Оральна фармацевтична дозована форма, захищена від несанкціонованого використання, що містить опіоїдний анальгетик
Номер патенту: 104745
Опубліковано: 11.03.2014
Автори: Манніон Річард Оуен, О'Доннелл Едвард Патрік, МакКенна Уільям Генрі, Хуанг Хеййонг Хуг


















































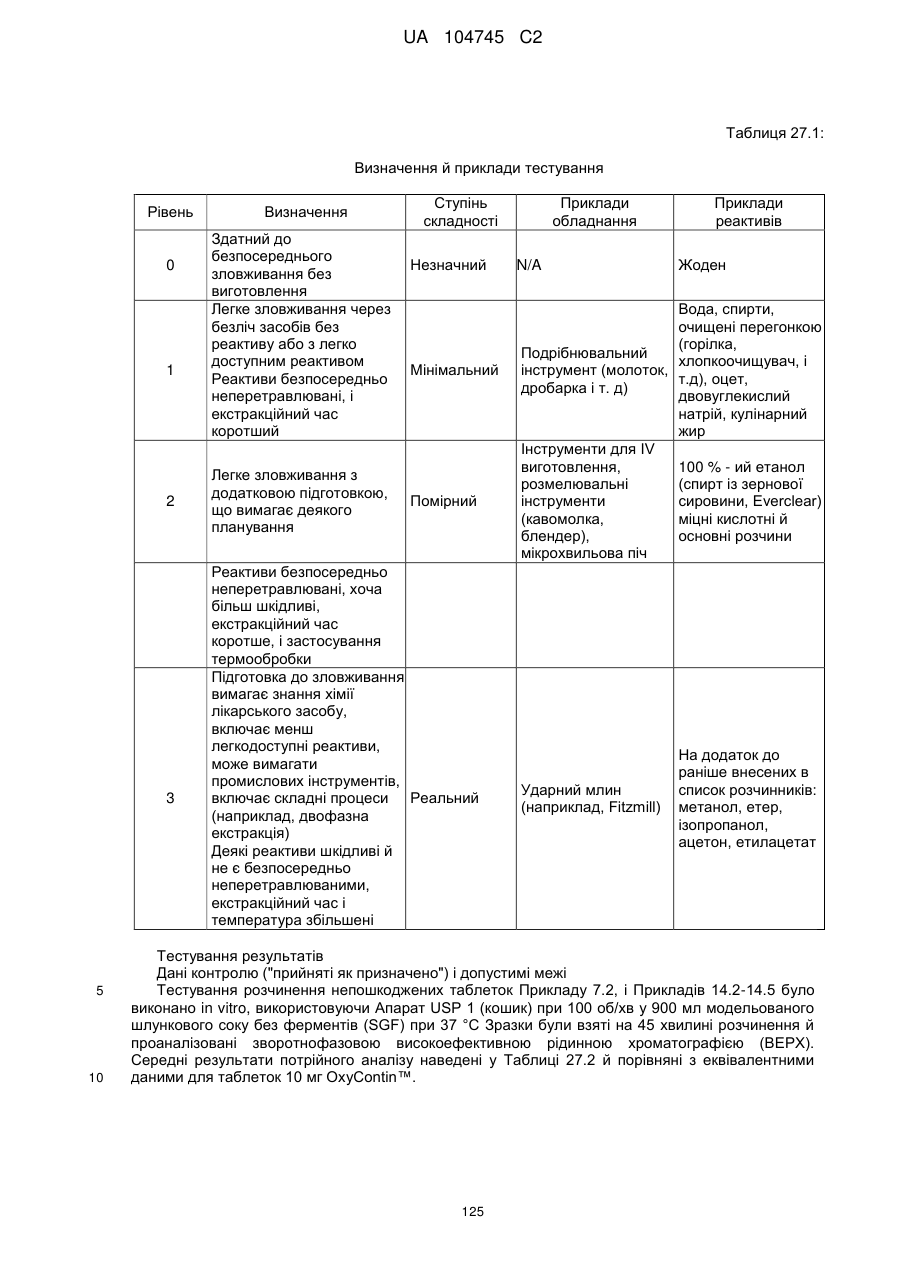
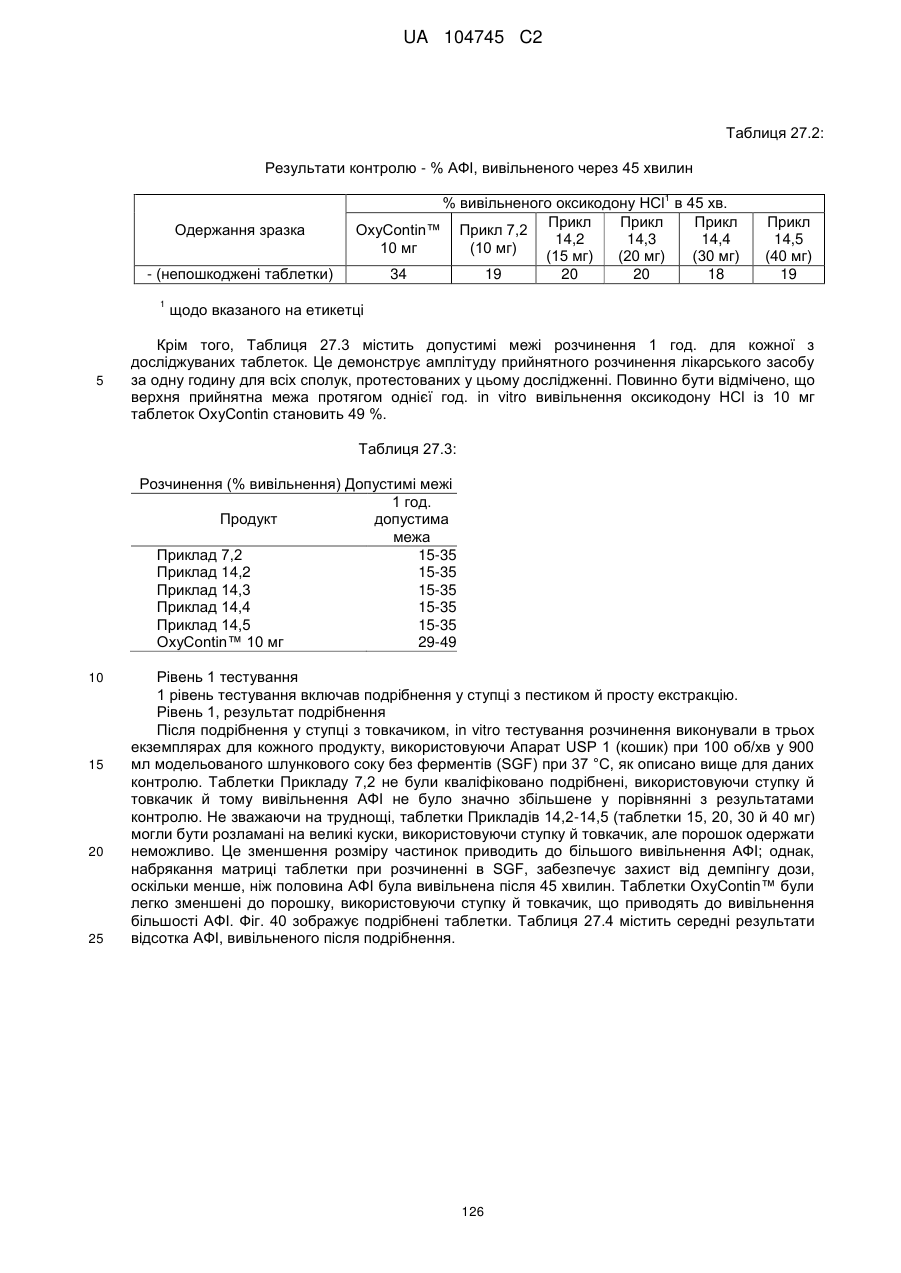
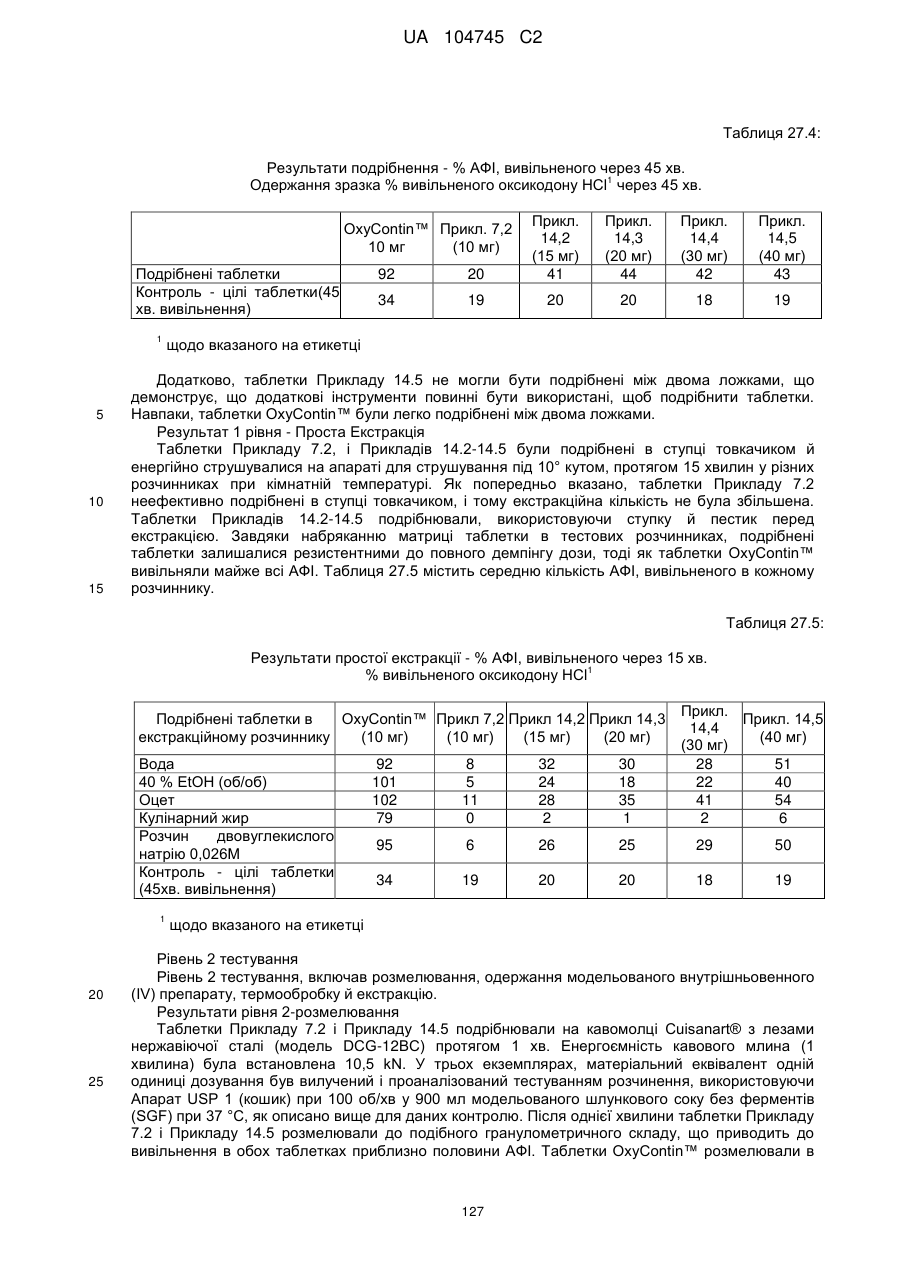
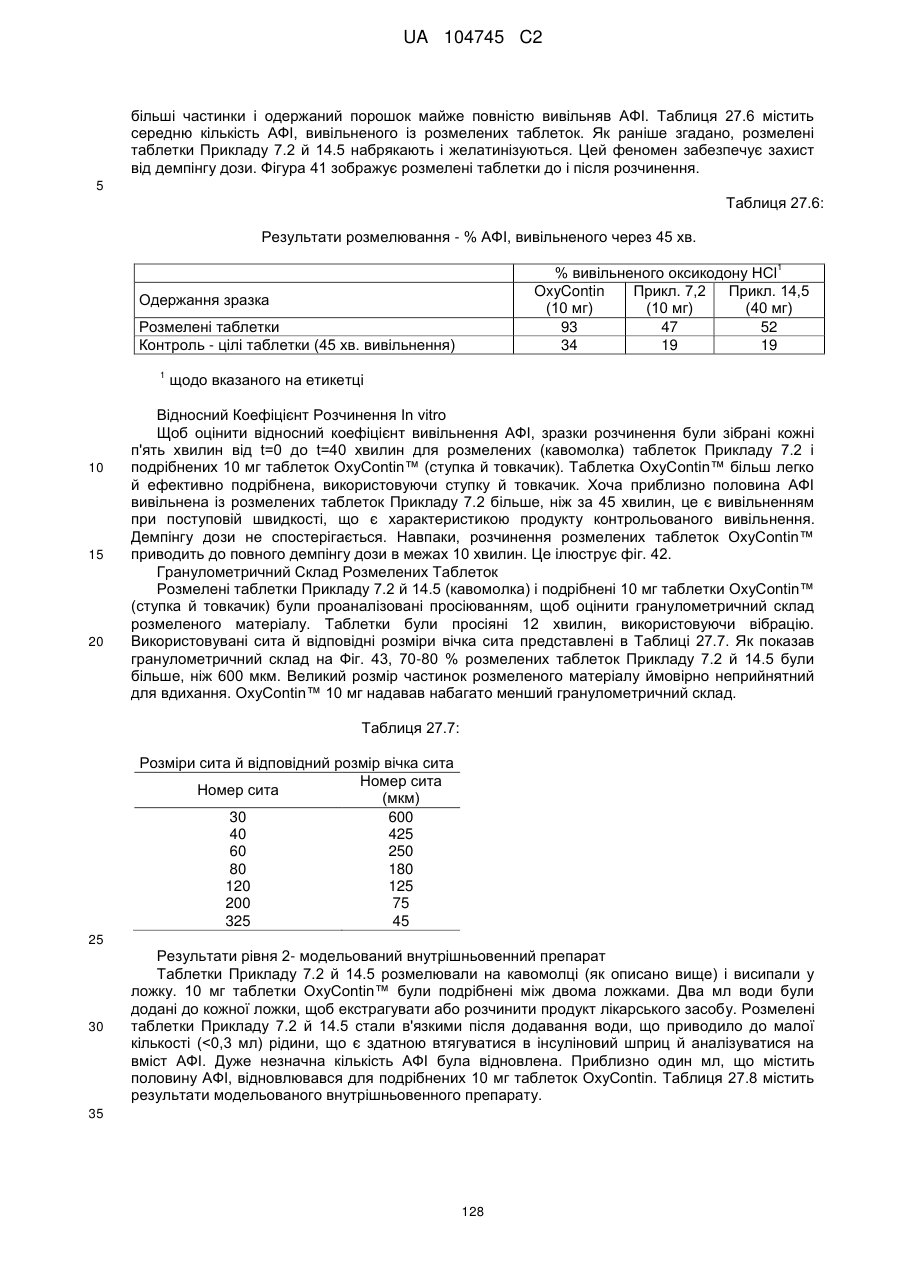












Формула / Реферат
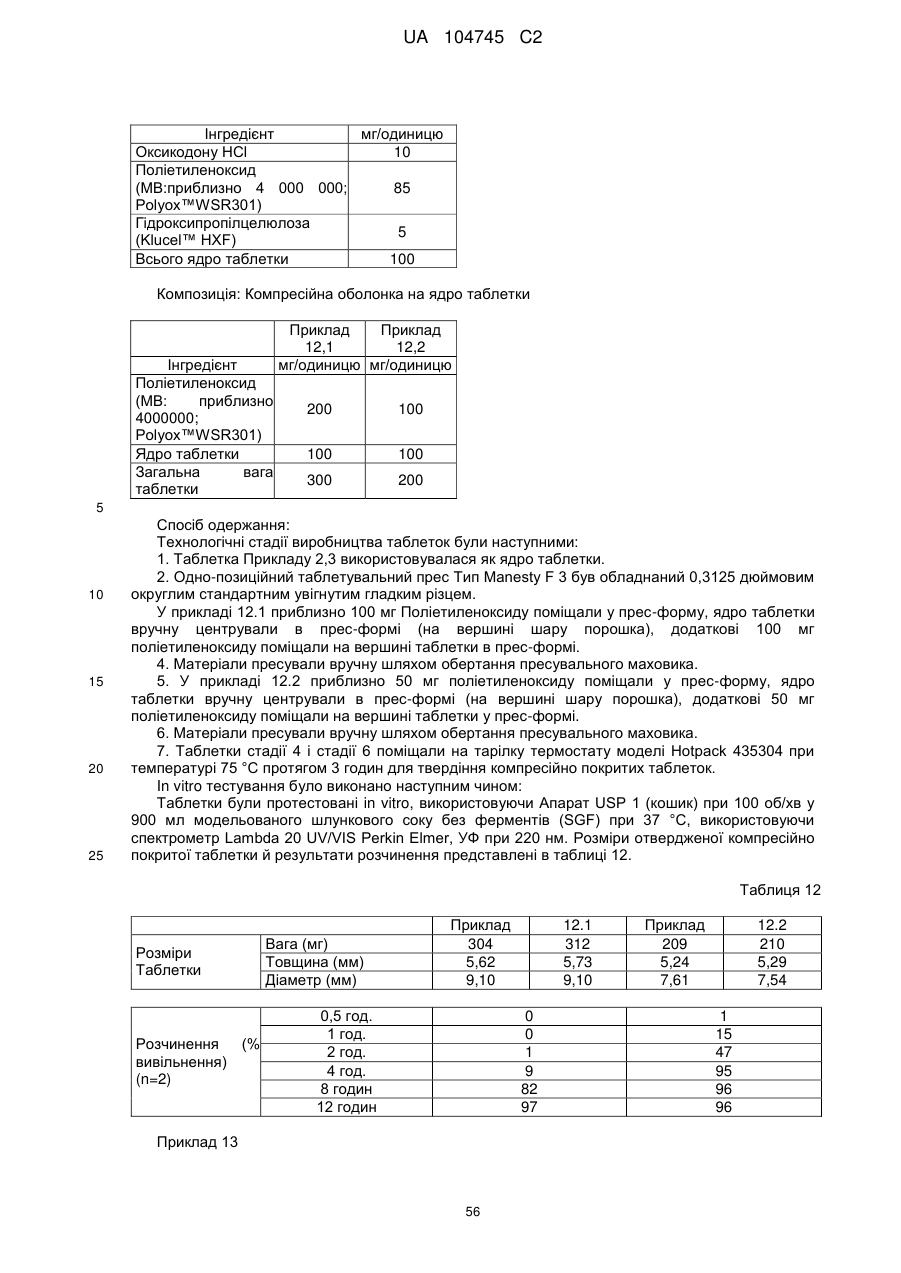
1. Тверда пероральна фармацевтична дозована форма пролонгованого вивільнення, що включає матричну композицію пролонгованого вивільнення, де матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні:
(1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і
(2) принаймні один активний агент, що вибирають з опіоїдних анальгетиків, де опіоїдним анальгетиком є оксикодону гідрохлорид і дозована форма містить від 5 мг до 500 мг оксикодону гідрохлориду; і
де композиція включає принаймні приблизно 80 мас. % поліетиленоксиду, що має базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000.
2. Тверда пероральна фармацевтична дозована форма пролонгованого вивільнення за пунктом 1, де опіоїдним анальгетиком є оксикодону гідрохлорид і композиція включає більше ніж 5 мас.% оксикодону гідрохлориду.
3. Тверда пероральна фармацевтична дозована форма пролонгованого вивільнення за пунктом 1, де композиція включає 10 мг оксикодону гідрохлориду і принаймні приблизно 85 мас. % поліетиленоксиду.
4. Тверда пероральна фармацевтична дозована форма пролонгованого вивільнення за пунктом 1, де композиція включає 15 мг або 20 мг оксикодону гідрохлориду.
