Спосіб одержання піранохінолінів або їх фармацевтично прийнятних солей





Формула / Реферат
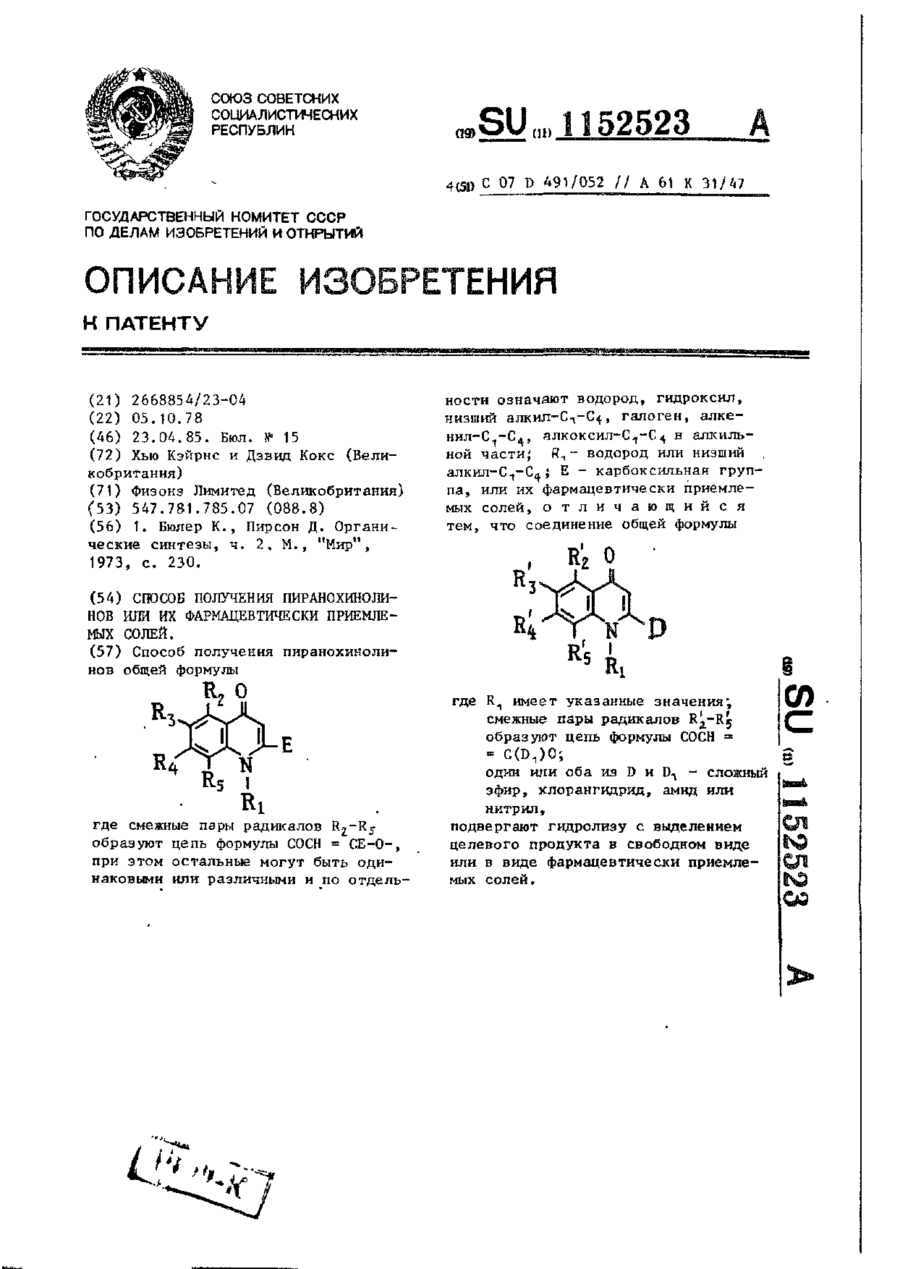
Способ получения пиранохинолинов общей формулы
где смежные пары радикалов R2—R5 образуют цепь формулы СОСН == СЕ-О-, при этом остальные могут быть одинаковыми или различными и по отдельности означают водород, гидроксил, низший алкил - С1-С4, галоген, алкенил - С1-С4, алкоксил - С1-С4 в алкильной части; r1 — водород или низший алкил - С1-С4; Е — карбоксильная группа, или их фармацевтически приемлемых солей, отличающийся тем, что соединение общей формулы
где r1 имеет указанные значения; смежные пары радикалов R'2 — R'5 образуют цепь формулы СОСН = C(D1)О; один или оба из D и D1 — сложный эфир, хлорангидрид, амид или нитрил, подвергают гидролизу с выделением целевого продукта в свободном виде или в виде фармацевтически приемлемых солей.
