Похідні бензаміду для інгібування активності abl1, abl2 та bcr-abl1
Номер патенту: 113208
Опубліковано: 26.12.2016
Автори: Менлі Пол, Шопфер Йозеф, Фуре Паскаль, Марцінцік Андреас, Джонс Дерріл Брінлі, Гротцфельд Роберт Мартін, Салем Баха, Янке Вольфганг, Пелле Ксав'є Франсуа Андре, Додд Стефані Кей







Формула / Реферат
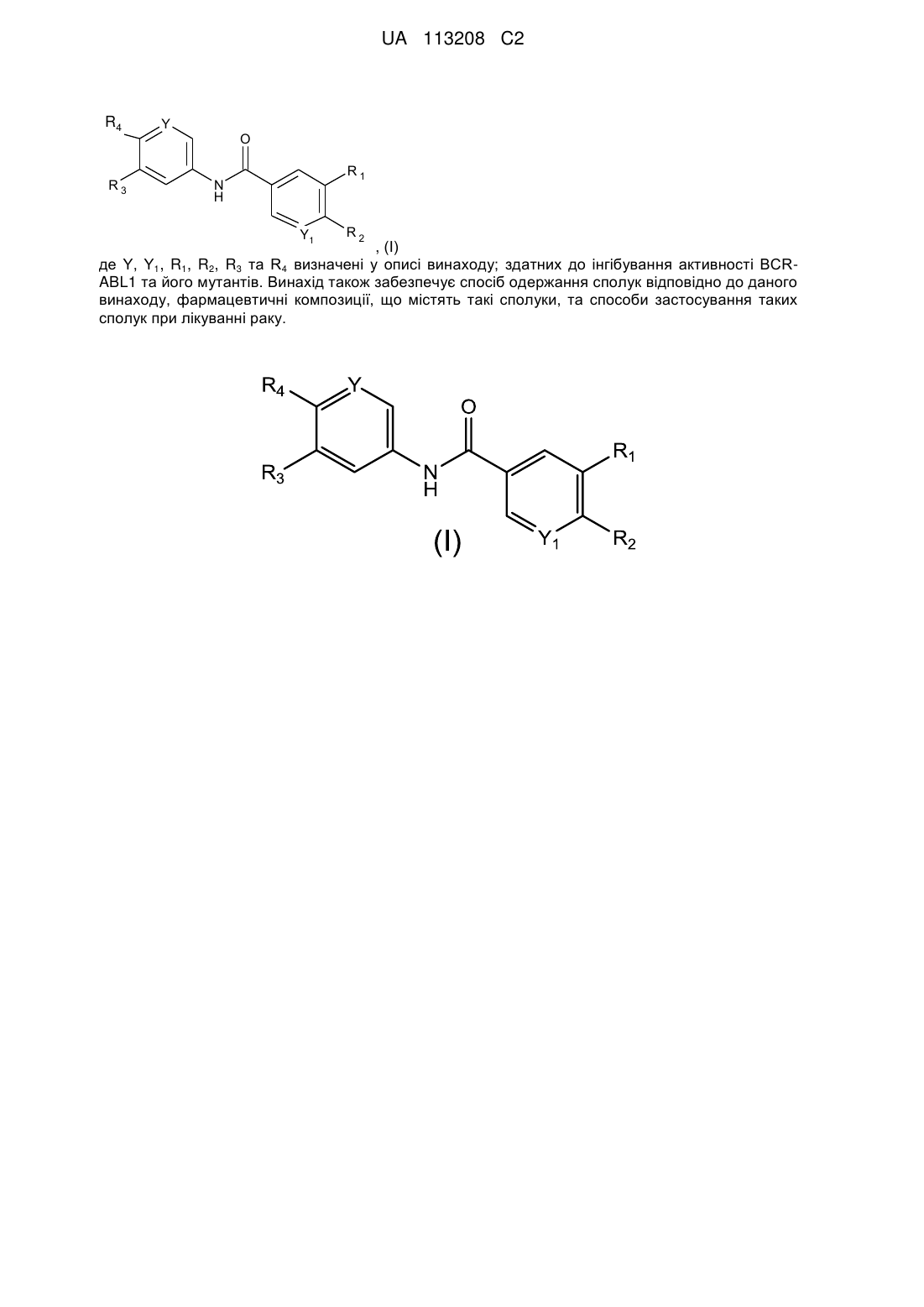
1. Сполука формули (І):
 , (І)
, (І)
де:
R1 являє собою піразоліл; де зазначений піразоліл є незаміщеним або заміщений 1-2 групами R6;
R2 являє собою піролідиніл; де зазначений піролідиніл заміщений однією групою R7;
R3 вибирають з водню та галогену;
R4 вибирають з -SF5 та -Y2-CF2-Y3;
R6 у кожному випадку незалежно вибирають з водню, гідроксигрупи, метилу, метоксигрупи, ціаногрупи, трифторметилу, гідроксиметилу, галогену, аміногрупи, фторетилу, етилу та циклопропілу;
R7 вибирають з гідроксигрупи, метилу, галогену, метоксигрупи, гідроксиметилу, аміногрупи, метиламіногрупи, амінометилу, трифторметилу, 2-гідроксипропан-2-ілу, метилкарбоніламіногрупи, диметиламіногрупи, 2-аміно-3-метилбутаноїлоксигрупи, карбоксигрупи, метоксикарбонілу, фосфонооксигрупи, ціаногрупи та амінокарбонілу;
Y вибирають з СН та N;
Y1 вибирають з СН та N;
Y2 вибирають з CF2, О та S(O)0-2; та
Y3 вибирають з водню, хлору, фтору, метилу, дифторметилу та трифторметилу;
або її фармацевтично прийнятні солі.
2. Сполука за п. 1 формули (Іb):
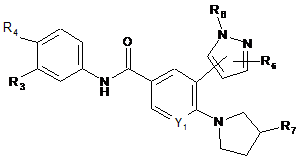 , (Ib)
, (Ib)
де:
R3 вибирають з водню та галогену;
R4 вибирають з -SF5 та -Y2-CF2-Y3;
R6, коли зв'язаний з азотом піразолільного кільця, вибирають з водню, метилу, гідроксіетилу, фторетилу, етилу та циклопропілу; та R6, коли зв'язаний з атомом вуглецю піразолільного кільця, вибирають з водню, гідроксигрупи, метилу, метоксигрупи, ціаногрупи, трифторметилу, гідроксиметилу, галогену, аміногрупи, фторетилу, етилу та циклопропілу;
R7 вибирають з гідроксигрупи, метилу, галогену, метоксигрупи, гідроксиметилу, аміногрупи, метиламіногрупи, амінометилу, трифторметилу, 2-гідроксипропан-2-ілу, метилкарбоніламіногрупи, диметиламіногрупи, 2-аміно-3-метилбутаноїлоксигрупи, карбоксигрупи, метоксикарбонілу, фосфонооксигрупи, ціаногрупи та амінокарбонілу;
Y1 вибирають з СН та N;
Y2 вибирають з CF2, О та S(O)0-2;
Y3 вибирають з водню, фтору, хлору, метилу, дифторметилу та трифторметилу;
або її фармацевтично прийнятні солі.
3. Сполука за п. 2 формули (Іс):
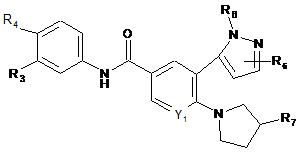 , (Іс)
, (Іс)
де:
R3 вибирають з водню та галогену;
R4 вибирають з -SF5 та -Y2-CF2-Y3;
R6, коли зв'язаний з азотом піразолільного кільця, вибирають з водню, метилу, гідроксіетилу, фторетилу, етилу та циклопропілу; та R6, коли зв'язаний з атомом вуглецю піразолільного кільця, вибирають з водню, гідроксигрупи, метилу, метоксигрупи, ціаногрупи, трифторметилу, гідроксиметилу, галогену, аміногрупи, фторетилу, етилу та циклопропілу;
R7 вибирають з гідроксигрупи, метилу, галогену, метоксигрупи, гідроксиметилу, аміногрупи, метиламіногрупи, амінометилу, трифторметилу, 2-гідроксипропан-2-ілу, метилкарбоніламіногрупи, диметиламіногрупи, 2-аміно-3-метилбутаноїлоксигрупи, карбоксигрупи, метоксикарбонілу, фосфонооксигрупи, ціаногрупи та амінокарбонілу;
Y1 вибирають з СН та N;
Y2 вибирають з CF2, О та S(O)0-2;
Y3 вибирають з водню, фтору, хлору, метилу, дифторметилу та трифторметилу;
або її фармацевтично прийнятні солі.
4. Сполука за п. 3 або її фармацевтично прийнятна сіль, вибрана з:
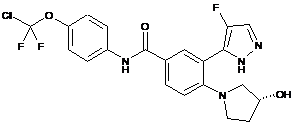 ,
,
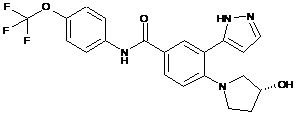 ,
,
 ,
,
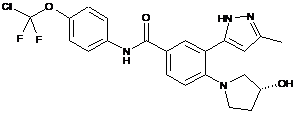 .
.
5. Сполука за п. 3 або її фармацевтично прийнятна сіль, вибрана з:
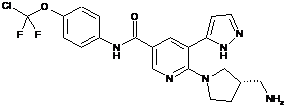 ,
,
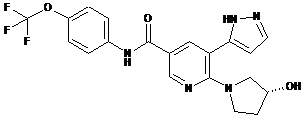 ,
,
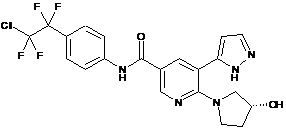 ,
,
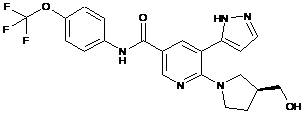 ,
,
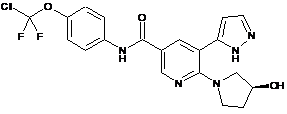 ,
,
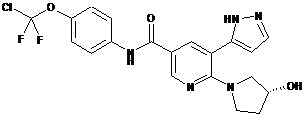 ,
,
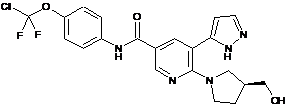 ,
,
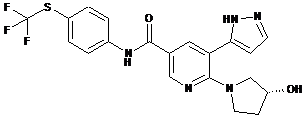 ,
,
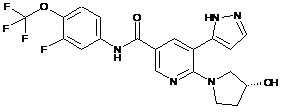 ,
,
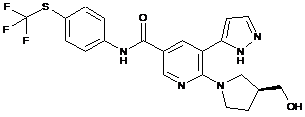 ,
,
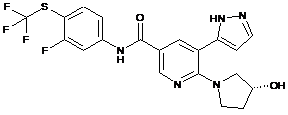 ,
,
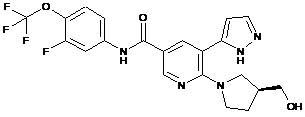 ,
,
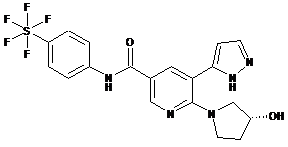 ,
,
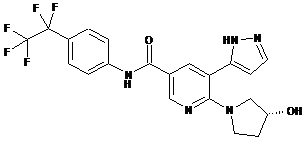 ,
,
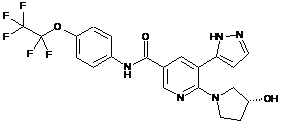 ,
,
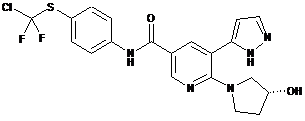 ,
,
 ,
,
 ,
,
 ,
,
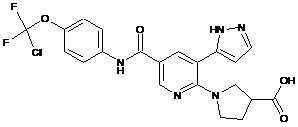 ,
,
 ,
,
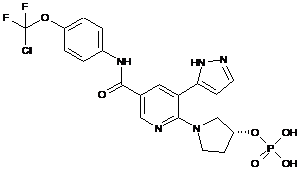 ,
,
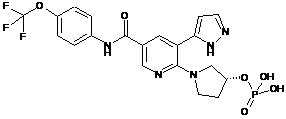 .
.
6. Сполука за п. 3 або її фармацевтично прийнятна сіль, вибрана з:
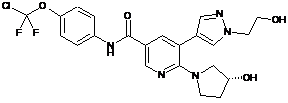 ,
,
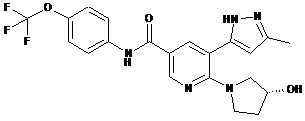 ,
,
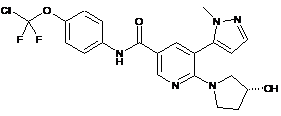 ,
,
 ,
,
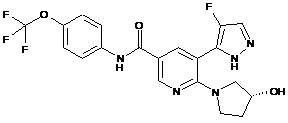 ,
,
 ,
,
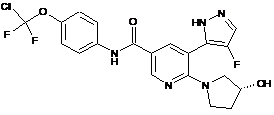 ,
,
 ,
,
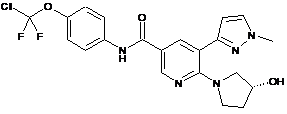 ,
,
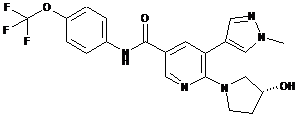 ,
,
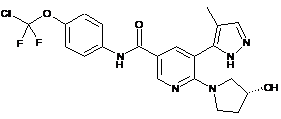 ,
,
 .
.
7. Сполука за п. 1 або її фармацевтично прийнятна сіль, яка являє собою:
 .
.
8. Сполука, вибрана з:
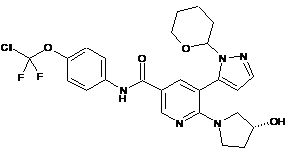 ,
,
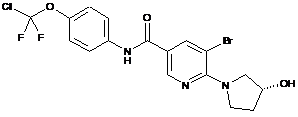 ,
,
 ,
,
 ,
,
 ,
,
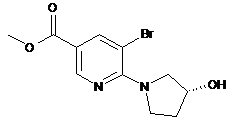 .
.
9. Сполука за п. 1, яка являє собою (R)-N-(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1Н-піразол-5-іл)нікотинамід або його фармацевтично прийнятну сіль.
10. Фармацевтична композиція, що містить аморфну дисперсію (R)-N-(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1Н-піразол-5-іл)нікотинаміду та 1-2 ексципієнти, вибрані з PVP VA64 та Pharmacoat 603.
11. Композиція за п. 10, де відсотковий вміст Pharmacoat 603 знаходиться у діапазоні від 30 до 45 %, відсотковий вміст PVP VA64 знаходиться у діапазоні від 30 до 45 %, та відсотковий вміст (R)-N-(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1Н-піразол-5-іл)нікотинаміду знаходиться у діапазоні від 20 до 30 %.
12. Композиція за п. 11, де відсотковий вміст Pharmacoat 603 становить 37,5 %, відсотковий вміст PVP VA64 становить 37,5 %, та відсотковий вміст (R)-N-(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1Н-піразол-5-іл)нікотинаміду становить 25 %.
13. Спосіб лікування пацієнта, що має лейкоз, вибраний з хронічного мієлоїдного лейкозу (CML) та гострого лімфобластного лейкозу (ALL), що включає введення зазначеному пацієнту терапевтично ефективної кількості (R)-N-(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1Н-піразол-5-іл)нікотинаміду або його фармацевтично прийнятної солі та необов'язково послідовне або одночасне введення терапевтично ефективної кількості сполуки, вибраної з іматинібу, нілотинібу, дазатинібу, босутинібу, понатинібу та бафетинібу.
14. Спосіб за п. 13, що включає введення зазначеному пацієнту терапевтично ефективної кількості (R)-N-(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1Н-піразол-5-іл)нікотинаміду або його фармацевтично прийнятної солі.
15. Спосіб за п. 13, що включає послідовне введення терапевтично ефективної кількості сполуки (R)-N-(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1Н-піразол-5-іл)нікотинаміду або його фармацевтично прийнятної солі та послідовне введення терапевтично ефективної кількості сполуки, вибраної з іматинібу, нілотинібу, дазатинібу, босутинібу, понатинібу та бафетинібу.
16. Спосіб за п. 13, що включає введення зазначеному пацієнту терапевтично ефективної кількості (R)-N-(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1Н-піразол-5-іл)нікотинаміду або його фармацевтично прийнятної солі та одночасне введення терапевтично ефективної кількості сполуки, вибраної з іматинібу, нілотинібу, дазатинібу, босутинібу, понатинібу та бафетинібу.
17. Спосіб за п. 16, де (R)-N-(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1Н-піразол-5-іл)нікотинамід вводять при дозі у межах 90-130 мг/кг.
18. Спосіб за п. 17, де нілотиніб вводять у дозі 10-50 мг/кг.
19. Спосіб за п. 18, де іматиніб вводять у дозі 50-200 мг/кг.
20. Застосування сполуки формули (І) або її фармацевтично прийнятної солі за будь-яким з пп. 1-9 для лікування раку.
21. Застосування за п. 20, де рак являє собою лейкоз, вибраний з хронічного мієлоїдного лейкозу та гострого лімфобластного лейкозу.
22. Застосування за п. 20 або 21 разом з додатковою сполукою, вибраною з іматинібу, нілотинібу, дазатинібу, босутинібу, понатинібу та бафетинібу.
23. Застосування за п. 22 для послідовного або одночасного введення із зазначеною додатковою сполукою, де зазначена додаткова сполука являє собою нілотиніб.
24. Застосування за будь-яким з пп. 20-23, де сполука являє собою (R)-N-(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1Н-піразол-5-іл)нікотинамід або його фармацевтично прийнятну сіль.
25. Застосування сполуки формули (І) або її фармацевтично прийнятної солі у одержанні лікарського засобу для лікування раку.
26. Застосування за п. 25, де рак являє собою лейкоз, вибраний з хронічного мієлоїдного лейкозу та гострого лімфобластного лейкозу.