5. Тверда пероральна фармацевтична дозована форма пролонгованого вивільнення за будь-яким з пунктів 1-4, де густина матричної композиції пролонгованого вивільнення дорівнює або менше, ніж приблизно 1,20 г/см3, переважно дорівнює або менше, ніж приблизно 1,19 г/см3.
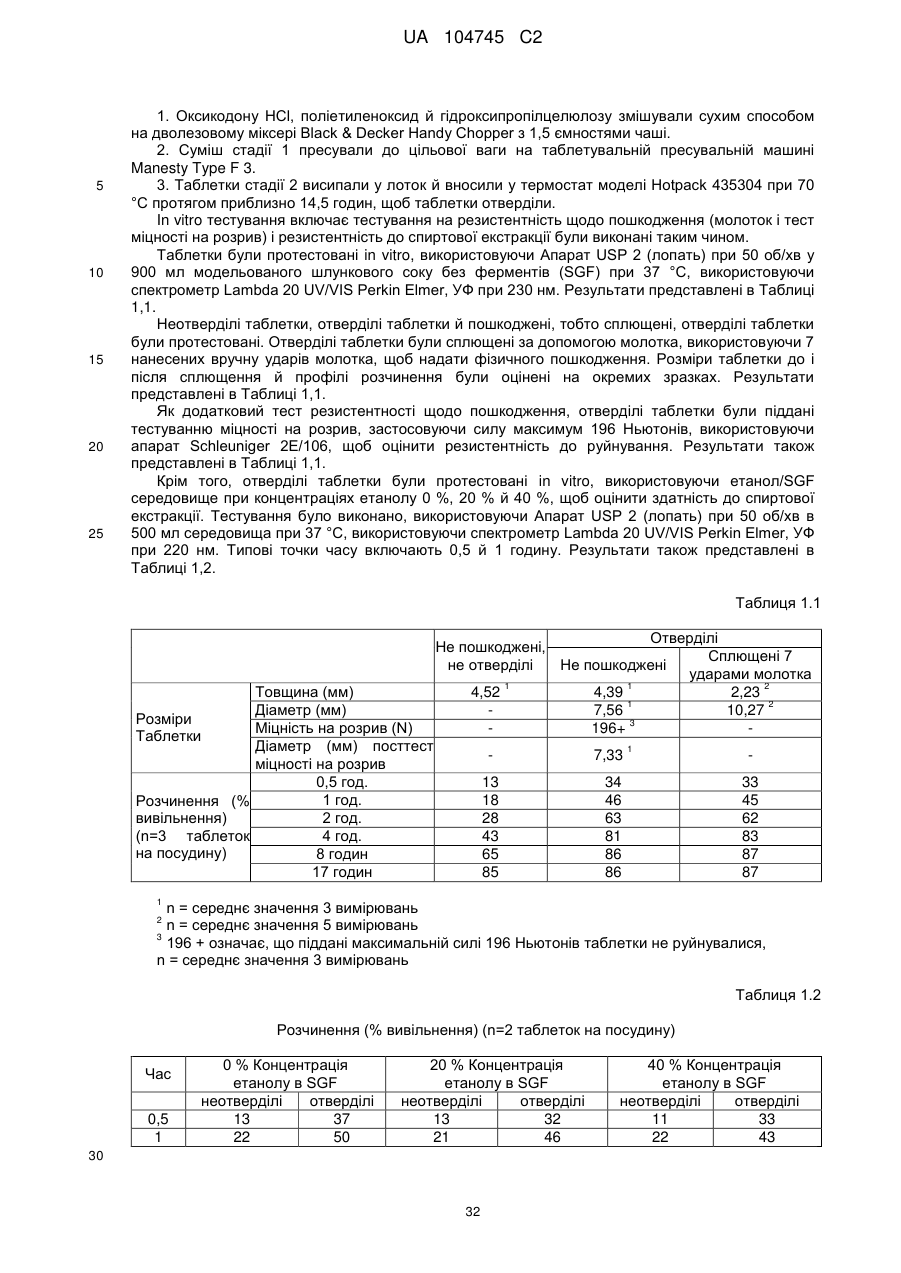
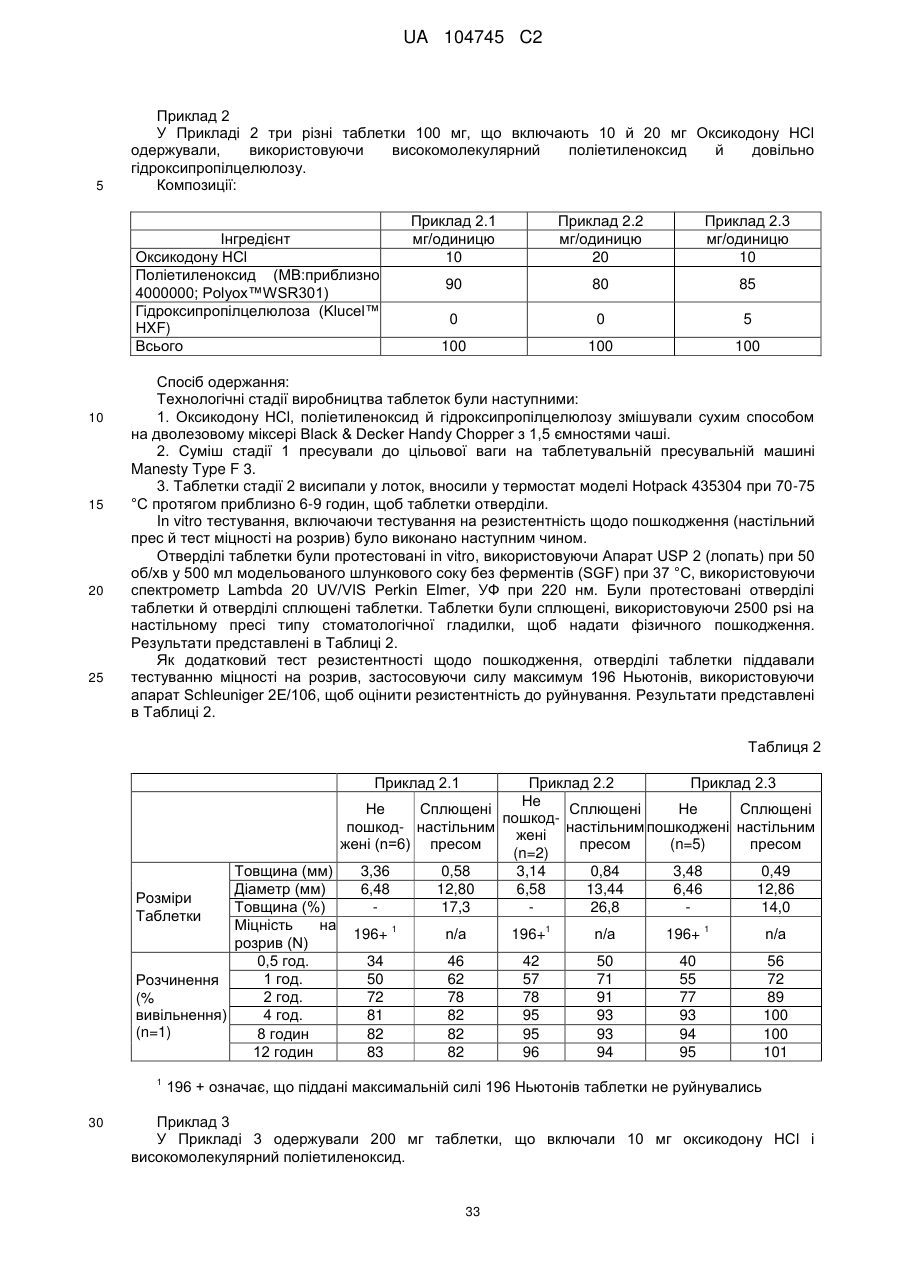
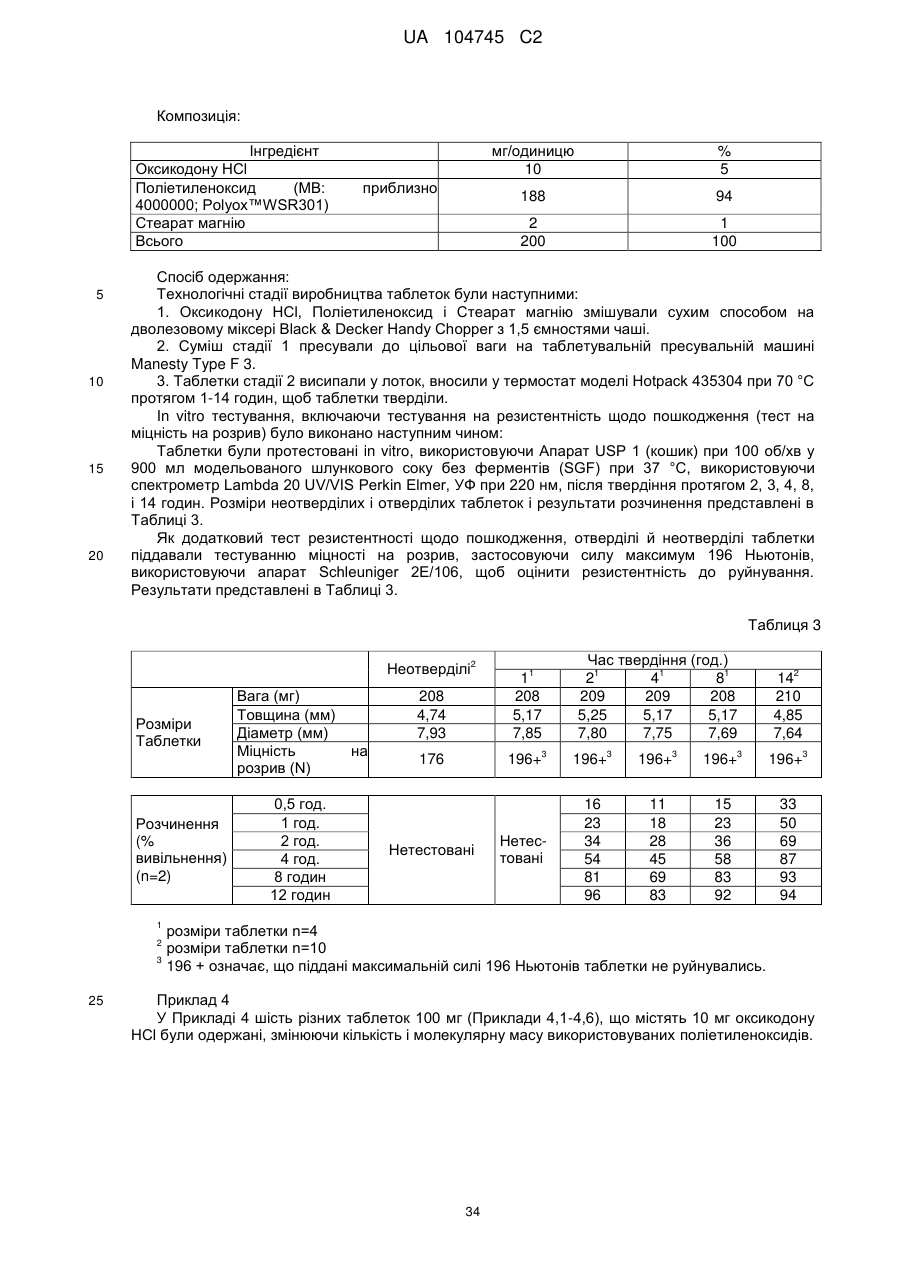
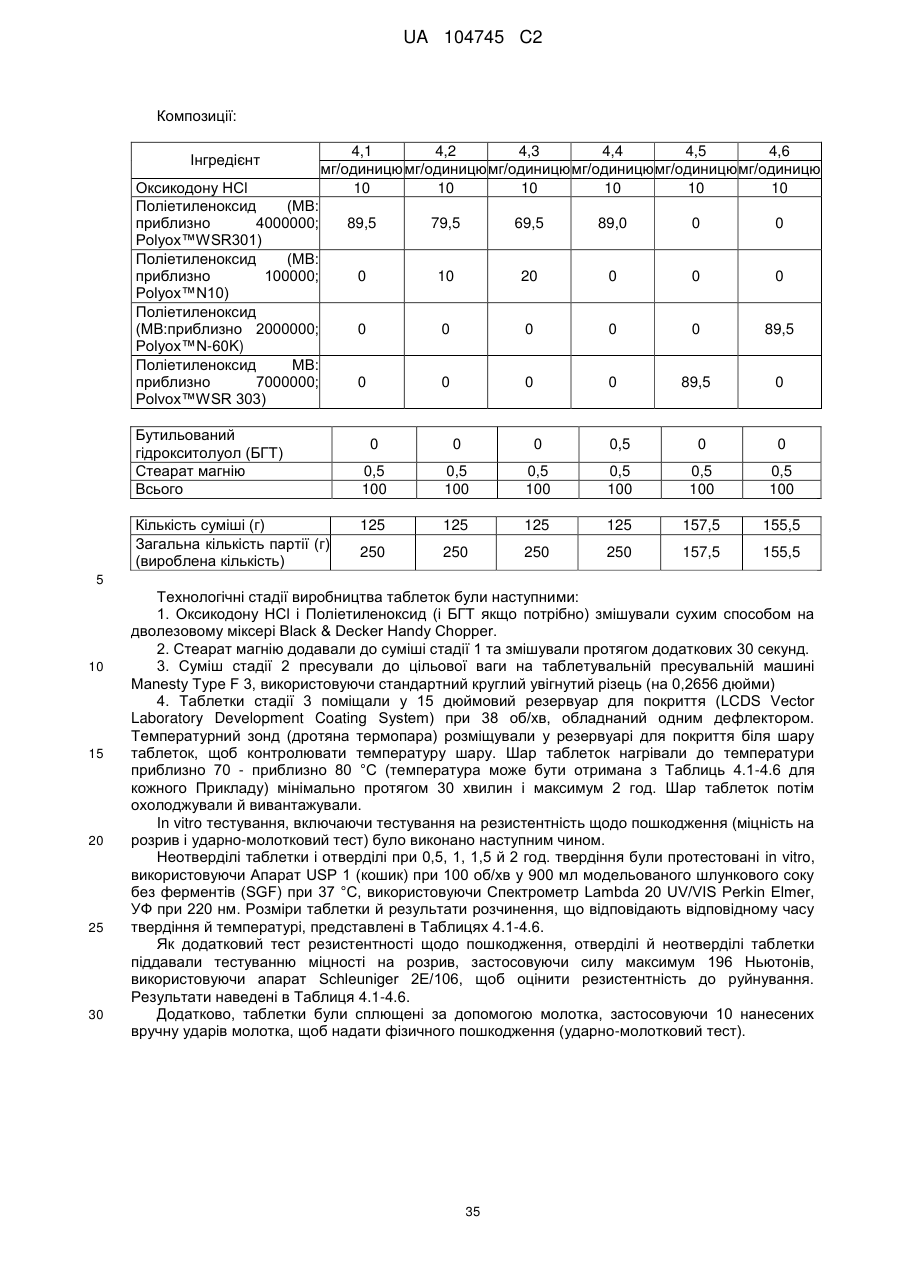
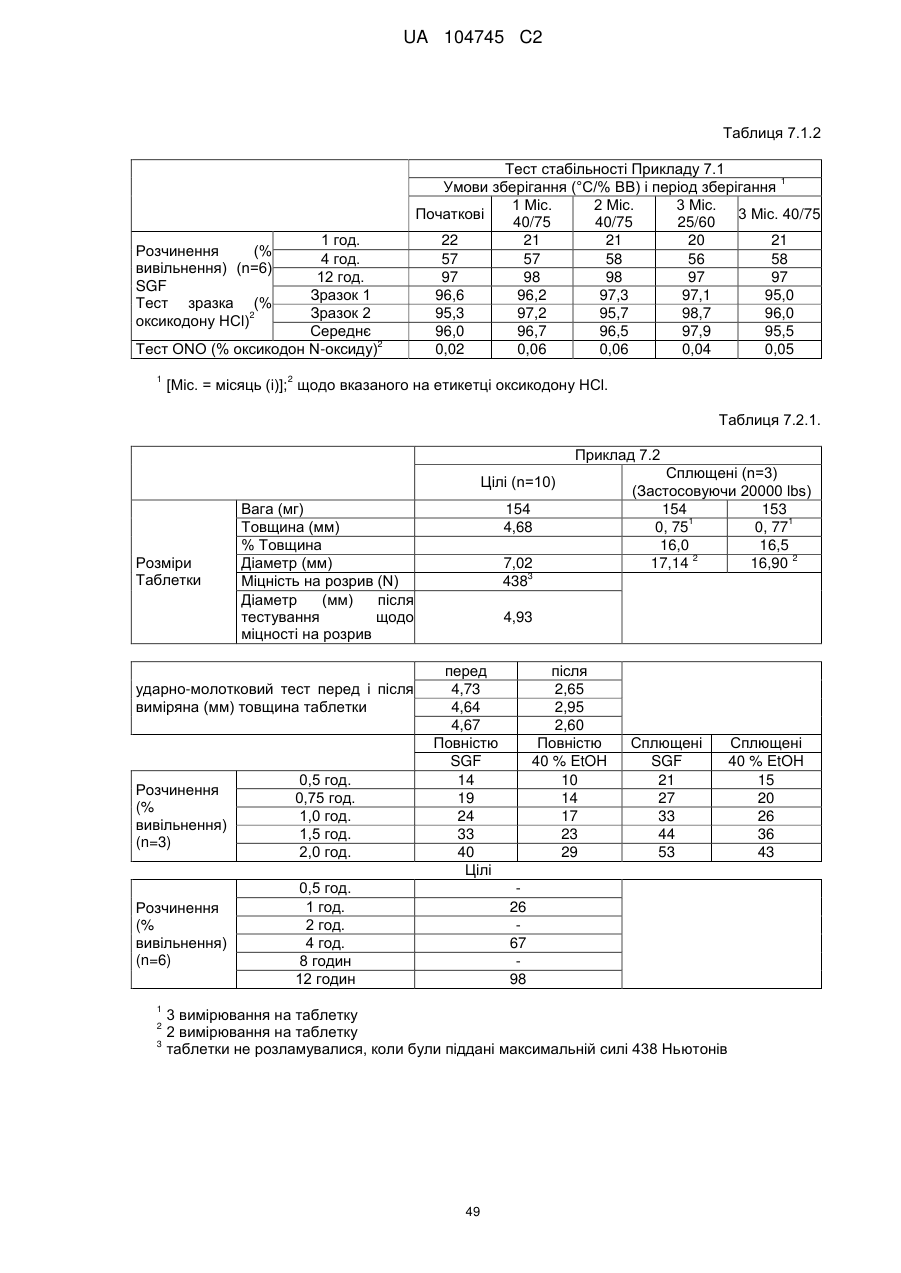
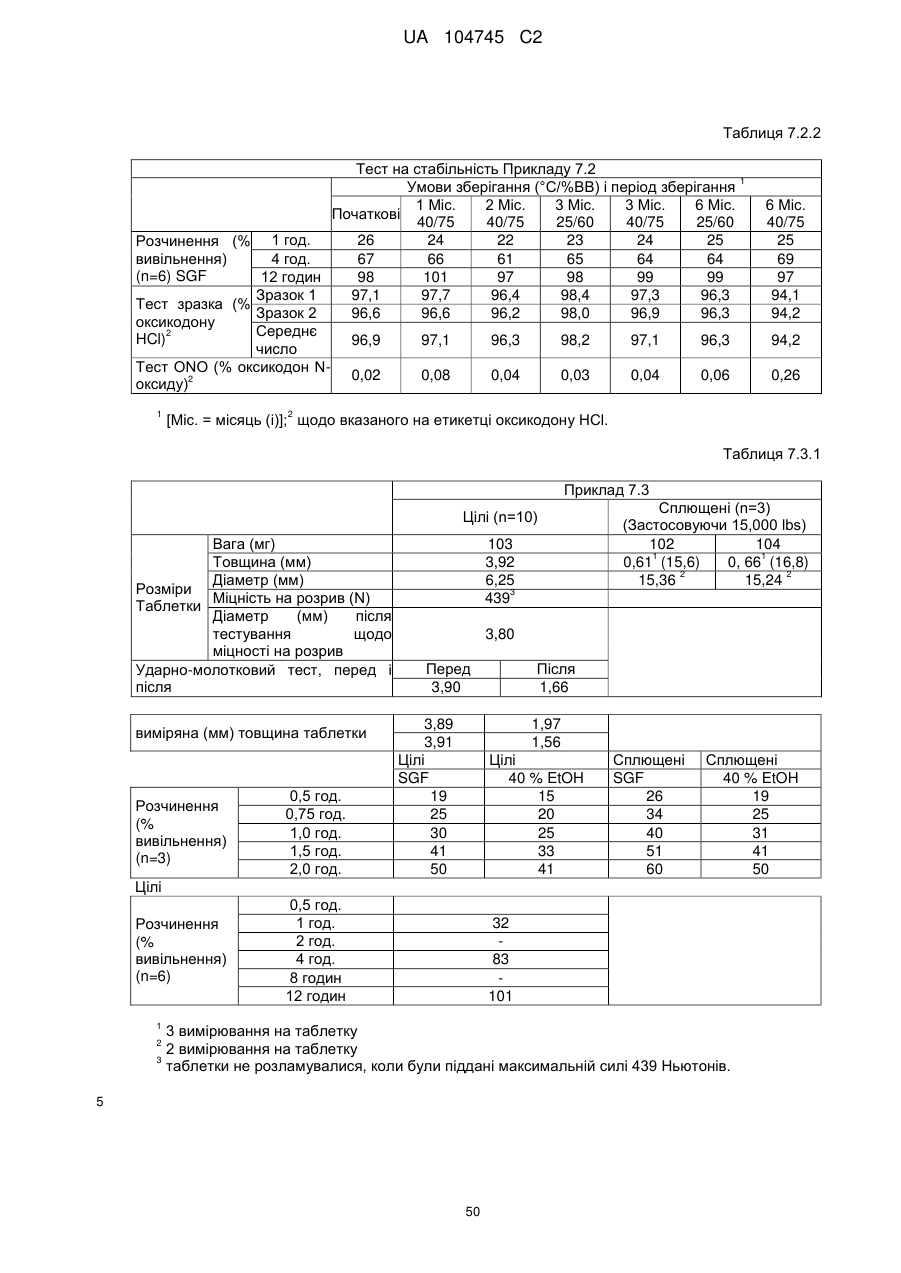
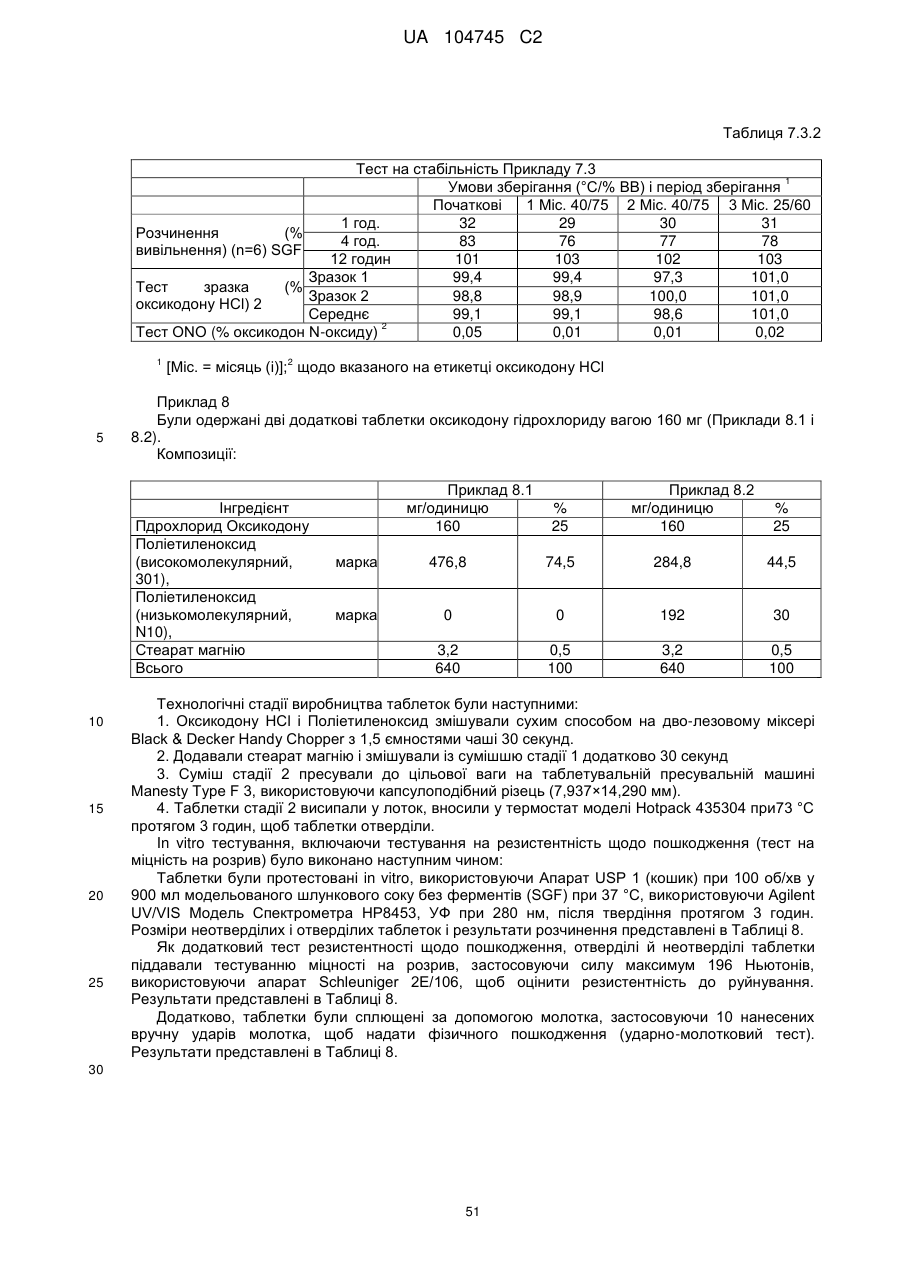
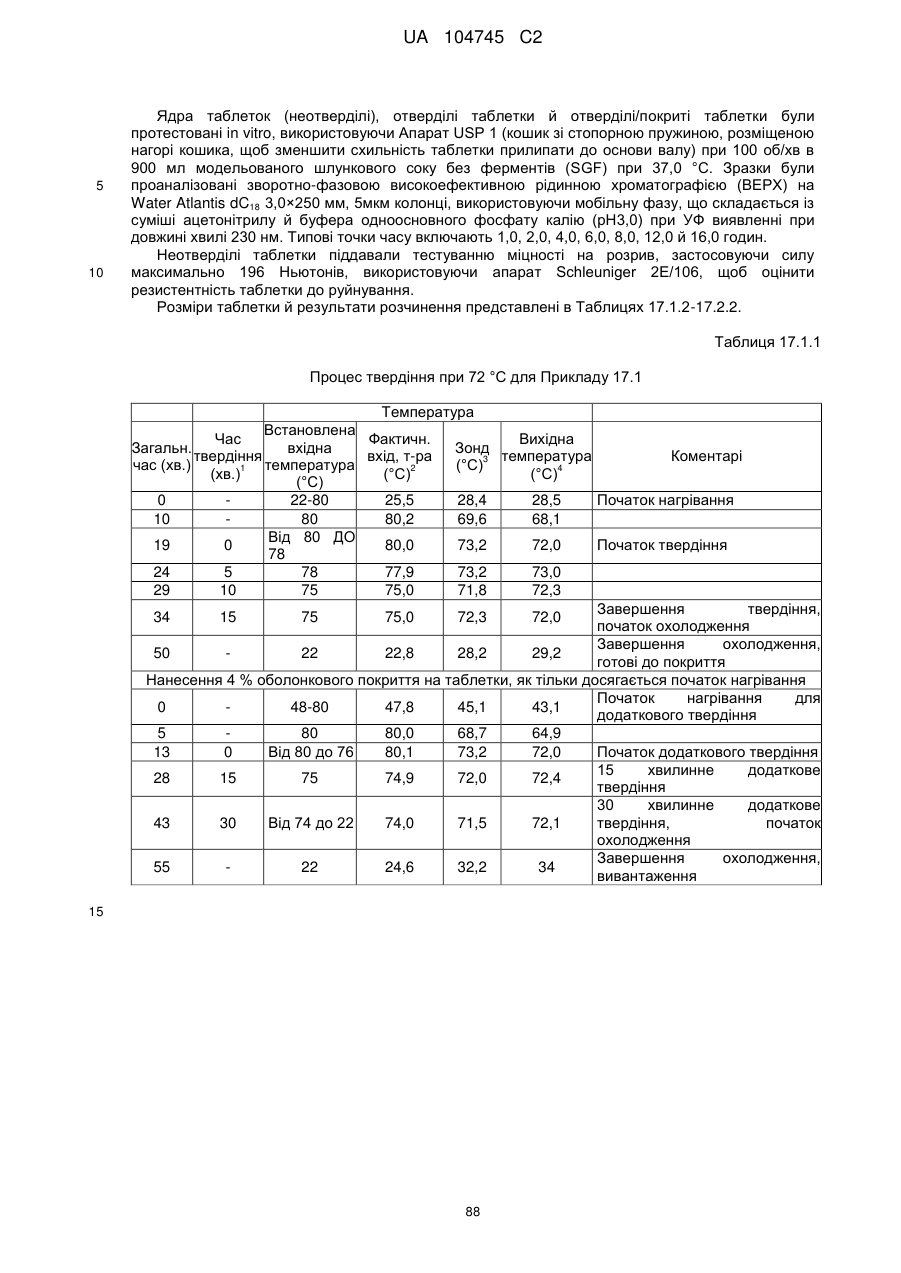
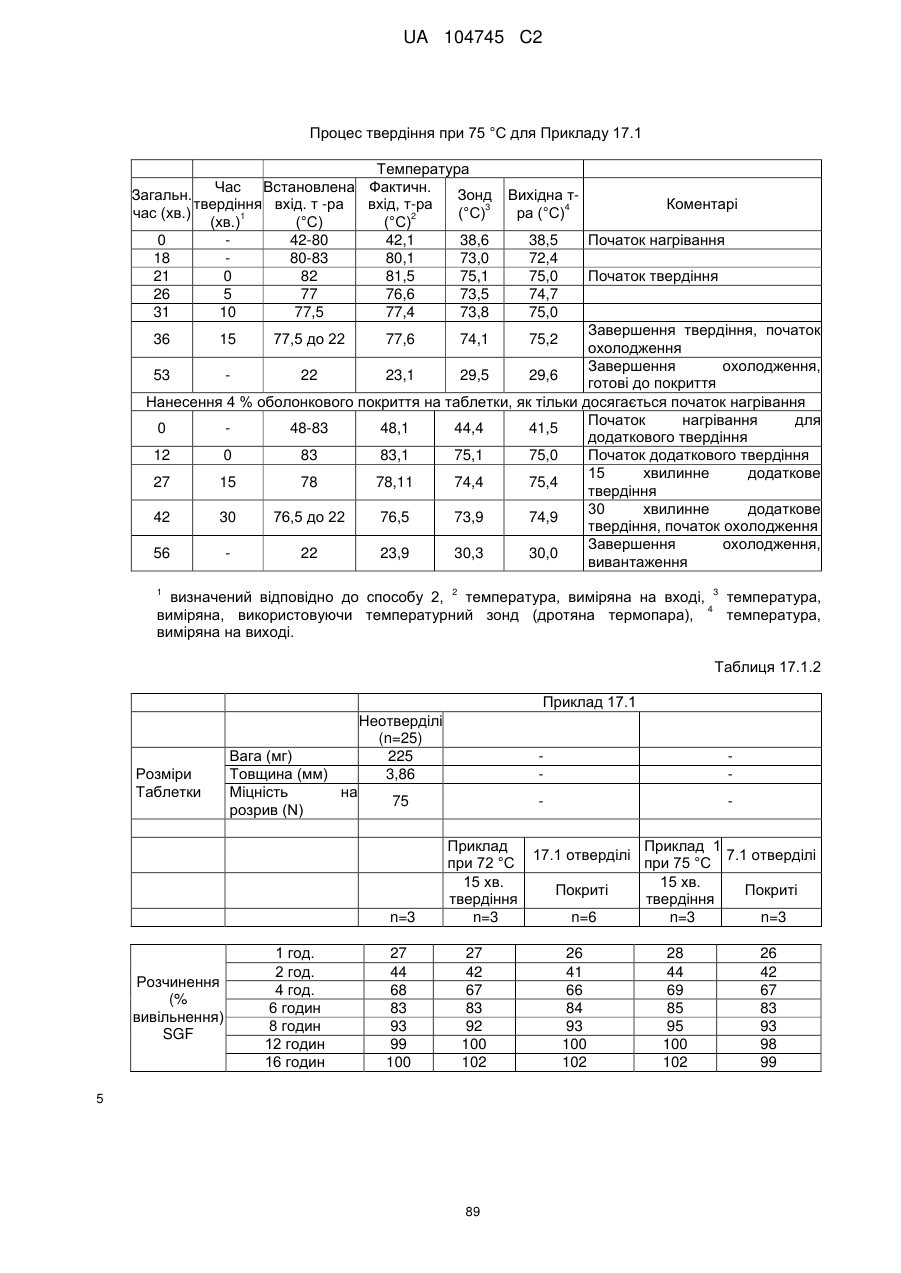
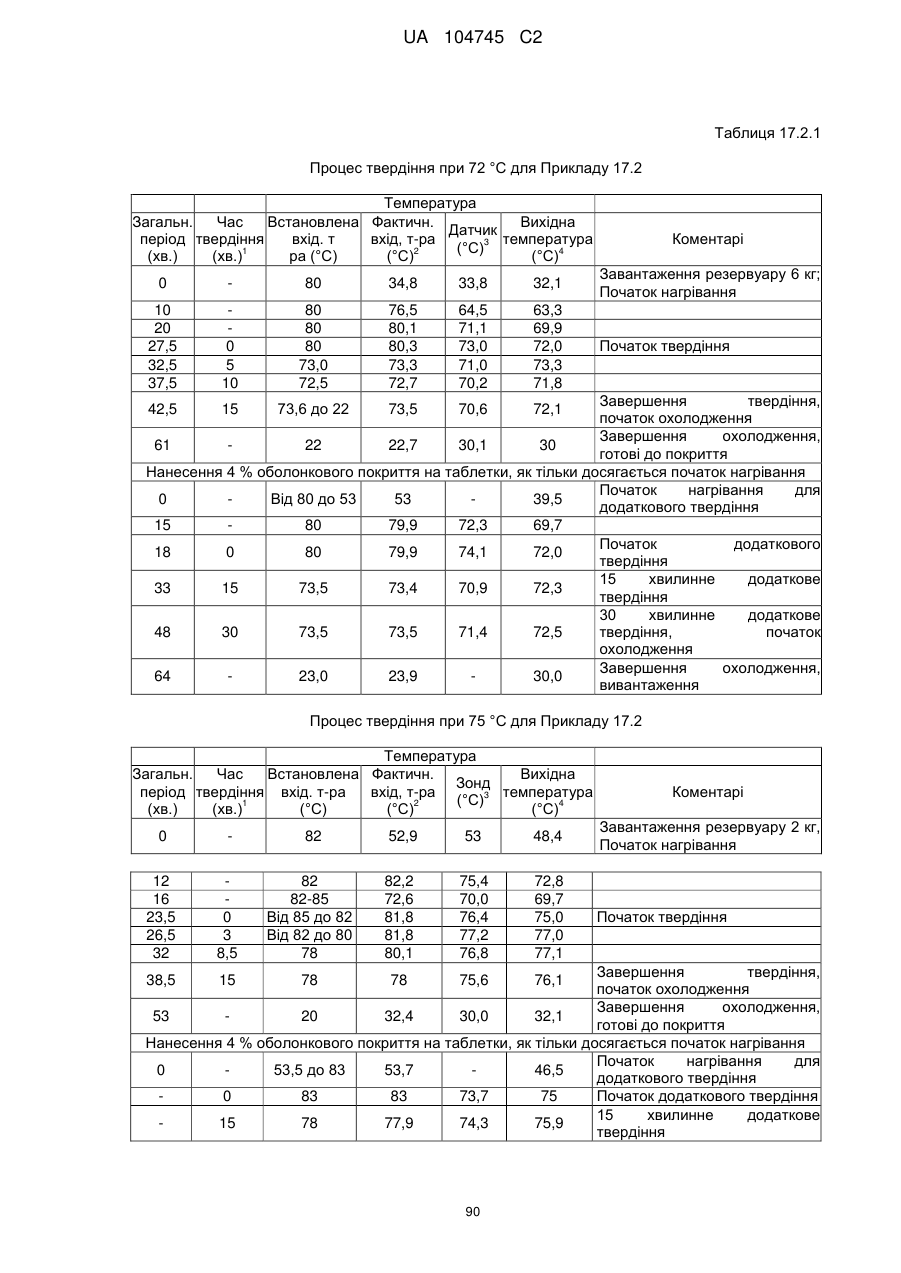
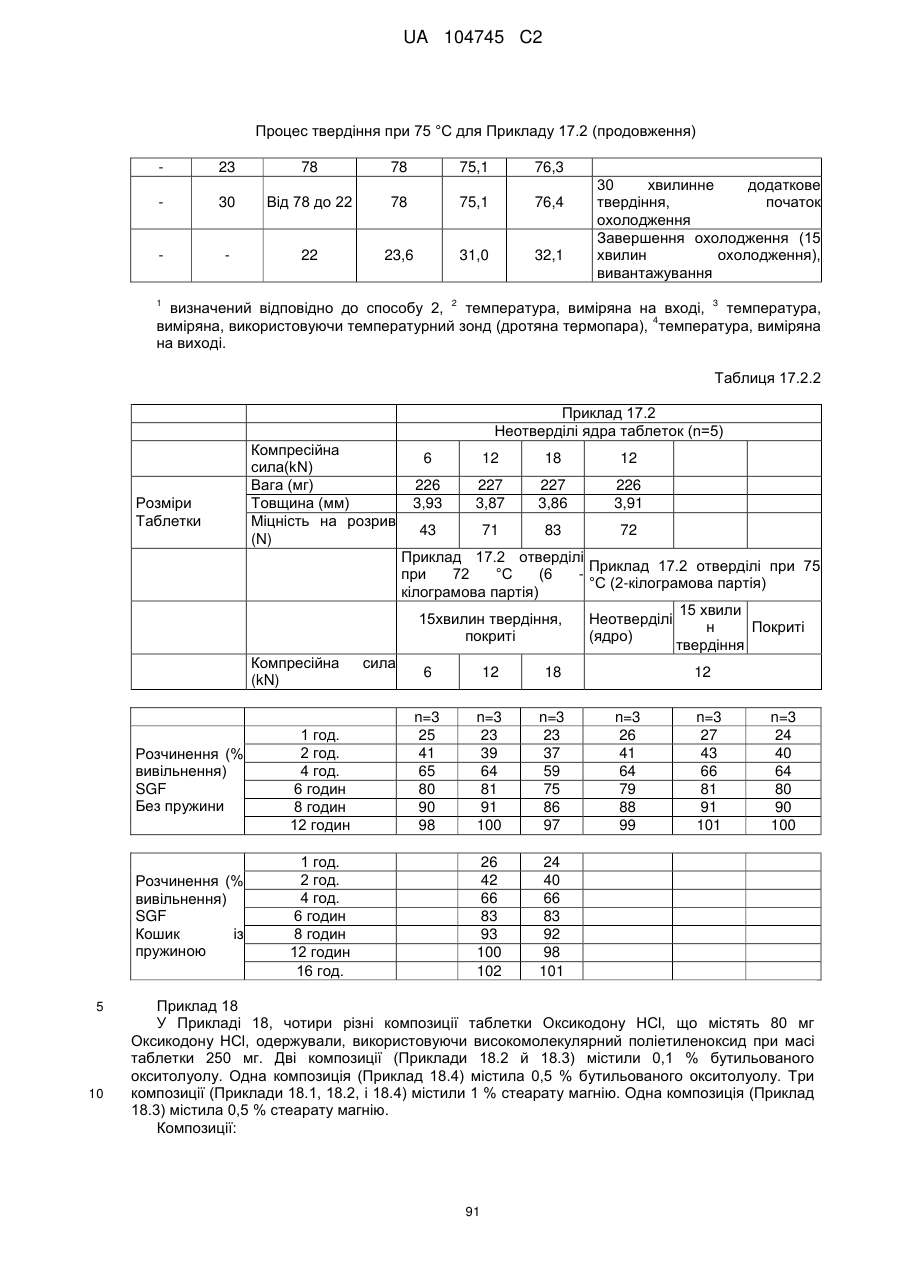
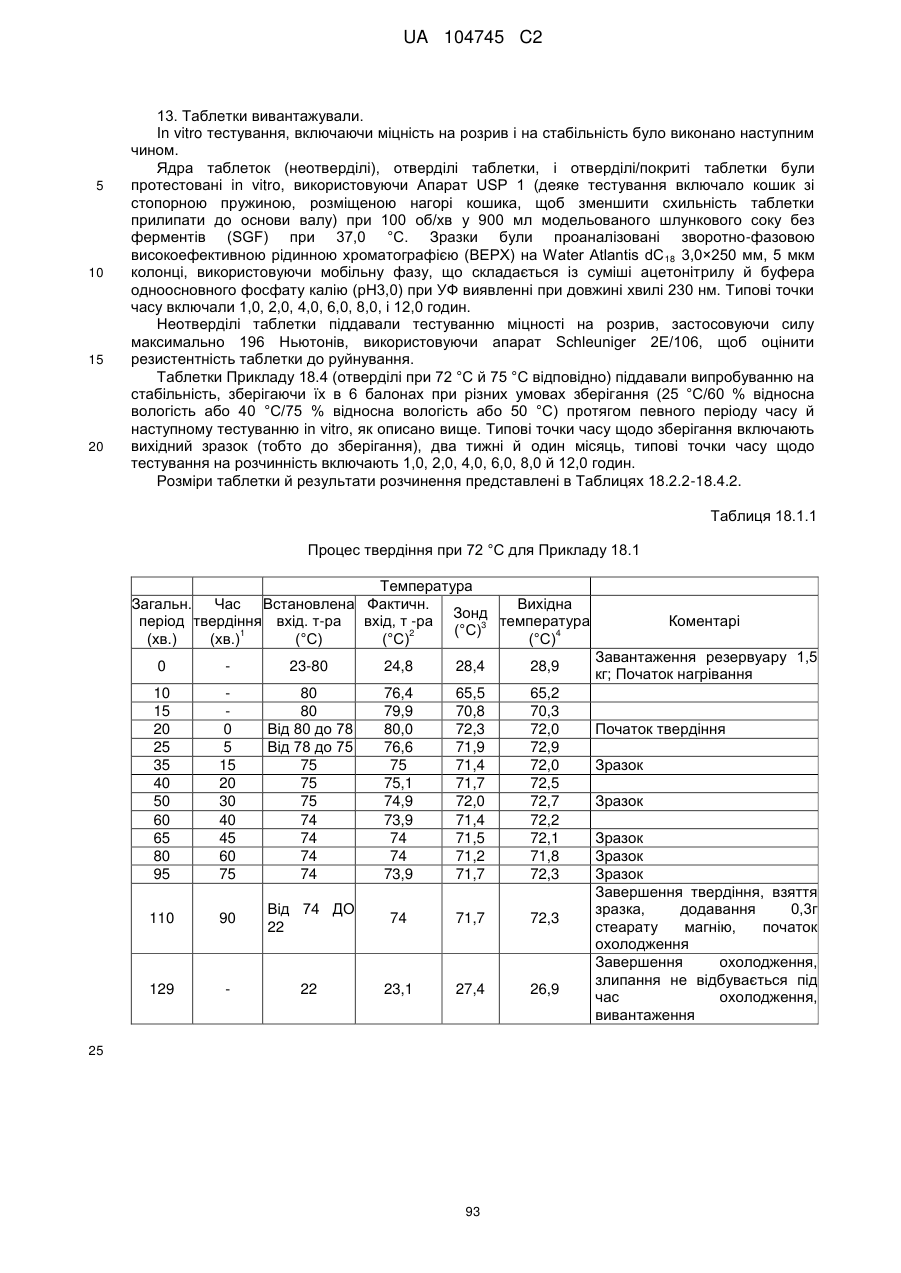
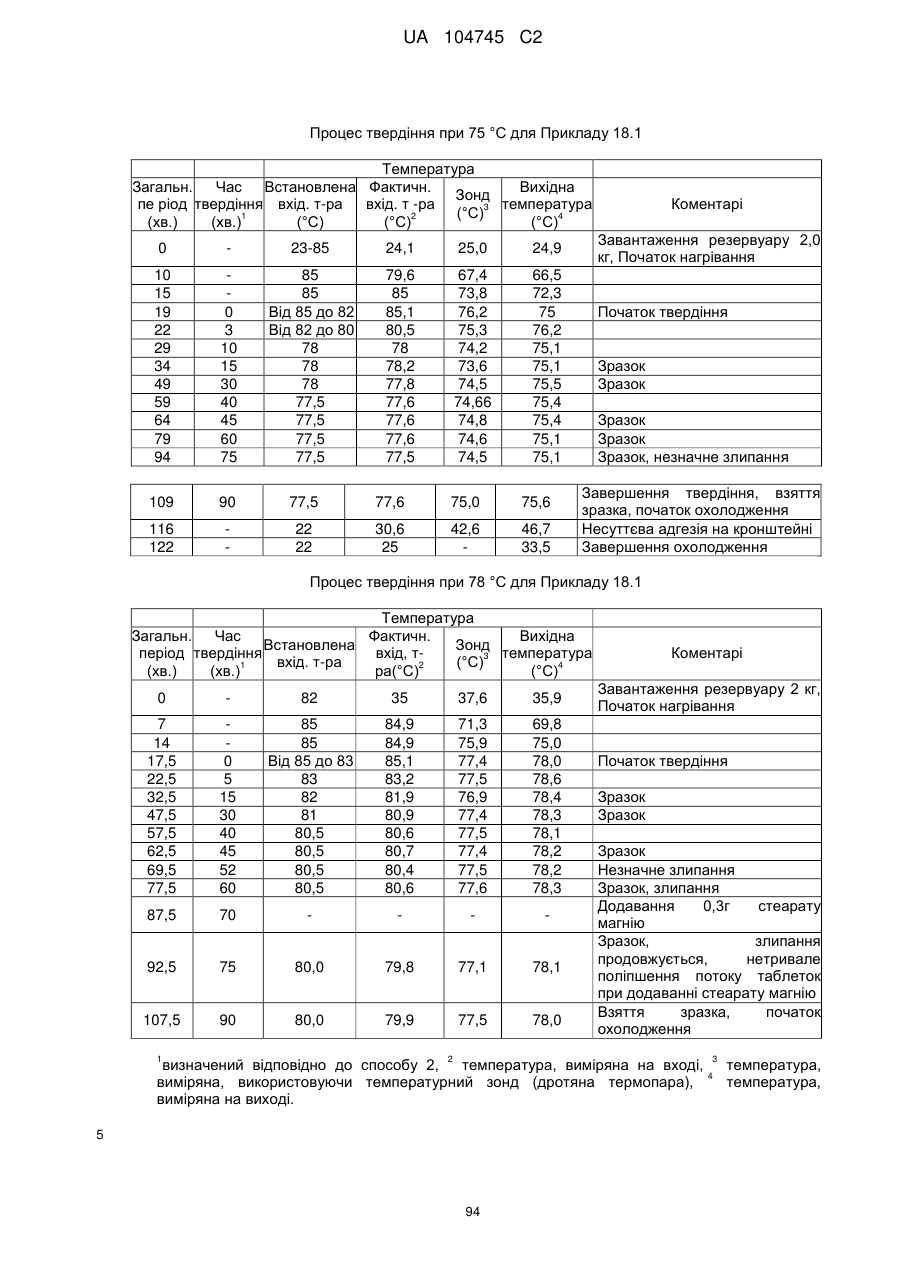
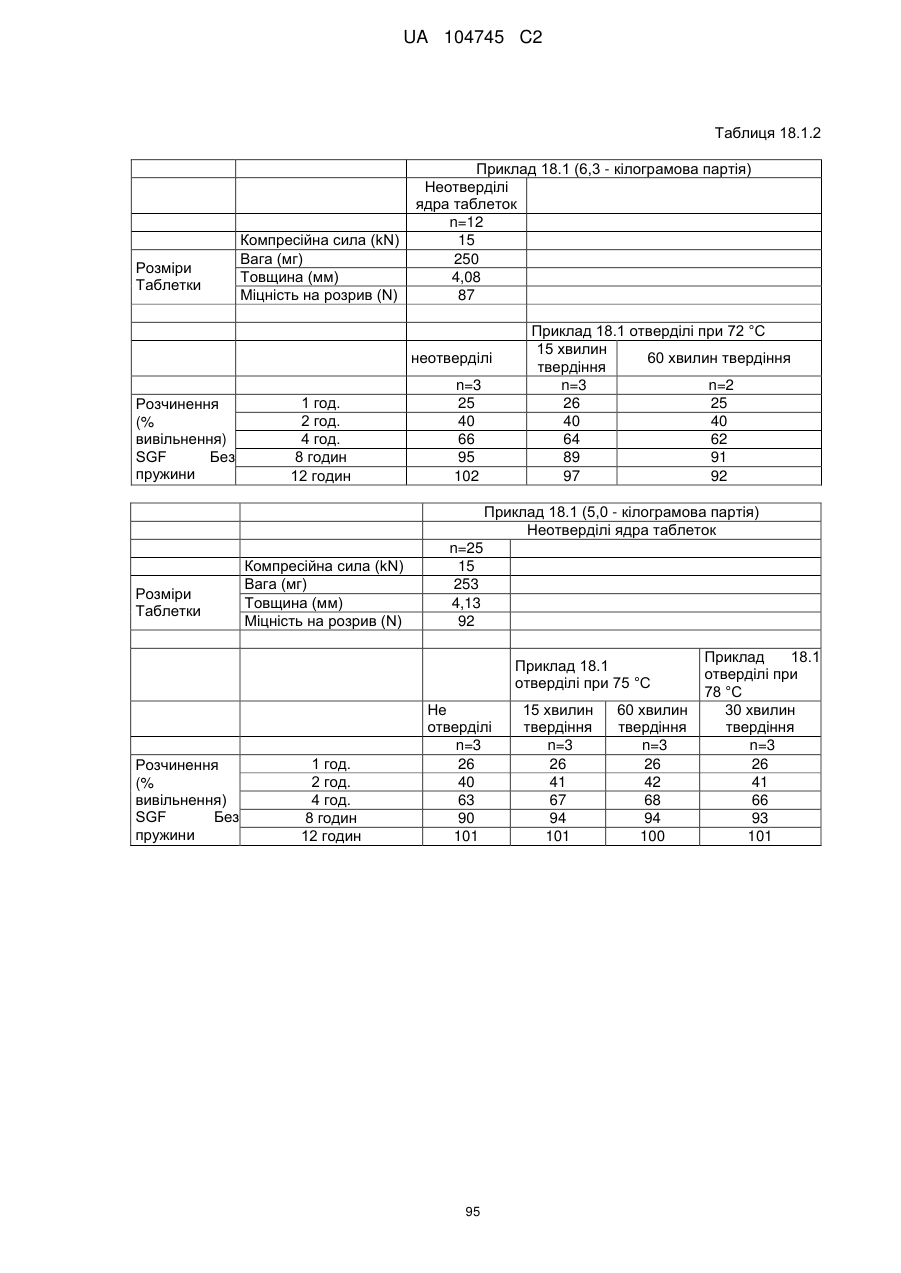
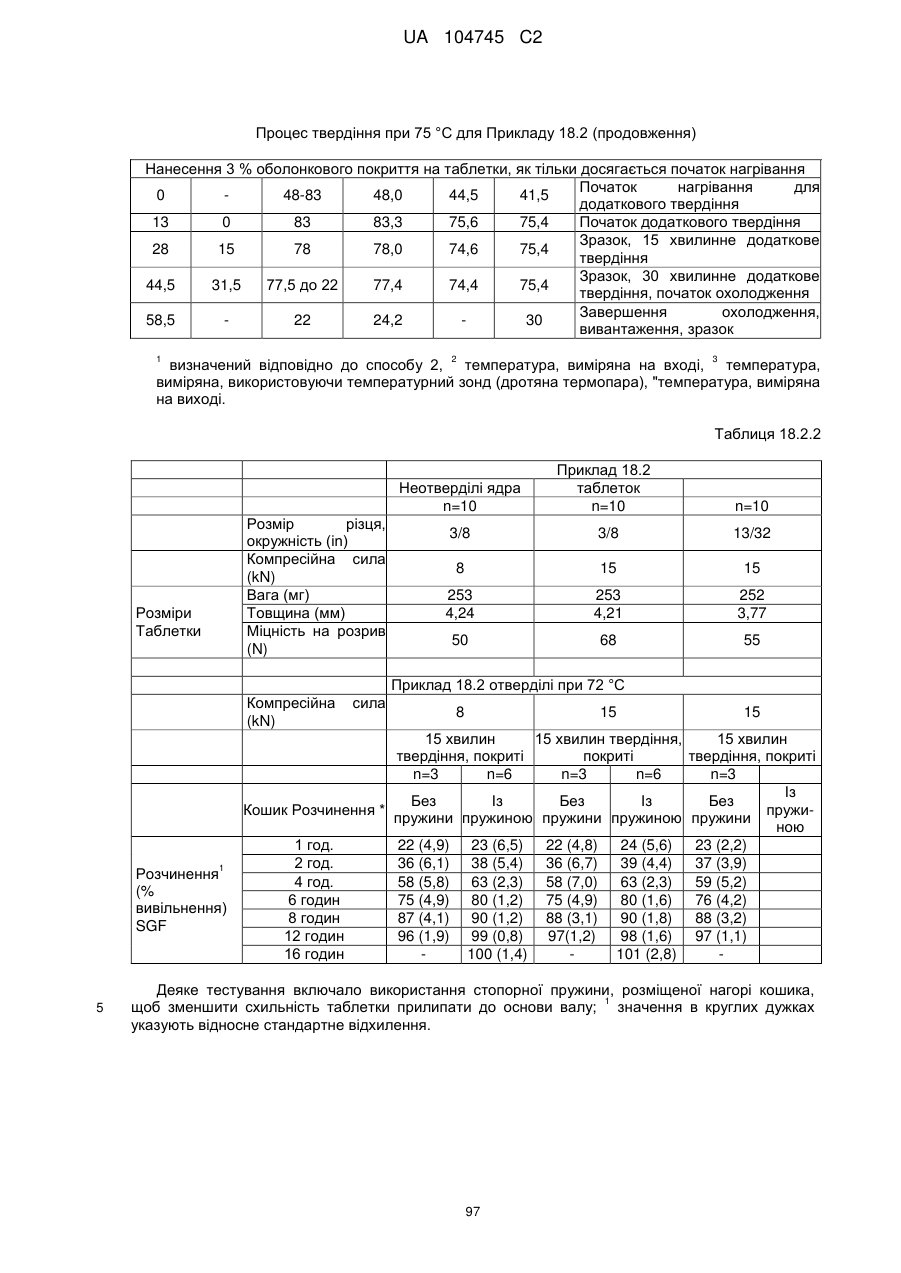
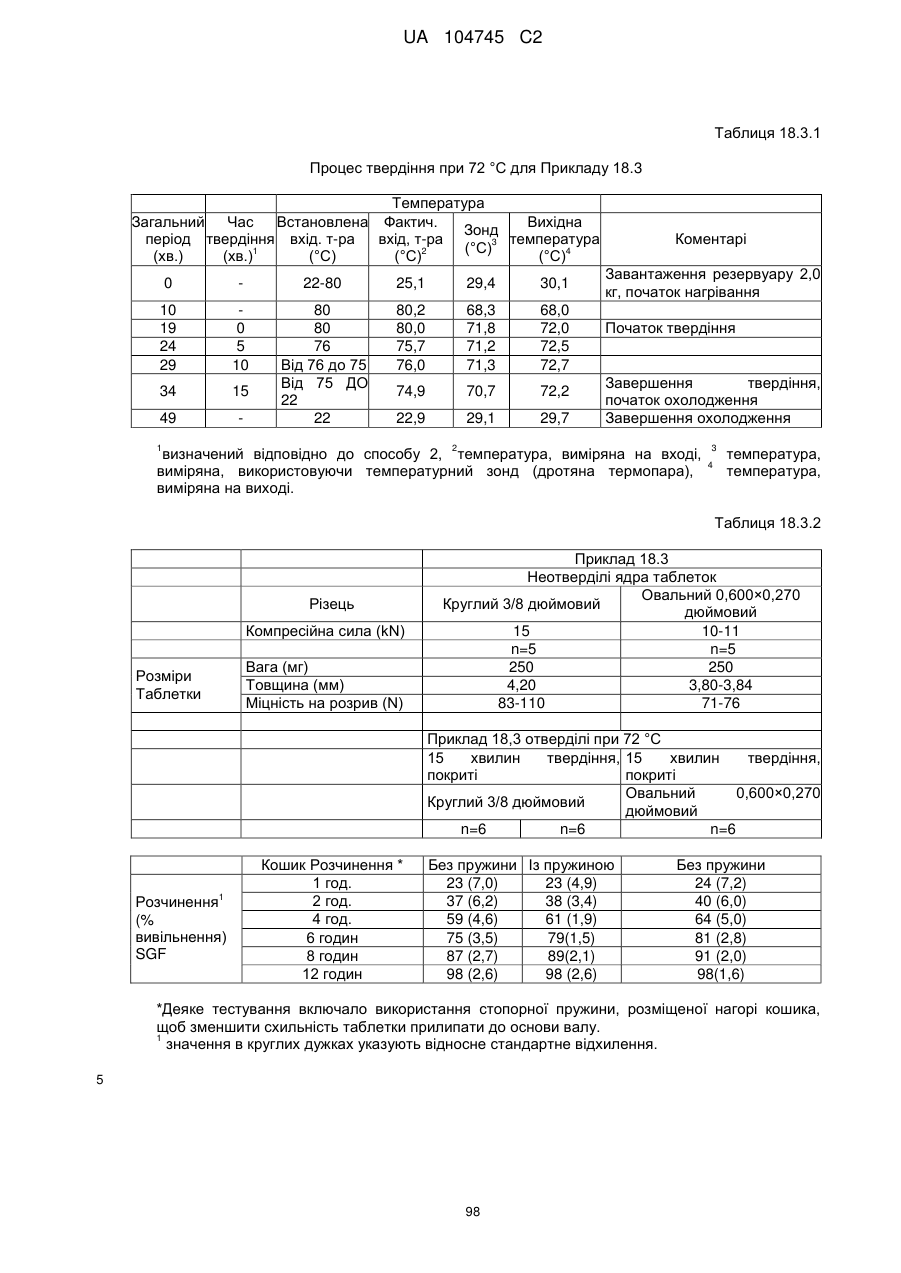
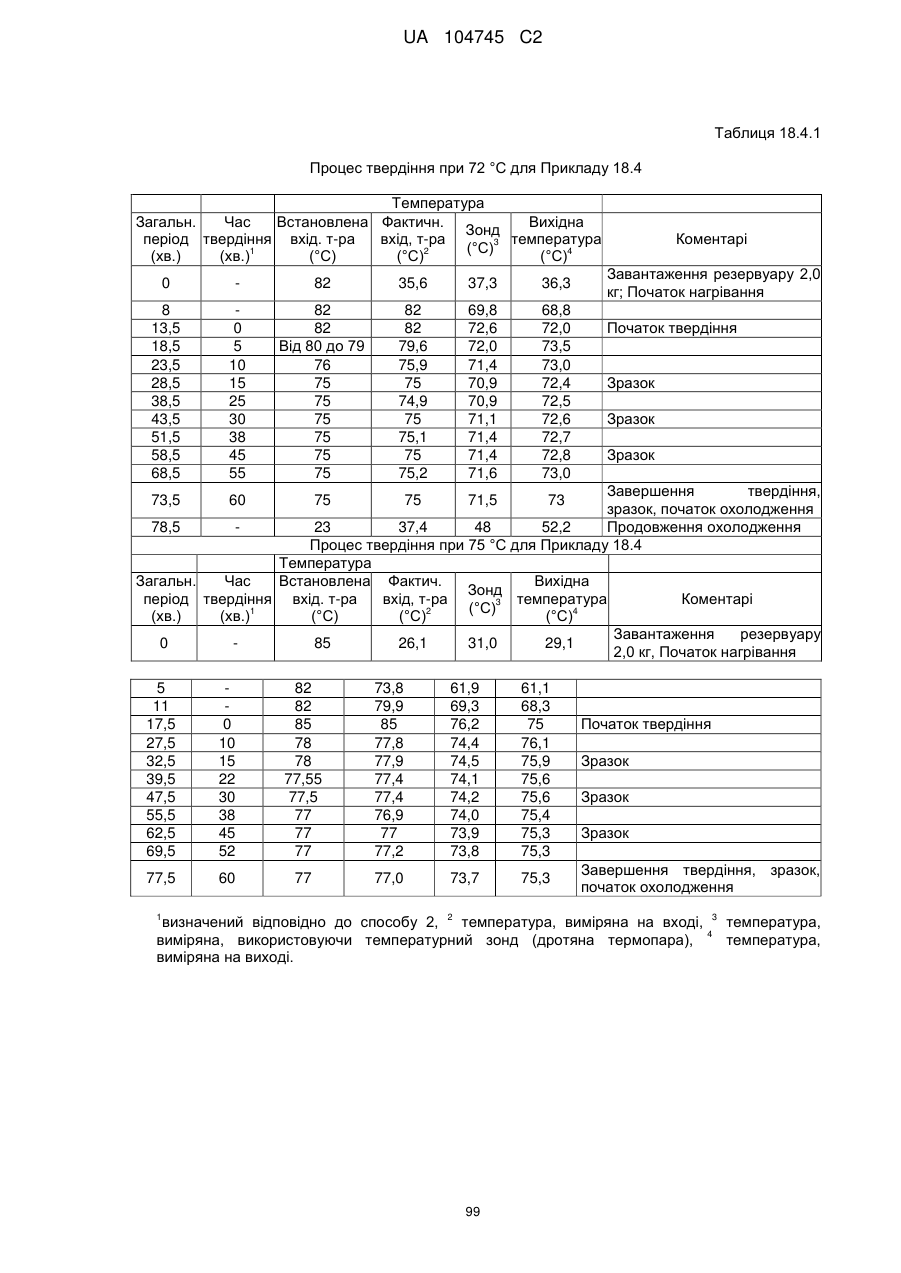
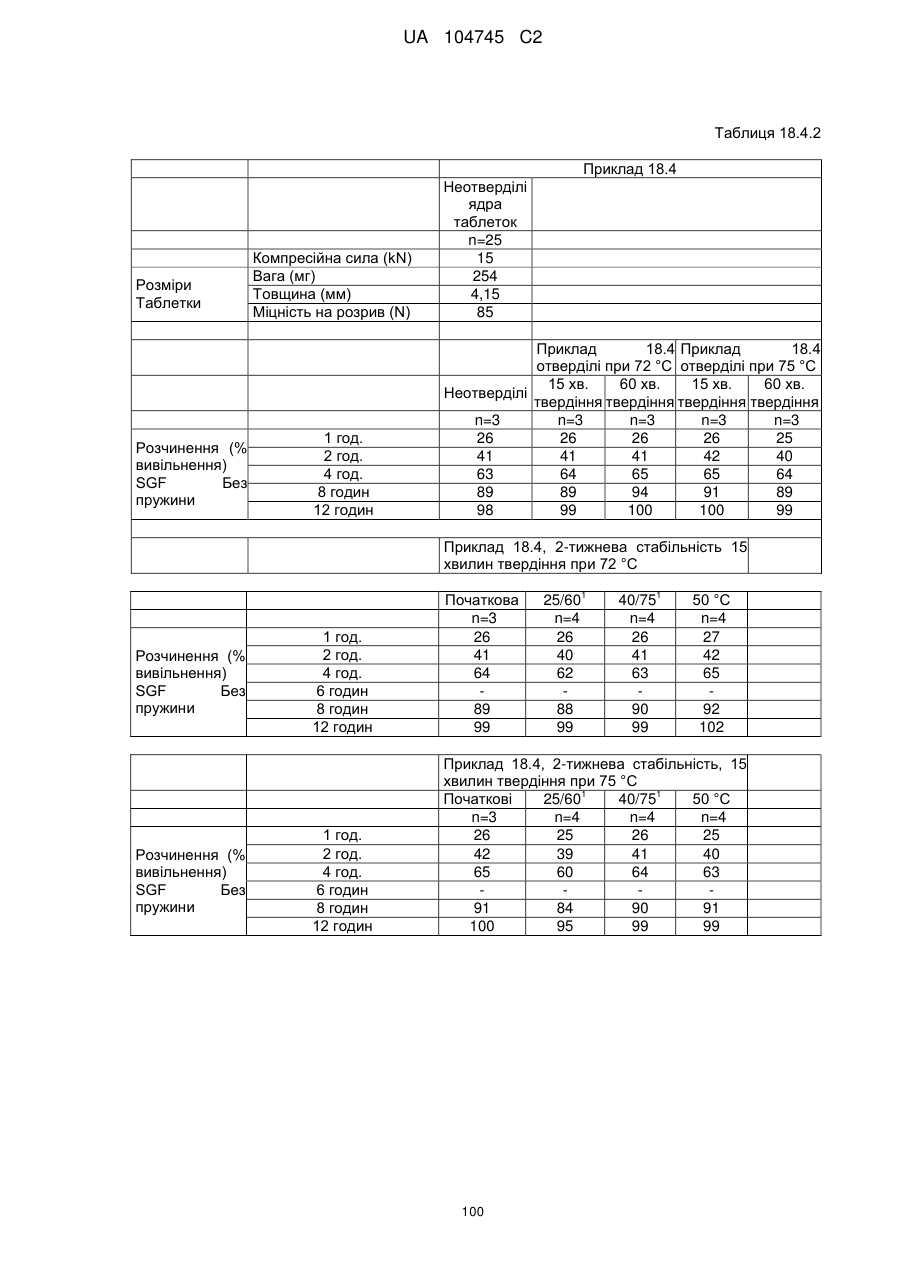
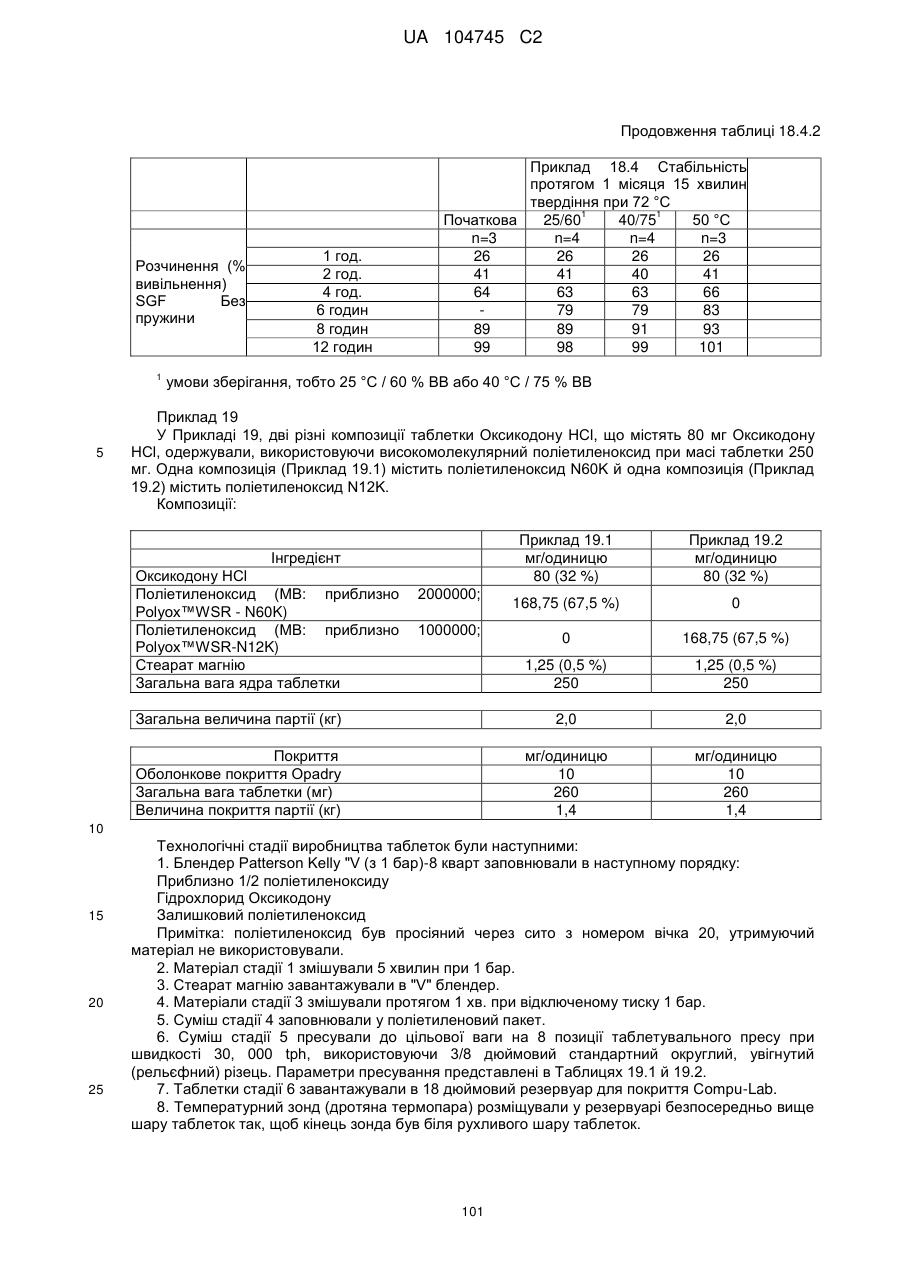
6. Тверда пероральна фармацевтична дозована форма пролонгованого вивільнення за будь-яким з пунктів 1-5, де матрична композиція пролонгованого вивільнення, після зберігання при 25 °С і 60 % відносній вологості (ВВ) протягом принаймні 1 місяця, забезпечує швидкість розчинення, як виміряється на апараті USP 1 (кошик) при 100 об./хв. в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °С, що характеризується кількістю відсотка активного агента, вивільненого за 1, 4 і 12 годин розчинення, що відхиляється не більше, ніж приблизно на 15 % пунктів від відповідної in vitro швидкості розчинення еталонної композиції до зберігання.
