Спосіб одержання прегабаліну та проміжних сполук, проміжні сполуки
Номер патенту: 75584
Опубліковано: 15.05.2006
Автори: Гекстра Марвін Саймон, Гоел Ом Пракаш, Міч Томас Фредерік, Берк Марк Джозеф, Малгерн Томас Артур, Ремсден Джеймс Ендрю












Формула / Реферат
1. Спосіб одержання похідної (S)-3-ціано-5-метилгексанової кислоти формули
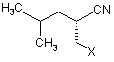 ,
,
де Χ є СО2Н або СО2-Y, a Y є катіоном,
що включає асиметричне каталітичне гідрогенування алкену формули
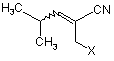
у присутності хірального каталізатора.
2. Спосіб за п. 1, який відрізняється тим, що Χ є СО2-Y.
3. Спосіб за п. 1, який відрізняється тим, що хіральним каталізатором є родієвий комплекс (R,R)-DuPHOS ліганду, який має формулу
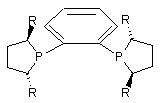 ,
,
де R є С1-С6 алкілом.
4. Спосіб за п. 3, який відрізняється тим, що хіральним каталізатором є [Rh(ліганд)(СOD)]ВF4.
5. Спосіб за п. 3, який відрізняється тим, що R є метилом або етилом.
6. Спосіб за п. 1, який відрізняється тим, що алкеном є Е ізомер або Ζ ізомер, або суміш Е та Ζ ізомерів.
7. Спосіб за п. 1, який відрізняється тим, що катіоном є лужний метал або лужноземельний метал.
8. Спосіб за п. 7, який відрізняється тим, що лужним металом є калій.
9. Спосіб за п. 1, який відрізняється тим, що катіоном є протонований первинний амін або протонований вторинний амін.
10. Спосіб за п. 9, який відрізняється тим, що аміном є трет-бутиламін.
11. Спосіб за п. 1, який відрізняється тим, що додатково включає спочатку перетворення карбоксильного естеру формули
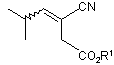 ,
,
де R1 є С1-С6 алкілом,
на карбоксильну сіль формули
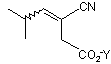 ,
,
де Υ є катіоном.
12. Спосіб за п. 11, який відрізняється тим, що R1 є етилом.
13. Спосіб за п. 11, який відрізняється тим, що карбоксильну сіль виділяють перед гідрогенуванням.
14. Спосіб за п. 11, який відрізняється тим, що карбоксильну сіль одержують in situ перед гідрогенуванням.
15. Спосіб за п. 1, який відрізняється тим, що включає окиснення карбоксильної солі (S)-3-ціано-5-метилгексанової кислоти для утворення (S)-3-ціано-5-метилгексанової кислоти.
16. Спосіб за п. 1, який відрізняється тим, що катіон Y вибирають з протонованого первинного аміну, протонованого вторинного аміну, лужного металу і лужноземельного металу.
17. Сполука формули
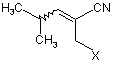 ,
,
де Χ є СО2Н або СО2-Y, a Y є катіоном.
18. Спосіб одержання прегабаліну, що включає взаємодію алкену формули
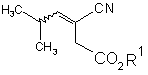
у присутності хірального каталізатора з гідрогеном з одержанням естеру карбоксильної кислоти формули
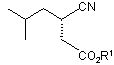 ,
,
де R1 є С1-С6 алкілом;
перетворення естеру карбоксильної кислоти на карбоксильну сіль формули 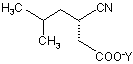 ,
,
де Y є катіоном;
відновлення ціаногрупи карбоксильної солі з утворенням аміносполуки та, коли Y є іншим, ніж Н+, аміносполуку піддають дії кислоти.
19. Спосіб за п. 18, який відрізняється тим, що хіральним каталізатором є родієвий комплекс (S,S)-DuPHOS ліганду, який має формулу
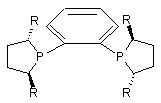 ,
,
де R є С1-С6 алкілом.
20. Спосіб за п. 19, який відрізняється тим, що хіральним каталізатором є [Rh(ліганд)(СOD)]ВF4.
21. Спосіб за п. 19, який відрізняється тим, що R є метилом або етилом.
22. Спосіб за п. 21, який відрізняється тим, що R1 є етилом.
23. Сполука формули
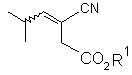 ,
,
де R1 є С1-С6 алкілом.
24. Сполука формули
,
де Υ є катіоном.
25. Спосіб одержання прегабаліну, який включає відновлення ціаногрупи похідної (S)-3-ціано-5-метилгексанової кислоти, одержаної за способом за п. 1, з утворенням аміногрупи, та, коли Υ є іншим, ніж Н+, протонування взаємодією з кислотою.
26. Спосіб одержання прегабаліну, який включає асиметричне гідрогенування
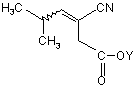 ,
,
де Υ є катіоном,
у присутності хірального каталізатора;
наступне відновлення ціаногрупи та протонування до вільної кислоти.
Текст
1. Спосіб одержання похідної (S)-3-ціано-5метилгексанової кислоти формули 2 (19) 1 3 75584 4 20. Спосіб за п.19, який відрізняється тим, що ну, протонованого вторинного аміну, лужного металу і лужноземельного металу. хіральним каталізатором є [Rh(ліганд)(СOD)]ВF4. 21. Спосіб за п.19, який відрізняється тим, що R є 17. Сполука формули H3C CN метилом або етилом. 22. Спосіб за п.21, який відрізняється тим, що R1 CH3 є етилом. X , 23. Сполука формули де Χ є СО2Н або СО2-Y, a Y є катіоном. CN 18. Спосіб одержання прегабаліну, що включає взаємодію алкену формули 1 CN H3C CO2 R , де R1 є С1-С6 алкілом. CH3 1 24. Сполука формули CO2 R у присутності хірального каталізатора з гідрогеном з одержанням естеру карбоксильної кислоти формули CN 1 CO2R1, де R є С1-С6 алкілом; перетворення естеру карбоксильної кислоти на карбоксильну сіль формули H3C CN CH3 COO-Y, де Y є катіоном; відновлення ціаногрупи карбоксильної солі з утворенням аміносполуки та, коли Y є іншим, ніж Н+, аміносполуку піддають дії кислоти. 19. Спосіб за п.18, який відрізняється тим, що хіральним каталізатором є родієвий комплекс (S,S)-DuPHOS ліганду, який має формулу R P R H3C CN CH3 COO-Y , де Υ є катіоном. 25. Спосіб одержання прегабаліну, який включає відновлення ціаногрупи похідної (S)-3-ціано-5метилгексанової кислоти, одержаної за способом за п.1, з утворенням аміногрупи, та, коли Υ є іншим, ніж Н+, протонування взаємодією з кислотою. 