Спосіб одержання похідних 3-хінолонкарбонової кислоти
Номер патенту: 41323
Опубліковано: 17.09.2001
Автори: Герхард Франковіак, Херберт Діль, Рудольф Цербес, Пауль Нааб





Формула / Реферат
1. Способ получения производных 3-хинолонкарбоновой кислоты общей формулы (I):
где
R1 и R2 вместе с атомом азота, с которым они связаны, образуют пирролидиновое кольцо, замещенное в положении 3 аминогруппой, пиперазинильное кольцо, замещенное у атома азота в положении 4 низшим алкилом, или 2,8-диазабицикло[4.3.0]нонил,
А - СН, CF, CCl,
включающий стадии взаимодействия галогенангидрида кислоты с эстером карбоновой кислоты в среде растворителя в присутствии основания, циклизации в присутствии основного агента, взаимодействия с гетероциклическим амином, щелочного омыления и выделения целевого продукта в свободной форме или в виде соли, отличающийся тем, что галогенангидрид кислоты общей формулы (II):
где
Hal, X и X1 означают фтор или хлор, а А имеет вышеуказанные значения,
подвергают взаимодействию с эстером аминоакриловой кислоты общей формулы (III):
где
R3 означает алкил с 1 - 4 атомами углерода,
или же сначала с эстером диметиламиноакриловой кислоты общей формулы (IV):
где
R3 имеет вышеуказанное значение,
и затем с циклопропиламином формулы (V):
получаемое при этом соединение общей формулы (VI):
где
X, X1, А и R3 имеют вышеуказанные значения,
подвергают циклизации в присутствии основного агента с последующим взаимодействием получаемого при этом соединения общей формулы (VII):
где
X1, A, R3 имеют вышеуказанные значения,
с гетероциклическим амином общей формулы (VIII):
где
R1 и R2 имеют вышеуказанные значения,
и щелочным омылением получаемого при этом соедиения формулы (IX):
где
R1 - R3 и А имеют вышеуказанные значения,
причем каждую стадию проводят без предварительного выделения соответствующих промежуточных продуктов.
2. Способ по п. 1, отличающийся тем, что в качестве гетероциклического амина формулы (VIII) используют соединение формулы
3. Способ по п. 1 или 2, отличающийся тем, что циклизацию соединения формулы (VI) проводят в присутствии карбоната калия в качестве основного агента.
Текст
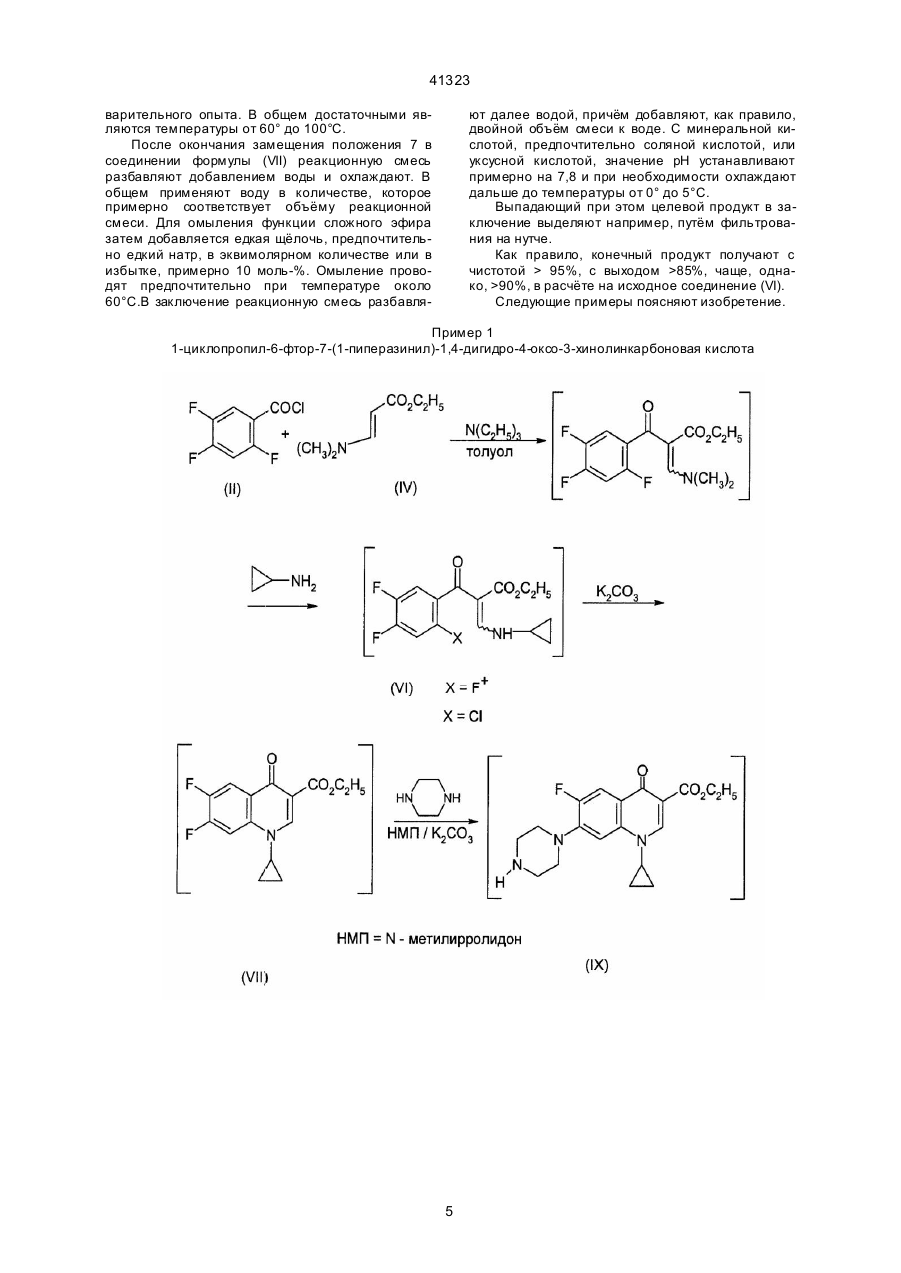
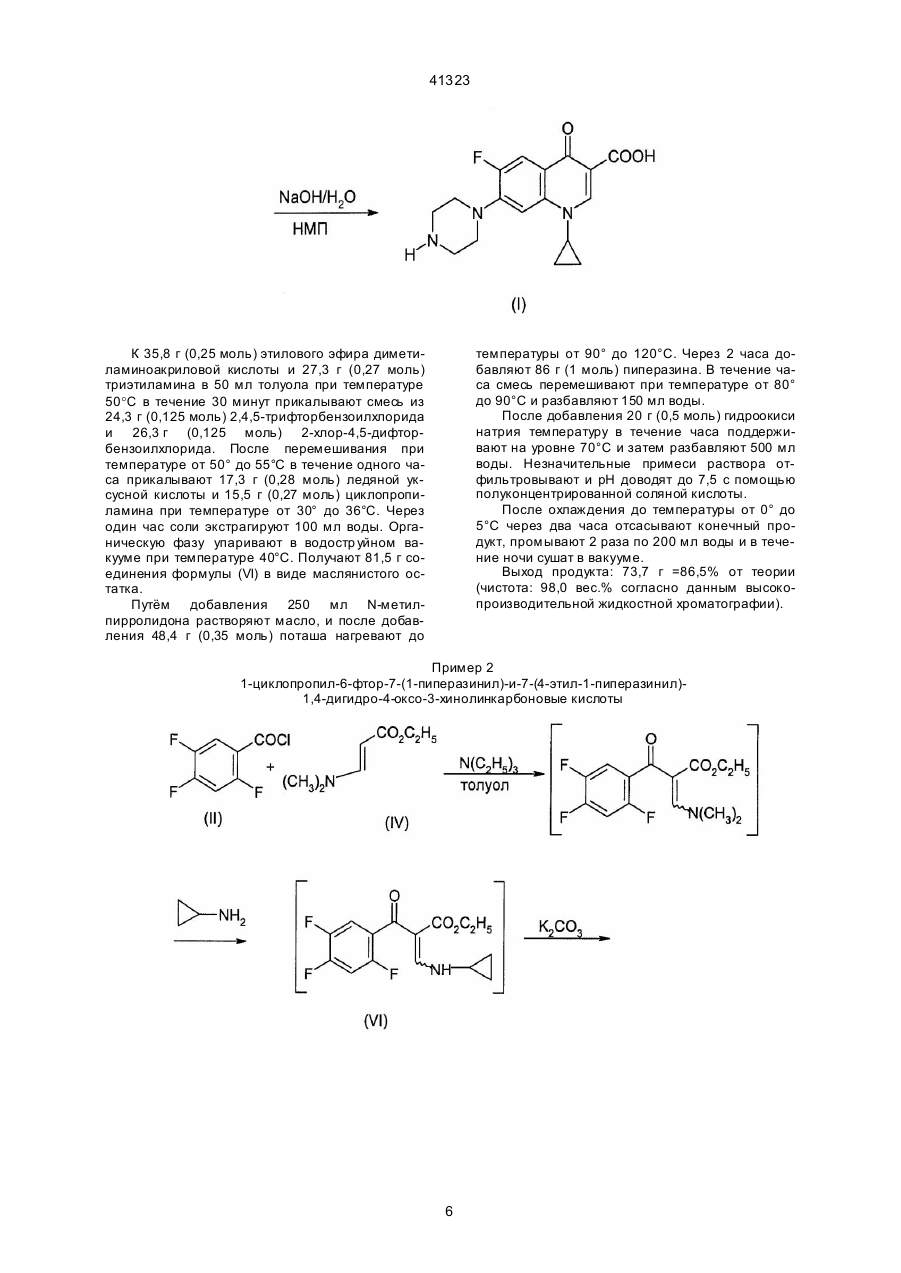
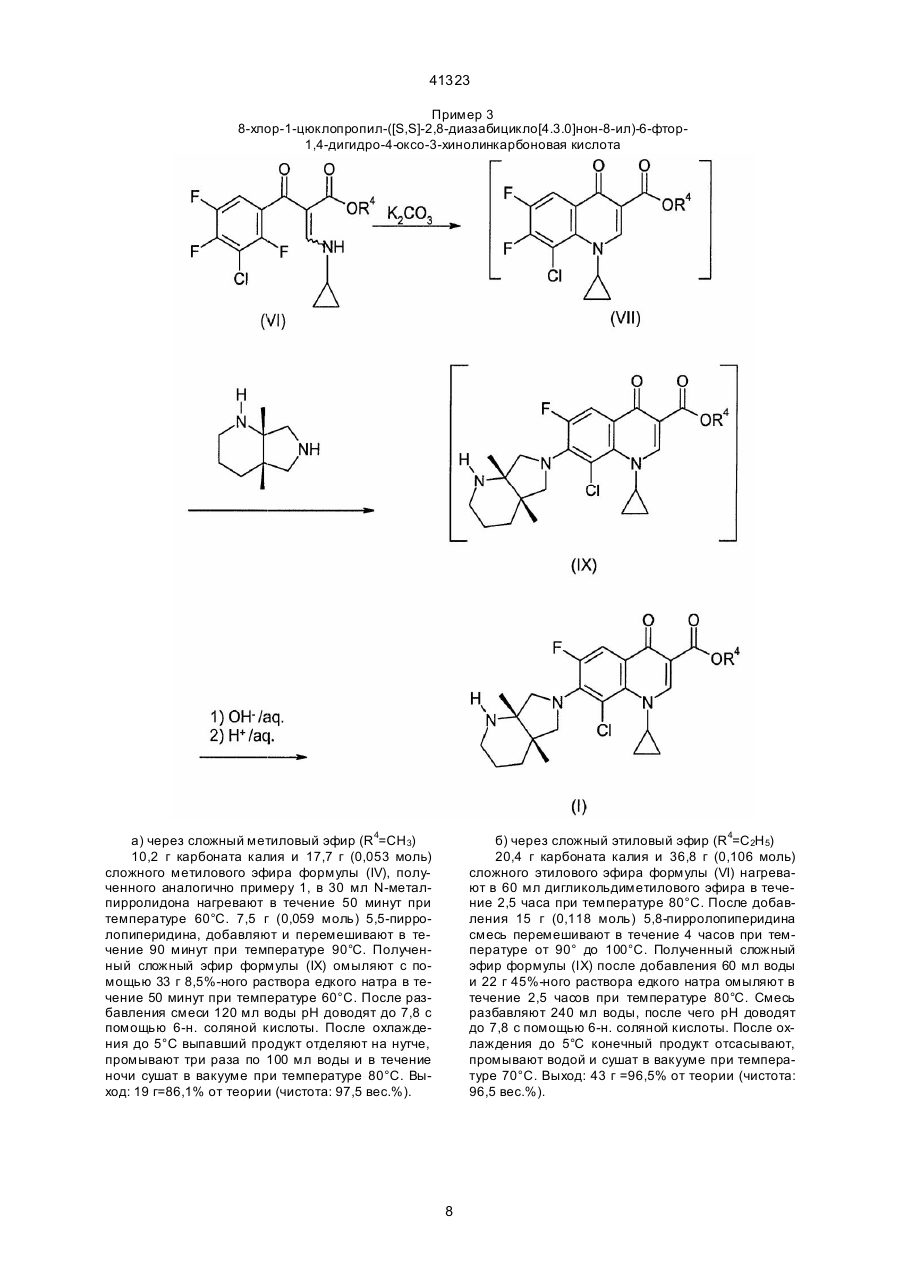
1. Способ получения производных 3-хинолонкарбоновой кислоты общей формулы (I): где R3 означает алкил с 1-4 атомами углерода, или же сначала с эстером диметиламиноакриловой кислоты общей формулы (IV): где R3 имеет вышеуказанное значение, и затем с циклопропиламином формулы (V): (II), где Hal, X и X1 означают фтор или хлор, а А имеет вышеуказанные значения, подвергают взаимодействию с эстером аминоакриловой кислоты общей формулы (III): (VII), где X1 , A, R3 имеют выше указанные значения, с гетероциклическим амином общей формулы (VIII): (VIII), (13) 41323 UA (11) (VI), где X, X1, А и R3 имеют вышеуказанные значения, подвергают циклизации в присутствии основного агента с последующим взаимодействием получаемого при этом соединения общей формулы (VII): (19) (I), где R1 и R2 вместе с атомом азота, с которым они связаны, образуют пирролидиновое кольцо, замещенное в положении 3 аминогруппой, пиперазинильное кольцо, замещенное у атома азота в положении 4 низшим алкилом, или 2,8-диазабицикло[4.3.0]нонил, А - СН, CF, CCl, включающий стадии взаимодействия галогенангидрида кислоты с эстером карбоновой кислоты в среде растворителя в присутствии основания, циклизации в присутствии основного агента, взаимодействия с гетероциклическим амином, щелочного омыления и выделения целевого продукта в свободной форме или в виде соли, отличающийся тем, что галогенангидрид кислоты общей формулы (II): C2 получаемое при этом соединение общей формулы (VI): 41323 где R1 и R2 имеют выше указанные значения, и щелочным омылением получаемого при этом соединения формулы (IX): причем каждую стадию проводят без предварительного выделения соответствующи х промежуточных продуктов. 