Похідні піримідину для лікування вірусних інфекцій
Номер патенту: 113956
Опубліковано: 10.04.2017
Автори: Влач Яромір, Мк Гован Давід, Джонкерс Тім Хьюго Марія, Ласт Стефан Джульєн, Пітерс Серж Марія Алойсіс, Ембрехтс Вернер, Ребойсон П'єр Жан-Марія Бернард






Формула / Реферат
1. Сполука формули (І)
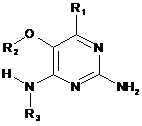
або її фармацевтично прийнятна сіль, таутомер (таутомери) або сольват, де
R1 являє собою водень, С1-4алкіл, циклопропіл або С1-6алкокси, галоген, гідроксил, трифторметил,
R2 являє собою С1-8алкіл, (С1-4)алкоксі-(С1-4)алкіл, С3-7циклоалкіл, С4-7гетероцикл, ароматичний, біциклічний гетероцикл, арилалкіл, гетероарил, гетероарилалкіл, кожен з яких необов'язково є заміщеним одним або декількома замісниками, незалежно вибраними з галогену, гідроксилу, аміно, С1-6алкілу, ді-(С1-6)алкіламіно, С1-6алкіламіно, С1-6алкілу, С1-6алкокси, С3-6циклоалкілу, карбонової кислоти, ефіру карбонової кислоти, аміду карбонової кислоти, гетероциклу, арилу, алкенілу, алкінілу, арилалкілу, гетероарилу, гетероарилалкілу, нітрилу,
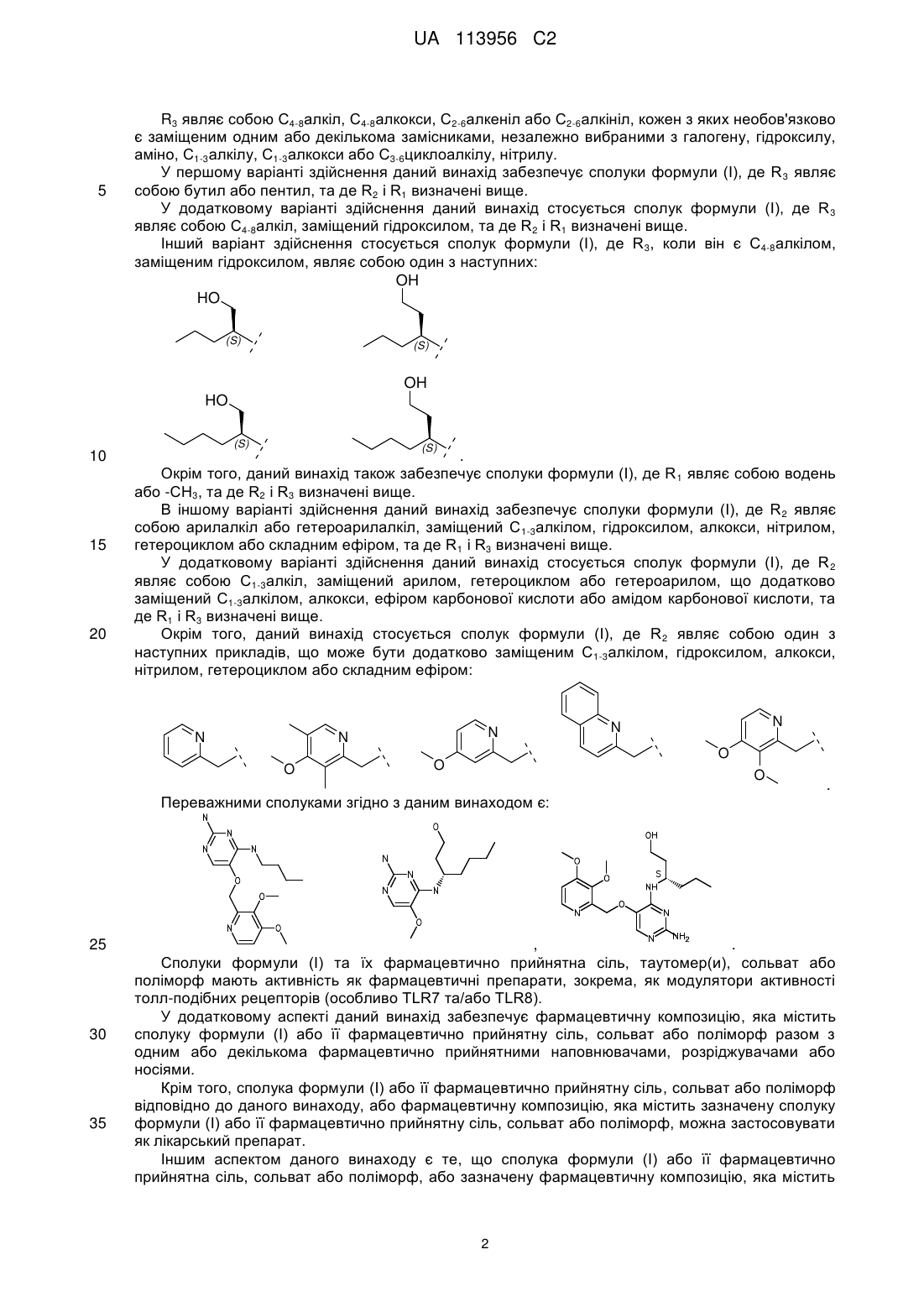
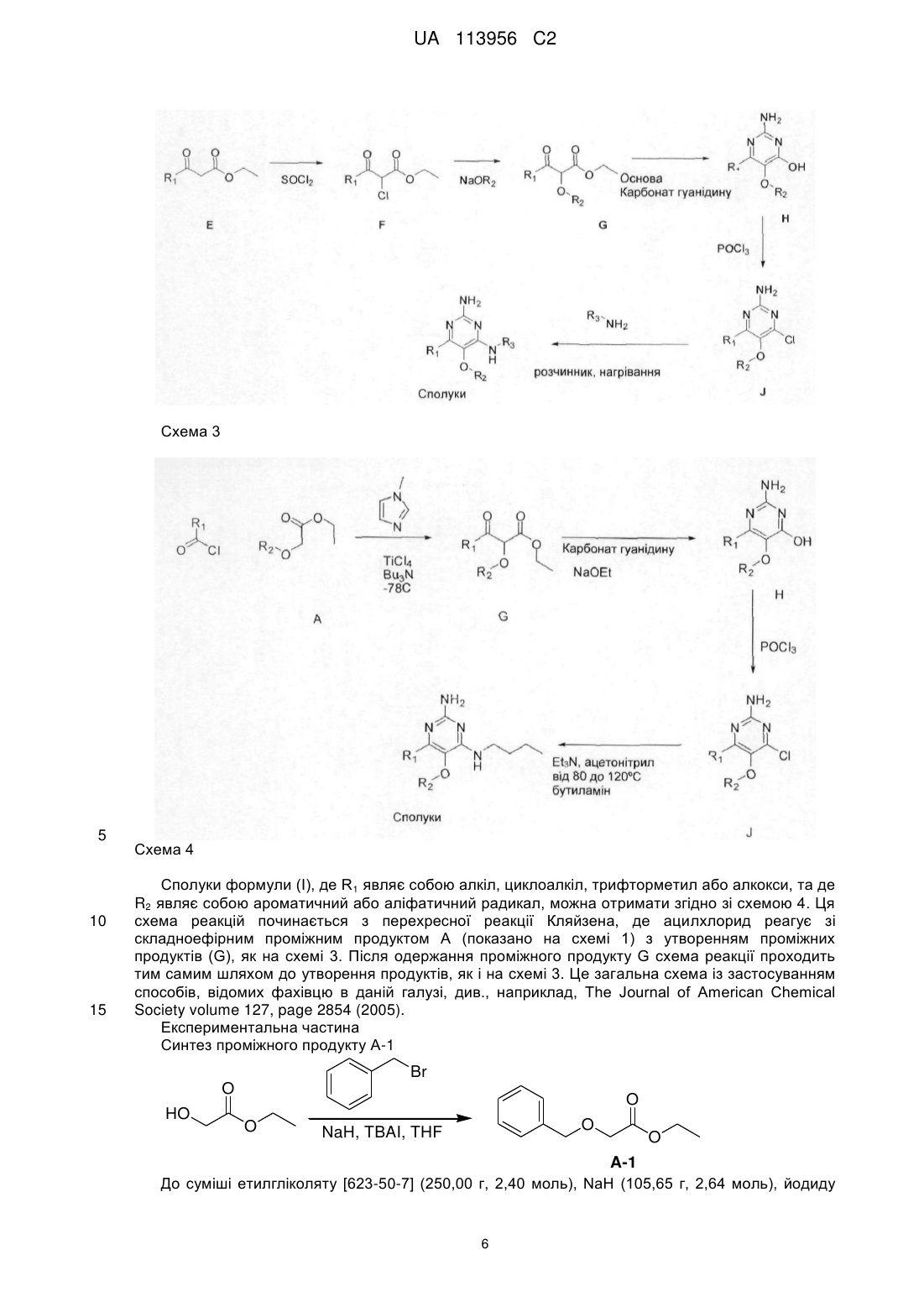
R3 являє собою С4-8алкіл, заміщений гідроксилом, вибраний з групи, яка складається з:
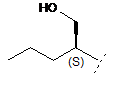 ,
, 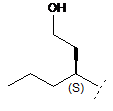 ,
,
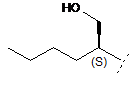 ,
, 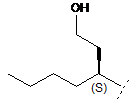 .
.
2. Сполука формули (І) за п. 1, де R2 являє собою арилалкіл або гетероарилалкіл, заміщений С1-3алкілом, гідроксилом, алкокси, нітрилом, гетероциклом або складним ефіром, і де R1 та R3 визначені вище.
3. Сполука формули (І) за п. 1, де R2 являє собою С1-3алкіл, заміщений арилом, гетероциклом або гетероарилом, який додатково заміщений С1-3алкілом, алкокси, ефіром карбонової кислоти або амідом карбонової кислоти, і де R1 та R3 визначені вище.
4. Сполука формули (І) за п. 1, де R2 являє собою
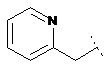 ,
, 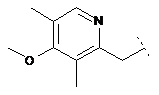 ,
, 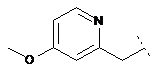 ,
,
 ,
, 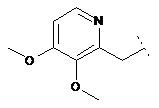 ,
,
кожен з яких може бути додатково заміщений С1-6алкілом, гідроксилом, С1-6алкокси, нітрилом, гетероциклом або складним ефіром, і де R1 та R3 визначені вище.
5. Сполука за п. 1, яка має формулу
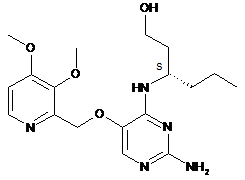 ,
, 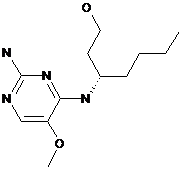
або
 .
.
6. Фармацевтична композиція для лікування порушення або захворювань, в яке залучена модуляція TLR7 і/або TLR8, що містить сполуку формули (І) або її фармацевтично прийнятну сіль, таутомер (таутомери) або сольват за будь-яким з пп. 1-5 разом з одним або декількома фармацевтично прийнятними наповнювачами, розріджувачами або носіями.
7. Сполука формули (І) або її фармацевтично прийнятна сіль, таутомер (таутомери) або сольват за будь-яким з пп. 1-5, або фармацевтична композиція за п. 6 для застосування як лікарського препарату.