26. Спосіб одержання прегабаліну, який включає асиметричне гідрогенування H3 C CN CH3 C OY O , де Υ є катіоном, у присутності хірального каталізатора; наступне відновлення ціаногрупи та протонування до вільної кислоти. R P R , де R є С1-С6 алкілом. Винахід стосується способу одержання (S)-(+)3-(амінометил)-5-метилгексанової кислоти (прегабаліну) в асиметричному синтезі. Прегабалін придатний длялікування і запобігання приступів, болю та психотичних розладів. (S)-(+)-3-(амінометил)-5-метилгексанова кислота відома в загальному як прегабалін. Цю сполуку також називають (S)-(+)- -ізобутил- аміномасляна кислота (ГАМК), (S)-ізобутил-ГАМК, та СІ-1008. Прегабалін пов'язаний з ендогенним інгібіторним нейротрансмітером -аміномасляною кислотою або ГАМК, яка приймає участь у регуляції нейронної активності мозку. Прегабалін має активність, що стримує приступи, [як описано Silverman et al, Патент США №5, 563, 175]. Інші ознаки прегабаліну також було нещодавно виявлено [див., наприклад, Guglietta et al., Патент США №6,127,418; та Singh et al., Патент США №6, 001, 876]. Приступ - це надлишкова несинхронізована нейронна активність, що руйнує нормальну функцію мозку. Раніше вважали, що приступи можна контролювати регуляцією концентрації нейротрансмітера ГАМК. Коли концентрація ГАМК знижується нижче порогового рівня у мозку, починаються приступи [Karlsson et al., Biochem. Pharmacol, 1974; 23:3053], коли рівень ГАМК підвищується у мозку під час конвульсії, приступи завершуються [Hayashi. Physiol (London), 1959; 145: 570]. Через важливість ГАМК як нейротрансмітеру, і через її дію на конвульсивні стани, та інші моторні дисфункції, різноманітні способи було отримано для підвищення концентрації ГАМК у мозку. Згідно з одним з підходів, сполуки, що активують декарбоксилазу L-глютамінової кислоти (ДГК), було використано для підвищення концентрації ГАМК, оскільки концентрації ДГК та ГАМК змінюються паралельно, і в результаті підвищення концентрацій ДГК підвищуються концентрації ГАМК [Janssens de Varebeke et al., Biochem. Pharmacol, 1983; 32:2751; Loscher, Biochem. Pharmacol, 1982; 31:837; Phillips et al., Biochem. Pharmacol., 1982; 31:2257]. Наприклад, рацемічна сполука (±)-3(амінометил)-5-метилгексанова кислота (рацеміч 5 75584 6 на ізобутил-ГАМК), яка є активатором ДГК, може його синтезу у великих масштабах виробництва. стримувати приступи, уникаючи небажаної побічДля того, щоб він став доступним для комерційноної дії атаксії. го виготовлення, такий спосіб має бути більш енаАнтиконвульсивна дія рацемічної ізобутилнтіоселективним, наприклад, якщо продукт отриГАМК є першою ознакою S-енантіомеру (прегабамано з суттєвим надлишком коректованого ліну). Ось чому, S-енантіомер ізобутил-ГАМК проенантіомеру. Ціль даного винаходу - забезпечити являє кращу антиконвульсивну активність, ніж Rтакий спосіб, а саме, спосіб асиметричного гідруенантіомер [див., наприклад, Yuen et al., Bioorganic вання. & Medicinal Chemistry Letters, 1994; 4:823]. Таким Способи асиметричного гідрування відомі для чином, комерційна придатність прегабаліну потредеяких сполук. [Burk et al., в WO 99/31041 та WO бує ефективного способу для отримання S99/52852] описують асиметричне гідрування похіденантіомеру, по суті вільного від R-енантіомеру. них β-заміщеної та β,β-дизаміщеної ітаконової киДекілька способів було використано для одерслоти для отримання похідних енантіомерно збажання прегабаліну. Звичайно, рацемічну суміш гаченої 2-заміщеної янтарної кислоти. Ітаконові синтезують і тоді розкладають на її R- та Sсубстрати мають дві карбоксильні групи, які забезенантіомери [дивись патент США №5, 563, 175], печують необхідну стеричну та електронну конфідля синтезу через азидний інтермедіат. Згідно з гурацію для того, щоб направити гідрування на іншим способом використовують потенційно неотримання збагаченого енантіомеру. В описі опистабільні нітросполуки, включаючи нітрометан, та сується, що форми солі формули інтермедіат, який відновлено до аміну в потенційRR'C=C(CO2Me)CH2CO2''Y+ необхідні для одерно екзотермічній та небезпечній реакції. Для синжання продуктів гідрування, що мають щонайментезу також використовують ше 95% енантіомерний надлишок. б/с(триметилсиліламід)літію в реакції, яку мають Згідно [з патентом США №4,939,288] асиметпроводити при -78°С (Andruszkiewicz et al.. ричне гідрування не спрацьовує на субстратах, що Synthesis, 1989: 953). Нещодавно, рацемат було мають ізобутилову групу. Ми нещодавно виявили, одержано "малонатним» синтезом та синтезом що ізобутилціано карбонова кислота, сіль або есГофмана [Патенти США №№5, 840, 956; 5, 637, тер субстрату формули iPrCH=C(CN)CH2CO2R 767; 5, 629, 447; та 5, 616, 793]. Класичний спосіб можна селективно гідрувати для отримання енанрозділення рацемату використовують для одертіомерно збагаченого похідного нітрилу, який можжання прегабаліну згідно з цими способами. Клана потім гідрувати для отримання по суті чистого сичне розділення полягає у виготовленні солі за прегабаліну. Ця селективність є особливо несподідопомоги хірального розділювального засобу для ваною тим, що дає ефективну різницю у стеричній виділення та очищення бажаного S-енантіомеру. конфігурації та індуктивній дії нітрильної групи, Це потребує затрат на значну обробку, а також порівняно з карбоксильною групою. Насправді, з значних додаткових витрат, пов'язаних з розділюрівня техніки невідомо про успішне асиметричне вальним засобом. Часткове відтворення отримагідрування будь-якого ціанозаміщеного карбоксиного розділювального засобу можливе, але це льного олефіну цього типу. потребує додаткової обробки й витрат, а також Даний винахід стосується ефективного спосопов'язаного з цим утворення відходів. Більш того, бу одержання (S)-3-(амінометил)-5небажаний R-енантіомер не можна ефективно метилгексанової кислоти (прегабаліну). Спосіб відтворювати і тому його в кінці необхідно відкинуполягає в асиметричному гідруванні ціаноти як відходи. Максимальний теоретичний вихід заміщеного олефіну для отримання ціанопрегабаліну є 50%, оскількилише половина рацепопередника (S)-3-(амінометил)-5-метилгексанової мату є бажаним продуктом. Це зменшує ефективкислоти. Спосіб, крім того, полягає у реакції для ну продуктивність способу (кількість, яку можна перетворення ціано-інтермедіату у (S)-3отримати у наявному об'ємі реактору), яка є ком(амінометил)-5-метилгексанову кислоту. Асиметпонентом виробничих витрат та продуктивності. ричний синтез (S)-3-(амінометил)-5Прегабалін було синтезовано безпосередньо метилгексанової кислоти, описаної тут, призводить різними схемами синтезу. Один спосіб полягає у до суттєвого збагачення прегабаліну, на відміну використанні н-бутиллітію при низьких температувід небажаної (R)-3-(амінометил)-5рах (99,9% (S)-енантіомеру простою перекристалізафільтрують, та концентрують ротаційним випарюцією з, наприклад, води/ізопропілового спирту. вання для отримання 226г етилового-(2-ціано-1Прегабалін, що містить вищі рівні (до 3,5%) (R)ізопропіл-алілового) діестеру карбонової кислоти у енантіомеру, також можна збагатити простою певигляді масла жовтого кольору, яке можна викорирекристалізацією з, наприклад, востати на наступному етапі, без подальшого очиди/ізопропілового