Спосіб отримання похідних акрилової кислоти






Формула / Реферат
Формула изобретения
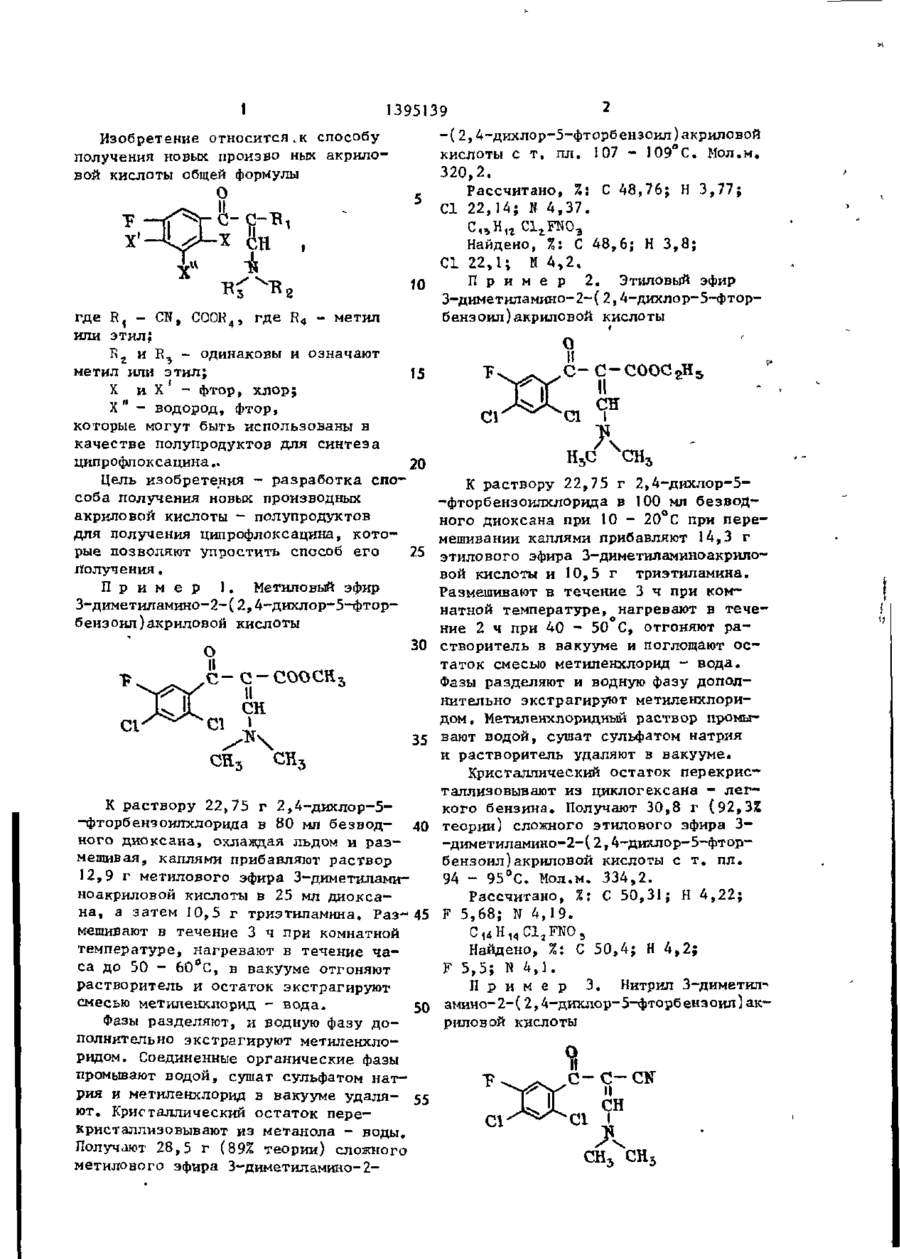
Способ получения производных акриловой кислоты общей формулы
где R1 - СN, - COOR4, где R4 - метил или этил;
R2 и R3 - одинаковы, метил или этил;
X и X’’ - фтор, хлор;
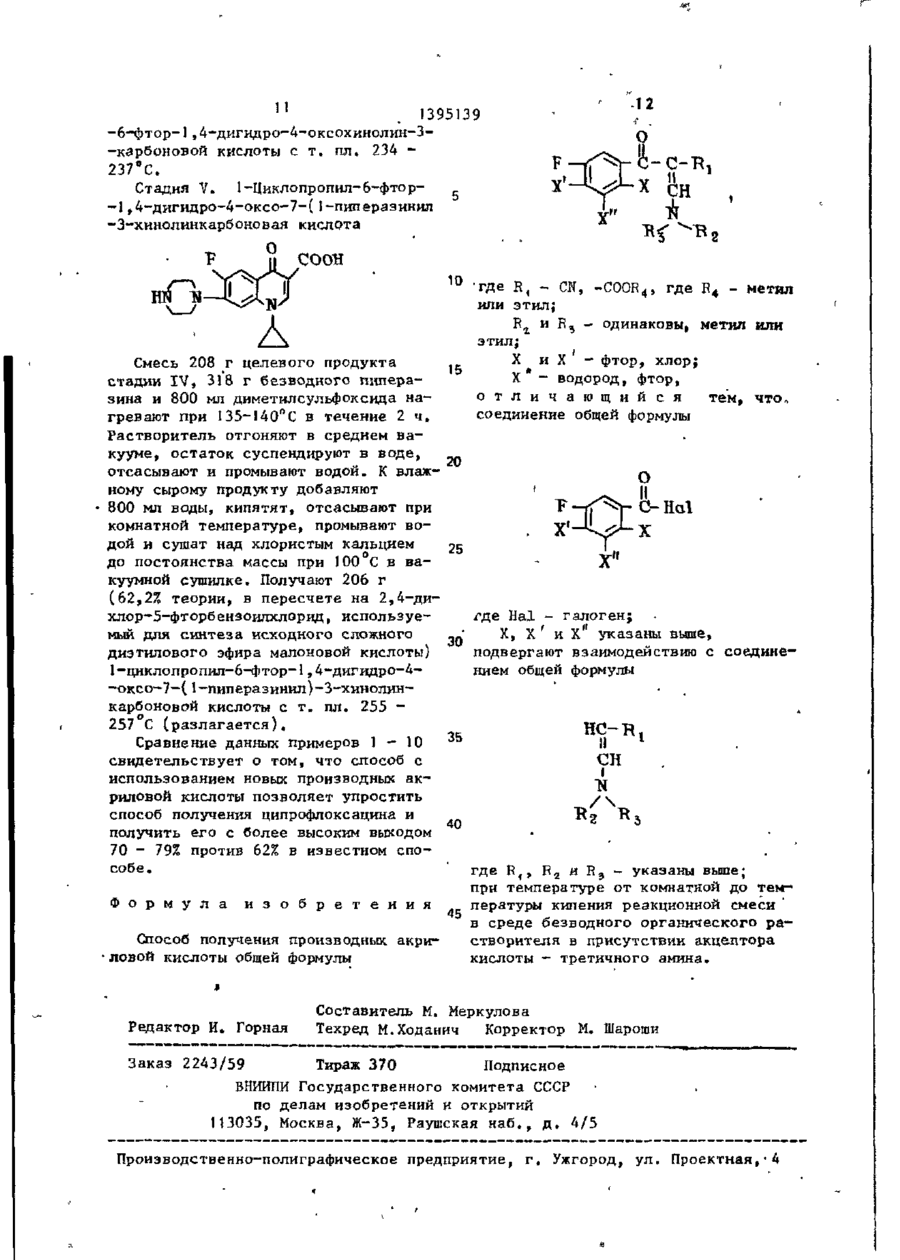
X’ - водород, фтор, отличающийся тем, что, соединение общей формулы
где Hal - галоген;
X, X’ и X’’ указаны выше, подвергают взаимодействию с соединением общей формулы
где R1, R2 и R3 - указаны выше;
при температуре от комнатной до температуры кипения реакционной смеси в среде безводного органического растворителя в присутствии акцептора кислоты — третичного амина.