2. Способ по п. 1, отличающийся тем, что в качестве гетероциклического амина формулы (VIII) используют соединение формулы (IX), 3. Способ по п. 1 или 2, отличающийся тем, что циклизацию соединения формулы (VI) проводят в присутствии карбоната калия в качестве основного агента. где R1-R3 и А имеют выше указанные значения, татки COR a, CN, SO2Rb причём Ra представляет собой водород, линейный или разветвлённый алкил с 1-4 атомами углерода, незамещённый или замещённый 1-3 заместителями из группы, включающей амино, алкоксикарбонил с 1-3 атомами углерода в алкильной части, карбоксил, алкокси с 1-3 атомами углерода и галоген, например, хлор, бром, фтор, алкокси с 1-4 атомами углерода, амино, алкиламино или диалкиламино с 1-5 атомами углерода в алкильной части, и R представляет собой линейный или разветвлённый алкил с 1-3 атомами углерода, и их фармацевтически приемлемых гидратов, кислотно-аддитивных солей, щелочных, щелочноземельных и гуанидиниевых солей, состоящий в том, что соединение общей формулы (2) Изобретение относится к технологии получения известных производных хинолонкарбоновой кислоты, в частности к способу получения производных 3-хинолонкарбоновой кислоты. Известен способ получения производных 3-хинолонкарбоновой кислоты общей формулы (1) где X' и X" являются одинаковыми или различными и означают хлор или фтор, но не могут являться одновременно фтором, и R' и R" вместе с атомом азота, с которым они связаны, образовывают пяти- или шестичленное гетероциклическое кольцо, которое в качестве члена кольца может содержать дополнительно атомы групп -О-, -S-, -SO-, >N-R'" или -CO-N-R"' и которое у атомов углерода может быть замещено одно-, дву- или трёхкратно алкилом, содержащим 1-4 атома углерода, фенилом или циклогексилом, незамещёнными или замещёнными фтором, хлором, бромом, метилом, фенилом, гидроксильной группой, бензилоксигруппой, нитро- или пиперидино-группой, 2тиенилом, гидроксильной группой, алкоксигруппой, содержащей от одного до трёх атомов углерода, амино-, метиламино- или этиламиногруппой, причём R'" означает водород, линейную или разветвлённую алкильную, алкенильную или алкинильную группу, содержащую от одного до шести атомов углерода, которые могут быть замещены одной или двумя гидроксильными, алкоксильными, алкиламино или диалкиламиногруппами, содержащими от одного до трёх атомов углерода в алкильном остатке, цианогруппой, алкоксикарбонильной группой, содержащей от одного до четырёх атомов углерода в алифатической части, фенилалкил с 1-4 атомами углерода в алкильной части, незамещённый или одно- или двукратно замещённый в фенильном остатке, фенацильный остаток, незамещённый или одно-, дву- или трёхкратно замещенный гидроксильной группой, метоксильной группой, хлором или фтором, оксалкильный остаток с количеством атомов углерода до шести, ос где X' и X" имеют выше указанное значение, и X'" и Hal означает галоген, подвергают взаимодействию с диэтиловым эфиром малоновой кислоты формулы (3) в среде растворителя в присутствии метилата магния, получаемое при этом соединение общей формулы (4) где X', X" и X"' имеют вышеуказанное значение, подвергают частичному омылению и декарбоксилированию в водной среде в присутствии каталитического количества серной кислоты или п-толуолфульфокислоты, получаемое при этом соединение общей формулы (5) 2 41323 дии, кроме циклизации, проводят при температуре в диапазоне от 20° до 150°С (см. заявку ЕР № 0167763 А1, МКИ: С07D215/56,1986 г.). Недостаток известного способа заключается в том, что общий выход является неудовлетворительным. При этом при щелочном омылении сложного эфира общей формулы (8) могут возникнуть побочные продукты, в которых остаток Z'" замещён гидроксилом или алкоксилом, а также олиго- или полимеры, в частности, если Z'" означает фтор. Если омыление проводят в кислых условиях, то выделяется фтористый водород, что ведёт к коррозии производственной установки и загрязнению продукта комплексными фторидами металлов. Это происходит, в частности, в том случае, когда после циклизации соединения общей формулы (7) до соединения общей формулы (8), при которой образуется фтористый водород, связываемый с основанием, реакционная смесь без предварительного выделения сложного эфира общей формулы (8) подаётся на кислый гидролиз, при котором снова высвобождается связанный ранее с основанием фтористый водород. Другой недостаток известного способа состоит в том, что для получения целевого продукта необходимо осуществить 5 стадий. Производные 3-хинолонкарбоновой кислоты общей формулы (1), в которой R' и R" вместе с атомом азота, с которым они связаны, образуют бициклический гетероцикл, кольца которого могут содержать ещё другие гетероатомы из числа азота, кислорода и серы, и который может быть замещён, описаны в заявке ЕР № 0350733 А2 (МКИ: С07D401/04, 1990 г.). Они могут быть получены таким