спирту, хоча подальші перекрищення. сталізації звичайно потрібні для досягнення 2-ціано-1-ізопропіл-аліловий естер оцтової ки>99,9% (S)-енантіомеру. "По суті чистий" прегабаслоти (використовуючи ацетилхлорид) лін, як використовуваний тут, означає щонайменше приблизно 95% (за масою) S-енантіомер, і не більше ніж приблизно 5% R-енантіомер. Наступні детально викладені приклади, крім того, ілюструють певні втілення винаходу. Ці прикУ тригорлу круглодонну колбу на 5л, при пелади не повинні виходити за рамки винаходу. Виремішуванні азотом, додають 50 г (0,4моль) 3хідні матеріали та різноманітні інтермедіати можна гідрокси-4-метил-2-метиленпентаннітрилу, 0,4л отримати з комерційних джерел, виготовити з кометиленхлориду та 80мл (1моль) піридину. Розчин мерційно доступних сполук, або одержати, викоохолоджують при 10-15°С у льодяній бані. Викориристовуючи добре відомі фахівцям з області оргастовуючи градуйовану крапельну воронку на нічної хімії синтетичні способи. 500мл, суміш 100мл метиленхлориду та 43мл Приготування Вихідних Матеріалів (0,6моль) ацетилхлориду додають повільно, поки 3-Гідрокси-4-метил-2-метиленпентаннітрил підтримуючи температуру реакції при 25°С±5°С. Реакційну суміш перемішують при 22°С±3°С протягом приблизно однієї додаткової години. Реакційний розчин виливають у розділювальну воронку У тригорлу круглодонну колбу на 250мл, пена 4л, яка містить 85мл (1,0моль) гідрохлоридної ремішуючи повітрям, додають 0,36г (1,6ммоль) кислоти та 750мл води. Нижчий органічний шар 2,6-ди-трет-бутил-4-метилфенолу, 37г (0,33моль) промивають знову розчином 20мл (0,2моль) НСІ 1,4-діазабіцикло[2,2,2]октану, 60мл (0,66моль) ізота 250мл води. Органічний шар висушують над бутиральдегіду, 52мл (0,79моль) акрилонітрилу, та безводним сульфатом магнію (20г), фільтрують та 7,2мл (0,4моль) води. Реакційну суміш перемішуконцентрують ротаційним випарювання для отриють при 50°С протягом 24 годин, охолоджують до мання 66г 2-ціано-1-ізопропіл-алілового естеру 25°С, і, гасять, переносячи у розчин 33мл оцтової кислоти у вигляді масла жовтого кольору, (0,38моль) гідрохлоридної кислоти та 100мл води. яке можна використати на наступному етапі, без Продукт екстрагують 120мл метиленхлориду. Воподальшого очищення. дний кислотний шар знову екстрагують 25мл ме2-ціано-1-ізопропіл-аліловий естер оцтової китиленхлориду. Поєднані шари метиленхлориду слоти (використовуючи оцтовий ангідрид) концентрують ротаційним випарюванням для У чотиригорлу, круглодонну колбу на 500мл, отримання 79,9г (96,7%) 3-гідрокси-4-метил-2оснащену насадженою зверху мішалкою, темпераметиленпентаннітрилу у вигляді масла жовтого турним датчиком, зворотним холодильником та кольору (яке може прийняти тверду форму білого вхідним отвором для пропускання азоту, додають кольору при відстоюванні), 96,7% (площа під криоцтовий ангідрид (40мл, 0,45моль). Розчин нагрівою) аналізом високоефективної рідинної хромавають до 50°С, а розчин 3-гідрокси-4-метил-2тографії (ВЕРХ), яке можна використати на настуметиленпентаннітрилу (50г, 0,40моль) та 4пному етапі, без подальшого очищення. (диметиламіно)піридину (1,5г) у тетрагідрофурані Етиловий-(2-ціано-1-ізопропіл-аліловий) діес(ТГФ) (25мл) додають через 35 хвилин. Температер карбонової кислоти туру 50-63°С підтримують, без зовнішнього нагрівання. Після того, як закінчили додавати, реакційну суміш нагрівають при 60°С протягом 75 хвилин. Розчин охолоджують до 30°С, а тоді охолоджену реакційну суміш розріджують 30мл третУ тригорлу круглодонну колбу на 5л, при пебутилметилового етеру (МТБЕ) та 25мл води. Суремішуванні азотом, додають 150г (1,2моль) 3міш охолоджують до 10°С, а розчин 50% водного гідрокси-4-метил-2-метиленпентаннітрилу, 1,0л гідроксиду натрію (37г, 0,46моль), розрідженого метиленхлориду, та 170мл (2,1моль) піридину. 45мл води, додають з охолодженням так, що темРозчин охолоджують при 10-15°С ульодяній бані. пература підтримується приблизно 15°С. Для кінВикористовуючи градуйовану крапельну воронку цевого регулювання рівня рН9,4 додають крапляна 1л, суміш 0,5л метиленхлориду та 200мл ми 9,8г 50% водного гідроксиду натрію (0,12моль). (2,1моль) етилхлорформіату додають повільно, 13 75584 14 Після додавання 10мл води та 10-15мл МТБЕ, нолу. Монооксид карбону уводять при тиску 40реакційну суміш фазують та розділяють. Вищий 50фунт/кв.дюйм, і суміш нагрівають при 50°С проорганічний шар продукту відділяють і промивають тягом 24 годин з перемішуванням. Коричневий 25мл соляного розчину, висушують над сульфарозчин фільтрують через сито для видалення тветом магнію, та концентрують у вакуумі для отрирдих частинок. Фільтрат концентрують ротаційним мання 63,7г (95%) 2-ціано-1-ізопропіл-алілового випарюванням. Концентровану реакційну суміш естеру оцтової кислоти у вигляді масла блідорозріджують 150мл метил-трет-бутилового етеру і жовтого кольору. промивають водою. Розчинник видаляють на роЕтилу 3-ціано-5-метил гекс-3-еноат таційному випарювачі для отримання 7,7г сирого масляного продукту жовтого кольору, етил-3ціано-5-метил гекс-3-еноат, (85% за площею при аналізі ГХ). Сирий продукт можна використовувати без подальшого очищення, або, як варіант, очисУ реактор високого тиску, перемішуючи повіттити вакуумною дистиляцією (0,6-1,0мл Нg при 60рям, завантажують 3,0г (13,4ммоль) ацетату пала70°С). дію, 7,0г (26,8ммоль) трифенілфосфіну, та 226г ПРИКЛАД 1 (0,92моль) сирого масла, до складу якого входить Синтез солей 3-ціано-5-метилгекс-3-енової киетиловий-(2-ціано-1-ізопропіл-аліловий) діестер слоти карбонової кислоти, та 500мл етанолу. Монооксид Атрет-Бутиламонієва сіль 3-ціано-5-метилгекскарбону уводять при тиску 280-300фунт/кв.