Спосіб одержання 4,4′-(1-метил-1,2-етандіїл)-біс-(2,6-піперазиндіону)








Формула / Реферат
1. Спосіб одержання сполуки формули (І)
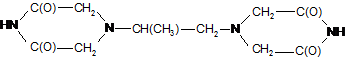 , (I)
, (I)
який відрізняється тим, що тетраестер формули (II)
(ROOCCH2)2N-CHCH3-CH2-N(CH2COOR)2, (II)
де R означає алкіл,
циклізують з аміаком у формаміді.
2. Спосіб за п. 1, який відрізняється тим, що R означає (С1-С3)-алкіл.
3. Застосування сполуки формули (II)
(ROOCCH2)2N-CHCH3-CH2-N(CH2COOR)2 (II)
для одержання сполуки формули (І)
, (I)
де R відповідно означає алкіл, бажано (С1-С3)-алкіл.
4. Спосіб одержання сполуки формули (І)
, (I)
який відрізняється тим, що здійснюють
(a) взаємодію (S)-1,2-діамінопропану або його прийнятної солі з хлороцтовою кислотою;
(b) обробку реакційного продукту, отриманого на стадії (а), у алкіловому спирті сильною кислотою, бажано неорганічною,
(c) циклізацію сполуки формули (II)
(ROOCCH2)2N-CHCH3-CH2-N(CH2COOR)2 (II)
з аміаком у формаміді, де R відповідно означає алкіл.
5. Спосіб за п. 4, який відрізняється тим, що R означає (С1-С3)-алкіл.
6. Спосіб за п. 4 або 5, який відрізняється тим, що перед стадією (с) сполуку формули (II) очищують від неорганічних солей шляхом екстракції органічним розчинником, нерозчинним у воді, з води.
7. Спосіб за п. 4 або 5, який відрізняється тим, що сполуку формули (II) застосовують на стадії (с) без попереднього відділення або очищення.
Текст
Реферат: Даний винахід стосується нового способу одержання сполук формули (І) шляхом кристалізації алкілового естеру тетраоцтової кислоти формули (II) у присутності аміаку та формаміду, а також сполук формули (II), які застосовують у цьому способі. UA 102509 C2 (12) UA 102509 C2 UA 102509 C2 5 10 15 20 25 30 35 40 45 50 Цей винахід стосується нового способу одержання 4,4'-(1-метил-1,2-етандіїл)-біс-(2,6піперазиндіону). Зокрема даний винахід стосується нового способу одержання 4,4'-(1-метил-1,2етандіїл)-біс-(2,6-піперазиндіону) високої якості та виходу. Більш того даний винахід стосується нових проміжних сполук, які застосовують у даному способі. 4,4'- (1-метил-1,2-етандіїл)-біс-(2,6-піперазиндіон) загальної формули (I) Сполука формули (I) існує у формі двох енантіомерів як (S)-(+)-4,4'-(1-метил-1,2-етандіїл)біс-(2,6-піперазиндіон), який також називають Дексразоксан, та як (R) - (-) -4, 4 ' – (1-метил-1,2етандіїл)-біс- (2,6-піперазиндіон), який також називають Леворазоксан, та у формі рацемату, (S, R)-4,4'-(1-метил-1,2-етандіїл)-біс-(2,6-піперазиндіон), який також називають Разоксан. У контексті даного винаходу "сполука формули (I)" або "4,4'-(1-метил-1,2-етандіїл)-біс-(2,6-ппіперазиндіон)", відповідно, відноситься до S-енантіомеру, а рацемат представлено Rенантіомером. Не зважаючи на стереохімію сполука формули (I) має протираковий вплив. Раніше, Sенантіомер сполуки формули (I), Дексразоксан, який є ефективним проти пухлин та інших форм раку та який також застосовують як сінергіст у комбінації з іншими протираковими агентами, має особливе значення. Особливо по відношенню до саркоми, лімфосаркоми та лейкемії, виявили, що Дексразоксан проявляє активність та є особливо ефективним, коли його застосовують разом із Адріаміцином. У попередньому рівні техніки протягом довгого часу було відомо декілька способів одержання сполуки формули (І). Наприклад, у патентах США № 3.941.790 та № 4.275.063, Creighton, описано три способи одержання бісдикетопіперазинів, включаючи і сполуки формули (I). У першому способі, (S)-1,2-діамінопропан взаємодіє із хлороцтовою кислотою для утворення (S)-1,2-діамінопропан-тетраоцтової кислоти. Після цього тетраоцтова кислота взаємодіє із формамідом у атмосфері азоту при підвищеній температурі до утворення відповідної сполуки формули (І). У другому способі отримують тетраоцтову кислоту, як описано вище, та перетворюють у відповідний амід тетраоцтової кислоти шляхом взаємодії із аміаком та наступною циклізацією у поліфосфорну кислоту або фенол шляхом нагрівання. Вважається, що цей спосіб має особливі переваги, коли тетраоцтова