Спосіб визначення вмісту теофіліну
Номер патенту: 113543
Опубліковано: 10.02.2017
Автори: Аль Насир Ейяд, Дзяк Георгій Вікторович, Дроздов Олексій Леонідович, Білоножко Максим Васильович, Адаб Мухамед






Формула / Реферат
Спосіб визначення вмісту теофіліну, що включає відбір проби досліджуваної субстанції, її кислотну обробку еквімолярною кількістю 6 % хлорної кислоти, струшування, екстрагування сумішшю хлороформу з ізопропанолом, при співвідношенні 95:5 мас. частин, центрифугування, відокремлення органічного шару, фільтрування, випаровування, розчинення сухого залишку, його рідинне хроматографування з використанням суміші ацетонітрилу з водою, при співвідношенні 20:80 мас. частин, як елюенту, ідентифікацію, визначення площі хроматографічного піку на заданій довжині хвилі аналізатора як показника концентрації, який відрізняється тим, що додатково випаровування проводять у випарних чашках під витяжним повітрям, до сухого залишку теофіліну додають 500 мкл ацетонітрилу, ідентифікацію теофіліну в ультрафіолетовому діапазоні здійснюють за спектральними максимумами хвильового діапазону 203, 272 нм і проймою між ними на довжині 241 нм, площу хроматографічного піку визначають за спектральними піками сигналів І203, І272, при співвідношенні 2,5 їх інтенсивностей, а хроматографування здійснюють шляхом діодно-матричного детектування.
Текст
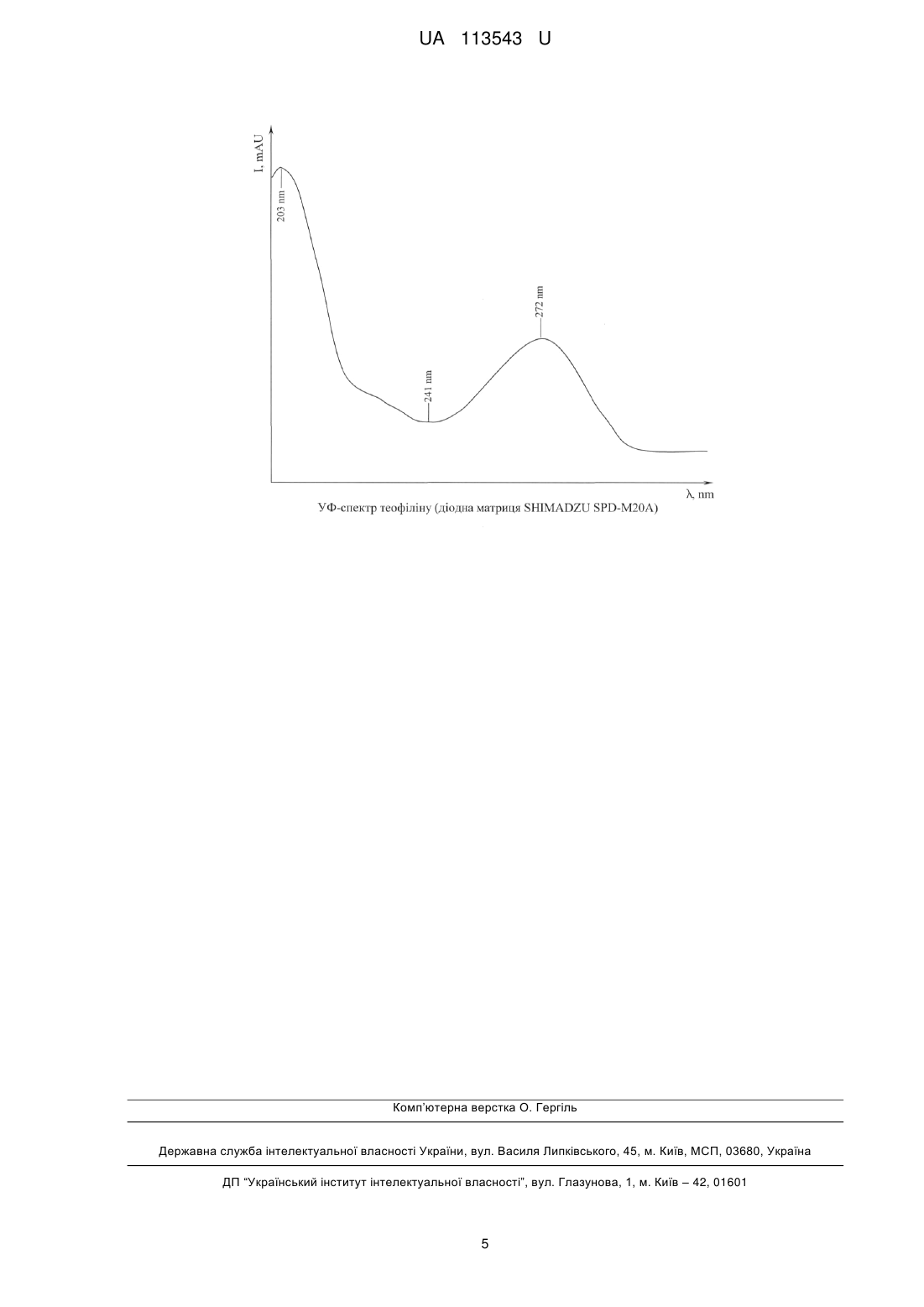
Реферат: Спосіб визначення вмісту теофіліну включає відбір проби досліджуваної субстанції, її кислотну обробку еквімолярною кількістю 6 % хлорної кислоти, струшування, екстрагування сумішшю хлороформу з ізопропанолом, при співвідношенні 95:5 мас. частин, центрифугування, відокремлення органічного шару, фільтрування, випаровування, розчинення сухого залишку, його рідинне хроматографування з використанням суміші ацетонітрилу з водою, при співвідношенні 20:80 мас. частин, як елюенту, ідентифікацію, визначення площі хроматографічного піку на заданій довжині хвилі аналізатора як показника концентрації. Додатково випаровування проводять у випарних чашках під витяжним повітрям, до сухого залишку теофіліну додають 500 мкл ацетонітрилу, ідентифікацію теофіліну в ультрафіолетовому діапазоні здійснюють за спектральними максимумами хвильового діапазону 203, 272 нм і проймою між ними на довжині 241 нм, площу хроматографічного піку визначають за спектральними піками сигналів І203, І272, при співвідношенні 2,5 їх інтенсивностей, а хроматографування здійснюють шляхом діодно-матричного детектування. UA 113543 U (12) UA 113543 U UA 113543 U 5 10 15 20 25 30 35 40 45 50 55 60 Корисна модель належить до досліджень матеріалів шляхом їх розділення на складові, переважно до рідинної колонкової хроматографії, аналізу медичних препаратів, і може бути використаною у сфері контролювання їхньої якості. Відомий спосіб одночасного визначення вітамінів "А" і "Е" в крові, що включає узяття проби, відділення сироватки, її екстракцію гексаном, хроматографування, ультрафіолетову (УФ) детекцію, ідентифікацію за часом утримання та визначення концентрації за площами піків на хроматограмах, де насухо випаровують аліквоту гексанового екстракту в потоці інертного газу, розчиняють