Спосіб сульфоксидування біологічно активних сполук
Номер патенту: 98792
Опубліковано: 25.06.2012
Автори: Гурджар Мукунд Кешав, Майкап Голакчандра Сударшан, Раджпут Махеш Рамсінг, Мехта Сатіш Раманлал, Гхарпур Мілінд Морешвар, Махал Раджендра Дагесінг, Гаварі Прашант Садашив







Формула / Реферат
1. Спосіб одержання сполуки формули (І), в якому здійснюють стадії взаємодії сполуки формули (Іа) з оксазиридином формули (VII) в присутності розчинника і основи і подальшого виділення сполуки формули (І)
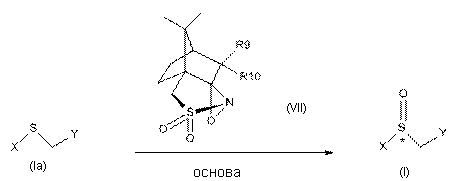 ,
,
 ,
,
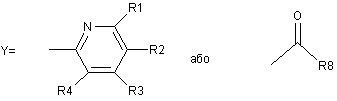 ,
,
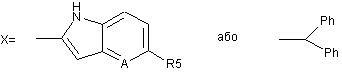
де R1=H; R2=H, CH3; R3=H, -OCH3, -OCH2CF3, -OCH2CH2OCH3, -O(CH2)3OCH3;
R4=H, CH3, -OCH3; і R5=H, -OCH3, -OCHF2; A=CH, N; R8=ОH, NH2, OR'9, NHR'9; і R'9=С1-4-алкіл; R9=R10=H, О, галоген, -О-С1-4-алкіл, -О-С=О, -О-(СН2)n-О-, де n=2 або 3;
і де розчинник не є іонним розчинником.
2. Спосіб за п. 1, в якому
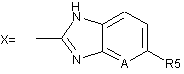 ,
,
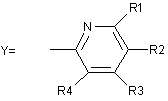 ,
,
де R1=H; R2 і R3=H, CH3, -OCH3, -OCH2CF3, ОСН2(СН2)ОСН3, -О(СН2)3ОСН3; R4=H, СН3; і R5=H, -ОСН3, -OCHF2; A=CH, N.
3. Спосіб за пунктом 2, в якому сполуку формули (І) вибирають з групи, що включає оптично активні пантопразол, лансопразол, рабепразол, тенатопразол, парипразол і омепразол.
4. Спосіб за п. 1, в якому
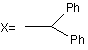 ,
,
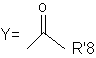 ,
,
де R'8=OH, NH2; OR'9, NHR'9; і R'9 =С1-4-алкіл.
5. Спосіб за п. 4, в якому сполука формули (І) являє собою армодафініл.
6. Спосіб за п. 1, в якому (2R,8aS)-кaмфopилcyльфoнiлoкcaзиpидин формули (XII) являє собою або правообертальний, або лівообертальний ізомер.
7. Спосіб за п. 1, в якому основа являє собою або органічну, або неорганічну основу.
8. Спосіб за п. 7, в якому основа переважно являє собою органічну основу.
9. Спосіб за п. 8, в якому органічну основу вибирають з групи, що включає 1,8-діазабіцикло[5.4.0]ундец-7-ен, діізопропілетиламін, гексаметилентетрамін і триетиламін.
10. Спосіб за п. 9, в якому органічна основа являє собою 1,8-діазабіцикло[5.4.0]ундец-7-ен.
11. Спосіб за п. 7, в якому неорганічна основа являє собою гідроксид лужного металу.
12. Спосіб за п. 11, в якому неорганічна основа являє собою гідроксид натрію або гідроксид калію.
13. Спосіб за п. 1, який здійснюють в присутності розчинника, вибраного з спирту, ароматичного вуглеводню, простого ефіру, складного ефіру, аміду, нітрилу, води або їх сумішей.
14. Спосіб за п. 13, в якому розчинник являє собою спирт або ароматичні вуглеводні.
15. Спосіб за п. 13, в якому розчинник вибирають з групи, що включає метанол, етанол, ізопропанол, бутанол, простий діізопропіловий ефір, толуол, воду, тетрагідрофуран, ацетонітрил, диметилформамід, діетилформамід, диметоксіетан або їх комбінації.
16. Спосіб за п. 14, в якому розчинник являє собою ізопропанол або толуол.
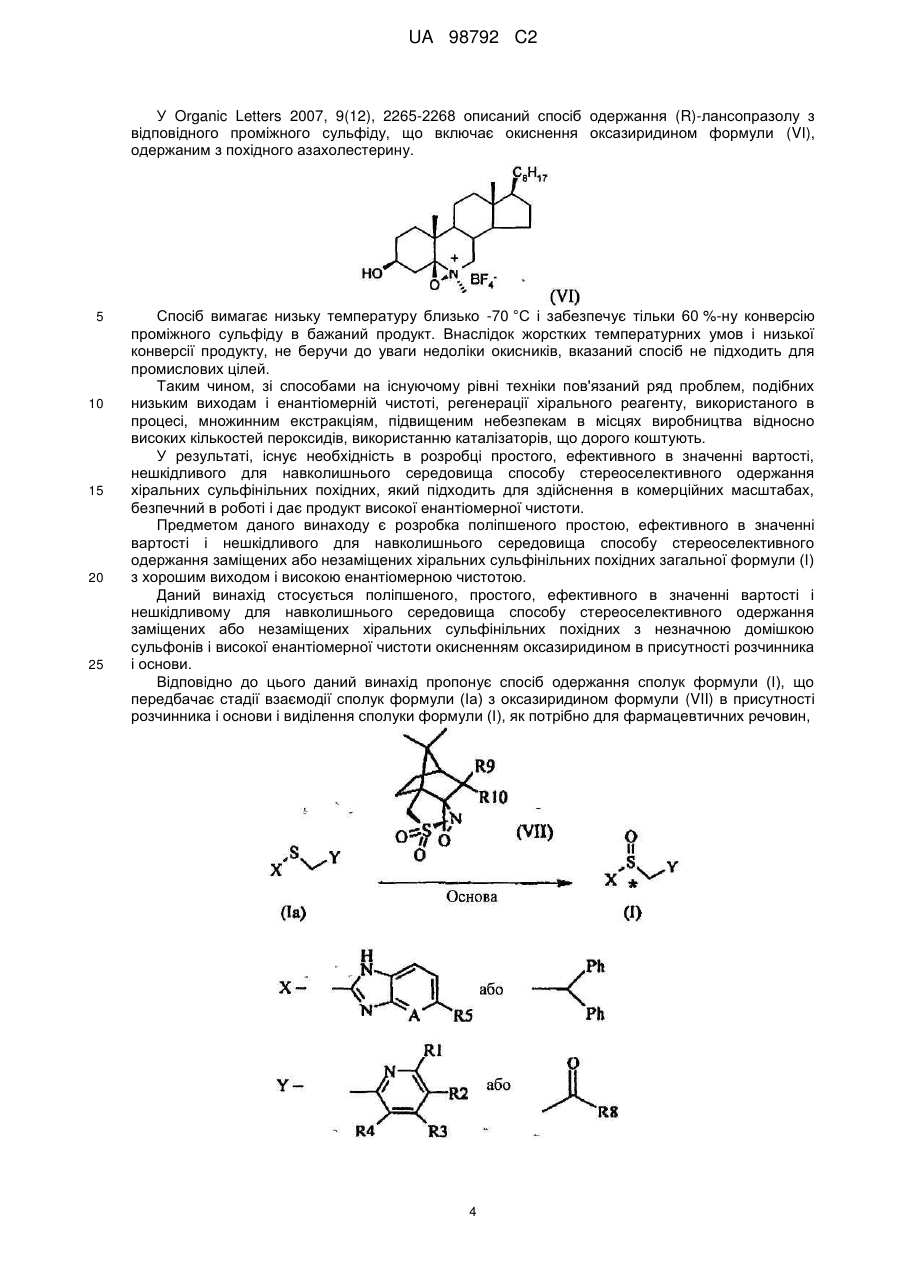
Текст
Реферат: Даний винахід стосується нового способу одержання сульфоксидів, переважно стереоселективного одержання заміщених або незаміщених хіральних сульфінільних похідних UA 98792 C2 (12) UA 98792 C2 2-(2-піридилметил)сульфініл-1Н-бензімідазолу відповідного розчинника і основи. окисненням оксазиридином в присутності UA 98792 C2 Даний винахід стосується поліпшеного способу одержання сульфоксидів і, більш конкретно, винахід передбачає стереоселективне одержання заміщених або незаміщених хіральних сульфінільних похідних загальної формули (І) окисненням оксазиридином в присутності відповідного розчинника і основи. 5 10 15 20 25 30 35 40 45 R1=H; R2 і R3=H, CH3, -OCH3, -OCH2CF3, -ОСН2СН2ОСН3, -О(СН2)3ОСН3 R4=H, СН3 і R5=H, ОСН3-OCHF2, R'8=OH, NH2, OR'9, NHR'9, і R'9=C1-4-алкіл; А=СН, N. Сульфоксиди мають асиметричний центр у атома сірки і тому існують у вигляді двох оптичних ізомерів, тобто енантіомерів. Бажано одержувати сполуки з поліпшеними фармакокінетичними і метаболічними властивостями, які дадуть поліпшений терапевтичний показник, такий як меншу міру міжіндивідуальних коливань. Сполуки загальної формули (І) звичайно одержують з відповідних проміжних сульфідів окисненням простої тіоефірної групи з використанням різних окисників. Вказані попередні способи звичайно супроводжуються утворенням відповідних похідних сульфонів як забруднюючих домішок. Більш того, в кінцевому продукті як забруднюючі домішки можуть також бути присутніми сульфіди, що не прореагували. Вельми схожі фізико-хімічні властивості супроводжуючих забруднюючих домішок ускладнюють їх відділення, тим самим роблячи продукт, що представляє інтерес, схильним до забруднень. Домішка сульфону утворюється під час конверсії сульфоксиду, роблячи завершення окиснення небезпечним. Домішка сульфону викликається головним чином надокисненням, яке, в свою чергу, пов'язано з окисниками. Таким чином, окиснення потрібно контролювати. У патенті США 5929244 запропонований спосіб одержання езомепразолу шляхом використання (3'S,2R)-(-)-N-(фенілсульфоніл)-(3,3-дихлоркамфорил)оксазиридину в присутності триетиламіну як основи і чотирьоххлористого вуглецю як розчинника. Оптична чистота становить 94 %, але одержаний вихід тільки 22 %, що унеможливлює спосіб для комерційного застосування. Він також пов'язаний з використанням чотирьоххлористого вуглецю як розчинника, що заборонено в промисловому масштабі. У патенті США 5948789 