Текст
Реферат: Даний винахід стосується похідних піримідину, способів їх одержання, фармацевтичних композицій та їх застосування в лікуванні вірусних інфекцій, таких як спричинювані HCV або HBV. R1 O R2 N H N N NH2 R3 UA 113956 C2 (12) UA 113956 C2 UA 113956 C2 5 10 15 20 25 30 35 40 45 50 55 Даний винахід стосується похідних піримідину, способів їх одержання, фармацевтичних композицій та їх застосування в лікуванні вірусних інфекцій, таких як спричинювані HBV або HCV. Даний винахід стосується застосування похідних піримідину в лікуванні вірусних інфекцій, імунних або запальних порушень, у яке залучено модуляцію або агонізм толл-подібних рецепторів (TLR). Толл-подібні рецептори являють собою найважливіші трансмембранні білки, що характеризуються позаклітинним багатим на лейцин доменом і цитоплазматичним розширенням, яке містить консервативну ділянку. Уроджена імунна система може розпізнавати патоген-асоційовані молекулярні патерни за допомогою цих TLR, що експресуються на поверхні певних типів імунних клітин. Розпізнавання чужорідних патогенів активує вироблення цитокінів та регуляцію спільно стимулюючих молекул на фагоцитах на підвищеному рівні. Це зумовлює модуляцію поведінки Т-клітин. Було підраховано, що більшість видів ссавців мають від десяти до п'ятнадцяти типів толлподібних рецепторів. У цілому в людини та мишей виявили тринадцять TLR (які мають назву TLR1-TLR13), а в інших видів ссавців виявили еквівалентні форми багатьох з них. Однак еквіваленти певних TLR, виявлених у людини, присутні не у всіх ссавців. Наприклад, ген, що кодує білок, аналогічний TLR10 людини, є присутнім у мишей, але, очевидно, у деякий момент у минулому він був ушкоджений ретровірусом. З іншого боку, у мишей експресуються TLR 11, 12 і 13, жоден з яких не представлений у людей. В інших ссавців можуть експресуватися TLR, які не виявляють у людини. Інші види, що не належать до ссавців, можуть мати TLR, відмінні від таких у ссавців, як показано стосовно TLR14, що виявляється в голкобрюха з роду Takifugu. Це може ускладнити спосіб застосування піддослідних тварин в якості моделей уродженого імунітету людини. Для докладного огляду толл-подібних рецепторів див. наступні журнальні статті: Hoffmann, J.A., Nature, 426, p33-38, 2003; Akira, S., Takeda, K., and Kaisho, T., Annual Rev. Immunology, 21, p335-376, 2003; Ulevitch, R. J., Nature Reviews: Immunology, 4, p512-520, 2004. Раніше були описані сполуки, що проявляють активність стосовно толл-подібних рецепторів, такі як похідні пурину в WO 2006/117670, похідні аденіну в WO 98/01448 і WO 99/28321 та піримідини в WO 2009/067081. Проте, існує гостра потреба в нових модуляторах толл-подібних рецепторів, які мають переважну селективність, вищу ефективність, вищу метаболічну стабільність, а також поліпшений профіль безпеки порівняно зі сполуками з відомого рівня техніки. Під час лікування деяких вірусних інфекцій можна вводити інтерферон (IFNα) у формі регулярних ін'єкцій, як і у випадку з вірусом гепатиту C (HCV) (Fried et. al. Peginterferon-alfa plus ribavirin for chronic hepatitis C virus infection, N Engl J Med 2002; 347: 975-82). Низькомолекулярні індуктори IFN, доступні для перорального застосування, надають потенційні переваги, що полягають у зниженій імуногенності та зручності введення. Таким чином, нові індуктори IFN являють собою потенційно ефективний новий клас лікарських засобів для лікування вірусних інфекцій. В якості літературного прикладу низькомолекулярного індуктора IFN, що має противірусний ефект, див. De Clercq, E.; Descamps, J.; De Somer, P. Science 1978, 200, 563-565. Під час лікування деяких видів раку IFNα також дають у комбінації з іншими лікарськими засобами (Eur. J. Cancer 46, 2849–57 і Cancer Res. 1992, 52, 1056). Агоністи TLR 7/8 також становлять інтерес в якості ад'ювантів у вакцинах через їхню здатність індукувати виражену Th1-відповідь (Hum. Vaccines 2010, 6, 1-14; Hum. Vaccines 2009, 5, 381-394). Відповідно до даного винаходу забезпечується сполука формули (I), R1 O R2 N H N N NH2 R3 (I), або її фармацевтично прийнятна сіль, таутомер(и), сольват або поліморф, де R1 являє собою водень, метил, C1-2алкіл, циклопропіл, метокси, галоген, гідроксил, трифторметил або дифторметил, R2 являє собою C1-8алкіл, C1-4алкокси-C1-4алкіл, C3-7циклоалкіл, C4-7гетероцикл, ароматичний, біциклічний гетероцикл, арилалкіл, гетероарил, гетероарилалкіл, кожен з яких необов'язково є заміщеним одним або декількома замісниками, незалежно вибраними з галогену, гідроксилу, аміно, C1-6алкілу, ди-C1-6алкіламіно, C1-6алкіламіно, C1-6алкілу, C1-6алкокси, C3-6циклоалкілу, карбонової кислоти, ефіру карбонової кислоти, аміду карбонової кислоти, гетероциклу, арилу, алкенілу, алкінілу, арилалкілу, гетероарилу, гетероарилалкілу, нітрилу, і 1 UA 113956 C2 5 R3 являє собою C4-8алкіл, C4-8алкокси, C2-6алкеніл або C2-6алкініл, кожен з яких необов'язково є заміщеним одним або декількома замісниками, незалежно вибраними з галогену, гідроксилу, аміно, C1-3алкілу, C1-3алкокси або C3-6циклоалкілу, нітрилу. У першому варіанті здійснення даний винахід забезпечує сполуки формули (I), де R 3 являє собою бутил або пентил, та де R2 і R1 визначені вище. У додатковому варіанті здійснення даний винахід стосується сполук формули (I), де R 3 являє собою C4-8алкіл, заміщений гідроксилом, та де R2 і R1 визначені вище. Інший варіант здійснення стосується сполук формули (I), де R 3, коли він є C4-8алкілом, заміщеним гідроксилом, являє собою один з наступних: OH HO (S) (S) OH HO (S) 10 15 20 (S) . Окрім того, даний винахід також забезпечує сполуки формули (I), де R 1 являє собою водень або -СН3, та де R2 і R3 визначені вище. В іншому варіанті здійснення даний винахід забезпечує сполуки формули (I), де R 2 являє собою арилалкіл або гетероарилалкіл, заміщений C 1-3алкілом, гідроксилом, алкокси, нітрилом, гетероциклом або складним ефіром, та де R1 і R3 визначені вище. У додатковому варіанті здійснення даний винахід стосується сполук формули (I), де R 2 являє собою C1-3алкіл, заміщений арилом, гетероциклом або гетероарилом, що додатково заміщений C1-3алкілом, алкокси, ефіром карбонової кислоти або амідом карбонової кислоти, та де R1 і R3 визначені вище. Окрім того, даний винахід стосується сполук формули (I), де R 2 являє собою один з наступних прикладів, що може бути додатково заміщеним C 1-3алкілом, гідроксилом, алкокси, нітрилом, гетероциклом або складним ефіром: N N N O N N O O O . Переважними сполуками згідно з даним винаходом є: 25 30 35 , . Сполуки формули (I) та їх фармацевтично прийнятна сіль, таутомер(и), сольват або поліморф мають активність як фармацевтичні препарати, зокрема, як модулятори активності толл-подібних рецепторів (особливо TLR7 та/або TLR8). У додатковому аспекті даний винахід забезпечує фармацевтичну композицію, яка містить сполуку формули (I) або її фармацевтично прийнятну сіль, сольват або поліморф разом з одним або декількома фармацевтично прийнятними наповнювачами, розріджувачами або носіями. Крім того, сполука формули (I) або її фармацевтично прийнятну сіль, сольват або поліморф відповідно до даного винаходу, або фармацевтичну композицію, яка містить зазначену сполуку формули (I) або її фармацевтично прийнятну сіль, сольват або поліморф, можна застосовувати як лікарський препарат. Іншим аспектом даного винаходу є те, що сполука формули (I) або її фармацевтично прийнятна сіль, сольват або поліморф, або зазначену фармацевтичну композицію, яка містить 2 UA 113956 C2 5 10 15 20 25 30 35 40 45 50 55 60 зазначену сполуку формули (I) або її фармацевтично прийнятну сіль, сольват або поліморф, можна відповідно застосовувати в лікуванні порушення або захворювання, у яке залучено модуляцію TLR7 та/або TLR8. Вираз "алкіл" стосується насиченого аліфатичного вуглеводню з нерозгалуженим або розгалуженим ланцюгом, що містить установлене число атомів вуглецю. Вираз "галоген" стосується фтору, хлору, брому або йоду. Вираз "алкеніл" стосується алкілу в значенні, визначеному вище, який складається щонайменше з двох атомів вуглецю та щонайменше з одного вуглець-вуглецевого подвійного зв'язку. Вираз "алкініл" стосується алкілу в значенні, визначеному вище, який складається щонайменше з двох атомів вуглецю та щонайменше з одного вуглець-вуглецевого потрійного зв'язку. Вираз "циклоалкіл" стосується карбоциклічного кільця, яке містить установлене число атомів вуглецю. Вираз "гетероарил" означає ароматичну кільцеву структуру, визначену стосовно виразу "арил", яка містить щонайменше 1 гетероатом, вибраний з N, O і S, зокрема, з N і О. Вираз "арил" означає ароматичну кільцеву структуру, яка необов'язково містить один або два гетероатоми, вибрані з N, O і S, зокрема, з N і О. Зазначена ароматична кільцева структура може містити 4, 5, 6 або 7 кільцевих атомів. Зокрема, зазначена ароматична кільцева структура може містити 5 або 6 кільцевих атомів. Вираз "біциклічний гетероцикл" означає ароматичну кільцеву структуру, визначену стосовно виразу "арил", яка складається з двох конденсованих ароматичних кілець. Кожне кільце необов'язково містить гетероатом, вибраний з N, O і S, зокрема, з N і О. Вираз "арилалкіл" означає ароматичну кільцеву структуру, визначену стосовно виразу "арил", необов'язково заміщену алкільною групою. Вираз "гетероарилалкіл" означає ароматичну кільцеву структуру, визначену стосовно виразу "гетероарил", необов'язково заміщену алкільною групою. Вираз "алкокси" стосується алкільної (з вуглецевим та водневим ланцюгом) групи, зв'язаної за допомогою одинарного зв'язку з киснем, такої як, наприклад, метоксигрупа або етоксигрупа. Гетероцикл стосується молекул, які є насиченими або частково насиченими та включають етилоксид, тетрагідрофуран, діоксан або інші циклічні прості ефіри. Гетероцикли, які містять азот, включають, наприклад, азетидин, морфолін, піперидин, піперазин, піролідин тощо. Інші гетероцикли включають, наприклад, тіоморфолін, діоксолініл та циклічні сульфони. Гетероарильні групи являють собою гетероциклічні групи, які є ароматичними за своєю природою. Вони є моноциклічними, біциклічними або поліциклічними, які містять один або декілька гетероатомів, вибраних з N, О або S. Гетероарильними групами