Текст
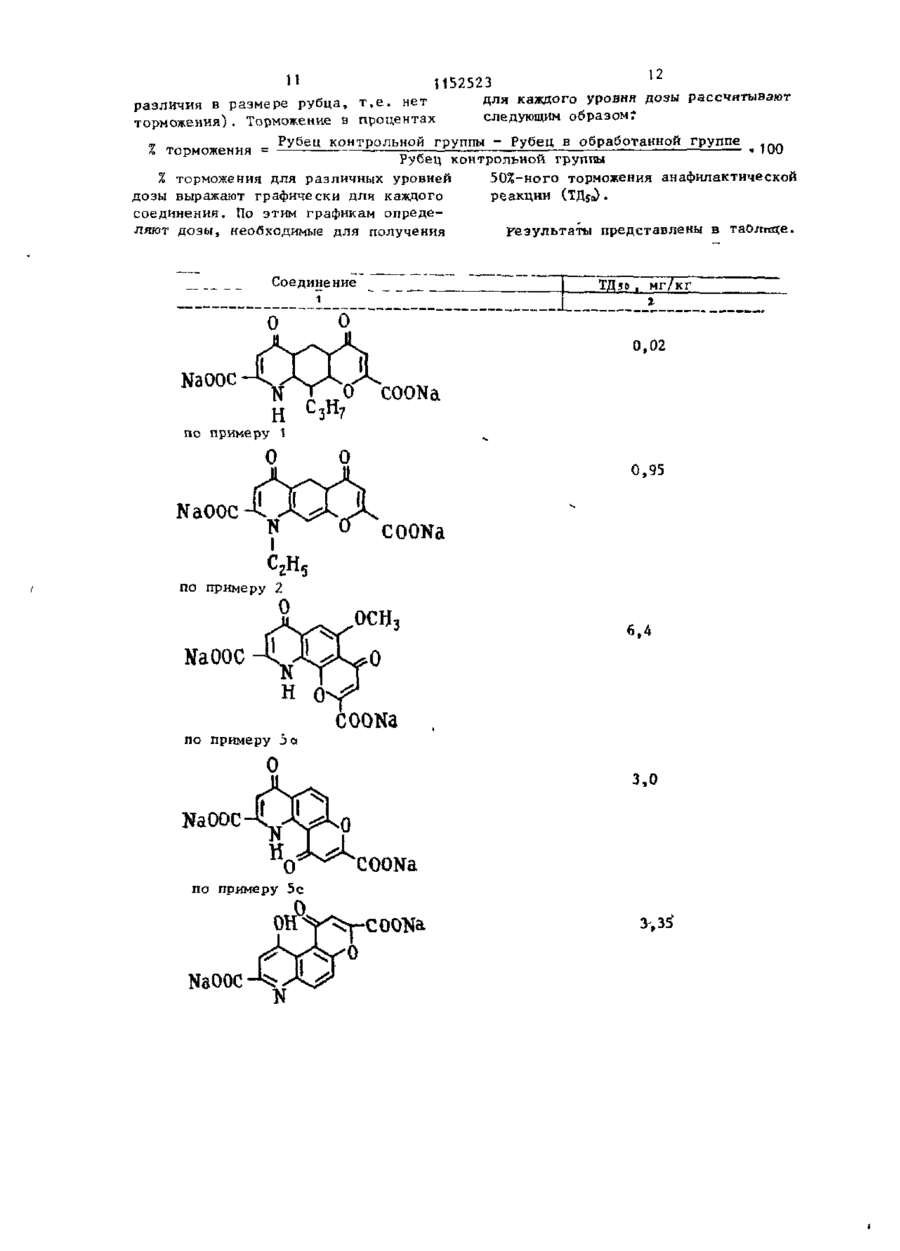
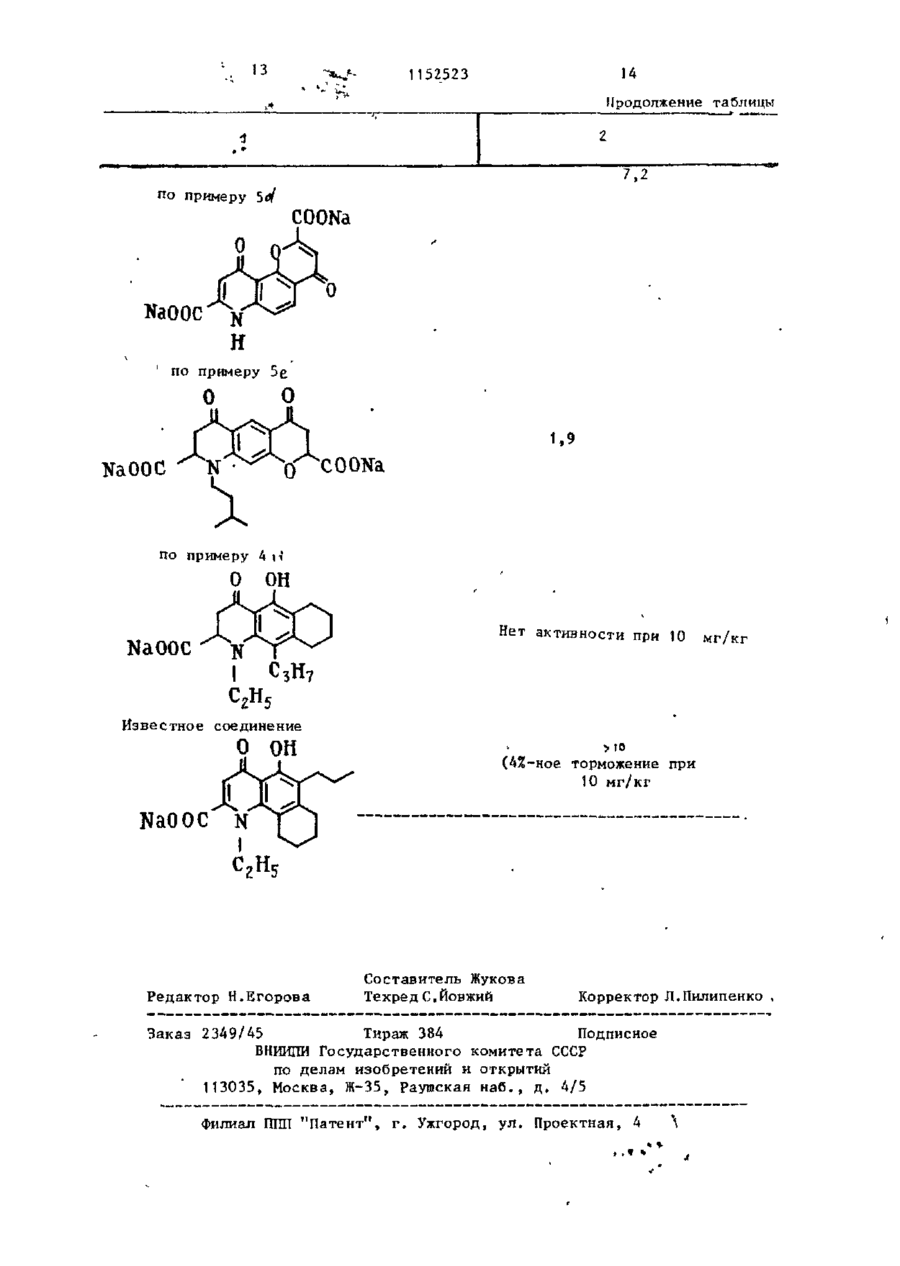
Способ получения пиранохинолинов общей формулы R, О где смежные пары радикалов R^-Rj образуют цепь формулы С С = СЕ-О-, ОН при этом остальные могут быть одинаковыми или различными и по отдель '>** ности означают водород, гидроксил, низший алкил-Ст-С^, галоген, алкенил-С - С . , алкоксил-С^-Г.4 в алкильной части; R., - водород или низший алкил-С,-(Ц; Е - карбоксильная груп па, или их фармацевтически приемлемых солей, о т л и ч а ю щ и й с я тем, что соединение общей формулы R где Rn имеет указанные значения; смежные пары радикалов R^-R^ образуют цепь формулы С С = ОН = C(D 1 )0; один или оба из D и Ьл - сложный эфир, хлорангкдрид, амид или нитрил, подвергают гидролизу с выделением целевого продукта в свободном виде или в виде фармацевтически приемлемых солей. со 1 1152523 Ї Изобретение относится к способам П р и м е р 1. 4,6-Диоксо-Юполучения новых пиранохинолинов о б -пропил~4Н,бН-пирано(3,2-?)хинолинщей ФОРМУЛЫ -2,8-дикарбоновая кислота. «, З-Ацетамидо-2-аллилоксиацетофеR О нон. 5 4.-Адетамидо-2-оксиацетофенон (19,3 г ) , аллилбромид (12,1 мл) и (I) безводный карбонат калия (21,5 г) перемешивают в сухом диметилформамиде 10 (250 мл) при комнатной температуре в течение 24 ч . Реакционную смесь где смежные пары радикалов R,-R^ сливают в воду и продукт экстрагиобразуют цепь формулы СОСН = СЕ-О-, руют с этилацетатом. Органический при этом остальные могут быть одираствор затем хорошо промывают вонаковыми или различными и по от15 дой, сушат над сульфатом магния и дельности означают водород, гкдрокупаривают досуха. Продукт получасил, низший алкил-С^-С^ , галоген, ют в виде шлифованного окрашенного алкенил-С 1 -С + , алкоксил с С , - С 4 в твердого вещества (20,5 г ) . Структуалкильной части,И^ - водород или ра продукта подтверждена ЯМР- и низший алкил-С^-С^, Е - карбоксиль- 20 масс-спектроскопией. ная группа, Ъ 4-Ацетамндо-3-аллил-2-оксиацетоили их фармацевтически приемлемых фенон. солей, обладающих биологической акУказанный аллиловый эфир (18,4 г) тивностью. нагревают при 200-210°С в течение Известна реакция образования 25 А ч . 17,1 г термически перегруппикарбоксильной группы путем гидролированного продукта этой стадии полуза сложноэфирноЙ группы, соответстчают в виде коричневого твердого в е вующего хлорангидрида амида или щества. Структура подтверждена ЯМРнитрила. Условия проведения реакции и масс-спектроскопией. зависят от характера исходных соедиt, b-Ацетамидо-2-ок си-3-пропил-аце30 нений D ] . тофенон. Цель изобретения - синтез новых Продукт стадии Ь(17 г) растворяпиранохинолинов, обладающих ценными ют в ледяной уксусной кислоте и гидфармакологическими свойствами. рируют в присутствии катализатора Поставленная цель достигается Адамса до тех пор, пока не прекратитпредлагаемым способом получения со- 35 ся принятие водорода. Катализатор единений общей формулы I , основанный фильтруют через кизельгур, и фильтна известной реакции гидролиза и рат упаривают с получением 13,0 г заключающимся в том, что соединение почти бесцветного твердого вещества. формулы ( Масс- и ЯМР-спектры подтвердили 40 структуру продукта. R, О d. Этил~7-ацетамидо-4-оксо-8-пропил-4Н-1-бензопиран-2- ь карбоксилат. 