запропонований енантіоселективний синтез єдиного енантіомера 2(2-піридинілметилсульфініл)-1H-бензімідазолу, також як і інших структурно родинних сульфоксидів. Процес окиснення ведуть в присутності основи, окисника, хірального комплексу титану і розчинника. Однак вказаний спосіб страждає основним недоліком: високим вмістом домішки сульфону, що утворюється в процесі. Більше того, вказані способи володіють очевидними недоліками і вони вимагають (застосування) ізопропілату титану, який не може бути регенерований і небезпечний для навколишнього середовища. Крім того, реагент, використаний для хіральної індукції, являє собою діетилтартрат, який важко регенерувати внаслідок-за епімеризації і гідролізу під час обробки. Реагент, використаний для окиснення, являє собою гідропероксид кумола, який є вибуховим і небезпечним, не кажучи вже про його дорожнечу. У патенті США 6919459 зроблене побічне посилання на використання Nсульфонілоксазиридинів для окиснення сульфідів до сульфоксидів. Однак у вказаному патенті не доведений і не запропонований спосіб такого окиснення N-сульфонілоксазиридинами. У Journal of the Chemical Society, Perkin Transactions 1, 1988, 1753-7 описаний спосіб одержання хіральних сульфоксидів із застосуванням (+)-(3оксокамфорсульфоніл)оксазиридину. (При цьому) спостерігали енантіомерний надлишок максимум близько 60 %, що є обмеженням для промислових цілей. 1 UA 98792 C2 5 10 15 20 25 30 35 40 У Chemical Sciences (1990), 45(12), 1689-94 запропонований спосіб окиснення сульфідів до сульфоксидів камфорлактон-сульфонілоксазиридином, 3-ендобромкамфорсульфонілоксазиридином і т.д. Переважний розчинник являв собою канцерогенний розчинник, подібний до чотирьоххлористого вуглецю, оскільки він давав найбільший енантіомерний надлишок (до 85 %). Інше обмеження "стандартної концентрації" вимагає більш високого розведення для переважного розчинника, запропонованого в єдиному прикладі. Однак чотирьоххлористий вуглець не використовують в фармацевтичній промисловості внаслідок його канцерогенності і високої токсичності. Вказаний посередній енантіомерний надлишок можливий для субстратів, що мають значні відмінності з обох сторін сульфідної групи. Для субстратів, таких як бензилфенілсульфід, енантіомерний надлишок недопустимо бідний (36 %). Обмеження пов'язане з використанням чотирьоххлористого вуглецю. Однак використання оксазиридину не показане для таких субстратів, як фенілбензилсульфід. Необхідний час для можливої взаємодії може бути навіть 160 годин. При меншому часі взаємодії енантіомерний надлишок вельми бідний, що нижче за обмеження, застосовні в промисловості. У Journal of Organic Chemistry 1992, 57(26), 7274-7285 запропонований спосіб одержання [{8,8-дигалогенокамфорил)сульфоніл]оксазиридинів і спосіб окиснення сульфідів до сульфоксидів з використанням дейтерованого хлороформу. Використання дейтерованих розчинників в промисловому масштабі недоступно дорого коштує. Таким чином, вказане посилання пропонує використання як розчинника чотирьоххлористого вуглецю, який промислово недопустимий, як вказано раніше. Більше того, енантіомерний надлишок одержаного бажаного ізомеру знаходиться в інтервалі 1-75 %, що дуже мало для одержання оптично чистої фармацевтичної сполуки. Спроба одержання вказаного енантіомерного надлишку також вимагає для звичайного розчинника точної і недоступної температури взаємодії -78 °C. Інший спосіб проведення енантіоселективного синтезу передбачає використання хіральних оксазиридинів (III) (Journal of American Chemical Society 1992, 114, 1428-1437). При використанні хіральних оксазиридинів енантіомерний надлишок є досяжним для прохіральних сульфідів, наприклад, якщо прохіральний сульфід має вельми об'ємний замісник, такий як 9-антрил, і інший (замісник), такий як метил. Взаємодія також залежить від розчинника і дає більший енантіомерний надлишок в ССl4, який взагалі не сприятливий для навколишнього середовища. Хіральні оксирани, використані для окиснення, являють собою Енантіомерний надлишок більше, якщо X являє собою галоген або водень. Таким чином, використання хіральних оксиранів для одержання енантіомерно чистих PPIs невідоме на сучасному рівні техніки і в той же час обмежено внаслідок: а) розміру груп прохірального сульфіду, b) заміщення хіральних оксазиридинів, с) розчинника і т.д. У Tetrahedron Asymmetry 1995, 6(12), 2911-2914 запропонований спосіб окиснення сульфідів до сульфоксидів із застосуванням [(3,3-диметоксикамфорил)сульфоніл]оксазиридину (IV) в 2 UA 98792 C2 присутності пероксиду водню. Використання вказаного оксазиридину дає сульфоксиди з неароматичних сульфідів з доброю енантіоселективністю. 