кислота має тенденцію до декарбоксилування протягом нагрівання. Третій спосіб представлено взаємодією тетранитрилу з амідом натрію у формаміді та наступне оброблення отриманого продукту хлоридом водню у метанолі. Згідно з Creighton цей альтернативний спосіб має таку перевагу як його здійснення при низькій температурі. Усі ці способи є стерео селективними, тобто усі проміжні сполуки у формі тетраоцтової кислоти, тетраміду або тетранітрилу мають бути наявними у стереохімічній конфігурації, бажаної для сполуки формули (I). Проміжні сполуки, такі як тетраоцтова кислота, які застосовують у вищезгаданих способах, одержують різними способами. Окрім вищезгаданих способів одержання, наприклад, у Британському патенті № 978,724 J.R., Geigy AG, описано спосіб одержання тетраоцтової кислоти, де діаміни взаємодіють з формальдегідом та ціанідом водню для утворення тетранитрилу, який омилюється. У патенті США № 2.461.519, Bersworth et al., описано спосіб одержання 1,2-діамінопропан-тетракарбоксильної кислоти шляхом взаємодії 1,2-діамінопропану із формальдегідом та ціанідом натрію при лужному значенні рН. Головною проблемою одержання сполуки формули (І) є очищення проміжних сполук, яке є дорогим та складним у промисловому виробництві. За допомогою багаточисельних способів, наприклад, проміжні сполуки, такі як тетраоцтова кислота, одержували разом із великою кількістю солей лужних металів як побічних продуктів, які потрібно було відділяти перед циклізацією у сполуку формули (І). Ці проблеми вищевказаних способів загалом основані на тому факті, що використані тетраоцтова кислота як тетрааміди, тетранітрили та сполука формули (I) самі по собі є дуже полярними сполуками та утворюють солі із сильними основами, як цього потребує спосіб. З цієї причини це завжди призводить до труднощів відділення попередників сполук, що не провзаємодіяли, та отриманих побічних продуктів. 1 UA 102509 C2 5 10 15 20 25 30 35 40 45 50 55 60 Проблеми, які виникають у зв'язку із та протягом очистки попередників сполуки у відомих способах одержання, детально описані у міжнародній заявці № 93/08172, P-L. MacDonald. Таким чином, для вирішення даних проблем запропонували спосіб одержання сполуки формули (I), Дераксозану, де останній одержують з високим виходом, перед циклізацією у дексразоксан, без проведення очищення проміжної тетраоцтової кислоти. Однак, за допомогою цього способу Дексразоксан одержують разом із великою кількістю побічних продуктів, що призводить до труднощів одержання Дексразоксану, вільного від солей. Окрім способів одержання сполук формули (І) або аналогів таких сполук, де проміжними продуктами є тетраоцтова кислота, тетраамід або тетранітрил, у літературі також описано спосіб одержання цис- та транс-циклопропіл-біс-2,6-(піперазиндіону), де обидві сполуки є аналогами сполуки формули (I), у якому як попередник використовують відповідний метиловий естер тетраоцтової кислоти. У D.T. Witiak et al, Journal of Medicinal Chemistry, Bd. 20, Nr. 5, pp 630-635 (1977), та Journal of Medicinal Chemistry, Vol. 21, No. 12, pp 1194-1197 (1978), описано циклізацію відповідного метилового естеру тетраоцтової кислоти у форму гідрохлориду при надлишку аміаку та метоксиду натрію у метанолі для одержання транс-сполуки. Вихід бажаної транс-сполуки є низьким та досягає тільки 27 % до очищення. Згідно з даними авторів застосування цього способу для одержання відповідної цис-сполуки є невдалим: для одержання цис-сполуки, метиловий естер тетраоцтової кислоти циклізували із гідридом натрію та формамідом у DME. Вихід транс-сполуки дорівнює 36,5 %. Вітіак та ін. запропонували застосовувати метиловий естер тетраоцтової кислоти виключно для одержання вищевказаних сполук. Не існує досвіду застосування сполук метилового естеру тетраоцтової кислоти як попередника для одержання аналогічних сполук. Навпаки, труднощі одержання цис- та транс-ізомерів бажаної сполуки складаються з того, що застосування таких сполук як попередників не є можливим. Метою даного винаходу є забезпечення способом