дюйм, 3-енової кислоти та суміш нагрівають при 50°С протягом ночі з перемішуванням. Розчин червоно-коричневого кольору фільтрують через сито для видалення твердих частинок. Фільтрат концентрують ротаційним випарюванням для отримання 165г сирого масляМолекулярна ного продукту жовтого кольору, етил-3-ціано-5Матеріал Кількість ммоль маса метилгекс-3-еноат, в якому газовою хроматографіЕтил 3-ціано-5181,24 20,02г 110 єю (ГХ) визначили 84%-вміст (за площею) суміші Ε метилгекс-3-еноат та Ζ геометричних ізомерів. Сирий продукт можна LiOH Н2O 41,96 13,0г 310 використати, без подальшого очищення, або, як Тетрагідрофуран 75мл варіант, очистити вакуумною дистиляцією (0,6Вода 25мл 1,0мм Hg при 60-70°С) для отримання безбарвноГідрохлоридна кисЯкщо необлота (2N) хідно го масла з кількісним аналізом >95% (за площею) Якщо необГХ. Етилацетат хідно Етилу 3-ціано-5-метил гекс-3-еноат (використрет-бутиламін 73,14 9,27г 127 товуючи KВr) У реактор високого тиску, перемішуючи повітЕтил 3-ціано-5-метилгекс-3-еноат (суміш Ε та рям, завантажують паладієвий ацетат (0,52г, Ζ ізомерів) та гідрат гідроксидулітію суспендують у 2,3ммоль), трифенілфосфін (0,65г, 2,3ммоль), суміші тетрагідрофурану та води. Кашицю сильно бромід калію (5,5г, 4,8ммоль), сире масло, що місперемішують протягом 4 годин при кімнатній темтить етиловий-(2-ціано-1-ізопропіл-аліловий) діеспературі. Суміш підкислюють до рН2 (3Ν НСІ) та тер карбонової кислоти (240г, 1,2ммоль), триетиекстрагують у етилацетат (3 150мл). Поєднані ламін (2,2г, 22ммоль), етанол 2В (45мл), та органічні шари висушують (MgSO4) і розчинник ацетонітрил (200мл). Монооксид карбону уводять видаляють у вакуумі для отримання сирої 3-ціанопри тиску 50фунт/кв.дюйм та суміш нагрівають при 5-метилгекс-3-енової кислоти. Сиру кислоту роз50°С протягом ночі з перемішуванням. Тиск у реачиняють в етилацетаті (400мл), і розчин треткторі зменшують до 10-15фунт/кв.дюйм через прибутиламіну в етилацетаті (20мл) додають. Темпеблизно 1,3-6 годин і знову завантажують моноократуру розчину підвищують приблизно до 10°С, сид карбону до тиску 40-50фунт/кв.дюйм. коли маса білої кристалічної твердої форми осаРеакційну суміш фільтрують через сито для видаджується. Продукт збирають фільтруванням і вилення твердих частинок. Фільтрат концентрують у сушують у вакуумі. Вихід 22,15г, 97,9ммоль, 89%. вакуумі та 800мл гексану додають. Отриману суА1. 3-ціано-5-метилгекс-3-еноат третміш промивають двічі 500мл води, та гексан видабутиламонію (альтернативний спосіб) ляють у вакуумі для отримання 147г сирого етил 3У тригорлу круглодонну колбу відповідного роціано-5-метил гекс-3-еноату у вигляді масла. Цей зміру додають 50г масла, яке містить етилу 3сирий продукт очищують фракційною дистиляцією ціано-5-метилгекс-3-еноат (29,9г, 165ммоль). Роз(0,7мм Hg при 60-70°С). чин КОН (91%, 10,2г, 165,1ммоль) в 50мл води Етилу 3-ціано-5-метил гекс-3-еноат (викорисдодають до естерового розчину через 20 хвилин, і товуючи NaBr) розчин продовжують перемішувати протягом одніУ реактор з високим тиском, перемішуючи поєї додаткової години. Воду (50мл) додають, і розвітрям, завантажують 0,5г (0,5ммоль) чин концентрують до 80мл у вакуумі. Водний розтрис(дибензиліденацетон)дипаладію (0), 0,5г чин промивають МТБЕ (100мл), і водний шар, що (2,0ммоль) трифенілфосфіну, 0,5г (5,0ммоль) містить продукт окислюють до рН1 концентроваброміду натрію, 4,5мл (25,0ммоль) диізопропіленою гідрохлоридною кислотою (20мл). Отриману тиламіну, 8,35г (50,0ммоль) 2-ціано-1-ізопропілкислоту екстрагують в МТБЕ (100мл). Розчин алілового естеру оцтової кислоти, та 100мл ета 15 75584 16 МолекулярМТБЕ, що містить продукт концентрують у вакуумі. Матеріал Кількість ммоль на маса Отримане масло розчиняють в ізопропіловому трет-Бутиламонієва сіль 3спирті (58мл) та гептані (85мл), і цей розчин фільціано-5-метилгекс-3-енової трують через сито. Осад на фільтрі промивають кислоти 226,33 19,0г 84 сумішшю ізопропілового спирту (58мл) та гептану [(R,R)+ (85мл). трет-бутиламін завантажують у розчин для MeDuPHOS]Rh(COD) BF4 604 49,6мг 0,082 утворення густої гелеподібної кашиці. Кашицю Метанол 32 200мл нагрівають під зворотним холодильником для 44фунт/кв. отримання розчину, який продовжують повільно Водень 2 дюйм охолоджувати до кімнатної температури. Отрима(3бар) ну кашицю охолоджують до 0-5°С протягом 1,5 години, а тоді фільтрують і промивають сумішшю У круглодонну колбу додають третізопропілового спирту (50мл) та гептану (150мл). бутиламонієву сіль 3-ціано-5-метилгекс-3-енової Тверду речовину висушують під вакуумом при 45кислоти (з Прикладу 1А) та [(R,R)50°С для отримання 23,1г (62%) 3-ціано-5MeDuPHOS]Rh(COD)+BF4 в атмосфері азоту. Деметилгекс-3-еноату трет-бутиламонію у вигляді зоксигенований метанол уводять шприцом, і розтвердої речовини білого кольору, яка є сумішшю Ε чин дезоксигенують повторною частковою відкачта Ζ ізомерів. Ζ ізомер можна отримати з ізомеркою, а тоді знову уводять азот. Апарат Парра на ною чистотою більше 99%, перекристалізацією з 600мл продувають воднем, створюючи високий ізопропілового спирту та гептану. тиск та спорожнюючи його тричі. Тоді цей апарат В. Калієва сіль 3-ціано-5-метилгекс-3-енової нагрівають до 55°С. Розчин субстрату та каталізакислоти тору переносять до реактора канюлею, і препарат знову продувають воднем, створюючи там кінцевий високий тиск 3бар (44фунт/кв.дюйм). Починають перемішувати і водень поглинається. У препараті повторно створюють тиск до 3 бар, поки не закінчиться поглинання водню (-45хв.). Після перемішування під тиском при 55°С протягом додатМолекулярна КільМатеріал Джерело ммоль кової години, нагрівання закінчують. Як тільки реамаса кість ктор охолодили до кімнатної температури, тиск Етилу 3-ціано-5PD водню зменшують, апарат продувають азотом, і метилгекс-3-еноат 61966X130 181,24 90,8 г 501 85% гідроксид реакційну суміш переносять у круглодонну колбу. калію Aldrich 56,11 33,1 г 501 Розчинник видаляють у вакуумі для отримання Метанол Fisher 90мл сирого продукту. Малу пробу відбирають та переттретворюють у (S)-3-ціано-5-метилгексанову кислоту Бутилметиловий Fisher 900мл обробкою водною гідрохлоридною кислотою та етер екстрагуванням у дихлорметан. Аналіз ГХ показує 100% перетворення у відновлений ціаноалкан з Гідроксид калію розчиняють в метанолі (70мл) 95,0% е.з (енантіомерний надлишок) (S). і додають до етилу 3-ціано-5-метилгекс-3-еноату В. Калієва сіль (S)-3-ціано-5-метилгексанової (суміш Ε та Ζ геометричних ізомерів) при перемікислоти (співвідношення субстрату до каталізатошуванні з такою швидкістю, щоб підтримувати теру (С/К) 1000/1) мпературу нижче 45°С. Залишковий метанольний гідроксид калію промивають у суміш з надлишком метанолу (2 10мл). Суміш нагрівають при 45°С протягом 1 години, а тоді дають охолонути до кімнатної температури, і протягом цього часу утворюМолеється кристалічний твердий продукт, третМатеріал кулярна Кіль-кість ммоль бутилметиловий етер (600мл) повільно додають маса Калієва сіль 3-ціано-5до суміші з сильним перемішуванням. метилгекс-3-енової кислоти 191,3 11,3г 57,7 Тверді частинки збирають через фритовий [(R,R)11мг в 18,2 10фільтр, промивають трет-бутил-метиловим етером + MeDuPHOS]Rh(COD) BF4 604 10мл 3 (3 100мл), і висушують для отримання названої С/К=000 сполуки. Вихід 83,9г, 439ммоль, 88%. МеОН за масою ПРИКЛАД 2 Метанол 32 100мл Асиметричне гідрування солей 3-ціано-560фунт/к метилгекс-3-енової кислоти Водень 2 в. дюйм А. трет-Бутиламонієва сіль (S)-3-ціано-5(4бар) метилгексанової кислоти У скляну гільзу завантажують калієву сіль 3ціано-5-метилгекс-3-енової кислоти (з Прикладу 1В) та метанол, а тоді поміщають її у заповнений воднем сосуд Парра на 600мл. Сосуд продувають азотом, а тоді воднем при тиску 60фунт/кв.