Аналог інсуліну, аналог інсуліну, що має здатність знижувати рівень цукру в крові
Номер патенту: 27299
Опубліковано: 15.09.2000
Автори: Чанс Рональд Юджин, Дімарчі Річард Денніс, Шілдз Джеймс Едвін, Френк Брюс Хілл








Формула / Реферат
(57) 1. Аналог инсулина формулы (І) 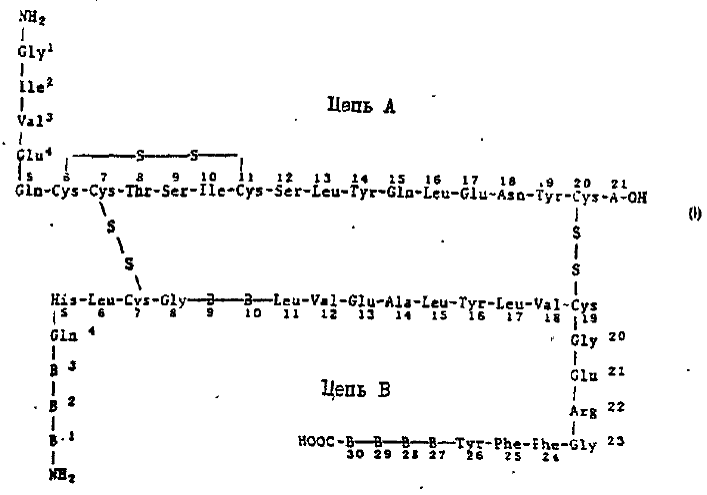
или его фармацевтически приемлемая соль, отличающийся тем, что А21 является аспарагином, В1 является фенилаланином, В2 является валином, ВЗ является аспарагином, В9 является сери-ном, В10 является гистидином или асларагиновой кислотой, В27 является треонином, В28 является любой природной L-аминокислотой, В29 является L-пролином, В30 является треонином
2. Аналог инсулина по п. 1, отличающийся тем, что В28 является аспаргиновой кислотой, валином, лейцином, иэолейцином, норлейцином, про-лином, аргинином, гистидином, цитруллином, орнитином, лизином, фенилаланином, аланином или глицином
3. Аналог инсулина по пп. 1 или 2, отличающийся тем, что В28 является аспарагиновои кислотой, валином, лейцином, изолейцином, иорлейцином, пролином, аргинином, гістидином, орнитином или лизином
4. Аналог инсулина по любому из пп.. 1-3, отличающийся тем, что В28 является лизином
5. Аналог инсулина по любому из пп.. 1-4, отличающийся тем, что В10 является аспарагиновой кислотой.
6 .Аналог инсулина по п 1, отличающийся тем, что А21 является аспарагином, В1 является фени-лаланином, В2 является валином, ВЗ является аспарагином, В9 является серимом, В10 является гистидином, В27 - треонином, В28 - лизином, В29 является L - пролином и В30 - треонином
7. Аналог инсулина формулы (I), обладающий активностью снижения уровня глюкозы в крови.
Текст
Настоящее изобретение касается аналогов инсулина, модифицированного в положении 29 аминокислоты В-цепи естественного человеческого инсулина и при желании в других положениях. Эти аналоги инсулина в меньшей сте пени склонны к димеризации или самопроизвольной ассоциации с более высокомолекулярными формами, в результате че го они более быстро повышают свою активность, сохраняя при этом биологическую активность естественного человеческого инсулина. Настоящее изобретение охватывает аналоги инсулина следующей формулы: в которой А21 представляет собой аланин, аспарагин, аспарагиновую кислоту, глутамин, глутаминовую кислоту, глицин, треонин, или серин; В1 представляет собой фенилаланин или аспарагиновую кислоту или отсутствует; В2 представляет собой валин или может отсутствовать при отсутствии В1; В3 представляет собой аспарагин или аспарагиновую кислоту; В9 представляет собой серин или аспарагиновую кислоту; В 10 представляет собой гисти дин или аспарагиновую кислоту; В 28 представляет собой любую аминокислоту; В29 представляет собой L-пролин, D-пролин, D-оксипролин, L-оксипролин, L-(N-метиллизин), D-лизин, L-(N-метиларгинин) или D-аргинин; В30 представляет собой аланин, треонин или отсутствуе т; Z представляет собой –OH, –NH2, –OCH 3, или –CH2CH3; X представляет собой Arg, Arg– Arg, L ys, Lys–Lys, Arg–Lys, Lys–Arg, или отсутствует; и Y может присутствовать лишь тогда, когда присутствует Х, и если он присутствуе т, то представляет собой Glu или аминокислотную последовательность, кото рая содержит всю или часть последовательности –Glu–Ala–Glu–Asp–Leu–Gln–Val–Gly –Gln–Val–Glu–Leu–Gly–Gly–Gly–Pro– Gly– Ala–Gly–Ser– Leu–Gln–Pro–Leu–Ala–Leu–Glu–Gly–Ser– Leu–Gln–Lys–Ary и который начинается у N-терминального Glu этой последовательности. Описан и заявлен также способ лечения гипергликемии путем ввода в организм пациента, нуждающе гося в таком лечении, эффективного количества аналога инсулина формулы I. Кроме того, описаны и заявлены фармацевтические композиции, содержащие эффективное количество аналога инсулина формулы I в комбинации с одним или несколькими фармацевтически пригодными эксципиентами. Приложенные к описанию рисунки не даются в точно выдержанном масштабе. Фиг. 1 – Ограничительная точка и функциональная карта плазмиды рКС 283. Фиг. 2 – Ограничительная точка и функциональная карта плазмиды рКС 283 РХ. Фиг. 3 – Ограничительная точка и функциональная карта плазмиды рКС 283–L. Фиг. 4 – Ограничительная точка и функциональная карта плазмиды рКС 283–LB. Фиг. 5 – Ограничительная точка и функциональная карта плазмиды рКС 283–PRS. Фиг. 6 – Ограничительная точка и функциональная карта плазмиды pL 32. Фиг. 7 – Ограничительная точка и функциональная карта плазмиды pNM 789. Фиг. 8 – С хематическое представление построения плазмиды 120. Фиг. 9 – Ограничительная точка и функциональная карта плазмиды pL 47. Фиг. 10 – Ограничительная точка и функциональная карта плазмиды pPR 12. Фиг. 11 – Ограничительная точка и функциональная карта плазмиды pPR 12 PI. Фиг. 12 – Ограничительная точка и функциональная карта плазмиды pL 110. Фиг. 13 – Схе матическое представление построения плазмиды pL 110 C. Фиг. 14 – Ограничительная точка и функциональная карта плазмиды pCZP 126 S. Фиг. 15 – Нук леотидная последовательность гена синтезированного че ловеческого проинсулина. Фиг. 16 – Ограничительная точка и функциональная карта плазмиды pP 145. Фиг. 17 – Ограничительная точка и функциональная карта плазмиды pRB 164 A. Фиг. 18 – Ограничительная точка и функциональная карта плазмиды pRB 172. Фиг. 19 – Ограничительная точка и функциональная карта плазмиды pRB 173. Фиг. 20 – Ограничительная точка и функциональная карта плазмиды pRB 175. Формула I воспроизводит с помощью сокращенных названий аминокислотную последовательность аналогов инсулина, отвечающи х данному изобретению. Сокращенные названия аминокислот могут быть расши фрованы, как указано ниже. Аббревиатура Аминокислота Aba Аминомасляная кислота Ala Ала нин Arg Ар гинин Asn Аспарагин Asp Аспарагиновая кислота Cys Цистеиновая кислота Cys Цистеины Gln Глутамин Gln Глутаминовая кислота Gly Глицин His Гистидин Il Изолейцин Leu Лейцин Lys Лизин Met Ме тионин Nle Норлейцин Nva Норвалин Orn Орнитин Phe Фенилаланин Pro Пролин Ser Серин Thr Треонин Trp Триптофан Tyr Тирозин Val Валин В28 может представлять собой любую аминокислоту природного или неприродного происхождения. Эта аминокислота является предпочтительно аспарагиновой кислотой, ва лином, лейцином, изолейцином, норлейцином, пролином, аргинином, гистидином, цитруллином, орнитином, лизином, фенилаланином, аланином или глицином. Из указанных выше аминокислот особенно предпочтительно подгруппой является аспарагиновая кислота, ва лин, лейцин, изолейцин, норлейцин, пролин, аргинин, гистадин, орнитин или лизин. Лизин является самой предпочтительной аминокислотой для В28. В29 представляет собой предпочтительно L-пролин, D-пролин, D-оксипролин или L-оксипролин. Особенно предпочти тельным аналогом инсулина согласно настоящему изобретению является такой, в котором В28 представляет собой лизин и В29 представляет собой пролин, то есть инверсию аминокислотной последовательности естественного человеческого инсулина в положениях 28 и 29 В-цепи. Согласно следующей особенности данного изобрете ния, как указыва лось выше, когда В28 представляет собой L-пролин, то положение В29 представляет собой предпочтительно L-(N-метиллизин), D-лизин, L-(N-метиларгинин) или D-аргинин. Предпочтительным аналогом согласно данной особенности является такой, в котором В28 представляет собой L-пролин, и В29 представляет собой L-(N-метиллизин) или D-лизин. Ниже рассматриваются также другие модифи кации аналогов инсулина согласно данному изобретению, то есть модифи кации аналогов инсулина в положениях иных, чем положения В28 и В29. В частности может быть желаемой замена группы Asn в положении 21 цепи А (то есть карбоксильной концевой группы) груп пами Ala, Asp, Gln, Glu, Gly, Thr или Ser, и при такой замене предпочтительной замещающей груп пой является Ala. Аналогично этому может быть желательным замена группы в положении 3 цепи -В аспарагиновой кислотой (Asp). Такие возможные замены приводят к уве личению стабильности аналогов при экстремальных значениях рН, поскольку Asn особенно чувствите лен к реакциям деамидирования и перегруп пировки как при низком, так и при высоком значении рН. Для специалистов в данной области должно быть ясно также, что глутаминовые группы аналогов инсулина могут быть одинаковым образом чувствительны к реакциям деамидирования и перегруппировки. Таким образом, замеще ние их глутаминовой кислотой также охватывается объемом настояще го изобретения. Возможные другие модифи кации аналогов инсулина согласно настояще му изобретению включают (в любой комбинации) замену гистидиновой гр уппы в положении В10 аспарагиновой аминокислотой; замену фенилаланиновой группы в положении А1 аспарагиновой кислотой; замену треониновой груп пы в положении В30 аланином; замену се риновой группы в положении В9 аспарагиновой кислотой; делецию аминокислот в положении В1 (des–В1) либо как таковых ли бо в комбинации с делецией в положении В2 (des–В2); и делецию треонина из положения В–30 (des–В30). Аналоги инсулина согласно данному изобретению могут быть также модифи цированы в концевых положениях В30 путем ввода любых из следующи х аминокислот или дипептидов: Arg, Arg–Arg, L ys, Lys–Lys, Arg–Lys или Lys–Arg. В случае их присутствия они обозначаются в формуле I как Х. Предпочтительным из них является Arg–Arg. Кроме того, в случае наличия этих добавленных звеньев в положении В30, аналог может быть дополнительно модифи цирован путем добавления к образуемой В30 – удлиненной концевой группы глутаминовой кислоты (Glu) или аминокислотной последовательности, включающей всю или часть последовательности –Glu–Ala–Glu–Asp–Leu–Gln–Val–Gly –Gln–Val–Glu–Leu–Gly–Gly–Gly–Pro– Gly– Ala–Gly–Ser– Leu–Gln– Pro–Leu–Ala–Leu–Glu–Gly–Ser– Leu–Gln–Lys–Arg которая начинается в ее N-терминальной Glu. Эта аминокислота или последовательность, в случае ее присутствия, обозначена в формуле I группой Y. Когда эта последовательность представляет лишь часть указанной выше, то она может представлять собой любую из тех частей, которые начинаются у N-конца данной последовательности, то есть у груп пы глутаминовой кислоты (Glu). Х или комбинация Х и Y, которые определены выше, представляет собой полностью или часть соединяющего пептида, обнаруживаемого в человеческом проинсулине, молекуле, которая является биологическим источником образования естественного человеческого инсулина. Предпочти тельными последовательностями для Y являются: –Glu–Ala–Glu–Asp–Leu–Gln–Val–Gly–Gln–Val–Glu–Leu–Gly–Gly–Gly–Pro–Gly– Ala–Gly–Ser–Leu–Gln– Pro–Leu–Ala-Leu–Glu–Gly–Ser–Leu–Gln–Lys–Arg; –Glu–Ala–Glu–Asp–Leu–Gln–Val–Gly–Gln–Val–Glu–Leu–Gly–Gly–Gly–Pro–Gly– Ala–Gly–Ser–Leu–Gln– Pro–Leu–Ala-Leu–Glu–Gly–Ser–Leu–Gln–Lys; –Glu–Ala–Glu–Asp–Leu–Gln–Val–Gly–Gln–Val–Glu–Leu–Gly–Gly–Gly–Pro–Gly– Ala–Gly–Ser–Leu–Gln– Pro–Leu–Ala-Leu–Glu–Gly–Ser–Leu–Gln; –Glu–Ala–Glu–Asp–Leu–Gln–Val–Gly–Gln–Val–Glu–Leu–Gly–Gly–Gly–Pro–Gly– Ala–Gly–Ser–Leu–Gln– Pro–Leu–Ala-Leu–Glu; –Glu–Ala–Glu–Asp–Leu–Gln–Val–Gly–Gln–Val–Glu–Leu–Gly–Gly–Gly–Pro–Gly– Ala–Gly–Ser–Leu–Gln– Pro–Leu–Ala; и –Glu–Ala–Glu–Asp–Leu–Gln–Val–Gly–Gln–Val–Glu–. Кроме того, независимо от того, присутствует или отсутствует Х отдельно или присутствуют или отсутствуют Х и Y совместно, концевая группа Z может быть любой из числа следующи х: –ОН, –NH2, –OCH 3 или –ОСН2СН3. Предпочтительным значением группы Z является –ОН. Как уже упоминалось выше, данное изобретение охваты вает фармацевтически пригодные соли аналогов инсулина. Такими предпочтительными солями являются соли цинка, натрия, калия, магния и кальция. Аналоги инсулина, отвечающие данному изобретению, могут быть получены одним из известных приемов синтеза пептидов, включая классические способы (в растворе), способы в твердофазных системах, способы полусинтеза и известные с недавнего времени способы рекомбинантной ДНК. В способах с использованием твердофазных систем аминокислотная последовательность составляется из исходной нерастворимой С-терминальной аминокислоты со смоляным носителем. Эти твердофазные способы описываются J. Stewart и др., Твердофазный синтез пептидов, Freeman and Co., Сан Франциско, 1969 г. Обычно при осуществлении твердофазного способа аминокислота, соответствующая С-терминальной аминокислотной группе желаемого пептида, закрепляется на смоляном носителе и далее формируется пептидная цепь, начинающаяся с нанесенной на смолу С-терминальной аминокислоты. Отдельные аминокислоты вводятся последовательно до тех пор, пока не получается желаемая аминокислотная последовательность. Как возможный вариант, могут быть получены небольшие пептидные фрагменты, кото рые могут быть введены в пептидную цепь в желаемом порядке. Пептидная цепь остается скрепленной со смолой в процессе синтеза и при завершении построения цепи данный пептид отщепляется от смолы. Пептидная цепь закрепляется с полистирольной смолой посредством сложноэфирной связи, образующейся между карбоксильной группой С-терминального звена и специфической метиленовой груп пой, присутствующей в смоляной матрице, являющейся точкой такого скрепления. Аминокислоты связываются сопряженными связями хорошо известными в данной области приемами образования пептидных свя зей. Один такой прием заключается в конверсии аминокислоты в производное, которое придает карбоксильной группе более высокую чувствительность к реакции со свободной N-терминальной аминогруппой пептидного фер мента. Так, например, аминокислота может быть превращена в смешанный ангидрид путем реакции защищенной аминокислоты с этилхлорформатом, фенилхлорформатом, вторбутилхлорформатом или изобутилхлорформатом. Как возможный вариант, аминокислота может быть превраще на в активный сложный эфир, такой как 1,4,5-трихлорфе ниловый сложный эфир, пентахлорфе ниловый сложный эфир, пара-нитрофениловый сложный эфир, N-оксисукцинимидный сложный эфир, или сложный эфир, образуемый из I-оксибензотриазола. Другой прием сопряжения заключает в себе использование подходяще го агента, такого как N,N'-дихлоргексилкарбодиимид (ДСС) или N,N'-диизопропилкарбодиимид (DIC). Другими подхо дящими сопрягающи ми агентами являются соединения, которые уже известны для специалистов в дан ной области. Смотри работу Schroder и Lubke "Пептиды", Academic Press 1965 г., Глава III, которая рассматривается здесь как ссылочный материал. Следуе т иметь в вид, что a-аминогруппа каждой аминокислоты, используемой в синтезе пептидов, должна быть защи щена в хо де реакции сопряжения для предотвращения побочных реакций, в кото рых принимает участие a -аминовая функциональная группа. Следует также иметь в виду, что некото рые ами нокислоты содержат боковые цепи, являющиеся функциональными группами (например, сульфгидрил, eамино, b и g -карбоксил, имидазол, гуанидино, и гидроксил), и что также функциональные группы также должны быть защище ны в начальном и в последующих этапах реакции сопряжения. Подхо дящие защитные группы уже известны в данной области. Смотри, например, публикацию "Защитные группы в органической химии", M. Mc Omie Ред. Plenum Press, N. Y. 1973, и патент США № 4 617 149, кото рые приводятся здесь как ссылочные материалы. При выборе типа защитной группы следует соблюдать определенные условия. Защитная для альфаамино группа: (1) должна придавать функциональной альфа-аминогруппе инертность в условиях, имеющи х место в реакции сопряжения, (2) должна быть легко удаляемой после реакции сопряжения в таких условиях, в которых не удаляются защи тные группы боковой цепи, и не должна изменять структуру пептидного фрагмента, и (3) должна исключать возможность рацемизации при активации непосредственно перед реакцией сопряжения. Защитная группа боковой цепи (1) должна придавать этой функциональной боковой цепи инертность в условиях, используемых при проте кании реакции сопряжения, (2) должна быть устойчивой в условиях, имеющи х место при удалении защитной для a -амино группы, и (3) должна легко удаляться при завершении построения желаемой аминокислотной последовательности в таких условиях реакции, которые не изменяют структуру пептидной цепи. Для специалистов в данной области должно быть ясно, что защитные группы, которые уже известны как группы, применяемые для синте за пептидов, будут по разному активны к агентам, используе мым для их уда ления. Так, например, некоторые защи тные группы, такие как трифе нилметил и 2-(пара-бифенил) изопропилоксикарбонил очень неустойчивы и могут расщепляться в слабо кислотных условиях. Др угие защитные группы, такие как трет-бути локсикарбонил, трет-амилоксикарбонил, адаматнилоксикарбонил и пара-метоксибензилоксикарбонил, менее устойчивы и требуют использования умеренно сильных кислот, таких как трифторуксусная кислота, соляная кислота, или трифтор бора в уксусной кислоте, для их удаления. Некоторые защитные группы, такие как бензилоксикарбонил, галоилбензилоксикарбонил, пара-нитробензилоксикарбонил, циклоалкилоксикарбонил и изопропилоксикарбонил, еще менее устойчивы и для их удаления требуется использование еще более сильных кислот, таких как фтористоводородная кислота, бромистоводородная кислота или трифторацетат бо ра в трифторуксусной кислоте. При завершении желаемого построения последовательности пептида, защищенный пептид должен быть отщеплен от смоляного носителя, и все защи щающие груп пы должны быть удалены. Реакция отщепления и удаление защитных групп могут осуществляться одновременно или поэтапно. Когда смоляной носитель представляет собой хлорметилированную полисти роловую смолу, то связь, скрепляющая пептид со смолой, является сложноэфирной связью, образуемой между сво бодной карбоксильной группой С-терминального звена и одной из многих хлорметильных групп, присутствующи х в смоляной матрице. Следуе т иметь в виду, что указанная скрепляющая связь может быть расщеп лена посредством реагентов, которые, как уже известно, способны разрывать сложноэфирную связь и проникать в смоляную матрицу. Осо бенно легко осуществимым способом является обработка жидким безводным фтористым водородом. Этот реагент не только отщепляет пептид от смолы, но и удаляет все защитные группы. Следовательно, использование этого реагента может обеспечить получение полностью лишенного защи ты пептида. Когда желательно удаление пептида без удаления защитных групп, то защи щенный пептид – смола может подвергаться метанолизу, в ре зультате чего получается защищенный пептид, в котором С-терминальная карбоксильная группа подвергнута метилированию. Этот сложный метиловый эфир затем может быть гидролизован в слабо щелочных условиях и в результате будет получен свободный С-терминальный карбоксил. За щитные группы пептидной цепи могут быть уда лены путем обработки сильной кислотой, такой как жидкая фтористоводородная кислота. Осо бенно интересный способ метанолиза описывается в работе G. Moore и др., Пептиды, Тр уды 5-го Американского симпозиума по пептидам, М. Goodman и J. Meinhofer ред. John Wiley, N. Y., 1977., стр. 518–521, где защи щенный пептид-смола обрабатывается метанолом и цианидом калия в присутствии простого кроун эфира. Следующий способ отщепления защи щенного пепти да от смолы заключается в аминолизе или обработке гидразином. При желании образуемый С-терминальный амид или гидразид может быть гидролизован до свободного С-терминального карбоксила, и защитные группы могут быть уда лены общепринятым образом. Следуе т также иметь в виду, что защитные группы, нахо дящиеся у N-терминальной a -аминогруппы, уда ляются предпочти тельно либо до, либо одновременно с отщеплением защищен ного пептида от смоляного носителя. А и В цепи аналогов инсулина, отвечающие данному изобретению, также могут быть получены методом рекомбинантной ДНК. При этом получается