одержання сполуки формули (I), який дозволяє одержання цієї сполуки з високим виходом, у промисловому масштабі та, який би вирішував технічні труднощі відомих способів. Цю мету досягають за допомогою способу даного винаходу, який включає стадію циклізації алкілового естеру 1,2-діамінопропан-N, N,N',N'-тетраоцтової кислоти (тут та надалі посилаються як на "алкіловий естер тетраоцтової кислоти") із аміаком у формаміді, де "алкіл" бажано є (C1C3)-алкілом, та включає обидва "Сз-алкіл" n-пропіл, ізопропіл, також як і ізопропіл. Спосіб даного винаходу основано на спостереженнях вийняткових якостей алкілових естерів тетраоцтової кислоти, таких як понижена полярність та гідрофільність, у порівнянні із відомими проміжними сполуками, які застосовують для покращення способу одержання сполуки формули (I). Більш того, завдяки більшій реактивності цих естерів, замикання кільця сполуки формули (I) досягають у більш простих умовах, як по відношенню до кількості стадій та критеріїв, так і по відношенню до реакційних умов, потрібних для цього. Наступними перевагами способу даного винаходу є застосування аміаку та формаміду, двох загальних хімічних речовин, де, у способі даного винаходу формамід також застосовують як розчинник. Метанол, отриманий протягом циклізації, видаляють з реакційної суміші шляхом простої дистиляції. Подальші деталі способу даного винаходу та його бажаний варіант здійснення наведено у наступних прикладах. Винахід також стосується алкілових естерів тетраоцтової кислоти, застосованих у способі даного винаходу, наступної формули (II) (ROOCCH2)2N-CHCH3-CH2-N(CH2COQR)2 (II), де R означає алкіл. Бажано R означає (C1-C3)-алкіл, такий як метил, етил або пропіл. Сполуки формули (II), які є попередниками сполук формули (I), є новими сполуками окрім метилового естеру тетраоцтової кислоти, описаними E. H. Herman et al. у Research Communications in Chemical Pathology and Pharmacology, Vol. 48, No, 1, pp 39-55 (1985). Алкіловий естер тетраоцтової кислоти одержують відомими способами, також описаними у наступних прикладах. Бажаний спосіб одержання алкілового естеру тетраоцтової кислоти згідно з даним винаходом, включає взаємодію діаміну формули (III) H2N-CHCH3-CH2-NH2 (III) або його придатної солі із хлороцтовою кислотою та наступна обробка алкіловим спиртом для утворення алкілового естеру тетраоцтової кислоти формули (II): (ROОCCH2)2N-CHCH3-CH2-N(CH2COOR)2 (II), де R означає алкіл, бажано (C1-C3)-алкіл, такий як метил, етил або пропіл. Таким чином отримані алкілові естери тетраоцтової кислоти піддівали циклізації у присутності аміаку та формаміду з метою одержання сполука формули (I). 2 UA 102509 C2 5 10 15 20 25 30 35 40 45 50 55 60 До циклізації у сполуку формули (I), алкілові естери тетраоцтової кислоти формули (II) у разі потреби піддавали очищенню, наприклад, їх піддавали розподіленню між розчинником, що не змішується з водою, та водою для відділення солей лужних металів. Зокрема як розчинники, що не змішуються з водою, використовували етилацетат та ізопропілацетат. Однак, алкілові естери тетраоцтової кислоти формули (II) можна циклізувати у сполуку формули (I) також без попереднього очищення. Цей варіант способу представляє собою особливо бажаний варіант способу одержання сполуки формули (I). У особливо бажаному варіанті здіснення способу винаходу, отримують сполуку формули (I) із більшим виходом та достатнім рівнем очистки, у порівнянні із відомими способами. Окрім цього, не потрібно додаткове очищення та відокремлення алкілових естерів тетраоцтової кислоти, які використовують як попередники. У способі даного винаходу, як і у бажаному варіанті здійснення способу, розкладання сполуки формули (I) внаслідок гідролізу протягом здійснення способу зведено до мінімуму. Відділення іонних матеріалів (таких як солі лужних металів) повністю та легко здійснюють шляхом розподілення алкілових естерів тетраоцтової кислоти формули (II) між розчинником, що не змішується із водою, таким як етилацетат, ізопропілацетат, та водою. Спосіб згідно з