дюйм та перемішують протягом 10хв. для забезпечення через співвідношення газів та зменшення тиску 17 75584 18 50-65 п'ять циклів. Сосуд нагрівають до 45°С, а розчин Водень 2 фунт/кв. [(R,R)-MeDuPHOSJRh(COD)+BF4- в дезоксигеновадюйм ному метанолі (11мг в 10мл) додають шприцом. Сосуд знову продувають воднем, а тоді створюють У скляну гільзу завантажують 3-ціано-5у ньому високий тиск 60фунт/кв. дюйм з перемішуметилгекс-3-еноат трет-бутиламонію та метанол ванням. Періодично уводять водень для підтри(1000мл). Гільзу поміщають у заповнений воднем мання тиску 50-65фунт/кв. дюйм. Через 120хв. сосуд Парра на 2л. Сосуд продувають азотом, а поглинання водню закінчилося. Через 2 години тоді воднем, уведенням до 60фунт/кв. дюйм та суміш охолоджують до кімнатної температури, зменшенням тиску через 5 циклів. Тоді сосуд натиск зменшують, а розчинник видаляють для грівали до 45°С. Розчин [(R.R)отримання сирого продукту. Малу пробу відбираMeDuPHOS]Rh(COD)+BF4- у дезоксигенованому ють та окислюють 1N НСІ для отримання (S)-Sметанолі (15мл) додавали шприцом. Сосуд знову ціано-5-метилгексанової кислоти. Аналіз ГХ покапродували воднем тричі, тоді створювали високий зує >99% перетворення у S ізомер з 96.7% е.з. тиск 65 фунт/кв. дюйм та починали перемішувати. С Калієва сіль (S)-3-ціано-5-метилгексанової Періодично уводили водень для підтримання тиску кислоти (співвідношення субстрат до каталізатору 50-65 фунт кв. дюйм. Через 4 години закінчилося (С/К) 3200/1, 640ммоль) поглинання водню, тоді через ще 1 годину сосуд охолодили до кімнатної температури. Тиск зменшився, суміш перенесли у колбу, а розчинник видалили у вакуумі для отримання продукту. Малу пробу відібрали і перетворили у (5)-3-ціано-5МолекулярМатеріал Кількість ммоль метилгекс-3-еноат метилу реакцією з метанолом на маса та 1N НСІ. Аналіз ГХ показав >99% перетворення 3-ціано-5-метилгекс-3еноат калію 181.2 123 г 640 з 97.7% е.з. [(R,R)Е. Калієва сіль 3-ціано-5-метилгексанової кис+ MeDuPHOS]Rh(COD) BF4 604 123 мг 0.204 лоти, генерованої in situ з 3-ціано-5-метилгекс-3Метанол 32 1015мл еноату етилу Водень 2 60фунт/кв. дюйм (4бар) У скляну гільзу завантажують 3-ціано-5метилгекс-3-еноат калію (з Прикладу 1В) та метанол (1000мл). Гільзу поміщають у заповнений воднем сосуд Парра на 2л. Сосуд продувають азотом, а тоді воднем, уведенням до 60фунт/кв. дюйм та зменшенням тиску через 5 циклів. Тоді сосуд нагрівали до 45°С. Розчин [(R,R)MeDuPHOS]Rh(COD)+BF4- у дезоксигенованому метанолі (15мл) додавали шприцом. Сосуд знову продували воднем тричі, а тоді створювали високий тиск до 65 фунт/кв. дюйм та починали перемішувати. Періодично уводили водень для підтримання тиску 50-65фунт/кв. дюйм. Через 2½ години поглинання водню закінчилося, сосуд охолодили до кімнатної температури та продовжували перемішувати протягом ночі. Тиск зменшився, суміш перенесли у колбу, а розчинник видалили у вакуумі для отримання продукту. Малу пробу відібрали та перетворили у метил (S)-3-ціано-5-метилгекс-3еноат. Аналіз ГХ показав >99% перетворення з 97.5% е.з. D. трет-Бутиламонієва сіль (S)-ціано-5метилгексанової кислоти (співвідношення С/К 2700/1, 557ммоль) Матеріал 3-ціано-5-метилгекс-3-еноат трет-бутиламонію [(R,R)+ MeDuPHOS]Rh(COD) BF4 Метанол МолекулярКількість ммоль на маса 226.33 604 32 125.8г 557 125мг 0.082 200мл Матеріал Молекулярна Кількість маса Етил 3-ціано-5-метилгекс-3еноат 181.2 Гідроксид калію [(R,R)+ MeDuPHOS]Rh(COD) BF4 604 Метанол 32 Вода 18 Водень 2 10,81г 11,68мл 120мл 120мл 18мл 60фунт/кв. дюйм (4бар) ммоль 59.7 58.4 29.8 103 У скляну гільзу завантажують етилу 3-ціано-5метилгекс-3-еноат (вихідний матеріал, одержаний вище), метанол (100мл), та воду (18мл). Гідроксид калію додають з перемішуванням. Гільзу поміщають у заповнений воднем сосуд Парра на 600мл. Сосуд продувають азотом, а тоді воднем, уведенням до 60фунт/кв. дюйм та зменшенням тиску після 5 циклів. Сосуд нагрівають до 55°С. Розчин [(R,R)-MeDuPHOS]Rh(COD)+BF4- у дезоксигенованому метанолі (18,0мг у 20мл) додають шприцом. Сосуд знову продувають воднем, а тоді створюють високий тиск до 60фунт/кв. дюйм з перемішуванням. Періодично уводять водень для підтримання тиску 50-60фунт/кв. дюйм. Через 5 годин закінчується поглинання водню. Через ще одну годину суміш охолоджують до кімнатної температури, тиск зменшується. Суміш переносять у колбу, а розчинник видаляють у вакуумі для отримання продукту. Малу пробу відбирають та перетворюють у (S)-3-ціано-5-метилгексанову кислоту реакцією з 1N гідрохлоридною кислотою. Аналіз ГХ 19 75584 20 показує 98.7% перетворення у бажану ціаноалкаколбу Парра, що містить губчатий нікелевий катанову сіль S ізомером з 96.6% е.з. лізатор (А-7000, Activated Metals and Chemicals, ПРИКЛАД 3 Inc., P.O. Box 4130, Severville, TN 37864. 5г, змочеГідрування етилу 3-ціано-5-метилгекс-3-еноату ний водою). Кашицю струшують у вібраторі Парра під тиском 50фунт/кв. дюйм водню при кімнатній температурі протягом ночі. Кашицю фільтрують через шар Supercel. Осад на фільтрі промивають водою (20мл) та 2В ЕtOН Молеку(7мл). Поєднаний фільтрат змішують зльодяною Матеріал Кількість ммоль лярна маса оцтовою кислотою (2,4мл, 2.5г, 41,6ммоль) та наЕтилу 3-ціано-5грівають при 70°С протягом 30хв. Суміш охолометилгекс-3-еноат 181 0.36г 2.00 джують до 0°С і твердий продукт збирають фільт[(R,R)-Me+ руванням, промивають ізопропанолом (50мл), та DuPHOS]Rh(COD) BF4 604 1.2мг 2 103 висушують для отримання 3,2г продукту (20ммоль, Метанол 5мл 49% вихід). Аналіз ВЕРХ матеріалу показує 99.7% 60 (площа під кривою) 3-ізобутил-ГАМК. Енантіомерфунт/кв. Водень ний аналіз (ВЕРХ) вказує на 3-ізобутил-ГАМК як на дюйм (4бар) суміш ізомерів: 97.82% є бажаним S-ізомером (прегабалін), та 2.18% є небажаним R-ізомером. A. Реакцію