Текст
Изобретение относится к способу получения произвбдных акриловой кислоты (АК) формулы где R, - QN, -COOR 4 , где R, - метил или этил; Е 2 и R } - одинаковыми означают метил или этил; X и X' - F, С1; X - Н, F, которые являются полу* продуктом для синтеза цкпрофлоксацина. Получение целевых соединений ведут из галоидангидрнда и производного амина при температуре от комнатной до температуры кипения реакционной смеси в среде безводного органического растворителя в присутствии акцептора кислоты - третичного амина. Способ обеспечивает повышение выхода до 70-79% (против 62%). СО СО СП 50 РП ч J t 1395139 -(2,4-дихлор-5~фторбензоил)акриловой кислоты с т. пл. 107 - 109°С. Мол.м, 320,2. рассчитано, %: С 48,76; Н 3,77; С1 22,14; К 4 , 3 7 . Изобретение относится.к способу получения новых произво ных акриловой кислоты общей формулы о » п г 3 Найдено, %: С 48,6; Н 3,8; С1 2 2 , 1 ; N 4,2. 10 Пример 2. Этиловый эфир 3-диметиламино-2-(2,4-дихлор-5-фторбензоил)акриловой кислоты где R1 - CN, СООН4, где R4 - метил или этил; Т\г H R , - одинаковы и означают метил или этил; 15 X и X - фтор, хлор; Х" - водород, фтор, которые могут быть использованы в качестве полупродуктов для синтеза цилрофлоксацина.. 20 Цель изобретения - разработка способа получения новых производных акриловой кислоты - полупродуктов для получения ципрофлоксацина, которые позволяют упростить способ его 25 получения. П р и м е р 1. Метиловый эфир 3-диметиламино-2-(2,4-дихлор-5-фторбензоил)акриловой кислоты 30 35 СЇЇ: К раствору 22,75 г 2,4-дихлор-5-фторбензоилхлорида в 80 мл безвод- 40 ного диоксана, охлаждая льдом и размешивая, каплями прибавляют раствор 12,9 г метилового эфира 3-диметиламиноакриловой кислоты в 25 мл диоксана, а затем 10,5 г триэтиламина. Раз-45 мешивают в течение 3 ч при комнатной температуре, нагревают в течение часа до 50 - 60 С, в вакууме отгоняют растворитель и остаток экстрагируют смесью метиленхлорид - вода. 50 Фазы разделяют, и водную фазу дополнительно экстрагируют метиленхлоридом. Соединенные органические фазы промывают водой, сушат сульфатом натрия и метиленхлорид в вакууме удаля- 55 ют. Кристаллический остаток перекристаллизовывают из метанола - воды. Получают 28,5 г (89% теории) сложного метилового эфира З-диметиламино-2 К раствору 22,75 г 2,4-дихлор-5-фторбензоилхлорида в 100 мл безводного диоксана при 10 - 20°С при перемешивании каплями прибавляют 14,3 г этилового эфира 3-диметиламиноакриловой кислоты и 10,5 г триэтиламина. Размешивают в течение 3 ч при комнатной температуре, нагревают в течение 2 ч при 40 - 50 С, отгоняют растворитель в вакууме и поглощают остаток смесью метиленхлорид - вода. Фазы разделяют и водную фазу дополнительно экстрагируют метиленхлоридом. Метиленхлоридный раствор промывают водой, сушат сульфатом натрия и растворитель удаляют в вакууме. Кристаллический остаток перекристаллизовьгаают из циклогексана - легкого бензина. Получают 30,8 г (92,32 теории) сложного этилового эфира 3-диметиламино-2-(2,4-дихлор-5-фторбензоил)акриловой кислоты с т. пл. 94 - 95°С. Мол.м. 334,2. Рассчитано, Z: С 50,31; Н 4,22; F 5,68; N 4,19. C i 4 H M Cl a FNO s Найдено, %: С 50,4; Н 4,2; F 5,5; И 4 , 1 . Пример 3. Нитрил 3-диметиламино- 2-(2,4-дихлор-5-фторб ензоил)акриловой кислоты Ч с-с-от II сн А сн, 395139 К раствору 22,75 г 2,4-дихлор-5Рассчитано, %: С 5 3 , 0 5 ; Н 5,00; -фторбензоилхлорида в 100 мл безводС1 19,57; N 3,86. ного толуола, охлаждая льдом и разС1ЬН1вС1гГЖ), мешивая, каплями прибавляют 9,6 г Найдено, %: С 52,9; Н 4 , 9 ; 3-диметиламиноакрилонитрила и 10,5 г С1 19,6; N 3 , 8 . триэтиламина. Размешивают в течение П р и м е р 5. Нитрил-3-диметилчаса при комнатной температуре и заамино-2-( 2,3,4,5-тетрафторб ензоил} а к тем в течение 4 ч кипятят с обратным риловой кислоты холодильником. Затем растворитель отгоняют в вакууме и остаток экстрагируют смесью метиленхлорид - вода, Метиленхлоридную фазу промывают водой, сушат сульфатом натрия и концент трируют в вакууме. Кристаллический 15 остаток перекристаллизовывают из этанола. Получают 26,4 г (92% теории) нитрила 3-диметиламино-2~(2,4-дихлор-5-фторбензоил)акриловой кислоты с К раствору 21,25 г 2,3,4,5-тетра- . т. пл. 138 - 139°СС Мол.