можуть бути, наприклад, імідазоліл, ізоксазоліл, фурил, оксазоліл, піроліл, піридоніл, піридил, піридазиніл або піразиніл. Фармацевтично прийнятні солі сполук формули (I) включають їх солі приєднання кислоти та основні солі. Придатні солі приєднання кислоти утворюються з кислот, які утворюють нетоксичні солі. Придатні основні солі утворюються з основ, які утворюють нетоксичні солі. Сполуки згідно з даним винаходом можуть також існувати в несольватованих та сольватованих формах. Вираз "сольват" застосовується в даному документі для опису молекулярного комплексу, який містить сполуку згідно з даним винаходом та одну або декілька молекул фармацевтично прийнятного розчинника, наприклад, етанолу. Вираз "поліморф" стосується здатності сполуки згідно з даним винаходом існувати більш ніж в одній формі або кристалічній структурі. Сполуки згідно з даним винаходом можна вводити у формі кристалічних або аморфних продуктів. Їх можна отримати, наприклад, у формі твердої пресованої маси, порошків або плівок за допомогою таких способів, як осадження, кристалізація, сублімаційне сушіння, розпилювальне сушіння або сушіння випарюванням. Їх можна вводити окремо, або в комбінації з однією або декількома іншими сполуками згідно з даним винаходом, або в комбінації з одним або декількома іншими лікарськими засобами. Як правило, їх уводять у формі складу в комбінації з одним або декількома фармацевтично прийнятними наповнювачами. Вираз "наповнювач" застосовується в даному документі для опису будь-якого інгредієнта, відмінного від сполуки (сполук) згідно з даним винаходом. Вибір наповнювача багато в чому залежить від таких чинників, як конкретний спосіб уведення, ефект наповнювача щодо розчинності та стабільності та природи лікарської форми. Сполуки згідно з даним винаходом або будь-яку їхню підгрупу можна скласти в різні фармацевтичні форми для введення. Як приклад придатних композицій можна навести всі 3 UA 113956 C2 5 10 15 20 25 30 35 40 45 50 композиції, зазвичай використовувані для системного введення лікарських засобів. Для одержання фармацевтичних композицій згідно з даним винаходом ефективну кількість конкретної сполуки, у деяких випадках у формі солі приєднання, об'єднують як активний інгредієнт в однорідну суміш із фармацевтично прийнятним носієм, при цьому носій може мати широку різноманітність форм залежно від форми препарату, бажаного для введення. Ці фармацевтичні композиції бажано перебувають у лікарській формі з одноразовим дозуванням, придатній, наприклад, для перорального, ректального введення або введення крізь шкіру. Наприклад, під час одержання композицій у пероральній лікарській формі можна використовувати будь-яке зі звичайних фармацевтичних середовищ, таких як, наприклад, вода, гліколі, олії, спирти тощо у випадку пероральних рідких препаратів, таких як суспензії, сиропи, еліксири, емульсії та розчини; або тверді носії, такі як крохмалі, цукри, каолін, розріджувачі, змащувальні речовини, зв'язувальні речовини, засоби для поліпшення розпадання тощо у випадку порошків, пігулок, капсул і таблеток. Внаслідок легкості введення таблетки та капсули представляють найбільш переважні одиничні лікарські форми для перорального введення, при цьому, очевидно, використовують тверді фармацевтичні носії. Також сюди включають препарати у твердій формі, які можна перетворити на рідкі форми безпосередньо перед застосуванням. У композиціях, придатних для введення крізь шкіру, носій необов'язково включає засіб, що підсилює проникнення, та/або придатний змочувальний засіб, необов'язково в комбінації з придатними добавками будь-якого походження в незначних кількостях, при цьому ці добавки не виявляють значний негативний ефект щодо шкіри. Зазначені добавки можуть полегшувати введення в шкіру та/або можуть бути корисними при одержанні бажаних композицій. Ці композиції можна вводити в різні способи, наприклад, за допомогою трансдермального пластиру, за допомогою точкового нанесення, за допомогою мазі. Сполуки згідно з даним винаходом можна також уводити шляхом інгаляції або інсуфляції за допомогою способів та сполук, використовуваних у даній галузі для введення таким шляхом. Таким чином, у цілому сполуки згідно з даним винаходом можна вводити в легені у формі розчину, суспензії або сухого порошку. Особливо переважним є складання зазначених вище фармацевтичних композицій у лікарській формі з одноразовим дозуванням для простоти введення та рівномірності дозування. Лікарська форма з одноразовим дозуванням, як застосовується в даному документі, стосується фізично дискретних одиниць, придатних в якості одиничних доз, при цьому кожна одиниця дози містить задану кількість активного інгредієнта, розраховану для одержання бажаного терапевтичного ефекту в комбінації з необхідним фармацевтичним носієм. Прикладами таких лікарських форм із одноразовим дозуванням слугують таблетки (включаючи ділені таблетки або таблетки, вкриті оболонкою), капсули, пігулки, пакетики з порошком, облатки, супозиторії, ін'єкційні розчини або суспензії тощо, а також їх розділені кратні одиниці. Фахівці в галузі лікування інфекційних захворювань зможуть визначити ефективну кількість за результатами тестів, представленими нижче. У цілому передбачається, що ефективна добова кількість повинна складати від 0,01 мг/кг до 50 мг/кг маси тіла, більш переважно від 0,1 мг/кг до 10 мг/кг маси тіла. Уведення необхідної дози у формі двох, трьох, чотирьох або більше частин дози через відповідні проміжки часу протягом доби може бути доцільним. Зазначені частини дози можна скласти як лікарські форми з одноразовим дозуванням, наприклад, такі, що містять від 1 до 1000 мг, зокрема, від 5 до 200 мг активного інгредієнта на лікарську форму з одноразовим дозуванням. Точне дозування і частота введення залежать від конкретної застосовуваної сполуки формули (I), конкретного стану, який лікують, тяжкості стану, який лікують, віку, ваги і загального фізичного стану конкретного пацієнта, а також від інших ліків, які може приймати особа, що добре відомо фахівцям у даній галузі. Крім того, очевидно, що можна зменшити або збільшити ефективну кількість залежно від реакції суб'єкта, якого лікують, та/або залежно від оцінки лікаря, який призначає сполуки згідно з даним винаходом. Отже, згадані вище діапазони ефективної кількості є тільки загальними вказівками та жодною мірою не припускають обмеження обсягу або застосування даного винаходу. Одержання сполук Сполуки формули (I), де R1 являє собою атом водню, отримують відповідно до схеми 1. 55 4 UA 113956 C2 5 10 Схема 1 Сполуки типу А отримують за схемою 1 у результаті однієї із двох реакцій. (i) Реакція гетероциклічного спирту з галогензаміщеним складним ефіром та підходящою основою, наприклад, карбонатом калію, карбонатом цезію або гідридом натрію. Приклад показано на схемі 2а. (ii) Реакція спирту або складного гідроксіефіру, наприклад, 2-гідроксіетилацетату, з алкілгалогенідом із застосуванням відповідної основи, наприклад, гідриду натрію. Приклад показано на схемі 2b. O Br O OH O O N NaH, DMF N O Схема 2a Br O HO 15 20 25 O O O NaH, TBAI, THF O Схема 2b Сполуки формули (I), де R1 являє собою алкіл, циклоалкіл, трифторметил або алкокси, та де R2 являє собою арил або гетероарил, отримують так, як показано на схемі 3 нижче. Бетакетоефір (Е) можна хлорувати за допомогою, наприклад, тіонілхлориду з одержанням проміжного продукту 2-хлор-бета-кетоефіру (F). Фенол або гетероароматичний спирт (R2OH) об'єднують з водним гідроксидом натрію в еквімолярному співвідношенні. Розчинники потім видаляють при зниженому тиску з одержанням фенолу або солі гетероароматичного спирту R2. Цю сіль об'єднують із проміжним продуктом 2-хлор-β-кетоефіром (F) з одержанням проміжного продукту G відповідно до способу, описаного в літературі. Потім проміжний продукт G об'єднують, з основою або без неї, з карбонатом гуанідину у відповідному розчиннику, наприклад, в етанолі. Потім проміжний продукт H уводять у реакцію з оксихлоридом фосфору з утворенням проміжного продукту хлорпіримідину (J). Потім у результаті нагрівання (J) в присутності надлишкової кількості аміну та необов'язково надлишкової кількості органічної основи, наприклад, триетиламіну, при підвищеній температурі утворюються продукти. Це загальна схема із застосуванням способів, відомих фахівцю в даній галузі, див., наприклад, Organic Syntheses volume 33, p.43 (1953). 30 5 UA 113956 C2 Схема 3 5 10 15 Схема 4 Сполуки формули (I), де R1 являє собою алкіл, циклоалкіл, трифторметил або алкокси, та де R2 являє собою ароматичний або аліфатичний радикал, можна отримати згідно зі схемою 4. Ця схема реакцій починається з перехресної реакції Кляйзена, де ацилхлорид реагує зі складноефірним проміжним продуктом А (показано на схемі 1) з утворенням проміжних продуктів (G), як на схемі 3. Після одержання проміжного продукту G схема реакції проходить тим самим шляхом до утворення продуктів, як і на схемі 3. Це загальна схема із застосуванням способів, відомих фахівцю в даній галузі, див., наприклад, The Journal of American Chemical Society volume 127, page 2854 (2005). Експериментальна частина Синтез проміжного продукту A-1 Br O HO O O O NaH, TBAI, THF O A-1 До суміші етилгліколяту [623-50-7] (250,00 г, 2,40 моль), NaH (105,65 г, 2,64 моль), йодиду 6 UA 113956 C2 5 10 15 20 25 30 тетрабутиламонію (TBAI) (88,70 г, 240,14 ммоль) у безводному THF (2 л) краплинами додавали бензилбромід (451,80 г, 2,64 моль) за 0 °C. одержану в результаті суміш перемішували при 25 °C протягом 16 годин. Реакційну суміш гасили насиченим водним хлоридом амонію (1 л), а водний шар екстрагували етилацетатом (3 × 1 л). Об'єднані органічні шари промивали розсолом (1 л), висушували над сульфатом магнію, тверді речовини видаляли за допомогою фільтрації, а розчинники фільтрату концентрували при зниженому тиску. Залишок очищали за допомогою колонкової хроматографії на силікагелі (петролейний ефір:етилацетат = 6:1) з одержанням проміжного продукту А-1 (200 г). 