7. Тверда пероральна фармацевтична дозована форма пролонгованого вивільнення за пунктом 6, де матричну композицію пролонгованого вивільнення зберігають при 40 °С і 75 % відносній вологості (ВВ).
8. Тверда пероральна фармацевтична дозована форма пролонгованого вивільнення за будь-яким з пунктів 1-5, де матрична композиція пролонгованого вивільнення, після зберігання при 25 °С і 60 % відносній вологості (ВВ) протягом принаймні 1 місяця, містить кількість принаймні одного активного агента в мас. % щодо вказаного на етикетці активного агента матричної композиції пролонгованого вивільнення, що відхиляється не більше, ніж приблизно на 10 % пунктів від відповідної кількості активного агента в мас. % щодо вказаного на етикетці активного агента матричної композиції пролонгованого вивільнення еталонної композиції до зберігання.
9. Тверда пероральна фармацевтична дозована форма пролонгованого вивільнення за пунктом 8, де матричну композицію пролонгованого вивільнення зберігають при 40 °С і 75 % відносній вологості (ВВ).
10. Тверда пероральна фармацевтична дозована форма пролонгованого вивільнення за будь-яким з пунктів 1-5, де дозована форма забезпечує швидкість розчинення, виміряну на апараті USP 1 (кошик) при 100 об./хв. в 900 мл модельованого шлункового соку без ферментів (SGF) при 37°С, між 12,5 і 55 мас. % активного агента, вивільненого після 1 год., між 25 і 65 мас. % активного агента, вивільненого після 2 годин, між 45 і 85 мас. % активного агента, вивільненого після 4 годин і між 55 і 95 мас. % активного агента, вивільненого після 6 годин.
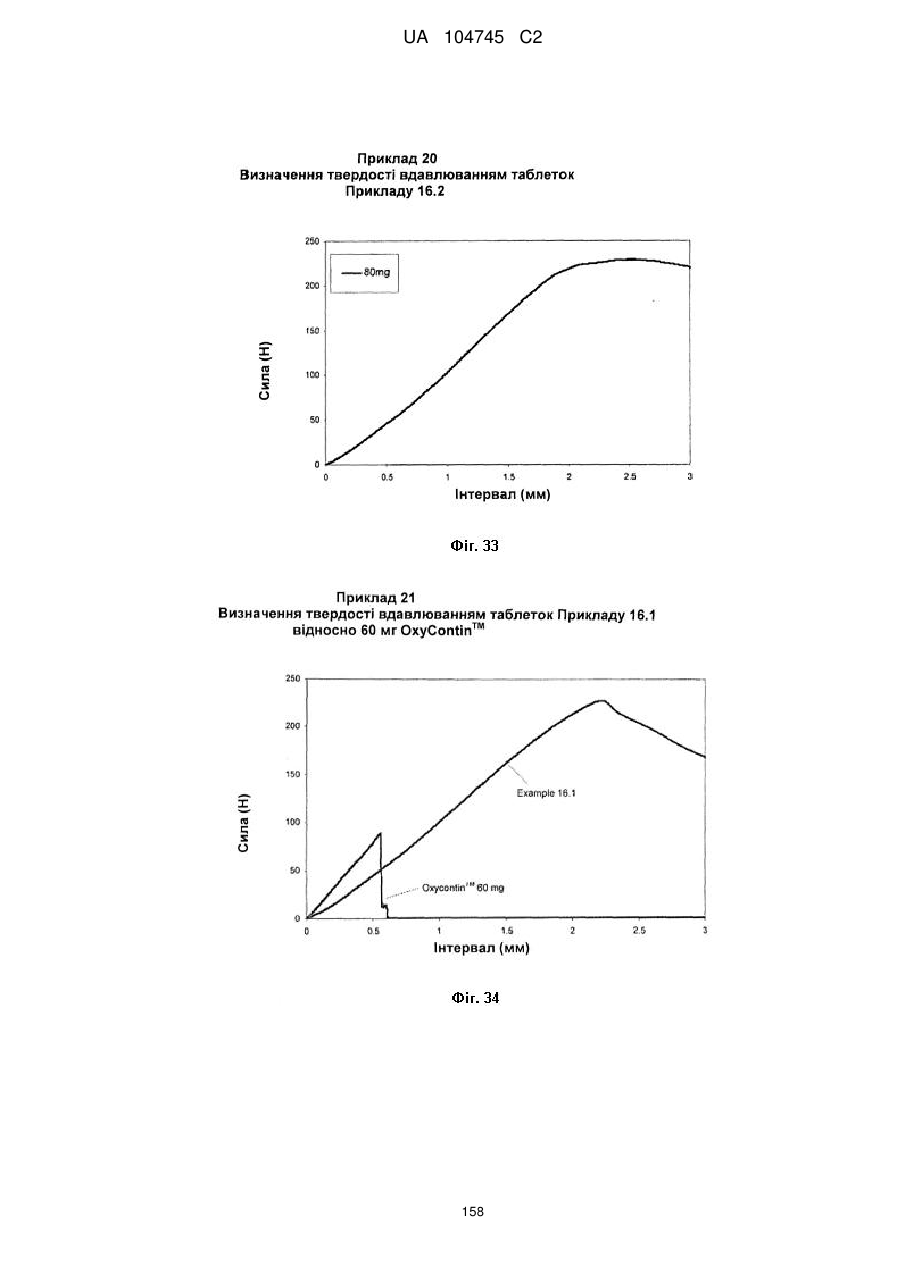
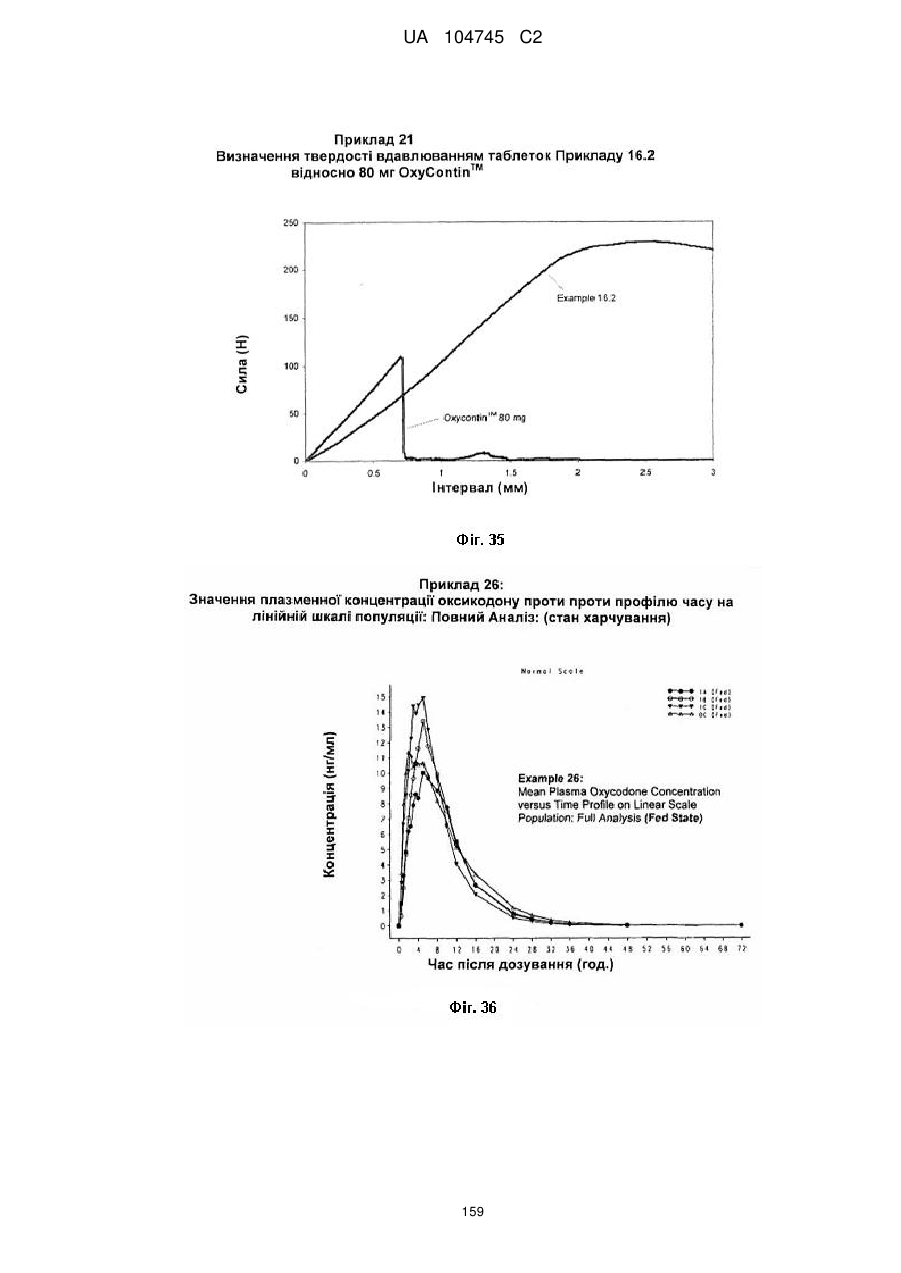
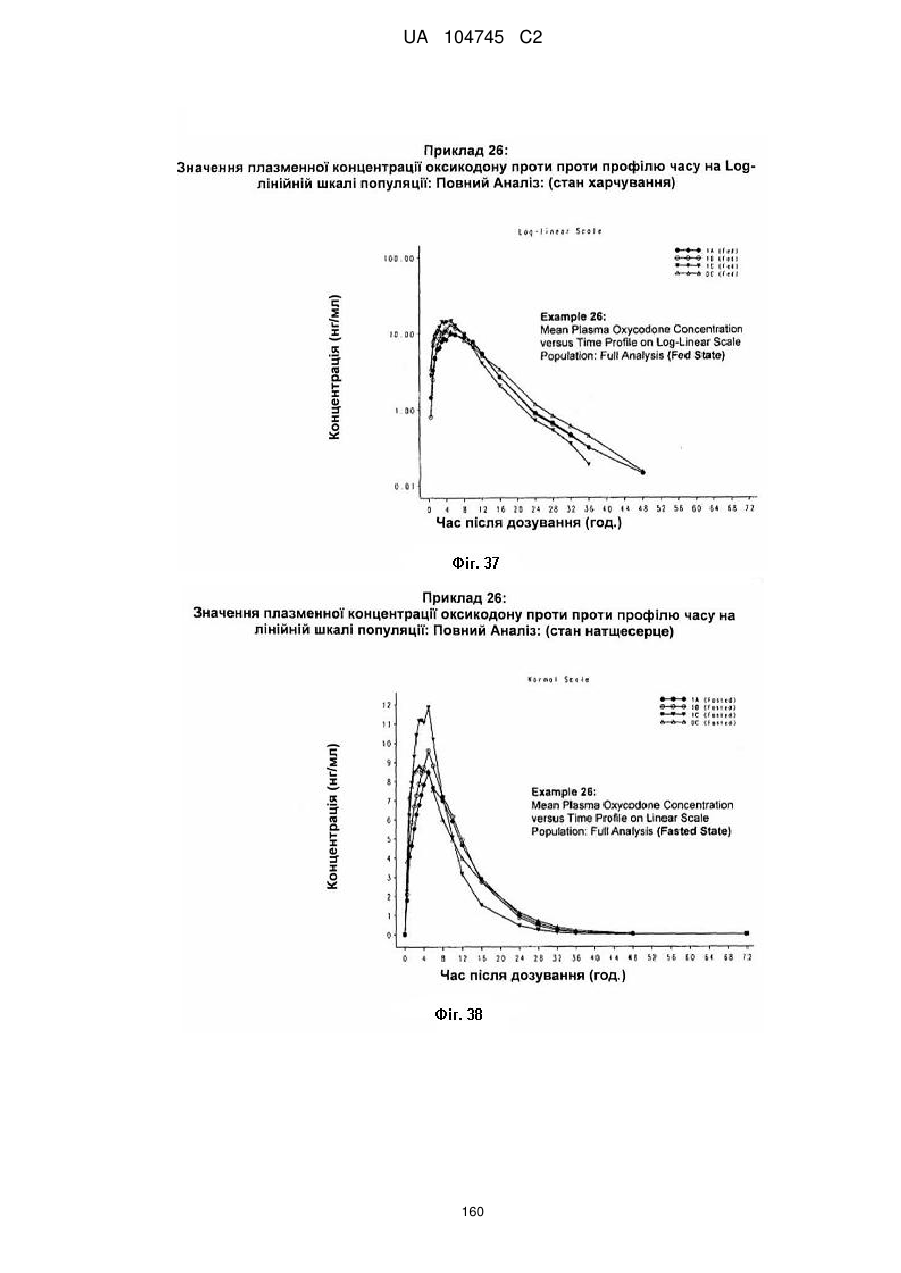
11. Тверда пероральна фармацевтична дозована форма пролонгованого вивільнення за будь-яким з пунктів 1-5, де активним агентом є оксикодону гідрохлорид і де дозована форма при тестуванні в порівняльному клінічному дослідженні є біоеквівалентною комерційному продукту OxyContin™.
12. Тверда пероральна фармацевтична дозована форма пролонгованого вивільнення за будь-яким з пунктів 1-5, де активним агентом є оксикодону гідрохлорид і де дозована форма, що містить 10 мг оксикодону гідрохлориду при тестуванні в порівняльному клінічному дослідженні є біоеквівалентною еталонній таблетці, що містить 10 мг оксикодону гідрохлориду в матричній композиції, і містить:
a) гідрохлорид оксикодону: 10,0 мг/таблетку,
b) лактозу (висушену розпиленням): 69,25 мг/таблетку,
c) Повідон: 5,0 мг/таблетку,
d) Eudragit ® RS 30D (твердий): 10,0 мг/таблетку,
e) Triacetin®: 2,0 мг/таблетку,
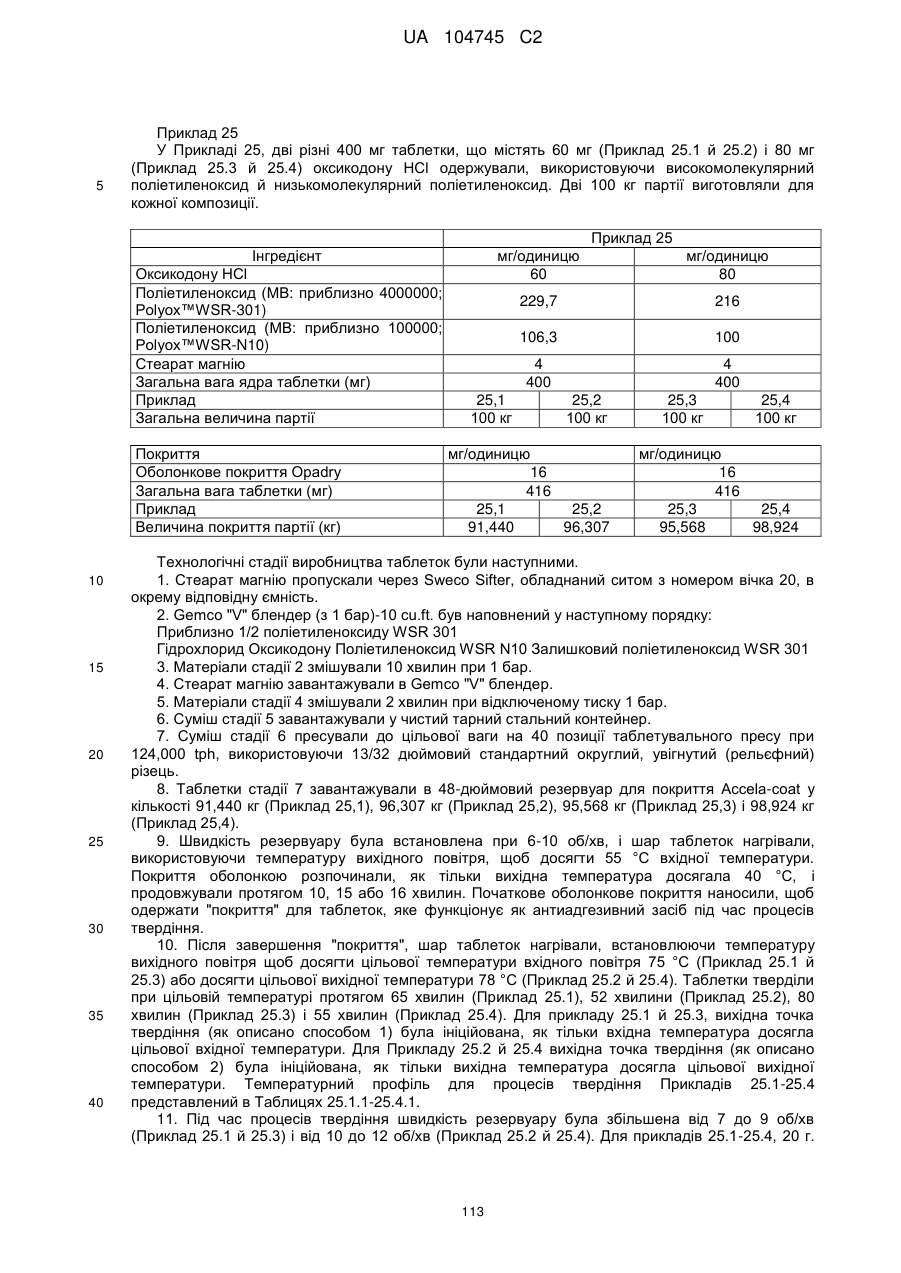
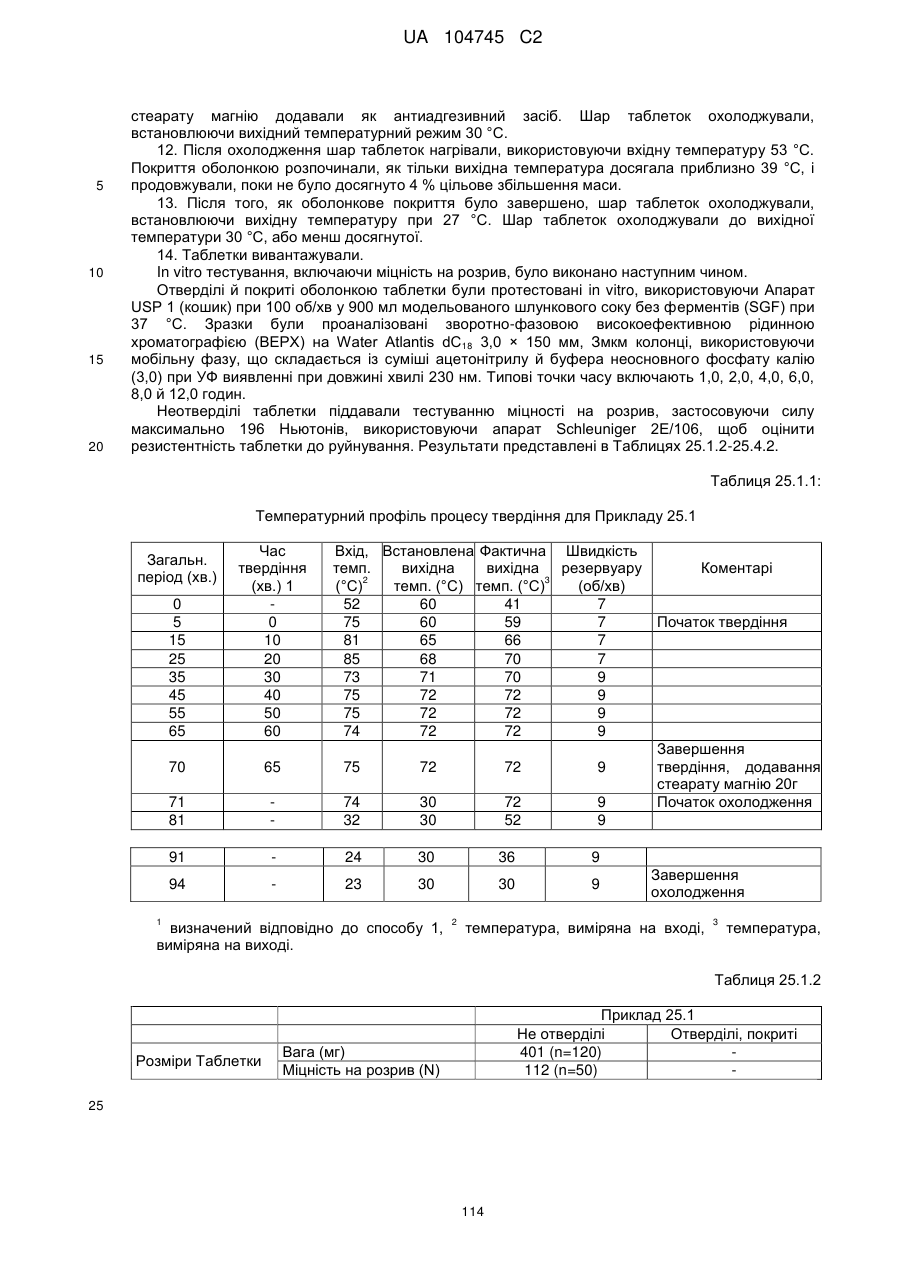
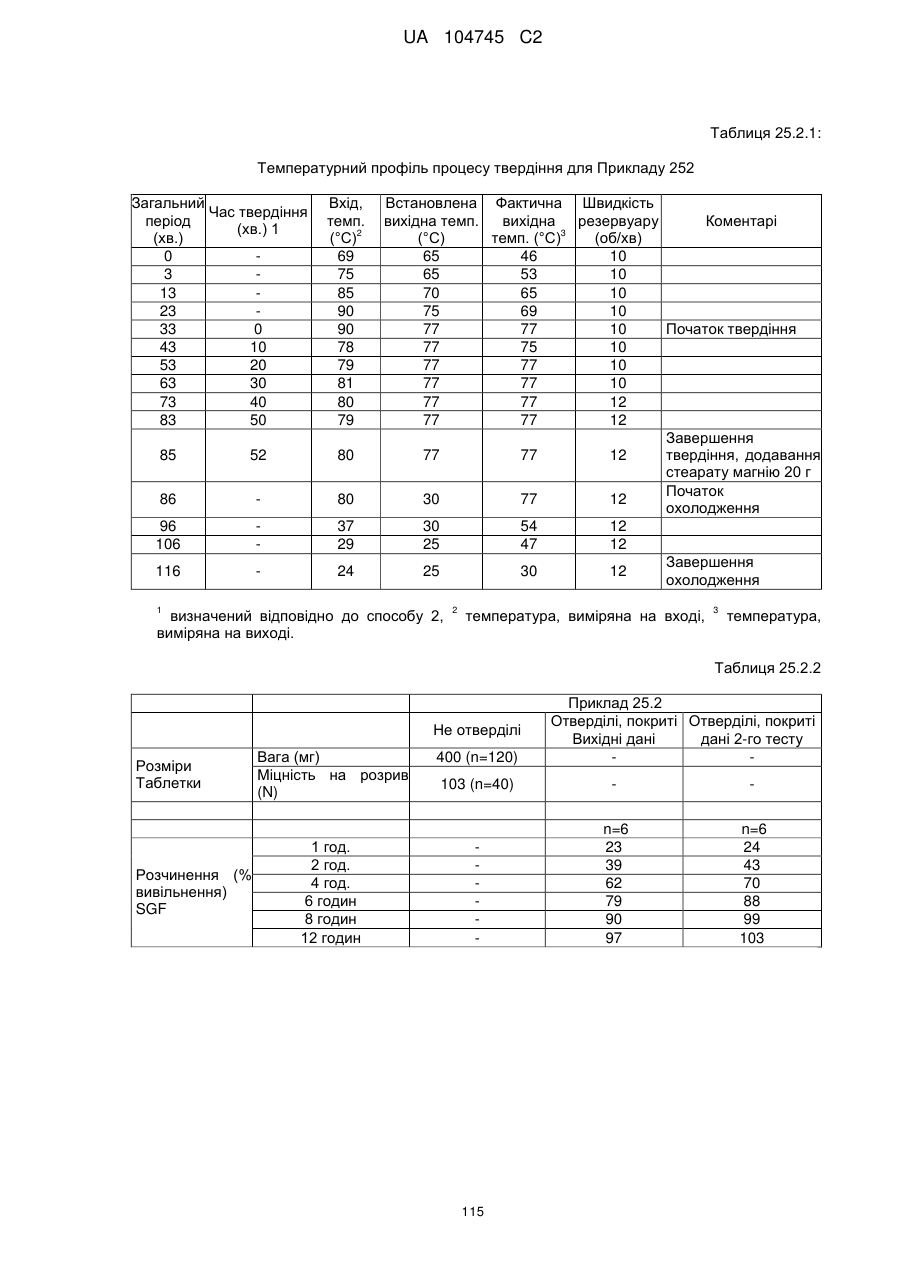
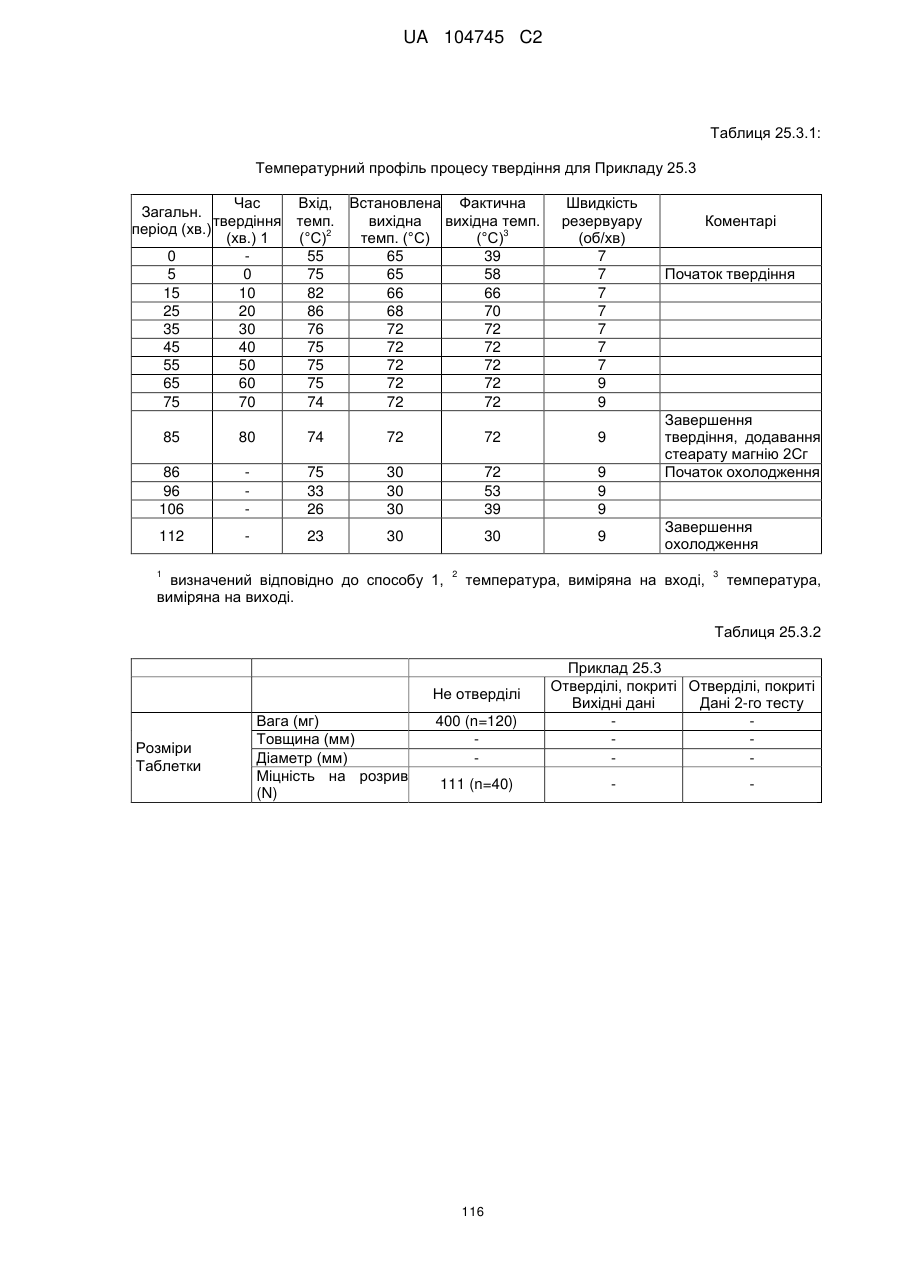
f) стеариловий спирт: 25,0 мг/таблетку,
g) тальк: 2,5 мг/таблетку,
h) стеарат магнію: 1,25 мг/таблетку;
і, де еталонна таблетка виготовлена використовуючи наступні стадії:
1) Eudragit® RS 30D і Triacetin® комбінували, пропускаючи через сито 60 меш, і змішували ножовим міксером приблизно 5 хвилин або до утворення однорідної дисперсії,
2) оксикодону HCl, лактозу і повідон поміщали в камеру гранулятора/сушарки псевдозрідженого шару (FBD), і суспензію розпилювали на порошок у псевдозрідженому шарі,
3) після розпилення гранулят пропускали через #12 сито, якщо необхідно зменшити грудкування,
4) сухий гранулят поміщали в міксер,
5) тим часом, необхідну кількість стеарилового спирту розплавляли при температурі приблизно 70 °С,
6) розплавлений стеариловий спирт вводили у гранулят перемішуванням,
7) вощений гранулят переносили до гранулятора/сушарки псевдозрідженого шару або контейнерів і дозволяли охолодитись до кімнатної температури або нижче,
8) охолоджений гранулят потім пропускали через #12 сито,
9) вощений гранулят поміщали в міксер/блендер і змащували необхідною кількістю тальку і стеарату магнію приблизно протягом 3 хвилин,
10) гранулят пресували у таблетки по 125 мг на відповідній таблетувальній машині.
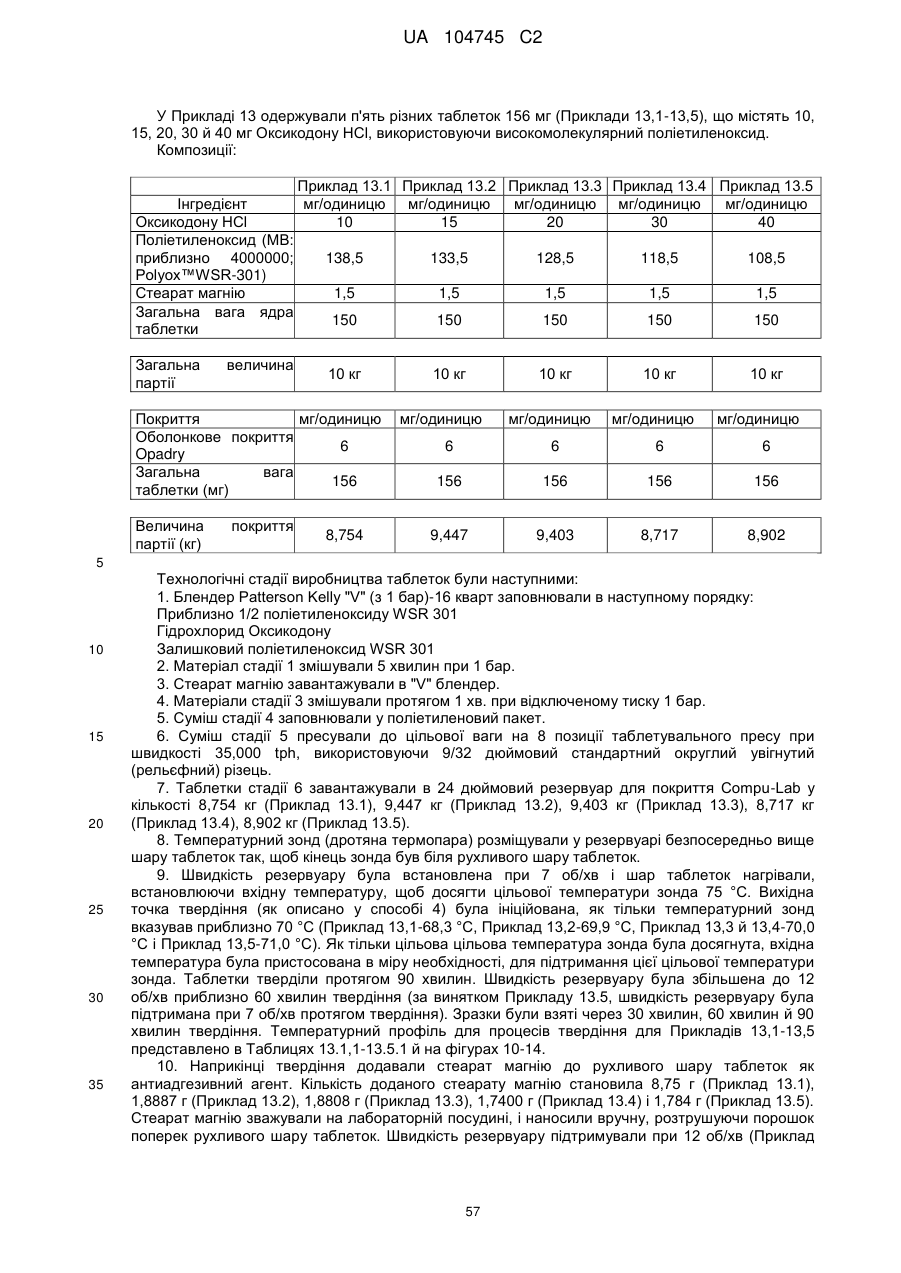
13. Дозована форма пролонгованого вивільнення за будь-яким з пунктів 1-12, де дозована форма містить 5 мг, 7,5 мг, 10 мг, 15 мг, 20 мг, 30 мг, 40 мг, 45 мг, 60 мг, 80 мг, 90 мг, 120 мг або 160 мг оксикодону гідрохлориду.
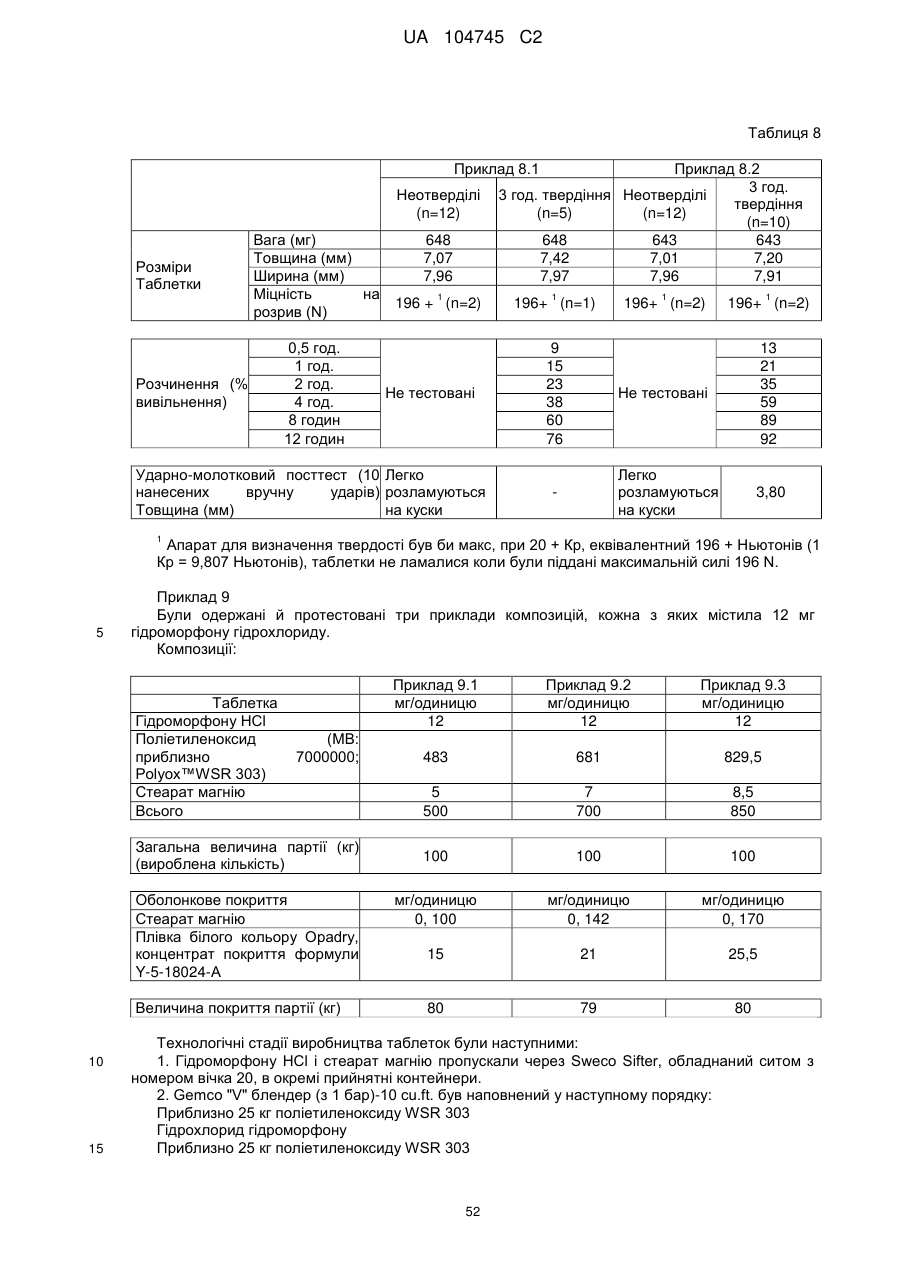
14. Дозована форма пролонгованого вивільнення за будь-яким з пунктів 1-13, де опіоїдним анальгетиком є оксикодону гідрохлорид, що має рівень 14-гідроксикодеїнону менше, ніж приблизно 25 млн. ч., переважно менше, ніж приблизно 15 млн. ч, менше, ніж приблизно 10 млн. ч., або менше, ніж приблизно 5 млн. ч.