даним винаходом, як і його бажаний варіант здійснення, є стереоселективними способами, тобто, сполука попередник повинна бути доступною у конфігурації бажаної для сполуки формули (I). Подальші аспекти даного винаходу наведені у наступних прикладах, які слугують для ілюстрації та не обмежують рамки винаходу. Фахівцям у галузі зрозуміло, що деталі способу, описані у наступних прикладах, можуть бути зміненими у рамках даного винаходу. Наприклад, згідно з способом у прикладі 5, який описано для S-енантіомеру, Дексарозоксану, Rенантіомеру, Леворазоксану, також можно одержувати рацемат. Доки не вказано протилежне або, якщо це не буде зрозуміло у контексті, проценти стосуються ваги. Приклади Приклад 1 Одержання (S)-(+)-1, 2-діамінопропан-N, N,N',N'-тетраоцтової кислоти 150,0 г (1,02 М) (S)-(-)-1,2-діамінопропан дигідрохлориду вводять у 780,0 г деіонізованої води при кімнатній температурі, 578,4 г (6,12 M) хлороцтової кислоти та 1785,0 г 32 %-го (14,28 M) гідроксиду натрію додавали до цього розчину протягом 45 хв. при охолодженні (при 15 °C). Після того як закінчували додавання, реакційну суміш нагрівали до 40 °C, де починаючи з 40 °C, реакція була екзотермічною та температуру підтримували на рівні 40-45 °C охолодженням. Після ослаблення екзотермічної реакції струшування здійснювали протягом 90 годин при 4045 °C. Зменшували об'єм лужної, безкольорової та прозорої рідини при вакуумі на бані, при температурі 70 °C приблизно в 2,5 рази. Маслянисту кристалічну суспензію змішували з 1,2 л метанолу, охолоджували при 20 °C, солі відфільтровували та залишок на фільтрі промивали 2 × 300 мл метанолу. Об'єднані розчини метанолу випаровували у вакуумі на бані при температурі 70 °C. Залишок перегонки високої в'язкості змішували з 300 мл деіонізованої води при 70 °C та охолоджували до 0 °C. При охолодженні значення рН доводили до 1,5 шляхом додавання 343,8 г 95 % сірчаної кислоти, та після реагування густу кристалічну суспензію змішували з 900 мл деіонізованої води. Кристалічну суспензію струшували протягом ночі при 0 °C з 2 л ацетону. Кристали відфільтровували та промивали 2 × 250 мл суміші вода/ацетон у співвідношенні 1:2 та 2 × 500 мл чистого ацетону. Об'єднаний органічний розчин повністю випаровують у вакуумі при температурі бані 70 °C, в'язкий залишок, що залишився, змішують з 600 мл концентрованої оцтової кислоти та продукт осаджують шляхом додавання 5 л ацетону при кімнатній температурі. Суспензію охолоджують до 5 °C, продукт відфільтровують, промивають 550 мл концентрованої оцтової кислоти/ацетон у співвідношенні 1:10 та 2 × 500 мл ацетону та висушують при 20 °C у вакуумі. Вихід: 272,1 г Приклад 2 Одержання метилового естеру (S)-(+)-1,2-діамінопропан-N, N,N',N'-тетраоцтової кислоти Етерифікацію здійснювали з використанням відокремленої (S)-(+)-1,2-діамінопропан-N, N,N',N'-тетраоцтової кислоти наступним чином: 37,5 г (S)-(+)-1,2-діамінопропан-N, N,N',N'-тетраоцтової кислоти разом з 756 мл метанолу та 22,5 г 95 % сірчаної кислоти нагрівають із зворотнім холодильником протягом 20 годин. Охолоджений розчин нейтралізували 41,5 г гідрокарбонату натрію та дистилювали до сухості у вакуумі. Залишок розподіляли між 300 мл деіонізованої води та 300 мл трет-бутилметилетеру, та водну фазу екстрагували 2 × 150 мл трет-бутилметилетеру. Об'єднані органічні фази 3 UA 102509 C2 5 висушували над сульфатом натрію, відфільтровували та розчинник відпаровували до сухості у вакуумі (вихід неочищеного продукту: 20,7 г). Неочищений продукт розчиняли у 300 мл суміші трет-бутилметилетеру/ петролейний ефір 60/95 у співвідношенні 1:2, змішували із 45 г силікагелю 0,06-0,2 мм протягом 30 хв. та відфільтровували. Залишок промивали 2 × 50 мл вищевказаної суміші розчинників та фільтрат випаровували до сухості у вакуумі. Вихід: 6,9 г безбарвної олії (метиловий естер) Дані аналізу: Аналіз елементів: C15H26N2О8 10 15 20 25 С розраховано: виявлено: Н N O 49,72 49,84 7,23 7,39 7,73 7,47 5,32 20 Повертання [α]d (C=4; метанол): +3,1° 1H-ЯМР: 0,97 (д, 3H; -CH-CH3); 2,49 (, 