Фармацевтична композиція для лікування патологічних станів, пов’язаних з активністю лейкотриєнів
Номер патенту: 27711
Опубліковано: 16.10.2000
Автори: Тімко Роберт Джозеф, Клементс Арлін, Едврдс Ієуан Джон, Бредвей Ренді Джон, Холохан Джеймс Джозеф




Формула / Реферат
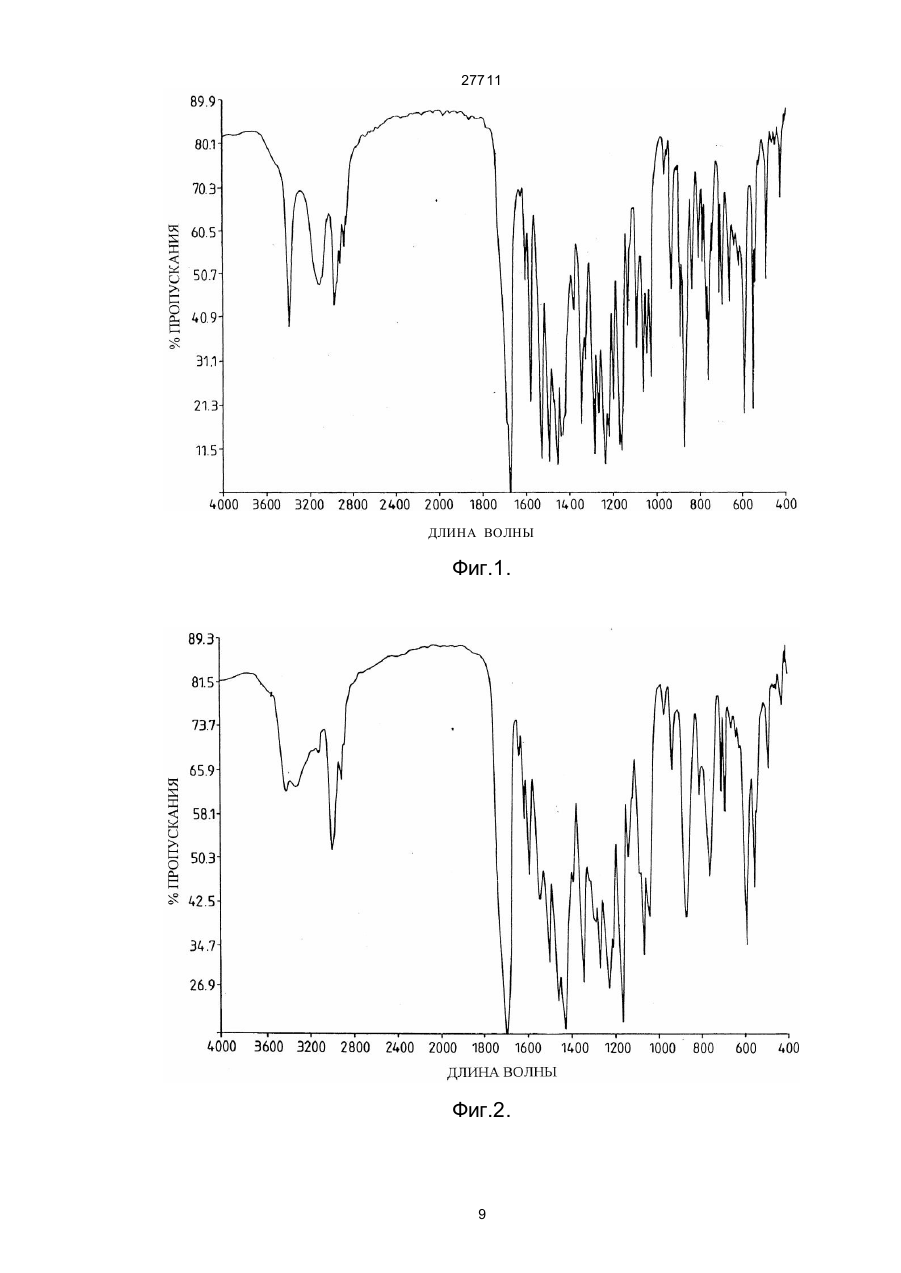
(57) 1 .Фармацевтическая композиция для лечения патологических состояний, связанных с активностью лейкотриенов, содержащая активный ингредиент и целевые добавки, отличающаяся тем, что в качестве активного ингредиента она содержит физическую форму М-[4-[5-(циклопентилокси~карбонил)амино-1-метилиндол-3-илметил]-3-мето-ксибензоил]-2-метилбензолсульфонамида, по существу свободную от других физических форм, характеризующуюся ИК-спектром (0,5% в КВг),имеющую четкие пики поглощения при: 1690,1530,1490, 1420, 1155, 1060, 862 и 550 см'1, в фармацевтически эффективном количестве и поливи-нилпирролидон.
2. Композиция по п. 1, отличающаяся тем, что она содержит фармацевтически приемлемый носитель.
3. Композиция по п. 1 или 2, отличающаяся тем, что она содержит активный ингредиент в количестве от 1 до 90% по весу в расчете на общий вес композиции.
4. Композиция по любому из лп. 1-3, отличающаяся тем, что она содержит поливинилпирроли-дон в количестве от 1 до 20% от общего веса композиции.
5. Композиция по любому из пп. 1-4, отличающаяся тем, что она содержит фармацевтически приемлемый носитель, выбранный из манкита, лактозы, сорбита, глюкозы, сахарозы, декстрозы, фруктозы, ксилита, микрокристаллической целлюлозы, порошкообразной целлюлозы и гидрокси-пропилметилцеллюлозы.
6. Композиция по любому из пп. 1-5, отличающаяся тем, что она дополнительно содержитадъюванты, способствующие процессу приготовления препарата, выбранные из чатрийкроскар-мелозы крахмального гликолята натрия, крахмала, стеарата магния, стеариновой кислоты, талька и порошкообразного растительного стеарина.
7. Композиция по любому из пп 1-6, отличающаяся тем, что она представлена в форме таблеток.
