сполуки для перорального введення повинні бути в дозуванні, прийнятному для такого введення. [0085] Для буккального введення композиції можуть бути виконані звичайним способом у формі таблеток або пастилок. [0086] Для введення інгаляцією композиція може бути звичайним чином доставлена у формі аерозолю з балончика під тиском або розпилювача, з використанням прийнятного пропеленту, наприклад, дихлордифторметану, трихлорфторметану, дихлортетрафторетану, вуглекислого газу або іншого прийнятного газу. У випадку аерозолю під тиском дозована одиниця може бути визначена присутністю клапана для доставки фіксованої кількості. Капсули й картриджі, наприклад, з желатину для використання в інгаляторі або інсуфляторі можуть містити порошкову суміш сполуки й прийнятної основи, наприклад, лактозу або крохмаль. [0087] Також описувані тут різні фармацевтичні композиції включають добре відомі у фармацевтичній галузі інтраокулярну, інтраназальну й інтрааурикулярну доставку. Прийнятні проникаючі агенти для цього застосування добре відомі в рівні техніки. Такі прийнятні фармацевтичні форми найчастіше й переважніше складають таким чином, щоб вони були стерильні, ізотонічні й буферні для стабільності й комфорту. Фармацевтичні композиції для інтраназальної доставки можуть також включати краплі й спреї, часто підготовлені для імітації назальної секреції для того, щоб гарантувати підтримку нормальної циліарної дії. Як описано в Remington's Pharmaceutical Sciences, 18th Ed., Mack Publishing Co., Easton, PA (1990), який наводиться тут для посилання у всій його цілісності й добре відомий фахівцям у даній галузі, прийнятні фармацевтичні форми є найчастіше й переважніше за все ізотонічними, буферними для збереження pН 5,5-6,5, і найчастіше й переважно містять антимікробні консерванти й 12 UA 114068 C2 5 10 15 20 25 30 35 40 45 50 55 60 відповідні стабілізатори лікарських засобів. Фармацевтичні форми для інтрааурикулярної доставки включають суспензії й мазі для місцевого застосування у вусі. Найпоширенішими розчинниками для таких вушних форм є гліцерин і вода. [0088] Композиції можуть також являти собою ректальні композиції, такі як супозиторії або утримуюча клізма, наприклад, що містить звичайну основу для супозиторію, таку як масло какао або інші гліцериди. [0089] Додатково до попередньо описаних форм композиції можуть бути також складені у вигляді форм із речовинами уповільненого всмоктування. Такі тривало діючі форми можуть бути введені імплантацією (наприклад, підшкірно або внутрішньом'язово) або внутрішнм'язовою ін'єкцією. При цьому, наприклад, сполуки можуть бути змішані з прийнятними полімерними або гідрофобними матеріалами (наприклад, у вигляді емульсії в прийнятному маслі) або іон обмінними смолами або важко розчинними похідними, наприклад, у вигляді важкорозчинної солі. [0090] Для гідрофобних сполук прийнятний фармацевтичний носій може являти собою систему співрозчинників, що містить бензиловий спирт, неполярну поверхнево-активну речовину (ПАР), водорозчинний органічний полімер і водну фазу. Розповсюджена система являє собою систему співрозчинників VPD, яка містить розчин 3 % вага/об'єм бензилового спирту, 8 % вага/об'єм неполярного ПАР POLYSORBATE 80™ і 65 % вага/об'єм поліетиленгліколю 300, доведених до об'єму абсолютним етанолом. Природньо пропорції системи співрозчинників можуть значно змінюватися без порушення характеристик їх розчинності й токсичності. Більше того, ідентичність компонентів співрозчинників може змінюватися: наприклад, інші низько токсичні ПАР можуть використовуватися замість POLYSORBATE 80™; кількість поліетиленгліколю може змінюватися; інші біосумісні полімери можуть замінити поліетиленгліколь, наприклад, полівінілпіролідон та інші цукри або полісахариди можуть замінити декстрозу. [0091] Способи лікування бактеріальних інфекцій можуть включати введення терапевтично ефективної кількості описаної тут сполуки. Лікування бактеріальної інфекці може також включати профілактичне введення терапевтичних сполук для запобігання інфекції або поширення інфекці у пацієнта при неминучому ризику, наприклад, у пацієнта, який щойно переніс хірургічне втручання або у пацієнта з послабленим імунітетом або пацієнта в зоні ризику, якщо сполука не була введена. Сполуки демонструють інгібіторну активність проти широкого спектру бактерій, проти метицилін резистентної Staphylococcus aureus (MRSA) і ванкоміцин резистентної Enterococci (VRE) і мають відносну антибіотичну активність із відносно низькою концентрацією або in vivo. Також сполуки за даним винаходом можуть проявляти потенційно антибактеріальну активність проти різних людських і тваринних патогенів, включаючи Грам-позитивні бактерії, такі як Staphylococi, Enterococci і Streptococi, анаеробні мікроорганізми, такі як бактеріоди й Clostridia, і кислотостійкі мікроорганізми, такі як Mycobacterium tuberculosis і Mycobacterium avium. В одному варіанті здійснення винаходу бактеріальна інфекція, яка може бути піддана