Спосіб одержання полімерних кон’югатів доксорубіцину з рн-контрольованим вивільненням лікарської речовини
Номер патенту: 94594
Опубліковано: 25.05.2011
Автори: Ульбріх Карел, Студеновскі Мартін, Пехар Міхал, Рігова Бланка, Етріх Томас, Хитіл Петер





Формула / Реферат
1. Спосіб одержання полімерних кон'югатів N-(2-гідроксипропіл)метакриламіду і метакрилоїламіноацилгідразону з доксорубіцином з рН-контрольованим вивільненням лікарської речовини, який відрізняється тим, що здійснюють наступні три стадії синтезу:
a) одержання мономерного метакрилоїламіноацилгідразину, де аміноацил - похідна амінокислоти або олігопептиду, реакцією метакрилоїлгаліду з відповідним пептидом, амінокислотою або їх похідними і подальшим гідразинолізом,
b) синтез полімерного прекурсору безпосередньою співполімеризацією N-(2-гідроксипропіл)метакриламіду з метакрилоїламіноацилгідразином, і
с) зв'язування доксорубіцину з полімерним прекурсором реакцією їх з гідрохлоридом доксорубіцину.
2. Спосіб за пунктом 1, який відрізняється тим, що ацилування на стадії (Іа) здійснюють реакцією гідрохлориду метилового естеру відповідної амінокислоти або олігопептиду з метакрилоїлхлоридом в хлорованому вуглеводні у присутності безводного карбонату натрію.
3. Спосіб за пунктом 1 або 2, який відрізняється тим, що гідразиноліз здійснюють реакцією метилового естеру метакроїлованої амінокислоти або олігопептиду з гідразингідратом у присутності сильної основи.
4. Спосіб за будь-яким з попередніх пунктів, який відрізняється тим, що на стадії (Іb) радикальну співполімеризацію N-(2-гідроксипропіл)метакриламіду з метакрилоїламіноацилгідразином здійснюють з ініціюванням здатними до терморозкладання ініціаторами на основі азо- або пероксиініціаторів, переважно азобіс(ізобутиронітрилом), азобіс(ізоціановалеріановою кислотою) або діізопропілперкарбонатом.
5. Спосіб за пунктом 4, який відрізняється тим, що полімеризацію здійснюють в розчиннику, вибраному з нижчих С1-С5 спиртів або апротонного полярного розчинника.
6. Спосіб за пунктом 5, який відрізняється тим, що розчинник вибирають з метанолу, етанолу, диметилформаміду або диметилсульфоксиду.
7. Спосіб за пунктом 6, який відрізняється тим, що, коли ініціатор вибраний із азобіс(ізобутиронітрилу) або азобіс(ізоціановалеріанової кислоти), полімеризацію здійснюють при 45-70 °С, а коли як ініціатор використовують діізопропілперкарбонат, полімеризацію здійснюють при 30-60 °С.
8. Спосіб за будь-яким з попередніх пунктів, який відрізняється тим, що реакцію полімерного прекурсору на стадії (Іс) здійснюють у розчиннику, вибраному з безводних С1-С5 спиртів або полярних апротонних розчинників, з каталізом оцтовою кислотою, і одержаний кон'югат осаджують етилацетатом.
9. Спосіб за пунктом 8, який відрізняється тим, що розчинник вибирають із метанолу, сухого етанолу, диметилформаміду або диметилсульфоксиду.
10. Спосіб за пунктом 9, який відрізняється тим, що вихідну концентрацію полімеру вибирають в діапазоні 100-190 мг/мл і концентрацію оцтової кислоти - в діапазоні 30-80 мг/мл.
11. Спосіб за пунктом 10, який відрізняється тим, що концентрація полімеру складає 170 мг/мл і оцтової кислоти - 55 мг/мл при 25 °С.
12. Спосіб за будь-яким з попередніхпунктів, який відрізняється тим, що одержаний продукт очищують гель-фільтрацією.