сухий залишок в метанолі, а хроматографування здійснюють шляхом градієнтного елюювання на зворотно-фазній колонці, при співвідношенні 9:1 мас. частин метанолу й води, та з використанням 100 % метанолу, шляхом двохвильової фотометричної детекції, на довжинах хвиль 300 і 320 нм для вітаміну "А", 270 і 290 нм для вітаміну "Е", ідентифікуючи вітаміни по спектральних співвідношеннях [1]. Недоліком процесу є недостатня точність кінцевого результату. Це зумовлене нестабільністю хроматографічної системи через проведення градієнтного елюювання, використанням рухливої фази метанолу як модифікатора, що призводить до розмивання хроматографічних піків на хроматограмах, зниження ефективності хроматографічної системи у теоретичних тарілках, використанням двохвильової фотометричної детекції, що стримує контролювання пройми між максимумами спектрів, виключає одержання "чистого" хроматографічного піку, неадекватністю хвильового діапазону до теофіліну та його поганою розчинністю у гексані. Серед відомих, точнішим є спосіб визначення еуфіліну у водному розчині, що включає попереднє готування першого, другого й третього буферних розчинів, фільтрацію, змішування проби з третім буферним розчином, як активатором субстанції, центрифугування суміші в заданому режимі, розміщення капілярної трубки між пробою й другим буферним розчином, як електролітичним середовищем, з можливістю підтримки заданого тиску, комутацію потенціалів джерела високовольтного живлення, реєстрацію оптичної щільності субстанції при її переміщенні по капілярній трубці, ідентифікацію та визначення вмісту шляхом перетворення даних в аналітичний сигнал [2]. Відомий аналог допускає збільшення екстракції аналізату, чутливості засобів детекції до теофіліну (еуфілін включає 80 % теофіліну і 20 % етилендіаміну [3]) й деяке підвищення точності. Подальше збільшення точності в умовах капілярного електрофорезу за наведеним способом обмежене. Це пояснюється утримуванням одним з буферних розчинів додецилсульфату нагрію як іон-парного агенту до теофіліну, від точності дозування котрого визначається можливість відтворення процесу. Помилки приготування останнього істотно впливають на стабільність системи та дисперсію часу утримання електрофоретичних піків. В'язкість проб збільшує вплив раптових похибок на точність. Поряд із цим, зняття аналітичного сигналу у спектральній точці між максимумом і мінімумом поглинання відбувається неселективно, що утрудняє ідентифікацію та погіршує чутливість засобів детекції. Це зумовлене й залученням фотометра з постійною довжиною хвилі 254 нм, тоді як спектральні максимуми поглинання теофіліну і пройма між ними з'являються на довжинах 203, 272 і 241 нм хвильового діапазону, відповідно. Додатково, відомому об'єкту бракує експлуатаційних зручностей, з причин приготування складних буферних розчинів, необхідності підтримки заданого технологічного тиску впродовж встановленого часу, під час введення проби в систему, та застосування джерела високовольтного живлення як фактора небезпечності. Найближчим аналогом до корисної моделі, що заявляється, серед об'єктів аналогічного призначення за найбільшою кількістю істотних ознак, є спосіб