Текст
Реферат: Даний винахід належить до сполук формули (І): UA 113208 C2 (12) UA 113208 C2 R4 R3 Y O R1 N H Y1 R2 , (І) де Y, Y1, R1, R2, R3 та R4 визначені у описі винаходу; здатних до інгібування активності BCRABL1 та його мутантів. Винахід також забезпечує спосіб одержання сполук відповідно до даного винаходу, фармацевтичні композиції, що містять такі сполуки, та способи застосування таких сполук при лікуванні раку. UA 113208 C2 5 10 15 20 25 30 35 40 45 50 ПЕРЕХРЕСНІ ПОСИЛАННЯ НА СПОРІДНЕНІ ЗАЯВКИ Дана заявка заявляє пріоритет до попередньої заявки США № 61/647174, поданої 15 травня 2012 року, та попередньої заявки США № 61/790967, поданої 15 березня 2013 року, кожна з яких включена у дану заявку як посилання у повному обсязі. ГАЛУЗЬ ТЕХНІКИ, ДО ЯКОЇ ВІДНОСИТЬСЯ ВИНАХІД Даний винахід відноситься до сполук, здатних до інгібування тирозинкіназної ферментативної активності білку Абельсона (ABL1), Абельсон-спорідненого білку (ABL2) та споріднених химерних білків, зокрема, BCR-ABL1. Винахід також забезпечує спосіб одержання сполук відповідно до даного винаходу, фармацевтичних препаратів, що містять такі сполуки, та способи застосування таких сполук при лікуванні раку. ПЕРЕДУМОВИ СТВОРЕННЯ ВИНАХОДУ Тирозинкіназна активність ABL1 білку звичайно жорстко регулюється N-кінцевою кепіруючою областю SH3 домену, що грає важливу роль. Один регуляторний механізм включає міристоілювання гліцин-2 залишку на N-кінцевому кеп-сайті та потім взаємодію з міристатзв'язуючим сайтом у SH1 каталітичному домені. Відмітною ознакою хронічного мієлогенного лейкозу (CML) є хромосома Philadelphia (Ph), утворена у результаті t(9,22) реципрокальної хромосомної транслокації у гемтатопоетичній стовбуровій клітині. Ця хромосома несе BCRABL1 онкоген, який кодує химерний BCR-ABL1 білок, який не містить N-кінцевий кеп та містить конститутивно активний тирозинкіназний домен. Хоча лікарські засоби, які інгібують тирозинкіназну активність BCR-ABL1 через ATPконкурентний механізм, такі як Gleevec®/Glivec® (іматиніб), Tasigna® (нілотиніб) та Sprycel® (дасатиніб), є ефективними для лікування CML, у деяких пацієнтів виникає рецидив через виникнення лікарсько-резистентних клонів, у яких мутації у SH1 домені заважають зв'язуванню з інгібітором. Хоча Tasigna® та Sprycel® зберігають ефективність у відношенні багатьох Gleevecрезистентних мутантних форм BCR-ABL1, мутація, у якій треонін-315 залишок замінений ізолейцином (T315I), залишається нечутливою до всіх трьох лікарських засобів та може привести до розвитку резистентності до терапії у CML пацієнтів. Тому інгібування BCR-ABL1 мутацій, таких як T315I, залишається незадоволеною медичною потребою. Окрім CML, BCRABL1 гібридні білки є причиною, що викликає певний відсоток гострих лімфоцитарних лейкозів, та лікарські засоби, прицільно діючі на ABL кіназну активність, також є корисними для цього показання. Засоби, що прицільно діють на міристоїл-зв'язуючий сайт (так звані алостеричні інгібітори), мають потенціал для лікування BCR-ABL1 розладів (J. Zhang, F. J. Adrian, W. Jahnke, S. W. Cowan-Jacob, A. G. Li, R. E. Iacob, T. Sim, J. Powers, C. Dierks, F. Sun, G.-R. Guo, Q. Ding, B. Okram, Y. Choi, A. Wojciechowski, X. Deng, G. Liu, G. Fendrich, A. Strauss, N. Vajpai, S. Grzesiek, T. Tuntland, Y. Liu, B. Bursulaya, M. Azam, P. W. Manley, J. R. Engen, G. Q. Daley, M. Warmuth., N.S. Gray. Targeting BCR-ABL by combining allosteric with ATP-binding-site inhibitors. Nature 2010; 463:501-6). Для попередження виникнення лікарської резистентності результаті використання ATP інгібітору та/або алостеричного інгібітору може бути розроблене комбіноване лікування з використанням обох типів інгібітору для лікування BCR-ABL1-зв'язаних розладів. Зокрема, існує потреба у невеликих молекулах або їх комбінаціях, які інгібують активність BCR-ABL1 та BCRABL1-мутації через ATP зв'язуючий сайт, міристоїл-зв'язуючий сайт або комбінацію обох сайтів. Крім того, сполуки відповідно до даного винаходу як інгібітори ABL1 кіназної активності мають потенціал для використання як терапевтичні засоби для лікування метастатичних інвазивних карцином та вірусних інфекцій, таких як покс-вірус та вірус Ебола. Сполуки відповідно до даного винаходу також мають потенціал для лікування або профілактики захворювань або розладів, асоційованих з аномально активованою кіназною активністю ABL1 дикого типу, включаючи незлоякісні захворювання або розлади, такі як захворювання ЦНС, зокрема, нейродегенеративні захворювання (наприклад, хвороба Альцгеймера, хвороба Паркінсона), захворювання рухових нейронів (аміотрофічний бічний склероз), м'язові дистрофії, аутоімунні та запальні захворювання (діабет та фіброз легенів), вірусні інфекції, пріонові захворювання. КОРОТКИЙ ОПИС ВИНАХОДУ У одному аспекті даний винахід забезпечує сполуки формули (I): 55 1 UA 113208 C2 , 5 10 15 20 25 30 35 40 45 50 де: R1 являє собою піразоліл; де зазначений піразоліл є незаміщеним або заміщений 1-2 групами R6; R2 являє собою піролідиніл; де зазначений піролідиніл заміщений однією групою R 7; R3 вибирають з водню та галогену; R4 вибирають з -SF5 та -Y2-CF2-Y3; R6 у кожному випадку незалежно вибирають з водню, гідрокси групи, метилу, метокси групи, ціано групи, трифторметилу, гідроксиметилу, галогену, аміно групи, фторетилу, етилу та циклопропілу; R7 вибирають з гідрокси групи, метилу, галогену, метокси групи, гідроксиметилу, аміно групи, метил-аміно групи, аміно-метилу, трифторметилу, 2-гідроксипропан-2-ілу, метил-карбоніл-аміно групи, диметил-аміно групи, 2-аміно-3-метилбутаноїл)окси групи, карбокси групи, метоксикарбонілу, фосфоноокси групи, ціано групи та амінокарбонілу; Y вибирають з CH та N; Y1 вибирають з CH та N; Y2 вибирають з CF2, O та S(O)0-2; та Y3 вибирають з водню, хлору, фтору, метилу, дифторметилу та трифторметилу. У другому аспекті даний винахід забезпечує фармацевтичну композицію, яка містить сполуку формули (I) або її N-оксидну похідну, індивідуальні ізомери та суміш ізомерів, або її фармацевтично прийнятну сіль у суміші з одним або декількома підходящими ексципієтами. У третьому аспекті даний винахід забезпечує спосіб лікування захворювання у тварини, де модуляція BCR-ABL1 активності може попереджати, інгібувати або полегшувати патологію та/або симптоматологію захворювань, при цьому спосіб включає введення тварині терапевтично ефективної кількості сполуки формули (I) або її N-оксидної похідної, індивідуальних ізомерів та суміші ізомерів, або її фармацевтично прийнятної солі. У четвертому аспекті даний винахід забезпечує застосування сполуки формули (I) для одержання лікарського засобу для лікування захворювання у тварини, у якому BCR-ABL1 активність сприяє патології та/або симптоматології захворювання. У п'ятому аспекті даний винахід забезпечує сполуку формули I для застосування у терапії для тварини, у якому BCR-ABL1 активність сприяє патології та/або симптоматології захворювання. У шостому аспекті даний винахід забезпечує спосіб одержання сполук формули (I) та їх Nоксидних похідних, пролікарських похідних, захищених похідних, індивідуальних ізомерів та суміші ізомерів та їх фармацевтично прийнятних солей. КОРОТКИЙ ОПИС КРЕСЛЕНЬ Фіг.1: порошкова рентгенівська дифрактограма (з використанням мідного джерела (лямбда = 1,54A) для вимірювання) для аморфної твердодисперсної композиції прикладу 9 (див. приклад 41), з 25 % навантаженням сполуки прикладу 9 з PVP VA64 (37,5 %) та Pharmacoat 603 (37,5 %). Фіг.2: тварини з підшкірними KCL-22 ксенотрансплантатами, які отримували щоденне лікування сполукою прикладу 9. Продемонстрована дозозалежна протипухлинна активність. Фіг.3: KCL-22 клітини вирощували як підшкірні ксенотрансплантати, та чотири тварини отримували дозу 75 мг/кг Нілотинібу BID (два рази на день). Коли у пухлин розвивалась резистентність до лікування Нілотинібом, дозування змінювали до 30 мг/кг сполуки прикладу 9 два рази на день. Лікування нілотиніб-резистентних пухлин сполукою прикладу 9 привело до регресії пухлин. Кожна лінія представляє окрему тварину. Фіг.4: тваринам з підшкірними KCL-22 ксенотрансплантатами вводили дозу у комбінації 30 мг/кг сполуки прикладу 9 два рази на день та 75 мг/кг Нілотинібу два рази на день. Кожна лінія представляє окрему тварину. Повну регресію пухлини спостерігали у всіх тварин та підтримували до кінця дослідження. Визначення 2 UA 113208 C2 5 10 15 20 25 30 35 Загальні терміни, використовувані вище та нижче у даній заявці, переважно мають у контексті даного розкриття наступні значення, якщо не зазначене інше, при цьому більш загальні терміни, де б вони не використовувалися, незалежно один від іншого, можуть бути замінені більш конкретними визначеннями або зберігатися, у такий спосіб визначаючи більш докладні варіанти втілення даного винаходу. "Алкіл" відноситься до розгалужених або нерозгалужених вуглеводневих груп, що містять від 1 до 7 атомів вуглецю (C1-7алкіл) або від 1 до 4 атомів вуглецю (C1-4алкіл). Репрезентативні приклади алкілу включають, але не обмежуються цим, метил, етил, н-пропіл, ізо-пропіл, нбутил, втор-бутил, ізо-бутил, трет-бутил, н-пентил, ізопентил, неопентил, н-гексил, 3метилгексил, 2,2-диметилпентил, 2,3-диметилпентил, н-гептил, н-октил, н-ноніл, н-децил та подібні. Заміщений алкіл являє собою алкільну групу, яка містить один або декілька, наприклад, один, два або три замісники, вибрані з галогену, гідрокси групи або алкоксигрупи. Галогензаміщений алкіл та галогензаміщена алкокси група може бути або лінійною, або розгалуженою та включає метокси групу, етокси групу, дифторметил, трифторметил, пентафторетил, дифторметокси групу, трифторметокси групу та подібні. "Арил" означає моноциклічну або конденсовану біциклічну ароматичну кільцеву структуру, що містить від шести до десяти кільцевих атомів вуглецю. Наприклад, арил може являти собою феніл або нафтил, переважно феніл. "Арилен" означає двовалентний радикал, утворений з арильної групи. "BCR-ABL1" відноситься до гібридного білку, утвореного з N-кінцевих екзонів гену, що включає область з кластером точкових розривів (BCR), та основної C-кінцевої частини (екзони 2-11) Abelson(ABL1) гену. Найбільш розповсюджені гібридні транскрипти кодують 210-кДа білок (p210BCR-ABL1), хоча більш рідкі транскрипти кодують 190-кДа білок (p190BCR-ABL1) та 230кДа білок (p230BCR-ABL1). ABL1 послідовності цих білків містять ABL1 тирозинкіназний домен, який є жорстко регульованим у білку дикого типу, але конститутивно активованим у BCR-ABL1 гібридних білках. Ця тирозинкіназа з порушеною регуляцією взаємодіє з декількома клітинними сигнальними путями, приводячи до трансформації та порушеної регуляції проліферації клітин. "BCR-ABL1 мутанти" відносяться до численних моносайтових мутацій у BCR-ABL1, що включають: Glu255→Лізин, Glu255→Валін, Thr315→Ізолейцин, Met244→Val, Phe317→Leu, Leu248→Val, Met343→Thr, Gly250→Ala, Met351→Thr, Gly250→Glu, Glu355→Gly, Gln252→His, Phe358→Ala, Gln252→Arg, Phe359→Val, Tyr253→His, Val379→Ile, Tyr253→Phe, Phe382→Leu, Glu255→Lys, Leu387→Met, Glu255→Val, His396→Pro, Phe311→Ile, His396→Arg, Phe311→Leu, Ser417→Tyr, Thr315→Ile, Glu459→Lys та Phe486→Ser. Сполуки відповідно до даного винаходу є чутливими до заміщення на R 3/R4 заміщеному кільці у положенні, яке є орто відносно точки приєднання NHC(O) групи. Для порівняння представлені, наприклад, наступні сполуки формули (I). Значення IC 50 прикладу 2 становить 1 нМ у порівнянні з хлор- або метилзаміщенням, де значення IC50 становить 1,6 та 1,8 мкМ, відповідно: Caliper ABL1 (64-515) IC50 [мкM] Сполука формули (I) 0,001 Приклад 2 3 UA 113208 C2 1,6 1,8 5 10 15 20 25 30 35 40 "Гетероарил" має значення, визначене для арилу вище, де один або декілька з кільцевих членів являє собою гетероатом. Наприклад, 5-8-членний гетероарил містить мінімум 5 членів кільця, вибраних з вуглецю, азоту, кисню та сірки. Отже, 5-8-членний гетероарил включає піридил, індоліл, індазоліл, хіноксалініл, хінолініл, бензофураніл, бензопіраніл, бензотіопіраніл, бензо[1,3]діоксол, імідазоліл, бензоімідазоліл, піримідиніл, фураніл, оксазоліл, ізоксазоліл, триазоліл, тетразоліл, піразоліл, тієніл та т. д. "Циклоалкіл" означає насичену, моноциклічну, біциклічну або зв'язану містковим зв'язком поліциклічну кільцеву структуру, що містить зазначену кількість кільцевих атомів. Наприклад, C3-10циклоалкіл включає циклопропіл, циклобутил, циклопентил, циклогексил та т. д. Частково ненасичений циклоалкіл означає циклоалкіл, як визначено вище, з щонайменше одним подвійним зв'язком. "Гетероциклоалкіл" означає циклоалкіл, як він визначений у даній заявці, за умови, що один або декілька із зазначених кільцевих атомів вуглецю заміщені групою, вибраною з -O-, -N=, -NR-, -C(O)-, -S-, -S(O) - або -S(O)2-, де R являє собою водень, C1-4алкіл або азот-захисну групу (наприклад, карбобензилокси групу, пара-метоксибензилкарбоніл, трет-бутилоксикарбоніл, ацетил, бензоїл, бензил, пара-метокси-бензил, пара-метокси-феніл, 3,4-диметоксибензил та подібні). Наприклад, 3-8-членний гетероциклоалкіл включає морфоліно, піролідиніл, піролідиніл2-он, піперазиніл, піперидиніл, піперидинілон, 1,4-діокса-8-аза-спіро[4,5]дец-8-ил, тіоморфоліно, сульфаноморфоліно, сульфономорфоліно тощо. "Галоген" (або гало) переважно являє собою хлор або фтор, але також може бути бромом або йодом. GLEEVEC® (іматиніб мезилати) показаний для лікування пацієнтів з KIT (CD117)позитивними неоперабельними та/або метастатичними злоякісними гастроінтестинальними стромальними пухлинами (GIST). Він також показаний для лікування дорослих пацієнтів після повної макроскопічної резекції KIT (CD117)-позитивних GIST. Він також показаний для лікування вперше діагностованих дорослих пацієнтів та пацієнтів дитячого віку з філадельфійська хромосома-позитивним хронічним мієлоцитарним лейкозом з (Ph+CML) у хронічній фазі та пацієнтів з Ph+CML у бластному кризі (BC), фазі акселерації (AP) або у хронічній фазі (CP) після невдачі застосування інтерферон-альфа терапії. Він може також бути використаний як прицільно діючий лікарський засіб для лікування наступних рідкісних захворювань з обмеженими властивостями лікування: рецидивного або резистентного філадельфійська хромосома-позитивного гострого лімфобластного лейкозу (Ph+ALL); мієлодиспластичних/мієлопроліферативних захворювань (MDS/MPD), зв'язаних з перегрупуванням генів рецепторів тромбоцитарного фактору росту (PDGFR); агресивного системного мастоцитозу (ASM) без D816V c-KIT мутації або з невідомим мутаційним статусом cKIT; гіпереозинофільного синдрому/хронічного еозинофільного лейкозу (HES/CEL) з FIP1L1PDGFRα гібридною кіназою (мутаційний аналіз або FISH демонстрація делеції CHIC2 алелю) та для пацієнтів з HES та/або CEL, які є FIP1L1-PDGFRα гібридна кіназа-негативними або це невідомо; та неоперабельних, рецидивуючих та/або метастатичних випираючих дерматофібросарком (DFSP). TASIGNA® (нілотиніб) показаний для лікування дорослих пацієнтів з вперше діагностованим філадельфійська хромосома-позитивним хронічним мієлоцитарним лейкозом (Ph+CML) у 4 UA 113208 C2 5 10 15 20 25 хронічній фазі. Він може бути використаний для лікування дорослих пацієнтів, які більше не отримують позитивного результату або не переносять інші терапії, включаючи іматиніб (GLEEVEC®), або отримували інші терапії, включаючи іматиніб (GLEEVEC), але не можуть переносити їх. SPRYCEL® (дазатиніб) являє собою рецептурний лікарський засіб для лікування дорослих пацієнтів з вперше діагностованим філадельфійська хромосома-позитивним (Ph+) хронічним мієлоцитарним лейкозом (CML) у хронічній фазі та для лікування дорослих пацієнтів, які більше не отримують позитивного результату або не переносять інші терапії, а також для пацієнтів з ALL. BOSULIF® (Босутиніб) являє собою рецептурний лікарський засіб для лікування дорослих пацієнтів з вперше діагностованим філадельфійська хромосома-позитивним (Ph+) хронічним мієлоцитарним лейкозом (CML) у хронічній фазі та для лікування дорослих пацієнтів, які більше не отримують позитивного результату або не переносять інші терапії, а також для пацієнтів з ALL. Сполуки формули (I) можуть мати різні ізомерні форми. Наприклад, будь-який асиметричний атом вуглецю може бути присутній у (R)-, (S)- або (R, S)-конфігурації, переважно у (R)- або (S)конфігурації. Замісники на подвійному зв'язку або особливо на кільці можуть бути присутніми у цис- (=Z-) або транс- (=E-) формі. Сполуки, таким чином, можуть бути присутніми у вигляді сумішей ізомерів або переважно у вигляді чистих ізомерів, переважно у вигляді чистих діастереомерів або чистих енантіомерів. Наступні сполуки формули (I) можуть існувати у таутомерній формі: Для ілюстрації таутомерії з наступними конкретними прикладами (R)-N-(4(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1H-піразол-5-іл)нікотинамід (права структура, нижче) являє собою таутомер (R)-N-(4-(хлордифторметокси)феніл)-6-(3гідроксипіролідин-1-іл)-5-(1H-піразол-3-іл)нікотинаміда (ліва структура, нижче), та навпаки: 30 35 Коли використовується форма множини (наприклад, сполуки, солі), вона включає і однину (наприклад, одна сполука, одна сіль). "Сполука" не виключає, що присутня (наприклад, у фармацевтичній композиції) більше ніж одна сполука формули (I) (або її сіль). Форму однини, таким чином, переважно слід розуміти як "один або декілька", менш переважно, альтернативно, як "один". 