лікуванню або стан пацієнта із цією інфекцією може бути поліпшений, являє собою MRSA. [0092] Композиції або фармацевтичні композиції, описані тут, можуть бути введені пацієнтові без яких-небудь прийнятних засобів. Приклади, що не обмежують обсяг домагань, включають серед іншого (а) уведення через пероральні способи, коли вводиться капсула, таблетка, гранула, спрей, сироп або інші такі форми; (b) уведення не пероральними способами, такі як ректальний, вагінальний, інтрауретральний, інтраокулярний, інтраназальний або інтрааурикулярний, де вводиться водна суспензія або масляна форма або тому подібне або краплі, спрей, супозиторій, бальзам, мазь або тому подібне; (c) уведення через ін'єкцію, підшкірно, інтраперитонеально (внутрішньочеревно), внутрішньовенно, внутрішньом'язово, внутрішньошкірно, інтраорбітально, внутрішньосуглобно, інтраспинально, інтрастернально або тому подібне, включаючи доставку інфузійним насосом; також (d) локальне введення; як зрозуміло фахівцеві в даній галузі для приведення активної сполуки в контакт із живою тканиною. [0093] Фармацевтичні композиції, прийнятні для введення, включають композиції, де активні інгредієнти присутні в кількості ефективній для досягнення заявленого призначення. Терапевтично ефективна кількість сполук, описана тут і необхідна в якості дози, буде залежати від способу введення, виду тварини, включаючи людину, яку необхідно піддати лікуванню, й фізичних характеристик конкретної розглянутої тварини. Дозування може бути підібране для досягнення бажаного ефекту, але воно також буде залежати від таких факторів, як вага, дієта, паралельний медичний курс й інші фактори, які фахівець у даній галузі легко виявить. Зокрема, терапевтично ефективна кількість означає кількість сполуки, ефективну для запобігання або 13 UA 114068 C2 5 10 15 20 25 30 35 40 45 50 55 60 полегшення симптомів захворювання або поліпшення стану або продовження життя пацієнта. Визначення терапевтично ефективної кількості перебуває в рамках можливостей фахівця в даній галузі, зокрема у світлі детального опису, представленого тут. [0094] Як очевидно фахівцеві в даній галузі, корисна in vivo доза для введення й конкретний режим уведення будуть змінюватися залежно від віку, ваги й виду ссавців, конкретних використовуваних сполук і конкретного призначення, для якого ці сполуки використовуються. Визначення рівня ефективної дози, необхідної для досягнення бажаного результату, може бути здійснене фахівцем у даній галузі з використанням звичайних фармакологічних способів. Звичайно клінічні випробування на людині починають із низьким рівнем доз, з поступовим збільшенням до досягнення бажаного ефекту. Альтернативно, прийнятні дослідження in vitro можуть бути використані для встановлення корисних доз і способів уведення композицій, що ідентифікуються справжніми методами з використанням встановлених фармакологічних методів. [0095] У тварин (не людини), випробування потенційних продуктів здійснюють із дозами більш високого рівня, з поступовим зниженням доз до рівня, доки бажаний ефект більше не буде досягатися, а протилежний ефект не зникне. Дозування може змінюватися досить широко, залежно від бажаних ефектів і терапевтичного призначення. Звичайно дозування можуть дорівнювати приблизно 10 мікрограм/кг до приблизно 100 мг/кг ваги тіла, переважно 100 мікрограм/кг до приблизно 10 мг/кг ваги тіла. Дозування можуть бути засновані й розраховані виходячи із площі поверхні пацієнта, що зрозуміло фахівцеві в даній галузі. [0096] Точна форма, спосіб уведення й дозування фармацевтичної композиції за даним винаходом можуть бути обрані індивідуальним медичним фахівцем з огляду на стан пацієнта. (Дивитися, наприклад, Fingl і інші, 1975, в "The Pharmacological Basis of Therapeutics", яка наводиться тут для посилання у всій її цілісності, з конкретним відсиланням на Главу 1, сторінку 1). Звичайно діапазон доз композиції, що вводиться пацієнтові, може становити приблизно від 0,5 до 1000 мг/кг ваги тіла пацієнта. Дозування може бути одиничним або являти собою серію із двох або більш прийомів протягом одного або більше днів, як необхідно пацієнтові. У випадку, коли дозування для людини вже встановлено, принаймні, для деякого стану, даний винахід буде використовувати ті ж самі дози або дози, які становлять від 0,1 % до приблизно 500 %, більш переважно від 25 % до 250 % установленої людської дози. Там, де доза для людини не встановлена, оскільки бувають випадки для нових фармацевтичних композицій, прийнятне людське дозування може бути підібране від ED 50 або ID50 значень або інших прийнятних значень, виведених з in vitro або in vivo досліджень, як визначено дослідженнями токсичності й ефективності у тварин. [0097] Необхідно відзначити, що практикуючий лікар знає, як і коли припинити, перервати або змінити лікування через токсичність або дисфункцію органів. Навпаки практикуючий лікар також буде знати, як змінити лікування до більш високого рівня доз, якщо клінічна відповідь