Текст
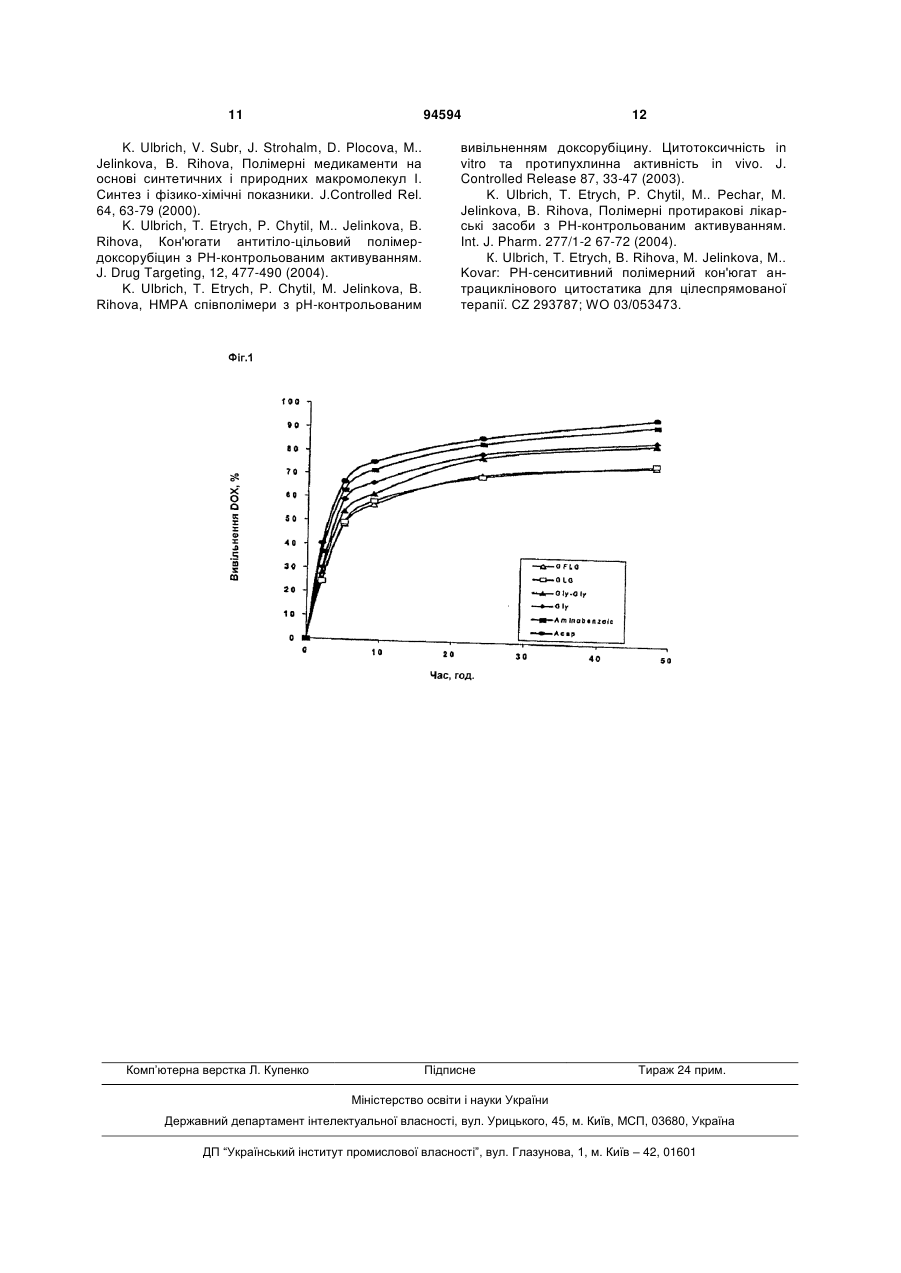
1. Спосіб одержання полімерних кон'югатів N(2-гідроксипропіл)метакриламіду і метакрилоїламіноацилгідразону з доксорубіцином з рНконтрольованим вивільненням лікарської речовини, який відрізняється тим, що здійснюють наступні три стадії синтезу: a) одержання мономерного метакрилоїламіноацилгідразину, де аміноацил - похідна амінокислоти або олігопептиду, реакцією метакрилоїлгаліду з відповідним пептидом, амінокислотою або їх похідними і подальшим гідразинолізом, b) синтез полімерного прекурсору безпосередньою співполімеризацією N-(2гідроксипропіл)метакриламіду з метакрилоїламіноацилгідразином, і с) зв'язування доксорубіцину з полімерним прекурсором реакцією їх з гідрохлоридом доксорубіцину. 2. Спосіб за пунктом 1, який відрізняється тим, що ацилування на стадії (Іа) здійснюють реакцією гідрохлориду метилового естеру відповідної амінокислоти або олігопептиду з метакрилоїлхлоридом в хлорованому вуглеводні у присутності безводного карбонату натрію. 3. Спосіб за пунктом 1 або 2, який відрізняється тим, що гідразиноліз здійснюють реакцією метилового естеру метакроїлованої амінокислоти або олігопептиду з гідразингідратом у присутності сильної основи. 2 (19) 1 3 Винахід відноситься до способу одержання водорозчинних полімерних протиракових лікарських засобів, здатних до цільової доставки і контрольованого вивільнення цитостатиків в організмі, переважно в тканині і клітинах пухлини. Використання полімерних кон'югатів зосереджено на цілеспрямованій терапії пухлинних захворювань у людей. Розвиток нових фармакологічно-активних субстанцій, особливо протиракових лікарських засобів, все більш і більш був зосереджений на таких формах, які забезпечують можливість специфічної дії активної субстанції тільки в специфічній тканині, або навіть тільки в специфічному типі клітин. Природні або синтетичні макромолекули - полімери - все більш і більш використовувані для одержання таких субстанцій. Було одержано і досліджено багато протиракових полімерних кон'югатів і показано, що в більшості випадків необхідно, щоб цитотоксична субстанція вивільнялась з її полімерної форми, якщо полімерна форма лікарського засобу повинна бути фармакологічно-ефективною. Крім того, показано, що прийнятна молекулярна вага полімерного носія може гарантувати переважне розташування полімерної лікарської речовини в тканині багатьох солідних пухлин (так званий EPR ефект) [Maeda та інші. 2000]. Вивільнення цитостатичного агента із його полімерного носія може бути забезпечене з використанням здатного до біодеструкції зв'язку, що використовується для зв'язування лікарської речовини з полімером, деструкція якого в цільовій тканині приводить до цільового і контрольованого активування лікарської речовини переважно в згадуваній тканині. Полімерні лікарські засоби на основі співполімерів N-(2гідроксипропіл)метакриламіду (НРМА) - важлива група таких медикаментів. Дуже хороший короткий огляд результатів дослідження в цій галузі аж до представлених може бути знайдений монографії G. S. Kwon і в публікації J. Kopecek та інші. [Коресек та інші. 2000, Kwon 2005]. Недавно були опубліковані дослідження активності полімерних лікарських засобів, в яких протиракова лікарська речовина - доксорубіцин, зв'язана з полімерним носієм на основі НМРА співполімерів за допомогою гідролітично нестабільного гідразонового зв'язку [Etrych та інші. 2001 і 2002, Rihova та інші. 2001, Ulbrich та інші. 2003, 2004], і ці субстанції були запатентовані [Ulbrich та інші.]. Ці лікарські засоби показали істотне зменшення побічних, особливо токсичних, ефектів на здоровий організм, водночас збільшуючи протираковий ефект, порівняно із зазвичай використовуваними цитостатиками [Rihova та інші. 2001, Kovar та інші. 2004, Hovorka та інші. 2002]. Синтез таких кон'югатів вперше здійснювався полімер-аналогічною реакцією полімерних естерів 4-нітрофенілу (ONp) з гідразином, і пізніше співполімеризацією НРМА з NBoc (трет-бутилоксикарбонілом), захищеним метакрилоїлованими гідразидами. Жоден із способів не приводив до утворення цілком визначених препа 94594 4 ратів (у разі естерів ONp, мають місце реакції передачі ланцюга і гідролізу частини груп ONp в процесі гідразинолізу; у разі Вос-гідразидів відбуваються реакції деструкції зустрічаються протягом депротектування гідразидних груп), і жоден з способів не здатний підібрати широкий інтервал молекулярної ваги полімерного ланцюга; синтез не зробив можливим одержання великих партій, і відтворюваність