нуклеотидная последовательность, кодирующая желаемый пептид А или В цепи, с использованием общепринятой технологии для такого синтеза. Эти методы обычно заключают в се бе получение олигонуклеоти дов, кодирующи х как фрагменты желаемой кодирующей последовательности, так и их полную последовательность. Олигонуклеотиды обеспечивают перекрытие одного фрагмента кодирующей последова тельности двумя фрагментами дополняющей последовательности и наоборот. Эти олигонуклеотиды конъюгируют и связываются с образованием в конечном итоге желаемой генной последовательности. Эта последовательность вставляется в клонирующий вектор в точке, которая допускает возможность выражения пептидного продук та, который он кодирует. Подходящий клонирующий вектор включает в себе, по меньшей мере, часть контрольной последовательности вы ражения гена. Цепи А и В аналогов ин сулина, согласно настоящему изобретению могут быть также получены посредством исходной молекулы проинсулиново го типа с использова нием мето дов рекомбинантной ДНК. Смотри Frank и др. Пептиды; Синтез – Структура – Функции. Труды 7-го Американского симпозиума по пептидам, Ред. D. Rich и E. Gross (1981 г.), которая (публикация) рассматривается здесь как ссылочный материал. Однако этап комбинирования отдельных получаемых цепей А и В может осуществляться способом, описанным в работе Chance и др., Пептиды: Синтез, структура и функции: Труды 7-го Американского симпозиума по пептидам, 1981 г. (приводится здесь как ссылочный материал). Для иллюстрации данного изобретения даются нижеследующие примеры, которые, однако, не служат для ограничения объема изобрете ния. Пример 1. Человеческий инсулин Pro (B29) Lys (B28). А. Получение образованной от рекомбинанты А цепи. А-цепь человеческого инсулина получается методом рекомбинантной ДНК путем химического синтеза гена, кодирующе го А-цепь и его выражения в E. coli. Ген для А-цепи синтезируется из различных тринуклеоти дов в синтети ческие фрагменты, составляющие по длине от дека до пентадекануклеотидов, посредством метода получения блок фосфотриэфи ра. Этот ген имеет однониточный когезионный конец для ограничительных эндонуклеаз Eco RI и Bam HI. Вставка его в подходящий выраженный вектор, содержащий b-галактозный ген (b -gae) приводит к хи мерной плазмиде, содержащей А-цепь, связанную с ге ном bgae через метиониновый кодон. Для достижения более высоких уровней выражения используется предпочтительно ген триптофан синтетазы (trp LE') в качестве промотора, а не b-gae ген. Хи мерная плазмида трансфор мируется в E. coli, приво дящей в результате к выражению исходного белка (белка предшественника), то есть b-gae-met- А-цепи (или trp LE'-met- А-цепи, когда используется система промотора trp LE'). Обработка исходного белка цианогенбромидом приводит к расщеплению метиониновой связи с получением после очистки А-цепи человеческого инсулина. Окислительный сульфи толиз приводит к образованию S-сульфо нированной А-цепи, которая используется для комбинации с В-цепью (S-сульфо нат) как описано ниже. Подробное описание химического синтеза генов для А-цепи человеческого инсулина дается в публикации Crea и др., Proc. Natl., Aead. Sci., США, 75, 5 765–5 769, 1978, и публикации, которые даются здесь как ссылочный материал. Для получения полных сведений о выражении в E. coli химически синте зированного гена для А-цепи человеческого инсулина смотри публикацию Goeddel и др. Proc. Natl., Aead. Sci., США, 76, 101–110, 1979, и публикации, приведенные здесь, как ссылочный материал. В. Получение аналога В-цепи [Lys (B28), Pro (B29)]. Для получения сырой пептидиловой смолы используется синтезатор пептида Applied Biosystems 430 А (вк лючая модификацию А.4). Используется 0,5 миллимоля (мМол) исходной твердофазной смолы (tBOC-Thr (BZl) OCH 2 Pam смолы) (0,76 мМол/г х 0,658 г). Все аминокислоты являются защи щенными ВОС, и кроме глутаминовой кислоты и гистидина, все аминокислоты используются непосредственно в том виде, как они получены (то есть в упаковках, содержащих примерно от 2 ммоля защи щенной аминокислоты, поставляемых фирмой Applied Biosystems Inc. Глутаминовая кислота и гисти дин поставляются фирмой Peptides International Corporation и они заключаются в та кие упаковки, что каждая содержит примерно 2 мМоля желаемой защищен ной аминокислоты. После сушки сырой пепти диловой смолы (в вакууме при комнатной температуре в течение ночи) определяется ее вес и сопоставляется с начальным весом для гарантии нужного приве са. Небольшая часть образца подвергается аминокислотному анализу для га рантии того, что желаемые аминокислоты вво дятся в точно заданных количествах. Пептид отщепляется от пептидиловой смолы, и защита с боковой цепи удаляется в результа те перемешивания примерно в те чение 1 часа при 0оС в растворе 10 частей (об/вес) HF (со держащего 5% об/об этилмеркапта на и 5% об/об мета-крезола) с 1 частью пептидиловой смолы. После вакуумной отгонки большей части HF пептид осаждается в простом этиловом эфире. После нескольких промывок простым этиловым эфиром с последующей вакуум ной фильтрацией пептид растворяется примерно в 200 мл 7 Мол. деионизированной воды, содержащей 0,1 Мол. TRIS, 0,1 Мол. Na2SO3 и 0,01 Мол. Na2S4 O6 . Величина рН раствора доводится до 8,5 посредством 5 нормальной NaOH, и этот раствор интенсивно перемеши вается в течение ночи при температуре 4оС. Образующийся S-суль фонированный пептидный раствор вводится в колонку размерами 5 х 215 см, наполненную Sephadex G-25 (тонкоизмельченным) при комнатной температуре. Образец элюируется 50 мМол бикарбонатом аммония с расхо дом его 20 мл/мин при комнатной температуре. Выхо дящий поток анализируется (при 276 нм). Фракции по 20 мл собираются и получают объем желаемых фракций, который затем очищается дополнительно мето дом жидкостной хроматографии высокого разреше ния (HPLC), как описано ниже. Слитые желаемые фракции нагнетаются в колонку 2,5 х 30 см с Iu Pont С8, 9–12 мкм (колонка HPLC) и осуществляется элюирование с использованием линейного ингредиента с уве личивающей ся концентрацией ацетонитрила в 100 мл бикарбоната аммония при комнатной температуре (2,6 мл/мин). Выходящий из колонки поток анализируется (280 нм). Фракции по 25 мл собираются. Выбранные фракции HPLC подвергаются анализу для определения того, какие фракции остаются. Желаемые фракции собираются и лиофилизируются и используются в последующей комбинации с А-цепью, полученной как описано выше. С. Получение человеческого инсулина Pro (B29), Lys (B28). Комбинация цепи А и В осуществляется согласно процедуре, описанной в публикации Chance и др., см. выше. 700 мг S-сульфоната А-це пи, образованного от рекомбинантной ДНК и 140 мг синтетического S сульфо ната В-цепи Lys (В28), Pro (29) (оба получены как описано выше) каждый растворяются в 70 мл и 14 мл, соответственно, 0,1 Мос буфер ного раствора глицина и величина рН каждого доводится до 10,5 посредством 4 н NaOH и затем они охлаждаются до 5оС. Приготавливается 6 мл раствора дитиотреитола (ДТТ) концентрацией 10,5 мг/мл в 0,1 Мол. глициновом буферном раство ре при комнатной температуре, величина рН этого раство ра доводится до 10,5 посредством 5-нормальной NaOH и затем этот раствор охлаждается до 5оС. Эти растворы А-цепи и В-цепи смешиваются друг с другом, после чего в эту смесь быстро вводится 5,21 мл раствора ДТТ (SHISSO3- = 0,90). Этот реакционный раствор перемешивается при температуре 5оС в открытых склянках 200 миллилитровых пробирках для центрифуги в течение 2,5 часов при 5 оС. Вводится 45 мл ледяной уксусной кислоты, и раствор выстаивается при температуре 5оС в течение ночи. Полученная смесь с осадком центрифугируется в течение 20 минут со скоростью 2000 об/мин. при температуре 5оС. Поверхностный слой смешивается с 1 мол. уксусной кислотой, промывается от твердых частиц, и вводится в колонку 5 х 200 см с Sephadex G-50 (сверхтонкого) в 1-молярной уксусной кислоте при 5оС, и элюируется при ускорении силы тяжести. Двадцати минутные фракции элюирования собираются в течение трех дней. Эти фракции анализируются (при 276 нм) и некоторые образцы анализируются методом аналити ческой HPLC. Фракции, содержащие последовательность Lys (B28), Pro (B29) аналога инсулина, сливаются и лиофи лизируются, давая 125 мг образец. Этот образец дополнительно очищается методом жидкостной хроматографии высокого разреше ния (HPLC) с обратимой фазой (с использованием колонки 2,12 х 25 см с Iu Pont СВ при элюирова нии при комнатной температуре со скоростью 2,6 мл/мин с использованием линейного градиента с увеличивающейся концентрацией ацетонитрила в 0,1 Мол NaH 2PO4 , рН = 2,2). Выхо дящий поток анализируется (при 276 нм). Выбранные фракции анализируются аналитическим мето дом HPLC. Желаемые фракции собираются и дополнительно очищаются с использованием рН 7 НР С, как описано ниже. Слитый продукт от HPLC низкого рН разбавляется примерно в два раза в ледяной бане 0,1 молярным (NH4)2HPO4. Величина рН доводится до 7 посредством холодной 2 нормальной NaOH в ледяной бане. Этот образец вводится в ту же колонку и элюируется из нее в тех же условиях, что и при приготовлении образца с низким рН с той разницей, что элюирующий буфер ный раствор в данном случае представляет собой 0,1 Мол (рН 7) (NH 4)2HPO4/ацетонитрил. Слитый продукт от HPLC с рН 7 быстро охлаждается в ледяной бане и в два раза разбавляется 0,1% водным раствором трифто руксусной кислоты (TFA). Вво дится 1 нормальная HCl (холодная, образец в ледяной бане) для снижения величины рН до 3. Образец вводится в колонку Vydae C4 или как возможный вариант в колонку с Iu Pont С8 РН С (2,12 х 25 см) и осуществляется элюирование линейным градиентом с повышающей ся концентрацией ацетонитрила в 0,1% воднoм растворе TFA. Вы хо дящий из колонки продукт анализируется (при 214 нм или 276 нм). Желаемые фракции сливаются и лиофи лизируются, давая образец в количестве 41 мг желаемого аналога со степенью чистоты более 97%, достигаемой путем HPLC с обрати мой фазой. Пример 2. Человеческий инсулин Pro (B29), Lys (B28). Второй способ получения человеческого инсулина Pro (B29), Lys (B28) заключается в использовании ферментного полусинтеза (обратимый протеолиз) для комбинирования des октапептидного инсулина (А1–21 – В1–22) с синтетическим октапепти дом. des-Октапептидный инсулин получается в результате триптического переваривания естественного свиного или человеческого инсулина, как описано в работе Bromer и Chance "Получение и свойства Ies-октапентид-инсулина", Biochem, Biophys Acta 133, 219–223 (1967 г.), которая рассматривается здесь как ссылочная публикация. Синтетический октапептид, Gl y–Phe–Phe–Tyr–Tyr–Lys– Pro–Thr получается методом автомати ческого твердофазного синтеза таким образом, как описано выше. Ies-октапептидный инсулин (435 мг) и синтетический октапептид Gly–Phe–Phe–Tyr–Thr –Lys–Pro–Thr (465 мг) смеши ваются в 15 мл раствора, содержащего 1 часть диметилсульфоксида, 2 части 1,4-бутандиола и 1 часть 0,25 Мол буферного раствора Трис-ацета та, рН=7,3. Пептиды полностью растворяются путем нагревания раство ра на горячей плите. За тем раствор инкубируется при 37оС и добавляется 90 мг свиного трипсина. Раствор время от времени перемешивается в течение 90 минут при температуре 37 оС. Реакция прекращается путем смеши вания раствора с 135 мл 0,05 нормальной HCl. Указанный в назва нии аналог инсулина очищается путем ввода подкисленного раствора, содержаще го этот аналог, в колонку HPLC размерами 2,5 х 25 см, наполненную С-8 Zorbax, и элюирования линейным градиентом с повышающейся концентрацией ацетонитрила в буфер ном растворе 0,1 Мол. одноосновного фосфа та натрия, рН=2,2. Выхо дящий из колонки продукт анализируется при 276 нм). Желаемые фракции сливаются, в два раза разбавляются водой и вводятся в колонку HPLC размерами 1 х 25 см с С-8 Ultrasphere. Этот аналог элюируется линейным градиентом с увеличивающейся концентрацией ацетонитрила в 0,5% водном растворе TFA. Вы ходящий из колонки продукт анализируется (при 276 нм). Желаемые фракции снова сливаются и лиофи лизируются, давая 125 мг очищенного аналога. Данная структура подтверждается аминокислотным анализом (таблица 1) и масс-спектроскопией (MS). MS:5809,2 (Теорети чески: 5808,7). Осуществляется анализ методом Быстрой Атомной бомбардировкой – Масс-спектроскопии с использованием масс-спектрометра с двойной фокусировкой VG–AB–25E, с разрешением примерно 1500. Аналоги человеческого инсулина растворяются в смеси глицерина и триглицерина с содержанием щавелевой кислоты. Для калибровки измерительного прибора, который осуществляет магнитное сканирование в диа пазоне от m/z 5300 до m/z 6500, используется иодид цезия. Полученные данные представлены как среднемассовое значение + 1. Пример 3. Человеческий инсулин Pro (B29), Aba (B28). Свиной des-октапентидный инсулин (384 мг) и синтетический окта пентид Gly–Phe–Phe–Tyr–Thr–Aba– Pro–Thr (362 мг) смеши ваются в 13 мл раствора, содержащего диметилсульфоксид, две части 1,4-бутандиола и одну часть буферного раствора 0,25 Мол Тpис рН 7,3 при температуре 37оС. Вводится свиной трипсин (75 мг). Раствор тщательно смеши вается и время от времени перемешивается в те чение 120 минут при т-ре 37оС. В этот момент времени реакция прекращается в результате вво да этой смеси в 137 мл 0,05 нормальной HCl. Весь этот раствор вво дится в колонку размерами 21 х 250 мм с С-8 Zorbax и продукты элюируются при небольшом ацето нитриловом градиенте в буфер ном растворе 0,1 Мол одноосновно фосфат натрия, рН=2. Соответствующие фракции, которые определены методом аналитической HPLC, сливаются, в два раза разбавляются водой и вводятся в колонку 25 х 300 мм с С-8 Vydac. Желаемый белок элюируется из колонки ацетонитрильным градиентом в 0,5% трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются с получением выхо да 59 мг. Указанная структура подтверждается аминокислотным анализом (таблица 1) и масс-спектроскопией (MS). MS : 5765,7 (Теоретически: 5765,6). Пример 4. Человеческий инсулин Pro (B29), Ala (B29). Свиной des-окта пентидный инсулин (290 мг) и систематический октапептид Gly–Phe–Phe–Tyr–Thr– Ala–Pro–Thr (310 мг) смеши ваются в 10 мл раствора, содержащего одну часть диметилсуль фоксида, две части 1,4-бутандиола и одну часть буферного раствора 0,25 Мол. Трис рН=7,3. Вводится свиной трипсин (60 мг). Раствор тщательно смеши вается и время от времени перемешивается в течение 60 минут при 37оС. В этот момент времени реакция прекращается за счет ввода смеси в 90 мл 0,05 нормальной HCl. Весь раствор вводится в колонку размерами 21 х 250 мм с С-8 Zorbax, и продукты элюируются при небольшом ацетонитриловом градиенте в буферном растворе 0,1 мол. одноосновного фосфата натрия, рН=2. Соответствующие фракции, которые определены методом аналитической HPLC, сливаются, в два раза разбавляются водой и вводятся в колонку размерами 10 х 250 мм с С-8 Ultrasphere. Желаемый белок элюируется из колонки c использованием ацетонитрильного градиента в 0,5%-ной трифторуксусной кислоте. Фракции, содержащие очи щенный аналог инсулина, сливаются и лиофи лизируются, с получением выхо да 59 мг. Указанная структура подтверждается аминокислотным анализом (таблица 1), и масс-спектроскопией (MS). MS : 5752,3 (Теоретически: 5751,6). Пример 5. Человеческий инсулин Pro (B29), Arg (B28). Свиной des-октапептидный инсулин (290 мг) и синтетический октапептид Gly–Phe–Phe–Tyr–Thr–Ala– Pro–Thr (310 мг) смеши ваются в 10 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола, и одну часть буфер ного раствора 0,25 Мол. Трис рН=7,3 при т-ре 37оС. Вводится свиной инсулин (60 мг). Раствор смешивается и время от времени тщательно перемешивается в течение 60 минут при т-ре 37оС. В этот момент времени реакция прекращается в результате ввода этой смеси в 90 мл 0,05 нормальной HCl. Весь раствор вводится в колонку размерами 21 х 250 мм с С-8, и продукты элюируются при небольшом ацетонитриловом градиенте в буферном растворе 0,1 Мол. одноосновного фосфата натрия, рН=2. Соответствующие фракции, которые определены методом аналитической HPLC, сливаются, в два раза разбавляются водой и вводятся в колонку размерами 10 х 250 мм с С-8 Ultrasphere. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5%-ной трифто руксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, и получается 103 мг продукта. Указанная структура подтверждается аминокислотным анализом (таблица 1), и масс-спектроскопическим анализом (MS). MS : 5836,1 (теоретически: 5836,7). Пример 6. Человеческий инсулин Pro (B29), Asn (B28). Свиной des-октапептидный инсулин (490 мг) и синтети ческий октапентид Gly–Phe–Phe–Tyr–Thr–Asn– Pro–Thr (398 мг) смеши ваются в 14 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола и одну часть буферного раствора 0,25 Мол. Трис, рН=7,9, при т-ре 37оС. Вводят свиной трипсин (81 мг). Раствор смеши вается и время от времени тщательно перемешивается в те чение 120 минут при т-ре 37оС. В этот момент времени реакция прекращается за счет ввода смеси в 136 мл 0,05 нормальной HCl. Весь раствор вливается в колонку размерами 21 х 250 мм с С-8 Zorbax, и продукты элюируются при небольшом ацетонитрильном градиенте в буферном раство ре 0,2 Мол. одноосновного фосфата натрия, рН=2. Соответствующие фракции, которые определены посредством аналитической HPLC, сливаются, в два раза разбавляются водой и вводятся в колонку размерами 25 х 300 мм с С-18 Vydae. Желаемый белок элюируется из колонки ацетонитрильным градиентом в 0,5%-ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, и в результате получается 56 мг продукта. Указанная структура подтверждается аминокислотным анализом (таблица 1), и масс-спектроскопией (MS). MS : 5794,7 (Теоретически: 5794,6). Пpимеp 7. Человеческий инсулин Pro (B29), Asp (B28) Свиной des-октапептидный инсулин (400 мг) и синте тический октапептид Gly–Phe–Phe–Tyr–Thr–Asp– Pro–Thr (388 мг) смеши ваются в 13 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола и одну часть буферного раствора 0,25 Мол. Трис, рН=7,3. при 37оС Вводится свиной трипсин (78 мг). Раствор тщательно смешивается и время от времени перемеши вается в течение 120 минут при 37оС. Реакция прекращается в данный момент времени путем ввода этой смеси в 137 мл 0,05 нормальной HCl. Весь раствор вво дится в колонку 21 х 250 мм, с С-8 Zorbax, и продук ты элюируются небольшим ацетонитрильным градиентом в буфер ном растворе 0,1 Мол. одноосновного фосфа та натрия, рН=2. Соответствующие фракции, которые определены аналитической HPLC, сливаются, разбавляются в четыре раза водой и вводятся в колонку размерами 25 х 300 мм с С-18 Vydae. Желаемый белок элюируется из колонки c ацетонитрильным градиентом в 0,1%-ной трифто руксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофи лизируются, и в результа те получается 85 мг продукта. Указанная структура подтверждается аминокислотным анализом (таблица 1), и масс-спектроскопией (MS). MS : 5795,7 (Теоретически: 5795,6). Пример 8. Человеческий инсулин Pro (B29), Asp (B10), Lys (B28) А. Получение В-цепи человеческого инсулина Pro (B29), Asp (B10), Lys (B28). Для получения сырой пептидной смолы используется пептидный синтезатор Applied Biosystems 430 А (включая модифи кацию 1.4). Используется 0,5 Миллимолей (мМол) исходной твердофазной смолы (tBOC-Thr (BZl) OCH 2 Pam смолы) (0,72 мМол/г х 0,705 г). Все используе мые аминокислоты представляют собой защи щенные ВОС, и за исключением глута миновой кислоты, аспарагиновой кислоты и гистидина, все аминокислоты используются в том виде, как они получены (то есть в упаковках, поставляемых фирмой Applied Biosystems Inc.; каждая упаковка содержит примерно 