5 10 15 20 25 30 35 40 Недоліком вказаного способу є той факт, що якщо окиснюють арилсульфіди, енантіомерний надлишок бажаного ізомеру становить тільки 61 %, тому вказаний спосіб промислово нездійсненний для окиснення фармацевтично активних інгредієнтів, що містять ароматичні замісники. У Indian Journal of Chemistry 2001,40В, 1132-1133 вказане (схема II), що алкілалкілтіометилсульфіди і арил-арилтіометилсульфіди можуть бути окиснені- до відповідних сульфоксидів 10-камфорсульфонілоксазиридином з виходом 70-80 % і енантіоселективністю 90-95 %. Щоб одержати такого роду продукт необхідної міри чистоти, використовують препаративну тонкошарову хроматографію. Однак в промисловому масштабі таке очищення неможливе. Спосіб стосується окиснення сульфідної групи, з одного боку якого знаходиться арильна або алкільна група, а з іншого боку алкільна група. Не існує прикладу окиснення, де у сульфидної групи з іншого боку знаходиться об'ємна група, оскільки об'ємна група прагне змінити енантіомерну селективність і існує імовірність, що буде одержана низька енантіомерна селективність. У Tetrahedron Asymmetry 2003, 14, 407-410 запропонований спосіб одержання (R)лансопразола із застосуванням гетерогенної каталітичної системи WO3, 30 %-ний Н2О2 і хінних алкалоїдів. Промислове використання вказаної системи вельми обмежене внаслідок застосування реагентів, що дорого коштують, таких як WO3 і хінні алкалоїди. Додаткове обмеження викликане також небезпеками (використання) Н2О2. Ternois James і інш. (Tetrahedron Asymmetry 18, 2007, 2959-2964) пропонують спосіб енантіоселективного окиснення сульфідної групи до сульфоксидної з використанням оксазиридину формули (V), для одержання деяких фармацевтичних сполук. Однак, як вказано там, з вказаним способом пов'язано декілька недоліків. У способі використовують чотирьоххлористий вуглець або іонні рідини і час взаємодії складає близько 48 годин, що зменшує ефективність процесу. Застосування чотирьоххлористого вуглецю має декілька шкідливих впливів на здоров'я, (оскільки) чотирьоххлористий вуглець являє собою канцерогенний розчинник, агент, що виснажує озон. Тривала дія чотирьоххлористого вуглецю може впливати на центральну нервову систему, викликати пошкодження печінки і нирок і в результаті привести до раку. Далі, використання іонної рідини, подібної до гексафторфосфату 1-бутил-3-метилімідазолію, яка відома своєї нелеткістю, створює серйозні проблеми під час сушіння фармацевтично активних інгредієнтів. Вказані іонні рідини можуть бути використані як розчинник для їх одержання внаслідок їх нелеткості; крім того, вказані іонні рідини вимагають застосування ультразвукового пристрою для розщеплення розчинів іонних рідин на базі імідазолію з пероксидом водню і оцтовою кислотою до відносно нешкідливих сполук. Більше того, енантіомерний надлишок, одержаний при використанні оксазиридинового реагенту, складає до 78 %, що є недостатнім. 3 UA 98792 C2 У Organic Letters 2007, 9(12), 2265-2268 описаний спосіб одержання (R)-лансопразолу з відповідного проміжного сульфіду, що включає окиснення оксазиридином формули (VI), одержаним з похідного азахолестерину. 5 10 15 20 25 Спосіб вимагає низьку температуру близько -70 °C і забезпечує тільки 60 %-ну конверсію проміжного сульфіду в бажаний продукт. Внаслідок жорстких температурних умов і низької конверсії продукту, не беручи до уваги недоліки окисників, вказаний спосіб не підходить для промислових цілей. Таким чином, зі способами на існуючому рівні техніки пов'язаний ряд проблем, подібних низьким виходам і енантіомерній чистоті, регенерації хірального реагенту, використаного в процесі, множинним екстракціям, підвищеним небезпекам в місцях виробництва відносно високих кількостей пероксидів, використанню каталізаторів, що дорого коштують. У результаті, існує необхідність в розробці простого, ефективного в значенні вартості, нешкідливого для навколишнього середовища способу стереоселективного одержання хіральних сульфінільних похідних, який підходить для здійснення в комерційних масштабах, безпечний в роботі і дає продукт високої енантіомерної чистоти. Предметом даного винаходу є розробка поліпшеного простою, ефективного в значенні вартості і нешкідливого для навколишнього середовища способу стереоселективного одержання заміщених або незаміщених хіральних сульфінільних похідних загальної формули (І) з хорошим виходом і високою енантіомерною чистотою. Даний винахід стосується поліпшеного, простого, ефективного в значенні вартості і нешкідливому для навколишнього середовища способу стереоселективного одержання заміщених або незаміщених хіральних сульфінільних похідних з незначною домішкою сульфонів і високої енантіомерної чистоти окисненням оксазиридином в присутності розчинника і основи. Відповідно до цього даний винахід пропонує спосіб одержання сполук формули (І), що передбачає стадії взаємодії сполук формули (Іа) з оксазиридином формули (VII) в присутності розчинника і основи і виділення сполуки формули (І), як потрібно для фармацевтичних речовин, 4 UA 98792 C2 5 10 15 20 25 30 R1=H; R2=H, CH3; R3=H, -OCH3, OCH2CF3, -ОСН2СН2ОСН3, -О-(СН2)3- ОСН3 R4=H, CH3, OCH3 і R5=H, -OCH3, -OCHF2; A=CH, N; R8=OH, NH2, OR'9, NHR'9 і R'9=C1-4алкіл; R9=R10=H, О, галоген, -О-С1-4алкіл, -O-C=O, -O-(CH2)n-O-j де n=2 або 3. Спосіб, як розглянуто вище, спричиняє асиметричне окиснення прохірального сульфіду для одержання енантіомерно збагаченої форми відповідного сульфоксиду. Атом сірки сульфіду не має асиметрії, тоді як внаслідок стереоселективного окиснення сульфоксид, що утворився, являє собою хіральну сполуку. Шляхом вказаного стереоселективного окиснення один з енантіомерів може бути одержаний в енантіомерному надлишку в порівнянні з іншим. Для цілей даного винаходу, якщо де R1=H; R2 і R3=H, CH3, -OCH3, -OCH2CF3, -ОСН2СН2ОСН3, -О(СН2)3ОСН3; R4=H, CH3 і R5=H, -ОСН3, -OCHF2, А=СН, N, то сполука формули (І) може бути вибрана з групи, що включає оптично активні празоли, такі як пантопразол, лансопразол, рабепразол, тенатопразол, парипразол і омепразол, або сполуку, запропоновану в патенті США 5776765. Для цілей даного винаходу, якщо де