визначення вмісту еуфіліну шляхом рідинної хроматографії, що включає відбір проби досліджуваної субстанції, її кислотну обробку еквімолярною кількістю 6 % хлорної кислоти, струшування, екстрагування сумішшю хлороформу з ізопропанолом, при співвідношенні 95:5 мас. частин, центрифугування, відокремлення органічного шару, фільтрування, випаровування, розчинення сухого залишку, його рідинне хроматографування з використанням суміші ацетонітрилу з водою, при співвідношенні 20:80 мас. частин, як елюенту, ідентифікацію, визначення площі хроматографічного піку на заданій довжині хвилі аналізатора, як показника концентрації, де випаровування сухого залишку проводять у вакуумних умовах, з використанням роторного випаровувача, сухий залишок досліджуваної субстанції розчиняють 0,03Н розчином соляної кислоти, а хроматографування здійснюють шляхом спектрофотометрії. За цих умов чутливість методу до теофіліну становить 80 нг/мл у воді, а ступінь його екстракції-0,95 [4]. У зазначеному вигляді відомий аналог задачі допускає деяке підвищення точності, завдяки підвищенню селективності хроматографічного розділення, опрацюванню елюенту, перебігу супровідних реакцій, покращення експлуатаційних зручностей за рахунок виключення складних буферних розчинів, підтримки заданого технологічного тиску та застосування джерела високовольтного живлення. Недоліком найближчого аналогу залишається недостатня точність кінцевого 1 UA 113543 U 5 10 15 20 25 30 35 40 45 50 55 результату. Це зумовлене забрудненням проб, стандартних зразків і обладнання під час випаровування екстракту проби у роторному випаровувачі. Його використання призводить до перехресного забруднення проб і стандартних зразків, з причини залучення одного й того ж обладнання. Забруднення обладнання посилюється викидами аналізату з випарної колби у чисту зону випаровувача від бурного кипіння, з-поза недосконалого контролю Т°С режиму. Залишки проби, стандартного зразку у випаровувачі можуть забруднювати усі наступні проби після викиду. Ризик забруднення роторного випаровувача підвищується й водою, внаслідок використання водоструминного вакуумного насоса при нестабільному тиску води у водопроводі. Неточність результатів, яка зумовлена використанням роторного випаровувача, в ряді випадків супроводжується отриманням артефактів. Поряд із цим, розчинення сухого залишку 0,03Н розчином соляної кислоти, перед введенням у хроматографічну колонку, призводить до зниження ефективності хроматографічної системи, адже октадецилсиланове щеплення до силікагелю у кислотному або лужному середовищі сильно руйнується, як основа оберненофазової рідинної хроматографії, що викривляє як час виходу теофіліну, так і площу піку на хроматограмі. Хроматографування розчину сухого залишку теофіліну шляхом спектрофотометрії, на єдиній довжини хвилі 270 нм, без урахування профілю його УФ спектра, де спектральні максимуми його поглинання і пройма між ними з'являються на довжинах 203, 272 і 241 нм хвильового діапазону, відповідно, відбувається без контролю за проймою між максимумами спектра, що запобігає оцінці "чистоти" хроматографічного піку, погіршує ідентифікацію, селективність хроматографічної системи і