2 мМол. защи щенной аминокислоты). Глутаминовая кислота, аспарагиновая кислота и гистидин являются промышленно получаемыми кислотами, и они заключаются в упаковки таким образом, что каждая упаковка содержит примерно 2 ммоля желаемой защи щенной аминокислоты. Сырая пептидиловая смола высуши вается в вакууме при комнатной температуре в течение ночи и ее вес сопоставляется с весом исходной смолы для гарантии нужного привеса. Небольшая часть образца подвергается аминокислотному анализу для гарантии того, что аминокислоты вводятся в правильных пропорциях. Пептид отщепляется от пепти диловой смолы, и защита боковой цепи удаляется в результате перемешивания в течение примерно одного часа при 0оС в растворе 10 частей (об/вес) HF (содержаще го 5% об/об пара-тиокрезола и 5% об/об мета-крезола) с 1 частью пептидиловой смолы. После удаления наибольшей части HF при вакуумной отгонке пептид осаждается в простом этиловом эфи ре. После нескольких промывок простым этиловым эфиром с последующей вакуумной фильтрацией пептид растворяется примерно в 120 мл 8 Мол гуанидина в HCl, рН=11, содержащего 0,1 Мол. Трис, 35 мг/мл Na2SO3 и 25 мг/мл Na2S4O6. Ве личина рН раствора доводится до 8,8 посредством 5 нормальной NaOH, и раствор интенсивно перемеши вается в те чение 3 часов при комнатной температуре. Полученный S-сульфо нированный пептидный раствор вводится в колонку 5 х 215 см с Sephadex G25 при комнатной температуре. Образец элюируется со скоростью 21 мл/мин при комнатной температуре, с использованием 50 мМол бикарбоната аммония. Выхо дящий из колонки продукт анализируется (при 276 нм). Фракции по 25 мл собираются и сливаются и собранный продукт из желаемых фракций дополнительно очищается путем жидкостной хроматографии вы сокого разрешения (HPLC), как описано ниже. Слитый продукт, состоящий из желаемых фракций, вводится в хроматографическую колонку 2,5 х 30 см с Iu Pont С8, 9–12 мкм и элюирование осуществляется с использованием линейного градиента с уве личивающейся концентрацией ацетонитрила в 100 мМол. бикарбоната аммония при комнатной температуре (2,6 мл/мин). Выхо дящий из колонки продукт анализируется при (280 нм). Фракции (по 25 мл) собираются и сливаются. Отобранные фракции подвергаются анализу методом аналитической HPLC для определения того, какие фракции сохраняются. Желаемые фракции сливаются и лиофи лизируются и используются в указанной ниже комбинации с А-цепью. В. Комбинация В-цепи человеческого инсулина Pro (B29) Lys (B28), Asp (B10) c А-цепью человеческого инсулина. Комбинация А и В цепей осуществляется согласно процедуре, описанной в работе Chance и др., см. выше. Два грамма S-сульфоната А-цепи, образованного от рекомбинантной ДНК, и 400 мг синтетического S-сульфо ната В-цепи Pro (В29), Asp (В10), Lys (В28) каждого растворяются в 200 мл и 40 мл соответственно 0,1 Мол. буферного раствора глицина при комнатной температуре, величина рН каждого доводится до 10,5 посредством 5 нормальной NaOH и затем каждый охлаждается до 5оС. Приготавливается раствор дитиотреито ла (ДТТ) концентрацией 15,5 мг/мл в 0,1 Мол. буферном растворе глицина при комнатной температуре, величина рН доводится до 10,5 посредством 5-нормальной NaOH и затем раствор охлаждается до 5оС. Растворы А-цепи и В-цепи смеши ваются и затем в эту смесь быстро вводится 15,9 мл раствора ДТТ (SU/SSO3- = 1,0). Реакционный раствор перемешивается при 4 оС в открыты х 200-миллилитровых стеклянных пробирках для центрифугирования в течение 19,6 часа при 4оС. Вводится ледяная уксусная кислота (129 мл), и раствор выстаивается при т-ре 4оС в те чение одного часа. Полученная смесь с выпавшим осадком центрифугируется в те чение 30 минут со скоростью 2000 об/мин при т-ре 4оС. Поверхностный слой смеши вается с 292 мл милли- Q-воды и 73 мл ацетонитрила и эта смесь дополнительно очищается посредством HPLC с обратимой фазой (с использованием колонки 2,5 х 30 см с Vydae С18 с элюи рованием при комнатной температуре со скоростью 2,6 мл/мин с использованием линейного ингредиента с уве личивающейся концентрацией ацетонитрила в 0,1 Мол NaH2PO4 рН=2,1). Выхо дящий из колонки продукт анализируется при 280 нм. Выбранные фракции анализируются методом аналитической HPLC, и желаемые фракции сливаются и разбавляются в два раза 0,1%-м водным раствором трифторуксусной кислоты (TFA), затем слитый продукт вво дится в хроматографи ческую колонку (0,1 x 25 см) с Ultrasphere, и осуществляется элюирова ние линейным градиентом с уве личивающейся концентрацией ацетонитрила в 0,1% водном растворе TFA. Выхо дящий из колонки продукт анализируется при 280 нм. Выбранные фракции анализируются методом аналитической HPLC, и желаемые фракции сливаются и повторно очищаются с использованием той же колонки и в тех же условиях, что указаны выше при несколько отличающемся градиенте ацето нитрила. Соответствующие фракции сливаются и лиофи лизируются и в ре зульта те получается 65 мг аналога инсулина со степенью чистоты бо лее чем 93% в результате HPLC с обратимой фазой. Указанная структура подтверждается аминокислотным анализом (таблица 1) и методом масс-спектроскопии (MS). MS: 5786,1 (Теоретически: 5786,7). Пример 9. Человеческий инсулин Pro (B29), Cya (B28). Осуществляется окисление надмуравьиной кислоты с целью превраще ния цистеина октапептида в форму цистеиновой кислоты. Синтетический октапептид Gly–Phe–Phe–Tyr–Lys–Pro–Thr (363 мг) растворяется в 36 мл свежеприготовленной надмуравьиной кислоты в охлажденной льдом колбе, и раствор осторожно перемеши вается в те чение одного часа. Окисленный мате риал разбавляется в де сять раз водой и лиофи лизируется. В данном полусинтезе используется этот лиофилизат. Свиной des-октапептидный инсулин (222 мг) и синтетический октапептид Gly–Phe–Phe–Tyr–Thr–Cya– Pro–Thr (225 мг) смеши ваются в 18 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола и одну часть буферного раствора 0,25 Ммол. Трис, рН=7,3, при т-ре 37оС. Вводится свиной трипсин (45 мг). Раствор тщательно смеши вается и время от времени перемешивается в те чение 120 минут при т-ре 37оС. В этот момент времени реакция прекращается в результате вво да этой смеси в 242 мл 0,05 нормальной HCl. Весь раствор вводится в колонку 21 х 250 см, с С–8 Zorbax, и продукты элюируются слабым ацетонитрильным градиентом в буферном растворе 0,1 Мол. одноосновного фосфа та натрия, рН=2. Подхо дящие фракции, которые определены методом аналитической HPLC, сливаются, разбавляются в два раза водой и вводятся в колонку 25 х 300 мм, с С–18 Vydae. Обессоленный белок элюируется из колонки с использованием ацетонитрильного градиента в 0,5%-ной трифто руксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, и в результате получается выход продукта 16 мг. Указанная структура продукта подтверждается аминокислотным анализом (таблица 1) и методом масс-спектроскопии (MS). MS: 5831,5 (Теоретически: 5831,7). Пример 10. Человеческий инсулин Pro (B29), Cln (B28). Свиной des-октапептидный инсулин (290 мг) и синте тический окта пептид Gl y–Phe–Phe–Tyr–Thr–Cln– Pro–Thr (310 мг) смеши ваются в 10 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола, и одну часть буферного раствора 0,25 Ммол. Трис, рН=7,3, при т-ре 37оС. Вво дится свиной трипсин (60 мг). Раствор тщательно смешивается и время от времени перемеши вается в течение 60 минут при 37оС. В этот момент времени реакция прекращается в результате вво да смеси в 90 мл 0,05 н HCl. Весь раствор вводится в колонку 21 х 250 мм, с С–8 Zorbax, и продукты элюируются с использованием слабого ацетонитрильного градиента в буфер ном растворе 0,1 Мол. одноосновного фосфа та натрия, рН=2. Подхо дящие фракции, которые определены путем аналитической HPLC, сливаются, разбавляются в два раза водой, вводятся в колонку размерами 10 х 250 мм, с С–8 Ultraphase. Обессоленный белок элюируется из колонки ацето нитрильным градиентом в 0,5% трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, и получается выход продукта 87 мг. Указанная структура подтверждается аминокислотным анализом (таблица 1) и масс-спектроскопией (MS). MS: 5809,4 (Теоретически: 5808,6). Пример 11. Человеческий инсулин Pro (B29), Glu (B28). Свиной des-октапептидный инсулин (402 мг) и синтетический октапептид Gly–Phe–Phe–Tyr–Thr–Glu– Pro–Thr (398 мг) смешивается в 14 мл раствора, содержащего одну часть диметилсульфоксида, 2 части 1,4-бутандиола и одну часть буферного раствора 0,25 Ммол. Трис, при т-ре 37оС. Вводится свиной трипсин (80 мг). Раствор тща тельно смешивается и время от времени перемешивается в течение 120 минут при 37оС. В этот момент времени реакция прекращается путем ввода этой смеси в 136 мл 0,05 нормальной HCl. Весь раствор вводится в колонку 21 х 250 мм с С–8 Zorbax, и продукты элюируются в слабом ацетонитрильном градиенте в буферном растворе 0,1 Мол. одноосновного фосфа та натрия, рН=2. Подхо дящие фракции, которые определены путем аналитической HPLC, сливаются, разбавляются в четыре раза водой и вводятся в колонку 25 х 300 мм, с С–18 Vydae. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5%-ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, и в ре зультате получается выход продук та 59 мг. Данная структура подтверждается аминокислотным анализом (таблица 1) и методом масс-спектроскопией (MS). MS: 5809,6 (Теоретически: 5809,6). Пример 12. Человеческий инсулин Pro (B29), Gly (B28). Свиной des-октапептидный инсулин (412 мг) и синте тический окта пептид Gl y–Phe–Phe–Tyr–Thr–Gly– Pro–Thr (376 мг) смеши ваются в 13 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола и одну часть буфер ного раствора 0,25 Мол. Трис, рН=7,3. Вводится свиной трипсин (79 мг). Раствор тщательно смеши вается и время от времени перемешивается в течение 180 минут при т-ре 37оС. В этот момент времени реакция прекращается путем ввода этой смеси в 147 мл 0,05 нормальной HCl. Весь раствор вводится в колонку 21 х 250 мм с С–8 Zorbax, и продук ты элюируются слабым ацетонитрильным градиентом в 0,1 Мол. буферном растворе одноосновного фосфа та натрия, рН=2. Подхо дящие фракции, которые определены аналитической HPLC, сливаются, разбавляются в четыре раза водой и вводятся в колонку 25 х 300 мм, с С–18 Vydae. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,1%-ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, и в результате получается 11 мг продукта. Данная структура продукта подтверждается аминокислотным анализом (таблица 1) и масс-спектpогpафией (MS). MS: 5737,2 (Теоретически: 6,537,6). Пример 13. Человеческий инсулин Pro (B29), His (B28). Свиной des-октапептидный инсулин (400 мг) и синтетический октапептид Gly–Phe–Phe–Tyr–Thr–His– Pro–Thr (398 мг) смеши ваются в 13 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола и одну часть буферного раствора 0,25 Мол. Трис, рН=7,3, при т-ре 37оС. Вводится свиной трипсин (79 мг). Раствор тщательно смеши вается и время от времени перемешивается в те чение 120 минут при 37оС. В этот момент времени реакция прекращается путем ввода смеси в 237 мл 0,05 нормальной HCl. Весь раствор вводится в колонку 21 х 250 мм С–8 Zorbax, и продукты элюируются в слабом ацетонитрильном градиенте в буферном растворе 0,1 Мол. одноосновного фосфа та натрия, рН=2. Подхо дящие фракции, которые определены аналитической HPLC, сливаются, разбавляются в четыре раза водой, вводятся в колонку 25 х 300 мм С–18 Vydae. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,1%-ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофи лизируются, и в результате получается выход продукта 79 мг. Данная структура подтверждается аминокислотным анализом. (Таблица 1) и масс-спектроскопией (MS). MS: 5816,9 (Теоретически: 5817,7). Пример 14. Человеческий инсулин Pro (B29), Jle (B28). Свиной des-октапептидный инсулин (409 мг) и синте тический октапептид Gly–Phe–Phe–Tyr–Thr–Jle– Pro–Thr (398 мг) смеши ваются в 13 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола, и одну часть буферного раствора 0,25 Мол. Трис, рН=7,3, при 37оС. Вводится свиной трипсин (81 мг). Раствор тщательно смешивается и время от времени перемеши вается в течение 120 минут при т-ре 37оС. В этот момент времени реакция прекращается путем ввода смеси в 136 мл 0,05 нормальной HCl. Весь раствор вводится в колонку 21 х 250 мм с С–8 Zorbax, и продукты элюируются в слабом ацетонитрильном градиенте в буфер ном растворе 0,1 Мол. одноосновного фосфа та натрия, рН=2. Подхо дящие фракции, определяемые аналитической HPLC, сливаются, разбавляются в два раза водой, и вводятся в колонку 25 х 300 мм, с С–18 Vydae. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5%-ной трифторуксусной кислоте. Фракции, содержащие очищенный инсулиновый аналог, сливаются и лиофи лизируются, и в результате получается выход продукта 57 мг. Данная структура подтверждается аминокислотным анализом (таблица 1) и масс-спектроскопии (MS). MS: 5793,7 (Теоретически: 5793,7). Пример 15. Человеческий инсулин Pro (B29), Leu (B28). Свиной des-октапептидный инсулин (418 мг) и синтетический октапептид Gly–Phe–Phe–Tyr–Thr–Leu– Pro–Thr (410 мг) смеши ваются в 14 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола и одну часть буферного раствора 0,25 Мол. Трис, рН=7,3, при т-ре 37оС. Вводится свиной трипсин (83 мг). Раствор тщательно смеши вается и время от времени перемешивается в те чение 120 минут при 37оС. В этот момент реакция прекращается путем ввода этой смеси в 136 мл 0,05 нормальной HCl. Весь раствор вводится в колонку 21 х 250 мм с С–8 Zorbax, и продукты элюируются в слабом ацетонитрильном градиенте в 0,1 Мол. буферном растворе одноосновного фосфата натрия, рН=2. Соответствующие фракции, определяемые аналитической HPLC, сливаются, разбавляются в четыре раза водой и вводятся в колонку 25 х 300 мм, с С–18 Vydae. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5%-ной трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, и в результате получается выход продукта 74 мг. Структура этого продукта подтверждается аминокислотным анализом (таблица 1) и масс-спектроскопией (MS). MS: 5793,8 (Теоретически: 5793,7). Пример 16. Человеческий инсулин Pro (B29), Nle (B28). Свиной des-октапептидный инсулин (290 мг) и синте тический окта пептид Gl y–Phe–Phe–Tyr–Thr–Nle– Pro–Thr (310 мг) смеши ваются в 10 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола, и одну часть буферного раство ра 0,25 Мол. Трис, рН=7,3, при т-ре 37оС. Вводят свиной трипсин (60 мг). Раствор тщательно смешивается и время от времени перемеши вается в течение 60 минут при 37оС. В этот момент реакция прекращается путем ввода смеси в 90 мл 0,05 нормальной HCl. Весь раствор вводится в колонку 21 х 250 мм с С–8 Zorbax, и продукты элюируются в слабом ацетонитрильном градиенте в 0,1 Мол. буферном растворе одноосновного фосфа та натрия, рН=2. Подхо дящие фракции, которые определены аналити ческой HPLC, сливаются, разбавляются в два ра за водой, и вво дятся в колонку 10 х 250 мм с С–8 Ultrasphere. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5%-ной трифторуксусной кислоте. Фракции, содержащие очищенный инсулиновый аналог, сливаются и лиофи лизируются, и в результа те получается выход продук та 54 мг. Указанная структура подтверждается аминокислотным анализом (таблица 1) и масс-спектроскопией (MS). MS: 5794,6 (Теоретически: 5793,7). Пpимеp 17. Человеческий инсулин J–Lys (B29). Свиной des-октапептидный инсулин (392 мг) и синтетический октапептид Gly–Phe–Phe–Tyr–Thr–Pro– J–Lys–Thr (387 мг) смеши ваются в 13,5 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола, и одну часть буферного раствора 0,25 Мол. Трис, рН=7,3, при т-ре 37оС. Вводятя свиной трипсин (78 мг). Раствор тщательно смеши вается и время от времени перемеши вается в те чение 120 минут при 37оС. В этот момент реакция прекращается путем ввода смеси в 136,5 мл 0,05 нормальной HCl. Весь раствор вводится в колонку 21 х 250 мм с С–8 Zorbax, и продукты элюируются в слабом ацетонитрильном градиенте в 0,1 Мол. буферном раство ре одноосновного суль фата натрия, рН=2. Подхо дящие фракции, определяемые аналитической HPLC, сливаются, разбавляются в четыре раза водой и вводятся в колонку 25 х 300 мм, с С–18 Vydae. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5% трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, и в результате получается 94 мг продукта. Данная структура подтверждается аминокислотным анализом (таблица 1) и масс-спектроскопией (MS). MS: 5809,0 (Теоретически: 5808,7). Пpимеp 18. Человеческий инсулин Pro (B29), Met (B28). Свиной des-октапептидный инсулин (350 мг) и синтетический октапептид Gly–Phe–Phe–Tyr–Thr–Met– Pro–Thr (366 мг) смеши ваются в 12 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола и одну часть буферного раствора 0,25 Мол. Трис, рН=7, при 37оС. Вводится свиной трипсин (71 мг). Раствор тща тельно смешивается и время от времени перемеши вается в течение 120 минут при тре 37оС. В данный момент времени реакция прекращается путем ввода смеси в 118 мл 0,05 нормальной HCl. Весь раствор вводится в колонку 21 х 250 мм с С–8 Zorbax, и продукты элюируются в слабом ацетонитрильном градиенте в 0,1 Мол. буферном растворе одноосновного фосфа та натрия, рН=2. Подхо дящие фракции, определяемые аналитической HPLC, сливаются, разбавляются в четыре раза водой и вводятся в колонку 25 х 300 мм С–18 Vydae. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,1%-ной трифторуксусной кислоте. Фракции, содержащие очищен ный аналог инсулина, сливаются и лиофилизируются, и в результате получается 72 мг продук та. Указанная структура подтверждается аминокислотным анализом (таблица 1) и масс-спектроскопией (MS). MS: 5811 (Теоретически: 5811,7). Пример 19. Человеческий инсулин Pro (B29), Orn (B28). Свиной des-октапептидный инсулин (290 мг) и синтетический октапептид Gly–Phe–Phe–Tyr–Thr–Orn– Pro–Thr (310 мг) смеши ваются в 10 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола, и одну часть буферного раствора 0,25 Мол. Трис, рН=7,3, при 37оС. Вводится свиной трипсин (60 мг). Раствор тщательно смешивается и время от времени перемеши вается в течение 90 минут при т-ре 37оС. В этот момент времени реакция прекращается путем ввода данной смеси в 90 мл 0,05 нормальной HCl. Весь раствор вводится в колонку 21 х 250 мм с С–8 Zorbax, и продукты элюируются в слабом ацетонитрильном градиенте в 0,1 Мол. буферном растворе одноосновного фосфа та натрия, рН=2. Подхо дящие фракции, определяемые аналитической HPLC, сливаются, разбавляются в четыре раза водой и вводятся в колонку 10 х 250 мм, С–8 Ultraphase. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5% трифторуксусной кислоте. Фракции, содержащие очищенный инсулиновый аналог, сливаются и лиофи лизируются и в результате получается 89 мг продукта. Структура продук та подтверждается аминокислотным анализом (таблица 1) и масс-спектроскопией (MS). MS: 5795,2 (Теоретически: 5794,7). Пример 20. Человеческий инсулин Pro (B29), Phe (B28). Свиной des-октапептидный инсулин (290 мг) и синтетический октапептид Gly–Phe–Phe–Tyr–Thr–Phe– Pro–Thr (310 мг) смеши ваются в 10 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола и одну часть буферного раство ра 0,25 Мол. Трис, рН=7,3, при 37оС. Вводится свиной трипсин (60 мг). Раствор тщательно смешивается и время от времени перемеши вается в течение 30 минут при 37оС. В этот момент реакция прекращается путем ввода смеси в 90 мл 0,05 нормальной HCl. Весь раствор вводится в колонку 21 х 250 мм С–8 Zorbax, и продукты элюируются в слабом ацетонитрильном градиенте в 0,1 Мол. буферном растворе одноосновного фосфа та натрия, рН=2. Подхо дящие фракции, определяемые аналитической HPLC, сливаются, разбавляются в четыре раза водой и вводятся в колонку 25 х 300 мм, с С–18 Vydae. