Спосіб одержання (r)-(-)-5-(2-амінопропіл)-2-метоксибензолсульфонаміду




Формула / Реферат
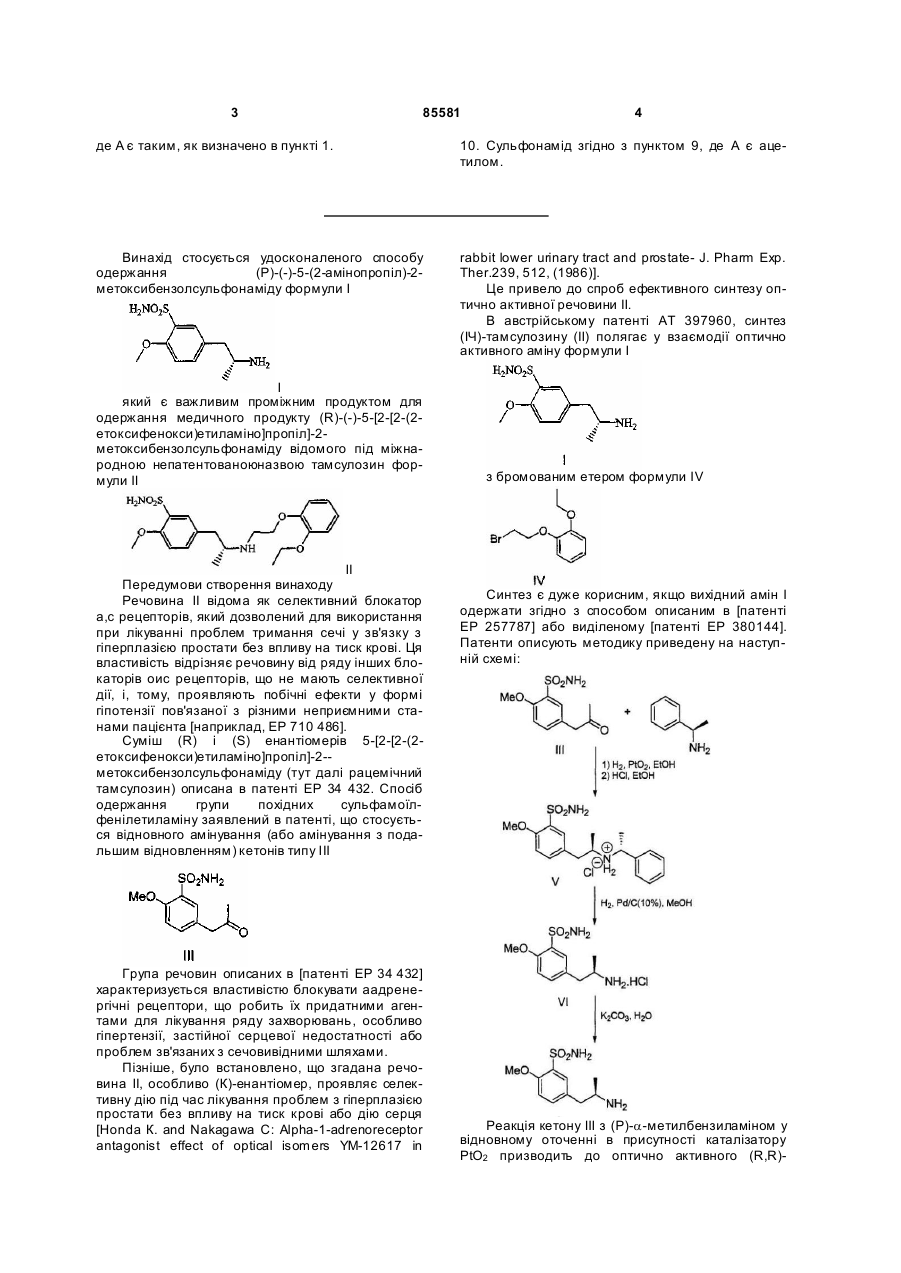
1. Спосіб одержання (R)-(-)-5-(2-амінопропіл)-2-метоксибензолсульфонаміду формули І
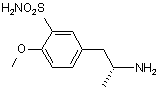 , І
, І
який відрізняється тим, що
а) в N-[(1R)-2-(4-метоксифеніл)-1-метилетил]-N-[(1R)-1-фенілетил)]амiн формули VIII
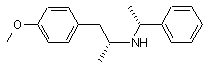 VIII
VIII
вводять захисну групу, одержуючи амід формули IX
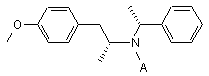 , IX
, IX
де А може бути ацилом з 2-8 атомами вуглецю,
б) після чого амід формули IX піддають хлорсульфонілуванню і одержаний сульфохлорид перетворюють у сульфонамід формули X
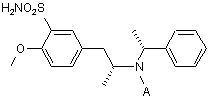 , X
, X
де А є таким, як визначено вище, і
в) сульфонамід формули X гідрують і деацилюють, одержуючи сполуку формули І.