стримує збільшення точності. В основу корисної моделі поставлена задача, що полягає у збільшенні точності шляхом проведення тривимірного діодно-матричного детектування та опрацювання технологічного режиму. Поставлена задача вирішується тим, що при здійсненні у відомому способі визначення вмісту теофіліну, що включає відбір проби досліджуваної субстанції, її кислотну обробку еквімолярною кількістю 6 % хлорної кислоти, струшування, екстрагування сумішшю хлороформу з ізопропанолом, при співвідношенні 95:5 мас. частин, центрифугування, відокремлення органічного шару, фільтрування, випаровування, розчинення сухого залишку, його рідинне хроматографування з використанням суміші ацетонітрилу з водою, при співвідношенні 20:80 мас. частин, як елюенту, ідентифікацію, визначення площі хроматографічного піку на заданій довжині хвилі аналізатора як показника концентрації, в якому згідно з корисною моделлю, додатково випаровування проводять у випарних чашках під витяжним повітрям, до сухого залишку теофіліну додають 500 мкл ацетонітрилу, ідентифікацію теофіліну в ультрафіолетовому діапазоні здійснюють за спектральними максимумами хвильового діапазону 203, 272 нм та проймою між ними на довжині 241 нм, площу хроматографічного піку визначають за спектральними піками сигналів І203, І272, при співвідношенні 2,5 їх інтенсивностей, а хроматографування здійснюють шляхом тривимірного діодно-матричного детектування. У запропонованому вигляді тривимірне діодно-матричне детектування (за часом утримання речовини хроматографічною колонкою, інтенсивністю аналітичного сигналу і довжиною хвилі) та опрацювання технологічного режиму дозволяють підвищити чутливість методу для теофіліну до 2-8 нг/мл у воді, а ступінь екстракції з крові до 0,51 од., що у 10-40 і ~1,9 разів вище відносно найближчого аналогу. Причинно-наслідковий зв'язок сукупності ознак запропонованої корисної моделі з вищезазначеним технічним результатом полягає в наступному. Випаровування субстанції у випарних чашках під витяжним повітрям компенсує залучення роторного випаровувача, виключаючи забруднення проб, стандартних зразків, обладнання, ризики їх забруднення водою та отримання артефактів, що сприяє збільшенню точності. При вирішенні поставленої задачі додання 500 мкл ацетонітрилу до сухого залишку теофіліну, в умовах кислотного середовища хроматографічної системи, збільшує ефективність за рахунок оптимізації октадецилсиланової адаптації до силікагелю та нормалізації як часу виходу, так і площ піків теофіліну, обмежених спектральними піками сигналів І203, І272, при середньостатистичному співвідношенні 2,5 їх інтенсивностей, як відгуків аналітичного сигналу (див. креслення). Хроматографічний пік теофіліну в УФ спектрі, у вигляді спектрального профілю (зрізу, де час утримання відповідає теофіліну), що складається з максимумів його поглинання і пройми між ними, які з'являються, відповідно, на довжинах 203, 272 і 241 нм хвильового діапазону, спостережувані на хроматограмі, збільшують достовірність ідентифікації та відрізнення теофіліну у широкому спектрі "шумів" з боку решти хімічних агентів. Контролювання пройми між 2 UA 113543 U 5 10 15 20 25 30 35 40 45 50 55 60 