5 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 Термін "та/або її N-оксид, її таутомер та/або її (переважно фармацевтично прийнятна) сіль", головним чином, означає, що сполука формули (I) може бути присутня як така або у суміші з її N-оксидом, у вигляді таутомеру (наприклад, у результаті кето-енольної, лактам-лактимної, амідімідокислотної або енамін-імінової таутомерії), або у вигляді (наприклад, викликаною еквівалентною реакцією) суміші з її таутомером, або у вигляді солі сполуки формули (I) та/або будь-якої з цих форм або сумішей двох або більше таких форм. Будь-які формули, представлені у даній заявці, призначені також для представлення немічених форм, а також ізотопномічених форм сполук. Ізотопномічені сполуки мають структури, відображені формулами, приведеними у даній заявці, за виключенням того, що один або декілька атомів замінені атомом, що має вибрану атомну масу або масове число. Приклади ізотопів, які можуть бути включені у сполуки відповідно до даного винаходу, включають ізотопи 2 3 11 13 14 15 18 31 водню, вуглецю, азоту, кисню, фосфору, фтору та хлору, такі як H, H, C, C, C, N, F, P, 32 35 36 123 124 125 P, S, Cl, I, I, I, відповідно. Винахід включає різні ізотопномічені сполуки, як визначено 3 14 у даній заявці, наприклад, ті, у яких радіоактивні ізотопи, такі як H та C, або ті, у яких присутні 2 13 нерадіоактивні ізотопи, такі як H та C. Такі ізотопномічені сполуки корисні у метаболічних 14 2 3 дослідженнях (з C), у дослідженнях кінетики реакцій (наприклад, з H або H), методах детекції або візуалізації, таких як позитронно-емісійна томографія (PET) або однофотонна емісійна комп'ютерна томографія (SPECT), включаючи аналізи розподілу лікарських засобів або тканин 18 субстрату або у радіоактивній терапії пацієнтів. Зокрема, F або мічена сполука можуть бути особливо бажаними для PET або SPECT досліджень. Ізотопномічені сполуки відповідно до даного винаходу, як правило, можуть бути отримані за звичайними способами, відомими спеціалістам у даній галузі техніки, або за способами, аналогічними тим, які описані у Прикладах, що додаються, з використанням підходящих ізотопномічених реагентів. 2 Крім того, заміщення більш важкими ізотопами, особливо дейтерієм (тобто H або D), може давати певні терапевтичні переваги у результаті більш високої метаболічної стабільності, наприклад, більший період напівжиття in vivo або зниження рівня необхідних доз або покращення терапевтичного індексу. Зрозуміло, що дейтерій у цьому контексті розглядається як замісник сполуки відповідно до даного винаходу. Концентрація такого важкого ізотопу, особливо дейтерію, може бути визначена за допомогою ізотопного коефіцієнту збагачення. Термін "ізотопний коефіцієнт збагачення", використовуваний у даній заявці, означає співвідношення між розповсюдженістю ізотопу та природною розповсюдженістю конкретного ізотопу. Якщо замісник у сполуці відповідно до даного винаходу позначений як дейтерій, така сполука має ізотопний коефіцієнт збагачення для кожного зазначеного атому дейтерію щонайменше 3500 (52,5 % включення дейтерію у кожному певному атомі дейтерію), щонайменше 4000 (60 % включення дейтерію), щонайменше 4500 (67,5 % включення дейтерію), щонайменше 5000 (75 % включення дейтерію), щонайменше 5500 (82,5 % включення дейтерію), щонайменше 6000 (90 % включення дейтерію), щонайменше 6333,3 (95 % включення дейтерію), щонайменше 6466,7 (97 % включення дейтерію), щонайменше 6600 (99 % включення дейтерію) або щонайменше 6633,3 (99,5 % включення дейтерію). Наприклад, сполука формули Ib, представлена у даній заявці, де R 3 являє собою водень та Y являє собою CH, може включати дейтерій на піролідинільному кільці, як показано: Ця дейтерована форма менш схильна до метаболічної трансформації (зліва вище) у порівнянні з недейтерованою формою (справа вище). Опис кращих варіантів втілення Даний винахід відноситься до сполук, здатних до інгібування активності BCR-ABL1 або мутантів BCR-ABL1 через алостеричний міристоїл-зв'язуючий сайт. У одному варіанті втілення, що відноситься до сполук відповідно до даного винаходу, представлені сполуки формули (Ib): 6 UA 113208 C2 , 5 10 де: R3 вибирають з водню та галогену; R4 вибирають з -SF5 та -Y2-CF2-Y3; R6, коли зв'язаний з азотом піразолільного кільця, вибирають з водню, метилу, гідроксиетилу, фторетилу, етилу та циклопропілу; та R6, коли зв'язаний з атомом вуглецю піразолільного кільця, вибирають з водню, гідрокси групи, метилу, метокси групи, ціано групи, трифторметилу, гідроксиметилу, галогену, аміно групи, фторетилу, етилу та циклопропілу; R 7 вибирають з гідрокси групи, метилу, галогену, метокси групи, гідроксиметилу, аміно групи, метил-аміно групи, аміно-метилу, трифторметилу, 2-гідроксипропан-2-ілу, метил-карбоніл-аміно групи, диметил-аміно групи, 2аміно-3-метилбутаноїл)окси групи, карбокси групи, метоксикарбонілу, фосфоноокси групи, ціано групи та амінокарбонілу; Y1 вибирають з CH та N; Y2 вибирають з CF2, O та S(O)0-2; Y3 вибирають з водню, фтору, хлору, метилу, дифторметилу та трифторметилу; або їх фармацевтично прийнятні солі. У наступному варіанті втілення представлені сполуки формули (Ic): , 15 20 25 30 35 де: R3 вибирають з водню та галогену; R4 вибирають з -SF5 та -Y2-CF2-Y3; R6, коли зв'язаний з азотом піразолільного кільця, вибирають з водню, метилу, гідроксиетилу, фторетилу, етилу та циклопропілу; та R6, коли зв'язаний з атомом вуглецю піразолільного кільця, вибирають з водню, гідрокси групи, метилу, метокси групи, ціано групи, трифторметилу, гідроксиметилу, галогену, аміно групи, фторетилу, етилу та циклопропілу; R 7 вибирають з гідрокси групи, метилу, галогену, метокси групи, гідроксиметилу, аміно групи, метил-аміно групи, аміно-метилу, трифторметилу, 2-гідроксипропан-2-ілу, метил-карбоніл-аміно групи, диметил-аміно групи, 2аміно-3-метилбутаноїл)окси групи, карбокси групи, метоксикарбонілу, фосфоноокси групи, ціано групи та амінокарбонілу; Y1 вибирають з CH та N; Y2 вибирають з CF2, O та S(O)0-2; Y3 вибирають з водню, фтору, хлору, метилу, дифторметилу та трифторметилу; або їх фармацевтично прийнятні солі. У іншому варіанті втілення представлені сполуки формули (I) або їх фармацевтично прийнятні солі, де R1 являє собою піразоліл; де зазначений піразоліл є незаміщеним або заміщений від 1 до 2 груп R6. У наступному варіанті втілення R1 являє собою незаміщений піразоліл. У наступному варіанті втілення R1 являє собою піразоліл, заміщений однією групою R6. У наступному варіанті втілення R1 являє собою піразоліл, заміщений двома групами R6. У іншому варіанті втілення R2 являє собою піролідин-1-іл, заміщений однією групою R7. У іншому варіанті втілення Y вибирають з CH та N. У наступному варіанті втілення Y являє собою N. У наступному варіанті втілення Y являє собою CH. У іншому варіанті втілення Y1 вибирають з CH та N. У наступному варіанті втілення Y1 являє собою N. 7 UA 113208 C2 5 10 15 20 25 30 У наступному варіанті втілення Y1 являє собою CH. Наступні додаткові варіанти відносяться до сполук будь-якої однієї з формул (I), (Ib) або (Ic) або їх фармацевтично прийнятних солей. У іншому варіанті втілення R3 вибирають з водню та галогену. У іншому варіанті втілення R4 вибирають з -SF5 та -Y2-CF2-Y3. У наступному варіанті втілення R4 являє собою хлордифторметокси групу. У наступному варіанті втілення R4 являє собою трифторметокси групу. У іншому варіанті втілення R6 у кожному випадку незалежно вибирають з водню, гідрокси групи, метилу, метокси групи, ціано групи, трифторметилу, гідроксиметилу, галогену, аміно групи, фторетилу, етилу та циклопропілу. У наступному варіанті втілення R6, коли зв'язаний з азотом піразолільного кільця, вибирають з водню, метилу, гідроксиетилу, фторетилу, етилу та циклопропілу. У наступному варіанті втілення R6, коли зв'язаний з атомом вуглецю піразолільного кільця, вибирають з водню, гідрокси групи, метилу, метокси групи, ціано групи, трифторметилу, гідроксиметилу, галогену, аміно групи, фторетилу, етилу та циклопропілу. У іншому варіанті втілення R7 вибирають з гідрокси групи, метилу, галогену, метокси групи, гідроксиметилу, аміно групи, метил-аміно групи, аміно-метилу, трифторметилу, 2гідроксипропан-2-ілу, метил-карбоніл-аміно групи, диметил-аміно групи, 2-аміно-3метилбутаноїл)окси групи, карбокси групи, метоксикарбонілу, фосфоноокси групи, ціано групи та амінокарбонілу. У іншому варіанті втілення Y2 вибирають з CF2, O та S(O)0-2. У наступному варіанті втілення Y2 являє собою O. У наступному варіанті втілення Y2 являє собою CF2. У наступному варіанті втілення Y2 являє собою S(O)0-2. У іншому варіанті втілення Y3 вибирають з водню, хлору, фтору, метилу, дифторметилу та трифторметилу. У наступному варіанті втілення Y3 являє собою хлор. У наступному варіанті втілення Y3 являє собою фтор. У наступному варіанті втілення представлені сполуки або їх фармацевтично прийнятні солі, вибрані з: У іншому варіанті втілення представлені сполуки або їх фармацевтично прийнятні солі, вибрані з: 35 8 UA 113208 C2 9 UA 113208 C2 У іншому варіанті втілення представлені сполуки або їх фармацевтично прийнятні солі, вибрані з: 5 10 UA 113208 C2 У іншому варіанті втілення представлена сполука або її фармацевтично прийнятна сіль, яка являє собою: 5 У іншому варіанті втілення представлені сполуки, вибрані з: 10 15 Фармакологія та корисність На основі досліджень інгібування, описаних у розділі "Аналіз" нижче, сполука формули (I) у відповідності з даним винаходом демонструє терапевтичну ефективність, особливо проти розладів, залежних від BCR-ABL1 активності. Зокрема, сполуки відповідно до даного винаходу інгібують алостеричний або міристоїл-зв'язуючий сайт BCR-ABL1 (включаючи BCR-ABL1 дикого типу та/або його мутації). Об'єднання ATP-конкурентного інгібітору BCR-ABL1 з алостеричним інгібітором BCR-ABL1 уповільнює набуту резистентність у BCR-ABL1+KCL-22 клітинах in vitro. На диво, BCRABL1+KCL-22 клітини, які обробляли через кожні 3-4 дні сполукою відповідно до даного 11 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 60 винаходу, показали набуту резистентність після приблизно 28 днів, тоді як ті ж самі клітини, які обробляли через кожні 3-4 дні нілотинібом або дасатинібом, показали набутну резистентність вже через 18-21 день. Ще більш дивно, коли BCR-ABL1+KCL-22 клітини обробляли через кожні 3-4 дні комбінацією сполуки відповідно до даного винаходу з або нілотинібом, або дасатинібом, не спостерігали ніякої набутої резистентності щонайменше протягом перших 60 днів. Тому сполуки відповідно до даного винаходу, що зв'язуються з міристоїл-зв'язуючим сайтом, у комбінації з інгібіторами BCR-ABL1, які зв'язуються із сайтом зв'язування ATP, мають особливо важливе значення для лікування проліферативних захворювань, у яких має місце позитивна регуляція ABL1 кіназної активності, як у випадку з BCR-ABL1 гібридними білками у CML та підвидах інших гематологічних злоякісних захворювань, таких як ALL та AML. Клітини карциноми використовують invapodia для руйнування позаклітинного матриксу у процесі інвазії та метастазування пухлини. ABL кіназна активність необхідна для SRCіндукованого утворення invapodia, регуляції окремих стадій складання та функції invapodia. Тому сполуки відповідно до даного винаходу, як інгібітори ABL, мають потенціал для використання як терапевтичні засоби для лікування метастатичних інвазивних карцином. Алостеричний інгібітор ABL1 кінази можна використовувати для лікування раку головного мозку: включаючи гліобластому, яка є найпоширенішою та найбільш агресивною злоякісною первинною пухлиною головного мозку, у якій експресія ABL1 імуногістохімічно визначається у субпопуляції пацієнтів (Haberler C, Gelpi E, Marosi C, Rоssler K, Birner P, Budka H, Hainfellner JA. Immunohistochemical analysis of platelet-derived growth factor receptor-alpha, -beta, c-KIT, ABL1 та ABL2 proteins in glioblastoma: possible implications for patient selection for imatinib mesylate therapy. J Neurooncol. 2006 Jan; 76(2):105-9). Однак клінічні випробування з використанням Gleevec® не були успішними у пацієнтів з гліобластомою (Reardon DA, Dresemann G, Taillibert S, Campone M, van den Bent M, Clement P, Blomquist E, Gordower L, Schultz H, Raizer J, Hau P, Easaw J, Gil M, Tonn J, Gijtenbeek A, Schlegel U, Bergstrom P, Green S, Weir A, Nikolova Z. Multicentre phase II studies evaluating imatinib plus hydroxyurea in patients with progressive glioblastoma. Br J Cancer. 2009 Dec 15; 101(12):1995-2004; Razis E, Selviaridis P, Labropoulos S, Norris JL, Zhu MJ, Song DD, Kalebic T, Torrens M, Kalogera-Fountzila A, Karkavelas G, Karanastasi S, Fletcher JA, Fountzilas G. Phase II study of neoadjuvant imatinib in glioblastoma: evaluation of clinical and molecular effects of the treatment. Clin Cancer Res. 2009 Oct 1; 15(19):6258-66; Dresemann G. Imatinib and hydroxyurea in pretreated progressive glioblastoma multiforme: a patient series. Ann Oncol. 2005 Oct; 16(10):1702-8), можливо через поганий внутрішньопухлинний вплив лікарського засобу у головному мозку та за відсутності порушеного гемато-енцефалічного бар'єру (Holdhoff et al, J Neurooncol. 2010; 97(2):241-5). Дійсно, у передклінічних дослідженнях було показано, що транспорт Gleevec® через гемато-енцефалічний бар'єр обмежується активним транспортом речовин, що витікають, таких як P-глікопротеїн. Це також відноситься і до дазатинібу (Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J Pharmacol Exp Ther. 2009 Sep; 330(3):956-63). Відомо, що опромінення підсилює відкриття гемато-енцефалічного бар'єру. У мишачих моделях, відповідь мультиформної гліобластоми на Gleevec® співвідносилася зі збільшенням затримки пухлинного росту та підвищенням виживання, коли Gleevec® вводили у комбінації із щоденним опроміненням (Geng L, Shinohara ET, Kim D, Tan J, Osusky K, Shyr Y, Hallahan DE. STI571 (Gleevec) improves tumor growth delay and survival in irradiated mouse models of glioblastoma. Int J Radiat Oncol Biol Phys. 2006 Jan 1; 64(1):263-71). Тому новий інгібітор ABL1 з високою експозицією у головному мозку являє собою серйозний терапевтичний підхід для лікування гліобластоми та інших пухлин головного мозку. ЦНС-CML: повідомлялось про ЦНС бласткризу та несприятливий результат у деяких CML пацієнтів, яких лікували препаратом Gleevec®, та це можна пояснити низькою експозицією Gleevec® у головному мозку (Kim HJ, Jung CW, Kim K, Ahn JS, Kim WS, Park K, Ko YH, Kang WK, Park K. Isolated blast crisis in CNS in a patient with chronic myelogenous leukemia maintaining major cytogenetic response after imatinib. J Clin Oncol. 2006 Aug 20; 24(24):4028-9; Radhika N, Minakshi M, Rajesh M, Manas BR, Deepak Kumar M.Central nervous system blast crisis in chronic myeloid leukemia on imatinib mesylate therapy: report of two cases. Indian J Hematol Blood Transfus. 2011 Mar; 27(1):51-4). Дійсно, у CML пацієнтів концентрація Gleevec® насправді набагато нижче (у ~100 разів) у ЦНС, ніж у плазмі (Leis JF, Stepan DE, Curtin PT, Ford JM, Peng B, Schubach S, Druker BJ, Maziarz RT. Central nervous system failure in patients with chronic myelogenous leukemia lymphoid blast crisis and Philadelphia chromosome positive acute lymphoblastic leukemia treated with imatinib (STI-571). Leuk Lymphoma. 