1H; N-CH-CH2-); 2,83 (дд, 2H; N-CH-CH2-); 3,5 (с, 4H; N-CH2-CO); 3,55 (с, 4H; N-CH2-CO); 3,61 (a, 12H; COO-CH3) 13С-ЯМР: 15,0 (к; -CH-CH3); 51,22 (к; O-CH3); 51,38 (к; O-CH3); 52,07 (т; N-CH2-); 54,99 (т; NCH2-); 55,96 (д; N-CH-); 58,08 (т; CH-CH2-N); 171,69 (с; -CO-); 172,31 (с; -CO-) Приклад 3a Одержання етилового естеру (S)-(+)-1,2-діамінопропан-N, N,N',N'-тетраоцтової кислоти Естерифікацію здійснювали з використанням ізольованого (S)-(+)-l, 2-діамінопропан-N, N,N',N'-тетраоцтової кислоти наступним чином: 25,0 г (S)-(+)-l, 2-діамінопропан-N, N,N',N'-тетраоцтової кислоти разом із 725 мл етанолу та 15,0 г 95 % сірчаної кислоти нагрівали із зворотнім холодильником протягом 120 г. Охолоджений розчин нейтралізували 27,5 г гідрокарбонату натрію та випаровували до сухості у вакуумі. Залишок розподіляли між 200 мл деіонізованої води та 200 мл трет-бутилметилетеру, та водну фазу екстрагували 2 × 100 мл трет-бутилметилетеру. Об'єднані органічні фази висушують над сульфатом натрію, відфільтровують та розчинник випаровують до сухості у вакуумі (вихід не очищеного продукту: 19,7 г). Неочищений продукт розчиняють у 300 мл петролейного ефіру 60/95, перемішують із 40 г силікагелю 0,06-0,2 мм протягом 30 хв., відфільтровують, залишок промивають 2 × 50 мл розчинника та фільтрат випаровують до сухості у вакуумі. Вихід: 7,1 г безбарвної олії (етиловий естер) 30 Дані аналізу: Аналіз елементів: C19H34N2О8 С розраховано: виявлено: Н N O 54,53 54,51 8,19 8,36 6,69 6,56 30,58 20 35 40 45 50 Повертання [α]d (C=4; метанол): +1,1° 1H-ЯМР: 1,08 (д, 3H; -CH-CH3); 1,15-1,35 (дд, 12H; -CH2-CH3); 2,5 (м, 1H, N-CH-CH2); 2,853,15 (м, 2H; N-CH-CH2-); 3,5 (с, 4H; N-CH2-CO); 3,6 (с, 4H; N-CH2-CO); 4,0-4,3 (м, 8H; COO-CH2CH3) 13C-ЯМР: 13,96 (к; -CH2-CH3); 14,0 (к; -CH2-CH3); 15,12 (к; -CH-CH3); 52,27 (т; N-CH2-CO); 55,28 (т; N-CH2-CO); 56,0 (д; N-CH-CH2-); 58,2 (т; CH-CH2-N); 60,08; 60,15 2x(т, - COO-CH2-); 171,22 (с; CO); 171,87 (с; CO) Приклад 3b (бажаний) Одержання етилового естеру (S)-(+)-1,2-діамінопропан-N/N, N',N'-тетраоцтової кислоти 50 г (S)-(-)-діамінопропан дигідрохлориду та 192,8 г хлороцтової кислоти у 321 мл води піддавали взаємодії з 190,4 г гідроксиду натрію у 343 мл води та обробляли протягом 132 г при 45 °C. Воду випаровували та отриману густу суспензію змішували з 100 мл етанолу та знову повністю випаровували. Залишок поміщали у 900 мл метанолу, обробляли 90 мл концентрованої сірчаної кислоти та кип'ятили із зворотнім холодильником протягом 46 годин. Реакційну суміш охолоджували до температури навколишнього середовища та кислоту нейтралізували шляхом додавання 240 г карбонату натрію. Осад відфільтровують, промивають 150 мл етанолу, фільтрат випаровують та масляний залишок суспендують у 250 мл толуолу. Після достатньої екстракції 2 N хлорводневою кислотою водну фазу нейтралізували твердим карбонатом натрію (приблизно 75 г) та екстрагували приблизно 375 мл толуолу. В результаті 4 UA 102509 C2 повного випаровуванні розчинників отримали 134 г етилового естеру як світло-жовтої олії. Один аналітичний зразок отримали шляхом очищення хроматографічною колонкою на силікагелі. Дані аналізу: Аналіз елементів: C19H34N2О8 С розраховано: виявлено: 5 10 15 20 25 30 45 50 O 54,53 54,18 8,19 8,36 6,69 6,59 30,58 20 С розраховано: виявлено: 40 N Повертання [α]d (C=10; метанол): +8,6° 1H-ЯМР: 1,02 (д, 3H; -CН-CH3); 1,21-1,27 (дд, 12H; -CH2-CH3); 2,5 (м, 1H, N-CH-CH2); 2,853,07 (м, 2H; N-CH-CH2-); 3,5 (с, 4H; N-CH2-CO); 3,6 (с, 4H; N-CH2-CO); 4,05-4,15 (м, 8H; COO-CH2CH3) 13C-ЯМР: 14,27 (к; -CH2-CH3); 14,30 (к; -CH2-CH3); 15,41 (к; -CH-CH3); 52,77 (т; N-CH2-CO); 55,60 (т; N-CH2-CO); 56,31 (д; N-CH-CH2-); 58,51 (т; CH-CH2-N); 60,44; 60,52 2x(т; COO-CH2-); 171,56 (с; CO); 172,22 (с; CO) Приклад 4a Одержання ізопропілового естеру (S)-(-)-1,2-діамінопропан-N, N,N',N'-тетраоцтової кислоти Естерифікацію здійснювали з використанням ізольованого (S)-(+)-1,2-діамінопропан-N, N,N',N'-тетраоцтової кислоти наступним чином: 25,0 г (S)-(+)-1,2-діамінопропан-N, N,N',N'-тетраоцтової