не є очікуваною (крім токсичності). Кількість дози, що вводиться, буде змінюватися залежно від серйозності стану хворого й способу введення. Серйозність стану може, наприклад, бути оцінена частково, стандартними прогностичними методами оцінки. Доза й можливо частота доз будуть також змінюватися згідно з віком, вагою тіла й реакції індивідуального пацієнта. Програма, порівнянна з обговорюваною вище, може бути використана у ветеринарії. [0098] Хоча точне дозування буде визначено тільки залежно від конкретного лікарського засобу, у більшості випадків можуть бути сформульовані деякі загальні правила, що стосуються дозування. Режим щоденного приймання для дорослого пацієнта може являти, наприклад, пероральну дозу приблизно від 0,1 мг - 2000 мг кожного активного інгредієнта, переважно від 1 мг до 500 мг, наприклад, від 5 до 200 мг. В інших варіантах здійснення винаходу, внутрішньовенна, підшкірна або внутрішнм'язова доза кожного активного інгредієнта становить приблизно від 0,01 мг - 100 мг, переважно від 0,1 мг до 60 мг, наприклад, від 1 до 40 мг. У випадках уведення фармацевтично прийнятної солі дозування можуть бути розраховані для вільної основи. У деяких варіантах здійснення винаходу композиція вводиться від 1 до 4 раз на день. Альтернативно композиції за винаходом можуть бути введені безперервним внутрішньовенним уливанням, переважно з дозою кожного активного інгредієнта до 1000 мг на день. Як зрозуміло фахівцеві в даній галузі, у деяких ситуаціях необхідно ввести описані тут сполуки у кількостях, які перевищують або навіть значно перевищують зазначений вище кращий діапазон доз для того, щоб ефективно й інтенсивно вилікувати конкретне захворювання або інфекцію. У деяких варіантах здійснення винаходу сполуки будуть уводитися протягом періоду безперервної терапії, наприклад, протягом тижня або більше або місяців або років. [0099] Кількість та інтервал можуть бути підібрані індивідуально для забезпечення рівнів плазми активного фрагменту, які є достатніми для підтримки модулюючих ефектів або 14 UA 114068 C2 5 10 15 20 25 30 35 40 45 50 55 мінімальної ефективної концентрації (MEК). MEК буде змінюватися для кожної сполуки, але може бути оцінена на підставі даних in vitro. Дози, необхідні для досягнення MEК, будуть залежати від індивідуальних характеристик і способу введення. Однак ВЕРХ дослідження або біодослідження можуть бути використані для визначення концентрацій у плазмі. [0100] Діапазон доз може бути також визначений з використанням значення MEК. Композиції повинні бути введені з використанням режиму, який підтримує рівні в плазмі вище MEК протягом 10-90 % часу, переважно між 30-90 % і найбільше переважно 50-90 %. [0101] У випадках місцевого введення або селективного споживання, ефективна локальна концентрація лікарського засобу може не відноситися до концентрації в плазмі. [0102] Кількість уведеної композиції може залежати від пацієнта, від ваги пацієнта, серйозності інфекції, способу введення й призначення запропонованого лікарем. [0103] Композиції, описані тут, можуть бути оцінені на ефективність і токсичність з використанням відомих способів. Наприклад, токсикологія сполуки може бути встановлена визначенням in vitro токсичності стосовно клітинної лінії, наприклад, ссавця або переважно людини. Результати таких досліджень часто прогнозують токсичність у тварин, таких як ссавці або зокрема в людини. Альтернативно, токсичність конкретних сполук може бути визначена шляхом використання відомих способів на тваринній моделі, такій як миші, щури, кролики або мавпи. Ефективність конкретної сполуки може бути встановлена шляхом використання різних визнаних способів, таких як in vitro способи, тваринні моделі або клінічні дослідження на людині. Визнані in vitro моделі існують майже для кожного класу станів. Подібним чином, прийнятні тваринні моделі можуть бути використані для встановлення ефективності хімічних сполук для лікування таких станів. При виборі моделі для визначення ефективності фахівець зможе керуватися станом рівня техніки для вибору відповідної моделі, дози й способу введення, режиму. Звичайно, клінічні випробування на людині можуть бути використані для визначення ефективності сполуки на людині. [0104] Композиції, якщо бажано, можуть бути представлені в упаковці або дозаторі, який може містити одну або більше дозовану форму з активним інгредієнтом. Упаковка може містити, наприклад, металеву або пластикову фольгу, наприклад, блістерну упаковку. Упаковка або дозатор може містити інструкцію із уведення. Упаковка або дозатор може також містити повідомлення, пов'язане з контейнером у формі, затвердженій урядовим органом, що регулюює виробництво, використання або продаж лікарського засобу, це повідомлення підтверджує схвалення форми лікарського засобу для введення людині або його застосування у ветеринарії. Таке повідомлення, наприклад, може бути маркуванням, схваленим Адміністрацією США по нагляду в сфері ліків і харчових продуктів або схваленим