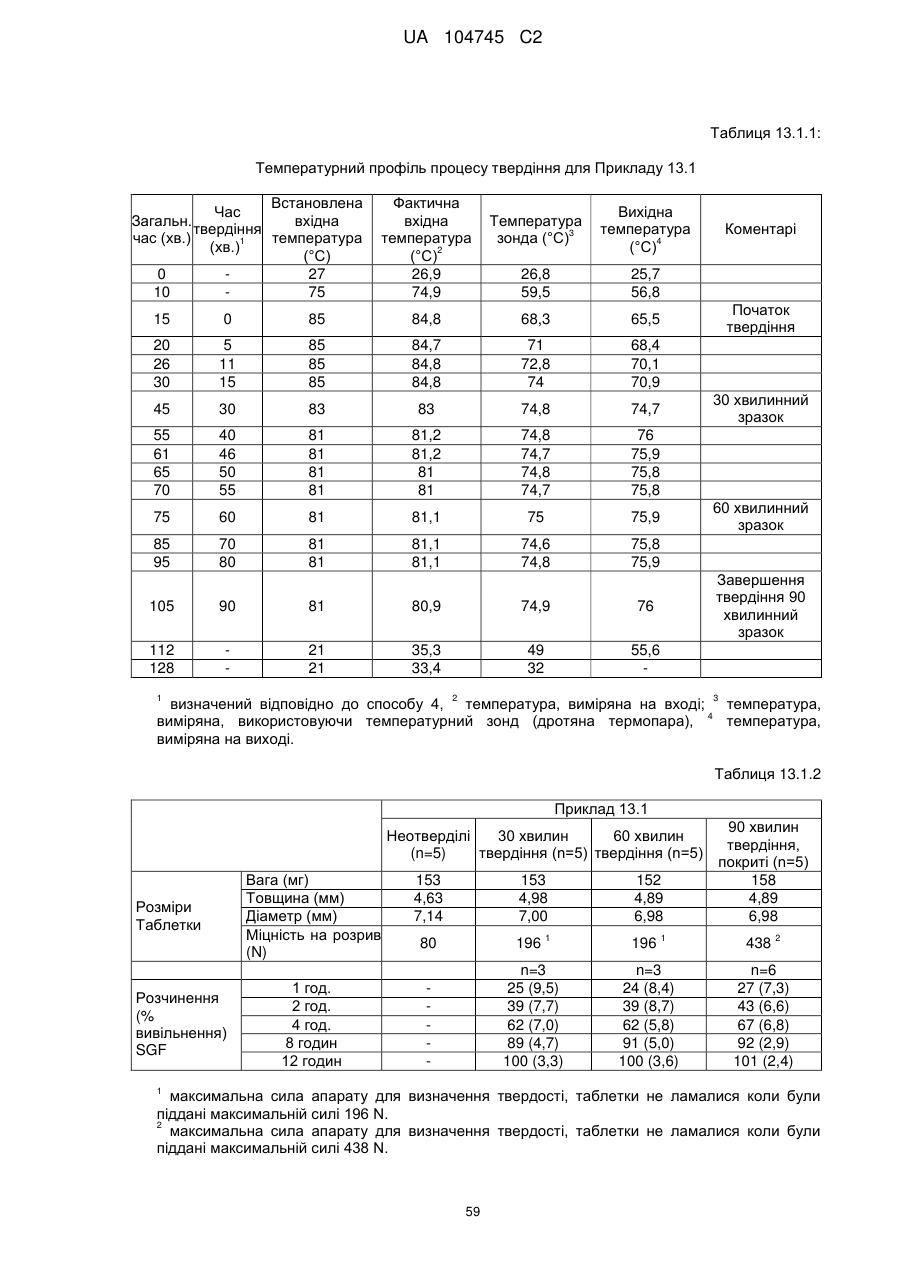
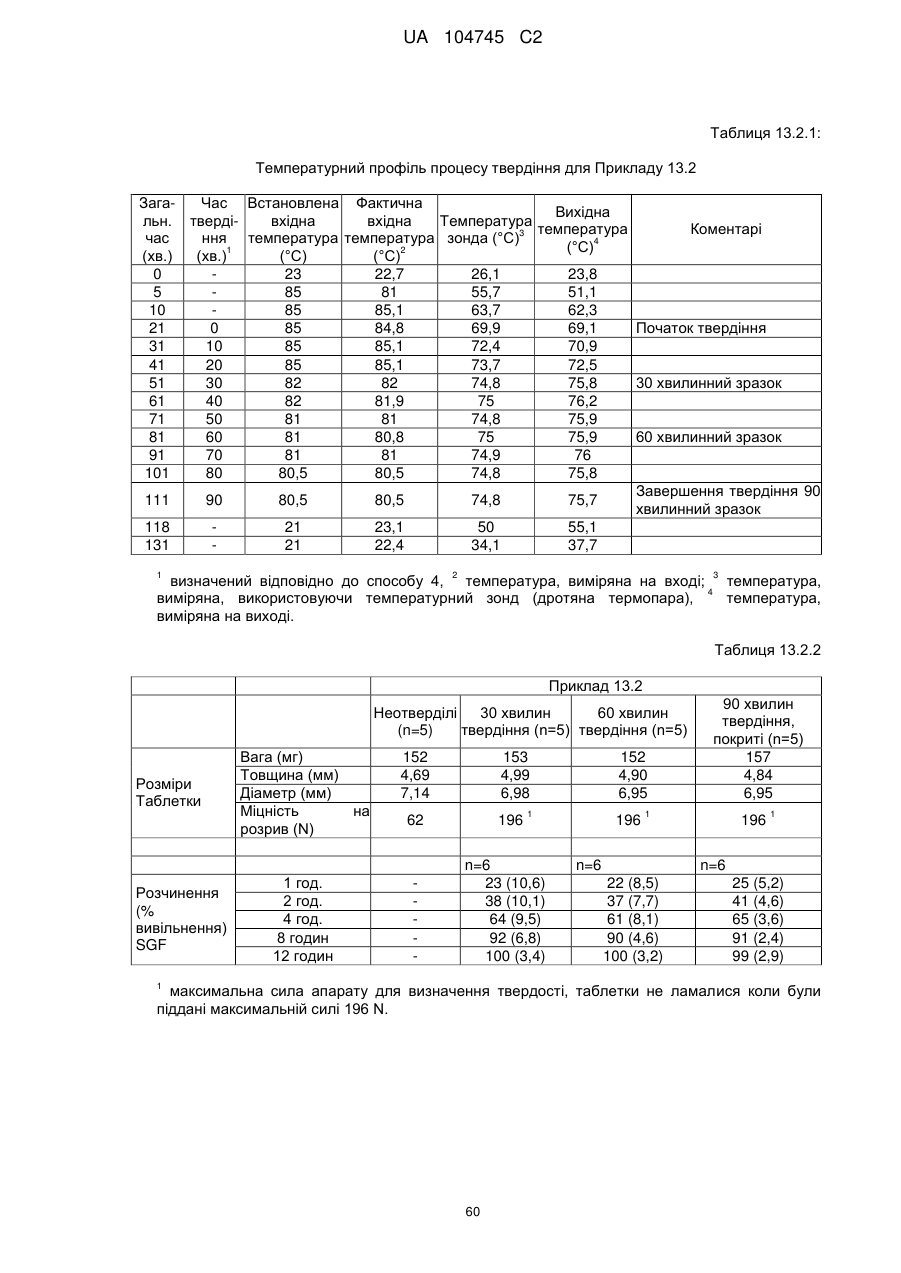
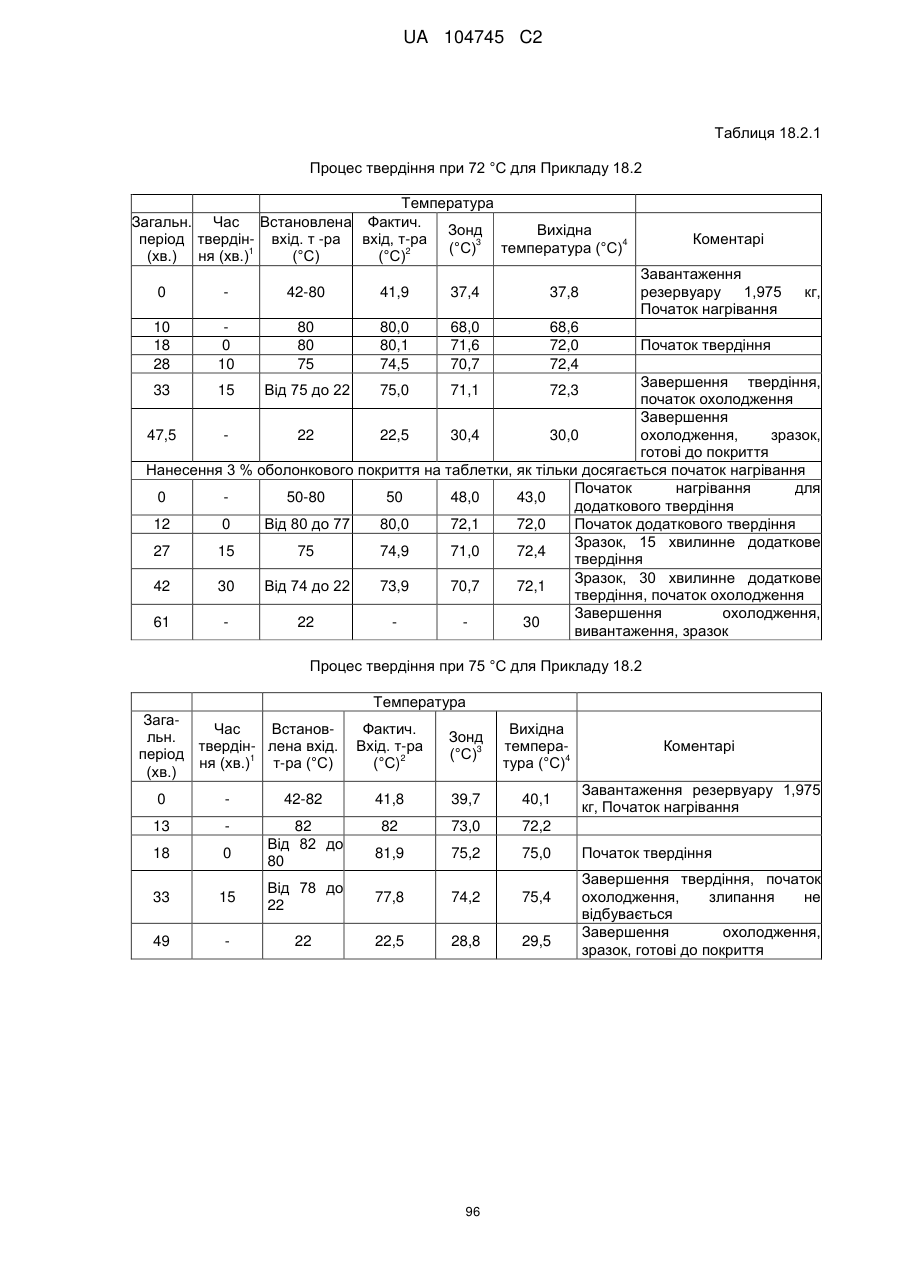
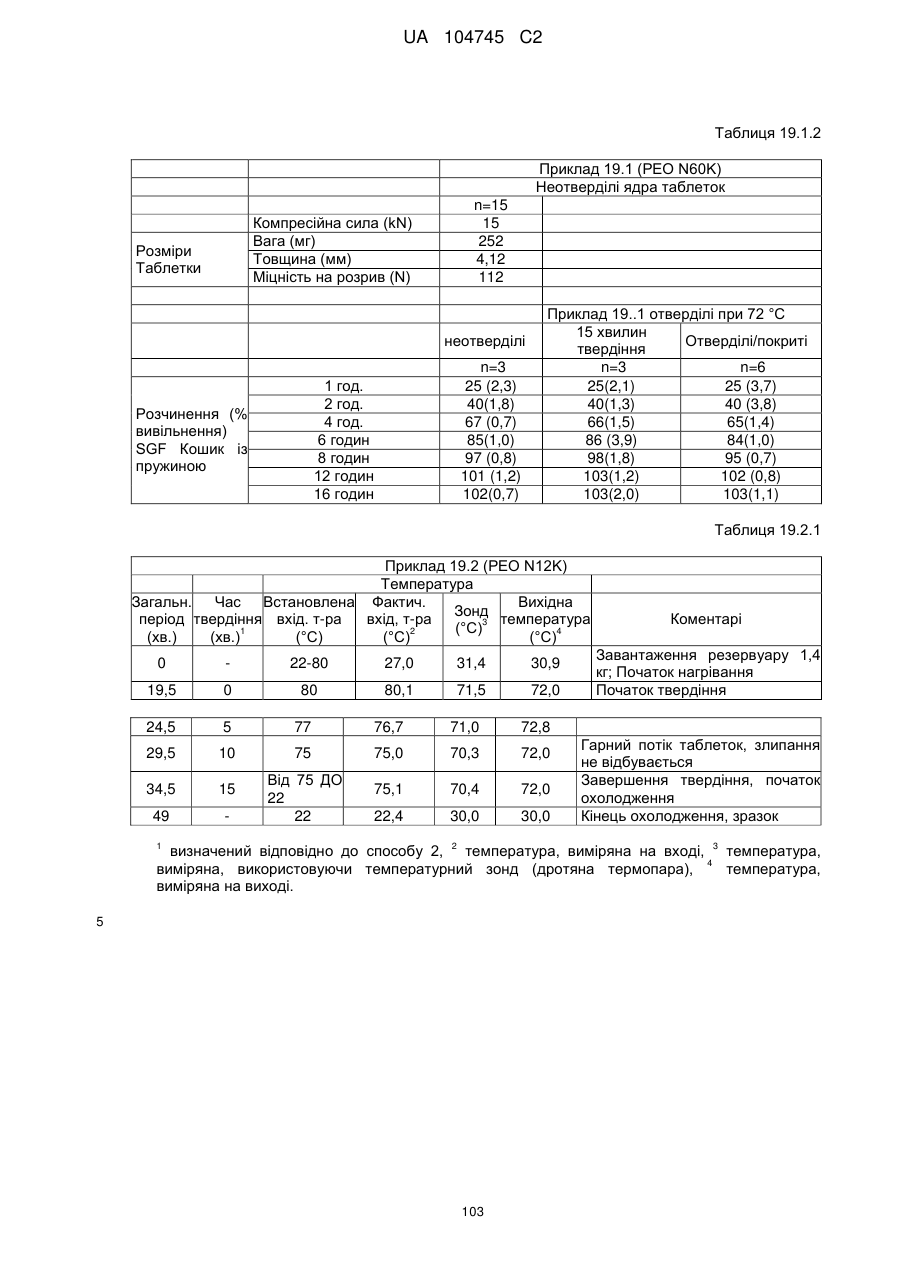
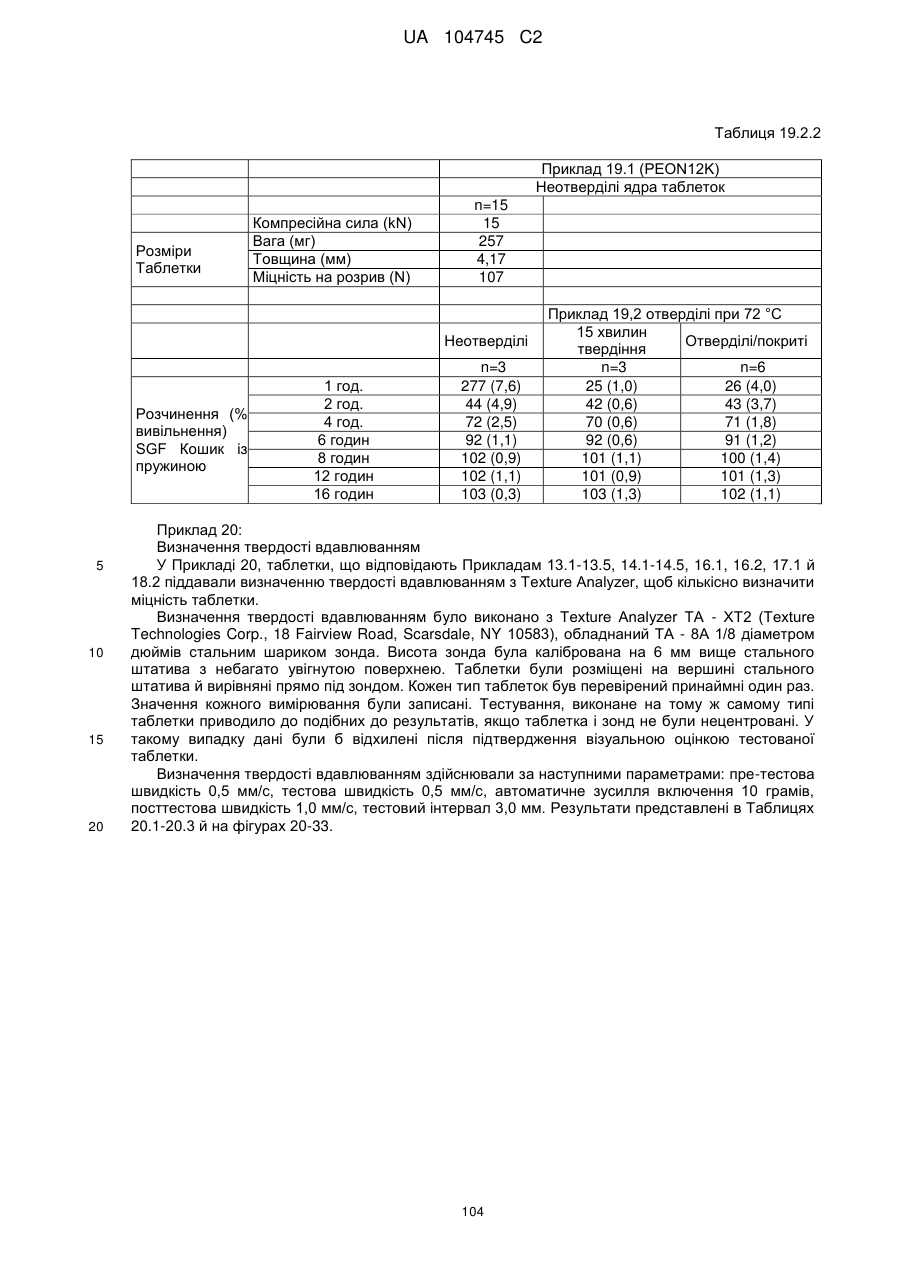
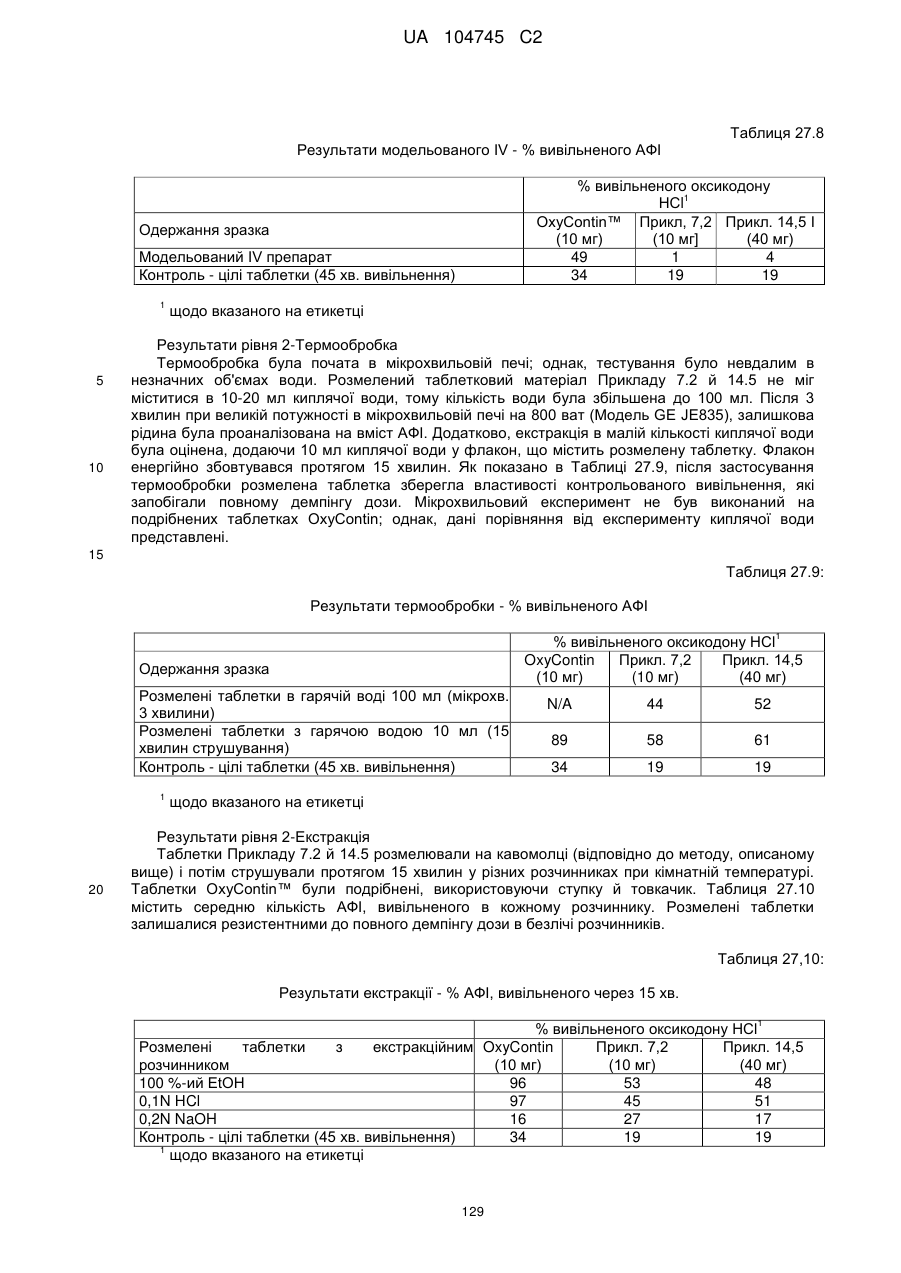
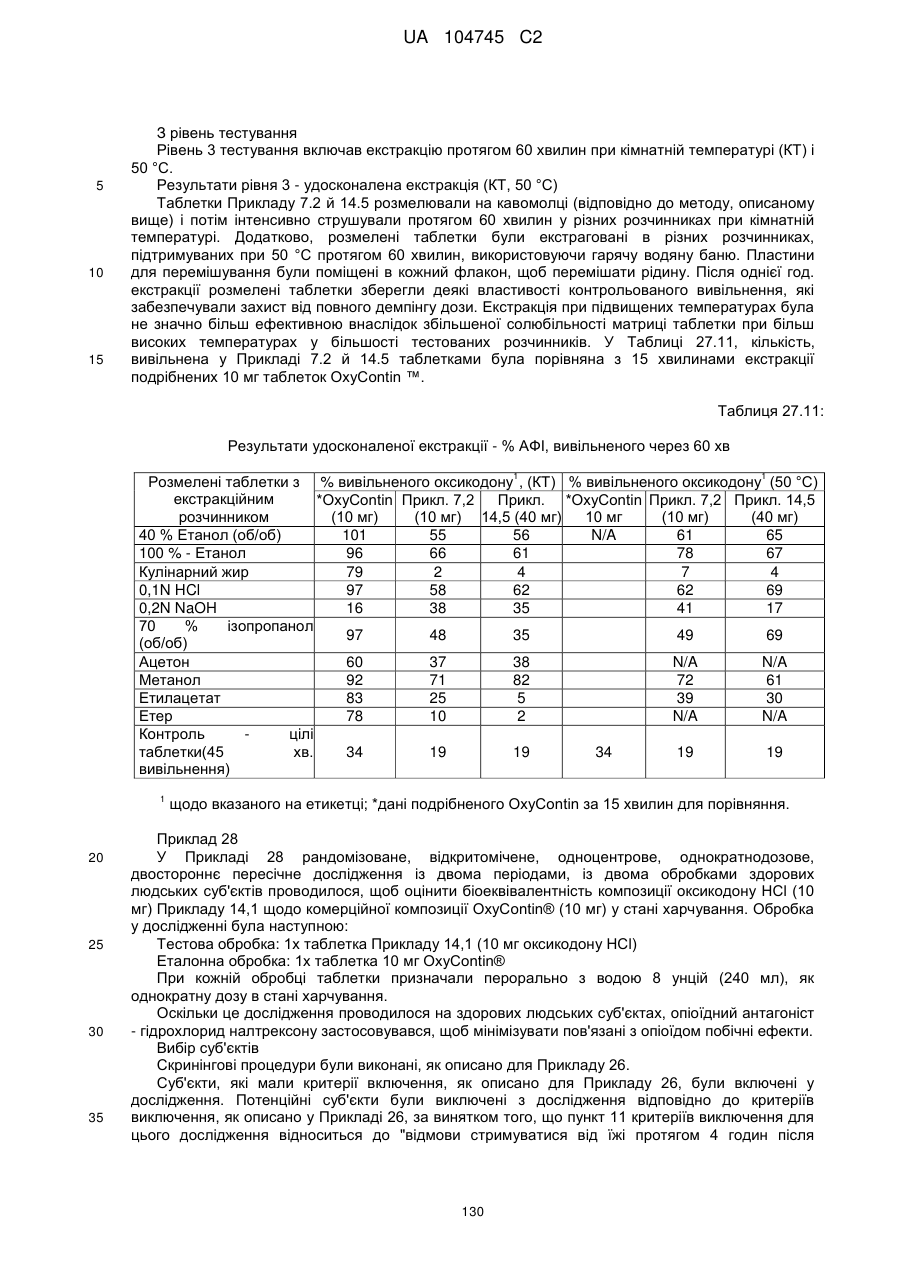
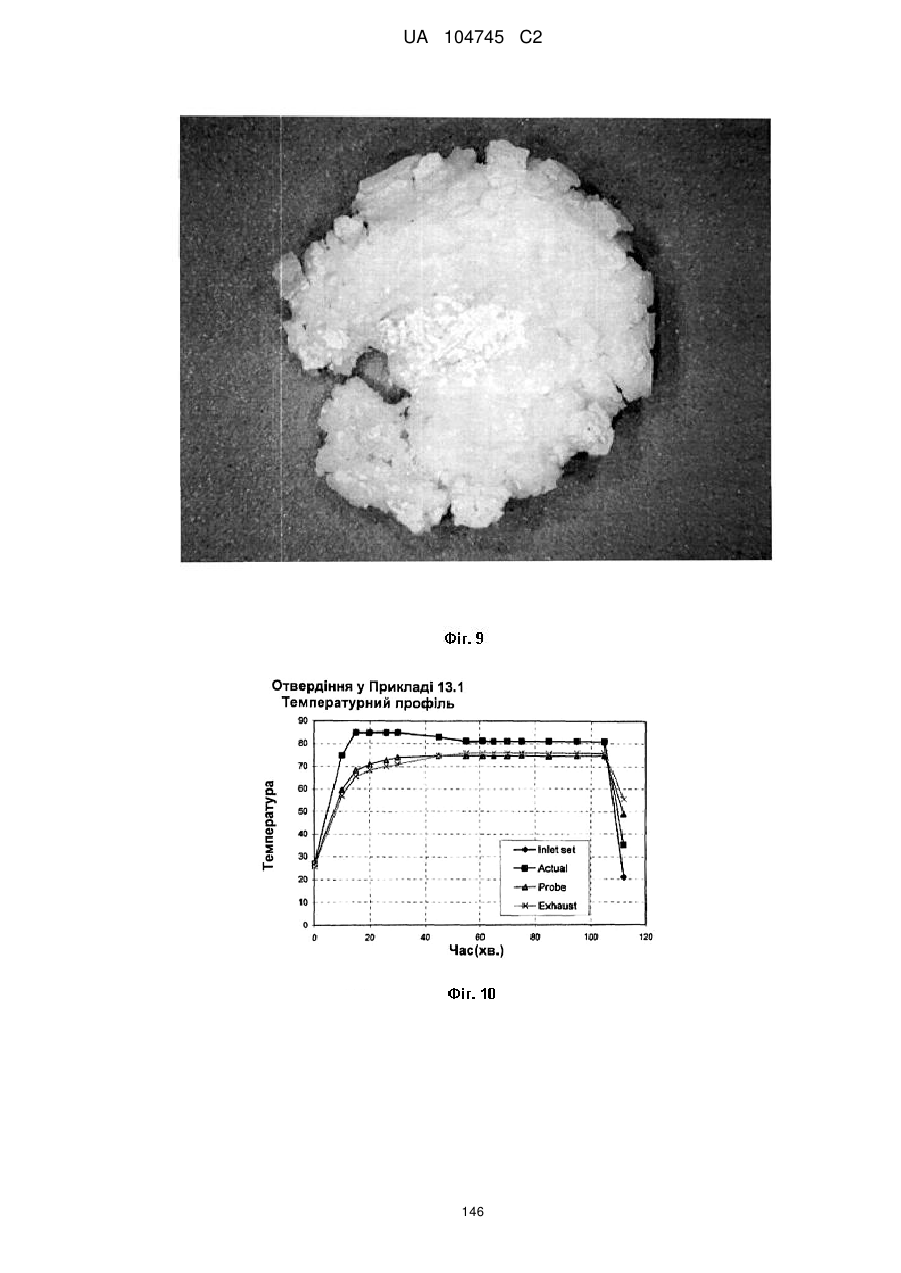
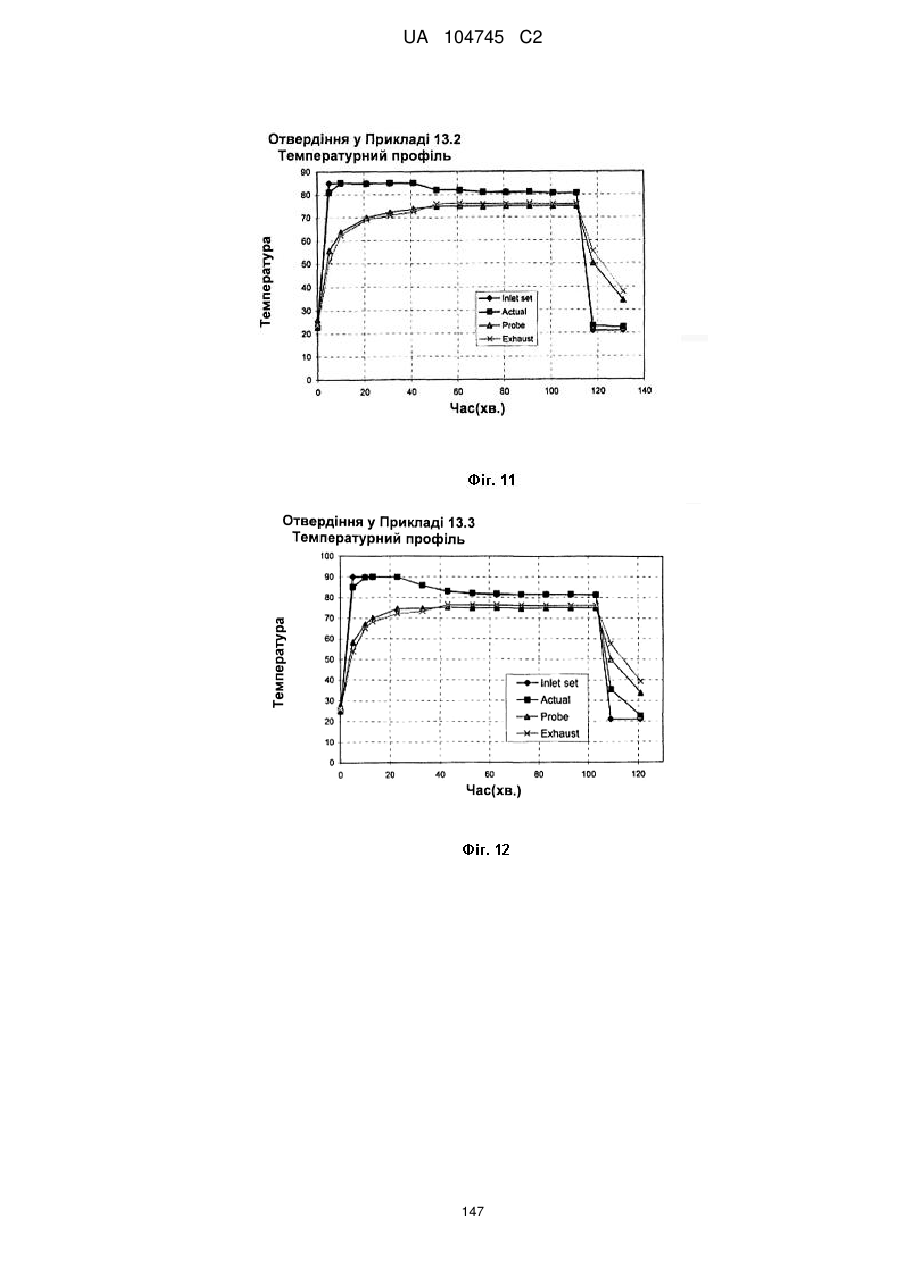
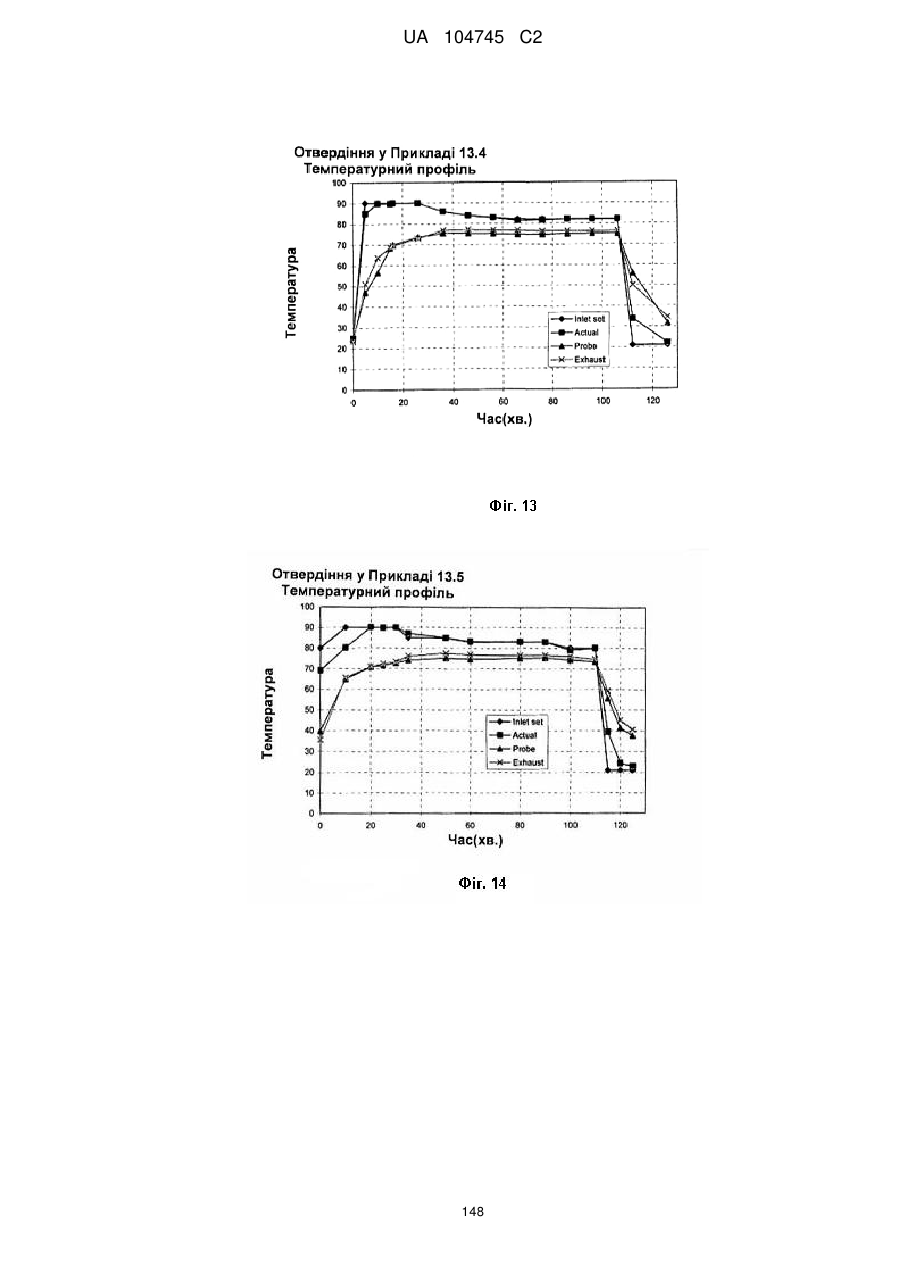
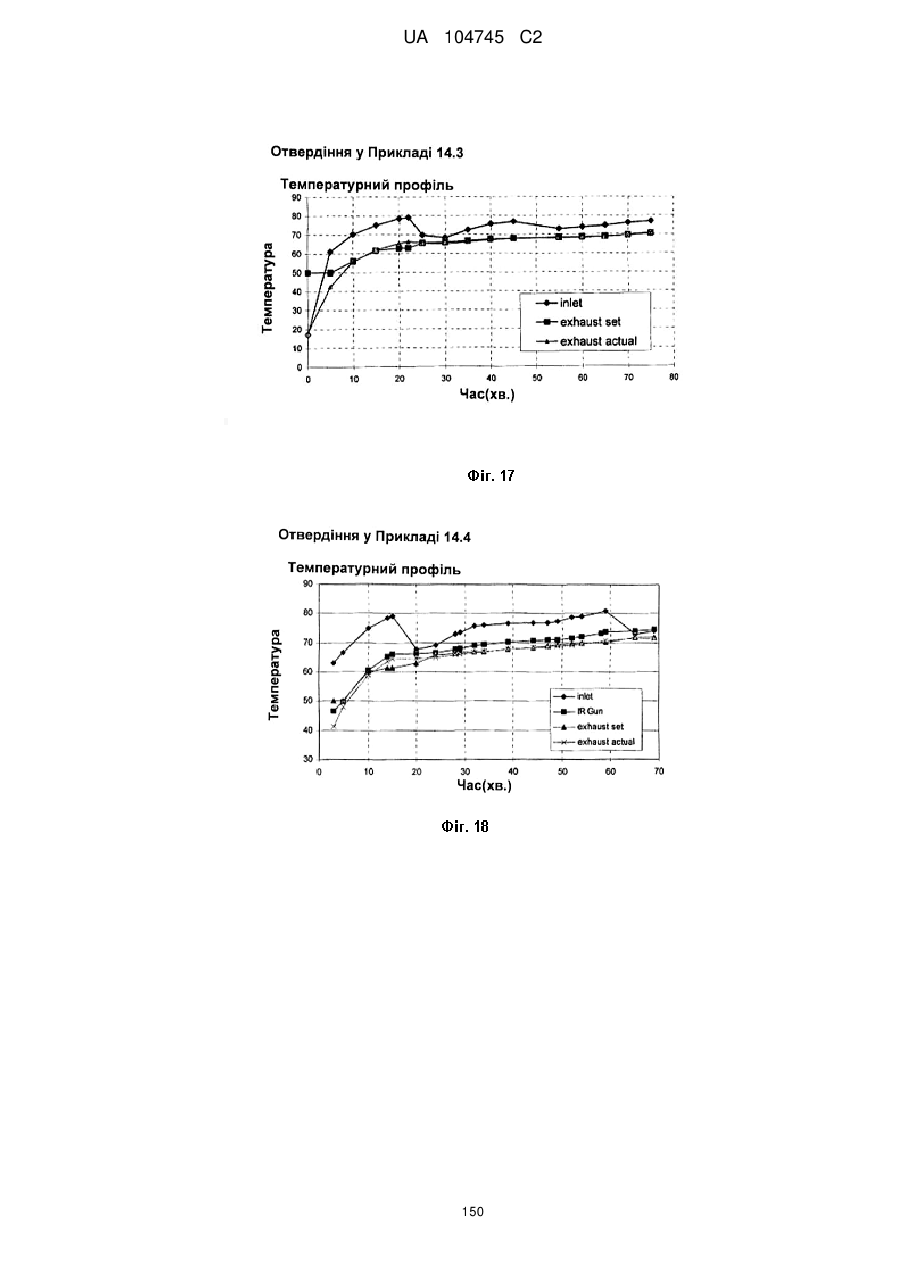
15. Дозована форма пролонгованого вивільнення за будь-яким з пунктів 1-14, що є у формі таблеток, одержаних безпосереднім пресуванням композиції і отвердженням, піддаючи згадувані таблетки температурі принаймні приблизно 60 °С або принаймні приблизно 62 °С протягом інтервалу часу принаймні приблизно 1 хв., переважно принаймні приблизно 5 хвилин або принаймні приблизно 15 хвилин.
16. Дозована форма пролонгованого вивільнення за будь-яким з пунктів 1-15, що перебуває у формі таблетки, яка є покритою шаром порошку поліетиленоксиду, з утворенням таблетки, яка має ядро і шар поліетиленоксиду, що оточує ядро таблетки.
17. Дозована форма пролонгованого вивільнення за будь-яким з пунктів 1-15, що перебуває у формі бі- або мультишарової таблетки, де один із шарів містить композицію пролонгованого вивільнення і один з інших шарів містить композицію швидкого вивільнення.
18. Дозована форма пролонгованого вивільнення за пунктом 17, де композиція пролонгованого вивільнення і композиція швидкого вивільнення містить ті ж самі або різні активні агенти.
19. Дозована форма пролонгованого вивільнення за пунктом 17, де композиція пролонгованого вивільнення включає опіоїдний анальгетик і композиція швидкого вивільнення включає неопіоїдний анальгетик.