одержання індивідуальних партій була відносно низькою. Окрім того, синтези включали декілька стадій, які збільшили їх час і фінансові витрати. Даний винахід забезпечує оптимізований і відтворюваний спосіб одержання полімерних цитостатиків на основі НМРА співполімерів, що містять доксорубіцин, зв'язаний рН-лабільним гідразоновим зв'язком з полімерним носієм, який виключає практично всі вищезазначені недоліки згаданих раніше способів одержання; зокрема, робить можливим збільшення виходу протягом синтезу як мономерів, так і полімерних прекурсорів, точне контролювання молекулярної ваги полімерних прекурсорів і кінцевого продукту; структура, завдяки переважним параметрам співполімеризації, є цілком визначеною, синтез є значно легшим і дешевшим, можливим збільшення його до великих партій, і відтворюваність синтезу є дуже хорошою. Протипухлинна активність полімерних цитостатиків, одержаних згідно з винаходом, така ж, або навіть краща, ніж, цитостатиків, одержаних попередніми способами. Предмет винаходу полягає в способі одержання полімерного кон'югату НМРА співполімеру, що містить зв'язаний з полімером доксорубіцин за допомогою різних зв'язків, що містять гідролітично розщеплювані гідразонові зв'язки. Спосіб одержання базується на трьох-етапному синтезі, що включає синтез мономерів, синтез полімерних прекурсорів і кінцеве зв'язування доксорубіцину з полімерним носієм ковалентним гідразоновим зв'язком. Синтез мономерів починається з синтезу мономера НРМА згідно описаному раніше способу [Ulbrich 2000]. Синтез метакрилоїл(аміноацил)гідразинів, що відрізняються структурою ацильного компонента, був дуже схожим для всіх одержаних мономерів і починається з метакрилоїлування гідрохлориду метилового естеру відповідної амінокислоти або олігопептиду з метакрилоїлхлоридом, здійснюваного в дихлорметані у присутності безводного карбонату натрію. Одержаний продукт перетворювали у метакрилоїлований аміноацилгідразин гідразинолізом відповідного метилового естеру з гідратом гідразину, здійснюваним у метанольному розчині у присутності NaOH. Гліцил, гліцилгліцил, β-аланіл, 6аміногексаноїл, 4-амінобензоїл або складний ацил, утворений з олігопептидів GlyPheGly, GlyLeuGly, або GlyPheLeuGly, переважно використовувався як аміноацил в метакрилоїл(аміноацил)гідразинах. Як приклади синтезу метакрило 5 їл(аміноацил)гідразину, Приклад 1 ілюструє синтез 6-метакроїл(аміногексаноїл)гідразину як приклад синтезу простого ацилу (спейсеру), метакроїлгліцилгліцилгідразину як мономера з дипептидним спейсером, і метакроїлгліцилфенілаланіллейцилгліцилгідразину як мономера з ензиматично дестрктуючим олігопептидом. Синтез полімерних прекурсорів - НМРА співполімерів з метакрилоїлованими аміноацилгідразинами - заснований на безпосередній радикальній співполімеризації НРМА з відповідними метакрилоїлованими гідразинами. Полімеризація здійснюється в розчині, використовуючи метанол, етанол, диметилсульфоксид або диметилформамід як середовище полімеризації. В обох випадках полімеризацію ініціюють здатними до терморозкладання ініціаторами радикальної полімеризації на основі азо- або перокси- ініціаторів. Переважно, використовувалися азобіс(ізобутиронітрил) (AIBN), азобіс(ізоціановалеріанова кислота) (АВІС), або діізопропілперкарбонатом (DIP). Температура полімеризації залежить від використовуваного ініціатора і розчинника (для AIBN, АВІС в метанолі, етанолі, DMF, і DMSO, температура складає 50 60 °С; для DIP - 40 - 50 °С). Полімеризація зазвичай триває 15-18 годин. Одержання всіх полімерних прекурсорів радикальною полімеризацією аналогічне; приклади співполімеризації НРМА з метакрилованими гідразидами надаються в Прикладах 2а - 2с. Порівняно з раніше використовуваним гідразинолізом реактивних естерів або співполімеризацією Вос-захищених гідразидів, пряма співполімеризація приводить до чистих і відтворювано-одержуваних полімерів, які можуть бути одержані у великих партіях і з високим виходом. Зв'язування доксорубіцину з полімерним прекурсором - результат реакції зв'язування гідрохлориду доксорубіцину з полімерним ацилгідразином, що приводить до гідразонового зв'язку. Реакція переважно здійснюється в метанолі, каталізується певною кількістю оцтової кислоти. Реакція може також здійснюватися в диметилсульфоксиді, диметилформаміді, сухому етанолі і диметилацетаміді. Використовуючи розчинники інші, ніж метанол, реакція відбувається добре, але вихід нижчий. Вплив структури зв'язку на хід реакції зв'язування мінімальний. Щоб досягти оптимального