2004 Apr; 45(4):695-8). Тому інгібітори ABL1 відповідно до даного винаходу, які демонструють високу експозицію у головному мозку, являють собою ефективний підхід для розробки терапевтичних засобів проти CML, включаючи ЦНС-CML. 12 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 60 Сполуки відповідно до даного винаходу можуть бути корисними у лікуванні вірусних інфекцій. Наприклад, вірусні інфекції можуть бути опосередковані ABL1 кіназною активністю, як у випадку покс-вірусів та вірусу Ебола. Було показано, що Gleevec® та Tasigna® зупиняють вивільнення частинок вірусу Ебола з інфікованих клітин, in vitro (Kalman, Daniel; Bornmann, William Gerard, Methods of use of non-ATP competitive tyrosine kinase inhibitors to treat pathogenic infection, PCT Int. Appl. 2007, WO 2007002441; Garcia Mayra; Cooper Arik; Shi Wei; Bornmann William; Carrion Ricardo; Kalman Daniel; Nabel Gary J. Productive Replication of Ebola Virus Is Regulated by the ABL1 Tyrosine Kinase. Science translational medicine 2012; 4:123ra24). Тому можна очікувати, що сполуки відповідно до даного винаходу, які інгібують ABL1 кіназу, будуть знижувати здатність патогену до реплікації. Сполуки відповідно до даного винаходу також можуть бути корисними у лікуванні нервової дегенерації. Хоча нативна ABL1 тирозинова кіназа залишається у відносному стані спокою у головному мозку здорової дорослої людини, вона може активуватися у головному мозку пацієнтів з ЦНС захворюваннями, включаючи нейродегенеративні захворювання, такі як хвороба Альцгеймера (AD), хвороба Паркінсона (AD), фронтотемпоральна деменція (FTD), хвороба Піка, хвороба Німанна-Піка C типу (NPC) та інші дегенеративні, запальні та аутоімунні захворювання та старіння. Хвороба Паркінсона є другим найбільшим розповсюдженим хронічним нейродегенеративним захворюванням, при цьому найбільше розповсюджена спадкова аутосомально-рецесивна форма викликана мутаціями у E3 убіхітинлігазі, parkin. Останні дослідження показали, що активований ABL1/ABL2 був виявлений у смугастому тілі у пацієнтів із спорадичною хворобою Паркінсона. Одночасно з цим, parkin був тирозин-фосфорильованим, викликаючи втрату його убіхітинлігази та цитозахисних дій, на що вказувала акумуляція parkin субстратів (Ko HS, Lee Y, Shin JH, Karuppagounder SS, Gadad BS, Koleske AJ, Pletnikova O, Troncoso JC, Dawson VL, Dawson TM. Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin's ubiquitination and protective function. Proc Natl Acad Sci U S A. 2010 Sep 21; 107(38):16691-6; Imam SZ, Zhou Q, Yamamoto A, Valente AJ, Ali SF, Bains M, Roberts JL, Kahle PJ, Clark RA, Li S. Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: implications for Parkinson's disease. J Neurosci. 2011 Jan 5; 31(1):157-63). Ці два дослідження також показали, що у клітині або тваринних моделях хвороби Паркінсона фармакологічне інгібування ABL1 кінази або генетичний ABL1 "нокдаун" перешкоджали фосфорилюванню тирозину білку parkin та відновлювали активність його E3 лігази та цитозахисну функцію як in vitro, так і in vivo. Ці результати показують, що ABL1-залежне фосфорилювання тирозину білку parkin являє собою основну після-трансляційну модифікацію, яка приводить до втрати функції parkin та прогресуванню захворювання у спорадичній PD. Тому здатність сполук відповідно до даного винаходу інгібувати міристатзв'язуючий сайт ABL1, як можна очікувати, відкриє нові терапевтичні можливості для блокування розвитку хвороби Паркінсона. Хвороба Альцгеймера характеризується двома основними особливостями: позаклітинні відкладення нейротоксичного амілоїду-β, що приводить до розвитку амілоїдних бляшок та внутрішньоклітинної акумуляції гіперфосфорильованого tau, що сприяє розвитку нейрофібрилярних сплетень (NFT). Рівень амілоїду-β знижується після інтратекального введення Gleevec® у головному мозку морських свинок дикого типу та у клітинних моделях (Netzer WJ, Dou F, Cai D, Veach D, Jean S, Li Y, Bornmann WG, Clarkson B, Xu H, Greengard P. Gleevec inhibits beta-amyloid production but not Notch cleavage. Proc Natl Acad Sci U S A. 2003 Oct 14; 100(21):12444-9). Цією ж групою було зроблено припущення, що Gleevec® досягає амілоїд-β-знижуючого ефекту через новий механізм, що перешкоджає GSAP взаємодії з гама-секретазним субстратом, APP-CTF (He G, Luo W, Li P, Remmers C, Netzer WJ, Hendrick J, Bettayeb K, Flajolet M, Gorelick F, Wennogle LP, Greengard P. Gamma-secretase activating protein is a therapeutic target for Alzheimer's disease. Nature. 2010 Sep 2; 467(7311):95-8). У цьому дослідженні ефект Gleevec®, такий як інгібування GSAP/APP-CTF, спостерігали тільки при мікромолярних концентраціях. Інша група показала, що фосфорилювання тирозину внутрішньоклітинного домену APP (тобто Tyr682) регулює процесинг амілоїдогенного APP, прискорюючи утворення амілоїду-β in vivo (Barbagallo AP, Weldon R, Tamayev R, Zhou D, Giliberto L, Foreman O, D'Adamio L. Tyr(682) in the intracellular domain of APP regulates amyloidogenic APP processing in vivo. PLoS One. 2010 Nov 16; 5(11):e15503). Інші дослідження показали, що APP є тирозин-фосфорильованим у клітинах, що експресують конститутивно активну форму ABL1 онкогену (Zambrano N, Bruni P, Minopoli G, Mosca R, Molino D, Russo C, Schettini G, Sudol M, Russo T. The beta-amyloid precursor protein APP is tyrosine-phosphorylated in cells expressing a constitutively active form of the Abl protoncogene. J 13 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 60 Biol Chem. 2001 Jun 8; 276(23):19787-92). Ці дані, взяті разом, припускають ABL 1-залежний процесинг амілоїдогенного APP для утворення токсичного амілоїд-β пептиду та наступних амілоїдних бляшок. Тому можна чекати, що інгібітор ABL1 буде знижувати утворення амілоїдних бляшок у пацієнтів із хворобою Альцгеймера. Було показано, що Tau фосфорилюється ABL1 кіназою по тирозинам 18, 197, 310 та 394 у клітинних моделях, та було показано, що tau pY394 присутня в ураженнях NFT у головному мозку AD пацієнтів. ABL1 активований у головному мозку пацієнтів із спорадичною хворобою Альцгеймера, як показано по його фосфорилюванню або по Y412, індикатору активації, який ко-локалізує грануловаскулярну дегенерацію, або по T735, який ко-локалізований з типовими ураженнями, амілоїдними бляшками, нейрофібрилярними сплетеннями (NFT), на додаток до GVD. Амілоїд-β та окисний стрес активують ABL1 кіназу у культурах нервових клітин, та інтрацеребральна ін'єкція фібрилярного амілоїдного пептиду приводить до підвищеної експресії ABL1 та далі на цьому шляху ефектору p73. Трансгенні миші (APP/Swe мишача модель AD) показали підвищені рівні ABL1 у головному мозку, та, коли цих мишей обробляли інгібітором ABL1 Gleevec®, tau фосфорилювання знижувалося у головному мозку тварин. Трансгенна мишача модель, що експресує конститутивно активний ABL1 у нейронах переднього мозку, демонструвала втрату нейронів, важке нейрозапалення та тирозин-фосфорильований tau у головному мозку (див. огляд у Schlatterer SD, Acker CM, Davies P. c-Abl in neurodegenerative disease. J Mol Neurosci. 2011 Nov; 45(3):445-52). На підставі всіх цих результатів існує доказ ролі ABL1 кінази у патогенезі хвороби Альцгеймера для розвитку обох уражень - амілоїдних бляшок та нейрофібрилярних сплетень. Крім того, активований ABL1 також є присутнім у інших таупатіях, крім спорадичної хвороби Альцгеймера, у тому числі у головному мозку пацієнтів із фронтотемпоральною деменцією з N279K та P301L мутаціями, хворобою Піка та Гуам Паркінсон-деменцією (Schlatterer SD, Acker CM, Davies P. c-abl in neurodegenerative disease. J Mol Neurosci. 2011 Nov; 45(3):445-52). Тому сполуки відповідно до даного винаходу, інгібуючі ABL1 у ЦНС, являють собою ефективний підхід для розробки терапевтичних засобів проти хвороби Альцгеймера, а також інших β-амілоїдозів, таких як судинна деменція та інші таупатії, такі як фронтотемпоральна деменція та хвороба Піка. Хвороба Німанна-Піка C типу (NPC) являє собою фатальне аутосомальний рецесивний розлад, що характеризується акумуляцією вільного холестерину та глікосфінголіпідів у ендосомальній-лізосомальній системі та прогресуючою загибеллю нейронів, особливо мозочкових нервових клітин Пуркін'є. У мишачій моделі NPC проапоптичний ABL1, що знаходиться далі по ходу транскрипції мішень, а також p73 гени-мішені експресуються у мозочку. Інгібування ABL1 за допомогою Gleevec® запобігало втраті нервових клітин Пуркін'є, поліпшувало неврологічні симптоми та підвищувало виживаність. Цей ефект провиживання Gleevec® співвідносився зі зниженими рівнями мРНК p73 проапоптичних генів-мішеней (Alvarez AR, Klein A, Castro J, Cancino GI, Amigo J, Mosqueira M, Vargas LM, Yеvenes LF, Bronfman FC, Zanlungo S. Imatinib therapy blocks cerebellar apoptosis and improves neurological symptoms in a mouse model of Niemann-Pick type C disease. FASEB J. 2008 Oct; 22(10):3617-27). Тому сполуки відповідно до даного винаходу, що інгібують ABL1 кіназу, являють собою ефективний підхід для розробки терапевтичних засобів проти захворювань, викликаних проапоптичним шляхом ABL1/p73, таких як NPC. У моделях пріонових захворювань Gleevec® показав сприятливі ефекти: він уповільнював пріонову нейроінвазію шляхом інгібування розповсюдження пріону з периферії у ЦНС (Yun SW, Ertmer A, Flechsig E, Gilch S, Riederer P, Gerlach M, Schаtzl HM, Klein MA. The tyrosine kinase inhibitor imatinib mesylate delays prion neuroinvasion by inhibiting prion propagation in the periphery. J Neurovirol. 2007 Aug; 13(4):328-37). Gleevec® та дефіцит ABL1 індукували клітинний кліренс PrPSc у пріон-інфікованих клітинах (Ertmer A, Gilch S, Yun SW, Flechsig E, Klebl B, Stein-Gerlach M, Klein MA, Schаtzl HM. The tyrosine kinase inhibitor STI571 induces cellular clearance of PrPSc in prion-infected cells. J Biol Chem. 2004 Oct 1; 279(40):41918-27). Тому, нові інгібітори ABL1 відповідно до даного винаходу також являють собою ефективний терапевтичний підхід для лікування пріонових захворювань, таких як хвороба Крейцфельда-Якоба. Причиною X-зв'язаної рецесивної м'язової дистрофії Емері-Дрейфуса є мутації емерину, ядерно-мембранного білку, що відіграє роль у ядерній архітектурі, генній регуляції та передачі сигналів. Нещодавно проведене дослідження показало, що емерин тирозин-фосфорилюється безпосередньо за допомогою ABL1 у клітинних моделях та що статус фосфорилювання емерину змінює зв'язування емерину з іншими білками, такими як BAF. Це, у свою чергу, може пояснити міслокалізацію мутантного емерину з ядерного у цитозольні компартменти та 14 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 відповідні зміни у розташованому далі на цьому шляху ефекторі та сигнальному інтеграторі для сигнального шляху (шляхів) на ядерній оболонці (Tifft KE, Bradbury KA, Wilson KL. Tyrosine phosphorylation of nuclear-membrane protein emerin by SRC, ABL1 and other kinases. J Cell Sci. 2009 Oct 15; 122(Pt 20):3780-90). Зміни у емерин-ламін взаємодіях як у фазі мітозу, так і у інтерфазі є важливими для патології м'язової дистрофії. Крім того, результати іншого дослідження показують, що Gleevec® ослаблює скелетно-м'язову дистрофію у mdx мишей (Huang P, Zhao XS, Fields M, Ransohoff RM, Zhou L. Imatinib attenuates skeletal muscle dystrophy in mdx mice. FASEB J. 2009 Aug; 23(8):2539-48). Тому, нові інгібітори ABL1 відповідно до даного винаходу також являють собою терапевтичні підходи для лікування скелетних та м'язових дистрофій. Крім того, ABL1 кіназа відіграє роль у запаленні та окисному стресі, двох механізмах, які залучені у різні захворювання людини, від гострих захворювань ЦНС, таких як удар та травматичні ураження головного мозку або спинного мозку, хронічних захворювань ЦНС, таких як хвороба Альцгеймера, хвороба Паркінсона, хвороба Гентінгтона та захворювання рухових нейронів, до не пов'язаних із ЦНС запальних та аутоімунних захворювань, таких як діабет, фіброз легенів. Наприклад, Gleevec® попереджає фіброз у різних передклінічних моделях системного склерозу та індукує регресію встановленого фіброзу (Akhmetshina A, Venalis P, Dees C, Busch N, Zwerina J, Schett G, Distler O, Distler JH. Treatment with imatinib prevents fibrosis in different preclinical models of systemic sclerosis and induces regression of established fibrosis. Arthritis Rheum. 2009 Jan; 60(1):219-24),та він демонструє антифібротичні ефекти у блеоміциніндукованому фіброзі легенів у мишей (Aono Y, Nishioka Y, Inayama M, Ugai M, Kishi J, Uehara H, Izumi K, Sone S. Imatinib as a novel antifibrotic agent in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Crit Care Med. 2005 Jun 1; 171(11):1279-85). Інше дослідження показало, що як іматиніб, так і нілотиніб зменшували блеоміцин-індуковане гостре легеневе ураження та фіброз легенів у мишей (Rhee CK, Lee SH, Yoon HK, Kim SC, Lee SY, Kwon SS, Kim YK, Kim KH, Kim TJ, Kim JW. Effect of nilotinib on bleomycin-induced acute lung injury and pulmonary fibrosis in mice. Respiration. 2011; 82(3):273-87). Хоча у цих дослідженнях автори були сфокусовані на участі механізму, пов'язаного з PDGFR, який представляв інтерес, у дослідженні Rhee et al. (Respiration. 2011; 82(3):273-87) нілотиніб, який є значно більш сильним інгібітором с-ABL, ніж іматиніб, показав більш високі терапевтичні антифібротичні ефекти, таким чином підтверджуючи терапевтичну застосовність інгібіторів с-ABL для лікування захворювань людини з легеневим запаленням. У іншому дослідженні гіпероксія, що викликалася у мишей, підвищувала активацію ABL1, яка необхідна для фосфорилювання динаміну 2 та продукції реактивних видів кисню та легеневого просочування (Singleton PA, Pendyala S, Gorshkova IA, Mambetsariev N, Moitra J, Garcia JG, Natarajan V. Dynamin 2 and с-Abl are novel regulators of hyperoxia-mediated NADPH oxidase activation and reactive oxygen species production in caveolinenriched microdomains of the endothelium. J Biol Chem. 2009 Dec 11; 284(50):34964-75). Тому ці дані показують, що нові інгібітори c-ABL відповідно до даного винаходу мають терапевтичну застосовність для лікування захворювань людини з легеневим запаленням. Активація ABL1 інсуліном, через модифікацію FAK відповіді, може грати важливу роль у напрямку мітогенного проти метаболічного сигналу інсулінового рецептору (Genua M, Pandini G, Cassarino MF, Messina RL, Frasca F. c-Abl and insulin receptor signalling. Vitam Horm. 2009; 80:77105). Було показано, що інгібітори c-Abl, такі як Gleevec®, забезпечують регресію діабету 1 типу у не страждаючих ожирінням діабетичних мишей (Louvet C, Szot GL, Lang J, Lee MR, Martinier N, Bollag G, Zhu S, Weiss A, Bluestone JA. Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci U S A. 2008 Dec 2; 105(48):18895-900). Полегшення важкості діабету за допомогою Gleevec® імітували шляхом siРНК-опосередкованого "нокдауну" ABL1 мРНК (Hаgerkvist R, Sandler S, Mokhtari D, Welsh N. Amelioration of diabetes by imatinib mesylate (Gleevec): role of beta-cell NF-kappaB activation and anti-apoptotic preconditioning. FASEB J. 2007 Feb; 21(2):618-28). Тому нові інгібітори ABL1 відповідно до даного винаходу мають терапевтичну застосовність для лікування діабету у людини. Інгібітор ABL1 відповідно до даного винаходу можна використовувати у комбінації з одним або декількома з існуючих лікувань зазначених вище захворювань: наприклад, інгібітор ABL1 відповідно до даного винаходу можна використовувати у комбінації з Levodopa або іншими LDOPA-вмісними лікарськими засобами або допаміновим агоністом для лікування хвороби Паркінсона, або у комбінації з інгібітором холінестерази, таким як Exelon капсула або крізьшкірний пластир, для лікування хвороби Альцгеймера. 15 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 У хронічному мієлогенному лейкозі (CML), реципрокна збалансована хромосомна транслокація у гематопоетичних стовбурових клітинах (HSC) утворює BCR-ABL1 гібридний ген. Цей ген кодує онкогенний BCR-ABL1 гібридний білок. Тоді як ABL1 кодує жорстко регульовану тирозинову протеїнкіназу, яка відіграє фундаментальну роль у регуляції клітинної проліферації, адгезії та апоптозу, BCR-ABL1 гібридний ген кодує як конститутивно активовану кіназу. Ця активована кіназа трансформує HSC з утворенням фенотипу, що демонструє порушену регуляцію клональної проліферації, знижену здатність до адгезії до строми кісткового мозку та знижену апоптичну відповідь на мутагенні стимули, приводячи до прогресивно більш злоякісних трансформацій. Отримані у результаті гранулоцити не можуть розвиватися у зрілі лімфоцити та вивільняються у кровоток, приводячи до дефіциту зрілих клітин та підвищеної сприйнятливості до інфекції. Було показано, що ATP-конкурентні інгібітори BCR-ABL1 перешкоджають кіназній активації мітогенного та антиапоптичного шляхів (наприклад, PI-3 кіназа та STAT5), приводячи до загибелі клітин з BCR-ABL1 фенотипом і, таким чином, забезпечуючи ефективну терапію проти CML. KCL-22 клітинна лінія (отримана від DSMZ, Ltibniz Institute, Germany) виділена із плевральної ефузії 32-річної жінки за допомогою апарату хромосомно-позитивного CML Philadelphia у бластному кризі у 1981 р. та описана для вмісту t(9; 22), що приводить до гібридного гену BCR-ABL1 та мутації р53. KCL-22 клітинна лінія може бути використана у моделі Xenograft для демонстрації in vivo ефективності сполук винаходу (див. Assay section, infra). Сполуки відповідно до даного винаходу, як інгібітори BCR-ABL1, включаючи його мутанти, є, таким чином, особливо підходящими для лікування захворювань, пов'язаних з його надмірною експресією, такихяк ALL або CML лейкози. Також було показано, що сполуки відповідно до даного винаходу мають протипухлинну активність in vitro: in vitro протипухлинну активність випробовують, наприклад, з використанням лейкозних клітинних ліній, таких як Ba/F3-BCR-ABL1, KCL-22, K-562, MEG-01, KYO-1, LAMA-84, KU812, EM-2, CML-T1, BV-173 або ALL-SIL. Даний винахід включає спосіб лікування раку, що включає введення суб'єктові, який потребує такого лікування, ефективної кількості сполуки відповідно до даного винаходу або фармацевтичній композиції. Наступний варіант втілення включає введення суб'єктові додаткового терапевтичного засобу. У наступному варіанті втілення додатковий терапевтичний засіб являє собою інший інгібітор BCR-ABL1, вибраний з іматинібу, нілотинібу, дасатинібу, досутинібу, радотинібу, понатинібу та бафетинібу. У іншому варіанті втілення представлений спосіб лікування стану, опосередкованого BCRABL1, що включає введення суб'єктові, який цього потребує, ефективної кількості сполуки відповідно до даного винаходу або фармацевтичної композиції. BCR-ABL1 містить одну або декілька мутацій. Приклади таких мутацій включають V299L, T315I, F317I, F317L, Y253F, Y253H, E255K, E255V, F359C та F359V (UJane F. Apperley. Part 1: Mechanism of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncology 2007; 8:1018). У наступному варіанті втілення представлений спосіб лікування стану, опосередкованого BCR-ABL1, де BCR-ABL1 містить одну або декілька мутацій, вибраних з V299L, T315I, F317I, F317L, Y253F, Y253H, E255K, E255V, F359C та F359V. У деяких варіантах втілення даний винахід відноситься до зазначеного вище способу, де зазначену сполуку вводять парентерально. У деяких варіантах втілення даний винахід відноситься до зазначеного вище способу, де зазначену сполуку вводять внутрішньом'язово, внутрішньовенно, підшкірно, перорально, внутрішньолегенево, інтратекально, місцево або інтраназально. У деяких варіантах втілення даний винахід відноситься до зазначеного вище способу, де зазначену сполуку вводять системно. У деяких варіантах втілення даний винахід відноситься до зазначеного вище способу, де зазначений пацієнт являє собою ссавця. У деяких варіантах втілення даний винахід відноситься до зазначеного вище способу, де зазначений пацієнт являє собою примата. У деяких варіантах втілення даний винахід відноситься до зазначеного вище способу, де зазначений пацієнт являє собою людину. У іншому аспекті даний винахід відноситься до способу лікування ABL1/BCR-ABL1опосредованного розладу, що включає наступну стадію: введення пацієнту, який цього потребує, терапевтично ефективної кількості хіміотерапевтичного засобу у комбінації з терапевтично ефективною кількістю сполуки формули (I). 