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,1% трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, и в результате получается 17 мг продукта. Структура продукта подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией (MS). MS: 5827,9 (Теоретически: 5827,7). Пример 21. Человеческий инсулин Pro (B29). Свиной des-октапептидный инсулин (339 мг) и синтетический октапептид Gly–Phe–Phe–Tyr–Thr–Pro– Pro–Thr (363 мг) смеши вают в 9 мл раствора, содержаще го одну часть ди метилсульфоксида, две части 1,4бутандиола и одну часть буферного раствора 0,25 Мол. Трис, рН=7,3, при 37оС. Вводится свиной трипсин (70 мг). Раствор тщательно смеши вается и время от времени перемешивается в течение 80 минут при 37оС. Данная реакция в этот момент времени прекращается путем ввода смеси в 108 мл 0,05 н. HCl. Весь раствор вводится в колонку 10 х 250 мм с С–8 Zorbax, и продукты элюируются слабым ацетонитрильным градиентом в 0,1 Мол. буфер ном раство ре одноосновного фосфата натрия, рН=2. Подхо дящие фракции, кото рые определены аналитической HPLC, сливаются, разбавляются в два раза водой и вводятся в колонку 10 х 250 мм с С–8 Ultrasphere. Обессоленный белок элюируется из колонны ацетонитрильным градиентом в 0,5%-ной трифто руксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются, и лиофи лизируются, и в результате получается 97 мг продукта. Структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией (MS). MS: 5778,6 (Теоретически: 5777,6). Пример 22. Человеческий инсулин Pro (B29), Ser (B28). Свиной des-октапептидный инсулин (412 мг) и синтетический октапептид Gly–Phe–Phe–Tyr–Thr–Ser– Pro–Thr (390 мг) смеши ваются в 13 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола и одну часть буферного раство ра 0,25 Мол. Трис, рН=7,3, при 37оС. Вводится свиной трипсин (80 мг). Раствор тщательно смешивается и время от времени перемеши вается в течение 120 минут при 37оС. В данный момент времени реакция прекращается путем ввода смеси в 1337 мг 0,05 н. HCl. Весь раствор вводится в колонку 21 х 250 мм с С–8 Zorbax, и продукты элюируются в слабом ацетонитрильном градиенте в 0,1 Мол. буферном растворе одноосновного фосфата натрия, рН=2. Подхо дящие фракции, определяемые аналитической HPLC, сливаются, разбавляются в два раза водой и вводятся в колонку 25 х 300 мм с С–18 Vydae. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5%-ной трифторуксусной кислоте. Фракции, содержащие очищен ный аналог инсулина, собираются и лиофилизируются с получением выхо да продукта 37 мг. Структура продукта подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией (MS). MS: 5768,1 (Теоретически: 5767,6). Пример 23. Человеческий инсулин Pro (B29), Thr (B28). Свиной des-октапептидный инсулин (437 мг) и синте тический окта пептид Gl y–Phe–Phe–Tyr–Thr–Thr– Pro–Thr (420 мг) смешиваются в 14,5 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола, и одну часть буферного раствора 0,25 Мол. Трис, рН=7,3, при 37оС. Вводится свиной трипсин (86 мг). Раствор тщательно смеши вается и время от времени перемешивается в те чение 120 минут при 37оС. В этот момент времени реакция прекращается путем ввода смеси в 135,5 мл 0,05 нормальной HCl. Весь раствор вводится в колонку 21 х 250 мм с С–8 Zorbax, и продукты элюируются небольшим ацетонитрильным градиентом в буферном растворе 0,1 Мол. одноосновного фосфа та натрия, рН=2. Подхо дящие фракции, определяемые аналитически HPLC, сливаются, в два ра за разбавляются водой и вводятся в колонку 25 х 300 мм с С–18 Vydae. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5% трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсу лина, сливаются и лиофилизируются, и получается выход продукта 78 мг. Данная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией (MS). MS: 5781,9 (Теоретически: 5781,6). Пример 24. Человеческий инсулин Pro (B29), Trp (В28). Свиной des-октапептидный инсулин (310 мг) и синте тический окта пептид Gl y–Phe–Phe–Tyr–Thr–Thr– Trp–Pro–Thr (325 мг) смеши ваются в 10,5 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола, и одну часть буферного раствора 0,28 Мол. Трис, рН=7,3, при 37оС. Вво дится свиной трипсин (64 мг). Раствор тщательно смеши вается и время от времени перемешивается в те чение 120 минут при 37оС. В этот момент времени реакция прекращается путем ввода смеси в 140 мл 0,05 нормальной HCl. Весь раствор вводится в колонку 21 х 250 мм с С–8 Zorbax, и продукты элюируются слабым ацетонитрильным градиентом в 0,1 Мол. буфер ном растворе одноосновного фосфата натрия, рН=2. Подхо дящие фракции, которые определены аналити ческой HPLC, сливаются, в два раза разбавляются водой и вводятся в колонку размерами 25 х 300 мм с С–18 Vydae. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5% трифторуксусной кислоте. Фракции, содержащие очищенный инсулин, сливаются и лиофилизируются, и в результате получается выход продукта 47 мг. Данная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией (MS). MS: 5866,2 (теоретически: 5866,7). Пример 25. Человеческий инсулин Pro (B29), Tyr (В28). Свиной des-октапептидный инсулин (391 мг) и синте тический окта пептид Gl y–Phe–Phe–Tyr–Thr–Thr– Tyr–Pro–Thr (400 мг) смеши ваются в 13 мл раствора, содержащего одну часть диметилсуль фоксида, две части 1,4-бутандиола и одну часть буфер ного раство ра 0,25 Мол. Трис, рН=7,3, при 37 оС. Вводится свиной трипсин (79 мг). Раствор тщательно смеши вается и время от времени перемешивается в те чение 120 минут при 37оС. В этот момент времени реакция прекращается путем ввода смеси в 137 мл 0,05 нормальной HCl. Весь раствор вводится в колонку размерами 21 х 250 мм с С–8 Zorbax, и продукты элюируются слабым ацетонитрильным градиентом в буферном растворе 0,1 Мол. одноосновного фосфата натрия, рН=2. Подхо дящие фракции, определяемые аналитической HPLC, сливаются, разбавляются в два раза водой, и вводятся в колонку размерами 25 х 300 мм с С–18 Vydae. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5% трифто руксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофи лизируются, и в результате получается выход продукта 30 мг. Данная структура подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией (MS). MS: 5843,7 (Теоретически: 5843,7). Пpимеp 26. Человеческий инсулин Pro (B29), Tyr (B28). Свиной des-окта пептидный инсулин (400 мг) и синте тический октапептид Gl y–Phe–Phe–Tyr–Tyr–Tyr– Pro–Thr (383 мг) смеши ваются в 12 мл раствора, содержаще го одну часть диметилсульфоксида, две части 1,4-бутандиола, и одну часть буферного раствора 0,25 Мол. Трис, рН=7,3, при 37оС. Вводится свиной трипсин (78 мг). Раствор тщательно смешивается и время от времени перемеши вается в течение 120 минут при 37оС. В этот момент времени реакция прекращается путем ввода смеси в 238 мл 0,05 нормальной HCl. Весь раствор вводится в колонку 21 х 250 мм с С–8 Zorbax, и продукты элюируются слабым ацетонитрильным градиентом в 0,1 Мол. буфер ном растворе одноосновного фосфата натрия, рН=2. Подхо дящие фракции, определяемые аналитической HPLC, сливаются, разбавляются в четыре раза водой и вводятся в колонку 25 х 300 мм с С–18 Vydae. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,1% трифторуксусной кислоте. Фракции, содержащие очищенный аналог инсулина, сливаются и лиофилизируются, и в результате получается выход продукта 74 мг. Структура продук та подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией (MS). MS: 5780,0 (Теоретически: 5799,6). Пример 27. Человеческий инсулин Pro (B29), Nva (B28). С ви н о й d e s -о к т а п е п ти д н ы й и н с ули н (292 мг) и синтетический октапептид Gly–Phe–Phe–Tyr–Thr–Nva–Pro–Thr (279 мг) смеши ваются в 10 мл раствора, содержащего одну часть диметилсульфоксида, две части 1,4-бутандиола, и одну часть буфер ного раствора 0,25 Мол. Трис, рН=7,3, при 37оС. Вво дится свиной трипсин (57 мг). Раствор тщательно смешивается и время от времени перемеши вается в те чение 120 минут при 37 оС. В этот момент времени реакция прекращается путем добавления смеси в 240 мл 0,05 нормальной HCl. Весь раствор вво дится в колонку 21 х 250 мм с С–8 Zorbax, и продук ты элюируются слабым ацетонитрильным градиентом в буферном растворе 0,1 Мол. одноосновного фосфа та натрия, рН=2. Подхо дящие фракции, кото рые определены аналитической HPLC, сливаются, разбавляются в два раза водой, и вводятся в колонку 25 х 30 мм с С–18 Vydae. Обессоленный белок элюируется из колонки ацетонитрильным градиентом в 0,5%-ной трифторуксусной кислоте. Фракции, содержащие очищен ный инсулиновый аналог, сливаются и лиофилизируются, в результате достигается выход продукта 51 мг. Структура продукта подтверждается аминокислотным анализом (табл. 1) и масс-спектроскопией (MS). MS: 5870,0 (Теоретически: 5779,6). Пример 28. Осуществляя процедуры так, как описано в данной заявке, получают нижеследующие дополнительные аналоги инсулина: (a) Человеческий инсулин Asp(BI), Lys(B28)m Pro(B29) (b) Человеческий инсулин des(Phe–BI), Lys(B28), Dro(B29) (c) Человеческий инсулин des(Phe–BI), Asp(B10), Lys(B28), Pro(B29) (d) Человеческий инсулин des(Phe–B1, Val–B2), Lys(B28), Pro(B29) (e) Человеческий инсулин des(Phe–B1,Val–B2),Asp(B10),Lys(B28) Pro(B29) (f) Человеческий инсулин Gly(A21), Asp(B10), Lys(B28), Pro(B29) (g) Человеческий инсулин Ala(A21), Asp(B10), Lys(B28), Pro(B29) (h) Человеческий инсулин des(Thr–B30), Lys(B28), Pro(B29) (i) Человеческий инсулин Asp(B10), Arg(B28), Pro(B29) (j) Человеческий инсулин Ala(A21), Arg(B28), Pro(B29) (k) Человеческий инсулин Asp(B1), Arg(B28), Pro(B29) Пpимеp 29. Человеческий инсулин Pro (B29), Lys (B28). А. Построение плазмиды рС Р126. 1. Выделение плазмиды рКС 283. Леофи лы E. coli К12 ВЕ1201/рКС 283 получены из Северной Региональной Научно-исследовательской Лаборато рии, Пеория, Иллинойс 61 604, где они имеют официальный номер NRRL В-15830. Эти лиофи лы деканти руются в пробирки, содержащие 10 мл среды LB (10 г Бакто-триптона, 5 г Бакто-дрожжевого экстракта и 10 г NaCl на литр; рН доводится до 7,5), и инкубируются в течение двух часов при т-ре 32оС; в течение этого времени культуры доводятся до концентрации 50 мкг/мл в ампициллине, и затем они инкубируются при 32оС в течение ночи. Клетки E. coli К12 ВЕ1201/рКС 283 выращи вают при 32оС, поскольку они содержат температурочувствительный подавляющий ген, интегрированный в клеточную ДНК. При использовании в процедурах выделения плазмиды (как описано в нижеследующих примерах), клеток, кото рые содержат подавляющий ген лямбда р дикого ти па, или не содержат промотора лямбда р, температура инкубации составляет 37оС. Небольшая часть выращенной за ночь культуры помещается в чашки с LB-агаром (среда LB с 15 г/л Бакто-агара), содержащие 50 мкг/мл ампициллина, так, чтобы получился один единственный изолят колонии E. coli К12 ВЕ1201/рКС 283. Эта единственная полученная колония инокулируется в 10 мл среды LB, содержащей 50 мкг/мл ампициллина, и инкубируется в течение ночи при 32оС при интенсивном встряхи вании. 10 мл ночной культуры инокулируется в 500 мл среды LB, содержащей 50 мкг/мл ампициллина и инкубируется при 32оС с интенсивным встряхиванием до тех пор, пока культура не дости гает ста ционарной фазы. Принята нижеследующая процедура, описанная в работе Мaniatis и др., 1982 г., Moulular Cloning (Cold Spring Harbor Laboratory). Клетки собираются путем центрифугирования с ускорением 4000 g в течение 10 минут при 4 оС, и поверхностный слой удаляется. Клеточный осадок промывается в 100 мл охлажденного льдом буферного раствора STE (0,1 Мол. NaCl; 10 мМол. Трис-HCl, рН=7,8; и 1 мМол. ЭДТА). После промывки клеточный осадок повторно суспензируется в 10 мл раствора 1 (50 мМол. глюкозы, 25 мМол. Трис-HCl, рН=8,0; и 10 мМол ЭДТА), со держащего 5 мг/мл лизозима и оставляется при комнатной температуре в те чение 10 минут. Затем в обработанные лизозимом клетки вводится 20 мл раство ра 2 (2-нормальная NaOH и 1% SDS), и раствор подвергается осторожному инверсионному перемеши ванию. Смесь инкубируется на льду в те чение 10 минут. Пятнадцать мл охлажденного льдом 5 Мол. ацетата калия, pH=4,8, вводится в ли зированную клеточную смесь, и раствор подвергается инверсионному перемешива нию. Приготавливается 5 Мол. раствор ацетата калия путем ввода 11,5 мл ледяной уксусной кислоты в 28,5 мл воды и 60 мл 5 Мол. ацета та калия; полученный раствор является 3 Мол. в отноше нии концентрации калия и 5 Молярным в отноше нии концентрации ацета та. Лизированная клеточная смесь центрифугируется в центрифуге Бекмана SW 27 (или его эквива ленте) со скоростью 20000 об/мин в течение 20 минут при температуре 4оС. Клеточная ДНК и клеточные продукты распада образуют осадок на дне пробирки. Извлекается примерно 36 мл поверхностного слоя, и вводится 0,6 объема изопропанола, осуществляется перемешивание, и полученный раствор выдерживается при комнатной температуре в течение 15 минут. Плазмидная ДНК извлекается путем центрифугирования с ускорением 12000 g в те чение 30 минут при комнатной температуре. Поверхностный слой декантируется, и осадок ДНК промывается 70%-ным этанолом при комнатной температуре. Затем этанольная промывка де кантируется и осадок высуши вается в эксикаторе. Этот осадок снова суспензируется в 8 мл буферного раствора ТЕ (10 мл Трис-HCl, рН=8,0 и 1 мл этилендиаминтетраацетата (ЭДТА)). В раствор ДНК вво дится 8 граммов CaCl. Вводится примерно 0,8 мл раство ра этилиденбромида в воде на каждые 10 мл раствора CaCl-ДНК. Конечная плотность этого раствора составляет примерно 1,55 г/мл, и концентрация этилиденбромида составляет примерно 600 мкг/мл. Данный раствор подается в пробирку для центрифугирования типа Бекмана 50, наполняется сверху парафи новым маслом, уплотняется и центрифугируется со скоростью 45000 об/мин в течение 24 часов при 20оС. После центрифугирования в обычном свете наблюдается два слоя ДНК. После удаления крышки с пробирки нижний слой ДНК удаляется посредством шприца с гиподермической иглой # 21, вставляемой в пробирку для центрифуги рования сбоку. Этиленбромид уда ляется путем нескольких экстракций 1-бута нолом, насыщенным водой. CaCl удаляется путем диализа с ТЕ буфе ром. После экстракций буферным раствором фенола, а затем хлороформом, ДНК осаждается, промывается 70%-ным этанолом и высуши вается. Получается примерно 1 мг плазмиды рКС 283, и она выдерживается при т-ре 4оС в буферном растворе ТЕ концентрацией примерно 1 мкг/мкл. Ограничительная точка и функциональная карта плазмиды рКС 283 представлена на фиг. 1 данной заявки. 2. Построение плазмиды рКС 283 РХ. Примерно 10 мкл ДНК плазмиды рКС 283, приготовленной, как описано в примере 1, смешивается с 20 мкл 10 х среднесолевого ограничительного буфера (500 мМол. NaCl; 100 мМол. Трис-HCl, рН=7,5; 100 мМол. MgCl2; и 10 мМол. ДТТ), 20 мкл BSA (1 мг/мл), 5 мкл ограничительного фер мента PVU II (~ 50 единиц, как определено в Лабораториях Bethesda Research Laboratories (BRL), из которой все эти ограничительные ферменты получены), и 145 мкл воды, и эта реакционная смесь инкубируется при 37оС в течение 2 часов. Реакции ограничительного фермента, описанные в данной заявке, завершаются обычным образом путем экстракции фенолом, а затем хло роформом, после чего происхо дит осаждение ДНК, который промывается этанолом и снова суспензируется в буферном растворе ТЕ. После заверше ния выпаривания с PVU, как описано выше, вы варенная с PVU II плазмидная рКС 283 ДНК осаждается и затем снова суспензируется в 5 мкл буфер ного раствора ТЕ. Примерно 500 пикомолей (рМ) связывающи х звеньев Xhol (5'CCTCGAGG3') киназируются в смеси, содержащей 10 мкл 5 х киназного буферного раствора (300 мМол. Трис-HCl, pH=7,8; 50 мМол. MgCl2; и 25 мМол ДТТ), 5 мкл 5 Мол. АТР, 24 мкл H2O, 0,5 мкл полинуклеоти докиназы Т4 (примерно 2,5 единиц, как определено в P-L Biochemicals), 5 мкл 1 мг/мл BSA и 5 мкл 10 мМол спермидина, путем инкубирования этой смеси при 37оС в течение 30 минут. Примерно 12,5 мкл киназированных связывающих звен ьев вводятся в 5 мкл вываренные с PVU II ДНК плазмиды рКС 283, и затем 2 мкл 10Х буферного раствора лигазы (300 мМол Трис-HCl, рН=7,6; 100 мМол MgCl2; и 50 мМол ДТТ), 2,5 мкл BSA концентрацией 1 мг/мл, 7 мкл 5 мМол АТР, 2,5 мкл лигазы Т4 ДНК примерно 2,5 ед. единиц как определено P-L Biochemicals), 2,5 мкл 10 мМол спермидина и 3 мкл воды вводятся в указанную ДНК. Полученная реакционная смесь сшивки инкубируется при 4оС в течение ночи. После реакции сшивки состав реакционной смеси регулируется таким образом, что бы получился высокосолевой буферный раствор (0,1 Мол. NaCl; 0,05 Мол. Трис-HCl, рН=7,5; 10,0 мМол MgCl2; и 1 мМол ДТТ). В смесь вводится примерно 10 мкл (100 единиц) ограничительного фермента Xhol, и полученная реакционная смесь инкубируется при 37оС в течение 2 часов. Реакция завершается, и вываренная с Xhol ДНК осаждается, повторно суспензируется, и сшивается, как описано выше, с той разницей, что никаких связывающих звен ьев Xhol не вводится в смесь сшивки. Сшитая ДНК состоит из желаемой плазмиды рКС 283 РХ. Ограничительная точка и функциональная карта плазмиды рКС 283 РХ. представлена на рисун ке 2 данной заявки. 3. Построение E. coli К 12 МО (l +)/ рКС 283 РХ. E. coli К 12 МО ( l +) может быть получена из Северных Ре гиональных Научных Лабораторий в лиофи лизированной форме, под официальным номером NRRL В15993. E. coli К 12 МО включая подавляющий ген лямбда дикого типа, так что транскрипции от гибридного промото ра pLI-pp, отвечающего данному изобрете нию, не происходит в клетках E. coli К 12 МО (l +). Осуществляется выделение лиофилов и перестроенных одиночных колоний МО (l +), и приготавливается 10 мл ночной культуры клеток МО (l +), в основном, согласно процедуре, описанной в примере 29 А1, с той разницей, что температура инкубации составляет 37оС, и в среде роста не используется никакого ампициллина. Пятьдесят мкл ночной культуры используется для инкуляции 5 мл среды LB, которая также содержит 2 мМол. MgSO4 и 10 мМол MgCl2. Данная культура инкубируется при 37оС в течение ночи при интенсивном встряхи вании. На следующее утро культура разбавляется до 200 мл средой LB, содержащей 10 мМол MgSO4 и 10 Ммол. MgCl2. Разбавленная культура инкубируется при 37оС при интенсивном встряхи вании до тех пор, пока поглоще ние при 550 мм (A550) не составит примерно 0,5, что показывает клеточную плотность примерно 1 х 10 клеток/мл. Эта культура охлаждается в те чение 10 минут в ледяной водяной бане, и затем клетки извлекаются путем центрифугирования при ускорении 4000 g в течение 10 минут при 4оС. Клеточный осадок на дне пробирки снова суспензируется в 100 мл холодного 10 мМол MgSO4 , и затем тотчас же повторно гранулируется путем центрифугирования. Клеточный осадок снова суспензируют в 100 мл 30 мМол. CaCl2 и инкубируется во льду в те чение 20 минут. Эти клетки снова собираются путем центрифугирования и снова суспензируются в 10 мл 30 мМол. CaCl2. Аликвота эти х клеток, составляющая пол-мл, вводится в сшитую ДНК, приготовленную как в примере 29 А2; Приготавливается ДНК концентрацией 30 мМол. в CaCl2. Смесь клетки-ДНК инкубируется во льду в те чение одного часа, нагревается при 42оС в течение 90 секунд и затем охлаждается на льду в те чение примерно двух минут. Смесь клетки-ДНК разбавляется в 10 мл среды LB в 125 миллилитровых колбах и инкубируется при 37оС в течение одного часа. Аликвоты в количестве 100 мкл высеваются в чашках с LBагаром, содержащих ампициллин, и инкубируются при 37оС до тех пор, пока не появляются колонии. Эти колонии индивидуально культивируются и ДНК плазмиды отдельных колоний подвергаются анализу на ограничительный фермент и гель-электрофорезу. Выделение ДНК плазмиды осуществляется в небольших количествах согласно процедуре примера 29 А1, но этап градиента CsCl не осуществляется до тех пор, пока инти фицируются E. coli К 12 МО (l +) рКС 283 РХ трансфор манты. Ограничительная точка и функциональная карта плазмиды рКС 283 РХ представлена на рисунке 2 данной заявки. 