виходу реакції зв'язування і мінімальної кількості незв'язаного доксорубіцину в продукті, важливо, у всіх випадках, підтримувати наступні концентрації полімеру і оцтової кислоти в реакційній суміші: концентрація полімеру 170 мг/мл, концентрація оцтової кислоти 55 мг/мл. Оптимальний час реакції складає 22 години при 25 °С. Полімерний лікарський засіб відокремлюється від реакційної суміші осаджуванням в етилацетаті і повторним осаджуванням з метанолу і знову в етилацетаті. Одержання полімерного доксорубіцину, зв'язаного з полімерним прекурсором - співполімером Полі(НРМА-Со-МА-АН-NНNН2) - гідразоновим зв'язком (PHPMA-AH-NH-N=DOX) надається в Прикладі 3. Одержання також включає, хоча це не є необхідним, кінцеве очищення кон'югату від незв'язаного вільної лікарської речовини фільтрацією на гелі, 94594 6 використовуючи Sephadex колонку LH-20 з метанолом як мобільною фазою. Фіг. 1 представляє графік, що показує вивільнення DOX з полімерних кон'югатів, що відрізняються структурою зв'язку між лікарською речовиною і полімером. Температура: 37 °С, фосфатний буфер, рН 5,5, GFLG - зв'язок, утворений послідовністю GlyPheLeuGly-, GLG є -GlyLeuGly-, амінобензойний - 4-амінобензоїл, і Асар є 6-аміногексаноїл. Приклади Приклад 1: Синтез мономерів НРМА одержували відповідно до попередньо описаного способу [Ulbrich та інші. 2000]. Елементний аналіз: Обчислено: С 58,8 %, Η 9,16 %, Ν 9,79 %. Одержано: С 58,98 %, Η 9,18 %, Ν 9,82 %. Продукт був хроматографічно чистий. 6-(Метакроїламіно)гексаноїлгідразин (ΜΑ-ΑΗΝΗΝΗ2) Метил (6-аміногексаноат)гідрохлорид (30 г, 0,165 моль) при інтенсивному перемішуванні при кімнатній температурі розчиняли в 350 мл дихлорметану з додаванням приблизно 100 мг гідрохінону. Розчин охолоджували до 10-15 °С, додавали безводний карбонат натрію (50 г, 0,48 моль), температуру знижували до 5 - 10 °С, і потім додавали краплями розчин метакроїлхлориду (17,3 г, 0,165 моль (екв.)) в 100 мл дихлорметану з такою швидкістю, щоб температура реакційної суміші не перевищувала 15 °С. Після того як увесь метакроїлхлорид вступав в реакцію, суміш перемішували при 15 - 20 °С протягом додаткових 45 хвилин, потім охолоджуючу ванну вилучали, суспензію перемішували ще 20 хвилин, відсмоктували на фільтрі із пористого скла № 3, промивали 300 мл дихлорметану і фільтрат випарювали до сухого залишку в ротаційному вакуумному випарнику. Залишок випарювання розчиняли в 150 мл метанолу, потім додавали гідразингідрат (13 мл, 13,4 г, 0,267 моль) і NaOH (1,5 г, 37,5 ммоль), і реакційну суміш перемішували при кімнатній температурі протягом 3 годин. Після того, як гідразиноліз був завершений, рН розчину коригували до 6,2 -6,5 додаванням 35% НСІ (приблизно 12 мл), додавали 300 г безводного сульфату натрію і суміш, включаючи десикант, випарювали до сухого залишку. Додавали до залишку випарювання 500 мл дихлорметану, суспензію інтенсивно перемішували протягом 2 годин, відсмоктували на фільтрі із пористого скла № 4, повністю промивали додатковими 500 мл дихлорметану, і фільтрат концентрували до 150 200 мл у випарнику. Розчин розбавляли 1500 мл етилацетату, концентрували для кристалізації до об'єму 300 - 350 мл у випарнику, і кристалізували в крижаному боксі протягом 24 годин. Продукт відсмоктували, промивали невеликою кількістю холодного етилацетату, висушували у вакуумі. Вихід після першої кристалізації склав 29,5 г продукту (84%) з температурою плавлення 79-81 °С; повторна кристалізація, використовуючи той же спосіб, надала 27,0 г продукту (77 %) з температурою плавлення 80-82 °С. Елементний аналіз: Обчислено: С 56,32 %, Η 8,98 %, Ν 19,70 %. Одержано: С 56,49 %, Η 8,63 %, Ν 19,83%. 7 Н-ЯМР 300 Мгц (CDCI3, 297 К): 1,35 м (2Н, CH2(CH2)2-N); 1,50-1,69 м (4Н, CH2CH2CH2CH2-N); 1,95 дд (3Н, СН3); 2,17 т (2Н, ((С=О)-СН2); 3,26 дт (2Н, N-CH2); 3,91 с (2Н, NH2); 5,30 т (1Η, С=СН2 Е); 5,67 т (1Н, С=СН2 Ζ); 6,10 с (1Н, ΝΗΝΗ2); 7,45 с (1Н, NH-CH2). Продукт був хроматографічно чистий, 1 пік на 4,72 хв (зворотно-фазова колонка Tessek SGX С18 (7 мкм, 125 4 мм), швидкість потоку 0,5 мл/хв, градієнт з 40 до 100 % розчину В за 35 хв (розчин А: 10 % метанол, 89,9 % вода, 0,1 % трифтороцтова кислота (TFA); розчин В: 89,9 % метанол; 10 % вода; 0,1 % TFA), ультрафіолетове виявлення). Метакроїлгліцилгліцилгідразин (MA-GlyGlyNHNH2) Одержання MA-GlyGly-NHNH2 здійснювали в подібних умовах, як для MA-AH-NHNH2. Метиловий естер гліцилгліцину (21,4 г, 0,1 моль) був розчинений в 250 мл дихлорметану з додаванням 60 мг гідрохінону. Після