16 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 60 У іншому аспекті представлена сполука формули I або будь-які конкретні варіанти її втілення, описані вище, для використання при лікуванні раку. У ще одному аспекті рак являє собою лейкоз, вибраний з хронічного мієлолейкозу (CML) та гострого лімфобластного лейкозу (ALL). У іншому аспекті представлена сполука формули I або будь-які конкретні варіанти її втілення для використання при лікуванні раку у комбінації з додатковою сполукою, вибраною з іматинібу, нілотинібу, дазатинібу, босутинібу, понатинібу та бафетинібу. У ще одному аспекті сполука формули I являє собою (R)-N-(4-(хлордифторметокси)феніл)6-(3-гідроксипіролідин-1-іл)-5-(1H-піразол-5-іл)-нікотинамід. У ще одному аспекті сполука формули I являє собою фармацевтично прийнятну сіль (R)-N(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1H-піразол-5-іл)нікотинаміду. У ще одному аспекті додаткову сполуку вводять послідовно. У ще одному аспекті додаткову сполуку вводять одночасно. У ще одному аспекті додаткова сполука являє собою нілотиніб. У ще одному аспекті додаткова сполука являє собою іматиніб. У ще одному аспекті додаткова сполука являє собою дазатиніб. У ще одному аспекті додаткова сполука являє собою босутиніб. У ще одному аспекті додаткова сполука являє собою понатиніб. У ще одному аспекті додаткова сполука являє собою бафетиніб. У іншому аспекті даний винахід відноситься до способу лікування ABL1/BCR-ABL1опосредованного розладу, що включає наступну стадію: введення пацієнту, який цього потребує, терапевтично ефективної кількості хіміотерапевтичного засобу у комбінації з терапевтично ефективною кількістю сполуки формули (I). Фармацевтичні композиції У іншому аспекті даний винахід забезпечує фармацевтично прийнятні композиції, які включають терапевтично ефективну кількість однієї або декількох сполук, описаних вище, сформульованих разом з одним або декількома фармацевтично прийнятними носіями (добавками) та/або розріджувачами. Як описано докладно нижче, фармацевтичні композиції відповідно до даного винаходу можуть бути спеціально сформульовані для введення у твердій або рідкій формі, включаючи композиції, призначені для наступного: (1) перорального введення, наприклад, змочувальні препарати (водні або неводні розчини або суспензії), таблетки, наприклад, призначені для букального, сублінгвального введення та системної абсорбції, болюси, порошки, гранули, пасти для нанесення на язик; (2) парентерального введення, наприклад, за допомогою підшкірної, внутрішньом'язової, внутрішньовенної або епідуральної ін'єкції, наприклад, у вигляді стерильного розчину або суспензії або композиції уповільненого вивільнення; (3) місцевого нанесення, наприклад, у вигляді крему, мазі або пластиру з контрольованим вивільненням або спрею, що наноситься на шкіру; (4) інтравагінального або інтраректального введення, наприклад, у вигляді песарію, крему або піни; (5) сублінгвального введення; (6) очного введення; (7) крізьшкірного введення; (8) назального введення; (9) внутрішньолегеневого введення; або (10) інтратекального введення. Фраза "терапевтично ефективна кількість", як це використовується у даній заявці, означає таку кількість сполуки, речовини або композиції, що включає сполуку відповідно до даного винаходу, яка є ефективною для одержання деякого бажаного терапевтичного ефекту у щонайменше субпопуляції клітин у тварини при розумному співвідношенні користь/ризик у застосуванні до будь-якого медичного лікування. Фраза "фармацевтично прийнятний", як це використовується у даній заявці, відноситься до тих сполук, речовин, композицій та/або лікарських форм, які, відповідно зваженій медичній оцінці, є підходящими для використання у контакті із тканинами людини та тварин, без надмірної токсичності, подразнення, алергійних реакцій або інших проблем або ускладнень, відповідаючи розумному співвідношенню користь/ризик. Фраза "фармацевтично прийнятний носій", як це використовується у даній заявці, означає фармацевтично прийнятну речовину, композицію або носій, такий як рідкий або твердий наповнювач, розріджувач, ексципієнт, технологічна добавка (наприклад, змащувальна речовина, тальк, стеарат магнію, кальцію або цинку або стеаринова кислота) або розчинювана інкапсулююча речовина, що бере участь у переносі або транспортуванні сполуки відповідно до даного винаходу з одного органу або частини тіла до іншого органу або частини тіла. Кожен носій повинен бути "прийнятним", у тому розумінні, що він повинен бути сумісним з іншими інгредієнтами композиції та не бути шкідливим для пацієнта. Деякі приклади речовин, які можуть служити як фармацевтично прийнятні носії, включають: (1) цукри, такі як лактоза, глюкоза та сахароза; (2) крохмалі, такі як кукурудзяний крохмаль та картопляний крохмаль; (3) 17 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 60 целюлозу та її похідні, такі як натрій карбоксиметилцелюлоза, етилцелюлоза та ацетат целюлози; (4) порошкоподібний трагакант; (5) солод; (6) желатин; (7) тальк; (8) ексципієнти, такі як масло какао та воски для супозиторіїв; (9) масло, таке як арахісове масло, масло насіння бавовнику, сафлорове масло, кунжутне масло, оливкове масло, кукурудзяне масло та соєве масло; (10) гліколі, такі як пропіленгліколь; (11) поліоли, такі як гліцерин, сорбіт, маніт та поліетиленгліколь; (12) складні ефіри, такі як етилолеат та етиллаурат; (13) агар; (14) буферні речовини, такі як гідроксид магнію та гідроксид алюмінію; (15) альгінову кислоту; (16) апірогенну воду; (17) ізотонічний сольовий розчин; (18) розчин Рінгера; (19) етиловий спирт; (20) pН буферні розчини; (21) складні поліефіри, полікарбонати та/або поліангідриди; та (22) інші нетоксичні сумісні речовини, використовувані у фармацевтичних композиціях. Як описано вище, деякі варіанти втілення сполук відповідно до даного винаходу можуть містити лужну функціональну групу, таку як аміно або алкіламіно, і, таким чином, можуть утворювати фармацевтично прийнятні солі з фармацевтично прийнятними кислотами. Термін "фармацевтично прийнятні солі", щодо цього, відноситься до відносно нетоксичних неорганічних та органічних кислотно-адитивних солей сполук відповідно до даного винаходу. Ці солі можна одержати in situ у процесі одержання носія для введення або лікарської форми, або шляхом окремої взаємодії очищеної сполуки відповідно до даного винаходу у формі вільної основи з підходящою органічною або неорганічною кислотою та виділення утвореної у такий спосіб солі у процесі наступного очищення. Репрезентативні солі включають гідробромід, гідрохлорид, сульфат, бісульфат, фосфат, нітрат, ацетат, валерат, олеат, пальмітат, стеарат, лаурат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, наптилат, мезилат, глюкогептаноат, лактобіонат та лаурилсульфонат та подібні (див., наприклад, Berge et al. (1977) "Pharmaceutical Salts", J. Pharm. Sci. 66:1-19). Фармацевтично прийнятні солі сполук відповідно до даного винаходу включають традиційні нетоксичні солі або четвертинні амонієві солі сполук, наприклад, утворені з нетоксичних органічних або неорганічних кислот. Наприклад, такі традиційні нетоксичні солі включають солі, утворені з неорганічних кислот, таких як хлористоводнева, бромистоводнева, сірчана, сульфамінова, фосфорна, азотна та подібні; та солі, утворені з органічних кислот, таких як оцтова, пропіонова, бурштинова, гліколева, стеаринова, молочна, яблучна, винна, лимонна, аскорбінова, пальмітинова, малеїнова, гідроксималеїнова, фенілоцтова, глутамінова, бензойна, саліцилова, сульфанілова, 2-ацетоксибензойна, фумарова, толуолсульфонова, метансульфонова, етандисульфонова, щавлева, ізотіонова та подібні. У інших випадках сполуки відповідно до даного винаходу можуть містити одну або кілька кислотних функціональних груп і, таким чином, можуть утворювати фармацевтично прийнятні солі з фармацевтично прийнятними основами. Термін "фармацевтично прийнятні солі" у цих випадках відноситься до відносно нетоксичних неорганічних та органічних основно-адитивних солей сполук відповідно до даного винаходу. Ці солі також можна одержати in situ у процесі одержання носія для введення або лікарської форми, або шляхом окремої взаємодії очищеної сполуки відповідно до даного винаходу у формі вільної кислоти з підходящою підставою, такою як гідроксид, карбонат або бікарбонат фармацевтично прийнятного металевого катіону, з аміаком або з фармацевтично прийнятним органічним первинним, вторинним або третинним аміном. Репрезентативні солі лужних або лужноземельних металів включають солі літію, натрію, калію, кальцію, магнію та алюмінію та подібні. Репрезентативні органічні аміни, корисні для утворення основно-адитивних солей, включають етиламін, діетиламін, етилендіамін, етаноламін, діетаноламін, піперазин та подібні (див., наприклад, Berge et al., supra) Змочувальні речовини, емульгатори та змащувальні речовини, такі як лаурилсульфат натрію та стеарат магнію, а також барвники, агенти вивільнення, речовини покриттів, підсолоджувачі, віддушки та ароматизатори, консерванти та антиоксиданти також можуть бути присутніми у композиціях. Приклади фармацевтично прийнятних антиоксидантів включають: (1) водорозчинні антиоксиданти, такі як аскорбінова кислота, цистеїн гідрохлорид, бісульфат натрію, метабісульфіт натрію, сульфіт натрію та подібні; (2) маслорозчинні антиоксиданти, такі як аскорбілпальмітат, бутильований гідроксианізол (BHA), бутильований гідрокситолуол (BHT), лецитин, пропілгалат, альфа-токоферол та подібні; та (3) метало-хелатні речовини, такі як лимонна кислота, етилендіамінтетраоцтова кислота (EDTA), сорбіт, винна кислота, фосфорна кислота та подібні. Фармацевтичні композиції відповідно до даного винаходу включають композиції, що підходять для перорального, назального введення, місцевого (включаючи букальне та сублінгвальне), ректального, вагінального та/або парентерального введення. Композиції зручним чином можуть бути представлені у стандартній лікарській формі та можуть бути 18 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 60 отримані будь-якими способами, добре відомими у фармацевтиці. Кількість активного інгредієнту, яка може бути об'єднана з речовиною-носієм для одержання однієї лікарської дози, варіюється залежно від хазяїна, який приймає лікування, конкретного способу введення. Кількість активного інгредієнта, яка може бути об'єднана з речовиною-носієм для одержання однієї лікарської дози, як правило, становить таку кількість сполуки, яка забезпечує терапевтичний ефект. Як правило, виходячи зі ста відсотків, ця кількість буде знаходитися у межах від близько 0,1 відсотка до близько дев'яноста дев'яти відсотків активного інгредієнту, переважно від близько 5 відсотків до близько 70 відсотків, найбільш переважно від близько 10 відсотків до близько 30 відсотків. У деяких варіантах втілення композиція відповідно до даного винаходу включає ексципієнт, вибраний із групи, що включає циклодекстрини, целюлози, ліпосоми, міцелоутворюючі речовини, наприклад, жовчні кислоти, та полімерні носії, наприклад, складні поліефіри та поліангідриди; та сполуку відповідно до даного винаходу. У деяких варіантах втілення зазначена вище композиція робить сполуку відповідно до даного винаходу перорально біодоступною. Способи одержання цих сполук або композицій включають стадію приведення сполуки відповідно до даного винаходу у асоціацію з носієм та, необов'язково, одним або декількома допоміжними інгредієнтами. Як правило, композиції одержують шляхом однорідного та гомогенного змішування для приведення у асоціацію сполуки відповідно до даного винаходу з рідкими носіями або тонкоподрібненими твердими носіями, або і тими і іншими, та потім, якщо це необхідно, формування продукту. Фармацевтичні композиції відповідно до даного винаходу, що підходять для перорального введення, можуть бути у формі капсул, саше, пігулок, таблеток, коржів (з використанням основи, що містить віддушки, звичайно такої як сахароза та аравійська камедь або трагакант), порошків, гранул або у вигляді розчину, суспензії або твердій дисперсії у водній або неводній рідині, або у вигляді рідкої емульсії масло-у-воді або вода-у-маслі, або у вигляді еліксиру або сиропу, або у вигляді пастилок (з використанням інертної основи, такої як желатин та гліцерин або сахароза та аравійська камедь), та/або у вигляді полоскань для ротової порожнини тощо, при цьому кожна така форма містить попередньо визначену кількість сполуки відповідно до даного винаходу як активний інгредієнт. Сполуку відповідно до даного винаходу також можна вводити у вигляді болюсу, електуарію або пасти. Фармацевтична композиція у вигляді твердої дисперсії відповідно до даного винаходу включає, наприклад, аморфну дисперсію сполуки відповідно до даного винаходу, ексципієнт (співполімери, такі як полівінілпіролідинон (PVP) VA64 (Kollidon® VA64 або Copovidone) та подібні). Тверда дисперсія може бути додатково посилена за допомогою гідроксилпропілметилцелюлоз (HPMC) з низькою в'язкістю (таких як Pharmacoat 603, Methocel E3 або подібні). Див. приклад 41 нижче для одержання більш детальної інформації для одержання фармацевтичної композиції у вигляді твердої дисперсії відповідно до даного винаходу. У одному варіанті втілення даного винаходу представлена фармацевтична композиція, що включає аморфну дисперсію (R)-N-(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5(1H-піразол-5-іл)нікотинаміду (приклад 9) та від 1 до 2 ексципієнтів; де ексципієнт вибирають із HPMC AS, Pharmacoat 603, Eudragit L100, PVP K30, PVP VA64 та Eudragit EPO. У наступному варіанті втілення ексципієнти являють собою PVP VA64 та Pharmacoat 603. У наступному варіанті втілення відсотковий вміст Pharmacoat 603 знаходиться у діапазоні від 30 % до 45 %, відсотковий вміст PVP VA64 знаходиться у діапазоні від 30 % до 45 %, та відсотковий вміст (R)-N-(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1Hпіразол-5-іл)нікотинаміду (приклад 9) знаходиться у діапазоні від 20 % до 30 %. У наступному варіанті втілення відсотковий вміст Pharmacoat 603 складає 37,5 %, відсотковий вміст PVP VA64 складає 37,5 %, та відсотковий вміст (R)-N-(4(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1H-піразол-5-іл)-нікотинаміду (приклад 9) складає 25 %. У твердих лікарських формах відповідно до даного винаходу для перорального введення (капсули, таблетки, пігулки, драже, порошки, гранули, коржі та подібне) активний інгредієнт змішано з одним або декількома фармацевтично прийнятними носіями, такими як цитрат натрію або дикальцій фосфат, та/або будь-яким з наступних: (1) наповнювачі або створюючі об'єм речовини, такі