4. Построение E. coli К 12 МО ( l +)/рКС 283 – L Десять микрограммов ДНК плазмиды рКС 283 РХ, полученной согласно процедуре примера 29 А1, растворяются в 20 мкл 10 х вы сокосолевого буферного раствора, 20 мкл 1 мг/мл BSA, 5 мкл (~ 50 единиц) ограничительного фер мента Bge 11,5 мкл (~ 50 единиц) ограничительного фер мента Xhol и 150 мкл воды, и полученная реакционная смесь инкубируется при 37оС в течение двух часов. Реакция прекращается и после охлаждения вываренного с Bge II – Xhol ДНК, эта ДНК снова суспензируется в 5 мкл буферного раствора ТЕ. Синтезируется и киназируется связывающее звено ДНК с однониточными концами ДНК, характерными для расщепления ограничительного фермента Bge II и Xhol. Это связывающее звено киназируется согласно процедуре примера 29 А2. Свя зывающее звено ДНК имеет следующую структуру: 5' 5' GATCTATTAACTCAATCTAGAC 3' ATAATTGAGTTAGATCTGAGCT 5' Приведенное выше связывающее звено синтезируется из однониточных деоксилигонуклеотидов согласно уже хорошо известным в данной области процедуры. Эти однониточные деоксилигонуклеотиды могут быть синтезирова ны посредством выпускаемых промышленностью инструментов, таких как Синтезатор ДНК 380 А, выпускаемых в продажу фирмой Applid Biosystems 1850 Lincoln Centre Irive, Foster City, (А 94 404), который используется в хи мии фосфо рамидитов. Известны также и другие процедуры синтеза ДНК. Обычный модифи цированный метод фосфотриэфи ра для синтеза донониточной ДНК описывается в работе Itakura и др., 1977, Science, 198 : 1 056 и Crea и др., 1978, Proc. Nat, Acad. Sci. USA 75: 5 765. Кроме того, особенно предпочти тельный способ синтеза ДНК описывается в работе Hsiung и др., 1983, Nucleic Acid Research, 11, 3 227; и в работе Narang и др., 1980, Methods in Enzymology 68 : 90. Это связывающее звено и вываренная с Bge II Xhol плазмида рКС 283 РХ сши ваются, в основном, согласно процедуре примера 29 А2. Сшитая ДНК образует желаемую плазмиду рКС 283-L. Ограничительная точка и функциональная карта плазмиды рКС 283-L представлена на рисун ке 3 данной патентной заявки. ДНК плазмиды рКС 283-L используется для трансформации E. coli К 12 МО (l+) и полученные трансформанты E. coli К 12 МО (l+)/рКС 283-L идентифицируются, в основном, согласно процедуре примера 29 А3. 5. Построение E. coli К 12 МО (l +)/рКС 283-LB. Примерно 10 мкг ДНК плазмиды рКС 283-L полученной согласно процедуре примера 29 А1, растворяются в 20 мкл 10 х высокосолевого буфер ного раствора, 20 мкл 1 мг/мл BSA, 5 мкл (~ 50 ед.) ограничительного фермента Xhol и 155 мкл H2O; и полученная реакционная смесь инкубируется при 37оС в течение двух ча сов. Вываренная с Xhol ДНК плазмиды рКС 283-L затем осаждается из реакционной смеси путем ввода трех объемов 95%-ного эта нола и 1/10 объема 3 Мол. ацета та натрия, инкубации в бане сухой лед этанол в те чение пяти минут, и центрифугирова ния. Полученный осадок ДНК промывается 70%-ным этанолом, высуши вается и повторно суспензируется в 2 мкл 10Х-Ник-трансляционного буферного раствора (0,5 Мол. Трис-HCl, рН=7,2; 0,1 Мол. MgSО 4; и 1 мМол ДТТ), 1 мкл раство ра 2 мМол. в каждом из деоксинуклеоти дотрифосфатов, 15 мкл Н2О, 1 мкл (~ 6 ед., как определено P-L Biochemical (фрагмента Кленова, который является большим фрагментом 1 ДНК полимеразы E. coli и 1 мкл 1 мг/мл BSA. Полученная реакционная смесь инкубируется при 25оС в течение 30 минут; реакция прекращается в результате инкубирования раствора при 70оС в те чение 5 минут. Связывающие звенья Bam HI (5'CCGGATCCCG-3') киназируются и сши ваются с вы варенной с Xhol, обработанной фрагментом Кленова ДНК плазмиды рКС 283-L, в основном, согласно процедуре примера 29 А2. После реакции сшивки ДНК вываривается и примерно с 100 ед. Bam HI в течение ~2 часов при 37оС в высокосолевом буферном растворе. После вываривания с ДНК подготавливается для сшивки согласно процедуре примера 29А2. Ограничительный фрагмент ~ 5,9 кб Bam HI замыкается в кольцо путем сшивки и трансформируется в E. coli К 12 МО ( l +) согласно процедуре примера 29 А2 и 29 А3. Трансформанты К 12 МО (l+)/рКС 283LB идентифицируется, и затем получается ДНК плазмиды рКС 283-LB согласно процедуре примера 29 А1. Ограничительная точка и функциональная карта плазмиды рКС 283-LB представлена на рис. 4 данной заявки. 6. Построение E. coli К 12 МО (l +)/pL 32 Примерно 10 мкг плазмиды рКС 283 РХ вываривается с ограничительным фрагментом Sal I в высокосолевом буферном растворе, обрабатывается фрагментом Кленова и сшивается со связывающими звеньями Eco RI (5'GAGGAATTCCTC-3'), в основном, согласно процедуре примера 29 A5, с той разницей, что используются исходная плазмида, ограничительные фрагменты и связывающие звенья. После вываривания с ограничительным фрагментом Eco RI, который образует в ре зульта те фрагмент ~ 2,1 kb от ДНК, огра ничительный фрагмент ~ 4,0 kb Eco RI замыкает кольцо путем сшивки с получением плазмиды рКС 283 PRS. Эта сшитая ДНК используется для трансформации E. coli К 12 МО (l +), в основном, согласно процедуре примера 293 А3. После идентификации трансформант E. coli К 12 МО (l +)/рКС 283 PRS, ДНК плазмиды рКС 283 PRS обрабатывается согласно процедуре примера 29 А1. Ограничительная точка и функциональная карта плазмиды рКС 283 PRS представлена на рис. 5 данной заявки. Примерно 10 мкг плазмиды рКС 283 PRS вываривается в 200 мкл высокосолевого буферного раствора примерно с 50 ед. каждого из ограничительных фрагментов Pst I и Sph I. После инкубирования реакционной смеси при 37оС в те чение примерно 2 часов реакционная смесь подвергается электрофорезу на 0,6% гель-агарозе с низкой температурой гелеобразования (Фирма Отделение Marine Colloids Division, Rockland, Maine 04 841) в течение 2 – 3 часов при ~ 130 Вольтах и ~ 75 миллиАмперах в Трис-Аце татном буферном растворе. Этот гель окраши вается в разбавленном растворе этилиденбромида и слой ДНК, составляющий ограничительный фрагмент ~0,85 кв Pst I- Sph I, который визуально обнаруживается в длинноволновых ультрафиолетовых лучах, отсекается от данного геля в небольшом сегменте. Объем этого сегмента определяется по весу и плотности, и в пробирку, содержащую данный сегмент, вводится равный объем 10 мМол Трис-HCl, рН=7,6. Затем этот сегмент расплавляется путем инкубации при 72оС. Получается примерно 1 мкг ограничительного фрагмента ~0,85 кв Pst I – Sph I плазмиды рКС 283 PRS, в объеме примерно 100 мкл. Аналогичным образом плазмида рКС 283-LB вываривается с ограничительными фермента ми Pst I и Sph I, и полученный ограничительный фрагмент ~3,0 кв изолируется путем электрофореза на агарозном геле и обрабатывается для подготовки к сшивке. Ограничительный фрагмент 0,85 кв Pst I- Sph I плазмиды рКС 283 PRS сшивается с ограничительным фрагментом ~0,3 кв Pst I Sph I плазмиды рКС 283 согласно процедуре примера 29 А2. Сшитая ДНК составляет желаемую плазмиду pL 32. Ограничительная точка и функциональная карта плазмиды pL 32 представлены на рис. 6 данной заявки. Плазмида pL 32 трансформируется в клетки E. coli К 12 МО (l +) в основном согласно процедуре примера 29 А3. ДНК плазмиды pL 32 приготавливается из трансформанта E. coli К 12 МО (l+)/pL 32 согласно процедуре, описанной в примере 29 А1. Анализ ДНК плазмиды pL 32 продемонстрировал, что более чем одно связывающее звено Eco RI прикрепляется к обработанным фрагментом Кленова концам Sal I плазмиды рКС 283 PX. Присутствие более чем одного связывающе го звена Eco RI не влияет на полезность плазмиды pL 32 или производных плазмиды pL 32 и может быть определено по присутствию ограничительной точки Xhol, кото рая образуется независимо от того, когда сшиваются оба связующие звена Eco RI вместе друг с др угом. Как возможный вариант, плазмида pL 32 может быть построена путем эксцизии фрагмента Sal I – Ero RI и сши вки согласно первому разделу данного примера, с описанием плазмиды рКС 283-LB. 7. Построение E. coli К 12 МО ( l+)/pL 47 E. coli К 12 RV 308/pNM 789 может быть получена из Северной региональной научно-исследовательской лаборатории в лиофи лизированной форме, под номером NRRLВ – 18 216. Ограничительная точка и функциональная карта pNM 789 представлена на фиг. 7 дан ной заявки. Плазмидная ДНК экстрагируется из данной культуры согласно описанию примера 1, с той разницей, что температура инкубирования составляет 37оС. Десять микрограммов pNM 789 суспензируются в 200 мл буферного раствора PVU II (50 мМол. Трис-HCl, рН=7,5, 60 мМол. NaCl и 6 мМол. MgCl2). Вводится одна единица PVU II и реакционная смесь инкубируется в те чение 5 минут при 37оС. Данный фермент инактивируется путем нагрева в те чение 10 минут при 65оС. Затем вводятся 30 мкл 10Х буферного раствора Bam HI (200 мМол. Трис-HCl (рН=8,0), 1 Мол. NaCl и 70 мМол. MgCl2), 70 мкл H2O и 10 ед. Bam HI, и реакционная смесь инкубируется в течение 1 часа при 37оС. После этого вводится 5 ед. щелочной фосфа тазы и осуществляется инкубирование в течение 1 часа при 65оС. Фрагменты ДНК отделяются на 1% агарозном геле, и осуществляется очистка фрагмента ДНК (фиг. 8) размером в единичный фрагментный отрезок. Связывающее звено ДНК с отсеченным концом и с Bam HI концом синтезируется, в основном, согласно процедуре примера 29 А4. Это связывающее звено (показано как 118 на фиг. 8) имеет следующую структуру: 5' CTGTGCCTTCTAG 3' 3' GACACGGAACATCCTAG 5' Это связывающее звено киназируется и сшивается в вываренную Bam HI – Pvu II плазмиду pNM 789, в основном, согласно процедуре примера 29 А2. Эта смесь сшивки используется как трансфор мация клеток E. coli К 12 RV 308, и вы деление плазмиды осуществляется с использованием данных трансформант согласно описанию примера 20 А3. Выбираются некоторые плазмиды, которые содержат фрагмент Pvu II (494 hp) подходящего размера и фрагмент Xba I – Bam HI (628 hp). Последовательность, по меньшей мере, двух из них определяется от последовательности от точки Bam HI к уникальной точке Sma I, и выбирается один клон с желаемой последовательностью. Эта промежуточная плазмида обозначается как плазмида 120. Схематическое изображение данной процедуры и ограничительная точка и функциональная карта плазмиды 120 представлена на фиг. 8 данной заявки. Для выделения кодирующей EK–BGH ДНК, примерно 10 мкг плазмиды 120 вываривается в 200 мкл высокосолевого буферного раствора, содержащего примерно 50 ед. каждого из ограничительных ферментов Xba I и Bam HI. Продук ты вываривания отделяются посредством электрофореза на агарозном геле, и ограничительный фермент ~0,6 кв Xba I – Bam HI, который колирует EK–BGH выделяется и препарируется для сшивки согласно процедуре, описанной в примере 29 А6. Плазмида pL 32 также вываривается с ограничительными ферментами Xba I и Bam HI, и ограничительный фрагмент ~3,9 кв извлекается и приготавливается для сшивки. Ограничительный фрагмент ~3,9 кв Xba I – Bam HI плазмиды pL 32 сшивается с ограничительным фрагментом ~0,6 кв Xba I – Bam HI плазмиды 120, в основном, согласно процедуре примера 29 А2, и в результате получается плазмида pL 47. Ограничительная точка и функциональная карта плазмиды pL 47 представлена на фиг. 9 данной заявки. Плазмида pL 47 трансформируется в E. coli К 12 МО (l +), в основном, согласно процедуре примера 29 А3, и идентифи цируются трансфор манты E. coli К 12 МО (l+)/pL 47. ДНК плазмиды pL 47 получается от данных трансфор мант, в основном, согласно процедурам примера 29 А1. 8. Построение E. coli К 12 Р 308/pPR 12 АР 1 Плазмида pPR 12 заключает в себе чувстви тельный к температуре подавляющий pL ген с 1857, и плазмида pBR 322 заключает ген, придающий стойкость к тетрациклину. Плазмида pPR 12 описана и заявлена в патенте США № 4436815, вы пущенном 13 марта 1984 года. Ограничительная точка и функциональная карта плазмиды pPR 12 представлена на фиг. 10 данной заявки. Примерно 10 мкг плазмиды pPR12 вываривается примерно с 50 ед. ограничительного фермента EcoRI в 200 мкл высокосолево го буферного раствора при 37оС в те чение 2 часов. Вываренная с EcoRI ДНК плазмиды pPR12 осаждается и обрабатывается фрагментом Кленова согласно процедуре примера 29 А5. После протекания реакции Кленова вываренная Eco RI, обработанная фрагментом Кленова ДНК плазмиды pPR12 снова замыкается в кольцо путем сшивки процедуре примера 29 А2. Эта сшитая ДНК, которая образует желаемую плазмиду pPR12 D RI, используется для трансфор мации E. coli К 12 RV 308, в основном, согласно процедуре примера 29 А3, с той разницей, что этот вы бор основан на стойкости к тетрациклину (5 мкг/мл), а не к ампициллину. E. coli К 12 R V 308 имеет официальный номер NRRL В – 15624. После идентифи кации трансфор мант E. coli К 12 RV 308/pPR 12 RI из данных трансформант получается ДНК плазмиды pPR12 D RI согласно процедуре примера 29 А11. Примерно 10 мкг плазмиды pPR 12 D RI вываривается примерно с 50 ед. ограничительного фермента АvaI в 200 мкл средне-солевого буфер ного раствора при 37оС в те чение 2 часов. Вы варенная с Ava I ДНК плазмиды pPR 12 D RI осаждается и обрабаты вается фрагментом Кленова согласно процедуре примера 29 А5. После реакции с фрагментом Кленова вываренная с Ava I, обработанная фрагментом Кленова ДНК плазмиды pPR 12 D RI сши вается со связывающи ми звеньями Eco RI (5'–GAGGAATTCCTC–3'), в основном, согласно процедуре примера 29 А2. После сшивки связывающим звеном, ДНК осаждается и затем повторно суспензируется примерно в 200 мкл высокосолевого буферного раствора, содержаще го примерно 50 ед. ограничительного фермента Eco RI. Полученная реакционная смесь инкубируется при 37оС в течение примерно 2 часов. После вываривания с Eco RI реакционная смесь помещается на агарозный гель, и ограничительный фрагмент ~5,1 кв Eco RI очищается, в основном, согласно процедуре примера 29 А6. Ограничительный фрагмент ~5,1 кв Eco RI снова замыкается в кольцо путем сшивки согласно процедуре примера 29 А2. Эта сшитая ДНК образует желаемую плазмиду pPR 12 AR1. Эта ДНК плазмиды pPR 12 ARI трансфор мируется в E. coli К 12 RV 308 согласно процедуре примера 29 А3, с той разницей, что выбор основывается на стойкости к тетрациклину, а не к ампициллину. После идентифи кации трансформант E. coli К 12 RV 308/pPR 12 ARI получается ДНК плазмиды pPR 12 ARI, в основном, согласно процедуре примера 29 А1. Ограничительная точка и функциональная карта плазмиды pPR 12 ARI представлена на фиг. 11 данной заявки. 9. Построение E. coli К 12 RV 308/pL 110 Примерно 10 мг ДНК плазмиды pPR 12 ARI суспензируется в примерно 200 мл высокосолевого буферного раствора, содержащего примерно 50 ед. каждого из ограничительных ферментов Pst I и Eco RI, и реакционная смесь вываривания инкубируется при 37оС в течение примерно 2 часов. Затем эта реакционная смесь помещается на агарозный гель, и ограничительный фрагмент ~2,9 кв Pst I – Eco RI плазмиды pPR 12 ARI вы деляется и подготавливается для сшивки, в основном, согласно процедуре примера 29 А6. Примерно 10 мкг плазмиды pL 47 вываривается с ограничительными фермента ми Pst I и Bam HI в 200 мкл высокосолевого буферного раствора при 37оС в течение 2 часов. Вываренная с Pst I – Bam HI ДНК помещается на агарозный гель, и выделяется ограничительный фрагмент ~2,7 кв Pst I – Bam HI, который является источником репликации и частью стойкого к ампициллину ге на, и кото рый подготавливается для сшивки согласно процедуре примера 29 А6. В реакции разделения примерно 10 мкг ДНК плазмиды pL 47 вываривается с ограничительными фрагментами Eco RI и Bam HI в 200 мкл высокосолевого буферного раствора при 37оС в течение двух часов, и вы деляется ограничительный фрагмент 1,03 кв Pst I – Bam HI, который заключает в себе новую транскрипционную и трансляционную акти вирующую последовательность, и кодирующую EK–BGH ДНК, и этот фрагмент подготавливается для сшивки согласно процедуре примера 29 А6. Примерно 2 мкг полученного ограничительного фрагмента ~1,03 кв Eco RI – Bam HI используются для построения плазмиды pL 110. Ограничительные фрагменты ~2,7 кв Pst I – Bam HI и 1,03 кв Eco RI – Bam HI плазмиды pL 47 сшиваются с ограничительным фрагментом ~2,9 кв Pst I – Eco RI плазмиды pPR 12 А RI для построения плазмиды pL 110, и сшитая ДНК используется для трансформации E. coli К 12 RV 308, в основном, согласно процедуре примеров 29 А2 и 29 А3, с той разницей, что основой для выбора трансфор мант является стойкость к тетрациклину, а не к ампициллину. В кодирующей зоне ЕК–BGH присутствуют две распознавательные точки ограничительного фрагмента Pst I, которые не отражаются на ограничительной точке и на функциональных карта х на рисун ках данной заявки. Ограничительная точка и функциональная карта плазмиды pL 110 представлена на фиг. 12 данной заявки. 10. Построение E. coli К 12 RV 308/pL 110 C а. Построение E. coli К 12 RV 308/pL 110 A Примерно 1 мкг ДНК плазмиды pL 110 вываривается с ограничительным ферментом Nde I в 20 мкл общего объема, содержащего 2 мкл 10Х вы сокосолевого буфер ного раствора (1,0 Мол NaCl; 0,50 Мол, Трис-HCl, рН=7,5; 0,10 Мол, MgCl2 и 10 мМол. дитиотреитола) и 3 ед. фермента Nde I, в течение 1 часа при 37оС. Реакционная смесь экстрагируется фенолом/хлорофор мом, и ДНК осаждается этанолом. Вываренная с Nde I ДНК плазмиды pL 110 растворяется в 50 мкл IX буферного раствора Кленова (40 мМол. KPO4, pH=7,5; 6,6 мМол MgCl2; 1,0 мМол 2-меркаптоэтанола; 33 мкМол d ATP; 33 мкМол dGTP; и 33 мкМол и 33 мкМол. ТТР). Два МКЛ (10 ед., New England Biolabs) крупных фрагмента ДНК полимеразы 1 E. coli, известных как фрагменты Кленова, вводятся в данный раствор и смешиваются с ДНК, и полученная реакционная смесь инкубируется при 16оС в течение 1 часа. Реакция завершается путем экстракции фенолом, и ДНК очищается обычными приемами. Вываренная с Nde I, обработанная фрагментом Кленова ДНК затем сшивается с Т4 ДНК лигазой при 4 оС в течение 16 часов. Полученная ДНК используется для обычной трансформации штамма E. coli К 12 RV 308 (NRRL B – 15624). Трансформанты отбираются на чашках с L-агаром, содержащи х 100 мкг/мл ампициллина, и плазмиды извлекаются из стойких колоний путем быстрой ще лочной экстракции, как описано в работе Birnboim и Doly. Отбирается плазмида (pL 110 А на фиг. 13), с отсутствием точки Nde I. б. Построение фага pL 110 B путем точечно специфи ческого мутагенеза. Осуществление процедуры уда ления точки Bam HI в гене, сообщающем стойкость к тетрациклину п утем точечно-специфи ческого мутаге неза представлено с правой сто роны фигуры 13 данной заявки. b(i) Построение фага M 13 Тс 3 Плазмида pL 110 служит в качестве источника придающего стойкость к тетрациклину ге на. Примерно 50 мкг плазмиды pL 110 в 50 мкл буферного раствора ТЕ вводится в 25 мкл буферного раствора 10Х Hind III и 170 мкл H2O. Примерно 5 мкл (~50 ед.) ограничительного фермента Hind III вводится в раствор ДНК плазмиды pL 110, и полученная реакционная смесь инкубируется при 37оС в течение 2 часов. Примерно 13 мкл 2 Мол. Трис-HCl, рН=7,4 и 5 мкл (~50 ед.) ограничительного фер мента Eco RI вво дятся в вы варенную с Hind III ДНК плазмиды pL 110, и реакционная смесь инкубируется в течение более 2 часов при 37оС. Реакционная прекращается в результате экстракции реакционной смеси насыщен ным ТЕ фенолом; фенол удаляется путем экстракций хлорофор мом. Вываренная с Eco RI – Hind III ДНК плазмиды pL 110 затем извлекается путем осаждения и центрифугирования, помещается на 1% агарозный гель, и крупный ограничительный фрагмент ~4,3 кв Eco RI – Hind III вы деляется и очищается. Примерно 5 мкг фага m 13 m p18 (New England Biolabs) растворяется в 50 мкл буферного раствора ТЕ и затем вываривается Hind III и Eco RI, как описано выше. Отсеченная Hind III – Eco RI ДНК фага М 13 m р18 очищается, как описано для pL 110, с той разницей, что осуществляется выделение и очистка ограничительного фрагмента ~7,25 кв. Примерно 100 нанограммов фрагмента ~4,3 кв Hind III – Eco RI плазмиды pL 110 смешивается с примерно 100 нанограммами фрагмента ~7,25 кв Hind III – Eco RI фага М 13 m р18, 2 мкл 10Х буферного раствора лигазы, 1 мкл (~ 100 ед.) Т4 ДНК лигазы и 14 мкл Н 2О. Реакционная смесь сшивки инкубируется при 15оС в течение полутора часов; Сши тая ДНК составляет ДНК желаемого фага m 13 Тс 3. Ограничительная точка и функциональная карта фа га m 13 Тс 3 представлена на фиг. 13 данной заявки. Один миллилитр ночной культуры E. coli К 12 JM 109 (E. coli K 12 JM 101, получена из New England Biolabs, может использоваться вместо E. coli К 12 JM 109), используется для инокулирования 50 мл питательного бульона L (50 мл), и полученная культура инкубируется при т-ре 37оС с аэрацией до тех пор, пока OD660 (оптич.