R'8=OH, NH2, OR'9, NHR'9 і R'9=C1-4алкіл, то сполука формули (І) може являти собою армодафініл, як запропоновано в патенті США 4927855. Модафініл має стереогенний центр біля атома сірки і, таким чином, існує у вигляді двох оптичних ізомерів, тобто енантіомерів. Модафініл, хімічно відомий як 2[(дифенілметил)сульфініл]ацетамід, тобто 2-(бензгідрилсульфініл)ацетамід (В), і який може бути одержаний з відповідного сульфіду (А), являє собою евгероїдний (eugeroic) лікарський препарат, звичайно призначений для лікування нарколепсії. R-енантіомер модафінілу, який є переважним енантіомером, відомий як армодафініл і має хімічну назву 2-[(R)-(дифенілметил)сульфініл]ацетамід. Таким чином, спосіб винаходу може бути використаний для одержання будь-якого рацемічного сульфоксиду, приклади яких описані вище. Для одержання рацемічних сульфоксидів можуть бути використані рацемічні оксазиридини. Використаний окисник являє собою сполуку формули (VIII), 5 UA 98792 C2 5 10 15 20 25 30 35 40 в якій один або більше з R6, R7 і R8 являють собою хіральні частини, що мають хіральні центри. Переважно він утворює циклічну систему. Переважно окисник, використаний в даному винаході, являє собою правообертальний або лівообертальний ізомер (2R, 8аS)-10(камфорилсульфоніл)оксазиридину, як зображено в формулі (VII). (+)-енантіомер, тобто (+)-(2R, 8aS)-10-(камфорсульфоніл)оксазиридин використовують для одержання відповідного енантіомеру сульфоксиду. (-)-Ізомер вказаної сполуки може також бути використаний для одержання енантіомерно збагаченої сполуки. i)R9=R10=H; іі) R9=R10=X=Cl, Br, І; ііі) R9=R10=-O-C1-4алкіл; iv) R9=R10=-О-(СН2)n-О-, де n = 2,3; v) R9=R10= кетону. Використаний згідно з способом хіральний окисник являє собою хіральний оксазиридин, який може бути одержаний без застосування комплексів металів, таких як тетраізопропілат титану. Тетраізопропілат титану приводить до величезного навантаження на промивання обладнання і тому несприятливий для навколишнього середовища. Реакцію даного винаходу проводять в присутності органічної або неорганічної основи. Неорганічну основу вибирають з групи, що включає гідроксиди, алкоголяти і бікарбонати, карбонати лужних або лужноземельних металів і переважні неорганічні основи являють собою гідроксид натрію або гідроксид калію. Однак переважною основою є органічна основа. Використану органічну основу вибирають з групи, що включає 1,8-діазабіцикло[5.4.0]ундец7-ен, діізопропілетиламін, гексаметилентетраамін, триетиламін і подібні. Переважно основою, використаною для окиснення, є 1,8-діазабіцикло[5.4.0]ундец-7-ен. Розчинник, використаний для даного винаходу, вибирають з групи органічних розчинників, що включає спирти, прості ефіри, складні ефіри, аміди, нітрили, ароматичні вуглеводні, воду і т.д., або їх комбінацій. Переважні розчинники вибирають з групи, що включає метанол, етанол, ізопропанол, бутанол, простий діізопропіловий ефір, толуол, воду, тетрагідрофуран, ацетонітрил, диметилформамід, діетилформамід, диметоксіетан або їх комбінації і т.д. Розчинник не охоплює іонних розчинників. Реакцію утворення сульфоксиду проводять при кімнатній температурі. Винахід ілюстрований наступними прикладами, які служать тільки для ілюстрації винаходу і не обмежують його. Всі варіанти здійснення, очевидні для способу на сучасному рівні техніки, вважаються такими, що потрапляють в рамки даного винаходу. Приклад 1 У колбі місткістю 250 мл 10 г рабепразолу суспендували в 70 мл ізопропілового спирту. (До суспензії) додавали 4,4 г 1,8-діазабіцикло[5.4.0]ундец-7-ену і охолоджували до від 10 до 15 °C. Потім додавали 6,6 г (+)-(2R, 8аS)-10-(камфорилсульфоніл)оксазиридину і перемішували до закінчення взаємодії сульфіду (близько 20 год.) при від 25 до 30 °C. Реакційну суміш фільтрували і тверду речовину промивали ізопропіловим спиртом, одержуючи 5,1 г (-)(камфорсульфоніл)іміну (витягання 78 %). До фільтрату додавали 1,1 г гідроксиду натрію і реакційну масу концентрували у вакуумі до утворення в'язкого масла. Потім додавали воду до одержання прозорого розчину. До водного 6 UA 98792 C2 5 10 15 20 25 30 35 40 45 50 55 60 шару додавали етилацетат (50 мл) і розбавленою оцтовою кислотою доводили рН до 10. Органічний шар відділяли і промивали 25 мл води. Органічний шар концентрували до одержання в'язкого масла. Потім додавали толуол (100 мл) до одержання прозорого розчину. Додавали розчин 1,2 г гідроксиду натрію в 2 мл води і перемішували протягом 15 хв. Тверду речовину відфільтровували і промивали 25 мл толуолу, одержуючи білу тверду речовину. Результат: Вихід: 8 г Хімічна чистота, включаючи R і S-ізомери: 97,67 % R-ізомер: 89,35 % S-ізомер: 10,65 %. Енантіомерна чистота будь-якого індивідуального ізомеру, наприклад, Rізомеру, може бути додатково збільшена перетворенням R-рабепразолу в R-рабепразол-натрій і/або розчиненням R-рабепразол-натрію у воді і встановленням рН оцтовою кислотою. Очищення R-ізомера: 8 г R-рабепразол-натрію, одержаного вище, розчиняли в 20 мл води і при (температурі) від 10 до 15 °C доводили рН до 9,5, використовуючи оцтову кислоту. Тверду речовину відфільтровували і промивали 10 мл води. Результат: R-ізомер: 95,09 % S-ізомер: 4,91 % Хімічна чистота, включаючи R- і S-ізомери: 99,22 %. Приклад 2 (армодафініл) У колбу місткістю 250 мл вміщували 10 г бензгідрилтіооцтової кислоти в 100 мл толуолу. Реакційну масу охолоджували до 0-5 °C. Потім до неї додавали5,9 