як крохмалі, лактоза, сахароза, глюкоза, маніт та/або кремнієва кислота; (2) сполучні речовини, такі як, наприклад, карбоксиметилцелюлоза, альгінати, желатин, полівінілпіролідон, сахароза та/або аравійська камедь; (3) зволожувачі, такі як гліцерин; (4) розпушувачі, такі як агар-агар, карбонат кальцію, картопляний або тапіоковий крохмаль, 19 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 60 альгінова кислота, деякі силікати та карбонат натрію; (5) уповільнювані розчинення речовини, такі як парафін; (6) прискорювачі абсорбції, такі як четвертинні амонієві сполуки, та поверхневоактивні речовини, такі як полоксамер та лаурилсульфат натрію; (7) змочувальні речовини, такі як, наприклад, цетиловий спирт, гліцеринмоностеарат та неіонні поверхнево-активні речовини; (8) абсорбенти, такі як каолін та бентонітова глина; (9) змащувальні речовини, такі як тальк, стеарат кальцію, стеарат магнію, тверді поліетиленгліколі, лаурилсульфат натрію, стеарат цинку, стеарат натрію, стеаринова кислота та їх суміші; (10) барвники; та (11) контролюючі вивільнення речовини, такі як кросповідон або етилцелюлоза. У випадку капсул, таблеток та пігулок, фармацевтичні композиції також можуть включати буферні речовини. Тверді композиції подібного типу також можна використовувати як наповнювачі у м'яких та твердих желатинових капсулах з використанням таких ексципієнтів, як лактоза або молочні цукри, а також високомолекулярні поліетиленгліколі та подібні. Таблетку можна одержати шляхом пресування або формування, необов'язково з використанням одного або декількох допоміжних інгредієнтів. Пресовані таблетки можна одержати з використанням сполучної речовини (наприклад, желатин або гідроксипропілметилцелюлоза), змащувальної речовини, інертного розріджувача, консерванту, розпушувача (наприклад, натрій крохмальгліколят або зшита натрій карбоксиметилцелюлоза), поверхнево-активної або диспергуючої речовини. Формовані таблетки можна одержати шляхом формування у підходящій машині суміші порошкоподібної сполуки, змоченої інертним рідким розріджувачем. Таблетки та інші тверді лікарські форми фармацевтичних композицій відповідно до даного винаходу, такі як драже, капсули, пігулки та гранули, необов'язково можуть мати насічку або можуть бути отримані з покриттями та оболонками, такими як ентеросолюбільні покриття та інші покриття, добре відомі у галузі фармацевтичного формулювання. Вони також можуть бути сформульовані таким чином, щоб забезпечувати повільне або контрольоване вивільнення активного інгредієнта, що міститься у них, з використанням, наприклад, гідроксипропілметилцелюлози у різних пропорціях для забезпечення бажаного профілю вивільнення, інших полімерних матриць, ліпосом та/або мікросфер. Вони можуть бути сформульовані для швидкого вивільнення, наприклад, з використанням сушіння заморожуванням. Вони можуть бути стерилізовані, наприклад, шляхом фільтрування через утримуючий бактерії фільтр або шляхом включення стерилізуючих засобів у формі стерильних твердих композицій, які можна розчинити у стерильній воді або якому-небудь іншому стерильному середовищі для ін'єкцій безпосередньо перед використанням. Ці композиції також можуть, але необов'язково, містити речовини, що роблять композицію непрозорою, та можуть мати таку композицію, яка забезпечує вивільнення активного інгредієнту (інгредієнтів) тільки або переважно у певній частині шлунково-кишкового тракту, необов'язково уповільненим чином. Приклади композицій, призначених для включення у них лікарського засобу, які можна використовувати, включають полімерні речовини та воски. Активний інгредієнт також може бути у мікроінкапсульованій формі, якщо це є підходящим, з одним або декількома описаними вище ексципієнтами. Рідкі лікарські форми для перорального введення сполук відповідно до даного винаходу включають фармацевтично прийнятні емульсії, мікроемульсії, розчини, суспензії, сиропи та еліксири. Крім активного інгредієнта рідкі лікарські форми можуть містити інертні розріджувачі, звичайно використовувані у даній галузі техніки, такі як, наприклад, вода або інші розчинники, солюбілізуючі речовини та емульгатори, такі як етиловий спирт, ізопропіловий спирт, етилкарбонат, етилацетат, бензиловий спирт, бензилбензоат, пропіленгліколь, 1,3бутиленгліколь, масла (зокрема, масло насіння бавовнику, масло земляного горіха, кукурудзяне масло, масло із проростків насіння, оливкове, касторове та кунжутне масло), гліцерин, тетрагідрофуриловий спирт, поліетиленгліколі та складні ефіри жирних кислот сорбітану та їх суміші. Крім інертних розріджувачів пероральні композиції також можуть включати ад'юванти, такі як змочувальні речовини, емульгатори та суспендуючі речовини, підсолоджувачі, віддушки, барвники, ароматизатори та консерванти. Суспензії, крім активних сполук, можуть містити суспендуючі речовини, наприклад, етоксильовані ізостеарилові спирти, поліоксиетиленсорбіт та складні ефіри сорбітану, мікрокристалічну целюлозу, метагідроксид алюмінію, бентоніт, агар-агар та трагакант та їх суміші. Фармацевтичні композиції відповідно до даного винаходу для ректального або вагінального введення можуть бути представлені у вигляді супозиторію, який можна одержати шляхом змішування однієї або декількох сполук відповідно до даного винаходу з одним або декількома 20 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 підходящими неподразнюючими ексципієнтами або носіями, включаючи, наприклад, масло какао, поліетиленгліколь, віск для супозиторіїв або саліцилат, та який є твердим при кімнатній температурі, але рідким при температурі тіла та тому буде плавитися у прямій кишці або вагінальній порожнині та вивільнятиактивну сполуку. Фармацевтичні композиції відповідно до даного винаходу, які є підходящими для вагінального введення, також включають песарії, тампони, креми, гелі, пасти, піни або композиції спреїв, що містять такі носії, які відомі з рівня техніки як підходящі. Лікарські форми для місцевого або крізьшкірного введення сполуки відповідно до даного винаходу включають порошки, спреї, мазі, пасти, креми, лосьйони, гелі, розчини, пластири та препарати для інгаляцій. Активна сполука може бути змішана у стерильних умовах з фармацевтично прийнятним носієм та з будь-якими консервантами, буферами або пропелентами, які можуть знадобитися. Мазі, пасти, креми та гелі можуть містити, крім активної сполуки відповідно до даного винаходу, ексципієнти, такі як тваринні та рослинні жири, масла, воски, парафіни, крохмаль, трагакант, похідні целюлози, поліетиленгліколі, силікони, бентоніти, кремінна кислота, тальк та оксид цинку або їх суміші. Порошки та спреї можуть містити, крім сполуки відповідно до даного винаходу, ексципієнти, такі як лактоза, тальк, кремінна кислота, гідроксид алюмінію, силікати кальцію та поліамідний порошок або суміші таких речовин. Спреї додатково можуть містити традиційні пропеленти, такі як хлорфторвуглеводні та леткі незаміщені вуглеводні, такі як бутан та пропан. Крізьшкірні пластири мають додаткову перевагу, забезпечуючи контрольовану доставку сполуки відповідно до даного винаходу у організм. Такі лікарські форми можна одержати шляхом розчинення або диспергування сполуки у підходящому середовищі. Підсилювачі абсорбції також можна використовувати для збільшення потоку сполуки через шкіру. Швидкість такого потоку можна контролювати або шляхом забезпечення контролюючої швидкість мембрани, або шляхом диспергування сполуки у полімерній матриці або гелі. Офтальмічні композиції, очні мазі, порошки, розчини тощо також передбачаються як такі, що охоплюються обсягом даного винаходу. Фармацевтичні композиції відповідно до даного винаходу, що підходять для парентерального введення, включають одну або кілька сполук відповідно до даного винаходу у об'єднанні з одним або декількома фармацевтично прийнятними стерильними ізотонічними водними або неводними розчинами, дисперсіями, суспензіями або емульсіями або стерильними порошками, які можуть бути реструктуровані з одержанням стерильних розчинів або дисперсій для ін'єкцій безпосередньо перед використанням, які можуть містити цукри, спирти, антиоксиданти, буфери, бактеріостатичні речовини, розчинені речовини, які роблять композицію ізотонічною з кров'ю передбачуваного реципієнта, або суспендуючі речовини або згущувачі. Приклади підходящих водних та неводних носіїв, які можна використовувати у фармацевтичних композиціях відповідно до даного винаходу, включають воду, етанол, поліоли (такі як гліцерин, пропіленгліколь, поліетиленгліколь та подібні) та підходящі суміші таких речовин, рослинні олії, такі як маслинове масло, та органічні складні ефіри для ін'єкцій, такі як етилолеат. Підходящу текучість можна підтримувати, наприклад, з використанням речовин для покриття, таких як лецитин, шляхом підтримки необхідного розміру часток у випадку дисперсій та шляхом використання поверхнево-активних речовин. Ці композиції також можуть містити ад'юванти, такі як консерванти, змочуючі речовини, емульгатори та диспергуючі речовини. Запобігання дії мікроорганізмів на сполуки відповідно до даного винаходу можна забезпечити шляхом включення різних антибактеріальних та протигрибкових засобів, наприклад, парабену, хлорбутанолу, фенолсорбінової кислоти та подібних. Також може бути бажаним включення у композиції ізотонічних речовин, таких як цукри, хлорид натрію та подібні. Крім того, пролонговану абсорбцію фармацевтичної форми для ін'єкцій можна одержати шляхом включення речовин, які уповільнюють абсорбцію, таких як моностеарат алюмінію та желатин. У деяких випадках для пролонгування дії лікарського засобу бажано уповільнити абсорбцію лікарського засобу з підшкірної або внутрішньом'язової ін'єкції. Це можна здійснити з використанням рідкої суспензії кристалічної або аморфної речовини, що має погану водорозчинність. Швидкість абсорбції лікарського засобу у цьому випадку залежить від швидкості його розчинення, яка, у свою чергу, може залежати від розміру кристала та кристалічної форми. Альтернативно, уповільнену абсорбцію лікарської форми, що вводиться парентерально, одержують шляхом розчинення або суспендування лікарського засобу у масляному носії. 21 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 60 Депо форми для ін'єкцій одержують шляхом формування матриці для мікроінкапсулювання сполук відповідно до даного винаходу у полімерах, що біорозкладаються, таких як полілактидполігліколід. Залежно від відношення лікарського засобу до полімеру та природи конкретного використовуваного полімеру швидкість вивільнення лікарського засобу можна контролювати. Приклади інших полімерів, що біорозкладаються, включають полі(ортоефіри) та полі(ангідриди). Депо форми для ін'єкцій також одержують шляхом включення лікарського засобу у ліпосоми або мікроемульсії, які сумісні із тканиною організму. Коли сполуки відповідно до даного винаходу вводять у вигляді фармацевтичних засобів людині та тваринам, їх можна вводити per se або у вигляді фармацевтичної композиції, що містить, наприклад, від 0,1 до 99 % (більш переважно, від 10 до 30 %) активного інгредієнту у комбінації з фармацевтично прийнятним носієм. Препарати відповідно до даного винаходу можна вводити перорально, парентерально, місцево або ректально. Їх, безумовно, вводять у формах, що підходять для кожного шляху введення. Наприклад, їх вводять у формі таблеток або капсул, шляхом ін'єкції, інгаляції, у вигляді очного лосьйону, мазі, супозиторію тощо, введення здійснюють шляхом ін'єкції, інфузії або інгаляції; місцевим шляхом з використанням лосьйону або мазі; та ректальним шляхом з використанням супозиторіїв. Пероральні введення є кращими. Фрази "парентеральне введення" та "що вводиться парентерально", як це використовується у даній заявці, означають способи введення, відмінні від ентерального та місцевого введення, звичайно шляхом ін'єкції, та включають, без обмеження, внутрішньовенну, внутрішньом'язову, інтраартеріальну, інтратекальну, інтракапсулярну, інтраорбітальну, інтракардіальну, внутрішньошкірну, інтраперитонеальну, транстрахеальну, підшкірну, субкутикулярну, інтраартикулярну, субкапсулярну, субарахіноїдальну, інтраспінальну та інтрастернальну ін'єкцію та інфузію. Фрази "системне введення", "що вводиться системно", "периферійне введення" та "що вводиться периферійно", як це використовується у даній заявці, означають введення сполуки, лікарського засобу або іншої речовини, відмінне від введення безпосередньо у центральну нервову систему, таким чином, щоб вона надходила у систему пацієнта й, таким чином, зазнала метаболізму та інших подібних процесів, наприклад, підшкірне введення. Ці сполуки можна вводити людині та іншим тваринам для лікування будь-яким підходящим шляхом введення, у тому числі перорально, назально, наприклад, у вигляді спрею, ректально, інтравагінально, парентерально, інтрацистернально та місцево, наприклад, у формі порошків, мазей або крапель, включаючи букальне та сублінгвальне введення. Незалежно від обраного шляху введення, сполуки відповідно до даного винаходу, які можна використовувати у підходящій гідратованій формі, та/або фармацевтичні композиції відповідно до даного винаходу формулюють у фармацевтично прийнятні лікарські форми за традиційними способами, відомими фахівцям у даній галузі. Фактичні рівні доз активних інгредієнтів у фармацевтичних композиціях відповідно до даного винаходу можуть варіюватися таким чином, щоб забезпечувати кількість активного інгредієнту, яка є ефективною для досягнення бажаної терапевтичної відповіді для конкретного пацієнта, композиції та шляху введення, не будучи при цьому токсичними для пацієнта. Вибраний рівень доз буде залежати від різних факторів, включаючи активність конкретної використовуваної сполуки відповідно до даного винаходу або її складного ефіру, солі або аміду, шлях введення, час введення, швидкість екскреції або метаболізму конкретної використовуваної сполуки, швидкість та ступінь абсорбції, тривалість лікування, інші лікарські засоби, сполуки та/або речовини, використовувані у комбінації з конкретною використовуваною сполукою, вік, стать, масу тіла, стан, загальний стан здоров'я та попередню медичну історію пацієнта, якого лікують, та подібні фактори, добре відомі у медицині. Лікар або ветеринар із середньою кваліфікацією легко зможе визначити та прописати ефективну кількість фармацевтичної композиції, яка потрібно. Наприклад, лікар або ветеринар може почати з доз сполук відповідно до даного винаходу, використовуваних у фармацевтичній композиції, на рівнях нижче тих, які необхідні для досягнення бажаного терапевтичного ефекту, та поступово збільшувати дозу аж до досягнення бажаного ефекту. Як правило, підходяща добова доза сполуки відповідно до даного винаходу буде являти собою таку кількість сполуки, яка являє собою найнижчу дозу, ефективну для забезпечення терапевтичного ефекту. Така ефективна доза, як правило, залежить від факторів, описаних вище. Як правило, пероральні, внутрішньовенні, інтрацеребровентрикулярні та підшкірні дози сполук відповідно до даного винаходу для пацієнта, коли їх використовують для показаних аналгетичних ефектів, знаходяться у межах від близько 0,0001 до близько 100 мг на кілограм маси тіла на день. 22 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 60 Якщо бажано, ефективну добову дозу активної сполуки можна вводити у вигляді двох, трьох, чотирьох, п'яти, шести або більше дробових доз, що вводяться роздільно з підходящими інтервалами часу протягом дня, необов'язково, у стандартних лікарських формах. PK параметри in vivo можуть бути використані для оцінки PK параметрів людини. Із застосуванням різних способів, відомих у даній галузі для прогнозування PK людини, можна оцінити прогнозований кліренс людини. Наприклад, (R)-N-(4-(хлордифторметокси)феніл)-6-(3гідроксипіролідин-1-іл)-5-(1H-піразол-5-іл)-нікотинамід (приклад 9) за оцінками становить 3 мл/хвил./кг, та об'єм розподілу оцінюється як 1 л/кг. Прогнозована ефективна добова доза для людини для сполуки прикладу 9, тому, була визначена як така, що знаходиться у межах від 90 до 130 мг/день. Хоча сполуку відповідно до даного винаходу можна вводити окремо, кращим є введення сполуки у вигляді фармацевтичного препарату (композиції). Сполуки відповідно до даного винаходу можуть бути сформульовані для введення будьяким зручним шляхом для застосування у лікуванні людини або у ветеринарії, аналогічно іншим фармацевтичним засобам. У іншому аспекті даний винахід забезпечує фармацевтично прийнятні композиції, які включають терапевтично ефективну кількість однієї або декількох сполук відповідно до даного винаходу, описаних вище, сформульованих разом з одним або декількома фармацевтично прийнятними носіями (добавками) та/або розріджувачами. Як докладно описано нижче, фармацевтичні композиції відповідно до даного винаходу можуть бути спеціально сформульовані