плотность) не будет составлять от 0,3 до 0,4. Клетки повторно суспензируются в 25 мл 10 мМол NaCl, инкубируются на льду в те чение 10 минут и извлекаются путем центрифугирования. 200 мкл аликвота этих клеток удаляется, вводится в 10 мкл сшитой ДНК, полученной как описано выше, и инкубируется на льду в те чение ~40 минут. За тем смесь клетки – ДНК инкубируется при 92оС в течение 2 минут и различные аликвоты (1,10 и 100 мкл) удаляются и вводятся в 3 мл верхнего агара (L бульон с 0,5% агаром, поддерживаемом в расплавленном состоянии при 45оС), который содержит также 50 мкл 2% X–Gal, 50 мкл 100 мМол IPTC, и 200 мкл E. coli К 12 JM 109 в фазе логарифмического роста. Затем верхняя клеточная часть агаровой смеси высевается в чашках с L-агаром, содержащи х 40 мкг/мл X–Gal (5-бромо-4-хлоро-3индолил- D-тиогалактозид) и 0,1 мМол IPTG (изопропил- b -D-тиогалакто зид) и чашки инкубируются в течение ночи при 37оС. На следующее утро несколько прозрачных, но не синих колоний используются для инокулирования 2 мл L-бульона, и образующиеся в результа те культуры инкубируются при 37оС с аэрацией в те чение 2 часов. Отсутствие синего окраши вания показывает, что происхо дит делаемая вставка ДНК. Затем данные культуры центрифугируются, и 200 мкл полученного поверхностного слоя вводятся в 10 мл культур (O.D.550=0,5) E. coli К 12 JM 109, выращенных при 37оС с аэрацией. Эти культуры инкубируются еще в течение 30 минут при 37оС, затем эти клетки выпадают в осадок в результате центрифугирования, и используются для получения репликаторной формы рекомбинантного фага, который они содержат. Из этих клеток выделяется двухниточная репликаторная форма фаговой ДНК с использованием процедур (в меньших объемах), описанных в примере 1. Трансформанты, содержащие фаговую m 13 Tc / ДНК, идентифи цируются путем анализа на ограничительный фермент их фа говой ДНК. b (ii) Получение Однониточной ДНК фага m 13 Tc 3. Полтора миллилитра ночной культуры E. coli К 12 JM 109/ m 13 Tc 3 центрифугируется и 100 мкл верхнего слоя, содержаще го фаг m 13 Tc 3, используется для инокуляции 25 мл культуры E. coli М 109 с оптической плотностью O.D.660 примерно 0,4–0,5. Данная культура инкубируется примерно в течение 6 ча сов при 37оС с аэрацией, и затем культура центрифугируется; и образованный верхний слой, примерно 20 мл, переносится в новую пробирку. Примерно 2 мл раствора, содержаще го 20% полиэтиленгликоля (РЕС) 6000 и 14,6% NaCl вводится в поверхностный слой, который затем инкубируется на льду в те чение 20 минут. Поверхностный слой центрифуги руется в те чение 25 минут со скоростью 7000 об/мин, и образующийся осадок, который содержит однониточную ДНК фа га m 13 Tc 3, снова суспензируется в 500 мкл буферного раствора ТЕ. Раствор ДНК двукратно экстрагируется насыщен ным ТЕ фе нолом и двукратно хлорофор мом. Затем однониточная ДНК осаждается с использованием NaOAc и этанола и центрифугируется. Полученный осадок промывается 70%-ным этанолом, высушивается и затем растворяется в 60 мкл Н2О. b (iii) Мута генез. Фрагмент однониточной ДНК, используемый в процессе мутагенеза, синте зируется в авто мати ческом синте заторе ДНК. Этот фрагмент имеет последовательность 5'–CCCGTCCTCTGGATACT CTACGCCGA–3', и является гомологом зоны, окружающей точку Bam HI (5'–GGATCC–3'), в придающем стойкость к тетрациклину гене от плазмиды pBR 322, с той разницей, что А гр уппа вторая от 5' конца (или третья от 3' конца) является С в плазмиде pBR 322. Такое видоизменение не меняет аминокислотный состав белка, придающего стойкость к тетрациклину, но исключает точку Bam HI. Примерно 10 пикомолей мута генной затравки и универсальной затравки М 13 (Bethesda Research Laboratories (BRL) D.O. Box 6009, Гатесбург, М D 20760) обрабатываются по отдельности 10-ю единицами (BRL) полинуклеотидокиназы Т4 в 20 мкл буферного раствора киназы 1Х (60 мМол. Трис-HCl, рН=7,8; 15 мМол 2-меркаптоэтанола; 10 мМол. MgCl2; и 0,41 мкМол. АТР) в течение 30 минут при 37оС. Обработанные киназой ДНК используются в описанной ниже процедуре мутагенеза. Реакция ренатурации (аннилирования) осуществляeтся путем смешивания друг с др угом 300 нанограммов (1,2 мкл) однониточного фа га m 13 Tc 3 пикомоля (2 мкл) универсальной затравки, 1 пикомоля (2 мкл) мутагенной затравки, 2 мкл 10Х ренатурирующе го буфер ного раствора (100 мМол. Трис-HCl, рН=7,5; 1 мМол. ЭДТА; и 500 мМол NaCl), и 12,8 мкл Н2О. Реакционная смесь инкубируется при 80оС в течение 2 минут, при т-ре 50оС в те чение 5 минут, а затем охлаждается до комнатной температуры. Реакция удлинения цепи осуществляется путем ввода 5 мкл 10Х буфер ного раствора вытягивания (или удлинения) (500 мМол Трис-HСl, рН=8; 1 мМол ЭДТА; и 120 мМол MgCl2); 5 мкл 2 мМол dATP; 1 мкл раствора 6 мМол. каждого из dGTP, ТТР и dCTP; 1 мкл (~ 2 ед., Pharmacia. P–L Biochemicals, 800 Centennial Avenue Piseataway, NJ 08854) фермента Кленова; 1 мкл (100 ед.) Т4 ДНК лигазы; и 17 мкл Н 2О в смесь ренатурированной ДНК. Реакционная смесь удлинения цепи инкубируется при комнатной температуре в течение 1 часа, затем при 37оС в течение 2,5 часов, и затем в течение ночи при 4оС. Реакция прекращается путем осуществления двух экстракций насыщен ным ТЕ фенолом, после которых осуществляются две экстракции CHCl3. ДНК осаждается этанолом и ацетатом натрия (NaOAc). ДНК извлекается путем центрифугирования и снова суспензируется в 50 мкл Н 2О, и затем 6 мкл 10Х буферного раствора SI, вво дятся в раствор ДНК. Этот раствор ДНК распределяется поровну в трех пробирках. В две из эти х пробирок вводится примерно 200 ед. (Miles Labotratories) нуклеазы 1. Одна реакционная смесь SI инкубируется при комнатной температуре в те чение 5 минут, др угая в те чение 10 минут. Реакции прекращаются в результате экстракции реакционной смеси два раза насыщен ным ТЕ фе нолом. После фенолиных экстракций осуществляются две экстракции хлороформом; затем из данной реакционной смеси осаждается ДНК ацетатом натрия (NaOAc) и этанолом. Необработанный образец ДНК служит в качестве отрицательного контрольного образца. Обработанные образцы сохраняются изолированными друг от др уга в хо де последующей процедуры, но результаты получаются одинаковые. Выпавшие осадки ДНК повторно суспензируются в 20 мкл Н 2О, и используется 10 мкл полученного раствора для трансформации E. coli К 12 JM 109 (E. coli К 12 JM 101 также может быть использован) согласно процедуре, осуществляемой для построения фага m 13 Tc 3, с той разницей, что в чашки не вводится ни IPTG, ни X–Gal. Двухниточная репликаторная форма ДНК от примерно 48 колоний выделяется, как описано выше, и отбирается на присутствие ограничительной точки Bam HI. Изоляты без точки Bam HI дополнительно отбираются с получением однониточной ДНК, как описано выше. Аминокислотная последовательность однониточной ДНК определяется с использованием метода определения дидеокси последовательности (J.H. Smith, Методы в энзимологии, 65: 560–580). Желаемый изолят обозначается как pL 110 В (фиг. 13). С. Построение плазмиды pL 110 C. Примерно 50 мкг репликаторной формы фага pL 110 В ДНК вываривается в 250 мкл IХ буферного раствора NheI (50 мМол. NaCl2; 6 мМол. Трис-HCl, рН=7,5; 6 мМол MgCl2; и 6 мМол b -меркаптоэтанола), содержащего ~50 ед. ограничительного фермента NheI при 37оС в течение 2 часов. Пять мкл 5 Мол. NaCl затем вводится в вываренную с NheI фа говую pL 110 В ДНК, с последующим вводом 5 мкл (~50 ед.) ограничительного фрагмента Sal. Вываривание продолжается в те чение 2 часов при 37оС. Затем желаемый фрагмент ~422 вр NheI – SalI, содержащий мутированную зону ге на, придающего стой кость к тетрациклину, выделяется из акриламидного гена, согласно известным стандартным процедурам. ДНК плазмиды pL 110 А вы варивается с NheI и SalI и идентичных условиях с той разницей, что вместо плазмиды pL 110 В используется плазмида pL 110 А. Ограничительный фрагмент ~ 6,1 кв NheI – SalI плазмиды pL 110 А очищается от агарозы. Построение желаемой плазмиды pL 110 С осуществляется путем сшивки друг с другом по 100 нанограммов каждого из NheI – SalI фрагментов pL 110 А (~ 6,1 кв) и pL 110 В (~ 422 вр) путем общеизвестных процедур. Ограничительная точка и функциональная карта плазмиды pL 110 C представлена на рис. 13 данной заявки. Желаемая плазмида pL 110 C сообщает стойкость к тетрациклину для 10 мкг/мл тетрациклина в E. coli, но в сообщающем стойкость к тетрациклину ге не отсутствует точка Bam HI. II. Построение плазмиды pCZR III Плазмида pL 110 С содержат единственную ограничительную точку Cla I, которая удаляется при протекании нижеследующих реакций. Примерно 1 мкг плазмиды pL 110 C вываривается с Cla I согласно процедуре примера 29 А2, с той разницей, что используются ограничительный фермент Cla I и 10Х буферный раствор Cla I (500 мМол. NaCl, 100 мМол. Трис-HCl, рН=7,9 и 100 мМол. MgCl2). Вываренная с Cla I ДНК затем обрабатывается фрагментом Кленова согласно описанию примера 29 А5, с той разницей, что вво дится лишь dCTP, а не все четыре dNTP. Затем ДНК осаждается и повторно суспензируется в 50 мкл буферного раствора бобовой нуклеазы Mung (50 мМол. Ацета та натрия pH=5,0, 30 мМол. NaCl и 1 мМол ZnSO4). Одна единица бобовой нуклеазы Mung (промышленно выпускается New England Biolabs) вводится в эту суспензию, и реакционная смесь инкубируется при 30оС в течение 30 минут. За тем пробирка помещается в лед и NaCl вводится в до концентрации 0,2 Мол., после чего смесь экстрагируется фенолом (хлороформом, осаждается этанолом и повторно суспензируется в 10 мМол. Трис-HCl (рН=8,0). Затем ДНК подвергается самосшивке и трансформируется в клетки E. coli согласно описанию примеров 29 А3 и 29 А4. Полученная плазмида представляет собой вываренную плазмиду pCZR III. 12. Построение плазмиды pCZR 126 Примерно 26 мкг плазмиды pCZR III вываривается с Xba I указанным ниже образом. Буферный раствор 10Х Xba I состоит 600 мМол. Трис-HCl, 100 мМол. MgCl2, 1 Мол. NaCl и 10 мМол 2-меркаптоэтанола, рН=7,5 (при 37оС). 50 мкл буферного раствора 10Х Xba I, 15 мкл Xba I (10 ед/мкл) и 185 мкл Н2О вводятся в 250 мкл воды, содержащей примерно 25 мкг плазмиды pL 110. Вываривание осуществляется при 37оС в течение 1 часа. pL 110, вываренная с Xba I, затем экстрагируется в феноле, вво дится 1/10 объема этанола; смесь инкубируется в бане сухой лед-эта нол в те чение 5 минут, и затем центрифугируется. Осажденная ДНК повторно суспензируется в 50 мкл H2O. Вываренная с Xba I плазмида pCZR III вываривается с Bam HI следующим образом. 0,2 мкл Bam HI (10 ед/мкл), 10 мкл буферного раствора Bam HI (100 мМол. Трис-HCl, 50 мМол MgCl2. 1 Мол. NaCl и 10 мМол 2-меркаптоэтанола) рН=8,0 при 37оС) и 90 мкл Н2О вводятся в 50 мкл pL 110, вываренного с Xba I, полученного как указано выше. Вы варивание осуществляется в течение 5 минут при 37оС. Вываренная pCZR III экстраги руется в феноле, вводится 1/10 объема CH3COO а, после чего вво дится 3 объема этанола. Осажденная ДНК снова суспензируется в 50 мкл 10 мМол. Трис, 1 мМол. ЭДТА, рН=8,0, буферном растворе. pCZR III, вываренная с Xba I и Bam HI, затем помещается на агарозный гель, и выделяется слой ДНК примерно 5,8 кв. Плазмида pCZR 126 S получается путем сшивки фрагмента ~5,8 к pCZR III со связывающи ми звеньями Xba I и Nde I и синте тическим геном, кодирующим ЕК-бычий гормон роста, который содержит точку Nde I у его 5'-конца и точку Bam HI у его 3'-конца. Последовательность Xba I до Nde I получается с использованием метода, построения стандартной олиго нуклеотидной последовательности, и она заключает в се бе следующую последовательность: Указанная выше последовательность построена путем хи мического синте за обеих нитей с последующим смеши ванием с целью гибридизации. Кодирующий ЕКb CН ген построен из 16 хи мически синтезированных сегментов однониточной ДНК, заключающих в себе от 71 до 83 нуклеотидов, которые совместно друг с др угом составляют дополняющие нити всего гена. Синтез осуществляется в устройстве Applied Biosystems (АВS) и вк лючает указанную ниже последовательность: Построение плазмиды pCZR126S осуществляется путем сшивки нижеследующих компонентов: ~ 0,28 мкг фрагмента 5,8 кв полученного из плазмиды pL 110 после полного вываривания с Xba I и частичного вываривания с Bam III в общем объеме 2 мкл, ~ 0,18 мкг синтети ческого гена, кодирующего производное фактора бычьего роста, который имеет конец 5', соответствующий точке Xba I, и конец 3', соответствующий точке Bam HI в общем объеме 2,5 мкл, 8,75 пикомолей химически синтезированного Xba I – Nde I связывающе го звена в 1 мкл. Эти компоненты плазмиды вво дятся в 6 мкл 5Х буферного раствора сшивки: 250 мМол Тpис-НCl, 50 мМол MgCl2, 5 мМол АТР, 5 мМол ДТТ 25% об/об полиэтиленгликоля 8000, рН=7,6, 2 мкл лигазы и 16,5 мкл Н 2О. Эта смесь сшивки инкубируется в течение ночи при 16оС. Далее замкнутая в кольцо плазмида pCZR126S используется для трансформации клеток E. coli RV 308 согласно способу примера 29 А3. Ограничительная точка и функциональная карта плазмиды pCZP126S представлена на фиг. 14 данной заявки. В. Построение выраженной плазмиды человеческого проинсулина pRB172. 1. Построение плазмиды pRB145. Ген человеческого проинсулина сначала синтезируется обычным образом и клонируется в плазмиду PuC18 (промышленно выпускаемая BRL). Этот ген синте зируется стандартными приемами и заключает в себе последовательность: Один из клонов, имеющий правильную последовательность, выбирается для продуцирования очищенной от хлорида цезия ДНК. Данная плазмида извлекается путем стандартной процедуры (Смотри, например, публикацию "Мо лекулярное клонирование, Лабораторное пособие, 1982 г., ed. Maniatis, T.; Fritsch, E.F. и Sambrook. J., Cold Spring Hcorbour Laboratory Publications, Нью-Йорк, кото рая приведена здесь как ссылочный материал). Примерно 6 мкл (20 мкг) этой ДНК плазмиды вво дится в 20 мкл 10Х буферного раствора Nde I (150 мм NaCl, 10 мМол. Трис-HCl, pH=7,8, 6 мМол MgCl2, 6 м Мол. 2-меркаптоэтанола, 100 мкг/мл BSA), 5 мкл ограничительного фермента Nde I (~40 ед.), и 169 мкл H2O. После перемеши вания реакционная смесь инкубируется при т-ре 37оС в течение 2 часов. ДНК осаждается путем приготовления 0,3 Мол. NaOAc, ввода трех объемов этанола, перемеши вания и охлаждения до -70оС и центрифугирования. Осажденная ДНК промывается 70% этанолом (1 мл), высуши вается и растворяется в 20 мкл 10Х Bam HI буфе ра (150 мм NaCl, 6 мМол. Трис-HCl, рН=7,9, 6 мМол. MgCl2, 100 мкг/мл BSA), 2 мкл ограничительного фермента Bam HI (40 ед.) и 178 мкл Н 2О. После слабого перемешивания реакционная смесь инкубируется при 37оС в течение 2 часов. ДНК снова осаждается тремя объемами этанола, как определено выше, и подвергается электрофо резу на 1% расплавленном агарозном геле (FMC, морская агароза). Желаемый фрагмент ДНК, соответствующий примерно 270 вр, срезается с геля и затем ДНК извлекается путем расплавления агарозы и пропускания через колонку Elutip–d (Schlicher and Schuell, Kune N.H.) согласно процедуре, рекомендуе мой продавцом. После осаждения и сушки ДНК выдерживается в 30 мкл 10 мМол. Трис-HCl, рН=8,0. Примерно 15 мкг плазмиды pCZR1266 (из примера 29 А 12) суспензируются в 20 мкл 10Х буферного раствора Nde I, 5 мкл ограничительного фермента Nde I (40 ед.) и H 2O (175 мкл), слабо перемеши вается и смесь инкубируется при 37оС в течение 2 часов. После инкубации ДНК осаждается тремя объемами этанола, как и выше, вы сушивается и затем повторно суспензируется в 20 мкл 10Х буферного раствора Bam HI, 2 мкл ограничительного фермента Bam HI (40 ед.) и 178 мкл воды. После слабого перемешивания реакционная смесь инкубируется при 37оС в течение 2 часов. ДНК снова осаждается тремя объемами этанолa, и осуществляется электрофорез на 1% низкоплавком агарозном геле. Наиболее крупный фрагмент, соответствующий векторной ДНК, срезается с этого геля и ДНК извлекается путем осуществления процедуры в колонке Elutip–d как описано выше. После осаждения и сушки векторная ДНК выдерживается в 35 мкл 10 мМол. Трис-HCl, рН=8,0. Примерно 2,5 мкл векторной ДНК смешивается с 12 мкл генного фрагмента очищен ного человеческого инсулина, 4 мкл 10 мМол. АТР, 0,5 мкл и 1 Мол дитиотреитола, 5 мкл 10Х буферного раствора лигазы (50 мМол. Трис-HCl, рН=7,6 и 100 мМол MgCl2), 26 мкл воды и 0,5 мкл Т4–ДНК лигазы (Pharmacia, 3,5 ед.). Эта реакционная смесь инкубируется при 4оС в течение 16 часов. Сшитая смесь разбавляется 50-ю мкл 10 мМол. Трис-HCl (рН=7,6) и 3 мкл 1 Мол. CaCl 2 и затем последовательно трансформируется в E. coli К 12 RV 308 согласно описанию примера 29 А3. Клетки высеваются на чашках ТУ, содержащих 5 мкг/мл тетрациклина, и затем инкубируются в те чение ночи при 32оС. Плазмиды от 3 мл культур вы деляются из стойких к тетрациклину колоний путем быстрой щелочной экстракции, как описано в публикации "Молекулярное клонирование, Лабораторное пособие, 1982 г., ред. Maniatis T.; Fritsch E.F. и Sambrook J., Cold Spring Harbour Publication Нью-Йорк, стр. 368–369. Присутствие нужного генного фрагмента человеческого проинсулина определяется путем процедуры миниотбора с использованием электрофо реза на полиакриламидном геле для анализа выва ренного с Xba I/Bam HI фрагмента. Плазмиды с правильными по размеру вставки (примерно 315 bp) отбираются для амплифи кации и очистки. Плазмида, содержащая ген челове ческого проинсулина, представляет собой pRB 145. Ограничительная точка и функциональная карта плазмиды pRB 145 представлена на фиг. 16 данной заявки. 