1 H-ЯМР (CDCl3, 400 МГц) δ частин на мільйон 7,37-7,27 (m, 5 H); 4,62 (s, 2 H), 4,24-4,19 (q, J=6,8 Гц, 2 H); 4,07 (s, 2 H); 1,29-1,25 (t, J=6,8 Гц, 3 H). Спосіб одержання проміжного продукту B-1 До перемішаної суспензії NaH (45,30 г, 1,13 моль) у безводному THF (1,2 л) додавали етилформіат (114,42 г, 1,54 моль). Суспензію охолоджували в льодяній бані, а потім за допомогою крапельної лійки краплинами додавали сполуку А-1 (200 г, 1,03 моль) у безводному THF (300 мл). Білу суміш перемішували при від 0 °C до кімнатної температури протягом 5 годин. Протягом цього часу реакція була екзотермічною, і реакційна суміш пожовтіла. В окремій колбі карбонат гуанідину [593-85-1] (111,31 г, 0,618 моль) обробили свіжоприготовленим розчином етоксиду натрію, одержаним у результаті обережного додавання Na (28,41 г, 1,24 моль) до безводного етанолу (750 мл) при кімнатній температурі. Потім білувату завись, одержану після перемішування протягом 1 години, додавали до жовтого розчину, одержаного вище. одержану в результаті блідо-жовту реакційну суміш нагрівали зі зворотним холодильником протягом 15 годин. Розчинник видаляли, а потім неочищений залишок розчиняли у воді (1,5 л). Суміш доводили до рН=5 за допомогою оцтової кислоти. Тверду речовину збирали, ретельно промивали водою та етанолом з одержанням проміжного продукту B-1 (160 г). 1 H-ЯМР (400 МГц, DMSO-d6) δ частин на мільйон 4,90 (s, 2 H), 6,33 (br. s., 2 H), 7,25 (s, 1 H), 7,29 – 7,42 (m, 5 H), 11,21 (br. s., 1 H). Спосіб одержання проміжного продукту C-1 Схема реакції Ph Ph O HO POCl3 N N NH2 O Cl 40 N NH2 C-1 B-1 35 N Суспензію проміжного продукту B-1 (160 г, 0,74 моль) в POCl3 (900 мл) нагрівали до 100 °C в атмосфері N2 при перемішуванні протягом 5 годин. Реакційну суміш охолоджували до кімнатної температури. Надлишкову кількість POCl3 видаляли при зниженому тиску, олійний залишок вливали в холодний насич. водн. NaHCO3 (2 л), який перемішували протягом 30 хвилин. Суміш екстрагували етилацетатом (3 × 1,5 л). Об'єднані органічні шари розділяли і промивали розсолом (1 л), висушували над сульфатом натрію, тверді речовини видаляли за допомогою фільтрації, а розчинники фільтрату концентрували з одержанням проміжного продукту C-1 (70 г) у формі жовтої твердої речовини. Продукт застосовували на наступному етапі без додаткового очищення. 7 UA 113956 C2 Спосіб одержання сполуки 1 Ph O O H2N N Et3N, EtOH, 80oC Cl 5 10 N N N H NH2 N NH2 C-1 1 До суспензії C-1 (70,00 г, 297,03 ммоль) в етанолі (1,4 л) додавали н-бутиламін (217,24 г, 2,97 моль) та триетиламін (60,11 г, 594,05 ммоль). Реакційну суміш нагрівали зі зворотним холодильником протягом 16 годин. Реакційну суміш охолоджували до кімнатної температури, а розчинники видаляли при зниженому тиску. Залишок очищали за допомогою флешхроматографії на силікагелі, застосовуючи градієнт петролейний ефір/етилацетат, з одержанням 1 (26 г) у формі блідо-жовтої твердої речовини. 1 H-ЯМР (400 МГц, МЕТАНОЛ-d4) δ частин на мільйон 0,96 (t, J=7,3 Гц, 3 H), 1,32 – 1,43 (m, 2 H), 1,52 – 1,61 (m, 2 H), 3,38 (t, J=7,2 Гц, 2 H), 5,01 (s, 2 H), 7,28 (s, 1 H), 7,31 – 7,46 (m, 5 H). O N H O N N NH2 Ac2O N H N N O N O 15 20 D-1 1 Одержання проміжного продукту D-1 У круглодонну колбу на 100 мл, оснащену магнітною мішалкою, поміщали 1 (1 г, 3,67 ммоль) в оцтовому ангідриді (40 мл). Жовтий розчин залишали перемішуватися зі зворотним холодильником протягом 15 годин. Розчинники видаляли при зниженому тиску. Неочищений продукточищали за допомогою гель-хроматографії на силікагелі, застосовуючи градієнт гептан/етилацетат. Кращі фракції збирали, а розчинники видаляли при зниженому тиску з одержанням білої твердої речовини, D-1. + LC-MS: аналіт. розрах. для C19H24N4O3: 356,19; знайдене значення 357 [M+H] . 1 H-ЯМР (400 МГц, ХЛОРОФОРМ-d) δ частин на мільйон 0,94 (t, J=7,4 Гц, 3 H), 1,31 – 1,45 (m, 2 H), 1,50 – 1,67 (m, 2 H), 2,31 (s, 6 H), 3,44 (m, J=6,0 Гц, 2 H), 5,12 (s, 2 H), 5,41 – 5,52 (m, 1 H), 7,43 (m, J=1,5 Гц, 5 H), 7,79 (s, 1 H). Одержання проміжного продукту D-2 O N H N N HO O N H2, 10% Pd/C O 25 30 35 N H N N O N O D-1 D-2 Спосіб А. У колбу Ерленмейєра на 250 мл, оснащену магнітною мішалкою, поміщали проміжний продукт D-1 (1 г) та етанол (100 мл). Колбу барботували азотом з наступним додаванням 10 % Pd на вугіллі (100 мг). Колбу запаювали, а повітря витісняли і заміняли на водень. Реакційній суміші дозволяли перемішуватися при кімнатній температурі протягом 15 годин. Гетерогенну суміш фільтрували крізь ущільнений целіт, а розчинники фільтрату видаляли при зниженому тиску з одержанням кількісного виходу D-2. Спосіб В. 0,1 М розчин вихідного матеріалу в метанолі пропускали крізь Н-куб, обладнаний картриджем з 10 % Pd/C, при 0,5 мл/хв. та при тиску водню 30 бар. LC-MS показала повне перетворення. Розчинники видаляли при зниженому тиску. Неочищений продукт очищали за допомогою хроматографії на силікагелі, застосовуючи градієнт дихлорметан/10 % метанол у дихлорметані. Кращі фракції зводили воєдино; розчинники видаляли при зниженому тиску з одержанням білої твердої речовини, D-2. 8 UA 113956 C2 + 5 10 15 LC-MS: аналіт. розрах. для C12H18N4O3: 266,14; знайдене значення 267 [M+H] . 1 H-ЯМР (400 МГц, DMSO-d6) δ частин на мільйон 0,87 (t, J=7,4 Гц, 3 H), 1,28 (dd, J=14,9, 7,4 Гц, 2 H), 1,49 (t, J=7,2 Гц, 2 H), 2,15 (s, 6 H), 3,20 – 3,37 (m, 2 H), 7,02 – 7,12 (m, 1 H), 7,58 (s, 1 H), 10,27 (br. s., 1 H). Одержання проміжного продукту D-3 У круглодонну колбу на 100 мл поміщали 1 (1 г, 3,67 ммоль), ди-трет-бутилдикарбонат (7,5 г) і ацетонітрил (50 мл). Жовтий розчин перемішували зі зворотним холодильником протягом 16 годин. Розчинники видаляли при зниженому тиску. Залишок очищали за допомогою хроматографії на силікагелі, застосовуючи колонку, попередньо заповнену 80 г силікагелю, і градієнт гептан/етилацетат з автоматичним збиранням при 254 нм. Кращі фракції зводили воєдино з одержанням жовтого масла D-3. + LC-MS: аналіт. розрах. для C25H36N4O5: 472,269; знайдене значення 473 [M+H] . 1 H-ЯМР (400 МГц, ХЛОРОФОРМ-d) δ частин на мільйон 0,94 (t, J=7,4 Гц, 3 H), 1,33 – 1,42 (m, 2 H), 1,46 (s, 18 H), 1,50 – 1,65 (m, 2 H), 3,35 – 3,51 (m, 2 H), 5,09 (s, 2 H), 5,31 – 5,38 (m, 1 H), 7,36 – 7,48 (m, 5 H), 7,75 (s, 1 H). Одержання проміжного продукту D-4 O N H N N N O 20 25 30 HO O N H O O N N H 2 , Pd/C O N O O O D-4 D-3 Проміжний продукт D-4 отримують згідно зі способом одержання проміжного продукту D-2 з використанням або способу А, або способу В. + LC-MS: аналіт. розрах. для C18H30N4O5: 382,222; знайдене значення 383 [M+H] . 1 H-ЯМР (400 МГц, ХЛОРОФОРМ-d) δ частин на мільйон 0,95 (t, J=7,3 Гц, 3 H), 1,39 (s, 18 H), 1,40 – 1,45 (m, 2 H), 1,53 – 1,64 (m, 2 H), 3,42 – 3,51 (m, 2 H), 5,66 (s, 1 H), 7,43 (s, 1 H). Одержання сполуки 2 У флакон на 30 мл поміщали проміжний продукт D-4 (200 мг, 0,52 ммоль), DMF (5 мл), 1-(3бромпропіл)-4-метоксибензен (130 мг, 0,57 ммоль) і карбонат цезію (508 мг, 1,56 ммоль). 9 UA 113956 C2 5 Реакційній суміші дозволяли перемішуватися протягом 15 годин при кімнатній температурі. Тверді речовини видаляли за допомогою фільтрації. Розчинники фільтрату видаляли при зниженому тиску, а неочищений продукт відновлювали в метанолі і до нього додавали HCl (6 М у ізопропанолі), а також реакційній суміші дозволяли перемішуватися протягом 15 годин при кімнатній температурі. Розчинники видаляли при зниженому тиску, а неочищений продукт очищали за допомогою розділення на оберненій фазі з одержанням 2 у формі вільної основи. Одержання проміжного продукту G-1 10 15 20 До перемішаного розчину А-1 (60 г, 309 ммоль, 1 екв.) та 1-метилімідазолу (30,4 г, 370 ммоль, 1,2 екв.) у CH2Cl2 (1 л) додавали ацетилхлорид (24,3 г, 309 ммоль, 1 екв.) за -45 °C у атмосфері N2. Після перемішування протягом 20 хв. до суміші додавали TiCl 4 (210 г, 1,08 моль, 3,5 екв.) та трибутиламін (230 г, 1,24 моль, 4 екв.) за -45 °C у атмосфері N2 і продовжували перемішувати протягом 50 хвилин при -45 °C у атмосфері N2. Після завершення додавали воду та етилацетат. Органічний шар відокремлювали, а водний шар двічі екстрагували етилацетатом. Органічний шар промивали розсолом і висушували над сульфатом натрію. Тверді речовини видаляли за допомогою фільтрації, а розчинники фільтрату видаляли при зниженому тиску. Неочищений продукт очищали за допомогою колонкової хроматографії на силікагелі, застосовуючи градієнт гептан/етилацетат, з одержанням блідо-жовтої олії, G-1. 1 H-ЯМР (400 MГц, ХЛОРОФОРМ-d) δ частин на мільйон 1,30 (t, J=7,2 Гц, 3 H), 2,28 (s, 3 H), 4,27 (q, J=7,2 Гц, 2 H), 4,41 (s, 1 H), 4,58 (d, J=11,8 Гц, 1 H), 4,75 (d, J=11,8 Гц, 1 H), 7,32 – 7,43 (m, 5 H). Одержання проміжного продукту H-1 25 30 35 У флакон на 20 мл для реакцій під дією мікрохвильового випромінювання поміщали проміжний продукт G-1 (500 мг, 2,12 ммоль), етанол (5 мл) і карбонат гуанідину (200 мг, 2,22 ммоль). Флакон запаювали і витримували при 120 °C, перемішуючи протягом 4 годин. Розчинники видаляли при зниженому тиску. Додавали воду (25 мл). Суміш доводили до рН=5 за допомогою обережного додавання оцтової кислоти. Осад виділяли за допомогою фільтрування з одержанням білої твердої речовини, Н-1. 