Текст
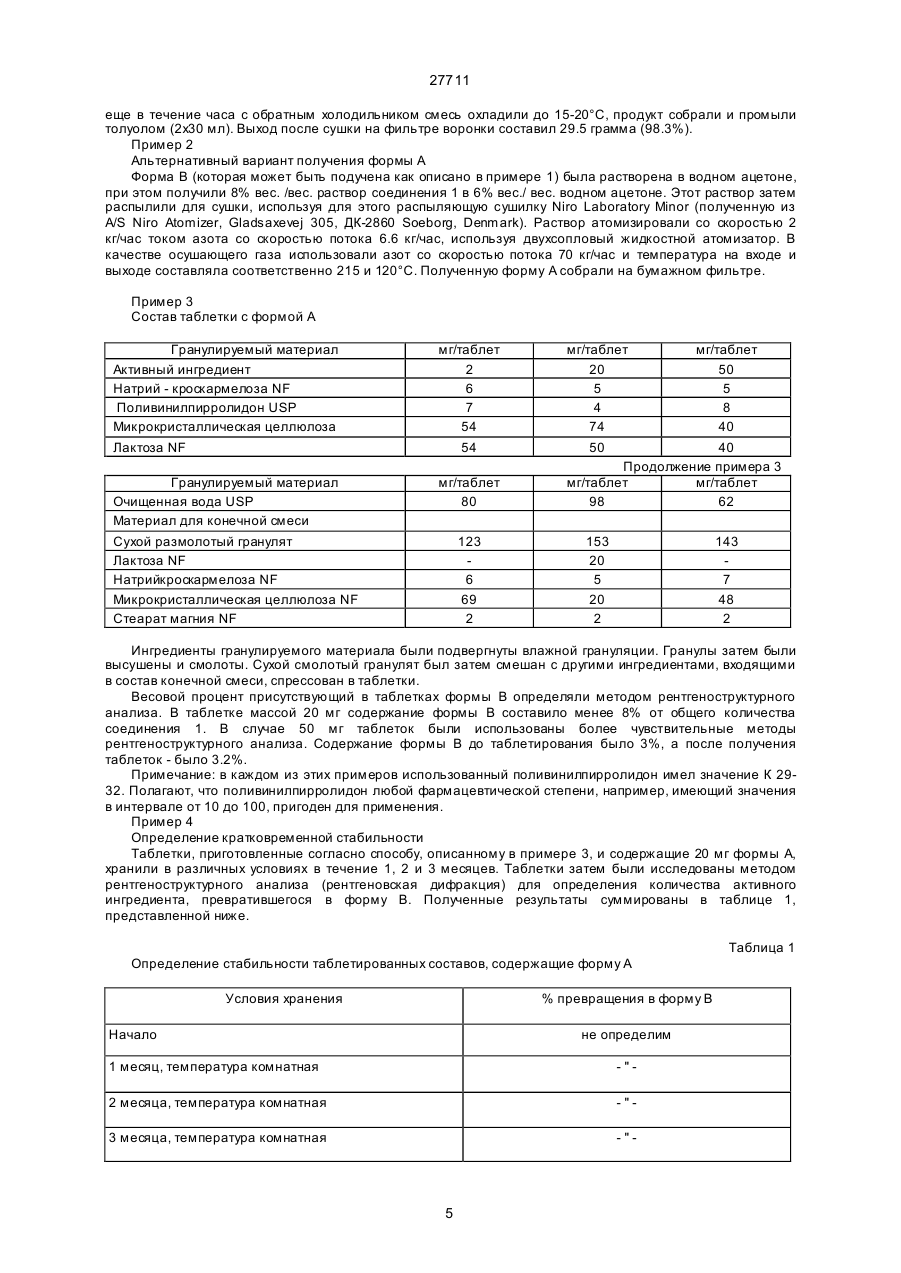
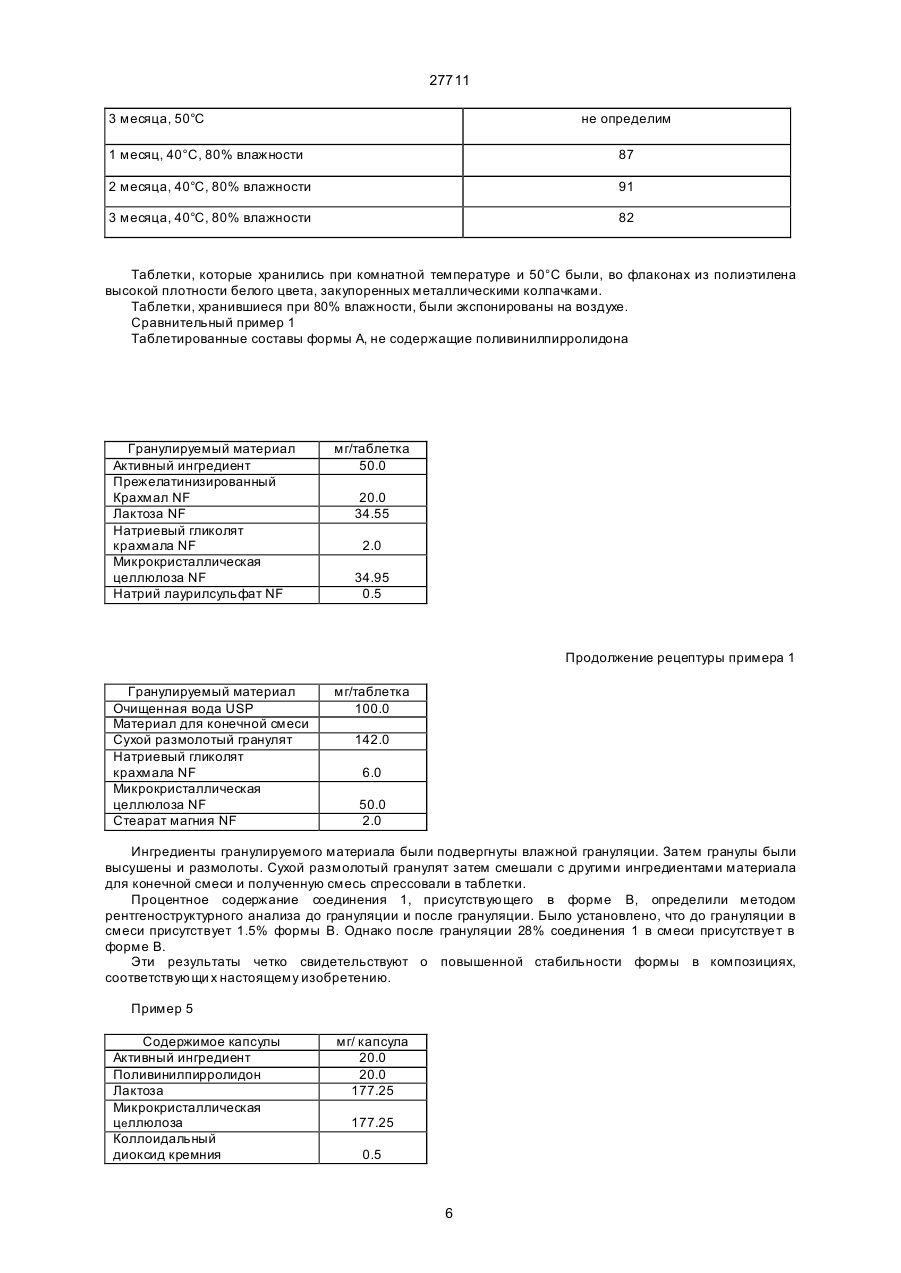
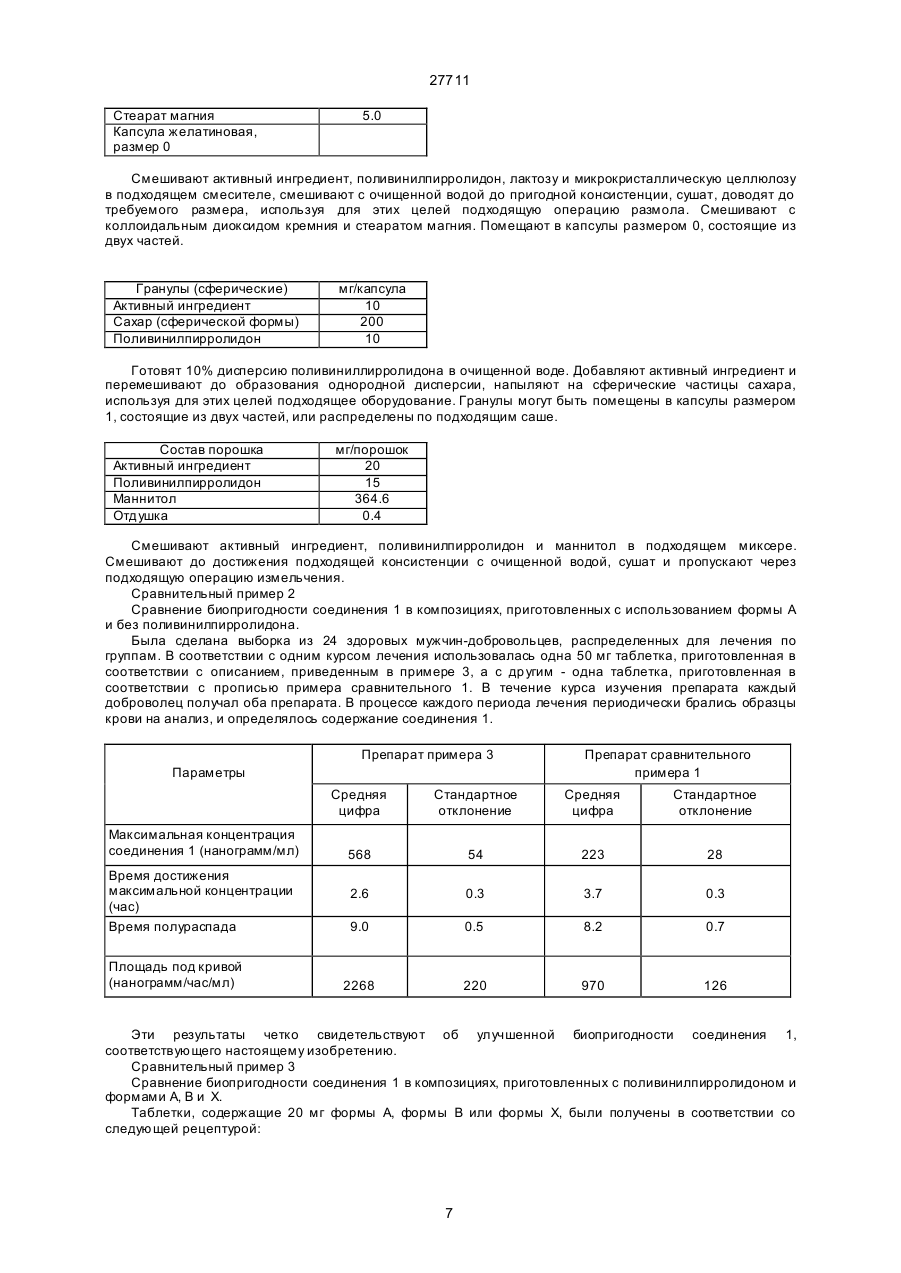
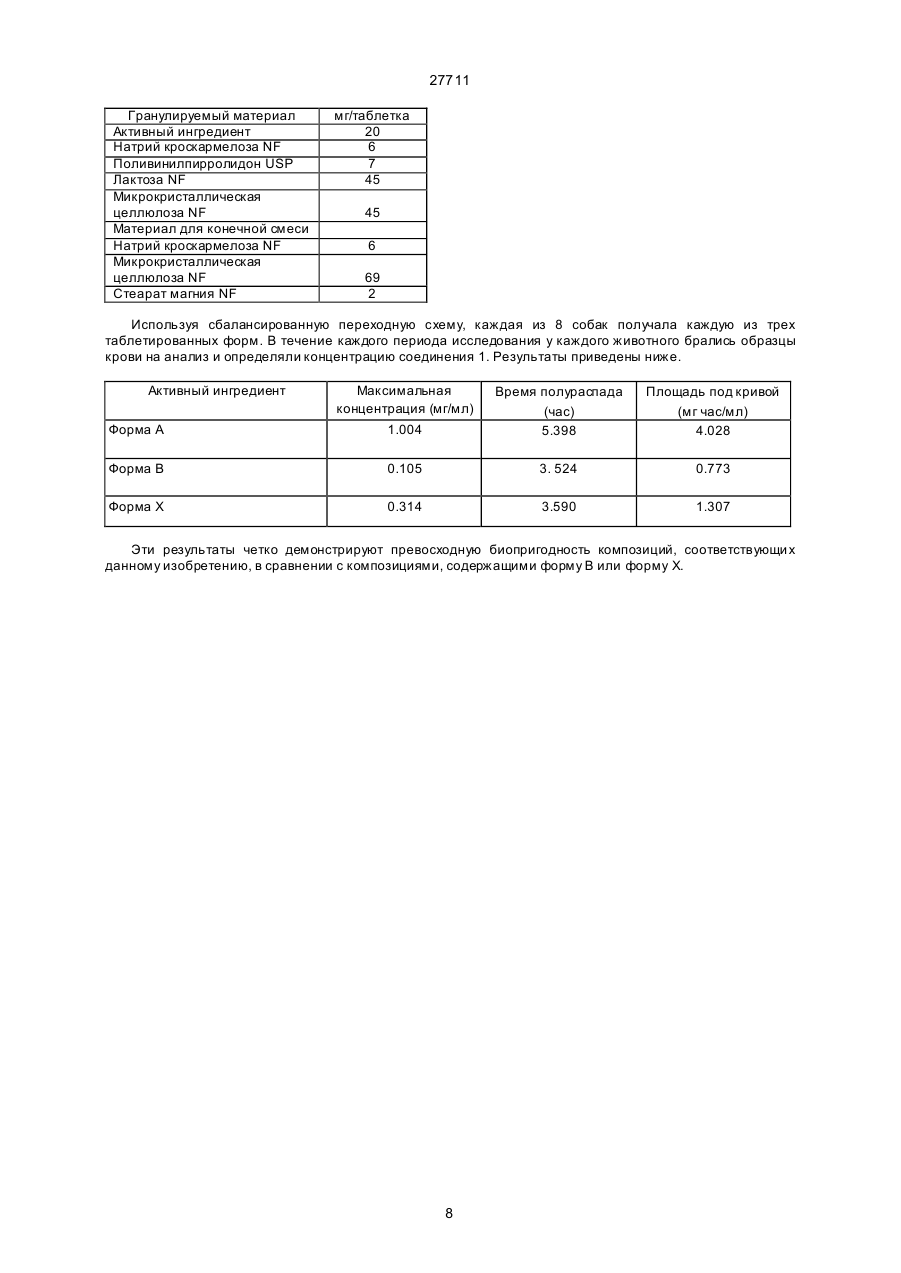
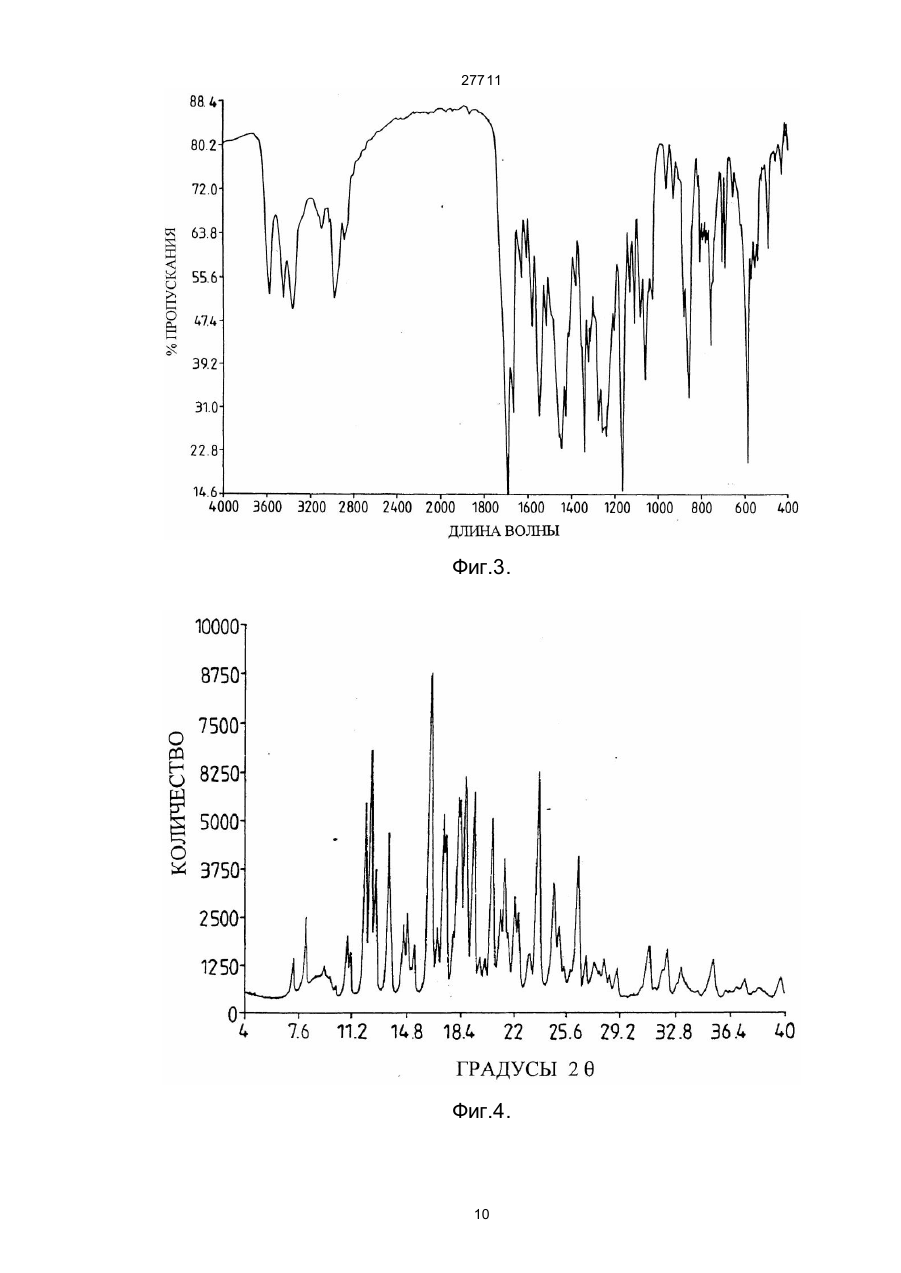
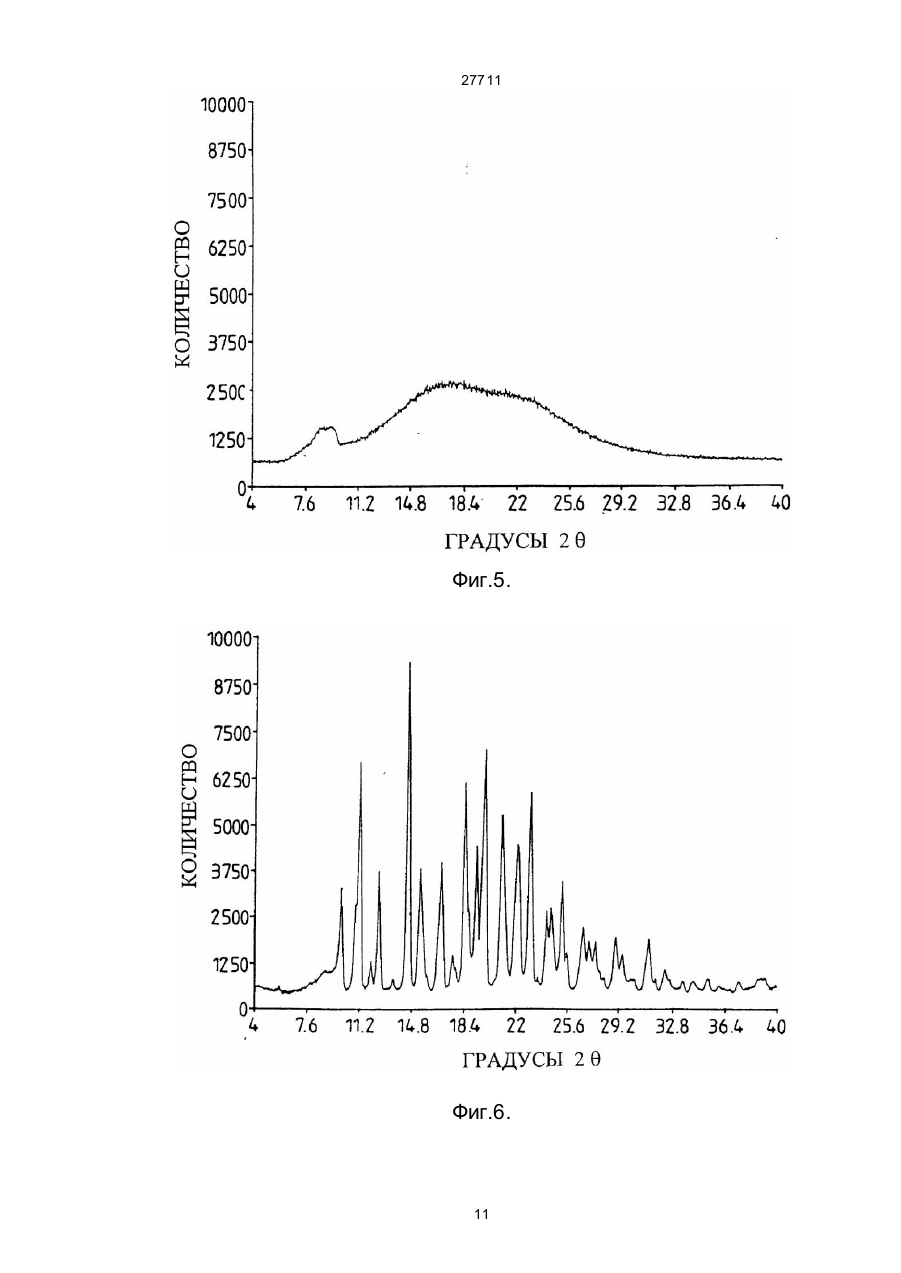
27711 Настоящее изобретение относится к фармацевтическим средствам. Более конкретно, оно относится к фармацевтической композиции, содержащей, в частности, физическую форму производного гетероциклического амида, способам получения этой физической формы, и к другим физическим формам гетероциклического амида, используемым при получении первой упомянутой физической формы. В публикации Европейской патентной заявки ЕР-А2-0199543 раскрываются некоторые производные гетероциклического амида, которые являются антагонистами одного или более метаболитов арахидоновой кислоты, известных под названием лейкотриены, например, С4, D4, и/или Е4, которые, как известно, должны быть сильными спазмогенами (в частности, легких), увеличивая сосудистую проницаемость и вовлекая в патогенез астмы и воспаления (см. J.L.Marx, Science 1982, 215, I380-I383), также как и эндотоксического шока (см. J.A.Cook и др., Pharmacol. Exp. The2, 1985, 230,330). Таким образом, эти соединения полезны при лечении заболеваний, в которых замешаны лейкотриены, и антагонизм действия по отношению к которым полезен. Эти заболевания включают, например, аллергические легочные расстройства, такие как астма, сенная лихорадка и аллергические риниты и некоторые воспалительные заболевания, такие как бронхиты, эктопические и атопические экземы, псориазы, также как вазоспастическое кардиоваскулярное заболевание и эндотоксичные и травматические шоковые условия. Одним из производных гетероциклического амида, раскрытого в ЕР-А2-0199543, является N-[4-[5-(циклопентилоксикарбонил)амино - 1-метилиндол-3-илметил]-3-метоксибензоил] - 2 - метилбензолсульфонамид. Это соединение описано в примере 105 патентной спецификации и будет обозначаться здесь и далее, как соединение 1. Недавно при клинических испытаниях было установлено, что соединение 1 эффективно при лечении астмы при оральном введении препарата. Эта способность соединения 1 проявлять эффективное действие при оральном ведении необычна и очень желательна. Фармацевтическая композиция, использованная в вышеупомянутом клиническом испытании, не полностью удовлетворила, и необходимо было думать об улучшенном составе. Обнаружено, что соединение 1 обладает относительно слабой растворимостью в воде. Имеется потребность в фармацевтической композиции, подходящей для орального приема, которая бы включала соединение 1 в твердом состоянии. Было обнаружено, что соединение 1 может быть получено в твердом состоянии как материал, имеющий диапазон различных физических свойств, зависящи х от способа его выделения и последующей обработки. Было установлено, что эта способность обусловлена тем фактом, что соединение 1 может существова ть в более чем одной физической форме, по крайней мере, одна из которых - физически не стабильна, и физические формы этого соединения 1 могут быть получены в смеси. Было также установлено, что различные образцы соединения 1 в твердом состоянии обладают различной биопригодностью. Нежелательно разрабатывать препарат, содержащий смесь физических форм соединения, обладающих различной биопригодностью, особенно когда одна из форм, входящи х в смесь, физически нестабильна, поскольку в этом случае нельзя точно контролировать эффективную дозу соединения. Следовательно, возникла необходимость в поиске методов для получения физических форм соединения 1, преимущественно свободных от др уги х физических форм. Методы получения трех физических форм соединения 1, преимущественно свободных от други х физических форм, были обнаружены, физическая стабильность и биопригодность этих тре х форм была исследована. Две из эти х форм, обозначаемые здесь и далее как формы В и X, как установлено, физически стабильны, но не очень биопригодны. Третья из этих форм, обозначенная здесь и далее как форма А, обладает вполне приемлемой биопригодностью. Однако было обнаружено, что эта форма имеет тенденцию превращаться в присутствии воды в форму В. Это свойство является недостатком для материала, который собираются вводить в твердую композицию, поскольку на стадии грануляции предусмотрено использование воды, как адьюванта в процессе смешения. Таблетки, использованные в вышеупомянутом клиническом испытании, были получены способом влажной грануляции из формы А, и было установлено, что форма В содержится в препарате в количестве, варьирующемся от примерно 25 до примерно 30% по массе соединения 1. Таким образом, существует необходимость в фармацевтической композиции, подходящей для орального введения, которая бы включала соединение 1 в одной физической форме, преимущественно свободной от других физических форм, композиции, которая была бы физически стабильна, могла бы быть воспроизводимой и обладать хорошей биопригодностью. Удивительно, но было установлено, что фармацевтические составы, удовлетворяющие этим требованиям, могут быть получены путем выбора в качестве активного ингредиента формы А и поливинилпирролидона - в качестве соингредиента. Итак, настоящее изобретение обеспечивает фармацевтическую композицию, которая включает в качестве активного ингредиента