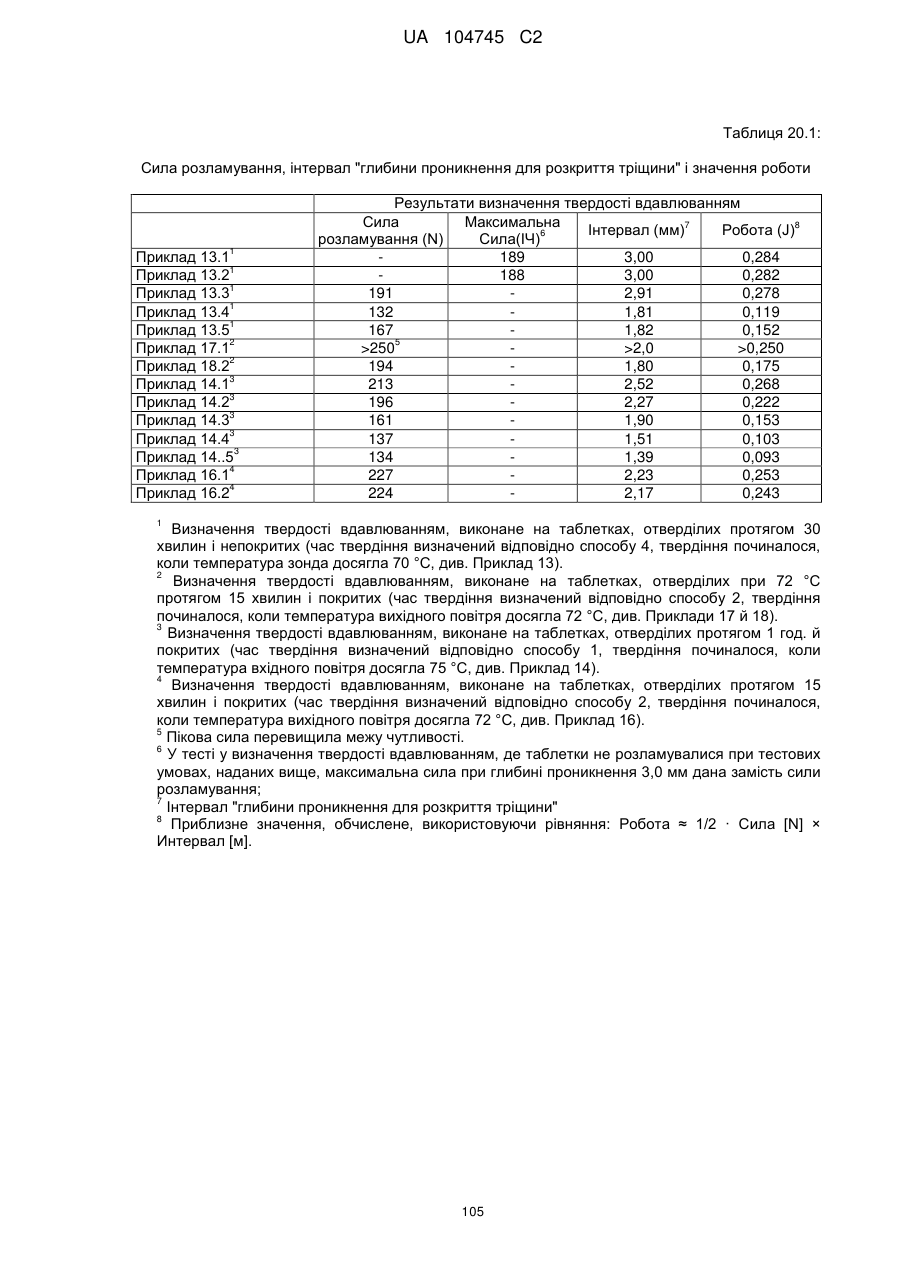
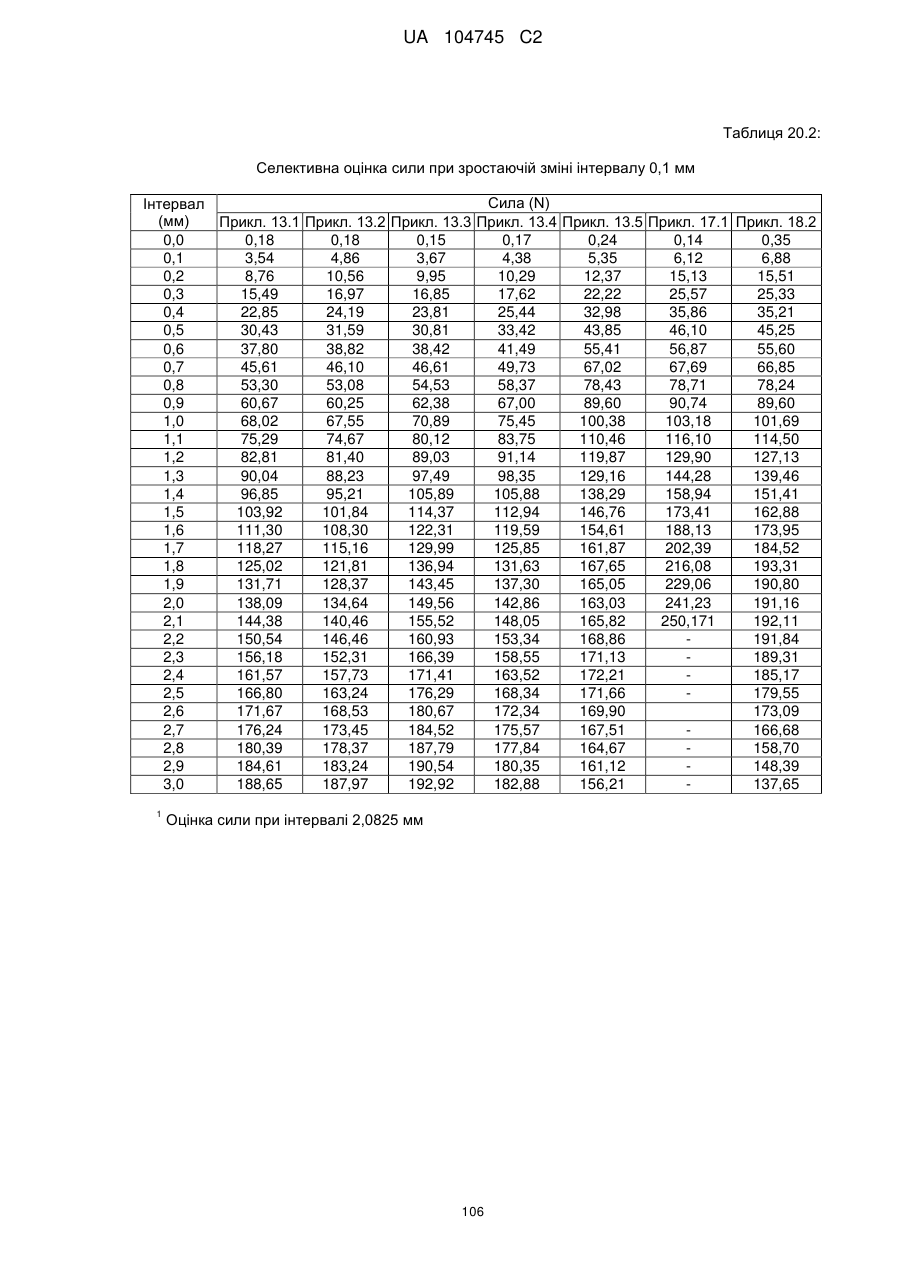
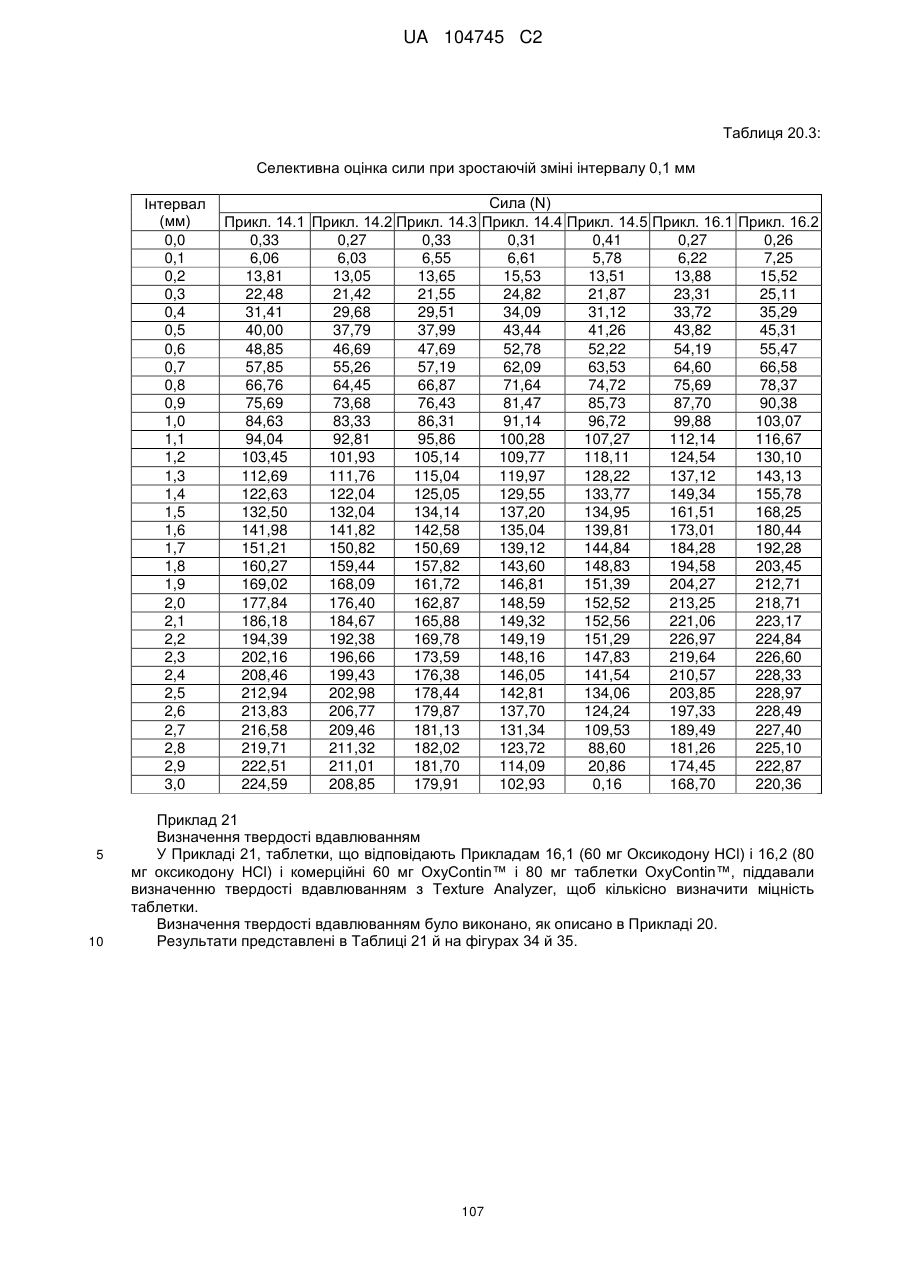
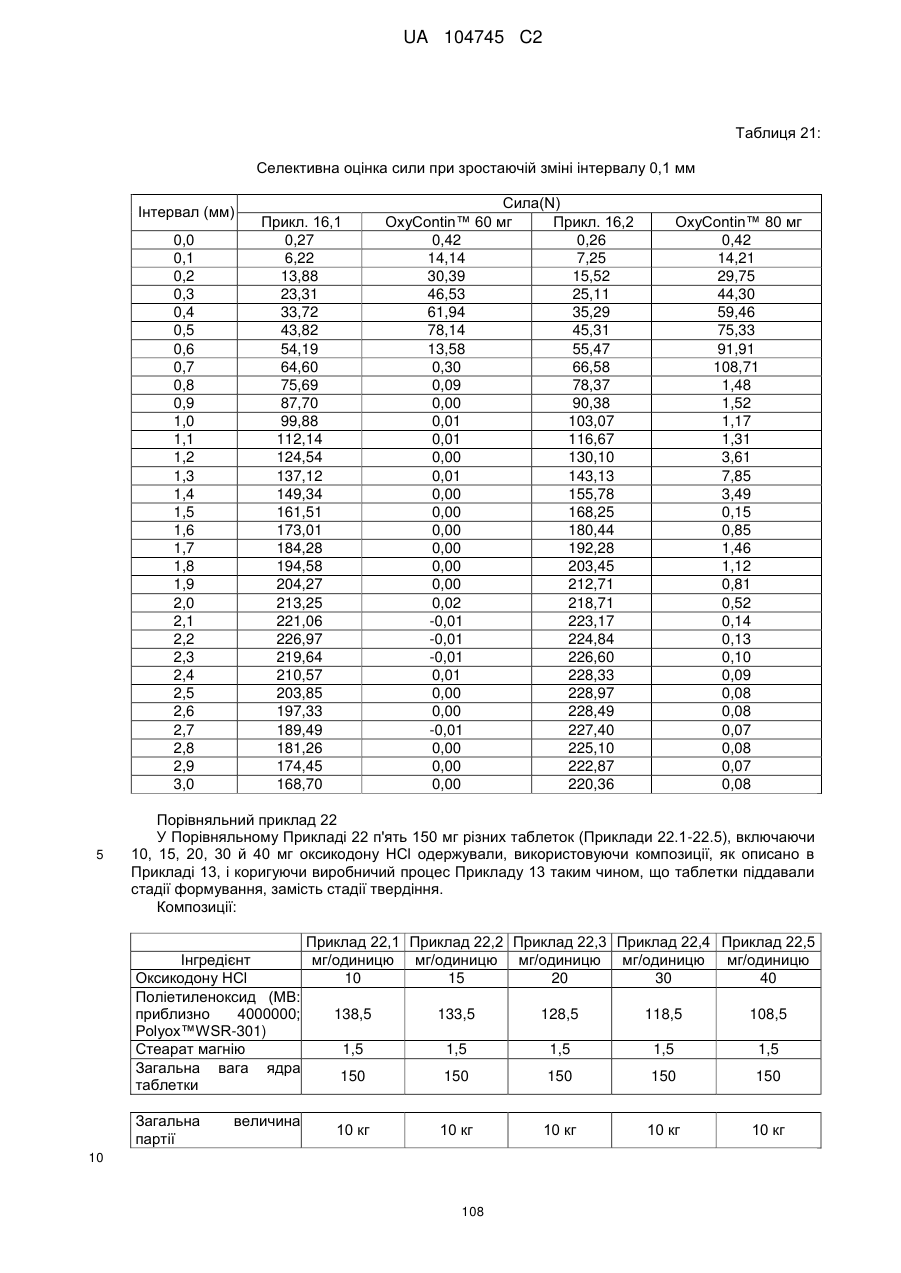
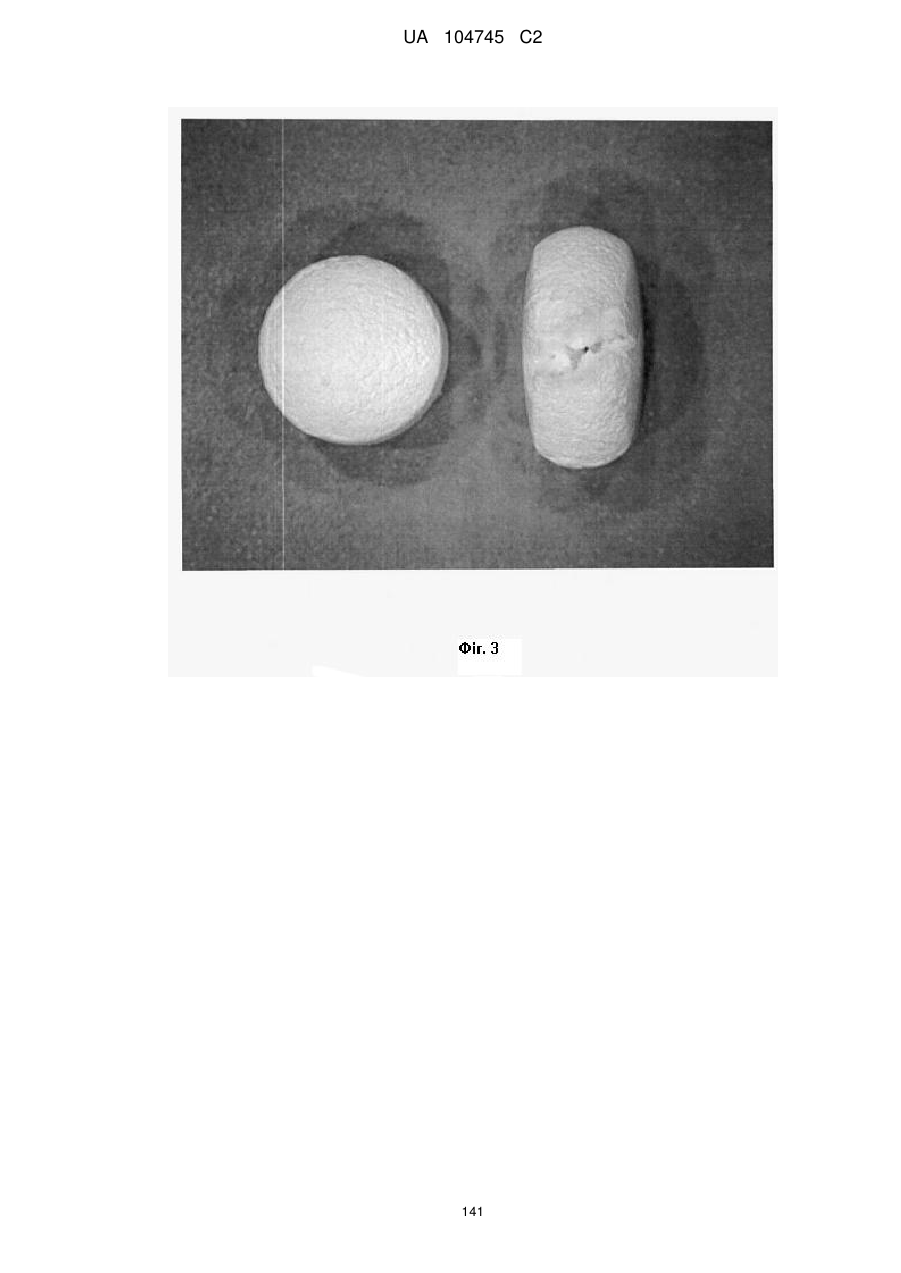
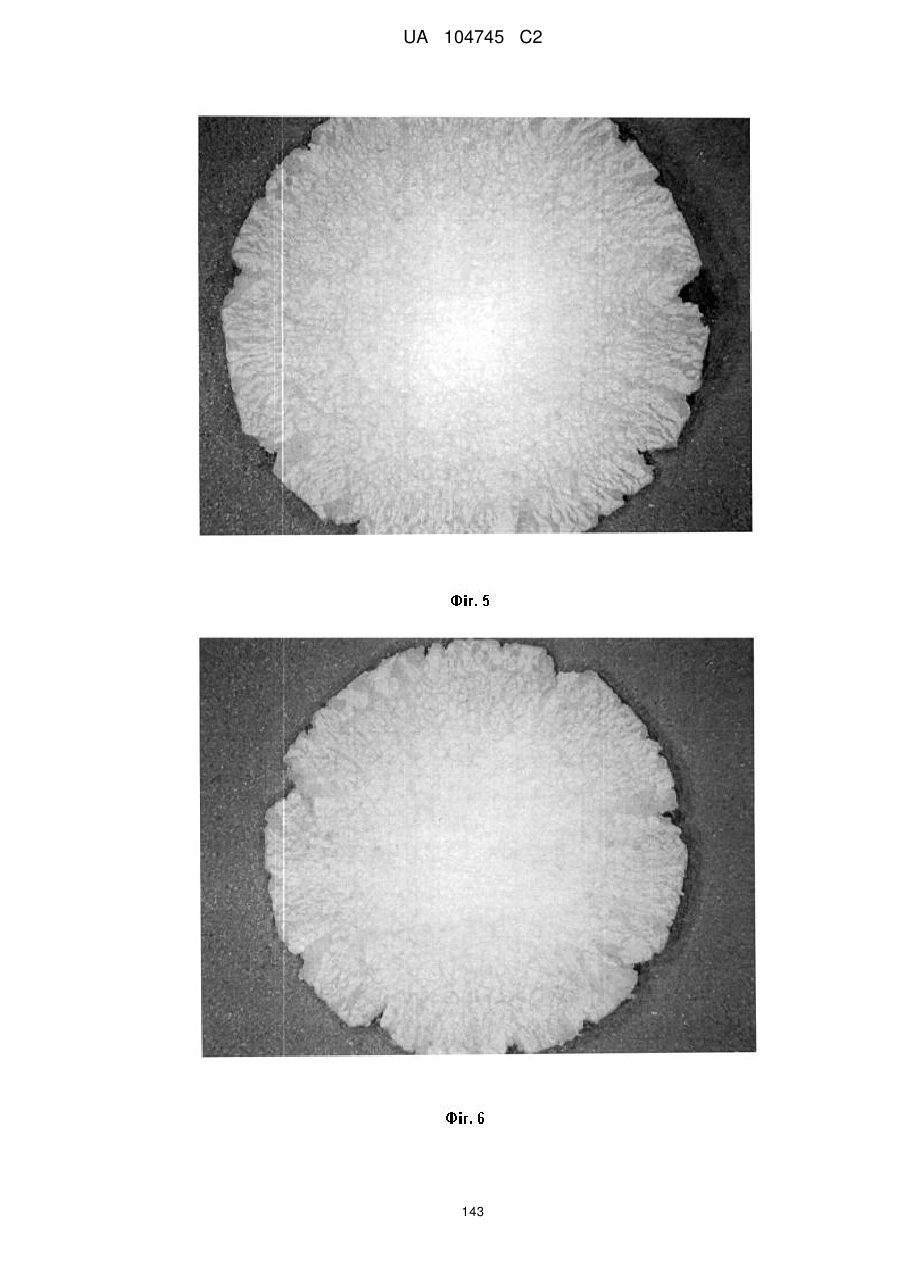
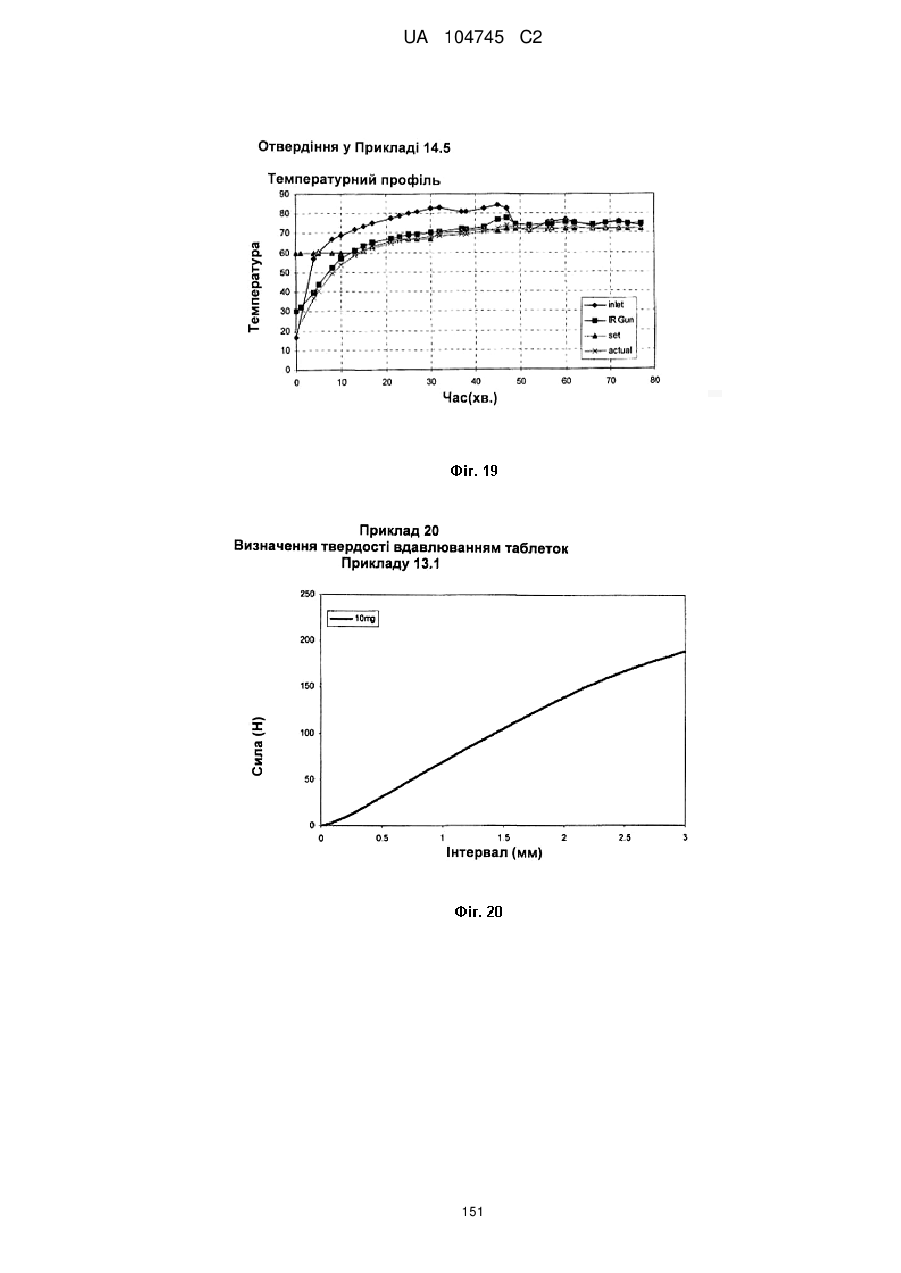
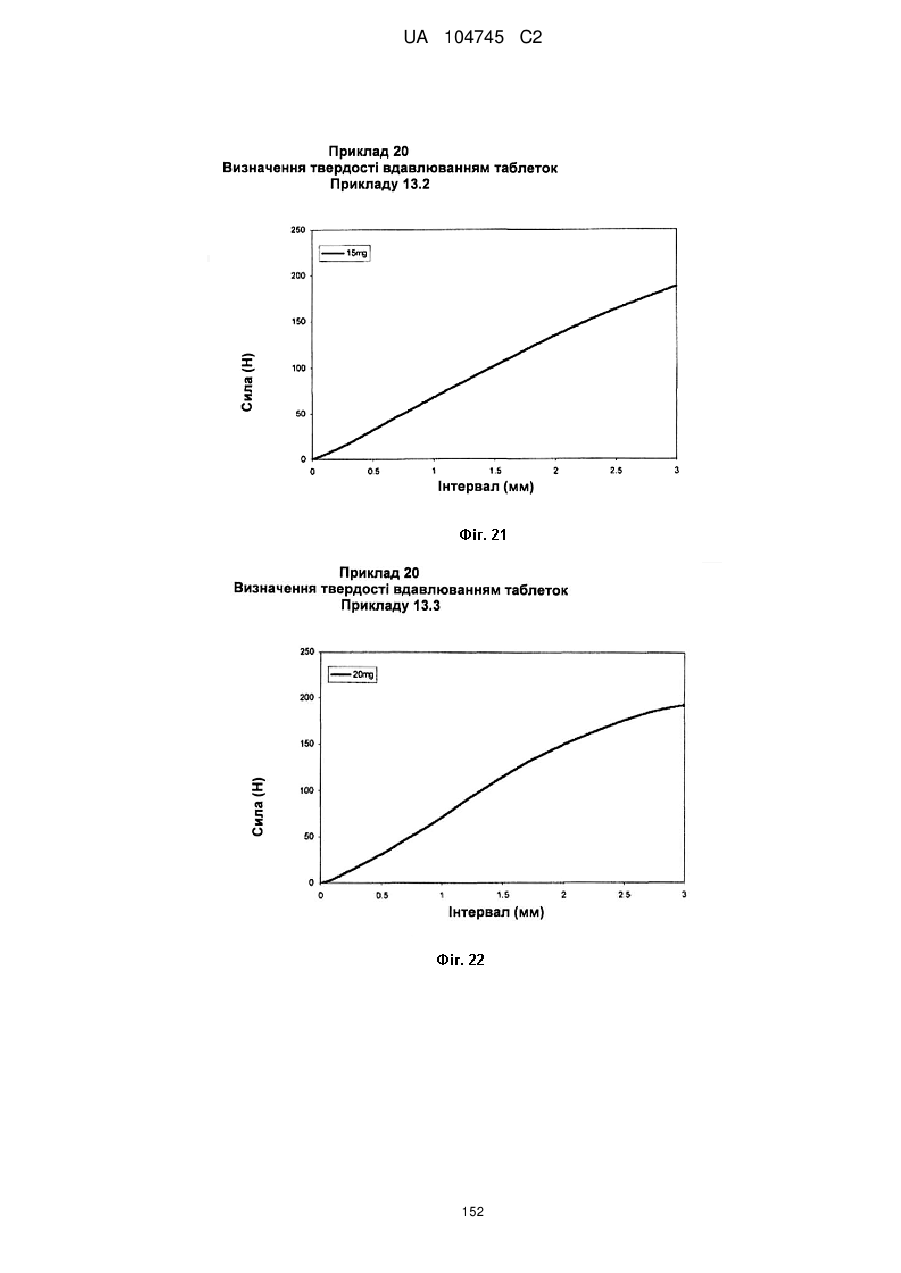
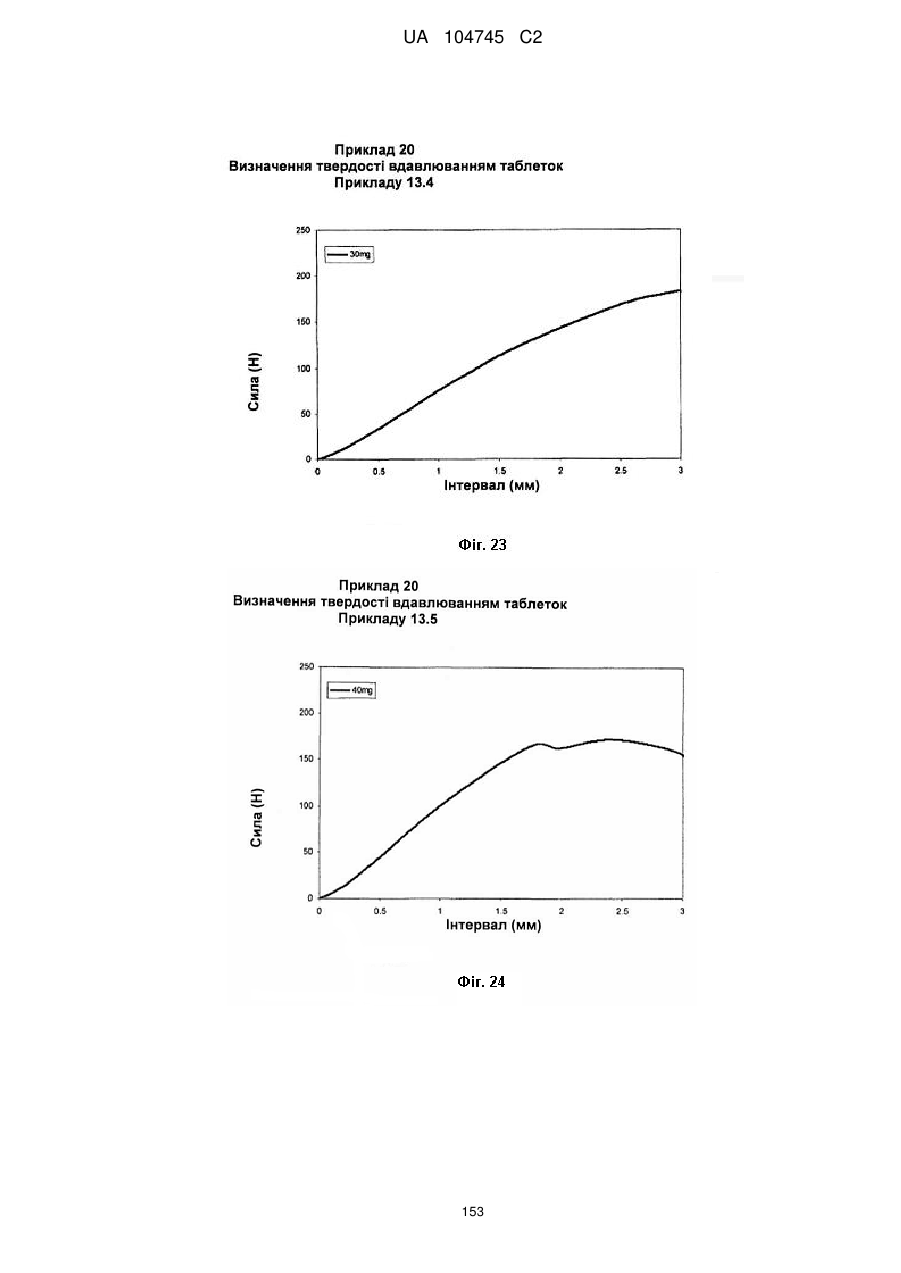
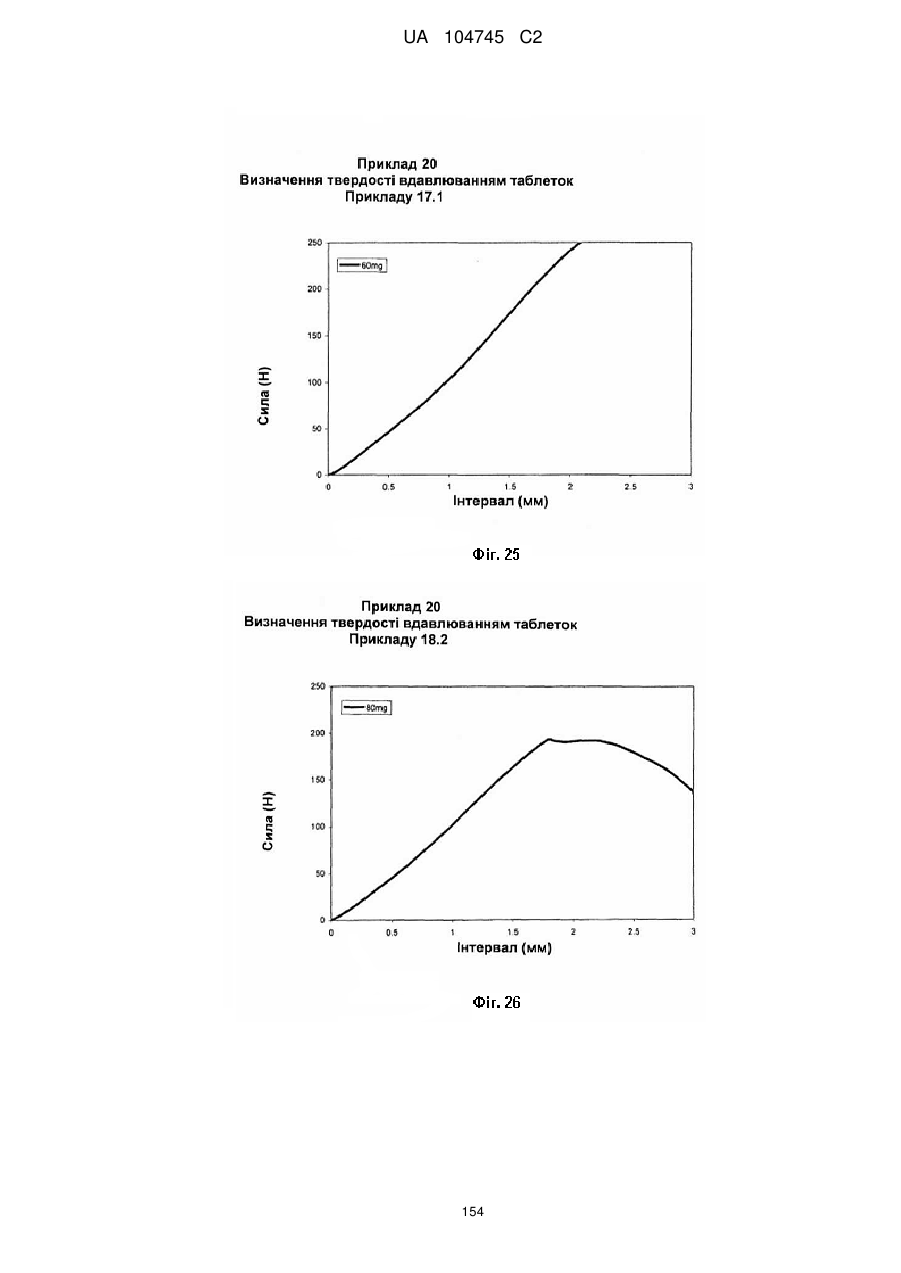
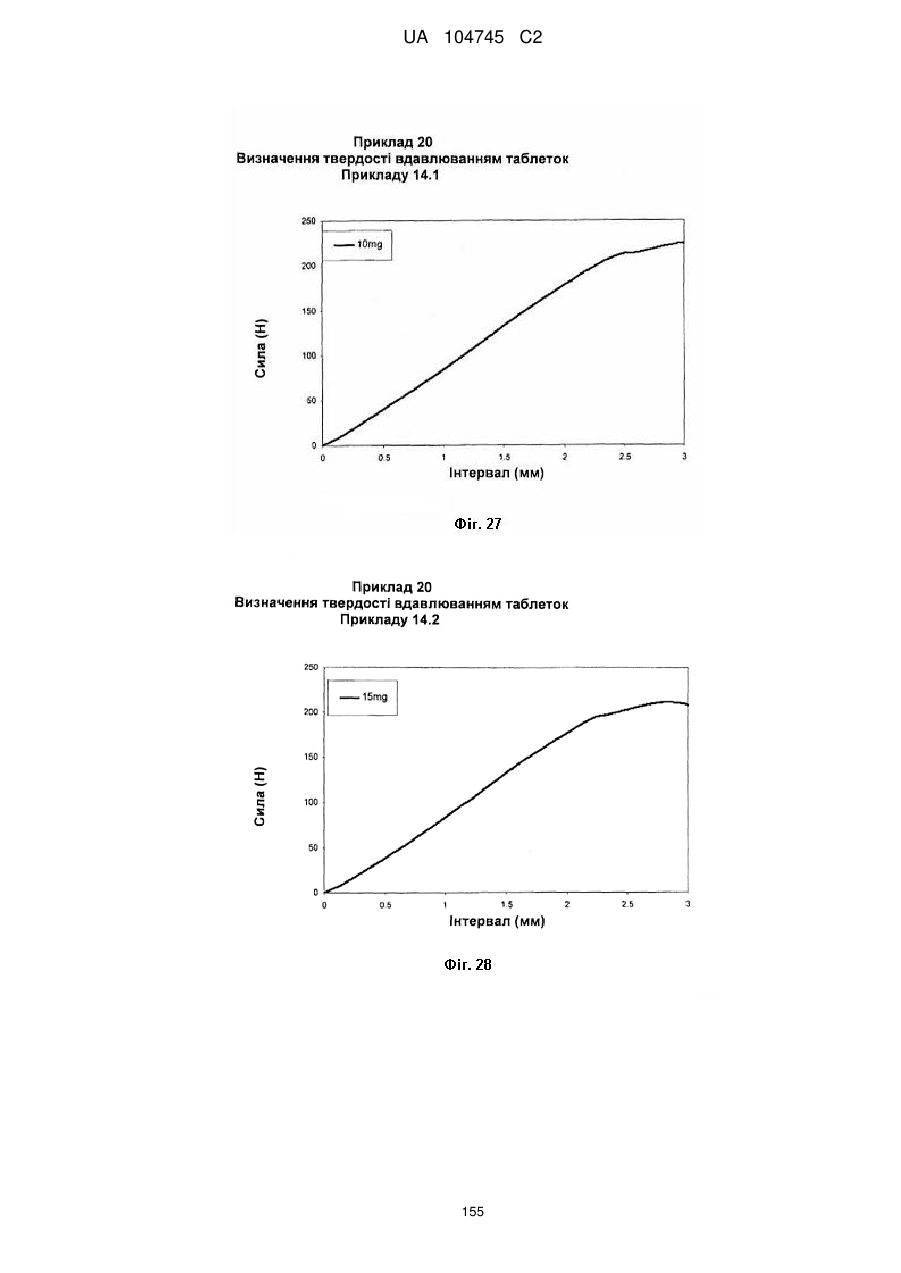
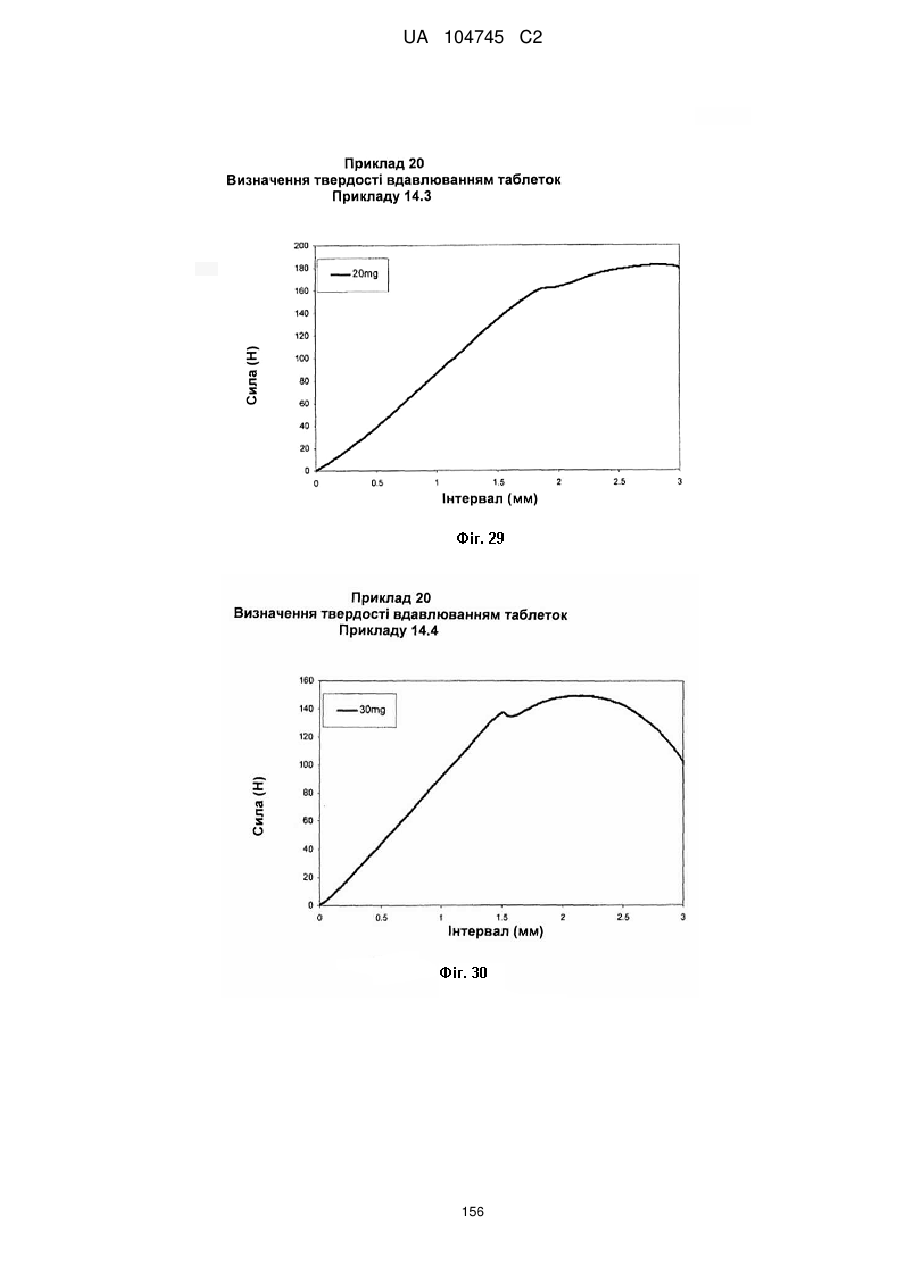
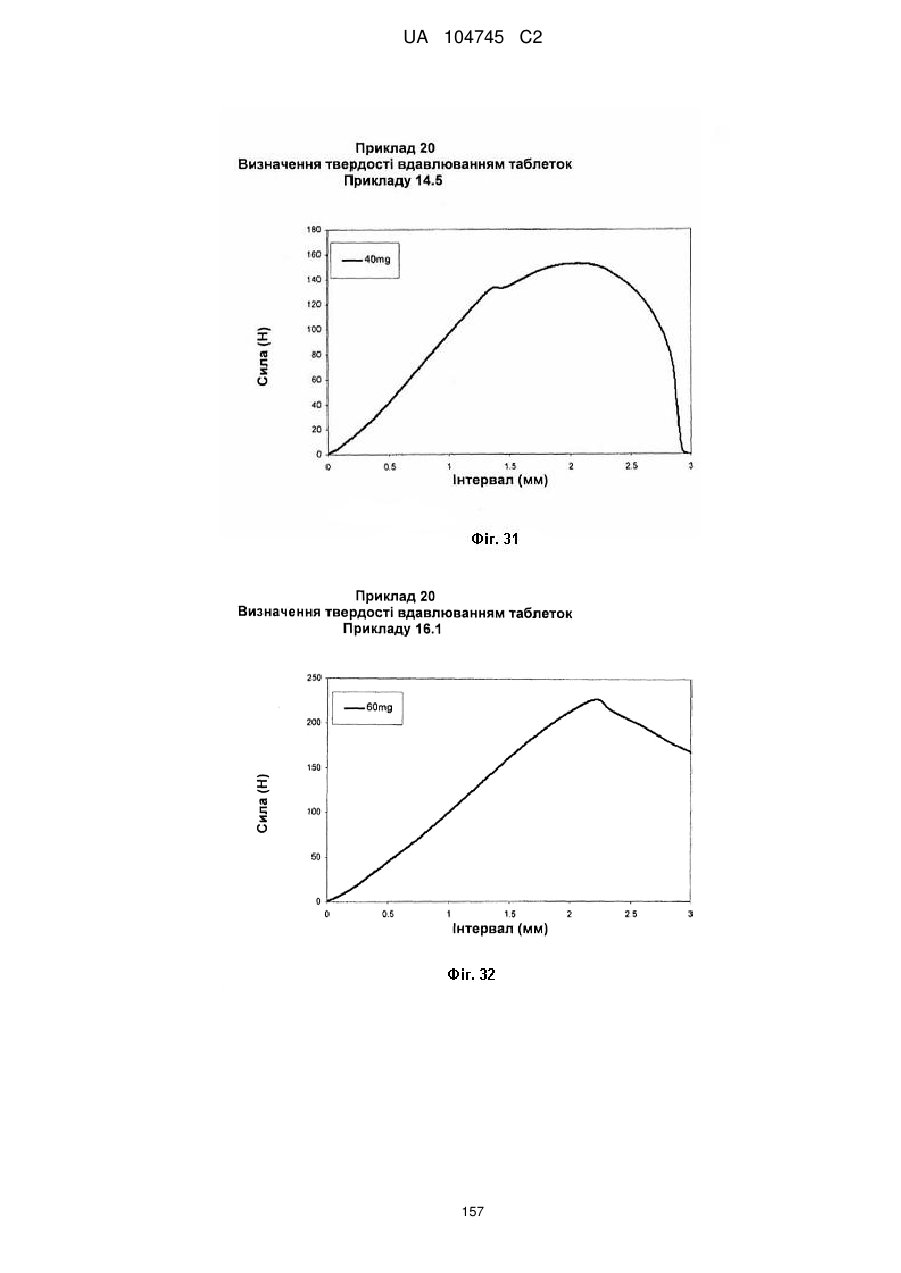
20. Дозована форма пролонгованого вивільнення за будь-яким з пунктів 1-19, що є у формі фармацевтичної таблетки, що має силу розтріскування принаймні 110 N, переважно 120 N, більш переважно 130 N, і навіть більш переважно 140 N, коли піддається тесту на вдавлювання.