45 где R,, имеет указанные значения; смежные пары радикалов R^-H^ образуют цепь формулы СОСН = 50 один или оба из D и D1 - сложный эфир, хлорангидрид, амид или нитрил , 55 подвергают гидролизу с выделением целевого продукта в свободном виде или в виде фармацевтически приемлемых солей. Смесь диэтилоксалата (19,3 г, 17,9 мл) и продукт стадии с (12,4 г) в сухом этаноле (100 мл) добавляют к перемешанному раствору этоксида натрия в этаноле,полученного путем растворения натрия (6,1 г) в сухом э т а ноле (200 м л ) . Реакционную смесь нагревают с обратным холодильником 3 ч и затем сливают в разбавленную соляную кислоту и хлороформ. Слой хлороформа отделяют, промывают водой и сушат. Раствор упаривают с получением коричневого твердого в е щества, которое растворяют в ттаноле (300 м л ) , содержащем концентри З 1152523 рованную соляную кислоту (3 мл) , и ,• « Найдено,7: С 6?,0; Н 5,1;N 3,7. все это нагревают с обратным холодильником 1 ч. Реакционную смесь Вычислено,%: С 6 2 , 3 , Н 4 , 9 ; N 3 , 6 . сливают в зоду, а продукт экстраги% 4,6-Диоксо-10-пропил-4Н,6Н-пира%. руют в этилацетате, загем промывают 5 но(3,2-$)хиноли:і-2,8-дикарбоновая водой и сушат .Растворитель упаривают кислота. с получением 10 г липкого твердого веУказанный бис-эфир (2,5 г) н а г р е щества ,которое имеет структуру ожидаевают с обратным холодильником с бимого вещества (данные масс- и ЯМРкарбонатом «а-'рил (1,64 г) в этаноле -спектроскопии). to (100 мл) и воде (50 мл) в течение 5. Этил-7-амино-4-оксо-8-пропил~4Н1,5 ч. Все это сливают в воду и под- 1-бензогшран-2-карбоксила г. кисляют с осаждением желатинового Раствор амида стадии d (10 г) в твердого вещества. Его собирают при этаноле (300 мл), содержащий конпомощи фильтрации, нагревают с обратцентрированную соляную кислоту 15 ным холодильником с этанолом, и про(5 мл), нагревают с обратным холодукт отделяют центрифугированием fl дильником 8 ч. Реакционную смесь (1,4 г) т . п л . 303-304 C ( р а з л . ) . разбавляют водой и экстрагируют в Структура продукта подтверждена массэтилацетате. Экстракт промывают вои ЯМР-спектром. дой, сушат, раствор упаривают с по- 20 К Динатрий-4,6-диоксо-10-пропиллучением темного коричневого полу-4Н,6Н-тшрано(3,2-£)хинолин-2,8 твердого продукта. Его подвергают -дикарбоксилат. хроматографии на силикагеле с исбис-Кислоту стадии i^l ( 1, 35 г) и бипользованием эфира в качестве разкарбонат натрия (0,661 г) в воде бавителя. Получают 4,8 г требуемого 25 (150 мл) нагревают, перемешивают до продукта. Структура подтверждена тех пор, пока раствор станет прозрачмасс- и ЯМР-спектром, т.пл. 84-87°С. ным. Этот раствор фильтруют ,а фильтрат £. 8-Этоксикарбонил-2-метоксикарбосушат замораживанием с получением нил-4,б-диокси-10-пропил-4Н,бН-пира1,43 г требуемой динатриевой соли. но(3,2-£)хинолин. Найдено Д : С 4 6 , 1 ; Н 4,0-, N 2 , 9 . за С 1 7 Н ,.,Ж)^а 2 12,5% Н 2 0 Амииобензопиран стадии е ( 2 , 0 г) ВычисленоД: С 4 6 , 1 ; Н 3 , 8 ; и диметилацетилендикарбоксилат N 3,15. (1,24 г, 1,01 мл) нагревают в этаноП р и м е р 2. А,6-Диоксо-1ле (30 мл) в течение 2 6 ч. Реакцион-этил-10-Т1ропил-4Н , бН-пирано (3 „ 2- %)~ ную смесь охлаждают до 0 °С, нераст- 35 хинолин-2,8-дикарбоновая кислота. воримое желто-коричневое твердое a, 4-(N-Ацетил-W-этил)-амино-2-алвещество собирают при помощи фильтлилоксиацетофенон, рации, промывают небольшим количест4-(Ы-Лцетил-М-этнл)-амино-2-аллилвом этанола и сушат с получением ^ оксйацетофенон (92,6 г ) , аллилбромид 2 , 0 г п р о д у к т а , который я в л я е т с я (51 мл) и безводный карбонат калия смесью малеинового и фумарового эфи(90,4 г) перемешивают в сухом димер о в , полученных путем д о б а в л е н и я тилформамиде (500 мл) в течение по Мишелю амина к а ц е т и л е н у . 