охолоджування до приблизно 12 °С додавали безводний карбонат натрію (30,2 г, 0,29 моль), і при охолоджуванні до 5-10 °С додавали повільно краплями розчин метакроїлхлориду (10,4 г, 0,1 моль) в 60 мл дихлорметану. Після завершення реакції, фільтрування і промивання осаду з приблизно 230 мл дихлорметану, розчинник повністю випарювали. Залишок випарювання розчиняли в 120 мл метанолу і піддавали гідразинолізу гідразингідратом (7,9 мл, 8,1 г, 0,166 моль) в присутності NaOH (0,91 г, 22,7 ммоль). Після завершення гідразинолізу, рН розчину коригували до 6,2 - 6,5 додаванням 35% НСІ (приблизно 12 мл), додавали 230 г безводного сульфату натрію, і суміш, включаючи десикант, випарювали до сухого залишку. Після екстрагування з 300 мл дихлорметану, суспензію відсмоктували на фільтрі із пористого скла, промивали додатково 300 мл дихлорметану, і фільтрат концентрували до приблизно 120 мл. Розчин розбавляли 900 мл етилацетату і після концентрування до 250 мл у випарнику, кристалізували в морозильному боксі. Після перекристалізації продукт відокремлювали фільтрацією, промивали невеликою кількістю холодного етилацетату і сушили у вакуумі. Вихід продукту становив 15,0 г (70 %), температура плавлення 172-174 °С. Елементний аналіз: Обчислено: С 44,86 %, Η 6,54 %, Ν 26,16 %. Одержано: С 45,01 %, Η 6,57 %, Ν 26,02 %. Продукт був хроматографічно чистий, один пік на 21,97 хв. Метакроїлгліцилфенілаланіллейцилгліцилгідразин (MA-Gly-D,L-PheLeuGlyNHNH2). Синтез цього мономера здійснювався аналогічним шляхом, як в обох попередніх випадках (див. вище). Склад реакційної суміші і умови були, як вказано нижче: метиловий естер гліцилфенілаланіллейцилгліцину (22,32 г, 0,05 моль), дихлорметан 510 мл (270 + 240 мл), гідрохінон (60 мг), карбонат натрію (15,1 г, 0,145 моль) і метакроїлхлорид 5,2 г (0,05 моль) використовували для одержання метакрилованого метилового естеру. Для гідразинолізу використовували 4,1 г (0,085 моль) гідразингідрату в 120 мл метанолу і 0,46 г (11,3 ммоль) NaOH. Для висушування використовували 210 г безводного сульфату натрію, і 2 94594 8 290 мл дихлорметану використовували для екстрагування. Продукт кристалізували із суміші дихлорметан-етилацетат. Вихід продукту становив 60 %, температура плавлення 139-140 °С. Елементний аналіз: Обчислено: С 58,10 %, Η 7,36 %, Ν 17,68 %. Одержано: С 58,21 %, Η 7,39 %, Ν 17,54 %. Продукт був хроматографічно чистий, два піки, що мають однакову площу при 19,39 (L-Phe, що містить мономер) і 19,91 хв (D-Phe). Приклад 2а: Синтез полімерного прекурсора співполімера НРМА з 6-(метакроїламіно)гексаноїлгідразином (полі(НРМА-спів-МА-АНМНМН2)) Співполімер полі(HPMA-спів-MA-AH-NHNH2) одержували радикальною співполімеризацією розчину НРМА і MA-AH-NHNH2, ініційованою AIBN в метанолі при 60 °С. 122,8 г НРМА і 13,94 г MA-AHNHNH2 (18 мас% мономерів) розчиняли в 780 мл метанолу і додавали до розчину 6,06 г AIBN (0,8 мас%). Після фільтрації полімеризаційну суміш поміщали в атмосфері аргону в реактор полімеризації (об'ємом 1,5 л), розташований в термостаті. Полімеризаційну суміш перемішували при швидкому обертанні (близько 100 обертів у хвилину). Азот вводили над поверхнею протягом ще декількох хвилин. Температуру полімеризаційної суміші доводили до 60 °С, і полімеризацію продовжували при перемішуванні (50 об./хв.) в атмосфері азоту. Азот видаляли через барботувальний пристрій. Через 17 годин полімеризаційну суміш виймали з термостату, охолоджували у ванні до кімнатної температури і полімер відокремлювали осадженням в етилацетаті (8 л в цілому). Осаджений полімер піддавали седиментації приблизно 0,5 годин, розчин над осадом видаляли відсмоктуванням і полімер відокремлювали фільтрацією через фільтр із пористого скла S4. Осад промивали етилацетатом, переносили у великі чашки Петрі, і висушували при кімнатній температурі в вакуумі, використовуючи мембранний вакуумний насос, приблизно 1 годину. Використовуючи ультразвук, полімер розчиняли в 550 мл метанолу (однолітрова колба Erlenmeyer) і осаджували 7,5 л етилацетату таким же чином, як протягом першого виділення. Осаджений полімер після седиментації протягом приблизно 0,5 годин виділяли фільтрацією через фільтр із пористого скла S4, промивали етилацетатом і висушували до постійної ваги, використовуючи мембранний вакуумний насос (до приблизно 5 годин), і сушильний процес завершували з використанням масляно-дифузійного насосу. Характеристика співполімера: Вихід 114 г (83 %), вміст гідразидних груп 5,83 моль %, молекуляра вага Mw = 28500 г/моль, індекс