2. Спосіб згідно з пунктом 1, який відрізняється тим, що захисною групою А є ацил, переважно ацетил.
3. Спосіб згідно з пунктом 2, який відрізняється тим, що як ацилюючий агент використовують ацетангідрид при 50-100 °С.
4. Спосіб згідно з пунктом 1, який відрізняється тим, що сульфохлорид, одержаний після хлорсульфонілування, не виділяють, а безпосередньо перетворюють у сульфонамід, використовуючи аміак.
5. Спосіб згідно з пунктом 4, який відрізняється тим, що хлорсульфонілування проводять в метиленхлориді при -30 - +30 °С.
6. Спосіб згідно з пунктом 1, який відрізняється тим, що гідрування проводять в присутності паладієвого каталізатора.
7. Спосіб згідно з пунктом 6, який відрізняється тим, що каталізатором є 3 % Pd/C з 50 % вмістом води при тиску 2 МПа і температурі 80-85 °С.
8. Спосіб згідно з пунктом 1, який відрізняється тим, що продукт формули І реагує зі сполукою формули IV
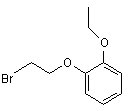 IV
IV
з утворенням кінцевого продукту формули II
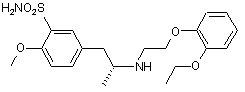 II.
II.
9. Сульфонамід формули X
, X
де А є таким, як визначено в пункті 1.
10. Сульфонамід згідно з пунктом 9, де А є ацетилом.
Текст
1. Спосіб одержання (R)-(-)-5-(2-амінопропіл)2-метоксибензолсульфонаміду формули І C2 2 (13) 1 H2NO2S O O NH O II. 9. Сульфонамід формули X H2NO2S O O N N A де А є таким, як визначено вище, і ,X A ,X (19) , IX де А може бути ацилом з 2-8 атомами вуглецю, б) після чого амід формули IX піддають хлорсульфонілуванню і одержаний сульфо хлорид перетворюють у сульфонамід формули X UA H2NO 2S A 3 85581 4 де А є таким, як визначено в пункті 1. 10. Сульфонамід згідно з пунктом 9, де А є ацетилом. Винахід стосується удосконаленого способу одержання (Р)-(-)-5-(2-амінопропіл)-2метоксибензолсульфонаміду формули І rabbit lower urinary tract and prostate- J. Pharm Exp. Ther.239, 512, (1986)]. Це привело до спроб ефективного синтезу оптично активної речовини II. В австрійському патенті AT 397960, синтез (ІЧ)-тамсулозину (II) полягає у взаємодії оптично активного аміну формули І І який є важливим проміжним продуктом для одержання медичного продукту (R)-(-)-5-[2-[2-(2етоксифенокси)етиламіно]пропіл]-2метоксибензолсульфонаміду відомого під міжнародною непатентованоюназвою тамсулозин формули II II Передумови створення винаходу Речовина II відома як селективний блокатор а,с рецепторів, який дозволений для використання при лікуванні проблем тримання сечі у зв'язку з гіперплазією простати без впливу на тиск крові. Ця властивість відрізняє речовину від ряду інших блокаторів оис рецепторів, що не мають селективної дії, і, тому, проявляють побічні ефекти у формі гіпотензії пов'язаної з різними неприємними станами пацієнта [наприклад, ЕР 710 486]. Суміш (R) і (S) енантіомерів 5-[2-[2-(2етоксифенокси)етиламіно]пропіл]-2-метоксибензолсульфонаміду (тут далі рацемічний тамсулозин) описана в патенті ЕР 34 432. Спосіб одержання групи похідних сульфамоїлфенілетиламіну заявлений в патенті, що стосується відновного амінування (або амінування з подальшим відновленням) кетонів типу III Група речовин описаних в [патенті ЕР 34 432] характеризується властивістю блокувати аадренергічні рецептори, що робить їх придатними агентами для лікування ряду захворювань, особливо гіпертензії, застійної серцевої недостатності або проблем зв'язаних з сечовивідними шляхами. Пізніше, було встановлено, що згадана речовина II, особливо (К)-енантіомер, проявляє селективну дію під час лікування проблем з гіперплазією простати без впливу на тиск крові або дію серця [Honda К. and Nakagawa C: Alpha-1-adrenoreceptor antagonist effect of optical isomers YM-12617 in з бромованим етером формули IV Синтез є дуже корисним, якщо вихідний амін І одержати згідно з способом описаним в [патенті ЕР 257787] або виділеному [патенті ЕР 380144]. Патенти описують методику приведену на наступній схемі: Реакція кетону III з (Р)-a-метилбензиламіном у відновному оточенні в присутності каталізатору PtO2 призводить до оптично активного (R,R) 5 85581 діастереоізомеру V, що має високу оптичну чистоту (приблизно 92%). Цей проміжний продукт надалі перекристалізують одержуючи високо оптичночистий продукт, який перетворюють шляхом гідрогенолізу у оптично активний (R)-гідрохлорид VI. Потім перетворюють у (R)-амін формули І шляхом дії основи. Однак, повторення цієї методики показало, що вихід R аміну І знаходиться в інтервалі від 10 до 15% і що одержання тамсулозину II складає тільки приблизно 4 - 5% від теорії. Вихід останньої стадії синтезу, тобто, одержання речовини II з речовини І, було значно покращено порівняно з [патентом CZ 291802]. Однак, все ще існує проблема обумовлена низьким виходом важливої проміжної сполуки І. Однак, неочікувано, було знайдено такий спосіб синтезу, що забезпечує навіть більше ніж двократний вихід. Вихідною речовиною для одержання аміну формули І є метилбензилкетон (VII), з якого шляхом відновного амінування одержують гідрохлорид N-[(1R)-2-(4-метоксифеніл)-1-метилетил]-М-[(1R)1-фенілетил)]аміну формули VIII Гідрування проводять при нормальному тиску протягом приблизно 12 годин з додаванням каталізатору в кількості 0,6г/1моль метилбензилкетону, який розходується приблизно на 60% згідно з [патентом ЕР 257787]. Враховуючи, що вартість каталізатору складає значну частину вартості сировини, значна його частина зберігається. Також усувається багатократна очистка в ацетоні і водоацетоновій суміші; в більшості випадків, достатньо повторити очищення в ацетоні тільки 1 - 3 рази. Зменшення вимог до оптичної чистоти від межі 0,2% до задовільного вмісту в 0,4% R,Sенантіомеру не спричиняє будь-якого впливу на високу оптичну чистоту кінцевого продукту формули II. Ви ходи реакцій становлять приблизно 50%. Введення захисної групи А, де А може бути ацилом, що має 2 - 8 атоми вуглецю, таким як, наприклад, ацетил, пропіоніл, гексаноїл або бензоїл, за допомогою реакції з безводною кислотою, галоїдом або ангідридом відповідної кислоти, дає амід формули IX Після N-ацетилювання ацетангідридом одержують N-[(1R)-2-(4-метоксифеніл)-1-метилетил]-N[(1R)-1-фенілетил]ацетамід. Реакція протікає за 4 6 годин при температурі 50 -100, переважно 60 70°С з високим виходом приблизно 100%. Амід формули IX потім хлорсульфонілюють і одержаний сульфохлорид перетворюють у сульфонамід. Реакцію з хлорсульфоновою кислотою 6 переважно проводять в дихлорметані при охолодженні до -30 - +30°С і корисно потім вилити реакційну суміш для розкладення в суміш льоду і 25% водного розчину аміаку. Ця методика безпосередньо дає N-{(1R)-2-[3-(аміносульфоніл)-4метоксифеніл]-1-метилетил}-N-[(1R)-1фентетил]ацетамід формули X Таким чином досягається виділення одного проміжного продукту, також як і випадку використання в синтезі іншого розчинника (тетрагідрофуран). Реакція забезпечує неочікувано високі виходи до 96%. Гідрування