17 ч. Реакционную смесь сливают в Смесь эфиров ( 2 , 0 г) о б р а б а т ы в а ю т 4 5 воду и продукт экстрагируют эфиром. Органический раствор -затем промываполифосфорной кислотой (80 мл) и н а ют хорошо водой, сушат над сульфатом гревают на паровой бане при п е р е м е магния и упаривают досуха. Продукт шивании в течение 20 мин. Р е а к ц и о н 'получают в виде масла (102,5 г ) . ную смесь сливают з а т е м на л е д и п е ремешивают с этил а ц е т а т о м . Органичес-50 Структура продукта подтверждена ЯМР- и масс-спектроскопией. кий слой отделяют, промывают водой b. 4-(Ы~Ацетил-Н-этил)-амино-3-про- і и сушат. Р а с т в о р и т е л ь упаривают с пил-2-оксиацетофенон. , > получением 1,6 г желто-оранжевого Аллиловый эфир стадии а (100,5 г) т в е р д о г о в е щ е с т в а . При п е р е к р и с т а л лизации э т о г о т в е р д о г о вещества 5$ нагревают с обратным холодильником в диэтиланилине (300 мл) в течениеиз э т и л а ц е т а т а получают требуемый 3 ч. Реакционную смесь охлаждают, продукт в виде пушистых оранжевых сливают в разбавленную соляную киси г о л о к , т . п л . 187-188 С 6 * 11 52523 лоту и экстрагируют эфиром, который Реакционную смесь охлаждают и упарипромывают разбавленной соляной кисвают досуха с получением ярко-краслотой и затем водой. Органический ного масла. Это масло подвергают раствор экстрагируют 10%-ным растхроматографии на силикагеле на колонвором гидроокиси натрия и затем 5 ке с использованием системы эфир/летподкисляют. Осажденный продукт экстролейный эфир 1:1 в качестве разбарагируют эфиром, который сушат над вителя с получением 19,1 г диметилсульфатом магния. Полученный эфир-1-(4~ацетил-3-окси-2~пропнлфенил)ный раствор упаривают досуха с полу-N-этиламиномалеата, т . п л . 83-87 С С. чением желто-коричневого масла 10 Малеиновый эфир (5 г) нагревают и (78,7 г ) . Это масло является смесью перемешивают в полифосфорной кислоте 4-(N-аце тил-К-э тил)-амино-3-аллштО00 мл) на паровой бане в течение -2-оксиацетофенона и 6-(N-aneTHn-N10 мин. Реакционную смесь охлаждают -этил)-амино-3-аллил-2-оксиацетофеи сливают на смесь ледяной воды и нона. 15 этилацетата. Органический раствор Эту смесь растворяют в этаноле отделяют, промывают водой и сушат (500 мл) и ледяной уксусной кислоте над сульфатом магния. Растворитель (20 мл) и гидрируют в присутствии упаривают досуха с получением бледнокатализатора Адамса до тех пор,"пока желтого твердого вещества. Этот проне прекратится принятие водорода. 20 дукт очищают при помощи жидкой хроКатализатор отфильтровывают через матографии высокого давления с полукизельгур, и фильтрат упаривают до чением 2,6 г соединения этой стадии, коричневого масла (79,9 г ) . Коричнет.пл. 121-123°С. вое масло является смесью и разделяНайдено,%: С 6 5 , 5 ; Н 6,6; N 4 , 2 . ется (при помощи хроматографии с 2 5 жидкостью высокого давления, раствоВычислено,%: С 6 5 , 3 ; Н 6,34; ритель - система эфир/петролейный N 4,23. эфир 1:1) с получением 44,2 г При очистке бледно-желтого твердосоединения этой стадии и 23,8 г го вещества получают метил-6-ацетил30 -1-этил-5-окси-4-оксо-4Н- хинолин-2-2-оксиацетофенона. -карбоксилат. с. 4~М-Этиламино-3-лропил-2-оксиацеє. Диэтил-4,6-диоксо-1-этил-10-пропилтофенон. -4Н,6Н-пирано(3,2-^)хннолин-2,8-дикарбоксилат. Оксикетон стадии d (1,0 г) и ди4-(Ы-Ацетил-И-этил)-амино-3-про- 35 этилоксалат (3,3 мл) в сухом диметилпил-2-оксиацетофенон (44 г) нагреформамиде (25 мл) добавляют в эфир, вают с обратным холодильником в промытый 50%-ным гидридом натрия 48%-ном бромистом водороде в ледяной (0,581 г в сухом диметилформамиде уксусной кислоте (100 м л ) , ледяной уксусной кислоте (500 мл) и воде 40 20 м л ) , и реакционную смесь перемешивают 4 ч. Реакционную смесь сливают (20 мл) в течение 6 ч . Реакционную в воду, подкисляют и экстрагируют смесь сливают на ледяную воду и экстэтилацегатом,который затем промывают рагируют этилацетатом, который проводой и сушат над сульфатом магния. мывают водой, раствором бикарбоната Растворитель упаривают досуха с полунатрия, затем опять водой и сушат чением масла, которое растворено в над сульфатом магния. Органический этаноле (100 мл) ,и добавляют