21. Дозована форма пролонгованого вивільнення за будь-яким з пунктів 1-20, що є у формі фармацевтичної таблетки, що має глибину розкриття тріщини принаймні 1,0 MM, переважно 1,2 мм, більш переважно 1,4 мм і навіть більш переважно 1,6 мм, коли піддається тесту на вдавлювання.
22. Дозована форма пролонгованого вивільнення за будь-яким з пунктів 1-21, що є у формі фармацевтичної таблетки, що здатна витримувати навантаження принаймні 0,06 Дж без розтріскування.
23. Дозована форма пролонгованого вивільнення за будь-яким з пунктів 1-22, що є у формі фармацевтичної таблетки, що має (а) силу розтріскування принаймні 110 N, переважно 120 N, більш переважно 130 N, і навіть більш переважно 140 N, коли піддається тесту на вдавлювання; (b) має глибину розкриття тріщини принаймні 1,0 мм, переважно 1,2 мм, більш переважно 1,4 мм і навіть більш переважно 1,6 мм, коли піддається тесту на вдавлювання; і (с) здатна витримувати навантаження принаймні 0,06 Дж без розтріскування.
24. Дозована форма пролонгованого вивільнення за будь-яким з пунктів 20-23, що має густину менше, ніж приблизно 1,20 г/см3 переважно менше, ніж приблизно 1,19 г/см3.
25. Застосування дозованої форми за будь-яким з пунктів 1-24, для виготовлення медикаменту для лікування болю, де дозована форма містить оксикодону гідрохлорид.
26. Застосування високомолекулярного поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, молекулярну масу принаймні 1000000, як матеріалу, що утворює матрицю, у виробництві твердої пероральної дозованої форми пролонгованого вивільнення, що містить активний агент, вибраний з опіоїдів, для надання пероральній твердій дозованій формі пролонгованого вивільнення резистентності щодо спиртової екстракції.
Текст
Реферат: Винахід належить до галузі фармацевтики і стосується твердих пероральних фармацевтичних дозованих форм пролонгованого вивільнення, що включають матричну композицію пролонгованого вивільнення, яка містить опіоїдний анальгетик, зокрема оксикодону гідрохлорид, та принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000. Винахід також стосується застосування таких дозованих форм та застосування високомолекулярного поліетиленоксиду, для надання пероральній твердій дозованій формі пролонгованого вивільнення резистентності щодо спиртової екстракції. UA 104745 C2 (12) UA 104745 C2 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 Ця заявка заявляє пріоритет заявки на попередній патент США № 60/840,244, поданій 25 серпня 2006, розкриття якої тим самим включено посиланням. Даний винахід відноситься до фармацевтичних дозованих форм, наприклад до резистентних щодо пошкодження дозованих форм, що містять опіоїдний анальгетик, і способу їх одержання, застосування і способів лікування. Фармацевтичні продукти іноді є суб'єктом зловживання. Наприклад, певна доза опіоїдного агоністу може бути більш сильнодіючою, коли застосовується парентерально в порівнянні з тією же самою дозою, застосовуваною перорально. У деякі композиції можливо втручатися, щоб одержати опіоїдний агоніст, що міститься там для незаконного використання. Композиції контрольованого вивільнення опіоїдного агоністу іноді подрібнюються, або піддаються екстракцій з розчинниками (напр. етанол) наркоманами, щоб одержати опіоїд, що міститься там, для безпосереднього вивільнення при пероральному або парентеральному введенні. Дозовані форми опіоїдного агоністу контрольованого вивільнення, які можуть вивільнити частину опіоїду під впливом етанолу, можуть також приводити до того, що пацієнт отримує дозу швидше, ніж передбачено, якщо пацієнт ігнорує інструкції щодо використання й паралельно вживає алкоголь із дозованою формою. Продовжує існувати потреба в даній галузі в пероральних фармацевтичних дозованих формах, що містять опіоїдний агоніст без значно змінених властивостей вивільнення опіоїду при контактуванні з алкоголем та/або із резистентністю щодо подрібнення. Об'єктом певних варіантів реалізації представленого винаходу є одержання пероральної дозованої форми пролонгованого вивільнення, що містить активний агент, такий як опіоїдний анальгетик, що резистентною щодо втручання. Об'єктом певних варіантів реалізації представленого винаходу, є одержання пероральної дозованої форми пролонгованого вивільнення, що містить активний агент, такий, як опіоїдний анальгетик, що є резистентною щодо подрібнення. Об'єктом певних варіантів реалізації представленого винаходу, є одержання пероральної дозованої форми пролонгованого вивільнення, що містить активний агент, такий, як опіоїдний анальгетик, що є резистентним щодо спиртової екстракції й демпінгу дози, коли використовується паралельно з або при контактуванні з алкоголем. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення у формі таблеток або мультичастинок, де таблетка або індивідуальні мультичастинки можуть бути принаймні сплющені без руйнування, що характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 60 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувана сплющена таблетка або сплющені мультичастинки забезпечують коефіцієнт розчинності in vitro при вимірюванні на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C, що характеризуються відсотком вивільнення кількості активного інгредієнта на 0,5 год. розчинення, що відхиляється не більше, ніж приблизно на 20 % пунктів від відповідного in vitro коефіцієнта розчинності несплющеної еталонної таблетки або еталонних мультичастинок. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення у формі таблеток або мультичастинок, де таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 60 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувані сплющені таблетки або сплющені мультичастинки, забезпечують коефіцієнт розчинності in vitro при вимірюванні на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF), що містить 40 % етанолу при 37 °C, що характеризуються відсотком вивільнення кількості активного інгредієнта на 0,5 год. розчинення, що відхиляється не більше, ніж приблизно на 20 % пунктів від відповідного in vitro коефіцієнта розчинності, вимірюваного на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C без етанолу, використовуючи сплющені та несплющені еталонні таблетки або сплющені та несплющені еталонні мультичастинки, відповідно. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: 1 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) принаймні один активний інгредієнт; і де композиція містить принаймні приблизно 80 ваг. % поліетиленоксиду. Відповідно до певних таких варіантів реалізації винаходу, активний інгредієнт - оксикодону гідрохлорид, і композиція включає більше, ніж приблизно 5 ваг. % оксикодону гідрохлориду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає: (1) принаймні один активний інгредієнт; (2) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (3) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, молекулярну масу менше, ніж 1000000, У певних варіантах реалізації даний винахід спрямований на спосіб одержання твердої пероральної фармацевтичної дозованої форми пролонгованого вивільнення, що включає принаймні наступні стадії: (a) комбінування принаймні: (1) принаймні одного поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000, і (2) принаймні одного активного інгредієнта, щоб утворити композицію; (b) формування композиції, щоб утворити матричну композицію пролонгованого вивільнення; і с) твердіння згадуваної матричної композиції пролонгованого вивільнення, що включає принаймні стадію твердіння матричної композиції пролонгованого вивільнення до температури, що є принаймні температурою розм'якшення згадуваного поліетиленоксиду протягом періоду часу принаймні приблизно 1 хв. У певних варіантах реалізації даний винахід спрямований на спосіб одержання твердої пероральної фармацевтичної дозованої форми пролонгованого вивільнення, що включає принаймні наступні стадії (a) комбінування принаймні: (1) принаймні одного поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000, і (2) принаймні одного активного інгредієнта, щоб утворити композицію; (b) формування композиції, щоб утворити матричну композицію пролонгованого вивільнення; і с) твердіння згадуваної матричної композиції пролонгованого вивільнення, що включає принаймні стадію твердіння, де згадуваний поліетиленоксид принаймні частково плавиться. У певних варіантах реалізації, даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що включає матричну композицію пролонгованого вивільнення, яка містить активний агент у формі таблеток або мультичастинок, де таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 60 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувана сплющена таблетка або сплющені мультичастинки забезпечують коефіцієнт розчинності in vitro при вимірюванні на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C, що характеризуються кількістю відсотка вивільнення активного агента, вивільненого на 0,5 год. розчинення, що відхиляється не більше, ніжприблизно на 20 % пунктів від відповідного in vitro коефіцієнта розчинності несплющеної еталонної таблетки або еталонних мультичастинок. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що включає матричну композицію пролонгованого вивільнення, яка містить активний агент у формі таблеток або мультичастинок, де таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 60 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувана сплющена таблетка або сплющені мультичастинки й несплющена еталонна таблетка або еталонні 2 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 мультичастинки забезпечують коефіцієнт розчинності in vitro, виміряний на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C, між приблизно 5 і приблизно 40 ваг. % активного інгредієнта, вивільненого після 0,5 годин. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що включає матричну композицію пролонгованого вивільнення, яка містить активний агент у формі таблеток або мультичастинок, де таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 60 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувані сплющені таблетки або сплющені мультичастинки забезпечують коефіцієнт розчинності in vitro при вимірюванні на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF), що містить 40 % етанолу при 37 °C, що характеризуються кількістю відсотка вивільнення активного агента, вивільненого на 0,5 год. розчинення, що відхиляється не більше, ніж приблизно на 20 % пунктів від відповідного in vitro коефіцієнта розчинності, вимірюваного на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C без етанолу, використовуючи сплющені та несплющені еталонні таблетки або сплющені та несплющені еталонні мультичастинки, відповідно. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що включає матричну композицію пролонгованого вивільнення, яка містить активний агент у формі таблеток або мультичастинок, де таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 60 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувані сплющені таблетки або сплющені мультичастинки, забезпечують in vitro коефіцієнт розчинення, виміряний на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF), що містить 40 % або 0 % етанолу при 37 °C, між приблизно 5 і приблизно 40 ваг. % активного інгредієнта, вивільненого після 0,5 годин. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) принаймні один активний інгредієнт, вибраний з опіоїдних анальгетиків; і де композиція містить принаймні приблизно 80 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) 10 мг оксикодону гідрохлориду; і де композиція містить принаймні приблизно 85 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) 15 мг або 20 мг оксикодону гідрохлориду; і де композиція містить принаймні приблизно 80 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) 40 мг оксикодону гідрохлориду; і 3 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 де композиція містить принаймні приблизно 65 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) 60 мг або 80 мг оксикодону гідрохлориду; і де композиція містить принаймні приблизно 60 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) 8 мг гідроморфону гідрохлориду; і де композиція містить принаймні приблизно 94 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) 12 мг гідроморфону гідрохлориду; і де композиція містить принаймні приблизно 92 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) 32 мг гідроморфону гідрохлориду; і де композиція містить принаймні приблизно 90 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення включає композицію, що містить принаймні: (1) принаймні один активний інгредієнт вибраний з опіоїдних анальгетиків; (2) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (3) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу менше, ніж 1000000, У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, молекулярну масу принаймні 800 000; і (2) принаймні один активний інгредієнт, вибраний з опіоїдних анальгетиків; і де композиція містить принаймні приблизно 80 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) принаймні один активний інгредієнта; і де матрична композиція пролонгованого вивільнення, коли підлягає тестуванню на твердість вдавлюванням, має силу розтріскування принаймні приблизно 110 N. 4 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) принаймні один активний інгредієнт; і де матрична композиція пролонгованого вивільнення, коли підлягає тестуванню на твердість вдавлюванням має "глибину проникнення для розкриття тріщини" принаймні приблизно 1,0 мм. У певних варіантах реалізації даний винахід спрямований на спосіб лікування де, дозована форма відповідно до винаходу, що містить опіоїдний анальгетик, призначена для лікування болю у пацієнта, який потребує цього. У певних варіантах реалізації даний винахід спрямований на використання дозованої форми відповідно до винаходу, що містить опіоїдний анальгетик для виробництва медикаменту для лікування болю. У певних варіантах реалізації даний винахід спрямований на використання високомолекулярного поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000, як матеріалу, що утворює матрицю у виробництві твердої пероральної пролонгованого вивільнення дозованої форми, що містить активний інгредієнт, вибраний з опіоїдів для надання пероральній твердій дозованій формі пролонгованого вивільнення резистентності щодо спиртової екстракції. У певних варіантах реалізації даний винахід спрямований на спосіб одержання твердої пероральної фармацевтичної дозованої форми пролонгованого вивільнення, що включає принаймні наступні стадії (а) комбінування принаймні: принаймні одного поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, молекулярну вагу принаймні 1000000, і (2) принаймні одного активного інгредієнта, щоб утворити композицію; b) формування композиції, щоб утворити матричну композицію пролонгованого вивільнення; c) твердіння згадуваної матричної композиції пролонгованого вивільнення, яка включає принаймні стадію твердіння матричної композиції пролонгованого вивільнення при температурі, що є принаймні температурою розм'якшення згадуваного поліетиленоксиду протягом періоду часу принаймні 5 хвилин. Відповідно до певних варіантів реалізації винаходу тверда фармацевтична дозована форма пролонгованого вивільнення використовується як супозиторій. Термін "пролонговане вивільнення" визначений для цілей представленого винаходу як такий, що відноситься до виробів, які утворені, щоб одержати біодоступність лікарського засобу протягом пролонгованого періоду після внутрішнього прийому, що, таким чином дозволяє знизити частоту дозування в порівнянні з лікарським засобом, представленим як звичайна дозована форма (наприклад, як розчин або дозована форма швидкого вивільнення). Термін "швидке вивільнення" визначений для цілей представленого винаходу як такий, що відноситься до виробів, які утворені, щоб дозволити лікарському засобу розчинятися в шлунково-кишковому тракті без наміру затримати або продовжити розчинення або абсорбцію лікарського засобу. Термін "тверда пероральна фармацевтична дозована форма пролонгованого вивільнення" відноситься до форми введення, що містить одиничну дозу активного інгредієнта у формі пролонгованого вивільнення, такій як "матрична композиція пролонгованого вивільнення" і, довільно, інші ад'юванти й добавки, загальноприйнятні у даній галузі, такі як захисне покриття або капсула й т.п., і, довільно, будь-які інші додаткові засоби або компоненти, які використовуються в дозованих формах. Якщо конкретно не зазначено, термін "тверда пероральна фармацевтична дозована форма пролонгованого вивільнення" відноситься до згадуваної дозованої форми в інтактній формі, тобто перед будь-яким втручанням. Фармацевтична дозована форма пролонгованого вивільнення може, наприклад бути таблеткою, що містить матричну композицію пролонгованого вивільнення або капсулу, що містить матричну композицію пролонгованого вивільнення у формі мультичастинок. "Фармацевтична дозована форма пролонгованого вивільнення" може включати частину активного інгредієнта у формі пролонгованого вивільнення й іншу частину активного інгредієнта у формі швидкого вивільнення, наприклад як шар активного інгредієнта 5 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 швидкого вивільнення, що оточує дозовану форму або компонент швидкого вивільнення, включений в дозовану форму. Термін "матрична композиція пролонгованого вивільнення" визначений для цілей представленого винаходу як формована тверда форма композиції, що містить принаймні один активний інгредієнт і принаймні один засіб пролонгованого вивільнення, такий як матричний матеріал пролонгованого вивільнення, такий як наприклад, високомолекулярний поліетиленоксид. Композиція може довільно включати більше, ніж ці дві сполуки, а саме, додаткові активні інгредієнти й додаткові пролонгатори та/або інші матеріали, включаючи, але не обмежуючись низькомолекулярним поліетиленоксидом й іншими ад'ювантами й добавками, загальноприйнятними в даній галузі. Термін "біоеквівалент/біоеквівалентність" визначений для цілей представленого винаходу, як такий, що відноситься до дозованої форми, що забезпечує середнє геометричне значення Cmax, AUCt, і AUCinf для активного інгредієнта, де 90 % довірчі інтервали, оцінені для співвідношення (тест/еталон), знаходяться в діапазоні від 80,00 % до 125,00 %. Переважно, середні значення Cmax, AUCt, і AUCinf знаходяться в діапазоні від 80,00 % до 125,00 % як визначено в стані натщесерце й після їжі. Термін "поліетиленоксид" визначений для цілей представленого як такий, що має молекулярну масу принаймні 25000, визначену, як прийнятно в даній галузі і переважно має молекулярну вагу принаймні 100000, Композиції з більш низькою молекулярною вагою звичайно відносяться до поліетиленгліколів. Термін "високомолекулярний поліетиленоксид" визначений для цілей представленого винаходу як такий, що має приблизну молекулярну масу принаймні 1000000. З метою цього винаходу приблизна молекулярна вага базується на реологічних вимірюваннях. Поліетиленоксид, як вважають, має приблизну молекулярну масу 1000000, коли 2 ваг. % водний розчин згадуваного поліетиленоксиду, використовуючи віскозиметр Brookfield модель RVF, шпиндель номер 1, при 10 об/хв, при 25 °C показує в'язкість у діапазоні 400-800 мПа с (сР). Поліетиленоксид, як вважають, має приблизну молекулярну масу 2000000, коли 2 ваг. % водний розчин згадуваного поліетиленоксиду, використовуючи віскозиметр Brookfield модель RVF, шпиндель номер 3, при 10 об/хв, при 25 °C, показує в'язкість у діапазоні 2000-4000 мПа с (сР). Поліетиленоксид, як вважають, має приблизну молекулярну масу 4000000, коли 1 ваг. % водний розчин згадуваного поліетиленоксиду, використовуючи віскозиметр Brookfield модель RVF, шпиндель номер 2, при 2 об/хв при 25 °C показує в'язкість у діапазоні 1650-5500 мПа (сР). Поліетиленоксид, як вважають, має приблизну молекулярну масу 5000000, коли 1 ваг. % водний розчин згадуваного поліетиленоксиду, використовуючи віскозиметр Brookfield модель RVF, шпиндель номер 2, при 2 об/хв, при 25 °C показує в'язкість у діапазоні 5500-7500 мПа (сР). Поліетиленоксид, як вважають, має приблизну молекулярну масу 7000000 коли 1 ваг. % водний розчин згадуваного поліетиленоксиду, використовуючи віскозиметр Brookfield модель RVF, шпиндель номер 2, при 2 об/хв, при 25 °C показує в'язкість у діапазоні 7500-10 000 мПа (сР). Поліетиленоксид, як вважають, має приблизну молекулярну масу 8000000, коли 1 ваг. % водний розчин згадуваного поліетиленоксиду, використовуючи віскозиметр Brookfield модель RVF, шпиндель номер 2, при 2 об/хв, при 25 °C показує в'язкість у діапазоні 10 000-15 000 мПа (сР). Щодо поліетиленоксидів більш низької молекулярної ваги; Поліетиленоксид, як вважають, має приблизну молекулярну масу 100 000, коли 5 ваг. % водний розчин згадуваного поліетиленоксиду, використовуючи модель віскозиметра Brookfield RVT, шпиндель номер 1, при 50 об/хв, при 25 °C показує в'язкість у діапазоні 30-50 мПа (сР) і поліетиленоксид, як вважають, має приблизну молекулярну масу 900,000, коли 5 ваг. % водний розчин згадуваного поліетиленоксиду, використовуючи віскозиметр Brookfield модель RVF, шпиндель номер 2, при 2 об/хв, при 25 °C показує в'язкість у діапазоні 8800-17 600 мПа (сР). Термін "низькомолекулярний поліетиленоксид" визначений для цілей представленого винаходу як такий, що має, базуючись на реологічних вимірюваннях, наведених вище, приблизну молекулярну масу менше, ніж 1000000. Термін "безпосереднє пресування" визначений для цілей представленого винаходу як такий, що відноситься до процесу таблетування де, таблетка або будь-яка інша пресована дозована форма одержані способом, що включає стадії сухого змішування сполук й пресування сухої суміші, щоб утворити дозовану форму, наприклад, використовуючи дифузійне змішування та/або спосіб конвекційного змішування (Guidance for Industry, SUPAC - IR/MR: Immediate Release and Modified Release Solid Oral Dosage Forms, Manufacturing Equipment Addendum). Термін "шар вільнотекучих таблеток" визначений для цілей представленого винаходу як такий, що відноситься до серії таблеток, які зберігаються в русі відносно одна одної, як наприклад, у резервуарі для покриття, встановленому на відповідній швидкості обертання або в 6 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 псевдозрідженому шарі таблеток. Шар вільнотекучих таблеток переважно зменшує або запобігає злипанню таблеток. Термін "сплющення" і пов'язані терміни, як використовується в контексті сплющених таблеток або інших дозованих форм відповідно до представленого винаходу означає, що таблетку піддають дії, застосовуючи осьову силу по суті перпендикулярно діаметра й товщини, наприклад, таблетки. Сила може бути накладена настільним пресом типу стоматологічної гладилки (якщо явно не згадано інакше) до необхідного ступеня, щоб досягти цільової сплющеності/зменшеної товщини. Відповідно до певних варіантів реалізації винаходу сплющення не закінчується розламуванням таблетки на частини, однак, можуть зустрічатися гострокутні уламки й тріщини. Сплющеність описана в термінах густини сплющеної таблетки в порівнянні з густиною несплющеної таблетки, вираженої у % товщини, базуючись на товщині несплющеної таблетки. Крім таблеток, сплющення може бути застосоване до будь-якої формованої дозованої форми, де осьова сила накладена лінійно найменшого діаметра (тобто, товщини) форми, коли форма є іншою, ніж сферичною й в будь-якому напрямку, коли форма є сферичною. Сплющеність потім описана в термінах товщина/найменший діаметр сплющеної форми в порівнянні з товщиною/найменшим діаметром несплющеної форми, вираженої у % товщини, базуючись на товщині/найменшому діаметрі несплющеної форми, коли початкова форма не є сферичною, або % товщини, базуючись на діаметрі несплющеної форми, коли початкова форма є сферичною. Товщину вимірювали, використовуючи товщиномір (наприклад, цифровий товщиномір або цифровий штангенциркуль) На фігурах 4-6 показані таблетки, що були сплющені, використовуючи настільний прес. Початкова форма таблеток показана на фігурах 1-3 з лівої сторони на фотографії. У певних варіантах реалізації винаходу, крім використання настільного пресу, може використовуватися молоток для сплющення таблетованих дозованих форм. При такому способі сплющення удари молотка наносять вручну безпосередньо по суті лінійно товщині, наприклад, таблетки. Сплющеність також описана в термінах товщина/найменший діаметр сплющеної форми в порівнянні з несплющеною формою, вираженої у % товщини, базуючись на товщині/найменшому діаметрі несплющеної форми, коли початкова форма не є сферичною, або % товщини, базуючись на діаметрі несплющеної форми, коли початкова форма є сферичною. Товщину вимірювали, використовуючи товщиномір (наприклад, цифровий товщиномір або цифровий штангенциркуль). На відміну від цього, при проведенні тестування міцності на розрив або визначенні твердості таблетки, як описано у Remington's Pharmaceutical Sciences, 18 видання, 1990, Глава 89 "Пероральні тверді дозовані форми", стор. 1633-1665, що включено тут посиланням, використовуючи Апарат Schleuniger, таблетку/дозовану форму поміщали між парою плоских пластин, розташованих паралельно, і пресували за допомогою плоских пластин, таким чином, що сила накладена по суті перпендикулярно товщині і по суті лінійно діаметра таблетки, таким чином зменшуючи діаметр у тому напрямку. Цей зменшений діаметр описаний як % діаметра, базуючись на діаметрі таблетки перш, ніж провести тестування міцності на розрив. Міцність на розрив або твердість таблетки визначена як сила, при якій тестована таблетка/дозована форма ламається. Таблетки/дозовані форми, які не ламаються, але які деформовані внаслідок застосованої сили, як вважають, є резистеними щодо розриву при певній силі. Додатковий тест, щоб кількісно визначити міцність таблеток/дозованих форм є визначення твердості вдавлюванням, використовуючи Texture Analyzer, такий як ТА - ХТ2 Texture Analyzer (Texture Technologies Corp., 18 Fairview Road, Scarsdale, Нью-Йорк 10583). У цьому способі таблетки/дозовані форми розміщені на вершині стального штатива з незначно увігнутою поверхнею й згодом проходять спадний датчик Texture Analyzer, такий як ТА - 8А шарик датчика, діаметром 1/8 дюйма із нержавіючої сталі. Перед початком вимірювання, таблетки розташовують лінійнобезпосередньо на датчику, таким чином що спадний датчик проникне через таблетку центрально, тобто в центр таблетки, і в таким чином сила спадного датчика є застосовною по суті перпендикулярно діаметра й лінійно товщині таблетки. По-перше, датчик Texture Analyzer запускає рух зразка таблетки при претестовій швидкості. Коли датчик входить у контакт із поверхнею таблетки, і зусилля включення досягнуте, датчик продовжує рухатися з іспитовою швидкістю й проникає через таблетку. Для кожної глибини проникнення датчика, що буде надалі згадуватися як "дистанція», була виміряна відповідна сила і зібрані дані. Коли датчик досяг бажаної максимальної глибини проникнення, він змінює напрямок і рухається назад з посттестовою швидкістю, у той час як подальші дані можуть бути зібрані. Сила розтріскування визначена, як сила першого локального максимуму, що досягнений у відповідній діаграмі сила/дистанція й обчислений, використовуючи наприклад програмне забезпечення Texture Analyzer "Texture Expert Exceed, Version 2,64 English". He бажаючи бути прив'язаним до 7 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 будь-якої теорії, вважають, що в цьому пункті, деяке структурне ушкодження форми таблетки/дозування зустрічається у формі розламу. Однак, розламані таблетки/дозовані форми відповідно до певних варіантів реалізації представленого винаходу залишаються здатними до зчеплення, як доведено постійною резистентністю до спадного датчика. Відповідна дистанція у першому локальному максимумі надалі згадується як "глибина проникнення для розкриття тріщини". Для цілей певних варіантів реалізації представленого винаходу термін "міцність на розрив" відноситься до твердості таблеток/дозованих форм, що переважно виміряна, використовуючи апарат Schleuniger, потім як термін "сила розламування" відбиває міцність таблеток/дозованих форм, що переважно виміряна визначенням твердості вдавлюванням, використовуючи Texture Analyzer. Подальший параметр матричних композицій пролонгованого вивільнення, які можуть бути отримані від тесту визначення твердості вдавлюванням, як описано вище, є роботою матричної композиції пролонгованого вивільнення, підданій визначенню твердості вдавлюванням, як описано вище. Значення роботи відповідає інтегралу сили на дистанції. Термін "резистентний до подрібнення", визначений для цілей певних варіантів реалізації представленого винаходу відноситься до дозованих форм, які можуть принаймні бути сплющені на настільному пресі, як описано вище, без розриву до не більше, ніж приблизно 60 % товщини, переважно не більше, ніж приблизно 50 % товщини, більш переважно не більше, ніж приблизно 40 % товщини, переважніше не більше, ніж приблизно 30 % товщини й найбільш переважно не більше, ніж приблизно 20 % товщини, 10 % товщини або 5 % товщини. Для цілей певних варіантів реалізації даного винаходу, дозовані форми розцінені як "резистентні щодо спиртової екстракції", коли відповідна дозована форма забезпечує коефіцієнт розчинності in vitro при вимірюванні на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF), що містить 40 % етанолу при 37 °C, характеризується відсотком кількості вивільненого активного інгредієнта за 0,5 годин, переважно за 0,5 й 0,75 годин, більш переважно за 0,5, 0,75 й 1 год., переважніше за 0,5, 0,75, 1 й 1,5 год. й найбільш переважно за 0,75, 1, 1,5 й 2 год. розчинення, що відхиляється не більше, ніж приблизно на 20 % пунктів або переважно не більше, ніж приблизно на 15 % пунктів у кожному із згадуваних пунктів часу від відповідного in vitro коефіцієнта розчинності, вимірюваного на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C без етанолу. Термін "резистентний до пошкодження" для цілей представленого винаходу відноситься до дозованих форм, які принаймні забезпечують резистентність до подрібнення або резистентність до спиртової екстракції, переважно обох, як визначено вище й можуть мати додаткові характеристики резистентності щодо пошкодження. З метою представленого винаходу, термін "активний інгредієнт" визначений як фармацевтично активна субстанція, що містить без обмеження опіоїдні анальгетики. Для цілей представленого винаходу термін "опіоїдний анальгетик" включає одиничні сполуки й композиції сполук, вибрані із групи опіоїдів й які забезпечують аналгетичний ефект, такі як один, взятий окремо опіоїдний агоніст або комбінація опіоїдних агоністів, один, взятий окремо змішаний опіоїдний антагоніст - агоніст або комбінація змішаних опіоїдних антагоністів агоністів, або один, взятий окремо частковий опіоїдний агоніст або комбінація часткових опіоїдних агоністів й комбінації опіоїдних агоністів, змішані опіоїдні антагоністи - агоністи й часткові опіоїдні агоністи з одним або більше опіоїдним антагоністом, стереоізомери, етери або естери, солі, гідрати і їх сольвати, будь-які вищезгадані композиції і т.п. Даний винахід, розкритий тут, зокрема призначений щоб охопити використання опіоїдного анальгетика у формі будь-якої його фармацевтично прийнятної солі. Фармацевтично прийнятні солі включають, але не обмежуються наступними: солі неорганічної кислоти, такі як гідрохлорид, бромгідрат, сульфат, фосфат і т.п.; солі органічної кислоти, такі як форміат, ацетат, трифторацетат, малеат, тартрат і т.п.; сульфонати, такі як метансульфонат, бензолсульфонат, п - толуолсульфонат і т.п.; солі амінокислот, такі як аргінат, аспаргінат, глутамат і т.п., і солі металу, такі як натрієва сіль, калієва сіль, сіль цезію й т.п.; солі лужно-земельних металів, такі як сіль кальцію, магнієва сіль і т.п.; солі органічних амінів, такі як сіль триетиламіну, сіль піридину, сіль піколіну, сіль етаноламіну, сіль триетаноламіну, сіль дициклогексиламіну, N,N'-дибензилетиленамінова сіль і т.п. Опіоїди, використовувані відповідно до представленого винаходу, можуть містити один або більше центрів асиметрії й можуть дати початок дзеркальним ізомерам, діастереомерам, або іншим стереоізомерним формам. Даний винахід також призначений, щоб охопити використання всіх таких можливих форм, так само як їх рацемічних і розділених, і їх композицій. Коли 8 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 композиції, описані тут, містять олефінові подвійні зв'язки або інші центри геометричної асиметрії, мається на увазі, що включають обидва - Е і Z геометричні ізомери. Всі таутомери включені відповідно до представленого винаходу також. Як використовується тут, термін "стереоізомери" є загальним терміном для всіх ізомерів індивідуальних молекул, які відрізняються тільки орієнтацією атомів у просторі. Він включає дзеркальні ізомери й ізомери сполук з більше, ніж одним хіральним центром, які не є дзеркальними відображеннями один одного (діастереомери). Термін "хіральний центр" відноситься до атома вуглецю, до якого приєднані чотири різні групи. Термін "енантіомер" або "енантіомерний" відноситься до молекули, що не накладається на її дзеркальне відображення й отже оптично активний, де енантіомер обертає площину поляризованого світла в одному напрямку, і його дзеркальне відображення обертає площину поляризованого світла в протилежному напрямку. Термін "рацемічний" відноситься до суміші рівних частин енантіомерів й, які є оптично неактивні. Термін "розділення" відноситься до відокремлювання або концентрування, або зменшення однієї із двох енантіомерних форм молекули. Опіоїдні агоністи, корисні в представленому винаході, включають, але не обмежуються наступними: алфентаніл, алілпродин, альфапродин, анілеридин, бензилморфін, безитрамід, бупренорфін, буторфанол, клонітазен, кодеїн, дезоморфін, декстроморамід, дезоцин, діапромід, діаморфон, дигідрокодеїн, дигідроморфін, дименоксадол, димефептанол, диметилтіамбутен, діоксагептилу бутират, дипіпанол, ептазоцин, етогептазин, етилметилтіамбутен, етилморфін, етонітазен, еторфін, дигідроеторфін, фентаніл і похідні, гідрокодон, гідроморфон, гідроксипетидин, ізометадон, кетобемідон, леворфанол, левофенацикломорфан, лофенантил, меперидин, мептазинол, метазоцин, метадон, метопон, морфін, мірофін, нарцеїн, нікоморфін, норлеворфанол, норметадон, налорфін, налбуфен, норморфін, норпіпанон, опій, оксикодон, оксиморфон, пантопон, пентазоцин, фенадоксон, феноморфан, феназоцин, феноперидин, пімінодин, піритрамід, профетазин, промедол, проперидин, пропоксифен, суфенантил, тилідин, трамадол, їх фармацевтично прийнятні солі, гідрати й сольвати, суміші будь-якого з вищевказаних, і т.п. Опіоїдні агоністи, корисні в комбінації з опіоїдними агоністами, як описано вище, є наприклад, налоксон, налтрексон й налмефен або їх фармацевтично прийнятні солі, гідрати й сольвати, сумішами кожного з попередніх, і т.п. У певних варіантах реалізації, наприклад використовується комбінація оксикодону HCl і налоксону HCl у співвідношенні 2:1. У певних варіантах реалізації, опіоїдний анальгетик вибраний з кодеїну, морфіну, оксикодону, гідрокодону, гідроморфону, або оксиморфону або їх фармацевтично прийнятних солей, гідратів і сольватів, сумішей будь-якого з попередніх, і т.п. У певних варіантах реалізації опіоїдний анальгетик - оксикодон, гідроморфон або оксиморфон або їх сіль, така, як наприклад, гідрохлорид. Дозована форма включає приблизно від 5 мг приблизно до 500 мг оксикодону гідрохлориду, приблизно від 1 мг приблизно до 100 мг гідроморфону гідрохлориду, або приблизно від 5 мг до приблизно 500 мг гідрохлориду оксиморфону. Якщо використовуються інші солі, похідні або форми, може використовуватися еквімолярна кількість будь-якої іншої фармацевтично прийнятної солі або похідної, або форми, що включає, але не обмежується гідратами й сольватами, або вільною основою. Дозована форма включає, наприклад, 5 мг, 7,5 мг, 10 мг, 15 мг, 20 мг, 30 мг, 40 мг, 45 мг, 60 мг, або 80 мг, 90 мг, 120 мг або 160 мг оксикодону гідрохлориду або еквімолярну кількість будь-якої іншої фармацевтично прийнятної солі, похідної або форми, включаючи, але не обмежуючись гідратами й сольватами або вільною основою. Дозована форма включає, наприклад, 5 мг, 7,5 мг, 10 мг, 15 мг, 20 мг, 30, мг, 40 мг, 45 мг, 60 мг, або 80 мг, 90 мг гідрохлориду оксиморфону або 160 мг, або 120 мг, або еквімолярну кількість будь-якої іншої фармацевтично прийнятної солі, похідної або форми, включаючи, але не обмежуючись гідратами й сольватами або вільною основою. Дозована форма включає, наприклад, 2 мг, 4 мг, 8 мг, 12 мг, 16 мг, 24 мг, 32 мг, 48 мг або 64 мг гідроморфону гідрохлориду або еквімолярну кількість будь-якої іншої фармацевтично прийнятної солі, похідної або форми, включаючи, але не обмежуючись гідратами й сольватами або вільною основою. WO 2005/097801 А1, US 7 129 248 В2 й US 2006/0173029 А1, усі з яких тим самим включені посиланням, описують спосіб одержання оксикодону гідрохлориду, що має рівень 14гідроксикодеїнону менше, ніж приблизно 25 млн. ч., переважно менше, ніж приблизно 15 млн.ч., менше, ніж приблизно 10 млн. ч., або менше, ніж приблизно 5 млн. ч., більш переважно менше, 9 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 ніж приблизно 2 млн. ч., менше, ніж приблизно 1 млн. ч., менше, ніж приблизно 0,5 млн. ч. або менше, ніж приблизно 0,25 млн. ч. Термін "млн. ч." як використовується тут, означає "частин на мільйон". Відносно 14гідроксикодеїнону, "млн. ч." означає частин на мільйон 14-гідроксикодеїнону у певному зразку виробу. Рівень 14-гідроксикодеїнону може бути визначений будь-яким способом, відомим у даній галузі, переважно ВЕРХ аналізом, використовуючи ультрафіолетове виявлення. У певних варіантах реалізації представленого винаходу, де активний інгредієнт - оксикодону гідрохлорид, використовується оксикодону гідрохлорид, що має рівень 14-гідроксикодеїнону менше, ніж приблизно 25 млн. ч., переважно менше, ніж приблизно 15 млн. ч., менше, ніж приблизно 10 млн. ч., або менше, ніж приблизно 5 млн. ч., більш переважно менше, ніж приблизно 2 млн. ч., менше, ніж приблизно 1 млн. ч., менше, ніж приблизно 0,5 млн. ч. або менше, ніж приблизно 0,25 млн. ч. У певних інших варіантах реалізації, інші терапевтично активні агенти можуть використовуватися відповідно до представленого винаходу, або в комбінації з опіоїдами або замість опіоїдів. Приклади таких терапевтично активних агентів включають антигістамінні (наприклад, дименгідринат, дифенілгідрамін, хлорфенірамін й дексхлорфеніраміну малеат), нестероїдні протизапальні засоби (наприклад, напроксен, диклофенак, індометацин, ібупрофен, суліндак, інгібітори ЦОК-2) і ацетамінофен, антиеметики (наприклад, метоклопрамід, метилналтрексон), антиепілептики (наприклад, фенітоїн, мепробамат, нітразепам), вазодилататори (наприклад, ніфедипін, папаверин, дилтіазем й нікардипін), протикашльові засоби й відхаркувальні засоби (наприклад, фосфат кодеїну), антиастматики (наприклад, теофілін), антациди, спазмолітики (наприклад, атропін, скополамін), антидіабетичні засоби (наприклад, інсулін), сечогінні засоби (наприклад, етакринова кислота, бендрофлутіазид), антигіпотензивні препарати (наприклад, пропранолол, клонідин), гіпотензивні засоби (наприклад, клонідин, метилдофа), бронходилататори (наприклад, альбутерол), стероїди (наприклад, гідрокортизон, тріамцинолон, преднизон), антибіотики (наприклад, тетрациклін), антигемороїдальні, снотворні засоби, психотропні засоби, антидіарейні засоби, муколітики, седативні засоби, деконгенсанти (наприклад псевдоефедрин), проносні засоби, вітаміни, стимулятори (включаючи засоби, що пригнічують апетит, такі як фенілпропаноламін) і канабіноїди, так само, як їх фармацевтично прийнятні солі, гідрати і сольвати. У певних варіантах реалізації винахід відноситься до використання інгібіторів ЦОК-2, як активних агентів у комбінації з опіоїдними анальгетиками або замість опіоїдних анальгетиків, наприклад використовуючи інгібітори ЦОК-2, такі як мелоксикам (4-гідрокси-2-метил-N-(5-метил2-тіазоліл)-2Н-1,2-бензотіазин-3-карбоксамід-1,1-діоксид), як розкрито в патентах US № 10/056,347 й 11/825,938, що тим самим включений посиланням, набуметон (4-(6-метокси-2нафтил)-2-бутанон), як розкрито в US No. 10/056,348, що тим самим включений посиланням, целекоксиб (4-[5-(4-метилфеніл)-3-(трифторметил)-1Н-піразол-1-іл] бензолсульфонамід), як розкрито в патенті US № 11/698,394, що тим самим включений посиланням, німесулід (N-(4нітро-2-феноксифеніл) метансульфонамід), як розкрито в патенті US № 10/057,630, що тим самим включений посиланням, і N-[3-(форміламіно)-4-оксо-6-фенокси-4Н-1-бензопіран-7іл]метансульфонамід (Т - 614), як розкрито в патенті US № 10/057,632, що тим самим включений посиланням. Даний винахід також відноситься до дозованих форм, що використовують активні агенти такі, як, наприклад, бензодіазепіни, барбітурати або амфетаміни. Вони можуть бути скомбіновані з відповідними антагоністами. Термін "бензодіазепіни" відноситься до бензодіазепінів і препаратів, які є похідними бензодіазепіну, які здатні до пригнічення центральної нервової системи. Бензодіазепіни включають, але не обмежуються наступними: алпразолам, бромазепам, хлордіазепоксид, хлоразепат, діазепам, естазолам, флуразепам, галазепам, кетазолам, лоразепам, нітразепам, оксазепам, празепам, квазепам, темазепам, тріазолам, метилфенідат, так само як їх фармацевтично прийнятні солі, гідрати і сольвати, й суміші. Антагоністи бензодіазепіну, які можуть використовуватися в представленому винаході, включають, але не обмежуються наступними: флумазеніл, так само як фармацевтично прийнятні солі, гідрати, і сольвати. Барбітурати відносяться до седативно-снодійних препаратів, отриманих з барбітурової кислоти (2,4,6,-тріоксогексагідропіримідин). Барбітурати включають, але не обмежуються наступними: амобарбітал, апробарботал, бутабарбітал, буталбітал, метогекситал, мефобарбітал, метарбітал, пентобарбітал, фенобарбітал, секобарбітал і, так само як їх фармацевтично прийнятні солі, гідрати і суміші сольватів. Антагоністи барбітуратів, які можуть використовуватися в представленому винаході, включають, але не обмежуються наступними: амфетаміни, так само як фармацевтично прийнятні солі, гідрати, і сольвати. 