над паладієм ва оцтовій кислоті з додаванням розведеної хлор водневої кислоти при температурі 80 - 85 °С і тиску 2 МПа забезпечує відщеплення етилбензолу з одержанням N-{(1R)-2[3-(аміносульфоніл)-4-метоксифеніл]-1метилетил}ацетаміду (XI). В гідр уванні використовували значно дешевший і менш витрачаємий каталізатор-3% Pd/C з 50% вмістом води в кількості 1/10 ваги використовуваної речовини формули X. Виходи реакцій наближатись до 100%. Кип'ятіння в 5% водному розчині НСІ призводить до деацетилювання проміжної сполуки (XI), а додавання карбонату до випадіння в осад (R)-(-)-5(2-aмінопропіл)-2-метоксибензолсульфонаміду формули І, який перекристалізують з води. Вихід реакції після кристалізації становить приблизно 80%. Приклади В експериментальній частині, всі дані 1Н-ЯМР знімали використовуючи пристрій BRUKER 250DPX (250,13 і 62,9МГц), d в м.ч.; J в Гц). Приклад 1 Одержання гідрохлориду N-[(1R)-2-(4метоксифеніл)1-метоксифеніл]-N-[(1R)-1фенілетил)]аміну VIII 4-Метоксибензилметилкетон (VII, 43,2г), R(+)метилбензиламін (32,0г) і каталізатор PtO2 (0,16г) відважували у 2-л колбу і додавали метанол (1000мл). Гідрування проводили до зупинки реакції, протягом приблизно 12 годин при 60°С. Після завершення реакції, каталізатор відфільтровують. Фільтрат упарюють і до жовтого масла (69,47г) додають 300 мл етанолу; одержують жовтий розчин. При охолодженні на бані з льодом по краплям протягом 30 хвилин додають 50мл HCI/EtOH; розчин поступово стає червоним. Реакційну суміш перемішують при кімнатній температурі протягом 1 години. Кристали, що випали в осад, відсмоктують і промивають 50мл етанолу. Кристали, що випали в осад, одержані з двох фракцій кип'ятять в 500мл ацетону протягом 1 години. Після охолодження, суспензію перемішують протягом 2 годин при охолодженні водою, кристали, що випали в осад, відсмоктують і промивають 50мл ацетону. 7 85581 Вихід становить 48,18г (59,9%). Необхідний аналіз на ГХ хімічної і оптичної чистоти і вміст R,Sдіастереоізомеру повинен бути нижче 0,4%. Якщо вміст вищий, повторюють очистку в ацетоні. 1 Н ЯМР: δ 1,41 (д, J=6,5, ЗН, СН2СН(СН3)); 1,95(д, J=6,8, ЗН, РhСН(СН 3)); 2,81 (дд, J=10,0, J=12,9, 1Н, СНг); 3,00(шм, 1Н, СН2СН(СН3)); 3,37(дд, J=4,3, J=13,0, 1Н, CH2): 3,76(с, 3Н, СН3О); 4,37(шм, 1Н, РпСН(СН3)); 6,78(м, 2Н, СНаром ); 6,93(м, 2Н, СНаром ); 7,45(м, 3Н, СНаром ); 7,69(м, 2Н, СНаром ); 9,80(ш, ΝΗ, НСІ); 10,26(ш, NH, HCI).(CD3OD, 30°С) Приклад 2 Одержання N-[(1R)-2-(4-метоксифеніл)-1метилетил]-N-[(1R)-1-фенілетил]ацетаміду IX Проміжну сполуку VIII (48г) відважують у 1-л колбу, додають етилацетат (450мл) і по краплям до одержаної суспензії при перемішуванні і помірному охолодженні водою кімнатної температури додають 10 в.-% водний розчин NaOH (120мл). Нерозчинені кристали повільно розчиняються утворюючи жовтий розчин і реакційну суміш перемішують при кімнатній температурі протягом 0,5г. Органічну фазу відокремлюють, водну фазу збовтують з 1x100мл етилацетату і об'єднані органічні фракції упарюють. Ацетангідрид (200мл) додають до упареного залишку основи і перемішування продовжували із зворотнім холодильником з трубкой, що містить хлорид кальцію при 60-70°С протягом 4-6 годин. Наявність вихідної сполуки контролюють за допомогою ТШХ і після завершення реакції, реакційну суміш упарюють. До упареного залишку при перемішуванні і охолодженні водою по краплям додають насичений розчин гідрокарбонату натрію (100 мл). Реакційну суміш екстрагують етилацетатом (200мл), органічну фаз у відокремлюють і водний шар екстрагують 1x100мл етилацетату. Об'єднані органічні фракції екстрагують 1x50 мл розсолу і упарюють. Вихід становить 53,1г (~100%)· 1 Н ЯМР: δ 1,27(д, J=6,2, 3Н, СН2СН(СН3)); 1,59(д, J=7,0, 3Н, PhCH(CH3)); 1,82(ш, 1Н, СН2); 2,28(с, 3Н, СОСН3); 2,31 (шм, 1Н, СШ); 3,13(шм, 1Н, СН2СН(СН3)); 3,72(с, 3Н, СН3О); 5,08(м, 1Н, PhCH(CH 3)); 6,42(м, 2Н, СНаром ); 6,63(м, 2Н, СНаром ); 7,38(м, 5Н, СН аром СDСІ3, 30°С) Приклад 3 Одержання N-{(1R)-2-[3-(аміносульфоніл)-4метоксифеніл]1-метилетил}-N-[(1R)-1фенілетил]ацетаміду X. Дихлорметан (50мл) додають до упареного залишку проміжної сполуки IX (25,39г), перемішують і охолоджують до -5- -10°С. По краплям при охолодженні повільно додають хлорсульфонову кислоту (55мл), так що температура реакційної суміші не перевищує максимально +5°С. Додавання проводять протягом 45хв і потім реакційну суміш перемішують при 0-10°С протягом 1 години. Реакційну суміш повільно виливають, при перемішуванні і охолодженні, в охолоджену суміш 25% водного розчину аміаку (210мл) і льоду (210г). Піс 8 ля завершення розкладення, додають етилацетат (420мл) і реакційну суміш перемішують протягом 5хв до розчинення кристалів, що випали в осад, і витримують при кімнатній температурі протягом максимум 8 годин. Після завершення реакції, органічну фракцію відокремлюють, водний шар збовтують з 1x210мл етилацетату. Об'єднані органічні фракції упарюють. Вихід становить 28,82г (90,6%). 1 Н ЯМР: δ 1,26(д, J=6,5, 3Н, СН2СН(СН3)); 1,58(д, J=6,9, 3Н, PhCH(CH3)); 1,80(ш, 1Н, СН2); 2,30(с, 3Н, СОСН 3); 2,32(шм, 1Н, СН2); 3,13(шм, 1Н, СН2СН(СН3)); 3,93(с, 3Н, СН3О); 5,11(шм, 1Н, PhCH(CH 3)); 6,77(м, 2Н, СНаром ); 7,02(шс, 1Н, СНаром ); 7,37(м, 5Н, СН аром ).(CDCI3, 30°С) Приклад 4 Одержання N-{(1R)-2-[3-(аміносульфоніл)-4метоксифеніл]-1-метилетил}ацетаміду XI. Проміжну сполуку X (3г) розчиняють в оцтовій кислоті (160мл), розводять НСІ 1:1 (10мл) і додають 3% Pd/C з вмістом води 50% (0,3г). Гідрують при 80-85°С і тиску 2МПа протягом 10-15 годин. Після завершення гідрування, розчин фільтрують і упарюють. Вихід становить 2,55г (-100%). 1 Н ЯМР: δ 1,10(д, J=6,6, 3Н, СН(СН 3)); 1,87(с, 3Н, СОСН3); 2,71 (д, J=6,9, 2H, СН2); 3,30(ш, ΝH2); 3,95(с, 3Н, СН3О); 4,05(м, 1Н, СН(СН 3)); 7,11(д, J=8,5, СНаром ); 7,40(дд, J=8,6, J=2,4, 1Н, СНаром ); 7,68(д, J=2,2, 1Н, СНаром ); 7,96(шд, NH). (CD3OD, 30°С). Приклад 5 Одержання (R)-(-)-5-(2-амінопропіл)-2-метоксибензолсульфонаміду І Проміжну сполуку VII (10,5г) кип'ятять в 5% НСІ (250мл) із зворотнім холодильником протягом 16-18г. Протікання реакції контролюють використовуючи ТШХ, детектуючи вихідн у речовину. Після завершення реакції, реакційну суміш концентрують до приблизно 1/3 її об'єму, і потім повільно по краплям при перемішуванні додають насичений розчин карбонату натрію (50мл). Після додавання, визначають рН~10 і реакційну суміш перемішують протягом 0,5 години, і залишають кристалізуватись при 0°С. Кристали, що випали в осад, відсмоктують і фільтрат концентрують до 1/2 його об'єму і залишають кристалізуватись при 0°С. Обидві фракції (4г+8г) білих - коричнюватих кристалів об'єднують і перекристалізують з води. Вихід становить 7,52г (80%). 