концентрастворитель упаривают досуха с порированную соляную кислоту (нескольлучением продукта этой стадии в ко к а п е л ь ) . Раствор нагревают с обвиде красного масла (34 г ) . Структу50 ратным холодильником в течение 0,5 ч , ра подтверждена ЯМР- и масс-спектохлаждают, сливают в воду и экстрагироскопией. руют этилацетатом, который промывают rf. Метил-6~ацетил-1-эгил~7-окси-4водой и сушат над сульфатом магния. -оксо-8-пропил-4Н-хинолин-2-карбокРастворитель упаривают досуха с посилат. лучением масла, которое отверждается 53 . Амин стадии с ( 1 7 г) и диметилпри растирании с 40-60°-ным петролейацетилендикарбоксилат (11,3 мл) наным эфиром (1,2 г ) . Структура соедигревают в этаноле (300 мл) с обратнения подтверждается ЯМР-спектром. ным холодильником в течение 17 ч . 8 1 152523 (2,З-f)хинолин-2,8-ди-(N-тетразол£. 4,6-Диокси-1-этил-10-пропил-5~ил)карбоксамид. -4Н,6Н-пирано(3,2-$)хинолин-2,8-диvii. 10-Бром-4,6-диоксо-2,8-дикарбоновая кислота. -(тетразол-5-ил)-4Н,6Н-пирано(3,2-^)Указанный бис-эфнр (1,0 г) и бис ХИНОЛИН. карбонат натрия (0,787 г) в этаноле П р и м е р 4. Предлагаемым (85 мл) и воде (32 мл) нагревают в способом могут быть получены также течение 4 ч. Реакционную смесь слиследующие соединения: вают в воду, подкисляют, осадок соі. 7-(3-Метилбутил)-4,10-диокбирают фильтрацией и сушат. Продукт 10 со-4Н,10Н-пирано(2,3-І)хинолин-2,8очищают при помощи растирания в -дикарбоновая кислота, т.пл. 246порошок с кипящим этанолом, затем 248°С ( р а з л . ) . дважды - с кипящим ацетоном. После її, 9-(3-Метилбутил)-4,6-диоксокаждого растирания смесь центрифу-4Н,6Н-лирано(2,3-^)-хинолин-2,8гируют и всплывающую жидкость удаля-дикарбоновая кислота, т . п л . 302ют декантацией. Оставшееся твердое (5 304°С ( р а з л . ) . вещество сушат с получением 0,547 г і і і . Динатрий-7-этил-4,10-диоксотребуемой ди-кислоты в виде желтого порошка, т . п л . 298-300°С(разл.). -4Н,10Н-пирано(2,3-І)хиколин-2,8-дикарбоксилат. Найдено,%: С 61,3, Н 5,0, N 3,6. Найдено,Z: С 4 4 , 1 ; Н 3 . 5 ; N 3,3. С191Ц7№\ 20 Вычислено,%: С 61,5-, Н 4,6; C i 6 H 3 N N a 2 0 7 14,4% Н 2 0 . N 3,79. Вычислено,%: С 4 4 , 1 ; Н 3 , 7 ; $. Динатрий-4,6-диоксо-1-этил-10N 3,2. -пропил-4Н,6Н-пирано(3,2-£)хинолинП р и м е р 5 . Предлагаемым с п о -2,8-дикарбоксилат. 25 бом м о г у т быть п о л у ч е н ы т а к ж е с л е Указанную ди-кислоту (4,098 г ) , дующие с о е д и н е н и я : суспендированную в воде (100 мл), а. 5~Метокси-4,7-диокси-4Н,7Нобрабатывают бикарбонатом натрия -пирано(3,2-Ь)хинолин-2,9-дикарбоно(1,82 г ) . Полученный раствор фильтвая кислота, т.пл. 294-6°С ( р а з л . ) . руют, и фильтрат обрабатывают аце^.4,б-Диоксо-10-(проп-2-енил),гоном до тех пор, пока не произой-4Н,6Н-пирано(3,2-^)хинолин-2,8-дидет полного осаждения продукта. Трекарбоновая кислота, структура подбуемую динатриевую соль фильтруют тверждена ЯМР-спектром. и сушат с получением 3,39 г бледнос. 4,10-Диоксо-4Н,10Н-пираножелтого порошка. (2,3-S)хинолин-2,8-дикарбоновая кисНайдено,%: С 5 1 , 1 , Н 4 , 3 , N 3,0. 35 лота, т . п л . 200°С. C 1 3 H , s N N a 2 0 l - 6 , 9 % H 2 0. J. 10-Окси-1-оксо-1Н-пирано(3,2-і) Вычислено,^: С 5 1 , 1 , Н 4 , 1 ; хинолин-3,8-дикарбоновая кислота, N 3,1. динатриевая соль. НайденоД: С 45,74; Н 2,45;N 3,6. 40 С 1 4 Н TE K'Na 2 0 7 -6,11 H 2 0 П р и м е р 3. Предлагаемым споВычислено,%: С 45,74, Н 2 , 1 ; собом могут быть получены следуюN 3,8. щие соединения: Є. 4,10-Диоксо-4Н,ЮН-пирано і . 5-Этил-4,8-диоксо-10-пропил(2,3-£)хинолин-2,8-дикарбоновая кис-4Н,8Н-пирано(2,3-L)хинолин-2,6лота, динатриевая соль. -дикарбоновая кислота. Найдено,%: С 44,36, Н 2,18, і і . 4,10-ДИОКСО-4Н,ЮН-пираноN 3,54. (2,3-£)хинолин-2,8-дикарбоновая кисV 2H,0 лота. Вычислено,%: С 44,1-, Н 2,37; і і і . 