10 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 Стимулятори відносяться до препаратів, які стимулюють центральну нервову систему. Стимулятори включають, але не обмежуються наступними: амфетаміни, такі як амфетамін, декстроамфетаміновий полімерний комплекс, декстроамфетамін, метамфетамін, метилфенідат, так само як їх фармацевтично прийнятні солі, гідрати і сольвати, й суміші. Антагоністи стимуляторів, які можуть використовуватися в представленому винаході, включають, але не обмежуються наступними: бензодіазепіни, так само як фармацевтично прийнятні солі, гідрати і сольвати, як описано тут. Короткий опис фігур. Фіг. 1 - фотографія, що зображує вид зверху (проекція відповідно товщини таблетки) таблеток Прикладу 7,1 перед (ліва сторона) і після (права сторона) тесту міцності на розрив, використовуючи апарат Schleuniger, модель 6D. Фіг. 2 - фотографія, що зображує вид зверху (проекція відповідно товщини таблетки) таблеток Прикладу 7,2 перед (ліва сторона) і після (права сторона) тесту міцності на розрив, використовуючи апарат Schleuniger, модель 6D. Фіг. 3 - фотографія, що зображує вид зверху (проекція відповідно товщини таблетки) таблеток Прикладу 7,3 перед (ліва сторона) і після (права сторона) тесту міцності на розрив, використовуючи апарат Schleuniger, модель 6D. Фіг. 4 - фотографія, що зображує вид зверху (проекція відповідно товщини таблетки) таблетки Прикладу 7,1 після сплющення на ручному настільному пресі Carver (гідравлічний пристрій, модель #3912). Фіг. 5 - фотографія, що зображує вид зверху (проекція відповідно товщини таблетки) таблетки Прикладу 7,2 після сплющення на ручному настільному пресі Carver (гідравлічний пристрій, модель #3912). Фіг. 6 - фотографія, що зображує вид зверху (проекція відповідно товщини таблетки) таблетки Прикладу 7,3 після сплющення на ручному настільному пресі Carver (гідравлічний пристрій, модель #3912). Фіг. 7 - фотографія, що зображує вид зверху (проекція відповідно товщини таблетки) таблетки Прикладу 7,1 після 10 нанесених вручну ударів молотка. Фіг. 8 - фотографія, що зображує вид зверху (проекція відповідно товщини таблетки) таблетки Прикладу 7,2 після 10 нанесених вручну ударів молотка. Фіг. 9 - фотографія, що зображує вид зверху (проекція відповідно товщини таблетки) таблетки Прикладу 7,3 після 10 нанесених вручну ударів молотка. Фіг. 10 - діаграма, що зображує температурний профіль процесів твердіння Прикладу 13.1. Фіг. 11 - діаграма, що зображує температурний профіль процесів твердіння Прикладу 13.2. Фіг. 12 - діаграма, що зображує температурний профіль процесів твердіння Прикладу 13,3. Фіг. 13 - діаграма, що зображує температурний профіль процесів твердіння Прикладу 13.4. Фіг. 14 - діаграма, що зображує температурний профіль процесів твердіння Прикладу 13.5. Фіг. 15 - діаграма, що зображує температурний профіль процесів твердіння Прикладу 14.1. Фіг. 16 - діаграма, що зображує температурний профіль процесів твердіння Прикладу 14.2. Фіг. 17 - діаграма, що зображує температурний профіль процесів твердіння Прикладу 14.3. Фіг. 18 - діаграма, що зображує температурний профіль процесів твердіння Прикладу 14.4. Фіг. 19 - діаграма, що зображує температурний профіль процесів твердіння Прикладу 14.5. Фіг. 20 - діаграма Прикладу 20 визначення твердості вдавлюванням, виконаного на таблетках Прикладу 13.1 (отверділі протягом 30 хвилин, непокриті). Фіг. 21 - діаграма Прикладу 20 визначення твердості вдавлюванням, виконаного на таблетках Прикладу 13.2 (отверділі протягом 30 хвилин, непокриті). Фіг. 22 - діаграма Прикладу 20 визначення твердості вдавлюванням, виконаного на таблетках Прикладу 13,3 (отверділі протягом 30 хвилин, непокриті). Фіг. 23 - діаграма Прикладу 20 визначення твердості вдавлюванням, виконаного на таблетках Прикладу 13,4 (отверділі протягом 30 хвилин, непокриті). Фіг. 24 - діаграма Прикладу 20 визначення твердості вдавлюванням, виконаного на таблетках Прикладу 13,5 (отверділі протягом 30 хвилин, непокриті). Фіг. 25 - діаграма Прикладу 20 визначення твердості вдавлюванням, виконаного на таблетках Прикладу 17.1 (отверділі протягом 15 хвилин при 72 °C, покриті). Фіг. 26 - діаграма Прикладу 20 визначення твердості вдавлюванням, виконаного на таблетках Прикладу 18.2 (отверділі протягом 15 хвилин при 72 °C, покриті). Фіг. 27 - діаграма Прикладу 20 визначення твердості вдавлюванням, виконаного на таблетках Прикладу 14,1 (отверділі протягом 1 год., покриті). Фіг. 28 - діаграма Прикладу 20 визначення твердості вдавлюванням, виконаного на таблетках Прикладу 14,2 (отверділі протягом 1 год., покриті). 11 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 Фіг. 29 - діаграма Прикладу 20 визначення твердості вдавлюванням, виконаного на таблетках Прикладу 14,3 (отверділі протягом 1 год., покриті). Фіг. 30 - діаграма Прикладу 20 визначення твердості вдавлюванням, виконаного на таблетках Прикладу 14,4 (отверділі протягом 1 год., покриті). Фіг. 31 - діаграма Прикладу 20 визначення твердості вдавлюванням, виконаного на таблетках Прикладу 14,5 (отверділі протягом 1 год., покриті). Фіг. 32 - діаграма Прикладу 20 визначення твердості вдавлюванням, виконаного на таблетках Прикладу 16.1 (отверділі протягом 15 хвилин, покриті). Фіг. 33 - діаграма Прикладу 20 визначення твердості вдавлюванням, виконаного на таблетках Прикладу 16.2 (отверділі протягом 15 хвилин, покриті). Фіг. 34 - діаграма Прикладу 21 визначення твердості вдавлюванням, виконаного на таблетках Прикладу 16.1 (отверділі протягом 15 хвилин, покриті) і комерційних 60 мг таблетках OxyContin™. Фіг. 35 - діаграма Прикладу 21 визначення твердості вдавлюванням, виконаного на таблетках Прикладу16,2 (отверділі протягом 15 хвилин, покриті) і комерційних таблетках 80 мг OxyContin™. Фіг. 36 демонструє середні плазменні концентрації оксикодону проти профілю часу на лінійній шкалі [Популяція: Повний Аналіз (стан харчування)] відповідно до Прикладу 26. Фіг. 37 демонструє середні плазменні концентрації оксикодону проти профілю часу на logлінійній шкалі [Популяція: Повний Аналіз (стан харчування)] відповідно до Прикладу 26. Фіг. 38 демонструє середні плазменні концентрації оксикодону проти профілю часу на лінійній шкалі [Популяція: Повний Аналіз (стан голодування)] відповідно до Прикладу 26. Фіг. 39 демонструє середні плазменні концентрації оксикодону проти профілю часу на logлінійній шкалі [Популяція: Повний Аналіз (Стан голодування)] відповідно до Прикладу 26. Фіг. 40 демонструє характерні зображення подрібнених таблеток 10 мг OxyContin™ і подрібнених таблеток Прикладу 7.2, відповідно до Прикладу 27. Фіг. 41 демонструє характерні зображення розмелених таблеток Прикладу 7,2 і 10 мг таблеток OxyContin ™ до і після 45 хвилин розчинення, відповідно до Прикладу 27. Фіг. 42 демонструє характерні зображення розмелених таблеток Прикладу 7,2 і подрібнених 10 мг таблеток OxyContin™, відповідно до Прикладу 27. Фіг. 43 демонструє діаграми гранулометричного складу розмелених таблеток (OxyContin™ 10 мг, таблетки Прикладу 7.2 і Прикладу 14,5) відповідно до Прикладу 27. Деталізований опис. У певних варіантах реалізації, даний винахід спрямований на спосіб одержання твердої пероральної фармацевтичної дозованої форми пролонгованого вивільнення, що включає принаймні наступні стадії: (a) комбінування принаймні: (1) принаймні одного поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000, і (2) принаймні одного активного інгредієнта, щоб утворити композицію; (b) формування композиції, щоб утворити матричну композицію пролонгованого вивільнення; і с) твердіння згадуваної матричної композиції пролонгованого вивільнення, що включає принаймні стадію твердіння матричної композиції пролонгованого вивільнення до температури, що є принаймні температурою розм'якшення згадуваного поліетиленоксиду протягом періоду часу принаймні приблизно 1 хв. Переважно, твердіння проводиться при атмосферному тиску. У певному варіанті реалізації, даний винахід відноситься до способу одержання твердої пероральної фармацевтичної дозованої форми пролонгованого вивільнення, що включає принаймні наступні стадії: (а) комбінування принаймні: (1) принаймні одного поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, молекулярну масу принаймні 1000000; і (2) принаймні одного активного інгредієнта, щоб утворити композицію; b) формування композиції, щоб утворити матричну композицію пролонгованого вивільнення; і c) твердіння згадуваної матричної композиції пролонгованого вивільнення, яка включає принаймні стадію твердіння матричної композиції пролонгованого вивільнення при температурі, 12 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 що є принаймні температурою розм'якшення згадуваного поліетиленоксиду протягом періоду часу принаймні 5 хвилин. Переважно, твердіння проводиться при атмосферному тиску. У певних варіантах реалізації даний винахід спрямований на спосіб одержання твердої пероральної фармацевтичної дозованої форми пролонгованого вивільнення, що включає принаймні наступні стадії (а) комбінування принаймні: (1) принаймні одного поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000, і (2) принаймні одного активного інгредієнта, щоб утворити композицію; (b) формування композиції, щоб утворити матричну композицію пролонгованого вивільнення; і с) твердіння згадуваної матричної композиції пролонгованого вивільнення, що включає принаймні стадію твердіння де згадуваний поліетиленоксид принаймні частково плавиться. Переважно, твердіння проводиться при атмосферному тиску. У певних варіантах реалізації композицію формують на стадії b), щоб утворити матричну композицію пролонгованого вивільнення у формі таблеток. Для формування матричної композиції пролонгованого вивільнення у таблетовану форму може використовуватися спосіб безпосередньої компресії. Безпосереднє пресування - ефективний і простий спосіб формування таблеток, уникаючи технологічної стадії, подібній вологому гранулюванню. Однак, будь-які інші способи виробництва таблеток, відомі у даній галузі можуть використовуватися, такі як вологе гранулювання й наступне пресування гранул, щоб утворити таблетки. В одному варіанті реалізації, твердіння матричної композиції пролонгованого вивільнення на стадії с) включає принаймні стадію твердіння де, високомолекулярний поліетиленоксид в матричній композиції пролонгованого вивільнення принаймні частково плавиться. Наприклад, принаймні приблизно 20 % або принаймні приблизно 30 % високомолекулярного поліетиленоксиду плавиться в матричних композиціях пролонгованого вивільнення. Переважно, принаймні приблизно 40 % або принаймні приблизно 50 %, більш переважно принаймні приблизно 60 %, принаймні приблизно 75 % або принаймні приблизно 90 % високомолекулярного поліетиленоксиду плавиться в матричних композиціях пролонгованого вивільнення. У переважному варіанті реалізації, розплавляється приблизно 100 % високомолекулярного поліетиленоксиду. В інших варіантах реалізації твердіння матричної композиції пролонгованого вивільнення на стадії с) включає принаймні стадію твердіння де, матричну композицію пролонгованого вивільнення піддають дії підвищеної температури протягом певного проміжку часу. У таких варіантах реалізації температура, використовувана на стадії с), тобто температура твердіння, принаймні настільки ж висока, як температура розм'якшення високомолекулярного поліетиленоксиду. Не бажаючи бути прив'язаним до теорії вважається, що твердіння при температурі, що принаймні настільки ж висока як температура розм'якшення високомолекулярного поліетиленоксиду, заподіює принаймні адгезування частинок поліетиленоксиду однієї до іншої або навіть сплавлення. Відповідно до деяких варіантів реалізації температура твердіння - принаймні приблизно 60 °C або принаймні приблизно 62 °C, або в діапазоні приблизно від 62 °C приблизно до 90 °C, або приблизно від 62 °C приблизно до 85 °C, або приблизно від 62 °C приблизно до 80 °C, або приблизно від 65 °C приблизно до 90 °C, або приблизно від 65 °C приблизно до 85 °C, або приблизно від 65 °C приблизно до 80 °C. Температура твердіння переважно знаходиться в діапазоні приблизно від 68 °C приблизно до 90 °C, або приблизно від 68 °C приблизно до 85 °C або приблизно від 68 °C приблизно до 80 °C, більш переважно приблизно від 70 °C приблизно до 90 °C або приблизно від 70 °C приблизно до 85 °C, або приблизно від 70 °C приблизно до 80 °C, найбільш переважно приблизно від 72 °C приблизно до 90 °C, або приблизно від 72 °C приблизно до 85 °C, або приблизно від 72 °C приблизно до 80 °C. Температура твердіння може бути принаймні приблизно 60 °C, або принаймні приблизно 62 °C, але менше, ніж приблизно 90 °C, або менше, ніж приблизно 80 °C. Переважно, перебуває у діапазоні приблизно від 62 °С приблизно до 72 °C, особливо приблизно від 68 °C приблизно до 72 °C. Переважно, температура твердіння принаймні настільки ж висока, як нижня межа температурного діапазону розм'якшення високомолекулярного поліетиленоксиду або принаймні приблизно 62 °C, або принаймні приблизно 68 °C. Більш переважно, в межах температурного діапазону розм'якшення високомолекулярного поліетиленоксиду або принаймні приблизно 70 °C. Ще більш переважно, температура твердіння принаймні настільки ж висока, як верхня межа температурного діапазону розм'якшення високомолекулярного поліетиленоксиду або принаймні приблизно 72 13 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 °C. В альтернативному варіанті реалізації, температура твердіння вище, ніж верхня межа температурного діапазону розм'якшення високомолекулярного поліетиленоксиду, наприклад температура твердіння - принаймні приблизно 75 °C або принаймні приблизно 80 °C. У тих варіантах реалізації, де твердіння матричної композиції пролонгованого вивільнення на стадії с) включає принаймні стадію твердіння де, матричну композицію пролонгованого вивільнення піддають дії підвищеної температури протягом певного проміжку часу, цей проміжок часу надалі згадується як час твердіння. Для вимірювання часу твердіння визначали вихідну точку й кінцеву точку стадії твердіння. Для цілей представленого винаходу вихідна точка стадії твердіння визначена, щоб бути точкою часу, коли температура твердіння досягнута. У певних варіантах реалізації, температурний профіль протягом стадії твердіння показувала платоподібна форма між вихідною точкою й кінцевою точкою твердіння. У таких варіантах реалізації кінцева точка стадії твердіння визначена, щоб бути точкою часу, коли нагрівання зупиняють або принаймні зменшують, наприклад, завершуючи або зменшуючи нагрівання та/або запускаючи наступну стадію охолодження, і поступово знижується нижче температури твердіння на більше, ніж приблизно 10 °C та/або нижче нижньої межі температури розм'якшення високомолекулярного поліетиленоксиду, наприклад нижче приблизно 62 °C. Коли температура твердіння досягнута, і стадія твердіння таким чином запущена, відхилення від температури твердіння в ході стадії твердіння можуть зустрічатися. Такі відхилення допустимі, поки вони не перевищують значення приблизно ±10 °C, переважно приблизно ±6 °C, і більш переважно приблизно ±3 °C. Наприклад, якщо температура твердіння повинна бути підтримана принаймні приблизно 75 °C, вимірювана температура може тимчасово збільшитися до значення приблизно 85 °C, переважно приблизно 81 °C і більш переважно приблизно 78 °C, і вимірювана температура може також тимчасово падати до значення приблизно 65 °C, переважно приблизно 69 °C і більш переважно приблизно 72 °C. У випадках більшого зменшення температури та/або у випадку, що температура знижується нижче нижньої межі температури розм'якшення високомолекулярного поліетиленоксиду, наприклад нижче приблизно 62 °C, стадія твердіння припинена, тобто кінцева точка досягнута. Твердіння може бути повторно запущено, знову досягаючи температури твердіння. В інших варіантах реалізації, температурний профіль протягом стадії твердіння показувала параболічна або трикутна форма між вихідною точкою й кінцевою точкою твердіння. Це означає, що після вихідної точки, тобто точки часу, коли температура твердіння досягнута, подальші збільшення температури досягають максимуму, і потім зменшуються. У таких варіантах реалізації кінцева точка стадії твердіння визначена, щоб бути точкою часу, коли температура знижується нижче температури твердіння. У цьому контексті повинно бути відмічено, що залежно від використовуваного для твердіння апарату, що надалі названий як пристрій для твердіння, різні види температур у межах пристрою твердіння можуть бути виміряні, щоб характеризувати температуру твердіння. У певних варіантах реалізації, стадія твердіння може мати місце в термостаті. У таких варіантах реалізації температуру вимірюють у внутрішній частині термостату. Базуючись на тому, коли стадію твердіння проводять в термостаті, температура твердіння визначена, щоб бути цільовою внутрішньої частини термостату, і вихідна точка стадії твердіння визначена, щоб бути точкою часу, коли внутрішня температура термостату досягає температури твердіння. Кінцева точка стадії твердіння визначена, щоб бути (1) точкою часу, коли нагрівання зупиняють або принаймні зменшують й температура у внутрішній частині термостату поступово знижується нижче температури твердіння на більше, ніж приблизно 10 °C та/або нижче нижньої межі температури розм'якшення високомолекулярного поліетиленоксиду, наприклад нижче приблизно 62 °С, на платоподібному температурному профілі або (2) точкою часу, коли температура внутрішньої частини термостату знижується нижче температури твердіння в параболічному або трикутному температурному профілі. Переважно, стадія твердіння починається, коли температура в термостаті досягає температури твердіння принаймні приблизно 62 °C, принаймні приблизно 68 °C або принаймні приблизно 70 °C, більш переважно принаймні приблизно 72 °C або принаймні приблизно 75 °C. У переважних варіантах реалізації, температурний профіль протягом стадії твердіння показувала плато-подібна форма, де температура твердіння, тобто внутрішня температура термостату, є переважно принаймні приблизно 68 °C, наприклад, приблизно 70 °C або приблизно 72 °C, або приблизно 73 °C, або перебуває в межах в діапазоні приблизно від 70 °C приблизно до 75 °C, і час твердіння перебуває переважно в діапазоні приблизно від 30 хвилин приблизно до 20 годин, більш переважно приблизно від 30 хвилин приблизно до 15 годин, або приблизно від 30 хвилин приблизно до 4 годин, або приблизно від 30 хвилин приблизно до 2 годин. Найбільш переважно, час твердіння перебуває в діапазоні приблизно від 30 хвилин приблизно до 90 хвилин. 14 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 У певних інших варіантах реалізації твердіння має місце в пристроях для твердіння, що нагріваються потоком повітря й включають подачу гарячого повітря (вхідний отвір) і вихлопну трубу, як наприклад резервуар для нанесення покриття або псевдозріджений шар. Такі пристрої для твердіння надалі називатимуться конвекційними пристроями для твердіння. У таких пристроях для твердіння можливо вимірювати температуру вхідного повітря, тобто температуру гарячого повітря, що надходить у конвекційний пристрій для твердіння та/або температуру вихідного повітря, тобто температуру повітря, що залишає конвекційний пристрій для твердіння. Також можливо визначити або принаймні оцінити температуру композицій у внутрішній частині конвекційного пристрою для твердіння під час стадії твердіння, наприклад при використанні інфрачервоних інструментів вимірювання температури, таких як інфрачервоний датчик, або вимірюючи температуру, використовуючи температурний зонд, що розміщували у внутрішній частині пристрою для твердіння біля матричних композицій пролонгованого вивільнення. Базуючись на тому, коли стадію твердіння проводять в конвекційному пристрої для твердіння, може бути визначена температура і час твердіння наступним чином. В одному варіанті реалізації, де час твердіння, вимірюваний відповідно до методу 1, температура твердіння визначена, щоб бути цільовою температурою вхідного повітря, і вихідна точка стадії твердіння визначена, щоб бути точкою часу, коли температура вхідного повітря досягає температури твердіння. Кінцева точка стадії твердіння визначена, щоб бути (1) точкою часу, коли нагрівання зупиняють або принаймні зменшують й температура вхідного повітря послідовно зменшується нижче температури твердіння на більше, ніж приблизно 10 °C та/або нижче нижньої межі температури розм'якшення високомолекулярного поліетиленоксиду, наприклад нижче приблизно 62 °С, на платоподібному температурному профілі або (2) точкою часу, коли температура вхідного повітря знижується нижче температури твердіння в параболічному або трикутному температурному профілі. Переважно, стадія твердіння починається відповідно до методу 1, коли температура вхідного повітря досягає температури твердіння принаймні приблизно 62 °C, принаймні приблизно 68 °C або принаймні приблизно 70 °C, більш переважно, принаймні приблизно 72 °C або принаймні приблизно 75 °C. У переважному варіанті реалізації, температурний профіль протягом стадії твердіння показувала плато-подібна форма, де температура твердіння, тобто цільова температура вхідного повітря, є переважно принаймні приблизно 72 °C, наприклад приблизно 75 °C, і час твердіння, вимірюваний відповідно до методу 1, перебуває переважно в діапазоні приблизно від 15 хвилин приблизно до 2 годин, наприклад приблизно 30 хвилин або приблизно 1 год. В іншому варіанті реалізації, де час твердіння, вимірюваний відповідно до методу 2, температура твердіння визначена, щоб бути цільовою температурою вихідного повітря, і вихідна точка стадії твердіння визначена, щоб бути точкою часу, коли температура вихідного повітря досягає температури твердіння. Кінцева точка стадії твердіння визначена, щоб бути (1) точкою часу, коли нагрівання зупиняють або принаймні зменшують й температура вихідного повітря послідовно зменшується нижче температури твердіння на більше, ніж приблизно 10 °C та/або нижче нижньої межі температури розм'якшення високомолекулярного поліетиленоксиду, наприклад нижче приблизно 62 °C, на платоподібному температурному профілі або (2) точкою часу, коли температура вихідного повітря знижується нижче температури твердіння в параболічному або трикутному температурному профілі. Переважно, стадія твердіння починається відповідно до методу 2, коли температура вихідного повітря досягає температури твердіння принаймні приблизно 62 °C, принаймні приблизно 68 °C або принаймні приблизно 70 °C, більш переважно, принаймні приблизно 72 °C або принаймні приблизно 75 °C. У переважних варіантах реалізації, температурний профіль протягом стадії твердіння показувала платоподібна форма, де температура твердіння, тобто цільова температура вихідного повітря, переважно принаймні приблизно 68 °C, принаймні приблизно 70 °C або принаймні приблизно 72 °C, наприклад цільова температура вихідного повітря - приблизно 68 °C, приблизно 70 °С, приблизно 72 °C, приблизно 75 °C або приблизно 78 °С, і час твердіння, що вимірюваний відповідно до методу 2, перебуває переважно в діапазоні приблизно від 1 хв. приблизно до 2 годин, переважно приблизно від 5 хвилин приблизно до 90 хвилин, наприклад час твердіння приблизно 5 хвилин, приблизно 10 хвилин, приблизно 15 хвилин, приблизно 30 хвилин, приблизно 60 хвилин, приблизно 70 хвилин, приблизно 75 хвилин або приблизно 90 хвилин. У більш переважному варіанті реалізації, час твердіння, що вимірюваний відповідно до методу 2, перебуває в діапазоні приблизно від 15 хвилин приблизно до 1 год. У подальшому варіанті реалізації, де час твердіння, вимірюваний відповідно до методу 3, температура твердіння визначена, щоб бути цільовою температурою матричних композицій пролонгованого вивільнення, і вихідна точка стадії твердіння визначена, щоб бути точкою часу, коли температура матричних композицій пролонгованого вивільнення, які можуть бути виміряні, 15 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 наприклад, 14 датчиком, досягає температури твердіння. Кінцева точка стадії твердіння визначена, щоб бути (1) точкою часу, коли нагрівання зупиняють або принаймні зменшують й температура матричних композицій пролонгованого вивільнення послідовно зменшується нижче температури твердіння на більше, ніж приблизно 10 °C та/або нижче нижньої межі температури розм'якшення високомолекулярного поліетиленоксиду, наприклад нижче приблизно 62 °C, на платоподібному температурному профілі або (2) точкою часу, коли температура матричних композицій пролонгованого вивільнення нижче температури твердіння в параболічному або трикутному температурному профілі. Переважно, стадія твердіння починається відповідно до методу 3, коли температура матричних композицій пролонгованого вивільнення досягає температури твердіння принаймні приблизно 62 °C, принаймні приблизно 68 °C або принаймні приблизно 70 °C, більш переважно, принаймні приблизно 72 °C або принаймні приблизно 75 °C. Ще в одному іншому варіанті реалізації, де час твердіння, вимірюваний відповідно до методу 4, температура твердіння визначена, щоб бути цільовою температурою, виміряною, використовуючи температурний зонд, такий як дротяна термопара, що була розміщена в пристрої для твердіння біля матричних композицій пролонгованого вивільнення, і вихідна точка стадії твердіння визначена, щоб бути точкою часу, коли температура, виміряна використовуючи температурний зонд, що розміщували у пристрої для твердіння біля матричних композицій пролонгованого вивільнення, що досягли температури твердіння. Кінцева точка стадії твердіння визначена, щоб бути (1) точкою часу, коли нагрівання зупиняють або принаймні зменшують, і температура, виміряна використовуючи температурний зонд, послідовно зменшується нижче температури твердіння на більше, ніж приблизно 10 °C та/або нижче нижньої межі температури розм'якшення високомолекулярного поліетиленоксиду, наприклад нижче приблизно 62 °C, на платоподібному температурному профілі або (2) точкою часу, коли температура, виміряна використовуючи температурний зонд, поступово зменшується нижче температури твердіння в параболічному або трикутному температурному профілі. Переважно, стадія твердіння починається відповідно до методу 4, коли температура, виміряна використовуючи температурний зонд, що розміщували у пристрої для твердіння біля матричних композицій С пролонгованого вивільнення, що досягли температури твердіння принаймні приблизно 62 С, принаймні приблизно 68 °C або принаймні приблизно 70 °C, більш переважно, принаймні приблизно 72 °C або принаймні приблизно 75 °C. У переважному варіанті реалізації, температурний профіль протягом стадії твердіння показувала плато-подібна форма, де температура твердіння, тобто цільова температура, виміряна використовуючи температурний зонд, що розміщували у пристрої для твердіння біля матричних композицій пролонгованого вивільнення, переважно принаймні приблизно 68 °C, наприклад, приблизно 70 °C, і час твердіння, що вимірюваний відповідно до методу 4, перебуває переважно в діапазоні приблизно від 15 хвилин приблизно до 2 годин, наприклад час твердіння - приблизно 60 хвилин або приблизно 90 хвилин. Якщо твердіння має місце в конвекційному пристрої для твердіння, час твердіння може бути вимірюваний кожним із способів 1, 2, 3 або 4, У переважному варіанті реалізації час твердіння, вимірюваний відповідно до методу 2. У певних варіантах реалізації, температура твердіння визначена як цільова гранична температура, наприклад температура твердіння визначена як цільова гранична температура вхідного повітря або цільова гранична температура вихідного повітря. У таких варіантах реалізації вихідна точка стадії твердіння визначена, щоб бути точкою часу, коли досягнута нижня межа цільової граничної температури, і кінцева точка стадії твердіння визначена, щоб бути точкою часу, коли нагрівання зупиняють або принаймні зменшують, і температура послідовно зменшується нижче нижньої межі цільової граничної температури більше, ніж приблизно 10 °C та/або нижче нижньої межі температури розм'якшення високомолекулярного поліетиленоксиду, наприклад нижче приблизно 62 °C. Час твердіння, тобто інтервал часу, коли матричну композицію пролонгованого вивільнення піддають дії температури твердіння, що може, наприклад бути виміряна відповідно до способів 1, 2, 3 й 4, як описано вище, є принаймні приблизно 1 хвилиною або принаймні приблизно 5 хвилинами. Час твердіння може змінитися приблизно від 1 хв. приблизно до 24 годин, або приблизно від 5 хвилин приблизно до 20 годин або приблизно від 10 хвилин приблизно до 15 годин, або приблизно від 15 хвилин приблизно до 10 годин, або приблизно від 30 хвилин приблизно до 5 годин залежно від конкретної композиції й складу й температури твердіння. Параметр композиції, час твердіння й температура твердіння вибрані, щоб досягти резистентності щодо пошкодження, як описано тут. Відповідно до певних варіантів реалізації, час твердіння змінюється приблизно від 15 хвилин приблизно до 30 хвилин. Відповідно до 16 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 подальших варіантів реалізації, де температура твердіння - принаймні приблизно 60 °C або принаймні приблизно 62 °C, переважно принаймні приблизно 68 °C, принаймні приблизно 70 °C, принаймні приблизно 72 °C або принаймні приблизно 75 °C або змінюється приблизно від 62 °C приблизно до 85 °C, або приблизно від 65 °C приблизно до 85 °C, час твердіння - переважно принаймні приблизно 15 хвилин, принаймні приблизно 30 хвилин, принаймні приблизно 60 хвилин, принаймні приблизно 75 хвилин, принаймні приблизно 90 хвилин або приблизно 120 хвилин. У переважних варіантах реалізації, де температура твердіння наприклад принаймні приблизно 62 °C, принаймні приблизно 68 °C або принаймні приблизно 70 °C, переважно принаймні приблизно 72 °C або принаймні приблизно 75 °C, або розташовується приблизно від 62 °C приблизно до 80 °C, приблизно від 65 °C приблизно до 80 °C, приблизно від 68 °C приблизно до 80 °C, приблизно від 70 °C приблизно до 80 °C або приблизно від 72 °C приблизно до 80 °C, час твердіння - переважно принаймні приблизно 1 хвилини або принаймні приблизно 5 хвилин. Більш переважно, час твердіння - принаймні приблизно 10 хвилин, принаймні приблизно 15 хвилин або принаймні приблизно 30 хвилин. У таких певних втіленнях, час твердіння може бути вибраний, щоб бути настільки ж коротким, наскільки можливо, усе ще досягаючи бажаної резистентності щодо пошкодження. Наприклад, час твердіння переважно не перевищує приблизно 5 годин, більш переважно не перевищує приблизно 3 год., і найбільш переважно не перевищує приблизно 2 год. Переважно, час твердіння перебуває в діапазоні приблизно від 1 хв. приблизно до 5 годин, приблизно від 5 хвилин приблизно до 3 годин, приблизно від 15 хвилин приблизно до 2 годин або приблизно від 15 хвилин приблизно до 1 год. Будь-яка комбінація температур твердіння й часу твердіння, як розкрито тут знаходиться в межах представленого винаходу. У певних варіантах реалізації, композиція піддана температурі твердіння, поки високомолекулярний поліетиленоксид, присутній в матричній композиції пролонгованого вивільнення не досяг своєї температури розм'якшення та/або принаймні частково плавиться. У таких певних втіленнях, час твердіння може бути менше, ніж приблизно 5 хвилин, наприклад час твердіння може змінитися приблизно від 0 хвилин приблизно до 3 годин або приблизно від 1 хв. приблизно до 2 годин або приблизно від 2 хвилин приблизно до 1 год. Миттєве твердіння можливо, вибираючи пристрій твердіння, що дозволяє високомолеулярному поліетиленоксиду швидко нагрітися в матричній композиції пролонгованого вивільнення до принаймні його температури розм'якшення, так, щоб високомолекулярний поліетиленоксид принаймні частково плавився. Такі пристрої для твердіння - наприклад мікрохвильові печі, ультразвукові пристрої, апарат світлового опромінення, такий як апарат ультрафіолетового опромінення, апарат УВЧ або будь-який спосіб, відомий фахівцю в даній галузі. Фахівець у даній галузі знає, що розмір матричної композиції пролонгованого вивільнення може визначатися заданим часом твердіння й температурою твердіння, щоб досягти бажаної резистентності щодо пошкодження. Не бажаючи бути прив'язаним до будь-якої теорії, вважають, що у випадку великої матричної композиції пролонгованого вивільнення, такої як велика таблетка, необхідний довший час твердіння, щоб провести теплоту у внутрішню частину композиції, ніж у випадку відповідної композиції меншого розміру. Більш висока температура збільшує коефіцієнт теплопровідності й таким чином зменшує заданий час твердіння. Стадія твердіння с) може мати місце в термостаті. Переважно, стадія твердіння с) має місце в шарі вільно-текучих матричних композицій пролонгованого вивільнення як, наприклад у резервуарі для покриття. Резервуар для нанесення покриття дозволяє відбутися стадії твердіння ефективним способом, з наступною стадією покриття без потреби переміщення дозованих форм, наприклад таблеток. Такий процес може включати стадії: (а) комбінування принаймні: (1) принаймні одного поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну вагу принаймні 1000000, і (2) принаймні одного активного інгредієнта, щоб утворити композицію; b) формування згадуваної композиції, щоб утворити матричну композицію пролонгованого вивільнення у формі таблеток безпосереднім пресуванням; c) твердіння згадуваної таблетки шляхом - піддавання дії шару вільно-текучих таблеток до температури приблизно від 62 °C приблизно до 90 °C, переважно від приблизно 70 °C до приблизно 90 °C протягом інтервалу часу принаймні приблизно 1 хв. або принаймні приблизно 5 хвилин, переважно принаймні приблизно 30 хвилин, у резервуарі для покриття й - наступного охолодження шару вільно-текучих таблеток до температури нижче приблизно 50 °C; і наступного (d) покриття дозованої форми в згадуваному резервуарі для покриття. 17 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 У певних варіантах реалізації, додаткова стадія твердіння може бути здійснена після стадії d) покриття дозованих форм. Додаткова стадія твердіння може бути здійснена, як описано для стадії твердіння с). У таких певних втіленнях, температура твердіння додаткової стадії твердіння - переважно принаймні приблизно 70 °C, принаймні приблизно 72 °C або принаймні приблизно 75 °C, і час твердіння перебуває переважно в діапазоні приблизно від 15 хвилин приблизно до 1 год., наприклад приблизно 30 хвилин. У певних варіантах реалізації антиоксидант, наприклад, БГТ (бутильований гідрокситолуол) додавали до композиції. У певних варіантах реалізації стадія твердіння с) приводить до зменшення в густині матричної композиції пролонгованого вивільнення, таким чином, що густина отвердженої матричної композиції пролонгованого вивільнення нижче, ніж густина матричної композиції пролонгованого вивільнення до стадії твердіння с). Переважно, густина отвердженої матричної композиції пролонгованого вивільнення в порівнянні із густиною неотвердженої матричної композиції пролонгованого вивільнення зменшується принаймні на приблизно 0,5 %. Більш переважно, густина отвердженої матричної композиції пролонгованого вивільнення в порівнянні із густиною неотвердженої матричної композиції пролонгованого вивільнення зменшується принаймні на приблизно 0,7 %, принаймні приблизно 0,8 %, принаймні приблизно 1,0 %, принаймні приблизно 2,0 % або принаймні приблизно 2,5 %. Не бажаючи бути прив'язаним до будь-якої теорії, вважають, що матрична композиція пролонгованого вивільнення, внаслідок відсутності підвищеного тиску під час стадії твердіння с), збільшується в об'ємі, що приводить до зменшення густини. Відповідно до подальшого аспекту винаходу, густина матричної композиції пролонгованого вивільнення у твердій пероральній фармацевтичній дозованій формі пролонгованого вивільнення, переважно у дозованій формі, що містить оксикодону HCl як активний інгредієнт, 3 дорівнює або менше, ніж приблизно 1,20 г/см . Переважно, дорівнює або менше, ніж приблизно 3 3 1,19 г/см , дорівнює або менше, ніж приблизно 1,18 г/см , або дорівнює або менше, ніж 3 приблизно 1,17 г/см . Наприклад, густина матричної композиції пролонгованого вивільнення 3 3 3 перебуває в діапазоні приблизно від 1,10 г/см приблизно до 1,20 г/см , приблизно від 1,11 г/см 3 3 3 приблизно до 1,20 г/см , або приблизно від 1,11 г/см приблизно до 1,19 г/см . Переважно 3 3 перебуває у діапазоні приблизно від 1,12 г/см приблизно до 1,19 г/см або приблизно від 1,13 3 3 3 г/см приблизно до 1,19 г/см , більш переважно приблизно від 1,13 г/см приблизно до 1,18 3 г/см . Густина матричної композиції пролонгованого вивільнення переважно визначена Правилом Архімеда, використовуючи рідину відомої густини (р 0). Матричну композицію пролонгованого вивільнення спочатку зважували у повітрі й потім занурювали у рідину й зважували. З цих двох зважувань густина р матричної композиції пролонгованого вивільнення може бути визначена рівнянням: A 0 , A B де ρ - густина матричної композиції пролонгованого вивільнення, А - вага матричної композиції пролонгованого вивільнення в повітрі, В - вага матричної композиції пролонгованого вивільнення зануреної у рідину, і ρ0 - густина рідини при даній температурі. Відповідна рідина відомої густини ρ0 є наприклад гексаном. Переважно, густина матричної композиції пролонгованого вивільнення вимірюють, використовуючи ваги верхнього завантаження моделі Mettler Толедо # АВ 135 - S/FACT, Серія # 1127430072 і набір для визначення густини 33360. Переважно, гексан використовується як рідина відомої густини ρ0. Значення густини всюди у цьому документі відповідають густини матричної композиції пролонгованого вивільнення при кімнатній температурі. Густина матричної композиції пролонгованого вивільнення переважно відноситься до густини непокритої композиції, наприклад, до густини ядра таблетки. У тих варіантах реалізації, де матрична композиція пролонгованого вивільнення є вкритою, наприклад де, матрична композиція пролонгованого вивільнення піддана стадії покриття d) після стадії твердіння с), густина матричної композиції пролонгованого вивільнення переважно виміряна до виконання стадії покриття, або шляхом видалення покриття з покритої матричної композиції пролонгованого вивільнення й наступним вимірюванням густини непокритої матричної композиції пролонгованого вивільнення. У вищезгаданих варіантах реалізації, високомолекулярний поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу від 2000000 до 15000000 або від 2000000 до 8000000, може використовуватися. 18 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 Зокрема, можуть використовуватися поліетиленоксиди, що мають, базуючись на реологічних вимірюваннях приблизну молекулярну масу 2000000, 4000000, 7000000 або 8000000. Зокрема, поліетиленоксиди, що мають, базуючись на реологічних вимірюваннях, приблизну молекулярну масу 4000000, можуть використовуватися. У варіантах реалізації, де композиція додатково включає, принаймні один низькомолекулярний поліетиленоксид є використовуваним поліетиленоксидом, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу менше, ніж 1000000, такий, як поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу від 100000 до 900,000 може використовуватися. Додатково, такі низькомолекулярні поліетиленоксиди можуть використовуватися, щоб спроектувати швидкість вивільнення, оскільки поліпшена швидкість вивільнення композиції, що інакше забезпечує швидкість вивільнення, щоб сповільнити для конкретної мети. У таких варіантах реалізації принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну вагу 100000, може використовуватися. У таких певних втіленнях композиція містить принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000 і принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу менше, ніж 1000000, де композиція містить принаймні приблизно 10 ваг. % або принаймні приблизно 20 ваг. % поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу менше, ніж 1000000. У таких певних втіленнях температура твердіння - менше, ніж приблизно 80 °C або навіть менше, ніж приблизно 77 °C. У певних варіантах реалізації загальний вміст поліетиленоксиду в композиції становить принаймні приблизно 80 ваг. %. Не бажаючи бути прив'язаним до теорії вважається, що високий вміст поліетиленоксиду забезпечує резистентність щодо пошкодження як описано тут, таку як міцність на розрив і резистентність до спиртової екстракції. Відповідно до певних таких варіантів реалізації винаходу активний інгредієнт - оксикодону гідрохлорид, і композиція включає більше, ніж приблизно 5 ваг. % оксикодону гідрохлориду. У таких певних втіленнях вміст у композиції принаймні одного поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000, становить принаймні приблизно 80 ваг. %. У певних варіантах реалізації вміст у композиції принаймні одного поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000, становить принаймні приблизно 85 % або принаймні приблизно 90 ваг. %. У таких варіантах реалізації поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 4000000 або принаймні 7000000 може використовуватися. У таких певних втіленнях активний інгредієнт оксикодону гідрохлорид або гідроморфону гідрохлорид, хоча інші активні агенти можуть також використовуватися відповідно до цього аспекту винаходу, і композиція включає більше, ніж приблизно 5 ваг. % оксикодону гідрохлориду або гідроморфону гідрохлориду. У певних варіантах реалізації, де кількість лікарського засобу в композиції становить принаймні приблизно 20 ваг. %, вміст поліетиленоксиду може бути настільки ж низьким, як приблизно 75 ваг. %. В іншому варіанті реалізації, де кількість лікарського засобу в композиції перебуває в діапазоні приблизно від 25 ваг. % приблизно до 35 ваг. %, вміст поліетиленоксиду може бути в діапазоні приблизно від 65 ваг. % приблизно до 75 ваг. %. Наприклад, у варіантах реалізації, де кількість лікарського засобу в композиції становить приблизно 32 ваг. %, вміст поліетиленоксиду може скласти приблизно 67 ваг. %. У певних варіантах реалізації винаходу, стеарат магнію додають під час або після процесу твердіння/стадії твердіння, щоб уникнути злипання таблеток. У таких певних втіленнях стеарат магнію доданий наприкінці процесу твердіння/стадії твердіння, перед охолодженням таблеток або під час охолодження таблеток. Інші антиадгезивні засоби, які могли б використовуватися, є тальком, кремнієм, тонкодисперсним кремнієм, колоїдним діоксидом кремнію, стеаратом кальцію, карнаубським воском, довголанцюговими спиртами жирного ряду й воском, таким як стеаринова кислота й стеариловий спирт, мінеральне масло, парафін, мікрокристалічна целюлоза, гліцерин, пропіленгліколь і поліетиленгліколь. Додатково або альтернативно покриття може бути запущене при високій температурі. У певних варіантах реалізації, де стадія твердіння с) виконана у резервуарі для покриття, злипання таблеток можна уникнути, або липкі таблетки можуть бути відділені, збільшуючи швидкість резервуару під час стадії твердіння або після стадії твердіння, в останньому випадку, наприклад, перед або під час охолодження таблеток. Швидкість резервуару збільшена до швидкості, коли всі таблетки відділені, або не відбувається злипання. 