1 Н ЯМР: δ 0,99(д, J=6,2, 3Н,СН 3); 2,54(дд, J=13,6 J=6,8, 1Н, CH2); 2,59 (дд, J=13,6; J= 6,7; 1Н, СН2); 3,00(секс, J=6,4; 1Н, СН); 3,53(шс, ΝH2): 3,92(с, ЗН, СН3О); 7,16(д, J=8,4; 1Н, СНаром ); 7,41 (дд, J=8,4; J= 2,2; 1Н, СНаром ); 7,59(д, J=2,2; 1Н, СНаром ). (CD3SOCD3, 30°С) Приклад 6 - порівняння Спосіб одержання тамсулозину (II) згідно з попереднім рівнем техніки у порівнянні з способом, що включає стадії згідно з винаходом 1. Спосіб згідно з попереднім рівнем техніки 1.1. Хлорсульфонування в дві стадії згідно з [US 4731478 (1988)] 9 85581 10 1.2. Гідрування на платині згідно з [ЕР 0 257 787 (1987)] Каталізатор використовують в кількості 1г/1моль кетону, гідрування проводять в метанолі при нормальному тиску протягом 20 годин. Для очистки до бажаної оптичної чистоти (нижче 0,2%), речовину очищають 4х в ацетоні і 3х в суміші води і ацетону. 1.3. Гідрування на паладії згідно з [ЕР 0 257 787] Гідрують в метанолі при нормальному тиску, каталізатор -10% паладій на вугіллі в кількості 10% від ваги вихідної речовини, час реакції не є специфічними. 1.4. Перетворення основи Кип'ятять в етанолі протягом 16 годин, використовують подвійну кількість аміну, очищають хроматографією, вихід приведений для неочищеної основи, вихід гідрохлориду не приводиться. 2. Новий синтез згідно з винаходом 2.1.Гідрування на платині У автоклав завантажують: 23,1г (0,141моль) 4метоксибензилметилкетону VII, 17,2г R(+) метилбензиламіну (0,142 моль), каталізатор PtO2 (0,05г) і додають метанол (450мл). Гідрування 11 85581 12 проводять при нормальному тиску при 60-62°С протягом 12 годин. Після обробки реакційної суміші етанольним розчином НСІ, одержують проміжну сполуку VIII, яку очищають в ацетоні, що кипить. Вихід становить 24,62г (57%). Гідрування проводять при нормальному тиску протягом приблизно 12 годин, каталізатор викори стовують в кількості 0,6г/1моль кетону. Очищення в ацетоні повторюють 1- 3х до бажаної оптичної чистоти, обмеження становить 0,4% небажаного діастереоізомеру. 2.2.Одержання ацетилпохідного Проміжну сполуку VIII, 6377г (20,85моль), перетворюють у основу використовуючи 10% водний розчин NaOH (15,9л), екстрагують етилацетатом, розчинник упарюють і упарений залишок нагріва ють в ацетангідриді (26,57л) при 65-70°С протягом 6 годин. Після упарювання, одержують 6693г (~ 100%) проміжної сполуки IX. 2.3. Хлорсульфонування Упарений залишок IX з попередньої стадії, 6690г (~20,85моль), перемішують в дихлорметані (13,17л) і по краплям при -5-+2°С додають хлорсульфонову кислоту (13,75л). Через 1 годину, реакційну суміш розкладають в суміші льоду (65,5кг) і 25% водного розчину аміаку (65,5л). Продукт X екстрагують етилацетатом і вихід після упарювання складає 8068г (96,2%) проміжної сполуки X. Сульфохлорид, одержаний після хлорсульфонування, переважно не виділяють, але безпосередньо перетворюють у сульфонамід X шляхом розкладання реакційної суміші в водному розчині аміаку. В цьому способі проведення реакції уникають використання тетрагідрофурану як іншого розчинника синтезу. Вихід є надзвичайно високим в цьому типі реакцій. 2.4. Гідрування на паладії 24,62г (0,063моль) проміжної сполуки X гідрують в автоклаві з каталізатором 3% Pd/C з вмістом води 50% (2,5г) в оцтовій кислоті (160мл) з додаванням розведеної хлорводневої кислоти (1:1; 19мл) при 80-85°С і тиску 2 МПа протягом 12 годин. Після завершення реакції, вихід становить 22,13г (~100%) проміжної сполуки XI. Гідрування проводять при 80-85°С і тиску 2МПа протягом 15 годин в оцтовій кислоті з дода ванням розведеної хлорводневої кислоти. Каталізатором, що використовується в кількості 1/10 ваги вихідної речовини X, є 3% Pd/C з вмістом води 50%, який значно дешевший і економічніший ніж 10% Pd/C, який дуже легко згоряє. Виходи віртуально складають 100%. 