кислоти разом із 950 мл ізопропанолу та 15,0 г 95 % сірчаної кислоти нагрівали із зворотнім холодильником протягом 162 годин. Охолоджений розчин нейтралізували 27,5 г гідрокарбонату натрію та випаровували до сухості у вакуумі. Залишок розподіляли між 200 мл деіонізованої води та 200 мл третбутилметилетеру, та водну фазу екстрагували 1 × 100 мл трет-бутилметилетеру. Об'єднані органічні фази висушували над сульфатом натрію, відфільтровували, та розчинник випаровували до сухості у вакуумі (вихід не очищеного продукту: 21,2 г). Неочищений продукт розчиняють у 300 мл петролейного ефіру 40/65, перемішують із 40 г силікагелю 0,06-0,2 мм протягом 30 хв., відфільтровують, залишок промивають 2 × 50 мл розчинника та фільтрат випаровують до сухості у вакуумі. Вихід: 10,8 г світло-жовтої олії (ізопропіловий естер) Дані аналізу: Аналіз елементів: C23H42N2О8 35 Н Н N O 54,21 58,12 8,92 9,08 5,90 5,70 26,97 20 Повертання [α]d (C=4; метанол): -2,6° 1H-ЯМР: 1,05 (д, ЗН; -CH-CH3); 1,15-1,35 (дд, 24H; iPr-CH-(CH3)2); 2,5 (м, 1H, N-CH-CH2-); 2,85-3,15 (м, 2H; N-CH-CH2-); 3,5 (2с, 2 × 4H; N-CH2-CО); 5,0 (2к, 4H; iPr-CH-(CH3)2). 13C-ЯМР: 15,44 (к; -CH2-CH3); 21,79 (к; -CH-(CH3)2); 21,85 (к; -CH-(CH3)2); 52,72 (т; N-CH2CO); 55,88 (т; N-CH2-CO); 56,25 (д; N-CH-CH2-); 58,53 (т; CH-CH2-N); 67,77; 67,79 2x(т; COO-CН-); 170,99 (с; CO); 171,67 (с; CO). Приклад 4b (бажаний) Одержання ізопропілового естеру (S)-(+)-1,2-діамінопропан-N, N,N',N'-тетраоцтової кислоти 50 г (S)-(-)-діамінопропан дигідрохлориду та 192,8 г хлороцтової кислоти у 321 мл води піддавали взаємодії з 190,4 г гідроксиду натрію у 343 мл води та обробляли протягом 114 г при 45 °C. Воду випаровували та отриману густу суспензію кіп'ятили із зворотнім холодильником із сумішшю з 90 мл концентрованої сірчаної кислоти у 1500 мл 2-пропанолу протягом 41 години. Реакційну суміш охолоджували до температури навколишнього середовища та кислоту нейтралізували шляхом додавання 240 г карбонату натрію. Осад відфільтровують, промивають 150 мл етанолу, фільтрат випаровують та масляний залишок суспендують у 250 мл толуолу. Після достатньої екстракції 2 N хлорводневою кислотою водну фазу нейтралізували твердим карбонатом натрію (приблизно 75 г) та екстрагували приблизно 375 мл толуолу. В результаті повного випаровуванні розчинників отримали 41 г ізопропілового естеру як світло-жовтої олії. Аналітичний зразок отримали шляхом повторення екстракції та наступного очищення хроматографічною колонкою на силікагелі, Дані аналізу: 5 UA 102509 C2 Аналіз елементів: C23H42N2О8 С N O 58,21 58,09 розраховано: виявлено: Н 8,92 9,06 5,90 5,88 26,97 20 5 10 15 20 25 30 Повертання [α]d (C=10; метанол): 0,5° 1H-ЯМР: 1,05 (д, 3H; -CH-CH3); 1,20-1,22 (дд, 24H, -iPr-CH-(CH3)2); 2,49 (м, 1H, N-CH-CH2-); 2,90, 3,04 (м, 2H; N-CH-CH2-); 3,50, 3,53 (2с, 2 × 4H; N-CH2-CO); 4,99 (2к, 4H; iPr-CH-(CH3)2). 13C-ЯМР: 15,53 (к; -CH-CH3); 21,92 (к; -CH-(CH3)2); 21,98 (к; -СН-(СНз)2); 52,85 (т; N-CH2-CO); 56,00 (т; N-CH2-CO); 56,36 (д; N-CH-CH2-); 58,63 (т; CH-CH2-N); 67,92; 67,94 2x(т; СOO-CH-); 171,10 (с; CO); 171,79 (с; CO). Приклад 5 Одержання (S)-(+)-4,4’-(1-метил-1,2-етандіїл)-біс-(2,6-піперазиндіон) (Дексразоксан) (I) 5.1. Одержання тетраметилового естеру (S)-1,2-діамінопропан-N-N, N',N'-тетраоцтової кислоти (II) 10 кг (S)-(-)-діамінопропан дигідрохлориду та 38,5 кг хлор водневої кислоти у 65 л води піддавали взаємодії із 38 кг гідроксиду натрію у 69 л води, та реакція тривала протягом часу від 70 дo 100 годин при 45 °C. Воду випаровували та отриману густу суспензію змішували із 80 л метанолу, фільтрували та осад промивали метанолом. Фільтрат повністю випаровували та залишок поміщали у 180 л метанолу, обробляли 18 л концентрованої сірчаної кислоти та кіп'ятили із зворотнім холодильником протягом 6 годин. Реакційну суміш охолоджували до температури навколишнього середовища та кислоту нейтралізували шляхом додавання від 20 до 25 кг гідрокарбонату натрію. Осад відфільтровували, фільтрат випаровували та масляний залишок розчиняли у 50 мл етил ацетат. Після достатньої екстракції 2 N хлорводневою кислотою, водну фазу нейтралізували твердим карбонатом натрію та екстрагували приблизно 100 л етил ацетату. Після повного випаровування розчинників отримали від приблизно 13,5 кг до 17,3 кг метилового естеру, який застосовують у наступній стадії без подальшого очищення. 