м. 287,1. фторбенэоилхлорида в 75 мл безводного 20 диоксана, охлаждая льдом, после размеРассчитано, %: С 50,19; Н 3,15; шивания приблизительно при 10 — 15 Ct С1 24,70; N 9,75. каплями прибавляют 9,7 г 3-диметиламиноакрилонитрила и затем 10,5 г триНайдено, %: С 50,3; Н 3,1; 25 этиламина. В течение 4 ч кипятят с С1 24,6; И 9,8. обратным холодильником и затем отгоП р и м е р 4. Этиловый эфир няют растворитель в вакууме и оста3-диэтиламино-2-(2,4-дихлор-5-фторток экстрагируют смесью метиленхлобензоил)акриловой кислоты рид - вода. Фазы разделяют и водную 30 дополнительно экстрагируют метиленI хлоридом. Соединенные органические с-о-соос 2 н 5 фазы промывают водой, сушат сульфаI/ том натрия и метиленхлорид удаляют в С1 С1 вакууме. После перекристаллизации а кристаллического остатка из этанола " 35 получают 23,5 г (86,4% теории) нитС2Н 2Н5 рила 3-диметиламино-2-(2,3,4,5-тетрафторбензоил)акриловой кислоты с К раствору 22,75 г 2,4-дихлор-5т. лл. 149 - 15J°С. Мол.м. 258,2,-фторбензоилхлорида в 80 мл безводРассчитано, %: С 55,81; Н 3,12; ного диоксана, охлаждая льдом и раз- 40 F 29,43; И 5,42. мешивая, каплями прибавляют 1791 г этилового эфира 3-диэтиламиноакриловой кислоты и затем !0,5 г триэтилНайдено, %х С 55,9; Н 3,1; амина. Размешивают в течение часа при F 29,2; N 5,3. ' комнатной температуре, в течение 45 Пример 6. Процесс проводят 45 мин кипятят с обратным холодильнианалогично примеру 1, но в среде ком, отгоняют растворитель в вакууме безводного толуола в присутствии пии затем маслянистый остаток экстраридина» При этом получают 29 г гируют смесью метиленхлорид — вода. (90,6% теории) сложного метилового Фазы разделяют и водный раствор доэфира 3-диметиламино-2-(2,4-дихлорполнительно экстрагируют метиленхло-5-фторбензоил)акриловой кислоты с ридом. Соединенные органические фазы т. пл. 107 - Ю9°С. промывают водой, сушат сульфатом натрия и метиленхлорид удаляют в вагсуПример 7. Процесс проводят уме. Получают 29 г (80,1% теории) аналогично примеру 2, но в среде без55 сложного этилового эфира 3-диэтиламиводного толуола в присутствии пирино-2-(2,4-днхлор-5-фторбензоил)акридина или в среде безводного ацетона ловой кислоты" в качестве коричневого в присутствии пиридина, или в среде масла. Мол.м. 362,2. безводного толуола в присутствии 4 395139 4-метилпиперидина, или в среде безКаОН и кипятят в течение 90 мин. Тепводного толуола в присутствии метиллый раствор фильтруют и промывают морфолина, или в среде безводного водой. При охлаждении льдом фильтрат этилацетата в присутствии триэтилами- г подкисляют до рН I - 2 полуконцентри-: на. рованной соляной кислотой, осадок отсасывают, промывают водой и сушат при При этом целевой сложный этиловый 100°С в вакууме. Получают 26,2 г 7эфир получают выходом соответственно -хлор-1- циклопропил 6-фтор-],4-дигид31,5 г (94,4% теории), или 30,8 г (92,3% теории), или 31,4 г (94,12 Ю ро-4-оксохинолин-З-карбоновой кислоты с т. пл. 234 - 237°С. теории), или 31,2 г (93,5% теории), или 31 г (92,9% теории). Стадия I I I , 1-Циклопропил-б-фторВо всех случаях целевой продукт -1,4-дигидро-4-оксо-7-(1-пиперазинил)имеет т. пл. 94 - 95°С. -3-хинолинкарбоновая кислота (ципроПример 8. Синтез ципрофдок* J J флоксацин) сацина. Стадия I . Сложный этиловый эфир 3-циклопропиламино-2-(2,4-дихлор-5соон -фторбензоил)акриловой кислоты. > О Я Смесь 26,2 г целевого продукта стастадии I I , А г безводного пипераэика О и 100 мл диметилсульфоксида нагрева25 < ют при 135 - 140°С в течение 2 ч . РаСмесь 30,8 г сложного этилового створитель отгоняют в среднем вакууме , эфира 3-диметиламино-2-(2,4-дихлоростаток суспендируют в воде, отсасы-5-фторбензоил)акриловой кислоты и вают н промывают водойм К влажному 6,5 г циклопропиламина в среде 100 мл 30 сырому продукту добавляют 100 мл воды, кипятят, отсасывают при комнаттолуола кипятят в течение 1 ч, после ной температуре, промывают водой и чего выделение газа прекращается. сушат над хлористым кальцием при Толуол отгоняют в вакууме и твердый 100 С до постоянства веса в вакуумной остаток кристаллизуют из легкого бенсушилке. Получают 26,1 г (79,2% теозина. Получают 30 г сложного этиловории в пересчете на 2,4-дихлор-5-фторго эфира 3-циклопропиламино-2-(2,4бензоилхлорид, используемый для син-дихлор-5-фторбензоил)акриловой кистеза исходного сложного этилового лоты с т. пл. 90 - 91°С. эфира стадии 1)1-циклопропил-6-фторСтадия I I . 7-Хлор-1-циклопропил-1,4-дигидро-4-оксо-7-(1-пиперазиннл)-6-фтор-1,4-ДИГИДРО-4-ОКСО-3-ХИНОЛИН-3-хинолинкарбоновой кислоты с т . пл. карбоновой кислоты 255 - 257°С.(разлагается). - О Пример 9. Синтез ципрофлоксацина. СООН Р Стадия I . Нитрил 3-циклопропил45 С1 амино-2- (2,4-дихлор~5-фторб ензоил) акриловой кислоты сн К раствору 30 г целевого продукта стадии І в 100 мл безводного диоксана порциями добавляют 3,25 г 80%-ного 1 гидрида натрия при охлаждении льдом и перемешивании. Затем, размешивают при комнатной температуре в течение 30 мин и при температуре кипения с м е - " си в течение 2 ч. После отгонки диоксана в вакууме остаток суспендируют в 150 мл воды, смешивают с 6,25 г Смесь 26,4 г нитрила 3-диметиламино-2~(2,4-днхлор-5-фторбенз оил)акриловой кислоты и 5,9 г циклопропиламина в 80 мл толуола нагревают с обратным холодильником в течение 30 мин, 395139 8 нил)-3~хинолинкарбоновая кисло і а после чего выделение газа законче(ципрофлоксацин) но. Толуол отгоняют в вакууме и остаток перекристяллиэовывают из смеси этанола с легким бензином. Получают 25,4 г нитрила 3-циклопропилами СООЇІ но-2-С 2,4-дихлор-5-фторбенэоил)акриловой кислоты с т . пл. 94 - 95°С. Стадия I I . Нитрил 7-хлор-і-цикло 1 пропил-6-фтор-1,4-дигидро-4-оксо-3Ї0 -хинолинкарбоновой кислоты Смесь 23,2 г целевого продукта О стадии I I I , 35,5 г безводного пнперазина и 100 мл диметилсульфоксида нагревают при 135 - І40°С в течение 15 2 ч. Растворитель отгоняют в среднем вакууме, остаток суспендируют в воде и промывают водой. К влажному сырому К раствору 25,4 г целевого продук продукту добавляют 100 мл воды, кипята стадии І в 70 мл диметилформамида тят, отсасывают при комнатной темпедобавляют 13,4 г карбоната калия и 20 ратуре, промывают водой и сушат над нагревают при 150°С в течение 3 ч. хлористым кальцием при 100°С до поРеакционную смесь наливают на лед, стоянства веса в вакуумной сушилке. осадок отсасывают и промывают водой. Получают 23,1 г (70,1% теории, в пеПосле сушки в вакууме при 100 С поресчете tia 2,4-дихлор-5-фторбензоиллучают 24,3 г нитрила 7-хлор-1-цикло - 25 хлорид, используемый для синтеза испропил-6-фтор-1,4~дигидро-4-оксо-3ходного нитрила стадии I !-циклопро-хинолинкарбоновой кислоты с т. пл. пил-6-фтор-1,4-дигидро-4-оксо-7-(1239 - 241 °С. -пиперазинил)-3-хинолинкарбоновой . Стадия I I I . 