продуктом вкладишем. Композиції, що містять сполуку за винаходом, можуть бути також отримані із сумісним фармацевтичним носієм, поміщені у відповідний контейнер, також має бути нанесене маркування для лікування зазначеного стану. А. Приклади 1, Використання інструментів, приладів [0105] Мікроскопія Рамана була здійснена на Renishaw System 1000, зі стабілізованим діодним лазером 385 нм збудженням і ближньою ІК-областю з охолодженням за допомогою ефекту Пелт'є й камерою на приладі із зарядовим зв'язком у якості детектора. Вимірювання були здійснені з 50x або більше робочої відстані на 20x об'єктиві з діапазоном частоти 2000-100 -1, см [0106] Фур'є-Раман спектр був отриманий на Bruker RFS100 спектрометрі з Nd:YAG 1064 нм збудженням, 100 мВ лазерною потужністю й Ge детектором. Було зареєстровано 64 сканограми -1 -1, в діапазоні 25-3500 см , при розділовій здатності 2 см [0107] Bruker D8; Bragg-Brentano, геометрія відбиття; Copper K(alpha) випромінювання, 40 кВ/ 40 мA; змінна дивергенційна щілина; Lynxeye детектор з 3° віконцем; крок, 0,02-°2; час, 37 с. Зразки оберталися (0,5 обороту на секунду) під час вимірювання. [0108] Підготовка зразка: Зразки були підготовлені без спеціальної обробки, крім застосування слабкого тиску для одержання гладкої поверхні. Види зразків власників із силіконового монокристала: a) стандартний тримач для перевірки поліморфів, 0,1 мм глибини, необхідний зразок менше 20 мг; b) 0,5 мм глибина, 12 мм діаметр порожнини, необхідний зразок 40 мг; c) 1,0 мм глибини, 12 мм діаметр порожнини, необхідний зразок 80 мг. Звичайно зразки вимірювалися непокритими. Наявність ковпачків з каптонової плівки або PMMA "купола" завжди вказувалося на дифрактограмі з ідентифікацією зразка. 1. Одержання кристалічної вільної кислоти 1 (R=PO(OH)2) Приклад 1 15 UA 114068 C2 5 10 15 20 25 30 35 40 45 50 55 60 [0109] Для одержання тонкої суспензії, яка після додавання тетрагідрофурана (ТГФ) була перемішана й відфільтрована, був підготовлений Розчин 1 (R=PO(ONa) 2) в H2O, і була додана 1 M HСl. Отриманий кристалічний твердий осад 1 (R=PO(OH) 2) був висушений у вакуумі й охарактеризований Фур'є-Раман спектром (FTR) (Фігура 1), рентгенограмою (XRPD, Malvern Mastersizer) (Фігура 2), діаграмою, отриманою методами термогравіметрії та інфрачервоної спектроскопії (TG-FTIR) і термограмою диференціальної скануючої калориметрії (DSC). Вимірювання DSC показало температуру плавлення в 256,9 °C, потім почалося розкладання зразка (Фігура 3). Приклад 2 [0110] До 1 (R=PO(ONa)2) (2 г), розчиненого в 10 мл H2O, була повільно додана HCI (6 мл; 1 M) з одержанням тонкої суспензії ясно-жовтої твердої речовини. Після додавання додаткової кількості 5 мл H2O і 20 мл ТГФ, суспензія була відфільтрована й висушена під вакуумом. Приклад 3 [0111] До 1 (R=PO(ONa)2) (2 г), розчиненого в 10 мл H2O була повільно додана HСl (8 мл; 1 M) з одержанням тонкої суспензії ясно-жовтої твердої речовини, до якої ще було додано 25 мл H2O. Тверда речовина була відфільтрована, промита 10 мл 0,1 M HCI і 100 мл води й висушена під вакуумом. Приклад 4 [0112] До 1 (R=PO(ONa)2) (5 г), розчиненого в 30 мл води, було додано 15 мл HCI (1 M) і 30 мл ТГФ для одержання ясно-жовтої суспензії, яка була перемішана протягом 30 хв при кімнатній температурі й відфільтрована. Отриманий твердий осад був суспендований в 150 мл води й перемішаний протягом 60 хвилин при кімнатній температурі. Потім було додано 50 мл ТГФ, і суспензія перемішувалася протягом 18 годин. Суспензія була відфільтрована, і твердий осад був промитий 10 мл HСl (0,1 M) і 100 мл води й висушений під вакуумом (15 годин). Приклад 5 [0113] До 1 (R=PO(ONa)2) (2 г), розчиненого в 15 мл води, була повільно додана HСl (6 мл; 1 M) з одержанням ясно-жовтої суспензії. Після додавання 20 мл ТГФ і 60 мл води, суспензія перемішувалася протягом 18 годин, була відфільтрована, і тверда речовина перемішувалася знову в 6 мл HCI (1M) протягом 15 хвилин. Потім суспензія була відфільтрована, і тверда речовина була висушена під вакуумом. Приклад 6 [0114] До 1 (R=PO(ONa)2) (3 г), розчиненого в 35 мл води, була додана HCI (9 мол; 1M) з одержанням ясно-жовтої суспензії. Після додавання 20 мл ТГФ суспензія перемішувалася протягом 30 хвилин при кімнатній температурі й потім була відфільтрована. Отримана тверда речовина була промита 20 мл HСl (0,1M) і водою, і висушена під вакуумом. Приклад 7 [0115] Твердий диводень фосфат додавався до об'єму DMSO або N-метилпіролідону при 50 °C доти, доки сіль не перестала розчинятися. Розчин, що містив суспендовану сіль, нагрівався, доки залишки твердої речовини не розчинилися, розчин був відфільтрований у гарячому стані й залишений охолоджуватися, спостерігалася поява кристалів диводень фосфату. Приклад 8 [0116] Розчин диводень фосфату був підготовлений у DMSO або N-метилпіролідоні й відфільтрований. До відфільтрованого розчину був доданий етанол з перемішуванням, доки розчин не став мутним. Перемішування було зупинено, і шар етанолу був