10-Бром-4,6-ДИОКСО-4Н,6НN 3,67. пирано(3,2~ % хинолин-2,8-дикарбоно) І . 1О-Метил-4,6-ДИОКСО-4Н,6Н-пивая кислота. рано(3,2- £) хинолин-2,8-дикарбоновая i v . 5-Окси-4,6-диоксо-10-пропил-4Н,бН-пирано(3,2-1>)хинолин-2 ,8-дикислота, т . п л . 302 С. карбоновая кислота. 55 П р и м е р 6. 4,6-Диоксо-Юv. 4, 9-Диоксо-4Н, 9Н-пирано(2 ,3~О -пропил-4Н,бН-пирано(3,2-^)хинолинхинолин-2,7-дикарбоновая кислота. -2,8-дикарбоновая кислота. v i . 4,10-Диоксо-4Н,10Н-пирано 9 1152523 10 Раствор 4,6-диоксо-10-пропил30 мин для удаления клеток из плазмы крови. Сыворотку собирают и исполь-4Н,6Н-пирано(3,2-^)хинолин-2,8-дизуют для приготовления сыворотки, карбонилхлорида (4,0 г) в безводном содержащей антитела N . b r a a i l i e n s i s . дихлорметане (30 мл) по каплям приОсуществляют пробное испытание на бавляют в воду (200 мл) при 5°С при чувствительность для определения энергичном перемешивании. После з а наименьшего количества сыворотки, вершения добавления перемешивание необходимого для получения рубца продолжают в течение часа. Дихлормена коже у контрольных животных тан удаляют отгонкой в вакууме, и диаметром 2 см» в описанном тесте. указ энное соединение отфильтровывают to Оптимальная чувствительность у крыс в виде бледно-желтого порошка (3,6 г)9 весом 100-130 г может быть получет . п л . ЗО2-ЗО4°С(в р а з л . ) . на при использовании сыворотки с 8 П р и м е р 7. 4,6-ДиоксоН-этилчастями физиологического раствора. -10-пропил-4Н, бН-пирано (3,2-^-) хиноЭтот разбавленный раствор - сыволин--2,8-дикарбоновая кислота. ^ ротка Л антител. 4,6-Диоксо-1-этил-10-пропил-4М t6Hпирано(3,2-%)хинолин-2,8-дикарбоксамид (3,60 г) растворяют в уксусной кислоте (50 мл), содержащей концентрированную бромистоводородную 20 к и с л о т у (48%-ную , 5 м л ) , и н а г р е вают при т е м п е р а т у р е д е ф л е г м а ц и и и перемешивании в т е ч е н и е 24 ч» Р е а к ционную с м е с ь охлаждают и у к а з а н н о е с о е д и н е н и е отфильтровывают в в и д е 25 шлифовального порошка ( 2 , 3 г ) , т.пл. 298-ЗОО°С (с р а з л . ) . П р и м е р 8І 4,6-Диоксо-Ю-ПРОПИЛ-4Н,6Н-пирано(3,2- % хинолин) -2,8-дикарбоновая кислота. 30 4,6-Диок.со~10-пропил-4Н,бН-пиран о ( 3 , 2 - £ • ) х и н о л и н - 2 ,8-динитриЛч (3,75 г) растворяют в уксусной к и с л о т е ( 5 0 м л ) , содержащий к о н ц е н т рированную бромистоводородную к и с л о - 35 ту (48%-ную, 5 м л ) , и н а г р е в а ю т при т е м п е р а т у р е д е ф л е г м а ц и и и перемешив а н и и в т е ч е н и е 24 ч . Реакционную смесь охлаждают и указанное соединение собирают фильтрованием в виде 40 ж е л т о г о порошка ( 2 , 5 г ) ; т , п л . 3 0 1 6 304 С (с р а з л . ) . Результаты испытания на пассивную анафилактическую реакцию кожи (ПАРК) у крыс. 45 Приплод крыс Ч а р л ь з а Р н в е р а Франс а - ф и з о н с а (мужские и женские о с о би) с в е с о м т е л а 1 0 0 - 1 5 0 г з а р а ж а ют подкожно с и н т е р в а л а м и в одну неделю личинками N . b r a s i l i e n s i s в $1 возрастающих дозах (от 2000 до 12000 личинок на животное) для того, чтобы усилить инфекцию. Через 8 недель крыс умерщвляют уколом в сердце,и у каждого животного отби55 рают J5-20 мл крови. Затем образцы крови подвергают центрифугированию со скоростью 3500 об/мин в течение Ан тиге н для ре акции с ан тителами в сыворотке А готовят, удалив червей N . b r a s i l i e n s i s из кишки зараженных крыс, подвергнув центрифугированию гомогенат и собрав всплывшую жидкость. Эту жидкость разбавляют физиологическим раствором до получения содержания протеина 1 мг/мл, 'Этот раствор называют раствором В. Приплод крыс Чарльза Ривера Франса-Физонса весом 100-130 г сенсибилизируют при помощи вкутрикожного введения 0,2 мл