проводять у мікрореакторі на 50мл, В. Перетворення трет-бутиламонієвої солі (S)який оснащений перегородкою та клапаном для 3-ціано-5-метилгексанової кислоти у прегабалін уведення проби. Мікрореактор використовують S-ціанокислоту, трет-бутиламонієву сіль (одеразом з скляною футерівкою. Метанол дезоксигержану, як описано у Прикладі 2А, 97% S-ізомер, нують 4 циклами часткового видалення та знову 8,0г. 35,0ммоль) завантажують разом з гідроксиуведення азоту при перемішуванні. У гільзу завандом калію (91% шар, 2,2г маса, 2,0г вага, тажують етилу 3-ціано-5-метилгекс-3-еноат, магні35,6ммоль), водою (15мл), та 2В ЕtOН (11мл) у тну мішалку поміщають у мікрореактор, а потім сосуд Парра, що містить губчатий нікелевий катамікрореактор компонують. Атмосферу водню лізатор (А-7000, 5г, маса води). Кашицю струшустворюють трьома циклами уведення у сосуд водють у вібраторі PARR під тиском водню 50фунт/кв. ню та зменшення тиску. Метанол (4мл) додають, а дюйм при кімнатній температурі протягом ночі. сосуд тоді поміщають у масляну баню на гарячу Кашицю фільтрують через шар Supercel. Осад пластину мішалки при 60°С і дають дійти до терміна фільтрі промивають водою (20мл) та 2В ЕtOН чної рівноваги (зовнішня темп.~45°С). У малу тубу (етанол, денатурований толуолом) (7мл). ПоєднаШленка завантажують [(R,R)-Meний фільтрат завантажують зльодяною оцтовою DuPHOS]Rh(COD)+BF4- та атмосферу азоту встакислотою (4,1мл, 4,3г, 71,6ммоль). Отриманий новлюють чотирма циклами часткового видалення розчин нагрівають до 70°С, а тоді дають охолонути і знову уведення азоту. Каталізатор розчиняють в повільно до кімнатної температури. Реакційну каметанолі так, щоб отримати розчин, який містить шицю тоді перемішують при 0-5°С протягом 6 го1,2мг каталізатору в 1мл розчиннику. 1мл розчину дин і фільтрують. Твердий продукт промивають з каталізатором додають шприцом у мікрореактор. ізофталевою кислотою (50мл) та висушують проСосуд знову продувають воднем, створенням витягом 2 днів у вакуумній печі для отримання тверсокого тиску до 60фунт/кв. дюйм та зменшенням дого продукт, що має масу 3,4г (61,0% загальний тиску через наступні 4 цикли. У сосуд тоді заванвихід). Аналіз ВЕРХ визначає продукт як 97,20% тажують під дією тису 60фунт/кв. дюйм та перемі(за площею) 3-ізобутил ГАМК, 99.92% якої є бажашують, доки не закінчиться поглинання водню (~3 ним S-ізомером (прегабалін). години). Реактор видаляють з масляної бані і даУ реактор на 600мл, під тиском аргону, заванють охолонути. Тиск тоді зменшується і розчинник тажують 3-ціано-5-метилгекс-3-еноат третвидаляють у вакуумі. Аналіз ГХ показує 99% перебутиламонію (одержаний як описано у Прикладі 1А творення. 22.7% е.з. (R). 36г, 159,1ммоль) та B. Слідуючи загальної процедури з Прикладу [(R,R)MeDUPHOS]Rh(COD)+BF4(0,054г, 3.1, 200мг (1,190ммоль) 3-ціано-5-метил-гекс-30,0894ммоль). Реактор продувають під тиском еноату метилу розчинили в 3мл метанолу і він аргоном, (3 50фунт/кв. дюйм). У реактор на прореагував з воднем (60фунт/кв. дюйм) у присут1000мл завантажують 360мл метанолу. Метанол ності 43мг (0,06ммоль) [(R,R)-Etпродувають під тиском аргоном (3 50фунт/кв. DuPHOS]Rh(COD)+BF4- для виходу 10% перетводюйм). Тоді метанол завантажують у реактор, що рення у метил 3-ціано-5-метилгексаноат, що має містить субстрат та каталізатор. Розчин продува33% е.з. (R). ють під тиском аргоном (3 50фунт/кв. дюйм), і тоді ПРИКЛАД 4 у реакторі створюють високий тиск до 50фунт/кв. Синтез Прегабаліну дюйм воднем і перемішують протягом ночі при 27А. Перетворення калієвої солі (S)-3-ціано-533°С. Тиск гідрогену зменшується, і розчин продуметилгексанової кислоти у прегабалін вають аргоном. Розчин переносять у сосуд, що S-ціанокислоту, калієву сіль (одержану у Примістить розчин гідроксид калію (91%, 10,3г, кладі 2В, 94.9% S-ізомер, 8,0г, 41.4ммоль) заван167ммоль) в 90мл води. Розчин концентрують до тажують разом з гідроксидом калію (91%, 44,0мг приблизно 180мл у вакуумі. Концентрований розбрутто, 40г нетто, 0,7ммоль), водою (15мл), та 2В чин переносять у реактор високого тиску, що місЕtOН (тобто, денатурований толуолом) (10мл) у тить губчатий нікель А-7000 (12,0г, 50% за масою 21 75584 22 170л. Розчин швидко охолоджують до 50°С, а тоді води). Розчин продувають аргоном (3 50фунт/кв. до -5°С ±5°С через приблизно 3,5 годин. Кашицю дюйм) і тоді у реакторі створюють високий тиск до утримують при -5°С ±5°С протягом приблизно 16 50фунт/кв. дюйм воднем та перемішують протягом годин. Твердий продукт фільтрують і промивають ночі. Тиск водню зменшується. Розчин продувають ізопропіловим спиртом (10л). Твердий продукт аргоном і фільтрують. Осад на фільтрі промивависушують у вакуумі при 45°С протягом 3 днів для ють 90мл метанолу. Фільтрат концентрують у ваотримання 4,0кг (57%) прегабаліну як твердого куумі для видалення метанолу, і 72мл ізопропілопродукту білого кольору (99.84% S). вого спирту завантажують. Розчин нагрівають до ПРИКЛАД 5 65°С.льодяну оцтову кислоту (9,4мл, 171ммоль) Гідрування 3-ціано-5-метилгекс-3-енової кисзавантажують, і розчин нагрівають до 73°С. Розлоти (вільна кислота) чин швидко охолоджують до 50°С, тоді повільно охолоджують до кімнатної температури. Кашицю охолоджують до 0-5°С протягом 3,5 години. Кашицю фільтрують, і осад промивають ізопропіловим спиртом. Твердий продукт висушують у вакуумі при 45°С для отримання 18,4г (73%) прегабалін як твердого продукту білого кольору (99.89% S). МолекуУ продутий аргоном реактор на 700мл заванМатеріал лярна Кількість ммоль тажують 3-ціано-5-метилгекс-3-еноат третмаса 3-Ціано-5-метилгекс-3бутиламонію (10кг, 44,2моль, одержаного як опиенова кислота 153 200мг 1,307 сано у Приклад 1А) і [(S,S)-Me0,0327 [(R,R)MeDuPHOS]Rh(COD)+BF4(0,015кг, + BPE]Rh(COD) BF4 618,48 20мг (2,5моль%) 0,0025моль). Реактор продувають під тиском аргоМетанол 4мл ном (3 50фунт/кв. дюйм). У дистилятор на 170л 50фунт/кв. Водень завантажують 100л метанолу. Реактор відкачують, дюйм (4бар) створюючи вакуум, і тоді запускають аргон. У дистиляторі створюють високий тиск до 50фунт/кв. А. Вільну гексанову кислоту розчинили в медюйм аргоном і тоді закривають. Цю процедуру танолі, а хіральний каталізатор додали до розчипродування повторюють двічі. Метанол завантану. Суміш струшували при 24°С протягом 19 годин жують у реактор, що містить субстрат та каталізав атмосфері азоту при тиску 50фунт/ кв. дюйм. тор. Розчин продувають під тиском аргоном Пробу аналізували протонним ЯМР, і було визна(3 50фунт/кв. дюйм), і тоді у сосуді створюють чено, що реакція є на 24% закінченою, з ціаногеквисокий тиск 50фунт/кв. дюйм воднем і перемішусановою кислотою, що має 95% е.