2.5. Деацетилювання [згідно з US 4,731,478, 1988] 13 85581 14 Проміжну сполуку XI (28,2г; 0,063моль) кип'ятять в 5% водному НСІ (660мл) протягом 18 годин, після концентрування продукт І осаджують додаючи насичений розчин карбонату калію (140мл) двома фракціями (10,4 г+37,3г). Після перекристалізації з води, вихід був 17,1г (80%). 2.6. Тамсулозин 220г (0,88моль) проміжної сполуки IV і 84г (0,79моль) карбонату натрію і Ν,Νдиметилформаміду (1500мл) додають до 208г (0,85моль) проміжної сполуки І. Реакційну суміш перемішують при 70°С протягом 5 годин. До реакційної суміші додають воду і продукт II екстрагують етилацетатом. Упарений залишок перемішують в етанолі і потім відсмоктують, вихід становить 173,9г (50%) неочищеної основи II. Спосіб згідно з [CZ 291802]. Вихід, для порівняння, також розраховується на неочищену основу. Реакцію проводять при 60 - 70 °С протягом 5 годин і продукт не очищають з використанням колонкової хроматографії. 3. Висновок Порівняння загальних виходів пров ідиться починаючи з 4-метоксифенілацетону VII, хоча в [ЕР 257787], описується одержанн і тільки починаючи з проміжної сполуки III. А) Ви хід (R)-(-)-5-(2-амінопропіл)-2метоксибензолсульфонаміду І без кінцевого конденсування у тамсулозин II, тобто реакції 1.1. 1.4., становить 12,38%; а наш спосіб -реакції 2 1.2.5. - виходи 38,40%, тобто, ви хід майже в три рази вищий. Б) Якщо порівняти також і загальний вихід неочищеної основи останньої стадії синтезу тамсулозину з [патенту ЕР 257787] -4,63%, з нашими 19,20%, вихід буде вищим майже в чотири рази. Комп’ютерна в ерстка В. Клюкін Підписне Тираж 28 прим. Міністерство осв іт и і науки України Держав ний департамент інтелектуальної в ласності, вул. Урицького, 45, м. Київ , МСП, 03680, Україна ДП “Український інститут промислов ої в ласності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for the preparation of (r)-(-)-5(2-aminopropyl)-2-methoxybenzenesulfonamide
Автори англійськоюHajicek Josef, Slavikova Marketa
Назва патенту російськоюСпособ получения (r)-(-)-5-(2-аминопропил)-2-метоксибензолсульфонамида
Автори російськоюХаицек Йозеф, Славикова Маркета
МПК / Мітки
МПК: C07C 303/00, C07C 311/37
Мітки: одержання, r)-(-)-5-(2-амінопропіл)-2-метоксибензолсульфонаміду, спосіб
Код посилання
<a href="https://ua.patents.su/7-85581-sposib-oderzhannya-r-5-2-aminopropil-2-metoksibenzolsulfonamidu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання (r)-(-)-5-(2-амінопропіл)-2-метоксибензолсульфонаміду</a>
Похідні 3-деоксидесмікозину та спосіб їх одержання

Номер патенту: 66927
Опубліковано: 15.06.2004
Автори: НАРАНДЬЯ Амалія, Дєрек Марко, Павловіч Дражен, ЛОПОТАР Невенка
МПК: C07H 17/08, A61P 31/04, A61K 31/7048, A61P 31/00
Мітки: 3-деоксидесмікозину, спосіб, одержання, похідні
Формула / Реферат:
1. Похідні 3-деоксі-3-оксодесмікозину формули І, (I)деR являє собою СНО або СН(ОСН3)2, R1 і R2 означають Н або ацетил, R3 являє собою Н або ОН, R4 являє собою N(CH3)2 або N-O(СН3)2, лінія - - - являє собою одинарний або подвійний зв'язок, лінія ....... являє собою або подвійний чи одинарний зв'язок, і лініяявляє собою подвійний чи одинарний зв'язок, і похідні 3-деокси-2,3-дидегідродесмікозину формули II,...