1 H-ЯМР (400 MГц, ХЛОРОФОРМ-d) δ частин на мільйон 1,88 (s, 3 H), 4,85 (s, 2 H), 6,38 (br. s., 2 H), 7,24 – 7,49 (m, 5 H), 11,16 (s, 1 H). Одержання проміжного продукту G-2 10 UA 113956 C2 O O Cl 10 O NaOPh O 5 O O O Ph F-1 G-2 Етап 1. Фенолят натрію отримували в результаті випарювання на роторному випарнику еквімолярних кількостей фенолу в гідроксиду натрію в круглодонній колбі на 1 л. Толуен застосовували під час азеотропного видалення води. Етап 2. Фенолят натрію (116 г, 1 моль), одержаний на етапі 1, та толуен (1 л) поміщали в тригорлу колбу на 2 л, оснащену механічною мішалкою, крапельною лійкою та зворотним холодильником з осушувальним патроном. Суспензію нагрівали зі зворотним холодильником, потім за допомогою крапельної лійки під час перемішування додавали α-хлорацетоацетат (165 г, 1 моль), при цьому реакційну суміш продовжували нагрівати зі зворотним холодильником протягом 4 годин. Світло-коричневу суспензію охолоджували до кімнатної температури, екстрагували водою (2 × 500 мл) та висушували (за допомогою безводного сульфату магнію). Тверді речовини видаляли за допомогою фільтрації, а розчинники фільтрату видаляли при зниженому тиску. Неочищений продукт застосовували на наступному етапі без очищення. Одержання проміжного продукту H-2 15 20 25 У круглодонну колбу на 100 мл, оснащену магнітною мішалкою і зворотним холодильником, додавали проміжний продукт G-2 (1 г, 4,5 ммоль), етанол (50 мл) і карбонат гуанідину [593-85-1] (203 мг, 2,25 ммоль). Реакційну суміш нагрівали зі зворотним холодильником протягом 15 годин. Розчинник видаляли при зниженому тиску. Додавали воду (25 мл). Суміш доводили до рН=5 за допомогою обережного додавання оцтової кислоти. Осад виділяли за допомогою фільтрації з одержанням білої твердої речовини, Н-2. Її застосовували без додаткового очищення на наступному етапі. Одержання проміжного продукту J-1 O HO 30 O N N NH2 N POCl3 Cl N NH2 H-2 J-1 У круглодонну колбу на 50 мл, оснащену магнітною мішалкою і зворотним холодильником, додавали проміжний продукт H-2 (500 мг, 2,3 ммоль) і POCl3 (20 мл). Суспензію нагрівали зі зворотним холодильником під час перемішування протягом 6 годин. Розчинники видаляли при зниженому тиску з одержанням неочищеного коричневого масла J-1. Додаткове очищення не проводили. Сполуку застосовували як таку на наступному етапі. Одержання 3 11 UA 113956 C2 5 10 15 20 25 30 35 У запаяну трубку на 50 мл, оснащену магнітною мішалкою, поміщали проміжний продукт J-1 (150 мг, 0,64 ммоль), н-бутиламін (70 мг, 0,96 ммоль), основний оксид алюмінію (100 мг) та діоксан (10 мл). Трубку запаювали, поміщали в масляну баню при 120ºC, а реакційну суміш нагрівали під час перемішування протягом 15 годин. Посудину охолоджували до кімнатної температури і акуратно знімали кришку. Вміст вливали в круглодонну колбу, при цьому видаляли розчинники при зниженому тиску. Неочищений продукт очищали за допомогою колонкової хроматографії на силікагелі, застосовуючи градієнт дихлорметан/5 % метанол у дихлорметані. Кращі фракції зводили воєдино, а розчинники видаляли при зниженому тиску з одержанням 3. + LC-MS: аналіт. розрах. для C15H20N4O: 272,16; знайдене значення 273 [M+H] . 1 H-ЯМР (300 МГц, ХЛОРОФОРМ-d) δ частин на мільйон 0,80 (t, J=7,3 Гц, 3 H), 1,20 (dq, J=15,0, 7,3 Гц, 2 H), 1,33 – 1,47 (m, 2 H), 1,98 (s, 3 H), 3,20 – 3,34 (m, 2 H), 4,74 (br. s., 2 H), 4,79 (br. s., 1 H), 6,78 – 6,84 (m, 2 H), 6,91 – 7,01 (m, 1 H), 7,18 – 7,28 (m, 2 H). Одержання 4 Етап 1 У флакон на 20 мл для реакцій під дією мікрохвильового випромінювання додавали комерційно доступний 2,4-дихлор-5-метоксипіримідин (300 мг, 1,68 ммоль), етанол (5 мл) та нбутиламін (0,166 мл, 1,68 ммоль). Флакон запаювали, потім нагрівали під дією мікрохвильового випромінювання протягом 10 хвилин при температурі 120 °C. LC-MS показала повне перетворення. Розчинники видаляли при зниженому тиску. Неочищений продукт застосовували як такий на етапі 2. Етап 2 Сполуку з етапу 1 поміщали в посудину високого тиску на 20 мл з водним аміаком (10 мл) і до цієї суміші додавали бікарбонат амонію (200 мг, 2,6 ммоль) та CuO (24 мг, 0,17 ммоль, 0,1 екв.). Посудину запаювали і суміш нагрівали до 120 °C під час перемішування протягом 24 годин. Реакційну суміш 3 рази екстрагували за допомогою 5 мл суміші дихлорметан:метанол (9:1), а леткі речовини видаляли при зниженому тиску. Сполуку фільтрували крізь силікагель, при цьому елюючи сумішшю дихлорметан:метанол (9:1), а леткі речовини видаляли при зниженому тиску. Залишок очищали за допомогою обернено-фазової хроматографії. + LC/MS: аналіт. розрах. для C9H16N4O: 196,13; знайдене значення 197 [M+H] . 1 H-ЯМР (400 MГц, ХЛОРОФОРМ-d) δ частин на мільйон 0,97 (t, J=7,3 Гц, 3 H), 1,35 – 1,48 (m, 2 H), 1,56 – 1,68 (m, 2 H), 3,44 – 3,52 (m, 2 H), 3,80 (s, 3 H), 5,86 (s, 1 H), 5,97 (s, 2 H), 7,07 – 7,14 (m, 1 H). 12 UA 113956 C2 Одержання 5 5 10 15 20 25 Етап 1 У тестову пробірку 16×100 поміщали проміжний продукт D-2 (180 мг, 0,66 ммоль), DMF (5 мл), пропілйодид (111 мг, 0,656 ммоль) і карбонат цезію (320 мг, 0,98 ммоль). Реакційній суміші дозволяли перемішуватися при кімнатній температурі протягом 15 годин. Тверді речовини видаляли за допомогою фільтрації, розчинники фільтрату видаляли при зниженому тиску. Неочищений продукт очищали за допомогою хроматографії на силікагелі, застосовуючи градієнт елюенту дихлорметан/10 % метанол у дихлорметані. Кращі фракції об'єднували, розчинники видаляли при зниженому тиску з одержанням білої твердої речовини. Етап 2 У флакон на 10 мл для реакцій під дією мікрохвильового випромінювання поміщали описану вище білу тверду речовину (100 мг), гідроксид амонію (1 мл) і етанол (1 мл). Флакон запаювали і нагрівали під час перемішування до 175 °C протягом 10 хвилин. LC-MS показала повне перетворення на продукт. Розчинники видаляли при зниженому тиску. Неочищений продукт очищали за допомогою хроматографії на силікагелі, застосовуючи градієнт елюенту дихлорметан/10 % метанол у дихлорметані. Кращі фракції об'єднували, розчинники видаляли при зниженому тиску з одержанням безбарвного масла. Додавання одного еквівалента HCl (при застосуванні від 5 до 6 н HCl в ізопропанолі) дало білу тверду речовину 5. + LC-MS: аналіт. розрах. для C11H20N4O: 224,16; знайдене значення 225 [M+H] . 1 H-ЯМР (400 МГц, DMSO-d6) δ частин на мільйон 0,90 (t, J=7,3 Гц, 3 H), 0,98 (t, J=7,4 Гц, 3 H), 1,20 – 1,35 (m, 2 H), 1,54 (t, J=7,2 Гц, 2 H), 1,69 – 1,75 (m, 2 H), 3,40 (d, J=7,0 Гц, 2 H), 3,87 (t, J=6,5 Гц, 2 H), 7,39 (d, J=5,5 Гц, 1 H), 7,46 (br. s., 2 H), 8,28 – 8,37 (m, 1 H). Схема синтезу для одержання AA-9 30 13 UA 113956 C2 Синтез проміжного продукту AA-3 5 10 До розчину валеріанового альдегіду (43 г, 500 ммоль) в THF (1 л) додавали AA-2 (200 г, 532 ммоль) і реакційну суміш перемішували протягом 16 годин при кімнатній температурі. Розчинники випарювали, а залишок розчиняли в петролейному ефірі та відфільтровували. Розчинники фільтрату видаляли при зниженому тиску, а залишок очищали за допомогою хроматографії на силікагелі, застосовуючи градієнт елюенту петролейний ефір/3 % етилацетат у петролейному ефірі, з одержанням AA-3 (90 г) у вигляді безбарвного масла. 1 H-ЯМР (400 МГц, CDCl3): δ частин на мільйон 6,81 – 6,77 (m, 1 H), 5,68 – 5,64 (td, J=1,2 Гц, 15,6 Гц, 1 H), 2,11 – 2,09 (m, 2 H), 1,406 (s, 9 H), 1,38 – 1,26 (m, 4 H), 0,85 – 0,81 (t, J=7,2 Гц, 3 H). Синтез сполуки AA-5 15 20 25 До перемішаного розчину АА-4 (165 г, 781 ммоль) в THF (800 мл) додавали н-бутиллітій (290 мл, 725 ммоль, 1,5 екв.) при -78 °C. Реакційну суміш перемішували протягом 30 хвилин, потім додавали AA-3 (90 г, 488,4 ммоль) в THF (400 мл), і реакційну суміш перемішували протягом 2 годин при -78 °C. Суміш гасили насич. водн. розчином NH4Cl і нагрівали до кімнатної температури. Продукт розподіляли між етилацетатом і водою. Органічну фазу промивали розсолом, висушували і випарювали. Залишок очищали за допомогою колонкової хроматографії, елюючи 5 % етилацетатом у петролейному ефірі, з одержанням безбарвного масла, AA-5 (132 г). 1 H-ЯМР (400 МГц, CDCl3): δ частин на мільйон 7,36 – 7,16 (m, 10 H), 3,75 – 3,70 (m, 2 H), 3,43 – 3,39 (d, J=15,2 Гц, 1 H), 3,33 – 3,15 (m, 1 H), 1,86 – 1,80 (m, 2 H), 1,47 – 1,37 (m, 2 H), 1,32 (s, 9 H), 1,26 – 1,17 (m, 7 H), 0,83 – 0,79 (t, J=7,2 Гц, 3 H). Синтез AA-6 30 35 AA-5 (130 г, 328 ммоль) розчиняли в THF (1,5 л) і додавали LAH (20 г, 526 ммоль) невеликими порціями при 0 °C. одержану в результаті суміш перемішували при тій самій температурі протягом 2 годин і потім їй дозволяли нагрітися до кімнатної температури. Суміш гасили насич. водн. розчином NH4Cl. Продукт розподіляли між етилацетатом і водою. Органічну фазу промивали розсолом, висушували і випарювали. Об'єднані органічні шари висушували над сульфатом натрію, тверді речовини видаляли за допомогою фільтрації і концентрували з 14 UA 113956 C2 5 10 15 20 25 одержанням AA-6 (100 г), застосовуваного на наступному етапі без додаткового очищення. 1 H-ЯМР (400 МГц, CDCl3): δ частин на мільйон 7,33-7,14 (m, 10 H), 3,91 – 3,86 (m, 1 H), 3,80 – 3,77 (d, J=13,6 Гц, 1 H), 3,63 – 3,60 (d, J=13,6 Гц, 1 H), 3,43 – 3,42 (m, 1 H), 3,15 – 3,10 (m, 1 H), 2,70 – 2,63 (m, 2 H), 1,65 – 1,28 (m, 10 H), 0,89 – 0,81 (m, 3 H). Синтез AA-9 Розчин AA-6 (38 г, 116,75 ммоль) та 10 % Pd/C у метанолі (200 мл) гідрували при тиску водню 50 фунтів/кв.дюйм при 50˚C протягом 24 годин. Реакційну суміш відфільтровували, а розчинник випарювали з одержанням неочищеного продукту AA- 7 (17 г). Неочищений продукт розчиняли в дихлорметані (200 мл), при 0 °C додавали триетиламін (26,17г, 259,1 ммоль) та ди-трет-бутилдикарбонат (84,7 г, 194,4 ммоль). одержану в результаті суміш перемішували при кімнатній температурі протягом 16 годин. Суміш розподіляли між дихлорметаном і водою. Органічну фазу промивали розсолом, висушували і випарювали. Залишок очищали за допомогою хроматографії на силікагелі, елюючи 20 % етилацетатом у петролейному ефірі, з одержанням AA-8 (13 г) у формі безбарвної олії. 1 H-ЯМР (400 МГц, CDCl3): δ частин на мільйон 4,08 – 4,03 (br, 1 H), 3,68 (m, 1 H), 3,58 – 3,55 (m, 2 H), 3,20 – 2,90(br, 1 H), 1,80 – 1,73 (m, 1 H), 1,42 – 1,17 (m, 15 H), 0,85 – 0,82 (t, J=6,8 Гц, 3 H). AA-8 (42 г, 0,182 моль) розчиняли в діоксані (200 мл) і при 0 °C додавали діоксан/HCl (4 М, 200 мл). одержану в результаті суміш перемішували при кімнатній температурі протягом 2 год. Розчинник випарювали з одержанням неочищеного продукту. До неочищеного продукту додавали суміш дихлорметану/петролейного ефіру (50 мл, 1:1, об./об.) і надосадову рідину декантували. Цей спосіб повторювали двічі з одержанням масла AA-9 (26,6 г). 1 H-ЯМР (400 МГц, DMSO-d6): δ частин на мільйон 8,04 (s, 3 H), 3,60 – 3,49 (m, 2 H), 3,16 – 3,15 (m, 1 H), 1,71 – 1,67 (m, 2 H), 1,60 – 1,55 (m, 2 H), 1,33 – 1,26 (m, 4 H), 0,90 – 0,87 (t, J=6,8 Гц, 3 H). Одержання AA-10 HO NH2 HCl (S) 30 35 AA-10 AA-10 отримували згідно з методом одержання AA-9, застосовуючи масляний альдегід замість валеріанового. 1 H-ЯМР (400 МГц, DMSO-d6): δ частин на мільйон 8,07 (s, 3 H), 4,85 (br, 1 H), 3,57 – 3,45 (m, 2 H), 3,14 – 3,12 (m, 1 H), 1,70 – 1,64 (m, 2 H), 1,56 – 1,49 (m, 2 H), 1,38 – 1,30 (m, 2 H), 0,90 – 0,80 (t, J=6,8 Гц, 3 H). 15 UA 113956 C2 Одержання 74 5 10 15 20 Етап 1. 