для введення у твердій або рідкій формі, включаючи призначені для наступного: (1) перорального введення, наприклад, змочувальні препарати (водні або неводні розчини або суспензії), таблетки, болюси, порошки, гранули, пасти для нанесення на язик; (2) парентерального введення, наприклад, за допомогою підшкірної, внутрішньом'язової або внутрішньовенної ін'єкції, наприклад, у вигляді стерильного розчину або суспензії; (3) місцевого нанесення, наприклад, у вигляді крему, мазі або спрею для нанесення на шкіру, легені або слизові оболонки; (4) інтравагінального або інтраректального введення, наприклад, у вигляді песарію, крему або піни; (5) сублінгвального або букального введення; (6) очного введення; (7) крізьшкірного введення; (8) назального введення. Термін "лікування" призначений для охоплення також профілактики, терапії та лікування. Пацієнт, що приймає таке лікування, являє собою будь-яку тварину, яка цього потребує, включаючи приматів, зокрема людину, та інших ссавців, таких як кінь, велика рогата худоба, свині та вівці; а також домашню птицю та свійських тварин у цілому. Технологія мікроемульгування може покращити біодоступність деяких ліпофільних (не розчинних у воді) фармацевтичних засобів. Приклади включають Триметрин (Dordunoo, S. K., et al., Drug Development and Industrial Pharmacy, 17(12), 1685-1713, 1991) та REV 5901 (Sheen, P. C., et al., J Pharm Sci 80(7), 712-714, 1991). Окрім іншого, мікроемульгування забезпечує підвищену біодоступність шляхом переважного спрямовування абсорбції на лімфатичну систему замість системи кровотоку, яка, таким чином, обходить печінку та попереджає розкладання сполук у гепатобіліарному кровообігу. Хоча передбачаються всі підходящі амфіфільні носії, у даному винаході кращими носіями в основному є такі, які мають офіційне маркування "Визнаний нешкідливим" (Generally-recognizedas-safe (GRAS)) та які можуть як розчиняти сполуку відповідно до даного винаходу, так і мікроемульгувати її на більш пізній стадії, коли розчин приходить у контакт із комплексною водною фазою (такою як у шлунково-кишковому тракті людини). Звичайно амфіфільні інгредієнти, які задовольняють цим вимогам, мають HLB (гідрофільно-ліпофільний баланс) значення 2-20, та їх структури містять аліфатичні радикали з лінійним ланцюгом у межах від C-6 до C-20. Прикладами є поліетилен-гліколизьовані гліцериди жирних кислот та поліетиленгліколі. Особливо передбачаються комерційно доступні амфіфільні носії, включаючи ряд Гелуцирових носіїв, Лабрафіл, Лабразол або Лаурогліколь (виробник та постачальник усіх перерахованих вище Gattefosse Corporation, Saint Priest, France), PEG-моно-олеат, PEG-діолеат, PEG-моно-лаурат та ди-лаурат, Лецитин, Полісорбат 80 та т. п. (виробляються та поставляються низкою компаній у США та по всьому світу). Гідрофільні полімери, що підходять для використання у даньому винаході, являють собою такі, які є легко водорозчинними, можуть бути ковалентно зв'язані з везикулоутворюючим ліпідом та які є переносимими in vivo без токсичних ефектів (тобто є біосумісними). Підходящі полімери включають поліетиленгліколь (PEG), полімолочну кислоту (що також називають полілактидом), полігліколеву кислоту (що також називають полігліколідом), cпівполімер полімолочної-полігліколевої кислоти та полівініловий спирт. Кращі полімери являють собою такі, які мають молекулярну масу від близько 100 або 120 дальтон до близько 5000 або 10000 23 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 60 дальтон та більш переважно від близько 300 дальтон до близько 5000 дальтон. У особливо кращому варіанті втілення полімер являє собою поліетиленгліколь, що має молекулярну масу від близько 100 до близько 5000 дальтон та більш переважно має молекулярну масу від близько 300 до близько 5000 дальтон. У особливо кращому варіанті втілення полімер являє собою поліетиленгліколь із молекулярною масою 750 дальтон (PEG(750)). Полімери також можуть визначатися за кількістю мономерів у них; у кращому варіанті втілення даного винаходу використовують полімери, що складаються із щонайменше близько трьох мономерів, такі PEG полімери, що складаються із трьох мономерів (приблизно 150 дальтон). Інші гідрофільні полімери, які можуть бути підходящими для використання у даному винаході, включають полівінілпіролідон, поліметоксазолін, поліетилоксазолін, полігідроксипропіл метакриламід, поліметакриламід, полідиметилакриламід та похідні целюлози, такі як гідроксиметилцелюлоза або гідроксиетилцелюлоза. У деяких варіантах втілення композиція відповідно до даного винаходу включає біосуміний полімер, вибраний з групи, яка включає поліаміди, полікарбонати, поліалкілени, полімери складних ефірів акрилової та метакрилової кислот, полівінілові полімери, полігліколіди, полісилоксани, поліуретани та їх співполімери, целюлози, поліпропілен, поліетилени, полістирол, полімери молочної кислоти та гліколевої кислоти, поліангідриди, полі(орто)ефіри, полі(масляну кислоту), полі(валеріанову кислоту), полі(лактид-со-капролактон), полісахариди, білки, полігіалуронові кислоти, поліціаноакрилати та їх композиції, суміші або cпівполімери. Циклодекстрини являють собою циклічні олігосахариди, що складаються з 6, 7 або 8 глюкозних ланок, які позначаються грецькими літерами альфа, бета або гама, відповідно. Невідомо про існування циклодекстринів, що містять менше шести глюкозних ланок. Глюкозні ланки зв'язані альфа-1,4-глюкозидними зв'язками. Як наслідок конформації крісла цукрових ланок, усі вторинні гідроксильні групи (по C-2, C-3) розташовані на одному боці кільця, тоді як усі первинні гідроксильні групи по C-6 розташовані на іншому боці. Як результат, зовнішні сторони є гідрофільними, роблячи циклодекстрини водорозчинними. На відміну від цього, порожнини циклодекстринів є гідрофобними, оскільки вони вистелені воднем атомів C-3 та C-5 та ефір-подібними киснями. Ці матриці уможливлюють комплексоутворення з різними відносно гідрофобними сполуками, включаючи, наприклад, стероїдні сполуки, такі як бета-естрадіол (див., наприклад, van Uden et al. Plant Cell Tiss. Org. Cult. 38:1-3-113 (1994)). Комплексоутворення відбувається за допомогою ван-дер-ваальсових взаємодій та шляхом утворення водневих зв'язків. Загальний огляд хімії циклодекстринів див. у Wenz, Agnew. Chem. Int. Ed. Engl., 33:803-822 (1994). Фізико-хімічні властивості циклодекстринових похідних сильно залежать від типу та ступеню заміщення. Наприклад, їхня розчинність у воді знаходиться у діапазоні від нерозчинних (наприклад, триацетил-бета-циклодекстрин) до розчинності 147 % (мас/об) (G-2-бетациклодекстрин). Крім того, вони розчинні у багатьох органічних розчинниках. Властивості циклодекстринів дозволяють контролювати розчинність різних компонентів композиції шляхом збільшення або зменшення їх розчинності. Були описані різні циклодекстрини та способи їх одержання. Наприклад, Parmeter (I), et al. (Патент США № 3453259) та Gramera, et al. (Патент США № 3459731) описують електронейтральні циклодекстрини. Інші похідні включають циклодекстрини з катіонними властивостями [Parmeter (II), Патент США № 3453257], нерозчинні зшиті циклодекстрини (Solms, Патент США№ 3420788) та циклодекстрини з аніонними властивостями [Parmeter (III), Патент США № 3426011]. Із циклодекстринових похідних з аніонними властивостями можна вказати такі, де карбонові кислоти, фосфористі кислоти, фосфінові кислоти, фосфонові кислоти, фосфорні кислоти, тіофосфонові кислоти, тіосульфінові кислоти та сульфонові кислоти приєднані до батьківського циклодекстрину [див. Parmeter (III), supra]. Крім того, сульфоалкілефірні похідні циклодекстринів описані Stella, et al. (Патент США № 5134127). Ліпосоми складаються із щонайменше однієї ліпідної бішарової оболонки, що містить у собі водний внутрішній компартмент. Ліпосоми можуть бути охарактеризовані по типу та по розміру оболонки. Невеликі одношарові везикули (SUV) мають одну оболонку та типовий діаметр у межах від 0,02 до 0,05 мкм; великі одношарові везикули (LUVS) типово більше, ніж 0,05 мкм. Оліголамелярні великі везикули та мультиламелярні везикули мають декілька, звичайно концентричних, шарів оболонки, та вони типово більше ніж 0,1 мкм. Ліпосоми з декількома неконцентричними оболонками, тобто декількома більш дрібними везикулами, що містяться у більшій везикулі, називають мультивезикулярними везикулами. Один аспект даного винаходу відноситься до композицій, що включають ліпосоми, що містять сполуку відповідно до даного винаходу, де оболонка ліпосоми сформульована для забезпечення ліпосоми з підвищеною несучою здатністю. Альтернативно або на додаток до 24 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 60 цього, сполука відповідно до даного винаходу може міститися у, або бути адсорбована на, ліпосомному бішарі ліпосоми. Сполука відповідно до даного винаходу може бути агрегована з ліпідною поверхнево-активною речовиною та може транспортуватися, перебуваючи у внутрішньому просторі ліпосоми; у цих випадках ліпосомна оболонка сформульована таким чином, щоб бути стійкою до руйнівних ефектів агрегату активний засіб-поверхнево-активна речовина. Відповідно з одним варіантом втілення даного винаходу ліпідний бішар ліпосоми містить ліпіди, дериватизовані поліетиленгліколем (PEG) таким чином, щоб PEG ланцюги простиралися від внутрішньої поверхні ліпідного бішару у внутрішній простір, інкапсульований ліпосомою, та простиралися від зовнішньої поверхні ліпідного бішару у оточуюче середовище. Активні речовини, що містяться у ліпосомах відповідно до даного винаходу, знаходяться у солюбілізованій формі. Агрегати, що складаються з поверхнево-активної речовини та активної речовини (такі як емульсії або міцели, що містять активну речовину, що представляє інтерес), можуть бути поміщені у внутрішньому просторі ліпосом відповідно до даного винаходу. Поверхнево-активна речовина діє як агент, що диспергує та солюбілізує активну речовину, та може бути вибрана з будь-якої підходящої аліфатичної, циклоаліфатичної або ароматичної поверхнево-активної речовини, включаючи, але не обмежуючись цим, біосумісні лізофосфатидилхоліни (LPC) з різною довжиною ланцюгів (наприклад, від близько C 14 до близько C20). Полімер-дериватизовані ліпіди, такі як PEG-ліпіди, також можна використовувати для міцелоутворення, оскільки їх дія спрямована на інгібування злиття міцела/оболонка, та оскільки приєднання полімеру до молекул поверхнево-активної речовини знижує CMC поверхнево-активної речовини та сприяє міцелоутворенню. Кращими є поверхнево-активні речовини з CMC у мікромолярному діапазоні; поверхнево-активні речовини з більш високим CMC можна використовувати для одержання міцел, поміщених у ліпосоми відповідно до даного винаходу, однак міцелярні поверхнево-активні мономери можуть впливати на стабільність ліпосомного бішару та можуть бути рушійної силою у розробці ліпосом бажаної стабільності. Ліпосоми у відповідності з даним винаходом можна отримати будь-яким з різних способів, які відомі з рівня техніки. Див., наприклад, Патент США № 4235871; опубліковані заявки PCT WO 96/14057; New RRC, Liposomes: A practical approach, IRL Press, Oxford (1990), pages 33-104; Lasic DD, Liposomes from physics to applications, Elsevier Science Publishers BV, Amsterdam, 1993. Наприклад, ліпосоми відповідно до даного винаходу можна одержати шляхом дифундування ліпіду, дериватизованого гідрофільним полімером, у попередньо сформовані ліпосоми, наприклад, піддаючи попередньо сформовані ліпосоми контактуванню з міцелами, утвореними з ліпідпривитих полімерів, при концентраціях ліпідів, що відповідають кінцевому мольному відсотку дериватизованого ліпіду, який є бажаним, у ліпосомі. Ліпосоми, що містять гідрофільний полімер, також можуть бути утворені шляхом гомогенізації, гідратування ліпідної області або методами екструзії, як це відомо з рівня техніки. У одному аспекті даного винаходу ліпосоми одержують таким чином, щоб вони мали по суті гомогенні розміри у обраному розмірному діапазоні. Один ефективний спосіб класифікації за розміром включає екструдування водної суспензії ліпосом через ряд полікарбонатних мембран, що мають вибраний однаковий розмір пор; розмір пор мембрани повинен приблизно відповідати найбільшим розмірам ліпосом, які одержують шляхом екструзії через таку мембрану. Див., наприклад, Патент США № 4737323 (12 квітня 1988 р.). Характеристики вивільнення препарату відповідно до даного винаходу залежать від інкапсулюючої речовини, концентрації інкапсульованого лікарського засобу та присутності модифікаторів вивільнення. Наприклад, вивільненням можна маніпулювати, оскільки воно є pН залежним, наприклад, з використанням pН-чутливого покриття, яке вивільняє тільки при низькому pН, як, наприклад, у шлунку, або при більш високому pН, як, наприклад, у кишечнику. Ентеросолюбільне покриття можна використовувати для запобігання вивільнення доти, поки препарат не пройде через шлунок. Можна використовувати кілька покриттів або суміші ціанаміду, інкапсульованого у різних речовинах, для одержання початкового вивільнення у шлунку з наступним більш пізнім вивільненням у кишечнику. Вивільненням також можна маніпулювати шляхом включення солей або пороутворюючих речовин, які можуть підвищувати водопоглинення або вивільняти лікарський засіб за допомогою дифузії з капсули. Ексципієнти, які модифікують розчинність лікарського засобу, також можна використовувати для контролювання швидкості вивільнення. Речовини, які підсилюють руйнування матриці або вивільняють із матриці, також можуть бути включені. Вони можуть бути додані до лікарського засобу, додані у вигляді окремої фази (тобто у вигляді дрібних часток) або можуть бути спільно розчинені у полімерній фазі, залежно від сполуки. У всіх випадках кількість повинна знаходитися 25 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 у межах від 0,1 до тридцяти відсотків (мас./мас. полімеру). Типи речовин, які підсилюють руйнування, включають неорганічні солі, такі як сульфат амонію та хлорид амонію, органічні кислоти, такі як лимонна кислота, бензойна кислота та аскорбінова кислота, неорганічні основи, такі як карбонат натрію, карбонат калію, карбонат кальцію, карбонат цинку та гідроксид цинку, та органічні основи, такі як протамінсульфат, спермін, холін, етаноламін, діетаноламін та триетаноламін, та поверхнево-активні речовини, такі як Tween® та Pluronic®. Пороутворюючі речовини, які додають мікроструктуру матрицям (тобто водорозчинні сполуки, такі як неорганічні солі та цукри), додають у вигляді твердих часток. Діапазон повинен становити від одного до тридцяти відсотків (мас/мас полімеру). Поглинанням також можна маніпулювати шляхом зміни часу знаходження часток у кишечнику. Це досягається, наприклад, шляхом покриття часток мукозально-адгезивним полімером або вибору його в якості інкапсулюючої речовини. Приклади включають більшість полімерів з вільними карбоксильними групами, такі як хітозан, целюлози та особливо поліакрилати (як використовується у даній заявці, поліакрилати відносяться до полімерів, що включають акрилатні групи та модифіковані акрилатні групи, таких як ціаноакрилати та метакрилати). Фармацевтичні комбінації Винахід, головним чином, відноситься до застосування сполуки формули (I) (або фармацевтичної композиції, що включає сполуку формули (I)) у лікуванні одного або декількох захворювань, зазначених у даній заявці; де відповідь на лікування є сприятливою, як показано, наприклад, шляхом часткового або повного усунення одного або декількох симптомів захворювання, аж до повного лікування або ремісії. Філадельфійська хромосома-позитивний (Ph+) ALL становить 15-30 % від ALL дорослих та до 5 % від педіатричних ALL (Faderl S, Garcia-manero G, Thomas D, et al. Philadelphia Chromosome Positive Acute Lymphoblastic Leukemia-Current Concepts and Future Perspectives. Rev Clin Exp Hematol 2002; 6:142-160). Педіатричний Ph+ALL характеризується більш старшим віком (у середньому 9-10 років проти приблизно 4 років для всіх ALL пацієнтів) та більш високим числом WBC при постановці діагнозу. Як у дорослих, так і у дітей Ph+ALL характеризується реципрокальною транслокацією між хромосомами 9 та 22 (t(9; 22)(q34; q11)), що приводить до злиття BCR гену на хромосомі 22 з ABL генними послідовностями, транслокованими із хромосоми 9, приводячи до експресії BCR-ABL1 білку. Існують 2 основних варіанти BCR-ABL1, p190BCR-ABL1, обумовлений у приблизно 85 % Ph+ALL пацієнтів, та p210 BCR-ABL1, типовий для CML, ідентифікований у приблизно 15 % Ph+ALL пацієнтів (Dombret H, Galbert J, Boiron J, et al. Outcome of Treatment in Adults with Philadelphia chromosome-posititve acute lymphoblastic leukemia-Results of the prospective multicenter LALA-94 trial. Blood 2002; 100:2357-2366; Faderl S, Garcia-manero G, Thomas D, et al. Philadelphia Chromosome Positive Acute Lymphoblastic Leukemia-Current Concepts and Future Perspectives. Rev Clin Exp Hematol 2002; 6:142-160). Лікування ALL основане на класифікації ризику для кожного пацієнта, з підвищенням інтенсивності лікування для пацієнтів, які мають більший ризик рецидиву; ця стратегія максимізує швидкості ремісії, при цьому обмежуючи непотрібні токсичності. Прогрес поступово збільшувався, від введення комбінованої хіміотерапії та лікування передсимптоматичного лейкозу центральної нервової системи до більш нових інтенсивних схем лікування для пацієнтів, які мають більший ризик рецидиву (C. H. Pui and W. E. Evans. Acute Lymphoblastic Leukemia New Engl J Med 1998; 339:605-615). До розробки іматинібу Ph+ALL пацієнтів лікували за допомогою інтенсивної хіміотерапії з наступною трансплантацією гематопоетичних стовбурових клітин (HSCT), ідеально з підходящим родинним донором, оскільки було показано, що це приводить до поліпшеного EFS проти будь-якого HSCT з іншими донорами або хіміотерапії, використовуваної окремо. У цілому та на відміну від більшості педіатричних пацієнтів з ALL, пацієнти з Ph+ALL мали погані прогнози з низьким відсотком виживання без несприятливих подій (EFS) (Arico M, Valsecchi M G, Camitta B, Schrappe M, Chessells J, Baruchel A, Gaynon P, Silverman L, Janka-Schaub G, Kamps W, et al. New Engl J Med 2000; 342:998-1006). TM Існуючі терапії (такі як GLEEVEC®, TASIGNA®, SPRYCEL®, BOSULIF®, ICLUSIG та подібні) зв'язуються з АТР-зв'язуючим сайтом кіназного домену. На відміну від цього, сполуки відповідно до даного винаходу є потужними інгібіторами BCR-ABL1, ABL1 та ABL2, які зв'язуються із сайтом на кіназному домені, який відрізняється від АТР-зв'язуючого сайту. Тому сполуки відповідно до даного винаходу з їхнім новим, алостеричним механізмом дії можуть бути використані як самостійна терапія або можуть бути використані послідовно або у комбінації з існуючими терапіями, вибраними з GLEEVEC®, TASIGNA®, SPRYCEL®, BOSULIF® TM та ICLUSIG . 