полідисперсності Іn = 1,9. Приклад 2b: Синтез полімерного прекурсора співполімера ΗΡΜА з метакроїлгліцилгліцилгідразином (полі(HPMA-спів-MA-GlyGly-NHNH2)) Конфігурація і процедура полімеризації були тими ж, як в Прикладі 2а, різниця була в складі полімеризаційної суміші. Склад полімеризаційної суміші був таким, як вказано нижче: НРМА 10 г, (70 ммоль), MA-GlyGly-NHNH2 1,5 г (7 ммоль), діізопропілперкарбонатом 1,15 г (0,91 мас%), і димети 9 лформамід 115 мл. Температура полімеризації 50 °С, і полімеризація відбулася за 16 годин. Полімеризаційний розчин перед осаджуванням в надлишку етилацетату концентрували до приблизно 2/3 його початкового об'єму у вакуумному випарнику, і осадження полімерного продукту здійснювали в 20-кратному об'ємі осаджувача. Полімер позбавляли від низькомолекулярних сумішей осаджуванням з метанолу в етилацетаті. Вихід склав 8,5 г (70 %), вміст гідразидних груп - 9,5 моль %, молекулярна вага Mw = 41700 г/моль, індекс полідисперсності Іn = 2,1. Приклад 2с: Синтез полімерного прекурсора співполімера НРМА з метакроїлгліцилфенілаланіллейцилгліцил гідразином (полі(НРМА-спів-МАСІуРheLеuСІу-NНNН2)). Процедура полімеризації була тією ж, як в Прикладі 2а, різниця була знову тільки в складі полімеризаційної суміші. Склад полімеризаційної суміші був таким, як вказано нижче: НРМА 10 г, (70 ммоль), HPMA-спів-MA-GlyPheLeuGly-NHNH2 2,5 г, (5,6 ммоль), азобіс(ізоціановалеріанова кислота) 1,125 г (1 мас%), і диметилсульфоксид 100 мл. Полімеризацію здійснювали при 55 °С і завершували через 18 годин. Полімер виділяли із полімеризаційної суміші осаджуванням в 20-кратному надлишку етилацетату. Полімер очищували повторним осаджуванням з метанолу в етилацетаті. Вихід склав 9,75 г (78 %), вміст гідразидних груп 5,5 моль %, молекулярна вага Mw = 43200 г/моль, індекс полідисперсності Іn = 2,1. Приклад 3: Одержання полімерного кон'югату PHPMA-AH-NH-N=DOX Співполімери з DOX, зв'язаним з РНРМА носієм гідролітичним розщепленням гідразонового зв'язку, були одержані реакцією гідразидмістких груп співполімерів полі(НРМА-спів-MA-AH-NHNH2) з DOX.HCI в метанолі, каталізованою оцтовою кислотою. Розчин 15,384 г співполімера полі(НРМА-співМА-АН-NНNН2) в 92,1 мл метанолу (167 мг полімеру/мл) поміщали в кювету термостату, в якому було розміщено 2,5 г DOX.HCI (4,3 ммоль). Неоднорідну суспензію перемішували в темноті при 25 °С і через одну хвилину додавали 4,9 мл оцтової кислоти (загальний об'єм 116 мл). Суспензію поступово розчиняли в ході реакції, і через 22 години реакції полімерний продукт виділяли із гомогенного розчину осаджуванням в 1 л етилацетату; осад полімерного медикаменту відокремлювали фільтрацією через фільтр із пористого скла S4, промивали 150 мл етилацетату і висушували до постійної ваги. Загальну кількість DOX визначали спектрально. Mw і Мn визначали рідинною хроматографією (LC АКТА) з аналізом світлорозсіювання (DAWN DSP багато ракурсний детектор, Wyatt). Характеристика полімерного медикаменту. Загальний вихід реакції зв'язування медикаменту: 17,2 г (96 %), загальний вміст DOX: 11,3 мас%, вільний DOX: 1,52 % поза загальним вмістом DOX. Приклад 4: Вивільнення доксорубіцину з полімерних кон'югатів. Вивільнення доксорубіцину з кон'югатів, відмінних за структурою зв'язку між лікарською речовиною і полімером, здійснювали інкубуванням в 94594 10 0,1 Μ фосфатному буфері, що містив 0,15 Μ NaCI при 37 °С. рН буфера коригували до умов ендосоми клітини, тобто до злегка кислотного середовища з рН 5,5. Аліквотні частини інкубувального середовища випробовували з відповідними проміжками, і вміст DOX визначали - після додавання карбонатного буфера (0,1 Μ Na2CO3 + 4 Μ NaCI), після екстрагування у хлороформі і після випаровування розчинника і переносу у метанольний розчин - за допомогою ВЕРХ (Shimadzu VP), використовуючи зворотно-фазову колонку (Tessek SGX С18, 7 рм, 125 4 мм), швидкість потоку елюента: 0,5 мл/хв, градієнт з 40 до 100 % розчину В за 35 хвилин (розчин А: 10 % метанол, 89,9 % вода, 0,1 % трифтороцтова кислота (TFA); розчин В: 89,9 % метанол; 10 % вода; 0,1 % TFA). Використовували для визначення флуоресцентний детектор (Shimadzu RF-IOAXL) (ехс = 480 нм, еm = 560 ran). Криву калібрування будували, використовуючи доксорубіцин. Результати вимірювання вивільнення медикаменту із кон'югатів, відмінних за структурою використовуваного зв'язку (спейсера), показані на Фіг. 1. Джерела інформації: Т. Etrych, M. Jelinkova, Β. Rihova, К. Ulbrich, Нові співполімери НРМА, що містять зв'язаний доксорубіцин через рН сенситивний зв'язок. Синтез, біологічні властивості in vitro і in vivo. J, Controlled Rel. 73, 89-102 (2001) T. Etrych; P. Chytil, M. Jelinkova, B. Rihova, K. Ulbrich, Синтез НМРА співполімерів, що містять доксорубіцин, зв'язаний через гідразоновий зв'язок. Вплив спейсера на вивільнення медикаменту і цитотоксичність in vitro. Macromolecular Biosci. 