обережно поміщений на поверхню мутного розчину, який був залишений, спостерігалася поява кристалів диводень фосфату. Приклад 9 [0117] Розчин диводень фосфату був підготовлений у DMSO або N-метилпіролідоні й відфільтрований. Відфільтрований розчин був підданий впливу парів етанолу, наприклад, шляхом розміщення відкритого контейнера розчину й відкритого контейнера з етанолом разом у герметичній посудині так, щоб обидва контейнера розділяли загальний простір усередині посудини. Через якийсь термін у контейнері з розчином з'явилися кристали диводень фосфату. Приклад 10 [0118] Був підготовлений розчин солі диводень фосфату, такий як моно- або динатрій фосфат. Такий розчин може бути підготовлений такими методами, як звичайне розчинення зразка твердого фосфату динатрію у воді або додаванням диводень фосфату до водяного розчину основи, досить сильного депротонування диводень фосфату. Ідентифікація відповідної основи є звичайною справою для практикуючого хіміка. Звичайно отриманий розчин солі диводень фосфату відфільтровується, до фільтрату додається кислота для репротонування 16 UA 114068 C2 5 10 15 20 25 солі й для початку кристалізації диводень фосфату. У звичайному прикладі, диводень фосфат додають до водяного розчину, що містить NaОН або Na2CO3 з одержанням розчину динатрій фосфату, до якого після фільтрації додають водний або газоподібний HСl для відновлення диводень фосфату, який випадає у вигляді кристалів. [0119] Для фармацевтичних цілей переважно використовувати фармацевтично прийнятні кислоти й основи в цьому процесі, наприклад, ті, які зазначені в Handbook of Pharmaceutical Salts: Properties, Selection and Use. (P. Heinrich Stahl and Camille G. Wermuth, eds.) InternatiONal Union of Pure and Applied Chemistry, Wiley-vch 2002 and L.D. Bighley, S.M. Berge, D.C. Monkhouse, в "Encyclopedia of Pharmaceutical Technology". Eds. J. Swarbrick and J.C. Boylan, Vol. 13, Marcel Dekker, Inc., New York, Basel, Hong Kong 1995, стор. 453-499, такі солі обговорюються детально. [0120] Як зрозуміло фахівцеві в даній галузі, елементи цих способів можуть бути комбіновані. Наприклад, розчин диводень фосфату в DMSO або N-метилпіролідоні може бути приготовлений при одній температурі, другий розчинник, такий як, етанол може бути доданий, і отриманий розчин залишений охолоджуватися. Подібним чином можуть бути використані суміші розчинників замість чистих розчинників, що також добре відомо фахівцям із кристалізації сполук. Більше того, також можуть бути використані інші розчинники й суміші. [0121] Елементний аналіз для C17H16FN6O6P (виміряне/розраховане) C 43,9 (44,8); H 3,6 (3,7); N 18,1 (18,4); O 21,2 (22,1); F 4,2 (4,2); P 6,7 (6,8). Приклад 11 [0122] Розмір частинок був виміряний, використовуючи Malvern Mastersizer. Були виконані інструкції з підготовки зразків, які збігалися з інструкціями виробника приладу. Зразок був підготовлений суспендуванням в 1-2 мл деіонізованої води й обробкою ультразвуком протягом 3 хвилин. [0123] Приблизний розподіл частинок за розмірами кристалічного матеріалу, такого як описано в Прикладах 1-10 вище, показаний на Фігурі 10 і в Таблиці 3 нижче: Таблиця 3 Звичайна гранулометрична сполука (неконтрольований процес) Лот 02090054 Середній 30 35 40 45 50 D10 (мкм) 0,28 D50 (мкм) 0,79 D90 (мкм) 44 Приклад 12 Експеримент по Регулюванню Розміру Частинок [0124] До 22-л реактору була завантажена 1 M HСl (1,95 л, 2,2 еквівалента) і етанол (1,6 л, 4 об'єму), і розчин був нагрітий до 70 °C. В окремий 12-л реактор, оснащений барботером для відстеження виділення газу, був завантажений TR-701FA [0,4 кг, AMRI лот # DUG-AH-166(2)], вода (2,8 л, 7 об'ємів) і етанол (0,4 л, 1 об'єм). Суспензія була перемішана при температурі навколишнього середовища, і за 30 хвилин через перистальтичний насос був доданий 5 % за вагою водяний розчин NaНСО3. Піноутворення не спостерігалося, однак виділення газу через барботер було дуже інтенсивним. По завершенню додавання, pН чистого жовтого розчину склало 6,6, За 90 хвилин до етанол/HСl розчину через перистальтичний насос був доданий водяний розчин TR-701. По завершенню додавання pН реакційної суміші було 1,9, суміш була охолоджена до 30 °C. Зразок суспензії був відібраний для аналізу оптичною мікроскопією. Суспензія була відфільтрована через поліпропіленову фільтруючу тканину, реактор і осад на фільтрі були промиті водою (5 об'ємів) і ацетоном (5 об'ємів). Загальний час фільтрації, включаючи промивання, склав 12 хвилин. Твердий осад був висушений під глибоким вакуумом 1 при 50 °C з одержанням 391,7 г повторно обложеного TR-701FA (98 % вихід). Аналіз H NMR відповідав зазначеній структурі. ВЕРХ аналіз (Спосіб A): 98,8 % (AUC) tr=5,2 хв. Рівень 1 залишкового етанолу H NMR аналізом склав 0,03 %, зміст води був 0,15 % за титруванням Карла Фішера, і зміст натрію був 5 мд. [0125] Розмір частинок був виміряний, використовуючи Malvern Mastersizer лазерну розсіюючу мікроскопію. Були чітко дотримані інструкції з підготовки зразків, що відповідають інструкціям виробника інструменту. Зразок був отриманий суспендуванням в 1-2 мл деіонізованої води й обробкою ультразвуком протягом 3 хвилин. Дані по лазерній дифракції представлено на Фігурі 11 і в Таблиці 4 нижче. 