г 1,8діазабіцикло[5.4.0]ундец-7-ену і охолоджували до 0-5 °C. Реакційну масу перемішували протягом 30 хвилин. Після цього додавали 8,8 г (+)-(2R, 8aS)-10(камфорилсульфоніл)оксазиридину і перемішували при (температурі) від 25 до 30 °C до повного завершення взаємодії. Реакційну суміш фільтрували і тверду речовину промивали водою, одержуючи 8 г хірально чистої армодафінової кислоти. Вихід: 75,25 % Оптична чистота: R-ізомер: 82,17 % S-ізомер: 17,83 % Приклад 3 До ізопропілового спирту (7 мл) в колбі місткістю 100 мл додавали рабепразолу сульфід (1г, 0,0029 моля). Колбу охолоджували до (температури) від 15 до 20 °C. Потім додавали 1,8діазабіцикло[5.4.0]ундец-7-ен (0,44 г, 0,0029 моля) і перемішували протягом 10 хв. Після цього реакційну масу охолоджували до (температури) від 10 до 15 °C і при від 10 до 15 °C завантажували простий діізопропіловий ефір (3 мл) разом з (+)-(2R, 8aS)-)(10(камфорилсульфоніл)оксазиридином (0,66 г, 0,0029 моля). Реакційну масу перемішували (при вказаній температурі) протягом від 10 до 15 хвилин і потім додатково перемішували при від 25 до 30 °C. Хід реакції контролювали за допомогою ВЕРХ до завершення взаємодії. Хімічна чистота: 96,20 % R-ізомер: 91,34 %. S-ізомер: 8,66 %. Приклад 4 До води (7 мл) в колбі додавали рабепразолу сульфід (1г, 0,0029 моля) і охолоджували до від 15 до 20 °C. Потім додавали 1,8-діазабіцикло[5.4.0]ундец-7-ен (0,44 г, 0,0029 моля) і перемішували протягом 5 хв. Після цього реакційну масу охолоджували до від 10 до 15 °C і додавали (+)-(2R, 8aS)-10-(камфорилсульфоніл)оксазиридин (0,66 г, 0,0029 моля). Реакційну масу протягом від 10 до 15 хвилин перемішували (при вказаній температурі) протягом 30 хвилин і потім додатково перемішували при (температурі) від 25 до 30 °C до завершення взаємодії, що було проконтрольовано за допомогою ВЕРХ. Хімічна чистота: 62,54 % R-ізомер: 70,45 %. S-ізомер: 29,54 %. Приклад 5 До диметилформаміду (7 мл) в колбі додавали рабепразолу сульфід (1г, 0,0029 моля) і охолоджували до (температури) від 15 до 20 °C. Потім додавали 1,8-діазабіцикло[5.4.0]ундец-7ен (0,44 г, 0,0029 моля) і перемішували протягом 5 хв. Після цього реакційну масу охолоджували до (температури) від 10 до 15 °C і додавали (+)-(2R, 8аS)-10 7 UA 98792 C2 5 10 15 20 25 30 35 40 45 50 55 60 (камфорилсульфоніл)оксазиридин (0,66 г, 0,0029 моля) при 10 °C. Реакційну масу протягом 30 хвилин перемішували при від 10 до 15 °C і потім при від 25 до 30 °C до завершення взаємодії, що було проконтрольовано за допомогою ВЕРХ. Хімічна чистота: 91,92 % R-ізомер: 83,04 %. S-ізомер: 16,96 %. Приклад 6 До дихлорметану (7 мл) в колбі додавали пантопразолу сульфід (1г, 0,0027 моля) і охолоджували до (температури) від 15 до 20 °C. Потім додавали 1,8-діазабіцикло[5.4.0]ундец-7ен (0,41 г, 0,0027 моля) і перемішували протягом 10 хв. Після цього реакційну суміш охолоджували до від 10 до 15 °C і додавали (+)-(2К, 8а8)-10-(камфорилсульфоніл)оксазиридин (0,63 г, 0,0027 моля). Реакційну суміш протягом 30 хвилин перемішували при (температурі) від 10 до 15 °C і далі додатково перемішували при від 25 до 30 °C. Реакційну суміш контролювали за допомогою ВЕРХ до завершення взаємодії, а потім гасили водним гідроксидом натрію і екстрагували MDC. Органічний шар відділяли і концентрували, одержуючи продукт. Хімічна чистота: 83,75 % R-ізомер: 80,30 %. S-ізомер: 19,70 %. Приклад 7 (S-пантопразол: діізопропілетішамін як основа і метанол як розчинник) Пантопразолу сульфід (1г, 0,0027 моля) додавали до метанолу (7 мл) в колбі місткістю 100 мл і охолоджували до від 15 до 20 °C. Потім додавали діізопропілетиламін (0,35 г, 0,0027 моля) і перемішували протягом 10 хвилин. Після цього реакційну суміш охолоджували до від 10 до 15 °C і додавали (+)-(2R, 8аS)-10-(камфорилсульфоніл)оксазиридин (0,63 г, 0,0027 моля). Реакційну суміш перемішували при від 25 до 30 °C до завершення взаємодії за даними ТШХ. Потім реакційну суміш гасили розбавленим гідроксидом натрію і екстрагували дихлорметаном. Органічний шар відділяли і концентрували. Хімічна чистота: 98,24 % R-ізомер: 74,67 %. S-ізомер: 25,33 %. Приклад 8 (пантопразол, використовуючи неорганічну основу в органічному розчиннику) Пантопразолу сульфід (1 г, 0,0027 моля) додавали до метанолу (4 мл) в колбі місткістю 100 мл. Потім колбу охолоджували до від 15 до 20 °C, додавали розчин гідроксиду натрію (0,108 г, 0,0027 моля), перемішували при від 10 до 15 °C і завантажували (+)-(2R, 8аS)-10камфорилсульфонілоксазиридин (0,63 г, 0,0027 моля). Реакційну суміш перемішували при від 25 до 30 °C до завершення взаємодії за даними ТШХ і ВЕРХ. Потім до реакційної суміші додавали дихлорметан (5 мл) і відділяли органічний шар, який потім концентрували, одержуючи продукт. Хімічна чистота: 98,36 %, R-ізомер: 69,58 %. S-ізомер: 30,53 %. Приклад 9 (інший молярний еквівалент (+)-(2R, 8aS)-10-(камфорилсульфонілоксазиридину) Рабепразолу сульфід (40 г, 0,116 моля) додавали до ізопропілового спирту (7 мл) в колбі місткістю 100 мл і охолоджували до (температури) від 15 до 20 °C. Потім додавали 1,8діазабіцикло[5.4.0]ундец-7-ен (17,9 г, 0,116 моля) і перемішували при від 10 до 15 °C. Завантажували (+)-(2R, 8aS)-10-камфорилсульфонілоксазиридин (25,3 г, 0,110 моля). Реакційну масу перемішували при від 25 до 30 °C до завершення взаємодії. Реакційну масу фільтрували і фільтрат концентрували при зниженому тиску. До залишку додавали воду (200 мл) і