2. Построение плазмиды pRB 164A. Примерно 30 мкг плазмиды pRB 145 суспензируется в 20 мкл 10Х буферного раствора Nde I, 5 мкл органичительного фер мента Nde I (New England Biolabs, 40 ед.) и 175 мкл Н 2О с осторожным перемеши ванием, и смесь инкубируется при 37оС в течение 1 часа Затем в реакционную смесь вводится два мкл ограничительного фер мента Bam HI (New England Biolabs, 40 ед.), и осуществляется инкубирование при 37оС в течение еще 2 часов. ДНК осаждается тремя объемами этанола и 0,3 Мол NaOAc, и подвергается электрофо резу на 1% низкоплавком агарозном геле. Более мелкий ограничительный фрагмент (примерно 270 вр) Nde I/Bam HI, кодирующий ген человеческого проинсулина, срезается с ге ля, и ДНК извлекается путем прохождения через колонку Elutip–d, как описано выше. После осаждения и сушки, ДНК выдерживается в 30 мкл 10 мМол. Трис, pH=8,0. Затем к этой ДНК (30 мкл) добавляется 20 мкл 10Х буферного раствора Ava II (50 мМол. NaCI, 6 мМол Трис-HCl, pH=8,0, 10 мМол MgCl2, 6 мМол 2-меркаптоэтанола, 100 мкг/мл BSA), 5 мкл ограничительного фермента Ava II (New England Biolabs, 20 ед), и 175 мкл Н 2О. После осторожного перемешивания данная реакционная смесь инкубируется при 37оС в течение 2 часов. ДНК осаждается тремя объемами этанола и 3 Мол. NaOAc (20 мкл), и затем подвергается электрофо резу на 1,2% низкоплавком агарозном геле. Более крупный ограничительный фрагмент Ava II/Bam HI (примерно 156 hp) срезается с геля и затем ДНК извлекается путем прохождения через колонку Elutip–d, как описано выше. После осаждения и сушки ДНК выдерживается в 30 мкл 10 мМол Трис, рН=8,0. ДНК (~ 115 hp), соответствующая ограничительному фрагменту Nde I/Ava II гена человеческого проинсулина, получается путем синтеза. Первый этап заключается в синте зе четырех однониточных деоксирибоолигонуклеоти дов посредством синтезатора ДНК Applied Biosystems, модели 380 В). Нуклеотидные последовательности этих четы рех олигонуклеотидов являются следующи ми: 1. TATGCGTATGТ TTGTTAACC AACACCTGTGCGGCTCCCACCTG GTGGAAGCTCTGTACCT (60 мер ) 2. GGTGTGCGGTGAACGTGGCTTCTTCTAC ACCAAGCCGACC CGCCGTGAGGC AGAG (55 мер ) 3. CACCAGGTAC AGAGCTTCCACC AGGTGGGAGCC GCAC AGGTGTTGGTTAAC AAAC ATACGC A (62 мер ) 4. GTCCTCTGCCTCACGGCGGGTCGGCTTGGTGTAGAA GAAGCCACGTTC ACCGCA (54 мер ) После очистки каждого олигонуклеотида путем электрофореза на полиакриламидном геле олигонуклеоти ды два и три фосфорилируются согласно описанию публикаций Brown E.L., Belagaje R. Kyan M.J. и Khorana H.G. (1979.). Методы в энзимологии, Ред. Wu, R., Academie Press Нью–Йорк, 68, 109–151, которая проводится здесь как ссылочный материал. Фосфорилированные олигонуклеоти ды два и три (~ 715 пикомолей каждого) затем смешиваются с олигонуклеотидами один и два (~ 860 пикомолей), ренатурируются и сшиваются в буферном растворе (200 мкл), содержащем 50 мМол, Трис-HCl (рН=7,6), 10 мМол. MgCl2, 10 мМол ДТТ, 50 мкМол, АТР и 20 ед. Т4 ДНК ли газин (Fharmacia) в течение 16 часов при 4оС. Продукт сшивки очищается на 15%-ном полиакриламидном геле. ДНК извлекается из гелевого слоя путем электрофо реза, с последующим обессоливанием в колонке с Sephadex G–50. Выход желаемого сши того продукта составляет 485 пикомолей. Примерно 100 пикомолей этой ДНК фосфо рилируется в буферном раство ре (50 мкл), содержащем 50 мМол. Трис. (рН=7,6), 10 мМол. MgCl2, 10 мМол. ДТТ и АТР, как описано в публикации Brown E.L. и др., 1979 г. Ме тоды в энзимологии, 68, 109–151, которая приводится здесь как ссылочный материал. После фильтрации через колонку Sephadex G–50 ДНК вы держивается в 50 мкл 10 мМол, Трис, рН=8,0. Примерно 2,5 мкл векторной ДНК (вываренной с Nde I–Bam HI pczR 126) смешивается с 18 мкл ограничительного фрагмента Ava III /Bam HI от плазмиды pzB 145 и 10 мкл (10 пикомолей) синтетического связывающе го звена Nde I/AAva II в буферном растворе (50 мкл), содержащем 50 мМол Трис (рН=7,6), 10 мМол MgCl2, 10 мМол ДТТ, 800 мкл АТР и 3,5 ед. Т4 ДНК лигазы. Реакционная смесь инкубируется при 4оС в те чение ночи и затем трансфор мируется в E.coli K12 RV 308 согласно процедуре примера 29А3. Желаемая трансформанта E.coli K12 RN 308/pRB 164A идентифи цируется путем анализа ее плазмидной ДНК. Нужная трансформанта выращи вается при 30оС в среде Т4, содержащей 5 мкг/мл тетрациклина до достижения О.Д550 примерно 0,2 (ранняя логарифмическая фаза роста) и затем при 42оС в течение 3–3,5 часа для индуцирования синте за человеческого проинсулина. Клетки выпадают в осадок и лизируются путем добавления буфе ра (0,125 Мол. Трис-HCl, рН=6,8, 2% SDS, 30% Глицерина, 1 Мол. 2-меркаптоэтанола, 6 Мол. мочевины). Образцы нагреваются до 97оС в течение 2 минут до помеще ния их на акриламидный гель. Слои легко визуально просматриваются путем окрашивания красителем Кумаси Брильянтовым синим. Для количественного определения процента клеточного белка используется сканирующий гелевый денситометр. Ограничительная точка и функциональная карта плазмиды pRB 164A представлена на фиг. 17 данной заявки. 3. Построение плазмиды pRB172 Примерно 25 мкг плазмиды pRB 145 суспензируется в 15 мкл 10Х буферного раство ра Dra III (200 мМол NaCl, 50 мМол, Трис-HCl, рН=7,5, 1 мМол ДТТ, 10 мкг/мл BSA), 6 мкл ограничительного фермента Dra III (Bochringer Meinnheim 27 ед.) и 129 мкл Н 2О, осто рожно перемешивается и инкубируется при 37оС в течение 6 часов. После инкубации ДНК осаждается, высушивается и повторно суспензируется в 10 мкл 10Х буфера XbaI (150 мМол NaCl, 6 мМол Трис-HCl, рН=7,9; 6 мМол MgCl2, 7 мМол 2-меркаптоэтанола, 100 мкг/мл BSA), 3 мкл ограничительного фермента XbaI (Bochringer Mainnheim 36 ед) и 77 мкл Н2О. Реакционная смесь осторожно перемеши вается и инкубируется при 37оС в течение 4 часов. ДНК снова осаждается тремя объемами этанола и ацетата натрия, подвергается электрофо резу на 1% низкоплавком агарозном геле. Нижний слой, соответствующий примерно 85bp ограничительного фермента XbaI/Dra III ге на человеческого проинсулина, срезается с геля и ДНК извлекается путем осуществления процедуры в колонке Elutip–d. После осаждения и сушки ДНК выдерживается в 30 мкл 10 мМол Трис, рН=8,0. Примерно 15 мкг плазмида pRB 164А отсекается с ограничительным ферментом Dra III и Xba I согласно описанной выше процедуре. Верхний слой, соответствующий векторному фрагменту Xba I/Dra III, выделяется из агарозного геля путем осуществления процедуры в колонке Elutip–d. После осаждения и сушки ДНК выдерживается в 30 мкл 10 мМол. Трис, рН=8,0. Примерно 5 мкл векторной ДНК pRB 164А, вываренной с XbaI/Dra III- cмешивается с 5 мкл ограничительного фрагмента XbaI/Dra III от плазмиды pВR 145 в буфер ном растворе (50 мкл), содержащем 50 мМол, Трис–HCl, рН = 7,6, 10 мМол MgCl2, 10 мМоль, DTT, 800 мМол АТР и 6 ед. Т4 ДНК лигазы. Реакционная смесь инкубируется при 4оС в течение ночи и используется для трансформации клеток E.coli K12/RV308, которые прекращаются в компетентные клетки путем стандартной обработки CaCl2. Трансфор манты выбираются на ТУ агаровых ча шках, содержащих 5 мкг/мл тетрациклина. Плазмиды выделяются из стойких к тетрациклину колоний и анализируются путем вываривания с ограничительными эндонуклеазами Xba I и Bam НI. Выстраивается аминокислотная последовательность ДНК от положительных клонов с использованием системы построения последовательности (U. S. Biochemicals). Плазмиды с желаемой правильной последовательностью отбираются для амплифи кации и очистки. Таким образом изолируется трансформанта (E.coli К12 RV 308/pRB 172. Выражение и аккумуляция человеческого проинсулина, заключающего в себе данную плазмиду, анализируются путем визуализации всего клеточного белка с последующим электрофо ретическим отделением в 15% полиакриламидной матрице. Ограничительная точка и функциональная карта плазмиды представлена на фигуре 18 данной заявки. С. Построение Escherichia coli RV 308pRB 173 и pRB 174 Примерно 20 мкг плазмиды pRB 172 или pRB 145 суспензируются в 20 мкг 10Х буфера Xba I (150 мМол NaCl, 6 мМол, Трис-НСl, рН = 9,6 мМол, MgCl2, 7 мМол 2-меркаптоэтанола, 100 мкг/мл BSA), вво дится 2 мкл ограничительного фермента XBa I (Bochringer Mannheim, 24 ед.), смесь осто рожно перемеши вается и инкубируется при 37оС в течение 4 часов. После инкубирования ДНК осаждается, высушивается и снова суспензируется в 10 мкл 10Х буферного раствора Bam HI (100 мМол, NaCl, 10 мМол Трис-HCl, рН = 8,0,5 мМол. MgCl2, 1 мМол. 2-меркаптоэтанола, 100 мкг/мл BSA), 5 мкл ограничительного фермента Bam HI (Bochringer Mannheim, 40 ед.), и 75 мкл H2O. Реакционная смесь осторожно перемешивается и инкубируется при 37оС в течение 4 часов. ДНК снова осаждается этанолом и ацетатом натрия и подвергается электрофо резу на 1 % низкоплавком агарозном геле. Ограничительный фермент Xba I/Bam HI примерно 320 bp, соответствующий гену человеческого проинсулина, извлекается из геля путем осуществления процедуры в колонке Elutip–d. После осаждения и сушки ДНК снова суспензируется в 20 мкл 10Х буфер ного раствора Xma I (25 мМол. NaCl, 10 мМол. Трис–HCl, рН=7,5, 10 мМол. MgCl2, 10 мМол 2-меркаптоэтанола, 100 мкг/мл BSA), 10 мкл ограничительного фермента Xma I (New England Biolabs 10 ед.) и 170 мкл воды. Реакционная смесь осторожно перемешивается и инкубируется при 37оС в течение 4 часов. После инкубирования ДНК осаждается этанолом ацетатом натрия и подвергается электрофорезу на 1 % низкоплавком агарозном геле. Более крупная ДНК, соответствующая ограничительному фрагменту Xba I/Xma I примерно 200 bp, срезается с геля, и эта ДНК выделяется путем осуществления процедуры в колонке Elutip–d, как описано выше. Эта ДНК выдерживается в 30 мкл 10 мМол, Трис, рН = 8,0. ДНК на 51 bp, соответствующая ограничительному фрагменту XmaI/Bam HI гена человеческого проинсулина, выравнивается в последовательность, которая определена ниже. Два однониточных деоксирибоолигонуклеотида: CCGGGTGC AGGC AGCCTGC AGCCGCTGGCCCTGGAGGGTTCCCTGCAGTAG (51 мер ) и GATCCTACTGCAGGGAACCCTCCAGGGCCAGCGGCTGC AGGCTGCCTGC AC (51 мер ) синте зируется с помощью синтезатора ДНК, Applied Biosystems (модели 380 В) и очищается путем электрофо реза на полиакриламидном геле в присутствии 7 Мол мочевины. После изоляции оба нуклеотида фосфо рилируются согласно процедуре в описаннии Brown E.L.; Belagaje R.; Ryan M.J. и Khorana H.G. (1979 г.). Ме тоды энзимологии. Ред. Wu.R. Academie Press, N.J. 68, 109–151, и ренатурируются с образованием желаемого свя зывающего звена. Примерно 10 мкг плазмиды pCZR 126 S (из примера 29 А 12) суспензируется в 20 скл 10Х буферного раствора Xba I (150 мМол NaCl, 6 мМол. Трис-HCl, рН = 7,9, 6 мМол MgCl2, 7 мМол 2-меркаптоэтанола, 100 мкг/мл BSA), 2 мкг ограничительного фер мента Xba I (Bochringer Mannheim, 24 ед.) и 178 мкл воды. Реакционная смесь осторожно перемешивается и инкубируется при 37оС в течение 2 часов. После этого инкубирования вводится 4 мкл 5 Mол. NaCl и 2 мкл ограничительного фер мента Bam HI (New England Biolabs, 20 ед.), и инкубирование продолжается еще в течение 2 часов при 37оС. Затем ДНК осаждается этанолом и ацета том натрия стандартным приемом, и подвергается электрофо резу на 1% низкоплавком агарозном геле. Верхний слой, соответствующий векторной ДНК Xba I/Bam HI выделяется путем осуществления процедуры в колонке Elutip–d. После осаждения и сушки ДНК выдерживается 30 мкл 10 мМол. Трис, рН = 8,0. Примерно 5 мкл векторной ДНК pCZR 126S, выверенной с Xba I/Bam HI, смешивается с 10 мкл ограничительного фрагмента Xba I/Bam HI от pRB 145 или pRB 172 и 4 мкл киназированного связующе го Xma I/Bam HI, приго товленного как указано выше в буферном растворе (50 мкл), содержащем 50 мМoл Трис (рН = 7,6), 10 мМол MgCl2, 10 мМол DTT, 800 мкМол АТР и 6 ед. Т4 ДНК лигазы. Реакционная смесь инкубируется при 4оС в течение ночи и используется для трансформации штамма E.coli К 12 RV 308 согласно процедуре примера 29 А3. Желаемые трансформанты E.coli К 12 RV 308/pRB 173 (если они образованы от векторного фрагмента pRB 172) и E.coli К 12 RV 308/pRB 174 (если образованы от векторного фрагмента pRB 145) идентифи цируется путем анализа их плазмидной ДНК и белка до и после индукции при 42оС. Ограничительная точка и функциональная карта плазмиды pRB 173 представлена на фиг. 19 данной заявки. Примерно 200 нанограммов ДНК плазмиды от мини-препаративной обработки, трансфор мируется в клетки E.coli L 201 и трансформанты отбираются на чашках с 2 х ТУ ага ром, содержащи х 15 мкг/мл тетрациклина. Белки, выраженные этими трансфор мантами, анализируются и желаемые трансформанты подтверждаются по продуцированию челове ческого инсулина. Таким образом выделяется E.coli L 20/ pRB 173 и E.coli L 201/pRB 174. Д. Построение Escherichia coli K 12 RV 308/pRB 175, pRB 176, pRB 177 и pRB 178. Примерно 12 мкг плазмиды pRB 172 суспензируется в 15 мкл 10Х буфера Nde I (100 Мол NaCl, 50 мМол Трис-HCl, рН = 7,5, 10 мМол MgCl2, 1 мМол DTT, 100 мкг/мл BSA), 2,5 мкл ограничительного фермента Nde I (20 ед.) и 133 мкл воды. Реакционная смесь осторожно перемешивается и инкубируется при 37оС в течение ночи. После инкубирования ДНК осаждается этанолом и ацета том натрия стандартными приемами, высуши вается и повторно суспензируется в 15 мкл 10Х буфе ра Dra III (50 мМол Трис-HCl, рН = 7,5, 20 мМол NaCl, 10 мМол MgCl2, 1 мМол DTT), 5 мкл ограничительного фермента Dra III (20 ед.) и 130 мкл H2O. После осторожного перемешивания реакционная смесь инкубируется при 37оС в течение ночи. ДНК снова осаждается этанолом и натрийацетатом, как указано выше, и подвергается электрофо резу на 15% низкоплавком агарозном геле. Желаемый крупный ограничительный фрагмент срезается с геля и ДНК извлекается путем осуществления процедуры в колонке Elutip–d ДНК выдерживается в 30 мкл 10 мМол Трис-HCl, рН = 8,0. Нижеследующие связывающие звенья ДНК химически синтезируются с помощью синтезатора Applied Biosystems (модели 380 В): (i) 5' 3' TATGTACGACC AACACCTGTGCGGCTCCCATCTG 3' AC ATGCTGGTTGTGGAC ACGCCGAGGGTA 5' для pRB 175 (ii) 5' 3' TATGTACGACGTTAACCAAC ACCTGTGCGGCTCCCATCTG 3' AC ATGCTGCAATTGGTTGTGGACACGCCGAGGGTA 5' для pRB 176 (iii) 5' 3' TATGTATAACCAAC ACCTGTGCGGCTCCCATCTG 3' AC ATATTGGTTGTGGAC ACGCCGAGGGTA 5' для pRB 177 (iv) 5' TATGTACGTTAACCAAC ACCTGTGCGGCTCCCATCTG 3' 3' AC ATGC AATTGGTTGTGGAC ACGCCGAGGGTA 5' дл я pRB 178 После очистки каждого олигонуклеотида путем электрофореза на акриламидном геле, они фосфорилируются согласно описанной выше процедуре и ренатурируются для ускорения сшивки и построения ДНК фрагментов, кодирующих аналог гена человеческого проинсулина. Связывающие звенья от (i) до (iv) используютс я для построения плазмид pRB 175, pRB 176, pRB 177 и pRB 178, соответственно. Примерно 3 мкл фра гмента ДНК век то ра pRB 172, вы ва рен ного с Nde I/D ra III, смеши вается с 4 мкл каждого из кина зирован ных свя зывающи х з вен ьев (i–i v) в буфер ном раство ре (50 мкл), со дер жащем 50 мк Мо л. Три с-HCl , р Н = 7 ,6 , 10 м Мо л Mg Cl2 , 10 м Мол D TT, 800 мкМол АТР и 6 ед. Т 4 ДНК ли га зы. Реак ционная смесь инкуби руе тся при т-ре 4 оС в те че ние ночи , и эта сме сь используе тся для трансфор мации клеток E.coli K 12 R V 308 со гласно проце дуре при мера 29 А3. Же лае мые трансфор манты E.co li K12 R V 308 /pR B 175 , E.coli K 1 2 R V 30 8/pR B 176 , E R V 308 /pR B 177 и E.coli K 12 R V 308/pR B 178 иден ти фи цируе тся п утем а на лиза и х ДНК плаз миды и про дуциро вания бе лка до и пос ле ин дук ции кле ток при 42 оС . Ограничи тель ная точка и функцио нальная карта плаз миды pRB 175 предста влена на фи г. 20 дан ной заявки. Е. Исходная культура E.coli K 12 P 308/pRB 172 приготавливается в фор ме перманентной исходной культуры следующим образом. Выделенн ые колон ии высева ются на нижеследующи х фе ноти пны х ча шка х: L – а гар + 5 мкг/м л Тс (те трациклин а), L-а гар + Sm (стр ептомицинсульфа т), а гарова я среда М–9 (солевая сре да , содержаща я Mg SO 4 , тиамин , НУ С АС Е и глюкоз у), и а гар овая среда М–9 , содержащая глюкоз у и не содержащая лактоз у. Одн а ча шка с L -а гаром /Тс инк убир уе тся при 42 оС , и оста льные ча шки инк убир уются п ри 30 о С в течение 24 часо в. Фен отипно прави льная колония , о тобра нная с ча шки L -а гар /Тс пр и 30 о С , и спо льз уе тся для инок улировани я в ко лбе, соде ржаще й 50 м л L-п итате льн ого бульон а (с соде ржан ием триптона , дрожжевого экстракта , NaC l и глюкозы ) п люс 5 мкг/м л среды Тс. Э та колба инк убир уе тся при 3 0 оС со встряхиван ием в течени е 7 часо в. Матери ал в ко лбе (1 ,2 мл) ра спределяе тся в би о-замор аживающи е амп улы и замор аживае тся в пар офазн ом жидком азо те . Затем мате риа л о ттаива е т, и и спольз уе тся в количестве 1 мл для инок уляц ии в колбе , содер жаще й 50 м л L-бульон а плюс 5 мкг/м л Тс. Э та колба и нк убир уе тся п ри 30 о С со встряхиван ием в течен ие 4 часо в. Две ко лбы, содержащи е 5 00 мл среды ВС–7 (содержи т MgSO 4 , тиан ин , глюкоз у, Тс и и зотопны е соли ), ин ок улир уются с 1 м л каждой из указанн ы х выше сре д, и эти к олбы инк уби р уются со взб алты ванием в течени е 9 часо в пр и 30 оС . 150-литровый ферментер, содержащий 80 литров среды (содержит лимонную кислоту, суль фат железа-аммония, К2HРO4 NH4Cl, NaH2PO4, CaCl2, MgSO4, глюкозу и изотопные неорганические соли) инокулируется 1 литром питательного бульона ВС–7, указанного выше. Ферментер функционирует при 30 оС до тех пор, пока скорость выделения двуокиси углерода не достигнет значения 1,0 мМол/л/мин. В этот момент 50 литров пита тельного бульона вводится в фер ментер для получения клеточного инокулума. 2500-литровый ферментер, содержащий 1500 литров той же среды, что используется в 150-литровом ферментере, функционирует при 30оС до тех пор, пока скорость выделения двуокиси углерода не достигнет значения 1,0 мМол/л/мин, и затем температура повышается до 40оС. Функционирование ферментера продолжается еще в те чение 8 часов. За тем пита тельный бульон инактивируется нагревом при 61оС в течение 7 минут. Из инактивированного нагреванием бульона получаются нижеследующие выхо ды: Выход клеток: 13,1 г/л на сухой вес; эффективность продукта: 273 мкг Des-формилата/мл питательного бульона; удельная активность (например граммов продукта/грамм клеток), 2,1 %; процент формилированного продукта, 13%. Бульон брожения подается в сборник из нержавеющей стали, в котором температура поддерживается от 2 до 8оС. Затем весь питательный бульон центрифугируется или фильтруется для сбора клеток E.coli, так чтобы выход твердых продуктов в расчете на сухой вес составляет 15-20%. Твердый продукт, составляющий собранные клетки, снова суспензируется в технологической воде. Затем клетки разрываются с использованием гомогенизации под высоким давлением или других подхо дящи х мето дов, так чтобы лизис клеток составлял 90–99%. Гомоге низированная суспензия разбавляется до 5 % сухой массы те хнологической водой. Величина рН разбавленной разорванной клеточной суспензии доводится до значения в пределах от 7 до 10 путем ввода гидрата окиси натрия. Инклюзионные образования (гранулы) отделяются от клеточных осколков путем дифференциального центрифугирования. Полученный концентрат содержит от 15 до 30% твердых ве ществ. Эти инклюзионные образования (гранулы) солюбилизируются при щелочном значении рН в присутствии цистеина и мочевины. Эти солюбилизированные гранулы подвергаются катионообменной хроматографии на те кучей Сефа розе SP для отделения смешанной дисульфидной формы проинсулинового аналога от обще го объема гранулированного материала. Ме тионил-тирозиновое удлинение аминового конца аналога проинсулина удаляется путем инкубирования с катепсином С, дипентидиламинопептидазой. Реакция катепсинового расщепления завершается путем сульфи толиза тетратионатом калия и цистеином при рН = 8,8. После вытеснения растворителем через Sephadex C–25 полученный S-сульфонат проинсулинового аналога подвергается перегибу п утем инкубации при величине рН равной 10,6 в присутствии цистеина. Процесс перегиба подавляется путем доведения величины рН до 2,4. Проинсулиновый аналог с должным перегибом отделяется от материала с неправильным перегибом, а также от загрязнений, оставшихся после этапа расщепления катепсина С, путем хроматографи ческого разделения с гидрофобным взаимодействием на смоле SP20SS. Это разделение происходит с использованием ацетонового градиента, так что органический растворитель должен быть удален из основного потока, путем выпаривания перед дальнейшей операцией. После выпаривания органического растворителя проинсулиновый агент с правильным перегибом