физическую форму N-[4-[5-(циклопентилоксикарбонил)амино-1метилиндол-3-илметил]- 3 - метоксибензоил]-2 - метилбензолсульфонамид (обозначенную здесь ранее как форма А), преимущественно свободную от др уги х физических форм, эта физическая форма имеет следующие параметры ИК-спектра (0.5% КВR): четкие полосы поглощения при 1690, 1530, 1490, 1420, 1155, 1060, 862 и 550 см -1, а также включает поливинилпирролидон. На дифракционной рентгеновской диаграмме порошка формы А отсутствуют различимые пики, что свидетельствует о его аморфности. Установлено, что композиции, соответствующие данному изобретению, обладают приемлемой 1 27711 физической стабильностью и могут быть приготовлены с воспроизводимыми характеристиками, а также обладают удивительно высокой биопригодностью. Когда речь идет в данной спецификации о том, что форма А преимущественно свободна от други х физических форм, то это предпочтительно означает, что, по крайней мере, 90% массы соединения 1 присутствует в этой физической форме, более предпочтительно, когда в этой физической форме присутствует, по крайней мере, 95% , например, по крайней мере 96%, 97%, 98% или 99%. Композиция, соответствующая данному изобретению, может находиться в любой стандартной форме, подходящей для перорального приема, например, в виде таблеток, капсул, гранул или порошка. Предпочтительно, когда она выпускается в форме таблеток. В композиции, соответствующей данному изобретению, активный ингредиент обычно присутствуе т в количестве от 1 до 90% по массе в расчете на общую массу композиции, например, от 10 до 50%. Количество поливинилпирролидона составляет, по крайней мере, 1% в расчете на общую массу композиции. Он может вместе с активным ингредиентом составлять всю массу композиции. Однако, как правило, более характерно, когда композиция включает кроме того, по крайней мере, один фармацевтически приемлемый носитель. Например, поливинилпирролидон может присутствовать в количестве от 1 до 20% от массы общей композиции, предпочтительно от 2 до 6% по массе. Примеры подходящих фармацевтически приемлемых носителей включают, например, производные сахаров, такие как, манитол, лактоза, сорбитол, глюкоза, сахароза, декстроза, фруктоза, ксилитол, а также производные целлюлозы, такие как микрокристаллическая целлюлоза, порошкообразная целлюлоза, гидроксипропилметилцеллюлоза. Предпочтительно, когда композиция включает производное сахара, особенно лактозу, и производное целлюлозы, особенно, микрокристаллическую целлюлозу. Количество производного сахара, присутствующего в композиции, может составлять, например, от 10 до 30% от массы композиции. Количество производного целлюлозы, например, может варьироваться от 25 до 75% от массы композиции. Далее композиция может включать одну или более добавок, способствующи х процессу приготовления препарата, таких как дезинтеграторы, например, натрий кроскармелоза, крахмал и натрия гликолят крахмала, а также лабриканты, например, стеарат магния, стеариновую кислоту, тальк, порошкообразный растительный стеарин. Дезинтегратор может присутствовать, например, в количестве от 1 до 10% по массе от массы композиции. Количество лабриканта может, например, варьироваться от 0.25 до 2% от массы композиции. Композиция может быть приготовлена при смешении ингредиентов, согласно традиционно существующим способам, например, в процессе грануляции. Соответственно, еще одной целью настоящего изобретения является создание способа приготовления фармацевтической композиции, которая включает смешение формы А, преимущественно свободной от други х физических форм, с поливинилпирролидоном и водой и высушивание полученной смеси. Количество использованной воды зависит от типа фармацевтической композиции, которую необходимо получить (например, таблетки, капсулы, порошок или гранулы), и природы других ингредиентов, которые включают в состав композиции. Традиционно использование такого количества воды, когда весовое отношение воды к форме А находится в интервале от 0.1 до 100:1. Когда композиция изготовлена в виде таблеток, то вес таблетки может варьироваться от 25 до 500 мг, например, от 25 до 250 мг или, например, от 100 до 200 мг. Таблетки могут быть с покрытием или без покрытия. Таблетки могут иметь покрытие из стандартного материала, который может наноситься стандартным способом. Еще одна цель настоящего изобретения состоит в обеспечении способов получения формы А, преимущественно свободной от других физических форм соединения 1. Итак, настоящее изобретение обеспечивает способ получения формы А, преимущественно свободной от други х физических форм, который включает нагревание другой физической формы N-[4-[5(циклопентилоксикарбонил)амино-1-метил-индол-3-ил-метил]-3-метоксибензоил]-2-метилбензолсульфонамида (обозначенного здесь ранее, как форма В), преимущественно свободной от других кристаллических форм, физическая форма которого представляет моногидрат N-[4-[5(циклопентилоксикарбонил)амино - 1-метилиндол-3-илметил] - 3 -метоксибензоил]-2-метилбензолсульфонамид, являющийся кристаллическим образованием, и имеет следующие параметры ИК-спектра (0,5% в КВR): четкие полосы поглощения при 3560, 1690, 1660, 1540, 1440, 1165, 880 и 858см -1, а рентгенограмма порошка имеет пики при 20 =10.0, 11.2, 14.6, 19.8 и 23.0°, в температурном интервале от 90 