тверду речовину відділяли фільтруванням. Фільтрат при перемішуванні обробляли водним розчином гідроксиду натрію (12 г в 15 мл води). Потім при від 15 до 20 °C оцтовою кислотою (25 %-ою) встановлювали рН 11. До суміші додавали етилацетат і доводили рН до 9,5, використовуючи оцтову кислоту (25 %-ну). Органічний шар відділяли і частково концентрували при зниженому тиску, а залишок розбавляли толуолом (120 мл). Одержану суміш при від 10 до 15 °C перемішували з розчином гідроксиду натрію (4,65 г в 5 мл води) і тверду речовину відділяли фільтруванням, знов розчиняли у воді (200 мл) і промивали дихлорметаном (160 мл). Об'єднаний органічний шар екстрагували розчином гідроксиду натрію (2,5 г в 80 мл води). Водний шар перемішували у вакуумі і доводили рН до 9,5 оцтовою кислотою. Додавали етилацетат і розчин перемішували протягом 30 хв. Одержану тверду речовину відфільтровували і двічі промивали водою (по 40 мл). Вологу тверду речовину вміщували в колбу місткістю 500 мл з 200 мл води і колбу охолоджували до (температури) від 10 до 15 °C. Потім при від 10 до 15 °C в масу, що перемішується, завантажували розчин гідроксиду натрію 8 UA 98792 C2 5 10 15 20 25 30 35 40 45 50 55 60 (4,65 г в 5 мл води). Оцтовою кислотою доводили рН до 9,5. Реакційну масу витримували протягом 30 хв. при від 10 до 15 °C, одержуючи в процесі витримування тверду речовину. Тверду речовину відфільтровували і двічі промивали водою (по 200 мл). Після сушіння і хірального очищення одержували білий продукт, визначений як R-ізомер 98,9 % ее. Хімічна чистота: 99,60 %. R-ізомер: 99,00 %. S-ізомер: 1,0 %. Приклад 10 (взаємодія будь-якого празолу в будь-якому розчиннику без неорганічної або органічної основи в будь-якому розчиннику) Рабепразолу сульфід (1г, 0,0029 моля) додавали до ізопропілового спирту (7 мл) в колбі місткістю 100 мл і охолоджували до від 10 до 15 °C. Додавали (+)-(2R, 8аS)-10камфорилсульфонілоксазиридин (0,66 г, 0,0029 моля) і реакційну суміш перемішували при від 25 до 30 °C до завершення взаємодії, як показано ТШХ і ВЕРХ. До реакційної суміші додавали розчин гідроксиду натрію і потім дихлорметан (5 мл). Органічний шар відділяли і концентрували, одержуючи продукт. Хімічна чистота: 28,47 % R-ізомер: 56,14 %. S-ізомер: 43,86 %. Приклад 11 (тенатопразол) Тенатопразолу сульфід (415 г, 1,25 моля) додавали до ізопропілового спирту (4980 мл) в колбі. Потім в колбу додавали 1,8-діазабіцикло[5.4.0]ундец-7-ен (191,3 г, 1,25 моля) і охолоджували до від 10 до 15 °C. Далі в колбу додавали 1(R)-(-)(камфорилсульфоніл)оксазиридин (303 г, 1,31 моля) і перемішували при від 25 до 30 °C до завершення взаємодії за даними ТШХ і ВЕРХ. Реакційну масу фільтрували і фільтрат концентрували при зниженому тиску. До залишку додавали воду (2075 мл) і тверду речовину відділяли фільтруванням. Фільтрат охолоджували до від 15 до 20 °C і оцтовою кислотою доводили рН до 7,5. Реакційну масу перемішували при від 15 до 20 °C і тверду речовину, що випала відфільтровували. Вологий коржик суспендували у воді (3735 мл). У колбу додавали розчин гідроксиду натрію (55,2 г в 415 мл води) і перемішували протягом 30 хвилин. Одержану тверду речовину відфільтровували; фільтрат екстрагували дихлорметаном (830 мл). Водний шар відділяли і доводили рН до 7,45 50 %-ним розчином оцтової кислоти. Водний шар відділяли і екстрагували дихлорметаном (830 мл). Органічний шар відділяли, частково концентрували і розбавляли етилацетатом (2905 мл). Суміш частково концентрували при зниженому тиску і охолоджували до від 25 до 30 °C. Тверду речовину, що випала, відфільтровували, промивали етилацетатом (1660 мл) і сушили. Вихід: 81,15 % Хімічна чистота: 99,38 % S-ізомер: 88,81 %. Приклад 12А (взаємодія для одержання езомепразолу) Омепразолу сульфід (50 г; 0,152 моля) додавали до ізопропілового спирту (350 мл). До суміші при 10-15 °C додавали 1,8-діазабіцикло[5.4.0]ундец-7-ен (23,1 г; 0,152 моля). Потім до суміші додавали 1(R)-(-)-(камфорилсульфоніл)оксазиридин (34,8 г; 0,151 моля) і залишали перемішуватися протягом 20 годин до завершення взаємодії за даними ТШХ. Реакційну (суміш) фільтрували і фільтрат концентрували при зниженому тиску. До залишку додавали воду (250 мл) і доводили рН приблизно до 8,5 оцтовою кислотою. Далі додавали етилацетат, органічний шар відділяли і концентрували у вакуумі. До суміші додавали ацетон (150 мл) і фільтрували. Одержану суміш концентрували і залишок розбавляли толуолом (8 мл) і метанолом (75 мл). До суміші додавали метилат натрію (8,2 г) і перемішували протягом 14 годин. Реакційну суміш фільтрували і фільтрат концентрували при зниженому тиску. До залишку додавали простий діізопропіловий ефір (150 мл) і реакційну суміш відфільтровували, одержуючи езомепразол-натрій. Вихід: 23,4 г. Вихід в %: 46,8 %. Хіральна чистота натрієвої солі (S)-езомепразолу: 99,64 %. Приклад 12В: одержання езопразол-магнію Езопразол-натрій (5,0 г; 0,1636 моля) розчиняли у воді (30 мл) і при кімнатній температурі по краплях додавали до розчину хлориду магнію (0,54 г; 0,0068 моля) у воді (30 мл). Одержану суміш перемішували протягом 1 години і фільтрували. Вологий коржик промивали водою (30 мл) і сушили. Вихід: 4,87 г. Вихід в %: 96 %. (S)- омепразол: 99,64 % (Я)-омепразол: 0,36 %. Приклад 13 (лансопразол; DBU як основа) 9 UA 98792 C2 5 10 15 20 25 30 35 40 45 Лансопразолу сульфід (1,0 г; 0,028 моля) при перемішуванні додавали до ізопропілового