17 UA 114068 C2 Таблиця 4 Лот JAS-I-45 Середній D10 (мкм) 0,45 D50 (мкм) 14,13 D90 (мкм) 38,42 [0126] В іншому експерименті розподіл за розмірами частинок з використанням способу з контролем, такого як описаний у цьому прикладі, представлено на Фігурі 12 і в Таблиці 5 нижче: 5 Таблиця 5: Звичайний розподіл за розмірами частинок (з використанням контрольного процесу розподілу частинок за розмірами) Лот 0209118 Середній Діапазон застосування 10 15 D10 (мкм) 3,3 1-5 D50 (мкм) 27 1-40 D90 (мкм) 66 45-80 [0127] Лікарська форма зі швидким вивільненням і форма для внутрішньовенної ін'єкції, описані в Прикладах 13-14 нижче, були підготовлені, використовуючи кристалічну вільну кислоту, де контролювався розмір частинок. Приклад 13 Форма зі швидким вивільненням [0128] Якісна й кількісна сполука таблеток зі швидким вивільненням (R)-3-(4-(2-(2метилтетразол- 5-іл) піридин-5-іл)- 3-фторфеніл)- 5-гідроксиметил оксазолідин-2-он диводень фосфату 1 (R=PO(OH)2) ("Таблетки Торезолід Фосфату"), 200 мг, представлена в Таблиці 6. Усі компоненти, що використовуються в підготовці, перераховані із вказівкою стандарту якості, їх функції, відсотка за вагою для кожного індивідуального компонента. Список містить усі матеріали, що використавуються при одержанні лікарського засобу, незалежно від їхньої присутності в кінцевому продукті. Таблиця 6 Склад Таблеток Торезолід Фосфату, 200 мг Інгредієнт Торезолід Фосфат 1 Мікрокристалічна целюлоза (Avicel PH101) 2 Манітол (Mannogen® EZ сухий спрей) Повідон (Plasdone K-29/32) Кросповідон (Kollidon® CL) Очищена вода 2 Стеарат магнію (Hyqual®) рослинне джерело 1 Загальна вага ядра таблеткиt Опадрай (гідроксипропілметилцелюлоза) II Жовтий Очищена вода 3 Стандарт якості Власна Активний інгредієнт розробка 200 50,0 NF Дилюєнт 78,0 19,5 NF NF NF. Дилюєнт Зв'язувальна речовина Розпушувач Гранулююче середовище 78,0 16,0 24,0 19,5 4,0 6,0 - - Лубрикант 4,0 1,0 400,0 100,0 Барвник оболонки 14,0 3,4 Середовище для оболонки - - 414,0 103,4 USP NF USP Загальна вага 20 200 мг Таблетка Вага % (за (мг/од) вагою) Функція Абревіатури: NF = Національний Формуляр; USP = Фармакопея США 1 Реальну кількість торезолід фосфату встановлюють на використовуваного лота лікарської речовини. 18 підставі активності UA 114068 C2 2 5 10 15 Реальну кількість манітолу встановлюють на підставі кількості використовуваної лікарської речовини. 3 Видалення під час обробки. Приклад 14 Порошок і форма для ін'єкції [0129] (R)-3-(4-(2-(2-метилтетразол-5-іл)піридин-5-іл)-3-фторфеніл)-5-гідроксиметил оксазолідин-2-он диводень фосфат 1 (R=PO(OH)2) ("Торезолід Фосфат для ін'єкції" або "TR- 701 FA для ін'єкції"), 200 мг/ампула, був отриманий у вигляді стерильного ліофілізованого порошку для ін'єкції. TR- 701 FA для ін'єкції підготовляють in situ у вигляді динатрійової солі, використовуючи гідроксид натрію через його більш високу розчинність у воді (> 130 мг/мл). [0130] TR- 701 FA для ін'єкції, 200 мг/ампула, відновлюють 4 мл стерильної води для ін'єкції (WFI), USP з одержанням 50 мг/мл розчину. Відповідний об'єм клінічної дози відбирають із ампули й додають до нон- ди(2-етилгексил)фталатного (DEHP) контейнера для внутрішньовенного (IV) уведення утримуючий або 0,9 % ін'єкцію хлориду натрію, USP (сольовий розчин) або 5 % ін'єкцію декстрози, USP (декстроза). Отриманий IV розчин уводять, використовуючи комплект нон- DEHP розчину з 0,22 мкм вбудованим фільтром. [0131] Сполука TR- 701 FA розчину для ліофілізації представлена в Таблиці 7 і сполука TR701 FA для ін'єкції, 200 мг/ампула представлена в Таблиці 8, Таблиця 7 Сполука TR- 701 FA розчину для ліофілізації Компонент TR- 701 FA Манітол, порошок, USP Функція лікарська речовина наповнювач для утворення солі in-situ, регулювання pН Гідроксид натрію, USP Соляна кислота, NF регулювання pН Вода для ін'єкції, USP/EP Розчинник Теоретична кількість 100 мг/мл 50 мг/мл кількість, достатня для регулювання pН до 7,75 кількість, достатня для регулювання pН до 7,75 кількість до 1,0 мл Таблиця 8 Сполука TR- 701 FA для ін'єкції, 200 мг/ампулу Компонент TR- 701 FA Манітол, порошок, USP Гідроксид натрію, USP Функція лікарська речовина наповнювач для утворення солі in-situ, регулювання pН Соляна кислота, NF регулювання pН Вода для ін'єкції, USP/Epb Розчинник Теоретична Кількість a 210 мг 105 мг кількість, достатнє для регулювання ph до 7,75 кількість, достатня для регулювання pН до 7,75 кількість до 2,1 мл a Об'ємний еквівалент 210 мг TR- 701 FA поміщають у кожну ампулу так, щоб відновлення ампули з 4,0 мл води для ін'єкції (фінальний об'єм в 4,2 мл одержують через витиснення об'єму нерозчинними твердими речовинами) давало розчин TR- 701 FA 50 мг/мл, що дозволить відмовитися від зазначення вмісту компонентів на етикетці. b Вода для ін'єкції по суті видаляється під час ліофілізації. 