до 125°С при пониженном давлении. Дегидратирование формы В предпочтительно проводить в температурном интервале от 115 до 122°С. Давление в процессе дегидратации формы В предпочтительно не должно превышать 100 миллибар (10 кПа), более предпочтительно - не должно превышать 50 миллибар (5 кПа). Например, давление может изменяться в пределах от 0.5 до 5 кПа. Форма В может быть получена преимущественно свободной от други х кристаллических форм путем перекристаллизации из неводного ацетона. В частности, она может быть получена путем растворения источника соединения 1 в водном ацетоне при повышенных температурах, с последующим добавлением воды и выдержкой полученной смеси до охлаждения. Предпочтительно, когда воду добавляют быстро, так, чтобы соединение 1 в начале выделилось в виде масла. Полученный таким образом материал соответствует форме А в состоянии высокой степени морфологической чистоты. Кристаллический продукт может быть высушен при повышенной температуре, например, при 60°С или ниже. Если необходимо начинать работу с источником соединения 1, содержащим примеси, то было 2 27711 найдено, что лучше вначале этот исходный источник, содержащий примеси, растереть с горячей смесью толуола и этилацетата. Следует учесть, что если форму В сушили при высокой температуре, например, выше 60°С, то могло произойти частичное превращение формы В в форму А. Материал, полученный при сушке формы при температуре ниже 60°С или примерно равной, преимущественно свободен от других физических форм, характерных для соединения 1. Полагают, что форма В - это неизвестная ранее форма соединения 1. Изобретение обеспечивает таким образом форму В, преимущественно свободную от др уги х кристаллических форм. Изобретение также обеспечивает другой способ получения формы, преимущественно свободной от други х физических форм соединения 1, который включает быстрое выпаривание растворителя из раствора соединения 1. Например, она может быть получена путем сушки распылением раствора соединения 1. Растворителем может служить любое жидкое вещество, способное растворять соединение 1, и кипеть при температуре ниже точки плавления формы А соединения 1. К таким растворителям относятся, например, кетоны, такие как ацетон, и нитрилы, такие как ацетонитрил, возможно в смеси с водой. Было найдено, что конкретно водный раствор ацетона является подходящим растворителем. Температура испарения растворителя должна быть ниже точки плавления формы А. Как правило, она ниже 125°С, предпочтительно, когда температура ниже 120°. Было установлено, что в тех случаях, когда в качестве растворителя используется ацетон, значительное количество кристаллического материала получается тогда, когда температура ниже 100°С. Следовательно, температура испарения растворителя может быть в интервале от 100 до 125°С. Раствор соединения 1, как правило, получают путем растворения кристаллической формы соединения 1, такой как форма В, в растворителе. Раствор, полученный таким путем должен содержать минимальное количество нелетучи х примесей. Из вышесказанного следует учесть, что водный ацетон является, в частности, предпочтительным растворителем для использования при получении формы А, преимущественно свободной от других физических форм соединения 1. Также было установлено, что содержание органическогорастворителя в форме, полученной из формы В с использованием такого раствора, очень мало. Итак, кроме всего, настоящее изобретение обеспечивает раствор соединения 1 в водном ацетоне. Этот раствор может содержать, например, от 5 до 15% соединения 1, предпочтительно от 6 до 13%. Растворитель может содержать, например, от 3 до 9% воды, предпочтительно от 4 до 8%. Преимущества таких композиций, соответствующих настоящему изобретению, может быть проиллюстрировано путем сопоставления их свойств со свойствами композиций, в которых форма А замещена на форму В или форму Х и с композицией, содержащей форму А, но не содержащей поливинилпирролидона. Форма Х представляет физическую форму соединения 1, это кристаллическая форма, рентгенограмма порошка этой формы имеет характерные пики при 2 q = 8.1, 13.7, 16.4, 20.5 и 23.7 и ИК- спектр этой формы Х имеет следующие параметры (0.5% в КВR ): четкие полосы поглощения при 3370, 1670, 1525, 1490, 1280, 890, 870 и 550 см -1. Форма Х может быть получена преимущественно свободной от других физических форм следующим путем: растворение источника, содержащего соединение 1 в горячем водном ацетоне, упаривание полученного раствора, прибавление толуола к оставшемуся объему и затем вновь упаривание раствора. Если в качестве источника соединения 1 используется материал, содержащий примеси, то лучше такой материал в начале растереть с горячей смесью толуола и этилацетата до проведения стадии кристаллизации. Каждая из этих трех форм А, В и Х может быть характеризована стандартными методами, например, путем получения рентгенограмм порошков каждой из форм или путем снятия ИК-спектров. В данной спецификации для получения ИК- спектров были получены таблетки, содержащие 0.5% исследуемого материала в бромиде калия, спектр снимали в диапазоне длин волн от 4000 до 400см -1. Примеры ИК-спектров каждой из форм Х, А и В приведены на рис.1, 2 и 3, прилагаемых здесь далее. Для получения рентгенограмм были использованы образцы, масса которых