7-Хлор-1-циклопрокислоты с т. пл, 255 - 257°С (разлапил-6~фтор-1,4-дигидро-4~оксо-3-хино 30 гается) , линкарбоновая кислота П р и м е р 10 (сравнительный) в Синтез ципрофлоксацин-а по известному COOK способу. А. Получение исходного сложного 35 диэтилового эфира 2,4~дихлор-5-фторбензоилмалоновой кислоты формулы Смесь 24,3 г целевого продукта стадии I I , 90 мл концентрированной 40 серной кислоты и ПО мл воды нагревают при 130 - 135°С в течение 3 ч . Горячий раствор наливают на лед, ососадок отсасывают, промывают водой и при нагревании растворяют в 250 мл 45 10%-ного натрового щелока и 250 мл воды. Горячий раствор фильтруют, промывают горячей водой и добавлением соляной кислоты ( i l l ) доводят до рН 1 Осадок отсасывают, промывают водой и сушат при 100 С в вакууме. После пе- 50 рекристаллизации из смеси монометилового эфира, гликоля и этанола получают 23,2 г 7-хлор-1-циклопропил-6-фтор-!,4-дигидро-4-оксо-3-хинолин55 карбоновой кислоты с т. пл. 240 241°С. Стадия IV» І-Циклопропил-6-фтор -1,4-дигидро-4-оксо-7-(1-пиперази-_ СООС 2 Н 5 23,3 г магниевых стружек суспен-' днруют в 50 мл безводного этанола. Добавляют 5 мл четыреххлористого углерода и после начала реакции каплями добавляют 160 г сложного диэтило— вого эфира малоновой кислоты, 100 мл абсолютного этанола и 400 мл безводного простого эфира. При этом наблюдается интенсивная флегма. После окончания реакции нагревают с обратным холодильником еще в течение 2 ч , охлаждают до (-5) - (-10) С при помощи сухого льда и ацетона и при этой температуре каплями медленно добавляют раствор 227,5 г 2,4-дихлор-5-фторбензоилхлорида в 100 мл абсолютного 10 9 13951 39 простого эфира. Размешивают при 0 В качестве остатка получают 260,4 г (-5)°С в течение 1 ч , смеси дают насырого сложного этилового эфира 2— греться до комнатной температуры в -(2 | 4-дихлор-5-фторбензоил -3-этокситечение ночи н затем при охлаждении _ акриловой)кислоты. льдом добавляют смесь 400 мл ледяной Стадия I I I , Сложный этиловый эфир воды и 25 мл концентрированной с е р 2-(2,4-дихлор-5-фторбензоил)-3-цикпоной кислоты. Фазы разделяют и два пропиламиноакриловой кислоты раза экстрагируют простым эфиром. О Объединенные эфирные экстракты про- ю И мывают насыщенным раствором хлористоС - С-СООС2Н5 го натрия, сушат над сульфатом натрия СН и растворитель отгоняют в вакууме. С1 Получают 349,5 г сырого сложного д и HN этилового эфира 2,4-дихлор-5-фторбен- 15 зоилмалоновой кислоты. К раствору 260,4 г целевого проВ. Синтез ципрофлоксацина. дукта стадии I I в 750 мл этанола Стадия I , Сложный диэтиловый каплями добавляют 45 г циклопропилэфир 2,4-дихлор-5-фторбензоилуксусамина при охлаждении льдом и переменой кислоты 20 шивании. После окончания экзотермической реакции перемешивают еще 1 ч при комнатной температуре, растворитель отгоняют в вакууме и остаток - сн 2 соос 2 н 5 перекристаллизовывают из смеси цик2 5 логексана с простым петролейным эфиЭмульсию 349,5 г сырого сложного ром. Получают 239,5 г сложного этилоэфира согласно А в 500 мл воды смешивого эфира 2-(2,4-дихлор-5-фторбенвают с 1,5 г п—толуолсульфокислоты. зоил)-3-циклопропиламиноакриловой При интенсивном размешивании нагревакислоты с т . пл. 89 - 90°С. ют с обратным холодильником в течение Стадия IV. 7-Хлор-1-циклопропил3 ч , охлажденную эмульсию пять раз 30 -б-фтор-1,4-ДИГИДРО-4-ОКСО-3-ХИНОЛИНэкстрагируют хлористым метиленом, карбоновая кислота объединенные фазы хлористого метилена промывают насыщенным раствором хлоО ристого натрия, сушат над сульфатом натрия и растворитель отгоняют в в а - 35 кууме. После фракционирования остатка в среднем вакууме получают 218 г сложного этилового эфира 2,4-дихлор-5-фторбензоилуксусной кислоты с т . кип, 40 при 0,09 мбар 127 - 142°С. К раствору 239,5 г целевого проСтадия I I . Сложный диэтиловый дукта стадии I I I в 700 мл безводноэфир 2-(2,4-дихлор-5-фторб енз о и л ) - 3 го диоксана порциями добавляют 25,8 г -этоксиакриловой кислоты 80%-ного гидрида натрия при охлажде -4 ід. с~с-соос г н 5 СІ С1 СН I - ос 2 н 5 Смесь 218 г целевого продукта с т а дии I , 172 г сложного этилового эфира о-муравьиной кислоты и 192 г ацетангидрида нагревают при 150 С в течение 2 ч . Летучие компоненты последовательно отгоняют в вакууме, созданном водоструйным насосом, и в среднем вакууме при температуре ванны 120°С. 45 нии льдом и размешивании. Затем р а з мешивают при комнатной температуре в течение 30 мин и при температуре к и пения смеси в течение 2 ч . Диоксан отгоняют в вакууме, остаток суспендируют в 1000 мл воды, добавляют 50 г 50 натрового щелока и нагревают с обратньм холодильником в течение 90 мин. Теплый раствор фильтруют и промывают водой. Затем при охлаждении льдом рН среды доводят до 1 - 2 добавлением полуконцентрированной соляной к и с лоты, осадок отсасывают, промывают водой и сушат в вакууме при 100°С. Получают 208 г 7-хлор-1-циклолропнл 11 . 