20 [0132] Звичайна виробнича формула для ін'єкції TR- 701 FA, 200 мг/ампула представлена в Таблиці 9, 19 UA 114068 C2 Таблиця 9 Звичайна виробнича формула для ін'єкції TR- 701 FA, 200 мг/ампулу Матеріал TR- 701 FA Манітол, порошок, USP Гідроксид натрію, NF Соляна кислота, NF Вода для ін'єкції, USP/EP Усього Теоретична кількість a 400 г 200 г кількість, достатня для регулювання pН до 7,75 кількість, достатня для регулювання pР до 7,75 достатня кількість 4276 г 4000 мл (~1900 ампул) a Реальна кількість лікарської речовини TR- 701 FA для зважування буде регулюватися активністю. 5 10 15 20 25 30 [0133] Короткий опис виробничого процесу представлений нижче, схематичне зображення виробничого процесу для одержання розчину, стерильної фільтрації, наповнення й ліофілізації презентовано на Фігурах 8 і 9. Одержання розчину [0134] Розчин одержують у відповідності з наступною схемою: [0135] Додати приблизно 50 % загальної кількості води для ін'єкції в посудину із цільовою сполукою. [0136] Додати TR- 701 FA і повільно нейтралізувати розчином гідроксиду натрію при перемішуванні. [0137] Додати й розчинити манітол при перемішуванні. [0138] Виміряти pН отриманого розчину. Якщо розчин перебуває за межами цільового діапазону pН 7,70-7,80, довести pН, використовуючи або 1н гідроксид натрію або 1н соляну кислоту. [0139] Додати воду для ін'єкції до фінального об'єму й перемішати. Стерильне Фільтрування/Наповнення/Ліофілізація [0140] Відфільтрувати отриманий розчин через 2 перевірених на відсутність ушкоджень 0,22 мкм фільтри послідовно в стерильну посудину. [0141] Наповнити цільовим розчином 20 мл ампули в асептичних умовах. [0142] Частково ввести ліофілізовані пробки в ампули. [0143] Провести ліофілізацію ампул відповідно до певного циклу. [0144] Наприкінці циклу ліофілізації, заповнити камеру азотом і закрити ампули під частковим вакуумом. [0145] Герметично закрити ампули ковпачками фліп-офф (обтискний ковпачок з гумовою пробкою, яка вкрита знімною пластиковою кришкою для забезпечення місця проколу). Приклад 15 [0146] Зразок кристалічної вільної кислоти, який був отриманий у відповідності зі способом одержання вільної кислоти, описаним у заявці на патент США № 12/577,089, права по якій передані тому ж правовласникові, що й за цією заявкою, був кристалізований у відповідності зі способами, описаними в цій заявці, і був охарактеризований, використовуючи ВЕРХ, зразок містить різні кількості домішок, такі як описано в таблиці 10 нижче: Таблиця 10 не більше 0,5 % Rx600013 не більше 0,5 % Rx600024 не більше 0,5 % Ідентифікуючі ВЕРХ Rx600014 не більше 0,2 % Rx600023 не більше 0,5 % Rx600025 не окремі (TM.1911) більше 0,5 % Rx600020 не більше 2,0 % Rx600001 не більше 1,5 % домішки Rx600022 35 40 [0147] Додатково, по суті чистий зразок кристалічної вільної кислоти, який був отриманий відповідно до процесів, не розкритих в опублікованій заявці на патент США № 20070155798 і був кристалізований у відповідності зі способами, описаними тут (тут і далі іменовані як "наші") був порівняний зі зразком матеріалу, отриманим Dong-A Pharm. Co. (тут і далі іменований як "Dong-A матеріал"), який був наданий компанії Траюс Терап'ютікс Інк. приблизно в 2007. Вміст активної речовини в Dong-A матеріалі склав приблизно 84 % за вагою зразка в порівнянні із 20 UA 114068 C2 чистим зразком, однак, чистота кристалічної вільної кислоти була 94,1 % за вагою матеріалу, ідентифікованого ВЕРХ, як зазначено нижче. Таким чином, приблизно 10 % домішок в Dong-A матеріалі не було ідентифіковано ВЕРХ. Порівняння характеристик чистоти презентовано в Таблиці 11 нижче: 5 Таблиця 11 Відсоток площі Найменування домішки 600011 600013** невідомо невідомо невідомо 600024 невідомо невідомо 600012 600023 невідомо невідомо 600025** невідомо невідомо невідомо невідомо невідомо 600020 невідомо 600001* 600022 600042** 600043** 600026* невідомо невідомо * ** Відносний час утримання 0,54 0,56 0,65 0,77 0,86-0,88 0,91 0,94 0,95 1 1,08 1,1 1,14 1,15 1,2 1,21 1,31 1,39 1,47 1,5-1,51 1,56 1,67-1,688 1,72-1,73 1,79 1,8 2,27-2,28 2,34 2,4 наші Dong A 0,12 немає даних 0,07 0,34 0,07 0,22 0,07 0,05 94,1 0,14 0,06 0,05 ND 0,07 0,05 0,05 0,26 0,35 0,2 ~0,05 1,12 0,28 немає даних немає даних 2,17 0,05 0,06 ID немає даних 0,08 немає даних немає даних 0,03 0,12 немає даних немає даних 97,1 немає даних немає даних немає даних 0,27 немає даних немає даних немає даних 0,04 немає даних 0,08 немає даних 0,63 1,2 0,12 0,15 0,06 немає даних немає даних DA-1димер diphos Des-Me Пірофосфат API N-1 Фосфорильований Над-алкільована пара Димер TR-700 Біс OA-700 змішаний ді ефір OA-700 змішаний ді ефір Хлор наші
ДивитисяДодаткова інформація
Автори російськоюReichenbacher, Katharina, Duguid, Robert, J., Simson, Jacqueline, A., Phillipson, Douglas
МПК / Мітки
МПК: A61K 31/675, C07D 413/14
Мітки: r)-3-(4-(2-(2-метилтетразол-5-іл)піридин-5-іл)-3-фторфеніл)-5-гідроксиметилоксазолідин-2-ондиводеньфосфату, кристалічні, частинки