3,4-Диметоксикоричну кислоту (5 г, 24 ммоль) розчиняли в THF (100 мл). До цього розчину в атмосфері N2 додавали нікель Ренея. Реакційну суміш піддавали впливу атмосфери водню і перемішували протягом 15 годин при кімнатній температурі. Реакційну суміш відфільтровували крізь картридж, наповнений діатомовою землею і розчинник фільтрату видаляли при зниженому тиску. Залишок застосовували як такий на наступному етапі. LC-MS: аналіт. розрах. для C11H14O4: 210,09; знайдене значення 209 [M-H]. Етап 2. 3-(3,4-Диметоксифеніл)пропанову кислоту розчиняли в THF (100 мл). Додавали комплекс боран-диметилсульфід (2 М у діетиловому ефірі, 20 мл, 40 ммоль). Реакційну суміш перемішували протягом ночі при кімнатній температурі. Повільно додавали метанол, щоб загасити реакційну суміш, потім додавали кремнезем, і леткі речовини видаляли при зниженому тиску. Залишок очищали на кремнеземі, застосовуючи градієнт елюенту гептан/етилацетат, отримуючи продукт у формі масла. Його застосовували як такий на наступному етапі. LC-MS: аналіт. розрах. для C11H16O3: 196,11; знайдене значення 195 [M-H]. MsCl, TEA, ACN 25 Етап 3. 3-(3,4-диметоксифеніл)пропан-1-ол (3,8 г, 19,5 ммоль) та триетиламін (3,8 мл, 27,3 ммоль) розчиняли в ацетонітрилі (15 мл) і потім додавали метансульфонілхлорид (1,5 мл, 19,5 ммоль). Реакційну суміш струшували протягом ночі при кімнатній температурі. Леткі речовини видаляли при зниженому тиску і залишок очищали за допомогою хроматографії на силікагелі, застосовуючи градієнт елюенту гептан/етилацетат, отримуючи продукт у формі прозорої олії. 1 H-ЯМР (400 МГц, DMSO-d6) δ частин на мільйон 1,91 – 2,01 (m, 2 H), 2,58 – 2,64 (m, 2 H), 3,17 (s, 3 H), 3,72 (s, 3 H), 3,75 (s, 3 H), 4,19 (t, J=6,4 Гц, 2 H), 6,71 – 6,76 (m, 1 H), 6,81 – 6,89 (m, 2 H). 30 16 UA 113956 C2 5 Етап 4. Розчин D-4 (400 мг, 1 ммоль), карбонату цезію (511 мг, 1,6 ммоль) та 3-(3,4диметоксифеніл)пропілметансульфонату (430 мг, 1,6 ммоль) в ацетоні (50 мл) нагрівали до 50 °C протягом 15 годин. Реакційну суміш поміщали в центрифугу і надосадову рідину декантували, потім випарювали насухо. Залишок очищали за допомогою колонкової хроматографії на силікагелі, застосовуючи градієнт елюенту гептан/етилацетат. Фракції, що містять продукт, зводили воєдино, і розчинники видаляли при зниженому тиску з одержанням D5. + LC-MS: аналіт. розрах. для C29H44N4O7: 560,32; знайдене значення 561 [M+H] . 10 15 Етап 5. Сполуку з boc-захисною групою розчиняли в дихлорметані (5 мл) і додавали 6 M HCl в ізопропанолі (3 мл). Реакційну суміш перемішували протягом 15 годин при кімнатній температурі. Леткі речовини видаляли при зниженому тиску. Додавали ефір (5 мл) і утворений осад, 74, виділяли за допомогою фільтрації, а потім висушували у вакуумній печі протягом 15 годин. Одержання 75 O O H2 N O N HO ( S) N N OH N DBU, BOP, ACN NH 2 O HN N N NH2 OH B-2 20 25 30 AA-9 H2 N Cl OH (S) O N N C-1 35 75 Етап 1. Проміжний продукт B-2 отримували згідно зі способом, описаним для одержання проміжного продукту B-1. Етап 2. До розчину B-2 (1 г, 3,62 ммоль) та DBU (5,4 мл, 36 ммоль) в ацетонітрилі (20 мл) додавали BOP (2,08 г, 4,71 ммоль), при цьому реакційна суміш стала прозорою, і її перемішували протягом 15 хвилин при кімнатній температурі. Додавали AA-9 (910 мг, 5,43 ммоль) і реакційну суміш перемішували протягом 2 діб при 50 °C. Леткі речовини видаляли при зниженому тиску і залишок очищали за допомогою хроматографії на силікагелі, застосовуючи градієнт елюенту дихлорметан/10 % метанол у дихлорметані. Кращі фракції зводили воєдино і розчинники видаляли при зниженому тиску. Неочищений продукт відновлювали в дихлорметані (2 мл), потім додавали HCl у діетиловому ефірі з утворенням солі HCl. Осад виділяли за допомогою фільтрації та висушували у вакуумній печі з одержанням сполуки 75. Одержання 76 HO O N DIPEA, ACN NH 2 (S) N H N NH2 D-6 Етап 1. С-1 (2 г, 8,49 ммоль), L-норвалін (1,75 г, 17 ммоль) та діізопропілетиламін (5,85 мл, 34 ммоль) розчиняли в ацетонітрилі (200 мл) у посудині високого тиску на 500 мл з тефлоновим покриттям і нагрівали до 130ºC протягом 15 годин. Суміші дозволяли охолонути до кімнатної температури, леткі речовини видаляли при зниженому тиску та неочищений продукт очищали 17 UA 113956 C2 за допомогою колонкової хроматографії на силікагелі, застосовуючи градієнт елюенту дихлорметан/10 % метанол у дихлорметані. Кращі фракції зводили воєдино і розчинники видаляли при зниженому тиску з одержанням проміжного продукту D-6. + LC-MS: аналіт. розрах. для C16H22N4O2: 302,17; знайдене значення 303 [M+H] . 5 10 Етап 2. D-6 (2 г, 6,61 ммоль) нагрівали зі зворотним холодильником протягом 4 годин в оцтовому ангідриді (100 мл) у круглодонній колбі на 250 мл. Леткі речовини видаляли при зниженому тиску і залишок очищали за допомогою колонкової хроматографії на силікагелі, застосовуючи градієнт елюенту гептан/етилацетат, отримуючи жовте масло D-7. + LC-MS: аналіт. розрах. для C22H28N4O5: 428,21; знайдене значення 429 [M+H] . O O O O (S) N H N N O O N Pd/C, H2 , MeOH HO (S) N H N N N O D-7 15 20 O O D-8 Етап 3. D-8 отримували згідно зі способом одержання проміжного продукту D-2. + LC-MS: аналіт. розрах. для C15H22N4O5: 338,16; знайдене значення 339 [M+H] . Етап 4. Проміжний продукт D-9 отримували згідно зі способом, описаним у прикладі 75 для проміжного продукту D-4. + LC-MS: аналіт. розрах. для C15H22N4O5: 338,16; знайдене значення 339 [M+H] . 18 UA 113956 C2 Етап 5. Зняття захисних груп з D-9 проводили згідно зі способом, описаним в етапі 2 щодо сполуки 5, з одержанням 76. Одержання сполуки 77 HO N H N N O O N O Br O O O O Cs 2 CO 3, DMF D-4 5 10 O O N H N N O N O O O D-10 Етап 1. D-10 отримували з D-4 згідно зі способом одержання прикладу 5 з очищенням за допомогою колонкової хроматографії на капілярній кварцовій колонці із градієнтом елюенту гептан/етилацетат. + LC-MS: аналіт. розрах. для C27H38N4O7: 530,27; знайдене значення 531 [M+H] . 1 H-ЯМР (400 МГц, ХЛОРОФОРМ-d) δ частин на мільйон 0,93 (t, J=7,3 Гц, 3 H), 1,37 (dd, J=14,9, 7,4 Гц, 2 H), 1,53 – 1,62 (m, 2 H), 3,40 – 3,50 (m, 2 H), 3,92 – 3,95 (m, 3 H), 5,13 (s, 2 H), 5,33 (s, 1 H), 7,46 – 7,52 (m, 1 H), 7,56 – 7,62 (m, 1 H), 7,73 (s, 1 H), 8,05 (dt, J=7,7, 1,4 Гц, 1 H), 8,09 (d, J=1,5 Гц, 1 H). 15 20 Етап 2. D-10 (2,14 г, 3,91 ммоль) розчиняли в безводному THF (250 мл). Алюмогідрид літію (1 М у THF, 5,87 мл, 5,87 ммоль) додавали краплинами і реакційну суміш перемішували протягом 3 годин при кімнатній температурі. NH 4Cl (насич., водн.) додавали краплинами до реакційної суміші, а солі, що випали в осад, видаляли за допомогою фільтрації та промивали THF. Фільтрат випарювали насухо і неочищений продукт D-11 застосовували як такий на наступному етапі. + LC-MS: аналіт. розрах. для C21H30N4O4: 402,23; знайдене значення 403 [M+H] . 25 30 Етап 3. D-11 (1,57 г, 3,91 ммоль) розчиняли в дихлорметані (20 мл) і до нього додавали HCl (6 М у ізопропанолі, 50 мл). Реакційну суміш перемішували протягом 16 годин при кімнатній температурі. Леткі речовини видаляли при зниженому тиску, а неочищений продукт очищали за допомогою капілярної кварцової колонки, застосовуючи градієнт елюенту дихлорметан/10 % дихлорметан у метанолі, отримуючи 77 у формі масла, що твердне під час витримки. 19 UA 113956 C2 Одержання 78 N HO N H N N O O OH N O N O N N O DIAD, PPh3 , THF 10 N O N O O O D-12 D-4 5 N N H Етап 1. Розчин D-4 (0,5 г, 1,31 ммоль), 3-піридазинілметанолу (158 мг, 1,44 ммоль) та трифенілфосфіну (377 мг, 1,44 ммоль) у безводному THF (4 мл) охолоджували до 0 °C і краплинами додавали розчин DIAD (0,28 мл, 1,44 ммоль) при 0 °C. Після додавання реакційну суміш перемішували протягом 3 годин при температурі навколишнього середовища. Розчинник гасили водою (10 мл), перемішували протягом 10 хвилин і леткі речовини видаляли при зниженому тиску. Водний шар екстрагували дихлорметаном, органічні шари об'єднували і розчинник видаляли при зниженому тиску. Неочищений продукт очищали за допомогою колонкової хроматографії на силікагелі, застосовуючи градієнт елюенту гептан/етилацетат. Кращі фракції об'єднували, розчинники видаляли при зниженому тиску з одержанням D-12. + LC-MS: аналіт. розрах. для C23H34N6O5: 474,26; знайдене значення 475 [M+H] . 15 20 Етап 2. D-11 (620 мг, 1,31 ммоль) розчиняли в дихлорметані (10 мл) і до нього додавали HCl (6 М у ізопропанолі, 10 мл). Реакційну суміш перемішували протягом 15 годин при кімнатній температурі. Леткі речовини видаляли при зниженому тиску і залишок очищали задопомогою обернено-фазової хроматографії з одержанням 78. Одержання 79 OH O OH O N N NH2 Ac2O, H2SO4 N O N H B-5 B-1 25 N Етап 1. У колбі на 500 мл нагрівали суміш B-1 (30 г, 138 ммоль) і сірчаної кислоти (3 мл) в оцтовому ангідриді (300 мл) до 90 °C протягом 3 годин. Реакційну суміш охолоджували до кімнатної температури і осад виділяли за допомогою фільтрації, промивали діізопропіловим ефіром і висушували in vacuo при 50 °C з одержанням білої твердої речовини B-5. OH O Cl N O O N O POCl3 , DIPEA, ACN N N H N N H C-2 B-5 Етап 2. Суміш B-5 (21,8 г, 84 ммоль) в ацетонітрилі (244 мл) перемішували при 30 °C у слабкому потоку азоту в реакторі MultiMAX на 400 мл. Фосфорилхлорид (18,14 мл, 195 ммоль) 20 UA 113956 C2 5 10 додавали краплинами протягом періоду 5 хвилин. Після додавання реакційну суміш нагрівали до 45 °C і суміш перемішували протягом 15 хвилин, потім повільно додавали DIPEA (33 мл, 195 ммоль) протягом періоду 1,5 годин. Реакційну суміш перемішували при 45 °C до завершення (що відслідковувалося за допомогою LC-MS). Розчин ацетату натрію (65 г) у воді (732 мл) нагрівали в колбі на 2 л до 35 °C і реакційну суміш порційно додавали до цього розчину протягом періоду 5 хвилин. Температуру підтримували на рівні 35-40 °C за допомогою зовнішньої охолоджувальної бані. Суміші дозволяли досягти температури навколишнього середовища і перемішування продовжували протягом 1 години. Осад виділяли за допомогою фільтра, промивали водою і висушували in vacuo при 50 °C з одержанням C-2 у формі твердої речовини. + LC-MS: аналіт. розрах. для C13H12ClN3O2: 277,06; знайдене значення 278 [M+H] . 