1395139 -6-фтор-1,4-ДИГИДРО-4-ОКСОХИНОЛИН-3-кзрбоновой кислоты с т . пл, 234 237°С. Стадия V. 1-Циклопропил-б-фтор-1,4-дигидро-4-оксо-7-(1-пипераэинил -3-хинолинкарбоновая кислота О соон 10 •где R, - CN, -COOR4, где R 4 - метил или этил; Г\_ И Л , - одинаковы, метил или этил; X и X ' - фтор, хлор; X — водород, фтор, о т л и ч а ю щ и й с я тем, что* соединение общей формулы Смесь 208 г целевого продукта стадии IV, 318 г безводного пиперазина и 800 мл диметилсульфоксида нагревают при І35-140°С в течение 2 ч . Растворитель отгоняют в среднем в а кууме, остаток суспендируют в воде, отсасывают и промывают водой. К влаж- 20 ному сырому продукту добавляют 800 мл воды, кипятят, отсасывают при комнатной температуре, промывают в о дой и сушат над хлористым кальцием 25 до постоянства массы при 100 С в в а куумной сушилке. Получают 206 г (62,2% теории, в пересчете на 2,4-дихлор-5-фторбензоилхлорид, используегде Hal - галоген; мый для синтеза исходного сложного X, X ' и х" указаны выше, диэтилового эфира малоновой кислоты) 30 подвергают взаимодействию с соедине1-циклопропил-6-фтор-1,4-дигидро-Днием общей формулы —оксо—7—(1-пиперазинил)-3-хинолинкарбоновой кислоты с т . пл. 255 257 С ( р а з л а г а е т с я ) . HC-R Сравнение данных примеров 1-10 I» свидетельствует о том, что способ с сн использованием новых производных акI риловой кислоты позволяет упростить "N способ получения ципрофлоксацина и получить его с более высоким выходом 40 70 - 79% против 62% в известном способе. где R f , R 2 и R 3 - указаны выше; при температуре от комнатной до темФ о р м у л а и з о б р е т е н и я пературы кипения реакционной смеси в среде безводного органического раСпособ получения производных акристворителя в присутствии акцептора ловой кислоты общей формулы кислоты - третичного амина. Редактор И, Горная Составитель М. Меркулова Техред М.Ходанич Корректор М. Шароши Заказ 2243/59 Тираж 370 Подписное БНИИПИ Государственного комитета СССР по делам изобретений и открытий 113035, Москва, Ж-35, Раушская наб., д. 4/5 Производственно-полиграфическое предприятие, г. Ужгород, ул. Проектная,-4
ДивитисяДодаткова інформація
МПК / Мітки
МПК: C07C 231/00, C07C 253/30, C07C 237/20, C07C 69/618, C07C 229/36, C07C 229/34, C07C 67/00, C07C 227/00, C07C 237/16, C07C 253/00, C07D 295/04, C07C 255/42, C07D 295/14
Мітки: похідних, отримання, кислоти, спосіб, акрилової
Код посилання
<a href="https://ua.patents.su/8-7018-sposib-otrimannya-pokhidnikh-akrilovo-kisloti.html" target="_blank" rel="follow" title="База патентів України">Спосіб отримання похідних акрилової кислоти</a>
Спосіб отримання похідних акрилової кислоти або їх стереоізомерів

Номер патенту: 6299
Опубліковано: 29.12.1994
Автори: Кевін Б'ютімент, Майкл Джон Бушелл, Вів'єнн Маргарет Ентоні, Поль Де Френ, Крістофер Річард Ейлс Годфрі, Джон Мартін Клаф
Мітки: акрилової, похідних, отримання, кислоти, спосіб, стереоізомерів
Формула / Реферат:
Формула изобретенияСпособ получения производных акриловой кислоты общей формулы где R1 и R2 - оба метил;X - в ортоположении по отношению к акрилатной группе - водород, фтор, хлор, бром, С1-С6-алкил, фенил-С1-С4-алкил, в котором фенильная группа возможно замещена на фтор, хлор или метокси-, диметоксиметил, этилендиметоксиметил, трифторметил, феноксиметил, возможно...