спирту (7 мл) при 25-30 °C. Потім додавали 1,8-діазабіцикло[5.4.0]ундец-7-ен (0,43 г, 0,0028 моля) і перемішували при 10-15 °C. Після цього додавали (-)-дихлороксазиридин [(-)-(8,8дихлоркамфорилсульфоніл)оксазиридин] (0,8 г; 0,027 моля) і реакційну суміш перемішували при 25-30 °C до завершення взаємодії. Потім реакцію гасили розбавленим розчином гідроксиду натрію і екстрагували дихлорметаном. Органічний шар концентрували, одержуючи продукт. Хімічна чистота: 92,43 % (S)- ізомер: 90,29 % (R)- ізомер: 9,71 % Приклад 14 (дихлороксазиридин без основи) Лансопразолу сульфід (1,0 г; 0,0028 моля) при перемішуванні додавали до ізопропілового спирту (7 мл) при 25-30 °C і охолоджували до 10-15 °C. Потім додавали [(-)-(8,8дихлоркамфорилсульфоніл)оксазиридин (0,8 г; 0,0027 моля) і реакційну суміш перемішували при 25-30 °C протягом 15 годин. Реакцію гасили розбавленим розчином гідроксиду натрію і екстрагували дихлорметаном. Органічний шар концентрували, одержуючи продукт. Хімічна чистота: 6,85 % (S)- ізомер: 68,29 % (R)-ізомер: 31,71 % Приклад 15 (S-пантопразолу сульфід) Пантопразолу сульфід (400 г; 1,089 моля) додавали до ізопропілового спирту (3600 мл). До суміші додавали 1,8-діазабіцикло[5.4.0]ундец-7-ен (164 г) і охолоджували до 10-15 °C. Потім додавали (R)-(-)-(камфорилсульфоніл)оксазиридин (259,70 г; 1,133 моля), підвищували температуру до 25-30 °C і перемішували до завершення взаємодії за даними ВЕРХ. Реакційну суміш відфільтровували і фільтрат частково концентрували при зниженому тиску. Після цього додавали воду (2000 мл) і залишок фільтрували. Оцтовою кислотою доводили рН фільтрату до 9,5 і розбавляли етилацетатом (2000 мл). Далі знов доводили оцтовою кислотою рН приблизно до 7,5 і відділяли органічний шар, який потім частково концентрували і розбавляли циклогексаном. Після цього суміш охолоджували до 10-15 °C і продукт відділяли фільтруванням. Вологий коржик додавали до етилацетату (2800 мл), нагрівали до 70 °C і частково концентрували при зниженому тиску. Залишок розбавляли циклогексаном (400 мл) і продукт відділяли фільтруванням при 10-15 °C. Вихід: 243 г Вихід в %: 60 %. Хімічна чистота: 99,95 %. Оптична чистота: 98,62 %. Переваги даного винаходу наведені нижче: А. Рентабельний і промислово здійсненний спосіб. В. Використання окисника, який може бути легко регенерований. C. Продукт, одержаний по закінченні взаємодії, має фармацевтично прийнятну міру чистоти. D. Застосування меншої кількості стадій очищення підвищує загальний вихід кінцевого продукту. Е. Екологічно нешкідливий, безпечний і простий спосіб стереоселективного одержання заміщених або незаміщених хіральних сульфінільних похідних з меншими домішками сульфонів і з високою енантіомерною чистотою. F. Може бути досягнута регенерація хірального реагенту. G. Уникнення множинних стадій перекристалізації. Н. Уникнення використання пероксидів/ферментів. ФОРМУЛА ВИНАХОДУ 50 1. Спосіб одержання сполуки формули (І), в якому здійснюють стадії взаємодії сполуки формули (Іа) з оксазиридином формули (VII) в присутності розчинника і основи і подальшого виділення сполуки формули (І) 10 UA 98792 C2 (VII) * (Ia) основа (I) H N , Ph або X= Ph R5 A , R1 O N R2 Y= 5 10 R4 або R8 R3 , де R1=H; R2=H, CH3; R3=H, -OCH3, -OCH2CF3, -OCH2CH2OCH3, -O(CH2)3OCH3; R4=H, CH3, -OCH3; і R5=H, -OCH3, -OCHF2; A=CH, N; R8=ОH, NH2, OR'9, NHR'9; і R'9=С1-4-алкіл; R9=R10=H, О, галоген, -О-С1-4-алкіл, -О-С=О, -О-(СН2)n-О-, де n=2 або 3; і де розчинник не є іонним розчинником. 2. Спосіб за п. 1, в якому H N X= N R5 A , R1 N R2 Y= R4 15 R3 , де R1=H; R2 і R3=H, CH3, -OCH3, -OCH2CF3, ОСН2(СН2)ОСН3, -О(СН2)3ОСН3; R4=H, СН3; і R5=H, -ОСН3, -OCHF2; A=CH, N. 3. Спосіб за пунктом 2, в якому сполуку формули (І) вибирають з групи, що включає оптично активні пантопразол, лансопразол, рабепразол, тенатопразол, парипразол і омепразол. 4. Спосіб за п. 1, в якому Ph X= Ph , O Y= 20 R'8 , де R'8=OH, NH2; OR'9, NHR'9; і R'9 =С1-4-алкіл. 5. Спосіб за п. 4, в якому сполука формули (І) являє собою армодафініл. 6. Спосіб за п. 1, в якому (2R,8aS)-кaмфopилcyльфoнiлoкcaзиpидин формули (XII) являє собою або правообертальний, або лівообертальний ізомер. 11 UA 98792 C2 5 10 15 7. Спосіб за п. 1, в якому основа являє собою або органічну, або неорганічну основу. 8. Спосіб за п. 7, в якому основа переважно являє собою органічну основу. 9. Спосіб за п. 8, в якому органічну основу вибирають з групи, що включає 1,8діазабіцикло[5.4.0]ундец-7-ен, діізопропілетиламін, гексаметилентетрамін і триетиламін. 10. Спосіб за п. 9, в якому органічна основа являє собою 1,8-діазабіцикло[5.4.0]ундец-7-ен. 11. Спосіб за п. 7, в якому неорганічна основа являє собою гідроксид лужного металу. 12. Спосіб за п. 11, в якому неорганічна основа являє собою гідроксид натрію або гідроксид калію. 13. Спосіб за п. 1, який здійснюють в присутності розчинника, вибраного з спирту, ароматичного вуглеводню, простого ефіру, складного ефіру, аміду, нітрилу, води або їх сумішей. 14. Спосіб за п. 13, в якому розчинник являє собою спирт або ароматичні вуглеводні. 15. Спосіб за п. 13, в якому розчинник вибирають з групи, що включає метанол, етанол, ізопропанол, бутанол, простий діізопропіловий ефір, толуол, воду, тетрагідрофуран, ацетонітрил, диметилформамід, діетилформамід, диметоксіетан або їх комбінації. 16. Спосіб за п. 14, в якому розчинник являє собою ізопропанол або толуол. Комп’ютерна верстка А. Крижанівський Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 12