1 H-ЯМР (400 МГц, DMSO-d6) δ частин на мільйон 2,11 (s, 3 H), 5,31 (s, 2 H), 7,33 – 7,39 (m, 1 H), 7,43 (t, J=7,2 Гц, 2 H), 7,46 – 7,51 (m, 2 H), 8,59 (s, 1 H), 10,65 (s, 1 H). O O N Cl N O O O NH 2 ( S) HN N H Et3 N, ACN C-2 20 N H (S) D-13 O HN N N O O (S) D-13 30 O Етап 3. Розчин проміжного продукту C-2 (5,9 г, 21,2 ммоль), метил-(2S)-2-аміногексаноату (5,79 г, 31,9 ммоль) і триетиламіну (14,8 мл, 106 ммоль) в ацетонітрилі (100 мл) нагрівали зі зворотним холодильником протягом 4 діб. Реакційну суміш охолоджували до кімнатної температури і розчинник видаляли при зниженому тиску. Залишок розчиняли в дихлорметані і промивали розсолом. Органічний шар висушували (сульфатом магнію), а потім прямо очищали за допомогою капілярної кварцової колонки, застосовуючи градієнт елюенту дихлорметан/10 % метанол у дихлорметані. Кращі фракції зводили воєдино і розчинники видаляли при зниженому тиску з одержанням D-13. + LC-MS: аналіт. розрах. для C20H26N4O4: 386,20; знайдене значення 387 [M+H] . O 25 N O O 15 N N O N H HN LiAlH4 , THF N O N H (S) OH D-14 Етап 2. D-13 (3,7 г, 9,57 ммоль) розчиняли в безводному THF (100 мл). Алюмогідрид літію (1 М у THF, 9,6 мл, 9,6 ммоль) додавали краплинами і реакційну суміш перемішували протягом 3 годин при кімнатній температурі. NH 4Cl (насич., водн.) додавали краплинами до реакційної суміші і солі, що випали в осад, видаляли за допомогою фільтрації і промивали THF. Фільтрат випарювали насухо і залишок очищали за допомогою колонкової хроматографії на силікагелі, застосовуючи градієнт елюенту дихлорметан/10 % метанол у дихлорметані. Кращі фракції об'єднували і розчинники видаляли при зниженому тиску з одержанням D-14. + LC-MS: аналіт. розрах. для C19H26N4O3: 358,20; знайдене значення 359 [M+H] . 21 UA 113956 C2 5 Етап 3. D-15 отримували згідно зі способом, описаним для проміжного продукту D-2. На наступному етапі його застосовували без очищення. + LC-MS: аналіт. розрах. для C12H20N4O3: 268,15; знайдене значення 269 [M+H] . Cl HO HN N N (S) OH O N N N H Cs 2CO3 , DMF N N O HN (S) N N O N H OH D-15 10 D-16 Етап 4. Суміш D-15 (210 мг, 0,78 ммоль) і карбонату цезію (765 мг, 2,35 ммоль) в DMF (25 мл) нагрівали до 60 °C під час перемішування, потім краплинами додавали розчин 5(хлорметил)-1,3-диметил-1Н-піразолу (113 мг, 0,78 ммоль) в DMF (10 мл). Реакційну суміш перемішували протягом 1 години при 60 °C. Тверді речовини видаляли за допомогою фільтрації, і розчинник видаляли при зниженому тиску. Неочищений продукт D-16 застосовували як такий на наступному етапі. + LC-MS: аналіт. розрах. для C18H28N6O3: 376,22; знайдене значення 377 [M+H] . 15 20 Етап 5. У скляну пробірку на 30 мл поміщали D-16 (295 мг, 0,78 ммоль), NaOCH3 (30 % у метанолі, 2 мл) та метанол (20 мл) і суміш перемішували при 60 °C протягом ночі. Реакційну суміш очищали за допомогою рідинної обернено-фазової хроматографії (Sunfire Prep C18 OBD 10 мм, 30 × 150 мм, рухома фаза: 0,25 % розчин NH4OAc у воді, метанол) з одержанням сполуки 79 у формі вільної основи. Одержання 80 22 UA 113956 C2 N N N N Cl HO N HN N O N O HN Cs2 CO 3, DMF, 60ºC N H N O N H OH OH D-17 D-15 Етап 1. Проміжний продукт D-17 отримували згідно з методом, застосовуваним для D-16, за допомогою алкілування D-15. + LC-MS: аналіт. розрах. для C19H24N6O3: 384,19; знайдене значення 385 [M+H] . N H2 N N N O O N HN N N H HN N NH2 OH OH 10 N O NaOMe, MeOH 5 O D-17 80 Етап 2. D-17 (301 мг, 0,78 ммоль) і NaOCH3 (30 % у метанолі, 2 мл) розчиняли в метанолі (20 мл) у скляній пробірці на 30 мл і перемішували при 60 °C протягом ночі. До реакційної суміші додавали 10 мл води і її перемішували протягом 2 годин при 60 °C. Реакційну суміш очищали за допомогою рідинної обернено-фазової хроматографії (Sunfire Prep C18 OBD 10 мм, 30 × 150 мм, рухома фаза: 0,25 % розчин NH4OAc у воді, метанол) з одержанням 80 у формі порошку. Одержання 81 OH (S) Cl O N N C-2 15 20 HN O O N H AA-9, Et3 N, ACN N N O N H D-18 Розчин проміжного продукту C-2 (2 г, 7,2 ммоль), AA-9 (3,02 г, 18 ммоль) і триетиламіну (5 мл, 36 ммоль) в ацетонітрилі (75 мл) нагрівали зі зворотним холодильником протягом 6 годин. Реакційну суміш охолоджували і розчинник видаляли при зниженому тиску. Залишок розчиняли в дихлорметані і промивали розсолом. Органічний шар завантажували в картридж із кремнеземом і застосовували градієнт елюенту дихлорметан/10 % метанол у дихлорметані. Фракції, що містять продукт, випарювали насухо з одержанням білого порошку, D-18. + LC-MS: аналіт. розрах. для C20H28N4O3: 372,22; знайдене значення 373 [M+H] . 1 H-ЯМР (400 МГц, DMSO-d6) δ частин на мільйон 0,77 – 0,92 (m, 3 H), 1,15 – 1,36 (m, 4 H), 1,42 – 1,72 (m, 4 H), 2,12 (s, 3 H), 3,35 – 3,42 (m, 2 H), 4,11 – 4,24 (m, 1 H), 4,35 – 4,52 (m, 1 H), 6,42 (d, J=8,80 Гц, 1 H), 7,42 (s, 1 H), 9,63 (br. s., 1 H). 23 UA 113956 C2 D-19 отримували з D-18 згідно зі способом, застосовуваним щодо проміжного продукту D-2. + LC-MS: аналіт. розрах. для C13H22N4O3: 282,1; знайдене значення 283 [M+H] . Cl OH N N (S) N N HN HO N N O N O HN Cs 2 CO 3/DMF N H HO N O N H ( S) D-19 D-20 D-20 отримували з D-19 згідно зі способом одержання D-17. + LC-MS: аналіт. розрах. для C19H30N6O3: 390,24; знайдене значення 391 [M+H] . 5 N N N N O N HN HO N O O NaOMe, MeOH N H HN HO (S) D-20 N N NH 2 (S) 81 81 отримували з D-20 згідно зі способом одержання сполуки 79. Одержання 82 10 15 Етап 1. Проміжний продукт B-3 отримували згідно зі способом, описаним стосовно до B-1. + LC-MS: аналіт. розрах. для C13H15N3O2: 245,12; знайдене значення 246 [M+H] . 1 H-ЯМР (400 МГц, DMSO-d6) δ частин на мільйон 1,79 – 1,93 (m, 2 H), 2,66 (t, J=7,8 Гц, 2 H), 3,76 (t, J=6,4 Гц, 2 H), 6,54 (br. s., 2 H), 7,11 – 7,21 (m, 3 H), 7,22 – 7,29 (m, 3 H), 11,46 (br. s., 1 H). O O HO N POCl3 Cl NH 2 N B-3 20 N N NH 2 C-3 Етап 2. Cуміш B-3 (15 г, 61,15 ммоль) в POCl3 (150 мл) нагрівали зі зворотним холодильником у круглодонній колбі на 250 мл і перемішували протягом 2 годин. Реакційній суміші дозволяли охолонути і розчинник видаляли при зниженому тиску. Залишкову фракцію розтирали на порошок з діізопропіловим ефіром. Осад, що утворювався, виділяли за допомогою фільтрації, промивали діізопропіловим ефіром і висушували під дією вакууму за 50 °C з одержанням твердої речовини C-3, яку застосовують як таку на наступному етапі. + LC-MS: аналіт. розрах. для C13H14ClN3O: 263,08; знайдене значення 264 [M+H] . O NH 2 O N O N ( S) Cl N NH 2 O DIPEA, CH 3CN HN N O (S) NH 2 O 25 30 C-3 82 Етап 3. У пробірку на 20 мл поміщали C-3 (0,45 г, 1,05 ммоль), HCl-сіль метилового ефіру L-2-аміногексанової кислоти (0,48 г, 2,62 ммоль), DIPEA (1,18 мл, 6,82 ммоль) і ацетонітрил (5 мл). Пробірку запаювали і нагрівали під дією мікрохвильового випромінювання протягом 1,5 годин при 120 °C. Реакційній суміші дозволяли охолонути і розчинник видаляли при зниженому тиску. 24 UA 113956 C2 5 Неочищену суміш очищали за допомогою препаративної HPLC на (RP Vydac Denali C18-10 мкм, 250 г, 5 см) з рухомою фазою (0,25 % розчин NH4OAc у воді, метанол), при цьому бажані фракції збирали і випарювали насухо. Залишкову фракцію розчиняли в суміші дихлорметан/метанол і пропускали крізь картридж для твердофазної екстракції (SCX) із застосуванням кислотно-модифікованих сорбентів. Продукт вивільняли за допомогою 7 н NH3 у метанолі. Зібраний розчин концентрували при зниженому тиску з одержанням бажаної твердої речовини 82. Одержання 83 10 15 Етап 1. Проміжний продукт B-4 отримували згідно з методом одержання B-1. + LC-MS: аналіт. розрах. для C14H17N3O3: 275,13; знайдене значення 276 [M+H] . 1 H-ЯМР (400 МГц, DMSO-d6) δ частин на мільйон 3,63 (dd, J=5,4, 3,9 Гц, 2 H), 3,95 (dd, J=5,4, 3,6 Гц, 2 H), 4,50 (s, 2 H), 6,33 (br. s., 2 H), 7,22 – 7,29 (m, 2 H), 7,30 – 7,36 (m, 4 H), 10,71 – 11,58 (m, 1 H). O O N N HO O O NH 2 Cl POCl3 B-4 20 NH 2 O N Cl N O DIPEA, CH3 CN NH2 N NH 2 83 Етап 3. У пробірки на 50 мл поміщали C-4 (10 г, 35,75 ммоль), н-бутиламін (10,6 мл, 107,25 ммоль) і DIPEA (30,8 мл, 178,75 ммоль) в ацетонітрилі (40 мл). Суміш нагрівали до 120 °C під впливом мікрохвильового опромінення протягом 3 годин. Об'єднані реакційні суміші концентрували при зниженому тиску і залишкове масло розчиняли в дихлорметані, і промивали 1 н HCl і водою. Органічний шар висушували (сульфатом магнію), тверді речовини видаляли за допомогою фільтрації, розчинник фільтрату видаляли при зниженому тиску з одержанням червоно-коричневої піни 83. Одержання 84 O O O O N N H N NH 2 N N H Boc 2O, CH3 CN N O N H O D-20 83 40 N N H C-4 35 NH 2 C-4 O 30 N Етап 2. У круглодонну колбу на 250 мл поміщали B-4 (10 г, 38,27 ммоль) і POCl3 (75 мл). Суміш нагрівали зі зворотним холодильником і перемішували протягом 5 годин. Реакційній суміші дозволяли досягти кімнатної температури і її перемішували протягом 15 годин. Розчинник видаляли при зниженому тиску. Неочищений продукт C-4 застосовували як такий на наступному етапі. + LC-MS: аналіт. розрах. для C12H12ClN3O2: 265,06; знайдене значення 266 [M+H] . O 25 N Етап 1. У круглодонну колбу на 500 мл поміщали 83 (13,5 г, 25,6 ммоль), Boc-ангідрид (27,94 г, 128 ммоль) і ацетонітрил (150 мл). Жовтий розчин перемішували зі зворотним холодильником протягом 16 годин. Розчинник видаляли при зниженому тиску. Залишкову фракцію розчиняли в дихлорметані і промивали насиченим водним розчином NaHCO 3 та водою. Органічний шар висушували (сульфатом магнію), тверді речовини видаляли за допомогою фільтрації і розчинники фільтрату видаляли при зниженому тиску з одержанням масла D-20. + LC-MS: аналіт. розрах. для C22H32N4O4: 416,24; знайдене значення 417 [M+H] . 