максимумами спектра допускає оцінку "чистоти" хроматографічних піків, покращує селективність. Висока достовірність (>0,98) формування профілю УФ спектра теофіліну за спектральними піками сигналів І203, І272, при співвідношенні 2,5 їх інтенсивностей, як відгуків аналітичного сигналу, підвищує точність визначення концентрації. Додаткова перевага запропонованого способу полягає у відсутності реакцій на вміст гепарину або тіопенталу в сироватці крові. Спосіб ілюструється хроматограмою теофіліну та зіставленням основних технічних характеристик. Для визначення вмісту теофіліну в розчині еуфіліну, плазмі крові, водному розчині чи у будьякій рідині залучають обладнання фірми "Shimadzu" (Японія): рідинну хроматографічну систему "Shimadzu LC-20AD", складену з двох плунжерних насосів "Shimadzu LC-20AD", з підтримкою швидкості потоку елюенту в межах 0,1-1,0 мл/хв., дегазатора "Shimadzu DGU-2OA3", системи градієнту високого тиску, хроматографічних колонок 100 мм довжини, з внутрішнім 02 мм, силікагелем зерненням 5 мкм, як обернено-фазовим сорбентом, хімічно привитим з октадецилсиланом "Kromasil С18", термостату "Shimadzu CTO-20A", спектрометричного діодноматричного детектора "Shimadzu SPD-M20A" для 180-800 нм хвильового діапазону, автоматичного інжектора "Shimadzu SPD-M20A Auto Sampler", а також елюент, у вигляді суміші ацетонітрилу з водою, при співвідношенні 20:80 мас. частин, також 6 % хлорну кислоту та суміш хлороформу з ізопропанолом, при співвідношенні 95:5 мас. частин. Спосіб визначення вмісту теофіліну включає відбір проби досліджуваної субстанції, її кислотну обробку еквімолярною кількістю 6 % хлорної кислоти, струшування, екстрагування сумішшю хлороформу з ізопропанолом, при співвідношенні 95:5 мас. частин, центрифугування, відокремлення органічного шару, фільтрування, випаровування у випарних чашках під витяжним повітрям, розчинення сухого залишку 500 мкл кількістю ацетонітрилу, рідинне хроматографування шляхом діодно-матричного детектування, з використанням суміші ацетонітрилу з водою, при співвідношенні 20:80 мас. частин, як елюенту, ідентифікацію теофіліну за спектральними максимумами хвильового діапазону 203, 272 нм та проймою між ними на довжині 241 нм в УФ діапазоні та визначення площі хроматографічного піку за спектральними піками сигналів І203, І272, при співвідношенні 2,5 їх інтенсивностей. Під час діодноматричного детектування, на відміну від спектрофотометричного, відбувалося зняття тривимірної хроматографи з отриманням спектрального профілю (зрізу) у проміжок часу, саме який відповідає часу утримання теофіліну. Ступінь співпадіння спектрального профілю у хроматограмі шуканої субстанції із спектральним профілем стандартного розчину теофіліну визначало за ступінь ідентифікації теофіліну у шуканій субстанції. Це дозволило збільшити чутливість до теофіліну та ступінь його екстракції у 10-40 і -1,9 разів, відповідно. Приклад 1. У центрифужну пробірку відбирали пробу плазми крові, додавали до її аліквоти еквімолярну кількість 6 % хлорної кислоти, короткочасно струшували. Вносили суміш хлороформу ізопропанолом, взятих при співвідношенні 95:5 мас. частин, струшували впродовж 5 хв. і центрифугували. Хлороформний