же способом, что и производные 3-хинолонкарбоновой кислоты по вышеназванной заявке ЕР № 0167763. Задачей данного изобретения являются повышение общего выхода известных производных 3-хинолонкарбоновой кислоты и упрощение процесса их получения при одновременном предотвращении образования корризионных продуктов распада и загрязнения целевого продукта. Эта задача решается в способе получения производных 3-хинолонкарбоновой кислоты общей формулы (I) где X', X" и X'" имеют вышеуказанное значение, подвергают взаимодействию с триэтиловым эфиром орто-муравьиновой кислоты в присутствии уксусного ангидрида, получаемое при этом соединение общей формулы (6) где X, X" и X'" имеют выше указанное значение, подвергают взаимодействию с циколпропиламином в среде растворителя, полученное при этом соединение общей формулы (7) где X1 , X" и X'" имеют вышеуказанное значение, подвергают циклизации в среде растворителя при температуре 60-300°С в присутствии связующего кислоту средства, получаемое при этом соединение общей формулы (8) где X', X" и X'" имеют вышеуказанное значение, подвергают щелочному или кислотному гидролизу сложноэфирной группы в среде растворителя и получаемое при этом соединение общей формулы (9) где R1 и R2 вместе с атомом азота, с которым они связаны, образуют пирролидиновое кольцо, замещенное в положении 3 аминогруппой, пиперазинильное кольцо, замещенное у атома азота в положении 4 низшим алкилом, или 2,8-диазабицикло[4.3.0]нонил, A-CH,CF,CCI, включающий стадии взаимодействия галогенангидрида кислоты эстером карбоновой кислоты в среде растворителя в присутствии основания, циклизации в присутствии основного агента, взаимодействия с гетероциклическим амином, щелоч где X', X" и X'" имеют вышеуказанное значение, подвергают взаимодействию с амином общей формулы (10) где R' и R" имеют выше указанное значение, в среде растворителя, при необходимости в присутствии связующего кислоту средства с последующим выделением целевого продукта в свободной форме или в виде соли, причем все ста 3 41323 ного омыления и выделения целевого продукта в свободной форме или в виде соли, за счет того, что галогенангидрид кислоты общей формулы (II) где R1-R3 и А имеют выше указанные значения, причем каждую стадию проводят без предварительного выделения соответствующи х промежуточных продуктов. Выделение целевого продукта происходит таким образом, что после щелочного омыления реакционную смесь нейтрализуют кислотой и отделяют выпавший продукт. В качестве гетероциклического амина формулы VIII предпочтительно используют соединение формулы где Hal, X и X1 означают фтор или хлор, а А имеет выше указанные значения, подвергают взаимодействию с эстером аминоакриловой кислоты общей формулы (III) где R3 означает алкил с 1-4 атомами углерода, или же сначала с эстером диметиламиноакриловой кислоты общей формулы (IV) где R3 имеет вышеуказанное значение, и затем с циклопропиламином формулы (V) . Циклизацию соединения формулы (VI) предпочтительно проводят в присутствии карбоната калия в качестве основного агента. Для проведения способа согласно изобретению могут быть применены доступные инертные органические растворители. В качестве примеров можно назвать диметилэтиленмочевину, диметилпропиленмочевину, N-метилкапролактам, третичный бутанол, тетраметилмочевину, тетраметилсульфон и диметоксиэтан. Предпочтительно применяются смешиваемые с водой растворители, как N-метилпирролидон, дигликольдиметиловый эфир, N,N'-диметилформамид или диоксан, особенно предпочтителен N-метилпирролидон. Взаимодействие галогенангидрида формулы (II) с эстером формулы (III) или (IV) проводят предпочтительно в присутствии щелочи. Циклизацию проводят при возможно низкой температуре. В общем, достаточной является температура от 60° до 100°С, что легко определяется путём проведения ориентировочных предварительных опытов с выбранной предварительной стадией в выбранной среде растворителя/основания и позволяет определить оптимальное количество растворителя. В качестве основания при циклизации в органическом синтезе могут применяться обычные акцепторы кислоты. В качестве такого акцептора могут быть названы третичный бутанолат калия, бутил-лития, литий-фенил, метилат натрия, гидрид натрия, карбонат натрия или калия и фторид натрия или калия. Может оказаться предпочтительным применять избыток до 10 мол-% основания, а при необходимости и выше. Как уже указывалось