25 UA 113956 C2 O O O N N H N HO O N H N N H Pd/C, H 2, CH 3OH O N D-20 5 HO D-21 N N H O S Cl O N O N H O O S O O O N N H Et3N, CH3 CN N D-21 15 O D-22 N N O N N H N N H O OH O O O N N H Cs 2 CO 3, CH 3CN D-22 25 O N H Етап 3. Розчин D-21 (8,7 г, 26,66 ммоль) і триетиламіну (7,41 мл, 53,31 ммоль) в ацетонітрилі (300 мл) перемішували в круглодонній колбі на 1 л при навколишній температурі і додавали метансульфонілхлорид (3,1 мл, 40 ммоль). Після додавання реакційну суміш перемішували протягом 1,5 годин при кімнатній температурі. Розчинник видаляли при зниженому тиску. Неочищений продукт розчиняли в етилацетаті і промивали насиченим водним NaHCO 3. Органічні шари об'єднували, висушували (сульфатом магнію), тверді речовини видаляли за допомогою фільтрації і розчинник фільтрату випарювали насухо з одержанням D-22 у формі олії. + LC-MS: аналіт. розрах. для C16H28N4O6S: 404,17; знайдене значення 405 [M+H] . O S O O 20 O Етап 2. У колбі Ерленмейєра на 1 л суспендували 10 % Pd/C (4 г) у метанолі (350 мл) під потоком газу N2, потім додавали D-20 (14,3 г, 34,33 ммоль). Суміш перемішували при 50 °C в атмосфері водню доки не було поглинено 1 еквівалент водню. Каталізатор видаляли шляхом фільтрації крізь ущільнений декаліт. Розчинник фільтрату видаляли при зниженому тиску з одержанням масла D-21. Залишок застосовували як такий на наступному етапі. + LC-MS: аналіт. розрах. для C15H26N4O4: 326,20; знайдене значення 327 [M+H] . O 10 O N H N O N H O D-23 Етап 4. У скляну пробірку на 30 мл поміщали суміш 4-гідроксипіридину (94 мг, 0,99 ммоль) і Cs2CO3 (0,8 г, 2,47 ммоль) в ацетонітрилі (10 мл). Флакон запаювали і струшували при температурі навколишнього середовища протягом 1 години. D-22 (400 мг, 0,99 ммоль) у формі розчину в ацетонітрилі (10 мл) додавали до реакційної суміші і струшували протягом додаткових 18 годин при кімнатній температурі. Додавали карбонат цезію (320 мг, 1 ммоль) і суміш струшували протягом 1 доби при кімнатній температурі. Розчинник видаляли при зниженому тиску і неочищений продукт обробляли сумішшю дихлорметан/метанол, 95/5, і струшували протягом 1 год., потім відфільтровували крізь 2 г ущільненого кремнезему. Фільтрат концентрували при зниженому тиску і D-23 застосовували як такий на наступному етапі. + LC-MS: аналіт. розрах. для C20H29N5O4: 403,22; знайдене значення 404 [M+H] . 30 35 HCl, кімнатна температура Етап 5. Зняття захисних груп з D-23 з одержанням 84 проводили згідно зі способом, застосовуваним для зняття захисних груп з 78. Одержання 85 26 UA 113956 C2 5 10 Етап 1. У круглодонну колбу на 250 мл, оснащену магнітною мішалкою, поміщали D-4 (0,35 г, 5,23 ммоль) і карбонат цезію (0,89 г, 2,75 ммоль) в ацетонітрилі (20 мл). Суміш перемішували при температурі навколишнього середовища протягом 30 хвилин. Додавали розчин алкілгалогеніду (0,19 г, 1 ммоль) в ацетонітрилі (5 мл) і реакційну суміш перемішували протягом 1 доби при кімнатній температурі. Реакцію завершували і солі видаляли за допомогою фільтрації. Фільтрат концентрували при зниженому тиску і неочищений продукт очищали за допомогою колонкової хроматографії на силікагелі, застосовуючи градієнт елюенту гептан/етилацетат, з одержанням проміжного продукту D-24. + LC-MS: аналіт. розрах. для C24H37N7O7: 535,28; знайдене значення 536 [M+H] . 15 20 Етап 2. У колбі Ерленмейєра на 100 мл під шаром газоподібного азоту суспендували Pt/C, 5 % (100 мг) у тіофені (0,25 мл) і метанолі (20 мл), потім додавали D-24 (130 мг, 0,24 ммоль). Реакційну суміш перемішували при 50 °C в атмосфері водню. Каталізатор видаляли за допомогою фільтрації крізь ущільнений декаліт. Розчинники фільтрату видаляли при зниженому тиску з одержанням D-25 у формі масла, що її застосовували як таку на наступному етапі. + LC-MS: аналіт. розрах. для C24H39N7O5: 505,30; знайдене значення 506 [M+H] . N N NH O NH 2 N N N N O N O NH O O HCl NH 2 N N NH 2 O 85 D-25 25 Етап 3. Зняття захисних груп із проміжного продукту D-25 з одержанням 85 проводили згідно зі способом, застосовуваним для одержання 78. Одержання 86 27 UA 113956 C2 5 Етап 1. У круглодонну колбу на 100 мл поміщали азид натрію (6,85 г, 103,76 ммоль) у воді (12,5 мл), потім хлорметилпівалат (10,6 г, 70,38 ммоль) і суміш енергійно перемішували при 90 °C протягом 16 годин. Реакційній суміші дозволяли охолонути до кімнатної температури і додавали дихлорметан (20 мл). Органічний шар відокремлювали, висушували над безводним сульфатом натрію, тверді речовини видаляли за допомогою фільтрації і розчинник фільтрату видаляли при зниженому тиску з одержанням А-2 у формі олії. + LC-MS: аналіт. розрах. для C6H11N3O2: 157,09; знайдене значення 158 [M+H] . 10 15 20 Етап 2. У пробірку на 25 мл поміщали D-26 (100 мг, 0,238 ммоль), A-2 (37,9 мг, 0,238 ммоль), трет-бутанол (2,5 мл) і воду (2,5 мл). Пробірку запаювали і суміш перемішували при температурі навколишнього середовища. Додавали пентагідрат сульфату міді (II) (3 мг, 0,012 ммоль) і натрієву сіль L-аскорбінової кислоти (15,5 мг, 0,079 ммоль). Реакційну суміш перемішували протягом 18 годин при кімнатній температурі, потім додавали воду (2,5 мл). Осад виділяли за допомогою фільтрації, промивали водою і висушували in vacuo при 60 °C з одержанням білого порошку D-27. + LC-MS: аналіт. розрах. для C27H43N7O7: 577,32; знайдене значення 578 [M+H] . N O N N NH N N 25 30 O N O D-27 N HN O O O N NH O HCl N N NH2 O 86 Етап 3. У круглодонній колбі на 100 мл перемішували суміш D-27 (0,1 г, 0,17 ммоль) у HCl (5 мл 6 М у ізопропанолі) і дихлорметану (5 мл) при температурі навколишнього середовища протягом 16 годин. Реакційну суміш нагрівали до 65 °C та перемішували протягом додаткових 16 годин. Розчинник видаляли при зниженому тиску. Неочищений продукт очищали за допомогою рідинної обернено-фазової хроматографії (RP Vydac Denali C18 – 10 мкм, 250 г, 5 см) з рухомою фазою (0,25 % розчин NH4HCO3 у воді, метанол), при цьому бажані фракції збирали, випарювали, розчиняли в метанолі і обробляли 2 М HCl в ефірі. Тверду речовину виділяли за допомогою фільтрації з одержанням 86 у формі солі HCl. 28
ДивитисяДодаткова інформація
Автори англійськоюMc Gowan, David, Raboisson, Peirre Jean-Marie Bernard, Embrechts Werner, Jonckers, Tim Hugo Maria, Last, Stefaabn, Julien, Pieters, Serge, Maria, Aloysius, Vlach, Jaromir
Автори російськоюМк Гован Давид, Ребойсон Пьер Жан-Мария Бернард, Эмбрехтс Вернер, Джонкерс Тим Хьюго Мария, Ласт Стэфан Джульен, Питэрс Серж Мария Алойсис, Влач Яромир
МПК / Мітки
МПК: C07D 239/48, A61K 31/505, A61K 31/506, C07D 471/04, C07D 405/12, C07D 401/12, C07D 413/12, C07D 403/12
Мітки: лікування, похідні, вірусних, піримідину, інфекцій
Код посилання
<a href="https://ua.patents.su/171-113956-pokhidni-pirimidinu-dlya-likuvannya-virusnikh-infekcijj.html" target="_blank" rel="follow" title="База патентів України">Похідні піримідину для лікування вірусних інфекцій</a>
Похідні піперидинопіримідину для лікування вірусних інфекцій

Номер патенту: 112668
Опубліковано: 10.10.2016
Автори: Йонкерс Тім Хьюго Марія, Рабуассон П'єр Жан-Марі Бернар, МакГован Девід Крейг, Даубі Хамлічі Мурад
МПК: C07D 471/04, A61K 31/519
Мітки: піперидинопіримідину, вірусних, похідні, лікування, інфекцій
Формула / Реферат:
1. Сполука формули (І) (I)або її фармацевтично прийнятна сіль, деА вибраний з групи, що складається з СН2 і NCOR2 в будь-якій стереохімічній конфігурації,В вибраний з групи, що складається з СН2 і NCOR4 в будь-якій стереохімічній конфігурації,за умови, що, якщо А являє собою NCOR2, тоді В не являє собою NCOR4,X вибраний з СН2 в...
Похідні пурину або деазапурину, корисні для лікування (серед інших) вірусних інфекцій

Номер патенту: 107805
Опубліковано: 25.02.2015
Автори: Ротл Пол А., Хелкомб Рендл Л.
МПК: A61K 31/522, C07D 473/16, C07D 473/18, A61P 31/12, C07D 473/24, C07D 473/34
Мітки: інфекцій, серед, деазапурину, похідні, інших, пурину, вірусних, лікування, корисні
Формула / Реферат:
1. Сполука Формули І Iабо її фармацевтично прийнятна сіль, у якій: L1 являє собою -О-; R1 являє собою Н, С1-6алкіл, С1-6гетероалкіл або С4-20гетероциклілалкіл, де кожна гетероалкільна група включає 1 або 2 гетероатоми, вибрані з О, N або S, і де кожна гетероциклільна група включає від 1 до 6...
Похідні 5,6-дихлорбензімідазолу, способи їх одержання, фармацевтичний склад та спосіб лікування вірусних інфекцій

Номер патенту: 66744
Опубліковано: 15.06.2004
Автори: Кошалка Джордж Волтер, Чемберлен Стенлі Доз
МПК: C07D 235/04, A61K 31/7052, A61P 31/12, C07H 19/052
Мітки: 5,6-дихлорбензімідазолу, інфекцій, одержання, вірусних, лікування, способи, склад, спосіб, фармацевтичний, похідні
Формула / Реферат:
1. Соединение формулы (I):, (I)в которой R - водород или галоген, или –NR1R2, гдеR1 и R2 одинаковы или различны и независимо выбраны из группы: водород, С1-6алкил, цианоС1-6алкил, гидроксиС1-6алкил, галогеноС1-6алкил, С3-7циклоалкил, С1-6алкилС3-7циклоалкил, С2-6алкенил, С3-7циклоалкилС1-6алкил, С2-6алкинил, арил, арилС1-6алкил, гетероциклический...
Похідні та аналоги 2-дезокси-2,3-дидегідро-n-ацетилнеурамінової кислоти, спосіб їх одержання, фармацевтична композиція, спосіб лікування вірусних інфекцій

Номер патенту: 41252
Опубліковано: 17.09.2001
Автори: Данілек Базіль, Ву Вен-Янг, Фан То Ван, Джін Бетті, Іцштейн Лоренс Марк фон
МПК: C07D 211/70, A61K 31/382, C07D 211/84, C07D 309/22, C07D 409/04, C07D 257/00, C07D 405/04, C07D 401/14, A61P 31/00, C07C 321/00, A61K 31/41, C07D 309/26, A61K 31/00, C07C 47/38, C07C 225/00, C07C 309/00, C07D 405/14, C07D 335/00, A61P 31/12, A61K 31/34, A61K 31/505, C07D 211/72, C07D 401/04, C07D 309/20, C07C 317/12, C07D 409/14, C07D 211/74, C07C 47/20, C07D 309/30, A61K 31/38, C07D 211/78, C07D 239/26, C07D 309/28, A61K 31/435
Мітки: похідні, інфекцій, лікування, спосіб, аналоги, композиція, кислоти, фармацевтична, одержання, вірусних, 2-дезокси-2,3-дидегідро-n-ацетилнеурамінової
Формула / Реферат:
1. Производные и аналоги 2-дезокси-2,3-дидегидро-N-ацетил-неураминовой кислоты формулы (I)где в общей формуле (I) А представляет кислород, углерод или серу,R1 обозначает СООН, Р(О)(ОН)2, NО3, SOOH, SО3Н, тетразол, СH2СНО, СНО или CH(CHO)2,R2 обозначает Н, OR6, F, Cl, Br, CN, NHR6, SR6 или CH2X, где Х представляет NHR6, галоген или OR6, иR6 представляет водород; ацильную группу, имеющую от 1 до 4 атомов...
Макроциклічні пурини для лікування вірусних інфекцій

Номер патенту: 113651
Опубліковано: 27.02.2017
Автори: Арну Ерік П'єр Александр, Рабуассон П'єр Жан-Марі Бернар, Мюллер Філіпп, Дубле Фредерік Марк Моріс, Бонфанті Жан-Франсуа, Фортен Жером Мішель Клод
МПК: C07D 487/16, C07D 498/16, A61K 31/52
Мітки: пурини, лікування, макроциклічні, інфекцій, вірусних
Формула / Реферат:
1. Сполука, що характеризується формулою (І), (I)де n=1-3,або її фармацевтично прийнятні солі, де X являє собою кисень, азот, сірку або,Y являє собою ароматичне кільце або гетероциклічне кільце, що містить щонайменше азот, необов'язково заміщене одним або декількома...
Наступний патент: Застосування емульгаторів в поєднанні з рослинними олеїнами в кормі для тварин
Випадковий патент: Похідні азаспіроалканів як інгібітори металопротеаз