2, 43-52 (2002). О. Hovorka, Т. Etrych, Μ. Subr, J. Strohalm, K. Ulbrich, B. Rihova, Відмінності у внутрішньоклітинній дії вільного і полімер-зв'язаного доксорубіцину. J. Controlled Release 80, 101-117(2002). J. Kopecek, P. Kopeckova, T. Minko, Z. Lu, Кон'югат НМРА співполімер-протиракова лікарська речовина: Дизайн, Активність і Механізм Дії. Europ. J. Pharm. Biopharm. 50, 61 - 81 (2000) Μ. Kovar, L. Kovar, V. Subr, T. Etrych, K. Ulbrich, T. Mrkvan, J. Loucka, і В. Rihova, НМРА співполімери, що містять доксорубіцин, зв'язаний протеолітично або гідролітично розшеплюваним зв'язком: порівняння біологічних властивостей in vitro. J. Controlled Release 99, 301- 314 (2004). G. S. Kwon, Полімерні системи доставки медикаменту, серії: Drugs and the Pharmaceutical Sciences, том 148, Dekker, Marcel Incorporated, 2005. H. Maeda, J. Wu, T. Sawa, Y. Matsumura, K. Hori, Судинна проникність пухлини і EPR ефект в макромолекулярній терапії: огляд. J Control Release 65, 271-284 (2000) В. Rihova, T. Etrych, M.. Pechar, Μ.. Jelinkova, Μ.. St'astny, Ο. Hovorka, M. Kovar, Κ. Ulbrich, Ефективність зв'язаного доксорубіцину з носієм НРМА співполімера через гідразоновий зв'язок на клітинній лінії раку з обмеженим вмістом лізосом. J. Controlled Release 74, 225-232 (2001). 11 94594 K. Ulbrich, V. Subr, J. Strohalm, D. Plocova, M.. Jelinkova, B. Rihova, Полімерні медикаменти на основі синтетичних і природних макромолекул І. Синтез і фізико-хімічні показники. J.Controlled Rel. 64, 63-79 (2000). K. Ulbrich, T. Etrych, P. Chytil, M.. Jelinkova, B. Rihova, Кон'югати антитіло-цільовий полімердоксорубіцин з РН-контрольованим активуванням. J. Drug Targeting, 12, 477-490 (2004). K. Ulbrich, Т. Etrych, P. Chytil, Μ. Jelinkova, В. Rihova, HMPA співполімери з рН-контрольованим Комп’ютерна верстка Л. Купенко 12 вивільненням доксорубіцину. Цитотоксичність in vitro та протипухлинна активність in vivo. J. Controlled Release 87, 33-47 (2003). K. Ulbrich, Т. Etrych, P. Chytil, M.. Pechar, M. Jelinkova, B. Rihova, Полімерні протиракові лікарські засоби з РН-контрольованим активуванням. Int. J. Pharm. 277/1-2 67-72 (2004). К. Ulbrich, Т. Etrych, В. Rihova, Μ. Jelinkova, Μ.. Kovar: РН-сенситивний полімерний кон'югат антрациклінового цитостатика для цілеспрямованої терапії. CZ 293787; WO 03/053473. Підписне Тираж 24 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for the preparation of polymeric conjugates of doxorubicin with ph-controlled release of the drug
Автори англійськоюEtrych Tomas, Chytil Petr, Studenovsky Martin, Pechar Michal, Ulbrich Karel, Rihova Blanka
Назва патенту російськоюСпособ получения полимерных конъюгатов доксорубицина с рн-контролированным высвобождением лекарственного вещества
Автори російськоюЭтрих Томас, Хитил Петер, Студеновски Мартин, Пехар Михал, Ульбрих Карел, Ригова Бланка
МПК / Мітки
МПК: A61K 47/48
Мітки: полімерних, лікарської, одержання, рн-контрольованим, вивільненням, кон'югатів, речовини, спосіб, доксорубіцину
Код посилання
<a href="https://ua.patents.su/6-94594-sposib-oderzhannya-polimernikh-konyugativ-doksorubicinu-z-rn-kontrolovanim-vivilnennyam-likarsko-rechovini.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання полімерних кон’югатів доксорубіцину з рн-контрольованим вивільненням лікарської речовини</a>
Спосіб одержання твердої лікарської форми з регульованим вивільненням активної речовини

Номер патенту: 50289
Опубліковано: 15.08.2005
Автори: Погорелий Валерій Костянтинович, Чуйко Олексій Олексійович, Барвінченко Валентина Миколаївна
МПК: A61K 9/22, A61K 36/00
Мітки: лікарської, форми, спосіб, вивільненням, регульованим, активної, речовини, одержання, твердої
Формула / Реферат:
Спосіб одержання твердої лікарської форми з регульованим вивільненням активної речовини шляхом обробки активною речовиною структуроутворювача - діоксиду кремнію, сушіння, наповнення в капсулу або пресування в таблетку, який відрізняється тим, що як активну речовину беруть сировину лікарських рослин і витяжки із лікарських рослин, обробку діоксиду кремнію ведуть спочатку витяжками із лікарських рослин, після сушіння порошкують, а потім...