выше, предпочтительно применение карбоната калия или натрия. После окончания циклизации при той же или при всё повышающейся температуре добавляют гетероциклический амин общей формулы (VIII). Оптимальная температура реакции зависит от реагентов, однако легко определится путём пред получаемое при этом соединение общей формулы (VI): где X, X1, А и R3 имеют вышеуказанные значения, подвергают циклизации в присутствии основного агента с последующим взаимодействием получаемого при этом соединения общей формулы (VII): где X1 , A, R3 имеют выше указанные значения, с гетероциклическим амином общей формулы (VIII) где R1 и R2 имеют выше указанные значения, и щелочным омылением получаемого при этом соедиения формулы (IX) 4 41323 варительного опыта. В общем достаточными являются температуры от 60° до 100°С. После окончания замещения положения 7 в соединении формулы (VII) реакционную смесь разбавляют добавлением воды и охлаждают. В общем применяют воду в количестве, которое примерно соответствует объёму реакционной смеси. Для омыления функции сложного эфира затем добавляется едкая щёлочь, предпочтительно едкий натр, в эквимолярном количестве или в избытке, примерно 10 моль-%. Омыление проводят предпочтительно при температуре около 60°С.В заключение реакционную смесь разбавля ют далее водой, причём добавляют, как правило, двойной объём смеси к воде. С минеральной кислотой, предпочтительно соляной кислотой, или уксусной кислотой, значение рН устанавливают примерно на 7,8 и при необходимости охлаждают дальше до температуры от 0° до 5°С. Выпадающий при этом целевой продукт в заключение выделяют например, путём фильтрования на нутче. Как правило, конечный продукт получают с чистотой > 95%, с выходом >85%, чаще, однако, >90%, в расчёте на исходное соединение (VI). Следующие примеры поясняют изобретение. Пример 1 1-циклопропил-6-фтор-7-(1-пиперазинил)-1,4-дигидро-4-оксо-3-хинолинкарбоновая кислота 5 41323 К 35,8 г (0,25 моль) этилового эфира диметиламиноакриловой кислоты и 27,3 г (0,27 моль) триэтиламина в 50 мл толуола при температуре 50°С в течение 30 минут прикалывают смесь из 24,3 г (0,125 моль) 2,4,5-трифторбензоилхлорида и 26,3 г (0,125 моль) 2-хлор-4,5-дифторбензоилхлорида. После перемешивания при температуре от 50° до 55°С в течение одного часа прикалывают 17,3 г (0,28 моль) ледяной уксусной кислоты и 15,5 г (0,27 моль) циклопропиламина при температуре от 30° до 36°С. Через один час соли экстрагируют 100 мл воды. Органическую фазу упаривают в водостр уйном вакууме при температуре 40°С. Получают 81,5 г соединения формулы (VI) в виде маслянистого остатка. Путём добавления 250 мл N-метилпирролидона растворяют масло, и после добавления 48,4 г (0,35 моль) поташа нагревают до температуры от 90° до 120°С. Через 2 часа добавляют 86 г (1 моль) пиперазина. В течение часа смесь перемешивают при температуре от 80° до 90°С и разбавляют 150 мл воды. После добавления 20 г (0,5 моль) гидроокиси натрия температуру в течение часа поддерживают на уровне 70°С и затем разбавляют 500 мл воды. Незначительные примеси раствора отфильтровывают и рН доводят до 7,5 с помощью полуконцентрированной соляной кислоты. После охлаждения до температуры от 0° до 5°С через два часа отсасывают конечный продукт, промывают 2 раза по 200 мл воды и в течение ночи сушат в вакууме. Выход продукта: 73,7 г =86,5% от теории (чистота: 98,0 вес.% согласно данным высокопроизводительной жидкостной хроматографии). Пример 2 1-циклопропил-6-фтор-7-(1-пиперазинил)-и-7-(4-этил-1-пиперазинил)1,4-дигидро-4-оксо-3-хинолинкарбоновые кислоты 6 41323 К 35,8 г (0,25 моль) этилового эфира диметиламиноакриловой кислоты и 27,3 г (0,27 моль) триэтиламина в 50 мл толуола при температуре 50°С в течение 30 минут прикалывают 48,6 г (0,25 моль) 2,4,5-трифторбензоилхлорида. После перемешивания в течение одного часа при температуре от 50° до 55°С прикалывают 17,3 г (0,28 моль) ледяной уксусной кислоты и 15,5 г (0,27 моль) циклопропиламина при температуре от 30° до 36°С. Через один час соли экстрагируют 100 мл воды. Органическую фазу упаривают в водоструйном вакууме при температуре 40°С. Получают 80,3 г соединения формулы (VI) в виде маслянистого остатка. Путём добавления 250 мл N-метилпирролидона масло растворяют и, добавив 48,4 г (0,35 моль) поташа, нагревают до 80°-90°С. Через один час добавляют 86 г (1 моль) пиперазина или 114 г (1 моль) N-этилпиперазина. Смесь переме шивают в течение часа при температуре от 80° до 90°С и разбавляют 150 мл воды. После добавления 20 г (0,5 моль) едкого натра температуру в течение одного часа поддерживают на уровне 70°С и затем разбавляют 500 мл воды. Незначительные примеси раствора отфильтровывают и рН доводят до 7,5 с помощью полуконцентрированной соляной кислоты. После охлаждения до температуры от 0° до 5°С через два часа конечный продукт отсасывают, промывают два раза по 200 мл воды и в течение ночи сушат в вакууме. Выход продукта: 71,4 г (84,5% от теории) конечного продукта с R=Н (чистота: 98,0 вес.