равнялась 2 г, эти образцы помещали в стандартный, глубоко расположенный, держатель установки марки Phillips, измерения проводили в области сканирования 4-40° 2 q, считая 4 секунды на точку с интервалом 0.02°, в результате получили запись зависимости интенсивности от расстояния для этой области. Примеры диффракционных рентгенограмм для каждой из форм Х, А и В приведены на рис.4, 5 и 6, прилагаемых далее. Температура плавления (точки плавления) каждой из форм А, В и Х зависят от степени их чистоты. Как правило, точка плавления формы Х лежит выше 190°С, например, примерно при 200°С. Точка плавления формы А плавает в пределах от 115 до 140°С, например, примерно от 124 до 132°С; и температура плавления формы В варьируется в пределах от примерно 140 до 160°С, например, от 145 до 155°С. Было обнаружено, что форма В теряет воду при температуре выше примерно 60°С, и поэтому не имеет резко выраженной температуры плавления, а плавится в интервале температур. Как утверждалось здесь ранее, форма А обладает приемлемой стабильностью в композициях, соответствующи х данному изобретению. Однако в условиях высокой влажности и при повышенных температурах, как было обнаружено, форма А может переходить в форму В. Соответственно, в определенных условиях желательно хранить фармацевтические составы, содержащие форму А, в присутствии подходящего осушителя, такого как силикагель. Также желательно хранить эти композиции в герметичных упаковках (контейнерах), таких как блистерная упаковка. 3 27711 Доза соединения 1, назначаемая больному, в композиции, соответствующей настоящему изобретению, зависит от тяжести заболевания, возраста и веса больного. Как правило, соединение следует назначать в дозе, величина которой варьируется в пределах от 0.1 до 10 мг/кг, например, от 0.2 до 5 мг/кг. Исследования по определению острой токсичности были проведены на соединении 1 с целью установления величины LD50. Например, для крыс и мышей было установлено, что значения LD50 для соединения 1 превышают 500 мг/кг. Приведенные ниже примеры иллюстрируют данное изобретение, не ограничивая его рамки. Пример 1 Получение формы А а) Получение источника соединения 1, содержащего примеси. Метил 3-метокси-4-/1-метил-5-нитроиндол-3-илметил/-бензоат/ полученный как описано в примере 4 ЕП-А2-0199543, был превращен в свободную кислоту путем обработки водным раствором гидроксида натрия. Свободную кислоту затем обработали в дихлорметане тионилхлоридом и получили хлорангидрид кислоты. Полученный хлорангидрид кислоты затем запустили в реакцию с о-толуолсульфонамидом в дихлорметане в присутствии 2.2 эквивалента 4-диметиламинопири-дина для того, чтобы получить соль диметиламинопиридина и 4-/1-метил-5-нитроиндол-3-илме-тил/-3 метоксибензоил -2метилбензолсульфона-мида. Раствор диметиламинопиридиниевой соли 4-/I-метил - 5 -нитроиндол- 3 -илметил /-3-метоксибензоил-2-метилбензолсульфонамида (30 грамм) в 2метоксиэтаноле (I30 мл) и концентрированный раствор гидроксида натрия (3.2 мл) загрузили в колбу, через которую продувается азот, содержащую 10% палладий на активированном угле (3.3 грамма 60.9% водой увлажненной пасты). Смесь затем перемешивали в атмосфере водорода под давлением 3 бара (З.00 кПа) в течение 2.5 часов. Смесь затем отфильтровали через диатомовую землю и промыли 2-метоксиэтанолом (37.5 мл). К объединенным растворам добавили циклопентилхлорформиат (9.2 мл), и смесь оставили перемешиваться в токе азота на ночь. Затем установили температур у 30-33°С, в течение 20 минут при энергичном перемешивании добавили 68 мл 0.8 М раствора хлористоводородной кислоты. Затем смесь охладили до 15-20°С и перемешивали еще час. Сырой кристаллический продукт отфильтровали, промыли водой и высушили при 50°С. Он был затем использован в следующей стадии. в) Растирание загрязненного соединения 1. 60 грамм (0.101 г-моля) продукта стадии а), 240мл толуола (4 объема) и 150 мл этилацетата (2.5 объема) медленно нагрели до кипения и отогнали 30 мл (0.5 объема) дистиллята, удалив при этом большую часть освобождающейся воды. Смесь нагревали в колбе с обратным холодильником в течение одного часа (88-90°С) и затем охладили до 10-15°С. После перемешивания в течение 3 часов при 10-I5°C отфильтровали твердый остаток через стеклянный спекшийся материал и промыли смесью 2:1 толуола (80 мл) и этилацетата (40 мл). Продукт затем высушивали до постоянного веса на фильтре, получив в результате 53.2 грамма сухого соединения 1 (выход 91.5%). с/ Получение формы В 30 грамм продукта стадии в), 210 мл ацетона и 12 мл воды загрузили в 500 мл реакционную колбу. Смесь затем нагревали с обратным холодильником в течение 15 минут и затем пропустили через слой диатомовой земли на стеклянном спекшемся материале при 45-50°С непосредственно в 500 мл реакционной колбе. Колбу и спекшийся материал промыли смесью ацетона (60 мл) и воды (З мл). Объединенные растворы затем перемешивали на водяной бане примерно при 40°С и затем в течение 5 минут добавили 120 мл воды. Сначала смесь выделилась в виде масла, но затем быстро закристаллизовалась. Смесь затем охладили до 20°С в течение одного часа, затем перемешивали при 1520°С в течение двух часов и затем отфильтровали. Продукт промыли водой (60 мл), высушили как можно быстрее на стеклянном спекшемся материале, а затем подсушили в воздушной печи