сыворотки А в правый бок. Чувствительность повышается в течение 24 ч. Затем крысам внутривенно вводят 1 мл/100 г веса тела смеси раствора В (0,25 мл) раствора голубого красителя Эванса (0,25 мл) и раствора испытываемого соединения (0,5 мл, изменяя процентное содержание активного ингредиента). Испытываемый раствор с каждой концентрацией активного ингредиента вводят 5 крысам. В каждом тесте 5 крыс составляют контрольную группу. Дозы испытываемого соединения выбирают таким образом, чтобы получить диапазон показателей торможения. Через 30 мин после введения раствора В крыс умерщвляют, снимают с них шкуру и выворачивают е е . Интенсивность анефилактической реакции оценивают путем сравнения размера характерных голубых рубцов, образованных проникновением голубого красителя Эвакса со стороны сенспбшшэирования., с размером рубца у контрольных животных. Размер рубца оценивают по шкале от 0 (рубец не определяется, ^ . е . 100%-ное торможение) до 4 (нет l2 H 1152523 различия в размере рубца, т.е. нет для каждого уровня дозы рассчитывают торможения). Торможение в процентах следующим образом: Рубец контрольной группы - Рубец в обработанной группе П ——— — « ]Л ии Рубец контрольной группы 50Х-ного торможения анафилактической % торможения для различных уровней дозы выражают графически для каждого реакции (ТДуо). соединения. По этим графикам опредеРезультаты представлены в таолпце. ляют дозы, необходимые для получения / торможения ~ о 7 Соединение ТД50. мг/кг 1 0,02 NaOOC 0" C00Na по примеру 1 о 0,95 NaOOC соока по примеру 2 6,4 cooNa по примеру За О NaOOC 3,0 О COONa по примеру 5с 0 он NaOOC С00№ 3-.35 13 14 1152523 Продолжение таблицы 7.2 по примеру COONa О по примеру 5е о о 1,9 NaOOC по примеру 4 її О ОН Нет активности при 10 NaOOC мг/ кг Известное соединение о он Редактор Н.Егорова (4%-ное торможение при 10 мг/кг Составитель Жукова Техред С.Йовжий Корректор Л.Пилипенко Заказ 2349/45 Тираж 384 Подписное ВНИИПИ Государственного комитета СССР по делам изобретений и открытий 113035, Москва, Ж-35, Раушская н а б . , д . 4/5 Филиал ППП " П а т е н т " , г. Ужгород, ул. Проектная, 4 \
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparation of pyranoquinolines or pharmaceutical acceptable salts thereof
Автори англійськоюKejrns Khyu, Koks Devid
Назва патенту російськоюСпособ получения пиранохинолинов или их фармацевтически приемлемых солей
Автори російськоюХью Кейрнс, Дэвид Кокс
МПК / Мітки
МПК: C07D 491/052
Мітки: фармацевтично, піранохінолінів, одержання, солей, спосіб, прийнятних
Код посилання
<a href="https://ua.patents.su/8-1784-sposib-oderzhannya-piranokhinoliniv-abo-kh-farmacevtichno-prijjnyatnikh-solejj.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання піранохінолінів або їх фармацевтично прийнятних солей</a>
Спосіб одержання цис, ендо-2-азабіцикло-/3,3,0/октан-3-карбонових кислот або їх кислотноадітивних солей

Номер патенту: 1780
Опубліковано: 25.10.1994
Автори: Хайнсйорг Урбах, Бернвард Шелькенс, Рольф Гайгер, Фолькер Тєєтц, Райнхард Беккер
МПК: A61K 31/40, C07C 229/22, C07C 229/36, A61P 43/00, C07D 451/02, A61K 38/00, C07K 1/02, A61K 31/403, C07C 229/28, C07K 5/06, C07K 14/81, C07C 229/16, A61P 9/12, C07D 209/52, A61K 38/55, A61K 31/405
Мітки: кислот, солей, одержання, спосіб, кислотноадітівних, цис
Формула / Реферат:
Способ получения производных цис, эндо-2-азабицикло-(3,3,0)-октан-3-карбоновых кислот общей формулыR2 — Н, С1-4 — алкил или бензил; Y — Н или ОН; Z — Н или Y и Z— вместе образуют кислород; Х — фенил, или их кислотно-аддитивных солей, отличающийся тем, что соединение общей формулыгде X, Y, Z, r1 и R2 имеют указанные значения, за исключением R2 — Н, подвергают взаимодействию с соединением общей...