% согласно данным высокопроизводительной жидкостной хроматографии) и 78,2 г (87,0% от теории) конечного продукта с R7= С2Н5 (чистота: 98,5 вес.% согласно данным высокопроизводительной жидкостной хроматографии). 7 41323 Пример 3 8-хлор-1-цюклопропил-([S,S]-2,8-диазабицикло[4.3.0]нон-8-ил)-6-фтор1,4-дигидро-4-оксо-3-хинолинкарбоновая кислота а) через сложный метиловый эфир (R4=СН3) 10,2 г карбоната калия и 17,7 г (0,053 моль) сложного метилового эфира формулы (IV), полученного аналогично примеру 1, в 30 мл N-металпирролидона нагревают в течение 50 минут при температуре 60°С. 7,5 г (0,059 моль) 5,5-пирролопиперидина, добавляют и перемешивают в течение 90 минут при температуре 90°С. Полученный сложный эфир формулы (IX) омыляют с помощью 33 г 8,5%-ного раствора едкого натра в течение 50 минут при температуре 60°С. После разбавления смеси 120 мл воды рН доводят до 7,8 с помощью 6-н. соляной кислоты. После охлаждения до 5°С выпавший продукт отделяют на нутче, промывают три раза по 100 мл воды и в течение ночи сушат в вакууме при температуре 80°С. Выход: 19 г=86,1% от теории (чистота: 97,5 вес.%). б) через сложный этиловый эфир (R4=C2H5) 20,4 г карбоната калия и 36,8 г (0,106 моль) сложного этилового эфира формулы (VI) нагревают в 60 мл дигликольдиметилового эфира в течение 2,5 часа при температуре 80°С. После добавления 15 г (0,118 моль) 5,8-пирролопиперидина смесь перемешивают в течение 4 часов при температуре от 90° до 100°С. Полученный сложный эфир формулы (IX) после добавления 60 мл воды и 22 г 45%-ного раствора едкого натра омыляют в течение 2,5 часов при температуре 80°С. Смесь разбавляют 240 мл воды, после чего рН доводят до 7,8 с помощью 6-н. соляной кислоты. После охлаждения до 5°С конечный продукт отсасывают, промывают водой и сушат в вакууме при температуре 70°С. Выход: 43 г =96,5% от теории (чистота: 96,5 вес.%). 8 41323 Пример 4 1-циклопропил-7-([S,S]-2,8-диазабицикло[4.3.0]нон-8-ил)-6,8-дифтор1,4-дигидро-4-оксо-3-хинолинкарбоновая кислота 20,4 г карбоната калия и 35,2 г (0,106 моль) сложного этилового эфира формулы (VI), полученного аналогично примеру 1, в 60 мл N-метилпирролидона нагревают в течение часа при температуре 60°С. (По истечении 40 минут по данным высокопроизводительной жидкостной хроматографии образуется 97,6% сложного этилового эфира формулы (VII).) Добавляют 15,5 г (0,12 моль) S,S-пирролипиперидина. Через два часа при температуре 80°С ещё имеется 0,2% этилового эфира форму лы (VII) (по данным высокопроизводительной жидкостной хроматографии). После добавления 60 мл воды и 12 г гидроокиси натрия полученный сложный эфир омыляют в течение четырёх часов при температуре 60°С до конечного продукта. Разбавляют 240 мл воды и рН доводят до 7,8 с помощью 6-н. соляной кислоты. Продукт отсасывают, промывают водой и суша т в вакууме при температуре 70°С. Выход: 38,1 г=91,4% от теории (чистота: 98,9 вес.%). 9 41323 Пример 5 1-циклопропил-6-фтор-7-(1-пиперазинил)-и-7-(4-этил-1-пиперазинил)1,4-дигидро-4-оксо-3-хинолинкарбоновые кислоты 82,4 г сложного этилового эфира формулы (VI), полученного аналогично примеру 1, растворяют в 250 мл диметилэтиленмочевины и после добавления 48,4 г (0,35 моль) поташа нагревают до температуры 100°-120°С. Через два часа прибавляют 86 г (1 моль) пиперазина или 114 г (1 моль) М-этилпиперазина. Смесь перемешивают на протяжении часа при температуре от 80° до 90°С и разбавляют 150 мл воды. После добавления 20 г (0,5 моль) гидроксида натрия температуру поддерживают на протяжении часа на уровне 70°С и далее разбавляют 500 мл воды. Незначи тельные примеси раствора отфильтровывают, и полуконцентрированной соляной кислотой устанавливают рН=7,5. После охлаждения до температуры от 0° до 5°С через два часа отсасывают конечный продукт, промывают два раза по 200 мл воды и продукт сушат в вакууме в течение ночи. Выход: 73,0 г (86,8% от теории) конечного продукта с R7=Н (чистота: 98,5 вес.