5.2. Циклізація до (S)-(+)-4,4'-(1-метил-1,2-етандіїл)-біс-(2,6-піперазиндіон) (Дексразоксан) (І) 4,7 кг аміаку додавали до розчину з 10 кг метилового естеру (S)-(+)-1,2-діамінопропан-N, N,N',N'-тетраоцтової кислоти, отриманої у вищенаведеному прикладі, у 34 л формаміду, та реакційну суміш витримували при 40-50 °C при максимальному тиску 5 бар протягом приблизно 12 годин. Після цього, реакційну суміш повільно підігрівали до 150 °C, отриманий метанол дистилювали протягом нагрівання та реакційну суміш витримували при 140-150 °C протягом 1012 г. Після цього розчинник дистилюють, масляний залишок кристалізують з метанолу з виходом 2,9-3,7 кг Дексразоксану, який потім очищають шляхом рекристалізування з 1,4діоксану. ФОРМУЛА ВИНАХОДУ 35 1. Спосіб одержання сполуки формули (І) C(O) CH2 HN C(O) 40 45 CH2 CH(CH3) CH2 C(O) CH2 N C(O) N CH2 NH , (I) який відрізняється тим, що тетраестер формули (II) (ROOCCH2)2N-CHCH3-CH2-N(CH2COOR)2, (II) де R означає алкіл, циклізують з аміаком у формаміді. 2. Спосіб за п. 1, який відрізняється тим, що R означає (С1-С3)-алкіл. 3. Застосування сполуки формули (II) (ROOCCH2)2N-CHCH3-CH2-N(CH2COOR)2 (II) для одержання сполуки формули (І) C(O) CH2 HN CH2 N C(O) CH2 CH(CH3) CH2 C(O) N NH CH2 C(O) 50 6 , (I) UA 102509 C2 де R відповідно означає алкіл, бажано (С1-С3)-алкіл. 4. Спосіб одержання сполуки формули (І) C(O) CH2 HN CH2 N C(O) CH2 CH(CH3) C(O) CH2 CH2 C(O) N NH , (I) 5 10 15 який відрізняється тим, що здійснюють (a) взаємодію (S)-1,2-діамінопропану або його прийнятної солі з хлороцтовою кислотою; (b) обробку реакційного продукту, отриманого на стадії (а), у алкіловому спирті сильною кислотою, бажано неорганічною, (c) циклізацію сполуки формули (II) (ROOCCH2)2N-CHCH3-CH2-N(CH2COOR)2 (II) з аміаком у формаміді, де R відповідно означає алкіл. 5. Спосіб за п. 4, який відрізняється тим, що R означає (С1-С3)-алкіл. 6. Спосіб за п. 4 або 5, який відрізняється тим, що перед стадією (с) сполуку формули (II) очищують від неорганічних солей шляхом екстракції органічним розчинником, нерозчинним у воді, з води. 7. Спосіб за п. 4 або 5, який відрізняється тим, що сполуку формули (II) застосовують на стадії (с) без попереднього відділення або очищення. Комп’ютерна верстка Г. Паяльніков Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 7
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for producing 4,4'-(1-methyl-1,2-ethanediyl)-bis-(2,6-piperazinedione)
Автори англійськоюKoch, Andreas, Neufellner, Erwin
Назва патенту російськоюСпособ получения 4,4'-(1-метил-1,2-этандиил)-бис-(2,6-пиперазиндиона)
Автори російськоюКох Андреас, Нойфелльнер Эрвин
МПК / Мітки
МПК: C07C 229/16, C07D 241/08
Мітки: одержання, 4,4'-(1-метил-1,2-етандіїл)-біс-(2,6-піперазиндіону, спосіб
Код посилання
<a href="https://ua.patents.su/9-102509-sposib-oderzhannya-44-1-metil-12-etandil-bis-26-piperazindionu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання 4,4′-(1-метил-1,2-етандіїл)-біс-(2,6-піперазиндіону)</a>
Спосіб одержання 4′-[[4-метил-6-(1-метил-1н-бензімідазол-2-іл)-2-пропіл-1н-бензімідазол-1-іл]метил]біфеніл-2-карбонової кислоти (телмісартан)

Номер патенту: 99140
Опубліковано: 25.07.2012
Автори: Стрелєц Іво, Стах Ян, Радл Станіслав, Цінібулк Йозеф, Яррах Камаль
МПК: C07D 235/18
Мітки: телмісартан, одержання, спосіб, кислоти, 4'-[[4-метил-6-(1-метил-1н-бензімідазол-2-іл)-2-пропіл-1н-бензімідазол-1-іл]метил]біфеніл-2-карбонової
Формула / Реферат:
1. Спосіб одержання кристалічної форми А телмісартану (І), (I) який відрізняється тим, що карбонову кислоту загальної формули R1COOH, де R1 є атом водню або С1-С4алкіл, додають до розчину калієвої солі телмісартану (VII) (VII)в спирті формули R2OH із...