з. (S). ють протягом ночі при 27-33°С. Тиск водню зменОдну еквівалентну кількість (0,18мл) триетишується, і розчин продувають азотом. Розчин філаміну було додано до реакційної суміші, і струшультрують у дистилятор на 170л, що містить розчин вання було продовжено протягом 5 додаткових гідроксиду калію (91%, 2,9кг, 46,4моль) в 25л води. годин (24°С, 50фунт/кв. дюйм. Реакційну суміш 5л промитого метанолу використовують для очивідфільтрували, і розчинник видалили випарюванщення автоматичноїлінії. Фільтрат концентрують ням. Продукт було аналізовано протонним ЯМР, до об'єму 50-60л вакуумною дистиляцією. Цей який показав вміст приблизно 43% бажаної (S)-3концентрований розчин переносять у реактор на ціано-5-метилгексанової кислоти, що має 95% е.з. 170л, що містить губчатий нікель А-7000 (5,0кг, для S-енантіомеру. 50% маси води). Розчин продувають азотом B. Після вищезазначеної процедури провели (3 50фунт/кв. дюйм). Тоді, у реакторі створюють реакцію 250мг (1,634ммоль) 3-ціано-5-метилгекс-3високий тиск 50фунт/кв. дюйм воднем і перемішуенової кислоти з воднем (50фунт/кв. дюйм) у приють протягом ночі. Тиск водню зменшується і розсутності 8мг (0,01634ммоль) [(S,S)-Etчин продувають азотом. Розчин фільтрують у дисBPE]Rh(COD)+BF4- та 0,023мл (0,1634ммоль; тиляторі на 170л, і фільтр ілінії промивають 30л 0.1екв.) триетиламіну в 5мл метанолу при 24°С метанолу. Фільтрат концентрують вакуумною диспротягом 40 годин. Реакційну суміш відфільтруватиляцією до об'єму 25-35л, і тоді 30л ізопропіловоли, розчинник видалили випарюванням, протонний го спирту завантажують. Розчин концентрують ЯМР показав, що продуктом є 71% (S)-3-ціано-5вакуумною дистиляцією до приблизно 18л. Ізопрометилгексанова кислота з 84% е.з. для Sпіловий спирт (20л) та воду (5л) завантажують, і енантіомеру. розчин нагрівають до 60-65°С. Льодяну оцтову C. Вищезазначену процедуру повторили, за кислоту (2,9кг, 47,7моль) завантажують, і розчин винятком того, що жодної основи не було до реакнагрівають під зворотним холодильником. Воду ційної суміші. Протонний ЯМР показав, що продук(8л) завантажують для отримання розчину. Розчин том є 26% (S)-3-ціано-5-метилгексанова кислота, швидко охолоджують до 50°С, а тоді до -5°С ±5°С що має 91% е.з. для S-енантіомеру. через приблизно 5,5 годин. Кашицю утримують D. Після вищезазначеної процедури було пропри -5°С ±5°С протягом приблизно 10 годин, а тоді ведено реакцію 200мг (1,307ммоль) 3-ціано-5фільтрують і промивають ізопропіловим спиртом метилгекс-3-еновї кислоти з воднем (50фунт/кв. (10л). Зволожений розчинником осад на фільтрі дюйм, 100 годин) у присутності 10мг завантажують у дистилятор на 170л, а потім воду (0,01307ммоль) [(S,S)-Et-DuPHOS]Rh(COD)+BF4-. (20л) та ізопропіловий спирт (40л). Кашицю нагріПротонний ЯМР показав, що продуктом є 82% (S)вають під зворотним холодильником для отриман3-ціано-5-метилгексанова кислота, що має 56% ня чистого розчину, який фільтрують у реактор на 23 75584 24 е.з. для S-енантіомеру. Прегабалін у твердій формі (117кг, 735моль), E. Процедуру з Прикладу 5D повторили, за що містить 0,6% (R)-енантіомеру, поєднують з вовинятком того, що 0,1екв. (0,02мл, 0,1307ммоль) дою (550л; 4,7л/кг прегабалін) та ізопропіловим триетиламіну було додано до реакційної суміші. спиртом (1100л: 9,4л/кг прегабалін). Суміш нагріРеакцію було зупинено через 16 годин, і було повають для розчинення твердих частинок (приблизказано, що продуктом є 86% (S)-3-ціано-5но 75°С ±5°С), фільтрують, поки нагрівають та метилгексанова кислота з 68% е.з. для Sохолоджують до 0°С ±5°С, для кристалізації проенантіомеру. дукту. F. Процедуру з Прикладу 5Е повторили, за виТвердий продукт збирають центрифугуванням нятком того, що 1екв. (0,18мл, 1,307ммоль) триеі промивають ізопропіловим спиртом. Вологий тиламіну було додано до реакційної суміші, і реактвердий продукт висушують під вакуумом при 35цію було зупинено через 16 годин. Протонний ЯМР 45°С, а тоді розмелюють для отримання 91,8кг показав, що продукт на 92% перетворився у (S)-3(78,5%) прегабаліну як кристалічного твердого ціано-5-метилгексанову кислоту, що має 56% е.з. продукту білого кольору. Співвідношення енантіодля S-енантіомеру. мерів є 99,94% (S)-енантіомер (прегабалін) і 0,06% G. Слідуючи загальної вищезазначеної проце(R)-енантіомер. дури, 250мг (1,634ммоль) 3-ціано-5-метилгекс-3Винахід і спосіб його створення та викорисенової кислоти прореагувало з воднем (50фунт/кв. тання описані зараз такими повними, ясними, чітдюйм, 16 годин, 24°С) у присутності 12мг кими та стислими термінами, що це дає можли(0,01634ммоль) [(R,R)MeDuPHOS]Rh(COD)+BF4- в вість будь-якому фахівцю в даній сфері метанолі (10мл) для отримання 51% 3-ціано-5створювати та використовувати теж саме. Має метилгексанової кислоти, що має 72% е.з. для Rбути зрозумілим, що вищевикладене описує бажаенантіомеру. ні втілення винаходу і модифікації можуть бути ПРИКЛАД 6 сюди внесені, але не виходячи за рамки винаходу, Рекристалізація Прегабалін як викладено у формулі. Комп’ютерна верстка Т. Чепелева Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparation of pregabalin and intermediate compounds, intermediate compounds
Назва патенту російськоюСпособ получения прегабалина и промежуточных соединений, промежуточные соединения
МПК / Мітки
МПК: C07C 255/19, C07C 227/00, C07C 253/30, C07C 255/23
Мітки: спосіб, одержання, сполук, сполуки, прегабаліну, проміжних, проміжні
Код посилання
<a href="https://ua.patents.su/12-75584-sposib-oderzhannya-pregabalinu-ta-promizhnikh-spoluk-promizhni-spoluki.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання прегабаліну та проміжних сполук, проміжні сполуки</a>
Спосіб одержання [(1s)-[1a,2b,3b,4a(s)]]-4-[7-[[1-(3-хлор-2-тієніл)метил] пропіл]аміно]-3н-імідазо[4,5-b]піридин-3-іл]-n-етил-2,3-дигідроксициклопентанкарбоксаміду, способи одержання проміжних сполук для його одержання та проміжні сполуки

Номер патенту: 54479
Опубліковано: 17.03.2003
Автори: Томпсон Майкл Д., Шах Харшавадан К., Паунер Торі Х., Вальтер Френсіс Л., Рейллі Лоренс, О'Брайєн Майкл К., Леон Патрік, Цуей Чінг Т., Гарсіа Ерве, Ванасс Бенуа Дж.