%) и 77,2 г (85,1% от теории) конечного продукта с R7= С2Н5 (чистота: 99,0 вес.%). 10 41323 Пример 6 7-(3-амино-1-пирродинил)-8-хлор-1-циклопропил-6-фтор1,4-дигидро-4-оксо-3-хинолинкарбоновая кислота 20,6 г сложного этилового эфира формулы (VI), полученного аналогично примеру 1, растворяют в 65 мл диметилэтиленмочевины и нагревают вместе с 12,2 г карбоната калия в течение двух часов при температуре от 100°-120°С. Добавляют 8,5 г 3-ацетамидопирролидина и перемешивают в течение часа при температуре от 80° до 90°С. Затем реакционную смесь разбавляют 40 мл воды и добавляют 10 г гидроксида натрия. Через четыре часа при температуре от 90° до 100°С разбавляют дополнительными 125 мл воды, фильтруют и нейтрализуют полуконцентрированной соляной кислотой. Твёрдое вещество отфильтровывают, промывают водой и изопропанолом и сушат в вакууме в течение ночи. Выход: 13,0 г (83,8% от теории) целевого соединения (чистота: 98,7 вес.% согласно данным высокопроизводительной жидкостной хроматографии). 11 41323 Пример 7 1-циклопропил-6-фтор-7-(4-этил-1-пиперазинил)-1,4-дигидро-4-оксо-3-хинолинкарбоновая кислота К 19,5 г (0,1 моль) 2,4,5-трифторбензоилхлорида в 100 мл диоксана прикалывают при 20°С в течение 30 минут 11 г (0,11 моль) триэтиламина и 17 г (0,11 моль) сложного этилового эфира циклопропиламиноакриловой кислоты и затем нагревают до температуры 40°С в течение часа. Растворитель упаривают в вакууме, остаток поглощают в 100 мл хлористого метилена и экстрагируют 75 мл воды. Органическую фазу упаривают в вакууме, в результате чего получают 30 г маслянистого остатка. После добавления 100 мл N-метилпирролидона маслянистый остаток смешивают с 20 г (0,145 моль) карбоната калия и нагревают до 80°90°С в течение часа. Затем добавляют 45,6 г (0,4 моль) N-этилпиперазина. Смесь перемешивают в течение часа при температуре от 80° до 90°С. После охлаждения до комнатной температуры разбавляют 75 мл воды, добавляют 8 г едкого натра и смесь нагревают в течение часа до 70°80°С. Затем реакционную смесь разбавляют 200 мл воды, фильтруют и рН доводят до 7,5 с помощью полуконцентрированной соляной кислоты. Осадок отсасывают, промывают два раза по 100 мл воды и сушат в вакууме при 50°С. Выход: 18,4 г (51% от теории). 12 41323 __________________________________________________________ ДП "Український інститут промислової власності" (Укрпатент) Україна, 01133, Київ-133, бульв. Лесі Українки, 26 (044) 295-81-42, 295-61-97 __________________________________________________________ Підписано до друку ________ 2002 р. Формат 60х84 1/8. Обсяг ______ обл.-вид. арк. Тираж 50 прим. Зам._______ ____________________________________________________________ УкрІНТЕІ, 03680, Київ-39 МСП, вул. Горького, 180. (044) 268-25-22 ___________________________________________________________ 13
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparing derivatives of 3- quinolonecarboxylic acid
Автори англійськоюRudolf Cerbes, Paul Naab, Gerhard Frankoviak, Herbert Diel
Назва патенту російськоюСпособ получения производных 3-хинолонкарбоновой кислоты
Автори російськоюРудольф Цербес, Пауль Нааб, Герхард Франковиак, Херберт Диль
МПК / Мітки
МПК: C07D 498/04, C07D 471/04, C07D 215/56, C07D 401/04, C07D 519/00
Мітки: одержання, похідних, спосіб, 3-хінолонкарбонової, кислоти
Код посилання
<a href="https://ua.patents.su/13-41323-sposib-oderzhannya-pokhidnikh-3-khinolonkarbonovo-kisloti.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання похідних 3-хінолонкарбонової кислоти</a>
Спосіб отримання похідних 4-оксіхінолінкарбонової кислоти

Номер патенту: 4898
Опубліковано: 28.12.1994
Автори: Франсуа Клеменс, Оділь Ле Мартре, Франсуаз Дельвалле
МПК: C07D 317/00, C07D 491/04, C07D 265/22, C07D 277/46, C07D 417/14, A61P 13/02, A61P 15/00, C07D 215/56, A61P 29/00, A61K 31/47, C07D 417/12
Мітки: отримання, кислоти, похідних, 4-оксіхінолінкарбонової, спосіб
Формула / Реферат:
Способ получения производных 4-оксихинолинкарбоновой кислоты общей формулыгде Х- трифторметил в положении 8 или галоген в положении 7, R1 - триазолил;R2 и R3 - одинаковые или различные и означают водород, С1-С4-алкилили O=C R5, где R5-С1-С4-алкил или арил, или R2 и R3 образуют остаток ацетонида;R4 - водород или С1-С4-алкил, отличающийся тем, что соединение общей формулыгде Х и R4 имеют...