Спосіб одержання солі 3-етил-5-метил-2-(2-аміноетоксиметил)-4-(2-хлорфеніл)-6-метил-1,4-дигідро-3,5-піридиндикарбоксилату і бензолсульфокислоти (безилату амлодипіну)
Номер патенту: 57722
Опубліковано: 15.07.2003
Автори: НЕМЕТ Норберт, Лакс Кованьі Дьйордьі, Блашко Габор, Немет Габор, Надь Кальман, Божінг Даніель, Томп Петер, Крацнаі Дьйордь, ШІМІГ Дьюла, ДОНАТ ВЕРЕЦКЕІ Дьйордьі
МПК: C07C 69/716, C07D 211/90, C07C 69/738
Мітки: одержання, безилату, амлодипіну, солі, спосіб, бензолсульфокислоти, 3-етил-5-метил-2-(2-аміноетоксиметил)-4-(2-хлорфеніл)-6-метил-1,4-дигідро-3,5-піридиндикарбоксилату
Формула / Реферат:
1. Спосіб одержання солі 3-етил-5-метил-2-(2-аміноетоксиметил)-4-(2-хлорфеніл)-6-метил-1,4-дигідро-3,5-піридиндикарбоксилату і бензолсульфокислоти (безилату амлодипіну) формули (I)(I),що включає реакцію сполуки формули (XI) I` (XI)з бензолсульфокислотою формули (XII)(XII).2. Спосіб згідно з п. 1, який включає реакцію сполуки формули XI з бензолсульфокислотою формули XII у суміші...
Спосіб одержання 5-(2-хлортіазол-5-ілметил)-3-метил-4-нітроімінопергідро-1,3,5-оксадіазину (тіаметоксаму)

Номер патенту: 73306
Опубліковано: 15.07.2005
Автори: Сейферт Готфрід, Рапольд Томас, Гізін Верена
МПК: C07C 257/00, C07C 277/00, C07B 61/00, C07D 277/36, C07D 417/06, C07C 279/36, C07D 277/32, C07D 213/38, C07C 261/00, C07C 243/00, C07D 277/20, C07D 213/40, C07D 277/28, C07D 401/06, C07D 307/14, C07D 213/61
Мітки: спосіб, тіаметоксаму, одержання, 5-(2-хлортіазол-5-ілметил)-3-метил-4-нітроімінопергідро-1,3,5-оксадіазину
Формула / Реферат:
1. Спосіб одержання сполуки формули,який відрізняється тим, що сполуку формулипіддають взаємодії зі сполукою формули,де Q означає хлор або бром, у присутності розчинника або розріджувача,...
2-(3,5-біс-трифторметилфеніл)-n-метил-(6-морфолін-4-іл-4-о-толілпіридин-3-іл)ізобутирамід, спосіб його одержання, фармацевтичний препарат і спосіб лікування захворювань, пов’язаних з рецептором нк-1

Номер патенту: 70326
Опубліковано: 15.10.2004
Автори: Слайт Ендрю, Хіггінс Гай Ендрю, Баллард Тереза Маріа, Полі Соня Маріа, Хоффманн Торстен
МПК: A61P 25/22, A61P 43/00, A61P 1/06, A61K 31/5377, A61P 25/24, C07D 213/75, A61P 1/08, C07D 213/74, A61P 25/00
Мітки: нк-1, фармацевтичний, захворювань, рецептором, лікування, препарат, 2-(3,5-біс-трифторметилфеніл)-n-метил-(6-морфолін-4-іл-4-о-толілпіридин-3-іл)ізобутирамід, одержання, спосіб, пов'язаних
Формула / Реферат:
1. 2-(3,5-біс-трифторметилфеніл)-N-метил-(6-морфолін-4-іл-4-о-толілпіридин-3-іл)ізобутирамід формули Iі його фармацевтичнo прийнятні кислі адитивні солі, що виявляють властивості антагоністів рецептора НК-1.2. Спосіб одержання сполуки формули І, який включає:реакцію сполуки формули
Спосіб одержання n-метил-n-[(1s)-1-феніл-2-((3s)-3-гідроксипіролідин-1-іл)етил]-2,2-дифенілацетаміду або n-метил-n-[(1r)-1-феніл-2-((3r)-3-гідроксипіролідин-1-іл)етил]-2,2-дифенілацетаміду

Номер патенту: 73472
Опубліковано: 15.08.2005
Автори: Бате Андрес, Будак Дженс, Стейн Інгеборг, Хелферт Берн, Акерманн Карл-Оугест, Готшліх Рудольф
МПК: A61K 31/40, C07D 207/12, A61P 29/00, A61P 7/10
Мітки: n-метил-n-[(1s)-1-феніл-2-((3s)-3-гідроксипіролідин-1-іл)етил]-2,2-дифенілацетаміду, одержання, n-метил-n-[(1r)-1-феніл-2-((3r)-3-гідроксипіролідин-1-іл)етил]-2,2-дифенілацетаміду, спосіб
Формула / Реферат:
1. Спосіб одержання N-метил-N-[(1S)-1-феніл-2-((3S)-3-гідроксипіролідин-1-іл)етил]-2,2-дифенілацетаміду або N-метил-N-[(1R)-1-феніл-2-((3R)-3-гідроксипіролідин-1-іл)етил]-2,2-дифенілацетаміду, який відрізняється тим, щоа) N-заміщену похідну фенілгліцину формули І, (І)в якійR означає OR1 або SR1,R1 означає А, арил, гетероарил, Si(R3)3,...
Попередній патент: Похідні індолу як агоністи рецептора s1p1
Наступний патент: Спосіб обробки потоку речовин
Випадковий патент: Спосіб отримання монокристалів agcd2gas4