Спосіб одержання пероральної мікрокапсульованої лікарської форми пролонгованої дії з регульованим вивільненням активної речовини незалежно від виду і кількості наповнення шлунка і травного тракту

Номер патенту: 73097
Опубліковано: 15.06.2005
Автори: Брендель Еріх, Шантрен Ернст, Вайземанн Клаус, Рупп Роланд, Каніканті Венката-Рангарао
Мітки: вивільненням, активної, пролонгованої, наповнення, речовини, лікарської, спосіб, дії, кількості, незалежно, мікрокапсульованої, одержання, травного, тракту, виду, перорально, шлунка, форми, регульованим
Формула / Реферат:
1. Спосіб одержання пероральної мікрокапсульованої лікарської форми пролонгованої дії з регульованим вивільненням активної речовини незалежно від виду і кількості наповнення шлунка і травного тракту, який відрізняється тим, що гідрофільний полімер гідроксипропілцелюлози з середньою молекулярною масою від 250000 до 1200000, в кількості 40-95% мас., у перерахунку на суміш активної речовини і полімеру, і молярним ступенем заміщення принаймні 3,...
Фармацевтична композиція з контрольованим вивільненням, що містить гідрохлорид трамадолу, і спосіб її одержання

Номер патенту: 76411
Опубліковано: 15.08.2006
Автори: Разус Любослав, Гаттнар Ондрей, Седларова Гелена, Варга Іван, Земанек Марьян
МПК: A61K 31/135, A61P 29/00, A61K 9/22
Мітки: одержання, вивільненням, контрольованим, гідрохлорид, композиція, спосіб, фармацевтична, трамадолу, містить
Формула / Реферат:
1. Фармацевтична композиція в таблетованій формі з контрольованим вивільненням, що містить гідрохлорид трамадолу, яка відрізняється тим, що вона містить від 100 до 200 мг активного інгредієнта в суміші з тонкоподрібненими складними ефірами гліцерину з вищими жирними кислотами в кількості від 10 до 53 % мас., солями фосфорної кислоти і лугів в кількості від 20 до 41 % мас., неіонними вінілпіролідоновими полімерами в кількості від 1,15 до 1,75...
Спосіб одержання твердої лікарської форми метопролола з регульованим його вивільненням

Номер патенту: 26211
Опубліковано: 19.07.1999
Автори: Улф Ерік Йонссон, Йон Альберт Сйогрен
МПК: A61K 9/22
Мітки: форми, твердої, спосіб, метопролола, вивільненням, лікарської, регульованим, одержання
Формула / Реферат:
Способ получения твердой лекарственной формы метопролола с регулируемым его высвобождением путем нанесения мембранообразующего раствора на ядро, отличающийся тем, что на ядро из двуокиси кремния с размером 0,15 - 2,00мм наносят соли метопролола, выбранные из группы, состоящей из сукцината или фумарата, или сорбата 1-энантиомера из раствора этанола, покрывают распылением мембранообразующим раствором, содержащим этилцеллюлозу и...
Композиція, що містить галантамін з контрольованим його вивільненням, спосіб її одержання, дозована лікарська форма та упаковка

Номер патенту: 79578
Опубліковано: 10.07.2007
Автори: ван Дікке Фредерік Анна Родольф, Макгі Джон Пауль, Гіліс Пауль Марі Віктор, де Веер Марк Моріс Жермен, де Конде Валентин Флорент Віктор, де Бруійн Герман Йоханнес Катеріна
МПК: A61P 25/28, A61K 31/55, A61K 9/54
Мітки: контрольованим, містить, упаковка, одержання, галантамін, дозована, композиція, лікарська, спосіб, форма, вивільненням
Формула / Реферат:
1. Композиція з контрольованим вивільненням, що містить як активний інгредієнт галантамін, яка відрізняється тим, що включає гранули, які складаються із серцевини з покриттям та мембранного покриття, причому серцевина з покриттям містить цукрові сфери у кількості від 45 мас. % до 75 мас. %; галантамін гідробромід (1:1) у кількості від 5 мас. % до 15 мас. %; водорозчинний плівкотвірний полімер, яким є...
Попередній патент: Антитромбозні подвійні інгібітори з біотиновою міткою
Наступний патент: Способи обробки та захисту матеріалу для размноження рослин, способи боротьби зі шкідниками, пестицидна композиція та матеріал для размноження рослин
Випадковий патент: Спосіб підвищення продуктивності лактуючих корів