Код посилання
<a href="https://ua.patents.su/38-114068-kristalichni-chastinki-r-3-4-2-2-metiltetrazol-5-ilpiridin-5-il-3-ftorfenil-5-gidroksimetiloksazolidin-2-ondivodenfosfatu.html" target="_blank" rel="follow" title="База патентів України">Кристалічні частинки (r)-3-(4-(2-(2-метилтетразол-5-іл)піридин-5-іл)-3-фторфеніл)-5-гідроксиметилоксазолідин-2-ондиводеньфосфату</a>
Кристалічні форми {2-[1-(3,5-бістрифторметилбензил)-5-піридин-4-іл-1н-[1,2,3]триазол-4-іл]-піридин-3-іл}-(2-хлорфеніл)метанону

Номер патенту: 82901
Опубліковано: 26.05.2008
Автори: Педерсен Стівен Уейн, Футман Памела Кей, Рутцель-Іденс Сюзан Марія, Коффі Дейвід Скотт, Вебер Карстен, Боргезе Альфіо, Тамезе Шелла Леньонга, Тімпе Карстен
МПК: C07D 401/04, A61P 25/00, C07D 213/46, A61K 31/44
Мітки: форми, кристалічні, 2-[1-(3,5-бістрифторметилбензил)-5-піридин-4-іл-1н-[1,2,3]триазол-4-іл]-піридин-3-іл}-(2-хлорфеніл)метанону
Формула / Реферат:
1. Кристалічна Форма IV {2-[1-(3,5-бістрифторметилбензил)-5-піридин-4-іл-1Н-[1,2,3]триазол-4-іл]-піридин-3-іл}-(2-хлорфеніл)метанону, яка характеризується щонайменше одним із нижченаведеного:a) спектром твердотільного ядерно-магнітного резонансу на 13С, що включає піки з такими значеннями хімічного зсуву, млн-1: 52,30,2 та 195,4
Кристалічні частинки, покриті міцелами

Номер патенту: 111850
Опубліковано: 24.06.2016
Автори: Томсон Найл Рей, Малквін Патрік Джозеф, Біггс Саймон Річард, Скенлон Шейн, Шаньє Неллі, Дюбуа Матьє Едмонд Рене, Саркер Продіп
МПК: A01N 43/12, A01N 25/26, A01N 43/90, A01N 51/00
Мітки: частинки, кристалічні, покриті, міцелами
Формула / Реферат:
1. Органічна кристалічна частинка, покрита щонайменше 10 міцелами, що самі по собі містять АВ-блок-співполімер, який містить (і) перший гідрофобний блок А, що містить полімер, вибраний з групи, що включає гомополімер з акрилатного або алкілакрилатного мономеру; співполімер, що містить два або три різні мономери, вибрані з акрилатних або алкілакрилатних мономерів; гомополімер з мономеру - похідної стиролу; співполімер, що містить два різні...
Кристалічні колоїдні масиви з високою відбивною здатністю, які містять частинки, що поглинають випромінювання

Номер патенту: 111087
Опубліковано: 25.03.2016
Автори: Ван'єр Ноел Р., Донелі Джон Т., Сюй Сянлін
МПК: C30B 29/54, C30B 29/60, C30B 7/00, C09C 1/28, C30B 29/16, C09D 5/33, C09C 1/04, C09D 5/02, B01J 13/00, C09C 1/36, C09C 1/16
Мітки: масиві, високою, містять, частинки, випромінювання, поглинають, здатністю, відбивною, колоїдні, кристалічні
Формула / Реферат:
1. Композитна композиція, яка відбиває й поглинає випромінювання, яка включає:велику кількість колоїдних кристалів, кожний з яких включає (1) частинки, які відбивають випромінювання, скомпоновані у вигляді колоїдного масиву, і (2) поглинаючі випромінювання частинки, дисперговані в згаданих кристалах, так, що згадана композиція розсіює випромінювання в одній смузі довжин хвиль власне у всіх напрямках і поглинає випромінювання в іншій...
Кристалічні солі 7-[4-(4-фторфеніл)-6-ізопропіл-2-[метил(метилсульфоніл)аміно]піримідин-5-іл]-(3r,5s)-3,5-дигідроксигепт-6-енової кислоти

Номер патенту: 74567
Опубліковано: 16.01.2006
Автори: Тейлор Найджел Філіп, Окада Тецуо
МПК: C07D 239/42, A61P 43/00, A61P 9/10, A61P 3/06, A61K 31/505
Мітки: кристалічні, солі, кислоти, 7-[4-(4-фторфеніл)-6-ізопропіл-2-[метил(метилсульфоніл)аміно]піримідин-5-іл]-(3r,5s)-3,5-дигідроксигепт-6-енової
Формула / Реферат:
1. Кристалічна сіль сполуки (Е)-7-[4-(4-фторфеніл)-6-ізопропіл-2-[метил(метилсульфоніл)аміно]піримідин-5-іл]-(3R,5S)-3,5-дигідроксигепт-6-енової кислоти, де сіль являє собою сіль амонію, метиламонію, етиламонію, діетаноламонію, трис(гідроксиметил)метиламонію, бензиламонію, 4-метоксибензиламонію, літію або магнію.2. Кристалічна сіль за п. 1, яка являє собою кристалічну амонієву сіль...
Кристалічні форми гідрохлориду (4a-r,9a-s)-1-(1н-бензоімідазол-5-карбоніл)-2,3,4,4а,9,9а-гексагідро-1н-індено[2,1-b]піридин-6-карбонітрилу та їх застосування як інгібіторів hsd 1

Номер патенту: 110807
Опубліковано: 25.02.2016
Автори: Зік Сандра, Екхардт Маттіас, Шюле Мартін, Ян Бін-Шіу, Мартін Ханс-Юрген
МПК: C07D 401/06, A61P 3/00, A61K 31/435
Мітки: гідрохлориду, застосування, інгібіторів, кристалічні, форми, 4a-r,9a-s)-1-(1н-бензоімідазол-5-карбоніл)-2,3,4,4а,9,9а-гексагідро-1н-індено[2,1-b]піридин-6-карбонітрилу
Формула / Реферат:
1. Кристалічна Форма І сполуки, що представлена наступною структурною формулою:яка характеризується піками порошкової рентгенівської дифракції при кутах 2θ, вибраних з 12,5°, 12,9°, 14,8°, 20,0°, 22,2° та 26,1°.2. Кристалічна Форма І за п. 1 для лікування або попередження хвороб або станів, на які можна впливати шляхом пригнічення ферменту...
Попередній патент: Водогрійний твердопаливний котел
Наступний патент: Маловитратні спосіб та система для обробки водних об’єктів, на які впливають бактерії та мікроводорості
Випадковий патент: Шарнірний робочий орган гвинтового конвеєра