19 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 У певних варіантах реалізації винаходу, початкове оболонкове покриття або фракція оболонкового покриття накладена до виконання стадії твердіння с). Це оболонкове покриття забезпечує "покриття" для матричних композицій пролонгованого вивільнення або таблеток, щоб функціонувати як антиадгезивний засіб, тобто, щоб уникнути злипання композицій або таблеток. У таких певних втіленнях оболонкове покриття, що накладено до стадії твердіння, є оболонковим покриттям Opadry. Після стадії твердіння с) може бути виконана наступна стадія оболонкового покриття. Даний винахід охоплює також будь-яку тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, одержану способом відповідно до будь-якого процесу, як описано вище. Незалежно, даний винахід також спрямований на тверді пероральні фармацевтичні дозовані форми пролонгованого вивільнення. У певних варіантах реалізації винахід спрямований на тверді пероральні фармацевтичні дозовані форми пролонгованого вивільнення, що включають матричну композицію пролонгованого вивільнення, яка містить активний агент у формі таблеток або мультичастинок, де таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 60 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувана сплющена таблетка або сплющені мультичастинки забезпечують коефіцієнт розчинності in vitro при вимірюванні на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C, характеризується відсотком кількості вивільненого активного інгредієнта за 0,5 годин або за 0,5 й 0,75 годин, або за 0,5, 0,75 й 1 год., або за 0,5, 0,75, 1 й 1,5 год. або за 0,5, 0,75, 1, 1,5 й 2 год. розчинення, що відхиляється не більше, ніж приблизно на 20 % пунктів у кожному із згадуваних пунктів часу від відповідного in vitro коефіцієнта розчинності несплющеної еталонної таблетки або еталонних мультичастинок. У таких певних втіленнях таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 50 %, або не більше, ніж приблизно 40 %, або не більше, ніж приблизно 30 %, або не більше, ніж приблизно 20 %, або не більше, ніж приблизно 16 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувана сплющена таблетка або сплющені мультичастинки забезпечують коефіцієнт розчинності in vitro при вимірюванні на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C, характеризується відсотком кількості вивільненого активного інгредієнта за 0,5 годин або за 0,5 й 0,75 годин, або за 0,5, 0,75 й 1 год., або за 0,5, 0,75, 1 й 1,5 год. або за 0,5, 0,75, 1, 1,5 й 2 год. розчинення, що відхиляється не більше, ніж приблизно на 20 % пунктів або не більше, ніж приблизно на 15 % пунктів у кожному із згадуваних пунктів часу від відповідного in vitro коефіцієнта розчинності несплющеної еталонної таблетки або еталонних мультичастинок. У певних варіантах реалізації, даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що включає матричну композицію пролонгованого вивільнення, яка містить активний агент у формі таблеток або мультичастинок, де таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 60 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувана сплющена таблетка або сплющені мультичастинки й несплющена еталонна таблетка або еталонні мультичастинки забезпечують коефіцієнт розчинності in vitro, виміряний на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C, між приблизно 5 і приблизно 40 ваг. % активного інгредієнта, вивільненого після 0,5 годин. У таких певних втіленнях, таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 50 %, або не більше, ніж приблизно 40 %, або не більше, ніж приблизно 30 %, або не більше, ніж приблизно 20 %, або не більше, ніж приблизно 16 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувана сплющена таблетка або сплющені мультичастинки й несплющена еталонна таблетка або еталонні мультичастинки забезпечують коефіцієнт розчинності in vitro, виміряний на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C, між приблизно 5 і приблизно 40 ваг. % активного інгредієнта, вивільненого після 0,5 годин, або між приблизно 5 і приблизно 30 ваг. % 20 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 активного інгредієнта, вивільненого після 0,5 годин, або між приблизно 5 і приблизно 20 ваг. % активного інгредієнта, вивільненого після 0,5 годин, або між приблизно 10 і приблизно 18 ваг. % активного інгредієнта, вивільненого після 0,5 годин. У певних варіантах реалізації винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що включає матричну композицію пролонгованого вивільнення, яка містить активний агент у формі таблеток або мультичастинок, де таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 60 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувані сплющені таблетки або сплющені мультичастинки, забезпечують коефіцієнт розчинності in vitro при вимірюванні на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF), що містить 40 % етанолу при 37 °C, характеризується відсотком кількості вивільненого активного інгредієнта за 0,5 годин або за 0,5 й 0,75 годин, або за 0,5, 0,75 й 1 год., або за 0,5, 0,75, 1 й 1,5 год. або за 0,5, 0,75, 1, 1,5 й 2 год. розчинення, що відхиляється не більше, ніж приблизно на 20 % в кожній точці часу від відповідного in vitro коефіцієнта розчинності, вимірюваного на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C без етанолу, використовуючи сплющені та несплющені еталонні таблетки, або сплющені та несплющені еталонні мультичастинки, відповідно. У таких певних втіленнях таблетка або мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 60 %, або не більше, ніж приблизно 50 %, або не більше, ніж приблизно 40 %, або не більше, ніж приблизно 30 %, або не більше, ніж приблизно 20 %, або не більше, ніж приблизно 16 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де сплющена або несплющена таблетка або індивідуальні мультичастинки забезпечують коефіцієнт розчинності in vitro при вимірюванні на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF), що містить 40 % етанолу при 37 °C, характеризується відсотком кількості вивільненого активного інгредієнта за 0,5 годин або за 0,5 й 0,75 годин, або за 0,5, 0,75 й 1 год., або за 0,5, 0,75, 1 й 1,5 год. або за 0,5, 0,75, 1, 1,5 й 2 год. розчинення, що відхиляється не більше, ніж приблизно на 20 % пунктів або не більше, ніж приблизно на 15 % пунктів у кожному із згадуваних пунктів часу від відповідного in vitro коефіцієнта розчинності, вимірюваного на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C без етанолу, використовуючи сплющену або несплющену еталонну таблетку або еталонні мультичастинки, відповідно. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що включає матричну композицію пролонгованого вивільнення, яка містить активний агент у формі таблеток або мультичастинок, де таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 60 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувані сплющені таблетки або сплющені мультичастинки, забезпечують in vitro коефіцієнт розчинення, виміряний на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF), що містить 40 % або 0 % етанолу при 37 °C, між приблизно 5 і приблизно 40 ваг. % активного інгредієнта, вивільненого після 0,5 годин. У таких певних втіленнях, таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 50 %, або не більше, ніж приблизно 40 %, або не більше, ніж приблизно 30 %, або не більше, ніж приблизно 20 %, або не більше, ніж приблизно 16 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувані сплющені таблетки або сплющені мультичастинки, забезпечують in vitro коефіцієнт розчинення, виміряний на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF), що містить 40 % або 0 % етанолу при 37 °C, між приблизно 5 і приблизно 40 ваг. % активного інгредієнта, вивільненого після 0,5 годин, або між приблизно 5 і приблизно 30 ваг. % активного інгредієнта, вивільненого після 0,5 годин, або між приблизно 5 і приблизно 20 ваг. % активного інгредієнта, вивільненого після 0,5 годин, або між приблизно 10 і приблизно 18 ваг. % активного інгредієнта, вивільненого після 0,5 годин. Такі дозовані форми можуть бути одержані, як описано вище. 21 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 У певних варіантах реалізації винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що включає матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну вагу принаймні 1000000; і (2) принаймні однин активний інгредієнт, переважно вибраний з опіоїдних анальгетиків; і, де композиція містить принаймні приблизно 80 ваг. % поліетиленоксиду. Композиція може також включати принаймні приблизно 85 або 90 ваг. % поліетиленоксиду. Відповідно до певних таких варіантів реалізації винаходу, де композиція містить принаймні приблизно 80 ваг. % поліетиленоксиду, активний інгредієнт - оксикодону гідрохлорид або гідроморфону гідрохлорид, і композиція включає більше, ніж приблизно 5 ваг. % оксикодону гідрохлориду або гідроморфону гідрохлориду. У таких певних втіленнях композиція містить принаймні приблизно 80 ваг. % поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) 10 мг оксикодону гідрохлориду; і де композиція містить принаймні приблизно 85 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) 15 мг або 20 мг оксикодону гідрохлориду; і де композиція містить принаймні приблизно 80 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) 40 мг оксикодону гідрохлориду; і де композиція містить принаймні приблизно 65 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що включає матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) 60 мг або 80 мг оксикодону гідрохлориду; і де композиція містить принаймні приблизно 60 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні одинполіетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) 8 мг гідроморфону гідрохлориду; і де композиція містить принаймні приблизно 94 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: 22 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) 12 мг гідроморфону гідрохлориду; і де композиція містить принаймні приблизно 92 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) 32 мг гідроморфону гідрохлориду; і де композиція містить принаймні приблизно 90 ваг. % поліетиленоксиду. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один активний інгредієнт, переважно вибраний з опіоїдних анальгетиків; (2) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну вагу принаймні 1000000; і (3) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу менше, ніж 1000000. У таких певних втіленнях композиція містить принаймні приблизно 80 ваг. % поліетиленоксиду. Композиція може також включати принаймні приблизно 85 або 90 ваг. % поліетиленоксиду. Відповідно до певних таких варіантів реалізації винаходу, де композиція містить принаймні приблизно 80 ваг. % поліетиленоксиду, активний інгредієнт - оксикодону гідрохлорид або гідроморфону гідрохлорид, і композиція включає більше, ніж приблизно 5 ваг. % оксикодону гідрохлориду або гідроморфону гідрохлориду. Композиція може також включати 15-30 ваг. % поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, молекулярну масу принаймні 1000000; і 65-80 ваг. % поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, молекулярну масу менше, ніж 1000000, або композиції можуть включати принаймні приблизно 20 ваг. % або принаймні приблизно 30 ваг. % або принаймні приблизно 50 ваг. % поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, молекулярну масу принаймні 1000000. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, молекулярну масу принаймні 800000 або принаймні 900,000; і (2) принаймні один активний інгредієнт, вибраний з опіоїдних анальгетиків; і де композиція містить принаймні приблизно 80 ваг. % поліетиленоксиду. У певних варіантах реалізації винаходу матриця пролонгованого вивільнення має густину, 3 що дорівнює або менше, ніж приблизно 1,20 г/см . У таких певних втіленнях, густина матричної 3 композиції пролонгованого вивільнення дорівнює або менше, ніж приблизно 1,19 г/см , 3 переважно дорівнює або менше, ніж приблизно 1,18 г/см або дорівнює або менше, ніж 3 приблизно 1,17 г/см . Наприклад, густина матричної композиції пролонгованого вивільнення 3 3 3 перебуває в діапазоні приблизно від 1,10 г/см приблизно до 1,20 г/см , приблизно від 1,11 г/см 3 3 3 приблизно до 1,20 г/см , або приблизно від 1,11 г/см приблизно до 1,19 г/см . Переважно 3 3 перебуває у діапазоні приблизно від 1,12 г/см приблизно до 1,19 г/см або приблизно від 1,13 3 3 3 г/см приблизно до 1,19 г/см , більш переважно приблизно від 1,13 г/см приблизно до 1,18 3 г/см . Переважно, густина визначена правилом Архімеда, як описано вище. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) принаймні один активний інгредієнт; і де матрична композиція пролонгованого вивільнення, коли підлягає тестуванню на твердість вдавлюванням має силу розтріскування принаймні приблизно 110 N. 23 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 У певних варіантах реалізації винаходу матрична композиція пролонгованого вивільнення має силу розтріскування принаймні приблизно 110 N, переважно принаймні приблизно 120 N, принаймні приблизно 130 N або принаймні приблизно 140 N, більш переважно принаймні приблизно 150 N, принаймні приблизно 160 N або принаймні приблизно 170 N, найбільш переважно принаймні приблизно 180 N, принаймні приблизно 190 N або принаймні приблизно 200 N. У певних варіантах реалізації даний винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, матрична композиція пролонгованого вивільнення містить композицію, що включає принаймні: (1) принаймні один поліетиленоксид, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000; і (2) принаймні один активний інгредієнт; і де матрична композиція пролонгованого вивільнення, коли підлягає тестуванню на твердість вдавлюванням має "глибину проникнення для розкриття тріщини" принаймні приблизно 1,0 мм. У певних варіантах реалізації винаходу матрична композиція пролонгованого вивільнення має інтервал "глибини проникнення для розкриття тріщини" принаймні приблизно 1,0 мм або принаймні приблизно 1,2 мм, переважно принаймні приблизно 1.4 мм, принаймні приблизно 1,5 мм або принаймні приблизно 1.6 мм, більш переважно принаймні приблизно 1.8 мм, принаймні приблизно 1.9 мм, або принаймні приблизно 2,0 мм, найбільш переважно принаймні приблизно 2,2 мм, принаймні приблизно 2,4 мм або принаймні приблизно 2,6 мм. У таких певних втіленнях винаходу матрична композиція пролонгованого вивільнення має силу розтріскування принаймні приблизно 110 N, переважно принаймні приблизно 120 N, принаймні приблизно 130 N або принаймні приблизно 140 N, більш переважно принаймні приблизно 150 N, принаймні приблизно 160 N або принаймні приблизно 170 N, найбільш переважно принаймні приблизно 180 N, принаймні приблизно 190 N або принаймні приблизно 200 N, та/або інтервал "глибини проникнення для розкриття тріщини" принаймні приблизно 1,0 мм або принаймні приблизно 1,2 мм, переважно принаймні приблизно 1.4 мм, принаймні приблизно 1,5 мм або принаймні приблизно 1.6 мм, більш переважно принаймні приблизно 1.8 мм, принаймні приблизно 1.9 мм або принаймні приблизно 2,0 мм, найбільш переважно принаймні приблизно 2,2 мм, принаймні приблизно 2,4 мм або принаймні приблизно 2,6 мм. Комбінація кожного з вищезгаданих значень сили розламування, і інтервал "глибини проникнення для розкриття тріщини" включений у межах представленого винаходу. У таких певних втіленнях матрична композиція пролонгованого вивільнення, коли підлягає тестуванню на твердість вдавлюванням, протистоїть роботі принаймні приблизно 0,06 J або принаймні приблизно 0,08 J, переважно принаймні приблизно 0,09 J, принаймні приблизно 0,11 J або принаймні приблизно 0,13 J, більш переважно принаймні приблизно 0,15 J, принаймні приблизно 0,17 J або принаймні приблизно 0,19 J, найбільш переважно принаймні приблизно 0,21 J, принаймні приблизно 0,23 J або принаймні приблизно 0,25 J, без розламування. Параметри "сила розтріскування", "глибина проникнення для розкриття тріщини" й "робота" визначені у тесті у визначення твердості вдавлюванням, як описано вище, використовуючи Texture Analyzer, такий як ТА - ХТ2 Texture Analyzer (Texture Technologies Corp., 18 Fairview Road, Scarsdale, Нью-Йорк 10583). Сила розтріскування та/або інтервал "глибини проникнення для розкриття тріщини" можуть бути визначені, використовуючи непокриту або покриту матричну композицію пролонгованого вивільнення. Переважно, сила розтріскування та/або інтервал "глибини проникнення для розкриття тріщини" визначені на непокритій матричній композиції пролонгованого вивільнення. Не бажаючи бути прив'язаним до будь-якої теорії, вважають, що покриття, таке як покриття, накладене на стадії d) виробничого процесу твердої пероральної фармацевтичної дозованої форми пролонгованого вивільнення як описано вище, значно не сприяє спостережуваній силі розтріскування та/або "інтервалу "глибини проникнення для розкриття тріщини". Тому, сила розтріскування та/або інтервал "глибини проникнення для розкриття тріщини", визначений для конкретної покритої матричної композиції пролонгованого вивільнення, як очікують, не зміняться в основному від значень, визначених для відповідної непокритої матричної композиції пролонгованого вивільнення. У певних варіантах реалізації матрична композиція пролонгованого вивільнення перебуває у формі таблеток або мультичастинок, і таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 60 % товщини таблетки або індивідуальних мультичастинок перед сплющенням. Переважно, таблетка або індивідуальні 24 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 50 %, або не більше, ніж приблизно 40 %, або не більше, ніж приблизно 30 %, або не більше, ніж приблизно 20 %, або не більше, ніж приблизно 16 % товщини таблетки або індивідуальних мультичастинок перед сплющенням. Переважно, сплющення таблеток або індивідуальних мультичастинок виконано на настільному пресі, такому, як лабораторний прес типу стоматологічної гладилки, або за допомогою молотка, як описано вище. У таких певних втіленнях матрична композиція пролонгованого вивільнення перебуває у формі таблеток або мультичастинок, і таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 60 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувана сплющена таблетка або сплющені мультичастинки забезпечують коефіцієнт розчинності in vitro при вимірюванні на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C, характеризується відсотком кількості вивільненого активного інгредієнта за 0,5 годин або за 0,5 й 0,75 годин, або за 0,5, 0,75 й 1 год., або за 0,5, 0,75, 1 й 1,5 год. або за 0,5, 0,75, 1, 1,5 й 2 год. розчинення, що відхиляється не більше, ніж приблизно на 20 % пунктів у кожному із згадуваних пунктів часу від відповідного in vitro коефіцієнта розчинності несплющеної еталонної таблетки або еталонних мультичастинок. Переважно, таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 50 %, або не більше, ніж приблизно 40 %, або не більше, ніж приблизно 30 %, або не більше, ніж приблизно 20 %, або не більше, ніж приблизно 16 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувана сплющена таблетка або сплющені мультичастинки забезпечують коефіцієнт розчинності in vitro при вимірюванні на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C, характеризується відсотком кількості вивільненого активного інгредієнта за 0,5 годин або за 0,5 й 0,75 годин, або за 0,5, 0,75 й 1 год., або за 0,5, 0,75, 1 й 1,5 год. або за 0,5, 0,75, 1, 1,5 й 2 год. розчинення, що відхиляється не більше, ніж приблизно на 20 % пунктів або не більше, ніж приблизно на 15 % пунктів у кожному із згадуваних пунктів часу від відповідного in vitro коефіцієнта розчинності несплющеної еталонної таблетки або еталонних мультичастинок. У певних варіантах реалізації винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення, що містить матричну композицію пролонгованого вивільнення, де матрична композиція пролонгованого вивільнення перебуває у формі таблеток або мультичастинок, і таблетка або індивідуальні мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 60 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де згадувані сплющені таблетки або сплющені мультичастинки, забезпечують коефіцієнт розчинності in vitro при вимірюванні на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF), що містить 40 % етанолу при 37 °C, характеризується відсотком кількості вивільненого активного інгредієнта за 0,5 годин або за 0,5 й 0,75 годин, або за 0,5, 0,75 й 1 год., або за 0,5, 0,75, 1 й 1,5 год. або за 0,5, 0,75, 1, 1,5 й 2 год. розчинення, що відхиляється не більше, ніж приблизно на 20 % в кожній точці часу від відповідного in vitro коефіцієнта розчинності, вимірюваного на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C без етанолу, використовуючи сплющені та несплющені еталонні таблетки або сплющені та несплющені еталонні мультичастинки, відповідно. Переважно, таблетка або мультичастинки можуть принаймні бути сплющені без руйнування, характеризуються товщиною таблетки або індивідуальних мультичастинок після сплющення, що відповідає не більше, ніж приблизно 60 %, або не більше, ніж приблизно 50 %, або не більше, ніж приблизно 40 %, або не більше, ніж приблизно 30 %, або не більше, ніж приблизно 20 %, або не більше, ніж приблизно 16 % товщини таблетки або індивідуальних мультичастинок перед сплющенням, і де сплющена або несплющена таблетка або індивідуальні мультичастинки забезпечують коефіцієнт розчинності in vitro при вимірюванні на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF), що містить 40 % етанолу при 37 °C, характеризуються відсотком кількості вивільненого активного інгредієнта за 0,5 годин або за 0,5 й 0,75 годин, або за 0,5, 0,75 й 1 год., або за 0,5, 0,75, 1 й 1,5 год. або за 0,5, 0,75, 1, 1,5 й 2 год. розчинення, що відхиляється не більше, ніж 25 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 60 приблизно на 20 % пунктів або не більше, ніж приблизно на 15 % пунктів у кожному із згадуваних пунктів часу від відповідного in vitro коефіцієнта розчинності, вимірюваного на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C без етанолу, використовуючи сплющену або несплющену еталонну таблетку або еталонних мультичастинок, відповідно. У таких певних втіленнях матрична композиція пролонгованого вивільнення, коли піддана максимальній силі приблизно 196 N або приблизно 439 N у визначенні твердості таблетки, не руйнується. Переважно, визначення твердості таблетки, щоб визначити міцність на розрив матричних композицій пролонгованого вивільнення виконано на апараті Schleuniger, як описано вище. Наприклад, міцність на розрив визначена, використовуючи Schleuniger 2Е/106 Апарат і застосовуючи силу максимально приблизно 196 N, або Модель 6D Апарату Schleuniger і застосовуючи силу максимально приблизно 439 N. Також спостерігали, що композиції представленого винаходу - стабільні при зберіганні, де матрична композиція пролонгованого вивільнення, після зберігання при 25 °C й 60 % відносній вологості (ВВ) або при 40 °C й 75 % відносній вологості (ВВ) протягом принаймні 1 місяця, більш переважно протягом принаймні 2 місяців, протягом принаймні 3 місяців або протягом принаймні 6 місяців, забезпечують коефіцієнт розчинення, як виміряно на Апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C, характеризується відсотком кількості вивільненого активного інгредієнта за 1 годину або за 1 й 2 год., або за 1 й 4 год., або за 1, 2 й 4 год., або за 1, 4 й 12 годин, або за 1, 2, 4 й 8 годин або за 1, 2, 4, 8 й 12 годин розчинення, що відхиляється не більше, ніж приблизно на 15 % пунктів, переважно не більше, ніж приблизно на 12 % пунктів або не більше, ніж приблизно на 10 % пунктів, більш переважно не більше, ніж приблизно на 8 % пунктів або не більше, ніж приблизно на 6 % пунктів, найбільш переважно не більше, ніж приблизно на 5 % пунктів у кожному із згадуваних пунктів часу від відповідного in vitro коефіцієнта розчинності еталонної композиції до зберігання. Переважно, матричну композицію пролонгованого вивільнення зберігають у кількості флаконів, наприклад, 100. Будь-яка комбінація вищезгаданих строків зберігання, пунктів часу розчинення й межі відхилень знаходяться в межах представленого винаходу. Відповідно до подальшого аспекту стабільності при зберіганні, матрична композиція пролонгованого вивільнення після зберігання при 25 °C й 60 % відносній вологості (ВВ) або при 40 °C й 75 % відносній вологості (ВВ) протягом принаймні 1 місяця, більш переважно протягом принаймні 2 місяців, протягом принаймні 3 місяців або протягом принаймні 6 місяців, містить кількість принаймні одного активного інгредієнта в ваг. % щодо вказаного на етикетці активного інгредієнта матричної композиції пролонгованого вивільнення, що відхиляється не більше, ніж приблизно на 10 % пунктів, переважно не більше, ніж приблизно на 8 % пунктів або не більше, ніж приблизно на 6 % пунктів, більш переважно не більше, ніж приблизно 5 % пунктів або не більше, ніж приблизно 4 % пунктів або не більше, ніж приблизно 3 % пунктів від відповідної кількості активного інгредієнта в ваг. % щодо вказаного на етикетці активного інгредієнта матричної композиції пролонгованого вивільнення еталонної композиції до зберігання. Переважно, матричну композицію пролонгованого вивільнення зберігають у кількості флаконів, наприклад, 100. Будь-яка комбінація вищезгаданих строків зберігання й межі відхилень знаходяться в межах представленого винаходу. Відповідно таким певним варіантам реалізацій активний інгредієнт - оксикодону гідрохлорид. Переважно, кількість принаймні одного активного інгредієнта в ваг. % щодо вказаного на етикетці активного інгредієнта матричної композиції пролонгованого вивільнення визначена екстрагуванням принаймні одного активного інгредієнта з матричної композиції пролонгованого вивільнення й послідовним аналізом, використовуючи високоефективну рідинну хроматографію. У певних варіантах реалізації, де принаймні один активний інгредієнт - оксикодону гідрохлорид, переважна кількість оксикодону гідрохлориду у ваг. % щодо вказаного на етикетці оксикодону гідрохлориду для матричної композиції пролонгованого вивільнення визначена екстрагуванням оксикодону гідрохлориду з матричної композиції пролонгованого вивільнення сумішшю 1:2 ацетонітрилу й модельованого шлункового соку без ферменту (SGF) при безперервному перемішуванні магнітною мішалкою, поки матрична композиція пролонгованого вивільнення повністю не диспергується або протягом ночі й послідовним аналізом, використовуючи високоефективну рідинну хроматографію, переважно високоефетивну рідинну хроматографію зворотної фази. У таких певних втіленнях, де матрична композиція пролонгованого вивільнення перебуває у формі таблеток, переважна кількість оксикодону гідрохлориду в ваг. % щодо вказаного на етикетці оксикодону гідрохлориду для таблеток визначена екстрагуваням оксикодону гідрохлориду із двох наборів десяти таблеток кожний з 900 мл суміші 1:2 26 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 ацетонітрилу й модельованого шлункового соку без ферменту (SGF) при безперервному перемішуванні магнітною мішалкою, поки таблетки повністю не диспергуються або протягом ночі й послідовним аналізом, використовуючи високоефективну рідинну хроматографію, переважно високоефетивну рідинну хроматографію зворотної фази. Переважно, результати тестів - середні значення двох вимірювань. У певних варіантах реалізації винахід спрямований на тверду пероральну фармацевтичну дозовану форму пролонгованого вивільнення де, дозована форма забезпечує коефіцієнт розчинення, виміряний на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C, між 12,5 й 55 ваг. % активного інгредієнта, вивільненого після 1 год., між 25 й 65 ваг. % активного інгредієнта, вивільненого після 2 годин, між 45 й 85 ваг. % активного інгредієнта, вивільненого після 4 годин і між 55 й 95 ваг. % активного інгредієнта, вивільненого після 6 годин, і, довільно, між 75 й 100 ваг. % активного інгредієнта, вивільненого після 8 годин. Переважно, дозована форма забезпечує коефіцієнт розчинення, виміряний на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C, між 15 й 45 ваг. % активного інгредієнта, вивільненого після 1 год., між 30 й 60 ваг. % активного інгредієнта, вивільненого після 2 годин, між 50 й 80 ваг. % активного інгредієнта, вивільненого після 4 годин і між 60 й 90 ваг. % активного інгредієнта, вивільненого після 6 годин і, довільно, між 80 й 100 ваг. % активного інгредієнта, вивільненого після 8 годин. Більш переважно, дозована форма забезпечує коефіцієнт розчинення, виміряний на апараті USP 1 (кошик) при 100 об/хв в 900 мл модельованого шлункового соку без ферментів (SGF) при 37 °C, між 17,5 й 35 ваг. % активного інгредієнта, вивільненого після 1 год., між 35 й 55 ваг. % активного інгредієнта, вивільненого після 2 годин, між 55 й 75 ваг. % активного інгредієнта, вивільненого після 4 годин і між 65 й 85 ваг. % активного інгредієнта, вивільненого після 6 годин і, довільно, між 85 й 100 ваг. % активного інгредієнта, вивільненого після 8 годин. У таких певних втіленнях активний інгредієнт - оксикодону гідрохлорид або гідроморфону гідрохлорид. Такі дозовані форми можуть бути одержані способом, як описано тут. У варіантах реалізації, як описано вище, таблетки можуть бути одержані безпосереднім пресуванням композиції й твердінням, принаймні піддаючи згадувану таблетку температурі принаймні приблизно 60 °C, принаймні приблизно 62 °C, принаймні приблизно 68 °C, принаймні приблизно 70 °C, принаймні приблизно 72 °C або принаймні приблизно 75 °C протягом інтервалу часу принаймні приблизно 1 хв., принаймні приблизно 5 хвилин або принаймні приблизно 15 хвилин. У певних варіантах реалізації винаходу таблетка, як описано вище може покрита шаром порошку поліетиленоксиду, шляхом нанесення на отверділу або неотверділу таблетку порошкового шару поліетиленоксиду, що оточує ядро, і твердіння покритої шаром порошку таблетки, як описано вище. Такий зовнішній шар поліетиленоксиду забезпечує час затримки перед початком вивільнення активного інгредієнта та/або більш повільне вивільнення активного інгредієнта. У певних варіантах реалізації винаходу одержана бі- або мультишарова таблетка, де принаймні один із шарів містить композицію пролонгованого вивільнення, як описано вище, і принаймні один з інших шарів містить композицію швидкого вивільнення активного інгредієнта, що міститься в композиції пролонгованого вивільнення або другий інший активний інгредієнт. У таких певних втіленнях таблетка - двошарова таблетка з шаром композиції пролонгованого вивільнення, як описано тут і шаром композиції швидкого вивільнення. У таких певних втіленнях, особливо двошарові таблетки, де опіоїдні анальгетики в шарі пролонгованого вивільнення й додатково неопіоїдні анальгетики містяться в шарі швидкого вивільнення. Неопіоїдні анальгетики можуть бути нестероїдними протизапальними засобами, але також і неопіоїдними анальгетиками, такими як ацетамінофен. Ацетамінофен може, наприклад, використовуватися в комбінації з гідрокодоном, як опіоїдним анальгетиком. Такі таблетки можуть бути одержані певними способами пресування таблетки, які дозволяють пресувати принаймні дві композиції, щоб утворити таблетки принаймні із двома різними шарами, кожен з яких містить одну із цих принаймні двох композицій. Наприклад, такі таблетки можуть бути одержані на таблетувальному пресі, заповнюючи компресійний інструмент першою композицією, і пресуючи згадувану першу композицію й наступним заповненням верхньої частини першої пресованої композиції другою композицією й наступним пресуванням цих двох композицій, щоб утворити готову таблетку. Композиція швидкого вивільнення може бути будьякою композицією, як відомо в даній галузі. 27 UA 104745 C2 5 10 15 20 25 30 35 40 45 50 55 Винахід також охоплює використання високомолекулярного поліетиленоксиду, що має, базуючись на реологічних вимірюваннях, приблизну молекулярну масу принаймні 1000000, як матеріалу, що утворює матрицю у виробництві твердої пероральної пролонгованого вивільнення дозованої форми, що містить активний інгредієнт, вибраний з опіоїдів для надання пероральній твердій дозованій формі пролонгованого вивільнення резистентності щодо спиртової екстракції. Використання може бути досягнуте, як описано тут щодо описаного процесу або описаних композицій або будь-яким іншим способом, як загальноприйнятий в даній галузі. Спостерігалося, що композиції представленого винаходу, що містять високомолекулярний поліетиленоксид, можуть бути сплющені до густини між приблизно 15 і приблизно 18 % несплющеної густини й що плоска таблетка відновляється частково або в основному відновляє свою початкову несплющену форму під час розчинення, зневажаючи набряканням, що також має місце під час розчинення, тобто густина таблетки збільшується й діаметр суттєво зменшується під час розчинення. Не бажаючи бути прив'язаним до теорії вважається, що високомолекулярний поліетиленоксид має пам'ять форми й здатність відновити початкову форму після деформації, наприклад після сплющення, у середовищі, що дозволяє відновлення, таке як водне середовище, використовуване у дослідженнях на розчинність. Ця здатність, як вважають, сприяє резистентності щодо пошкодження, особливо спирторезистентності дозованих форм представленого винаходу. Винахід також охоплює спосіб лікування, де дозована форма застосовується для лікування захворювання або певного стану пацієнта, що потребує лікування при певному болі й застосування дозованої форми відповідно до винаходу для виробництва медикаменту для лікування захворювання або певного стану пацієнта, що потребує лікування при певному болі. В одному аспекті представленого винаходу тверда пероральна фармацевтична дозована форма пролонгованого вивільнення двократного на день застосування, що забезпечує в середньому tmax від приблизно 2 приблизно до 6 год. або від приблизно 2,5 приблизно до 5,5 год., або від приблизно 2,5 приблизно до 5 год. після призначення при рівноважній концентрації або однократної дози людським суб'єктам. Дозована форма може містити оксикодон або його сіль, або гідроморфон, або його сіль. В одному аспекті представленого винаходу тверда пероральна фармацевтична дозована форма пролонгованого вивільнення однократного на день дозування, що забезпечує в середньому tmax від приблизно 3 приблизно до 10 год. або від приблизно 4 приблизно до 9 год., або від приблизно 5 приблизно до 8 год. після призначення при рівноважній концентрації або однократної дози людським суб'єктам. Дозована форма може містити оксикодон або його сіль, або гідроморфон або його сіль. В подальшому аспекті представленого винаходу забезпечена тверда пероральна фармацевтична дозована форма пролонгованого вивільнення двократного на день дозування, де дозована форма включає оксикодон або його сіль в кількості приблизно від 10 мг приблизно до 160 мг, і де дозована форма забезпечує в середньому максимальну плазменну концентрацію (Сmах) оксикодону приблизно до 240 нг/мл або приблизно від 6 нг/мл приблизно до 240 нг/мл після призначення при рівноважній концентрації або однократної дози людським суб'єктам. В подальшому аспекті представленого винаходу забезпечена тверда пероральна фармацевтична дозована форма пролонгованого вивільнення, де дозована форма включає оксикодон або його сіль в кількості приблизно від 10 мг приблизно до 40 мг, і де дозована форма забезпечує в середньому максимальну плазменну концентрацію (С mах) оксикодону приблизно від 6 нг/мл приблизно до 60 нг/мл після призначення при рівноважній концентрації або однократної дози людським суб'єктам. В подальшому аспекті винаходу забезпечена тверда пероральна фармацевтична дозована форма пролонгованого вивільнення, що є біоеквівалентною комерційному продукту OxyContin™. В подальшому аспекті винаходу забезпечена тверда пероральна фармацевтична дозована форма пролонгованого вивільнення, що є біоеквівалентною комерційному продукту Palladone™ який продавався в Сполучених Штатах в 2005. В подальшому аспекті винаходу забезпечена тверда пероральна фармацевтична дозована форма пролонгованого вивільнення, де активний інгредієнт - оксикодону гідрохлорид й де дозована форма містить 10 мг оксикодону гідрохлориду, при тестуванні в порівняльному клінічному дослідженні є біоеквівалентною еталонній таблетці, що містить 10 мг оксикодону гідрохлориду в матричній композиції, що містить: Гідрохлорид 10,0 мг/таблетку 28
ДивитисяДодаткова інформація
Автори англійськоюMannion, Richard, Owen, O'Donnell, Edward, Patrick, McKenna, William, Henry, Huang, Haiyong, Hugh
Автори російськоюМаннион Ричард Оуэн, О'Доннелл Эдвард Патрик, МакКенна Уильям Генри, Хуанг Хеййонг Хуг
МПК / Мітки
МПК: A61P 25/04, A61K 9/32, A61K 9/14, A61K 47/32, A61K 31/485
Мітки: опіоїдний, використання, форма, несанкціонованого, захищена, містить, оральна, анальгетик, дозована, фармацевтична
Код посилання
<a href="https://ua.patents.su/165-104745-oralna-farmacevtichna-dozovana-forma-zakhishhena-vid-nesankcionovanogo-vikoristannya-shho-mistit-opiodnijj-analgetik.html" target="_blank" rel="follow" title="База патентів України">Оральна фармацевтична дозована форма, захищена від несанкціонованого використання, що містить опіоїдний анальгетик</a>
Сублінгвальна таблетка з покриттям, що містить опіоїдний анальгетик

Номер патенту: 88509
Опубліковано: 26.10.2009
Автори: Урі Паскаль, Еррі Катрін, Бенуа Гійом, Дювошель Йозеф
МПК: A61K 31/485, A61P 25/04, A61K 9/28
Мітки: містить, покриттям, сублінгвальна, таблетка, анальгетик, опіоїдний
Формула / Реферат:
1. Сублінгвальна таблетка з покриттям, яка містить- спресовану серцевину, вільну від фармацевтично активної речовини, що містить один чи більше розріджувачів, та- покриття, яке містить щонайменше один опіоїдний анальгетик, придатний для сублінгвального введення.2. Сублінгвальна таблетка з покриттям за пунктом 1, де опіоїдний анальгетик вибраний з групи, яку складають бупренорфін, нор-бупренорфін, фентаніл, альфентаніл,...
Оральна дозована форма азитроміцину зі зниженими побічними ефектами

Номер патенту: 78793
Опубліковано: 25.04.2007
Автори: Ло Джуліан Белкнап, Фізен Дуейн Томас, Маккрей Скотт Болдуін, Хербіг Скотт Макс, Вест Джеймс Блер, Хаген Тімоті Артур, Томбр Евінеш Говінд, Лайон Кейт Девід, Аппель Лі Елізабет, Крю Маршалл Девід
МПК: A61K 9/14, A61P 31/00, A61K 31/7042
Мітки: ефектами, форма, азитроміцину, дозована, побічними, оральна, зниженими
Формула / Реферат:
1. Оральна дозована форма, що містить:(а) ефективну кількість підлужувального агента; і(б) мультичастинки, де згадані мультичастинки включають:(і) від приблизно 20% до приблизно 75% азитроміцину; і(іі) від приблизно 25% до приблизно 80% гліцериду, який містить гліцерилмонобехенат, гліцерилдибехенат і гліцерилтрибехенат або їх суміш; і(ііі) полоксамер.2. Оральна дозована форма за п. 1, де полоксамер...
Тверда фармацевтична дозована форма, що містить інгібітор віл протеази, спосіб її одержання

Номер патенту: 85564
Опубліковано: 10.02.2009
Автори: Алані Ламан, Ліпольд Бернд, Брайтенбах Йорг, Розенберг Йорг, Берндль Гунтер, Райнхольд Ульріх, Гхош Соумоджіт
МПК: A61P 31/18, A61K 9/10, A61K 31/425
Мітки: одержання, віл, тверда, містить, дозована, протеази, форма, інгібітор, фармацевтична, спосіб
Формула / Реферат:
1. Тверда фармацевтична дозована форма, яка включає тверду дисперсію принаймні одного інгібітора ВІЛ протеази у принаймні одному фармацевтично прийнятному водорозчинному полімері і принаймні одну фармацевтично прийнятну поверхнево-активну речовину, причому вказаний інгібітор ВІЛ протеази являє собою...
Фармацевтична дозована форма, яка містить метформін та піоглітазон як активні медикаменти

Номер патенту: 91852
Опубліковано: 10.09.2010
Автори: Кардінал Джек Р., Нангія Авінаш, Окохі Казухіро, Лодін Анчалі
МПК: A61K 31/4439, A61P 3/10, A61K 31/155
Мітки: фармацевтична, форма, медикаменти, яка, метформін, піоглітазон, активні, містить, дозована
Формула / Реферат:
1. Фармацевтична дозована форма, яка має перший та другий активний медикаменти, і складається з:a) ядра контрольованого вивільнення, оточеного оболонкою уповільненого вивільнення, яка контролює вивільнення медикаменту таким чином, що піковий рівень медикаменту у плазмі забезпечується за 6-12 годин після введення дозованої форми, після прийому їжі, та тільки одного медикаменту, який складається з метформіну або його фармацевтично...
Оральна дозована форма саквінавіру мезилату

Номер патенту: 81335
Опубліковано: 25.12.2007
Автори: Жанг Лін, Фуапредіт Уантані, Албано Антоніо А., Шах Навніт-Харговіндас, Інфелд Мартін Хауард
МПК: A61K 9/22, A61P 31/18, A61K 31/16
Мітки: оральна, форма, саквінавіру, дозована, мезилату
Формула / Реферат:
1. Тверда одинична оральна фармацевтична дозована форма саквінавіру мезилату, що містить від приблизно 60% до приблизно 80% мікронізованого саквінавіру мезилату, виходячи з солі мезилату, від приблизно 4% до приблизно 8% фармацевтично прийнятного водорозчинного зв'язуючого, фармацевтично прийнятний дезінтегратор та фармацевтично прийнятний носій, де кожен відсоток є відсотком ваги ядра фармацевтичної дозованої форми.2. Дозована форма...
Попередній патент: Спосіб карбонілювання з використанням морденітного каталізатора, нанесеного на неорганічні оксиди
Наступний патент: Заміщені похідні бензоксазолу, бензімідазолу, оксазолопіридину та імідазопіридину як модулятори гамма-секретази
Випадковий патент: Спосіб запобігання раптовим газовиділенням у виробці при підході до геологічних порушень