витяг відокремлювали та випаровували у випарних чашках під витяжним повітрям. Сухий залишок розчиняли 500 мкл ацетонітрилу, як рухливою фазою, та хроматографували шляхом діодно-матричного детектування, з використанням як елюенту суміші ацетонітрилу з водою, при співвідношенні 20:80 мас. частин. Хроматографічний пік теофіліну ідентифікували за спектральними максимумами хвильового діапазону 203, 272 нм та проймою між ними на довжині 241 нм УФ діапазону та часом утримання досліджуваної субстанції. Концентрацію теофіліну у плазмі крові визначали за спектральними піками сигналів І203, І272, при середньостатистичному співвідношенні 2,5 їх інтенсивностей, як відгуків аналітичного сигналу. Приклад 2. У центрифужну пробірку, від розчину еуфіліну невідомої концентрації, відбирали аліквоту об'ємом 500 мкл, до котрої додавали 200 мкл еквімолярної кількості 6 % хлорної кислоти, короткочасно струшували, до утворення сироподібної маси. Здійснювали екстрагування, вводячи у пробірку 500 мкл суміші хлороформу з ізопропанолом, при співвідношенні 95:5 мас. частин, піддавали струшуванню й піддавали центрифугуванню, з кутовою швидкістю ~1500 об./хв., впродовж 5 хв. Субнатант відбирали піпеточним дозатором, проколюючи сироподібну масу, фільтрували через мікролійку з маленьким шматочком знежиреної вати та збирали у випарну чашку. Хлороформний екстракт випаровували під витяжкою у потоці повітря без нагрівання. Сухий залишок розчиняли у 500 мкл ацетонітрилу та переносили у віалу хроматографа. За допомогою автодозатора 10 мкл екстракту вводили у колонку хроматографа та хроматографували його зі швидкістю потоку 0,2 мл/хв., з урахуванням тиску на вході колонки. Хроматографували діодно-матричним шляхом, в ізократичному режимі, 3 UA 113543 U 5 10 використовуючи як елюент суміш ацетонітрилу з водою при співвідношенні 20:80 мас. частин. Після повної екстракції лікарської форми теофіліну, ідентифікували його за спектральними максимумами хвильового діапазону 203, 272 нм, проймою між ними на довжині 241 нм УФ діапазону та часом його утримання. Концентрацію теофіліну в розчині еуфіліну визначали за спектральними піками сигналів І203, І272, при середньостатистичному співвідношенні 2,5 їх інтенсивностей. За наведеними прикладами була підтверджена можливість ідентифікації теофіліну та визначення його вмісту як у плазмі крові, так і розчині еуфіліну шляхом діодно-матричної детекції, в умовах істотного розширення діапазону лінійних хроматографічних вимірювань, без викривлення кількісно-якісних відбитків, одержання артефактів, з чутливістю та ступенем його екстракції, відповідно, у 10-40 і -1,9 разів вищими за найближчий аналог. Таблиця Основні технічні характеристики способу визначення вмісту теофіліну Показники Довжина хвилі, нм Чутливість, нг/мл Лінійність діапазону вимірюваних концентрацій, нг/мл 15 20 25 30 Пат. Пат. Пропонована корисна 45279 UA 45278 UA модель 254 270 203 272 30 80 2 8 від 2 до від 8 до 1000 1000 Джерела інформації: 1. Способ одновременного определения витаминов А и Е в крови: Заяв. 95110518 России, МПК G01N 33/48, G01N 33/52 / НИИ хирургии "Восточно-сибирский НЦ" СО РАМН (Россия); Г.А. Федорова, Л.Р. Макаева, И.