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess of sulfoxidation of biologically active compounds
Автори англійськоюGharpure, Milind, Moreshwar, Maikap, Golakchandra, Sudarshan, Mahale, Rajendra, Dagesing, Mehta, Satish, Ramanlal, Gurjar, Mukund, Keshav, Rajput, Mahesh, Ramsing, Gawari, Prashant, Sadashiv
Назва патенту російськоюСпособ сульфоксидирования биологически активных соединений
Автори російськоюГхарпур Милинд Морешвар, Майкап Голакчандра Сударшан, Махал Раджендра Дагесинг, Мехта Сатиш Раманлал, Гурджар Мукунд Кешав, Раджпут Махеш Рамсинг, Гавари Прашант Садашив
МПК / Мітки
МПК: C07D 471/04, C07D 401/12, C07C 315/02
Мітки: активних, сульфоксидування, спосіб, сполук, біологічно
Код посилання
<a href="https://ua.patents.su/14-98792-sposib-sulfoksiduvannya-biologichno-aktivnikh-spoluk.html" target="_blank" rel="follow" title="База патентів України">Спосіб сульфоксидування біологічно активних сполук</a>
Безвідходний спосіб одержання біологічно активних сполук прополісу

Номер патенту: 55059
Опубліковано: 17.01.2005
Автори: Тихонова Світлана Олександрівна, Ярних Тетяна Григорівна, ТИХОНОВ ОЛЕКСАНДР ІВАНОВИЧ
МПК: A61K 35/64
Мітки: одержання, активних, спосіб, сполук, прополісу, безвідходний, біологічно
Формула / Реферат:
1. Безвідходний спосіб одержання біологічно активних сполук прополісу, що включає обробку прополісу-сирцю водою підвищеної температури, який відрізняється тим, що прополіс-сирець обробляють водою при співвідношенні відповідно 1 : 2 - 1 : 10 при температурі 50 - 100 °С до одержання трьох фракцій: воску, водної витяжки та осаду, при додаванні до водної витяжки солюбілізатора одержують гідрофільні сполуки прополісу, при додаванні до осаду...
Мікрокапсула для біологічно активних сполук та спосіб її одержання

Номер патенту: 46025
Опубліковано: 15.05.2002
Автори: Шер Герберт Бенсон, Чен Дзін Лінг
МПК: A01N 25/04, B01J 13/06, A01N 25/22, A01N 53/00, A01N 25/28, A01N 25/30, B01J 13/02, B01J 13/04
Мітки: спосіб, сполук, одержання, активних, мікрокапсула, біологічно
Формула / Реферат:
1. Мікрокапсула для біологічно активних сполук, що містить органічну рідину, яка включає чутливий до ультрафіолетового світла біологічно активний матеріал та ефективну кількість засобу захисту від ультрафіолетового світла у вигляді часток, яка відрізняється тим, що засіб захисту від ультрафіолетового світла вибраний із групи, яка включає діоксид титану, оксид цинку та їх суміші, що суспендовані та ретельно дисперговані у рідини.2....
Спосіб оцінки ступеня цитотоксичності біологічно активних сполук та фармакологічних препаратів

Номер патенту: 15629
Опубліковано: 17.07.2006
Автори: Славіна Ніна Георгіївна, Бощенко Юрій Анатолійович, Лозицький Віктор Петрович, Віноходов Дмитро Олегович, Федчук Алла Семенівна, Григорашева Ірина Миколаївна, Гридіна Тетяна Леонідівна, Позднякова Людмила Іванівна, Полежаєв Фіал Ібрагімович
МПК: C12Q 1/18, G01N 33/15
Мітки: біологічно, активних, оцінки, сполук, фармакологічних, цитотоксичності, препаратів, ступеня, спосіб
Формула / Реферат:
1. Спосіб оцінки ступеня цитотоксичності біологічно активних сполук та фармакологічних препаратів, що полягає у створенні контакту водних розчинів досліджуваних препаратів у різних концентраціях з тест-клітинами, який відрізняється тим, що як тест-клітини використовують інфузорії Colpoda steinii, а визначення максимально переносимої концентрації досліджуваних біологічно активних сполук здійснюють за показниками їх життєздатності через три...
Спосіб одержання біологічно активних сполук з прополісу

Номер патенту: 1741
Опубліковано: 25.10.1994
Автори: Ярних Тетяна Григорівна, ТИХОНОВ ОЛЕКСАНДР ІВАНОВИЧ, Благой Юрій Павлович, Гончаров Володимир Григорович, Котенко Олександр Михайлович, Бутко Анатолій Єгорович
МПК: A61K 35/64
Мітки: активних, прополісу, біологічно, спосіб, одержання, сполук
Формула / Реферат:
Способ получения биологически активных соединений из прополиса путем его измельчения, экстракцииэтаноломифильтрования.отличающийся тем, что, с целью повышения выхода целевого продукта и сокращения времени способа, перед измельчением прополис охлаждают жидким азотом до температуры от —30 до—196 °C, измельчение проводят до размеров частиц 10—15 мкм, а экстрагирование ведут при температуре от—10 до 10 °C.
Спосіб тестування біологічно активних сполук на їх придатність як лікарських засобів

Номер патенту: 35037
Опубліковано: 26.08.2008
Автори: Гарманчук Людмила Василівна, Сидоренко Михайло Васильович, Перепелиціна Олена Михайлівна
МПК: G01N 33/15, C12N 5/00
Мітки: активних, біологічно, сполук, лікарських, придатність, тестування, засобів, спосіб
Формула / Реферат:
Спосіб тестування біологічно активних сполук на їх придатність як лікарських засобів шляхом інкубування сполук з культурами клітин ссавців в системі in vitro та з наступною оцінкою життєздатності та проліферації клітин МТТ-колориметричним методом, який відрізняється тим, що як культуру клітин використовують багатоклітинні сфероїди.
Попередній патент: Ослаблене спалювання
Наступний патент: Заміщені (оксазолідинон-5-ілметил)-2-тіофенкарбоксаміди і їх застосування в галузі коагуляції крові
Випадковий патент: Спосіб запобігання процесу нагромадження інкрустів, накипу на стінках труб, баків та інших конструкцій, що контактують з гарячими електролітами