26 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 60 У якості самостійної терапії сполуки відповідно до даного винаходу можуть бути використані для лікування BCR-ABL1, ABL1 та ABL 2-споріднених захворювань та розладів. BCR-ABL1 може бути дикого типу або мутантним BCR-ABL1, вибраним з V299L, T315I, F317I/L, Y253F/H, E255K/V та F359C/V. Сполуки відповідно до даного винаходу можуть бути використані для лікування пацієнтів, які не реагують на існуючі методи лікування у результаті мутацій, що виникають у АТР-зв'язуючому сайті. У якості комбінованої терапії сполуки відповідно до даного винаходу являють собою унікальну можливість для лікування пацієнтів з Ph+ лейкозом з використанням комбінації двох потужних, механістично різних інгібіторів BCR-ABL. Комбінований підхід у клініці може забезпечити пацієнтів з більш глибоким та більш стійким скороченням пухлинного навантаження зі зниженням ризику рецидиву. У іншому варіанті втілення даного винаходу представлений спосіб лікування теплокровної тварини, що має лейкоз, вибраний із хронічного мієлолейкозу (CML) та гострого лімфобластного лейкозу (ALL), що включає введення зазначеній тварині терапевтично ефективної кількості сполуки відповідно до даного винаходу або її фармацевтично прийнятної солі. У наступному варіанті втілення теплокровна тварина являє собою людину (пацієнта). У наступному варіанті втілення сполука відповідно до даного винаходу являє собою (R)-N(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1H-піразол-5-іл)нікотинамід (приклад 9) або його фармацевтично прийнятну сіль. У іншому варіанті втілення даного винаходу представлений спосіб лікування теплокровної тварини, що має лейкоз, вибраний з хронічного мієлолейкозу (CML) та гострого лімфобластного лейкозу (ALL), що включає послідовне введення зазначеній тварині терапевтично ефективної кількості сполуки відповідно до даного винаходу або її фармацевтично прийнятної солі та терапевтично ефективної кількості сполуки, вибраної з іматинібу, нілотинібу, дазатинібу, босутинібу, понатинібу та бафетинібу. У наступному варіанті втілення теплокровна тварина являє собою людину (пацієнта). У наступному варіанті втілення сполука відповідно до даного винаходу являє собою (R)-N(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1H-піразол-5-іл)нікотинамід (приклад 9) або його фармацевтично прийнятну сіль. У наступному варіанті втілення доза (R)-N-(4-(хлордифторметокси)феніл)-6-(3гідроксипіролідин-1-іл)-5-(1H-піразол-5-іл)нікотинаміду (приклад 9) складає 90-130 мг. У наступному варіанті втілення доза нілотинібу складає 10-50 мг/кг, іматинібу складає 50200 мг/кг, дазатинібу складає 5-20 мг/кг або понатинібу складає 2-10 мг/кг. У наступному варіанті втілення доза босутинібу складає 500 мг. У іншому варіанті втілення даного винаходу представлений спосіб лікування теплокровної тварини, що має лейкоз, вибраний з хронічного мієлолейкозу (CML) та гострого лімфобластного лейкозу (ALL), що включає одночасне введення зазначеній тварині терапевтично ефективної кількості сполуки відповідно до даного винаходу або її фармацевтично прийнятної солі та терапевтично ефективної кількості сполуки, вибраної з іматинібу, нілотинібу, дазатинібу, босутинібу, понатинібу та бафетинібу. У наступному варіанті втілення теплокровна тварина являє собою людину (пацієнта). У наступному варіанті втілення сполука відповідно до даного винаходу являє собою (R)-N(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1H-піразол-5-іл)нікотинамід (приклад 9) або його фармацевтично прийнятну сіль. У наступному варіанті втілення доза (R)-N-(4-(хлордифторметокси)феніл)-6-(3гідроксипіролідин-1-іл)-5-(1H-піразол-5-іл)нікотинаміду (приклад 9) складає 90-130 мг. У наступному варіанті втілення доза нілотинібу складає 10-50 мг/кг, іматинібу складає 50200 мг/кг, дазатинібу складає 5-20 мг/кг або понатинібу складає 2-10 мг/кг. У наступному варіанті втілення доза босутинібу складає 500 мг. У іншому варіанті втілення даного винаходу представлений спосіб лікування теплокровної тварини, що має лейкоз, вибраний з хронічного мієлолейкозу (CML) та гострого лімфобластного лейкозу (ALL), що включає одночасне введення зазначеній тварині терапевтично ефективної кількості (R)-N-(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1H-піразол-5іл)нікотинаміду (приклад 9) або його фармацевтично прийнятної солі та терапевтично ефективної кількості нілотинібу. У іншому варіанті втілення даного винаходу представлений спосіб лікування теплокровної тварини, що має лейкоз, вибраний з хронічного мієлолейкозу (CML) та гострого лімфобластного лейкозу (ALL), що включає одночасне введення зазначеній тварині терапевтично ефективної кількості (R)-N-(4-(хлордифторметокси)феніл)-6-(3-гідроксипіролідин-1-іл)-5-(1H-піразол-5іл)нікотинаміду (приклад 9) або його фармацевтично прийнятної солі та терапевтично ефективної кількості нілотинібу. 27 UA 113208 C2 5 10 15 20 25 30 35 40 45 50 55 60 Сполуку формули (I) також можна використовувати у комбінації з іншими протипухлинними сполуками. Такі сполуки включають, але не обмежуються цим, інгібітори рибонуклеотидредуктази, інгібітори топоізомерази I; JAK інгібітори, такі як руксолітиніб; інгібітори smoothened, такі як LDE225; інтерферон; інгібітори топоізомерази II; діючі на мікротрубочки активні сполуки; алкілуючі сполуки; інгібітори гістон деацетилази; інгібітори mTOR, такі як RAD001; протипухлинні антиметаболіти; сполуки платини; сполуки, що прицільно діють на/знижують протеїн- або ліпідкіназну активність, інгібітори метіонінамінопептидази; модифікатори біологічної відповіді; інгібітори Ras онкогенних ізоформ; інгібітори теломерази; інгібітори протеасоми; сполуки, що використовуються у лікуванні гематологічних злоякісних захворювань, такі як флударабін; сполуки, які прицільно діють на, знижують або інгібують активність PKC, такі як мідостаурин; інгібітори HSP90, такі як 17-AAG (17аліламіногелданаміцин, NSC330507), 17-DMAG (17-диметиламіноетиламіно-17-деметоксигелданаміцин, NSC707545), IPI-504, CNF1010, CNF2024, CNF1010 від компанії Conforma Therapeutics, HSP990 та AUY922; темозоломід (TEMODAL); інгібітори кінезинового веретенного білку, такі як SB715992 або SB743921 від компанії GlaxoSmithKline або пентамідин/хлорпромазин від компанії CombinatoRx; інгібітори PI3K, такі як BEZ235, BKM120 або BYL719; інгібітори MEK, такі як ARRY142886 від компанії Array PioPharma, AZD6244 від компанії AstraZeneca, PD181461 від компанії Pfizer, лейковорин, EDG зв'язуючі, антилейкемічні сполуки, інгібітори S-аденозилметіонін декарбоксилази, антипроліферативні антитіла або інші хіміотерапевтичні сполуки. Крім того, альтернативно або на додаток до цього, їх можна використовувати у комбінації з іонізуючим опроміненням. Крім того, альтернативно або на додаток, вони можуть бути використані у комбінації з JAK інгібіторами, такими як руксолітиніб. Крім того, альтернативно або на додаток, вони можуть бути використані у комбінації з інгібіторами smoothened, такими як LDE225. Крім того, альтернативно або на додаток вони можуть бути використані у комбінації з інтерфероном. Термін "інгібітори рибонуклеотидредуктази" відноситься до піримідинових або пуринових нуклеозидних аналогів, включаючи, але не обмежуючись цим, флударабін та/або цитозин арабінозид (ara-C), 6-тіогуанін, 5-фторурацил, кладрибін, 6-меркаптопурин (особливо у комбінації з ara-C проти ALL), клофарабін, неларабін (проліки 9-β-арабінофуранозилгуанін, araG), пентостатин, гідроксисечовину або 2-гідрокси-1H-ізоіндол-1,3-діонові похідні (Nandy et al., Acta Oncologica 1994; 33:953-961). Термін "інгібітор топоізомерази I", як це використовується у даній заявці, включає, але не обмежується цим, топотекан, гіматекан, іринотекан, камптотеціан та його аналоги, 9нітрокамптотецин та макромолекулярний камптотециновий кон'югат PNU-166148 (сполука A1 в WO99/17804). Іринотекан можна вводити, наприклад, у формі, як він поставляється на ринок, наприклад, під торговою маркою CAMPTOSAR. Топотекан можна вводити, наприклад, у формі, як він поставляється на ринок, наприклад, під торговою маркою HYCAMTIN. Термін "інгібітор топоізомерази II", як це використовується у даній заявці, включає, але не обмежується цим, антрацикліни, такі як доксорубіцин (включаючи ліпосомний препарат, наприклад, CAELYX), даунорубіцин, епірубіцин, ідарубіцин та неморубіцин, антрахінони мітоксантрон та лосоксантрон та подофілотоксини етопозид та теніпозид. Етопозид можна вводити, наприклад, у формі, як він поставляється на ринок, наприклад, під торговою маркою ETOPOPHOS. Теніпозид можна вводити, наприклад, у формі, як він поставляється на ринок, наприклад, під торговою маркою VM 26-BRISTOL. Доксорубіцин можна вводити, наприклад, у формі, як він поставляється на ринок, наприклад, під торговою маркою ADRIBLASTIN або ADRIAMYCIN. Епірубіцин можна вводити, наприклад, у формі, як він поставляється на ринок, під торговою маркою FARMORUBICIN. Ідарубіцин можна вводити, наприклад, у формі, як він поставляється на ринок, наприклад, під торговою маркою ZAVEDOS. Мітоксантрон можна вводити, наприклад, у формі, як він поставляється на ринок, наприклад, під торговою маркою NOVANTRON. Термін "діюча на мікротрубочки активна сполука" відноситься до стабілізуючих мікротрубочки, дестабілізуючих мікротрубочки сполук та інгібіторів полімеризації мікротубуліну, що включають, але не обмежуючись цим, таксани, наприклад, паклітаксел та доцетаксел, алкалоїди барвінку, наприклад, вінбластин, особливо вінбластин сульфат, вінкристин, особливо вінкристин сульфат, та вінорелбін, дискодермоліди, кохіцин та епотилони та їх похідні, наприклад, епотилон B або D або їх похідні. Паклітаксел можна вводити, наприклад, у формі, як він поставляється на ринок, наприклад, TAXOL. Доцетаксел можна вводити, наприклад, у формі, як він поставляється на ринок, наприклад, під торговою маркою TAXOTERE. Вінбластин сульфат можна вводити, наприклад, у формі, як він поставляється на ринок, наприклад, під 28
ДивитисяДодаткова інформація
Назва патенту англійськоюBenzamide derivatives for inhibiting the activity of abl1, abl2 and bcr-abl1
Автори англійськоюDodd, Stephanie Kay, Furet, Pascal, Grotzfeld, Robert Martin, Jahnke, Wolfgang, Jones, Darryl Brynley, Manley, Paul, Marzinzik, Andreas, Pelle, Xavier Francois Andre, Salem, Bahaa, Schoepfer, Joseph
Автори російськоюДодд Стэфани Кэй, Фурэ Паскаль, Гротцфельд Роберт Мартин, Янке Вольфганг, Джонс Дэррил Бринли, Мэнли Пол, Марцинцик Андреас, Пэллэ Ксавье Франсуа Андрэ, Салэм Баха, Шопфер Йозеф
МПК / Мітки
МПК: C07D 403/10, A61K 31/4439, C07D 213/82, C07D 401/14, C07D 401/04
Мітки: бензаміду, bcr-abl1, abl1, похідні, інгібування, активності
Код посилання
<a href="https://ua.patents.su/91-113208-pokhidni-benzamidu-dlya-ingibuvannya-aktivnosti-abl1-abl2-ta-bcr-abl1.html" target="_blank" rel="follow" title="База патентів України">Похідні бензаміду для інгібування активності abl1, abl2 та bcr-abl1</a>
Конденсовані піримідинові похідні для інгібування тирозинкіназної активності

Номер патенту: 111272
Опубліковано: 11.04.2016
Автори: Кім Мі Ра, Кім Маєнг Суп, Дзеон Дзи Янг, Лі Дзу Йєон, Сух Квеє Хіун, Дзо Міоунг Гі, Канг Сеок Дзонг, Квак Єун Дзоо, Ча Мі Янг, Ха Тає Хеє, Лі Кванг-Ок
МПК: A61P 35/00, A61K 31/4365, C07D 495/04, C07D 409/12
Мітки: інгібування, похідні, тирозинкіназної, конденсовані, активності, піримідинові
Формула / Реферат:
1. Сполука формули (I) або її фармацевтично прийнятна сіль: , (I)деW являє собою О;X являє собою О, NH, S, SO або SO2;Y являє собою атом водню, атом галогену, C1-6алкіл або C1-6алкокси;кожний з А і В незалежно являє собою атом водню, атом галогену або ді(C1-6алкіл)амінометил;Z являє собою арил або гетероарил, який містить...
Сполука для інгібування активності rho кінази, фармацевтична композиція, що містить таку сполуку, та спосіб інгібування активності rho кінази

Номер патенту: 96126
Опубліковано: 10.10.2011
Автори: Кірк Брайян, Сешадрі, Хемалатха, Рам Сія, Світнем Пол, Кемпбелл Стюарт, Фудулакіс Хоуп, Бартолоцці Алессандра
МПК: C07D 403/02
Мітки: таку, композиція, спосіб, сполука, кінази, інгібування, фармацевтична, містить, активності, сполуку
Формула / Реферат:
1. Сполука формули І: (І)або її фармацевтично прийнятна сіль або гідрат, де: R1 є вибраним з групи, що складається з: -(CH2)y-NR13R14, -O-(CH2)y-CO2R12, -O-(CH2)y-C(=O)NR13R14, -О-(СН2)у-гетероарилу, -О-(СН2)у-циклоалкілу, -O-C(=O)-(CH2)y-NR13R14, -O-(CH2)z-NR13R14, -NH-C(=O)-(CH2)y-NR13R14, -NH-C(=O)-X-R15,...
Конденсовані піримідинові похідні для інгібування тирозинкіназної активності

Номер патенту: 108889
Опубліковано: 25.06.2015
Автори: Ха Тає Хеє, Квак Єун Дзоо, Ча Мі Янг, Дзо Міоунг Гі, Дзеон Дзи Янг, Лі Дзу Йєон, Кім Маєнг Суп, Кім Мі Ра, Сух Квеє Хіун, Лі Кванг-Ок, Канг Сеок Дзонг
МПК: A61P 35/00, C07D 409/12, A61K 31/4365, C07D 495/04
Мітки: активності, тирозинкіназної, піримідинові, похідні, інгібування, конденсовані
Спосіб інгібування активності impdh, похідні сечовини, фармацевтична композиція для лікування або профілактики impdн-опосередкованих захворювань (варіанти), спосіб лікування або профілактики impdн-опосередкован

Номер патенту: 61074
Опубліковано: 17.11.2003
Автори: Новак Перрі М., Бадія Майкл.К, Беміс Гай В., Бетайл Ренді С., Ронкін Стівен М., Армістед Девід М., Франк Катаріна А., Саундерс Джеффрі О.
МПК: C07C 275/42, C07D 263/32, A61K 31/42, A61K 31/18, A61K 31/421, A61P 9/14, A61P 35/00, C07D 413/12, A61P 43/00, A61K 31/17, A61P 31/12, A61K 31/422, A61K 31/404, A61P 37/06, A61P 29/00
Мітки: варіанти, композиція, активності, лікування, impdн-опосередкован, фармацевтична, сечовини, похідні, захворювань, інгібування, профілактики, impdh, спосіб, impdн-опосередкованих
Формула / Реферат:
1. Спосіб інгібування активності IMPDH у ссавця, при якому вводять зазначеному ссавцеві сполуку формули:,у якій:В означає насичену, ненасичену або частково насичену моноциклічну або біциклічну кільцеву систему, яка не обов'язково має до 4 гетероатомів, вибраних із N, O або S, яку вибирають із сполук, виражених формулами:,де кожний Х означає кількість атомів водню, необхідних для досягнення потрібної...
Галогенізовані похідні бензаміду

Номер патенту: 90864
Опубліковано: 10.06.2010
Автор: Россіньоль Жан Франсуа
МПК: C07D 277/44, A61K 31/426
Мітки: бензаміду, похідні, галогенізовані
Формула / Реферат:
1. Сполука формули (III):, (ІІІ)деR1 - атом галогену, аR2-R6, незалежно, - гідроген, гідроксил, -С1-С4алкоксил, ацилоксил, -C(O)R7, де R7 - -С1-С4алкіл, або є ароматичними групами, у тому числі солі та гідрати цієї сполуки, де принаймні один з R2-R6 не є гідрогеном.2. Сполука за п. 1, де принаймні один з R2-R6 - гідроксил.3....
Попередній патент: Пасивна динамічна інерційна балансувальна система ротора для турбомашинного обладнання
Наступний патент: Термокаталітичний газоаналізатор випаровувань автозаправних станцій
Випадковий патент: Електроакустичний перетворювач