Б. Рудых, Э.Э. Кузнецова (Россия). - № 95110518/14; заявл. 20.06.95; опубл. 27.08.97. 2. Спосіб визначення вмісту еуфіліну у водному розчині: Пат. 45279 України, МПК G01N 30/00 Дніпропетровська державна медична академія (Україна); Дніпропетровський державний центр стандартизації, метрології та сертифікації (Україна); О.Л.Дроздов, О.К.Вяткін, Г.В.Дзяк, В.П.Маматов, A.M.Рудько, В.Г.Варченко (Україна). - № 2001107280; заявл. 15.10.01; опубл. 15.03.02. 3. Машковский М.Д. Лекарственные средства. М: "Медицина", 1987.-Т.1.-С.457. 4. Спосіб визначення вмістуеуфіліну шляхом рідинної хроматографії: Пат. 45278 України, МПК G01N 30/00, G01N 30/02 Дніпропетровська державна медична академія (Україна); Дніпропетровський державний центр стандартизації, метрології та сертифікації (Україна); Т.О.Перцева, В.Г.Варченко, O.K.Вяткін, О.Л.Дроздов, В.П.Маматов, О.Ю.Красновська (Україна). - № 2001107279; заявл. 25.10.01; опубл. 15.03.02. ФОРМУЛА КОРИСНОЇ МОДЕЛІ 35 40 45 Спосіб визначення вмісту теофіліну, що включає відбір проби досліджуваної субстанції, її кислотну обробку еквімолярною кількістю 6 % хлорної кислоти, струшування, екстрагування сумішшю хлороформу з ізопропанолом, при співвідношенні 95:5 мас. частин, центрифугування, відокремлення органічного шару, фільтрування, випаровування, розчинення сухого залишку, його рідинне хроматографування з використанням суміші ацетонітрилу з водою, при співвідношенні 20:80 мас. частин, як елюенту, ідентифікацію, визначення площі хроматографічного піку на заданій довжині хвилі аналізатора як показника концентрації, який відрізняється тим, що додатково випаровування проводять у випарних чашках під витяжним повітрям, до сухого залишку теофіліну додають 500 мкл ацетонітрилу, ідентифікацію теофіліну в ультрафіолетовому діапазоні здійснюють за спектральними максимумами хвильового діапазону 203, 272 нм і проймою між ними на довжині 241 нм, площу хроматографічного піку визначають за спектральними піками сигналів І203, І272, при співвідношенні 2,5 їх інтенсивностей, а хроматографування здійснюють шляхом діодно-матричного детектування. 4 UA 113543 U Комп’ютерна верстка О. Гергіль Державна служба інтелектуальної власності України, вул. Василя Липківського, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 5
ДивитисяДодаткова інформація
МПК / Мітки
МПК: G01N 30/02
Мітки: спосіб, вмісту, теофіліну, визначення
Код посилання
<a href="https://ua.patents.su/7-113543-sposib-viznachennya-vmistu-teofilinu.html" target="_blank" rel="follow" title="База патентів України">Спосіб визначення вмісту теофіліну</a>
Застосування способу визначення вмісту теофіліну як способу визначення концентрації німесуліду у водному розчині

Номер патенту: 107186
Опубліковано: 25.05.2016
Автори: Краснов Олександр Олександрович, Жилюк Володимир Іванович, Білоножко Максим Васильович, Марзан Олександр Олександрович, Дроздов Олексій Леонідович
МПК: G01N 33/15, G01N 30/00, G01N 30/02
Мітки: застосування, розчині, теофіліну, концентрації, визначення, німесуліду, вмісту, водному, способу
Формула / Реферат:
Застосуванням способу визначення вмісту теофіліну як способу визначення концентрації німесуліду у водному розчині.