при температуре 60°С (макс.). Выход составил формы В 30.0 грамм (97%). d/ Получение формы А. 15.0 грамм продукта стадии с) поместили в круглодонную колбу объемом 500 мл, которую затем откачали на роторном испарителе при давлении 20 миллибар (2.0 кПа). Затем колбу с содержимым погрузили в масляную баню, предварительно нагретую до 118°С, и медленно вращали при этой температуре в течение 6 часов. Содержимое выгрузили при охлаждении и получили в результате порошок белого цвета. В промышленных масштабах форма А может быть получена следующим образом: 30 кг продукта, полученного на стадии с), распылили равномерно на металлический поддон и нагрели при 120°С в вакуумной печи площадью 7 м 2, вакуумный нагрев длился в течение 24 часов. Характерное давление было равно примерно 20 миллибар (2.0 кПа). При охлаждении (до 40°С или ниже) материал выгрузили из печи и после помола получили требующуюся форму А. В случае необходимости форма А до употребления может быть микронизирована. Получение формы Х 30.0 грамм продукта стадии в) (0.0521 грамм моля) растворили в ацетоне (150 мл) и воде (4.7 мл) при умеренном нагревании до кипения, раствор отфильтровали через воронку с фильтром из спекшегося стекла. Фильтрат нагрели до температуры кипения и отобрали 90 мл дистиллята. Добавили 120 мл толуола и далее отобрали 75 мл дистиллята. Добавили еще 120 мл толуола и отобрали еще 75 мл дистиллята. После нагревания 4 27711 еще в течение часа с обратным холодильником смесь охладили до 15-20°С, продукт собрали и промыли толуолом (2х30 мл). Выход после сушки на фильтре воронки составил 29.5 грамма (98.3%). Пример 2 Альтернативный вариант получения формы А Форма В (которая может быть подучена как описано в примере 1) была растворена в водном ацетоне, при этом получили 8% вес. /вес. раствор соединения 1 в 6% вес./ вес. водном ацетоне. Этот раствор затем распылили для сушки, используя для этого распыляющую сушилку Niro Laboratory Minor (полученную из A/S Niro Atomizer, Gladsaxevej 305, ДК-2860 Soeborg, Denmark). Раствор атомизировали со скоростью 2 кг/час током азота со скоростью потока 6.6 кг/час, используя двухсопловый жидкостной атомизатор. В качестве осушающего газа использовали азот со скоростью потока 70 кг/час и температура на входе и выходе составляла соответственно 215 и 120°С. Полученную форму А собрали на бумажном фильтре. Пример 3 Состав таблетки с формой А Гранулируемый материал Активный ингредиент Натрий - кроскармелоза NF Поливинилпирролидон USP Микрокристаллическая целлюлоза Лактоза NF мг/таблет 2 6 7 54 54 Гранулируемый материал Очищенная вода USP Материал для конечной смеси Сухой размолотый гранулят Лактоза NF Натрийкроскармелоза NF Микрокристаллическая целлюлоза NF Стеарат магния NF мг/таблет 80 123 6 69 2 мг/таблет мг/таблет 20 50 5 5 4 8 74 40 50 40 Продолжение примера 3 мг/таблет мг/таблет 98 62 153 20 5 20 2 143 7 48 2 Ингредиенты гранулируемого материала были подвергнуты влажной грануляции. Гранулы затем были высушены и смолоты. Сухой смолотый гранулят был затем смешан с другими ингредиентами, входящими в состав конечной смеси, спрессован в таблетки. Весовой процент присутствующий в таблетках формы В определяли методом рентгеноструктурного анализа. В таблетке массой 20 мг содержание формы В составило менее 8% от общего количества соединения 1. В случае 50 мг таблеток были использованы более чувствительные методы рентгеноструктурного анализа. Содержание формы В до таблетирования было 3%, а после получения таблеток - было 3.2%. Примечание: в каждом из этих примеров использованный поливинилпирролидон имел значение К 2932. Полагают, что поливинилпирролидон любой фармацевтической степени, например, имеющий значения в интервале от 10 до 100, пригоден для применения. Пример 4 Определение кратковременной стабильности Таблетки, приготовленные согласно способу, описанному в примере 3, и содержащие 20 мг формы А, хранили в различных условиях в течение 1, 2 и 3 месяцев. Таблетки затем были исследованы методом рентгеноструктурного анализа (рентгеновская дифракция) для определения количества активного ингредиента, превратившегося в форму В. Полученные результаты суммированы в таблице 1, представленной ниже. Таблица 1 Определение стабильности таблетированных составов, содержащие форму А Условия хранения % превращения в форму В Начало не определим 1 месяц, температура комнатная -" 2 месяца, температура комнатная -" 3 месяца, температура комнатная -" 5 27711 3 месяца, 50°С не определим 1 месяц, 40°С, 80% влажности 87 2 месяца, 40°С, 80% влажности 91 3 месяца, 40°С, 80% влажности 82 Таблетки, которые хранились при комнатной температуре и 50°С были, во флаконах из полиэтилена высокой плотности белого цвета, закупоренных металлическими колпачками. Таблетки, хранившиеся при 80% влажности, были экспонированы на воздухе. Сравнительный пример 1 Таблетированные составы формы А, не содержащие поливинилпирролидона Гранулируемый материал Активный ингредиент Прежелатинизированный Крахмал NF Лактоза NF Натриевый гликолят крахмала NF Микрокристаллическая целлюлоза NF Натрий лаурилсульфат NF мг/таблетка 50.0 20.0 34.55 2.0 34.95 0.5 Продолжение рецептуры примера 1 Гранулируемый материал Очищенная вода USP Материал для конечной смеси Сухой размолотый гранулят Натриевый гликолят крахмала NF Микрокристаллическая