МПК: C07D 409/14, C07D 409/12, A61P 3/06, C07D 213/69, A61P 9/00, C07D 471/04, A61P 9/12
Мітки: 1s)-[1a,2b,3b,4a(s)]]-4-[7-[[1-(3-хлор-2-тієніл)метил, пропіл]аміно]-3н-імідазо[4,5-b]піридин-3-іл]-n-етил-2,3-дигідроксициклопентанкарбоксаміду, сполуки, проміжні, способи, проміжних, одержання, спосіб, сполук
Формула / Реферат:
1. Спосіб одержання [(1S)-[1a,2b,3b,4a(S*)]]-4-[7-[[1-(3-хлор-2-тієніл)метил]пропіл]аміно]-3H-імідазо[4,5-b]піридин-3-іл]-N-етил-2,3-дигідроксициклопентанкарбоксаміду, який включає взаємодію [(1S)-[1a,2b,3b,4a(S*)]]-4-[[3-аміно-4-[[1-(3-хлор-2-тієніл)метил]пропіл]аміно]-2-піридиніл]аміно]-N-етил-2,3-дигідроксициклопентанкарбоксаміду зі складним ефіром ортоформіату, ацетатом формамідину або диметилацеталем диметилформаміду.2. Спосіб за...
Спосіб одержання бензопіранових сполук, проміжні сполуки, спосіб одержання проміжних сполук
Номер патенту: 41904
Опубліковано: 15.10.2001
Автори: Манн Індерджіт Сінг, Гін Річард Грехем, Новак Ванс, Сміт Ніл, Джонсон Грехем
МПК: C07D 405/04, C07D 257/00
Мітки: одержання, сполуки, бензопіранових, спосіб, проміжних, сполук, проміжні
Формула / Реферат:
1. Способ получения бензопирановых соединений, общей формулы (I)в которой R1 представляет собойА представляет простую связь, Х - кислород, R2 - водород и R3 - водород, отличающийся тем, что осуществляют циклизацию соединения структуры (II)или его соли, гидрата или сольвата, причем в формуле R1, R2, R3, А и Х имеют значения, определенные для структуры (I), и при необходимости получают после этого соль,...
Азольні сполуки з протигрибковою активністю, спосіб одержання цих сполук, спосіб одержання проміжних сполук та фармацевтична композиція

Номер патенту: 55405
Опубліковано: 15.04.2003
Автори: Альбіні Енріко, Скіоппакассі Джованна, Фраіре Крістіна, Наполетано Мауро
МПК: A61P 31/10, A61K 31/4164, A61K 31/00, A61K 31/4196, A61K 31/41, C07D 521/00, C07D 249/08, C07D 233/60, A61K 31/415, A61P 31/00
Мітки: спосіб, протигрибковою, цих, азольні, сполуки, композиція, фармацевтична, сполук, одержання, проміжних, активністю
Формула / Реферат:
1. Сполука формули (ІІ-А), (ІІ-А)де:R1 являє собою хлор, фтор, бром або трифторметил;R2 являє собою водень, хлор, фтор, бром або трифторметил;Z являє собою СН або N;R3, R4, R5, які можуть бути однаковими або різними, являють собою водень або С1-С4 алкіл при умові, що R4 відрізняється від R5, якщо R3 - водень;Х являє собою О, S, SO або SO2;R6 являє собою С1-С5 поліфторалкільну групу, яка...
Хіральні біфосфінові сполуки, комплекси на їх основі, 4,16-дибром[2.2]парациклофани як проміжні сполуки для їх одержання та спосіб одержання хіральних біфосфінових сполук

Номер патенту: 57742
Опубліковано: 15.07.2003
Автори: Россен Кай, Воланте Ральф П., Пай Філіп
МПК: C07D 401/06, C07F 9/655, C07D 241/04, C07D 241/06, C07F 9/50, C07D 241/24, C07F 9/6553, C07C 67/31
Мітки: хіральних, основі, спосіб, біфосфінових, сполуки, біфосфінові, 4,16-дибром[2.2]парациклофани, одержання, сполук, проміжні, хіральні, комплекси
Формула / Реферат:
1. Соединение нижеприведенной формулы, представляющей собой хиральные бифосфиныили,где R представляет С1-4-алкил, С3-6-циклоалкил или арил, незамещенный или замещенный -F, -СН3, -СF3 или СН3О-, иX1 и X2 связывают два R2P-замещенные бензола и независимо образуют 2-4-членную...
Способи одержання проміжних сполук b-метилкарбапенему, проміжні cполуки

Номер патенту: 50705
Опубліковано: 15.11.2002
Автори: Воланте Ральф П., Шінкай Ічіро, Томпсон Ендрю С., Рейдер Пол Дж., Хамфрі Гай Р., Чой Ву-Баєг
МПК: C07D 405/04, C07D 205/00
Мітки: проміжних, одержання, cполуки, сполук, проміжні, b-метилкарбапенему, способи
Формула / Реферат:
1. Способ получения промежуточного соединения b-метилкарбапенема формулы (VI):, (VI)где R представляет собой:(a) водород,(b) метил, или(c) гидроксизащитную группу, иР' представляет собой азотзащитную группу,Nu представляет собой нуклеофильную группу, выбранную из –СН2СО2-т-Вu и -SR2,где R2 представляет собой водород, прямой или разветвленный C1-C10aлкил, прямой или разветвленный...
Попередній патент: Процес моделювання порушення прохідності ворітної вени
Наступний патент: Зернозбиральний комбайн