Застосування способу визначення вмісту теофіліну як способу визначення концентрації елетриптану у водному розчині

Номер патенту: 105659
Опубліковано: 25.03.2016
Автори: Жилюк Володимир Іванович, Свіргун Ілля Степанович, Білоножко Максим Васильович, Мамчур Віталій Йосипович, Дроздов Олексій Леонідович
МПК: G01N 30/02, G01N 33/15, G01N 30/00
Мітки: концентрації, застосування, теофіліну, елетриптану, визначення, розчині, способу, вмісту, водному
Формула / Реферат:
Застосування способу визначення вмісту теофіліну як способу визначення концентрації елетриптану у водному розчині.
Застосовування способу визначення вмісту теофіліну як способу вимірювання концентрації сіднокарбу у водному розчині

Номер патенту: 54648
Опубліковано: 25.11.2010
Автори: Аль Насир Ейяд, Дзяк Георгій Вікторович, Белоножко Максим Васильович, Вяткін Олександр Констянтинович, Кошелев Олег Станіславович, Дроздов Олексій Леонідович, Качанов Сергій Олександрович
МПК: G01N 30/02, G01N 30/00, G01N 33/15
Мітки: концентрації, вимірювання, водному, застосовування, теофіліну, визначення, способу, розчині, вмісту, сіднокарбу
Формула / Реферат:
Застосовування способу визначення вмісту теофіліну як способу вимірювання концентрації сіднокарбу у водному розчині.
Застосування способу визначення вмісту теофіліну як способу вимірювання концентрації доксофіліну у водному розчині

Номер патенту: 44980
Опубліковано: 26.10.2009
Автори: Дзяк Георгій Вікторович, Гашенова Катерина Юріївна, Дроздов Олексій Леонідович, Качанов Сергій Олександрович, Перцева Тетяна Олексіївна, Білоножко Максим Васильович, Вяткін Олександр Костянтинович
МПК: G01N 30/00, G01N 30/02, G01N 33/15
Мітки: застосування, вмісту, вимірювання, розчині, теофіліну, визначення, концентрації, водному, доксофіліну, способу
Формула / Реферат:
Застосування способу визначення вмісту теофіліну у водному розчині як способу вимірювання концентрації доксофіліну у водному розчині.
Спосіб визначення вмісту еуфіліну шляхом рідинної хроматографії

Номер патенту: 45278
Опубліковано: 15.03.2002
Автори: Онищенко Тетяна Станіславівна, Маматов Валерій Петрович, Дроздов Олексій Леонідович, Красновська Ольга Юрієвна, Варченко Віталій Григорович, Перцева Тетяна Олексіївна, Вяткін Олександр Костянтинович
МПК: G01N 30/02, G01N 30/00
Мітки: еуфіліну, визначення, рідинної, спосіб, хроматографії, шляхом, вмісту
Формула / Реферат:
Спосіб визначення вмісту еуфіліну шляхом рідинної хроматографії, який містить кислотну обробку проби, її екстрагування сумішшю хлороформу з ізопропанолом, взятих у співвідношенні 95:5, відділення органічного шару, фільтрацію, випаровування, розчинення сухого залишку 0,03 Н розчином соляної кислоти та хроматографування з використанням як елюенту суміші ацетонітрилу з водою при заданому співвідношенні частин, який відрізняється тим, що кислотну...
Попередній патент: Світлодіод білого світла з монокристалічним фосфором
Наступний патент: Установка для лазерної обробки отворів
Випадковий патент: Способи поширення і запобігання поширенню інформації в мережі