Спосіб одержання кристалічних солей ацетоацетамід-n-сульфофториду

Номер патенту: 1782
Опубліковано: 25.10.1994
Автори: Дітер Ройшлинг, Адольф Линкис
МПК: C07C 307/00, A23L 1/236, C07B 61/00, B01J 27/00, B01J 27/20, C07C 67/00, C07D 291/00, C07C 303/00, C07C 301/00, C07C 311/00
Мітки: кристалічних, солей, одержання, спосіб, ацетоацетамід-n-сульфофториду
Формула / Реферат:
Способ получения кристаллических солей ацетоацетамид-N-сульфофторида общей формулыгде М — литий, натрий или калий, отличающийся тем, что дикетен формулыподвергают взаимодействию с аминосульфоф-торидом формулы H2NSO2F и с карбонатом или бикарбонатом лития или натрия, или калия в среде ацетона, или ацетонитрила, или диметилформа-мида при температуре от—10 до 0 °С в течение 15—90 мин, а затем при 30—50 °C в течение...
Спосіб одержання трифторуксусної кислоти або її солей

Номер патенту: 791
Опубліковано: 15.12.1993
Автори: Тітов Володимир Євгенович, Кошечко Вячеслав Григорович, Седнєв Дмитро Вікторович, Походенко Віталій Дмитрович
МПК: C25B 3/00, C07C 53/18
Мітки: солей, спосіб, трифторуксусної, кислоти, одержання
Формула / Реферат:
Формула изобретенияСпособ получения трифторуксусной кислоты или ее солей путем электролиза смеси галогентрифторметана и диоксида углерода в присутствии четвертичной аммониевой соли в апротонном полярном растворителе в ячейке с катодом из инертного материала, отличающийся тем, что, с целью упрощения процесса, в качестве галогентрифторметана используют бромтрифторметан, в качестве анода - металл, способный к анодному растворению в данной...
Спосіб одержання солей трифторметансульфонової кислоти

Номер патенту: 790
Опубліковано: 15.12.1993
Автори: Тітов Володимир Євгенович, Кошечко Вячеслав Григорович, Походенко Віталій Дмитрович, Седнєв Дмитро Вікторович
МПК: C07C 309/00, C07C 303/00
Мітки: одержання, спосіб, трифторметансульфонової, кислоти, солей
Формула / Реферат:
Формула изобретенияСпособ получения солей трифторметансульфоновой кислоты восстановлением смеси бромтрифторметана и диоксида серы в апротонном полярном органическом растворителе с последующим окислением образующегося при этом трифторметансульфината, отличающийся тем, что, с целью увеличения выхода целевого продукта и упрощения процесса, восстановление проводят электрохимически в потенцио- или гальваностатическом режиме в герметичной...
Спосіб одержання 6-метил-3,4-дігідро-1,2,3-оксатіазін-4-он-2,2-діоксиду або його калієвої солі та спосіб одержання амоній ацетоацетамід-n-сульфонатів

Номер патенту: 1779
Опубліковано: 25.10.1994
Автори: Дітер Ройшлинг, Карл Клаус, Адольф Линкис
МПК: C07D 291/00, B01J 31/16, A23L 1/236, B01J 31/00, B01J 31/02, C07C 307/00, C07B 61/00, C07C 303/00
Мітки: солі, одержання, спосіб, ацетоацетамід-n-сульфонатів, калієвої, амоній, 6-метил-3,4-дігідро-1,2,3-оксатіазін-4-он-2,2-діоксиду
Формула / Реферат:
1. Способ получения 6-метил-3,4-дигидро-1,2,3- оксатиазин-4-он-2,2-диоксида или его калиевой соли взаимодействием производного амидосульфокислоты с ацетоацетилирующим средством в среде растворителя, такого как ледяная уксусная кислота, метиленхлорид или диметил-формамид, или их смеси при температуре от 0 до 30 °C, в случае необходимости в присутствии амина в качестве катализатора с последующей циклизацией образующегося ацетоацетамидного...
Попередній патент: Гербіцидний засіб
Наступний патент: Резервуар
Випадковий патент: Пристрій для наплавлення з термоциклюванням