инкубируется с трипсином и карбоксипептизадой В. Эти ферменты эксцизируют С пептид аналога проинсулина и образуют че ловеческий инсулин Lys (В28), Pro (B29). Этот аналог инсулина затем дополнительно очищается путем катионообменной хроматографии на S-Сефарозе, с последующей хро матографией высокого разреше ния с обратимой фазой на смоле С–8 (10 мкм). Ацетонитрил удаляется из основного потока обратимой фазы путем вытеснения растворителем на Sephadex C–25, и полученный продукт концентрируется с использованием спирально намотанной систе мы ультрафильтрации. Концентрированный продукт подвергается хроматографии исключения на смоле Sephadex C–50 и лиофи лизируется до сухого состояния. В нижеследующей таблице 2 представлены данные аминокислотного анализа соединений, отвечающи х изложенным выше примерам. Физиологические эффекты аналогов инсулина, отвечающи х настояще му изобретению, показаны в нижеследующей испытательной системе в условиях "ин ви во". В качестве испытательных животных используются обычные самцы крыс Spraque Dawley, которые получены из Charles River Laboratories. Полученные крысы имеют вес в диапазоне от 160 до 180 г и их выдерживают в течение одной недели в помеще нии при 75оF (24оС) с регулируе мым световым циклом (с включенным светом с 7,00 вечера до 7 утра и с выключенным светом – с 7 утра до 7 вечера). Рацион крыс составляет капуста Пурина 5001 ad libitum. Крысы, используе мые для каждого испытания, подвергаются голоданию в течение примерно 16 дней до их использования. При первом использовании их вес составляет примерно 200 граммов. При достижении веса примерно 275 граммов (в период за три недели) животных уже больше не используют. Одна группа из 10 крыс-самцов используется ежедневно для каждого испытываемого соединения (то есть биосинтезированный человеческий инсулин, свиной инсулин и аналог человеческого инсулина). В течение недели каждая группа используется лишь однократно. Десять крыс были разделены на две группы по пять крыс в каждой. Одна группа служит в качестве контрольной, и в нее вводится лишь носитель путем подкожной инъекции. Другая груп па, состоящая из пяти крыс, подвергается обработке испытываемым соединением. Белки растворяются в 0,05 нормальной HCl (рН = 1,6) и в результа те получается исходный раствор концентрацией 100 мкг на мл. Из него получают ряд разбавленных растворов в обычном солевом растворе, который вводится путем подкожной инъекции в организм крыс. 100 мкл образца крови берут из хвостовой вены у каждой крысы при нулевом моменте времени и через 30 минут, 1 час, 2 часа, 3 часа и 4 часа после ввода. Глюкозу определяют колориметрическим способом оксидазы глюкозы (Sigma Chemical Co.). Для каждой крысы рассчитывают изменение содержания глюкозы в крови от нулевого момента времени, и конечные результаты выражены как среднее изменение процента ± SEM для экспериментальной группы с корректировкой на среднее изменение для контрольной группы для того же дня. Кривая доза – реакция была построена по данным испытания с 4–7 различными концентрациями испытываемого соединения с использованием максимальной реакции в заданный момент времени (обычно один или два часа после ввода белка). Из данной кривой определяют значение ED50, как значение вводимой подкожной дозы белка, которая дает половину максимальной гипогликемической реакции. Результаты представлены в нижеследующей таблице 3. Как указывалось выше, аналоги инсулина, отвечающие данному изобрете нию, имеют пониженную способность к димеризации или иными словами спосо бно сть к самоа ссоциир ован и ю высоком олек улярн ы х фо рм . Так , пр и вводе о дн ого или не ско льки х ук азанн ы х ан алого в до стига е тся о чень бы стро е ин ициир ован ие акти вно сти . Ан алоги и нсулин а , о твеча ющи е да нном у из обр етен и ю, эффе кти вны пр и лече ни и гипе р гликеми и п ри воде в орган изм пац и ента , н уждающе го ся в этом лечен ии , эффекти вн ого количе ства аналога инсули на форм улы 1 . По д и спо льз уемым термин ом "эффе кти вно е количе ство " имее тся в виде один и ли н еск ольк о аналого в инсулин а , о твеча ющи х данн ом у и зобр ете ни ю, к отор ые не обхо димы для снижен ия и ли по ддержан ия ур о вня саха ра в кр ови , либо терап е вти ческ и , либо п рофи лактиче ски . Э то к оличе ство обы чно м оже т н ахо ди ться в п редела х прим ерн о о т 10 е д до 6 0 е д и ли более в де нь (или пр имерн о о т 0 ,3 до 2 мг, что со ста вляет пр имерн о 29 един иц на м и лли грамм ). Одн ако следуе т име ть в виду, что к оличе ство а нало га (а налого в) инсулин а , к отор ое фактиче ск и вводи тся в орга низм , до лжно о пределя ться леча щи м врачом в соо тве тстви и с обсто яте ль ствами , включа ющи м и со сто яни е з аболеван ия , то е сть п ричин у, вы зва вшую ги пе р гликеми ю), ти п и сп ольз уем ого аналога и нсулин а , кото ры й вы бр ан для ввода в о рган изм , выбранн ый п ар эн тера льн ый спосо б ввода , во зра ст пац иента , ве с и реакци ю каждого п ациента и о стр оту си мптом п ациента . С ледовате льно , указ анны е выше пределы доз ы н икак не м огут я вля ться о гр аниче ни ем объ ема да нного изо бр етен ия . Аналоги инсулина, отве чающие данному изобретению, вво дятся в организм пациента, нуж дающе гося в этом лечении (то есть пациента, страдающего от гипергликемии) с использованием фармацевтических композиций, содержащих эффективное количество по меньшей мере одного аналога инсулина формулы 1 в комбинации с одним или несколькими фармацевтически пригодными эксципиентами или носителями. Для этих це лей фармацевтические композиции могут быть приготовлены таким образом, что будут содержать примерно 100 ед. на мл или аналогичные концентрации с содержанием эффективного количества аналога (аналогов) инсулина. Эти композиции обычно (хотя и необязательно) парэнтеральны по своей природе и могут быть приготовлены любым способом с применением обычных эксципиентов или но сителей для вводимых парэнтерально продук тов, которые уже известны. Смотри, например, публикацию Remington's Pharmaceutical Scien ce , 17 -е из д. Ma ck Pu blish in g Comp an y, Eas ton PA, U SA 1985 , к ото р ая да е тся з де сь к ак ссыло чны й мате р иа л. Так , на прим ер , доз ир ованн ые фо рмы для па рэн те р альн ого ввода могут бы ть п риго то влен ы п утем сусп енз ирова ни я и ли ра створ ени я желаемого количе ства по м еньше й мер е о дн ого а налога инсулина фо рм улы I в нетокси чн ом жи дком н осите ле , пр иго дном для и нъекции , так ом к ак водн ая среда и стер илиз ующа я сусп енз ия или ра ство р . Как возможны й вар иа н т, и змер енно е количе ство соедин ен ия може т бы ть п омеще но в амп улу и амп ула и ее содержимо е стер илиз уются и зап аива ются . Амп ула и ли носитель могут бы ть подго то влены для целе й смеши ван ия до ввода в о рган изм . В фа рмаце вти че ски х комп озиц ия х, п ри го дн ы х для па рэн тер а льного ввода в о рга низм , и спо льз уются р азбавите ли , эк сцип ие нты и носи те ли , такие как вода и смеши ваем ые с водо й орган иче ски е ра створ ители , такие как глицер ин , сез амово е м асло , арахи сово е ма сло , во дн ый пр опиле н глико ль , N ,N '-димети лфо р мами д и т. д. П ример ами таки х фа рмаце вти че ски х комп озиц ий я вля ются стер и льны е из отон иче ские во дносо левы е р аство р ы ан алого в инсулин а фо рм улы I, котор ые могут бы ть п риго то влен ы в форм е бу фе рн ого ра створ а с фармаце вти че ски п риго дн ым буфе ром , и кото ры е свободн ы о т пир оге нны х веще ств. Кроме того , вводимая па рэн тер а льн о фа рмаце вти че ская к омпоз иция м оже т соде ржа ть п редо хран яющи е ср едства , такие как мета -кр езо л, и др уги е веще ства , для регулир ован ия вели чин ы р Н коне чн ого продук та , такие как гидр а т окиси н а тр ия и ли солян ая ки слота . Ан алоги и нсулин а , о тве чающи е данн ом у изо бр етен ию, м огут бы ть пр игото влены также в фо рме фа рмаце вти че ски х комп озиц и й , пр иго дны х для ввода чер ез но с . Так ие к омпоз иции опи сыва ются по др обн о в Евр оп ей ской п атен тно й зая вке 220 038 3 А3 , к отор ая пр иводи тся з де сь как ссыло чны й матер и ал. Такие к омпоз иции п ригота влива ются вм есте с о дним и ли н еск ольк ими фа рмаце вти че ски пр иго дным и разб авите лями , фармаце вти че ски при емлемым к оличе ство соли ще ло чного мета лла , соли аммон ия , и ли свободн ой к ислоты свободн ого о т цинк а инсулина , и пр и желан ии усилива ющи м а бсор бци ю количе ством по меньше й мер е одн ого уси лива ющего а бсо рбци ю а гента , вы бра нного из гр уппы , включа юще й : (1 ) олеин овую к ислоту и ли е е сложный эфи р , (2 ) жи дк ую форм у сложного эфира сорбита и жирной кислоты (3) жидкую форму полиоксиэтиленового производного сложного эфира сорбита и жирной кислоты, и (4) жидкую форму сополимера оксиполиоксиэтилен-полиоксипропилен-полиоксиэтилен. Для демонстрации эффективности внутриносового ввода аналога человеческого инсулина Lys (B28), Pro (В29) в сопоставлении с человеческим натриевым инсулином проводится нижеследующее исследование. Самцы коротконогих гончих собак весом от 10 до 12 кг поддерживаются в прекрасном физическом состоянии до и в хо де данного исследования. Собаки выдерживаются в режиме голодания в течение 16 часов, но они имеют доступ к воду для гарантии предотвраще ния непредвиденных колебаний содержания инсулина. В те чение всего исследования собак анестезируют пентабарбитолом натрия и температура их те ла сохраняется посредством нагревательных подушек для сведения к минимуму колебания содержания глюкозы. Испытательные образцы человеческого инсулина (то есть Lys (В28), Pro (В29) и человеческого натрийинсулина) растворяются в дистиллированной воде, и величина рН раствора доводится до 7,5 посредством гидрата окиси натрия. Конечная концентрация инсулина составляет 70 ед. на мл. Человеческий инсулин Lys (В28), Pro (B29) дозой 0,8 ед на кг вводится в каждую из восьми собак, и человеческий натрийинсулин вводится дозой 0,8 ед на кг в каждую из че тырех собак. Все дозы вводятся через нос путем ввода струи че рез ноздрю с помощью дозировочного распылителя и модифи цированного носового аппликатора. Образцы крови отбираются из шейной вены за 30, 15 и 0 минут до вво да и через 10, 20, 30, 45, 60, 90, 120, 180 и 240 минут после ввода для измерения снижения глюкозы в крови. Результаты данного исследования приводятся в таблице 4. При осуществлении описанной выше методики, сопоставляется профиль снижения глюкозы крови при внутриносовом вводе человеческого инсулина Lys (В28), Pro (B29) с профи лем снижения при внутривенном и подкожном вводе аналога. Для внутривенного вво да раствор инсулина, содержащий аналог человеческого инсулина Lys (B28), Pro (B29) вводится путем инъекции ударной дозы (0,1 ед на кг) в подкожную ве ну ноги каждой из четырех собак. Образцы крови бе рутся за 30, 15 и 0 минут до вво да и через 5, 10, 15, 20, 30, 45, 60, 90, 120, 180 и 240 минут после ввода. Для подкожного ввода раствор инсулина, содержащий аналог человеческого инсулина Lys (В28), Pro (B29), вводится под эпидермис (0,2 ед на кг) ругой боковой стороны живота каждой из шести собак. Образцы крови берутся таким же образом, как в случае носового вво да, как описано выше. Ре зультаты данного сопоставительного исследования приводятся в таблице 5. Таблица 1 Аналоги человеческого проинсулина Плазмида % белка Met-Tyr Аналог pRB 145 11.3 Met-Arg-Met-[L ys(B28), Pro(B29)HPI] pRB164A 10.5 Met-Tyr-[Lys(B28), Pro(B29)HPI] Met-Tyr-Met-[Lys(B28), Pro(B29)B-chain]-Q* pRB 172 pRB 173 7.3 ND pRB 174 ND Met-Tyr-[Des(BI), Des(B2), Asp(B3), Lys(B28, Pro(B29)HPI] pRB 175 8.5 Met-Tyr-[Asp(B1), Lys(B28), pRB 176 9.3 Pro(B29)HPI] Met-Tyr-(Des(B1), Des(B2), Lys(B28), pRB 177 9.4 pRB 178 8.5 Met-Tyr-[B-цепь ]-Q* Pro(B29)HPI] Met-Tyr-[Des(B1), Lys(B28), Pro(B29)HPI] *Q = -Arg-Arg-Glu-Ala-Glu-Asp-Leu-Gln-Val-Gly-Gln-Val-Glu- Leu-Gly-Gly-Gl y-Pro-Gl y-Ala-Gly-Ser-Leu-ClnPro-Leu-Ala-Leu-Glu -Gly-Ser-Leu-Gln-; Таблица 2 Аминокислотный анализ аналогов че ловеческого инсулина 1,2 Аминокислот Пример ная группа 2 3 4 5 6 7 8 9 10 Asp 3.00(3) 3.00(3) 3.00(3) 3.00(3) 4.00(4) 4.00(4) 4.00(4) 3.00(3) 3.00(3) Thr 2.82(3) 2.81(3) 2.64(3) 2.80(3) 2.80(3) 2.94(3) 2.81(3) 2.71(3) 2.73(3) Ser Glu 2.76(3) 7.16(7) 2.73(3) 7.05(7) 2.70(3) 6.93(7) 2.75(3) 7.08(7) 2.71(3) 7.01(7) 2.46(3) 6.69(7) 2.71(3) 7.18(7) 2.74(3) 6.99(7) 2.72(3) 7.94(8) Pro 1.02(1) 1.11(1) 0.92(1) 0.98(1) 1.01(1) 0.98(1) 1.02(1) 1.09(1) 0.98(1) Gly Ala 3.93(4) 1.01(1) 4.00(4) 1.04(1) 3.85(4) 1.93(2) 3.99(4) 1.02(2) 4.02(4) 1.05(1) 3.97(4) 1.05(1) 4.01(4) 1.03(1) 3.95(4) 1.03(1) 3.90(4) 1.02(1) Cys(1/2) 5.13(6) 5.13(6) 5.17(6) 5.32(6) 4.91(6) 5.53(6) 5.23(6) 5.33(6) Val Ile 3.45(4) 1.47(2) 3.44(4) 1.47(2) 3.49(4) 1.49(2) 3.51(4) 1.58(2) 3.41(4) 1.44(2) 3.67(4) 1.65(2) 3.54(4) 1.68(2) 3.45(4) 1.48(2) 3.48(4) 1.47(2) Leu 6.04(6) 6.01(6) 5.86(6) 6.19(6) 6.06(6) 5.67(6) 5.99(6) 6.02(6) 5.98(6) Tyr Phe 3.83(4) 2.84(3) 3.74(4) 2.85(3) 3.52(4) 2.64(3) 4.19(4) 2.85(3) 3.90(4)2.84(3) 3.55(4) 3.02(3) 3.82(4) 2.86(3) 3.73(4) 2.72(3) 3.58(4) 2.73(3) His 2.35(2) 2.10(2) 2.02(2) 2.14(2) 1.60(2) 2.09(2) 1.07(1) 2.02(2) 2.10(2) Lys Arg 0.97(1) 0.99(1) 0.81(1) 0.97(1) 0.91(1) 0.89(1) 1.98(1) -(6)3 0.90(1) 0.88(1) 1.96(2) 0.91(1) -(1)3 Aba Cya Nle 0.90(1) Orn Nva Met Trp Продолжение табл. 2 Аминокислотная группа 11 12 13 14 Пример 15 16 17 18 19 Asp Thr Ser Glu Pro Gly Ala Cys(1/2) Val Ile Leu Tyr Phe His 3.00(3) 2.76(3) 2.69(3) 8.04(7) 1.11(1) 4.04(4) 1.05(1) 5.26(6) 3.60(4) 1.62(2) 5.99(6) 3.73(4) 2.88(3) 2.08(2) 3.00(3) 2.59(3) 2.64(3) 6.84(7) 1.48(1) 5.70(5) 1.14(1) 4.54(6) 3.52(4) 1.82(2) 5.79(6) 3.89(4) 3.47(3) 2.24(2) 3.00(3) 2.80(3) 2.43(3) 6.89(7) 1.09(1) 3.89(4) 1.04(1) 4.57(6) 3.58(4) 1.70(2) 5.95 (6) 3.75(4) 3.01(3) 3.39(3) 3.00(3) 2.80(3) 2.78(3) 7.09(7) 1.12(1) 4.03(4) 1.05(1) 5.19(6) 3.43(4) 2.36(3) 5.98(6) 3.74(4) 2.85(3) 2.07(2) 3.00(3) 2.70(3) 2.87(3) 7.04(7) 0.93(1) 3.98(4) 1.08(1) 5.00(6) 3.41(4) 1.44(2) 6.89 (7) 3.64(4) 2.71(3) 2.30(2) 3.00(3) 2.83(3) 2.78(3) 7.08(7) 1.16(1) 4.00(4) 1.04 (1) 4.75(6) 3.51(4) 1.51(2) 6.10(6) 3.86 (4) 2.86(3) 2.48(2) 3.00(3) 2.85(3) 2.97(3) 6.94(7) 1.10(1) 4.12(4) 1.09(1) 5.20(6) 3.49(4) 1.52(2) 5.95(6) 3.74(4) 2.87(3) 2.20(2) 3.00(3) 3.03(3) 2.68(3) 6.99(7) 1.14(1) 4.08(4) 1.07(1) 4.82(6) 3.63(4) 1.75(2) 5.98(6) 3.80(4) 3.13(3) 2.29(2) 3.00(3) 2.80(3) 2.72(3) 7.01(7) 1.02(1) 3.92(4) 1.03(1) 5.30(6) 3.51(4) 1.55(2) 6.11(6) 3.84(4) 2.89(3) 2.10(2)
ДивитисяДодаткова інформація
Назва патенту англійськоюInsuline analogue, capable to reduce sugar level in blood
Автори англійськоюChance Ronald Eugene, Dimarchi Richard Dennis, Franc Bruce Khill, Shields James Edwin
Назва патенту російськоюАналог инсулина, способного снижать уровень сахара в крови
Автори російськоюЧанс Рональд Юджин, Димарчи Ричард Деннис, Френк Брюс Хилл, Шилдз Джеймс Едвин
МПК / Мітки
МПК: A61K 9/00, A61K 38/28, C12R 1/19, C12P 21/02, C07K 1/02, C12N 15/09, A61P 3/08, C07K 14/575, A61K 38/00, C07K 14/62
Мітки: здатність, знижувати, інсуліну, має, цукру, крові, аналог, рівень
Код посилання
<a href="https://ua.patents.su/43-27299-analog-insulinu-analog-insulinu-shho-maeh-zdatnist-znizhuvati-riven-cukru-v-krovi.html" target="_blank" rel="follow" title="База патентів України">Аналог інсуліну, аналог інсуліну, що має здатність знижувати рівень цукру в крові</a>
Спосіб одержання контррецепторного фактора, який блокує зв’язування інсуліну з його рецептором та має діабетогенну дію

Номер патенту: 13921
Опубліковано: 25.04.1997
Автори: Іванова Жана Вікторівна, Гуріна Наталія Марківна, Корпачев Вадим Валерійович
МПК: A61K 35/16
Мітки: інсуліну, одержання, контррецепторного, рецептором, фактора, блокує, дію, зв'язування, спосіб, діабетогенну, має
Формула / Реферат:
Способ получения контррецепторного фактора, блокирующего связывание инсулина с его рецептором и обладающего диабетогенным действием, предусматривающий обработку крови антикоагулянтом, отделение плазмы и ее фракционирование, отличающийся тем, что плазму крови обрабатывают активированным углем, покрытым декстраном, а фракционирование осуществляют методом афинной хроматографии на агарозе с иммобилизованными рецепторами инсулина, целевой...
Спосіб одержання контрінсулінового фактора, який блокує зв’язування інсуліну з його рецептором та має діабетогенну дію

Номер патенту: 13922
Опубліковано: 25.04.1997
Автори: Іванова Жана Вікторівна, Гуріна Наталія Марківна, Корпачев Вадим Валерійович
МПК: A61K 35/16
Мітки: спосіб, блокує, рецептором, діабетогенну, має, фактора, одержання, контрінсулінового, інсуліну, дію, зв'язування
Формула / Реферат:
Способ получения контринсулинового фактора, блокирующего связывание инсулина с его рецептором и обладающего диабетогенным действием, предусматривающий обработку крови антикоагулянтом, отделение плазмы и ее фракционирование, отличающийся тем, что плазму крови обрабатывают активированным углем, покрытым декстраном, а фракционирование осуществляют методом афинной хроматографии на агарозе с иммобилизованными рецепторами инсулина, целевой...
Спосіб отримання еталонного розчину для візуального контролю гемоглобіну в крові

Номер патенту: 14320
Опубліковано: 25.04.1997
Автори: Пучіхін Кім Олександрович, Гусаковська Ніна Семенівна, Носенко Петро Володимирович
МПК: G01N 33/72
Мітки: контролю, розчину, візуального, спосіб, еталонного, отримання, крові, гемоглобіну
Формула / Реферат:
Способ получения эталонного раствора для визуального контроля гемоглобина в крови путем экстракции растительного сырья смесью этилового спирта с глицерином и фильтрования, отличающийся тем, что, с целью повышения экономичности целевого продукта за счет замены дорогостоящего сырья, в качестве растительного сырья используют кору дуба (Quercus robur), а экстракцию ведут при соотношении компонентов, %: сырье 6-8, глицерин 41-42, этиловый спирт...
Склад для стабілізації плазми крові у процесі пастеризації, спосіб пастеризації плазми крові і засіб для терапії препаратом плазми

Номер патенту: 27102
Опубліковано: 28.02.2000
Автор: Бюрнуф Міріана
МПК: A61K 35/16, A01N 1/02
Мітки: препаратом, склад, засіб, крові, терапії, пастеризації, процесі, стабілізації, спосіб, плазми
Формула / Реферат:
1. Состав для стабилизации плазмы крови в процессе пастеризации, состоящий из смеси сорбита, препарата кальция и лизина, отличающийся тем, что он дополнительно содержит сахарозу, тринатрийцитрат и аргинин при следующем соотношении компонентов на 1 литр плазмы, г: Сорбит 800 – 1400 Сахароза 400 – 600 Глюконат кальция 3 – 5мМ ...
Спосіб одержання кристалічного ліз в28 про в29 інсуліну людини (варіанти)

Номер патенту: 26638
Опубліковано: 11.10.1999
Автори: Френк Брюс Хілл, Картер Ненсі Делорес, Бейкер Джеффрі Клейтон
МПК: C07K 14/62, C07K 1/14, C07B 63/00, A61P 5/00, A61P 5/48, A61K 38/28, C07K 14/575, A61K 38/00
Мітки: варіанти, кристалічного, ліз, спосіб, людини, інсуліну, одержання
Формула / Реферат:
1. Способ получения кристаллического ЛизВ28ПроВ29-инсулина человека, отличающийся тем, что включает: кристаллизацию ЛизВ28ПроВ29-инсулина человека из раствора, содержащего ЛизВ28ПроВ29-инсулин человека, цинк, по меньшей мере 0,3N органическую кислоту, выбранную из группы: уксусная кислота, лимонная кислота или глицин, и фенольное соединение при pH от 5,5 до 6,5.2. Способ по п.1, отличающийся тем, что концентрация ЛизВ28ПроВ29-инсулина...
Попередній патент: Спосіб регулювання сполучення гетеродина та преселектора
Наступний патент: Спосіб помелу матеріалу та млин для його здійснення
Випадковий патент: Вібраційний змішувач