целлюлоза NF Стеарат магния NF мг/таблетка 100.0 142.0 6.0 50.0 2.0 Ингредиенты гранулируемого материала были подвергнуты влажной грануляции. Затем гранулы были высушены и размолоты. Сухой размолотый гранулят затем смешали с другими ингредиентами материала для конечной смеси и полученную смесь спрессовали в таблетки. Процентное содержание соединения 1, присутствующего в форме В, определили методом рентгеноструктурного анализа до грануляции и после грануляции. Было установлено, что до грануляции в смеси присутствует 1.5% формы В. Однако после грануляции 28% соединения 1 в смеси присутствуе т в форме В. Эти результаты четко свидетельствуют о повышенной стабильности формы в композициях, соответствующи х настоящему изобретению. Пример 5 Содержимое капсулы Активный ингредиент Поливинилпирролидон Лактоза Микрокристаллическая це ллюлоза Коллоидальный диоксид кремния мг/ капсула 20.0 20.0 177.25 177.25 0.5 6 27711 Стеарат магния Капсула желатиновая, размер 0 5.0 Смешивают активный ингредиент, поливинилпирролидон, лактозу и микрокристаллическую целлюлозу в подходящем смесителе, смешивают с очищенной водой до пригодной консистенции, сушат, доводят до требуемого размера, используя для этих целей подходящую операцию размола. Смешивают с коллоидальным диоксидом кремния и стеаратом магния. Помещают в капсулы размером 0,состоящие из двух частей. Гранулы (сферические) Активный ингредиент Сахар (сферической формы) Поливинилпирролидон мг/капсула 10 200 10 Готовят 10% дисперсию поливиниллирролидона в очищенной воде. Добавляют активный ингредиент и перемешивают до образования однородной дисперсии, напыляют на сферические частицы сахара, используя для этих целей подходящее оборудование. Гранулы могут быть помещены в капсулы размером 1, состоящие из двух частей, или распределены по подходящим саше. Состав порошка Активный ингредиент Поливинилпирролидон Маннитол Отдушка мг/порошок 20 15 364.6 0.4 Смешивают активный ингредиент, поливинилпирролидон и маннитол в подходящем миксере. Смешивают до достижения подходящей консистенции с очищенной водой, сушат и пропускают через подходящую операцию измельчения. Сравнительный пример 2 Сравнение биопригодности соединения 1 в композициях, приготовленных с использованием формы А и без поливинилпирролидона. Была сделана выборка из 24 здоровых мужчин-добровольцев, распределенных для лечения по группам. В соответствии с одним курсом лечения использовалась одна 50 мг таблетка, приготовленная в соответствии с описанием, приведенным в примере 3, а с др угим - одна таблетка, приготовленная в соответствии с прописью примера сравнительного 1. В течение курса изучения препарата каждый доброволец получал оба препарата. В процессе каждого периода лечения периодически брались образцы крови на анализ, и определялось содержание соединения 1. Препарат примера 3 Параметры Препарат сравнительного примера 1 Средняя цифра Максимальная концентрация соединения 1 (нанограмм/мл) Время достижения максимальной концентрации (час) Время полураспада Площадь под кривой (нанограмм/час/мл) Стандартное отклонение Средняя цифра Стандартное отклонение 568 54 223 28 2.6 0.3 3.7 0.3 9.0 0.5 8.2 0.7 2268 220 970 126 Эти результаты четко свидетельствуют об улучшенной биопригодности соединения 1, соответствующего настоящему изобретению. Сравнительный пример 3 Сравнение биопригодности соединения 1 в композициях, приготовленных с поливинилпирролидоном и формами А, В и X. Таблетки, содержащие 20 мг формы А, формы В или формы Х, были получены в соответствии со следующей рецептурой: 7 27711 Гранулируемый материал Активный ингредиент Натрий кроскармелоза NF Поливинилпирролидон USP Лактоза NF Микрокристаллическая целлюлоза NF Материал для конечной смеси Натрий кроскармелоза NF Микрокристаллическая целлюлоза NF Стеарат магния NF мг/таблетка 20 6 7 45 45 6 69 2 Используя сбалансированную переходную схему, каждая из 8 собак получала каждую из трех таблетированных форм. В течение каждого периода исследования у каждого животного брались образцы крови на анализ и определяли концентрацию соединения 1. Результаты приведены ниже. Активный ингредиент Форма А Максимальная концентрация (мг/мл) 1.004 Bрeмя полураспада (час) 5.398 Площадь под кривой (мг час/мл) 4.028 Фopмa В 0.105 3. 524 0.773 Форма Х 0.314 3.590 1.307 Эти результаты четко демонстрируют превосходную биопригодность композиций, соответствующи х данному изобретению, в сравнении с композициями, содержащими форму В или форму X. 8 27711 ДЛИНА ВОЛНЫ Фиг.1. Фиг.2. 9 27711 Фиг.3. Фиг.4. 10 27711 Фиг.5. Фиг.6. 11
ДивитисяДодаткова інформація
Назва патенту англійськоюPharmaceutical composition useful in the treatment of diseases in which leukotrienes are implicated
Автори англійськоюHolohan James Joseph, Edwards Ieuan John, Timko Robert Joseph, Bradway Randy John, Clements Arlene
Назва патенту російськоюФармацевтическая композиция для лечения патологических состояний, связанных с активностью лейкотриенов
Автори російськоюХолохан Джеймс Джозеф, Едврдс Иєуан Джон, Тимко Роберт Джозеф, Бредвей Ренди Джон, Клементс Арлин
МПК / Мітки
МПК: A61K 31/13
Мітки: лейкотриєнів, лікування, станів, пов'язаних, патологічних, фармацевтична, композиція, активністю
Код посилання