Спосіб одержання похідних акрилової кислоти або їх ізомерів

Номер патенту: 6305
Опубліковано: 29.12.1994
Автори: Ян Фергусон, Пол Дефрейн, Вів'єнн Маргарет Ентоні, Патрік Джелф Кроулі, Джон Мартін Клаф, Майкл Гордон Хічінгс, Крістофер Річард Ейлз Годфрі
МПК: C07D 239/93, A01N 43/54, C07D 239/40, C07D 213/70, C07D 215/24
Мітки: похідних, кислоти, спосіб, ізомерів, акрилової, одержання
Формула / Реферат:
Способ получения производных акриловой кислоты общей формулыгде W - пирвдиинп или пиримидинил, возможно замещенные галогенами С1-С4-алкилом, который, в свою очередь, может быть замещен галогеном, фенвлом, С1-С4-алкоксигруппой, феноксигруппой, которая, в свою очередь, может быть замещена 1-метоксикарбонил-2-метоксиэтенилом, галогеном, циано- или нитрогругаюй, циано-, нитро-, амино-, формамидогруппой или N-оксидной группой, или...
Спосіб отримання похідних 1,5-дифенилпіразол-3-карбонової кислоти

Номер патенту: 5583
Опубліковано: 28.12.1994
Автори: Ханс Мозер, Вернер Фьорі, Беат Бьонер
Мітки: спосіб, 1,5-дифенилпіразол-3-карбонової, кислоти, отримання, похідних
Формула / Реферат:
Формула изобретенияСпособ получения производных 1,5-дифенилпиразол-3-карбоновой кислоты общей формулыгде Rd - галоген, С1-С5 - алкил;Rb - галоген, С1-С5 - алкил, метокси;n - 0, 1 или 2;R1 - водород или физиологически переносимый растением катион металла или аммония, С1-С18 - алкил, незамещенный или замещенный одним галогеном, нитро, С1-С4 - алкоксикарбонилом, триметилсилилом, амино, ди (С1-С4) –...
Спосіб отримання складного 1-етоксікарбонілоксіетілового ефіру 6-[d-(-)-2-аміно-2-фенілацетамідо]-пеніциланової кислоти у вигляді його адітивної солі з галоідводневою кислотою

Номер патенту: 3638
Опубліковано: 27.12.1994
Автори: Роберт Грахем Тисон, Луіджі Ратті, Дерек Регінальд Пальмер
МПК: A61P 31/04, C07D 499/00, C07D 501/00, A61K 31/43, C07C 69/96
Мітки: отримання, кислоти, галоідводневою, 6-[d-(-)-2-аміно-2-фенілацетамідо]-пеніциланової, 1-етоксікарбонілоксіетілового, вигляді, солі, спосіб, ефіру, кислотою, складного, адітивної
Формула / Реферат:
Способ получения сложного 1-этоксикарбонилоксиэтилового эфира 6-[D-(-)-2-амино-2-фенилацетамидо]-пенициллановой кислоты в виде его аддитивной соли с галоидводородной кислотой путем взаимодействия соли щелочного металла 6-[D-(-)-2-N-производного 2-фенилацетамино]-пенициллановой кислоты с а-галоиддиэтилкарбонатом формулы I где Наl - хлор, бром или под, в апротронном полярном растворителе...
Спосіб отримання похідних оксимів 1,2,5,6,-тетрагідропірідін-3-карбоксальдегіду

Номер патенту: 5167
Опубліковано: 28.12.1994
Автори: Карла Бонетті, Джуліо Галіані, Фернандо Барцагі, Еміліо Тоя
МПК: A61P 43/00, A61P 25/28, A61K 31/44, A61K 31/445, C07D 211/70
Мітки: спосіб, похідних, отримання, оксимів, 1,2,5,6,-тетрагідропірідін-3-карбоксальдегіду
Формула / Реферат:
Способ получения производных оксимов 1,2,5,6-тетрагидропиридин-3-карбоксальдегида общей формулыгде R1 - метил или 2-пропинил; R-С1-С4-алкил, прямой или разветвленный, возможно замещенный триметилсилильной группой, адамантил, фенил, возможно замещенный одной или двумя группами, выбранными из С1, изопропил, метоксифенилэтил или метилсульфонилэтил, отличающийся тем, что соединение общей формулыгде R1 имеет...
Попередній патент: Спосіб одержання похідних пірідазінона або їх водорозчинних солей з фармацевтично прийнятною кислотою
Наступний патент: Судовий дизельний агрегат
Випадковий патент: Спосіб діагностики та профілактики первинної дисменореї у дівчат пубертатного віку