Спосіб одержання амінометильних похідних 3-алкілхінолін-4-онів

Номер патенту: 34400
Опубліковано: 11.08.2008
Автори: Подольський Ілля Миколайович, Гриценко Іван Семенович, Зубков Вадим Олексійович
МПК: C07D 215/00
Мітки: 3-алкілхінолін-4-онів, амінометильних, одержання, спосіб, похідних
Формула / Реферат:
1. Спосіб одержання амінометильних похідних 3-алкілхінолін-4-онів загальної формули,де R=C3H7 або С6Н5, що включає утворення і виділення галогенпохідного проміжного продукту реакції з подальшою його взаємодією з аміном у рідкому середовищі полярного апротонного розчинника, який відрізняється тим, що як галогенпохідний проміжний продукт реакції одержують...
Спосіб одержання 2-флуоренілгліцидилового етеру

Номер патенту: 15250
Опубліковано: 15.06.2006
Автори: Гетьманчук Юрій Петрович, Шолудченко Людмила Іванівна, Гуменюк Людмила Миколаївна
МПК: C07D 303/00
Мітки: етеру, одержання, спосіб, 2-флуоренілгліцидилового
Формула / Реферат:
Спосіб одержання 2-флуоренілгліцидилового етеру формули ,що включає використання 2-оксифлуорену, який відрізняється тим, що оксифлуорен вводять у взаємодію з епіхлоргідрином і твердим лугом в спиртовому середовищі.
1,3-дизаміщені сечовини та спосіб їх одержання

Номер патенту: 61125
Опубліковано: 17.11.2003
Автори: Какалік Іван, Земанек Маріан, Шмаховскі Венделін, Фаберова Вєра, Шмідтова Людміла, Оремус Владімір
МПК: A61K 31/4409, C07C 275/36, A61P 43/00, A61K 31/4406, A61K 31/4402, C07D 213/75, C07C 323/44, A61K 31/17, C07D 213/74, A61P 3/06
Мітки: сечовини, одержання, 1,3-дизаміщені, спосіб
Формула / Реферат:
1. 1,3-Дизаміщені сечовини загальної формули І, IдеX = O, S,R2 = NO2, NH2,якщо R1 означає 2-фторфеніл, 2,4-дифторфеніл, 2,5-дифторфеніл, 2,6-дифторфеніл, 2-хлорфеніл, 2,3-дихлорфеніл, 2,6-дихлорфеніл, 3,5-дихлорфеніл, 2-метилфеніл, 4-метилфеніл, 2,4-диметилфеніл, 2,6-диметилфеніл, 3,5-диметилфеніл, 2,6-ди(метилетил)феніл, 2-трифторметилфеніл, 3-трифторметилфеніл, 4-трифторметилфеніл, 2-піридил, 3-піридил,...
Спосіб одержання олігомерних сульфідів

Номер патенту: 51119
Опубліковано: 15.11.2002
Автори: Коваль Іван Васильович, Олійник Тетяна Григорівна, Гринько Олександр Сергійович
МПК: C08G 75/00
Мітки: одержання, спосіб, сульфідів, олігомерних
Формула / Реферат:
Спосіб одержання олігомерних сульфідів конденсацією галогенопохідних, який відрізняється тим, що дигалогенопохідні взаємодіють із ізотіуронієвими солями в присутності 10 %-го розчину гідрооксиду натрію.