Спосіб одержання похідних хінолінкарбонової кислоти або її фармацевтично придатних солей та проміжні сполуки для їх одержання
Номер патенту: 26568
Опубліковано: 11.10.1999
Автори: Хермец Іштван, Балог Марія, Шіпош Юдіт, Хорват Агнеш, Вашварі Лелле, Пайор Аніко, Керестурі Геза, Рітлі Петер
МПК: C07D 215/56, A61P 31/04, A61K 31/495, C07D 401/04, C07F 5/00
Мітки: сполуки, придатних, хінолінкарбонової, кислоти, солей, похідних, проміжні, одержання, спосіб, фармацевтично
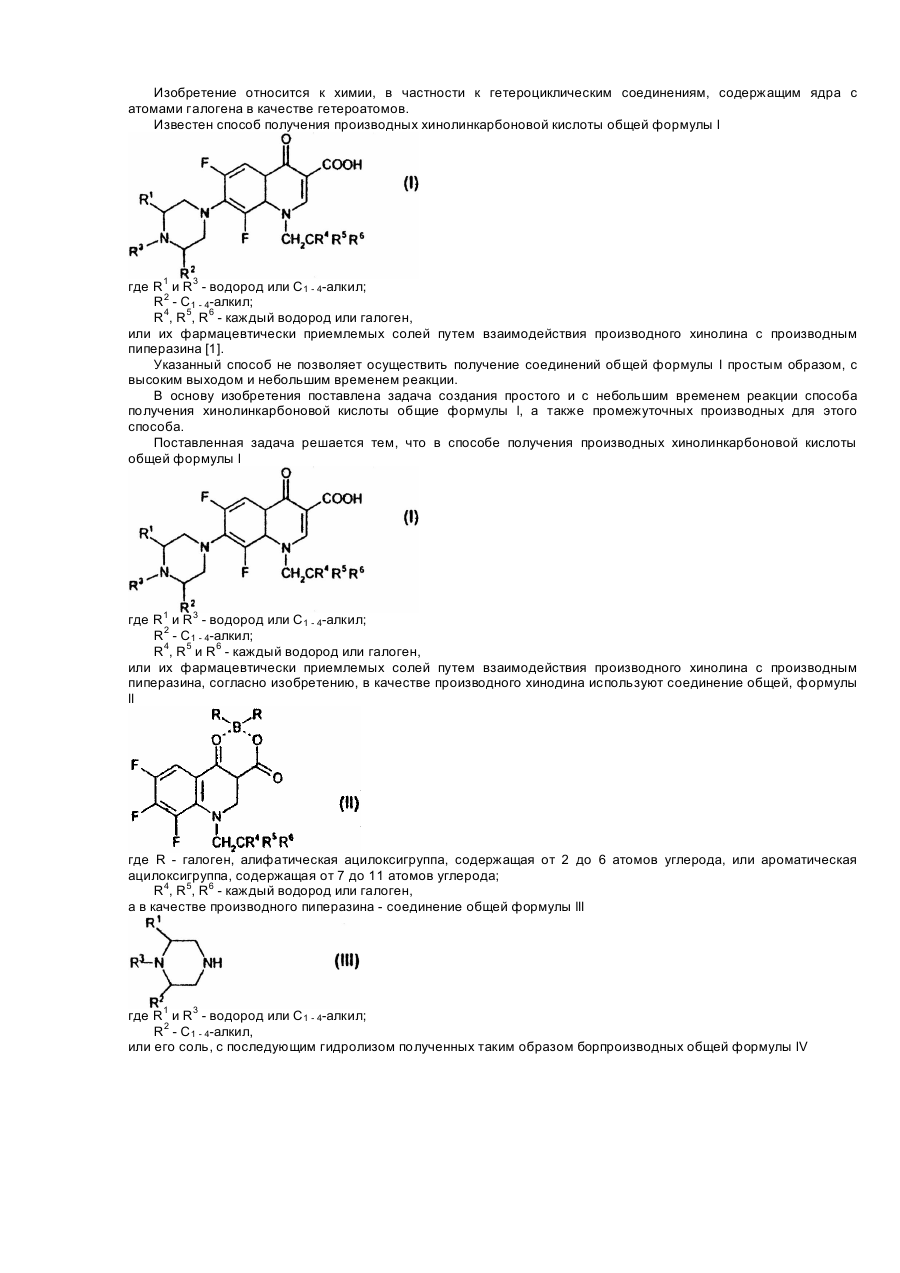
Формула / Реферат:
1. Способ получения производных хинолинкарбоновой кислоты общей формулы lгде R1 и R3 - водород или C1 - 4-алкил;R2 - C1 - 4-алкил;R4, R5 и R6 - каждый - водород или галоген,или ее фармацевтически приемлемых солей, взаимодействием производного хинолина с производным пиперазина, отличающийся тем, что в качестве производного хинолина используют соединение общей формулы llгде R - галоген,...
Спосіб одержання похідних хінолінкарбонової кислоти або її метансульфонатної солі

Номер патенту: 3538
Опубліковано: 27.12.1994
Автори: Гєза Керестурі, Марія Балог, Тамаш Сютш, Петер Рітлі, Леллє Вашварі, Габор Ковач, Аніке Пайор, Юдіт Шипош, Іштван Хермец, Агнеш Хорват
МПК: C07D 215/56, C07F 5/00, A61K 31/47, A61P 31/04
Мітки: хінолінкарбонової, похідних, спосіб, метансульфонатної, одержання, солі, кислоти
Формула / Реферат:
Способ получения производных хинолинкар-боновой кислоты общей формулы где R — водород или метил, или ее метансульфонатной соли путем взаимодействия борсодержащего эфира хинолинкарбоновой кислоты с пиперазином общей формулы где R — имеет указанные значения, в присутствии органического растворителя в щелочной среде с последующим гидролизом полученного при этом соединения, отличающийся тем, что, с целью...