<a href="https://ua.patents.su/11-27711-farmacevtichna-kompoziciya-dlya-likuvannya-patologichnikh-staniv-povyazanikh-z-aktivnistyu-lejjkotriehniv.html" target="_blank" rel="follow" title="База патентів України">Фармацевтична композиція для лікування патологічних станів, пов’язаних з активністю лейкотриєнів</a>
Біологічно активна речовина фармацевтичної композиції для лікування гіперглікемій та фармацевтична композиція з протигіперглікемічною активністю

Номер патенту: 27954
Опубліковано: 16.10.2000
Автори: КААН Ельберт, ЦІГЛЕР Дітер, Брюкнер Райнхард
МПК: A61K 31/505
Мітки: протигіперглікемічною, гіперглікемій, біологічно, активністю, композиція, речовина, активна, фармацевтично, композиції, фармацевтична, лікування
Текст:
...0,2 мг Моксонидина на таблетку и третья тест-гр уппа (=гр уппа 3) получает таблетки с содержанием 0,4 мг Моксонидина на таблетку В каждом случае за день до начала тест-фазы пациентов выдерживают в состоянии натощак и спустя 6 недель после последнего дня тест-фазы отбирают пробы крови В них определяют количества са хара в плазме крови в м г глюкозы на децилитр Для оценки измеряемых результатов для каждой из 4-х гр упп осуществляют...
Спосіб профілактики та лікування патологічних станів у дітей з обтяженим перинатальним амнезом

Номер патенту: 23911
Опубліковано: 31.08.1998
Автори: Лук'янова Олена Михайлівна, Радченко Ніна Олександрівна, Сапа Ірина Юріївна, Мозалевський Анатолій Феодосович, Омельченко Людмила Іванівна, Квашніна Людмила Вікторівна, Починок Тетяна Вікторівна
МПК: A61K 33/22
Мітки: станів, амнезом, обтяженим, профілактики, лікування, перинатальним, дітей, спосіб, патологічних
Формула / Реферат:
Способ профилактики и лечения патологических состояний у детей с отягощенным перинатальным анамнезом путем использования медикаментозных и немедикаментозных средств, отличающийся тем, что детям с отягощенным перинатальным анамнезом дифференцированно назначают видеин-3 в сочетании с глицерофосфатом кальция, пищевую добавку "Окарин" и рибомунил в зависимости от факторов риска и наличия основного заболевания.
Похідні 3,4-дигідроізохіноліну або їх фармацевтично прийнятні солі, спосіб їх одержання і фармацевтична композиція з інгібуючою трансмембранний перехід кальцію в клітинах активністю
Номер патенту: 27369
Опубліковано: 15.09.2000
Автори: Роос Отто, Арндтс Дітріх, Штреллер Ільзе, Кун Франц-Йозеф, Льозель Вальтер
МПК: A61K 31/445, A61K 31/44, A61P 7/02, A61K 31/4725, A61P 25/28, C07D 217/18, A61K 31/4741, A61P 29/00, A61P 11/06, C07D 217/12, A61K 31/475, A61K 31/5377, A61K 31/535, A61K 31/54, A61P 9/00, A61K 31/4365, A61K 31/4745, A61K 31/47, A61K 31/496, A61K 31/435, A61K 31/437, A61K 31/495, A61K 31/472
Мітки: трансмембранний, спосіб, 3,4-дигідроізохіноліну, одержання, перехід, фармацевтично, похідні, композиція, активністю, кальцію, інгібуючою, солі, фармацевтична, прийнятні, клітинах
Текст:
...FMLP), или изопреналином) "перегрузку кальцием" и гибель клетки (значение KTso (= концентрация торможения) находится в области 1 мкмоль). Измеряли полумаксимальную концентрацию торможений соединения примера, которую ингибируют индуцируемый тринуклеотид ом FMLP (1 наномоль) переход кальция на клетках HL-60 (10 6 клеток в суспензии}, содержащих FLUO3. Она составляет 5,4 • 106 моль. На отдельных клетках HL-60 с использованием...
Спосіб одержання біологічно активної речовини для профілактики і лікування патологічних станів, спосіб одержання фармакологічної композиції на основі біологічно активної речовини та спосіб лікування злоякісних

Номер патенту: 22549
Опубліковано: 17.03.1998
Автори: Атаманюк Віктор Петрович, Новик Анатолій Матвійович
МПК: A61K 36/899
Мітки: одержання, станів, фармакологічно, патологічних, лікування, спосіб, біологічно, профілактики, композиції, основі, речовини, активної, злоякісних
Формула / Реферат:
1. Способ получения биологически активного вещества для профилактики и лечения патологических состояний, в котором в качестве растительного сырья используют зеленые части злаковых растений семейства Gramineae, включающий выделение водорастворимых фракций и фильтрацию, отличающийся тем, что в качестве сырья используют зеленые части растений семейства Gramineae в виде смеси растений рода Calamfgrostis Adans и рода Deschamsla Beauv, собранных...
Фармацевтична композиція для перорального введення тваринам, спосіб її одержання та спосіб лікування захворювань, пов’язаних з секрецією кислоти в шлунку

Номер патенту: 27550
Опубліковано: 15.09.2000
Автори: Оловсон Стіг-Йоран Артур, Пільбрант Окє Гуннар
МПК: A61K 31/4188, A61P 1/04, A61K 31/4184
Мітки: лікування, одержання, пов'язаних, спосіб, захворювань, тваринам, перорального, введення, кислоти, композиція, фармацевтична, секрецією, шлунку
Текст:
...низшим алкилом или низшей алкокси-фуппой. Термин "низший алкил" в настоящем изобретении означает алкильную группу, имеющую 1 5 атомов углерода Термин "низшая алкокси-группа" в настоящем изобретении означает алкокси-группу, имеющую 1-5 атомов углерода Примерами ингибиторов протонных насосов, имеющих формулу 1, являются следующие соединения о 9 в NH осн, О СН, N S N Ингибиторы протонных насосов, входящие в состав композиций настоящего...
Попередній патент: Спосіб одержання гетерологічного поліпептиду
Наступний патент: Гірничопрохідницький комбайн
Випадковий патент: Композиція для лікування і профілактики післяімплантаційних ускладнень