Спосіб одержання похідних 5(6)-тіобензімідазолу
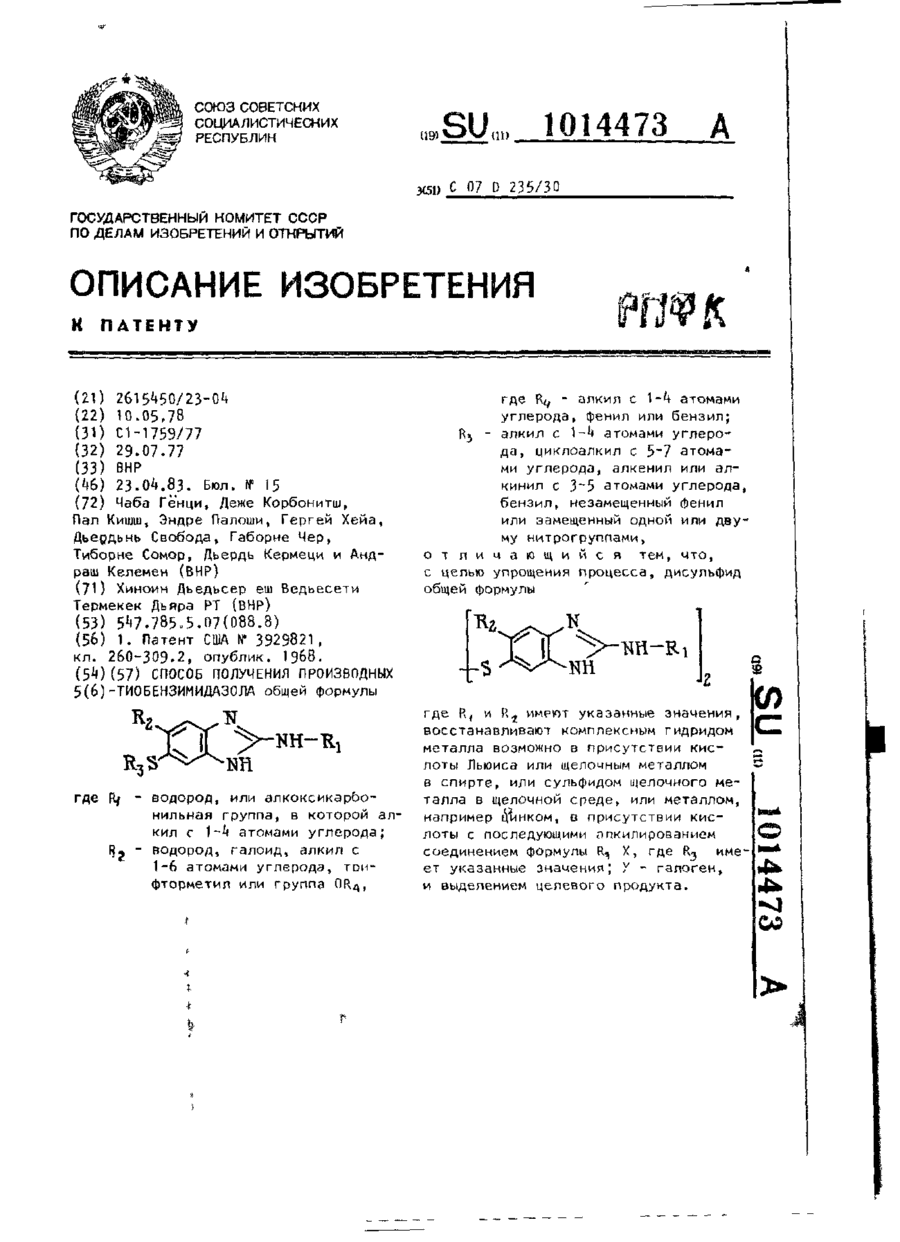
Номер патенту: 3535
Опубліковано: 27.12.1994
Автори: Деже Карбонітш, Тіборне Сомор, Андраш Келемен, Габорне Чер, Гергей Хейа, Пал Кішш, Дьордьнь Свобода, Дьордь Кермеці, Ендре Палоші, Чаба Генци
МПК: C07C 331/00, A61K 31/415, A61P 33/00, A01N 47/18, A61K 31/135, C07D 235/30, C07D 233/32, A01N 43/52, A61P 33/10, C07C 67/00, A61P 31/04, C07D 235/32, C07C 325/00, A61K 31/4184
Мітки: одержання, похідних, 5(6)-тіобензімідазолу, спосіб
Формула / Реферат:
Способ получения производных 5(6) -тиобензимидазола общей формулы где R1 - водород, или алкоксикарбонильная группа, в которой алкил с 1-4 атомами углерода; R2 - водород, галоид, алкил с 1-6 атомами углерода, трифторметил или группа ОR4, где R4 - алкил с 1-4 атомами углерода, фенил или бензил; R3 - алкил с 1-4 атомами углерода, циклоалкил с 5-7 атомами углерода, алкенил или алкинил с 3-5 атомами углерода, бензил,...
Спосіб одержання похідних 2-тієнілоксіуксусної кислоти чи їх фармацевтично прийнятних солей

Номер патенту: 4227
Опубліковано: 27.12.1994
Автори: Хуберт Петер Фербер, Франц Ровенсцкі, Дітер Біндер
МПК: A61P 43/00, A61P 9/00, A61K 31/381, A61P 7/02, A61P 11/00, C07D 333/32, A61K 31/38, A61P 29/00
Мітки: одержання, прийнятних, спосіб, кислоти, солей, 2-тієнілоксіуксусної, фармацевтично, похідних
Формула / Реферат:
1. Способ получения производных 2-тиенил-оксиуксусной кислоты общей формулы где R - незамещенный или замещенный галогеном фенил, или их фармацевтически применимых солей, отличающийся тем, что соединение общей формулыгде R имеет указанные значения, подвергают окислению окисью серебра в водно-щелочной среде с последующим выделением целевого продукта в свободном виде или в виде фармацевтически применимой если....
Наступний патент: Спосіб покращення росту рослин (варіанти), композиція, що покращує ріст рослин (варіанти)
Випадковий патент: Аерозольний генератор